

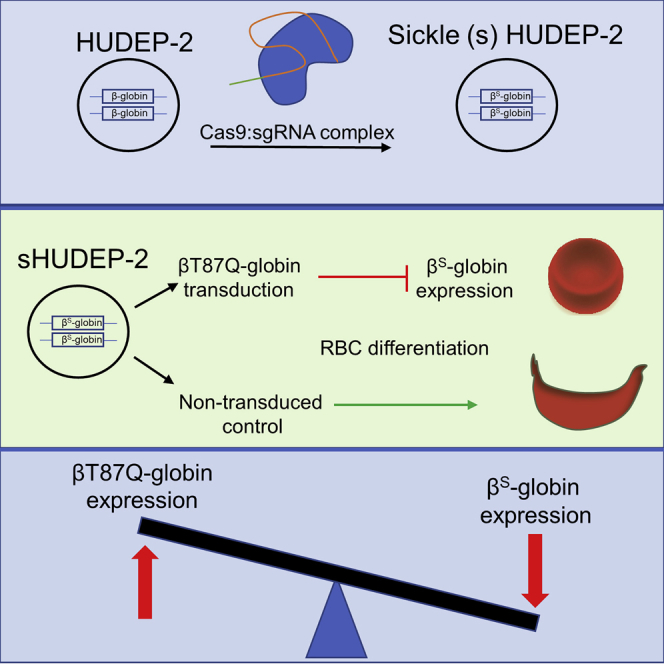
Abstract
Lentiviral addition of βT87Q-globin, a modified β-globin with an anti-sickling mutation, is currently being used in gene therapy trials for sickle cell disease (SCD) and β-thalassemia patients. βT87Q-globin interferes with sickle hemoglobin (HbS) polymerization. Here, we generated the SCD mutation in an immortalized human erythroid cell line (HUDEP-2) to investigate the anti-sickling activity of βT87Q-globin. Sickle HUDEP-2 (sHUDEP-2) cells produced robust HbS after differentiation and sickled under deoxygenated conditions, comparable with SCD CD34+ progeny. Lentiviral transduction provided 9.5–26.8 pg/cell βT87Q-globin (R2 = 0.83) in a vector copy number (VCN)-dependent manner, resulting in a significant reduction of sickling ratios (R2 = 0.92). Interestingly, βT87Q-globin transduction markedly reduced endogenous βS-globin (R2 = 0.84) to an undetectable level (0.4–16.8 pg/cell) in sHUDEP-2 cells, as well as endogenous β-globin in human CD34+ cell-derived erythroid cells. RNA sequencing (RNA-seq) analysis with βT87Q-transduced sHUDEP-2 and human CD34+-derived cells revealed activation of inflammation- and proliferation-related programs, suggesting minimal changes in background gene expression except for βT87Q-globin expression and endogenous β/βS-globin suppression. In summary, using sHUDEP-2 and CD34+-derived cells, we demonstrated that lentiviral addition of βT87Q-globin strongly reduced endogenous β-/βS-globin expression, resulting in an anti-sickling effect. Our findings should be helpful to understand the anti-sickling effects of therapeutic genes in SCD gene therapy.
Keywords: sickle cell disease, gene therapy, hemoglobin, immortalized erythrocytes
Graphical Abstract
Introduction
Sickle cell disease (SCD) is a monogenic red blood cell (RBC) disorder with approximately 300,000 new affected individuals globally per year, which is predicted to rise to 400,000 patients in 2050.1 A homozygous missense mutation (E6V, rs334) in the β-globin gene leads to polymerization of hemoglobin (Hb) under deoxygenated conditions that results in various secondary molecular and cellular changes, many of which disrupt circulation and contribute to major tissue damage.2 As of now, allogeneic hematopoietic stem cell (HSC) transplantation is the only curative option available to patients outside of clinical trials. Despite the procedure’s high success rate, a majority of sickle cell patients lack a histocompatible sibling donor, and there remains a significant risk for graft-versus-host disease (GVHD), graft rejection, and transplant-related mortality.3 Thus, the development of novel and efficient treatment modalities is of great interest to the biomedical community.
In SCD, sickle Hb (HbS) forms polymers under deoxygenated conditions that drastically change mechanical and rheological properties of RBCs, interrupting circulation and reducing oxygen transfer to vital organs. Efforts to develop gene therapy for SCD include various anti-sickling gene transfer approaches (including β-globin, γ-globin, and β/γ-globin hybrids) in SCD blood progenitor cells (reviewed in Demirci et al.4). However, insufficient globin expression was observed in early attempts because of ineffective transfer of large cassettes, instability of globin expression from cassettes in retroviral vectors, and a lack of long-term persistence of erythroid-specific globin expression.5, 6, 7
The inhibitory effect of β-globin on sickling is relatively low compared with γ-globin.8,9 Biochemical studies revealed that a critical difference at position 87 (Q replacing T) in γ-globin is one of the main reasons for its superior anti-sickling activity.10 Lentiviral delivery of βT87Q-globin containing this anti-sickling mutation has been tested in SCD mouse models; along with an erythroid-specific accumulation of anti-sickling protein, inhibition of sickling and correction of hematological parameters in mice were reported.11 βT87Q-globin transfer is now being evaluated in clinical trials for β-thalassemia and SCD12 (ClinicalTrials.gov: NCT03207009, NCT02906202, NCT01745120, and NCT02151526), and encouraging results have been reported. However, vector copy numbers (VCNs) and transgenic protein amounts are variable, and consensus on the protein level needed for therapeutic effects has not yet been established.
Rapid and remarkable advances in molecular biology, particularly genome-editing tools, have led to efficient editing of γ-globin regulatory elements13,14 and even correction of the SCD mutation15 in HSCs, generating excitement about the emergence of new therapeutic modalities. However, these approaches should be investigated, optimized, and verified in ex vivo SCD models regarding their molecular mechanism, efficacy, and safety before testing in animal models and subsequent clinical trials to enhance the success rate for early participants in those trials. In terms of speed and cost, ex vivo cell culture models have a great advantage over human primary cells for drug candidate screening and molecular mechanism evaluation. To the best of our knowledge, there is no publicly available SCD cell line for ex vivo research. Here, we introduced the SCD mutation into a previously generated immortalized erythroid progenitor cell line (HUDEP-2)16 using the CRISPR-Cas9 approach, allowing us to evaluate the anti-sickling activity of βT87Q-globin, as well as its potential mechanism of action using RNA sequencing (RNA-seq) in this cell line.
Results
Sickle HUDEP-2 (sHUDEP-2) Cells Produce the βS-Globin Protein
To introduce the SCD mutation into the adult β-globin gene in HUDEP-2 cells, we used the CRISPR-Cas9 approach. The electroporated bulk HUDEP-2 cell population was differentiated in order to determine whether there was any detectable HbS production. While wild-type HUDEP-2 cells mostly expressed adult Hb (HbA), edited cells (bulk) produced HbS and HbA (Figure 1A). To derive an SCD cell line clone, we cloned single cells from the bulk population and performed PCR-based genotyping to determine the editing status of the clones. The results revealed that the total editing ratio was 67% (49 out of 73 clones) and biallelic editing was 22% (16 out of 73 clones) with the homozygous SCD mutation (Figure 1B). To confirm HbS protein expression, homozygous gene-edited clones were differentiated and subjected to Hb electrophoresis. All homozygous gene-edited clones produced HbS protein expression (Figure 1C), indicating gene conversion was realized at the protein level. Because the seventh clone (hereafter referred to as sHUDEP-2) produced significant HbS protein amount without fetal globin (HbF) expression, it was selected for further characterization, anti-sickling, and RNA-seq experiments.
Figure 1.

Sickle HUDEP-2 (sHUDEP-2) Cells Produce Sickle Hemoglobin (HbS)
(A) Hemoglobin (Hb) electrophoresis of differentiated cells derived from wild-type HUDEP-2 and cells electroporated with ribonucleoprotein complex and donor template containing the sickle cell disease (SCD) mutation (Edited). (B) qRT-PCR analysis of single-cell cloned electroporated cells. (C) Hb electrophoresis for single-cell cloned sHUDEP-2 cells. (D and E) Cell number (D) and cell surface marker (GPA, CD71, and CD36) expression change (E) during red blood cell (RBC) differentiation of sHUDEP-2 cells (n = 3). (F) Giemsa-wright staining of sHUDEP-2 cells at day 10 of differentiation.
sHUDEP-2 cell numbers increased over the course of a 14-day differentiation period (Figure 1D), similar to the parental HUDEP-2 cell line as reported previously.16 There was a ∼13-fold increase in cell number at day 10 and a ∼24-fold increase at day 14 of differentiation. sHUDEP-2 cells were evaluated for erythrocyte marker (CD36, CD71, and glycophorin A [GPA]) expressions throughout differentiation. Most of the cells were already positive for CD36 (82.3% ± 2.8%), CD71 (68.0% ± 2.8%), and GPA (69.0% ± 2.9%) at day 0 (Figure 1E), similar to wild-type HUDEP-2 cells.16 Although there was a slight reduction in CD71 and CD36 expression during the early phase of differentiation, expression increased after 5 days. Moreover, GPA levels reached 99.2% ± 2.9% at day 10 of differentiation. Because GPA is a terminal marker for erythrocyte differentiation, we used 10 days for the differentiation experiments. Whereas GPA positivity was almost 100% at day 10 of differentiation, enucleation efficiency was only 6.5% ± 2.9% as determined by flow cytometric analysis of cells stained with Hoechst 23322 dye (Figure 1F), which is similar to the enucleation efficiency of wild-type HUDEP-2 cells (8.4%–10%).13,17 These observations indicate that sHUDEP-2 cells are similar to wild-type HUDEP-2 cells except for the SCD mutation.
βT87Q-Globin Addition Reduces βS-Globin Production and Sickling in sHUDEP-2 Cells
Next, we investigated whether sHUDEP-2 would sickle under deoxygenated conditions. Concurrently, we introduced βT87Q-globin gene into the sHUDEP-2 cells using lentiviral transduction at increasing multiplicities of infection (MOIs; 0.5, 1, 2, 5, 10, and 25) to evaluate its anti-sickling activity. Transduced cells were single-cell cloned to obtain a homogeneous VCN inside the cell population. Single-cell-derived clones were subjected to qPCR and 10 clones (with VCNs of 0.6, 1.12, 1.57, 2.45, 3.48, 4.46, 5.25, 6.02, 7.99, and 10.06) were selected for further evaluation. Hb electrophoresis and reverse-phase high-performance liquid chromatography (RP-HPLC) analysis demonstrated significant expression of βT87Q-globin in transduced sHUDEP-2 cells (Figures 2A and 2B). Up to a VCN of 3.48, the βT87Q-globin amount was 0%–36.7%, whereas the cells with a VCN of 3.48 or greater expressed 81.3%–89.2% βT87Q-globin and 1.16%–11.9% HbS. Absolute quantification of Hb in each clone showed that there is a significant polynomial correlation between VCN and βS-globin (R2 = 0.8423) and βT87Q-globin (R2 = 0.8338) protein levels (Figure 2C). It is also noteworthy that higher βT87Q-globin levels reduced the amount of βS-globin expression in sHUDEP-2 cells at the protein level, indicating an internal feedback mechanism for globin chain incorporation for this cell line or more rapid βT87Q-globin production from the vector construct than βS-globin.
Figure 2.

βT87Q-Globin Production Reduces Sickling in sHUDEP-2 Cells
(A) Hb electrophoresis for single-cell cloned sHUDEP-2 cells transduced with a βT87Q-globin-expressing vector. (B) β-like globin protein expression in single-cell clones by RP-HPLC (n = 3). (C) Correlation of βS-globin and βT87Q-globin protein amounts per cell with vector copy number in sHUDEP-2 cells (n = 3). (D) Sickling of adult Hb (HbA)/HbS (AS) or HbS/HbS (SS) human RBCs, and sHUDEP-2 cells with or without βT87Q-globin expression under deoxygenated conditions (5% O2 and 95% N2) (n = 8; see also Figure S1). Correlation of sickling fraction in sHUDEP-2 cells with (E) vector copy number and (F) βT87Q-globin protein amount (n = 3).
βT87Q-globin-expressing cells were evaluated for their sickling potential under deoxygenated conditions. RBCs derived from patients with a genotype of HbAS (AS) globin rarely sickled, whereas a significant proportion of RBCs with HbSS (SS) genotype sickled under low oxygenic conditions (Figure 2D). Interestingly, a large majority of deoxygenated sHUDEP-2 cells also sickled (51%, 47%, and 42% in 410, 354, and 298 mOsm conditions, respectively). There were clear signs of sickle fiber formation and deformation of the cell membrane, but sickling was irregular and distinct from that observed in patient-derived RBCs. However, the sickling fraction was significantly negatively correlated with VCN (R2 = 0.61–0.73) and βT87Q-globin protein amount (R2 = 0.92) (Figures 2E, 2F, and S1). Osmolarity of saline solutions strongly affects both the rate and the degree of deformation of deoxygenated sickle cells. Increased osmolarity leads to dehydration of RBCs and increased intracellular HbS concentration, thereby increasing the rate of polymerization under deoxygenated conditions. In this report, we evaluated the influence of VCN on the sickling of HUDEP-2 cells at different osmolarities, as can be seen in Figure 2E. The sickling ratio was 12%–17% for all osmolarity conditions in sHUDEP-2 with a VCN of 10 (Figure 2E), confirming the inhibitory effect of βT87Q-globin on sickling in sHUDEP-2 cells.
To evaluate globin chain RNA and protein levels in both sHUDEP-2 and human CD34+-derived cells, we first transduced the cells with a βT87Q-globin vector at different MOIs (Figure 3A). After differentiating transduced cells, RNA and protein levels (normalized to α-globin level) revealed that whereas the transcript copies for βT87Q-globin were as high as 3.9 ± 0.002 copies per α-globin transcript, the βs-globin transcript was as low as 0.25 ± 0.003 copies in sHUDEP-2 cells (Figure 3B). In addition, α-globin RNA levels were similar in βT87Q-globin vector-transduced cells and non-transduced mock control (Figure S2). In line with RNA levels, higher MOI correlated with enhanced βT87Q-globin protein levels (44.9%–87.5%) and reduced amount of HbS (7.3%–47.0%) in sHUDEP-2 cells (Figures 3C and 3D). Similar to sHUDEP-2 cells, healthy donor CD34+-derived cells transduced with βT87Q-globin-expressing lentivirus expressed fewer endogenous β-globin transcripts per α-globin transcript (0.52–0.86 copies per α-globin), while the non-transduced control expressed 1.02 ± 0.03 copies per α-globin (Figure 3E). Accordingly, transduced CD34+-derived cells expressed enhanced βT87Q-globin protein levels (13.3%–34.5%) and lower levels of endogenous β-globin (62.3%–83.0%), supporting the notion that βT87Q-globin expression affects endogenous β- or βs-globin expression both at the RNA and the protein levels.
Figure 3.

βT87Q-Globin Expression Lowers Sickle or Adult β-Globin in sHUDEP-2 or Human CD34+ Cells
(A) Schematic representation of sHUDEP-2 transduction with a βT87Q-globin-expressing vector. (B) Relative globin RNA determined by qRT-PCR in sHUDEP-2 cells transduced with different MOIs of the βT87Q-globin expressing vector (n = 3). Hb/β-like globin protein analysis by (C) Hb electrophoresis and (D) RP-HPLC. (E) Relative globin RNA determined by qRT-PCR in CD34+-derived cells transduced with different MOIs of βT87Q-globin-expressing lentivirus (n = 3). (F) β-like globin protein analysis by RP-HPLC.
Distinct Gene Expression Signatures in sHUDEP-2 Cells versus CD34+-Derived Cells Expressing βT87Q-Globin
We performed gene expression profiling by RNA-seq to determine which transcripts were affected by overexpression of βT87Q-globin. sHUDEP-2 cells and healthy donor mobilized CD34+-derived cells were transduced with control lentiviral (GFP) vector or βT87Q-globin vector. CD34+ cells were transduced at day 2 of our two-phase erythroid culture system,18 and sHUDEP-2 cells were transduced in expansion medium followed by erythrocyte differentiation as described above. At day 7 post-transduction (day 9 of culture for CD34+ cells and day 6 of culture for sHUDEP-2 cells), RNA was isolated for RNA-seq and differential gene expression profiling, and expression data were analyzed by ingenuity pathway analysis (IPA). To confirm the RNA-seq data, we analyzed expression of selected genes in sHUDEP-2 cells and mobilized PB CD34+-derived cells by qRT-PCR. Our results show that Smoothened, Frizzled Class Receptor (SMO), and Adenomatous Polyposis Coli Protein 2 (APC2) are upregulated in CD34+ cells transduced with βT87Q-globin lentiviral vector (Figure S3A), and phosphoserine aminotransferase 1 (PSAT1), phosphoglycerate dehydrogenase (PHGDH), and cystathionine gamma-lyase (CTH) are upregulated in sHUDEP-2 cells transduced with βT87Q-globin lentiviral vector (Figure S3B).
Using a 1.4-fold change in expression as a cutoff, RNA-seq identified 748 differentially expressed transcripts in sHUDEP-2 cells transduced with GFP expression control vector (VCN = 30.3) and 761 transcripts in sHUDEP-2 cells transduced with βT87Q-globin lentiviral vector (VCN = 11.2) compared with non-transduced (Mock) sHUDEP2 cells. Compared with non-transduced control CD34+ cells, 711 transcripts were differentially expressed in CD34+ cells transduced with GFP control vector (VCN = 1.4) and 725 transcripts in CD34+ cells transduced with βT87Q-globin lentiviral vector (VCN = 0.5), respectively (Tables S1A–S1D). Of these transcripts, 207 were common to both sHUDEP-2 transduced with GFP control vector or βT87Q-globin lentiviral vector (Table S1E), and 307 were common to CD34+ cells transduced with control vector or βT87Q-globin lentiviral vector (Table S1F). The differentially expressed transcripts common to sHUDEP-2 cells transduced with GFP vector or βT87Q-globin vector, or the transcripts common to CD34+ cells transduced with GFP vector or βT87Q-globin vector are most likely non-specific, i.e., an effect of the viral transduction. Interestingly, only seven genes were found to be common to all four groups, including the phospholipase D family member 4 (PLD4) gene, which is involved in inflammatory cytokine responses and regulation of nucleic acid sensing (Table S1G).19 This small overlap in commonly expressed genes among all four groups likely represents the biological difference between the sHUDEP-2 cell line and primary CD34+ cells. In addition, principal-component analysis (PCA) revealed that CD34+ and sHUDEP-2 cells cluster differently (Figure S3C), indicating the different transcriptomic structures of the cells.
Further analysis of the datasets showed that 554 transcripts were specific to sHUDEP-2 cells transduced with βT87Q-globin vector when compared with sHUDEP-2 transduced with GFP control vector (Table S1H), and 418 transcripts were specific to CD34+ cells transduced with T87Q-globin lentiviral vector (Table S1I) when compared with CD34+ cells transduced with GFP control vector. Our IPA of sHUDEP-2 cells showed a predominant cancer-related gene expression signature in both GFP control and βT87Q-globin lentiviral vector groups in comparison with mobilized CD34+-derived cells (Figure 4A). These signatures may reflect the status of the sHUDEP-2 cells because they are immortalized with an inducible HPV16 E6/E7 viral vector system.16 Analysis of genes specifically expressed in sHUDEP-2 cells transduced with βT87Q-globin vector showed enrichment of genes and pathways involved in amino acid metabolism and protein synthesis, whereas mobilized CD34+-derived cells transduced with βT87Q-globin vector showed preferential expression of genes involved in Wnt signaling (Figure 4B). Wnt signaling is critical in development and stem cell function20 but has not been shown to have a clear role in globin expression.
Figure 4.

βT87Q-Globin Vector-Transduced Cells Have Distinct Disease and Canonical Pathway Activation
(A) The most significant diseases and functions in sHUDEP-2 and human CD34+ cells transduced with a βT87Q-globin vector compared with GFP-expressing control cells. (B) The most significant canonical pathways in sHUDEP-2 and human CD34+ cells transduced with a βT87Q-globin vector compared with GFP-expressing control cells.
Further, the IPA shows that amino acid metabolism, cancer, and small-molecule biochemistry are the most significantly affected network in sHUDEP-2 cells transduced with the βT87Q-globin vector (Figure 5). According to the Bloodspot database, MAPK11, TRB3, and PSAT1 are expressed in erythroid cells, but the majority of the genes in the network do not appear to be normally significantly expressed in that lineage. In mobilized CD34+ cells transduced with the βT87Q-globin vector, the most significantly affected network is organismal injury and abnormalities, behavior, and gastrointestinal disease (Figure 4B). A significant fraction of the upregulated genes in the network is comprised of small nuclear ribonucleoproteins (snRNPs), which are involved in RNA splicing, i.e., removal of introns from pre-mRNA. Taken together, transduction of sHUDEP-2 cells and mobilized CD34+ cells with βT87Q-globin vector activates common gene expression programs (inflammation) and cancer-related (sHUDEP-2 cells) or developmental/stem cell-related (CD34+ cells) programs.
Figure 5.

Ingenuity Pathway Analysis (IPA) Identifies Amino Acid Metabolism, Cancer, and Small-Molecule Biochemistry as a Top Network in βT87Q-Globin-Transduced sHUDEP-2 Cells Compared with GFP-Expressing Cells and Basal Cell Carcinoma Signaling as a Top Network in CD34+ Cells Compared with GFP
sHUDEP-2 cells and mobilized CD34+ cells were transduced with GFP control vector (MOI 50) or βT87Q-globin vector (MOI 50) and RNA isolated at day 9 post-transduction. Red denotes upregulated genes, and green denotes downregulated genes.
Discussion
Immortalized cell lines are critical ex vivo models in the study of cellular responses to specific stimuli and basic cellular mechanisms, including differentiation and disease development, because they are cost-effective, easy to handle, unlimited in supply, not associated with ethical concerns, and provide consistent starting materials. Only a handful of cell lines are able to express adult β-globin and are available for RBC metabolism and disorder research, including the HUDEP-216 and BEL-A21 cell lines. In the present study, we introduced the sickle mutation into the widely studied HUDEP-2 cells using CRISPR-Cas9 technology to generate an HbS-expressing cell line. Cell proliferation is thought to enhance homology-directed repair (HDR) in genome editing,22 as well as HbF induction in erythroid progenitors. The HUDEP-2 cell line contains HbF-expressing subclones, which were selected using lower-concentration stem cell factor (SCF) when thawed from a frozen stock (data not shown). The SCF-oversensitive HbF-expressing clones seem to be more proliferative; thus, biallelic HDR might be more frequently observed in these HbF-expressing clones because of their increased proliferation (Figure 2B). The sickle cell line can be used not only for understanding erythropoiesis, but also to perform SCD mutation correction, HbF induction, and gene addition research. Here, we have shown proof of concept of their ex vivo research potential by investigating the anti-sickling effect of βT87Q-globin using this sickle cell line.
The anti-sickling βT87Q-globin was developed by Pawliuk et al.11 by introducing the anti-sickling amino acid found at position 87 of γ-globin into β-globin. Naturally occurring mutations (hereditary persistence of fetal Hb [HPFH] mutations) result in elevated (20%–30%) pancellular persistent expression of γ-globin that alleviates SCD complications, indicating ∼20% βT87Q-globin (pancellular expression) would reverse the disease outcome. However, the challenge of transducing HSCs23 and the requirement for larger vectors containing erythroid-specific regulatory elements means that transduction and protein expression are variable in the bulk transplantation product, making the resulting in vivo transgene levels and clinical outcome for each patient unpredictable. Therefore, higher VCNs are targeted in the current clinical trials to enhance the success rate. Although the same vector type and process are being used for each patient, the results and outcomes are variable.24 Determining the therapeutic threshold for βT87Q-globin, along with HSC transduction optimization, would advance the approach and improve prospects for all patients.
We have generated an SCD cell line (sHUDEP-2) and transduced it with βT87Q-globin-encoding lentivirus to evaluate its anti-sickling activity. The results showed that the anti-sickling activity of βT87Q-globin increased in a dose-dependent manner. A VCN of 3.48 or more provided a significant amount of βT87Q-globin protein levels (81%–89%) and reduced sickling activity under low oxygen conditions. Most interestingly, βT87Q-globin expression reduced endogenous βs- or β-globin expression at both RNA and protein levels in sHUDEP-2 or CD34+-derived cells, respectively. In β-thalassemia patients, unbound α-chains precipitate in RBC precursors leading to apoptosis and insufficient erythropoiesis.25 On the other hand, excessive β/βs-globin protein is degraded in α-thalassemia/SCD patients.26,27 Consistently, our results suggest predominantly degradation of excessive β/βs-globin, but also transcriptional regulatory mechanisms might be involved to provide globin chain balance. In the clinical setting, pre-processed Hb level measurements show a mixture of HbS and HbA (resulting from transfusion). This ex vivo model clearly demonstrates that increased βT87Q-globin levels reduce βS-globin protein levels, possibly as a result of endogenous controls on β-like globin levels. This globin expression regulation mechanism could be either through mRNA degradation or transcription control by a feedback mechanism. This observation needs to be evaluated further in other globin addition (i.e., γ-globin) approaches for confirmation. Interestingly, early data on our clinical trial testing βT87Q lentiviral gene transfer to HSCs of SCD patients yielded similar observations of a reciprocal decrease in HbS.28
To investigate the possible transcriptomic mechanism responsible for globin regulation, we conducted RNA-seq on sHUDEP-2 and CD34+-derived cells. Although we did not find significant changes in any major β-globin expression regulating transcription factors (TFs), including GATA1, NF-E2, KLF1, and SCL,29,30 many long non-coding RNAs (lncRNA), whose roles in erythropoiesis are unknown, were differentially expressed in βT87Q-globin-transduced cells. lncRNAs are known for their role in regulating protein-coding gene activity in cis and in trans.31 In addition to TFs and regulatory RNAs, epigenetic regulators also control globin expression. β-Globin expression is controlled at the DNA level by methylation and formation of the heterochromatin structure.32 Further detailed studies are warranted to fully elucidate the exact molecular mechanism for the maintenance and control of globin chain mRNA and protein levels.
Overall, we developed an SCD cell line that can be used in ex vivo optimization studies before translation of promising approaches to animal models and clinical studies. Evaluation of βT87Q-globin overexpressing sHUDEP-2 and CD34+-derived cells demonstrated marked reduction in β/βS-globin transcripts and protein levels in sHUDEP-2 cells in a dose-dependent manner. These results provide insight into globin chain expression regulation and can be used for optimization in clinical trials involving β-globin gene transfer. Finally, these results are important in evaluating the promise of this approach for the hemoglobinopathies.
Materials and Methods
Generation and Differentiation of the SCD Erythroid Cell Line
HUDEP-2 cells, an immortalized erythroid progenitor cell line (a gift from Drs. Yukio Nakamura and Ryo Kurita; RIKEN Tsukuba Branch, Ibaraki, Japan), were cultured and differentiated as previously described.16 In brief, cells were expanded in StemSpan II supplemented with 3 U/mL erythropoietin (EPO; AMGEN, Thousand Oaks, CA, USA), 50 ng/mL SCF (R&D Systems; Minneapolis, MN, USA), 10−6 M dexamethasone (DEX; VETone, Boise, ID, USA), and 1 μg/mL doxycycline (DOX; Sigma-Aldrich, Saint Louis, MO, USA). Cells were differentiated in Iscove’s modified Dulbecco’s media (IMDM; Sigma-Aldrich) supplemented with 50 ng/mL SCF, 10 μg/mL insulin (INS; Lilly, Indianapolis, IN, USA), 3 U/mL EPO, 400 μg/mL holo-transferrin (Sigma-Aldrich), 2 U/mL heparin (Sigma-Aldrich), 1 μg/mL DOX, and 5% human AB plasma (Rhode Island Blood Center, Providence, RI, USA) for 7 days.
For the SCD mutation introduction, the ribonucleoprotein complex (RNP) was first formed by incubation of guide RNA (200 pmol; Synthego, CA, USA) (5′-GUA ACG GCA GAC UUC UCC UC-3′) and Cas9 protein (200 pmol; UC Berkeley, Berkeley, CA, USA) at room temperature for 15 min. HUDEP-2 cells (5 × 104 cells) were washed with phosphate-buffered saline (PBS) solution (Thermo Fisher Scientific, Waltham, MA, USA) twice and suspended in 20 μL SF buffer solution (#V4XC-2032, Lonza, Basel, Switzerland). The cells were mixed with RNP and single-strand donor DNA (300 pmol, 100 bases of βs-globin gene around the SCD mutation; IDT, Coralville, IA, USA). Cells were electroporated with Amaxa 4D Nucleofector X Unit (Lonza) using the manufacturer-optimized FF-120 program. The electroporated cells were incubated at room temperature for 10 min and added to pre-warmed 80 μL expansion medium before transferring into 12-well cell culture plates (Corning, NY, USA). Granulocyte colony-stimulating factor-mobilized hematopoietic cells were collected from healthy donors and plerixafor-mobilized SCD patients enrolled in institutional review board-approved studies. Apheresis product underwent density gradient centrifugation, and CD34 cells were positively selected using a Miltenyi magnetic bead system. Flow cytometry was used to confirm and quantify CD34+ cell purity (>90% CD34+ and >90% viability).
Hb Electrophoresis
Hb types in differentiated cells (1–3 × 106 cells) (on day 7 of differentiation for sHUDEP-2 and on day 14 for CD34+ cells) were determined using cellulose acetate membranes and alkaline buffer solution according to the manufacturer’s instructions (Helena Laboratories, Beaumont, TX, USA).
qPCR and Lentiviral Transduction of sHUDEP-2 Cells
After determining the presence of HbS in the bulk population, the culture was single-cell cloned. The clones were subjected to qPCR analysis using PCR-based genotyping (Sickle FWD4 primer: 5′-GG CAG AGC CAT CTA TTG CTT AC-3′, Sickle REV2 primer: 5′-CCA ACT TCA TCC ACG TTC ACC-3′, and Sickle probe: 5′-FAM-CTG ACT CCT GTG GAG AA-3′). A selected sHUDEP-2 clone (2 × 105 cells) was pre-stimulated with fresh 1 mL expansion media overnight in a 12-well plate. The culture medium was renewed with a fresh 1 mL expansion medium containing a βT87Q-globin expressing lentiviral vector33 at different MOIs (0.5, 1, 2, 5, 10, and 25) and protamine (2 mg/mL; Sigma-Aldrich). The next day, transduced cells were split into two wells in 12-well plates in 1 mL fresh expansion media. Transduced cells at different MOIs were single-cell cloned, and genomic DNA from each clone was extracted using an automated DNA system following the manufacturer’s recommendation (Biomek 400; Beckman Coulter, Brea, CA, USA). VCN was determined for each clone by qPCR using the QuantStudio 6 Flex real-time PCR system (Thermo Fisher Scientific) as previously described.34 In addition, globin transcripts per α-globin was calculated as described previously.35
Flow Cytometry Analysis
Differentiated cells were characterized in terms of erythroid-specific cell surface markers using flow cytometry analysis. In brief, 1 × 105 sHUDEP-2 cells were washed with PBS three times and incubated with CD36 (#555454; Becton Dickinson Biosciences, Franklin Lakes, NJ, USA), CD71 (#551374; Becton Dickinson Biosciences), and GPA (#555570; Becton Dickinson Biosciences) antibodies for 30 min at 4°C. Cells were washed with PBS and analyzed using flow cytometry (FACSCalibur; Becton Dickinson). The results were evaluated using FlowJo software (FlowJo, Ashland, OR, USA).
Morphological Evaluation/Enucleation Analysis of Differentiated Cells
For morphological analysis, differentiated sHUDEP-2 cells (2 × 105 cells) were centrifuged onto Shandon EZSingle Cytofunnel (Thermo Fisher Scientific) at 2,000 rpm for 2 min. Air-dried slides were stained with Wright-Giemsa and examined with light microscopy (Olympus, Tokyo, Japan) equipped with Dual-CCDDP80 camera. To determine enucleation efficiency, we stained differentiated cells with Hoechst 23322 (5 μg/mL; BD Biosciences) for 30 min at 37°C and analyzed them using LSRFortessa (Becton Dickinson).
RP-HPLC
Globin chain analysis in differentiated cells (1–3 × 106 cells) was evaluated as previously described.36 In short, cells were washed with PBS (3×) and lysed in 100 μL HPLC-grade water (Sigma-Aldrich) by vortexing for 30 s (3×). The lysate was centrifuged at 16,000 × g for 10 min, and 10 μL Tris (2-carboxyethyl) phosphine hydrochloride (TCEP; 100 mmol; Thermo Fisher Scientific) was added to 90 μL supernatant. After 5-min incubation at room temperature, 85 μL of 0.1% trifluoroacetic acid (TFA)/32% acetonitrile was added, and the tube was vortexed briefly. A 10 μL aliquot of the solution was analyzed at a 0.7 mL/min flow rate for 50 min using the Agilent 1100 HPLC (Agilent Technologies, Santa Clara, CA, USA) equipped with a reverse-phase column, Aeris 3.6 μm Widepore C4 200 (250 × 4.6 mm; Phenomenex, Torrance, CA, USA) with two solvents: solvent A, 0.12% TFA in water; and solvent B, 0.08% TFA in acetonitrile. The globin chains were detected at 215 nm. Hb content per cell was calculated as described previously.37
Anti-sickling Analysis
A Lionheart FX automated microscope system (BioTek Instruments, Winooski, VT, USA) was employed in the sickling assay. The instrument contains a humidified chamber for the insertion of a polypropylene 384-well Greiner plate (Part no. 3770) with the percentage of nitrogen and oxygen in the chamber determined by a gas controller (CO2/O2 Gas Controller; BioTek Instruments). The cells were suspended in a PBS solution (20 mM sodium/potassium phosphate, 155 mM sodium chloride for 298 mOsm [additional NaCl was added to adjust the buffer solutions for higher osmolarity (354 and 410 mOsm)], 1 mg/mL dextrose, and 1 mg/mL albumin [pH 7.4]) at a concentration of 4 million cells/cc. 10 μL of the cell suspension at each osmolarity was inserted into each well (32 total). Images were collected for 8 h, with one image taken every 15–16 min with deoxygenation using a 95/5 nitrogen/oxygen mixture. Additional details, as well as the theoretical simulations of the assay, can be found in Dunkelberger et al.,38 with the only difference being that the final atmosphere used in our study was 0% oxygen.
Because a change in shape can only arise from formation of structures within the cell that distorts the cytoskeleton, we interpret any change in shape as resulting from the formation of HbS fibers and identify that cell as “sickled.” To determine whether or not a cell sickled, images of cells before and 8 h after the start of deoxygenation were compared. About 400 cells were counted to determine the fraction of cells sickled for each experimental condition, i.e., different osmolarity and VCN.
RNA-Seq and Data Analysis
The sequencing libraries were constructed from 100 to 1,000 ng total RNA from experimental groups (n = 1) using Illumina’s TruSeq Stranded Total RNA kit with Ribo-Zero Globin (Illumina, San Diego, CA, USA) following the manufacturer’s instruction. The fragment size of RNA-seq libraries was verified using the Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA), and the concentrations were determined using Qubit instrument (LifeTech, Waltham, MA, USA). Libraries were loaded onto the Illumina HiSeq 3000 for 2 × 75 bp paired-end read sequencing. Fastq files were generated using bcl2fastq software for further analysis. Quality control of paired-end reads was assessed using FastQC tools. Adaptor sequences were trimmed using Trimmomatic.39 Reads were aligned to the human reference genome (GRCh38) using HISAT2.40 FeatureCounts41 was used for gene-level abundance estimation using the GENCODE (v.29) comprehensive gene annotations.42 Genes were kept in the analysis if they had raw read counts >5 in at least one sample. Limma43 was used to process the read counts into log2 counts per million (log2cpm). The log2cpm values were normalized between samples using the trimmed mean of M values (TMM).44 The sHUDEP-2 and CD34+ samples were processed and normalized separately. Fold change ≥1.4 was used as a cutoff in the analysis. The datasets have been deposited in the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under the accession number GEO: GSE148529.
Statistical Analysis
The data were statistically analyzed with GraphPad Prism Software (v.6.05; GraphPad Software, USA) using one-way analysis of variance and Tukey’s post hoc test. Experiments were performed in triplicate (experimental replicates), and standard errors were represented as error bars. The p values <0.05 were considered to be statistically significant.
Author Contributions
S.D., J.F.T., and N.U. designed the experiments, analyzed and interpreted data, and wrote the paper. S.D., J.J.H.-M., T.N., C.D., M.Y., and J.G. conducted cell culture and molecular experiments. B.G. and F.S. performed RNA-seq data analysis. Q.L. performed ex vivo sickling experiments.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. William A. Eaton for his valuable comments on the article. In addition, we appreciate Dr. Mehdi Pirooznia’s help with RNA-seq data uploading to the database. We thank Dr. Ryo Kurita and Dr. Yukio Nakamura for providing the HUDEP-2 cell line. Furthermore, we would like to acknowledge NIH/NHLBI Biochemistry core facility for HPLC service and DNA core facility for RNA-seq service.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.013.
Supplemental Information
References
- 1.Piel F.B., Hay S.I., Gupta S., Weatherall D.J., Williams T.N. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10:e1001484. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bender M. Sickle cell disease. In: Adam M.P., editor. GeneReviews. University of Washington; 2017. pp. 5–13. [Google Scholar]
- 3.Norkin M., Wingard J.R. Recent advances in hematopoietic stem cell transplantation. F1000Res. 2017;6:870. doi: 10.12688/f1000research.11233.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demirci S., Uchida N., Tisdale J.F. Gene therapy for sickle cell disease: An update. Cytotherapy. 2018;20:899–910. doi: 10.1016/j.jcyt.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender M.A., Gelinas R.E., Miller A.D. A majority of mice show long-term expression of a human beta-globin gene after retrovirus transfer into hematopoietic stem cells. Mol. Cell. Biol. 1989;9:1426–1434. doi: 10.1128/mcb.9.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlsson S., Bodine D.M., Perry L., Papayannopoulou T., Nienhuis A.W. Expression of the human beta-globin gene following retroviral-mediated transfer into multipotential hematopoietic progenitors of mice. Proc. Natl. Acad. Sci. USA. 1988;85:6062–6066. doi: 10.1073/pnas.85.16.6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q., Emery D.W., Fernandez M., Han H., Stamatoyannopoulos G. Development of viral vectors for gene therapy of β-chain hemoglobinopathies: optimization of a γ-globin gene expression cassette. Blood. 1999;93:2208–2216. [PubMed] [Google Scholar]
- 8.Poillon W.N., Kim B.C., Rodgers G.P., Noguchi C.T., Schechter A.N. Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proc. Natl. Acad. Sci. USA. 1993;90:5039–5043. doi: 10.1073/pnas.90.11.5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton W.A., Hofrichter J. Sickle cell hemoglobin polymerization. In: Anfinsen C.B., Edsall J.T., Richards F.M., Eisenberg D.S., editors. Advances in Protein Chemistry. Volume 40. Elsevier; 1990. pp. 63–279. [DOI] [PubMed] [Google Scholar]
- 10.Srinivasulu S., Perumalsamy K., Upadhya R., Manjula B.N., Feiring S., Alami R., Bouhassira E., Fabry M.E., Nagel R.L., Acharya A.S. Pair-wise interactions of polymerization inhibitory contact site mutations of hemoglobin-S. Protein J. 2006;25:503–516. doi: 10.1007/s10930-006-9034-3. [DOI] [PubMed] [Google Scholar]
- 11.Pawliuk R., Westerman K.A., Fabry M.E., Payen E., Tighe R., Bouhassira E.E., Acharya S.A., Ellis J., London I.M., Eaves C.J. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 12.Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 13.Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 15.Hoban M.D., Cost G.J., Mendel M.C., Romero Z., Kaufman M.L., Joglekar A.V., Ho M., Lumaquin D., Gray D., Lill G.R. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Couch T., Murphy Z., Getman M., Kurita R., Nakamura Y., Steiner L.A. Human erythroblasts with c-Kit activating mutations have reduced cell culture costs and remain capable of terminal maturation. Exp. Hematol. 2019;74:19–24.e4. doi: 10.1016/j.exphem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Demirci S., Haro-Mora J.J., Leonard A., Hsieh M.M., Tisdale J.F. High-Efficiency Lentiviral Transduction of Human CD34+ Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019;13:187–196. doi: 10.1016/j.omtm.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavin A.L., Huang D., Huber C., Mårtensson A., Tardif V., Skog P.D., Blane T.R., Thinnes T.C., Osborn K., Chong H.S. PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat. Immunol. 2018;19:942–953. doi: 10.1038/s41590-018-0179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luis T.C., Ichii M., Brugman M.H., Kincade P., Staal F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia. 2012;26:414–421. doi: 10.1038/leu.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trakarnsanga K., Griffiths R.E., Wilson M.C., Blair A., Satchwell T.J., Meinders M., Cogan N., Kupzig S., Kurita R., Nakamura Y. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017;8:14750–14757. doi: 10.1038/ncomms14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S., Staahl B.T., Alla R.K., Doudna J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papanikolaou E., Anagnou N.P. Major challenges for gene therapy of thalassemia and sickle cell disease. Curr. Gene Ther. 2010;10:404–412. doi: 10.2174/156652310793180724. [DOI] [PubMed] [Google Scholar]
- 24.Magrin E., Semeraro M., Magnani A., Puy H., Miccio A., Hebert N., Diana J.-S., Lefrere F., Suarez F., Hermine O. Results from the Completed Hgb-205 Trial of Lentiglobin for β-Thalassemia and Lentiglobin for Sickle Cell Disease Gene Therapy. Blood. 2019;134(Suppl 1):3358. [Google Scholar]
- 25.Thein S.L. Pathophysiology of betaχ thalassemia—a guide to molecular therapies. Hematology Am. Soc. Hematol. Educ. Program. 2005;2005:31–37. doi: 10.1182/asheducation-2005.1.31. [DOI] [PubMed] [Google Scholar]
- 26.Sancar G.B., Cedeno M.M., Rieder R.F. Rapid destruction of newly synthesized excess beta-globin chains in HbH disease. Blood. 1981;57:967–971. [PubMed] [Google Scholar]
- 27.Shaeffer J.R., Kleve L.J., DeSimone J. betaS Chain turnover in reticulocytes of sickle trait individuals with high or low concentrations of haemoglobin S. Br. J. Haematol. 1976;32:365–372. doi: 10.1111/j.1365-2141.1976.tb00940.x. [DOI] [PubMed] [Google Scholar]
- 28.Bonner M., Kanter J., Macari E., Lane R., Lewis G., Coles P., Kassenaar S., Mynampati S., Schulze R., Hebert M., Walters M. The Relationships between Target Gene Transduction, Engraftment of HSCs and RBC Physiology in Sickle Cell Disease Gene Therapy. Blood. 2019;134:206. [Google Scholar]
- 29.Cantor A.B., Orkin S.H. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21:3368–3376. doi: 10.1038/sj.onc.1205326. [DOI] [PubMed] [Google Scholar]
- 30.Kim A., Dean A. Chromatin loop formation in the β-globin locus and its role in globin gene transcription. Mol. Cells. 2012;34:1–5. doi: 10.1007/s10059-012-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kornienko A.E., Guenzl P.M., Barlow D.P., Pauler F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mettananda S., Gibbons R.J., Higgs D.R. Understanding α-globin gene regulation and implications for the treatment of β-thalassemia. Ann. N Y Acad. Sci. 2016;1368:16–24. doi: 10.1111/nyas.12988. [DOI] [PubMed] [Google Scholar]
- 33.Uchida N., Hsieh M.M., Raines L., Haro-Mora J.J., Demirci S., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat. Commun. 2019;10:4479. doi: 10.1038/s41467-019-12456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uchida N., Evans M.E., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Sellers S.E., Dunbar C.E., Donahue R.E., Tisdale J.F. Integration-specific in vitro evaluation of lentivirally transduced rhesus CD34(+) cells correlates with in vivo vector copy number. Mol. Ther. Nucleic Acids. 2013;2:e122. doi: 10.1038/mtna.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida N., Haro-Mora J.J., Fujita A., Lee D.Y., Winkler T., Hsieh M.M., Tisdale J.F. Efficient Generation of β-Globin-Expressing Erythroid Cells Using Stromal Cell-Derived Induced Pluripotent Stem Cells from Patients with Sickle Cell Disease. Stem Cells. 2017;35:586–596. doi: 10.1002/stem.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demirci S., Bhardwaj S.K., Uchida N., Haro-Mora J.J., Ryu B., Blobel G.A., Tisdale J.F. Robust erythroid differentiation system for rhesus hematopoietic progenitor cells allowing preclinical screening of genetic treatment strategies for the hemoglobinopathies. Cytotherapy. 2018;20:1278–1287. doi: 10.1016/j.jcyt.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Uchida N., Haro-Mora J.J., Demirci S., Fujita A., Raines L., Hsieh M.M., Tisdale J.F. High-level embryonic globin production with efficient erythroid differentiation from a K562 erythroleukemia cell line. Exp. Hematol. 2018;62:7–16.e11. doi: 10.1016/j.exphem.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunkelberger E.B., Metaferia B., Cellmer T., Henry E.R. Theoretical Simulation of Red Cell Sickling Upon Deoxygenation Based on the Physical Chemistry of Sickle Hemoglobin Fiber Formation. J. Phys. Chem. B. 2018;122:11579–11590. doi: 10.1021/acs.jpcb.8b07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 42.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.