

The earliest evidence of life on Earth is from organosedimentary structures, termed microbialites, preserved in 3.481-billion-year-old (Ga) rocks. Phototrophic microbial mats form in association with an ∼700-km2 expanse of morphologically diverse microbialites in the hypersaline Great Salt Lake (GSL), Utah. Here, we show taxonomically similar microbial mat communities are associated with morphologically diverse microbialites across the lake. Metagenomic sequencing reveals an abundance and diversity of autotrophic and heterotrophic taxa capable of harvesting light energy to drive metabolism. The unexpected abundance of and diversity in the mechanisms of harvesting light energy observed in GSL mat populations likely function to minimize niche overlap among coinhabiting taxa, provide a mechanism(s) to increase energy yield and osmotic balance during salt stress, and enhance fitness. Together, these physiological benefits promote the formation of robust mats that, in turn, influence the formation of morphologically diverse microbialite structures that can be imprinted in the rock record.
KEYWORDS: microbialite, cyanobacteria, nitrogen fixation, aerobic anoxygenic photosynthesis, rhodopsin, retinal, photosystem, bacteriochlorophyll, Great Salt Lake, aerobic photoheterotroph
ABSTRACT
Microbial mat communities are associated with extensive (∼700 km2) and morphologically variable carbonate structures, termed microbialites, in the hypersaline Great Salt Lake (GSL), Utah. However, whether the composition of GSL mat communities covaries with microbialite morphology and lake environment is unknown. Moreover, the potential adaptations that allow the establishment of these extensive mat communities at high salinity (14% to 17% total salts) are poorly understood. To address these questions, microbial mats were sampled from seven locations in the south arm of GSL representing different lake environments and microbialite morphologies. Despite the morphological differences, microbialite-associated mats were taxonomically similar and were dominated by the cyanobacterium Euhalothece and several heterotrophic bacteria. Metagenomic sequencing of a representative mat revealed Euhalothece and subdominant Thiohalocapsa populations that harbor the Calvin cycle and nitrogenase, suggesting they supply fixed carbon and nitrogen to heterotrophic bacteria. Fifteen of the next sixteen most abundant taxa are inferred to be aerobic heterotrophs and, surprisingly, harbor reaction center, rhodopsin, and/or bacteriochlorophyll biosynthesis proteins, suggesting aerobic photoheterotrophic (APH) capabilities. Importantly, proteins involved in APH are enriched in the GSL community relative to that in microbialite mat communities from lower salinity environments. These findings indicate that the ability to integrate light into energy metabolism is a key adaptation allowing for robust mat development in the hypersaline GSL.
IMPORTANCE The earliest evidence of life on Earth is from organosedimentary structures, termed microbialites, preserved in 3.481-billion-year-old (Ga) rocks. Phototrophic microbial mats form in association with an ∼700-km2 expanse of morphologically diverse microbialites in the hypersaline Great Salt Lake (GSL), Utah. Here, we show taxonomically similar microbial mat communities are associated with morphologically diverse microbialites across the lake. Metagenomic sequencing reveals an abundance and diversity of autotrophic and heterotrophic taxa capable of harvesting light energy to drive metabolism. The unexpected abundance of and diversity in the mechanisms of harvesting light energy observed in GSL mat populations likely function to minimize niche overlap among coinhabiting taxa, provide a mechanism(s) to increase energy yield and osmotic balance during salt stress, and enhance fitness. Together, these physiological benefits promote the formation of robust mats that, in turn, influence the formation of morphologically diverse microbialite structures that can be imprinted in the rock record.
INTRODUCTION
The oldest identifiable fossil assemblages of Earth’s early biosphere (3.481 billion years ago [Ga]) are preserved in microbialites, specifically, those with laminated morphologies (1–4). Microbialites are organosedimentary structures that form in lacustrine, marine, and other aquatic environments due to the binding or trapping of sediment by microbial mat communities and/or the biomediated precipitation of carbonate minerals (5, 6). Modern microbialites often display structural and textural similarities to those identified in ancient rocks that, together with geochemical and isotopic evidence, have led to the interpretation of the latter structures as being biogenic (7).
While thought to have been widespread on early Earth (2, 6, 8), microbialites have a far more limited distribution today and are typically only found in environments with conditions that restrict higher order organisms that graze the microbial mats involved in their formation. Such environments where microbialites are found include saline marine systems such as in the Hamelin Pool of Shark Bay, Western Australia (9–11), Socompa Lake, Argentina (12), and in Highborne Cay, Bahamas (13, 14). In addition, microbialites can be found in modern lacustrine environments, including Pavilion Lake, Canada (15), and Alchichica, Mexico (16), among others. Perhaps the largest expanse of microbialites (∼700 km2) is the carbonate structures that form in shallow marginal areas in the hypersaline Great Salt Lake (GSL), United States (Fig. 1) (17–20). In areas of the lake with salinities averaging ∼12% (south arm, south of the railroad causeway), these structures are associated with robust phototrophic microbial mats that may account for close to one-half of the total production in the lake (21). However, the physiological adaptations that allow for the establishment of extensive phototrophic mats and associated microbialite structures in the shallow margins of hypersaline GSL are poorly understood.
FIG 1.

Location of study areas and examples of microbialite types. (A) Aerial image of Great Salt Lake and surrounding highlands from 2016 displaying estimated microbialite extent (17, 20). Lake bathymetry from Baskin and Allen 2005 (83) and Baskin and Turner 2006 (84). (B) Sample area 1 (samples windward east, windward west, Bridger Bay, Ridges, and Buffalo Point), northern Antelope Island; Google Earth image from July 2016. (C) Sample area 2 (samples Stansbury polygons, Stansbury interpolygons), northeast corner of Stansbury island; Google Earth image from July 2016. (D). Flat low-profile microbialites in Bridger Bay, Antelope Island (sample BB). (E and F). Large domal microbialites that form on the perimeter of megapolygons (sample SP) and low-profile elongate microbialites within the polygons (sample SI).
16S rRNA gene characterizations of GSL mats have been conducted on a single morphologic type of microbialite collected from Bridger Bay, near Antelope Island (22, 23). This work showed the GSL microbialite mat community was dominated by a phototroph that is closely affiliated with the halophilic cyanobacterial genus Euhalothece. However, GSL microbialites (20, 24), and microbialites in general (e.g., see references 2, 5, and 6), exhibit a variety of morphologies that may suggest differences in the compositions of communities and/or processes that are involved in their formation. For example, GSL microbialites were previously categorized into several distinct morphological groups, including ridge-like structures, composite rings, low-profile collapsed domes, and large-diameter domal mounds (20). One possible explanation for the various microbialite morphotypes in GSL is the differences in the average energy states and depositional environments where they form, both of which are controlled largely by protection from wave action (20, 24, 25). However, based on available data, it is unknown if differences in the taxonomic and functional compositions of the mat communities putatively involved in their formation also contribute to morphological differences among microbialites in GSL.
To begin to address these outstanding questions, seven morphologically distinct microbialites and their associated mats from two locations in GSL were characterized at the level of morphology, qualitative local environmental characteristics, and microbial community composition inferred from 16S rRNA gene sequence profiles. Using these data, a single mat sample that was representative of the seven mats was identified, and metagenomic sequencing and informatics analyses were conducted to reveal metabolic potentials of the major members. These results were then compared to those from microbialite mat communities from other sites to identify functionalities and adaptations that are characteristic of hypersaline mat assemblages and that allow for their persistence. Results are discussed in the context of the unexpected importance of light energy in supporting mat-forming populations in the hypersaline GSL and its implications in the formation of the extensive microbialites found in the lake today and in saline lakes across the world.
RESULTS AND DISCUSSION
Site and sample description.
Microbialite subsamples were collected from seven different locations in the south arm (SA) of GSL. These locations were chosen to sample a variety of microbialite morphologies and environment lake conditions present in the productive (shallow marginal) zone of GSL (Fig. 1), as reported previously (17, 20, 25, 26). Physical and chemical measurements of waters overlaying microbialites from these locations indicated little variation, with temperatures ranging from 22.0 to 30.1°C, pHs ranging from 8.1 to 8.2, and salinity ranging from 14.5% to 17.0% (Table 1).
TABLE 1.
Location and chemical/physical measurements made on waters overlying microbialites from specified sampling locations in Great Salt Lake, Utah
| Locationa | Date (mo/day/yr) | GPS (NAD83)b
|
Temp (°C) | pH | Salinity (%) | Cond.c (mS cm−1) | |
|---|---|---|---|---|---|---|---|
| UTM E | UTM W | ||||||
| WE | 8/15/2016 | 394399 | 4546259 | 22.0 | 8.23 | 14.5 | 166.0 |
| WW | 8/15/2016 | 393988 | 4546261 | 23.9 | 8.22 | 14.6 | 168.4 |
| BB | 8/15/2016 | 394518 | 4545827 | 25.2 | 8.15 | 15.5 | 178.2 |
| BP | 8/15/2016 | 392530 | 4543686 | 26.6 | 8.05 | 15.0 | 183.0 |
| R | 8/15/2016 | 392661 | 4542499 | 30.1 | 8.06 | 17.0 | 195.0 |
| SI | 8/16/2016 | 374474 | 4530603 | 25.2 | 8.12 | 15.6 | 138.3 |
| SP | 8/16/2016 | 374369 | 4530692 | 26.9 | 8.16 | 15.8 | 139.8 |
WE, windward east; WW, windward west; BB, Bridger Bay; BP, Buffalo Point; R, ridges; SI, Stansbury interpolygons; SP, Stansbury polygons.
GPS, global positioning system; UTM E, universal transverse Mercator east; UTM W, universal transverse Mercator west.
Cond., conductivity.
The seven microbialite sampling sites were all located in the SA of GSL, since mats and microbialites in the north arm are no longer thought to be growing due to an anthropogenic salinity increase up to 30% (22). Detailed descriptions of the variation in microbialite morphologies and the hydrological and geological characteristics of their local environment in GSL were reported previously (20). Briefly, the windward west (WW) and windward east (WE) sites were located on the northern shore of Ladyfinger Point, a peninsula off the north shore of Antelope Island (north of Bridger Bay), with WW on the west side and WE on the east side of a small spit (Fig. 1). Both WW and WE environments are characterized as being subject to substantial wave action (i.e., a high-wave-energy environment) and are on a shallow ramp distant from the bedrock outcrop. Microbialites in these locations are up to ∼1 m in diameter and are well cemented with carbonate (typically aragonite) mineral that has been suggested to be bioprecipitated (27, 28). Microbialites tend to form in linear accumulations at WE sites and form distinct clusters (up to 5 m in diameter) at WW sites.
The Bridger Bay (BB) samples were from the sheltered Bridger Bay on the south side of Ladyfinger Point, which is thought to minimize disruption from wave action (i.e., a low-wave-energy environment). Like those at WW and WE sites, microbialites in Bridger Bay formed on a shallow ramp that is distant from bedrock outcrops. However, the BB microbialites are low-profile domes, including collapsed domes (20), and are poorly cemented. The Buffalo Point (BP) and ridge (R) samples were collected on the west shore of Antelope Island near Buffalo Point, south of Bridger Bay, which are both high-wave-energy environments. The BP samples were collected along a steep margin, close to outcrops of bedrock, where the microbialites form taller, larger well-cemented domes that incorporate an abundance of lithic fragments into their structure. In contrast, the R sampling location was along a shallow margin but is subject to substantial wave action. Microbialites here form long linear structures related to cemented wave ripples (20).
The Stansbury Island sampling location was on the northeast side of the island in an area with moderate wave energy. The area is a shallow ramp, is far from a bedrock exposure, and contains extensive megapolygon formations (20). Large (often >1 m in diameter and up to 5 m) well-cemented microbialites (sample designation SP) tend to form on the perimeters of the large polygon structures and are thought to be related to groundwater seeps (20). Samples were also collected from the low-profile elongate microbialites found within the interiors of the polygons (sample designation SI). The morphological differences associated with the sampled GSL microbialites are consistent with previous data indicating that morphology is the expression of localized environmental conditions, including ramp geometry, wave energy, proximity to bedrock, availability of hard substrates, and proximity to possible groundwater seeps (20, 25, 29).
Microbialite 16S rRNA gene composition.
To begin to examine similarities and differences in the composition of microbialite-associated microbial mats from the seven sample locations, a total of 38,007 16S rRNA gene sequences were subsampled for each mat community, and these were profiled taxonomically. Subsampling of 16S rRNA genes resulted in similar coverages (range, 87% to 91%) of the predicted taxonomic diversity in these mat communities (see Table S1 in the supplemental material). Likewise, these communities hosted similarly diverse microbial communities as indicated by comparable inverse Simpson indices (range, 29.6 to 46.4) (Table S1).
The taxonomic compositions of the seven communities examined were also similar (Fig. 2), as indicated by Bray-Curtis pairwise community similarities of operational taxonomic unit (OTU) distributions among communities that ranged from 0.70 to 0.91 (see Table S2). The compositions of the communities sampled from BB in the present study and those sampled from BB in prior studies (22, 23) also had similar structures. Thus, despite significant morphological differences among microbialites sampled from the seven different GSL locations (Fig. 1), the taxonomic compositions of the communities thought to be involved in their formation (see references 22, 27, and 28) exhibited only minimal differences at the levels of OTU diversity and distribution. This finding is in agreement with those obtained from morphologically distinct microbialites from Shark Bay, Australia, where minimal taxonomic and functional differences were detected among mat communities associated with morphologically distinct microbialites (11). Together, these results and those of others (20, 25, 29) suggest that an interplay between local geology, the mats involved in trapping and binding of sediment grains, and the differential delivery and deposition of sediment grains, based on the direction and average wave energy of overriding water in the environment, shapes the morphology of microbialites in GSL and likely elsewhere.
FIG 2.

Compositions of 16S rRNA gene operational taxonomic units (OTUs) recovered from microbialite mat communities in seven different locations in the south arm of Great Salt Lake. Representative OTUs were binned at the order level, with orders that represented >0.5% of the total sequences in a given sample site included in the legend. Taxonomic bins that represented <0.5% of the total sequences from each assemblage were pooled and depicted as “other.” An unweighted pair group method with arithmetic mean (UPGMA) dendrogram of community composition differences was constructed using classical clustering and Bray-Curtis similarity indices (top left).
While the microbial mat communities were largely similar at the levels of 16S rRNA gene OTU diversity and distribution, several slight compositional differences were detected, in particular, among the abundance of the most abundant community members (Fig. 2). The most abundant OTU in five of the seven microbialite communities was affiliated with one of several putative bacterial heterotrophs. This included four communities (WE, BB, R, and SP) where the most abundant OTU (14.3% to 16.4% of the total reads) exhibited 98% sequence identity with the heterotroph and halophile Salisaeta within the phylum Rhodothermaeota. The most abundant OTU (15.3% of total reads) in the BP sample exhibited 98% sequence identity to the heterotroph Gracilimonas within the order Balneolales. Consistent with its detection in phototrophic mats in hypersaline GSL, halophilic Gracilimonas has been previously isolated from a variety of solar salterns (e.g., see reference 30).
In contrast, in two of the seven microbialite communities (WW and SI), the most abundant OTU (12.2% and 14.9% of total reads, respectively) exhibited 99% sequence identity to the halophilic and cyanobacterial phototroph Euhalothece of the order Chroococcales. Euhalothece was previously shown to be a dominant component of GSL microbialite mat communities from BB (22, 23). This OTU was also detected in the other five communities sampled in this study in abundances ranging from 9.2% to 12.8% of total reads. 16S rRNA gene OTUs affiliated with Archaea were not detected in abundances >0.5% in any community examined, consistent with previous 16S rRNA gene characterizations of these communities (22, 23). Thus, based on inference from 16S rRNA gene sequencing, the compositions of mat communities across the SA of GSL are largely similar and consist of a dominant primary producer and several dominant heterotrophic consumers. This “simplified” trophic structure is common among characterized phototrophic mat communities (e.g., see reference 31).
Microbialite metagenomic composition.
The average composition of the seven GSL microbialite mat communities was calculated at the level of taxonomic order, and this was used to calculate a Bray-Curtis metric of community similarity among this and the other seven communities. The average composition of the communities was most similar (Bray Curtis similarity = 0.92) to communities from BB, SI, and SP (see Table S3 in the supplemental material). This observation, combined with the extensive work conducted on microbialites from the BB sampling location (22, 23, 26, 28), led to the decision to generate a metagenome from this microbial mat to potentially uncover additional insights into adaptations that contribute to their extensive habitation of the hypersaline GSL environment.
Sequencing of the BB microbial mat metagenome yielded a total of 10.02 Gbp of reads, with assembly generating a total of 354.0 Mbp of contigs (see Table S4). Contigs were binned into metagenome-assembled genomes (MAGs), and raw reads were mapped to the MAGs to generate a rank abundance plot (Fig. 3A). A total of 82.4% of the quality-filtered metagenome reads were represented by the preliminary draft MAGs. Among the bins, a total of 38 MAGs were identified that met the criteria of medium- to high-quality draft MAGs (see Data Set S1) (32), and the 18 most abundant MAGs (>1.0% estimated relative abundance) represented 55.4% of the total community (Fig. 3A). The most abundant taxonomic orders associated with the 18 most abundant MAGs were broadly similar to the taxonomic orders identified in 16S rRNA gene surveys of the seven GSL microbialite communities (Fig. 2 and 3A).
FIG 3.

(A) Rank-abundance plot of GSL microbialite reconstructed population level metagenome-assembled genome (MAG) bins. Each vertical bar represents a reconstructed MAG that has an estimated completeness >50% (n = 38) (see Data Set S1 in the supplemental material). MAGs are arranged by relative abundance (as a percentage) in decreasing order, as determined by read mapping. The taxonomy of genome bins was based on BLASTp searches of housekeeping genes against homologs from cultivated representatives with available genomes (Data Set S1) and are color coded based on taxonomic order to match the scheme presented in Fig. 2. (B) PCO of a matrix describing the dissimilarity of KEGG orthologs (KOs) associated with energy metabolism pathways in each of the 18 most abundant MAGs. Each dot corresponds to the MAG indicated (Data Set S1) and is colored based on taxonomic order as in Fig. 2 and 3A. Numbers correspond with taxonomy presented in Table 2.
The distribution of KEGG orthologs (KOs) associated with energy metabolism pathways in each of the 18 most abundant MAGs (see Data Set S2) was determined, and this information was used to generate a principal-component ordination (PCO) plot describing the dissimilarity in their functional potentials. KOs associated with the most abundant MAG (12.2% of the estimated relative abundance), which is most closely related (97% DNA-directed RNA polymerase subunit beta [RpoB]) to the cyanobacterium Euhalothece (Chroococcales), clustered distinctly from the rest of the MAGs (Fig. 3B). KOs associated with the other 17 MAGs formed a cluster that was further subclustered into those associated with Proteobacteria and those associated with Bacteroidetes/Verrucomicrobia. Thus, the distribution of energy metabolism-related KOs was largely synchronous with phylum-level taxonomic classifications.
To identify functionalities associated with the broad patterns of clustering in the PCO, protein-coding genes in each of the 18 most abundant MAGs were subjected to annotation and metabolic pathway analysis. Detailed accounts of the distribution of target proteins and further descriptions of the inferred metabolisms of each of these 18 MAGs can be found in Data Set S2 and results in the supplemental material, respectively. The Euhalothece population is inferred to be an oxygenic phototroph based on the presence of homologs encoding a complete Calvin cycle and both reaction centers photosystem I (Psa) and II (Psb) (Table 2), all of which are characteristic of Cyanobacteria (33). This interpretation is also consistent with the physiology of closely related strains, which have been shown to grow autotrophically via oxygenic photosynthesis (e.g., Euhalothece sp. strain PCC 7418 [34]). Based on protein annotations, the GSL Euhalothece population is also likely capable of fermenting pyruvate into formate, lactate, and acetate and can convert dinitrogen (N2) to ammonia via molybdenum (Mo)-dependent nitrogenase. This characteristic is consistent with recent reports of N2 fixation in this genus (35) and indicates that Euhalothece likely supplies both fixed carbon and nitrogen for secondary consumers in GSL. As such, Euhalothece likely represents a keystone species in the GSL mat community and the broader ecosystem (23, 36).
TABLE 2.
Distribution of proteins involved in phototrophy in the 18 most abundant MAG bins in the Great Salt Lake microbial mat community
| Bin no. | Closest BLASTp hit | % IDa | Rhodopsinb | Reaction centersc
|
Bch synthesisd |
Calvin cyclee |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| PSI | PSII | DPOR | COR | RuBisCO | PRK | |||||
| 1 | Halothece sp. PCC 7418 | 97 | XR | PsaABD | PsbBCE | BchLNB | BchXYZ | CbbSL | PRK | |
| 2 | Fabibacter pacificus | 60 | NaR, HR | |||||||
| 3 | Longibacter salinarum | 97 | XR | |||||||
| 4 | Pelagibaca abyssi | 95 | PufLMH | BchLNB | BchXYZ | |||||
| 5 | Wenzhouxiangella sediminis | 96 | PufLMH | |||||||
| 6 | Arboricoccus pini | 80 | XR | |||||||
| 7 | Roseibaca calidilacus | 96 | PufHff | BchLNB | ||||||
| 8 | Longibacter salinarum | 84 | ||||||||
| 9 | Coraliomargarita akajimensis | 93 | XR, R | |||||||
| 10 | Psychroflexus sp. WDS2C27 | 93 | R | |||||||
| 11 | Henriciella sp. JN25 | 87 | XR | |||||||
| 12 | Rhodohalobacter halophilus | 91 | HRg | |||||||
| 13 | Rhodohalobacter halophilus | 88 | HR | |||||||
| 14 | Thiohalocapsa sp. ML1 | 93 | PufLMH | BchLNB | BchXYZ | CbbSL, CbbM | PRK | |||
| 15 | Coraliomargarita akajimensis | 81 | XR | |||||||
| 16 | Wenzhouxiangella sediminis | 95 | XRg | PufLMH | ||||||
| 17 | Rhodothermaceae bacterium | 77 | NaR, HR | |||||||
| 18 | Inquilinus limosus | 83 | XR | |||||||
ID, identity. RpoB amino acid identity to homologs from the nearest cultivar, with the exceptions of bin 4 (RplB), bin 7 (RplA), and bin 16 (RplS).
R, rhodopsin; NaR, sodium-transporting rhodopsin; HR, halorhodopsin; XR, xanthorhodopsin.
Homolog of PsaC or PsbF was not detected. PSI, photosystem I; PSII, photosystem II.
DPOR, dark-operative protochlorophyllide oxidoreductase; COR, chlorophyllide a oxidoreductase.
RuBisCO, ribulose-1,5-bisphosphate carboxylase oxygenase; PRK, phosphoribulokinase.
Homologs of PufMH were not detected.
Partial sequence.
Among the 17 next most abundant MAGs that cluster in the lower half of the PCO (Fig. 3B), only one was inferred to be capable of photosynthesis based on the detection of homologs of proteins involved in the Calvin cycle (CbbSL, CbbM), (bacterio)chlorophyll (Bch) biosynthesis (BchLNBXYZ), and reaction centers (PufLMH). This MAG exhibited 93% RpoB sequence identities to the anoxygenic phototroph Thiohalocapsa sp. strain ML1, a purple sulfur bacterium affiliated with the Chromatiales (37), and it comprised an estimated 1.3% relative abundance of the GSL mat community. This MAG also encodes molybdenum (Mo)-dependent nitrogenase suggesting that, like Euhalothece, it contributes both fixed carbon and nitrogen to the community. Thus, taxa capable of light-driven CO2 fixation and N2 fixation were represented in 2 of the 18 most abundant bins and together represented 13.5% of the total community.
Clustering of the remaining 16 MAGs based on dissimilarity in the distribution of KOs involved in energy metabolism (Fig. 3B) suggests at least partial overlap in encoded metabolic pathways. Based on the absence (16/16 MAGs) of protein homologs encoding the Calvin cycle (CbbSL or CbbM), the general absence (15/16 MAGs) of proteins allowing for anaerobic respiration (e.g., nitrate or bisulfate reductases), and the near uniform (15/16 MAGs) presence of homologs of cytochrome c oxidases (Data Set S2), these 16 MAGs (combined, 41.9% of total reads) are inferred to be from facultatively anaerobic or aerobic heterotrophic bacteria. The completeness of these MAGs ranged from 51.7 to 99.7% (average, 86.7%), indicating that the lack of genes encoding autotrophic pathways in MAGs is unlikely to be an artifact associated with poor sequencing, assembly, or binning. Thus, aerobic heterotrophic bacteria likely outnumber photoautotrophs in GSL mats, at least among the abundant members of the communities.
The MAGs from the 16 putatively aerobic heterotrophic bacteria were subjected to metabolic reconstruction to identify differences in pathways/KOs related to carbon metabolism that could explain their coexistence. Carbon degradation pathways differed among MAGs (Data Set S2) and included those involved in lipid, protein, organic acid, and/or sugar degradation, including both simple and complex carbohydrates. Sources of such organic compounds are inferred to be supplied by the dominant primary producer in both the mats and the water column, Euhalothece (22), and secondarily by Thiohalocapsa. Close relatives of Euhalothece have been shown to store photosynthate internally within carboxysomes (38). Furthermore, cultivated strains of halophilic Euhalothece have been shown to produce copious amounts of nutritionally rich extracellular polymeric substances (EPS) that typically comprise six to eight monosaccharides and that can contain acetyl, pyruvyl, and/or sulfate groups (39, 40). EPS or its degradation products could also support heterotrophic members of the GSL mat community.
An expanded characterization to include all pathways of energy metabolism of the 16 putative aerobic heterotrophic bacteria surprisingly revealed homologs of rhodopsins, reaction center proteins, and bacteriochlorophyll biosynthesis proteins in 15 of these MAGs. Thus, when combined with the Euhalothece (bin 1) and Thiohalocapsa (bin 14) MAGs, 17 of the 18 most abundant MAGs in the GSL mat community encode the potential for phototrophy. Among these 17 MAGs, 13 encoded homologs of one or more rhodopsins, including xanthorhodopsin (XR), sodium-transporting rhodopsin (NaR), halorhodopsin (HR), and/or rhodopsin (R); homologs of proteorhodopsin and heliorhodopsin were not detected (Table 2). Homologs of XR and R, both of which are retinal-containing proton pumps that function to convert light into electrochemical potential to energize cells (41), were identified in 8 and 2 MAGs, respectively. In addition to XR and R, homologs of NaR and HR, both of which use light energy to balance the osmotic potential across the membrane primarily with sodium or chloride ions, respectively (42, 43), were identified in 2 and 3 MAGs, respectively. These results suggest the importance of rhodopsins, and thus light energy, in supporting not only Euhalothece and Thiohalocapsa but also numerous putative aerobic heterotrophs in GSL mats. Moreover, given that various forms of rhodopsins and Bch absorb light of differing wavelengths (44), it is likely that their variable distribution among dominant taxa helps to minimize niche overlap, thereby enabling their coexistence.
XR from Salinibacter ruber has been shown to function in association with the carotenoid, salinixanthin (41). Intriguingly, homologs of β-carotene 15,15′-dioxygenase, a protein involved in the synthesis of salinixanthin (45), were detected in only 5 of the 8 XR-encoding MAGs: Longibacter (bin 3), Coraliomargarita (bin 9), Henriciella (bin 11), Wenzhouxiangella (bin 16), and Inquilinus (bin 18). One of the MAGs, Euhalothece (bin 1), encodes a homolog of apocarotenoid-15,15′ oxygenase that also functions in production of retinal from carotenals and carotenols (46); however, it is not known if the product of this enzyme can function with XR. Thus, the function of XR in the 3 MAGs with no evidence for salinixanthin biosynthesis is unknown. However, as pointed out previously for MAGs with XR and no apparent salinixanthin biosynthesis capability (47), it is possible that novel mechanisms of synthesizing this or a related pigment that functions with XR will be discovered. Intriguingly, it has also been suggested that salinixanthin produced by one member of the community may act as a community resource and thus represent an additional factor driving community assembly (48).
Additional evidence pointing toward the importance of light energy in supporting autotrophs and aerobic heterotrophs in GSL mats comes from the distribution of homologs of photosystem (PS) proteins and proteins involved in the synthesis of Bch. Among the 18 most abundant MAGs, only the most abundant Euhalothece bin encoded both PSI (PsaABD) and PSII (PsbBCE) found in oxygenic phototrophs (33). As expected, this MAG also encoded dark-operative protochlorophyllide oxidoreductase (DPOR; BchLNB) and chlorophyllide a oxidoreductase (COR; BchXYZ). DPOR reduces a double bond in protochlorophyllide to yield chlorophyllide a, a precursor for bacteriochlorophyll a, whereas COR reduces the double bond between the C-7 and C-8 carbons of chlorophyllide a during bacteriochlorophyll a biosynthesis (49). While we did not examine the distribution of additional protein homologs that could potentially further inform on the types of chlorophylls synthesized in these taxa, it is important to note that all major types of bacteriochlorophylls can be synthesized from the intermediate chlorophyllide a (49).
Among the 15 MAGs of putative aerobic heterotrophs in GSL mats, 4 encoded homologs of PSII protein complexes (PufLMH) associated with anoxygenic phototrophs; none of the MAGs encoded homologs of the other PSII complexes Psc or Psh associated with anoxygenic phototrophic green sulfur bacteria, heliobacteria, and Acidobacteria (Table 2). However, only two of these 4 MAGs (Pelagibaca [bin 4] and Roseibaca [bin 7]) encoded COR and/or DPOR, while the two Wenzhouxiangella-affiliated MAGs lacked homologs of COR and DPOR. The type strain of Wenzhouxiangella is a facultative heterotroph, and available genome sequences from this and other closely related strains do not reveal homologs of PufLMH or other photosynthetic gene cluster (PGC) homologs (50). Among available genomes, the PufLMH in the Wenzhouxiangella sediminis bins were most closely related to homologs among other Alphaproteobacteria. Since several recent studies suggest that PGC genes, including those encoding Puf or Bch biosynthesis genes, can be encoded on extrachromosomal elements, including plasmids (as reviewed in reference 51), unbinned contigs were examined for genes coding for Bch biosynthesis proteins that might be attributable to W. sediminis. However, a BLASTp search failed to identify homologs of BchLNBXYZ related to Alphaproteobacteria among unbinned contigs, discounting the probability that these proteins were unbinned or were present on extrachromosomal elements in W. sediminis. In the absence of an apparent ability to synthesize Bch, the function of these PS proteins is unclear. Nonetheless, these data suggest an ability to harvest light energy in 17 of the 18 dominant MAGs, with 15 of these being from putative aerobic heterotrophic bacterial taxa. This metabolism was previously termed aerobic photoheterotrophy (APH) (44).
The elevated abundance of putative APH in GSL mats suggests that this physiological strategy imparts a selective advantage for microbial inhabitants. To begin to evaluate this hypothesis, a comparison of the abundance of key proteins that are involved in light harvesting was conducted among the GSL metagenome and other mat system metagenomes (Fig. 4). Included in this comparison is Mushroom Spring, Yellowstone National Park, Wyoming, since it is among the most comprehensively characterized phototrophic mat ecosystems (52, 53), and mat communities have been suggested to harbor an abundance of taxa dependent on light energy based on the distribution of genes coding for PS, Bch biosynthesis, and rhodopsin proteins (47, 48). Also included in the analysis are metagenomes from microbialite-forming mats from four locations, including Socompa, Argentina (54), Highborne Cay, Bahamas (55), Alchichica, Mexico (56), and Shark Bay, Australia (11). The distribution of homologs of phototrophy-related proteins in assembled metagenomes was determined first (Table 3), and this information was used to calculate their level of enrichment in the GSL community relative to that in the other communities (Fig. 4).
FIG 4.
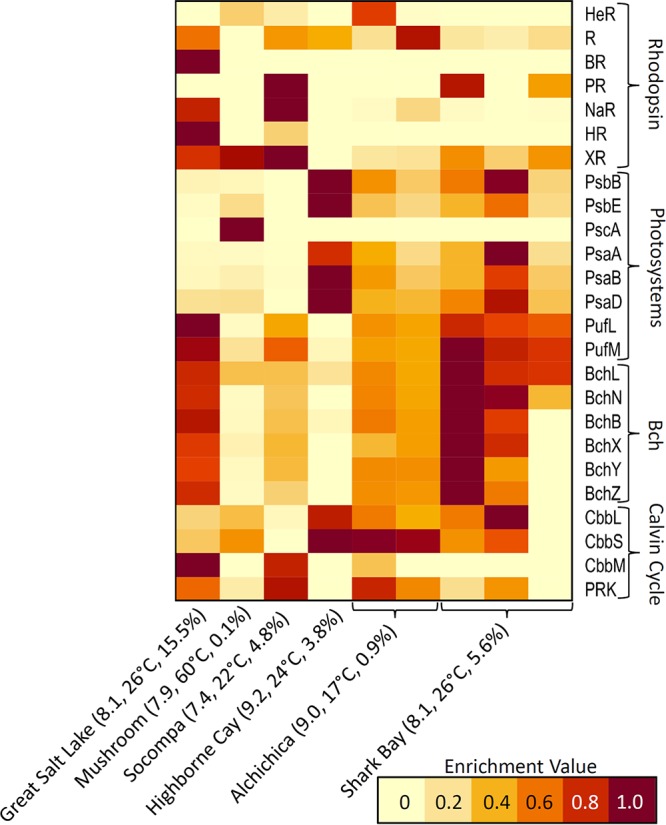
Enrichment of genes coding for specified proteins in microbial mats sampled from Great Salt Lake (GSL; this study), Mushroom Spring, Yellowstone National Park (48), Socompa, Argentina (54), Highborne Cay, Bahamas (55), Alchichica, Mexico (56), and Shark Bay, Australia (11). For Alchichica (AL) microbialite mat samples, the left column is for the north shore (ALN) metagenome, while the right column is for the average enrichment of homologs in three replicate metagenomes from the west shore (ALW) (56). For Shark Bay microbialite mat samples, the left column corresponds to a metagenome from postular A, the middle corresponds to a metagenome from coliform D, and the right column corresponds to a metagenome from smooth D (11). The pH, temperature, and salinity of the water column for GSL (this study), Mushroom Spring (48), Socompa (54), Highborne Cay (55), Alchichica (56), and Shark Bay (11) are indicated in parentheses. Abbreviations: HeR, heliorhodopsin; R, rhodopsin; BR, bacteriorhodopsin; PR, proteorhodopsin; NaR, sodium-transporting rhodopsin; HR halorhodopsin; XR, xanthorhodopsin; Bch, bacteriochlorophyll biosynthesis; PRK, phosphoribulokinase.
TABLE 3.
Relative enrichment of proteins in the metagenomes of specified communities
| Proteina | Enrichment (homologs/Gb of assembled sequence)b
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| GSL | Mushroom | Socompa | Highborne Cay | Alchichicac |
Shark Bay |
||||
| ALN1 | ALW | Postular A | Coliform D | Smooth D | |||||
| HeR | — | 4.5 | 2.3 | — | 11.4 | 0.6 | — | — | — |
| R | 16.9 | — | 13.8 | 11.8 | 5.7 | 24.7 | 4.9 | 4.0 | 6.4 |
| BR | 2.8 | — | — | — | — | — | — | — | — |
| PR | — | — | 2.3 | — | — | — | 2.0 | — | 1.1 |
| NaR | 28.2 | — | 34.5 | — | 1.9 | 8.5 | 2.0 | — | 1.1 |
| HR | 16.9 | — | 4.6 | — | — | — | — | — | — |
| XR | 42.4 | 49.5 | 55.3 | — | 9.5 | 11.0 | 28.5 | 15.9 | 27.6 |
| PsbB | 5.6 | 4.5 | — | 53.0 | 26.7 | 16.1 | 30.5 | 51.5 | 13.8 |
| PsbE | 2.8 | 13.5 | — | 58.9 | 20.0 | 14.2 | 22.6 | 35.7 | 13.8 |
| PsaA | 5.6 | 4.5 | 2.3 | 64.7 | 34.3 | 19.9 | 32.5 | 83.2 | 18.1 |
| PsaB | 5.6 | 9.0 | 2.3 | 70.6 | 33.3 | 21.9 | 27.5 | 51.5 | 21.2 |
| PsaD | 8.5 | 9.0 | — | 41.2 | 16.2 | 15.4 | 22.6 | 35.7 | 13.8 |
| PscA | — | 4.5 | — | — | — | — | — | — | — |
| PshA | — | — | — | — | — | — | — | — | — |
| PufL | 79.1 | 4.5 | 34.5 | — | 40.0 | 35.4 | 63.0 | 55.5 | 52.0 |
| PufM | 62.1 | 13.5 | 43.8 | 5.9 | 31.4 | 29.2 | 67.9 | 55.5 | 51.0 |
| BchL | 73.4 | 31.5 | 32.2 | 17.7 | 49.5 | 39.3 | 92.5 | 71.3 | 70.1 |
| BchN | 62.1 | 4.5 | 25.3 | — | 42.9 | 34.4 | 78.7 | 75.3 | 29.7 |
| BchB | 56.5 | 4.5 | 23.0 | 5.9 | 38.1 | 31.0 | 65.9 | 47.6 | — |
| BchX | 59.3 | 9.0 | 29.9 | — | 30.5 | 37.8 | 80.7 | 63.4 | — |
| BchY | 53.7 | 4.5 | 27.6 | — | 39.0 | 38.3 | 74.8 | 35.7 | — |
| BchZ | 59.3 | 4.5 | 20.7 | — | 39.0 | 36.6 | 75.7 | 43.6 | — |
| CbbL | 16.9 | 22.5 | 4.6 | 53.0 | 36.2 | 25.5 | 36.4 | 63.4 | — |
| CbbS | 5.6 | 9.0 | — | 17.7 | 17.1 | 16.3 | 8.9 | 11.9 | — |
| CbbM | 5.6 | — | 4.6 | — | 1.9 | — | — | — | — |
| PRK | 19.8 | 4.5 | 27.6 | — | 25.7 | 16.9 | 6.9 | 15.9 | — |
HeR, heliorhodopsin; R, rhodopsin; BR, bacteriorhodopsin; PR, proteorhodopsin; NaR, sodium-transporting rhodopsin; HR halorhodopsin; XR, xanthorhodopsin; PRK, phosphoribulokinase.
—, no enrichment relative to the average abundance of homologs.
Alchichica (AL) microbialite mat metagenomes from the north shore (ALN) and the west shore (ALW); the average enrichment of protein homologs in three replicate metagenomes is presented for ALW.
When homologs of all rhodopsin varieties are considered together, their abundance in GSL (107 homologs/Gb of assembled sequence) is nearly twice that of other communities, with the exception of mats from Socompa Lake (112 homologs) (Table 3; Fig. 4). Both GSL (15.5% salinity, 1,282-m elevation) and Socompa Lake (4.8% salinity, 3,570-m elevation) are hypersaline and are located at higher elevations than the other communities considered, aside from Mushroom Spring (∼2,700-m elevation). Thus, selective pressure to mitigate potentially deleterious effects associated with elevated salinity and to take advantage of increased solar radiation may lead to the assembly of resident communities including taxa that harbor rhodopsins. Potentially consistent with this hypothesis, homologs of proton-pumping XR and sodium-transporting NaR were abundant in mats from both GSL (42 and 28 homologs/Gbp, respectively) and Socompa Lake (55 and 35 homologs/Gbp, respectively) (Table 3; Fig. 4). The abundances of homologs of XR in GSL mats and Socompa Lake mats were similar to that of mats from Mushroom Spring, which is likely to be a high-radiation environment considering its close proximity (<10 m) to the high-radiation Octopus Spring (57). This finding is also consistent with a previous study that identified numerous rhodopsin homologs in Mushroom Spring, in particular, XR (47). Genes encoding HR and BR, albeit in low overall abundance (17 and 3 homologs/Gbp), were also enriched in GSL relative to that in the other considered communities.
The abundance of homologs of PS associated with oxygenic phototrophs (Psa and Psb) in GSL mats was similar to those from Socompa Lake and Mushroom Spring. The abundance of these proteins in these three communities was, however, low compared to that in most other communities (Table 3; Fig. 4). In contrast, the abundance of homologs of Puf (PS II), associated with anoxygenic phototrophs in GSL, exceeded all mats except those from Shark Bay. Mats from GSL and Shark Bay were also enriched (∼60 and 27 homologs/Gbp, respectively) in homologs of proteins involved in the synthesis of Bch. Importantly, the enrichment of PS and Bch biosynthesis proteins in GSL did not correlate with enrichment of homologs of type I ribulose bisphosphate carboxylase/oxygenase (RuBisCO; CbbSL) (∼11 homologs/Gbp) (Fig. 4). Likewise, the abundance of type II RuBisCO (CbbM), albeit enriched in the GSL metagenome, was low (∼6 homologs/Gbp) relative to that of Puf and Bch proteins (∼50 to 70 homologs/Gbp, respectively). Together, these results suggest that light harvesting via Puf and Bch is not necessarily associated with CO2 fixation in GSL. This form of APH is more specifically referred to as aerobic anoxygenic photosynthesis (AAPH) (58). Among the most abundant bins, Puf and Bch, but not CbbSL, were identified in MAGs associated with two aerobic heterotrophic populations in GSL, Pelagibaca (bin 4) and Roseibaca (bin 7), indicating the potential for AAPH in these taxa.
Collectively, these data indicate that phototrophic potential, whether in the form of oxygenic or anoxygenic photosynthesis, APH, or AAPH, is enriched in mat communities that form in hypersaline GSL (14.0 to 17.5% salinity), Socompa (4.8%), and Shark Bay (5.8%) environments. Photoheterotrophy via rhodopsin-based light harvesting has long been associated with enhanced fitness in a variety of environments, including those that are saline (59) and hypersaline (60). New data, however, indicate a much wider ecological distribution of rhodopsin-supported phototrophic organisms in aquatic systems (61), including those that are freshwater (62). Taking into account the phylogenetic, physiological, and ecological data, it has been suggested that rhodopsins can promote survival during nutrient stress, can stimulate growth and thus promote fitness, and can enhance metabolic efficiencies that together may influence community assembly (as reviewed in reference 61).
However, GSL is hypereutrophic (63) due to anthropogenic nutrient input, and this contributes to high levels of microbial production (21, 23). This suggests that nutrient stress is unlikely to be the primary characteristic responsible for the enrichment of rhodopsins. Rather, the potential benefits associated with capturing energy from high solar radiation at GSL is suggested to select for taxa that harbor rhodopsins to mitigate energetic stress imposed by hypersalinity. Furthermore, AAPH has been suggested to be a key adaptation allowing for enhanced fitness compared to that of nonphototrophic heterotrophic taxa in illuminated environments (64). Like rhodopsins, the potential enrichment of AAPH in GSL mat populations may enhance their fitness, potentially helping to explain the ∼700-km2 expanse of 1- to 2-cm thick phototrophic mats that form in shallow margins of the lake.
Conclusions.
The thick, widely distributed benthic microbial mats that form in GSL bind and stabilize sediment grains (20) and have been suggested to also promote the precipitation of carbonate minerals, including aragonite, that can act as cement during mat lithification (27, 28). Over extended periods of time, perhaps on the order of 2,500 to 20,000 years (24), numerous cycles of mat growth, sediment binding, and lithification lead to the formation of microbialites in GSL whose morphology largely reflects the average wave energy/direction and depositional setting of the local environment (20, 24, 25). The data presented herein indicate that the ability to harvest light either as an oxygenic phototroph, anoxygenic phototroph, APH, or AAPH is enriched in GSL mats and likely represents a key adaptation allowing for the establishment of robust phototrophic mats in this and perhaps other hypersaline locations. To this end, the unexpected abundance of and diversity in mechanisms of harvesting light energy observed in GSL mat populations likely functions to (i) minimize niche overlap among coinhabiting taxa thereby permitting their coexistence, (ii) provide a mechanism(s) to increase energy yield and osmotic balance during salt stress, and (iii) enhance the growth and fitness of taxa. Together, these physiological benefits promote the formation of robust mats that, in turn, influence the formation of microbialite structures that can be imprinted in the rock record.
MATERIALS AND METHODS
Description of sample locations.
Microbialites and associated photosynthetic mats were collected from seven locations in the south arm (SA) of GSL (Fig. 1; Table 1). Samples were collected from five locations near the northern end of Antelope Island (N 40.44.1172, W 111.52.288), Great Salt Lake (GSL), Utah, on 15 August 2016. This includes sites that are referred to as windward east (WE), windward west (WW), Bridger Bay (BB), Buffalo Point (BP), and ridges (R). Samples were also collected from two additional sites on the northeast side of Stansbury Island on 16 August 2016. These are referred to as Stansbury polygons (SP) and Stansbury interpolygons (SI).
Sample collection and in-field measurements.
Triplicate 5-g subsamples of microbial mat were collected from triplicate submerged microbialites at each location (9 subsamples collected for each sampling site). To sample only active mat communities, we selected well-hydrated microbialite structures that were submerged to a depth of ∼0.5 m. Replicate microbialites at each location were generally within 5 m of each other. Samples of microbial mat from the surface of each microbialite structure were collected with flame-sterilized spatulas and were placed in sterile 15-ml tubes. Tubes and their contents were immediately frozen on dry ice for transport to the laboratory at Montana State University for storage at −80°C. Concurrent with microbialite mat sample collection, geochemical and physical measurements were taken in overlaying waters. Temperature, pH, and conductivity were measured in situ in water directly above a single microbialite within each sampling site using a YSI Professional Plus (Pro Plus) multiparameter instrument (YSI Inc., Yellow Springs, OH). Salinity was measured on site on water sampled directly above each microbialite sampling location, using an AR200 digital refractometer (Reichert Instruments, Buffalo, NY).
DNA extraction, quantification, and 16S rRNA gene amplification and sequencing.
DNA was extracted from triplicate subsamples of microbial mat from three separate microbialites from each sampling site using the FastDNA Spin kit for soil (MP Biomedicals, Santa Ana, CA). The concentration of DNA in each extract was determined with the Qubit dsDNA HS assay kit (Molecular Probes). Equal volumes of each replicate extraction were combined for each of the three microbialites from each site. Consequently, a single DNA extract pool (combination of triplicate extracts) was generated for each of the three microbialites at each of the seven sites (21 extracts in total).
PCR amplification of 16S rRNA genes was conducted at the environmental sample preparation and sequencing facility at Argonne National Laboratory. Specifically, the V4 region of the 16S rRNA gene (515F-806R) was amplified with region-specific primers that included sequencer adapter sequences used in the Illumina MiSeq flow cell (65, 66). Each 25-μl PCR mixture contained 12 μl of DNA-free PCR water, 10 μl of 5 Prime HotMasterMix at 1× final concentration (QuantaBio, Beverly, MA), 1 μl Golay barcode-tagged forward primer (5 μM concentration, 200 pM final concentration), 1 μl reverse primer (5 μM concentration, 200 pM final concentration), and 50 ng of template DNA. PCR conditions were 94°C for 3 min, with 35 cycles at 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s, with a final extension of 10 min at 72°C. Amplicons were then quantified using PicoGreen (Invitrogen, Carlsbad, CA). Each product was pooled into a single tube, purified using the UltraClean PCR Clean-Up kit (Qiagen, Venlo, The Netherlands), and quantified using a fluorometer (Qubit, Invitrogen). After quantification, the pool was diluted to 2 nM, denatured, and then further diluted to a final concentration of 6.75 pM with a 10% PhiX spike for sequencing.
Sequence processing was performed with mothur (ver. 1.36.1) (67), as previously described (68), after merging the paired reads. A total of 1,939,054 paired-end 16S rRNA gene sequences were generated from sequencing on the MiSeq platform. Briefly, primers and adapters were removed from raw sequences and trimmed based on a Phred quality score of >25. The remaining sequences were trimmed to a minimum length of 250 bases and were subjected to a filtering step using the quality scores to remove sequences with any anomalous base calls. Unique sequences were aligned using the bacterial SILVA database (release 132) (69), and sequences were trimmed using defined start and end sites based on inclusion of 80% of the total sequences, as previously described (70). The resulting unique sequences were preclustered to mitigate amplification and sequencing errors, and chimeras were identified and removed using UCHIME (71). Operational taxonomic units (OTUs) were assigned at a sequence similarity of >97% using the nearest-neighbor method. The remaining sequences (1,784,275 quality-filtered 16S rRNA gene sequences) were randomly subsampled to normalize the total number of sequences in each sample library, and rarefaction analyses were used (at a depth of 38,000 sequences per sample, sampled in steps of 100 sequences) to compute the percent coverage of the predicted taxonomic richness for each library. The composition of each community was averaged for each triplicate microbialite sampling location, and this is what is presented. Representative sequences for each OTU were classified using the Bayesian classifier (72) and the Ribosomal Database Project as previously described (70).
Metagenomic sequencing.
Metagenomic sequence was generated from genomic DNA recovered from a single representative site, Bridger Bay (BB). DNA from mats from this site was chosen to represent the seven sampling locations based on 16S rRNA gene sequencing results and statistical analysis of these results, information that indicated it represented the “average” taxonomic composition of communities (see Results and Discussion). Equal volumes of triplicate genomic DNA extracts for each of the three microbialites at the BB sampling location were pooled and subjected to quantification as described above. Library preparation and sequencing were conducted at the genomics core facility at the University of Wisconsin—Madison using the paired-end (2 × 125 bp) Illumina HiSeq 2000 Platform. DNA fragments were prepared and quality controlled according to the manufacturer’s protocol using the Illumina Nextera DNA library preparation kit (Illumina, San Diego, CA, USA).
Quality-filtered reads were then assembled into contigs and subjected to metagenome-assembled genome (MAG) reconstructions, as described previously (73). Briefly, reads were quality trimmed from both ends, and Illumina adapters were removed using Trimmomatic v 0.35 (74). Quality-filtered reads were downsampled to improve assembly and were then assembled using MetaSPAdes (v.3.1.0) (75) over a range of k-mer lengths and using the default parameters. A final assembly was chosen for further analysis based on comparison of assembly statistics using the metaquast function of quast v.4.3 (76). Read depth was assessed by read mapping of nondownsampled quality-filtered reads to the final assembly with bowtie2 (77). Contigs greater than 2,500 bp from the assembly were binned by tetranucleotide word frequency distribution patterns and read-depth coverage profiles using MetaBAT (78).
Contigs from each bin were then assessed for quality, contamination, and completeness using CheckM v.1.0.5 (79) and curated using several different methods. Curation of the draft MAGs was conducted by first merging bins that represented putative partial complementary bins, while those likely representing multiple populations were separated using k-means cluster separation and removal of outlier contigs with tools implemented in the RefineM program v.0.0.23 (80). Outlier contigs were defined as being outside the distribution of 95% of each bin’s contig characteristic genomic profiles, as described previously (80). Curated bins were then reassessed using the same quality control metrics in CheckM described earlier.
After generating an initial draft MAG data set, the remaining unbinned contigs were then rebinned, evaluated for quality, and curated, as described above, thereby ultimately representing ∼82% of the total sequenced reads. The final complete moderate-to-high-quality MAG bin data set comprised 38 draft MAG bins.
The estimated genome size-corrected relative abundance of each population represented by the MAGs was then calculated from read-mapped coverage profiles in CheckM. Only those with estimated >1.0% relative abundance were subjected to further in-depth characterization. Gene predictions and annotations were conducted with Prokka v 1.11 (81). Annotations of proteins that were of specific interest in delineating the putative physiology of each MAG were further scrutinized using homology searches against the National Center for Biotechnology Information (NCBI) nonredundant (nr) database as reported in Data Set S2 in the supplemental material.
Comparative metagenomics analyses.
Since microbial mats are implicated in the formation of microbialite structures (as reviewed in reference 6), we compiled a metagenome data set combining data from other locations for comparative analysis with the GSL results. We selected sites based on the number and quality of available sequence reads. Raw sequence reads for microbialites from Alchichica Lake (56), Socompa Lake (54), Highborne Cay (55), and Shark Bay (11) were recovered from the NCBI SRA under accession numbers PRJNA315555, PRJNA317551, PRJNA197372, and PRJNA429237, respectively. In addition, raw sequence reads (NCBI SRA accession number PRJNA539623) from Mushroom Spring, Yellowstone National Park, Wyoming, were compiled for use in the comparative analysis due to evidence for enrichment of protein-coding genes involved in phototrophy in this community (47). The raw sequence reads of the compiled metagenomes were trimmed of adapters and quality-filtered using Trim Galore specifying default settings (https://www.bioinformatics.babraham.ac.uk/). Reads were then assembled with MetaSPAdes specifying a minimum contig length of 500 bp (75). The quality of assemblies was evaluated with MetaQUAST (82).
The assembled contigs of the reference metagenomes were subjected to gene prediction with Prokka, as described above. Custom databases were generated for each metagenome that comprised all predicted proteins for the assembled contigs. The databases, and GSL microbialite bins, were then subject to BLASTp analysis to identify homologs of key marker genes involved in phototrophy and carbon fixation. These included a variety of rhodopsins, reaction center proteins, proteins involved in bacteriochlorophyll biosynthesis, and proteins involved in the Calvin cycle (see Data Set S3). BLASTp results were filtered using an E value cutoff of 10e−30 and a query coverage of >50%. Owing to high similarity within protein families, BLASTp results were further filtered to include only protein homologs that exhibited >50% amino acid identity. Thus, the distribution of protein hits in each metagenome is likely to be a conservative estimate of protein family diversity and abundance.
The total number of hits for a protein identified by BLASTp analysis was normalized to the total number of giga base pairs of sequence in each assembled metagenome. This value was further normalized to the maximum abundance value for a specified protein as determined across all metagenomes. This yielded a scaled value (0 to 1) for the relative enrichment of each protein relative to the maximum enrichment observed in all metagenomes. This information was then used to generate a heat map with the R packages ggplots and rcolorbrewer.
Data availability.
Raw reads, quality scores, and mapping files for the 21 16S rRNA gene libraries have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject number PRJNA592167. The metagenomic assembly is available from the NCBI SRA archive under BioProject number PRJNA598870. The individual moderate-to-high-quality GSL microbialite MAGs (Data Set S1) discussed above are also available under this BioProject number and under individual biosample numbers SAMN13763250 to SAMN13763287. Access to the raw reads can be obtained through communication with the authors.
Supplementary Material
ACKNOWLEDGMENTS
This Precambrian Biosphere graduate class project was supported by the head of the Department of Microbiology and Immunology at Montana State University, Mark Jutila. The W.M. Keck Foundation provided support for metagenomic sequencing costs. E.S.B. is supported by the NASA Astrobiology Institute (grant number NNA15BB02A).
We thank Don Bryant for helpful discussions during the preparation of the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Walter MR, Buick R, Dunlop J. 1980. Stromatolites 3,400-3,500 Myr old from the North Pole area, Western Australia. Nature 284:443–445. doi: 10.1038/284443a0. [DOI] [Google Scholar]
- 2.Allwood AC, Walter MR, Kamber BS, Marshall CP, Burch IW. 2006. Stromatolite reef from the Early Archaean era of Australia. Nature 441:714–718. doi: 10.1038/nature04764. [DOI] [PubMed] [Google Scholar]
- 3.van Kranendonk MJ, Philippot P, Lepot K, Bodorkos S, Pirajno F. 2008. Geological setting of Earth’s oldest fossils in the ca. 3.5 Ga Dresser Formation, Pilbara Craton, Western Australia. Precambrian Res 167:93–124. doi: 10.1016/j.precamres.2008.07.003. [DOI] [Google Scholar]
- 4.Sugitani K, Mimura K, Takeuchi M, Lepot K, Ito S, Javaux E. 2015. Early evolution of large microorganisms with cytological complexity revealed by microanalyses of 3.4 Ga organic-walled microfossils. Geobiology 13:507–521. doi: 10.1111/gbi.12148. [DOI] [PubMed] [Google Scholar]
- 5.Burne RV, Moore LS. 1987. Microbialites; organosedimentary deposits of benthic microbial communities. Palaios 2:241–254. doi: 10.2307/3514674. [DOI] [Google Scholar]
- 6.Riding R. 2000. Microbial carbonates: the geological record of calcified bacterial-algal mats and biofilms. Sedimentology 47:179–214. doi: 10.1046/j.1365-3091.2000.00003.x. [DOI] [Google Scholar]
- 7.Laval B, Cady SL, Pollack JC, McKay CP, Bird JS, Grotzinger JP, Ford DC, Bohm HR. 2000. Modern freshwater microbialite analogues for ancient dendritic reef structures. Nature 407:626–629. doi: 10.1038/35036579. [DOI] [PubMed] [Google Scholar]
- 8.Osborne RH, Licari GR, Link MH. 1982. Modern lacustrine stromatolites, Walker Lake, Nevada. Sediment Geol 32:39–61. doi: 10.1016/0037-0738(82)90013-6. [DOI] [Google Scholar]
- 9.Logan BW. 1961. Cryptozoon and associate stromatolites from the recent, Shark Bay, Western Australia. J Geol 69:517–533. doi: 10.1086/626769. [DOI] [Google Scholar]
- 10.Pages A, Welsh DT, Teasdale PR, Grice K, Vacher M, Bennett WW, Visscher PT. 2014. Diel fluctuations in solute distributions and biogeochemical cycling in a hypersaline microbial mat from Shark Bay, WA. Mar Chem 167:102–112. doi: 10.1016/j.marchem.2014.05.003. [DOI] [Google Scholar]
- 11.Babilonia J, Conesa A, Casaburi G, Pereira C, Louyakis AS, Reid RP, Foster JS. 2018. Comparative metagenomics provides insight into the ecosystem functioning of the Shark Bay stromatolites. Front Microbiol 9:1359. doi: 10.3389/fmicb.2018.01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farías ME, Rascovan N, Toneatti DM, Albarracín VH, Flores MR, Poiré DG, Collavino MM, Aguilar OM, Vazquez MP, Polerecky L. 2013. The discovery of stromatolites developing at 3570 m above sea level in a high-altitude volcanic Lake Socompa, Argentinean Andes. PLoS One 8:e53497. doi: 10.1371/journal.pone.0053497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster JS, Green SJ, Ahrendt SR, Golubic S, Reid RP, Hetherington KL, Bebout L. 2009. Molecular and morphological characterization of cyanobacterial diversity in the stromatolites of Highborne Cay, Bahamas. ISME J 3:573–587. doi: 10.1038/ismej.2008.129. [DOI] [PubMed] [Google Scholar]
- 14.Myshrall KL, Mobberley JM, Green SJ, Visscher PT, Havemann SA, Reid RP, Foster JS. 2010. Biogeochemical cycling and microbial diversity in the thrombolitic microbialites of Highborne Cay, Bahamas. Geobiology 8:337–354. doi: 10.1111/j.1472-4669.2010.00245.x. [DOI] [PubMed] [Google Scholar]
- 15.Russell JA, Brady AL, Cardman Z, Slater GF, Lim DSS, Biddle JF. 2014. Prokaryote populations of extant microbialites along a depth gradient in Pavilion Lake, British Columbia, Canada. Geobiology 12:250–264. doi: 10.1111/gbi.12082. [DOI] [PubMed] [Google Scholar]
- 16.Couradeau E, Benzerara K, Moreira D, Gérard E, Kaźmierczak J, Tavera R, López-García P. 2011. Prokaryotic and eukaryotic community structure in field and cultured microbialites from the alkaline lake Alchichica (Mexico). PLoS One 6:e28767. doi: 10.1371/journal.pone.0028767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eardley AJ. 1939. Sediments of Great Salt Lake, Utah. Am Assoc Pet Geol Bull 22:1089–1090. doi: 10.1306/3D9330DE-16B1-11D7-8645000102C1865D. [DOI] [Google Scholar]
- 18.Baskin R, Wright V, Driscoll N, Kent G, and, Hepner C. 2012. Microbialite bioherms in Great Salt Lake, Utah: influence of active tectonics and anthropogenic effects. American Association of Petroleum Geologists Hedburg Conference, Houston, TX, 4 to 8 June 2012. [Google Scholar]
- 19.Baskin RL. 2014. Occurrence and spatial distribution of microbial bioherms in Great Salt Lake, Utah. The University of Utah, Salt Lake City, UT: MSc thesis. [Google Scholar]
- 20.Vanden Berg M. 2019. Domes, rings, ridges, and polygons: characteristics of microbialites from Utah’s Great Salt Lake. Sed Record 17:4–10. doi: 10.2110/sedred.2019.1.4. [DOI] [Google Scholar]
- 21.Wurtsbaugh WA, Gardberg J, Izdepski C. 2011. Biostrome communities and mercury and selenium bioaccumulation in the Great Salt Lake (Utah, USA). Sci Total Environ 409:4425–4434. doi: 10.1016/j.scitotenv.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 22.Lindsay MR, Anderson C, Fox N, Scofield G, Allen J, Anderson E, Bueter L, Poudel S, Sutherland K, Munson-McGee JH, Van Nostrand JD, Zhou J, Spear JR, Baxter BK, Lageson DR, Boyd ES. 2017. Microbialite response to an anthropogenic salinity gradient in Great Salt Lake, Utah. Geobiology 15:131–145. doi: 10.1111/gbi.12201. [DOI] [PubMed] [Google Scholar]
- 23.Lindsay MR, Johnston RE, Baxter BK, Boyd ES. 2019. Effects of salinity on microbialite-associated production in Great Salt Lake, Utah. Bull Ecol Soc Am 100:e01513. doi: 10.1002/bes2.1513. [DOI] [PubMed] [Google Scholar]
- 24.Bouton A, Vennin E, Boulle J, Pace A, Bourillot R, Thomazo C, Brayard A, Désaubliaux G, Goslar T, Yokoyama Y, Dupraz C, Visscher PT. 2016. Linking the distribution of microbial deposits from the Great Salt Lake (Utah, USA) to tectonic and climatic processes. Biogeosciences 13:5511–5526. doi: 10.5194/bg-13-5511-2016. [DOI] [Google Scholar]
- 25.Carozzi AV. 1962. Observations on algal biostromes in the Great Salt Lake, Utah. J Geol 70:246–252. doi: 10.1086/626814. [DOI] [Google Scholar]
- 26.Chidsey TC Jr, Vanden Berg MD, Eby DE. 2015. Petrography and characterization of microbial carbonates and associated facies from modern Great Salt Lake and Uinta Basin’s Eocene Green River Formation in Utah, USA, p 261–286. In Bosence DWJ, Gibbons KA, Le Heron DP, Morgan WA, Pritchard T, Vining BA (ed), Microbial carbonates in space and time: implications for global exploration and production. vol 418 Geological Society, London, United Kingdom. doi: 10.1144/SP418.6. [DOI] [Google Scholar]
- 27.Dunham EC, Fones EM, Lindsay MR, Steuer C, Fox N, Willis M, Walsh A, Colman DR, Baxter BK, Lageson DR, Mogk D, Rupke A, Xu H, Boyd ES. 2020. An ecological perspective on dolomite formation in Great Salt Lake, Utah. Front Earth Sci (Lausanne) 8:24. doi: 10.3389/feart.2020.00024. [DOI] [Google Scholar]
- 28.Pace A, Bourillot R, Bouton A, Vennin E, Galaup S, Bundeleva I, Patrier P, Dupraz C, Thomazo C, Sansjofre P, Yokoyama Y, Franceschi M, Anguy Y, Pigot L, Virgone A, Visscher PT. 2016. Microbial and diagenetic steps leading to the mineralisation of Great Salt Lake microbialites. Sci Rep 6:31495. doi: 10.1038/srep31495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanden Berg M, Lindsay MR, Boyd ES, Chidsey TC Jr, Eby DE. 2017. The microbialites of Utah’s Great Salt Lake: geology vs. biology. Abstr Geol Soc Am, paper 119-3. doi: 10.1130/abs/2017AM-298166. [DOI] [Google Scholar]
- 30.Lu DC, Xia J, Dunlap CA, Rooney AP, Du ZJ. 2017. Gracilimonas halophila sp. nov., isolated from a marine solar saltern. Int J Syst Evol Microbiol 67:3251–3255. doi: 10.1099/ijsem.0.002093. [DOI] [PubMed] [Google Scholar]
- 31.White IIR, Power I, Dipple G, Southam G, Suttle C. 2015. Metagenomic analysis reveals that modern microbialites and polar microbial mats have similar taxonomic and functional potential. Front Microbiol 6:966. doi: 10.3389/fmicb.2015.00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, The Genome Standards Consortium, Lapidus A, Meyer M, Yilmaz P, Parks DH, Eren AM, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen JP, Williams JC. 1998. Photosynthetic reaction centers. FEBS Lett 438:5–9. doi: 10.1016/S0014-5793(98)01245-9. [DOI] [PubMed] [Google Scholar]
- 34.Shih PM, Wu D, Latifi A, Axen SD, Fewer DP, Talla E, Calteau A, Cai F, Tandeau de Marsac N, Rippka R, Herdman M, Sivonen K, Coursin T, Laurent T, Goodwin L, Nolan M, Davenport KW, Han CS, Rubin EM, Eisen JA, Woyke T, Gugger M, Kerfeld CA. 2013. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc Natl Acad Sci U S A 110:1053–1058. doi: 10.1073/pnas.1217107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samylina OS, Namsaraev ZB, Grouzdev DS, Slobodova NV, Zelenev VV, Borisenko GV, Sorokin DY. 2019. The patterns of nitrogen fixation in haloalkaliphilic phototrophic communities of Kulunda Steppe soda lakes (Altai, Russia). FEMS Microbiol Ecol 95:fiz174. doi: 10.1093/femsec/fiz174. [DOI] [PubMed] [Google Scholar]
- 36.Lindsay MR, Dunham EC, Boyd ES. 2020. Microbialites of Great Salt Lake In Baxter BK, Butler JK (ed), Great Salt Lake: biology of a terminal lake in the age of change. Springer, New York, NY. [Google Scholar]
- 37.Hamilton TL, Bovee RJ, Thiel V, Sattin SR, Mohr W, Schaperdoth I, Vogl K, Gilhooly IIW, Lyons TW, Tomsho LP, Schuster SC, Overmann J, Bryant DA, Pearson A, Macalady JL. 2014. Coupled reductive and oxidative sulfur cycling in the phototrophic plate of a meromictic lake. Geobiology 12:451–468. doi: 10.1111/gbi.12092. [DOI] [PubMed] [Google Scholar]
- 38.Mogany T, Swalaha FM, Allam M, Mtshali PS, Ismail A, Kumari S, Bux F. 2018. Phenotypic and genotypic characterisation of an unique indigenous hypersaline unicellular cyanobacterium, Euhalothece sp. Microbiol Res 211:47–56. doi: 10.1016/j.micres.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 39.De Philippis R, Margheri MC, Pelosi E, Ventura S. 1993. Exopolysaccharide production by a unicellular cyanobacterium isolated from a hypersaline habitat. J Appl Phycol 5:387–394. doi: 10.1007/BF02182731. [DOI] [Google Scholar]
- 40.De Philippis R, Margheri MC, Materassi R, Vincenzini M. 1998. Potential of unicellular cyanobacteria from saline environments as exopolysaccharide producers. Appl Environ Microbiol 64:1130–1132. doi: 10.1128/AEM.64.3.1130-1132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balashov SP, Imasheva ES, Boichenko VA, Antón J, Wang JM, Lanyi JK. 2005. Xanthorhodopsin: a proton pump with a light-harvesting carotenoid antenna. Science 309:2061–2064. doi: 10.1126/science.1118046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schobert B, Lanyi JK. 1982. Halorhodopsin is a light-driven chloride pump. J Biol Chem 257:10306–10313. [PubMed] [Google Scholar]
- 43.Inoue K, Ono H, Abe-Yoshizumi R, Yoshizawa S, Ito H, Kogure K, Kandori H. 2013. A light-driven sodium ion pump in marine bacteria. Nat Commun 4:1678. doi: 10.1038/ncomms2689. [DOI] [PubMed] [Google Scholar]
- 44.Bryant DA, Frigaard NU. 2006. Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol 14:488–496. doi: 10.1016/j.tim.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Smith JJ, Tian L, Dellapenna D. 2009. The evolution and function of carotenoid hydroxylases in Arabidopsis. Plant Cell Physiol 50:463–479. doi: 10.1093/pcp/pcp005. [DOI] [PubMed] [Google Scholar]
- 46.Ruch S, Beyer P, Ernst H, Al-Babili S. 2005. Retinal biosynthesis in Eubacteria: in vitro characterization of a novel carotenoid oxygenase from Synechocystis sp. PCC 6803. Mol Microbiol 55:1015–1024. doi: 10.1111/j.1365-2958.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- 47.Thiel V, Hügler M, Ward DM, Bryant DA. 2017. The dark side of the Mushroom Spring microbial mat: life in the shadow of chlorophototrophs. II. Metabolic functions of abundant community members predicted from metagenomic analyses. Front Microbiol 8:943. doi: 10.3389/fmicb.2017.00943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiel V, Wood JM, Olsen MT, Tank M, Klatt CG, Ward DM, Bryant DA. 2016. The dark side of the Mushroom Spring microbial mat: life in the shadow of chlorophototrophs. I. Microbial diversity based on 16S rRNA gene amplicons and metagenomic sequencing. Front Microbiol 7:919. doi: 10.3389/fmicb.2016.00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chew AG, Bryant DA. 2007. Chlorophyll biosynthesis in bacteria: the origins of structural and functional diversity. Annu Rev Microbiol 61:113–129. doi: 10.1146/annurev.micro.61.080706.093242. [DOI] [PubMed] [Google Scholar]
- 50.Guo LY, Dunlap CA, Rooney AP, Chen GJ, Du ZJ. 2016. Wenzhouxiangella sediminis sp. nov., isolated from coastal sediment. Int J Syst Evol Microbiol 66:4575–4579. doi: 10.1099/ijsem.0.001393. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Zheng Q, Lin W, Jiao N. 2019. Characteristics and evolutionary analysis of photosynthetic gene clusters on extrachromosomal replicons: from streamlined plasmids to chromids. mSystems 4:e00358-19. doi: 10.1128/mSystems.00358-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward DM, Ferris MJ, Nold SC, Bateson MM. 1998. A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol Mol Biol Rev 62:1353–1370. doi: 10.1128/MMBR.62.4.1353-1370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klatt C, Inskeep W, Herrgard M, Jay Z, Rusch D, Tringe S, Parenteau MN, Ward D, Boomer S, Bryant D, Miller S. 2013. Community structure and function of high-temperature chlorophototrophic microbial mats inhabiting diverse geothermal environments. Front Microbiol 4:106. doi: 10.3389/fmicb.2013.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurth D, Amadio A, Ordoñez OF, Albarracín VH, Gärtner W, Farías ME. 2017. Arsenic metabolism in high altitude modern stromatolites revealed by metagenomic analysis. Sci Rep 7:1024. doi: 10.1038/s41598-017-00896-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khodadad CLM, Foster JS. 2012. Metagenomic and metabolic profiling of nonlithifying and lithifying stromatolitic mats of Highborne Cay, The Bahamas. PLoS One 7:e38229. doi: 10.1371/journal.pone.0038229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saghaï A, Zivanovic Y, Moreira D, Benzerara K, Bertolino P, Ragon M, Tavera R, López-Archilla AI, López-García P. 2016. Comparative metagenomics unveils functions and genome features of microbialite-associated communities along a depth gradient. Environ Microbiol 18:4990–5004. doi: 10.1111/1462-2920.13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller SR, Wingard CE, Castenholz RW. 1998. Effects of visible light and UV radiation on photosynthesis in a population of a hot spring cyanobacterium, a Synechococcus sp., subjected to high-temperature stress. Appl Environ Microbiol 64:3893–3899. doi: 10.1128/AEM.64.10.3893-3899.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yurkov VV, Beatty JT. 1998. Aerobic anoxygenic phototrophic bacteria. Microbiol Mol Biol Rev 62:695–724. doi: 10.1128/MMBR.62.3.695-724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Béjà O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, Spudich EN, DeLong EF. 2000. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 60.Oesterhelt D, Stoeckenius W. 1971. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat New Biol 233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 61.Pinhassi J, DeLong EF, Béjà O, González JM, Pedrós-Alió C. 2016. Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol Mol Biol Rev 80:929–954. doi: 10.1128/MMBR.00003-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atamna-Ismaeel N, Sabehi G, Sharon I, Witzel K-P, Labrenz M, Jürgens K, Barkay T, Stomp M, Huisman J, Beja O. 2008. Widespread distribution of proteorhodopsins in freshwater and brackish ecosystems. ISME J 2:656–662. doi: 10.1038/ismej.2008.27. [DOI] [PubMed] [Google Scholar]
- 63.Belovsky GE, Stephens D, Perschon C, Birdsey P, Paul D, Naftz D, Baskin R, Larson C, Mellison C, Luft J, Mosley R, Mahon H, Van Leeuwen J, Allen DV. 2011. The Great Salt Lake ecosystem (Utah, USA): long term data and a structural equation approach. Ecosphere 2:art33. doi: 10.1890/ES10-00091.1. [DOI] [Google Scholar]
- 64.Koblížek M. 2015. Ecology of aerobic anoxygenic phototrophs in aquatic environments. FEMS Microbiol Rev 39:854–870. doi: 10.1093/femsre/fuv032. [DOI] [PubMed] [Google Scholar]
- 65.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamilton TL, Peters JW, Skidmore ML, Boyd ES. 2013. Molecular evidence for an active endogenous microbiome beneath glacial ice. ISME J 7:1402–1412. doi: 10.1038/ismej.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lindsay MR, Amenabar MJ, Fecteau KM, Debes RVI, Fernandes Martins MC, Fristad KE, Xu H, Hoehler TM, Shock EL, Boyd ES. 2018. Subsurface processes influence oxidant availability and chemoautotrophic hydrogen metabolism in Yellowstone hot springs. Geobiology 16:674–692. doi: 10.1111/gbi.12308. [DOI] [PubMed] [Google Scholar]
- 71.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Payne D, Dunham EC, Mohr E, Miller I, Arnold A, Erickson R, Fones EM, Lindsay MR, Colman DR, Boyd ES. 2019. Geologic legacy spanning >90 years explains unique Yellowstone hot spring geochemistry and biodiversity. Environ Microbiol 21:4180–4195. doi: 10.1111/1462-2920.14775. [DOI] [PubMed] [Google Scholar]
- 74.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. 2017. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol 2:1533–1542. doi: 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 81.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 82.Mikheenko A, Saveliev V, Gurevich A. 2016. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32:1088–1090. doi: 10.1093/bioinformatics/btv697. [DOI] [PubMed] [Google Scholar]
- 83.Baskin RL, Allen DV. 2005. Bathymetric map of the south part of Great Salt Lake, Utah, 2005. U.S. Geological Survey scientific investigations, map 2894 USGS, Reston, VA. doi: 10.3133/sim2894. [DOI] [Google Scholar]
- 84.Baskin RL, Turner J. 2006. Bathymetric map of the north part of Great Salt Lake, Utah, 2006. U.S. Geological Survey scientific investigations, map 2954 USGS, Reston, VA. doi: 10.3133/sim2954. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads, quality scores, and mapping files for the 21 16S rRNA gene libraries have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject number PRJNA592167. The metagenomic assembly is available from the NCBI SRA archive under BioProject number PRJNA598870. The individual moderate-to-high-quality GSL microbialite MAGs (Data Set S1) discussed above are also available under this BioProject number and under individual biosample numbers SAMN13763250 to SAMN13763287. Access to the raw reads can be obtained through communication with the authors.