

Abstract
The transcription factors of the MYC family play pivotal roles in the initiation and progression of human cancers. High oncogenic level of MYC invades low‐affinity sites and enhancer sequences, which subsequently alters the transcriptome, causes metabolic imbalance, and induces stress response. The endoplasmic reticulum (ER) not only plays a central role in maintaining proteostasis, but also contributes to other key biological processes, including Ca2+ metabolism and the synthesis of lipids and glucose. Stress conditions, such as shortage in glucose or oxygen and disruption of Ca2+ homeostasis, may perturb proteostasis and induce the unfolded protein response (UPR), which either restores homeostasis or triggers cell death. Crucial roles of ER stress and UPR signaling have been implicated in various cancers, from oncogenesis to treatment response. Here, we summarize the current knowledge on the interaction between MYC and UPR signaling, and its contribution to cancer development. We also discuss the potential of targeting key UPR signaling nodes as novel synthetic lethal strategies in MYC‐driven cancers.
Keywords: cancer, ER stress, MYC, synthetic lethality, UPR
Subject Categories: Cancer,
This article reviews the current knowledge on the interactions between endoplasmic reticulum stress, unfolded protein response (UPR), and MYC family, and discusses recent publications that highlight the therapeutic potential of targeting key UPR signaling as a novel strategy in MYC‐driven cancers.
Glossary
- Apoptosis
Controlled cell death that occurs in response to a variety of cellular stressors and as part of developmental programs of multicellular organisms.
- Autophagy
Regulated mechanism used by the cells to maintain homeostasis and normal function through orderly degradation and recycling of unnecessary or dysfunctional components.
- Autophosphorylation
Phosphorylation of the protein kinase by itself, which plays an important role in the process of cell signal transduction.
- Dimerization
Chemical reaction that binds two molecular subunits, resulting in the formation of a single dimer.
- ER stress (Endoplasmic reticulum stress)
Stress caused by the accumulation of misfolded and unfolded proteins in the ER lumen or by Ca2+ balance disorders.
- ERAD (Endoplasmic reticulum‐associated degradation)
Umbrella term that covers a range of different mechanisms by which misfolded proteins are retained in the ER and delivered for proteasomal degradation after retrotranslocation into the cytosol.
- GEMM (Genetically engineered mouse model)
Mouse model for research on human diseases, in which the mouse genome is altered through the use of genetic engineering techniques.
- Gluconeogenesis
Metabolic process in which glucose is formed from non‐carbohydrate precursors.
- Metabolic reprogramming
Molecular adjustments in metabolic pathways that alter the bioenergetic profile and metabolism of the cell.
- PDX (Patient‐derived xenograft)
Mouse model based on transplantation and serial propagation of fresh human tumor biopsies in immunodeficient mice.
- Proteostasis
Homeostatic mechanisms controlling the biogenesis, trafficking, and degradation of proteins in cells. Its imbalances may lead to the aggregation of misfolded proteins, trigger stress responses, or excessive protein degradation.
- RIDD (Regulated IRE1α‐dependent decay)
Degradation of mRNAs encoding mostly ER‐targeted proteins by IRE1α, to reduce the load of incoming ER “client” proteins during ER stress.
- Tumor microenvironment
Cellular environment in which tumor cells reside. It consists of extracellular matrix and different populations of stromal cells, including endothelial cells, fibroblasts, and immune cells.
- UPR (Unfolded protein response)
Collection of phylogenetically conserved signaling pathways initiated by transmembrane stress sensors of the endoplasmic reticulum.
The MYC family and cancer
MYC gene encodes the basic helix–loop–helix/leucine zipper (bHLH‐LZ) transcription factor c‐Myc that belongs to the MYC family, together with L‐Myc and N‐Myc (encoded by MYCL and MYCN, respectively). These genes are differentially expressed during development, but the MYC proteins are functionally equivalent in most biological systems (Conacci‐Sorrell et al, 2014). c‐Myc heterodimerizes with MAX, another bHLH‐LZ protein, and the complex binds DNA sequences enriched in the promoters and enhancers to regulate gene expression. The canonical high‐affinity sites of c‐Myc‐MAX heterodimer are termed “E‐boxes” with a consensus sequence 5′‐CACGTG‐3′ (Blackwell et al, 1993; Fernandez et al, 2003). In malignant cells where c‐Myc expression exceeds normal level, c‐Myc can bind DNA sequences beyond E‐boxes (Wolf et al, 2015). Upon DNA binding, c‐Myc‐MAX recruits the positive transcription elongation factor complex, which subsequently phosphorylates RNA polymerase II to increase transcription rate (Rahl et al, 2010). In addition to its well‐established role as a transcriptional activator, c‐Myc can also repress expression of numerous target genes when transcriptional co‐repressors are recruited to the c‐Myc‐MAX complex (Kleine‐Kohlbrecher et al, 2006).
As a global transcriptional regulator, c‐Myc can bind to approximately 10‐15% of the genome and regulate the expression of both protein‐encoding genes and non‐coding RNAs, which have been implicated in various cellular processes such as proliferation, growth, apoptosis, energy metabolism, and diverse biosynthetic pathways (Kress et al, 2015; Hsieh & Dang, 2016). By acting on RNA polymerases, c‐Myc not only upregulates target gene expression, but also promotes the synthesis of rRNA and tRNA, thus stimulating both transcription and translation of various ribosomal proteins and eukaryotic translation initiation factors. c‐Myc thereby activates the entire protein synthetic apparatus required for cancer cell growth (Dunn & Cowling, 2015; Stine & Dang, 2015). Furthermore, c‐Myc reprograms the metabolic landscape to generate building blocks (such as amino acids and lipids) essential for increased biomass and growth of cancer cells (Stine et al, 2015).
Alterations in MYC oncogene are a hallmark of many human cancers (Beroukhim et al, 2010). Constitutive c‐Myc activation can result from diverse mechanisms, such as chromosomal translocation and rearrangements, which frequently occur in Burkitt's lymphoma and multiple myeloma (Dalla‐Favera et al, 1982; Shou et al, 2000). In tumors where MYC is not amplified, loss of the tumor suppressor adenomatous polyposis coli and activation of the WNT/β‐catenin pathway lead to transcriptional activation of MYC via TCF transcription factor, a phenomenon occasionally observed in colorectal and prostate cancers (He et al, 1998; Nandana & Chung, 2014). While wild‐type c‐Myc has a half‐life of 15‐20 min, mutations in c‐Myc residues (such as Thr58 and Ser62) increase protein stability and contribute to in vivo tumorigenesis (Wang et al, 2011). c‐Myc overexpression is observed in up to 70% viral and alcohol‐related hepatocellular carcinoma and is associated with an aggressive phenotype (Schlaeger et al, 2008; Lin et al, 2010). Similarly, MYCN is frequently deregulated in solid tumors of neuroendocrine and neuronal origin. In neuroblastoma, the most common extracranial pediatric solid tumor, MYCN amplification is an important clinical biomarker associated with poor prognosis (Grimmer & Weiss, 2006). Furthermore, N‐Myc is a critical driver of neuroendocrine prostate cancer, a subtype of castration‐resistant prostate cancer with neuroendocrine features (Wyatt & Gleave, 2015; Dardenne et al, 2016). Finally, L‐Myc is the least understood member of this oncoprotein family, with a much lower transforming capacity than c‐Myc or N‐Myc (Birrer et al, 1988; Barrett et al, 1992). However, MYCL amplification is detected in small‐cell lung cancer more frequently than MYC or MYCN amplification and is believed to play a tumorigenic role therein (Kim et al, 2016).
ER stress and UPR signaling
The endoplasmic reticulum (ER) contributes to the proper functioning of the secretory pathway by providing a complex network of chaperones, foldases, cofactors, and quality control mechanisms (Wang & Kaufman, 2014). It is also involved in metabolic processes including lipid synthesis, gluconeogenesis, and calcium metabolism (Schwarz & Blower, 2016). Perturbations in ER homeostasis, such as disrupted proteostasis, lead to accumulation of misfolded or unfolded proteins in the ER lumen. This stress triggers an adaptive mechanism named the unfolded protein response (UPR), which increases ER chaperone expression, improves the clearance of misfolded proteins via ER‐associated degradation (ERAD), and attenuates protein translation (Walter & Ron, 2011; Ruggiano et al, 2014; Hetz et al, 2015). On the other hand, the UPR initiates apoptotic signaling when the damage is irremediable (Kim et al, 2008). The canonical UPR is initiated by three ER transmembrane stress sensors: inositol‐requiring enzyme 1 (IRE1, IRE1α, and IRE1β), protein kinase R‐like ER kinase (PERK), and activating transcription factor 6 (ATF6, ATF6α, and ATF6β) (Fig 1). They are maintained inactive when their luminal domains are bound to the glucose‐regulated protein (GRP) 78 (Hotamisligil, 2010; Walter & Ron, 2011).
Figure 1. The three arms of the UPR pathways.
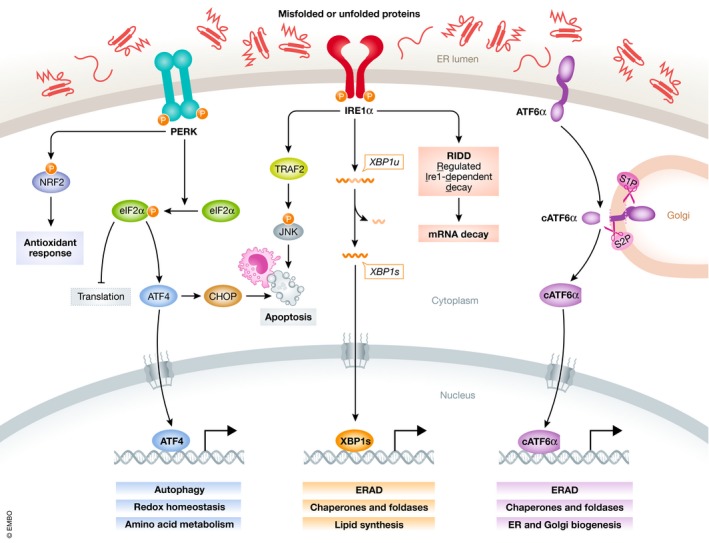
Accumulation of misfolded or unfolded proteins in the ER lumen activates the three UPR pathways initiated by PERK, IRE1α, and ATF6α. This leads to either the recovery of ER homeostasis by blocking protein translation and enhancing protein‐folding capacity and clearance of misfolded proteins, or apoptosis upon unresolved ER stress.
IRE1α comprises a kinase domain and an endoribonuclease domain on its cytosolic region. In response to the accumulation of unfolded or misfolded proteins in the ER lumen, IRE1α undergoes dimerization and trans‐autophosphorylation. This conformational change activates its RNase domain, which excises a 26‐nucleotide intron within the XBP1 mRNA (Yoshida et al, 2001; Calfon et al, 2002). This results in the expression of spliced XBP1 (XBP1s), a potent transcription factor that regulates numerous genes involved in protein folding, quality control, ERAD, and lipid synthesis (Karagoz et al, 2019). Under certain conditions, IRE1α also cleaves mRNAs, rRNAs, and miRNAs through its RNase domain via regulated IRE1α‐dependent decay (RIDD), which either preserves ER homeostasis or facilitates cell death (Hollien et al, 2009; Coelho & Domingos, 2014). When faced with unresolved stress, IRE1α may induce apoptosis by activating the c‐Jun N‐terminal kinase (JNK) signaling (Urano et al, 2000; Dhanasekaran & Reddy, 2008).
Once dissociated from GRP78, PERK undergoes dimerization and autophosphorylation, which activates its cytosolic kinase domain and phosphorylates Ser51 in eukaryotic translation initiation factor 2 (eIF2) α‐subunit (Liu et al, 2000; Holcik & Sonenberg, 2005). This transiently halts global translation and decreases the load of nascent proteins entering the ER (Wang & Kaufman, 2016). Meanwhile, it allows translation of a small subset of mRNAs with specific upstream open reading frames, such as ATF4 (Harding et al, 2000a). ATF4 is a key transcription factor that promotes adaptive response by regulating the expression of genes involved in protein folding, autophagy, and redox homeostasis (Wortel et al, 2017). It also transactivates the pro‐apoptotic protein C/EBP homologous protein (CHOP) under chronic ER stress and triggers apoptosis (Averous et al, 2004). Three additional kinases, protein kinase R, heme‐regulated eIF2α kinase, and general control nonderepressible 2 (GCN2), phosphorylate eIF2α at the same residue, which are collectively known as the “integrated stress response” (Pakos‐Zebrucka et al, 2016).
ATF6α translocates to the Golgi apparatus upon ER stress, where it is proteolytically processed by the site‐1 and site‐2 proteases (S1P and S2P), generating a cytosolic fragment that functions as a basic leucine zipper transcription factor (Haze et al, 1999). ATF6α transcriptionally upregulates the expression of many ER chaperones, as well as key UPR component genes such as XBP1 (Yoshida et al, 2001; Shoulders et al, 2013). It also plays a role in ERAD, for instance by forming heterodimers with XBP1s, and drives specific gene expression programs (Yamamoto et al, 2007).
UPR signaling in cancer
The UPR is often co‐opted by cancer cells to cope with the increased protein synthesis or the hostile tumor microenvironment (such as hypoxia and nutrient deprivation). Recently, several studies have provided comprehensive insights on the role of UPR in promoting different cancers (Clarke et al, 2014; Chevet et al, 2015; Storm et al, 2016; Urra et al, 2016; Madden et al, 2019; Wang et al, 2019).
For example, the IRE1α‐XBP1s arm helps triple‐negative breast cancer cells (TNBC) overcome hypoxic conditions by interacting with HIF1α and cooperatively regulating its transcriptional network (Chen et al, 2014). IRE1α‐XBP1s is also directly activated by androgen receptor signaling in prostate cancer cells and promotes their survival (Sheng et al, 2015). XBP1s rewires key metabolic pathways, which enables cancer cells to survive nutrient shortage conditions via transcriptional regulation of several rate‐limiting enzymes involved in hexosamine biosynthesis (Wang et al, 2014; Madden et al, 2019). In glioblastoma, XBP1 splicing promotes tumor stroma remodeling, angiogenesis, and invasion, whereas IRE1α‐mediated RIDD for miR‐17 displays anti‐angiogenic and antimigratory effects, suggesting a dual role of IRE1 RNase in glioblastoma aggressiveness (Lhomond et al, 2018). The function of PERK is also dependent on the context. PERK and eIF2α phosphorylation is suppressed in proliferative prostate cancer cells stimulated by androgens (Sheng et al, 2015), whereas ATF4 is essential for prostate cancer growth and survival (Pallmann et al, 2019). PERK activation is also shown to confer hypoxia tolerance and radiotherapy resistance to different tumor cells by upregulating expression of autophagy‐related genes via ATF4 and CHOP (Rouschop et al, 2010). Pharmacological inhibition of PERK kinase activity triggers robust antitumor effect in multiple preclinical models of pancreatic cancer and multiple myeloma (Atkins et al, 2013). ATF6α also appears to play a cytoprotective role, such as in TP53 mutant tumor cells (Sicari et al, 2019). It is required for tumor cell dormancy and contributes to resistance to chemotherapy and radiotherapy by activating mTOR and NOTCH signaling, respectively (Schewe & Aguirre‐Ghiso, 2008; Dadey et al, 2016). Additional critical functions of UPR signaling consist of reshaping the tumor stroma (Tyekucheva et al, 2017), especially that of cancer‐associated immune cells (Cubillos‐Ruiz et al, 2017). For instance, persistent activation of the IRE1α‐XBP1s axis in tumor‐associated dendritic cells and T cells disrupts their metabolic homeostasis, which results in impaired immunosuppression in ovarian cancer models (Cubillos‐Ruiz et al, 2015, 2017; Song et al, 2018).
Therefore, and contrary to what was originally thought, UPR signaling in cancer cells has a profound and complex impact on tumor initiation, progression, metastasis, and tumor microenvironment (Clarke et al, 2014; Dufey et al, 2015). Over the last few years, small molecules modulating the activity of specific UPR branches or components have been developed, and some of them are currently under clinical evaluation (Hetz et al, 2013, 2019; Jin & Saatcioglu, 2020). In line with this effort, identification of cancers potentially responsive to drugs targeting the UPR will be of great importance.
Interaction between MYC and UPR in cancer
During tumor development, protein synthesis rate is tightly regulated to sustain cell survival. Increased protein synthesis requires concomitant increased folding capacity to avoid proteotoxicity (Harding et al, 2000b). MYC activation constitutes an intrinsic stress that places further weight on protein synthesis and secretion (Tameire et al, 2015). While the ER constitutes a link between these intracellular processes and the changes in cellular biomass and growth, it has been underappreciated in the context of MYC‐hyperactivated cancers until recently.
We summarize below the direct and indirect connections found between MYC and UPR activation in different cancers and propose that MYC and UPR activation may work together to foster tumor progression. We also discuss the therapeutic potential of targeting UPR signaling in cancers with MYC overexpression.
Indirect regulation of UPR by MYC
Remarkably, UPR is induced in tumors with MYC alterations. For example, PERK‐eIF2α pathway is selectively activated in a mouse model of prostate cancer with MYC hyperactivation and is believed to hijack global protein synthesis required for cancer progression (Nguyen et al, 2018). Similarly, c‐Myc‐enhanced protein synthesis induces an adaptive ER stress response in mice with malignant rhabdoid tumors of the liver, while c‐Myc depletion decreases the levels of GRP78, ATF4, and CHOP (Carugo et al, 2019).
As a vital piece of the proteostasis system, autophagy is frequently activated to clear misfolded proteins following MYC‐induced proteotoxicity (Levy et al, 2017). In lymphoma cells, both c‐Myc and N‐Myc activate PERK‐eIF2α‐ATF4 signaling, which induces cytoprotective autophagy and attenuates ER Ca2+ release to support malignant transformation and survival (Hart et al, 2012). In Drosophila, Myc induces autophagy and cell overgrowth by activating another PERK effector, nuclear factor erythroid 2‐related factor 2 (Nrf2), a master transcription factor mediating the antioxidant responses (Cullinan et al, 2003; Ma, 2013; Nagy et al, 2013).
Furthermore, MYC direct targets also contribute to the regulation of ER stress and autophagy. As an example, N‐myc downstream‐regulated gene 1 (NDRG1) is transcriptionally repressed by both N‐Myc and c‐Myc, and inhibits PERK‐mediated autophagic pathway (Okuda & Kondoh, 1999; Sahni et al, 2014). A recent study further shows that NDRG1 inhibits IRE1α arm while facilitating ATF6α cleavage and inducing the expression of GRP78, calreticulin, and calnexin (Merlot et al, 2019). Thus, NDRG1 provides another molecular hub linking MYC with activation of UPR and autophagy.
On the other hand, MYC may suppress autophagy to induce ER stress. In non‐small‐cell lung cancer models, c‐Myc transcriptionally activates miR‐150, which blocks the fusion of autophagosomes and lysosomes through direct inhibition of EPG5. The miR‐150‐mediated autophagy defect further induces ER stress and promotes tumor growth (Li et al, 2019). Bioinformatics analysis predicts that miR‐214‐3p is c‐Myc‐regulated and likely controls the expression of XBP1 in B‐cell lymphoma, yet its function remains to be determined (Malpeli et al, 2018).
Another link between MYC and UPR in cancer is the rewired metabolism. Elevated ATF4 expression is a common feature of neuroblastoma cells with MYCN amplification and is responsible for the activation of the serine–glycine synthesis pathways essential for cell survival (Locasale, 2013; Liu et al, 2016). MYC also alters mitochondrial metabolism in these cells, making them vulnerable to glutamine deprivation. In this context, ATF4 is activated by GCN2‐eIF2α axis and promotes apoptosis by inducing PUMA, NOXA, and TRB3 expression (Qing et al, 2012). Likewise, blockade of essential amino acid transport triggers the GCN2‐eIF2α‐ATF4 pathway and inhibits neuroblastoma tumor growth, which is concomitant with attenuated translation of MYC and MYCN mRNAs (Yue et al, 2017). Therefore, the role of ATF4 in neuroblastoma cells with elevated MYC varies depending on the condition.
Notably, GCN2‐eIF2α‐ATF4 activation by MYC was recently described. By generating excess uncharged tRNAs, c‐Myc induces an optimal expression of ATF4. Then, c‐Myc and ATF4 cooperate to regulate a specific program of c‐Myc target genes, mainly involved in amino acid and protein synthesis (Tameire et al, 2019). One of these targets is eIF4E‐binding protein 1 (4E‐BP1), a repressor of eIF4F complex and mRNA translation (Gingras et al, 1999). Thus, these results provide additional mechanisms by which eIF2α phosphorylation regulates translation rate and maintains proteostasis in malignant cells with MYC overexpression.
In addition, both the RNase and kinase activities of IRE1α have been implicated in MYC‐hyperactivated tumors. In c‐Myc‐overexpressing endocrine‐resistant breast cancer cells, IRE1α activation turns on either JNK signaling for apoptosis or XBP1 splicing for survival (Shajahan‐Haq et al, 2014). In pancreatic ductal adenocarcinoma cells with activated c‐Myc, IRE1α induces the MKK4‐JNK signaling and the ATF2 transcriptional program, driving an adaptive response to the increased protein metabolism (Genovese et al, 2017). In contrast, XBP1s transactivates SIRT7 in liver cancer cells, which represses translation by cooperatively inhibiting transcription of genes encoding ribosomal proteins with c‐Myc (Shin et al, 2013). Therefore, indirect interaction between c‐Myc and IRE1α may also mitigate proteotoxicity and ER stress.
Direct regulation of UPR by MYC
Beside indirect regulation, recent studies have also shed light on the direct regulation of UPR by MYC. Zhao and colleagues have shown that c‐Myc is required for the activation of the IRE1α‐XBP1s pathway in TNBC models: Genetic knockdown of c‐Myc leads to a marked decrease in IRE1α and XBP1s, rescued by ectopic expression of c‐Myc. Chromatin immunoprecipitation (ChIP) and luciferase reporter assays further demonstrate that c‐Myc transactivates ERN1 gene expression by directly binding to multiple sites in its proximal promoter and enhancer (Zhao et al, 2018). Along these lines, another study in Burkitt's lymphoma cells reports that c‐Myc binds the E‐box sequences in the promoters of both ERN1 and XBP1 genes (Xie et al, 2018), establishing c‐Myc as a direct upstream regulator of the IRE1α‐XBP1s pathway.
At the protein level, c‐Myc physically interacts with XBP1s and enhances its transcriptional activity in TNBC models (Zhao et al, 2018). Furthermore, while the mechanism remains unknown, c‐Myc is crucial for IRE1α protein stability in Burkitt's lymphoma cells (Xie et al, 2018). As an example, IRE1α‐XBP1s mediates the oncogenic effect of c‐Myc by upregulating the expression of stearoyl‐CoA desaturase 1 (SCD1), which generates unsaturated lipids to maintain ER membrane homeostasis despite c‐Myc‐dependent proteotoxicity (Xie et al, 2018).
Several recent studies have also shed light on the direct regulation of PERK pathway components by MYC. c‐Myc binds and activates ATF4 promoter, which plays a role in anoikis resistance in human osteosarcoma cells (Mo et al, 2018) and in response to bortezomib in Elt3 rat leiomyoma cells (Babcock et al, 2013). Similarly, N‐Myc and ATF4 collectively drive the metabolic reprogramming in neuroblastoma cells, leading to dependency on the serine–glycine–one‐carbon metabolic pathway. Mechanistically, N‐Myc transactivates ATF4 expression while ATF4 contributes to the stabilization of N‐Myc protein by antagonizing its ubiquitination in a positive feedback loop (Xia et al, 2019). In addition, ATF3, an ATF4 target with critical functions in cell fate determination under stress conditions, is also directly regulated by c‐Myc and plays a role in mediating its proliferative effect (Tamura et al, 2005). c‐Myc‐mediated transcriptional repression plays a critical role in preventing cells from exiting cell cycle and in facilitating proliferation via inhibition of growth arrest and DNA damage (GADD) gene expression, such as GADD153 that encodes CHOP (Chen et al, 1996; Amundson et al, 1998). c‐Myc‐MAX complex binds to the minimal promoter region of GADD153 in vivo, where it prevents transcriptional activator c‐Myc‐interacting zinc finger protein 1 (Miz‐1) activity and impairs gene expression (Barsyte‐Lovejoy et al, 2004; Wiese et al, 2013). Taken together, these studies suggest that UPR signaling is tightly regulated by MYC and plays a key role in mediating its oncogenic effect.
Indirect regulation of MYC by UPR
Importantly, the link between MYC and UPR does not appear to be only one way, as ER stress signaling has also been shown to affect MYC expression. The calcium‐dependent serine/threonine phosphatase calcineurin is activated upon disruption in calcium homeostasis and ER stress, and activates a number of transcription factors, one of them being the nuclear factor of activated T cell (NFAT) (Bonilla et al, 2002). Activated NFAT directly binds to the proximal MYC promoter and stimulates its transcription, ultimately resulting in enhanced anchorage‐dependent and anchorage‐independent growth of pancreatic cancer cells (Buchholz et al, 2006). In multiple myeloma cells, c‐Myc protein level is maintained despite global decreased protein synthesis mediated by PERK‐eIF2α activation, owing to the upregulated activity of the MYC mRNA internal ribosome entry site upon ER stress (Shi et al, 2016).
Direct regulation of MYC by UPR
One of the most intriguing findings is that XBP1s also directly regulates MYC expression. Exogenous XBP1s has previously been shown to dose‐dependently enhance the reporter activity driven by MYC promoter (Chae et al, 2016). A similar phenomenon is also observed in colon cancer cells co‐transfected with XBP1s expression vector and MYC luciferase reporter. This is significantly reversed when Fbw7, a substrate recognition component of the SKP1‐Cullin‐F‐box‐type E3 ligase, is introduced, as Fbw7 interacts with XBP1 and facilitates its ubiquitination and degradation (Chae et al, 2019).
Consistently, our recent study in prostate cancer cells demonstrates that XBP1s directly transactivates MYC expression. Strikingly, c‐Myc and XBP1s transcriptional activities are positively correlated in multiple prostate cancer patient cohorts, underscoring the fact that these two critical transcription factors are often concurrently activated in prostate cancer (Sheng et al, 2019). Furthermore, a recent study shows that activities of c‐Myc and AR pathways are significantly correlated in prostate cancer, while c‐Myc depletion leads to decreased expression of full‐length AR, as well as of several AR splice variants involved in AR‐targeted therapy resistance (Bai et al, 2019). Therefore, androgen signaling, IRE1α‐XBP1s pathway, and c‐Myc may form a dynamic trio to support prostate cancer progression. Interestingly, this direct regulation of MYC by IRE1α‐XBP1s is not restricted to cancer cells, as a recent study reports that XBP1s also upregulates MYC expression to promote proliferation of natural killer cells (Dong et al, 2019). Taken together, these data reinforce the hypothesis of a positive feedback loop between MYC and IRE1α‐XBP1s pathway, which may be a critical driver of various MYC‐dependent cancers. The major findings on the interactions between MYC and PERK or IRE1 are summarized in Figs 2 and 3, respectively.
Figure 2. Interaction between MYC and PERK pathway.
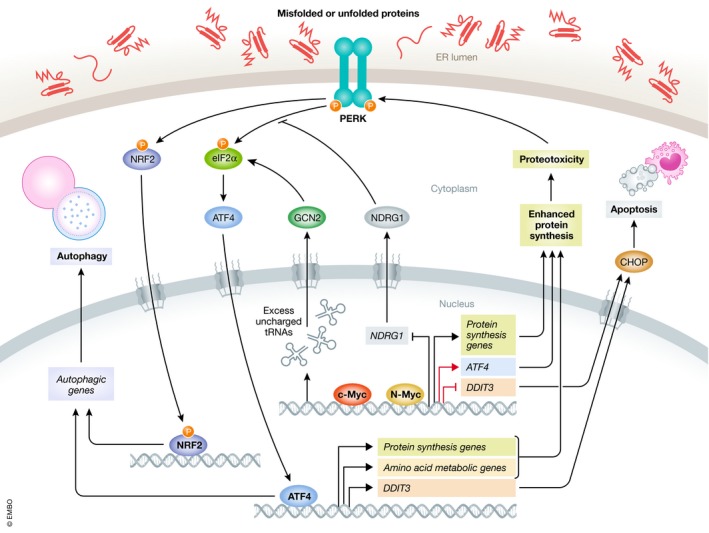
Oncogenic MYC upregulates the expression of genes involved in protein synthesis, such as ATF4, which may result in proteotoxicity. PERK‐eIF2α‐ATF4 pathway is often activated upon this intrinsic stress, which subsequently induces cytoprotective autophagy. Alternatively, PERK may activate autophagy by phosphorylating NRF2. Meanwhile, GCN2‐eIF2α‐ATF4 axis can be activated by c‐Myc‐induced excess tRNAs, resulting in metabolic reprogramming and enhanced protein synthesis. In addition, MYC mediates transcriptional repression on NDRG1 and DDIT3, which leads to enhanced cytoprotective autophagy and suppressed apoptosis, respectively. Red arrows indicate direct transcriptional regulation of PERK arm by MYC.
Figure 3. The positive feedback loop between MYC and IRE1α pathway.
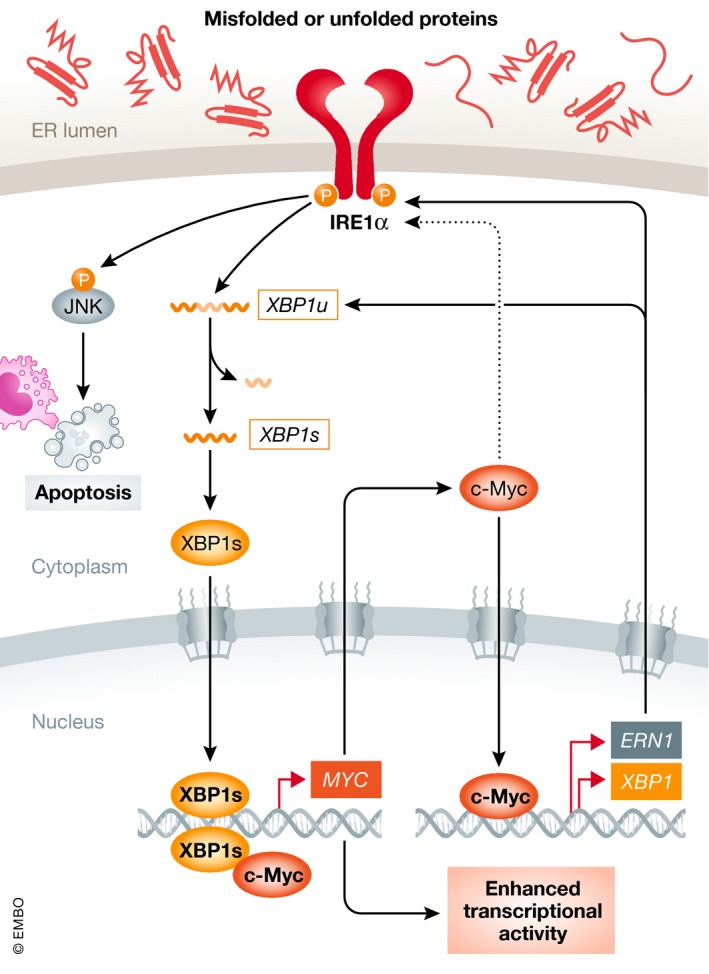
In MYC‐hyperactivated tumors, IRE1α‐XBP1s signaling and MYC likely engage in a positive feedback loop, as XBP1s transcriptionally upregulates MYC while c‐Myc directly induces ERN1 (encoding IRE1α) and XBP1 expression. c‐Myc also physically interacts with XBP1s and enhances its transcriptional activity. MYC is further shown to contribute to IRE1α protein stability via unknown mechanisms (denoted as dashed line). Red arrows indicate direct transcriptional regulation between MYC and IRE1α arm.
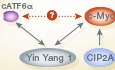
Comparatively, much less is known about the interaction between MYC and ATF6α in malignant conditions. Indirect evidence suggests that ATF6α promotes MYC activity. Indeed, ATF6α transcriptionally induces the expression of cancerous inhibitor of PP2A (CIP2A), which directly interacts with and stabilizes c‐Myc protein (Liu et al, 2018). ATF6α also induces XBP1 expression, which is capable of activating c‐Myc expression (Yoshida et al, 2001; Sheng et al, 2019). Furthermore, protein–protein interaction databases (such as BioGRID) indicate that the known ATF6α interactor Yin Yang 1 transcription factor associates with c‐Myc (Shrivastava et al, 1993; Li et al, 2000). Thus, it is worth investigating whether these proteins form a complex, and what would then be its functional significance (Fig 4). Lastly, it is reasonable to speculate that ATF6α‐mediated elevation in chaperone expression and ERAD is required for coping with the increased nascent protein load driven by MYC.
Figure 4. The interaction between MYC and ATF6α pathway.
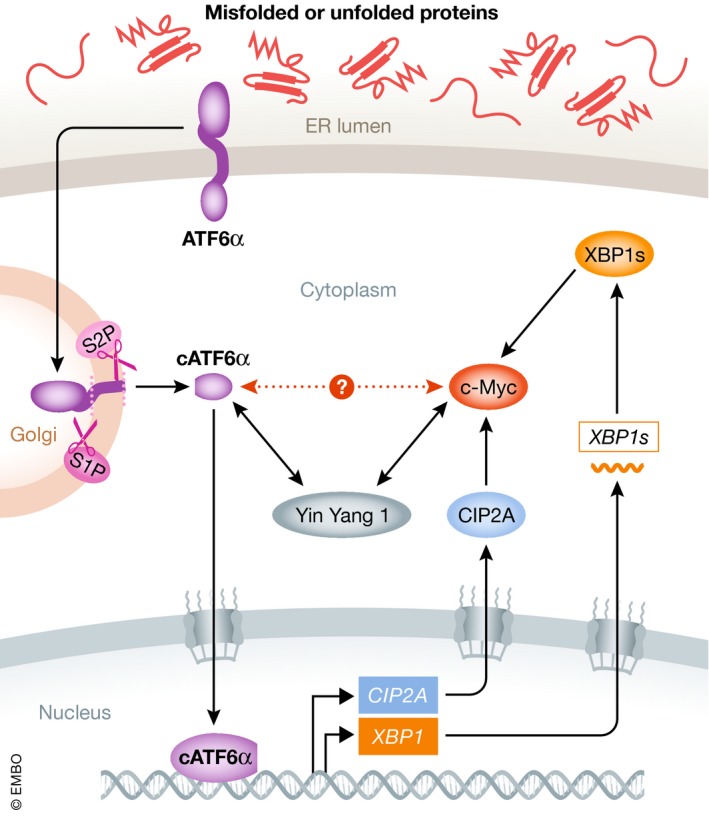
ATF6α directly induces the expression of CIP2A, which interacts with and stabilizes c‐Myc protein. As a direct target of ATF6α, XBP1 also contributes to sustain c‐Myc expression. Meanwhile, ATF6α interactor Yin Yang 1 transcription factor has been shown to associate with c‐Myc, but whether these two proteins interact with each other is not known (denoted as red dashed line).
Interestingly, a direct connection between MYC and ERAD was recently established, as c‐Myc activates ubiquitin fusion degradation 1 (UFD1) to promote progression of T‐cell acute lymphoblastic leukemia (Huiting et al, 2018). UFD1 is an E2 component of the ERAD complex and facilitates the elimination of misfolded proteins from the ER, whereas UFD1 knockdown exacerbates ER stress, activates PERK‐CHOP pathway, and induces apoptosis (Wolf & Stolz, 2012; Huiting et al, 2018). Nevertheless, the potential crosstalk between MYC and ATF6α signaling as well as ERAD remains to be explored.
Targeting UPR in MYC‐driven cancers
Building upon these critical findings, targeting the UPR has been proposed as a novel therapeutic strategy in tumors with hyperactivated MYC. Here, we highlight the application and efficacy of targeting UPR signaling in MYC‐hyperactivated cancers (Table 1).
Table 1.
Strategies and outcomes of targeting UPR in MYC‐hyperactivated cancer models
| UPR branch | Compound/Intervention | Target | Experimental models | Effect | Synergy | References |
|---|---|---|---|---|---|---|
| PERK | GSK2606414 | PERK kinase | Multiple neuroblastoma cell lines and xenografts | Reduce autophagy and inhibit growth | With GLI inhibitor GANT‐61 | Axten et al (2013), Wang et al (2018) |
| ISRIB | eIF2B | PCa mouse models and PDX | Impair cancer development, prolong survival, and inhibit metastases | / | Tsai et al (2018), Nguyen et al (2018) | |
| Genetic depletion | PERK | Transformed MEFs allografted in immunodeficient mice | Inhibit growth | / | Hart et al (2012) | |
| Genetic depletion | PERK | Drosophila fat body cells | Inhibit overgrowth | / | Nagy et al (2013) | |
| Genetic depletion | ATF4 | MEFs and lymphoma mouse models | Induce apoptosis and prolong tumor‐free and overall survival | / | Tameire et al (2019) | |
| IRE1α | MKC8866 | IRE1α RNase | TNBC PDX and GEMM | Inhibit growth | With docetaxel | Sanches et al (2014), Zhao et al (2018), Logue et al (2018) |
| MKC8866 | IRE1α RNase | Multiple PCa cell lines and xenografts | Inhibit growth | With cabazitaxel | Sheng et al (2019) | |
| B‐I09 | IRE1α RNase | Multiple BL cell lines and xenografts | Inhibit growth and induce apoptosis | With doxorubicin or vincristine | Xie et al (2018) | |
| Genetic depletion | ERN1 | 3D PDAC cell growth and orthotopic transplants | Inhibit growth and suppress tumorigenicity | / | Genovese et al (2017) |
BL, Burkitt's lymphoma; GEMM, genetically engineered mouse model; MEFs, mouse embryonic fibroblasts; PCa, prostate cancer; PDAC, pancreatic ductal adenocarcinoma; TNBC, triple‐negative breast cancer.
Genetic ablation of PERK significantly attenuates the growth of transformed mouse embryonic fibroblasts (MEFs) with induced c‐Myc expression allografted in immunodeficient mice (Hart et al, 2012). Similarly, PERK depletion prevents Myc‐induced overgrowth of fat body cell clones in Drosophila (Nagy et al, 2013). Furthermore, ATF4 ablation significantly reduces in vitro clonogenic survival of MEFs with high c‐Myc level and extends tumor‐free and overall survival in syngeneic mouse model of lymphoma with hyperactive c‐Myc (Tameire et al, 2019). PERK inhibition with an optimized kinase inhibitor, GSK2606414 (Axten et al, 2013), reduces autophagy in MYCN‐amplified neuroblastoma cells and further enhances the efficacy of GLI inhibitor in repressing the growth of these cells in vitro and in vivo (Wang et al, 2018). ISRIB is a small‐molecule compound that enhances the guanine nucleotide‐exchanging activity of eIF2B and its interaction with eIF2α, and thus re‐activates protein synthesis despite of eIF2α phosphorylation (Tsai et al, 2018). ISRIB impairs cancer development, prolongs survival of different prostate cancer mouse models, and decreases metastatic progression in an advanced castration‐resistant prostate cancer patient‐derived xenograft (PDX) model (Nguyen et al, 2018).
In parallel, genetic silencing of XBP1 selectively blocks the growth of c‐Myc‐hyperactivated TNBC cells. Pharmacological inhibition of IRE1α RNase activity using an optimized hydroxy‐aryl‐aldehyde compound MKC8866 counteracts the growth of c‐Myc‐overexpressing TNBC tumors in both PDX and genetically engineered mouse models (Sanches et al, 2014; Zhao et al, 2018). Similarly, pharmacological and genetic inhibition of XBP1 induce c‐Myc‐dependent apoptosis of Burkitt's lymphoma models, which is alleviated by exogenous unsaturated fatty acids (Xie et al, 2018). In the mesenchymal pancreatic ductal adenocarcinoma mouse models with activated c‐Myc, constitutive knockdown of Ern1 potently impairs 3D clonogenic cell growth and suppresses tumorigenicity in orthotopic transplants in vivo (Genovese et al, 2017). Likewise, disruption of the IRE1α‐XBP1s pathway by either RNA interference or small molecules targeting IRE1α RNase results in significant repression in the growth of multiple prostate cancer xenografted tumors (Sheng et al, 2015, 2019).
Of note, these studies also unanimously demonstrate that IRE1α RNase inhibition augments the effect of chemotherapy, a strategy with inferior therapeutic efficacy in MYC‐high tumors (Savage et al, 2009; Emadali et al, 2013; Lee et al, 2017). IRE1α RNase inhibition enhances the cytotoxic effect of doxorubicin or vincristine in different c‐Myc‐overexpressing Burkitt's lymphoma cells in vitro (Xie et al, 2018). In prostate cancer xenograft models, a strong synergistic tumor growth inhibition is observed when MKC8866 treatment is combined with cabazitaxel (Sheng et al, 2019). In TNBC, the same IRE1α RNase inhibitor substantially enhances the efficacy of docetaxel in PDX as well as syngeneic p53‐null transgenic mouse models with c‐Myc hyperactivation (Zhao et al, 2018). These findings coincide with a recent TNBC study showing that MKC8866 increases the effectiveness of xenografted tumors to paclitaxel, which may be due to the modulation of the tumor cell secretome (Logue et al, 2018). Nevertheless, these data certainly underline the potential of targeting IRE1α either as a monotherapy in MYC‐high tumors or in combination with chemotherapy in the future.
Conclusions
Direct pharmacological inhibition of MYC has proven to be challenging. Thus, alternative means, such as targeting MYC synthetic lethal partners, have raised interest. The reprogrammed growth, proliferation, and metabolism driven by oncogenic MYC render cancer cells more vulnerable to the disruption of certain biological processes on which they rely. MYC activation has been shown to be synthetically lethal with inhibition of translation, spliceosome, cell cycle, and metabolism (Stine & Dang, 2015; Hsieh & Dang, 2016). The ER stress response now takes its place among these synthetic lethal targets. However, despite exciting recent progress, further preclinical and clinical evaluation will be needed to establish rational therapeutic design. Importantly, biomarkers should also be identified to help discriminating patients that may benefit from different UPR inhibitors.
Conflict of interest
The authors declare that they have no conflict of interest.
Pending issues
-
(i)
Detailed knowledge of the interactions between MYC and the entire UPR network, depending on the context.
-
(ii)
Elucidating the potential direct interaction between MYC and ATF6α and its functional role in different cancers and cancer phases.
-
(iii)
In‐depth preclinical evaluation and optimization of strategies targeting the UPR in MYC‐hyperactivated cancers.
-
(iv)
Uncovering synergy and its underlying mechanism between compounds modulating UPR activity and clinical drugs, such as chemotherapeutic agents, in MYC‐high tumors.
-
(v)
Translation of the basic and preclinical knowledge into clinical application.
Acknowledgements
This work was supported by the Talent Introduction Fund, Huazhong University of Science and Technology (3004513124); Young Scholar Fund, National Natural Science Foundation of China (81802546); Southern Eastern Norway Regional Health Authority (2017043); and Sanming Project of Medicine in Shenzhen (No. SZSM201911003).
EMBO Mol Med (2020) 12: e11845
See the Glossary for abbreviations used in this article.
References
- Amundson SA, Zhan Q, Penn LZ, Fornace AJ Jr (1998) Myc suppresses induction of the growth arrest genes gadd34, gadd45, and gadd153 by DNA‐damaging agents. Oncogene 17: 2149–2154 [DOI] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N et al (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73: 1993–2002 [DOI] [PubMed] [Google Scholar]
- Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P (2004) Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem 279: 5288–5297 [DOI] [PubMed] [Google Scholar]
- Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E et al (2013) Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med Chem Lett 4: 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock JT, Nguyen HB, He Y, Hendricks JW, Wek RC, Quilliam LA (2013) Mammalian target of rapamycin complex 1 (mTORC1) enhances bortezomib‐induced death in tuberous sclerosis complex (TSC)‐null cells by a c‐MYC‐dependent induction of the unfolded protein response. J Biol Chem 288: 15687–15698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Cao S, Jin L, Kobelski M, Schouest B, Wang X, Ungerleider N, Baddoo M, Zhang W, Corey E et al (2019) A positive role of c‐Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 38: 4977–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J, Birrer MJ, Kato GJ, Dosaka‐Akita H, Dang CV (1992) Activation domains of L‐Myc and c‐Myc determine their transforming potencies in rat embryo cells. Mol Cell Biol 12: 3130–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsyte‐Lovejoy D, Mao DY, Penn LZ (2004) c‐Myc represses the proximal promoters of GADD45a and GADD153 by a post‐RNA polymerase II recruitment mechanism. Oncogene 23: 3481–3486 [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M et al (2010) The landscape of somatic copy‐number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrer MJ, Segal S, DeGreve JS, Kaye F, Sausville EA, Minna JD (1988) L‐myc cooperates with ras to transform primary rat embryo fibroblasts. Mol Cell Biol 8: 2668–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H (1993) Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol 13: 5216–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla M, Nastase KK, Cunningham KW (2002) Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J 21: 2343–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V (2006) Overexpression of c‐myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J 25: 3714–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415: 92–96 [DOI] [PubMed] [Google Scholar]
- Carugo A, Minelli R, Sapio L, Soeung M, Carbone F, Robinson FS, Tepper J, Chen Z, Lovisa S, Svelto M et al (2019) p53 is a master regulator of proteostasis in SMARCB1‐deficient malignant rhabdoid tumors. Cancer Cell 35: 204–220.e209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Nussenzweig A, Guo M, Kim D, Li GC, Ling CC (1996) Down‐regulation of gadd153 by c‐myc in rat fibroblasts and its effect on cell growth and radiation‐induced apoptosis. Oncogene 13: 1659–1665 [PubMed] [Google Scholar]
- Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y et al (2014) XBP1 promotes triple‐negative breast cancer by controlling the HIF1alpha pathway. Nature 508: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae U, Park SJ, Kim B, Wei S, Min JS, Lee JH, Park SH, Lee AH, Lu KP, Lee DS et al (2016) Critical role of XBP1 in cancer signalling is regulated by PIN1. Biochem J 473: 2603–2610 [DOI] [PubMed] [Google Scholar]
- Chae U, Lee H, Kim B, Jung H, Kim BM, Lee AH, Lee DS, Min SH (2019) A negative feedback loop between XBP1 and Fbw7 regulates cancer development. Oncogenesis 8: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevet E, Hetz C, Samali A (2015) Endoplasmic reticulum stress‐activated cell reprogramming in oncogenesis. Cancer Discov 5: 586–597 [DOI] [PubMed] [Google Scholar]
- Clarke HJ, Chambers JE, Liniker E, Marciniak SJ (2014) Endoplasmic reticulum stress in malignancy. Cancer Cell 25: 563–573 [DOI] [PubMed] [Google Scholar]
- Coelho DS, Domingos PM (2014) Physiological roles of regulated Ire1 dependent decay. Front Genet 5: 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci‐Sorrell M, McFerrin L, Eisenman RN (2014) An overview of MYC and its interactome. Cold Spring Harb Perspect Med 4: a014357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales‐Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K et al (2015) ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell 161: 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos‐Ruiz JR, Bettigole SE, Glimcher LH (2017) Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168: 692–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol 23: 7198–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadey DY, Kapoor V, Khudanyan A, Urano F, Kim AH, Thotala D, Hallahan DE (2016) The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 7: 2080–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla‐Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM (1982) Human c‐myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA 79: 7824–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T et al (2016) N‐Myc induces an EZH2‐mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30: 563–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27: 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, Chen X, Sun JC, Glimcher LH (2019) The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c‐Myc. Nat Immunol 20: 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufey E, Urra H, Hetz C (2015) ER proteostasis addiction in cancer biology: novel concepts. Semin Cancer Biol 33: 40–47 [DOI] [PubMed] [Google Scholar]
- Dunn S, Cowling VH (2015) Myc and mRNA capping. Biochem Biophys Acta 1849: 501–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emadali A, Rousseaux S, Bruder‐Costa J, Rome C, Duley S, Hamaidia S, Betton P, Debernardi A, Leroux D, Bernay B et al (2013) Identification of a novel BET bromodomain inhibitor‐sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers. EMBO Mol Med 5: 1180–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c‐Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, Nezi L, Corti D, Minelli R, Pettazzoni P et al (2017) Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 542: 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N (1999) Regulation of 4E‐BP1 phosphorylation: a novel two‐step mechanism. Genes Dev 13: 1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer MR, Weiss WA (2006) Childhood tumors of the nervous system as disorders of normal development. Curr Opin Pediatr 18: 634–638 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000a) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000b) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897–904 [DOI] [PubMed] [Google Scholar]
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M et al (2012) ER stress‐mediated autophagy promotes Myc‐dependent transformation and tumor growth. J Clin Investig 122: 4621–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c‐MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP (2013) Targeting the unfolded protein response in disease. Nat Rev Drug Discovery 12: 703–719 [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Oakes SA (2015) Proteostasis control by the unfolded protein response. Nat Cell Biol 17: 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Axten JM, Patterson JB (2019) Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 15: 764–775 [DOI] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6: 318–327 [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009) Regulated Ire1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AL, Dang CV (2016) MYC, metabolic synthetic lethality, and cancer. Recent Results Cancer Res 207: 73–91 [DOI] [PubMed] [Google Scholar]
- Huiting LN, Samaha Y, Zhang GL, Roderick JE, Li B, Anderson NM, Wang YW, Wang L, Laroche F, Choi JW et al (2018) UFD1 contributes to MYC‐mediated leukemia aggressiveness through suppression of the proapoptotic unfolded protein response. Leukemia 32: 2339–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Saatcioglu F (2020) Targeting the unfolded protein response in hormone‐regulated cancers. Trends Cancer 6: 160–171 [DOI] [PubMed] [Google Scholar]
- Karagoz GE, Acosta‐Alvear D, Walter P (2019) The unfolded protein response: detecting and responding to fluctuations in the protein‐folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol 11: a033886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery 7: 1013–1030 [DOI] [PubMed] [Google Scholar]
- Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, Dunn CT, Lee AY, Kim K, Lee CS et al (2016) Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev 30: 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine‐Kohlbrecher D, Adhikary S, Eilers M (2006) Mechanisms of transcriptional repression by Myc. Curr Top Microbiol Immunol 302: 51–62 [DOI] [PubMed] [Google Scholar]
- Kress TR, Sabo A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607 [DOI] [PubMed] [Google Scholar]
- Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero‐Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sanchez V, Sanders ME et al (2017) MYC and MCL1 cooperatively promote chemotherapy‐resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab 26: 633–647.e637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, Thorburn A (2017) Targeting autophagy in cancer. Nat Rev Cancer 17: 528–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhomond S, Avril T, Dejeans N, Voutetakis K, Doultsinos D, McMahon M, Pineau R, Obacz J, Papadodima O, Jouan F et al (2018) Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol Med 10: e7929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, Lee AS (2000) ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress‐induced changes and synergistic interactions with NF‐Y and YY1. Mol Cell Biol 20: 5096–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liu J, Cao W, Xiao X, Liang L, Liu‐Smith F, Wang W, Liu H, Zhou P, Ouyang R et al (2019) C‐myc/miR‐150/EPG5 axis mediated dysfunction of autophagy promotes development of non‐small cell lung cancer. Theranostics 9: 5134–5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CP, Liu CR, Lee CN, Chan TS, Liu HE (2010) Targeting c‐Myc as a novel approach for hepatocellular carcinoma. World J Hepatol 2: 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Schroder M, Kaufman RJ (2000) Ligand‐independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem 275: 24881–24885 [DOI] [PubMed] [Google Scholar]
- Liu M, Xia Y, Ding J, Ye B, Zhao E, Choi JH, Alptekin A, Yan C, Dong Z, Huang S et al (2016) Transcriptional profiling reveals a common metabolic program in high‐risk human neuroblastoma and mouse neuroblastoma sphere‐forming cells. Cell Rep 17: 609–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Hsu CC, Huang TT, Lee CH, Chen JL, Yang SH, Jiang JK, Chen WS, Lee KD, Teng HW (2018) ER stress‐related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol Oncol 12: 1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW (2013) Serine, glycine and one‐carbon units: cancer metabolism in full circle. Nat Rev Cancer 13: 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SE, McGrath EP, Cleary P, Greene S, Mnich K, Almanza A, Chevet E, Dwyer RM, Oommen A, Legembre P et al (2018) Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat Commun 9: 3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden E, Logue SE, Healy SJ, Manie S, Samali A (2019) The role of the unfolded protein response in cancer progression: from oncogenesis to chemoresistance. Biol Cell 111: 1–17 [DOI] [PubMed] [Google Scholar]
- Malpeli G, Barbi S, Tosadori G, Greco C, Zupo S, Pedron S, Brunelli M, Bertolaso A, Scupoli MT, Krampera M et al (2018) MYC‐related microRNAs signatures in non‐Hodgkin B‐cell lymphomas and their relationships with core cellular pathways. Oncotarget 9: 29753–29771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlot AM, Porter GM, Sahni S, Lim EG, Peres P, Richardson DR (2019) The metastasis suppressor, NDRG1, differentially modulates the endoplasmic reticulum stress response. Biochim Biophys Acta 1865: 2094–2110 [DOI] [PubMed] [Google Scholar]
- Mo H, Guan J, Mo L, He J, Wu Z, Lin X, Liu B, Yuan Z (2018) ATF4 regulated by MYC has an important function in anoikis resistance in human osteosarcoma cells. Mol Med Rep 17: 3658–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Varga A, Pircs K, Hegedus K, Juhasz G (2013) Myc‐driven overgrowth requires unfolded protein response‐mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLoS Genet 9: e1003664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandana S, Chung LW (2014) Prostate cancer progression and metastasis: potential regulatory pathways for therapeutic targeting. Am J Clin Exp Urol 2: 92–101 [PMC free article] [PubMed] [Google Scholar]
- Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F et al (2018) Development of a stress response therapy targeting aggressive prostate cancer. Sci Transl Med 10: eaar2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Kondoh H (1999) Identification of new genes ndr2 and ndr3 which are related to Ndr1/RTP/Drg1 but show distinct tissue specificity and response to N‐myc. Biochem Biophys Res Commun 266: 208–215 [DOI] [PubMed] [Google Scholar]
- Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM (2016) The integrated stress response. EMBO Rep 17: 1374–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallmann N, Livgard M, Tesikova M, Zeynep Nenseth H, Akkus E, Sikkeland J, Jin Y, Koc D, Kuzu OF, Pradhan M et al (2019) Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene 38: 6301–6318 [DOI] [PubMed] [Google Scholar]
- Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM et al (2012) ATF4 regulates MYC‐mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22: 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c‐Myc regulates transcriptional pause release. Cell 141: 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW et al (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, Carvalho P (2014) Quality control: ER‐associated degradation: protein quality control and beyond. J Cell Biol 204: 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni S, Bae DH, Lane DJ, Kovacevic Z, Kalinowski DS, Jansson PJ, Richardson DR (2014) The metastasis suppressor, N‐myc downstream‐regulated gene 1 (NDRG1), inhibits stress‐induced autophagy in cancer cells. J Biol Chem 289: 9692–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches M, Duffy NM, Talukdar M, Thevakumaran N, Chiovitti D, Canny MD, Lee K, Kurinov I, Uehling D, Al‐awar R et al (2014) Structure and mechanism of action of the hydroxy‐aryl‐aldehyde class of IRE1 endoribonuclease inhibitors. Nat Commun 5: 4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage KJ, Johnson NA, Ben‐Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD (2009) MYC gene rearrangements are associated with a poor prognosis in diffuse large B‐cell lymphoma patients treated with R‐CHOP chemotherapy. Blood 114: 3533–3537 [DOI] [PubMed] [Google Scholar]
- Schewe DM, Aguirre‐Ghiso JA (2008) ATF6alpha‐Rheb‐mTOR signaling promotes survival of dormant tumor cells in vivo . Proc Natl Acad Sci USA 105: 10519–10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R, Lichter P et al (2008) Etiology‐dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 47: 511–520 [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Blower MD (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73: 79–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan‐Haq AN, Cook KL, Schwartz‐Roberts JL, Eltayeb AE, Demas DM, Warri AM, Facey CO, Hilakivi‐Clarke LA, Clarke R (2014) MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer 13: 239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, Arnoldussen YJ, Storm M, Tesikova M, Nenseth HZ, Zhao S, Fazli L, Rennie P, Risberg B, Waehre H et al (2015) Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol Med 7: 788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, Nenseth HZ, Qu S, Kuzu OF, Frahnow T, Simon L, Greene S, Zeng Q, Fazli L, Rennie PS et al (2019) IRE1alpha‐XBP1s pathway promotes prostate cancer by activating c‐MYC signaling. Nat Commun 10: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yang Y, Hoang B, Bardeleben C, Holmes B, Gera J, Lichtenstein A (2016) Therapeutic potential of targeting IRES‐dependent c‐myc translation in multiple myeloma cells during ER stress. Oncogene 35: 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K et al (2013) SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM (2000) Diverse karyotypic abnormalities of the c‐myc locus associated with c‐myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA 97: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR III, Su AI, Kelly JW, Wiseman RL (2013) Stress‐independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3: 1279–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Saleque S, Kalpana GV, Artandi S, Goff SP, Calame K (1993) Inhibition of transcriptional regulator Yin‐Yang‐1 by association with c‐Myc. Science 262: 1889–1892 [DOI] [PubMed] [Google Scholar]
- Sicari D, Fantuz M, Bellazzo A, Valentino E, Apollonio M, Pontisso I, Di Cristino F, Dal Ferro M, Bicciato S, Del Sal G et al (2019) Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 38: 6184–6195 [DOI] [PubMed] [Google Scholar]
- Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C et al (2018) IRE1alpha‐XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562: 423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Dang CV (2015) Splicing and dicing MYC‐mediated synthetic lethality. Cancer Cell 28: 405–406 [DOI] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5: 1024–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm M, Sheng X, Arnoldussen YJ, Saatcioglu F (2016) Prostate cancer and the unfolded protein response. Oncotarget 7: 54051–54066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameire F, Verginadis II, Koumenis C (2015) Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: mechanisms and targets for therapy. Semin Cancer Biol 33: 3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy AM, Fu W et al (2019) ATF4 couples MYC‐dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 21: 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Hua B, Adachi S, Guney I, Kawauchi J, Morioka M, Tamamori‐Adachi M, Tanaka Y, Nakabeppu Y, Sunamori M et al (2005) Stress response gene ATF3 is a target of c‐myc in serum‐induced cell proliferation. EMBO J 24: 2590–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Miller‐Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, Walter P (2018) Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory‐enhancing molecule. Science 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyekucheva S, Bowden M, Bango C, Giunchi F, Huang Y, Zhou C, Bondi A, Lis R, Van Hemelrijck M, Andren O et al (2017) Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat Commun 8: 420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666 [DOI] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, Hetz C (2016) Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2: 252–262 [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC (2011) Phosphorylation regulates c‐Myc's oncogenic activity in the mammary gland. Can Res 71: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ (2014) The impact of the endoplasmic reticulum protein‐folding environment on cancer development. Nat Rev Cancer 14: 581–597 [DOI] [PubMed] [Google Scholar]
- Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N et al (2014) Spliced X‐box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156: 1179–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326–335 [DOI] [PubMed] [Google Scholar]
- Wang J, Huang S, Tian R, Chen J, Gao H, Xie C, Shan Y, Zhang Z, Gu S, Xu M (2018) The protective autophagy activated by GANT‐61 in MYCN amplified neuroblastoma cells is mediated by PERK. Oncotarget 9: 14413–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang K, Jin Y, Sheng X (2019) Endoplasmic reticulum proteostasis control and gastric cancer. Cancer Lett 449: 263–271 [DOI] [PubMed] [Google Scholar]
- Wiese KE, Walz S, von Eyss B, Wolf E, Athineos D, Sansom O, Eilers M (2013) The role of MIZ‐1 in MYC‐dependent tumorigenesis. Cold Spring Harb Perspect Med 3: a014290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DH, Stolz A (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochem Biophys Acta 1823: 117–124 [DOI] [PubMed] [Google Scholar]
- Wolf E, Lin CY, Eilers M, Levens DL (2015) Taming of the beast: shaping Myc‐dependent amplification. Trends Cell Biol 25: 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN (2017) Surviving stress: modulation of ATF4‐mediated stress responses in normal and malignant cells. Trends Endocrinol Metab 28: 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AW, Gleave ME (2015) Targeting the adaptive molecular landscape of castration‐resistant prostate cancer. EMBO Mol Med 7: 878–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ye B, Ding J, Yu Y, Alptekin A, Thangaraju M, Prasad PD, Ding ZC, Park EJ, Choi JH et al (2019) Metabolic reprogramming by MYCN confers dependence on the serine‐glycine‐one‐carbon biosynthetic pathway. Can Res 79: 3837–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Tang CH, Song JH, Mancuso A, Del Valle JR, Cao J, Xiang Y, Dang CV, Lan R, Sanchez DJ et al (2018) IRE1alpha RNase‐dependent lipid homeostasis promotes survival in Myc‐transformed cancers. J Clin Investig 128: 1300–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13: 365–376 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891 [DOI] [PubMed] [Google Scholar]
- Yue M, Jiang J, Gao P, Liu H, Qing G (2017) Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep 21: 3819–3832 [DOI] [PubMed] [Google Scholar]
- Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ et al (2018) Pharmacological targeting of MYC‐regulated IRE1/XBP1 pathway suppresses MYC‐driven breast cancer. J Clin Investig 128: 1283–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]