

Abstract
Fanconi anemia (FA) is a chromosome instability syndrome characterized by increased cancer predisposition. Specifically, the FA pathway functions to protect genome stability during DNA replication. The central FA pathway protein, FANCD2, locates to stalled replication forks and recruits homologous recombination (HR) factors such as CtBP interacting protein (CtIP) to promote replication fork restart while suppressing new origin firing. Here, we identify alpha-thalassemia retardation syndrome X-linked (ATRX) as a novel physical and functional interaction partner of FANCD2. ATRX is a chromatin remodeler that forms a complex with Death domain-associated protein 6 (DAXX) to deposit the histone variant H3.3 into specific genomic regions. Intriguingly, ATRX was recently implicated in replication fork recovery; however, the underlying mechanism(s) remained incompletely understood. Our findings demonstrate that ATRX forms a constitutive protein complex with FANCD2 and protects FANCD2 from proteasomal degradation. ATRX and FANCD2 localize to stalled replication forks where they cooperate to recruit CtIP and promote MRE11 exonuclease-dependent fork restart while suppressing the firing of new replication origins. Remarkably, replication restart requires the concerted histone H3 chaperone activities of ATRX/DAXX and FANCD2, demonstrating that coordinated histone H3 variant deposition is a crucial event during the reinitiation of replicative DNA synthesis. Lastly, ATRX also cooperates with FANCD2 to promote the HR-dependent repair of directly induced DNA double-stranded breaks. We propose that ATRX is a novel functional partner of FANCD2 to promote histone deposition-dependent HR mechanisms in S-phase.
Introduction
Fanconi anemia (FA) is an inherited genome instability disorder characterized by bone marrow failure and a strong predisposition to cancer, predominantly leukemia and squamous cell carcinoma (1, 2). Currently, 22 different FA genes have been identified and mutations in any one of them are sufficient to cause FA (2, 3). FA patient cells are highly sensitive to interstrand crosslink (ICL)-inducing agents such as mitomycin C (MMC). Moreover, FA cells exhibit spontaneous chromosomal aberrations that are further exacerbated upon treatment with replication inhibiting agents such as hydroxyurea (HU) or aphidicolin (APH) (4–7).
The FA ICL repair pathway operates predominantly in S-phase and consists of an upstream FA core complex (eight proteins), a central FANCD2-FANCI protein heterodimer and several downstream proteins including breast cancer-associated protein 2 (BRCA2/FANCD1) and the radiation sensitive 51 (RAD51/FANCR) recombinase (1, 8). Upon ICL detection, the FA core complex and the FANCD2-FANCI heterodimer are recruited to chromatin, followed by FA core complex-mediated FANCD2 monoubiquitination (FANCD2Ub) (9–11). FANCD2Ub then coordinates downstream FA nucleases to mediate incisions at the ICL, followed by homologous recombination (HR)-dependent repair of the newly generated DNA double-strand breaks (DSBs) (2, 12). HR repair of such DSBs requires the concerted actions of FA and non-FA proteins, such as BRCA2 and CtIP, respectively.
Recent studies have discovered novel roles for FANCD2 during the replication stress response. Upon replication fork stalling in the presence of HU or APH, FANCD2 is recruited to the stalled forks where it performs two roles. First, it protects the stalled replication forks from nucleolytic degradation (4, 13, 14). FANCD2 fulfills this role in concert with the upstream FA core complex and several downstream FA proteins such as BRCA2 (4, 15). Second, FANCD2 promotes the restart of the stalled replication forks while simultaneously suppressing the firing of new replication origins (13). Importantly, this fork recovery function of FANCD2 does not depend on FANCD2Ub formation. Instead, the non-ubiquitinated FANCD2 isoform binds stalled replication forks and cooperates with downstream FA proteins as well as non-FA HR factors such as CtIP or the meiotic recombination 11-like (MRE11) nuclease to promote fork restart (14, 16, 17). However, despite these advances in our knowledge, the molecular mechanisms that allow FANCD2 to promote replication fork recovery remain incompletely understood.
In the current study, we identify the chromatin remodeler ATRX as a novel physical and functional interactor of FANCD2. ATRX interacts directly with DAXX (18, 19) to form a histone chaperone complex. This complex deposits a specific histone H3 variant, H3.3, onto pericentric or telomeric chromatin regions to promote heterochromatin formation (20, 21). Intriguingly, recent studies reported a new function for ATRX in the protection of HU-stalled replication forks and the promotion of replication fork restart (22–24).
To investigate a putative functional cross talk between human FANCD2 and ATRX during the replication stress response, we generated human ATRX−/0, FANCD2−/− and ATRX−/0:FANCD2−/− knockout cells in an isogenic background (the human HCT116 cell line). We provide evidence that ATRX interacts constitutively with FANCD2 and stabilizes FANCD2 protein levels. In addition, ATRX and FANCD2 cooperate with the MRE11 exonuclease to promote the HR-dependent restart of HU-stalled replication forks; moreover, ATRX and FANCD2 are both required to recruit the key replication restart factor, CtIP. Strikingly, ATRX’s own histone H3.3 chaperone activity and FANCD2’s recently described histone H3 depositing activity (25, 26) are both required for efficient fork restart. Our study introduces the ATRX/DAXX histone chaperone as a novel FA-associated factor in the context of DNA replication stress and functionally connects the FA pathway to processes of nucleosome remodeling during replication fork recovery.
Results
ATRX interacts constitutively with FANCD2 and promotes FANCD2 protein stability
We initially identified ATRX as a novel FANCD2 interactor in Xenopus laevis egg extracts (data not shown). To confirm this interaction, we immunoprecipitated (IPed) ATRX from human wild type-like cells (PD20 + 3xFLAG-FANCD2, an FA-D2 patient-derived cell line complemented with triple FLAG-tagged, wild-type FANCD2) that were untreated or HU-treated to cause replication fork stalling and induce FANCD2 monoubiquitination. FANCD2 co-immunoprecipitated (co-IPed) with ATRX in the absence or presence of HU (Fig. 1A, compare lanes 4 and 5). Vice versa, ATRX co-IPed with FANCD2 in unperturbed conditions (Fig. 1B). Thus, the complex formation between ATRX and FANCD2 is constitutive and does not require DNA damage-induced FANCD2Ub formation. We also confirmed that both ATRX and FANCD2 interacted with the Nijmegen breakage syndrome 1 (NBS1) subunit of the MRE11-RAD50-NBS1 (MRN) nuclease complex in these cells (Supplementary Material, Fig. S1A and B), in agreement with previous reports of either an MRN–ATRX interaction (22, 23) or an MRN–FANCD2 interaction (27). Thus, ATRX is a novel FANCD2 interactor and may participate in a common protein complex with FANCD2 and MRN that forms independently of DNA damage induction.
Figure 1.
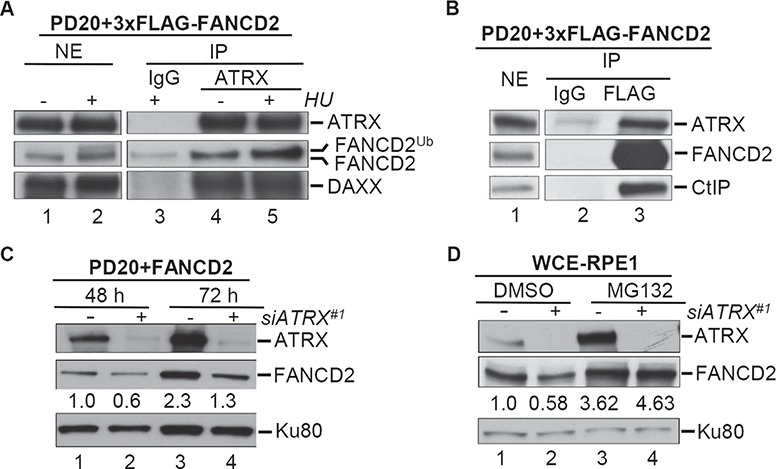
FANCD2 interacts with ATRX and requires ATRX to maintain protein stability. Immunoprecipitation (IP) experiments were performed from nuclear extracts (NEs) of PD20 + 3xFLAG-FANCD2 (wild-type) cells. (A) FANCD2 co-IPs with ATRX. NEs were either untreated or treated with 2 mm HU for 24 h (lanes 1 and 2) and subjected to IP with rabbit IgG (lane 3) or an anti-ATRX antibody (lanes 4 and 5). NE and IP samples were analyzed for the presence of ATRX, FANCD2 and DAXX (positive co-IP control for ATRX). (B) ATRX co-IPs with FANCD2. NEs (lane 1) were subjected to IP with mouse IgG (lane 2) or an anti-FLAG antibody (lane 3). NE and IP samples were analyzed for the presence of ATRX, FANCD2 and CtIP (positive co-IP control for FANCD2). (C–E) ATRX protects FANCD2 protein stability. (C) WCEs were prepared from PD20 + D2 cells that had been treated with control siRNA (lanes 1 and 3) or ATRX-#1 siRNA (lanes 2 and 4) for the indicated time points and analyzed for the presence of ATRX and FANCD2. Ku-80, loading control. (D) WCEs were prepared from human hTERT-RPE1 cells that had been treated with control siRNA (lanes 1 and 3) or ATRX-#1 siRNA (lanes 2 and 4) for 72 h. At 67 h, cells were additionally supplemented with DMSO (lanes 1 and 2) or 10 μm MG132 (lanes 3 and 4). WCEs were then analyzed for ATRX and FANCD2. Ku-80, loading control. Immunoblot signals for FANCD2 were analyzed by densitometry and normalized against Ku80 signals using ImageJ. The relative FANCD2 protein level values are provided underneath each corresponding lane.
FANCD2’s protein stability relies on its interaction partners CtIP and the MRN complex (17, 27). To test if FANCD2 and ATRX have a comparable relationship, we treated PD20 + D2 cells with control siRNA or two different siRNAs against ATRX (siATRX#1 and siATRX#2). ATRX depletion with either siRNA caused an approximately 40% reduction in FANCD2 protein levels, in contrast to the control cells (Fig. 1C and Supplementary Material, Fig. S1C). To confirm this result in another cell line, we treated hTERT immortalized retinal pigment epithelial-1 (RPE-1) cells with control siRNA or siATRX#1. Knockdown with siATRX#1 caused a significant reduction in FANCD2 protein levels in contrast to the control cells (Fig. 1D, compare lanes 1 and 2). Importantly, treatment with the proteasome inhibitor MG132 restored FANCD2 protein levels in ATRX-depleted RPE-1 cells (Fig. 1D, compare lanes 2 and 4). Thus, ATRX supports FANCD2 protein stability, likely by protecting it from proteasomal degradation.
Generation of human isogenic ATRX−/0, FANCD2−/− and ATRX−/0:FANCD2−/− cell lines
To dissect putative functional interactions between ATRX and FANCD2, we generated isogenic ATRX knockout (ATRX−/0), FANCD2 knockout (D2−/−) and ATRX/FANCD2 double knockout (ATRX−/0:FANCD2−/−; hereafter named AD2DKO) cells using HCT116 as the parental cell line. Note that ATRX is located on the X chromosome, and since HCT116 cells are derived from a male, this makes the ATRX gene hemizygous. To obtain the somatic knockout cell lines, we used a combination of clustered regularly interspersed palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) and recombinant adeno-associated virus (rAAV)-mediated gene targeting techniques. The generation of the HCT116 ATRX−/0 and D2−/− cells has been described (28, 29). To generate the AD2DKO cell line, we used essentially the same strategy (28) to target exon 9 of ATRX in the D2−/− cells (Supplementary Material, Fig. S1D). Western blot analysis of whole cell extracts (WCEs) prepared from WT, ATRX−0, D2−/− and AD2DKO cells confirmed that the genetically null cells lacked protein expression of ATRX, FANCD2 or both (Fig. 2A). In agreement with the siATRX knockdown experiments described previously, the ATRX−/0 cells exhibited reduced FANCD2 protein expression levels in contrast to the WT cells (Fig. 2A, compare lanes 1 with 3, and 5 with 7). In contrast, ATRX protein levels were comparable in wild-type and D2−/− cells, demonstrating that ATRX protein stability does not rely on FANCD2. Importantly, despite the reduced FANCD2 protein levels in the ATRX−/0 cells, the HU-induced FANCD2 monoubiquitination was robustly triggered in WT and ATRX−/0 cells (Fig. 2A, compare lanes 5 and 7), indicating that ATRX is dispensable for the DNA damage-inducible FANCD2 monoubiquitination.
Figure 2.
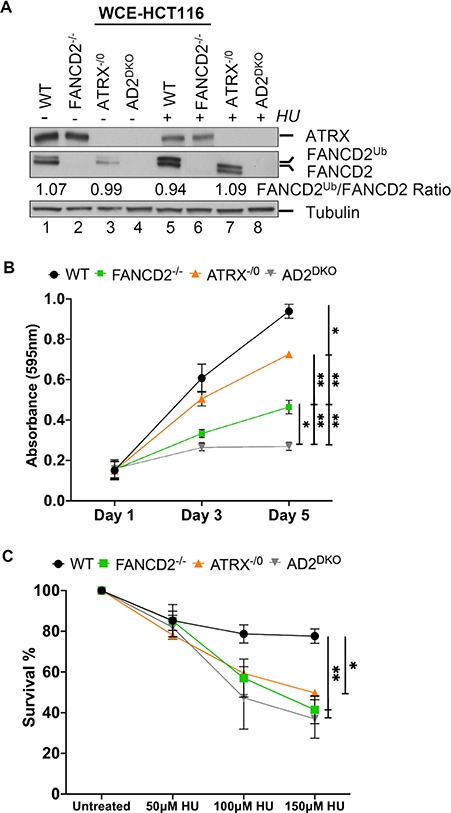
FANCD2 and ATRX can support normal cell proliferation independently, but cooperate to promote cellular HU resistance. (A) Analysis of ATRX and FANCD2 protein expressions in HCT116 WT, FANCD2−/−, ATRX−/0 and AD2DKO cells. WCEs were prepared in the absence or presence of HU and analyzed for FANCD2 and ATRX. Tubulin, loading control. (B) The absence of ATRX and FANCD2 affects cell proliferation synergistically. Proliferation rates of WT, FANCD2−/−, ATRX−/0 and AD2DKO cells were measured following plating at Days 1, 3 and 5 via an MTS assay. (C) ATRX and FANCD2 act in concert to promote HU resistance. WT, FANCD2−/−, ATRX−/0 and AD2DKO cells were untreated or treated with 50, 100 or 150 μm HU and assayed for their colony-forming ability after 10 days.
Human ATRX and FANCD2 can contribute separately to cell proliferation
Human FANCD2 supports normal somatic cell proliferation (29, 30). To test if human ATRX participates in this role, we analyzed the cellular growth of WT, ATRX−/0, D2−/− and AD2DKO cells using a cell proliferation assay. The three knockout cell lines exhibited significantly reduced cellular growth rates in contrast to the WT cells, indicating that FANCD2 and ATRX both contribute to cell proliferation. Importantly, the cellular growth rates of the three knockout cell lines were also significantly different from one another, with decreasing viabilities in the following order: ATRX−/0 > D2−/− > AD2DKO cells (Fig. 2B). These results indicate that FANCD2 and ATRX have at least partially non-overlapping roles to promote cellular proliferation under otherwise unperturbed conditions.
Human ATRX and FANCD2 cooperate to promote cell survival and replication fork restart following HU treatment
Previous studies reported independently that human ATRX and FANCD2 promoted cellular resistance to HU treatment (23, 31). To test if ATRX and FANCD2 act in concert to perform this function, WT, ATRX−/0, D2−/− and AD2DKO cells were plated and either left untreated or treated with increasing concentrations of HU. Colony formation ability of each cell line was determined after 12 to 14 days. Strikingly, the ATRX−/0, D2−/− and AD2DKO cells exhibited similarly increased HU sensitivities in contrast to the WT cells (Fig. 2C), suggesting that FANCD2 and ATRX act within the same pathway to mediate cellular HU resistance.
To investigate if ATRX and FANCD2 cooperate to promote the restart of stalled replication forks and suppress new replication origin firing in the presence of HU, we monitored replication events in WT, ATRX−/0, D2−/− and AD2DKO cells using a dual-labeling DNA fiber assay (13). Replication tracts were first labeled with DigU (red label) for 25 min, then untreated or treated with HU for 5 h to cause replication fork arrest, followed by second labeling with EdU (green label) for 40 min (Fig. 3A). The proportion of replication forks competent for restart was significantly and similarly reduced in the three knockout cell lines in contrast to WT cells (Fig. 3B). In parallel, the proportion of newly originated replication tracts increased significantly in the three knockout cell lines (Fig. 3C, ∼2-fold), in contrast to WT cells. To confirm these results in a different genetic background, we utilized FA patient-derived, FANCD2-deficient (PD20) fibroblasts and their complemented counterpart (PD20 + D2). To additionally deplete ATRX, the PD20 + D2 and PD20 cells were treated with either siControl or siATRX (Supplementary Material, Fig. S1C), followed by the dual-label DNA fiber analysis. In agreement with our observations in the HCT116 knockout cell lines, the ATRX-, FANCD2- and ATRX/FANCD2 double-depleted fibroblasts exhibited comparable defects in replication fork restart after HU treatment, accompanied by a significant upregulation of new origin firing (2–3-fold) in contrast to the complemented parental cells (Supplementary Material, Fig. S1E and F). Thus, FANCD2 and ATRX cooperate to promote the restart of HU-stalled replication forks while suppressing the firing of new replication origins.
Figure 3.
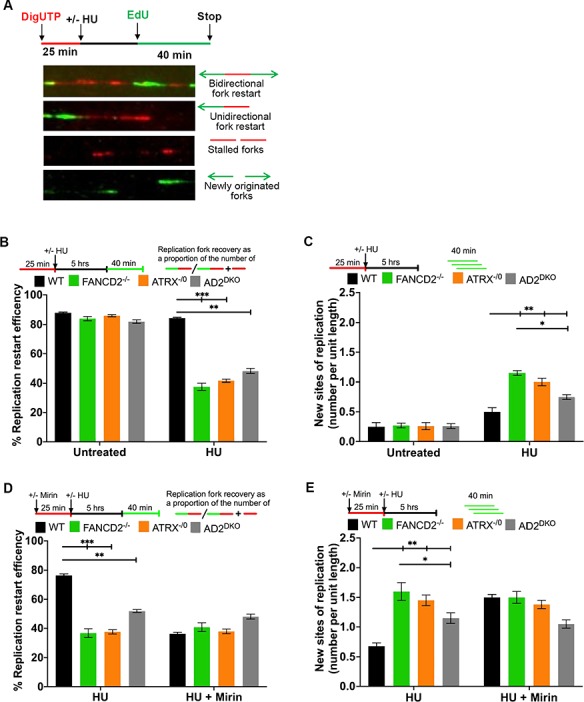
ATRX acts in concert with FANCD2 and the MRN exonuclease activity to mediate replication fork restart and suppression of new origin firing. (A) Schematic of the replication restart protocol with representative images of DNA fibers. Red tracks: DigU; green tracks: BioU. (B) ATRX and FANCD2 cooperate to mediate replication fork restart. Replication fork restart efficiencies were compared between HCT116 WT, FANCD2−/−, ATRX−/0 and AD2DKO cells. Replication restart efficiency was measured as the number of restarted forks after HU-mediated fork stalling (DigU → BioU tracts), in contrast with the total number of DigU-labeled tracts (DigU plus DigU → BioU). (C) ATRX and FANCD2 act in concert to suppress new origin firing during replication blockade. The fraction of new sites of replication originating during the 40 min recovery period after HU treatment was compared between WT, FANCD2−/−, ATRX−/0 and AD2DKO cells. Fractions were measured as the number of green-only (BioU) tracts in contrast with the total number of spreading replication tracts (BioU plus DigU→BioU). [N.B. The same DNA fiber assay analysis described in Fig. 3B and C for analyzing replication fork restart and new origin firing following HU treatment was used throughout this study.] (D) ATRX and FANCD2 cooperate during replication fork restart in an MRN exonuclease-dependent manner. Replication fork restart efficiencies were compared between in HCT116 WT, FANCD2−/−, ATRX−/0 and AD2DKO cells that had additionally been either untreated or supplemented with 50 μm mirin. (E) ATRX and FANCD2 cooperate with the MRN exonuclease to suppress new origin firing during replication blockade. The number of new replication sites originating during BioU labeling after HU treatment was compared between WT, FANCD2−/−, ATRX−/0 and AD2DKO cells that had additionally been either untreated or treated with 50 μm mirin.
ATRX and FANCD2 cooperate with the MRE11 exonuclease to promote fork restart
The MRN complex member MRE11 promotes the restart of HU-stalled replication forks, and restart relies on MRE11’s exonuclease activity (14, 32). To test if ATRX and FANCD2 work in concert with MRE11 to promote replication fork restart, we used mirin, a small molecule inhibitor of the MRE11 exonuclease activity. WT, ATRX−/0, D2−/− and AD2DKO cells were either untreated or preincubated with 50 μm mirin for 1 h. The cells were subsequently treated with HU for 5 h and subjected to DNA fiber analysis. Inhibition of the MRE11 exonuclease activity during HU-mediated fork stalling caused a severe reduction in fork restart events and a 2-fold increase in new origin firing in WT cells. In contrast, the addition of mirin did not further exacerbate the fork restart defects or new origin firing events in the three knockout cell lines (Fig. 3D and E), demonstrating that FANCD2, ATRX and the MRE11 exonuclease cooperate to promote fork restart.
FANCD2 and ATRX cooperate to recruit the HR factor CtIP to HU-stalled replication forks
ATRX and FANCD2 form a protein complex (Fig. 1A and B) and act in concert to recover HU-stalled replication forks (Fig. 3B). Thus, we tested if the recruitment of these two factors to stalled forks occurred interdependently. We analyzed FANCD2 foci formation in untreated or HU-treated WT and ATRX−/0 cells. ATRX−/0 cells were fully competent in supporting spontaneous and HU-inducible FANCD2 foci formation (Supplementary Material, Fig. S2A). Conversely, we analyzed ATRX foci formation in untreated or HU-treated HCT116 WT and D2−/− cells, as well as in FA patient-derived PD20 + D2 (FANCD2 proficient) and PD20 (FANCD2-deficient) cells. Spontaneous and HU-inducible ATRX foci formation was comparable between WT and FANCD2-deficient cells (Supplementary Material, Fig. S2B and C). Thus, ATRX and FANCD2 can form chromatin foci during normal replication and following HU-induced replication stress independently of one another. Next, we asked if ATRX shares roles with FANCD2 in the recruitment of the downstream HR factor CtIP to stalled replication forks. We analyzed CtIP foci formation in untreated or HU-treated WT, ATRX−/0, D2−/− and AD2DKO cells. The spontaneous CtIP foci formation observed during unperturbed DNA replication (untreated conditions) was already reduced in all three knockout cell lines in contrast to WT cells (Fig. 4A and B). Following HU treatment, WT cells showed a significant increase in CtIP foci numbers whereas all three knockout cell lines exhibited severe defects in CtIP foci formation (Fig. 4A and B). We further confirmed ATRX’s role in promoting CtIP foci formation in an independently generated ATRX knockout cell line (rAAV-ATRX−/0, clone #15.1 (28); Supplementary Fig. S2D). Taken together, these findings indicate that ATRX and FANCD2 are recruited to stalled replication forks independently of one another, but cooperate to recruit CtIP, which in turn promotes fork recovery events.
Figure 4.
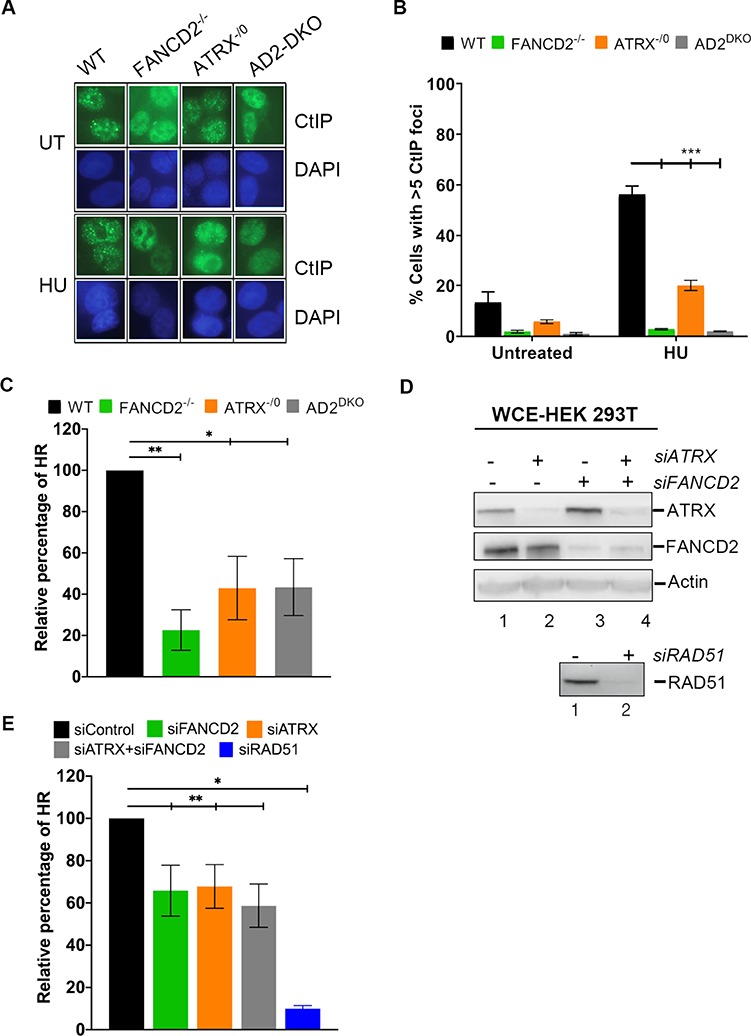
FANCD2 and ATRX function to recruit CtIP to stalled replication forks and cooperate to promote efficient HR repair. (A and B) ATRX and FANCD2 promote CtIP foci formation at HU-stalled replication forks. HCT116 WT, FANCD2−/−, ATRX−/0 and AD2DKO cells were untreated or treated with 2 mm HU for 20 h and cellular nuclei were analyzed for the presence of CtIP foci. Nuclei with > 5 foci were considered positive for CtIP foci. (A) Representative images; (B) graphic depiction of the calculated CtIP foci numbers. (C–E) ATRX and FANCD2 cooperate to promote HR-mediated DNA DSB repair in two different cell lines. (C) A plasmid-based GFP-HDR reporter assay was used to determine HDR efficiencies in HCT116 WT, FANCD2−/−, ATRX−/0 and AD2DKO cells. In this assay, I-SceI restriction enzyme digestion creates a DSB in the HR reporter plasmid (DR-GFP). HDR repair of the DSB restores GFP expression. The repair efficiency was determined by dual GFP and mCherry-positive cells divided by the mCherry-positive cells. Results were averaged from a minimum of three replicates and normalized to the average repair efficiency of the WT cells. (D) Upper panel: HEK293T cells harboring a chromosomally integrated GFP-HDR reporter system were treated with control siRNA (lane 1), siATRX (lane 2) siFANCD2 (lane 3) or siATRX/siFANCD2 (lane 4) and analyzed for the presence of ATRX and FANCD2 by WB. Actin, loading control. Lower panel: The HEK 293T cells were treated with either control siRNA (lane 1) or siRAD51 (right lane 2), and analyzed for the presence of RAD51 by WB. (E) Following siRNA treatment of the HEK293T cells (see D), HDR was initiated by induction of DSB following transient I-SceI expression. The repair efficiency was measured by dual GFP and dsRED-positive cells divided by the dsRED-positive cells. Results were averaged from three independent experiments and normalized to the average repair efficiency of the WT cells. siRAD51-treated cells were used as a control.
FANCD2 and ATRX cooperate to regulate HR-mediated DNA DSB repair
In addition to its HR functions at stalled replication forks, FANCD2 also plays a key role in promoting the HR-directed repair of directly induced DNA DSBs (33, 34). To test if ATRX supports FANCD2 in this role, we utilized a well-established reporter plasmid HR DNA repair assay (35). In this assay, a plasmid containing two non-functional green fluorescent protein (GFP) genes in tandem is transfected into cells. One non-functioning GFP gene contains an I-SceI restriction enzyme recognition site, and the other is truncated at both the 5′- and 3′-ends. Digestion of the plasmid with I-SceI creates a DNA DSB in the non-functional GFP gene that can subsequently use the truncated GFP gene as template to restore GFP expression by homology-directed gene conversion. Strikingly, the ATRX−/0, D2−/− and AD2DKO cells had severe, and comparable, defects in HR-mediated DNA DSB repair in contrast to WT cells (Fig. 4C).
To further support this finding, we repeated the assay in a second cell line, HEK293T, engineered to harbor a genomically integrated GFP cassette (36). We used siRNAs against ATRX, FANCD2 or both to generate ATRX-deficient, FANCD2-deficient or ATRX/FANCD2 double-deficient cells, respectively (Fig. 4D). Subsequently, the cells were transfected with an I-SceI expression plasmid and analyzed as described previously. Both the singly and doubly deficient cells exhibited severe and comparable defects in HR-mediated DNA DSB repair, in contrast to WT cells (Fig. 4E). Taken together, these findings indicate that ATRX and FANCD2 cooperate within the same pathway to promote the HR-dependent repair of induced DNA DSBs.
The ATRX interactor, DAXX, cooperates with FANCD2 to promote replication fork restart
ATRX and DAXX interact to function as a histone H3.3-specific deposition complex. Within this complex, DAXX represents the component that actually contains H3.3 histone chaperone activity (37–39). We therefore asked if DAXX—like ATRX—promotes the FANCD2-dependent restart of HU-stalled replication forks. To generate wild-type, DAXX-, FANCD2- and DAXX/FANCD2 double-deficient cells, we treated PD20 + D2 or PD20 cells with control siRNA or DAXX siRNA (Fig. 5A). DNA fiber analysis after HU-mediated replication fork stalling revealed that the proportion of restart-competent replication forks was significantly and similarly reduced in DAXX-, FANCD2- and DAXX/FANCD2 double-deficient cells (Fig. 5B). In parallel, we observed a significant and comparable increase in the number of newly fired replication origins in the singly or doubly deficient cells, in contrast to WT cells (Fig. 5C, 2- to 5-fold). These results indicate that both subunits of the ATRX/DAXX complex—and thus likely its H3.3 chaperone activity—cooperate with FANCD2 to mediate the restart of HU-stalled replication forks while suppressing the firing of new origins.
Figure 5.
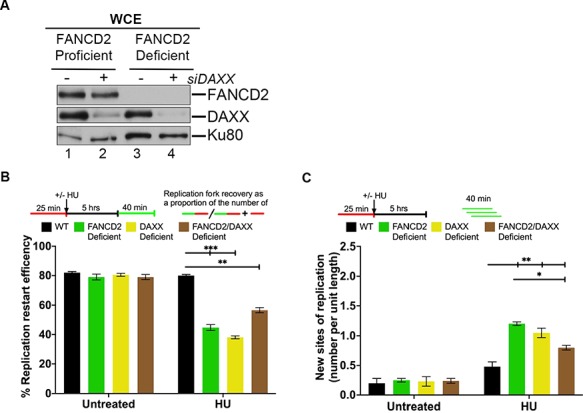
FANCD2 cooperates with DAXX to mediate replication fork restart. (A) Efficiency of siRNA-mediated DAXX knockdown. WCEs were prepared from human FANCD2-proficient cells (PD20 + D2, WT; lanes 1 and 2), or FANCD2-deficient cells (PD20, lanes 3 and 4) that had been untreated or treated with DAXX siRNA (siDAXX) and analyzed by WB for FANCD2 and DAXX. Ku80, loading control. (B) FANCD2 and DAXX cooperate to mediate replication fork restart. Replication fork restart efficiencies were compared between WT, FANCD2-, DAXX- or FANCD2/DAXX double-deficient cells. (C) FANCD2 and DAXX act in concert to suppress new origin firing during replication blockade. The number of new replication sites originating during BioU labeling after HU treatment was compared between WT, FANCD2-, DAXX- or FANCD2/DAXX double-deficient cells.
The histone chaperone activity of non-ubiquitinated FANCD2 is required for replication restart
FANCD2 has a histone H3 chaperone activity in vitro and promotes the chromatin deposition of at least one histone H3 variant, histone H3.1 (the canonical H3 variant), in vivo to support cellular DNA ICL resistance (25, 26, 40). Spurred by our findings suggesting that replication fork restart requires the ATRX/DAXX histone H3.3 chaperone activity (Fig. 5), we asked if FANCD2’s own histone chaperone activity is also required for mediating this process. We took advantage of a recently described patient-derived FANCD2 missense mutant, FANCD2L231R, that lacks histone H3.1 deposition activity (26) and reconstituted D2−/− cells with FANCD2WT (D2−/− + D2WT) or FANCD2L231R (D2−/− + D2L231R) (Fig. 6A). Importantly, the FANCD2L231R mutant is also known to exhibit significant monoubiquitination and chromatin-binding deficiencies (26), making it difficult to specifically tie this mutant’s histone chaperone defect to a given cellular phenotype. To bypass this obstacle, we analyzed FANCD2WT and FANCD2L231R protein expression and chromatin recruitment in several complemented D2−/− cell clones (Fig. 6A and B) and identified one D2−/− + D2L231R clone (#8) that strongly overexpressed FANCD2L231R in contrast to WT or D2−/− + D2WT cells (Fig. 6A, compare lane 8 with lanes 1 or 2). Overexpression of FANCD2L231R was able to compensate for its reduced chromatin affinity, resulting in equal chromatin-bound FANCD2 levels in D2−/− + D2L231R cells (clone #8) in contrast to D2−/− + D2WT cells (clone #3) (Fig. 6B, compare lanes 3 and 4 with lanes 1 and 2). As an additional control for our DNA fiber assay, we included an HCT116-derived FANCL−/− cell line that lacks FA core complex ubiquitin ligase activity and only expresses the non-ubiquitinated FANCD2 isoform. We previously used this cell line to demonstrate that FANCD2 monoubiquitination is dispensable for replication fork restart (16).
Figure 6.
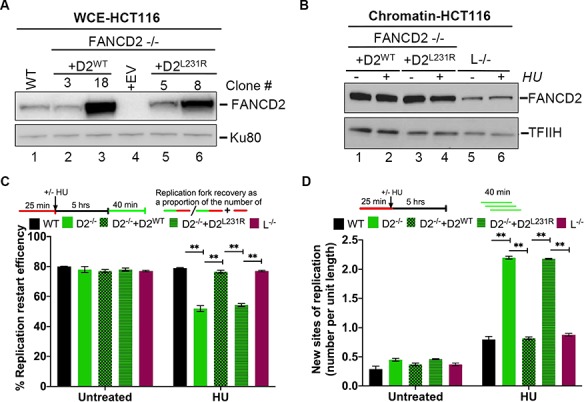
FANCD2’s histone H3 chaperone activity is required to mediate replication fork restart. (A) FANCD2 protein expression in different cell clones. WCEs were prepared from HCT116 WT and D2−/− cells, as well as from several complemented D2−/− + D2WT (#3, 18, 20) and D2−/− + D2L231R (#5, 7, 8) clones, and analyzed for FANCD2 protein levels by WB. Tubulin, loading control. (B) Chromatin-binding efficiencies of D2L231R versus D2WT. HCT116-derived D2−/− + D2WT (clone #3), D2−/− + D2L231R (clone #8) and FANCL−/− cells were either untreated or HU-treated as indicated. Chromatin fractions were prepared from the cells and analyzed for the presence of FANCD2 by WB. TFIIH loading control. Immunoblot signals for chromatin-bound FANCD2WT (lanes 1 and 2) and FANCD2L231R (lanes 3 and 4) were analyzed by densitometry and normalized against TFIIH signals using ImageJ. Relative chromatin-bound FANCD2 levels: lane 1, 1.0; lane 2, 1.0; lane 3, 1.7; lane 4, 1.2. (C) Replication fork restart requires the FANCD2 histone chaperone activity. Replication fork restart efficiencies were compared between WT, FANCD2−/−, FANCD2−/− + D2WT, D2−/− + D2L231R and L−/− cells. (D) The FANCD2 histone chaperone activity is required to suppress new origin firing during replication blockade. The number of new replication sites originating during BioU labeling after HU treatment was compared between WT, FANCD2−/−, FANCD2−/− + D2WT, D2−/− + D2L231R and L−/− cells.
Next, we compared replication fork restart efficiencies in WT, D2−/− (+ empty vector), D2−/− + D2WT (clone #3), D2−/− + D2L231R (clone #8) and FANCL−/− cells that had been untreated or treated with HU for 5 h. As expected, the FANCL−/− cells were fully competent for replication fork restart and new origin suppression (Fig. 6C and D). Strikingly, the expression of FANCD2WT, but not FANCD2L231R, restored replication restart efficiencies and the suppression of new origin firing in D2−/− cells (Fig. 6C and D). Since FANCD2L231R chromatin binding is WT-like in the D2−/− + D2L231R clone #8 (Fig. 6B), and FANCD2 monoubiquitination is dispensable for fork recovery (see the FANCL−/− data in Fig. 6C and D), these results strongly suggest that the FANCD2 histone H3 chaperone activity is crucial for the restart of HU-stalled replication forks and the simultaneous suppression of new origin firing. Thus, our findings identify a new role for FANCD2-dependent histone H3 deposition at HU-stalled replication forks, i.e. in order to promote fork recovery.
Discussion
We have identified the ATRX/DAXX histone chaperone complex as a novel physical and functional interactor of the central FA pathway member, FANCD2. Like other FANCD2 interactors such as FANCI (29, 41), CtIP (17) or the MRN complex (27), ATRX is crucial to stabilizing cellular FANCD2 protein levels, indicating that ATRX may make direct contact with FANCD2. Alternatively, since ATRX and FANCD2 both interact with the MRN complex (22, 23, 27), ATRX may simply strengthen the FANCD2–MRN interaction as part of an ATRX–MRN–FANCD2 ‘supercomplex’, in which loss of any one protein member causes accelerated FANCD2 degradation. In further support of the existence of a common ATRX–FANCD2 protein complex, ATRX and FANCD2 were recently shown to co-localize in chromatin foci in a replication stress (APH)-responsive manner (42). Notably, the full protein complex is likely formed after individual complex components bind chromatin, since ATRX and FANCD2 localize to such replication stress-induced foci independently of one another. We thus envision a model where the ATRX/DAXX–MRN–FANCD2 complex assembles on chromatin to promote the recruitment of the CtIP nuclease (17, 43) (Fig. 4A and B), which in turn enables DNA processing at stalled replication forks. Intriguingly, when considering the molecular weight of individual complex members FANCD2 and ATRX, and accounting for the fact that the basic MRN complex contains two MRE11, two RAD50 and one NBS1 molecule(s) (43), such a supercomplex would be predicted to weigh about 1.1 MDa, closely matching a previously identified, but as-of-yet uncharacterized, FANCD2-containing 1.2 MDa protein complex (5).
Supported by their close physical interactions, ATRX, the MRN exonuclease and FANCD2 act as a functional unit that promotes the HR-dependent restart of HU-stalled replication forks. This cooperation suggests that ATRX shares common roles with FANCD2 and MRE11 (reviewed in (43, 44)), both of which act during early HR steps at stalled replication forks to recruit downstream HR factors. Supportive of this idea, ATRX and FANCD2 are both required to recruit CtIP to stalled replication forks. Moreover, two previous studies reported independently that ATRX and FANCD2 promote the recruitment of the key HR factor RAD51 to stalled forks (24, 45). Since our results also show that ATRX acts in the same pathway as FANCD2 to support HR repair of DNA DSBs, it seems likely that ATRX generally cooperates with FANCD2 and MRN during the early steps of HR-associated DNA processing. Interestingly, a recent study analyzed ATRX’s role during the HR repair of DNA DSBs that had been induced by ionizing irradiation treatment of G2 phase cells (i.e. in a replication-independent manner). The authors reported that in this scenario, ATRX was crucial for the late steps of HR-mediated DNA DSB repair, downstream of RAD51 recruitment (46). Thus, ATRX may be able to act at several steps during HR repair, similar to other HR factors such as BRCA1 or members of the RAD51 paralog family (47, 48), and its roles may be dependent on the cell cycle stage and/or the type of DNA lesion.
One unexpected finding of our study was that ATRX−/0 and D2−/− cells were equally defective in HR-mediated replication fork restart, whereas the AD2DKO cells consistently exhibited a less severe fork restart deficiency in contrast to either of the singly null cell lines. While this finding warrants further investigation, we speculate that in the absence of several HR factors, i.e. ATRX and FANCD2, other DNA repair pathways such as the non-homologous end joining (NHEJ) machinery may be activated to promote fork recovery via homology-independent mechanisms. In agreement with this idea, the recruitment of NHEJ repair factors to sites of DNA lesions increases in cells lacking FANCD2 (49, 50) or ATRX (24). Moreover, members of the NHEJ machinery were recently shown to be capable of promoting replication fork restart as well (51, 52).
To our knowledge, the ATRX/DAXX complex is the first histone variant chaperone complex to be identified as a physical and functional FANCD2 interactor. Our observation that the functional cooperation between ATRX and FANCD2 requires both the H3.3 chaperone activity of ATRX/DAXX and the (less well defined) histone H3 chaperone activity of FANCD2 suggests that an appropriate deposition of certain histone variants at temporarily stalled replication forks represents a critical mechanism for fork rescue. In support of this idea, FANCD2’s H3 chaperone activity was recently shown to be required for the protection of stalled replication forks against nucleolytic degradation (40); moreover, histone H3.3 deposition promotes DNA replication under UV-triggered replication stress conditions (53). Furthermore, accumulating evidence has implicated additional histone variants such as H2AZ and macroH2A1.2 during the process of replication fork recovery (reviewed in (54)). Of note, while the previous studies showed that FANCD2 has histone H3.1 chaperone activity (25, 26, 40), they did not test whether FANCD2 harbors a preference towards the H3.1 variant, in contrast to other H3 variants. Further studies are necessary to address this question.
Based on our current knowledge of ATRX gene functions, the newly identified involvement of the ATRX complex in FANCD2-regulated, HR-mediated replication fork recovery and DSB repair provides an intriguing conundrum. On the one hand, several studies support a role for ATRX in replication fork protection and recovery and in the general maintenance of genome stability in somatic cells. In addition, spontaneous disease-driving ATRX mutations have been found in a number of different cancer types ((22–24) and reviewed in (55)). On the other hand, and very different from the FA syndrome, the ATRX syndrome (caused by inherited ATRX mutations) is typically characterized by alpha thalassemia and mental retardation, but not an increased cancer predisposition that one would predict in patients with inherited HR defects. Interestingly, very recent studies described for the first time a tumor development (osteosarcoma) in five ATRX syndrome patients from four different families (56–58). Therefore, future studies will need to address whether only certain inherited ATRX gene mutations—perhaps localized to distinct ATRX domains—cause cancer predisposition in humans.
In summary, we propose a working model (Supplementary Material, Fig. S3) where the ATRX/DAXX complex cooperates with FANCD2 and the MRN exonuclease at sites of stalled replication forks to support the deposition of histone variants H3.1 and H3.3, the recruitment of the key HR factor CtIP, and ultimately the HR-mediated restart of the stalled replication forks.
Materials and Methods
Cell lines and cell culture techniques
FANCD2-deficient (PD20) and FANCD2-complemented cells (PD20 + D2) were obtained from the FA cell repository at the Oregon Health & Science University. PD20 cells complemented with 3xFLAG-tagged FANCD2 (PD20 + 3xFLAG-FANCD2) were a kind gift from Dr Tosiyashu Taniguchi at the Fred Hutchinson Research Centre. FA patient-derived fibroblast lines and HEK293T cells were cultured in DMEM (GIBCO) with 10% FBS and 1% penicillin–streptomycin. All HCT116-derived cell lines were cultured in McCoy’s media (Corning) enriched with 10% FBS, 1% penicillin–streptomycin and 1% glutamine.
CRISPR/Cas9 generation of ATRX−/0, FANCD2−/− and ATRX−/0:FANCD2−/− (AD2DKO) cells
We have described the generation of the ATRX−/0 (28) and the FANCD2−/− cell lines (29). The same ATRX single guide RNA (sgRNA) containing CRISPR/Cas9 expression plasmid (targeting ATRX exon 9) was transfected into the FANCD2−/− cells using Lipofectamine 3000 (Life Technologies). Two days after transfection, the cells were sub-cloned, and individual sub-clones were screened for targeting by amplification of exon 9 and subsequent digestion with SmlI (New England BioLabs, Inc). Sub-clones that were resistant to digestion with SmlI were TOPO TA cloned (Life Technologies). Sanger sequencing of the TOPO TA clones confirmed targeted insertion of a single nucleotide that would induce a frameshift in FANCD2, generating an AD2DKO (double knockout) cell line. Western blot analysis further confirmed that these clones were null for FANCD2 and ATRX expression.
Complementation of D2−/− cells
For expression of hFANCD2, the human FANCD2 transcript 1 cDNA (NM_001018115) was optimized by silent mutagenesis, thereby introducing unique restriction enzyme recognition sites and removing potential prokaryotic instability motifs, cryptic splice sites, RNA instability motifs and polyA sites (Hanenberg et al., manuscript in preparation). The cDNA was purchased from GenScript (Piscataway, NJ). The D2−/− cells were complemented with the FANCD2WT or FANCD2L231R cDNA using the optimized human FANCD2 expression cassette that was Gateway-cloned into a PiggyBac transposition vector containing a cytomegaloviral/beta-actin/beta-globin (CAG) promoter and a neomycin resistance (NEO) selection cassette. G418-resistant clones were screened for FANCD2 expression by western blot analyses. FANCD2L231R mutagenesis primer sequences are available upon request.
Preparation of WCEs
For WCE preparation, cells were washed in PBS, re-suspended in lysis buffer (10 mm Tris pH 7.4, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mm EDTA, 1 mm DTT, 0.5 mg/ml pefabloc™ protease inhibitor) and incubated on ice for 20 min. Cell extracts were centrifuged for 15 min at 10000 rpm, and the supernatant was used for further analysis.
Immunoblotting
Protein samples were separated on gradient gels and transferred to Immobilon-P membranes (Millipore). After blocking in 5% milk, the membranes were incubated with the following primary antibodies: FANCD2 (1:1000), ATRX (1:2000), NBS1 (1:1500), Ku80 (1:5000) and tubulin (1:5000), A horseradish peroxidase-conjugated rabbit secondary antibody (Jackson Laboratories) or a mouse secondary antibody (Bio-Rad) was used at a dilution of 1:10000 or 1:3000, respectively. Protein bands were visualized using an EL Plus system (Amersham).
Antibodies
The following commercially available antibodies were used for western blotting: anti-FANCD2 (Santa Cruz, sc-20 022), anti-ATRX (GeneTex, GTX101310), anti-DAXX (Santa Cruz, sc-7152), anti-NBS1 (GeneTex, GTX70222), anti-Ku80 (Santa-Cruz, sc-5280), anti-cyclin B1 (Santa Cruz, sc-245) and anti-tubulin (Abcam, ab7291). For IP and immunofluorescence assays, we used anti-FLAG (Sigma M2 monoclonal), anti-ATRX (Santa Cruz, sc-15 408) and anti-CtIP (59) antibodies.
IP assay
All IPs were performed using nuclear extracts (NEs) prepared from PD20 + 3xFLAG FANCD2 cells. Freshly isolated cells were washed in PBS, and nuclei were isolated using CSK buffer (10 mm PIPES pH 6.8, 100 mm NaCl, 300 mm sucrose, 3 mm MgCl2, 0.1% Triton-X 100 and cOmplete Protease Inhibitor). The isolated nuclei were lysed in nuclear lysis buffer (10 mm HEPES pH 7.9, 100 mm NaCl, 1.5 mm MgCl2, 25% glycerol, 1 mm DTT and cOmplete Protease Inhibitor) using a Dounce homogenizer. The nuclear lysates were treated with 50 U/ml Benzonase for 1 h at 4°C and pelleted twice. The obtained supernatant (NE) was precleared with rabbit or mouse IgG and subjected to immunoprecipitation with anti-ATRX, anti-FLAG or IgG antibodies in the presence of EtBr (10 μg/ml) at 4°C overnight. One hundred microliters of Sepharose 4B beads (50% slurry) was added to the NE and rotated for 30 min at 4°C. The beads were pelleted from the solution, washed in nuclear lysis buffer, boiled in 1× NuPAGE buffer (Invitrogen) and analyzed by SDS–PAGE and western blotting.
Immunofluorescence analysis of chromatin foci
Indirect immunofluorescence was carried out essentially as described (60). Primary antibodies used were anti-FANCD2 (1:4000), anti-ATRX (1:2500) and anti-CtIP (mouse monoclonal, 1:400). The secondary antibodies used were Alexa Fluor 594-conjugated goat anti-rabbit (1:1000) and Alexa Fluor 488-conjugated goat anti-mouse (1:1000; Molecular Probes). For statistical analysis of nuclear foci formation, images were taken using a Leica DM LB2 microscope with a Hamamatsu ORCA-ER camera. Quantification of ATRX, FANCD2 and CtIP foci was carried out using ImageJ. At least 150 cells were counted per sample per experiment.
siRNA experiments
ATRX and FANCD2 siRNA duplexes were purchased from Dharmacon research (Thermo Scientific, MA, USA). The sequences of ATRX siRNA#1 and ATRX siRNA#2 are 5′GAGGAAACCUUCA AUUGUAUU-3′ and 5′-GCAGAGAAAUUCCUAAAGAUU-3′, respectively. The FANCD2 siRNA sequence is 5′-CAACAUACCUCGACUCAUUUU. The DAXX siRNA was obtained from Invitrogen (HSS102654). RAD51 siRNA was purchased from Dharmacon (L-003530-00). siGENOME Non-Targeting siRNA from Dharmacon was used as a control. Transfections were performed using DharmaFECT1 transfection reagent according to the manufacturer’s protocol.
Cell proliferation assay
Wild-type, knockout cells and complemented cells were plated in 96-well plates according to their plating efficiencies. Wild-type and ATRX−/0 cells were plated at 200 cells/well, FANCD2−/− cells were plated at 500 cells/well and AD2DKO cells were plated at 1000 cells/well. Cell growth was analyzed using an MTS assay (Promega, manufacturer’s instructions). Each assay was performed in triplicate on Days 1, 3 and 5 after seeding.
HU sensitivity assay
Cells were plated in 6-well tissue culture plates according to their plating efficiency. Wild-type and ATRX−/− cells were plated at 350 cells/well, FANCD2−/− cells were plated at 1000 cells/well and AD2DKO cells were plated at 1200 cells/well. On the following day, the media were removed and replaced with media containing 0, 50, 100 or 150 μm HU. Cells were allowed to grow for 10–12 days, and the resulting cell colonies were washed in PBS, fixed in 10% acetic acid/10% methanol and stained with crystal violet. Colonies reaching a minimum size of 50 cells were counted and normalized to the average colony number in the untreated wells. Each assay was performed in triplicate.
Homology-directed repair assay
Plasmid assay (in HCT116 cell-derivatives)
The homology-directed repair (HDR) reporter assay was performed with the DR-GFP plasmid as described (35). The DR-GFP reporter plasmid contains two mutated GFP genes in tandem, and one of the mutated GFP genes contains an I-SceI restriction enzyme recognition site. If HDR/gene conversion between the two mutated GFP genes is used to repair the DSB induced by I-SceI digestion, then GFP expression is restored.
Integrated reported assay
The HDR reporter assay was conducted with HEK 293T cells stably bearing DR-GFP as described (36). After 2 days, the transfected cells were seeded on a 12-well plate at 0.4 × 105/well. The following day, cells were co-transfected with 0.5 μg of either I-SceI expression vector or empty vector and 0.1 μg of dsRED vector (used as a transfection control) in 0.1 ml Opti-MEM containing 3 μl of Lipofectamine 3000 (Invitrogen). After 6 h, the media was removed and replaced with the growth media. Two days after I-SceI transfection was done, the percentage of GFP+ cells was analyzed by the Becton Dickinson FACSVerse flow cytometer. Experiments were repeated at least three times, and the average values are used.
DNA fiber assay
We used a previously described DNA fiber protocol (61). Moving replication forks were labeled with digoxigenin-dUTPs (DigU) for 25 min and then with EdU-dUTPs (BioU) for 40 min. To allow efficient incorporation of the DigUs, a hypotonic buffer treatment (10 mm HEPES, 30 mm KCl, pH 7.4) preceded the DigU-labeling step. To visualize labeled fibers, cells were mixed with a 10-fold excess of unlabeled cells and fixed and dropped onto slides. After cell lysis, DNA fibers were released and extended by tilting the slides. EdU-labeled DNA was conjugated to biotin using a click reaction with biotin azide. Incorporated dUTPs were visualized by immunofluorescence detection using anti-digoxigenin-rhodamine (Roche) and streptavidin-Alexa-Fluor-488 (Invitrogen). Images were captured using a DeltaVision microscope (Applied Precision) and analyzed using DeltaVision softWoRx 5.5 software. All shown DNA fiber results are the means of two or three independent experiments (300 DNA fibers/experiment). Error bars represent the standard error of the mean, and significance was determined by t test and Mann–Whitney test.
Statistical analysis
Error bars represent the standard deviation of the mean, and P values were determined using a Student’s t test. Statistical significances at P < 0.05, P < 0.01 and P < 0.001 were indicated as *, ** and ***, respectively. A summary of all P values for results shown in Figures 2–6 is provided in Supplementary Material, Table S1.
Funding
Work in the Sobeck laboratory was funded, in part, by the American Cancer Society (RSG-13-039-01-DMC) and the National Institutes of Health (CA194871). Work in the Hendrickson laboratory was funded, in part, by a grant from the National Institutes of Health (GM088351) and from a grant from the National Cancer Institute (CA190492). A portion of this work was also funded by a Brainstorm award, administered by the University of Minnesota’s NCI-designated Cancer Center. Work in the Bielinsky lab was funded by a grant from the National Institutes of General Medical Sciences (GM 074917). Work in the Yeo and Schärer laboratories was supported by the Korean Institute for Basic Science (IBS-R022-A1). Work in the Hoatlin laboratory was funded by the National Institutes of Health ().
Supplementary Material
Acknowledgements
We thank Dr Jeremy Stark (Beckman Research Institute, City of Hope) for kindly providing us with HEK 293T cells stably bearing a DR-GFP reporter. We are grateful to Dr Richard Baer (Columbia University, New York) for generously sharing a human CtIP antibody with us. We thank Dr Helmut Hanenberg (Heinrich Heine University, Düsseldorf, Germany) for sharing a codon-optimized human FANCD2 cDNA construct with us. We also thank Dr Andrew Deans (St. Vincent’s Institute of Medical Research, Australia) for providing NE preparation protocols. Last but not least, we would like to thank Dr Naoko Shima for her comments during the preparation of this manuscript.
Conflict of interest statement. E.A.H. declares that he is a member of the scientific advisory boards of Horizon Discovery, Ltd, and Intellia Therapeutics, companies that specialize in applying gene editing technology to basic research and therapeutics. The other authors declare no conflict of interest.
References
- 1. Ceccaldi R., Sarangi P. and D'Andrea A.D. (2016) The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell. Biol., 17, 337–349. [DOI] [PubMed] [Google Scholar]
- 2. Nalepa G. and Clapp D.W. (2018) Fanconi anaemia and cancer: an intricate relationship. Nat. Rev. Cancer, 18, 168–185. [DOI] [PubMed] [Google Scholar]
- 3. Knies K., Inano S., Ramirez M.J., Ishiai M., Surralles J., Takata M. and Schindler D. (2017) Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Invest., 127, 3013–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schlacher K., Wu H. and Jasin M. (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell, 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhi G., Chen X., Newcomb W., Brown J., Semmes O.J. and Kupfer G.M. (2010) Purification of FANCD2 sub-complexes. Br. J. Haematol., 150, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kee Y. and D'Andrea A.D. (2010) Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev., 24, 1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naim V. and Rosselli F. (2009) The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol., 11, 761–768. [DOI] [PubMed] [Google Scholar]
- 8. Kottemann M.C. and Smogorzewska A. (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 493, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meetei A.R., De Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., Van De Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E. et al. (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet, 35, 165–170. [DOI] [PubMed] [Google Scholar]
- 10. Taniguchi T., Garcia-Higuera I., Xu B., Andreassen P., Gregory R., Kim S., Lane W., Kastan M. and D'Andrea A. (2002) Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell, 109, 459–472. [DOI] [PubMed] [Google Scholar]
- 11. Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M. and D'Andrea A.D. (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell, 7, 249–262. [DOI] [PubMed] [Google Scholar]
- 12. Lopez-Martinez D., Liang C.C. and Cohn M.A. (2016) Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell. Mol. Life Sci., 73, 3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chaudhury I., Sareen A., Raghunandan M. and Sobeck A. (2013) FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res., 41, 6444–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaudhury I., Stroik D.R. and Sobeck A. (2014) FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol. Cell. Biol., 34, 3939–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schlacher K., Christ N., Siaud N., Egashira A., Wu H. and Jasin M. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raghunandan M., Chaudhury I., Kelich S.L., Hanenberg H. and Sobeck A. (2015) FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi anemia core complex. Cell Cycle, 14, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yeo J.E., Lee E.H., Hendrickson E.A. and Sobeck A. (2014) CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet., 23, 3695–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xue Y., Gibbons R., Yan Z., Yang D., McDowell T.L., Sechi S., Qin J., Zhou S., Higgs D. and Wang W. (2003) The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. U S A, 100, 10635–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang J., Wu S., Liu H., Stratt R., Barak O.G., Shiekhattar R., Picketts D.J. and Yang X. (2004) A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem., 279, 20369–20377. [DOI] [PubMed] [Google Scholar]
- 20. Voon H.P. and Wong L.H. (2016) New players in heterochromatin silencing: histone variant H3.3 and the ATRX/DAXX chaperone. Nucleic Acids Res., 44, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ratnakumar K. and Bernstein E. (2013) ATRX: the case of a peculiar chromatin remodeler. Epigenetics, 8, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clynes D., Jelinska C., Xella B., Ayyub H., Taylor S., Mitson M., Bachrati C.Z., Higgs D.R. and Gibbons R.J. (2014) ATRX dysfunction induces replication defects in primary mouse cells. PLoS One, 9, e92915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leung J.W., Ghosal G., Wang W., Shen X., Wang J., Li L. and Chen J. (2013) Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J. Biol. Chem., 288, 6342–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huh M.S., Ivanochko D., Hashem L.E., Curtin M., Delorme M., Goodall E., Yan K. and Picketts D.J. (2016) Stalled replication forks within heterochromatin require ATRX for protection. Cell Death. Dis., 7, e2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato K., Ishiai M., Toda K., Furukoshi S., Osakabe A., Tachiwana H., Takizawa Y., Kagawa W., Kitao H., Dohmae N. et al. (2012) Histone chaperone activity of Fanconi anemia proteins, FANCD2 and FANCI, is required for DNA crosslink repair. Embo J., 31, 3524–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato K., Ishiai M., Takata M. and Kurumizaka H. (2014) Defective FANCI binding by a fanconi anemia-related FANCD2 mutant. PLoS One, 9, e114752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roques C., Coulombe Y., Delannoy M., Vignard J., Grossi S., Brodeur I., Rodrigue A., Gautier J., Stasiak A.Z., Stasiak A. et al. (2009) MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. Embo J., 28, 2400–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Napier C.E., Huschtscha L.I., Harvey A., Bower K., Noble J.R., Hendrickson E.A. and Reddel R.R. (2015) ATRX represses alternative lengthening of telomeres. Oncotarget, 6, 16543–16558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson E.L., Yeo J.E., Lee E.A., Kan Y., Raghunandan M., Wiek C., Hanenberg H., Scharer O.D., Hendrickson E.A. and Sobeck A. (2017) FANCI and FANCD2 have common as well as independent functions during the cellular replication stress response. Nucleic Acids Res., 45, 11837–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian Y., Shen X., Wang R., Klages-Mundt N.L., Lynn E.J., Martin S.K., Ye Y., Gao M., Chen J., Schlacher K. et al. (2017) Constitutive role of the Fanconi anemia D2 gene in the replication stress response. J. Biol. Chem., 292, 20184–20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen X., Bosques L., Sung P. and Kupfer G.M. (2016) A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene, 35, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bryant H.E., Petermann E., Schultz N., Jemth A.S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P. and Helleday T. (2009) PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. Embo J., 28, 2601–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto K., Hirano S., Ishiai M., Morishima K., Kitao H., Namikoshi K., Kimura M., Matsushita N., Arakawa H., Buerstedde J.M. et al. (2005) Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol. Cell. Biol., 25, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakanishi K., Yang Y.G., Pierce A.J., Taniguchi T., Digweed M., D'Andrea A.D., Wang Z.Q. and Jasin M. (2005) Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. U S A, 102, 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pierce A.J., Johnson R.D., Thompson L.H. and Jasin M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev., 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gunn A. and Stark J.M. (2012) I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol., 920, 379–391. [DOI] [PubMed] [Google Scholar]
- 37. Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X. et al. (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell, 140, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Drane P., Ouararhni K., Depaux A., Shuaib M. and Hamiche A. (2010) The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev., 24, 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lewis P.W., Elsaesser S.J., Noh K.M., Stadler S.C. and Allis C.D. (2010) Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U S A, 107, 14075–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Higgs M.R., Sato K., Reynolds J.J., Begum S., Bayley R., Goula A., Vernet A., Paquin K.L., Skalnik D.G., Kobayashi W. et al. (2018) Histone methylation by SETD1A protects nascent DNA through the nucleosome chaperone activity of FANCD2. Mol. Cell, 71, e26, 25–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R. 3rd, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D'Andrea A.D. et al. (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell, 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pladevall-Morera D., Munk S., Ingham A., Garribba L., Albers E., Liu Y., Olsen J.V. and Lopez-Contreras A.J. (2019) Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res., 47, 8004–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Syed A. and Tainer J.A. (2018) The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem., 87, 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Michl J., Zimmer J. and Tarsounas M. (2016) Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J., 35, 909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sato K., Shimomuki M., Katsuki Y., Takahashi D., Kobayashi W., Ishiai M., Miyoshi H., Takata M. and Kurumizaka H. (2016) FANCI-FANCD2 stabilizes the RAD51-DNA complex by binding RAD51 and protects the 5'-DNA end. Nucleic Acids Res., 44, 10758–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Juhasz S., Elbakry A., Mathes A. and Lobrich M. (2018) ATRX promotes DNA repair synthesis and sister chromatid exchange during homologous recombination. Mol. Cell, 71(11–24), e17. [DOI] [PubMed] [Google Scholar]
- 47. Roy R., Chun J. and Powell S.N. (2011) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer, 12, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sullivan M.R. and Bernstein K.A. (2018) RAD-ical new insights into RAD51 regulation. Genes (Basel), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adamo A., Collis S.J., Adelman C.A., Silva N., Horejsi Z., Ward J.D., Martinez-Perez E., Boulton S.J. and La Volpe A. (2010) Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell, 39, 25–35. [DOI] [PubMed] [Google Scholar]
- 50. Pace P., Mosedale G., Hodskinson M.R., Rosado I.V., Sivasubramaniam M. and Patel K.J. (2010) Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science, 329, 219–223. [DOI] [PubMed] [Google Scholar]
- 51. Teixeira-Silva A., Ait Saada A., Hardy J., Iraqui I., Nocente M.C., Freon K. and Lambert S.A.E. (2017) The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun., 8, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Haro L.P., Wray J., Williamson E.A., Durant S.T., Corwin L., Gentry A.C., Osheroff N., Lee S.H., Hromas R. and Nickoloff J.A. (2010) Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res., 38, 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frey A., Listovsky T., Guilbaud G., Sarkies P. and Sale J.E. (2014) Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol., 24, 2195–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fournier L.A., Kumar A. and Stirling P.C. (2018) Chromatin as a platform for modulating the replication stress response. Genes (Basel), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Watson L.A., Goldberg H. and Berube N.G. (2015) Emerging roles of ATRX in cancer. Epigenomics, 7, 1365–1378. [DOI] [PubMed] [Google Scholar]
- 56. Smolle M.A., Heitzer E., Geigl J.B., Al Kaissi A., Liegl-Atzwanger B., Seidel M.G., Holzer L.A. and Leithner A. (2017) A novel mutation in ATRX associated with intellectual disability, syndromic features, and osteosarcoma. Pediatr. Blood Cancer, 64. [DOI] [PubMed] [Google Scholar]
- 57. Ji J., Quindipan C., Parham D., Shen L., Ruble D., Bootwalla M., Maglinte D.T., Gai X., Saitta S.C., Biegel J.A. et al. (2017) Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am. J. Med. Genet. A, 173, 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Masliah-Planchon J., Levy D., Heron D., Giuliano F., Badens C., Freneaux P., Galmiche L., Guinebretierre J.M., Cellier C., Waterfall J.J. et al. (2018) Does ATRX germline variation predispose to osteosarcoma? Three additional cases of osteosarcoma in two ATR-X syndrome patients. Eur. J. Hum. Genet., 26, 1217–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu X. and Baer R. (2000) Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem., 275, 18541–18549. [DOI] [PubMed] [Google Scholar]
- 60. Xu D., Guo R., Sobeck A., Bachrati C.Z., Yang J., Enomoto T., Brown G.W., Hoatlin M.E., Hickson I.D. and Wang W. (2008) RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev., 22, 2843–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sugimura K., Takebayashi S., Ogata S., Taguchi H. and Okumura K. (2007) Non-denaturing fluorescence in situ hybridization to find replication origins in a specific genome region on the DNA fiber. Biosci. Biotechnol. Biochem., 71, 627–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.