

Abstract
No single cancer immunotherapy will likely defeat all evasion mechanisms of solid tumors, including plasticity of tumor antigen expression and active immune suppression by the tumor environment. In this study, we increase the breadth, potency, and duration of anti-tumor activity of chimeric antigen receptor (CAR) T cells using an oncolytic virus (OV) that produces cytokine, checkpoint blockade, and a bispecific tumor-targeted T cell engager (BiTE) molecule. First, we constructed a BiTE molecule specific for CD44 variant 6 (CD44v6), since CD44v6 is widely expressed on tumor but not normal tissue, and a CD44v6 antibody has been safely administered to cancer patients. We then incorporated this BiTE sequence into an oncolytic-helper binary adenovirus (CAdDuo) encoding an immunostimulatory cytokine (interleukin [IL]-12) and an immune checkpoint blocker (PD-L1Ab) to form CAdTrio. CD44v6 BiTE from CAdTrio enabled HER2-specific CAR T cells to kill multiple CD44v6+ cancer cell lines and to produce more rapid and sustained disease control of orthotopic HER2+ and HER2−/− CD44v6+ tumors than any component alone. Thus, the combination of CAdTrio with HER2.CAR T cells ensures dual targeting of two tumor antigens by engagement of distinct classes of receptor (CAR and native T cell receptor [TCR]), and significantly improves tumor control and survival.
Keywords: oncolytic viro-immunotherapy, CAR T cell, BiTE molecule, cytokine, checkpoint inhibitor, CD44 variant 6
Graphical Abstract
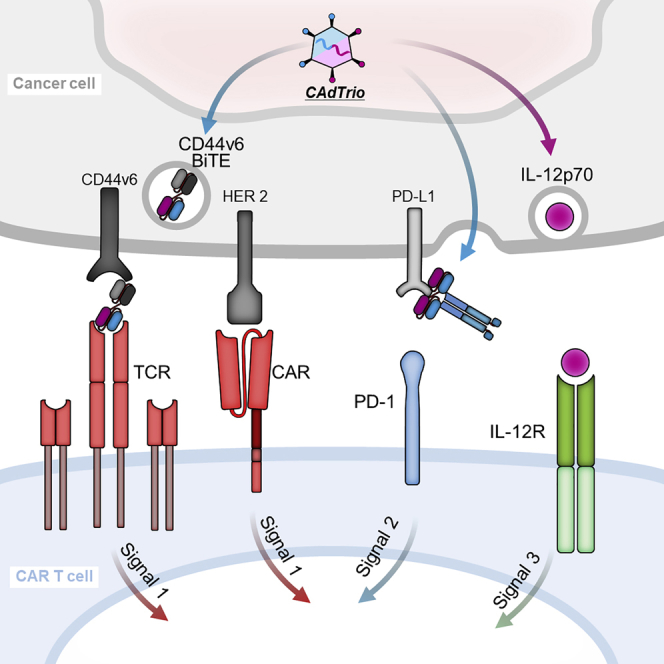
To enable HER2.CAR T cells to target two tumor antigens through both CAR and native TCR, Porter et al. developed a binary oncolytic-helper adenovirus expressing an engager molecule (BiTE), IL-12, and PD-L1 antibody. This combination therapy significantly improved tumor control and survival in models of HER2-positive and HER-negative cancer.
Introduction
Current clinical trials of immunotherapies combine multiple approaches to overcome solid tumor evasion strategies and plasticity of antigen expression. Our group has expertise creating oncolytic viruses (OVs) for solid tumor treatments,1 and in this study we investigated whether the therapeutic benefits of a tumor-targeting bispecific T cell engager (BiTE) can be augmented by combination with the immune-potentiating effects of an OV. Furthermore, we addressed whether it is feasible and beneficial to combine these approaches with the adoptive transfer of T cells engineered to express chimeric antigen receptors (CARs) directed to a different tumor antigen.
BiTE molecules engage T cells and target cells simultaneously, leading to T cell activation and specific target cell killing. BiTEs are composed of two single-chain variable fragments (scFvs) connected through a flexible peptide linker, one of which is specific for a tumor-associated antigen on target cells and the other for CD3 (signal 1).2 BiTEs, however, are rapidly catabolized and cleared from the circulation, leading to a short serum half-life.3 Furthermore, when used to target solid tumors, responses to BiTEs have been limited and toxicities high.4
OVs induce local pro-inflammatory lymphocytic and myelomonocytic responses and are an alternative means of constitutively expressing BiTEs. When OVs are used to deliver BiTEs, the greatest concentrations of these molecules will be at tumor sites, where viral replication occurs, and will favor recruitment of local tumor-infiltrating lymphocytes (TILs), including cells attracted by the inflammatory response.5 While BiTE-expressing OVs do indeed increase T cell activation and enhance the anti-tumor effects of TILs, benefits have been modest compared to OVs without BiTEs.6,7
In order to increase the potency of BiTE-expressing OVs by introducing additional costimulatory and cytokine stimuli (signals 2 and 3, respectively), we made use of our combinatorial Ad vector (CAd) system. In our CAd, co-infection of an oncolytic adenovirus (Onc.Ad) with helper-dependent Ad (HDAd), which can express multiple immunomodulatory molecules in a single vector (cargo capacity of up to 34 kb), replicates both Onc.Ad and HDAd through the Onc.Ad replication machinery. Infection with CAd leads to local oncolysis and expression of multiple transgene products without loss of oncolytic titer both in vitro and in vivo.1
We have shown that local CAd treatment simultaneously provides oncolysis while enhancing signal 2 by checkpoint inhibition (PD-L1 blocking antibody) and signal 3 by encoding the proinflammatory cytokine interleukin (IL)-12p70 (CAdDuo). In several xenograft models, CAdDuo significantly increased the anti-tumor activity of adoptively transferred HER2-specific CAR T cells.8,9 Here, we incorporated into CAdDuo a BiTE directed to the CD44 molecule, variant 6 isoform (CD44v6), which is highly expressed on many tumor cells but has no or low expression on normal tissues.10,11 We termed this new construct with all three molecules CAdTrio.
We show that CAdTrio enhances the anti-tumor effects of T cells that have been directed to a second tumor antigen (HER2neu, PSCA) by means of CAR expression. We used murine xenograft models to show that the combination of oncolysis, cytokine stimulation, checkpoint blockade, and dual targeting of two distinct antigens through both T cell receptor (TCR) (BiTE) and CAR engagement enables broad, potent, and durable anti-tumor activity.
Results
CD44v6 Is Highly Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC), and Other Human Tumors and Can Be Targeted by HDAd-Derived CD44v6 BiTE
We hypothesized that our HDAd could effectively express a BiTE molecule to enable CAR T cells to target HNSCC. Since the CD44v6 antibody (bivatuzumab) has been used safely to treat patients with HNSCC12,13 and CD44v6 targeting CAR T cells (CARTs) have demonstrated anti-tumor effects with limited toxicity in preclinical studies,10,14,15 we first evaluated CD44v6 positivity on HNSCC tissue microarrays (TMAs) (Figure 1A). We confirmed that most HNSCC tumors had significantly higher CD44v6 expression, both in intensity and proportion, than normal tissue. Next, we generated HDAds expressing CD44v6.BiTE or control CD19.BiTE (HDAdBiTE) (Figure 1B) and tested whether the secretion of the BiTE by HDAd-infected tumor cells led to specific target cell killing by non-transduced T cells (NTs). We used CRISPR/Cas9 to generate CD44 knockout FaDu (FaDuCD44−/−) (Figure S1) and infected FaDu or FaDuCD44−/− with CD19 and CD44v6 HDAdBiTE. By co-culturing these cancer cells with NTs (Figure 1B), we confirmed that T cells killed tumor cells in a CD44-dependent manner in the presence of CD44v6.BiTE, indicating that CD44v6.BiTE leads to antigen-dependent T cell recognition and killing.
Figure 1.

CD44v6 Is Highly Expressed on HNSCC Tumors, and CD44v6.BiTE Enables T Cell Killing of Multiple Cancer Cell Lines
(A) CD44v6 immunohistochemistry of larynx and salivary glands TMA. Data are presented as means ± SD. ∗∗∗∗p < 0.001. (B) Schematic structure of HDAd encoding a BiTE expression cassette. FaDu- and FaDuCD44−/−-expressing ffLuc cells were infected with 100 vp/cell of HDAds (n = 4 per group). Non-transduced T cells (NTs) were added with an effector-to-target ratio of 1:10 (E:T = 1:10) at 24 h post-infection. Cells were harvested 72 h post-co-culture with T cells, and viable cancer cells were analyzed by a luciferase assay. Data are presented as means ± SD. ∗p < 0.001. (C) CD44v6 expression was analyzed by flow cytometry on MDA-MB-231, CAPAN-1, SiHa, and PC-3 cells. These cells expressing ffLuc were infected with 100 vp/cell HDAds (n = 4 per group). NTs were added at 24 h post-infection (E:T = 1:10). Cells were harvested 72 h post-co-culture, and viable cancer cells were analyzed by a luciferase assay. Data are presented as means ± SD. ∗p < 0.001.
Since other solid tumors also express CD44v6,11,16, 17, 18, 19 we tested CD44v6.BiTE-dependent NT killing of prostate, breast, cervical, and pancreatic cancer cell lines and confirmed that CD44v6.BiTE induces T cell recognition and killing of all of these tumor types (Figure 1C). Hence, CD44v6.BiTE effectively targets not only HNSCC but also other solid tumors expressing the cognate antigen.
CD44v6.BiTE Expressed by HDAdTrio Enables T Cells to Kill Tumor Cells In Vitro
In order to test how the addition of signals 2 (PD-L1 blockade) and 3 (cytokine stimulation) contribute to BiTE-mediated killing in our system, we next constructed HDAd encoding CD44v6.BiTE, IL-12p70, and PD-L1 blocking mini-antibody expression cassettes (HDAdTrio). First, we confirmed that HDAdTrio expressed IL-12p70 and PD-L1 blocking antibody similarly to HDAdDuo (Figure 2A). Next, we compared the NT killing of target cells in the presence of HDAdBiTE alone and HDAdTrio. As a control, we used HDAdDuo. We found that HDAdBiTE induced CD44v6.BiTE-dependent killing, but these effects on NTs were greatly increased by the inclusion of the cytokine and checkpoint inhibitor present in HDAdTrio. These results indicate that incorporating an additional BiTE expression cassette into a single HDAd vector to create HDAdTrio does not interfere with the expression and function of the other transgenes, and it increases the anti-tumor effects.
Figure 2.

HDAd-Derived CD44v6.BiTE, IL-12p70, and PD-L1 Blocking Antibody Increase the Anti-tumor Effects of Non-transduced T Cell In Vitro
(A) Schematic structure of HDAd encoding CD44v6.BiTE, human IL-12p70, and anti-PD-L1 mini-antibody expression cassettes (HDAdTrio). FaDu- and MDA-MB-231-expressing ffLuc cells were infected with 100 vp/cell HDAdCD44v6.BiTE, HDAdDuo, or HDAdTrio (n = 4 per group). NTs were added at 24 h post-infection (E:T = 1:10). Cells were harvested 72 h post-co-culture with T cells, and viable cancer cells were analyzed by a luciferase assay. Data are presented as means ± SD. ∗p < 0.001. FaDu and MDA-MB-231 cells were infected with 200 vp/cell HDAdCD44v6.BiTE, HDAd12_PDL1, or HDAdTrio (n = 4 per group), and media samples collected 48 h post-infection were subjected to IL-12p70 ELISA and western blotting for PD-L1 mini-antibody, which was detected by anti-HA antibody. IL-12p70 data are presented as means ± SD. (B) FaDu- and MDA-MB-231-expressing ffLuc cells were infected with 100 vp/cell HDAd0 (no transgene), HDAdCD44v6.BiTE, HDAdDuo, or HDAdTrio (n = 5 per group). Either CD8 or CD4 NTs were added at 24 h post-infection (E:T = 1:10). Cells were harvested 72 h post-co-culture with T cells, and viable cancer cells were analyzed by a luciferase assay. Data are presented as means ± SD. ∗p < 0.001. (C) T cells were harvested 72 h post-co-culture, and CD25, PD-1, 4-1BB, and OX40 expression were analyzed by flow cytometry. (D) RNA was extracted from T cells 72 h post-co-culture, and gene expression was profiled with NanoString. Genes showing more than 75% coefficient of variation (CV) compared to pre-treatment T cells are shown.
To test whether BiTE induces target cell killing independent of the class of major histocompatibility complex (MHC) presentation, we examined which T cell subsets (CD4 or CD8 T cells) showed CD44v6.BiTE-dependent killing of target cells in the presence of cytokine and checkpoint inhibitor. We found that HDAdTrio improved the cytotoxicity of both CD8 and CD4 NTs compared to cells infected with HDAdBiTE or HDAdDuo (Figure 2B).
To address how BiTE, IL-12, and PD-L1 blocking antibody affect T cell signaling pathways, we first phenotyped NTs co-cultured with cancer cells infected with HDAdBiTE, HDAdDuo, or HDAdTrio by flow cytometry. HDAd-derived CD44v6.BiTE induced PD-1 expression on both CD4 and CD8 T cells and upregulated the activation markers CD25, 4-1BB, and OX40 (Figure 2C). Transcriptional profiling of these CD44v6.BiTE-stimulated T cells revealed upregulation of LAG3, TIM3 and CTLA-4 mRNA, as well as increased expression of T helper (Th)1-related genes (e.g., IFNG, granzyme B, CXCL9, CXCL10) in both CD4 and CD8 T cells (Figure 2D). Adding IL-12 and PD-L1 blocking antibody (HDAdTrio) further increased the expression of these Th1-related genes. Overall, these data indicate that HDAd-derived CD44v6.BiTE increases T cell activation but that this phenotype is also associated with T cell “exhaustion.”20
CD44v6.BiTE Expressed by HDAdTrio Increases the Anti-tumor Activity of HER2.CAR T Cells, and Co-infection of Onc.Ad to HDAd (CAd System) Increases Transgene Expression with Lytic Effect In Vitro
Since clinical experience has demonstrated the necessity of dual antigen targeting to eradicate heterogeneous solid tumors and produce sustained anti-tumor responses,21 we tested the in vitro effects of combining our HDAd-expressed BiTE molecule with T cells redirected by expression of a transgenic CAR to a second tumor-associated antigen. First, to confirm the ability of CARTs to kill target cells through a BiTE molecule (TCR), we co-cultured HER2.CARTs with the BiTE molecule either alone (from HDAdBiTE) or as a component of HDAdTrio. We found that expression of the BiTE in both contexts significantly improved the anti-tumor activity of HER2.CARTs against HER2+ and HER2−/− FaDu (CD44v6+) in vitro compared to HER2.CARTs alone (p < 0.001) (Figure S2A). We also phenotyped these CARTs and found no difference in activation markers in either CD8 or CD4 populations among groups (HDAd0, HDAdBiTE, HDAdDuo, and HDAdTrio) after co-culture with FaDu (HER2+ and CD44v6+). However, these markers were upregulated in a BiTE-dependent manner by both HDAdBiTE and HDAdTrio when HER2.CARTs were co-cultured with FaDuHER2−/−, similar to results obtained with NTs (Figure S2B).
Next, in order to evaluate the efficacy of our CAdTrio (co-infection with Onc.Ad and HDAdTrio that expresses IL-12, PD-L1 antibody, and BiTE), we confirmed that infecting target tumor cells (FaDu) with CAdTrio resulted in oncolysis and amplified expression of transgenes (IL-12 and PD-L1 antibody) (Figures S3A and S3B).1
Addition of BiTE to the CAd System Does Not Increase Anti-tumor Activity in the Subcutaneous Solid Tumor Model
To evaluate the synergistic benefits of CAdTrio and CARTs in vivo, we pre-treated mice with subcutaneous FaDu tumors with either CAdDuo or CAdTrio before infusing 1 × 106 HER2.CARTs 3 days later (Figure 3A). We were surprised to observe no difference in tumor growth between mice treated with HER2.CARTs and CAdDuo or CAdTrio. Indeed, although HER2.CARTs appeared at 7 days post-infusion to have expanded earlier at the tumor sites of mice treated with CAdTrio than mice treated with CAdDuo, by 14 days post-infusion it became clear that, overall, CARTs expanded less in mice treated with CAdTrio than CAdDuo.
Figure 3.

Addition of BiTE to CAdDuo Construct Does Not Increase Anti-tumor Activity in Subcutaneous Solid Tumor Models
(A) FaDu cells were transplanted into the right flank of NSG mice (n = 5 per group). A total of 1 × 108 vp of CAdDuo or CAdTrio (Onc:HD = 1:20) were injected into the tumor. A total of 1 × 106 HER2.CARTs expressing ffLuc were systemically administered 3 days post-injection of CAd. Tumor volumes and bioluminescence of HER2.CARTs were monitored at different time points. Data are presented as means ± SD, (B) CAPAN-1 cells were transplanted into the right flank of NSG mice (n = 5 per group). A total of 1 × 108 vp of CAdDuo or CAdTrio (Onc:HD = 1:20) were injected into the tumor. A total of 1 × 106 PSCA.CARTs expressing ffLuc were systemically administered 3 days post-injection of CAd. Tumor volumes and bioluminescence of PSCA.CARTs were monitored at different time points. Data are presented as means ± SD.
To address whether this effect was unique to either the target antigen (HER2.CARTs) or tumor (HNSCC), we evaluated the reproducibility of our results in a subcutaneous model of pancreatic cancer using cell line CAPAN-1. We pre-treated these mice with CAdDuo or CAdTrio and 3 days later infused 1 × 106 pre-clinically evaluated PSCA.CARTs22 (Figure 3B). Although both CAdDuo and CAdTrio improved the anti-tumor activity of adoptively transferred PSCA.CARTs, similar to results seen in our FaDu and HER2.CART model, we observed no difference in tumor growth between the treatment groups. Instead, as in our HNSCC model, we found that PSCA.CARTs expanded 50% less at tumor sites in mice treated with CAdTrio than CAdDuo. These results suggest that oncolysis with PD-L1 blockade and the proinflammatory cytokine IL-12 alone are sufficient to enhance the potency of adoptively transferred CARTs to control tumor growth in these subcutaneous models.
CAdTrio Enables Dual Targeting of HER2.CAR T Cells in Mice with Orthotopic FaDu Tumors
We next tested the anti-tumor activity of CARTs with and without BiTE in a more clinically relevant animal model producing both primary and metastatic disease similar to HNSCC patients, who have differences in tumor tissue location and phenotype.23 We transplanted ffLuc-labeled FaDu into the tongues of NSG mice and analyzed the anti-tumor effects of HER2.CARTs with CAdTrio compared to CAdDuo treatment in this model (Figure 4A). Three days after CAdDuo or CAdTrio treatment, we injected a sub-therapeutic dose (0.2 × 106) of HER2.CARTs intravenously (i.v.) and monitored tumor growth via ffLuc intensity. Tumors grew quickly in both the HER2.CART and CAdTrio only conditions. Although primary tumor growth was controlled in mice treated with CAdTrio alone, as a single therapy, the CAdTrio was unable to control metastatic tumor growth due to the limited distribution of the locally injected CAd.8
Figure 4.

CAd-Derived CD44v6.BiTE Induces Early Activation of HER2.CAR T Cells and Enhances Their Anti-tumor Effects in an Orthotopic HNSCC Model
(A) FaDu cells expressing ffLuc were transplanted into the tongues of NSG mice. A total of 1 × 108 vp of CAdDuo or CAdTrio (Onc:HD = 1:20) were injected into the tongue. A total of 0.2 × 106 HER2.CARTs were systemically administered 3 days post-injection of CAds. Bioluminescence of FaDu cells was monitored at different time points (n = 5 per group). Data are presented as means ± SD. ∗p = 0.002. (B) To monitor tumor growth (n = 5) and HER2.CART persistence (n = 5), serum samples were collected from mice at 0, 3, 10, 24, 45, 66, and 87 days post-injection of CAd, and IFNγ and IL-12p70 levels in serum were measured by ELISA. Data are presented as means ± SD. ∗p < 0.001. (C) T cells were isolated from tongue and lymph node sites at 8 and 105 days post-infusion of HER2.CARTs, and HER2.CAR, CD25, and PD-1 expression were analyzed by flow cytometry. (D) Kaplan-Meier survival curve after CAdDuo or CAdTrio administration in mice (n = 5–10). Data are presented as means ± SD. ∗p < 0.01.
Within 3 weeks post-treatment, the speed and durability of the anti-tumor effect were significantly greater in mice receiving CAdTrio and HER2.CARTs than in mice receiving CAdDuo and HER2.CARTs (p = 0.002). We next determined whether anti-tumor responses to CAdTrio were associated with increased HER2.CART expansion and activation. Although we detected a trend toward such associations, neither reached statistical significance. For example, Figure S4A shows that HER2.CART-derived signal at tumor sites in mice treated with CAdTrio increased earlier than in mice treated with CAdDuo (8 days versus 14 days post-infusion) (95% confidence interval [CI]: CARTs, 2.11 × 108 to 8.36 × 108; CAdDuo + CARTs, −2.06 × 107 to 1.23 × 108; CAdTrio + CARTs, −7.5 × 107 to 4.61 × 108). Similarly, Figure 4B shows that the level of interferon (IFN)γ in the blood of CAdTrio-treated mice trended higher than in mice treated with HER2.CARTs alone or in combination with CAdDuo at 7 days post-infusion. Although the changes shown in Figure S4A were modest, when taken together with the speedier activation (IFNγ expression) observed when HER2.CARTs were combined with CAdBiTE (expressing CD44v6.BiTE alone) (Figure S4C), these data suggest that the CAd-derived BiTE accelerates tumor recognition and activation of CARTs. Any early tumor control induced by CAdBiTE alone, however, was evidently of limited value, and was offset by subsequent loss of later anti-tumor effector activity. These results are in contrast to outcomes in mice treated with CAdDuo or CAdTrio, which respectively provided alternative and additional immunostimulatory signals (Figure S4C).
To discover whether CAdTrio altered the activation, expansion, or distribution of ffLuc-labeled HER2.CARTs at tumor sites compared to CAdDuo, we isolated and phenotyped TILs from the primary tumor at 8 days post-infusion (Figure 4C). We found that at this early time point HER2.CARTs from mice treated with CAdTrio trended toward higher expression of CD25 than did mice treated with CAdDuo. These results, coupled with higher circulating IFNγ levels (Figure 4B), suggest that BiTE induces early activation of HER2.CARTs. We also found that combinatorial treatment with CAdTrio and HER2.CARTs attenuated early mouse death from tumor growth-induced weight loss (Figure S4B).
However, from 3 weeks post-infusion until the end of experiment, mice treated with CAdDuo had similar HER2.CART persistence (Figure S4A) and levels of IFNγ (Figure 4B) as those treated with CAdTrio. Therefore, we also phenotyped TILs from these mice at the end of experiment (105 days post-infusion) and found no phenotypic differences (Figure 4C), suggesting that later HER2.CART phenotypes are dependent on IL-12 and PD-L1 antibody and not the presence of the BiTE. Mice treated with CAdBiTE also had significantly improved HER2.CART activity at 7 days post-infusion (assessed by circulating IFNγ levels: p < 0.001) compared to mice treated with HER2.CARTs alone. Although CAdBiTE prolonged median survival of HER2.CART-treated mice (CAdBiTE + CARTs [45 days] versus CARTs alone [18 days]) (Figure S4C), mice receiving CAdBiTE died significantly earlier than those pre-treated with CAdDuo (90 days) or CAdTrio (>105 days) (Figure 4D; Table 1). Overall, these data support a model in which initial control through the BiTE allows emergence of longer-term tumor control mediated by T cell stimulation and checkpoint inhibition through IL-12 and PD-L1.
Table 1.
Median Survival of Orthotopic Animal Models
| Control | CARTs | CAdDuo + CARTs | CAdTrio + CARTs | CAdTrio | |
|---|---|---|---|---|---|
| FaDu | 14 days | 18 days | 90 days | >105 days | 42 days |
| FaDuHER2−/− | 16 days | 18 days | 60 days | >90 days | 14 days |
CAdTrio Improves Anti-tumor Activity of HER2.CAR T Cells in FaDuHER2−/− Orthotopic Mice
Due to the intrinsic heterogeneity of antigen expression by human solid tumors,24 and their propensity to downregulate antigens and/or select negative variants,25 T cell therapies for solid tumors targeting a single antigen have had limited success in the clinic. To determine whether CD44v6.BiTE stimulation of CARTs through their TCR is sufficient for control of HER2−/− tumors in vivo, we transplanted FaDuHER2−/− labeled with ffLuc into the tongues of NSG mice. Three days after CAd treatment, we injected 1 × 106 HER2.CARTs i.v. and monitored tumor growth by ffLuc activity (Figure 5A). As expected, neither HER2.CARTs alone nor HER2.CARTs with CAdDuo produced significant anti-tumor activity compared to control (untreated) animals. Mice treated with CAdTrio and HER2.CARTs, however, had significant tumor control compared to mice treated with HER2.CARTs or CAdTrio alone (p = 0.012), indicating that CAd-derived CD44v6.BiTE can engage adoptively transferred HER2.CARTs sufficiently well to produce anti-tumor effects even against HER2−/− targets. Indeed, we saw a similar survival benefit in mice bearing HER2−/− tumors treated with CAdBiTE plus HER2.CARTs (Figure S5). These results show that CD44v6.BiTE was required for transferred HER2.CARTs to have any anti-tumor activity in animals with HER2− tumors (FaDu HER2−/−). Since the CARTs are active even against tumor cells lacking the target antigen for their CAR but that express the BiTE-specific antigen, sufficient BiTE must be produced from our CAd system to induce in vivo immunological activity.
Figure 5.

CAd-Derived CD44v6.BiTE Improves the Anti-tumor Effects of HER2.CARTs in an Orthotopic HER2−/− HNSCC model
(A) FaDuHER2−/− cells expressing ffLuc were transplanted into the tongues of NSG mice. A total of 1 × 108 vp of CAdDuo or CAdTrio (Onc:HD = 1:20) were injected into the tongue (n = 5 per group). A total of 1 × 106 HER2.CARTs were systemically administered 3 days post-injection of CAds. Bioluminescence of FaDuHER2−/− cells was monitored at different time points. Data are presented as means ± SD. ∗p < 0.01. (B) Serum samples were collected at 0, 3, 10, 24, 45, 66, and 87 days post-injection of CAds (n = 5 per group), and IFNγ and IL-12p70 levels in serum were measured by ELISA. Data are presented as means ± SD. ∗p < 0.001. (C) Kaplan-Meier survival curve after CAdDuo or CAdTrio administration. Data are presented as means ± SD. ∗p = 0.012.
Compared to CAdDuo, CAdTrio induced more rapid anti-tumor activity in mice with FaDu CD44v6+ HER2−/− tumors, similar to results seen in mice with parental orthotopic FaDu tumors (HER2+, CD44v6+), and this CAdTrio-dependent HER2.CART activity again correlated with an early increase in IFNγ levels (Figure 5B). Finally, mice treated with CAdTrio had improved median survival compared to mice treated with CAdBiTE (CAdTrio + CARTs [>90 days] versus CAdBiTE + CARTs [63 days]) (Figure 5C; Figure S5; Table 1), indicating that IL-12 and PD-L1 blockade are an additional requirement if long-lasting tumor control is to be attained by HER2.CARTs, irrespective of the presence of the HER2.CAR target antigen.
Overall, these studies suggest that CAdTrio-derived BiTE, PD-L1 blocking antibody, and IL-12 independently but additively increase the early and long-term anti-tumor efficacy of adoptively transferred HER2.CARTs and enhance activity against both HER2+ and HER2− subpopulations of HNSCC tumors, indicating that benefit would be obtained even when HER2 expression is heterogeneous.
Discussion
Overall, we demonstrate that CAd-derived expression of CD44v6.BiTE molecules, PD-L1 blockade, and the proinflammatory cytokine IL-12 allow HER2.CARTs to control tumor growth in several xenograft models, including orthotopic mouse models of both HER2+ and HER2− HNSCC.
One major limitation of any T cell therapy targeting a single antigen is the intrinsic heterogeneity of antigen expression by human solid tumors, and their propensity to select antigen-negative variants.21 Clinically, relapse with CD19-negative disease following CD19.CART treatment led to strategies for dual targeting CARTs (CD22 and CD19,26 CD19, and BCMA27), which have shown preclinical success against hematologic malignancies. In this study, we used a multimodal approach to provide T cells with two distinct targeting mechanisms to two different tumor-associated antigens. We incorporated BiTE molecules into our binary oncolytic adeno-immunotherapy CAd system (Onc.Ad plus HDAd) to enable CARTs to target two distinct tumor-associated antigens via two distinct mechanisms (HER2 through the CAR and CD44v6 through the BiTE) while also providing cytokine support and checkpoint blockade.
We found that the BiTE molecule increased the anti-tumor activity of HER2.CARTs against multiple solid tumor cell lines in vitro, indicating that the CARTs targeted HER2 through CAR and CD44v6 through BiTE-TCR. These data support a recent study that combined FRα.CARTs and Onc.Ad expressing EGFR.BiTE,28 which demonstrated that adoptively transferred CARTs could target cancer cells expressing both antigens at different levels in vitro and in vivo. One clinical study with TCR-directed T cell therapy targeting HPV16 E6 demonstrated MHC downregulation-dependent cancer cell escape and relapse,29 while BiTEs can ensure T cell engagement independent of MHC presentation. In addition, engagement through the TCR may result in physically distinct immune synapse structure and organization compared to CAR engagement, with subsequent modulation of T cell cytotoxic function and persistence.30 Hence, a combination of targeting by CAR and BiTE may offer additional advantages over targeting the same antigen with CAR alone.
Given the heterogeneity and plasticity of human solid tumors, oncolytic virotherapy should be most effective against both injected and metastatic/abscopal tumor sites when oncolysis is combined with relatively sustained immunostimulatory transgene expression.5 Our orthotopic animal models8 and previous clinical trials with local oncolytic adenovirus treatment31 suggest that potent oncolysis can prematurely terminate the recruitment of an effective immune response. Conversely, inadequate oncolysis does not disrupt the local immunosuppressive environment sufficiently to generate effective immune responses for the desired “virtuous circle” of continuing tumor destruction. In this study, we used our CAd system to amplify the therapeutic transgenes encoded in HDAd with lytic effects through the Onc.Ad replication machinery and confirmed that the ratio used maximally amplified the transgenes encoded in HDAd with oncolysis.1 Although we think that our CAd system provides a balance between oncolysis and immunostimulatory transgene expression, we currently do not know where the line between inadequate and excessive oncolysis lies. Additionally, even in the presence of abscopal effects, local oncolytic adeno-immunotherapy treatment was still insufficient to completely cure distant metastases on previous clinical trials.31 We thus think that the described combination of local CAd treatment with systemic CART treatment will meet these requirements, as each component alone has significant anti-tumor activity. Ultimately, clinical trials will reveal whether more potent oncolytic activity is necessary for the desired systemic anti-tumor effects, and analyses to address this question will be a major component of our phase I clinical trials with these agents that are currently under US Food and Drug Administration (FDA) review.
In this study, we found that combining IL-12 and PD-L1 blocking antibody with a BiTE molecule (CAdTrio) enhanced HER2.CART killing efficacy in vitro, indicating that BiTE and additional transgenic immunomodulatory molecules synergistically improve CART activity. However, in our subcutaneous animal models, inclusion of the BiTE molecule in CAdTrio did not improve the anti-tumor activity of CARTs compared to pre-treatment with CAdDuo (IL-12 + PD-L1). Since activated CARTs highly express PD-1 in vitro, and cancer cells highly express PD-L1 in the presence of IFNγ expressed by activated CARTs,8,9 it is possible that blockade of PD-1/PD-L1 interaction at the tumor site, coupled with provision of IL-12, is sufficient to sustain CART function in subcutaneous models.
In a more clinically relevant orthotopic HNSCC model with lymph node metastasis similar to HNSCC patients,23 however, we found that mice treated with CAdTrio showed early tumor control in the presence of adoptively transferred CARTs, and thus fewer mice died early (within 14 days of treatment) than in the CAdDuo cohort. Earlier death in the mice treated with CAdDuo was attributable to poor control of the primary orthotopic tumor, leading to severe inanition. Although we found that CAd-derived BiTE leads to immediate tumor recognition and activation of CARTs in vivo, the longer-term benefit from early tumor control induced by CAdBiTE (no IL-12 and PD-L1 antibody expression cassettes) in vivo was offset by subsequent loss of anti-tumor activity in our FaDu (HER2+, CD44v6+) orthotopic model. Although the combination of CAdTrio and HER2.CARTs significantly prolonged animal survival compared to mice treated with either CAdTrio or HER2.CARTs alone, we found residual tumor signals in some of these xenograft mice. Local oncolytic viro-immunotherapy is intended to augment the host anti-tumor responses by inducing potent abscopal immune responses,32, 33, 34 an effect that cannot be fully replicated in these immunodeficient models. Hence, we are developing approaches to measure the anti-tumor efficacy of our combinatorial treatment in mice reconstituted with human innate and adaptive immune cells to address this potential limitation.35
We were intrigued to find that BiTE molecules derived from CAdBiTE or CAdTrio improved the anti-tumor activity of HER2.CARTs even against HER2−/− HNSCC tumors in vivo. These results indicate that when “signal 1” is provided through CD44v6.BiTE, anti-tumor activity is increased even in the absence of the CART target antigen (HER2). A previous study found that an irrelevant CART (CD19.CART) with Onc.Ad-expressing EGFR.BiTE had transient anti-tumor activity against CD19−, EGFR+ pancreatic tumors.28 Unlike that report, however, in our HER2− HNSCC model when combined with CAdTrio, HER2.CARTs demonstrated long-term anti-tumor activity, increasing median survival to >90 days compared to 18 days with CARTs alone. These studies reinforce the requirement of antigen recognition (through BiTE), cytokine stimulation (IL-12), and PD-L1 blockade for long-term HER2.CART function.
In summary, we demonstrate that the local provision of CD44v6.BiTE and immunomodulatory molecules as a CAd package augments the anti-tumor effects of adoptively transferred HER2.CARTs, allowing them to control the growth of both HER2+ and HER2−/− HNSCC orthotopic tumors.
Materials and Methods
Adenoviral Vectors (HDAds and Onc.Ads)
HDAd without transgene (HDAd0) was produced as previously described.36 To generate the CD44v6.BiTE, the anti-human CD44v6 single-chain variable region of BIWA-4 was fused with anti-human CD3ε single-chain variable region with a sequence encoding a flexible glycine-serine (GS) linker. Control CD19.BiTE was produced as previously described.2 BiTE expression cassettes driven by the EF1 promoter (HDAdBiTE vector) were inserted into the pHDΔ28E4 vector. The CD44v6.BiTE, IL-12p70, and PD-L1 blocking mini-antibody expression cassettes (CD44v6.BiTE driven by EF1 promoter, IL-12p70 driven by hamster GRP78 promoter, and PD-L1 mini-antibody driven by human GRP94 promoter [InvivoGen]) were cloned into pHDΔ21E4 (HDAdTrio vector). HDAd encoding the same IL-12p70 and PD-L1 blocking mini-antibody expression cassettes was cloned into pHDΔ25E4 (HDAd12_PDL1 vector). After confirming the sequence and expression of IL12p70 (ELISA [BD Bioscience]) and PD-L1 blocking mini-antibody (western blotting with anti-HA IgG [Thermo Fisher Scientific]), all HDAds were rescued with chimeric helper virus 5/3 (knob replacing Ad serotype 3 from Ad serotype 5) as previously described.37,38 Infectious units (IU) of HDAds were measured using A549 cells as previously described.37 IU of HDAds used in this study are summarized in Table 2. Chimeric Onc.Ad5/3Δ24 (knob replacing Ad serotype 3 from Ad serotype 5) was produced as previously described.1,39
Table 2.
Infectious Units of HDAds
| HDAd0 | HDAd |
||||
|---|---|---|---|---|---|
| CD19.BiTE | CD44v6.BiTE | Duo | Trio | ||
| vp:IU | 100:4 | 100:5 | 100:10 | 100:7 | 100:11 |
Cell Lines
Human HNSCC line FaDu, human prostate cancer cell line PC-3, human cervical cancer cell line SiHa, and human breast cancer cell line MDA-MB-231 were obtained from ATCC (Manassas, VA, USA) in 2016. Cell lines were authenticated utilizing short tandem repeat (STR) profiling by ATCC. Cells were cultured under the conditions recommended.
To generate the HER2- or CD44-deficient FaDu cell lines (FaDuHER2−/−, FaDuCD44−/−), we removed the HER2 or CD44 gene using CRISPR/Cas9. FaDu cells were electroporated with 0.4 μg of guide RNA (gRNA) and 1 μg of Cas9 protein (Integrated DNA Technologies) with 0.25 × 106 FaDu cells using Neon transfection system (Thermo Fisher Scientific) in 10 μL of buffer R with 1,600 V and 10-ms pulses.40 Following electroporation and expansion, FaDuHER2−/− and FaDuCD44−/− cells were sorted using an SH800 cell sorter (Sony) after staining cells with anti-human HER2 or CD44 antibody (BD Biosciences, BioLegend).
To generate cell lines expressing the fusion protein EGFP-ffLuc, we infected cells with retrovirus encoding EGFP-ffLuc as previously described.8,9 EGFP+ cells were sorted using an SH800 cell sorter after 3 passages post-infection of retrovirus.
Primary Cells
Human peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque Plus according to the manufacturer’s instructions (Axis-Shield). The vector encoding the HER2-directed CAR incorporating the CD28 costimulatory endodomain (second-generation HER2.28ζ.CAR)41 or PSCA-directed CAR incorporating the CD28 costimulatory endodomain (second-generation PSCA.28ζ.CAR),22 the fusion protein EGFP-ffLuc, and the methodology for the production of retrovirus and CARTs have been described previously.42 Briefly, PBMCs were activated with OKT3 (1 mg/mL) (Ortho Biotech) and CD28 antibodies (1 mg/mL) (Becton Dickinson) and fed every 2 days, beginning the day after stimulation, with media supplemented with 100 U/mL recombinant human IL-2 (rIL-2, NIH). On day 2 post-OKT3/CD28 T blast generation, activated T cells (0.125 × 106/mL) were added to CAR retroviral-coated plates and centrifuged at 400 × g for 5 min. CAR-transduced T cells (HER2.CARTs, PSCA.CARTs) were expanded with media supplemented with 100 U/mL rIL-2.
Co-culture Experiments
ffLuc-expressing cancer cells, including knockout lines, were seeded in 48-well plates and infected with 200 viral particles (vp) (14–23 IU) per cell of HDAds 24 h later. Either non-transduced T cells or HER2.CARTs were added 24 h post-infection at ratios described in the figure legends and cultured for 72 h. For co-culture experiments with CD4 or CD8 T cell subsets, CD4 and CD8 T cells were separated using human CD8 or CD4 microbeads (Miltenyi Biotec) and checked by flow cytometry for purity (more than 95% enrichment) 1 day before co-culture. After a 72-h co-culture with T cells, we measured residual live cancer cells (ffLuc activity) using a luciferase assay system (Promega) and measured by plate reader (BMG Labtech).
Microarray Analysis
Total RNA was extracted from CD4 or CD8 T cells, either after co-culture with HDAd-infected FaDu cells or without stimulation, as described in the figure legends. Total RNA was extracted from T cells using the RNeasy Mini kit (QIAGEN) and quantified using the NanoDrop 2000 (Thermo Fisher Scientific). The Baylor Genomic & RNA Profiling Core performed RNA expression profiling with the nCounter human immunology v2 panel (NanoString Technologies). Data quality control, normalization, and analysis were performed using nSolver 4.0 analysis software following NanoString analysis guidelines.
Flow Cytometry
The following fluorochrome-conjugated monoclonal antibodies were used: anti-human CD3, CD4, CD8, CD25, CD69, CD134, CD137, CCR7, CD45RO, PD-1, PD-L1, HER2, CD44v6, recombinant human HER2-Fc chimera, and anti-Fc (for detection of HER2.CAR) (BD Biosciences, Beckman Coulter, BioLegend, R&D Systems). Adherent cells were harvested using 0.5 mM EDTA solution (Thermo Fisher Scientific) before staining.43 Cells were stained with these antibodies or the appropriate isotype controls antibodies for 30 min at 4°C. Live/dead discrimination was determined via exclusion of 7-aminoactinomycin D (7AAD)+ cells (BD Pharmingen). Stained cells were analyzed using a Gallios (Beckman Coulter) flow cytometer and analyzed with Kaluza software (BD Bioscience) according to the manufacturers’ instructions.
Animal Experiments
All animal experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
For the subcutaneous models, 1 × 106 FaDu cells or 2 × 106 CAPAN-1 cells were resuspended in a volume of 100 μL of PBS and injected into the right flank of 7- to 8-week-old NSG male or female mice. Fourteen days post-transplantation, a total of 1 × 108 vp of CAd (Onc.Ad/HDAd at 1:201) were injected in a volume of 20 μL into the tumor. The ratio of Onc.Ad to HDAd in CAd system was optimized to effectively propagate transgenes encoded in the co-injected HDAd with lytic effects in multiple animal models.1,8,9 Three days post-injection of CAds, mice received 1 × 106 HER2.CARTs (for FaDu model) or 1 × 106 PSCA.CARTs (for CAPAN-1 model) i.v. CARTs expressing ffluc were assessed using an in vivo imaging system (Xenogen).9 The endpoint was established at a tumor volume >1,500 mm3.
For the orthotopic models, 0.5 × 106 FaDu cells, ffLuc-expressing FaDu cells, or ffLuc-expressing FaDuHER2−/− cells were resuspended in a volume of 50 μL of PBS and injected into the tongues of 7- to 8-week-old NSG male or female mice. Six days post-transplantation, a total of 1 × 108 vp of CAd (Onc.Ad/HDAd at 1:201) were injected in a volume of 20 μL into the tongue. Three days post-injection of CAds, mice received 0.2 × 106 HER2.CARTs (for the FaDu model) or 1.0 × 106 HER2.CARTs (for the FaDuHER2−/− model) i.v. Tumor or HER2.CARTs expressing ffluc were assessed using an in vivo imaging system (Xenogen).9 For cytokine detection in serum samples, serum was collected on the dates described in Results. The endpoint was established as animal body weight <80%.
Isolation of Tumor-Infiltrating T Cells
After rinsing harvested tumors with PBS, tumors were minced and incubated in RPMI l640 media containing collagenase type IV (5 mg/mL) and type I (1 mg/mL) (Thermo Fisher Scientific) at 37°C for 2 h.44 Cells were passed through a 70-μm cell strainer (BD Pharmingen), and murine stroma cells were removed using a mouse cell depletion kit (Miltenyi Biotec). Human cells were stained with the antibodies described in Results.
Immunohistochemistry
The Human Tissue Acquisition and Pathology Core at Baylor College of Medicine stained and scored larynx and salivary gland TMAs with anti-human CD44v6 antibody (VFF-7; Thermo Fisher Scientific).
Statistical Analysis
Data were analyzed by one-way ANOVA followed by Rank’s protected least significant difference test (SigmaPlot).
Author Contributions
Conceptualization, M.S.; Methodology, C.E.P., A.R.S., Y.J., T.Y., and P.D.C; Investigation, C.E.P., A.R.S, Y.J., T.Y., P.D.C., M.M.I., and M.S.; Resources, A.S. and S.G.; Writing – Original Draft, C.E.P. and M.S.; Writing – Review & Editing, M.S. and M.K.B.; Supervision, M.S.; Funding Acquisition, M.K.B. and M.S.
Conflicts of Interest
M.S. is a consultant for Tessa Therapeutics Ltd. The remaining authors declare no competing interests.
Acknowledgments
The authors would like to thank Catherine Gillespie in the Center for Cell and Gene Therapy at Baylor College of Medicine for editing the paper, and Walter Mejia in the Center for Cell and Gene Therapy at Baylor College of Medicine for the graphical design of the abstract. This work was supported by Tessa Therapeutics Pte. This work was also supported by NIH P01 CA094237 to M.K.B. and NIH P30-CA125123 to the Human Tissue Acquisition and Pathology Core.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.02.016.
Supplemental Information
References
- 1.Farzad L., Cerullo V., Yagyu S., Bertin T., Hemminki A., Rooney C., Lee B., Suzuki M. Combinatorial treatment with oncolytic adenovirus and helper-dependent adenovirus augments adenoviral cancer gene therapy. Mol. Ther. Oncolytics. 2014;1:14008. doi: 10.1038/mto.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velasquez M.P., Torres D., Iwahori K., Kakarla S., Arber C., Rodriguez-Cruz T., Szoor A., Bonifant C.L., Gerken C., Cooper L.J. T cells expressing CD19-specific engager molecules for the immunotherapy of CD19-positive malignancies. Sci. Rep. 2016;6:27130. doi: 10.1038/srep27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu M., Wu B., Brandl C., Johnson J., Wolf A., Chow A., Doshi S. Blinatumomab, a bispecific T-cell Engager (BiTE®) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin. Pharmacokinet. 2016;55:1271–1288. doi: 10.1007/s40262-016-0405-4. [DOI] [PubMed] [Google Scholar]
- 4.Slaney C.Y., Wang P., Darcy P.K., Kershaw M.H. CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 2018;8:924–934. doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- 5.Harrington K., Freeman D.J., Kelly B., Harper J., Soria J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019;18:689–706. doi: 10.1038/s41573-019-0029-0. [DOI] [PubMed] [Google Scholar]
- 6.Fajardo C.A., Guedan S., Rojas L.A., Moreno R., Arias-Badia M., de Sostoa J., June C.H., Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77:2052–2063. doi: 10.1158/0008-5472.CAN-16-1708. [DOI] [PubMed] [Google Scholar]
- 7.Freedman J.D., Duffy M.R., Lei-Rossmann J., Muntzer A., Scott E.M., Hagel J., Campo L., Bryant R.J., Verrill C., Lambert A. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78:6852–6865. doi: 10.1158/0008-5472.CAN-18-1750. [DOI] [PubMed] [Google Scholar]
- 8.Rosewell Shaw A., Porter C.E., Watanabe N., Tanoue K., Sikora A., Gottschalk S., Brenner M.K., Suzuki M.S. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol. Ther. 2017;25:2440–2451. doi: 10.1016/j.ymthe.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanoue K., Rosewell Shaw A., Watanabe N., Porter C., Rana B., Gottschalk S., Brenner M., Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77:2040–2051. doi: 10.1158/0008-5472.CAN-16-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casucci M., Nicolis di Robilant B., Falcone L., Camisa B., Norelli M., Genovese P., Gentner B., Gullotta F., Ponzoni M., Bernardi M. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–3472. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 11.Misra S., Hascall V.C., Markwald R.R., Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015;6:201. doi: 10.3389/fimmu.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tijink B.M., Buter J., de Bree R., Giaccone G., Lang M.S., Staab A., Leemans C.R., Guus van Dongen G.A.M.S. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006;12:6064–6072. doi: 10.1158/1078-0432.CCR-06-0910. [DOI] [PubMed] [Google Scholar]
- 13.Rupp U., Schoendorf-Holland E., Eichbaum M., Schuetz F., Lauschner I., Schmidt P., Staab A., Hanft G., Huober J., Sinn H.P. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: final results of a phase I study. Anticancer Drugs. 2007;18:477–485. doi: 10.1097/CAD.0b013e32801403f4. [DOI] [PubMed] [Google Scholar]
- 14.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.-C., Naranjo A., Starr R., Wagner J., Wright C. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez J.R., Kim J.J., Verdone J.E., Liu X., Torga G., Pienta K.J., Mooney S.M. Alternative CD44 splicing identifies epithelial prostate cancer cells from the mesenchymal counterparts. Med. Oncol. 2015;32:159. doi: 10.1007/s12032-015-0593-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotoda T., Matsumura Y., Kondo H., Saitoh D., Shimada Y., Kosuge T., Kanai Y., Kakizoe T. Expression of CD44 variants and its association with survival in pancreatic cancer. Jpn. J. Cancer Res. 1998;89:1033–1040. doi: 10.1111/j.1349-7006.1998.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Götte M., Yip G.W. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. Cancer Res. 2006;66:10233–10237. doi: 10.1158/0008-5472.CAN-06-1464. [DOI] [PubMed] [Google Scholar]
- 19.Ayhan A., Baykal C., Al A., Ayhan A. Altered CD44 variant 6 expression in FIGO stage IB cervical carcinoma. Gynecol. Oncol. 2001;83:569–574. doi: 10.1006/gyno.2001.6406. [DOI] [PubMed] [Google Scholar]
- 20.Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 21.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe N., Bajgain P., Sukumaran S., Ansari S., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology. 2016;5:e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masood R., Hochstim C., Cervenka B., Zu S., Baniwal S.K., Patel V., Kobielak A., Sinha U.K. A novel orthotopic mouse model of head and neck cancer and lymph node metastasis. Oncogenesis. 2013;2:e68. doi: 10.1038/oncsis.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gengenbacher N., Singhal M., Augustin H.G. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat. Rev. Cancer. 2017;17:751–765. doi: 10.1038/nrc.2017.92. [DOI] [PubMed] [Google Scholar]
- 25.Guedan S., Calderon H., Posey A.D., Jr., Maus M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2018;12:145–156. doi: 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan Z., Cao J., Cheng H., Qiao J., Zhang H., Wang Y., Shi M., Lan J., Fei X., Jin L. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 2019;6:e521–e529. doi: 10.1016/S2352-3026(19)30115-2. [DOI] [PubMed] [Google Scholar]
- 28.Wing A., Fajardo C.A., Posey A.D., Jr., Shaw C., Da T., Young R.M., Alemany R., June C.H., Guedan S. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol. Res. 2018;6:605–616. doi: 10.1158/2326-6066.CIR-17-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doran S.L., Stevanović S., Adhikary S., Gartner J.J., Jia L., Kwong M.L.M., Faquin W.C., Hewitt S.M., Sherry R.M., Yang J.C. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J. Clin. Oncol. 2019;37:2759–2768. doi: 10.1200/JCO.18.02424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davenport A.J., Cross R.S., Watson K.A., Liao Y., Shi W., Prince H.M., Beavis P.A., Trapani J.A., Kershaw M.H., Ritchie D.S. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA. 2018;115:E2068–E2076. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw A.R., Suzuki M. Immunology of adenoviral vectors in cancer therapy. Mol. Ther. Methods Clin. Dev. 2019;15:418–429. doi: 10.1016/j.omtm.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuryk L., Møller A.W., Jaderberg M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J. Med. Virol. 2019;91:1702–1706. doi: 10.1002/jmv.25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chon H.J., Lee W.S., Yang H., Kong S.J., Lee N.K., Moon E.S., Choi J., Han E.C., Kim J.H., Ahn J.B. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin. Cancer Res. 2019;25:1612–1623. doi: 10.1158/1078-0432.CCR-18-1932. [DOI] [PubMed] [Google Scholar]
- 34.Leoni V., Vannini A., Gatta V., Rambaldi J., Sanapo M., Barboni C., Zaghini A., Nanni P., Lollini P.L., Casiraghi C., Campadelli-Fiume G. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018;14:e1007209. doi: 10.1371/journal.ppat.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theobald S.J., Khailaie S., Meyer-Hermann M., Volk V., Olbrich H., Danisch S., Gerasch L., Schneider A., Sinzger C., Schaudien D. Signatures of T and B cell development, functional responses and PD-1 upregulation after HCMV latent infections and reactivations in Nod.Rag.gamma mice humanized with cord blood CD34+ cells. Front. Immunol. 2018;9:2734. doi: 10.3389/fimmu.2018.02734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki M., Bertin T.K., Rogers G.L., Cela R.G., Zolotukhin I., Palmer D.J., Ng P., Herzog R.W., Lee B. Differential type I interferon-dependent transgene silencing of helper-dependent adenoviral vs. adeno-associated viral vectors in vivo. Mol. Ther. 2013;21:796–805. doi: 10.1038/mt.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki M., Cela R., Clarke C., Bertin T.K., Mouriño S., Lee B. Large-scale production of high-quality helper-dependent adenoviral vectors using adherent cells in cell factories. Hum. Gene Ther. 2010;21:120–126. doi: 10.1089/hum.2009.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guse K., Suzuki M., Sule G., Bertin T.K., Tyynismaa H., Ahola-Erkkilä S., Palmer D., Suomalainen A., Ng P., Cerullo V. Capsid-modified adenoviral vectors for improved muscle-directed gene therapy. Hum. Gene Ther. 2012;23:1065–1070. doi: 10.1089/hum.2012.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fueyo J., Gomez-Manzano C., Alemany R., Lee P.S., McDonnell T.J., Mitlianga P., Shi Y.X., Levin V.A., Yung W.K., Kyritsis A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 40.Gomes-Silva D., Srinivasan M., Sharma S., Lee C.M., Wagner D.L., Davis T.H., Rouce R.H., Bao G., Brenner M.K., Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmed N., Ratnayake M., Savoldo B., Perlaky L., Dotti G., Wels W.S., Bhattacharjee M.B., Gilbertson R.J., Shine H.D., Weiss H.L. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–5964. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biddle A., Gammon L., Fazil B., Mackenzie I.C. CD44 staining of cancer stem-like cells is influenced by down-regulation of CD44 variant isoforms and up-regulation of the standard CD44 isoform in the population of cells that have undergone epithelial-to-mesenchymal transition. PLoS ONE. 2013;8:e57314. doi: 10.1371/journal.pone.0057314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim M.P., Evans D.B., Wang H., Abbruzzese J.L., Fleming J.B., Gallick G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009;4:1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.