

Abstract
The molecular and pharmacological manipulation of the endogenous redox system is a promising therapy to limit myocardial damage after a heart attack; however, antioxidant therapies have failed to fully establish their cardioprotective effects, suggesting that additional factors, including antioxidant system interactions with other molecular pathways, may alter the pharmacological effects of antioxidants. Since gender differences in cardiovascular disease (CVD) are prevalent, and sex is an essential determinant of the response to oxidative stress, it is of particular interest to understand the effects of sex hormone signaling on the activity and expression of cellular antioxidants and the pharmacological actions of antioxidant therapies. In the present review, we briefly summarize the current understanding of testosterone effects on the modulation of the endogenous antioxidant systems in the CV system, cardiomyocytes, and the heart. We also review the latest research on redox balance and sexual dimorphism, with particular emphasis on the role of the natural antioxidant system glutathione (GSH) in the context of myocardial infarction, and the pro- and antioxidant effects of testosterone signaling via the androgen receptor (AR) on the heart. Finally, we discuss future perspectives regarding the potential of using combing antioxidant and testosterone replacement therapies to protect the aging myocardium.
Keywords: Oxidative stress, Testosterone, Androgen signaling, Glutathione, Heart, Cardiomyocytes
Graphical abstract
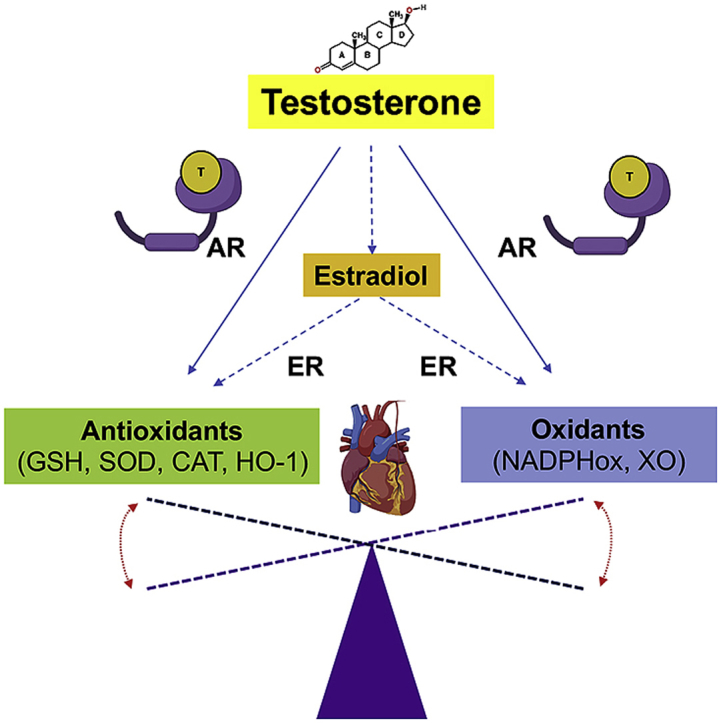
1. Introduction
Cardiovascular disease (CVD) is a leading cause of death worldwide, with heart disease accounting for more than 75% of all CVD-related deaths [1]. In the United States, the incidence of heart attacks as a consequence of coronary artery disease, heart arrhythmias, heart valve disease, and other risk factors is approximately one million attacks per year. The average age of first myocardial infarction (MI) is 65.6 years old for men, while for women it is 72.0 years old [1]. In addition to differences in the onset of MI for men and women, sex-specific differences in the presentation and outcomes in patients with MI are well-documented [[2], [3], [4]]. However, the pathophysiological mechanisms accounting for these sex-specific differences remain mostly uncharacterized.
The balance in the myocardium oxidative/reductive state is an essential determinant of the pathogenesis of MI [5]. Increased generation of reactive oxygen species (ROS), reactive nitric oxide and peroxynitrite species (RNS) and depletion of enzymatic and nonenzymatic antioxidant systems lead to increased oxidative stress [6], which results in adverse cellular effects in the myocardium, including impairments in regulatory pathways involved in Ca2+ homeostasis, alterations in DNA repair mechanisms, conformational changes in myofibrillar proteins and cell membrane properties, mitochondria dysfunction, and ultimately cell death [[6], [7], [8], [9], [10]]. Similarly, accumulation of reducing agents can lead to reductive stress, which is characterized by an elevation in antioxidant systems, such as glutathione (GSH) and dinucleotide (NADPH), and the overexpression of the endogenous antioxidant enzymatic system [11]. Increased reductive stress impairs cell growth, alters posttranslational modification processes and cellular metabolism, and has been associated with increased inflammation and proteotoxicity in the failing heart [11,12]. Therefore, myocardial oxidative/reductive balance is a critical mechanism involved in modulating the response to ischemia/reperfusion (I/R) injury.
Studies show that males are more susceptible to oxidative stress and have a lower antioxidant capacity than females [[13], [14], [15]]. These sex differences have been attributed to estrogen, which is known to promote the activation of antioxidant systems and to regulate the expression and activity of a number of antioxidant enzymes [14]. However, estrogen alone cannot explain gender differences in the susceptibility to oxidative stress. Thus, more research is needed to understand the mechanisms behind sexual dimorphism in maintaining redox balance. Herein, we will discuss the current understanding of sex hormone effects on endogenous antioxidant defense mechanisms in the heart. Since most reviews and research have focused on estrogen, this review will mainly discuss the interactions between the natural antioxidant systems and testosterone signaling in cardiac pathology, with particular focus on the GSH system and MI.
1.1. Sex-specific effects of oxidative stress on the heart
Studies in rodent models and humans have shown an association between oxidative stress and gender (Table 1). Females, in general, have been found to have lower levels of oxidative stress and ROS production than males [13,14,[16], [17], [18], [19]]. However, the data are still controversial since some studies show modest effects on the differential expression of antioxidant enzymes or no significant gender differences (Table 1) [15,20,21].
Table 1.
Sex differences in oxidative stress.
| Males | Females | |
|---|---|---|
| Cells of the cardiovascular system (superoxide dismutase, NADPH‐oxidases, thioredoxin catalase, glutathione reductase, glutathione peroxidase, glutathione S-transferase) | ||
| Myocardium | ⇑ (Ref. 14, 20, 23, 25 ) or ∅ | ⇓ |
| Endothelial cells | ⇑ (Ref. 14) or ∅ (Ref. 17, 18, 30) | ⇓ |
| Smooth muscle cells |
⇑ (Ref. 18) or ∅ (Ref. 16, 28) |
⇓ |
|
Circulating ROS (Plasma superoxide dismutase, catalase, and vitamin E levels) | ||
| Plasma |
⇑ (Ref. 15, 19) or ∅ (Ref. 22) |
⇓ |
|
Tissue markers of ROS generation (cyclooxygenase, xanthine oxidase, NADPH oxidase, catalase, glutathione reductase, glutathione peroxidase, and glutathione S-transferase enzymatic activity) | ||
| Renal cortex and kidney | ⇑ (Ref. 19, 23) | ⇓ |
| Pancreas | ⇑ (Ref. 21) | ⇓ |
| Brain | ⇑ or ∅ (Ref. 23, 24) | ⇓ |
| Lung | ⇑ (Ref. 23) | ⇓ |
| Blood vessels | ⇑ (Ref. 26) | ⇓ |
| Rat Aorta | ⇑ (Ref. 29) or ∅ (Ref. 16, 31) | ⇓ |
Oxidative stress is higher in male cardiovascular tissues than in female. The data regarding the expression of the antioxidant enzymes are still controversial with some studies reporting lower expression in male tissue, while others reported no differences. ROS: reactive oxygen species. Higher: ⇑; Lower: ⇓; and No change: ∅
Studies on myocardial tissue harvested from male rats showed that the male myocardium exhibited higher levels of glutathione peroxidase (GPx) (50%) and lower levels of superoxide dismutase (SOD, 14%) than the female myocardium [20]. The same study showed that ovariectomy leads to a significant increase in GPx levels and a decrease in the level of SOD in the female myocardium, while castration in males has no effects [20]. In agreement with these data, male rat aortas have been found to generate more superoxide radicals than female aortas [16,18]. Additionally, the systemic levels of oxidative stress markers are higher in hypertensive male rats than in their female counterparts [19]. Moreover, males are more responsive to the vascular effects of antioxidants than females [19]. These data correlate with clinical studies that suggest that men have a lower antioxidant potential than women [15].
Young men had higher plasma levels of oxidative stress markers such as thiobarbituric acid-reactive substances (TBARS, a marker of oxidative degradation of lipids by reactive oxygen species) and 8-isoprostaglandin F2alpha (8-iso-PGF2alpha, a marker of oxidative tissue damage) than premenopausal women [15]. Interestingly, the same study showed no differences in plasma levels of SOD [15], suggesting that variations in SOD activity are not responsible for the differences in systemic oxidative stress between genders in this particular study. However, a later study on premenopausal women that underwent total hysterectomy or bilateral salpingo-oophorectomy showed a reduction in the circulating levels of SOD, and estrogen replacement therapy countered this effect [22]. Additional studies have shown that SOD levels and activity are higher in females than in males in specific tissues such as the brain, lungs, and heart [20,23], but no differences have been found in other organs such as the kidney [23]. Additionally, while catalase activity is not sexually dimorphic in brain, lung, or heart, the kidneys do display sexual dimorphism in the activity of this antioxidant enzyme [23]. These results suggest that the sex-specific expression and activity of the antioxidant enzymes may also be tissue-specific, which will explain discrepancies found in studies.
Regarding the role of estrogen as the culprit for sex differences in the response to oxidative stress in CVD, data from C57BL/6 J mice infused with 17β-estradiol in conjunction with ANG II show that estradiol blocks ROS generation in males, whereas ovariectomy of female C57BL/6 J mice results in greater ANG II-mediated increases in cardiac NADPH oxidase-derived O2·− production relative to intact females [24,25]. In agreement with these findings, data from human male and female smooth muscle and endothelial cells also show that ROS production is higher in the vascular cells from males than in the cells from females. Additionally, a study comparing coronary artery disease (CAD) in post‐menopausal women with males demonstrated that even in the control groups that did not have CAD, post‐menopausal women were found to have higher oxidative stress levels than men [26]. Also, estrogen deficiency in ovariectomized mice induced the downregulation of extracellular super dismutase (ecSOD) and manganese super dismutase (MnSOD) expression, which was associated with increased production of vascular free radicals and prevented by estrogen replacement or treatment with superoxide dismutase-polyethylene glycol (PEG-SOD). Similar results were found using human-derived monocytes, in which increased estrogen levels led to enhanced ecSOD and MnSOD expression [27]. Together, these data suggest the possibility that the lower oxidative stress observed in females than males can be attributed perhaps to the estrogen-mediated regulation of antioxidant systems. Therefore, estrogen can at least in part explain the lower levels of oxidative stress observed in females [28]. However, some studies showed no differences in antioxidant enzyme activity levels between males and females [15,29]. Data on the effects of estrogen on the expression and activities of the NADPH oxidases, Nox1, Nox2, and Nox4, are conflicting [15,20,21]. Some studies show that Nox1 and Nox4 expression is higher in males than females, which correlates with higher production of superoxide in males than in females [16,30]. The same studies found no sex differences in Nox2 expression [16,30]. Another independent study using pig coronary arteries showed high expression levels of Nox1 and Nox2 in females, but Nox4 expression was significantly higher in males [31]. Therefore, no consensus has been reached regarding negative or positive effects of estrogen on the modulation of Nox subunit expression and whether or not sexual dimorphism in the expression of NADPH oxidases can explain the higher oxidative stress in males relative to females. Additional studies on tissue- and gender-specific differences in the expression of antioxidant enzymes are needed to fully understand sexual dimorphism in oxidative stress in CVD. To date, most studies regarding sex differences in oxidative stress have primarily focused on the effects of estrogen. However, other mechanisms likely are contributing to the higher oxidative stress observed in the male cardiovascular system relative to their female counterparts. The next two sections of this review we will discuss the latest research on the crosstalk between testosterone and the endogenous antioxidant systems, with a focus on glutathione and testosterone signaling in the heart.
1.2. GSH synthesis and regulation
Glutathione (GSH) is the principal endogenous antioxidant and reducing factor, plays a critical role in protecting against free radicals and is pivotal in the maintenance of antioxidant systems, including the antioxidant enzymes GPx, glutathione-s-transferase (GST), and glutathione reductase (GR). Fig. 1 illustrates the classical pathway for GSH synthesis and systemic effects. Intracellular GSH is predominantly maintained as a monomer in the reduced form and in the oxidized form as GSSG to a lesser degree [32]. The reduced and oxidized forms of GSH represent the primary cellular redox buffer, and under normal physiology, the concentration of GSH is higher relative to GSSG. Thus, the ratio of GSH and GSSG is considered a marker of oxidative stress.
Fig. 1.

Glutathione (GSH) Synthesis and role in maintaining systemic redox balance. (A) GSH is synthesized from glutamine, cysteine, and glycine in the cytosol by an ATP-dependent two-step process: 1) This step conjugates cysteine with glutamate, generating γ-glutamyl cysteine (γ-glu-cys) by the action of γ-glutamyl cysteine synthase (γ-GCS, also known as glutamate–cysteine ligase (GCL)) and 2) glutathione synthetase then catalyzes the addition of glycine to γ-glutamylcysteine to form γ-glutamylcysteinylglycine (γ-Glu-Cys-Gly) or GSH. Increased levels of GSH activates a negative feedback inhibition. GHS is then distributed to different areas in the cell, such as the endoplasmic reticulum (ER), the nucleus, and the mitochondria. (B) GSH functions as a detoxification system, antioxidant, and it is the major reserve for cysteine. GSH conjugates with electrophile compounds spontaneously or via enzymatically in reactions catalyzed by GSH-S-transferase. These conjugates are cleaved by γ-glutamyltranspeptidase leaving a cysteinyl-glycine conjugate (X-Cys-Gly). The cysteinyl-glycine bond is then cleaved by dipeptidase. The remaining cysteinyl conjugate (X-Cys) is acetylated by N-acetylase leading to the formation of a mercapturic acid (N-acetyl-Cys-X). This conjugate is then metabolized in the biliary tree, intestine, or kidney. As an antioxidant, when reactive oxygen species (ROS) are produced (mainly by mitochondria), GSH peroxidase or GSH-S-transferase catalyzes the conjugation of GSH to ROS. Oxidized GSH (GSSG) and water are then formed. GSSG can be reduced back to GSH by GSSG reductase. GSH also serves as a source for cysteine. Gamma-Glutamyl Transferase (GGT) catalyze the transfer of the γ-glutamyl moiety of GSH to an amino acid (aa) forming γ-glutamyl amino acid and cysteinylglycine, which is broken down by dipeptidase (DP) to generate cysteine and glycine, which are then transported back into the cell and use for protein synthesis or GSH regeneration.
GSH acts as a free radical scavenger, detoxifies xenobiotics and/or their metabolites, maintains the essential thiol status of proteins, and provides a reservoir for cysteine [32]; therefore, GSH is a critical modulator of cellular processes such as DNA synthesis, microtubular-related processes, and immune function [33], and variations in its levels are a hallmark of many pathological disorders, including cancer, metabolic abnormalities, and cardiovascular disease.
GSH synthesis is regulated by various factors, including l-cysteine and ATP availability, the activity of the rate-limiting enzymes glutamate-cysteine ligase (GLC) and glutathione synthetase (GS), and the availability of GSH, which in high levels inhibits the activity of GCL [34] (Fig. 1). The first step in GSH synthesis is the formation of γ-glutamylcysteine from glutamate and cysteine via GCL activity and consumption of one ATP molecule. Then, a molecule of glycine is added in a reaction catalyzed by GS, which requires the consumption of an additional ATP molecule [34]. Another mechanism to control GSH levels is via the action of glutathione reductase (GSR) and GSH peroxidase (GPx) and by a GSH-negative feedback loop [34]. In addition to these well-characterized mechanisms of GSH synthesis, additional factors have been shown to contribute to maintaining the cellular GSH/GSS ratio, including microRNAs and long noncoding RNAs (lncRNA), heat shock proteins, and NADPH oxidase-Nrf2 signaling [35]. These mediators and their effects on GSH antioxidant properties in the cardiovascular system have been recently discussed in an elegant review by Bajic VP et al. [35]. In the present review, we are only focusing on the association between low GSH levels, MI, and progression to heart failure.
In the context of the heart, systemic glutathione deficiency is associated with pathological cardiac remodeling and progression to heart failure in animal models and humans [36]. Early studies found increased oxidative stress in patients with dilated cardiomyopathic heart failure related to reduced circulating glutathione levels [37]. This study showed that patients with dilated cardiomyopathy were more prone to lipid peroxidation and oxidative damage than healthy controls, and this increase in oxidative stress correlated with decreased whole-blood GSH concentrations [37]. Another study by Shimizu H et al. also demonstrated that stroke and MI patients present a significant reduction in total GSH levels, suggesting that administration of GSH may be a potential therapeutic strategy to prevent negative cardiovascular events [38]. A more recent study by Damy T. et al. showed that GSH is significantly depleted in patients with coronary heart disease, and the decreased GSH levels correlate with the severity of the functional and structural heart abnormalities in these patients [36]. Interestingly, the same study suggested that since GSH levels are decreased in asymptomatic cardiac patients with structural abnormalities, determining blood GSH may be a novel test to detect heart disease before the onset of full-blown heart failure [36].
Animal models have also highlighted the role of GSH in heart disease and failure. Studies by Adamy et al. showed that 2-month postmyocardial infarction (MI) rats with chronic heart failure displayed a significant decrease in GSH levels in left ventricular tissue, and complementation with the GSH precursor N-acetylcysteine (NAC) normalized LV glutathione, improved LV contractile function and lessened adverse LV remodeling in 3-month post-MI rats [39]. In correlation with these findings, LV tissue harvested from patients undergoing heart transplantation for end-stage heart failure secondary to dilated cardiomyopathy or ischemic heart disease also displayed deficiency in GSH levels [39]. Recent clinical data showed that intravenous GSH administration during acute phase MI before coronary recanalization ameliorates reperfusion damage, suggesting that exogenous administration of GSH may improve the outcomes after MI and slow the progression of cardiac abnormalities to heart failure [40]. Therefore, a better understanding of the mechanism and factors regulating GSH levels, and the downstream signaling modulated by GSH, is critical to take full advantage of this endogenous cardioprotective system.
1.3. Effects of testosterone on endogenous antioxidant systems in the heart
Testosterone is the most predominant androgen in circulation and the primary sex hormone in males. In addition to playing a critical role in the growth and development of the male reproductive system and secondary sexual characteristics [[41], [42], [43], [44]], testosterone also affects the function of many other organ systems [45]. The testis synthesizes approximately 95% of testosterone from cholesterol, whereas 5% is produced by the zona reticularis of the adrenal glands [44].
Testosterone exerts its actions by binding the androgen receptor (AR), a member of the nuclear receptor family of transcription factors. Upon testosterone binding, the AR signals through genomic (translocation to the nucleus) and nongenomic (cytosolic interactions with various signaling pathways, including MAPK, PKC, nitric oxide, etc.) mechanisms [[46], [47], [48]]. Fig. 2 illustrates the canonical pathway for testosterone signaling via the AR. The structure and detailed mechanisms underlying AR actions are reviewed in detail elsewhere [[48], [49], [50], [51]].
Fig. 2.

Classical mechanisms of androgen signaling in a target cell. In circulation, testosterone is bound to the serum sex hormone-binding globulin (SHBG). Testosterone dissociates from its binding carrier protein and diffuses freely through the cell membrane. Once in the cytoplasm, testosterone can be converted to its active metabolite 5α-dihydrotestosterone (DHT). Testosterone or DHT can directly bind to the androgen receptor (AR) leading to the dissociation of heat shock proteins (hsp) from the inactive receptor. The ligand-bound receptor then exerts its effects by rapid nongenomic effects by modulating the activity of the Src/Raf-1/Erk-2 pathway or the phosphoinositide 3-kinase (PI3K)/AKT pathway, or by genomic mechanisms involving the activated AR translocation into the nucleus. In the nucleus the androgen receptor binds as homodimer to specific DNA elements present as enhancers in upstream promoter sequences of androgen target genes. Upon AR binding, coactivators are recruited and the basal transcription machinery (BTM) (e.g. RNA-polymerase II [RNA-Pol II], TATA box binding protein [TBP], TBP associating factors [TAF's], general transcription factors [GTF's]) is activated. The interactions between AR, coactivators and the BTM results in gene transcription.
AR is highly expressed by all cardiovascular cells, including vascular smooth muscle and endothelial cells, cardiomyocytes and cardiac fibroblasts, and its effects on gene expression regulation vary depending on the cell type, tissue, sex, and whether there is an underlying disease [[52], [53], [54], [55], [56], [57]]. These factors are important determinants of testosterone effects on the cardiovascular system.
Numerous studies associate testosterone with increased cardiovascular risk [[58], [59], [60], [61], [62], [63], [64], [65]]; however, a similar number of studies show that intact testosterone signaling is critical for cardiovascular health [[66], [67], [68], [69], [70], [71], [72], [73], [74]]. Therefore, the actions of testosterone in the cardiovascular system remain controversial, and perhaps the discrepancy in testosterone effects can be explained by various contributing factors, including the dose and duration of testosterone treatment (e.g., acute vs. chronic), nonmedical testosterone use, tissue and cell type, type of signaling (genomic or nongenomic), and metabolic and cellular status.
The cellular environment, in particular, the redox status, has been shown to influence testosterone effects on the cardiovascular system. High oxidative stress has been associated with negative testosterone effects, whereas low oxidative stress is correlated with testosterone cardioprotective effects [75] (Fig. 3). In a rat model of angiotensin II-induced hypertension, testosterone increased ROS generation by increasing the phosphorylation of c-Src, which is an upstream regulator of NADPH oxidase expression and activity, only in hypertensive rats, while no effects were observed in normotensive experimental animals [76]. In agreement with these results, testosterone exacerbated cardiac injury in angiotensin II-induced hypertensive rats by a mechanism involving the conversion of testosterone into 6β-hydroxytestosterone by cytochrome P–4501B1. 6β-hydroxytestosterone increased the sensitivity to angiotensin II induced hypertension by upregulating NADPH oxidase activity and ROS generation [77]. Boldenone, a synthetic derivative of testosterone causes cardiotoxicity, and in male rats it was also shown to increase levels of cardiac malondialdehyde, H2O2 production in heart tissues, and expression of NOX2 and NOX4 mRNA [78]. There is limited evidence for an effect of testosterone on XO in the heart, but one study showed that rats exposed to gestational testosterone show an elevated xanthine oxidase and decreased G6PD-dependent antioxidant defense [79].
Fig. 3.

Testosterone's effects as a pro- and antioxidant in cardiomyocytes. Testosterone has been reported to increase reactive oxygen species (ROS) production and oxidative stress in cardiomyocytes by signaling via the androgen receptor (AR) through genomic and nongenomic mechanisms (right panel). Activated AR translocates into the nucleus and increases the gene expression of pro-oxidant enzymes (e.g. NAD(P)H oxidases, Xanthine oxidase, and cyclooxygenase 2 (COX-2)) and by increasing the transcription of genes involved in the c-Src and PI3K/Akt pathways, which in turn further exacerbate the activation of pro-oxidant enzymes and increase ROS generation by altering mitochondria function. AR also upregulates the activity of the Proto-oncogene tyrosine kinase Src (c-Src) and phosphoinositide-3-kinase/protein kinase B (PI3K/Akt) pathways by direct protein-protein interactions in the cytosol (nongenomic mechanisms). Testosterone may also increase ROS, via its nongenomic action, through the G protein-coupled receptor, family C, group 6, member A, GPRC6A. Increased ROS levels lead to cardiomyocyte injury, inflammation, lipid accumulation, and eventually cell death, causing heart failure. Recent data suggest that testosterone exerts antioxidant effects, in particular in aging, by a mechanism independent of AR. Although, it is still controversial the proposed mechanism relies on the conversion of testosterone to estradiol, which in turn increases the levels of antioxidant enzymes and reduces oxidative stress in cardiomyocytes by genomic and nongenomic mechanisms (protein-protein interactions with ERKs, MAP and PI3 kinases in the cytoplasm or by binding to the G protein-coupled receptor, family C, group 30, GPR30) (left panel).
Testosterone induces vasodilation by increasing nitric oxide (NO) production [80], which positively regulates the vascular tone. In castrated male rats the typical I/R-induced elevation of HIF-1a in the myocardium was blunted. Testosterone treatment restored the high hypoxia-inducible factor-1a (HIF-1a) levels, although the antiandrogen futamide had only a modest impact on the testosterone effect. In other tissues, testosterone has been found to increase Nrf2-ARE nuclear factor erythroid 2-related factor 2-antioxidant response element (Nrf2-ARE) pathway in aged rats leading to increased HO-1 levels and restoration of redox balance [81]. In addition to altering redox pathways, testosterone can also protect against ROS-induced injury. Using siRNA to knockdown the canonical NF-κB (RelA/p50) signaling pathway in cardiomyocytes, Xiao FY et al., 2015 showed that testosterone attenuated H2O2 induced cell death through activation of NF-kB. This protective effect was mediated through the AR [82]. Therefore, testosterone interplay with ROS balance is still controversial. We would like to highlight that most of the studies on the effects of testosterone in the cardiovascular system have been performed on the vasculature, and there have only been a few studies investigating testosterone signaling on cardiomyocytes and the crosstalk between the antioxidant system and androgen signaling.
Androgens are known to modulate cardiac function by regulating the activity and expression of proteins involved in maintaining cardiac homeostasis [83]. However, as discussed above, whether the effects of testosterone on the heart are positive or negative is still under debate. Some data show that testosterone via aromatization to 17β-estradiol positively regulates the expression and activity of the antioxidant enzymes SOD and GSH-Px in isolated adult murine cardiomyocytes and decreases lipid peroxidation (a marker of oxidative damage) [75] (Fig. 3). Moreover, via this potential AR-independent pathway, testosterone has been shown to decrease aging-associated oxidative stress in cardiomyocytes [84] (Fig. 3). In two models of testosterone deprivation, castration and aging, heme oxygenase levels and activity in the left ventricle decreased alongside lower cGMP, GSH and GSSG concentrations. Testosterone therapy was effective in reversing the effects, thereby showing a role for testosterone in protecting redox balance, at least under conditions where low testosterone was the initiating factor for these imbalances [85]. However, while estrogen had important antioxidant effects in the heart in a model of doxorubicin-induced oxidative stress, exogenous testosterone administration does not prevent oxidative damage [86]. Furthermore, a recent study showed that testosterone treatment increases apoptosis and lipid accumulation in cardiomyocytes (Fig. 3) [87]. Together, these results suggest that endogenous testosterone has a potential antioxidant effect in the heart, but exogenous administration may not elicit the same effects.
GSH level and the activity of several of its enzymes involved in its metabolism are significantly affected by sex hormones. Estrogen has been shown to regulate the expression and activity of glutathione reductase and glutathione peroxidase in several tissues [22,88,89]. However, there is little explicit emphasis on the role of testosterone signaling in modulating GSH levels and activity. As discussed above high testosterone levels are associated with increased oxidative stress, but diminished testosterone levels also lead to redox imbalances. However, few studies have explored the significance of testosterone signaling in regulating GSH levels and its related enzymatic system in the heart. A recent study showed that testosterone increases the expression and activity of SOD and GSH-Px in male cardiomyocytes, which reduces oxidative stress [75,84] The molecular mechanisms responsible for these effects are not clear, but they seemed to be independent of AR signaling and dependent on testosterone conversion to estradiol [75,84]. These results correlate with the increased oxidative stress that normally occurs during aging and opens the possibility that testosterone cardioprotective effects depend upon testosterone conversion to estrogen. On the other hand, studies showed that testosterone creates an oxidative environment, induces inflammation and increases cell death via AR signaling effects on GSH levels and on ROS production by the mitochondria [90]. Therefore, testosterone positive and negative effects on the heart may depend on whether or not this hormone is acting through a nuclear receptor.
1.4. Testosterone and mitochondrial function
Testosterone can also have opposing effects on mitochondria in different cell types of the cardiovascular system. For example, testosterone has been shown to exert negative effects in the vasculature by regulating mitochondrial ROS generation and activation of apoptosis pathways via activation of the AR [91] (Fig. 3). In contrast, there is a growing body of evidence that testosterone is beneficial in the cardiomyocytes. For example, testosterone deprivation is sufficient to induce LV contractile dysfunction and cardiac sympathovagal imbalance in orchiectomized rats [92]. This is accompanied by increased mitochondrial ROS production, membrane depolarization and swelling of the mitochondria [93,94]. Mitochondrial DNA deletion mutations are also increased in cardiomyocytes from castrated male mice, and in sham-operated testicular feminized mice [75], models that also exhibited decreased antioxidant (SOD and GPx) activities, and that were treatable with testosterone. Furthermore, I/R-induced myocardial dysfunction is associated with reduced fission and increased fusion of mitochondria. The fission-fusion imbalance is exacerbated by castration of male rats, accompanied by increased levels of the mitochondrial fission protein, DRP1, downregulation of the mitochondrial fusion protein, MFN2, and impaired ATP generation that is reversible by testosterone treatment. These findings suggest that testosterone is important in preserving mitochondrial function under both physiological and pathophysiological conditions. Some human studies are in line with this observation, in that low levels of testosterone are seen in type 2 diabetes, a condition associated with mitochondrial function. Although not tested in the heart tissue, circulating testosterone levels positively correlated with mitochondrial membrane potential, and with antioxidant (SOD and GSH) levels in blood leukocytes, while negatively correlating with total and mitochondrial ROS generation by leukocytes [95,96]. In contrast, other studies contradict the protective effect of testosterone. For example, testosterone treatment of female-to-male transsexuals impairs mitochondrial oxygen consumption and membrane potential, decreases GSH levels and GSH:GSSG, and elevates ROS generation in peripheral blood PMNs [97,98].
Exactly how testosterone is mediating its actions is unclear. However, it is noteworthy that mitochondria are important targets of estradiol, and that ERβ expression is higher in mitochondria isolated from the left ventricle of females versus males [99]. While castration leads to an increased ERb expression in both sexes, the functional significance of the estrogen receptors in mitochondrial functional differences between sexes remains to be elucidated. Furthermore, Ballantyne et al. found that testosterone protects female embryonic heart H9c2 cells against severe metabolic stress by its conversion into metabolites that activate estrogen receptors and up-regulate a mitochondrial sulfonylurea receptor 2B intraexonics splice variant (IES SUR2B) [100].
2. Conclusions and future perspectives
The role of testosterone in regulating oxidative stress in the heart, in particular in cardiomyocytes, is far from clear, with testosterone acting as an antioxidant as well as a pro-oxidant. Several questions remain, such as, what factors influence testosterone signaling through AR? What are the mechanisms underlying testosterone impact on the endogenous antioxidant defense mechanisms? Could the redox environment determine whether testosterone is cardioprotective or cardiotoxic? Future studies need to focus on determining how the redox environment regulates testosterone actions in the heart, and in particular, it would be of great interest to test whether variation in GSH/GSS ratios (reductive or oxidative environment) impacts testosterone role in the heart, in particular in the setting of I/R. These studies will provide important information regarding the potential of combining antioxidant and testosterone replacement therapies to decrease the risk of MI in aging men.
Disclosures
The authors have nothing to disclose.
Declaration of competing interest
The author(s) declare(s) that there is no conflict of interest regarding the publication of this article.
Acknowledgments
We would like to extend our apologies to those colleagues whose work we were unable to cite owing to space limitations. We would like to thank and acknowledge these individuals for their contributions to our current understanding of sexual dimorphism in redox biology.
This research was supported by the Molecular and Cellular Physiology Department (D. CT, K.S), the Center for Cardiovascular Diseases and Sciences, and The Center for Redox Biology and Cardiovascular Disease (D. CT, K.Y.S, P.D) (NIH P20GM121307), and the NHLBI K01 HL144882-01A1 (D. CT).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101490.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., Delling F.N., Djousse L., Elkind M.S.V., Ferguson J.F., Fornage M., Jordan L.C., Khan S.S., Kissela B.M., Knutson K.L., Kwan T.W., Lackland D.T., Lewis T.T., Lichtman J.H., Longenecker C.T., Loop M.S., Lutsey P.L., Martin S.S., Matsushita K., Moran A.E., Mussolino M.E., O'Flaherty M., Pandey A., Perak A.M., Rosamond W.D., Roth G.A., Sampson U.K.A., Satou G.M., Schroeder E.B., Shah S.H., Spartano N.L., Stokes A., Tirschwell D.L., Tsao C.W., Turakhia M.P., VanWagner L.B., Wilkins J.T., Wong S.S., Virani S.S. American heart association council on E, prevention statistics C and stroke statistics S. Heart disease and stroke statistics-2019 update: a report from the American heart association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Yang H.Y., Huang J.H., Hsu C.Y., Chen Y.J. Gender differences and the trend in the acute myocardial infarction: a 10-year nationwide population-based analysis. Sci. World J. 2012;2012 doi: 10.1100/2012/184075. 184075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson R.D., Pepine C.J. Gender differences in the treatment for acute myocardial infarction: bias or biology? Circulation. 2007;115:823–826. doi: 10.1161/CIRCULATIONAHA.106.685859. [DOI] [PubMed] [Google Scholar]
- 4.Diercks D.B., Owen K.P., Kontos M.C., Blomkalns A., Chen A.Y., Miller C., Wiviott S., Peterson E.D. Gender differences in time to presentation for myocardial infarction before and after a national women's cardiovascular awareness campaign: a temporal analysis from the can rapid risk stratification of unstable Angina patients suppress ADverse outcomes with early implementation (CRUSADE) and the national cardiovascular data registry acute coronary treatment and intervention outcomes network-get with the guidelines (NCDR ACTION registry-GWTG) Am. Heart J. 2010;160:80–87 e3. doi: 10.1016/j.ahj.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 5.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurian G.A., Rajagopal R., Vedantham S., Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid Med Cell Longev. 2016;2016 doi: 10.1155/2016/1656450. 1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuteja N., Ahmad P., Panda B.B., Tuteja R. Genotoxic stress in plants: shedding light on DNA damage, repair and DNA repair helicases. Mutat. Res. 2009;681:134–149. doi: 10.1016/j.mrrev.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Di Filippo C., Cuzzocrea S., Rossi F., Marfella R., D'Amico M. Oxidative stress as the leading cause of acute myocardial infarction in diabetics. Cardiovasc. Drug Rev. 2006;24:77–87. doi: 10.1111/j.1527-3466.2006.00077.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y.S., He L., Liu B., Li N.S., Luo X.J., Hu C.P., Ma Q.L., Zhang G.G., Li Y.J., Peng J. A novel pathway of NADPH oxidase/vascular peroxidase 1 in mediating oxidative injury following ischemia-reperfusion. Basic Res. Cardiol. 2012;107:266. doi: 10.1007/s00395-012-0266-4. [DOI] [PubMed] [Google Scholar]
- 10.Johnston A.S., Lehnart S.E., Burgoyne J.R. Ca(2+) signaling in the myocardium by (redox) regulation of PKA/CaMKII. Front. Pharmacol. 2015;6:166. doi: 10.3389/fphar.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Torres I., Guarner-Lans V., Rubio-Ruiz M.E. Reductive stress in inflammation-associated diseases and the pro-oxidant effect of antioxidant agents. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christians E.S., Benjamin I.J. Proteostasis and REDOX state in the heart. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H24–37. doi: 10.1152/ajpheart.00903.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavoie J.C., Tremblay A. Sex-specificity of oxidative stress in newborns leading to a personalized antioxidant nutritive strategy. Antioxidants. 2018;7 doi: 10.3390/antiox7040049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kander M.C., Cui Y., Liu Z. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J. Cell Mol. Med. 2017;21:1024–1032. doi: 10.1111/jcmm.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ide T., Tsutsui H., Ohashi N., Hayashidani S., Suematsu N., Tsuchihashi M., Tamai H., Takeshita A. Greater oxidative stress in healthy young men compared with premenopausal women. Arterioscler. Thromb. Vasc. Biol. 2002;22:438–442. doi: 10.1161/hq0302.104515. [DOI] [PubMed] [Google Scholar]
- 16.Miller A.A., De Silva T.M., Jackman K.A., Sobey C.G. Effect of gender and sex hormones on vascular oxidative stress. Clin. Exp. Pharmacol. Physiol. 2007;34:1037–1043. doi: 10.1111/j.1440-1681.2007.04732.x. [DOI] [PubMed] [Google Scholar]
- 17.Lubrano V., Balzan S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J. Exp. Med. 2015;5:218–224. doi: 10.5493/wjem.v5.i4.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matarrese P., Colasanti T., Ascione B., Margutti P., Franconi F., Alessandri C., Conti F., Riccieri V., Rosano G., Ortona E., Malorni W. Gender disparity in susceptibility to oxidative stress and apoptosis induced by autoantibodies specific to RLIP76 in vascular cells. Antioxidants Redox Signal. 2011;15:2825–2836. doi: 10.1089/ars.2011.3942. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia K., Elmarakby A.A., El-Remessy A.B., Sullivan J.C. Oxidative stress contributes to sex differences in angiotensin II-mediated hypertension in spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;302:R274–R282. doi: 10.1152/ajpregu.00546.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barp J., Araujo A.S., Fernandes T.R., Rigatto K.V., Llesuy S., Bello-Klein A., Singal P. Myocardial antioxidant and oxidative stress changes due to sex hormones. Braz. J. Med. Biol. Res. 2002;35:1075–1081. doi: 10.1590/s0100-879x2002000900008. [DOI] [PubMed] [Google Scholar]
- 21.Gomez-Perez Y., Gianotti M., Llado I., Proenza A.M. Sex-dependent effects of high-fat-diet feeding on rat pancreas oxidative stress. Pancreas. 2011;40:682–688. doi: 10.1097/MPA.0b013e31821f2645. [DOI] [PubMed] [Google Scholar]
- 22.Bellanti F., Matteo M., Rollo T., De Rosario F., Greco P., Vendemiale G., Serviddio G. Sex hormones modulate circulating antioxidant enzymes: impact of estrogen therapy. Redox Biol. 2013;1:340–346. doi: 10.1016/j.redox.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y., Ji L.L., Liu T.Y., Wang Z.T. Evaluation of gender-related differences in various oxidative stress enzymes in mice. Chin. J. Physiol. 2011;54:385–390. doi: 10.4077/CJP.2011.AMM080. [DOI] [PubMed] [Google Scholar]
- 24.Xue B., Zhao Y., Johnson A.K., Hay M. Central estrogen inhibition of angiotensin II-induced hypertension in male mice and the role of reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H1025–H1032. doi: 10.1152/ajpheart.00021.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebrahimian T., He Y., Schiffrin E.L., Touyz R.M. Differential regulation of thioredoxin and NAD(P)H oxidase by angiotensin II in male and female mice. J. Hypertens. 2007;25:1263–1271. doi: 10.1097/HJH.0b013e3280acac60. [DOI] [PubMed] [Google Scholar]
- 26.Vassalle C., Sciarrino R., Bianchi S., Battaglia D., Mercuri A., Maffei S. Sex-related differences in association of oxidative stress status with coronary artery disease. Fertil. Steril. 2012;97:414–419. doi: 10.1016/j.fertnstert.2011.11.045. [DOI] [PubMed] [Google Scholar]
- 27.Vina J., Gambini J., Lopez-Grueso R., Abdelaziz K.M., Jove M., Borras C. Females live longer than males: role of oxidative stress. Curr. Pharmaceut. Des. 2011;17:3959–3965. doi: 10.2174/138161211798764942. [DOI] [PubMed] [Google Scholar]
- 28.Strehlow K., Rotter S., Wassmann S., Adam O., Grohe C., Laufs K., Bohm M., Nickenig G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res. 2003;93:170–177. doi: 10.1161/01.RES.0000082334.17947.11. [DOI] [PubMed] [Google Scholar]
- 29.Brandes R.P., Mugge A. Gender differences in the generation of superoxide anions in the rat aorta. Life Sci. 1997;60:391–396. doi: 10.1016/s0024-3205(96)00663-7. [DOI] [PubMed] [Google Scholar]
- 30.Zhang R., Thor D., Han X., Anderson L., Rahimian R. Sex differences in mesenteric endothelial function of streptozotocin-induced diabetic rats: a shift in the relative importance of EDRFs. Am. J. Physiol. Heart Circ. Physiol. 2012;303:H1183–H1198. doi: 10.1152/ajpheart.00327.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong P.S., Randall M.D., Roberts R.E. Sex differences in the role of NADPH oxidases in endothelium-dependent vasorelaxation in porcine isolated coronary arteries. Vasc. Pharmacol. 2015;72:83–92. doi: 10.1016/j.vph.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Meister A., Anderson M.E. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 33.Forman H.J., Zhang H., Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol. Aspect. Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu S.C. Regulation of glutathione synthesis. Mol. Aspect. Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajic V.P., Van Neste C., Obradovic M., Zafirovic S., Radak D., Bajic V.B., Essack M., Isenovic E.R. Glutathione "redox homeostasis" and its relation to cardiovascular disease. Oxid Med Cell Longev. 2019;2019 doi: 10.1155/2019/5028181. 5028181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Damy T., Kirsch M., Khouzami L., Caramelle P., Le Corvoisier P., Roudot-Thoraval F., Dubois-Rande J.L., Hittinger L., Pavoine C., Pecker F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PloS One. 2009;4 doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yucel D., Aydogdu S., Cehreli S., Saydam G., Canatan H., Senes M., Cigdem Topkaya B., Nebioglu S. Increased oxidative stress in dilated cardiomyopathic heart failure. Clin. Chem. 1998;44:148–154. [PubMed] [Google Scholar]
- 38.Shimizu H., Kiyohara Y., Kato I., Kitazono T., Tanizaki Y., Kubo M., Ueno H., Ibayashi S., Fujishima M., Iida M. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: the Hisayama study. Stroke. 2004;35:2072–2077. doi: 10.1161/01.STR.0000138022.86509.2d. [DOI] [PubMed] [Google Scholar]
- 39.Adamy C., Mulder P., Khouzami L., Andrieu-abadie N., Defer N., Candiani G., Pavoine C., Caramelle P., Souktani R., Le Corvoisier P., Perier M., Kirsch M., Damy T., Berdeaux A., Levade T., Thuillez C., Hittinger L., Pecker F. Neutral sphingomyelinase inhibition participates to the benefits of N-acetylcysteine treatment in post-myocardial infarction failing heart rats. J. Mol. Cell. Cardiol. 2007;43:344–353. doi: 10.1016/j.yjmcc.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 40.Truscelli G., Tanzilli G., Viceconte N., Dominici M., Arrivi A., Sommariva L., Granatelli A., Gaudio C., Mangieri E. Glutathione sodium salt as a novel adjunctive treatment for acute myocardial infarction. Med. Hypotheses. 2017;102:48–50. doi: 10.1016/j.mehy.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Mooradian A.D., Morley J.E., Korenman S.G. Biological actions of androgens. Endocr. Rev. 1987;8:1–28. doi: 10.1210/edrv-8-1-1. [DOI] [PubMed] [Google Scholar]
- 42.Morales A. Determining the role of testosterone deficiency in sexual function: the beginning of the end or the end of the beginning? Eur. Urol. 2014;65:113–114. doi: 10.1016/j.eururo.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 43.Morales A. Testosterone Deficiency Syndrome: an overview with emphasis on the diagnostic conundrum. Clin. Biochem. 2014;47:960–966. doi: 10.1016/j.clinbiochem.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 44.Morales A. The long and tortuous history of the discovery of testosterone and its clinical application. J. Sex. Med. 2013;10:1178–1183. doi: 10.1111/jsm.12081. [DOI] [PubMed] [Google Scholar]
- 45.Brinkmann A.O. Molecular mechanisms of androgen action--a historical perspective. Methods Mol. Biol. 2011;776:3–24. doi: 10.1007/978-1-61779-243-4_1. [DOI] [PubMed] [Google Scholar]
- 46.Foradori C.D., Weiser M.J., Handa R.J. Non-genomic actions of androgens. Front. Neuroendocrinol. 2008;29:169–181. doi: 10.1016/j.yfrne.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy A.K., Tyagi R.K., Song C.S., Lavrovsky Y., Ahn S.C., Oh T.S., Chatterjee B. Androgen receptor: structural domains and functional dynamics after ligand-receptor interaction. Ann. N. Y. Acad. Sci. 2001;949:44–57. doi: 10.1111/j.1749-6632.2001.tb04001.x. [DOI] [PubMed] [Google Scholar]
- 48.Davey R.A., Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin. Biochem. Rev. 2016;37:3–15. [PMC free article] [PubMed] [Google Scholar]
- 49.MacLean H.E., Warne G.L., Zajac J.D. Localization of functional domains in the androgen receptor. J. Steroid Biochem. Mol. Biol. 1997;62:233–242. doi: 10.1016/s0960-0760(97)00049-6. [DOI] [PubMed] [Google Scholar]
- 50.Heinlein C.A., Chang C. Androgen receptor (AR) coregulators: an overview. Endocr. Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 51.Heinlein C.A., Chang C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol. 2002;16:2181–2187. doi: 10.1210/me.2002-0070. [DOI] [PubMed] [Google Scholar]
- 52.Pedernera E., Gomora M.J., Meneses I., De Ita M., Mendez C. Androgen receptor is expressed in mouse cardiomyocytes at prenatal and early postnatal developmental stages. BMC Physiol. 2017;17:7. doi: 10.1186/s12899-017-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu D., Hadoke P.W., Wu J., Vesey A.T., Lerman D.A., Dweck M.R., Newby D.E., Smith L.B., MacRae V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016;6:24807. doi: 10.1038/srep24807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzales R.J., Duckles S.P., Krause D.N. Dihydrotestosterone stimulates cerebrovascular inflammation through NFkappaB, modulating contractile function. J. Cerebr. Blood Flow Metabol. 2009;29:244–253. doi: 10.1038/jcbfm.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsh J.D., Lehmann M.H., Ritchie R.H., Gwathmey J.K., Green G.E., Schiebinger R.J. Androgen receptors mediate hypertrophy in cardiac myocytes. Circulation. 1998;98:256–261. doi: 10.1161/01.cir.98.3.256. [DOI] [PubMed] [Google Scholar]
- 56.Dart D.A., Waxman J., Aboagye E.O., Bevan C.L. Visualising androgen receptor activity in male and female mice. PloS One. 2013;8 doi: 10.1371/journal.pone.0071694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai H.W., Taniguchi S., Samoza J., Ridder A. Age- and sex-dependent changes in androgen receptor expression in the developing mouse cortex and Hippocampus. Neuroscience J. 2015;2015:525369. doi: 10.1155/2015/525369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vigen R., O'Donnell C.I., Baron A.E., Grunwald G.K., Maddox T.M., Bradley S.M., Barqawi A., Woning G., Wierman M.E., Plomondon M.E., Rumsfeld J.S., Ho P.M. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. J. Am. Med. Assoc. 2013;310:1829–1836. doi: 10.1001/jama.2013.280386. [DOI] [PubMed] [Google Scholar]
- 59.Finkle W.D., Greenland S., Ridgeway G.K., Adams J.L., Frasco M.A., Cook M.B., Fraumeni J.F., Jr., Hoover R.N. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9 doi: 10.1371/journal.pone.0085805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fisher M., Appleby M., Rittoo D., Cotter L. Myocardial infarction with extensive intracoronary thrombus induced by anabolic steroids. Br. J. Clin. Pract. 1996;50:222–223. [PubMed] [Google Scholar]
- 61.Basaria S., Coviello A.D., Travison T.G., Storer T.W., Farwell W.R., Jette A.M., Eder R., Tennstedt S., Ulloor J., Zhang A., Choong K., Lakshman K.M., Mazer N.A., Miciek R., Krasnoff J., Elmi A., Knapp P.E., Brooks B., Appleman E., Aggarwal S., Bhasin G., Hede-Brierley L., Bhatia A., Collins L., LeBrasseur N., Fiore L.D., Bhasin S. Adverse events associated with testosterone administration. N. Engl. J. Med. 2010;363:109–122. doi: 10.1056/NEJMoa1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ment J., Ludman P.F. Coronary thrombus in a 23 year old anabolic steroid user. Heart. 2002;88:342. doi: 10.1136/heart.88.4.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldstein D.R., Dobbs T., Krull B., Plumb V.J. Clenbuterol and anabolic steroids: a previously unreported cause of myocardial infarction with normal coronary arteriograms. South. Med. J. 1998;91:780–784. [PubMed] [Google Scholar]
- 64.Hartgens F., Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Med. 2004;34:513–554. doi: 10.2165/00007256-200434080-00003. [DOI] [PubMed] [Google Scholar]
- 65.Hartgens F., Rietjens G., Keizer H.A., Kuipers H., Wolffenbuttel B.H. Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a) Br. J. Sports Med. 2004;38:253–259. doi: 10.1136/bjsm.2003.000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khaw K.T., Dowsett M., Folkerd E., Bingham S., Wareham N., Luben R., Welch A., Day N. Endogenous testosterone and mortality due to all causes, cardiovascular disease, and cancer in men: European prospective investigation into cancer in Norfolk (EPIC-Norfolk) Prospective Population Study. Circulation. 2007;116:2694–2701. doi: 10.1161/CIRCULATIONAHA.107.719005. [DOI] [PubMed] [Google Scholar]
- 67.Hyde Z., Norman P.E., Flicker L., Hankey G.J., Almeida O.P., McCaul K.A., Chubb S.A., Yeap B.B. Low free testosterone predicts mortality from cardiovascular disease but not other causes: the Health in Men Study. J. Clin. Endocrinol. Metab. 2012;97:179–189. doi: 10.1210/jc.2011-1617. [DOI] [PubMed] [Google Scholar]
- 68.Muraleedharan V., Marsh H., Kapoor D., Channer K.S., Jones T.H. Testosterone deficiency is associated with increased risk of mortality and testosterone replacement improves survival in men with type 2 diabetes. Eur. J. Endocrinol. 2013;169:725–733. doi: 10.1530/EJE-13-0321. [DOI] [PubMed] [Google Scholar]
- 69.Militaru C., Donoiu I., Dracea O., Ionescu D.D. Serum testosterone and short-term mortality in men with acute myocardial infarction. Cardiol. J. 2010;17:249–253. [PubMed] [Google Scholar]
- 70.Booth A., Johnson D.R., Granger D.A. Testosterone and men's depression: the role of social behavior. J. Health Soc. Behav. 1999;40:130–140. [PubMed] [Google Scholar]
- 71.Stanworth R.D., Jones T.H. Testosterone for the aging male; current evidence and recommended practice. Clin. Interv. Aging. 2008;3:25–44. doi: 10.2147/cia.s190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stanworth R.D., Kapoor D., Channer K.S., Jones T.H. Androgen receptor CAG repeat polymorphism is associated with serum testosterone levels, obesity and serum leptin in men with type 2 diabetes. Eur. J. Endocrinol. 2008;159:739–746. doi: 10.1530/EJE-08-0266. [DOI] [PubMed] [Google Scholar]
- 73.Toma M., McAlister F.A., Coglianese E.E., Vidi V., Vasaiwala S., Bakal J.A., Armstrong P.W., Ezekowitz J.A. Testosterone supplementation in heart failure: a meta-analysis. Circ Heart Fail. 2012;5:315–321. doi: 10.1161/CIRCHEARTFAILURE.111.965632. [DOI] [PubMed] [Google Scholar]
- 74.Sharma R., Oni O.A., Gupta K., Chen G., Sharma M., Dawn B., Sharma R., Parashara D., Savin V.J., Ambrose J.A., Barua R.S. Normalization of testosterone level is associated with reduced incidence of myocardial infarction and mortality in men. Eur. Heart J. 2015;36:2706–2715. doi: 10.1093/eurheartj/ehv346. [DOI] [PubMed] [Google Scholar]
- 75.Zhang L., Wu S., Ruan Y., Hong L., Xing X., Lai W. Testosterone suppresses oxidative stress via androgen receptor-independent pathway in murine cardiomyocytes. Mol. Med. Rep. 2011;4:1183–1188. doi: 10.3892/mmr.2011.539. [DOI] [PubMed] [Google Scholar]
- 76.Chignalia A.Z., Schuldt E.Z., Camargo L.L., Montezano A.C., Callera G.E., Laurindo F.R., Lopes L.R., Avellar M.C., Carvalho M.H., Fortes Z.B., Touyz R.M., Tostes R.C. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension. 2012;59:1263–1271. doi: 10.1161/HYPERTENSIONAHA.111.180620. [DOI] [PubMed] [Google Scholar]
- 77.Pingili A.K., Kara M., Khan N.S., Estes A.M., Lin Z., Li W., Gonzalez F.J., Malik K.U. 6beta-hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension. 2015;65:1279–1287. doi: 10.1161/HYPERTENSIONAHA.115.05396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tousson E., Elgharabawy R.M., Elmasry T.A. Grape seed proanthocyanidin ameliorates cardiac toxicity induced by boldenone undecylenate through inhibition of NADPH oxidase and reduction in the expression of NOX2 and NOX4. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/9434385. 9434385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olatunji L.A., Areola E.D., Badmus O.O. Endoglin inhibition by sodium acetate and flutamide ameliorates cardiac defective G6PD-dependent antioxidant defense in gestational testosterone-exposed rats. Biomed. Pharmacother. 2018;107:1641–1647. doi: 10.1016/j.biopha.2018.08.133. [DOI] [PubMed] [Google Scholar]
- 80.Lopes R.A., Neves K.B., Carneiro F.S., Tostes R.C. Testosterone and vascular function in aging. Front. Physiol. 2012;3:89. doi: 10.3389/fphys.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang G., Cui R., Kang Y., Qi C., Ji X., Zhang T., Guo Q., Cui H., Shi G. Testosterone propionate activated the Nrf2-ARE pathway in ageing rats and ameliorated the age-related changes in liver. Sci. Rep. 2019;9:18619. doi: 10.1038/s41598-019-55148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao F.Y., Nheu L., Komesaroff P., Ling S. Testosterone protects cardiac myocytes from superoxide injury via NF-kappaB signalling pathways. Life Sci. 2015;133:45–52. doi: 10.1016/j.lfs.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 83.Chistiakov D.A., Myasoedova V.A., Melnichenko A.A., Grechko A.V., Orekhov A.N. Role of androgens in cardiovascular pathology. Vasc. Health Risk Manag. 2018;14:283–290. doi: 10.2147/VHRM.S173259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang L., Lei D., Zhu G.P., Hong L., Wu S.Z. Physiological testosterone retards cardiomyocyte aging in Tfm mice via androgen receptor-independent pathway. Chin. Med. Sci. J. 2013;28:88–94. doi: 10.1016/s1001-9294(13)60028-0. [DOI] [PubMed] [Google Scholar]
- 85.Szabo R., Borzsei D., Kupai K., Hoffmann A., Gesztelyi R., Magyarine Berko A., Varga C., Posa A. Spotlight on a new heme oxygenase pathway: testosterone-induced shifts in cardiac oxidant/antioxidant status. Antioxidants. 2019;8 doi: 10.3390/antiox8080288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rattanasopa C., Kirk J.A., Bupha-Intr T., Papadaki M., de Tombe P.P., Wattanapermpool J. Estrogen but not testosterone preserves myofilament function from doxorubicin-induced cardiotoxicity by reducing oxidative modifications. Am. J. Physiol. Heart Circ. Physiol. 2019;316:H360–H370. doi: 10.1152/ajpheart.00428.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akdis D., Saguner A.M., Shah K., Wei C., Medeiros-Domingo A., von Eckardstein A., Luscher T.F., Brunckhorst C., Chen H.S.V., Duru F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: from a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017;38:1498–1508. doi: 10.1093/eurheartj/ehx011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suojanen J.N., Gay R.J., Hilf R. Influence of estrogen on glutathione levels and glutathione-metabolizing enzymes in uteri and R3230AC mammary tumors of rats. Biochim. Biophys. Acta. 1980;630:485–496. doi: 10.1016/0304-4165(80)90003-3. [DOI] [PubMed] [Google Scholar]
- 89.Borras C., Gambini J., Lopez-Grueso R., Pallardo F.V., Vina J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim. Biophys. Acta. 2010;1802:205–211. doi: 10.1016/j.bbadis.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 90.Tostes R.C., Carneiro F.S., Carvalho M.H., Reckelhoff J.F. Reactive oxygen species: players in the cardiovascular effects of testosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;310:R1–14. doi: 10.1152/ajpregu.00392.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lopes R.A., Neves K.B., Pestana C.R., Queiroz A.L., Zanotto C.Z., Chignalia A.Z., Valim Y.M., Silveira L.R., Curti C., Tostes R.C. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am. J. Physiol. Heart Circ. Physiol. 2014;306:H1485–H1494. doi: 10.1152/ajpheart.00809.2013. [DOI] [PubMed] [Google Scholar]
- 92.Pongkan W., Chattipakorn S.C., Chattipakorn N. Chronic testosterone replacement exerts cardioprotection against cardiac ischemia-reperfusion injury by attenuating mitochondrial dysfunction in testosterone-deprived rats. PloS One. 2015;10 doi: 10.1371/journal.pone.0122503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pongkan W., Pintana H., Sivasinprasasn S., Jaiwongkam T., Chattipakorn S.C., Chattipakorn N. Testosterone deprivation accelerates cardiac dysfunction in obese male rats. J. Endocrinol. 2016;229:209–220. doi: 10.1530/JOE-16-0002. [DOI] [PubMed] [Google Scholar]
- 94.Pongkan W., Pintana H., Jaiwongkam T., Kredphoo S., Sivasinprasasn S., Chattipakorn S.C., Chattipakorn N. Vildagliptin reduces cardiac ischemic-reperfusion injury in obese orchiectomized rats. J. Endocrinol. 2016;231:81–95. doi: 10.1530/JOE-16-0232. [DOI] [PubMed] [Google Scholar]
- 95.Rovira-Llopis S., Banuls C., Diaz-Morales N., Hernandez-Mijares A., Rocha M., Victor V.M. Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol. 2017;11:637–645. doi: 10.1016/j.redox.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rovira-Llopis S., Banuls C., de Maranon A.M., Diaz-Morales N., Jover A., Garzon S., Rocha M., Victor V.M., Hernandez-Mijares A. Low testosterone levels are related to oxidative stress, mitochondrial dysfunction and altered subclinical atherosclerotic markers in type 2 diabetic male patients. Free Radic. Biol. Med. 2017;108:155–162. doi: 10.1016/j.freeradbiomed.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 97.Victor V.M., Rocha M., Banuls C., Rovira-Llopis S., Gomez M., Hernandez-Mijares A. Mitochondrial impairment and oxidative stress in leukocytes after testosterone administration to female-to-male transsexuals. J. Sex. Med. 2014;11:454–461. doi: 10.1111/jsm.12376. [DOI] [PubMed] [Google Scholar]
- 98.Victor V.M., Rovira-Llopis S., Saiz-Alarcon V., Sanguesa M.C., Rojo-Bofill L., Banuls C., Falcon R., Castello R., Rojo L., Rocha M., Hernandez-Mijares A. Altered mitochondrial function and oxidative stress in leukocytes of anorexia nervosa patients. PloS One. 2014;9 doi: 10.1371/journal.pone.0106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pavon N., Martinez-Abundis E., Hernandez L., Gallardo-Perez J.C., Alvarez-Delgado C., Cerbon M., Perez-Torres I., Aranda A., Chavez E. Sexual hormones: effects on cardiac and mitochondrial activity after ischemia-reperfusion in adult rats. Gender difference. J. Steroid Biochem. Mol. Biol. 2012;132:135–146. doi: 10.1016/j.jsbmb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 100.Ballantyne T., Du Q., Jovanovic S., Neemo A., Holmes R., Sinha S., Jovanovic A. Testosterone protects female embryonic heart H9c2 cells against severe metabolic stress by activating estrogen receptors and up-regulating IES SUR2B. Int. J. Biochem. Cell Biol. 2013;45:283–291. doi: 10.1016/j.biocel.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.