

Abstract
Non-small cell lung cancer (NSCLC) carries a high mortality, and efficacious therapy is lacking. Therapy using chimeric antigen receptor (CAR) T cells has been used efficaciously against hematologic malignancies, but the curative effect against solid tumors is not satisfactory. A lack of antigen targets is one of the main reasons for this limited efficacy. Previously, we showed that lung-specific X (LUNX; also known as BPIFA1, PLUNC, and SPLUNC1) is overexpressed in lung cancer cells. Here, we constructed a CAR-T-cell-based strategy to target LunX (CARLunX T cells). CAR T cells were developed so that, upon specific recognition of LunX, they secreted cytokines and killed LunX-positive NSCLC cells. In vitro, CARLunX T cells displayed enhanced toxicity toward NSCLC lines and production of cytokines and showed specific LunX-dependent recognition of NSCLC cells. Adoptive transfer of CARLunX T cells induced regression of established metastatic lung cancer xenografts and prolonged survival. CARLunX T cells could infiltrate into the tumor. Also, we constructed a patient-derived xenograft model of lung cancer. After therapy with CARLunX T cells, tumor growth was suppressed, and survival was prolonged significantly. Together, our findings offer preclinical evidence of the immunotherapeutic targeting of LunX as a strategy to treat NSCLC.
Keywords: LunX, CAR-T cell therapy, PDX model, solid tumor
Graphical Abstract
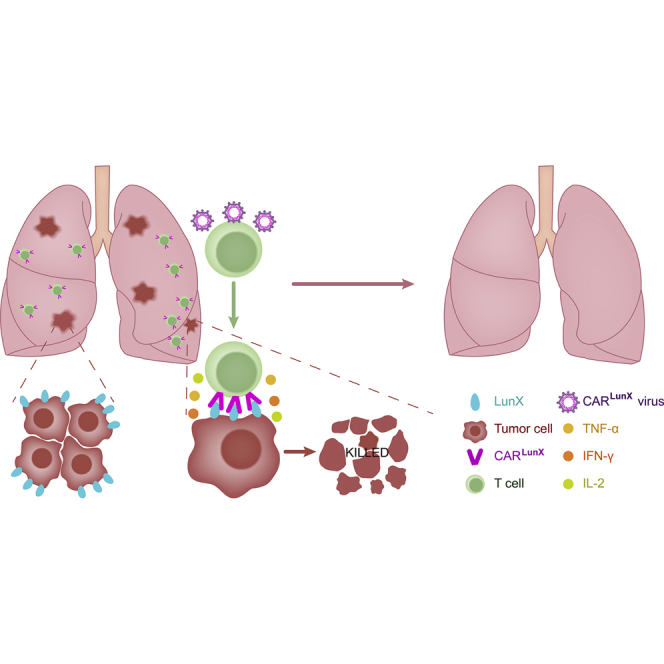
CAR-T cell therapy has been used for many kinds of cancers, but the specific target antigen for non-small cell lung cancer (NSCLC) is rare. Hu et al. report the CARLunX-T cell therapy for NSCLC. By treatment with CARLunX-T cells, the tumor growth was inhibited significantly in the mouse model.
Introduction
Non-small cell lung cancer (NSCLC) is an aggressive disease and an efficacious therapy is lacking.1 Therapy using chimeric antigen receptor (CAR) T cells has been developed against hematologic malignancies, but their curative effect against solid tumors has not been satisfactory. Also, targets for solid tumors (especially NSCLC) are limited.2
Several types of antigen are overexpressed on the surface of tumor cells. However, “on-target/off tumor” side effects can occur because of the high sensitivity of CAR T cells for low-level antigen expression. Thus, a good antigen target must be restricted only to tumor cells, and be expressed at very low levels in normal tissues.3
For treatment of lung cancer using CAR T cells, the candidate target antigens that have been reported recently are mesothelin,3, 4, 5, 6 disialoganglioside (GD2), human epidermal growth factor receptor (HER)2 and epidermal growth factor receptor (EGFR).3,5,7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20
Lung-specific X (LunX) is a member of the palate, lung, and nasal epithelium clone protein family.21 The latter shows high expression on NSCLC cells but shows no expression on human normal lung tissues or adipose tissues, or tissues from the liver, brain, pancreas, skeletal muscle, adrenal glands, kidneys, prostate gland, heart, stomach, spleen, mammary glands, or thyroid gland.22 Previously, we demonstrated that LunX antibody-based therapy could suppress the growth, metastasis, and invasion of LunX-positive tumor cells by inducing downregulation of expression of LunX and blockade of the downstream pathways of LunX.23,24 In another study by our research team using mice, we showed that treatment with LunX antibody led to no cases of sudden death, pulmonary inflammation, or lung injury, which demonstrated an absence of toxic side effects of LunX antibody.25
In the present study, we searched for a target antigen specific for lung cancer that has high expression among NSCLC patients. We constructed human T cells to express a CAR that was specific to LunX (CARLunX T cells), and we incorporated a CD28-4-1BB-CD3ζ co-stimulatory signaling domain. We evaluated the anti-NSCLC function of these cells in vitro and in an in vivo orthotopic xenograft and patient-derived xenograft (PDX) mouse model of NSCLC.
Our results showed that CARLunX T cells could eradicate LunX-expressing NSCLC cells specifically and efficiently in vitro and in vivo. Also, CARLunX T cells could infiltrate into a solid tumor, and this eradication was dependent upon LunX. Our data suggest that treatment using CARLunX T cells could be promising against NSCLC.
Results
Construction of a LunX-Targeted CAR
Previously, we demonstrated that LunX is overexpressed in NSCLC cells and that LunX mRNA could be a diagnostic marker for NSCLC. Furthermore, we selected one antibody (S-35-8) and showed that it could bind specifically to LunX.24 Here, we also used surface plasmon resonance to analyze kinetic affinity and confirmed that single chain variable fragment (scFv) does have strong affinity for LUNX (Figure 1A). Therefore, S-35-8 could be an “ideal” antibody to construct the CAR. It has been reported26,27 that, compared with the CD28 CD3ζ signaling domain, the 4-1BB CD3ζ signaling domain not only mediates the killing and proliferative capacity of CAR T cells, it also increases their persistence in vivo. We generated a specific CARLunX with a backbone of a PCDH lentiviral vector sequentially containing a signal peptide (SP), a heavy-chain variable region (VH), linker, light-chain variable region (VL), Myc tag, hinge, CD28 transmembrane part, 4-1BB, and CD3ζ (Figures 1B and 1C).28,29
Figure 1.

Construction of CARLunX
(A) Representative SPR affinity response curve of LunX antigen protein and the gradient concentration of s-35-8 antibody from 9.375 nM to 300 nM. (B) CARLunX cells used in functional experiments (schematic). VH, variable heavy chain; VL, variable light chain and the LunX-antigen specific CAR design. (C) The structure of each part of CARLunX. (D) Analyses of the sequence of s-35-8, CD137, and CD247 from the constructed plasmid by PCR. (E) Measurement of the mRNA expression of s-35-8, CD137, and CD247 from PCDH-MSCV-CARLunX-EF1-EGFP or empty vector-transfected 293T cells by RT-PCR. M represents the 100-bp DNA ladder and 1, 2, and 3 represent three repeated samples. One of three independent experiments is shown. (F) Immunofluorescence staining for c-myc tag of PCDH-MSCV-CARLunX-EF1-EGFP or empty vector-transfected 293T cells to analyze the transmembrane structure of the CAR. Scale bar, 100 μm.
To ensure that each part of the CAR was inserted into the backbone of the PCDH lentiviral vector and could express at the mRNA level, we carried out polymerase chain reaction (PCR) and reverse transcription polymerase chain reaction (RT-PCR) (Figures 1D and 1E). Notably, expression of S-35-8 scFv, CD137, and CD247 was increased remarkably in the PCDH-CARLunX lentiviral-vector backbone and transfected 293T cells compared with the empty vector. To detect the transmembrane part, we used the c-myc tag. Using immunofluorescence, C-myc was detected and localized on the cytomembrane of transduced 293T cells, which indicated that CARLunX had been constructed.
LunX Antigen Sensitizes CARLunX T Cells to Secrete Cytokines
Next, we stimulated normal human donor T cells with anti-CD3 and anti-CD28 monoclonal antibody (mAb)-coated beads and then transduced T cells with a lentiviral vector encoding CD19 CAR or LunX CAR. Forty-eight hours after transfection, the efficiency of transfection was observed by inverted fluorescence microscopy (Figure 2A) and measured by flow cytometry (Figure 2B). By measuring expression of EGFP and C-myc tag, high transfection efficiency was shown. Furthermore, immunofluorescence revealed that C-myc was localized on the membrane of CARLunX T cells, indicating that the CAR was expressed on the surface of T cells after transduction with the CAR construct. Next, we coated 96-well plates with LunX-antigen peptides (1,000 ng/mL), left the plates for 12 h at 4°C, and then co-cultured with 104 CARLunX T cells, CARCD19 T cells, or anti-CD3ε mAb as control (1 mg/mL) for 24 h; OKT3 was used as a positive control for T cell stimulation (Figure 2D). Expression of interferon (IFN)-γγ, interleukin (IL)-2, and tumor necrosis factor (TNF)-α was measured by enzyme-linked immunosorbent assays (ELISAs) after co-culturing the peptides of LunX antigen and CAR T cells (Figure 2E). As expected, CARLunX T cells secreted high quantities of IFN-γγ, IL-2, and TNF-α in response to the peptides of LunX antigen. In contrast, CARCD19 T cells demonstrated no reactivity to the peptides of LunX antigen but showed similar cytokine secretion in response to OKT3 stimulation. Collectively, we constructed CARLunX T cells that could produce cytokines specifically in response to the peptides of LunX antigen.
Figure 2.

LunX Antigen Induces Expression of the Immune-Function Molecules of CARLunX T Cells
(A) Microscopic appearance of enhanced GFP (EGFP) expressed by lentivirus-infected human primary T cells. Scale bar, 50 μm. (B) Expression of chimeric s-35-8 scFv on the surface of human primary T cells transduced with the LunX-CAR construct was measured by flow cytometry after cells had been stained with an anti-myc antibody or IgG1 isotype control. Data are representative of three experiments with similar results. (C) Immunofluorescence staining for c-myc tag and EGFP of human primary T cells transduced with the LunX-CAR construct. Scale bar, 5 μm. (D)The graphical representation of experimental protocol in (E). (E) Indirect ELISAs quantifying production of the cytokines IFN-γ, IL-2, or TNF-α in supernatants from LunX CAR T cells and CD19 CAR T cells cultured on peptide-bound plates for 24 h. LunX-antigen peptides were plated at 1,000 ng/mL. Antibody against OKT3 (10 μg/mL) but not transfected by lentivirus was used as a control stimulant of T cells. n = 3, and results are representative of three independent experiments. Data in (E) are the mean ± SEM. Unpaired t test, ∗∗∗∗p < 0.0001.
Targeted Killing of Lung Cancer Cells by CARLunX T Cells
After generation of CARLunX T cells, we constructed CARCD19 T cells as control cells. We determined if LunX-positive cells were killed more efficiently by CARlunX T cells than by CARCD19 T cells. We measured LunX expression in the NSCLC cell lines NCI-H292, NCI-H1650, and A549 by immunofluorescence. In accordance with previous work,24 all three NSCLC cell lines showed high expression of LunX. Conversely, the lung fibroblast cell line HFL1 showed no expression of LunX, which demonstrated that the antibody S-35-8 had good specificity (Figure 3A; Figure S1A). In long-term killing assays at a ratio of effector cell:target cell of 10:1, CARLunX T cells killed NCI-H292, NCI-H1650, and A549 cells more quickly than that observed using CARCD19 T cells. As the control, CARCD19 T cells did not show good killing ability (Figure 3B).
Figure 3.

CARLunX T Cells Are Toxic against LunX-Positive Lung Cancer Cells
(A) Immunofluorescent staining for LUNX in NSCLC (NCI-H292, NCI-H1650, A549, and NCI-H358) and lung fibroblasts (HFL1). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar, 50 μm. (B) Toxicity of LunX-based CAR T cells against the lung cancer cell lines NCI-H292, NCI-H1650, and A549 at a ratio of effector cell:target cell of 10:1. The control indicates that effector cells were not added. One of three independent experiments is shown. (C) Toxicity of CARLunX T cells and CARCD19 T cells against the lung cancer cell lines NCI-H292, NCI-H1650, and A549 at different ratios of effector cell:target cell for 40 h. One of three independent experiments is shown.
In addition, we measured toxicity with different ratios of effector cell:target cell of CAR T cells and NSCLC cell lines. After co-culture of CAR T cells and NSCLC cell lines for 20 h, CARLunX T cells exhibited better specific toxicity than that seen for CARCD19 T cells. However, CARCD19 T cells killed NSCLC cell lines at very low killing efficiency, so this toxicity may have been caused by T cells themselves (Figure 3C).
Specific Recognition of LunX-Positive Lung Cancer Cells Enhances the Immune Response of CARLunX T Cells
We showed above that LunX-antigen peptides could stimulate CARLunX T cells to secrete several cytokines. Next, we co-cultured CAR T cells and NSCLC cell lines to ascertain if LunX-positive cell lines could induce a stronger response from CARLunX T cells than from CARCD19 T cells. We analyzed CAR T cells after a 24-h challenge with LunX-positive cells; as controls, we used CARCD19 T cells. As anticipated, in the presence of LunX-positive tumor cells, CARLunX T showed a significant (p < 0.001) increase in expression of IFN-γγ, IL-2, and TNF-α. In contrast, CARCD19 T cells showed low expression of IFN-γ and IL-2 and no expression of TNF-α (Figures 4A–4C). When testing proliferation against NSCLC cell lines (NCI-H292, NCI-H1650, and A549), CARLunX T cells proliferated more (as determined by flow cytometry using cell trace violet staining) than CARCD19 T cells. The latter showed only slight proliferation in the presence of LunX-positive tumor cell lines (Figure 4D). Therefore, LunX-positive tumor cell lines could stimulate only CARLunX T cells (but not CARCD19 T cells) to secrete IFN-γγ, IL-2, and TNF-α and proliferate.
Figure 4.

Recognition of LunX-Positive Lung Cancer Cells Induces CARLunX T Cells to Secrete Cytokines and Proliferate
(A) CARLunX T cells and CARCD19 T cells were incubated with LunX-positive lung cancer lines NCI-H292, NCI-H1650, and A549 for 24 h. Then, the supernatant was harvested and expression of IFN-γ. (B and C) (B) IL-2, and (C) TNF-α was measured by ELISAs. n = 3, results are representative of three independent experiments. Data in (A)–(C) are the mean ± SEM. Unpaired t test, ∗∗∗∗p < 0.0001. (D) Proliferation assays undertaken on CARLunX T cells and CARCD19 T cells using cell trace violet staining following in vitro incubation with the LunX-positive cancer cell lines NCI-H292, NCI-H1650, and A549 demonstrated antigen-specific proliferation of CARLunX T cells. Experiments were repeated twice independently. ∗∗p < 0.01.
CARLunX T Cells Suppresses Lung Cancer Progression in Orthotopic Xenografts
To evaluate the efficacy of CARLunX T cells against LunX-positive lung cancer, we examined their antitumor activity in A549-xenografted B-NDG mice. Luciferin-expressing A549 cells (5 × 106) were injected (subcutaneously [s.c.]) into the tail veins of 6- to 8-week-old female B-NDG mice (n = 30). Three days after the xenograft model had been established, mice were divided into three groups of 10 randomly (CARLunX T, CARCD19 T, and phosphate-buffered saline [PBS]) and received first-time transfusions. We set three times of transfusion treatment for each group (Figure 5A). Tumor burden was measured using bioluminescence imaging. After three courses of treatment, abdominal organs showed metastatic colonization of CARCD19 T cells and PBS on day 17, but not CARLunX T cells. From day 28 to day 33, CARLunX T cells could suppress the growth of lung cancer cells and improve survival compared with that observed in the CARCD19 T cell group and PBS group (Figure 5B). Additionally, we analyzed the dynamic growth of tumors of each mouse in the three groups. Mice treated with CARLunX T cells showed reduced proliferation of tumor cells. In contrast, CARCD19 T cells or PBS in the respective mice groups could not suppress tumor growth, and the mice died (Figure 5C). Statistical analyses of the dynamic growth of tumors in each group demonstrated that CARLunX T cells suppressed tumor growth better than that in the other two groups throughout the experiment and did not cause toxicity by T cells themselves (Figure 5D). Mice treated with CARLunX T cells showed significantly (p < 0.0001) increased survival compared with that observed with control mice (Figure 5E). To investigate if CARLunX T cells could infiltrate into the tumor, we carried out immunofluorescence analyses using EGFP to indicate CAR T cells. Infiltration by CARLunX T cells was observed, whereas CARCD19 T cells showed little infiltration into the tumor 7 days after first treatment (Figure 5F; Figure S3). These results showed that treatment with CARLunX T cells not only suppressed LunX-positive tumor growth but also increased survival significantly among mice in the CARCD19 T cells group and PBS group.
Figure 5.

CARLunX T Cells Inhibit Growth of Lung Cancer Cells and Improve Survival in an Orthotopic Xenograft Model in Mice
(A) Experimental protocol for the A549-xenografted model used in (B)–(F). (B) Representative bioluminescence imaging of B-NDG mice xenografted with luciferase-expressing A549 injected via the tail vein (color map for all images—radiance, minimum = 108, maximum = 2 × 109) and infused (i.v.) with CARLunX T cells or CARCD19 T cells as the experimental protocol from day 7 to day 33. (C and D) Mean flux (D) and individual flux (C) of each group of tumor burden in A549-engrafted CARLunX T cells, CARCD19 T cells, and PBS-treated mice (n = 10 mice, respectively, on day 7) over time. (E) Survival of mice treated with CARLunX T cells (n = 10), CARCD19 T cells (n = 10), or PBS at various times (horizontal axis) after first-time treatment. The survival rate was analyzed by log rank tests. (F) Representative immunofluorescence images using confocal microscopy of tumor-infiltrating CARLunX T cell- and CARCD19 T cell-treated A549 cells. Partially infiltrating CAR-T cells were gated by the red frame. Scale bar, 50 μm.
CARLunX T Cells Inhibit Growth of Lung Cancer Cells in a PDX Model in Mice
PDX models have been shown to mimic the microenvironment of primary tumors. We obtained post-resection lung-cancer tissue from patients admitted to the First Affiliated Hospital of the University of Science and Technology of China (Hefei, China). Immunohistochemical analyses showed that the tumor tissue of patients had high expression of LunX (Figure 6A). To determine the ability of CARLunX T cells to target patient-derived tumors, we evaluated the effect of treatment on a LunX-positive PDX model. Patient-derived lung tumor tissue was separated into 25-mm3 portions and transplanted (s.c.) into B-NDG mice (n = 21). After 3 days, an identical mean tumor size was assigned to all three groups of seven mice randomly (CARLunX T cells, CARCD19 T cells, and PBS), and we undertook first-time adoptive transfer of CAR T cells or PBS treatment. Like the A549-xenografted model, we also treated mice of each group with adoptive transfer three times (Figure 6B). The bodyweight of each mouse from the three groups was measured dynamically. We observed bodyweight loss in mice from the CARCD19 T cell group and PBS group, but they had no statistic difference among three groups on day 15 (Figure 6C; Figure S2), which indicated that CARLunX T cells could not only slow down the symptoms of weight loss caused by the tumor, but also had no side effects on the bodyweight of mice. Under the condition that each group had an identical mean tumor size before adoptive transfer on day 3 (Figure 6D), we monitored the time to tumor growth. On day 12, data showed that treatment with CARLunX T cells reduced tumor growth significantly (p < 0.05) (Figure 6E) and that the differences in the mean tumor size between the three groups had increased (Figure 6F). Furthermore, improved survival was observed after treatment with CARLunX T cells (Figure 6G). To determine if CARLunX T cells could infiltrate into PDX tumor tissue, we carried out immunofluorescence analyses to detect GFP-positive CAR T cells. CARLunX T cells were located mainly at tumor edges, whereas CARCD19 T cells were not detected in the tumor (Figure 6F; Figure S4). These results showed that therapy using CARLunX T cells could suppress growth of lung cancer cells and improve survival in a PDX model in mice.
Figure 6.

CARLunX T Cells Suppress Growth of Lung Cancer Cells and Prolong Survival in a PDX Model in Mice
(A) Representative immunohistochemical-stained LunX-positive lung cancer tissue of patient number 1. Results are representative of three independent experiments. (B) Experimental protocol for the PDX-xenografted model used in (C)–(H). (C) Bodyweight of mice in (B) (n = 7 [CARLunX T cells], n = 7 [CARCD19 T cells], or n = 7 [PBS]) at various times (horizontal axis) after PDX tissue had been xenografted. (D) Tumor size in mice in (B) (n = 7 [CARLunX T cells], n = 7 [CARCD19 T cells], or n = 7 [PBS]) on days 3. (E) Tumor size in mice in (B) (n = 7 [CARLunX T cells], n = 7 [CARCD19 T cells], or n = 7 [PBS]) on day 12. (F) Tumor size in mice in (B) (n = 7 [CARLunX T cells], n = 7 [CARCD19 T cells], or n = 7 [PBS]) on day 15. (G) Survival of mice in (B) (n = 7 [CARLunX T cells], n = 7 [CARCD19 T cells], or n = 7 [PBS]) at various times (horizontal axis) after challenge. The survival rate was analyzed by log rank tests. (H) Representative immunofluorescence images using confocal microscopy of tumor-infiltrating CARLunX T cell- and CARCD19 T cell-treated LunX-positive PDX tumors. Partially infiltrating CAR-T cells were gated by the red frame. Scale bar, 50 μm. Each symbol (C and E–G) or line (D) represents an individual mouse. p values, one-way ANOVA followed by Tukey’s multiple-comparisons test (D–F).
Discussion
Here, we report that LunX-specific CARs can target LunX-positive lung cancer cells. CARLunX T cells could be stimulated by LunX and secrete a series of cytokines. Upon co-culture of CARLunX T cells with LunX-positive cells, excellent killing efficacy could be observed. After establishment of a metastatic xenograft model, CARLunX T cells were shown to inhibit growth of LunX-positive tumor cells and prolong the survival of mice. Also, tumor-infiltrating CARLunX T cells could be found within tumor niches. In addition, we measured LunX expression in patient-derived lung-tumor tissue and established a PDX model. Treatment with CARLunX T cells led to not only suppressed growth of LunX-positive tumors, but also prolonged survival in mice.
It is difficult for immune cells to infiltrate solid-tumor niches due to an absence of sufficient co-stimulated signals to activate immune cells.30 Therefore, targeted therapies have opened a new era of cancer treatment and improved the prognosis of patients.31 Therapy using CAR T cells has shown excellent outcomes against hematologic malignancies such as acute lymphocyte leukemia, non-Hodgkin’s lymphoma, and multiple myeloma.32 However, CAR T cells have shown limited efficacy against solid tumors in several preclinical studies.3,32 With regard to lung cancer, few candidate target antigens have been being investigated in clinical trials. In addition, the target antigens for solid tumors are limited and clinical treatment unsatisfactory.
Of the target antigens available for lung cancer, most show some shortcomings because the expression range of target proteins is not specific (e.g., mucin1, Her2). The best-known target antigen for NSCLC is the EGFR, but the proportion of patients with EGFR-positive lung cancer is low.7,11,20,33 Previously, we demonstrated that of 150 total samples of adenocarcinoma and squamous cell carcinoma, 80.6% (121 of 150) displayed increased expression of LUNX.24 Hence, therapy using CARLunX T cells could be promising treatment for NSCLC. In the present study, LunX represented a unique antigen for NSCLC and showed a considerable range of expression among NSCLC patients.
LunX has been reported to exert anti-inflammatory effects during microbial infections and agonist stimulation with Toll-like receptors. Also, studies have indicated that LunX expression is upregulated during infection in nasopharynx epithelium.34,35 Sung et al.36 used Northern blotting to measure PLUNC expression and found no signals in the brain, spleen, liver, muscles, kidneys, or testes. Iwao et al.22 demonstrated, using northern blotting, that LUNX mRNA was undetectable in the liver, brain, pancreas, skeletal muscles, adrenal glands, kidneys, prostate gland, heart, stomach, spleen, mammary glands, adipose tissue, or thyroid tissues but was highly expressed in lung cancer, in humans.
Previously, we showed that LunX was overexpressed in most samples of NSCLC and was high in lymph-node metastases. Importantly, benign lung disease and cancer in other organs (colon, liver, and breast) were not detected.24 LunX has been reported to be a multifunctional protein that is secreted by airway epithelia and has antimicrobial activity and regulates ion channels. Meanwhile, we also observed LunX expression in the membranes of NSCLC cells by immunofluorescence staining and flow cytometry. In our previous study, we generated a therapeutic LunX antibody, S-35-8, which slowed the growth and blocked the local invasion and metastasis of multiple established NSCLC xenografts without side effects. Taken together, LunX appears to be a promising target to construct CAR T cells.
A case report by Feng et al.33 provided quantitative analyses of EGFR-targeting CAR T cells in the circulation. They found that, after the second infusion, levels of CAR T cells were detectable for ≤ 24 weeks, compared with 8 weeks following the initial infusion of EGFR-targeting CAR T cells. Hence, their case report revealed that more than one dose of CAR T cells is necessary for treatment of solid tumors. In our study, we employed three doses of CARLunX T cells in the xenografted model and PDX model. We showed that LunX-positive tumor growth was suppressed and survival was increased after three infusions of CARLunX T cells.
Our data show that LunX is a promising target antigen for CAR-T-cell-based therapy against NSCLC. We demonstrated that CARLunX T cells could kill LunX-positive tumor cells and induce regression of established LunX xenografts and a PDX model. Thus, we propose CARLunX T cells are attractive and promising targeted therapy against NSCLC.
MATERIALS AND METHODS
Ethical Approval of the Study Protocol
All experimental procedures involving mice were approved by the ethics committee of the University of Science and Technology of China (Hefei, China).
Cell Lines
293T cells were obtained from the Shanghai Cell Bank (Chinese Academy of Sciences, Shanghai, China) and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum. A549, HFL1, NCIH292, NCI-H1299, and NCI-H358 lines were purchased from the Shanghai Cell Bank and passaged in our laboratory for fewer than 6 months after receipt or revival. DNA fingerprinting and the isoenzymes test were used to authenticate the cell lines maintained in RPMI 1640 medium with 10% fetal bovine serum.
Isolation of T Cells and Gene Transfer
Blood products were obtained from healthy donors recruited by the Hefei Central Blood Bank (Hefei, China). Peripheral blood mononuclear cells were isolated by density gradient centrifugation. T cells were isolated using a CD3+ MACS kit (Miltenyi Biotec, order no.130090874) and were stimulated with Dynabeads Human T Expander CD3/CD28 (Thermo Scientific, Waltham, MA, USA) at a ratio of T cell/bead of 1:3. Two days after activation, T cells were incubated with freshly prepared lentivirus at 32°C in the presence of 10 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO, USA). After centrifugation (780 × g, 2 h, room temperature), cells were cultured for 6 h at 37°C, followed by culture for an additional 7 days in complete 1640 medium with human recombinant IL-2 (50 U/mL) at 37°C.
Construction of a Lentiviral Vector and Virus Production
293T cells cultured in DMEM were co-transfected with PCDH-MSCV-LunX-scfv-CD137-CD3ζ or PCDH-MSCV-CD19-scfv-CD137-CD3ζ together with the packaging constructs, psPAX.2, and pMD2G. After 8 h, DMEM was replaced with DMEM containing 10% fetal bovine serum. Forty-eight hours after transfection, the medium containing lentivirus was harvested and passed through a 0.45-μm filter (Millipore, Billerica, MA, USA) to remove cell debris. Virus in the supernatant was concentrated by polyethylene glycol-8000.
Flow Cytometry
To assess the transfection efficiency of T cells, a single suspension of transduced T cells was incubated for 1 h at 4°C with anti-Myc tag mouse mAb 9E10 (Cell Signaling Technology, Danvers, MA, USA). Cells were washed thrice with PBS and incubated with PE-conjugated goat anti-mouse immunoglobulin (Ig)G1 secondary antibody (Invitrogen, Carlsbad, CA, USA). The transfection efficiency of T cells was also examined by GFP expression. LunX expression on the surface of lung cancer cells was assessed after labeling with fluorescein isothiocyanate-anti-LunX mAb (s-35-8) and examined by fluorescence-activated cell sorting using a LSRII analyzer (Beckton Dickinson, Franklin Lakes, NJ, USA). Data were analyzed using FlowJo (Tree Star, Ashland, OR, USA).
Cytokine Generation In Vitro and Cell Killing
LunX-antigen peptides were plated at 1,000 ng/mL. Control T cells were stimulated with OKT3 (10 μg/mL) for 12 h, but not transfected by lentivirus, and the previous culture medium was changed into the fresh one. After culturing control T cells for 4 days, we co-cultured them with LunX protein in the fresh culture medium. Human CAR T cells (105) were co-cultured with lung cancer cell lines (A549, NCI-H292, NCI-H1650; 105) or LunX. After 24 h, supernatants were harvested, and cytokine expression was measured using ELISAs for IL-2, IFN-γ, and TNF-α (Dakewe Biotech, Shenzhen, China).
Cytotoxicity assays were run on an xCELLigence real-time cell analysis instrument according to manufacturer’s (ACEA Biosciences, San Diego, CA, USA) instructions. In brief, lung cancer cells were seeded at 104 cells per well. After 20 h (when cells had reached exponential growth), CARlunx T cells or CARCD19 T cells were resuspended in fresh complete medium without IL-2 and added to target cells at different ratios of effector cell:target cell. Then, cell growth was measured.
Histology
Samples of lung cancer tissue from the First Affiliated Hospital of the University of Science and Technology of China were placed in 12% neutral buffered formalin overnight and then dehydrated and embedded in paraffin. Paraffin-embedded tissues were sectioned at a thickness of 7 μm, followed by incubation with primary antibodies to anti-LunX (S-35-8) and IgG1. The signal was detected using a DAB peroxidase substrate kit (SK-4100; Vector Laboratories, Burlingame, CA, USA).
Immunofluorescence Analyses
CAR T cells or frozen sections of lung tumor tissues were fixed with 4% paraformaldehyde for 15 min, followed by blockade with 5% goat serum for 1 h at room temperature. Then, sections were incubated with primary antibodies overnight at 4°C. Secondary antibodies were added, followed by staining with 4′,6-diamidino-2-phenylindole. Stained sections were imaged using a microscope (LSM 880; Zeiss, Oberkochen, Germany).
Generation of an A549 Cell Line that Expresses Luciferase Stably
Luciferase was PCR amplified from a pGL3-Basic plasmid (Promega, Fitchburg, WI, USA) and inserted into a PCDH lentiviral vector to generate a PCDH-luciferase construct. Lentivirus production and infection of A549 cells were undertaken using the methods described above.
Model of Metastatic Lung Cancer in Mice and Bioluminescence Imaging
Female B-NDG (NOD-PrkdcscidIL-2rgtm1/Bcgen) mice (6–8 weeks old; Jiangsu Biocytogen, Jiangsu, China) were used to establish a PDX model and metastatic lung cancer xenografts. The A549 line with stable expression of luciferase (5 × 106 cells) was injected in the tail vein of B-NDG mice. Tumors were allowed to develop for 3 days. Before treatment, the tumor burden was assessed through luminescence imaging in vivo by the standard circular regions of interest on mouse lungs to ensure that CARlunx-T cells, CARCD19 T cells, and PBS carried an equivalent initial tumor burden. CAR T cells were delivered at 107 transduced cells in 200 μL of PBS through injection into the tail veins of mice for the first treatment. After 7 days, mice received a second treatment with 5 × 106 CAR T cells. Then, 7 days later, mice received the final treatment of 5 × 106 CAR T cells.
In the PDX model, lung tumor tissues (25 mm3) were transplanted (s.c.) into B-NOG mice. Three days later, the tumor volume was calculated ([width2 × length]/2) to ensure that each group had an identical mean size of tumor. Then 107 CARlunx T cells, CARCD19 T cells, or PBS were adoptively transferred to PDX mice for the first treatment. Ten days later, mice received the second treatment of 5 × 106 CAR T cells. Then, 10 days later, mice received the final treatment of 5 × 106 CAR T cells. The size of tumors was measured every 3 days. Death was recorded when moribund animals were euthanized in accordance with the requirements of the IACUC protocol.
Statistical Analyses
Data are the mean ± SEM. Plots were generated using Prism 5 (GraphPad, San Diego, CA, USA). Differences between two groups were determined using two-tailed t tests. Differences among three or more groups were determined using one-way analysis of variance followed by two-tailed t tests. Survival curves were analyzed by log rank tests. p < 0.05 was considered significant.
Author Contributions
Conception and design, Z.H., X.Z., R.S., Z.T., and H.W.; Development of methodology, Z.H., X.Z., and H.W.; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.), Z.H., X.Z., R.S., and Z.T.; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis), Z.H., X.Z., Z.T., and H.W.; Writing, review, and/or revision of the manuscript, Z.H., X.Z., Z.T., and H.W.; Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases), Z.H., X.Z., D.J., Y.Z., R.S., Z.T., and H.W.; Study supervision, Z.T. and H.W.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the key project of the Natural Science Foundation of China (reference numbers 81872318 and 81602491).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.04.008.
Contributor Information
Baolong Wang, Email: wbl196555@163.com.
Zhigang Tian, Email: tzg@ustc.edu.cn.
Haiming Wei, Email: ustcwhm@ustc.edu.cn.
Supplemental Information
References
- 1.Reck M., Heigener D.F., Mok T., Soria J.C., Rabe K.F. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382:709–719. doi: 10.1016/S0140-6736(13)61502-0. [DOI] [PubMed] [Google Scholar]
- 2.Chen J., López-Moyado I.F., Seo H., Lio C.J., Hempleman L.J., Sekiya T., Yoshimura A., Scott-Browne J.P., Rao A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530–534. doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morello A., Sadelain M., Adusumilli P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye L., Lou Y., Lu L., Fan X. Mesothelin-targeted second generation CAR-T cells inhibit growth of mesothelin-expressing tumors in vivo. Exp. Ther. Med. 2019;17:739–747. doi: 10.3892/etm.2018.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G., Chew A., Zhao Y., Levine B.L., Albelda S.M. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D.R., Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H., Huang Y., Jiang D.Q., Cui L.Z., He Z., Wang C., Zhang Z.W., Zhu H.L., Ding Y.M., Li L.F. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018;9:177. doi: 10.1038/s41419-017-0238-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu W., Zhou Y., Tang Q., Wang M., Ji Y., Yan J., Yin D., Zhang S., Lu H., Shen J. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci. Trends. 2018;12:298–308. doi: 10.5582/bst.2018.01048. [DOI] [PubMed] [Google Scholar]
- 9.Marcinkowski B., Stevanović S., Helman S.R., Norberg S.M., Serna C., Jin B., Gkitsas N., Kadakia T., Warner A., Davis J.L. Cancer targeting by TCR gene-engineered T cells directed against Kita-Kyushu Lung Cancer Antigen-1. J. Immunother. Cancer. 2019;7:229. doi: 10.1186/s40425-019-0678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeltsman M., Dozier J., McGee E., Ngai D., Adusumilli P.S. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl. Res. 2017;187:1–10. doi: 10.1016/j.trsl.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng K., Guo Y., Dai H., Wang Y., Li X., Jia H., Han W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016;59:468–479. doi: 10.1007/s11427-016-5023-8. [DOI] [PubMed] [Google Scholar]
- 12.Li N., Liu S., Sun M., Chen W., Xu X., Zeng Z., Tang Y., Dong Y., Chang A.H., Zhao Q. Chimeric Antigen Receptor-Modified T Cells Redirected to EphA2 for the Immunotherapy of Non-Small Cell Lung Cancer. Transl. Oncol. 2018;11:11–17. doi: 10.1016/j.tranon.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owen D.H., Giffin M.J., Bailis J.M., Smit M.D., Carbone D.P., He K. DLL3: an emerging target in small cell lung cancer. J. Hematol. Oncol. 2019;12:61. doi: 10.1186/s13045-019-0745-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen N., Li X., Chintala N.K., Tano Z.E., Adusumilli P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018;51:103–110. doi: 10.1016/j.coi.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao J., Ly D., Dervovic D., Fang L., Lee J.B., Kang H., Wang Y.H., Pham N.A., Pan H., Tsao M.S., Zhang L. Human double negative T cells target lung cancer via ligand-dependent mechanisms that can be enhanced by IL-15. J. Immunother. Cancer. 2019;7:17. doi: 10.1186/s40425-019-0507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W., Haderk F., Bivona T.G. Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer. Cancers (Basel) 2017;9:E164. doi: 10.3390/cancers9120164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei X., Lai Y., Li J., Qin L., Xu Y., Zhao R., Li B., Lin S., Wang S., Wu Q. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. OncoImmunology. 2017;6:e1284722. doi: 10.1080/2162402X.2017.1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallstabe L., Göttlich C., Nelke L.C., Kühnemundt J., Schwarz T., Nerreter T., Einsele H., Walles H., Dandekar G., Nietzer S.L., Hudecek M. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight. 2019;4:126345. doi: 10.1172/jci.insight.126345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliveres H., Caglevic C., Passiglia F., Taverna S., Smits E., Rolfo C. Vaccine and immune cell therapy in non-small cell lung cancer. J. Thorac. Dis. 2018;10(Suppl 13):S1602–S1614. doi: 10.21037/jtd.2018.05.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., Jiang J., Wu X., Zhang M., Luo D., Zhang R., Li S., He Y., Bian H., Chen Z. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Front. Med. 2019;13:57–68. doi: 10.1007/s11684-019-0683-y. [DOI] [PubMed] [Google Scholar]
- 21.Bingle C.D., LeClair E.E., Havard S., Bingle L., Gillingham P., Craven C.J. Phylogenetic and evolutionary analysis of the PLUNC gene family. Protein Sci. 2004;13:422–430. doi: 10.1110/ps.03332704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwao K., Watanabe T., Fujiwara Y., Takami K., Kodama K., Higashiyama M., Yokouchi H., Ozaki K., Monden M., Tanigami A. Isolation of a novel human lung-specific gene, LUNX, a potential molecular marker for detection of micrometastasis in non-small-cell lung cancer. Int. J. Cancer. 2001;91:433–437. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1059>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 23.Zheng X., Tian Z., Wei H. Lung specific X protein as a novel therapeutic target for lung cancer. OncoImmunology. 2015;4:e1052931. doi: 10.1080/2162402X.2015.1052931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng X., Cheng M., Fu B., Fan X., Wang Q., Yu X., Sun R., Tian Z., Wei H. Targeting LUNX inhibits non-small cell lung cancer growth and metastasis. Cancer Res. 2015;75:1080–1090. doi: 10.1158/0008-5472.CAN-14-1831. [DOI] [PubMed] [Google Scholar]
- 25.Wu T., Huang J., Moore P.J., Little M.S., Walton W.G., Fellner R.C., Alexis N.E., Peter Di Y., Redinbo M.R., Tilley S.L., Tarran R. Identification of BPIFA1/SPLUNC1 as an epithelium-derived smooth muscle relaxing factor. Nat. Commun. 2017;8:14118. doi: 10.1038/ncomms14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velasquez M.P., Szoor A., Vaidya A., Thakkar A., Nguyen P., Wu M.F., Liu H., Gottschalk S. CD28 and 41BB Costimulation Enhances the Effector Function of CD19-Specific Engager T Cells. Cancer Immunol. Res. 2017;5:860–870. doi: 10.1158/2326-6066.CIR-17-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byrd T.T., Fousek K., Pignata A., Szot C., Samaha H., Seaman S., Dobrolecki L., Salsman V.S., Oo H.Z., Bielamowicz K. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018;78:489–500. doi: 10.1158/0008-5472.CAN-16-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng X., Liu X., Guo X., Jiang S., Chen T., Hu Z., Liu H., Bai Y., Xue M., Hu R. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564:130–135. doi: 10.1038/s41586-018-0756-0. [DOI] [PubMed] [Google Scholar]
- 31.Lv J., Zhao R., Wu D., Zheng D., Wu Z., Shi J., Wei X., Wu Q., Long Y., Lin S. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019;12:18. doi: 10.1186/s13045-019-0704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metzinger M.N., Verghese C., Hamouda D.M., Lenhard A., Choucair K., Senzer N., Brunicardi F.C., Dworkin L., Nemunaitis J. Chimeric Antigen Receptor T-Cell Therapy: Reach to Solid Tumor Experience. Oncology. 2019;97:59–74. doi: 10.1159/000500488. [DOI] [PubMed] [Google Scholar]
- 33.Feng K.C., Guo Y.L., Liu Y., Dai H.R., Wang Y., Lv H.Y., Huang J.H., Yang Q.M., Han W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017;10:4. doi: 10.1186/s13045-016-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukinskiene L., Liu Y., Reynolds S.D., Steele C., Stripp B.R., Leikauf G.D., Kolls K., Di Y.P. Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J. Immunol. 2011;187:382–390. doi: 10.4049/jimmunol.1001769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Caballero A., Rasmussen J.E., Gaillard E., Watson M.J., Olsen J.C., Donaldson S.H., Stutts M.J., Tarran R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. USA. 2009;106:11412–11417. doi: 10.1073/pnas.0903609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung Y.K., Moon C., Yoo J.Y., Moon C., Pearse D., Pevsner J., Ronnett G.V. Plunc, a member of the secretory gland protein family, is up-regulated in nasal respiratory epithelium after olfactory bulbectomy. J. Biol. Chem. 2002;277:12762–12769. doi: 10.1074/jbc.M106208200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.