

Abstract
Treatment of patients with triple-negative breast cancer (TNBC) is limited by a lack of effective molecular therapies targeting this disease. Recent studies have identified metabolic alterations in cancer cells that can be targeted to improve responses to standard-of-care chemotherapy regimens. Using MDA-MB-468 and SUM-159PT TNBC cells, along with LC-MS/MS and HPLC metabolomics profiling, we found here that exposure of TNBC cells to the cytotoxic chemotherapy drugs cisplatin and doxorubicin alter arginine and polyamine metabolites. This alteration was because of a reduction in the levels and activity of a rate-limiting polyamine biosynthetic enzyme, ornithine decarboxylase (ODC). Using gene silencing and inhibitor treatments, we determined that the reduction in ODC was mediated by its negative regulator antizyme, targeting ODC to the proteasome for degradation. Treatment with the ODC inhibitor difluoromethylornithine (DFMO) sensitized TNBC cells to chemotherapy, but this was not observed in receptor-positive breast cancer cells. Moreover, TNBC cell lines had greater sensitivity to single-agent DFMO, and ODC levels were elevated in TNBC patient samples. The alterations in polyamine metabolism in response to chemotherapy, as well as DFMO-induced preferential sensitization of TNBC cells to chemotherapy, reported here suggest that ODC may be a targetable metabolic vulnerability in TNBC.
Keywords: amino acid, breast cancer, metabolomics, polyamine, DNA damage, antizyme, difluoromethylornithine, ornithine decarboxylase (ODC), triple-negative breast cancer (TNBC), cell metabolism
Introduction
In the past two decades, a renewed interest in tumor metabolism has led to the identification of novel therapeutic vulnerabilities in a variety of cancers (1, 2). This is especially promising for tumor types that lack effective targeted therapies, such as triple-negative breast cancer (TNBC).3 TNBC composes 15–20% of breast cancer cases but accounts for a disproportionately high percentage of breast cancer–related deaths (3). This is in part because of a lack of targeted therapies for this molecular subtype of breast cancer, and the standard of care treatment for TNBC remains combination genotoxic chemotherapy regimens such as Adriamycin (doxorubicin), cyclophosphamide, paclitaxel (Taxol) (AC-T) (4, 5). Previous studies have identified metabolic vulnerabilities in breast cancer cells, including nucleotide metabolism, GSH biosynthesis, and glutamine metabolism, that improve response to chemotherapy drugs in pre-clinical settings (6–8).
The amino acid arginine has been extensively studied in the context of cancer metabolism and has been suggested to contribute to the development and progression of cancer (9). Arginine is involved in numerous cell growth control processes, including protein synthesis, nitric oxide (NO) production, and polyamine biosynthesis, as well as cellular energy production via the tricarboxylic acid cycle (10). Furthermore, arginine plays an important role in the immune system, as it is required for full activation of natural killer and T-cells (11, 12). A number of studies aimed at targeting arginine metabolism have focused on arginine auxotroph tumors, because they are sensitive to arginine depletion (13, 14). However, less than 10% of breast tumors lack the rate-limiting arginine synthesis enzyme argininosuccinate synthase (15). Therefore, additional branches of arginine metabolism may be altered in breast cancer and represent potential metabolic vulnerabilities.
The polyamines putrescine, spermidine, and spermine are cationic molecules that are synthesized from arginine following its conversion to ornithine and are essential for eukaryotic cell growth and differentiation (16). Functions attributed to polyamines include binding to nucleic acids and chromatin, stabilizing cellular membranes, regulating ion channels, and scavenging free radicals (17). Polyamines can form hydrogen bonds with anions, such as nucleic acids, proteins, and phospholipids and lead to condensation of DNA and chromatin (18, 19). Ornithine decarboxylase (ODC), the first rate-limiting enzyme in polyamine synthesis, is regulated transcriptionally, translationally, and posttranslationally (20). Moreover, ODC levels and activity are altered in response to extracellular stimuli such as hormones and growth factors, as well as changes in intracellular free polyamine levels (17, 20, 21).
Elevated polyamine levels have been detected in tumors, including breast tumors, and increased polyamine synthesis has been shown to promote tumor initiation and growth (16, 19). Combined inhibition of polyamine uptake and synthesis results in antiproliferative effects in breast cancer cell lines and mouse models, suggesting that targeting polyamine metabolism may be therapeutically effective in breast cancer patients (22, 23). Although drugs that target polyamine metabolism have not yet been approved for therapeutic use in cancer, clinical trials using polyamine uptake and synthesis inhibitors are underway for a number of indications, although there are no current trials in breast cancer (24). Here we show that polyamine synthesis is suppressed in response to DNA damaging chemotherapy through the proteasomal degradation of ODC. Further decreasing the polyamine pool using ODC inhibitors increases the sensitivity of TNBC to standard-of-care genotoxic drugs.
Results
Genotoxic chemotherapy alters levels of polyamines and related metabolites in TNBC
To evaluate the alterations in intracellular metabolites in breast cancer cells exposed to genotoxic chemotherapy drugs, we measured the levels of polar metabolites in MDA-MB-468 and SUM-159PT TNBC cells, which represent the Basal A and B subtypes according to the classification of Lehmann et al. (25), following treatment with cisplatin or doxorubicin for 8, 24, or 48 h. These drugs were selected because doxorubicin is a standard-of-care as part of AC-T therapy for TNBC patients, and neoadjuvant cisplatin has shown some therapeutic efficacy in a subset of TNBC patients, particularly those with hereditary BRCA1-mutant breast cancer (26–29). We first determined the concentration of both drugs required for the induction of DNA damage as measured by increased H2A.X phosphorylation, while remaining below the IC50 for cell death (Fig. 1, A and B). Based on this analysis, both cell lines were treated with 0.5 μm doxorubicin and 2.5 μm cisplatin, and targeted metabolomics profiling of 186 metabolites was performed using LC-MS/MS (Fig. 1C and Table S1). Increases in pyrimidine nucleotides were observed in response to DNA damage, consistent with previous reports (6).
Figure 1.
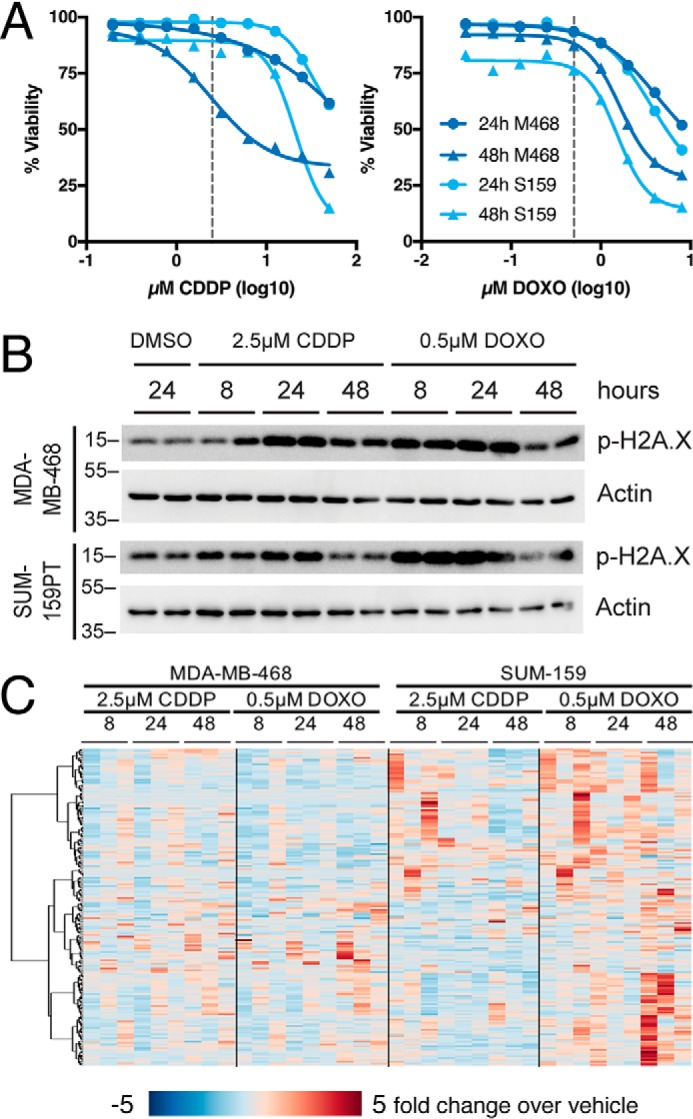
Genotoxic chemotherapy alters TNBC metabolism. A, viability measured by propidium iodide uptake following 24 or 48 h of exposure to chemotherapy agents. Selected concentrations (2.5 μm cisplatin, 0.5 μm doxorubicin) denoted by dashed lines. nonlinear curve fit by four parameter logistic regression. B, representative immunoblot of phospho–Ser-139 histone H2A.X (p-H2A.X) following exposure to chemotherapy agents at times and doses used for metabolite measurements. C, -fold change metabolite abundance over 24 h vehicle control for 186 metabolites measured by LC-MS/S in MDA-MB-468 and SUM-159PT cells treated with 2.5 μm cisplatin (CDDP) or 0.5 μm doxorubicin (DOXO).
We focused on metabolites involved in arginine metabolism, including the urea cycle, NO cycle, polyamine metabolism, proline metabolism, and creatine synthesis (Fig. 2A). Twelve metabolites involved in arginine metabolism were detected by our LC-MS/MS platform (Fig. 2, A and B). In response to genotoxic drugs, the most up-regulated metabolite in arginine metabolism was ornithine, and the most decreased was S-adenosylmethionine (SAM) (Fig. 2, B and C). Ornithine can be synthesized either from arginine by arginase, or from glutamine via ornithine aminotransferase, although previous reports have shown that in transformed cells, ornithine is derived exclusively from arginine (30). In MDA-MB-468 and SUM-159PT TNBC cells, carbons from 13C6-arginine, but not 13C5-glutamine, were incorporated into ornithine, even though both exogenous 13C6-arginine and 13C5-glutamine efficiently labeled their respective intracellular pools, and this was unaffected in doxorubicin-treated cells (Fig. 2D). Because both ornithine and SAM are required for the synthesis of polyamines, but polyamines were not detectable on our LC-MS/MS platform, we measured polyamine levels by conventional HPLC analysis (Fig. 2E). After 48 h of exposure to genotoxic drugs, putrescine and spermidine were significantly decreased by doxorubicin and also decreased in response to cisplatin. These data suggest that genotoxic drugs alter arginine metabolites involved in polyamine synthesis in TNBC cells.
Figure 2.

Genotoxic chemotherapy alters arginine and polyamine metabolism. A, arginine synthesis and degradation. Metabolites in black, genes in blue. Bold text indicates metabolites detected by LC-MS/MS. B, -fold change metabolite abundance over 24 h vehicle control for arginine metabolites in MDA-MB-468 and SUM-159PT cells treated with 2.5 μm cisplatin (CDDP) or 0.5 μm doxorubicin (DOXO) (n = 3). C, relative abundance of ornithine and SAM by LC-MS/MS; vehicle at 24 h (n = 3). D, fraction metabolite pool labeled as measured by LC-MS/MS following incubation with 13C6-arginine or 13C5-glutamine in the presence of vehicle or 0.5 μm doxorubicin for 48 h (n = 2 biological replicates; technical replicates denoted by shared symbol). E, relative abundance of polyamines by HPLC; vehicle control at 24 h (n = 3–5). All error bars represent S.E. *, p < 0.05; **, p < 0.01 by two-way ANOVA.
Chemotherapy decreases levels and activity of ornithine decarboxylase
We reasoned that alterations in polyamine metabolites were because of changes in enzyme levels or activity in response to chemotherapy exposure. ODC catalyzes the first rate-limiting step in polyamine synthesis, specifically the conversion of ornithine to putrescine (Fig. 2A). Decreased ODC levels or activity could account for the observed decreases in polyamines and increases in ornithine (Fig. 2, C and E), although ornithine could also be elevated by increased activity of arginase II (ARG2) (Fig. 2A). Cisplatin and doxorubicin increased total ODC protein at early time points (8 h), consistent with a stress response (24), but led to decreased ODC and increased ARG2 at later time points (Fig. 3A and Fig. S1). This pattern of ODC expression was also observed in multiple TNBC and non-TNBC breast cancer cell lines (Fig. 3, B–D). By contrast, alterations in ARG2 expression were not significant across all lines. We also confirmed a concomitant decrease in ODC activity after 24 and 48 h of exposure to cisplatin or doxorubicin (Fig. 4A). A significant decrease in putrescine at 72 and 96 h was also observed, in addition to significantly reduced spermidine after 96 h of doxorubicin treatment and a time-dependent decreasing trend in spermine concentration (Fig. 4B).
Figure 3.

Chemotherapy decreases ODC and increases ARG2 proteins. A–C, Representative immunoblots and quantification (n = 4) of total ornithine decarboxylase (ODC) and arginase II (ARG2) proteins, and phospho–Ser-139 histone H2A.X (p-H2A.X) following exposure to chemotherapy agents in MDA-MB-468 and SUM-159PT cells, (B) other TNBC cells, and (C) non-TNBC cells. “0 h” chemotherapy treatment indicates 24-h vehicle control. D, average quantification of n = 4 immunoblots of ODC and ARG2 from nine breast cancer cell lines in A–C following chemotherapy exposure. Band above 55 kD in ODC blots is nonspecific. All error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by two-way ANOVA.
Figure 4.

Chemotherapy decreases ODC activity and polyamine levels. A, ODC activity measured by CO2 release following exposure to 2.5 μm cisplatin or 0.5 μm doxorubicin; vehicle control at 24 h (n = 3). B, relative abundance of polyamines in MDA-MB-468 cells by HPLC; vehicle control at 72 h (n = 3). All error bars represent S.E. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by two-way ANOVA.
To investigate the mechanism by which ODC protein and activity are decreased following chemotherapy exposure, we first evaluated transcriptional regulation of ODC1, which is a well-characterized target of c-Myc (31). Depletion of c-Myc using siRNA did not alter the overall ODC response to chemotherapy (Fig. 5A), although ODC1 transcript was increased following chemotherapy exposure (Fig. 5B). ODC protein is posttranslationally regulated by the activity of antizyme, which binds ODC to promote its ubiquitin-independent degradation by the 26S proteasome (32). Pre-treatment with the proteasome inhibitor MG132 rescued the decrease in ODC protein following doxorubicin exposure (Fig. 5C). siRNA against antizyme decreased the corresponding transcript of OAZ1 by over 80% (Fig. 5D) and blocked the decrease in ODC in response to doxorubicin in both TNBC- and ER-positive cells (Fig. 5, E and F). Therefore, it appears that genotoxic drugs decrease polyamines by reducing ODC protein and activity, possibly through the negative regulator antizyme.
Figure 5.

Chemotherapy regulates ODC via proteasomal degradation. A, representative immunoblots of ODC and c-Myc in cells pre-treated for 24 h with 20 nm nontargeting siRNA or siRNA MYC, then treated with doxorubicin for indicated times (n = 2). B, qRT-PCR of ODC1 transcript following treatment with 2.5 μm cisplatin or 0.5 μm doxorubicin, relative to 24-h vehicle control. C, representative immunoblots of ODC in SUM-159PT cells pre-treated for 2 h with 0.5 μm proteasome inhibitor MG132 followed by addition of vehicle or doxorubicin (n = 3). D, qRT-PCR of OAZ1 transcript following treatment with 20 nm indicated siRNA (n = 3). E and F, representative immunoblots and quantification of ODC in (E) 20 μg/lane MDA-MB-468 or (F) 40 μg/lane ZR-75-1 cells pre-treated for 24 h with 20 nm siOAZ1 followed by 48-h addition of vehicle or 0.5 μm doxorubicin (n = 3). Band above 55 kD in ODC blots is nonspecific. All error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 by two-way ANOVA.
Targeting polyamine synthesis increases sensitivity to chemotherapy
Polyamines promote cell cycle progression (33) and depletion of ODC or polyamines induces cell cycle arrest at the G2/M phase (34–36), where cells are more sensitive to DNA damage induced by cisplatin and doxorubicin (37–39). Because we observed a decrease in polyamines and ODC activity following chemotherapy treatment, we reasoned that targeting ODC to further decrease polyamines could increase tumor cell killing. We proceeded with doxorubicin because it is a standard-of-care chemotherapeutic agent used for the majority of TNBC patients (26, 27). Treatment with the irreversible suicide inhibitor of ODC, α-difluoromethylornithine (DFMO), sensitized both MDA-MB-468 and SUM-159PT cells to doxorubicin (Fig. 6A). Addition of exogenous putrescine or spermidine did not rescue this sensitization (Fig. 6B). A previous study reported that DFMO can decrease colon cancer cell growth by increasing polyamine recycling, leading to a futile cycle that depletes SAM and nucleotides, and that this effect can be rescued by exogenous thymidine (40). However, in TNBC cells, thymidine addition, alone or in combination with putrescine or spermidine, did not rescue the sensitization to doxorubicin by DFMO (Fig. 6C). Treatment of MDA-MB-468 cells with the arginase inhibitor Nω-hydroxy-nor-arginine (NOHA) also increased sensitivity to doxorubicin (Fig. 6D), consistent with its ability to decrease polyamine levels (41, 42).
Figure 6.

ODC inhibition increases sensitivity to doxorubicin. A, viability measured by propidium iodide uptake following 72-h pre-treatment with 1 mm DFMO or vehicle and 72-h exposure to doxorubicin in the presence of DFMO or vehicle (n = 3). B, MDA = MB-468 viability following 72-h pre-treatment with 1 mm DFMO with or without 10 μm putrescine or 10 μm spermidine and 1 mm aminoguanidine, and addition of doxorubicin for 72 h (n = 2 and n = 3). C, viability following 72-h pre-treatment with 1 mm DFMO with or without 0.1 mm thymidine (T), 10 μm putrescine (P), or 10 μm spermidine (S), followed by addition of doxorubicin for 72 h (n = 2). 1 mm aminoguanidine was added with putrescine and spermidine. D, viability following 72-h pre-treatment with 0.5 mm Nω-hydroxy-nor-arginine (NOHA) and addition of doxorubicin for 72 h (n = 4). E, HPLC measurements of polyamines following 72-h treatment with 1 mm DFMO (n = 2). ND indicates not detected. F, top 10% most significantly altered polar metabolites measured by LC-MS/MS following treatment with 1 mm DFMO (n = 2). All error bars represent S.E. Nonlinear curve fit by four parameter logistic regression. p values by unpaired two-tailed t test.
To confirm the on-target effects of DFMO, we measured polyamine levels after 72 h of treatment with DFMO. As expected, putrescine was decreased in MDA-MB-468 cells and undetectable in SUM-159PT cells, and spermidine was also reduced (Fig. 6E). To further investigate the effects of DFMO, we measured polar metabolites following exposure to DFMO (Table S2). The putrescine metabolite 4-aminobutyrate was significantly decreased, and other metabolites related to nucleotide, one-carbon, and amino acid metabolism were also decreased (Fig. 6F). This suggests that these metabolites must be replenished to reverse the effects of DFMO and resulting sensitization to doxorubicin.
Across a panel of breast cancer cell lines representing both estrogen receptor (ER)–positive luminal A, HER2+, and TNBC subtypes, we observed that all TNBC cell lines tested were sensitized to doxorubicin by pre-treatment with DFMO, whereas most non-TNBC lines were not (Fig. 7, A and B). Overall, cell lines that displayed the greatest sensitization to doxorubicin by DFMO are classified as TNBC (Fig. 7C). These findings prompted us to determine whether there exists an intrinsic property of TNBC that makes this molecular subtype of breast cancer more dependent on ODC.
Figure 7.

ODC inhibition increases sensitivity of TNBC cells to doxorubicin. A and B, viability measured by propidium iodide uptake following 72-h pre-treatment with 1 mm DFMO or vehicle and 72-h exposure to doxorubicin in the presence of (A) DFMO or vehicle in TNBC and (B) non-TNBC cells (n = 3); p value by paired two-tailed t test. C, percent change doxorubicin IC50 for DFMO over vehicle control from Fig. 6A and Fig. 7, A and B; IC50 calculated by four parameter logistic nonlinear curve fit. All error bars represent S.E. *, p < 0.05 by unpaired two-tailed t test.
Ornithine decarboxylase is a metabolic vulnerability in TNBC
To investigate the role of arginine and polyamine metabolism in TNBC, we queried the alterations in the transcripts of 1270 genes in the KEGG “metabolic pathways” (KEGG:hsa01100) in the METABRIC breast cancer dataset (43–45). Of these transcripts, 1134 were analyzed in 1904 tumors (Table S3). We found that ODC1 is one of the top five most significantly enriched transcripts in TNBC samples (Fig. 8, A and B). ODC1 was also enriched in TNBC patient samples from the TCGA provisional breast dataset (Fig. 8C). Although we did not observe the same trends for transcript and protein levels of ODC in response to chemotherapy (Figs. 3A and 5B), baseline transcript and protein levels positively correlated in the TCGA breast samples for which protein MS data are available (Fig. 8D). ODC1 transcript levels were also enriched in the basal subtype of breast cancer, which is commonly associated with TNBC (Fig. 8E) (46).
Figure 8.

Ornithine decarboxylase levels are increased in TNBC tumors. A, comparison of mRNA z-scores from 1134 metabolic gene transcripts between 299 TNBC and 1605 non-TNBC patient samples from METABRIC breast cancer dataset. B and C, comparison of ODC1 mRNA z-scores between (B) METABRIC breast cancer patient samples and (C) TCGA provision breast cancer patient samples, separated by TNBC status. D, correlation of ODC1 mRNA z-scores and ODC protein expression CPTAC z-scores in TCGA provision breast cancer patient samples; dashed lines indicate 90% confidence interval. E, ODC1 mRNA z-scores in METABRIC samples grouped by PAM50 subtype. F, average copy number alterations of ODC1 in METABRIC and TCGA provisional breast cancer patient samples scored by type as −1, shallow deletion; 0, diploid; 1, gain; 2, amplification. G, comparison of ODC1 mRNA z-scores between METABRIC breast cancer patient samples separated by TNBC status and grouped by MYC copy number alterations. H, copy number alterations in METABRIC patient samples for antizyme genes, scored as in F. I, mRNA z-scores of antizyme transcripts in METABRIC samples grouped by TNBC status. J, copy number alterations in METABRIC patient samples for antizyme inhibitor gene AZIN1, scored as in F. All error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by Welch's t test (A–C and I), one-way ANOVA (E and G), or two-tailed unpaired t test (F, H, and J); linear curve fit, p value, and R2 by linear regression (D).
The increased ODC1 levels in TNBC could be because of changes in ODC1 copy number, because the average ODC1 copy number was higher in TNBC compared with non-TNBC (Fig. 8F). Because ODC1 is a target of c-Myc, we evaluated the relationship between MYC amplification and ODC1 transcript in TNBC and observed a significant correlation (Fig. 8G). ODC stability is regulated by antizyme and antizyme inhibitor (AZI), and we also observed decreased copy number and transcript levels for multiple antizyme genes and transcripts (OAZ1/2/3) in TNBC (Fig. 8, H and I), as well as increased copy number of AZI gene AZIN1 (Fig. 8J) (47).
Enrichment of ODC1 transcript was also observed in TNBC cells lines according to the Cancer Cell Line Encyclopedia (48) and confirmed by RT-PCR analysis of select breast cancer cell lines (Fig. 9, A and B), consistent with a previous study of four breast cancer cell lines (49). A trend toward higher baseline protein expression of ODC in TNBC cells was also observed (Fig. 9C). Moreover, DFMO treatment significantly reduced proliferation of breast cancer cell lines regardless of their ER/PR/HER2 status (Fig. 9, D and E). By contrast, DFMO treatment of TNBC cell lines resulted in increased cell death when compared with non-TNBC lines, regardless of doubling time (Fig. 9, F–H).
Figure 9.

Ornithine decarboxylase is a metabolic vulnerability in TNBC. A, comparison of ODC1 mRNA z-scores in 25 TNBC and 25 non-TNBC breast cancer cell lines (Cancer Cell Line Encyclopedia). B, comparison of ODC1 transcript levels measured by qRT-PCR. C, representative immunoblots and quantification (n = 3) of ODC protein in untreated breast cancer cell lines; band above 55 kD in ODC blots is nonspecific. D and E, growth of breast cancer cell lines measured as population size relative to vehicle after 72-h treatment with 2 mm DFMO. F and G, viability of breast cancer cell lines measured by propidium iodide uptake following 72-h treatment with 2 mm DFMO. H, doubling time of breast cancer cell lines compared with average viability from (F); R2 by linear regression. All error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by two-tailed unpaired t test (A–C, E, and G) or two-way ANOVA (D and F).
Discussion
Because of the poor prognosis for patients with TNBC, many new strategies have been proposed and evaluated for improved therapeutic efficacy. In this context, the implementation of poly ADP-ribose polymerase inhibitors in BRCA1-mutant TNBC and immune checkpoint therapy in TNBC patients with infiltrating lymphocytes and PD-L1 expression has provided considerable success (4, 50, 51). However, chemotherapy remains the only FDA-approved therapy for the majority of patients with TNBC (5). Advances in understanding the reprogramming of metabolic pathways in human cancers have led to the identification of druggable targets that have served as the basis for clinical trials, with the goal of targeting tumor-specific metabolic vulnerabilities. In breast cancer, alterations in GSH, nucleotide, and glutamine metabolism occur in response to genotoxic chemotherapy (6–8). In the present study, we investigated changes in arginine and polyamine-related metabolites in response to genotoxic chemotherapy in TNBC cells, with the goal of proposing novel therapeutic combinations. We found that genotoxic drugs decrease ODC levels and activity, with a corresponding reduction in the polyamines putrescine and spermidine. ODC expression is increased in TNBC patient samples and cell lines, to the extent that targeting ODC with the inhibitor DFMO sensitizes TNBC cells to doxorubicin.
TNBC cell lines exposed to chemotherapy showed a significant decrease in the expression and activity of ODC (Figs. 3 and 4). This decrease was not because of alterations in transcript levels nor is it mediated by c-Myc, a major transcriptional regulator of ODC (52). Instead, reduced ODC expression in response to chemotherapy was mediated through degradation by the 26S proteasome (Fig. 5). ODC has a half-life of 10 to 30 min, one of the shortest of any mammalian protein (53). ODC turnover is regulated by binding to antizyme, a protein that binds ODC and targets it for destruction by nonubiquitin-mediated degradation (32). Antizyme itself is translationally regulated through the binding of polyamines to its transcript OAZ1, thereby inducing a +1 ribosomal frameshift (32). Thus, antizyme levels are increased when intracellular polyamine concentrations are elevated. The observed initial increases in polyamines after 8 h of chemotherapy exposure may be sufficient to increase translation of OAZ1 and subsequently increase antizyme expression (Fig. 3), resulting in ODC degradation. One previous study demonstrated that doxorubicin increases antizyme expression mediated by c-Jun, although the mechanistic basis was not determined (54). An alternative mechanism that could account for ODC regulation is increased antizyme binding to ODC in response to chemotherapy. Moreover, because AZI binds antizyme with a greater affinity than ODC, thereby protecting ODC from antizyme-mediated degradation (17, 31), decreased AZI would free antizyme to bind ODC. Future studies are necessary to address which of these mechanisms is responsible for regulation of ODC in response to chemotherapy.
Treatment with the ODC inhibitor DFMO sensitizes TNBC cells to chemotherapy drugs, consistent with previous observations that polyamines bind and stabilize DNA and in turn occlude genotoxic agents (55). Conversely, depleting polyamines increases DNA damage and induces cell cycle arrest (56–58). Depletion of ODC or polyamines induces arrest in G2 (35), the cell cycle phase in which tumor cells are most sensitive to damage by cisplatin and doxorubicin (37–39). Depending on the cell type, DFMO arrests cells in either G1 or G2 (34, 59–61). Our observations are more consistent with a G2 arrest in response to ODC and DFMO treatment, because this is the phase when cells are most sensitive to genotoxic damage.
Targeting ODC and polyamine synthesis is an attractive antitumor target because high levels of polyamines have been measured in multiple tumor types and are necessary for transformation and progression (16, 19, 24). DFMO is approved for treatment of trypanosomiasis and facial hirsutism and is in clinical trials for treatment or prevention of various tumor types (24). DFMO is of particular chemotherapeutic interest in MYCN-amplified neuroblastoma, where Myc-driven overexpression of ODC contributes to tumor hyperproliferation (31). At the time of this study, the only studies of DFMO in combination with genotoxic chemotherapies are in brain tumors, and presently there are no clinical trials in breast cancer using DFMO as single agent therapy or in combination with other drugs. Clinical trials in the late 1990s established that DFMO reduces polyamines in breast cancer patients, but it did not reduce tumor burden as a single agent (62, 63). Application of DFMO in combination with doxorubicin to TNBC in pre-clinical models would lend further support for the efficacy of this combination, with likely promise because of the apparent low toxicity of DFMO. Combining DFMO with genotoxic chemotherapies could also be effective in other tumor types with elevated ODC activity or expression, including melanoma, esophageal, prostate, and colorectal cancers (35, 64–66).
The increased expression of ODC in TNBC could be because of changes in ODC1 copy number, but may also be because of other regulatory factors (Fig. 8). For example, MYC amplification also correlated with increased ODC1 transcript in TNBC, but not in non-TNBC patient samples, and copy number and transcript of antizyme and AZI were altered in TNBC patient samples in a manner that would lead to increased ODC protein stability. Changes in polyamine metabolism downstream of ODC also indicate that this pathway in general is of critical importance in TNBC etiology as well as advanced disease. The levels of spermine synthase transcript and its catabolic product diacetylspermine are elevated in TNBC patients and predictive of poor survival, and spermidine is elevated in breast tumors and increases further with tumor grade (67, 68). Determining which of these factors are predictive of DFMO sensitivity will be important to identify a biomarker that would identify which patients are most likely to benefit from combination therapy strategies that include DFMO.
The pronounced effects of DFMO as a single agent on TNBC cell viability (Fig. 9, F and G) indicate that this molecular subtype of breast cancer is especially reliant on de novo polyamine biosynthesis, because in many other cell types targeting polyamine synthesis is not effective unless combined with a polyamine uptake inhibitor (17). Although the specific polyamine transport mechanisms are not well-defined (24), it will be interesting to investigate the expression and function of polyamine transporters in TNBC, as deficiencies in transport systems may contribute to a more pronounced reliance on intracellular biosynthesis. Further characterization of polyamine metabolism and the effects of DFMO treatment in TNBC will help identify why this subtype is particularly sensitive.
To our knowledge, this is the first study to profile metabolic changes in response to DFMO in breast cancer. Adding exogenous polyamines or thymidine, alone or in combination, was insufficient to rescue the effects of DFMO in TNBC (Fig. 6). Importantly, it is evident that treatment of TNBC cells with DFMO also affects metabolites outside of polyamine metabolism, related to nucleotide, one-carbon, and other amino acid metabolism. Whereas in colon tumor cells the effects of DFMO were the result of depleting SAM and nucleotide pools and could be reversed with exogenous thymidine (40), this was not the case in TNBC cells (Fig. 6). Because TNBC cells are more sensitive to DFMO than non-TNBC cells, this implies that an intrinsic genetic or epigenetic property of TNBC renders these tumors more susceptible to ODC inhibition, at least in vitro. Whether this represents a metabolic vulnerability in TNBC that can be exploited therapeutically remains to be determined, but given the efficacy of DFMO used in other indications, this warrants exploration in pre-clinical models.
In summary, we have identified changes in arginine and polyamine metabolism in TNBC cells exposed to chemotherapy, because of a decrease of the polyamine synthesis enzyme ODC. By targeting ODC with the inhibitor DFMO, we show that we can sensitize TNBC cells to doxorubicin. Further studies on the mechanisms linking genotoxic damage to ODC degradation, mechanisms of sensitization, and evaluation of combinations in pre-clinical models will provide additional insight for eventual therapeutic applications.
Experimental procedures
Cell culture
SUM-159PT and SUM-149PT cells were obtained from Asterand Bioscience; all other cell lines were obtained from the ATCC. Cell lines were authenticated using short tandem repeat profiling, and no cell lines used in this study were found in the database of commonly misidentified cell lines, which is maintained by the International Cell Line Authentication Committee and National Center for Biotechnology Information BioSample. All cells were maintained in RPMI medium (Wisent Bioproducts) containing 10% FBS (Gibco). Cells were passaged for no more than 4 months and routinely assayed for mycoplasma contamination.
Chemotherapy agents and inhibitors
Doxorubicin was purchased from Cell Signaling Technology and dissolved in DMSO at 10 mm; dl-α-difluoromethylornithine (hydrochloride hydrate) and MG132 were purchased from Cayman Chemical and dissolved in DMSO at 50 mm or 10 mm, respectively; spermidine, thymidine, and aminoguanidine hydrochloride were purchased from Sigma and dissolved in water at 10 mm,100 mm, or 1 m, respectively; putrescine (dihydrochloride) was purchased from Santa Cruz Biotechnology and dissolved in water at 10 mm; N-ω-hydroxy-l-norarginine acetate salt was purchased from Bachem and dissolved in water at 50 mg/ml. Cisplatin was obtained from the Dana Farber Cancer Institute pharmacy at 1 mg/ml in PBS. DMSO for use as an organic solvent and vehicle control was purchased from Fisher Scientific.
Antibodies
ODC (MABS36, 1:100) was purchased from Millipore. ARG2 (ab137069, 1:1000) was purchased from Abcam. pH2A.XS139 (9718, 1:1000), beta-actin (4970, 1:1000), c-Myc (5605, 1:1000), and vinculin (13901, 1:1000) were purchased from Cell Signaling Technology. Antibodies were used at indicated dilutions in 5% milk (Andwin Scientific) in TBST buffer (Boston Bioproducts), except for p-H2A.X in 5% BSA (Boston Bioproducts) in TBST.
LC/MS-MS metabolomics profiling
Cells were maintained in RPMI + 10% FBS, and fresh medium was added at the time cells were treated. For metabolite extraction, medium was aspirated and ice-cold 80% (v/v) methanol was added. Cells and the metabolite-containing supernatants were collected. Insoluble material was pelleted by centrifugation at 20,000 × g for 5 min. The resulting supernatant was evaporated under nitrogen gas. Samples were resuspended using 20 μl HPLC-grade water for MS. For polar metabolite profiling, 5 μl from each sample were injected and analyzed using a 5500 QTRAP hybrid triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) with HILIC chromatography (Waters Amide XBridge) via selected reaction monitoring (SRM) with polarity switching. A total of 293 endogenous water-soluble metabolites were targeted for steady-state analyses. Electrospray source voltage was +4950 V in positive ion mode and −4500 V in negative ion mode. The dwell time was 3 ms per SRM transition (69). Peak areas from the total ion current for each metabolite were integrated using MultiQuant v2.1.1 software (AB/SCIEX). For 13C-labeled experiments, 5 μl from each sample (20 μl) were injected with similar methodology as above using a 6500 QTRAP (AB/SCIEX) and integrated using MultiQuant v3.0 software (70). SRM transitions were created for expected 13C incorporation in various forms. Metabolite total ion counts were the integrated total ion current from a single SRM transition and normalized by cellular protein content. Nonhierarchical clustering was performed by Metaboanalyst 4.0 using a Euclidean distance measure and Ward's clustering algorithm (71).
Polyamine concentration determinations and ODC activity assays
Cells were washed with PBS and harvested in ODC breaking buffer (25 mm Tris-HCl, pH 7.5, 0.1 mm EDTA, 2.5 mm DTT). Intracellular polyamine concentrations of cell lysates were determined by HPLC following acid extraction and dansylation of the supernatant, as originally described by Kabra et al. (72). Standards prepared for HPLC included diaminoheptane (internal standard), PUT, SPD, and SPM, all of which were purchased from Sigma-Aldrich. Enzyme activity assays were performed for ODC using radiolabeled substrates, as described previously (73). All enzyme activities and intracellular polyamine concentrations are presented relative to total cellular protein, as determined using Bio-Rad protein dye (Hercules, CA) with interpolation on a BSA standard curve.
Isotope labeling
RPMI powder lacking glutamine, arginine, tryptophan, and glucose was obtained from U.S. Biological Life Sciences and supplemented with 25 μm l-tryptophan (Sigma-Aldrich), 11.1 mm d-glucose (Gibco), and 10% dialyzed FBS (Gibco). For arginine labeling, 2 mm glutamine (Gibco) and 1.1 mm l-arginine-13C6 hydrochloride (Aldrich) were added; for glutamine labeling, 2 mm l-glutamine-13C5 (Cambridge Isotope Laboratories), and 1.1 mm l-arginine hydrochloride (Sigma) were added. Labeled medium, with or without chemotherapy agents, was added to cells, and cellular metabolites were extracted as described above after 48 h.
Immunoblotting
Cells were washed with ice-cold PBS (Boston Bioproducts) and lysed in radioimmunoprecipitation buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, protease inhibitor mixture, 50 nm calyculin A, 1 mm sodium pyrophosphate, and 20 mm sodium fluoride) for 15 min at 4 °C. Cell extracts were cleared by centrifugation at 14,000 rpm for 10 min at 4 °C, and protein concentration was measured with the Bio-Rad DC protein assay. Lysates were resolved on acrylamide gels with 20 μg per lane by SDS-PAGE and transferred electrophoretically to nitrocellulose membrane (Bio-Rad) at 100 V for 90 min. Blots were blocked in TBS buffer (10 mmol/L Tris-HCl, pH 8, 150 mmol/L NaCl; Boston Bioproducts) containing 5% (w/v) nonfat dry milk (Andwin Scientific). Membranes were incubated with near-IR dye-conjugated IRDye 800CW secondary antibodies (LiCor, 1:20,000 in 5% milk-TBST) and imaged on a LiCor Odyssey CLx.
Quantitative real-time PCR
Total RNA was isolated with the NucleoSpin RNA Plus (MACHEREY-NAGEL) according to the manufacturer's protocol. Reverse transcription was performed using the TaqMan Reverse Transcription Reagents (Applied Biosciences). Quantitative real-time PCR (qRT-PCR) was performed using a CFX384 Touch Real-Time PCR Detection System (Bio-Rad). Quantification of mRNA expression was calculated by the DCT method with 18S rRNA as the reference gene. ODC1 and 18S primers were designed using Primer3 (74); OAZ1 primers were published (75): ODC1 forward, 5′-CGCTCTGAGATTGTCACTGC-3′, reverse, 5′-ATCGAGGAAGTGGCAGTCAA-3′; OAZ1 forward, 5′-CGAGCCGACCATGTCTTCAT-3′, reverse, 5′-CCGGTCTCACAATCTCAAAG-3′; 18S forward, 5′-GTAACCCGTTGAACCCCATT-3′, reverse, 5′-CCATCCAATCGGTAGTAGCG-3′.
RNAi
siRNA ON-TARGETplus SMARTpools against human ODC1 (L-006668–00), OAZ1 (L-019216–00), Myc (L-003282–02), and ARG2 (L-009454–01) were purchased from Dharmacon and dissolved in siRNA buffer (Dharmacon). Cells were transfected with Lipofectamine RNAiMAX Reagent (Invitrogen) in Opti-MEM (Gibco) according to the manufacturer's protocol (Invitrogen, MAN0007825), with a final concentration of 20 nm siRNA and 3 μl/ml Lipofectamine.
Propidium iodide viability assay
Cell viability was assayed with a propidium iodide–based plate-reader assay, as described previously (76). Briefly, cells in 96-well plates were treated with a final concentration of 30 mm propidium iodide (Cayman Chemical) for 20 min at 37 °C. The initial fluorescence intensity was measured in a GENios FL (Tecan) at 560 nm excitation/635 nm emission. Digitonin (Millipore) was then added to each well at a final concentration of 600 mm. After incubating for 20 min at 37 °C, the final fluorescence intensity was measured. The fraction of dead cells was calculated by dividing the background-corrected initial fluorescence intensity by the final fluorescence intensity. Viability was calculated by (1 − fraction of dead cells).
Sulforhodamine B growth assay
Population size as cell confluency was assayed by sulforhodamine B staining, as described previously (77). Briefly, cells in 96-well plates were fixed in 8.3% final concentration of tricarboxylic acid of 1 h at 4 °C, washed three times with water, and stained for 30 min with 0.5% sulforhodamine B in 1% acetic acid. Plates were washed three times with 1% acetic acid and dye was solubilized in 10 mm Tris pH 10.5 before measuring absorbance at 510 nm on an Epoch plate reader (BioTek). Relative growth was determined as (treatment absorbance/control absorbance) following 72 h of growth. Doubling time was determined by fitting an exponential growth equation to population sizes over 4 days.
Analysis of public data
Data from METABRIC and CCLE were accessed through cBioPortal (RRID:SCR_014555). Data were analyzed using MATLAB R2017b and Prism 7.
Statistics and reproducibility
Sample sizes, reproducibility, and statistical tests for each figure are denoted in the figure legends. All replicates are biological unless otherwise noted. All error bars represent S.E., and significance between conditions is denoted as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Author contributions
R. C. G., R. A. C., and A. T. conceptualization; R. C. G. resources; R. C. G. data curation; R. C. G. software; R. C. G., R. A. C., and A. T. formal analysis; R. C. G., R. A. C., and A. T. supervision; R. C. G., R. A. C., and A. T. funding acquisition; R. C. G. validation; R. C. G., J. R. F., T. M. S., J. M. A., and A. T. investigation; R. C. G., J. R. F., T. M. S., J. M. A., and A. T. methodology; R. C. G., T. M. S., R. A. C., and A. T. writing-original draft; R. C. G., R. A. C., and A. T. project administration; R. C. G. and A. T. writing-review and editing.
Supplementary Material
Acknowledgments
We thank B. Manning (Harvard School of Public Health) for suggestions and critical reagents; J. Brugge (Harvard Medical School), N. Kalaany (Boston Children's Hospital), and members of the Toker lab (Beth Israel Deaconess Medical Center) for discussion; M. Yuan and C. Dibble (Beth Israel Deaconess Medical Center) for assistance with MS. The BIDMC Research Capital Fund provided funds for the mass spectrometry instrumentation (QTRAP 5500 and 6500).
This work was supported in part by National Institutes of Health Grants 1R01CA200671 (to A. T.); RO1CA204345 and RO1CA235863 (to R. A. C.); and 5P01CA120964, 5R35CA197459, and 5P30CA006516 (to J. M. A.) and the Ludwig Center at Harvard (A. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Tables S1–S3.
All data presented and discussed are contained within the manuscript.
- TNBC
- triple-negative breast cancer
- AC-T
- Adriamycin (doxorubicin), cyclophosphamide, paclitaxel (Taxol)
- ANOVA
- analysis of variance
- AZI
- antizyme inhibitor (protein)
- DFMO
- α-difluoromethylornithine
- ER
- estrogen receptor
- ODC
- ornithine decarboxylase
- SAM
- S-adenosylmethionine
- SRM
- selected reaction monitoring.
References
- 1. Vander Heiden M. G., and DeBerardinis R. J. (2017) Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeBerardinis R. J., and Chandel N. S. (2016) Fundamentals of cancer metabolism. Science Adv. 2, e1600200 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boyle P. (2012) Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 23, Suppl. 6, vi7–vi12 10.1093/annonc/mds187 [DOI] [PubMed] [Google Scholar]
- 4. Denkert C., Liedtke C., Tutt A., and von Minckwitz G. (2017) Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. Lancet 389, 2430–2442 10.1016/S0140-6736(16)32454-0 [DOI] [PubMed] [Google Scholar]
- 5. Waks A. G., and Winer E. P. (2019) Breast cancer treatment: A review. JAMA 321, 288–300 10.1001/jama.2018.19323 [DOI] [PubMed] [Google Scholar]
- 6. Brown K. K., Spinelli J. B., Asara J. M., and Toker A. (2017) Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 7, 391–399 10.1158/2159-8290.CD-16-0611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lien E. C., Lyssiotis C. A., Juvekar A., Hu H., Asara J. M., Cantley L. C., and Toker A. (2016) Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 18, 572–578 10.1038/ncb3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gross M. I., Demo S. D., Dennison J. B., Chen L., Chernov-Rogan T., Goyal B., Janes J. R., Laidig G. J., Lewis E. R., Li J., Mackinnon A. L., Parlati F., Rodriguez M. L. M., Shwonek P. J., Sjogren E. B., et al. (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 10.1158/1535-7163.MCT-13-0870 [DOI] [PubMed] [Google Scholar]
- 9. Szefel J., Danielak A., and Kruszewski W. J. (2019) Metabolic pathways of l-arginine and therapeutic consequences in tumors. Adv. Med. Sci. 64, 104–110 10.1016/j.advms.2018.08.018 [DOI] [PubMed] [Google Scholar]
- 10. Morris S. M. (2009) Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 157, 922–930 10.1111/j.1476-5381.2009.00278.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Werner A., Amann E., Schnitzius V., Habermeier A., Luckner-Minden C., Leuchtner N., Rupp J., Closs E. I., and Munder M. (2016) Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur. J. Immunol. 46, 92–103 10.1002/eji.201546047 [DOI] [PubMed] [Google Scholar]
- 12. Hoechst B., Voigtlaender T., Ormandy L., Gamrekelashvili J., Zhao F., Wedemeyer H., Lehner F., Manns M. P., Greten T. F., and Korangy F. (2009) Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50, 799–807 10.1002/hep.23054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jahani M., Noroznezhad F., and Mansouri K. (2018) Arginine: Challenges and opportunities of this two-faced molecule in cancer therapy. Biomed. Pharmacother. 102, 594–601 10.1016/j.biopha.2018.02.109 [DOI] [PubMed] [Google Scholar]
- 14. Qiu F., Huang J., and Sui M. (2015) Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett. 364, 1–7 10.1016/j.canlet.2015.04.020 [DOI] [PubMed] [Google Scholar]
- 15. Dillon B. J., Prieto V. G., Curley S. A., Ensor C. M., Holtsberg F. W., Bomalaski J. S., and Clark M. A. (2004) Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer 100, 826–833 10.1002/cncr.20057 [DOI] [PubMed] [Google Scholar]
- 16. Agostinelli E., Marques M. P. M., Calheiros R., Gil F. P. S. C., Tempera G., Viceconte N., Battaglia V., Grancara S., and Toninello A. (2010) Polyamines: Fundamental characters in chemistry and biology. Amino Acids 38, 393–403 10.1007/s00726-009-0396-7 [DOI] [PubMed] [Google Scholar]
- 17. Murray-Stewart T. R., Woster P. M., and Casero R. A. Jr. (2016) Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 473, 2937–2953 10.1042/BCJ20160383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Battaglia V., DeStefano Shields C., Murray-Stewart T., and Casero R. A. Jr. (2014) Polyamine catabolism in carcinogenesis: Potential targets for chemotherapy and chemoprevention. Amino Acids 46, 511–519 10.1007/s00726-013-1529-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nowotarski S. L., Woster P. M., and Casero R. A. Jr. (2013) Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 15, e3 10.1017/erm.2013.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez-Leal O., and Merali S. (2012) Regulation of polyamine metabolism by translational control. Amino Acids 42, 611–617 10.1007/s00726-011-1036-6 [DOI] [PubMed] [Google Scholar]
- 21. Murray Stewart T., Dunston T. T., Woster P. M., and Casero R. A. Jr. (2018) Polyamine catabolism and oxidative damage. J. Biol. Chem. 293, 18736–18745 10.1074/jbc.TM118.003337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burns M. R., Graminski G. F., Weeks R. S., Chen Y., and O'Brien T. G. (2009) Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a polyamine biosynthesis inhibitor. J. Med. Chem. 52, 1983–1993 10.1021/jm801580w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayes C. S., Shicora A. C., Keough M. P., Snook A. E., Burns M. R., and Gilmour S. K. (2014) Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2, 274–285 10.1158/2326-6066.CIR-13-0120-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casero R. A. Jr., Murray Stewart T., and Pegg A. E. (2018) Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 18, 681–695 10.1038/s41568-018-0050-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lehmann B. D., Bauer J. A., Chen X., Sanders M. E., Chakravarthy A. B., Shyr Y., and Pietenpol J. A. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lebert J. M., Lester R., Powell E., Seal M., and McCarthy J. (2018) Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 25, S142–S150 10.3747/co.25.3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao H., He G., Yan S., Chen C., Song L., Rosol T. J., and Deng X. (2017) Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 8, 1913–1924 10.18632/oncotarget.12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar P., and Aggarwal R. (2016) An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 293, 247–269 10.1007/s00404-015-3859-y [DOI] [PubMed] [Google Scholar]
- 29. Silver D. P., Richardson A. L., Eklund A. C., Wang Z. C., Szallasi Z., Li Q., Juul N., Leong C. O., Calogrias D., Buraimoh A., Fatima A., Gelman R. S., Ryan P. D., Tung N. M., De Nicolo A., et al. (2010) Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 28, 1145–1153 10.1200/JCO.2009.22.4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roci I., Watrous J. D., Lagerborg K. A., Lafranchi L., Lindqvist A., Jain M., and Nilsson R. (2019) Mapping metabolic events in the cancer cell cycle reveals arginine catabolism in the committed SG2M phase. Cell Rep. 26, 1691–1700.e5 10.1016/j.celrep.2019.01.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bachmann A. S., and Geerts D. (2018) Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem. 293, 18757–18769 10.1074/jbc.TM118.003336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kahana C. (2018) The antizyme family for regulating polyamines. J. Biol. Chem. 293, 18730–18735 10.1074/jbc.TM118.003339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oredsson S. M. (2003) Polyamine dependence of normal cell-cycle progression. Biochem. Soc. Trans. 31, 366–370 10.1042/bst0310366 [DOI] [PubMed] [Google Scholar]
- 34. Weicht R. R., Schultz C. R., Geerts D., Uhl K. L., and Bachmann A. S. (2018) Polyamine biosynthetic pathway as a drug target for osteosarcoma therapy. Med. Sci. 6, E65 10.3390/medsci6030065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He W., Roh E., Yao K., Liu K., Meng X., Liu F., Wang P., Bode A. M., and Dong Z. (2017) Targeting ornithine decarboxylase (ODC) inhibits esophageal squamous cell carcinoma progression. NPJ Precis. Oncol. 1, 13 10.1038/s41698-017-0014-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anehus S., Pohjanpelto P., Baldetorp B., Långström E., and Heby O. (1984) Polyamine starvation prolongs the S and G2 phases of polyamine-dependent (arginase-deficient) CHO cells. Mol. Cell Biol. 4, 915–922 10.1128/MCB.4.5.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mueller S., Schittenhelm M., Honecker F., Malenke E., Lauber K., Wesselborg S., Hartmann J. T., Bokemeyer C., and Mayer F. (2006) Cell-cycle progression and response of germ cell tumors to cisplatin in vitro. Int. J. Oncol. 29, 471–479 [PubMed] [Google Scholar]
- 38. Potter A. J., Gollahon K. A., Palanca B. J., Harbert M. J., Choi Y. M., Moskovitz A. H., Potter J. D., and Rabinovitch P. S. (2002) Flow cytometric analysis of the cell cycle phase specificity of DNA damage induced by radiation, hydrogen peroxide and doxorubicin. Carcinogenesis 23, 389–401 10.1093/carcin/23.3.389 [DOI] [PubMed] [Google Scholar]
- 39. Ling Y. H., el-Naggar A. K., Priebe W., and Perez-Soler R. (1996) Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol. Pharmacol. 49, 832–841 [PubMed] [Google Scholar]
- 40. Witherspoon M., Chen Q., Kopelovich L., Gross S. S., and Lipkin S. M. (2013) Unbiased metabolite profiling indicates that a diminished thymidine pool is the underlying mechanism of colon cancer chemoprevention by alpha-difluoromethylornithine. Cancer Discov. 3, 1072–1081 10.1158/2159-8290.CD-12-0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh R., Pervin S., Wu G., and Chaudhuri G. (2001) Activation of caspase-3 activity and apoptosis in MDA-MB-468 cells by Nω-hydroxy-l-arginine, an inhibitor of arginase, is not solely dependent on reduction in intracellular polyamines. Carcinogenesis 22, 1863–1869 10.1093/carcin/22.11.1863 [DOI] [PubMed] [Google Scholar]
- 42. Singh R., Pervin S., Karimi A., Cederbaum S., and Chaudhuri G. (2000) Arginase activity in human breast cancer cell lines: Nω-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 60, 3305–3312 [PubMed] [Google Scholar]
- 43. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pereira B., Chin S. F., Rueda O. M., Vollan H. K., Provenzano E., Bardwell H. A., Pugh M., Jones L., Russell R., Sammut S. J., Tsui D. W., Liu B., Dawson S. J., Abraham J., Northen H., et al. (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 10.1038/ncomms11479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayer I. A., Abramson V. G., Lehmann B. D., and Pietenpol J. A. (2014) New strategies for triple-negative breast cancer—deciphering the heterogeneity. Clin. Cancer Res. 20, 782–790 10.1158/1078-0432.CCR-13-0583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Keren-Paz A., Bercovich Z., and Kahana C. (2007) Antizyme inhibitor: A defective ornithine decarboxylase or a physiological regulator of polyamine biosynthesis and cellular proliferation. Biochem. Soc. Trans. 35, 311–313 10.1042/BST0350311 [DOI] [PubMed] [Google Scholar]
- 48. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thomas T., Kiang D. T., Jänne O. A., and Thomas T. J. (1991) Variations in amplification and expression of the ornithine decarboxylase gene in human breast cancer cells. Breast Cancer Res. Treat. 19, 257–267 10.1007/BF01961162 [DOI] [PubMed] [Google Scholar]
- 50. Lee A., and Djamgoz M. B. A. (2018) Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 62, 110–122 10.1016/j.ctrv.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 51. Bianchini G., Balko J. M., Mayer I. A., Sanders M. E., and Gianni L. (2016) Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 10.1038/nrclinonc.2016.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bello-Fernandez C., Packham G., and Cleveland J. L. (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 90, 7804–7808 10.1073/pnas.90.16.7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Casero R. A. Jr., and Marton L. J. (2007) Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 6, 373–390 10.1038/nrd2243 [DOI] [PubMed] [Google Scholar]
- 54. Dulloo I., Gopalan G., Melino G., and Sabapathy K. (2010) The antiapoptotic DeltaNp73 is degraded in a c-Jun-dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 4902–4907 10.1073/pnas.0906782107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nayvelt I., Hyvönen M. T., Alhonen L., Pandya I., Thomas T., Khomutov A. R., Vepsäläinen J., Patel R., Keinänen T. A., and Thomas T. J. (2010) DNA condensation by chiral α-methylated polyamine analogues and protection of cellular DNA from oxidative damage. Biomacromolecules 11, 97–105 10.1021/bm900958c [DOI] [PubMed] [Google Scholar]
- 56. Zahedi K., Bissler J. J., Wang Z., Josyula A., Lu L., Diegelman P., Kisiel N., Porter C. W., and Soleimani M. (2007) Spermidine/spermine N1-acetyltransferase overexpression in kidney epithelial cells disrupts polyamine homeostasis, leads to DNA damage, and causes G2 arrest. Am. J. Physiol. Cell Physiol. 293, C1204–C1215 10.1152/ajpcell.00451.2006 [DOI] [PubMed] [Google Scholar]
- 57. Johansson V. M., Oredsson S. M., and Alm K. (2008) Polyamine depletion with two different polyamine analogues causes DNA damage in human breast cancer cell lines. DNA Cell Biol. 27, 511–516 10.1089/dna.2008.0750 [DOI] [PubMed] [Google Scholar]
- 58. Courdi A., Milano G., Bouclier M., and Lalanne C. M. (1986) Radiosensitization of human tumor cells by alpha-difluoromethylornithine. Int. J. Cancer 38, 103–107 10.1002/ijc.2910380117 [DOI] [PubMed] [Google Scholar]
- 59. Seidenfeld J., Block A. L., Komar K. A., and Naujokas M. F. (1986) Altered cell cycle phase distributions in cultured human carcinoma cells partially depleted of polyamines by treatment with difluoromethylornithine. Cancer Res. 46, 47–53 [PubMed] [Google Scholar]
- 60. Saunders L. R., and Verdin E. (2006) Ornithine decarboxylase activity in tumor cell lines correlates with sensitivity to cell death induced by histone deacetylase inhibitors. Mol. Cancer Ther. 5, 2777–2785 10.1158/1535-7163.MCT-06-0298 [DOI] [PubMed] [Google Scholar]
- 61. Yamashita T., Nishimura K., Saiki R., Okudaira H., Tome M., Higashi K., Nakamura M., Terui Y., Fujiwara K., Kashiwagi K., and Igarashi K. (2013) Role of polyamines at the G1/S boundary and G2/M phase of the cell cycle. Int. J. Biochem. Cell Biol. 45, 1042–1050 10.1016/j.biocel.2013.02.021 [DOI] [PubMed] [Google Scholar]
- 62. O'Shaughnessy J. A., Demers L. M., Jones S. E., Arseneau J., Khandelwal P., George T., Gersh R., Mauger D., and Manni A. (1999) Alpha-difluoromethylornithine as treatment for metastatic breast cancer patients. Clin. Cancer Res. 5, 3438–3444 [PubMed] [Google Scholar]
- 63. Fabian C. J., Kimler B. F., Brady D. A., Mayo M. S., Chang C. H., Ferraro J. A., Zalles C. M., Stanton A. L., Masood S., Grizzle W. E., Boyd N. F., Arneson D. W., and Johnson K. A. (2002) A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: Breast tissue, imaging, and serum and urine biomarkers. Clin. Cancer Res. 8, 3105–3117 [PubMed] [Google Scholar]
- 64. Yang D., Hayashi H., Takii T., Mizutani Y., Inukai Y., and Onozaki K. (1997) Interleukin-1-induced growth inhibition of human melanoma cells. Interleukin-1-induced antizyme expression is responsible for ornithine decarboxylase activity down-regulation. J. Biol. Chem. 272, 3376–3383 10.1074/jbc.272.6.3376 [DOI] [PubMed] [Google Scholar]
- 65. Young L., Salomon R., Au W., Allan C., Russell P., and Dong Q. (2006) Ornithine decarboxylase (ODC) expression pattern in human prostate tissues and ODC transgenic mice. J Histochem. Cytochem. 54, 223–229 10.1369/jhc.5A6672.2005 [DOI] [PubMed] [Google Scholar]
- 66. Hu H. Y., Liu X. X., Jiang C. Y., Lu Y., Liu S. L., Bian J. F., Wang X. M., Geng Z., Zhang Y., and Zhang B. (2005) Ornithine decarboxylase gene is overexpressed in colorectal carcinoma. World J. Gastroenterol. 11, 2244–2248 10.3748/wjg.v11.i15.2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fahrmann J. F., Vykoukal J., Fleury A., Tripathi S., Dennison J. B., Murage E., Wang P., Yu C. Y., Capello M., Creighton C. J., Do K. A., Long J. P., Irajizad E., Peterson C., Katayama H., Disis M. L., Arun B., and Hanash S. (2019) Association between plasma diacetylspermine and tumor spermine synthase with outcome in triple negative breast cancer. J. Natl. Cancer Inst. Sept. 10, djz182 10.1093/jnci/djz182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reznik E., Luna A., Aksoy B. A., Liu E. M., La K., Ostrovnaya I., Creighton C. J., Hakimi A. A., and Sander C. (2018) A landscape of metabolic variation across tumor types. Cell Syst. 6, 301–313 10.1016/j.cels.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuan M., Breitkopf S. B., Yang X., and Asara J. M. (2012) A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 7, 872–881 10.1038/nprot.2012.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yuan M., Kremer D. M., Huang H., Breitkopf S. B., Ben-Sahra I., Manning B. D., Lyssiotis C. A., and Asara J. M. (2019) Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat. Protoc. 14, 313–330 10.1038/s41596-018-0102-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D. S., and Xia J. (2018) MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kabra P. M., Lee H. K., Lubich W. P., and Marton L. J. (1986) Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: Improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. J. Chromatogr. 380, 19–32 10.1016/S0378-4347(00)83621-X [DOI] [PubMed] [Google Scholar]
- 73. Seely J. E., and Pegg A. E. (1983) Ornithine decarboxylase (mouse kidney). Methods Enzymol. 94, 158–161 10.1016/S0076-6879(83)94025-9 [DOI] [PubMed] [Google Scholar]
- 74. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B. C., Remm M., and Rozen S. G. (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Do T. H., Gaboriau F., Morel I., Lepage S., Cannie I., Loréal O., and Lescoat G. (2013) Modulation of ethanol effect on hepatocyte proliferation by polyamines. Amino Acids 44, 869–877 10.1007/s00726-012-1413-9 [DOI] [PubMed] [Google Scholar]
- 76. Zhang L., Mizumoto K., Sato N., Ogawa T., Kusumoto M., Niiyama H., and Tanaka M. (1999) Quantitative determination of apoptotic death in cultured human pancreatic cancer cells by propidium iodide and digitonin. Cancer Lett. 142, 129–137 10.1016/S0304-3835(99)00107-X [DOI] [PubMed] [Google Scholar]
- 77. Vichai V., and Kirtikara K. (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116 10.1038/nprot.2006.179 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.