

Abstract
Gliomas are prime brain cancers which are initiated by malignant modification of neural stem cells, progenitor cells and differentiated glial cells such as astrocyte, oligodendrocyte as well as ependymal cells. Exchange proteins directly activated by cAMP (EPACs) are crucial cyclic adenosine 3’,5’-monophosphate (cAMP)-determined signaling pathways. Cyclic AMP-intermediated signaling events were utilized to transduce protein kinase A (PKA) leading to the detection of EPACs or cAMP-guanine exchange factors (cAMP-GEFs). EPACs have been detected as crucial proteins associated with the pathogenesis of neurological disorders as well as numerous human diseases. EPAC proteins have two isoforms. These isoforms are EPAC1 and EPAC2. EPAC2 also known as Rap guanine nucleotide exchange factor 4 (RAPGEF4) is generally expression in all neurites. Higher EAPC2 levels was detected in the cortex, hippocampus as well as striatum of adult mouse brain. Activation as well as over-secretion of EPAC2 triggers apoptosis in neurons and EPAC-triggered apoptosis was intermediated via the modulation of Bcl-2 interacting member protein (BIM). EPAC2 secretory levels has proven to be more in low-grade clinical glioma than high-grade clinical glioma. This review therefore explores the effects of EPAC2/RAPGEF4 on the pathogenesis of glioma instead of EPAC1 because EPAC2 and not EPAC1 is predominately expressed in the brain. Therefore, EPAC2 is most likely to modulate glioma pathogenesis rather than EPAC1.
Key words: Apoptosis, EPAC2, gliomas, inhibition, RAPGEF4, pathogenesis
Introduction
Gliomas are prime brain cancers which are initiated by malignant modification of neural stem cells, progenitor cells and differentiated glial cells such as astrocyte, oligodendrocyte as well as ependymal cells.1-4 These cancers are histologically categorized into Grades I- IV based on the World Health Organization (WHO) criteria.5,6 Most commonly, Grade I gliomas are found in children and they often have good outcomes.1
On the other hand, Grade II gliomas have 5-8-years average survival rate and are usually depicted with hypercellularity.1,7,8 Nevertheless, Grade III comprises of astrocytoma or anaplastic astrocytoma according to histological classification. They are depicted with hypercellularity, nuclear atypia as well as mitotic figures. The anaplastic astrocytoma has 3-years average survival rate.1,5,6,9-11 Glioblastoma multiforme (GBMs) constitutes Grade IV gliomas.1
However, the 2016 WHO classification of CNS tumors presents major restructuring of the diffuse gliomas, medulloblastomas as well as other embryonal tumors, and included new entities that are defined by both histology and molecular features, such as glioblastoma, IDH-wildtype and glioblastoma, IDH-mutant; diffuse midline glioma, H3 K27M-mutant; RELA fusion-positive ependymoma; medulloblastoma, WNT-activated as well as medulloblastoma, SHH-activated; and embryonal tumors with multilayered rosettes, C19MC-altered.12 GBMs are often depicted with hypercellularity, nuclear atypia, mitotic figures as well as angiogenesis and/or necrosis. GBM patients with ages >60 years often have very short survival rate while patients with age <60 years have average survival rates of 12-18 months.1,7,13-15 Also, patients’ outcomes are often poor because tumor cells invade normal brain tissue around the tumor which make surgery resection incomplete and often necessitating adjuvant therapy with irradiation as well as chemotherapy to lessen the remaining tumor cells.16,17
Cyclic adenosine monophosphate (cAMP) is normally produced from adenosine 5’-triphosphate by adenylate cyclases.4,18,19 It is a prototypic second messenger that partakes in cellular reactions. These reactions trigger several intermediates signaling pathways linked with many human diseases such as cancer, diabetes, and urinary dysfunction as well as immunological, central nervous and cardiac diseases.4,18,20 Exchange proteins directly activated by cAMP (EPAC) is a new and promising protein that is expressed in almost all mammals’ cells.18,21 It is one of the prime cAMPs’ effector proteins in mammals. EPAC proteins have two isoforms. These isoforms are EPAC1 and EPAC2.18,21 EPAC2 is also referred to as Rap guanine nucleotide exchange factor 4 (RAPGEF4).19,22 EPAC2 happens to be more regulated and restricted to the brain, pancreas, testes, as well as secretory cells.19,21,23-25 This review therefore explores the effects of EPAC2 on the pathogenesis of glioma instead of EPAC1 because EPAC2 and not EPAC1 is predominately expressed in the brain. Therefore, EPAC2 is most likely to modulate glioma pathogenesis rather than EPAC1.
Origin of EPAC
EPAC protein was discovered in 1998 during a database exploration aimed at detecting the machinery via which cAMP-dependent activation of GTPase Rap1 that was uninhibited by protein kinase A (PKA).21,26-28 EPAC protein is also referred to as cAMPguanine exchange factor (cAMP-GEF).26 Kawasaki et al. established that, EPAC1 also referred to as cAMPGEF-I and EPAC2 also referred to as cAMP-GEF-II where independently detected via a differential display screen for novel cyclic nucleotide binding domain-bearing proteins, which where augmented in the striatum. 25
EPAC proteins were discovered in Metazoa within the evolutionary hierarchy as single polypeptide molecules.21 De Rooij et al. established that EPAC1 is a novel cAMP sensor that intermediates the PKA-independent RAP1 stimulation in feedback reaction to cAMP27,28 while Ozaki et al. established that, EPAC2 is a cAMP sensor linked to the sulfonylurea receptor (SUR1) in a yeast twohybrid screen.29
EPAC protein is made-up of a C-terminal catalytic region and an N-terminal regulatory region.19,26 The C-terminal catalytic region triggers Rap1 but not Ras, Ral, or R-ras.21,27 This region contains the enzymatic GEF domain as well as the RAS exchange motif (REM), which are desired for stability of the GEF domain.21,26 The N-terminal segment of EPAC houses the disheveled, Egl-10 and pleckstrin (DEP) domain and a cAMP binding domain. The function of DEP domain is uncertain but the cAMP binding domain is analogous to the cAMP binding domains at the regulatory subunit of PKA.21 Also, the N-terminal region serves as an auto-inhibitory domain during activation of full-length EPAC in vitro via cAMP.19,21
Structure and function of EPAC2
Epac2, was coded by RAPGEF4 genes which comprised of 31 exons as well as 30 introns situated on chromosome 2q31.19 EPAC2 is a multi-domain protein with a molecular weight of ~116 kDa, containing a regulatory as well as catalytic components.4,26 NH2-terminal forms the regulatory segment while COOH-terminal form the catalytic segment.21,25 The amino terminal regulatory segment contains cNBD-A and cNBD-B cyclic nucleotide-binding domains as well as a DEP domain.4,19,30 Furthermore, an extra CNB domain expressed NH2 terminal to the DEP domain is wellknown in a complete EPAC2.21
It was affirmed that, EPAC2 CNB-A domain’s affinity for cAMP is much punier than that of CNB-B domain.18,23,26 Furthermore, isolated EPAC2 CNB-B domain was necessitous to inhibit GEF action of the EPAC2 catalytic part.21,26 However, EPAC2 CNB-A and DEP domains are not requisite for upholding EPAC2 in an autoinhibitory state.21,28 Also, EPAC2 catalytic segment was depicted with a Ras exchange motif (REM), a Ras-association (RA) domain, as well as a continuous CDC25 homology domain (CDC25-HD) which are conscientious to the nucleotide exchange activity of EPAC2.18,19,21 The continuous CDC25 -HD is also known as the GEF for Ras-like small GTPases (RasGEF) domain.21
The main function of EPAC2 is a GEF for Rap1 and Rap2 with a small GTPases cycle involving an inactive GDP-bound form as well as an active GTP-bound form. Rap1 and Rap2 are strictly modulated by GEFs and GTPase-activating proteins (GAPs), which are liable for triggering of GTP loading and catalysis of GTP hydrolysis, correspondingly.19,21,23,26 CDC25-HD of EPAC2 interrelates with GDP-bound Rap1. It is consequently stimulated by exchange of GDP for GTP resulting in down-regulated signaling via interface with its specific effector proteins. Studies have shown that, EPAC2 was more regulated and restricted to the brain, pancreas, testes, as well as secretory cells.24,25 EPAC2 was therefore straightforwardly linked to the pathogenesis of Glioma and several neurological disorders.4,19
Seo and Lee demonstrated that, EPAC2-inhibition compromised pituitary adenylate cyclase-activating peptide (PACAP)- triggered astrocytic differentiation of neural precursor cells without affecting neuronal differentiation.31 They stressed that, upsurge in intracellular calcium levels was critical in the PACAP-EPAC2 signaling pathway-triggered astrocytogenesis.31
EPAC and apoptosis
Cell survival as well as cell death are very crucial events in tissues with post-mitotic cells constitution.32 It was obvious that, cAMP is able to wield a definite effect on cell predisposition to apoptosis thereby safeguarding neuronal cells.32 Also, EPAC2 was triggered by 8-p-methoxyphenylthon-2-O-methyl-cAMP. Furthermore, over-secretion of EPAC2 expressively augmented DNA fragmentation as well as terminal deoxynucleotidyltransferase- mediated biotin nick end-labeling (TUNEL)-positive cell numbers in mouse cortical neurons. Thus, the effect of cAMP on cell death has been comprehensively studied.32
It was obvious that, EPAC2 triggers cAMP signals in neuronal cells leading to decrease in the rate of neuronal cell death. The experiment above were performed in variety of stresses like β- amyloid protein, sialoglycopeptide as well as low potassiuminduced neurotoxicity.32-35 Nevertheless, studies have demonstrated that, dopamine or prostanoid receptor-mediated cAMP generation stimulates neurotoxicity.36,37 It was affirmed that, the influence of cAMP signaling on apoptosis focused principally on PKA, a classic target molecule of cAMP.32
EPAC2 has demonstrated to modulate a diverse cellular activity such as cell proliferation, migration, secretion, as well as differentiation. 32,38 Studies have shown that, either EPAC2 alone or with PKA provides protection to immune cells against apoptosis.39,40 Suzuki et al. demonstrated that, activation as well as over-secretion of EPAC2 triggers apoptosis in neurons. Their study established that, EPAC-triggered apoptosis is intermediated via the modulation of Bcl-2 interacting member protein (BIM).32 BIM acts on mitochondria as a pro-apoptotic factor resulting in the distraction of mitochondrial membrane potential.32 Studies have demonstrated that, BIM binds to Bcl-2 and neutralizes its pro-survival role, leading to apoptosis in several cell types.41,42 EPAC2 is therefore a crucial protein to avert apoptosis during gliomagenesis. Further studies on the effect of apoptosis on glioma cells are still warranted to determine whether EPAC2 can prevent the progressing of glioma.
EPAC2 expressive levels
It was affirmed that, the cortex, hippocampus as well as striatum of adult mouse brain express EAPC221,43 (Figure 1A). It was further affirmed that, EPAC2 was unanimously expressed in all neurites.21 Analyses of EPAC2 protein levels in rat brain, spinal cord, as well as dorsal root ganglion (DRG) neurons at distinctive phases of growth revealed a developmental modulation of high EPAC2 in the rat nervous system21,44 (Figure 1A). Also, northernblot analyses revealed that full-length EPAC2 was highly expressed in the pituitary gland during transcription.21,44 EPAC2 mRNA was predominantly interconnected with the central nervous system (CNS) and adrenal gland with restricted quantities in heart, small intestine, as well as the testis.25 Moreover, a truncated transcription revealed EPAC2 expressive levels in liver with low levels in the lungs, kidneys as well as pancreatic islets.21,29,45
It was affirmed that; mature neurons raise the concentrations of EPAC2 to regulate dendrite stability and over growth. It was further demonstrated that, elevated levels of EPAC2 were limited to growth cones and neurites.21,46 It was also proven that, in the CNS, EPAC2 release was similar to neurotransmitter release. This form of EPAC2 expression is another form of exocytosis used by neurons to precipitously interconnect throughout the body.21,45 Furthermore, mossy fiber CA3 synapses in the hippocampus demonstrated genuine basal activity as well as short sequences of synaptic transmission when EPAC2 was inhibited, but lasting action as well as forskolin-dependent potentiation was compromised. 21,43 Moreover, during EPAC2 inhibition, the quantity of existing vesicles to express EPAC2 after prolonged synaptic activity were reduced. This affirms that, EPAC2 was fundamental in the modulation of the spare pool of vesicles as well as vesicle release in reaction to augmented cAMP quantities.21,43
Nevertheless, EPAC2 inhibited synapses also exhibited destruction of NMDA receptor related long-standing depression. 21,47 In comparison, it was established that normal brain tissues expressed more EPAC2 levels than clinical glioma tissues4 (Figure 1B). This therefore means that, the number of EPAC2- secreting cells may reduce during glioma pathogenesis.4 Further studies are still warranted to determine the expressive levels of EPAC2 in glioma microenvironment.
EPAC2 inhibition
EPAC2 inhibition is very crucial during laboratory experiments. It was established that, all compounds produced through bioisosteric substitution of tert-butyl isoxazole ring with tert-butyl phenyl group reserved EPAC2 inhibitory activities.48 Liu et al. formulated and synthesized novel series of 2-substituted phenyl-Nphenyl- 2-oxoacetohydrazonoyl cyanides as EPAC2 inhibitors through ingenuous chemistry with low-cost maiden material as well as synthetic affluence appropriate for scale up.48 ZL0524 was the utmost potent EPAC2 inhibitory (Figure 2) activities with IC50 values of 1.2 mM. Docking analyses of ZL0524 with triggered EPAC2 showed that it occupies the cAMP binding domain 2 (CBD2) hydrophobic domain, constitutes hydrogen bonds with Arg448 as well as stretched to the solvent region.48
Tsalkova et al demonstrated that ESI-05 as well as ESI-07 inhibited (Figure 2) cAMP-intermediated EPAC2 GEF activity with obvious IC50 of 0.43±0.05 as well as 0.7±0.1 μM, correspondingly. 49 It was affirmed that, the role of ESI-07 is to bind to the interfaces of two CBDs on EPAC2 as well as hairs the protein in its autoinhibitory conformation. This means that ESI-07 was completely specific for EPAC2.49 Tsalkova et al. indicated that, the likelihood that ESI-07 binds to alternative unknown allosteric site on EPAC2 cannot be entirely exclude. This binding process could have prohibited the triggering of EPAC2 by stabilizing the inactive conformation.49
Figure 1.
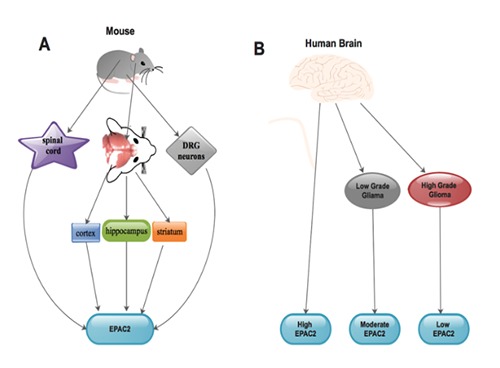
Expression of EPAC2 in adult mouse brain (A) and human normal brain/glioma tissues (B). A) The expressive levels of EPAC2 in brain (cortex, hippocampus and striatum), spinal cord as well as the dorsal root ganglion (DRG) neurons of adult mouse; B) the expressive levels of EPAC2 in normal brain tissues, low-grade glioma tissues as well as high-grade glioma tissues.
Rehmann indicated that, ESI-09 and HJC0197 do not function as selective inhibitors of EPAC250 (Figure 2). He indicated that, both compounds appear to have undefined protein denaturing influences. He further explained that, a weak influence of particular inhibition was covered by the typical protein denaturing influences, but both influences could occur in the uniform concentration range.50 ESI-09 as well as HJC0197 affect EPAC2 devoid of the cAMP concentration and even inhibit the cAMP neutral intrinsic action.50 It was established that, ESI-05 is a direct and selective inhibitor of EPAC251 (Figure 2). It was estimated that, ESI-05 binds EPAC2 with approximately 20-fold higher affinity than cAMP.51 Chen et al. established that, numerous EPAC2 inhibitors could be coin out of ESI-05.51 Also, ESI-05 as well as ESI-10 have been isolated as cAMP-mediated EPAC2 GEF inhibitors with IC50 values of 0.5 μM and 18 μM, correspondingly.51 Nevertheless, ESI-05 shows a selective antagonist affinity for EPAC2 while ESI- 10 is not completely specific for EPAC2.49
EPAC2 signaling pathways in the neurons
The development as well as conservation of dendrites are imperative to the routes of synaptic transformation or remodeling as well as plasticity of the brain.21 EPAC2 stimulation of Rap triggers synapse destabilization of the dendritic spines during synaptic transformation or remodeling.21 EPAC2 influences spine decline as well as internalization of α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) leading to decrease excitatory synaptic transmission. This process results in destabilization of the synapse but avoids synaptic eradication.21 Also, this action may be synchronized via interface between EPAC2 and neuroligin, which recruits EPAC2 to the membrane thereby enhancing GEF activity.21 Further studies on EPAC2 and neuroligin in the glioma microenvironment could lead to a novel therapeutic discovery.
The detection of a rare EPAC2 mutation in the RA domain in numerous autistic patients established that, the function of EPAC2 was maintenance of basal dendrites.52 This rare EPAC2 mutation as well as other forms of mutation could further be exploited in the glioma microenvironment. This mutation has proven to efficiently reduced the quantity as well as length of basal dendrites that was associated with the activities of EPAC2 via a Ras-intermediated pathway in pyramidal neurons.52 The ability of EPAC2 to astrocyte differentiation was evaluated in EPAC2 knockout mice. It was established that, EPAC2 needed PACAP to modulate external influx of calcium necessary for GFAP secretion as well as differentiation. 31
It was well established that, growth arrest was needed to bestow comprehensive differentiation though axonal growth is essential for the maturation of neurons.53 Therefore, a synchronized action between the cAMP-neutral nerve growth factor (NGF) triggering of extracellular-signal-regulated kinase (ERK) as well as EPAC2 stimulation of p38 mitogen activated protein kinase (MAPK) triggered growth arrest and neuritogenic consequences, correspondingly in neuroscreen-1 (NS-1) as well as PC12 cells.53 It was established that, EPAC2 alone could decrease the significant Rap1B activity as well as configuration of axons in cultured hippocampal neurons.21 It was also established that, triggering of EPAC2 augmented Rap and p38 stimulation to assemble intracellular calcium, which in turn triggers calcium-sensitive big potassium channels, ion channels normally linked with PKA stimulation in cerebellar granule cells.54,55 It was affirmed that; activation of these ion channels generates slight membrane hyperpolarization weakening neuron firing.54,55 Also, stimulation of EPAC2 triggered the phosphorylation of syntabulin, a microtubule-related as well as syntaxin-1-binding protein that binds syntaxin-filled vesicles to microtubules and kinesin I as well as intermediates anterograde transport of syntaxin-1 to neuronal processes in INS-1E cells.56 Ras-binding happens to be obligatory for the cAMP-determined stimulation of Rap1 through EPAC2 though binding of EPAC2 to Ras-GTP is devoid of cAMP.57 It was established that EPAC2 interrelates precisely with the nucleotide-binding fold-1 (NBF-1) of sulfonylurea receptor SUR1, a subunit of the ATP-sensitive potassium channel (KATP).58,59 This association in gliomagenesis is yet to be determined. Furthermore, EPAC2 binds Rim1α, resulting in its augmented linkage with Rab3 to expedient synaptic vesicles juxtaposition to the plasma membrane.58-60 Additionally, EPAC2-intermediated production of DAG via the PLC pathway may be associated with the stimulation as well as translocation of Munc13-1 binding to Rim1α following SNARE-intermediated synaptic vesicle expression.58,60 It was also perceived that, EPAC2’s binding to SUR1 influenced exocytosis in dentate granule presynaptic terminals. Its interface weakens the action of the neuronal-type KATP but boosted voltage-dependent Ca2+ channel activityh.61
Figure 2.
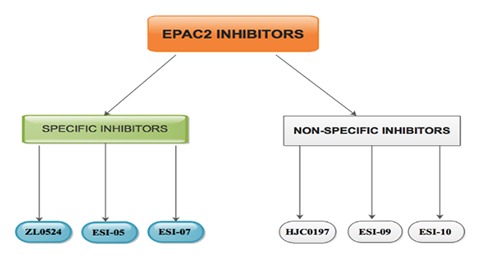
EPAC2 inhibitor: The inhibitors are classified into specific/selective and non-specific/non-selective.
EPAC2 and glioma
It was obvious that, EPAC2 was universally secreted in all neurites and the expression of EPAC2 was higher in normal brain tissues. 21 We established that EPAC2 secretory levels were more in low-grade clinical glioma than high-grade clinical glioma. Which means that, high-grade clinical glioma tissues have lesser quantities of EPAC2 glands unlike low-grade clinical glioma tissues4 (Figure 1B). Furthermore, using U251 and U87 cell lines, we demonstrated that, MMP-2 secretion was reduced subsequent to EPAC2 over-secretory plasmid transfection unlike normal control as well as plasmid control cells.4 Our results indicated that EPAC2 over-secretion resulted in diminished MMP-2 protein levels through the EPAC2/MMP-2 signaling pathway. We therefore concluded that, in the glioma microenvironment, MMP-2 triggers EPAC2 glands, which resulted in a trigger of higher levels of EPAC2 during glioma pathogenesis4 (Figure 3).
Seo and Lee established that, EPAC2 inhibition compromised proapoptotic caspase adaptor protein (PACAP)-triggered astrocytic differentiation of neural precursor cells devoid of neuronal differentiation. 31 They further proposed that an upsurge in intracellular calcium levels was essential in the PACAP/EPAC2 signaling pathway-triggered astrocytogenesis31 (Figure 3). Studies have proven that astrocytogenesis was primarily linked with PACAP/EPAC signaling pathway.4,62,63 We therefore advocated that further studies on signaling pathways through which EPAC2 secretion reduces in glioma specimens other than the EPAC2/MMP-2 signaling pathway4. It was further advocated that, EPAC2 could be of therapeutic valve in glioma since EPAC2 levels are drastically reduced in glioma microenvironment. Base on the hypothesis above, further studies on the up-regulatory effect of EPAC2 on glioma cells are still warranted.
EPAC2 and cAMP in glioma pathogenesis
It was established that, cAMP produced from adenosine triphosphate (ATP) by adenylyl cyclase, was a second messenger for intracellular signal transduction in numerous diverse organisms. 48,64 Several studies have demonstrated that, cAMP-intermediated signaling events were utilized to transduce PKA leading to the detection of EPACs or cAMP-GEF.25,27,34,64,65 It was affirmed that, brain region-specific transformations in cAMP levels have been associated with the pattern of gliomagenesis.66 Studies have proven that, low levels of cAMP triggered glioma configuration in neurofibromatosis-1 genetically engineered mouse models.66-68
Several studies have demonstrated that, cAMP is a ubiquitous modulator of inflammatory as well as immunological reactions. 69,70 Also, cAMP modifies several physiological activities through the stimulation PKA and EPAC2. Studies have shown that, PKA and EPAC2 are molecular competitors that are down-regulated by cAMP.70,71 Sugimoto et al. evaluated the consequences of cAMP on Ras as well as Akt signaling pathways in U87MG human malignant glioma cells.66 They indicated that cAMP inhibits p44/42 MAPK activity as well as proliferation in PTEN-depleted human glioblastoma cells in vitro via PKA/EPAC2 pathway.66
It was proven that cAMP suppress cell growth as well as p44/42 MAPK action via down-regulation of the Ras signaling pathway, but not Akt activity in a PKA and EPAC2-determined fashion.66 Also, higher levels of cAMP in the cell resulted in the stimulation of diverse cAMP targets such as PKA and EPAC2.66 As indicated earlier, PKA and EPAC2 are accountable for cAMPdependent p44/42 MAPK dephosphorylation which affirms that cAMP inhibits cell growth via PKA and EPC2 stimulation in U172 as well as U87MG human glioblastoma cells.72 Furthermore, cAMP exhibited inhibitory actions towards Akt via EPAC2-PTEN pathway stimulation in glial as well as osteosarcoma cells.71,73
Figure 3.
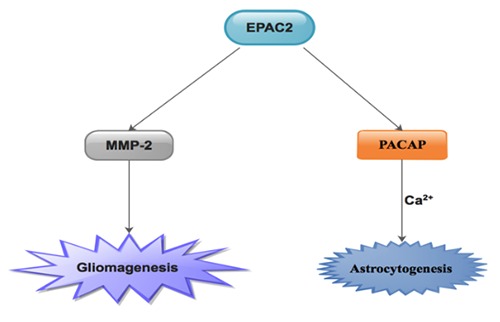
The known pathways via which EPAC2 participating in gliomagenesis as well as astrocytogenesis.
Conclusions
Cyclic AMP-intermediated signaling events were utilized to transduce PKA leading to the detection of EPACs or cAMP-GEF. EPAC2 inhibition compromised proapoptotic caspase adaptor protein (PACAP)-triggered astrocytic differentiation of neural precursor cells devoid of neuronal differentiation. Furthermore, EPAC2 over-secretion resulted in diminished MMP-2 protein levels through the EPAC2/MMP-2 signaling pathway. Further studies on signaling pathways through which EPAC2 secretion reduces in glioma specimens other than the EPAC2/MMP-2 signaling pathway are warranted. Compounds like ZL0524, ESI-05 as well as ESI-07 are absolutely specific inhibitors for EPAC2 while ESI-09, ESI-10 and HJC0197 do not function as selective inhibitors of EPAC2 (Figure 2). As a hypothesis, up-regulatory effect of EPAC2 on glioma cells could lead to discovery on the therapy of gliomas.
References
- 1.Gladson CL, Prayson RA, Liu WM. The pathobiology of glioma tumors. Annual Review of Pathological Mechanical Disease 2010;5:33-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol Mech Dis 2006;1:97-117. [DOI] [PubMed] [Google Scholar]
- 3.Seidu RA, Wu M, Su Z, Xu H. Paradoxical role of high mobility group box 1 in glioma: a suppressor or a promoter? Oncology Reviews 2017;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang M, Zhuang Y, Zu WC, Jiao L, Richard SA, Zhang S. Overexpression of EPAC2 reduces the invasion of glioma cells via MMP-2. Oncology letters 2019;17:5080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. Journal of Neuropathology & Experimental Neurology 2002;61:215-25. [DOI] [PubMed] [Google Scholar]
- 6.Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain pathology 1993;3:255-68. [DOI] [PubMed] [Google Scholar]
- 7.Wen PY, Kesari S. Malignant gliomas in adults. New England Journal of Medicine 2008;359:492-507. [DOI] [PubMed] [Google Scholar]
- 8.Pollack IF. Brain tumors in children. New England Journal of Medicine 1994;331:1500-7. [DOI] [PubMed] [Google Scholar]
- 9.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & development 2001;15:1913-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleihues P, Burger PC, Scheithauer BW. Histological typing of tumours of the central nervous system: Springer Science & Business Media; 2012. [Google Scholar]
- 11.Richard SA. The Therapeutic Potential of Resveratrol in Gliomas. Advances in Bioscience and Clinical Medicine 2019;7:44-59. [Google Scholar]
- 12.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta neuropathologica 2016;131:803-20. [DOI] [PubMed] [Google Scholar]
- 13.Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine 2005;352:987-96. [DOI] [PubMed] [Google Scholar]
- 14.Seidu A., Richard YY, Hao Li, Lu Ma, Chao You. Glioblastoma multiforme subterfuge as acute cerebral hemorrhage: A case report and literature review. Neurology International 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richard SA. Novel Pathogenic, Biomarker and Therapeutic Potentials of Foxm1 in Glioma. NeuroQuantology 2019;17. [Google Scholar]
- 16.Stangeland B, Mughal AA, Grieg Z, et al. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015;6:26192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helseth R, Helseth E, Johannesen T, et al. Overall survival, prognostic factors, and repeated surgery in a consecutive series of 516 patients with glioblastoma multiforme. Acta neurologica scandinavica 2010;122:159-67. [DOI] [PubMed] [Google Scholar]
- 18.Wang P, Liu Z, Chen H, Ye N, Cheng X, Zhou J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorganic & medicinal chemistry letters 2017;27:1633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4). Gene 2016;575:577-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wild CT, Zhu Y, Na Y, et al. Functionalized N, N-diphenylamines as potent and selective EPAC2 inhibitors. ACS medicinal chemistry letters 2016;7:460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robichaux WG, III, Cheng X. Intracellular cAMP sensor EPAC: physiology, pathophysiology, and therapeutics development. Physiological reviews 2018;98:919-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guček A, Gandasi NR, Omar-Hmeadi M, et al. Fusion pore regulation by cAMP/Epac2 controls cargo release during insulin exocytosis. Elife 2019;8:e41711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar N, Prasad P, Jash E, et al. Insights into exchange factor directly activated by cAMP (EPAC) as potential target for cancer treatment. Molecular and cellular biochemistry 2018;447:77-92. [DOI] [PubMed] [Google Scholar]
- 24.Parnell E, Palmer TM, Yarwood SJ. The future of EPAC-targeted therapies: agonism versus antagonism. Trends in pharmacological sciences 2015;36:203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawasaki H, Springett GM, Mochizuki N, et al. A family of cAMP-binding proteins that directly activate Rap1. Science 1998;282:2275-9. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacological reviews 2013;65:670-709. [DOI] [PubMed] [Google Scholar]
- 27.De Rooij J, Zwartkruis FJ, Verheijen MH, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998;396:474. [DOI] [PubMed] [Google Scholar]
- 28.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. Journal of Biological Chemistry 2000;275:20829-36. [DOI] [PubMed] [Google Scholar]
- 29.Ozaki N, Shibasaki T, Kashima Y, et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nature cell biology 2000;2:805. [DOI] [PubMed] [Google Scholar]
- 30.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends in biochemical sciences 2006;31:680-6. [DOI] [PubMed] [Google Scholar]
- 31.Seo H, Lee K. Epac2 contributes to PACAP-induced astrocytic differentiation through calcium ion influx in neural precursor cells. BMB reports 2016;49:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki S, Yokoyama U, Abe T, et al. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. Journal of Biological Chemistry 2010;285:24248-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proceedings of the National Academy of Sciences 1993;90:10989-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi Y, Shinozawa T. Effect of dibutyryl cAMP and several reagents on apoptosis in PC12 cells induced by a sialoglycopeptide from bovine brain. Brain research 1997;778:309-17. [DOI] [PubMed] [Google Scholar]
- 35.Parvathenani LK, Calandra V, Roberts SB, Posmantur R. cAMP delays beta-amyloid (25-35) induced cell death in rat cortical neurons. Neuroreport 2000;11:2293-7. [DOI] [PubMed] [Google Scholar]
- 36.Jonakait GM, Ni L. Prostaglandins compromise basal forebrain cholinergic neuron differentiation and survival: action at EP1/3 receptors results in AIF-induced death. Brain research 2009;1285:30-41. [DOI] [PubMed] [Google Scholar]
- 37.Porat S, Simantov R. Bcl-2 and p53: role in dopamine-induced apoptosis and differentiation. Annals of the New York Academy of Sciences 1999;893:372-5. [DOI] [PubMed] [Google Scholar]
- 38.Roscioni SS, Kistemaker LE, Menzen MH, et al. PKA and Epac cooperate to augment bradykinin-induced interleukin-8 release from human airway smooth muscle cells. Respiratory research 2009;10:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grandoch M, Bujok V, Fleckenstein D, Schmidt M, Fischer J, Weber AA. Epac inhibits apoptosis of human leukocytes. Journal of leukocyte biology 2009;86:847-9. [DOI] [PubMed] [Google Scholar]
- 40.Tiwari S, Felekkis K, Moon E-Y, Flies A, Sherr DH, Lerner A. Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood 2004;103:2661-7. [DOI] [PubMed] [Google Scholar]
- 41.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998;281:1322-6. [DOI] [PubMed] [Google Scholar]
- 42.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007;129:1337-49. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes HB, Riordan S, Nomura T, et al. Epac2 mediates cAMP-dependent potentiation of neurotransmission in the hippocampus. Journal of Neuroscience 2015;35:6544-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Molecular and Cellular Neuroscience 2008;38:578-88. [DOI] [PubMed] [Google Scholar]
- 45.Ueno H, Shibasaki T, Iwanaga T, et al. Characterization of the gene EPAC2: structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics 2001;78:91-8. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Z, Tanaka KF, Matsunaga S, et al. Photoactivated adenylyl cyclase (PAC) reveals novel mechanisms underlying cAMP-dependent axonal morphogenesis. Scientific reports 2016;6:19679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K, Kobayashi Y, Seo H, et al. Involvement of cAMP-guanine nucleotide exchange factor II in hippocampal long-term depression and behavioral flexibility. Molecular brain 2015;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, Zhu Y, Chen H, et al. Structure-activity relationships of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as novel antagonists of exchange proteins directly activated by cAMP (EPACs). Bioorganic & medicinal chemistry letters 2017;27:5163-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsalkova T, Mei FC, Li S, et al. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proceedings of the National Academy of Sciences 2012;109:18613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rehmann H. Epac-inhibitors: facts and artefacts. Scientific reports 2013;3:3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Tsalkova T, Chepurny OG, et al. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. Journal of medicinal chemistry 2013;56:952-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srivastava DP, Woolfrey KM, Jones KA, et al. An autism-associated variant of Epac2 reveals a role for Ras/Epac2 signaling in controlling basal dendrite maintenance in mice. PLoS biology 2012;10:e1001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emery AC, Eiden MV, Eiden LE. Separate cyclic AMP sensors for neuritogenesis, growth arrest, and survival of neuroendocrine cells. Journal of Biological Chemistry 2014;289:10126-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bos JL, Bockaert J, Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca 2-dependent K channels in cerebellar neurons. PNAS 2007;104:2519-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ster J, De Bock F, Guerineau NC, et al. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proceedings of the National Academy of Sciences 2007;104:2519-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su Q, Cai Q, Gerwin C, Smith CL, Sheng Z-H. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nature cell biology 2004;6:941. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, Takahashi M, Li Y, et al. Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Molecular and cellular biology 2008;28:7109-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartolome-Martin D, Torres M, Ciruela F. Protein Directly Activated by cAMP (Epac), Translocate Munc13-1, and Enhance the Rab3A-RIM1 Interaction to Potentiate Glutamate Release at C. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferrero JJ, Alvarez AM, Ramirez-Franco J, et al. β-Adrenergic receptors activate exchange protein directly activated by cAMP (Epac), translocate Munc13-1, and enhance the Rab3ARIM1α interaction to potentiate glutamate release at cerebrocortical nerve terminals. Journal of Biological Chemistry 2013;288:31370-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrero JJ, Ramirez-Franco J, Martin R, Bartolome-Martin D, Torres M, Sanchez-Prieto J. Cross-talk between metabotropic glutamate receptor 7 and beta adrenergic receptor signaling at cerebrocortical nerve terminals. Neuropharmacology 2016;101:412-25. [DOI] [PubMed] [Google Scholar]
- 61.Zhao K, Wen R, Wang X, et al. EPAC inhibition of SUR1 receptor increases glutamate release and seizure vulnerability. Journal of Neuroscience 2013;33:8861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cebolla B, Fernandez-Perez A, Perea G, Araque A, Vallejo M. DREAM mediates cAMP-dependent, Ca2+-induced stimulation of GFAP gene expression and regulates cortical astrogliogenesis. Journal of Neuroscience 2008;28:6703-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vallejo M. PACAP signaling to DREAM: a cAMP-dependent pathway that regulates cortical astrogliogenesis. Molecular neurobiology 2009;39:90-100. [DOI] [PubMed] [Google Scholar]
- 64.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta biochimica et biophysica Sinica 2008;40:651-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annual review of pharmacology and toxicology 2010;50:355-75. [DOI] [PubMed] [Google Scholar]
- 66.Sugimoto N, Miwa S, Tsuchiya H, et al. Targeted activation of PKA and Epac promotes glioblastoma regression in vitro. Molecular and clinical oncology 2013;1:281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis- 1 heterozygosity. Journal of Neuroscience 2010;30:5579-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warrington NM, Gianino SM, Jackson E, et al. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer research 2010;70:5717-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moore A, Willoughby D. The role of cAMP regulation in controlling inflammation. Clinical & Experimental Immunology 1995;101:387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saito T, Sugimoto N, Ohta K, et al. Phosphodiesterase Inhibitors Suppress Lactobacillus casei Cell-Wall-Induced NF- uuoB and MAPK Activations and Cell Proliferation through Protein Kinase A‚Aior Exchange Protein Activated by cAMPDependent Signal Pathway. The Scientific World Journal 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugimoto N, Miwa S, Ohno-Shosaku T, et al. Activation of tumor suppressor protein PTEN and induction of apoptosis are involved in cAMP-mediated inhibition of cell number in B92 glial cells. Neuroscience letters 2011;497:55-9. [DOI] [PubMed] [Google Scholar]
- 72.Moon E-Y, Lee G-H, Lee M-S, Kim H-M, Lee J-W. Phosphodiesterase inhibitors control A172 human glioblastoma cell death through cAMP-mediated activation of protein kinase A and Epac1/Rap1 pathways. Life sciences 2012;90:373-80. [DOI] [PubMed] [Google Scholar]
- 73.Miwa S, Sugimoto N, Shirai T, et al. Caffeine activates tumor suppressor PTEN in sarcoma cells. International journal of oncology 2011;39:465-72. [DOI] [PubMed] [Google Scholar]