

Abstract
The cardiac action potential is critical to the production of a synchronized heartbeat. This electrical impulse is governed by the intricate activity of cardiac ion channels, among them the cardiac voltage-gated potassium (Kv) channels KCNQ1 and hERG as well as the voltage-gated sodium (Nav) channel encoded by SCN5A. Each channel performs a highly distinct function, despite sharing a common topology and structural components. These three channels are also the primary proteins mutated in congenital long QT syndrome (LQTS), a genetic condition that predisposes to cardiac arrhythmia and sudden cardiac death due to impaired repolarization of the action potential and has a particular proclivity for reentrant ventricular arrhythmias. Recent cryo-electron microscopy structures of human KCNQ1 and hERG, along with the rat homolog of SCN5A and other mammalian sodium channels, provide atomic-level insight into the structure and function of these proteins that advance our understanding of their distinct functions in the cardiac action potential, as well as the molecular basis of LQTS. In this review, the gating, regulation, LQTS mechanisms, and pharmacological properties of KCNQ1, hERG, and SCN5A are discussed in light of these recent structural findings.
Keywords: cardiac action potential, long QT syndrome, KCNQ1, hERG, SCN5A, structural biology
Introduction
The cardiac action potential is critical to proper heart function. Beginning with the activation of “pacemaker” cells, the action potential propagates through the atria and into the ventricles in a unidirectional waveform of excitation and relaxation, resulting in the coordinated expansion and contraction of heart tissue (Nerbonne and Kass, 2005). The action potential is governed by an intricate series of ion channel activities (Grant, 2009), including those of the KCNQ1 (KVLQT1, KV7.1) and hERG (KCNH2, KV11.1) potassium channels and the SCN5A (NaV1.5) sodium channel. Mutations in these three channels are the most frequent cause of congenital long QT syndrome (LQTS), a cardiac arrhythmia disorder that is one of the primary causes of sudden arrhythmic death syndrome (SADS) (Skinner et al., 2019).
KCNQ1, hERG, and SCN5A each play a distinct role in generating the cardiac action potential (Figure 1), consequently producing distinct LQTS forms when mutated (Skinner et al., 2019). The initial upstroke is governed primarily by SCN5A, producing the INa current that amplifies membrane depolarization and propagates the action potential (Moss and Kass, 2005; Skinner et al., 2019). Mutation in SCN5A causes LQTS type 3 (LQT3). hERG shapes both the plateau and the repolarization phases of the action potential, along with KCNQ1 in complex with the KCNE1 accessory protein (Moss and Kass, 2005; George, 2013). hERG and the KCNQ1–KCNE1 complex produce the rapid (IKr) and slow (IKs) delayed rectifier currents (Figures 1B, C), respectively. KCNQ1 mutation causes LQT1, and hERG, LQT2 (Skinner et al., 2019). Additional ion channels and transporters also shape the cardiac action potential (Skinner et al., 2019), among them Cav1.2, NCX1, and Kir2.1. The roles of these channels in the cardiac action potential and cardiac arrhythmias have been reviewed elsewhere (Bidaud and Lory, 2011; Hancox et al., 2018; Meza et al., 2018; Vaidyanathan et al., 2018; Li et al., 2019). These channels are not typically causative for LQTS and will not be discussed here.
Figure 1.
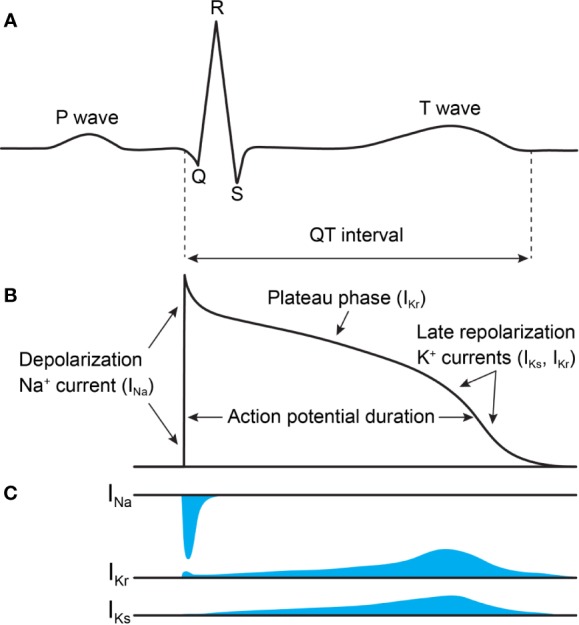
Voltage-activated Na+ and K+ currents define the ventricular action potential and QT interval of the ECG. (A) ECG trace. The rapid upstroke of the ventricular action potential gives rise to the QRS complex. The duration of the QT interval is determined by the time of the ventricular repolarization. (B) Trace of the ventricular action potential. The rapidly activating and inactivating INa current drives membrane depolarization. Two K+ currents, IKs and IKr, contribute most to the plateau phase and repolarization phase of the action potential, which reestablishes the membrane resting potential. (C) Time course of INa, IKr, and IKs currents (not drawn to scale). Currents of other ion channels contributing to the action potential (e.g. ICa,L, IKur, and INCX) are not shown for clarity. This figure is inspired by figures in Moss and Kass (2005) and George (2013).
Structurally, the KCNQ1, hERG, and SCN5A channels belong to the voltage-gated ion channel superfamily and share a general transmembrane topology (Bezanilla, 2005; Dehghani-Samani et al., 2019). The transmembrane channel domain is composed of six helices per subunit in hERG and KCNQ1 (Figures 2A, B) or a monomeric tetrad repeat of linked 6-helix domains for SCN5A, each repeat exhibiting varying sequences, lengths, and tertiary folds (Figures 2C, D). The assembled channel is tetrameric (pseudo-tetrameric for SCN5A), with each channel domain composed of four voltage sensing domains (VSDs) surrounding a central pore domain (PD) (Figure 2D). The VSD is comprised of the first four transmembrane helices (S1–S4) preceded by a small amphipathic helix—S0—in both KCNQ1 and SCN5A (Jiang et al., 2020; Sun and Mackinnon, 2020). The PD is formed by the tetramerization of S5 and S6 helices from each subunit/repeat (Figure 2D). A pore loop between S5 and S6 contains the selectivity filter (SF) that confers ion specificity. A linker helix between helices S4 and S5, termed the S4–S5 linker, connects the VSD to the PD (Bezanilla, 2005; Dehghani-Samani et al., 2019) (Figures 2A–C).
Figure 2.

Overall topology of KCNQ1, hERG, and SCN5A channels. (A) KCNQ1 topology. Transmembrane domain alpha helices are labeled S0–S6. PH indicates the pore helix. SF denotes the selectivity filter. S2–S3 indicates the S2–S3 linker. S4–S5 denotes the S4–S5 linker. Cytosolic alpha helices are labeled HA-HD. (B) hERG topology. PAS denotes the Per-ARNT-Sim domain. CNBHD indicates the C-terminal cyclic nucleotide-binding homology domain. (C) SCN5A topology. CTD indicates the C-terminal domain. (D) Top views of the human KCNQ1 (PDB ID: 6UZZ) (Sun and Mackinnon, 2020), human hERG (PDB ID: 5VA1) (Wang and Mackinnon, 2017), and rat SCN5A (PDB ID: 6UZ3) (Jiang et al., 2020) channels, respectively. Outer voltage-sensing domains (VSD) and the central pore domain (PD) are labeled.
Given these similarities in channel topology and components, how is it that KCNQ1, hERG, and SCN5A perform such distinct functions, and produce phenotypically distinct forms of LQTS? To explore this question, a number of high-resolution ion channel structures, including cryo-electron microscopy (cryo-EM) structures of frog and human KCNQ1 (Sun and Mackinnon, 2020) and of human hERG (Wang and Mackinnon, 2017), as well as the structures of the rat homolog of SCN5A (Jiang et al., 2020) and human Nav1 isoforms Nav1.4 and 1.7 (Pan et al., 2018; Shen et al., 2019; Xu et al., 2019), have been determined. Moreover, the frog KCNQ1 structure has been used to develop what is likely a reliable homology model for the human KCNQ1 channel in resting and fully active conformations (Kuenze et al., 2019). A homology model of human SCN5A in the resting state has also been devised (Kroncke et al., 2019). These structures and structural models reveal critical differences in the atomic details of KCNQ1, hERG, and SCN5A structures associated with their distinct functions and disease phenotypes. Notably, the subunits of KCNQ1 undergo domain swapping, with a similar arrangement observed in SCN5A but not in the hERG channel (Figure 2D). The monomeric sequence of SCN5A causes the channel to adopt an asymmetric three-dimensional fold, in contrast to the inherent symmetry of tetrameric hERG and KCNQ1 (Figure 2D). Additionally, the C-terminal domains contain distinct folds and mediate unique regulatory functions. These and other structural differences contribute to the varying properties of these three channels and to their distinct roles in the cardiac action potential.
The aim of this review is to compare and contrast the KCNQ1, hERG, and SCN5A channels using available structures and structural models as a guide. Through this lens, channel gating, regulation, LQTS mechanisms, and pharmacology will be discussed, in order to explore the molecular basis of these unique properties.
Structural Mechanisms of Channel Gating
KCNQ1, hERG, and SCN5A undergo conformational changes in response to changes in membrane potential that result in channel opening or closing. These responses confer specific gating properties including activation, deactivation, inactivation, and recovery from inactivation (Hosseini, 2018; Zhang et al., 2018). In activation, protein conformational changes result in channel pore opening from a resting state, while deactivation entails a return to the resting state (Zhang et al., 2018). Inactivation confers a third channel state distinct from the activated and resting states which inhibits current flow prior to full deactivation (Zhang et al., 2018). While all three channels share common structural elements that are responsible for producing these states, there are also elements that give rise to specific gating properties in each channel, as discussed below.
Activation and Ion Conduction
KCNQ1
Voltage-gated channels contain up to six positively-charged basic residues in the S4 helix, called gating charges, which move in the electric field of the membrane in response to voltage (Jiang et al., 2003). Gating charges are numbered according to their position in the S4 helix, from the extracellular to the intracellular side. In KCNQ1, S4 contains four arginine (R) gating charges that are conserved in other KV channels and confer voltage sensitivity. However, in KCNQ1 the canonical R3 is replaced by a neutral glutamine (Q234, Q3), and the fifth gating charge (K5 in Shaker) is replaced by histidine (H241, H5), which in the membrane environment is expected to be neutral at physiological pH (Figures 3A, B). Due to these substitutions at positions 3 and 5, the S4 helix of KCNQ1 has a lower net positive charge (+4) than Shaker-class K+ channels, such as KV1.2 (+6). The lower net positive charge may explain why KCNQ1 S4 mutations that result charge loss or reversal (Panaghie and Abbott, 2007; Wu et al., 2010b) result in constitutive channel activity.
Figure 3.
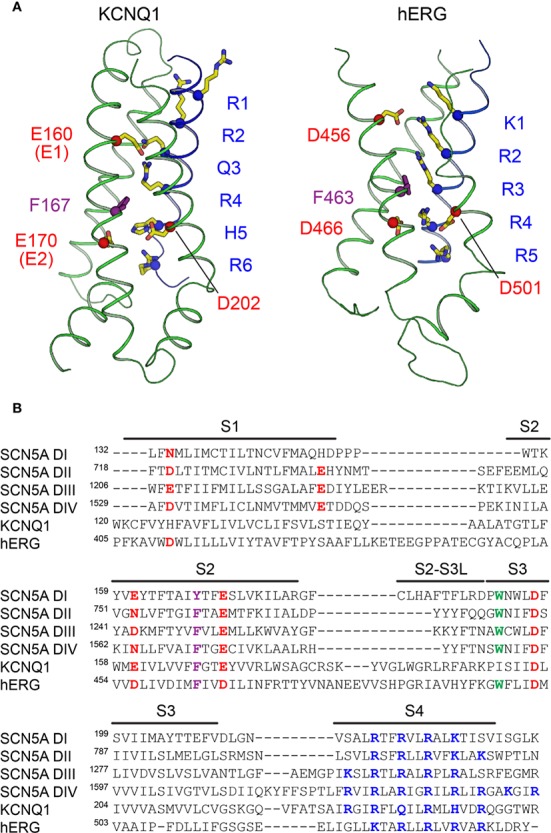
The voltage sensors of KCNQ1, hERG, and SCN5A. (A) Structure of the VSD from human KCNQ1 (left) (PDB: 6UZZ) (Sun and Mackinnon, 2020) and hERG (right) (PDB: 5VA2) (Wang and Mackinnon, 2017) in putative activated conformations. Basic residues on S4 (labeled in blue), acidic residues on S2 and S3 (labeled in red), and the phenylalanine residue in the gating charge transfer center (purple) are shown as sticks. The first four basic residues on S4 (R1–R4) in KCNQ1 and the first three (K1–R3) in hERG are located above the charge transfer center. (B) Multiple sequence alignment of VSDs I-IV of SCN5A with the VSDs of human KCNQ1 and hERG. Basic residues in S4 implicated with voltage sensing are colored blue, and acidic or polar residues in S1–S3 suggested to interact with S4 gating charges are colored red. The conserved aromatic residue at the gating charge transfer center in S2 is colored purple. Another tryptophan residue in S3, conserved in the VSDs of NaV channels and also present in hERG, is colored green.
Voltage-gated channels also feature a charge transfer center (CTC), formed by a bulky aromatic ring and two negatively charged residues, that facilitates S4 movement (Tao et al., 2010). The KCNQ1 CTC consists of E170 (E2) and F167 on S2, and D202 on S3, which work with E160 (E1) to define the S4 position. During activation, S4 moves towards the extracellular side of the membrane (Nakajo and Kubo, 2007; Rocheleau and Kobertz, 2008; Osteen et al., 2010; Ruscic et al., 2013; Barro-Soria et al., 2014; Nakajo and Kubo, 2014; Barro-Soria et al., 2017) through interactions between basic gating charges and E1 and E2 in the CTC (Figure 3A). These interactions change during the course of activation, permitting S4 translocation. E1 interacts with R1 (R228) or R4 (R237) in the resting and activated states of the VSD, respectively (Wu et al., 2010a). S4 motion occurs in two distinct steps, transitioning through a stable intermediate before reaching the activated state (Wu et al., 2010a; Barro-Soria et al., 2014; Zaydman et al., 2014). The intermediate state features salt bridge interactions between E1 and R2, distinct from the resting and activated VSD states, according to the recently-determined structure of the intermediate state KCNQ1 VSD (Taylor et al., 2020). Interestingly, the pore of KCNQ1 opens in both the intermediate state (IO) and fully activated (AO) states (Zaydman et al., 2014). These two open states possess distinct channel properties with differing opening probabilities and pharmacology (Hou et al., 2017), and have distinct pore structures (Zaydman et al., 2014). Importantly, ion conductance when the VSD is in either the intermediate or activated state appears to be unique to KCNQ1. However, formation of the KCNQ1–KCNE1 complex eliminates the conductance associated with the VSD intermediate state (Zaydman et al., 2014), such that IKs reflects only the fully activated state.
The ion conduction pathway in KCNQ1 is lined by the four S6 helices, with the SF on the extracellular side of the pore. Mutations in S6 cause changes in current amplitude and voltage dependence of activation (Wang et al., 1999; Seebohm et al., 2005; Panaghie et al., 2006; Hoosien et al., 2013). Comparison of S6 in the closed pore structure of human KCNQ1 (Sun and Mackinnon, 2020) with that of the open channel demonstrates that channel opening results from bending of S6 so that the cytosolic ends of the four S6 segments swing away from the central axis, enlarging the diameter of the pore to allow diffusion of K+ into the central cavity. The hinge responsible for this bending motion in S6 is the P343–A344–G345 (PAG) motif, which corresponds to PVP in Shaker K+ channels (Labro and Snyders, 2012). Additionally, A336 may also be important in the motion of the activation gate, as mutations at this position alter the voltage dependence of activation (Seebohm et al., 2006).
Once the intracellular gate is opened, K+ ions move through the pore along their electrochemical gradient. The backbone carbonyl oxygens of the TIGYG motif in the SF of KCNQ1 (TVGYG in KV1.2 and KcsA) (Figure 4A) and the sidechain of T312 form four evenly spaced K+ binding sites (Figure 4B) that facilitate K+ movement (Zhou et al., 2001). The arrangement of these oxygens mimics the displaced hydration shell of K+, which lowers the transfer energy from the aqueous cavity at the center of the channel to the SF, allowing conduction to occur at rates near the diffusion limit (Morais-Cabral et al., 2001; Zhou et al., 2001).
Figure 4.

The selectivity filters of K+ and Na+ channels. (A) Multiple sequence alignment of the SF region of selected K+ (left) and Na+ (right) channels. Conserved amino acids are highlighted in bold. Amino acids belonging to the DEKA signature motif in eukaryotic Na+ channels are colored red. (B) Side and top view of the SF of human KCNQ1 (PDB: 6UZZ) (Sun and Mackinnon, 2020). Two subunits are omitted for clarity in the left plot. (C) Side and top view of the SF of SCN5A (PDB: 6UZ3) (Jiang et al., 2020). Repeat II is omitted for clarity in the left plot.
hERG
The cryo-EM structure of hERG shows an open pore and activated VSDs, with the first three gating charges (K1–R3) of S4 located on the extracellular side of the CTC (Figure 3A). This is a translocation of one charge fewer than in the activated VSD of KCNQ1 (Sun and Mackinnon, 2017) and Shaker-like KV1.2–2.1 (Long et al., 2007) where four gating charges are located above the conserved phenylalanine in the CTC. This observation agrees with gating current measurements suggesting a total charge movement of only ~6 elementary charge units (movement of 1.5 positive charges per S4 helix) for hERG during activation (Zhang et al., 2004), compared to 8 to 9 (2 positive charges per helix) for KCNQ1 (Ruscic et al., 2013) and 12 to 16 (3–4 positive charges per helix) for Shaker-like KV channels (Schoppa et al., 1992; Aggarwal and Mackinnon, 1996; Seoh et al., 1996). However, the ca. 50% lower total gating charge movement for hERG relative to Shaker-like channels is not explained by differences in the number of S4 gating charges, since hERG has a total of five positively-charged residues on S4 and Shaker has six (Pless et al., 2011b). These combined structural and functional data thus imply that S4 translocates less in hERG during activation, resulting in smaller overall VSD conformational changes.
However, the activated VSD of hERG may not be fully defined. While the position of K1–R3 above the CTC in the cryo-EM structure of hERG is consistent with a depolarized VSD, some salt bridge interactions in the VSD have suboptimal geometry, particularly with R4. This is likely due to the limited resolution of the VSD in the final map (approximately 4.5–5.5 Å) (Wang and Mackinnon, 2017), impeding unambiguous determination of sidechain conformations. While cryo-EM has proven to be a powerful structural tool, the resolution is often lower in the periphery of protein structures (Herzik et al., 2019). This can prevent accurate modeling of functional features and lead to discrepancies with experimental data. Molecular dynamics may be a be useful tool in refining cryo-EM structures to mitigate these discrepancies, as is currently being carried out for the hERG structure (Khan et al., 2020).
While a structure of hERG with a closed PD has not yet been determined, we can gain insight into the conformational changes that occur during pore opening using the closed-pore structure of the closely related rat potassium voltage-gated subfamily H member 1 channel (EAG1 or KCNH1) (Whicher and Mackinnon, 2016). In hERG, the intracellular gate is likely constricted by the Q664 side chains in S6, since the radius of the cavity at Q664 is almost 6 Å in the open state hERG structure (Wang and Mackinnon, 2017), while at the corresponding position (Q476) in the EAG1 closed state structure, the pore is at its narrowest (< 1 Å) (Whicher and Mackinnon, 2016). Bending and displacement of the S6 helices is suggested by a glycine residue, located at the same position in both channels, acting as a gating hinge (G648 in hERG, G460 in EAG1).
The SF in hERG is unique among Kv channels, containing a GFG motif (Figure 4A) in place of the typical GYG motif (Long et al., 2005). The position of the phenylalanine residue in this motif is different from the corresponding tyrosine in other Kv channels of known structure (Wang and Mackinnon, 2017). This structural variation may have important implications for fast inactivation in hERG, as discussed below.
SCN5A
The mechanism of voltage sensing in NaV channels is thought to be similar to that of KV channels. The S4 helix is the key sensor of transmembrane voltage. Pairing of the positively-charged residues in S4 with polar or negatively-charged residues catalyzes S4 movement from its inward resting-state position to the outward activated state upon membrane depolarization. Recent cryo-EM structures of NaV channels with VSDs in activated (Yan et al., 2017b; Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020) and resting (Clairfeuille et al., 2019; Wisedchaisri et al., 2019; Xu et al., 2019) conformations uncover a remarkable 10 to 15 Å translation of S4 across the membrane, fully consistent with the “sliding helix” model of VSD activation (Catterall, 1986). A conserved aromatic residue (tyrosine in repeat I, phenylalanine II, III, and IV) on S2 serves as the hydrophobic plug that constricts the S4 gating canal and prevents ion leak through the VSD (Jiang et al., 2020). This hydrophobic plug is mechanistically identical to the corresponding residues in the CTC of KV channels (F167 in KCNQ1, F463 in hERG). However, in contrast to the VSD of KCNQ1 and hERG, the number of basic residues on S4 in SCN5A varies from four (repeat I) to six (repeat IV) (Jiang et al., 2020) (Figure 3B). The variation in the number of gating charges and the heterogenous distribution of acidic and polar residues on S1–S3 between KCNQ1, hERG, and SCN5A (Figure 3B) may be responsible for their distinct voltage sensitivities and kinetics of VSD activation. In NaV channels, the VSDs of repeats I, II, and III are mainly responsible for channel activation and pore opening, while the VSD of repeat IV is responsible for initiating and maintaining fast inactivation (Chanda and Bezanilla, 2002; Capes et al., 2013; Clairfeuille et al., 2019). This activation process has been studied in detail for the human skeletal muscle channel NaV1.4, giving rise to the “asynchronous gating model” (Chanda and Bezanilla, 2002; Capes et al., 2013; Goldschen-Ohm et al., 2013) wherein the S4 segments of repeats I, II, and III move quickly, permitting conductance before activation of VSDIV, while S4 movement in repeat IV is slower and represents the rate-limiting step for development of and recovery from inactivation.
The structure and function of the selectivity filter in SCN5A and other NaV channels differs fundamentally from that of KV channels such as KCNQ1 and hERG (Figure 4). The SF gate in NaV channels is wider to allow Na+ ions to pass in a partially hydrated state (Hille, 1971; Naylor et al., 2016). The extracellular vestibule of NaV channels is lined by negatively-charged residues that recruit Na+ ions to the SF. Coordination by both sidechain and backbone carbonyls contribute to the Na+ permeation mechanism (Chakrabarti et al., 2013; Ulmschneider et al., 2013; Naylor et al., 2016). Additionally, the SF of eukaryotic NaV channels is formed in a pseudo-symmetric fashion by four short helix-connecting turn motifs from each subunit (Figure 4A). Four distinct residues–DEKA, one in each repeat-form the signature motif for Na+ selectivity found in all human NaV channel pore-forming repeats (Figure 4C). The lysine residue in the DEKA motif (K1419 in SCN5A) confers selectivity for Na+ and prevents permeability of Ca2+ (Favre et al., 1996). The recent rat SCN5A structure suggests a mechanism by which K1419 contributes to this selectivity of Na+ over Ca2+, whrein lysine forms a charge delocalization network at a constriction point in the SF. Only Na+ ions, which have a compatible size and electric field strength, are able to pass through (Jiang et al., 2020).
Electromechanical Coupling
KCNQ1
In the absence of accessory subunits, KCNQ1 exhibits a constitutive current reflecting close-to-open state transitions even at very negative (~−120 mV) voltages (Ma et al., 2011). Analysis of a large group of KCNQ1 mutants suggests that gating follows an allosteric model (Ma et al., 2011). According to this model, the pore can open independently of the state of the VSD, but VSD activation increases the probability of pore opening. Furthermore, KCNQ1 opening does not require concerted VSD movements (Osteen et al., 2012). Indeed, the VSDs appear to move independently, and pore opening can occur before all VSDs are activated (Osteen et al., 2012), consistent with an allosteric gating model.
Allosteric coupling for KCNQ1 is thought to be mediated by interactions between the VSD and the PD that translate S4 movement to channel opening and closing (Figure 5). Studies of KCNQ1 (Boulet et al., 2007; Labro et al., 2011) and of other KV channels (Lu et al., 2001; Lu et al., 2002; Long et al., 2005) have pointed to the interface between the S4–S5 linker and the C-terminal end of S6 (S6C) as one important mediator of electromechanical coupling (Figure 5A). Certain mutations in the S4–S5 linker (Labro et al., 2011) and S6C (Boulet et al., 2007) slow the opening rate and shift channel activation to more depolarized voltages, while other mutations, specifically at V254 in the S4–S5 linker and at L353 in S6C, promote a constitutively open channel. Interestingly, a V254L/L353A double mutant rescued channel closing, suggesting that the S4–S5 linker interacts with S6C to stabilize the closed state. Relocation of the S4–S5 linker during gating abolishes this interaction, releasing tension on S6 and allowing it to kink at the PAG gating hinge in a cantilever-like fashion to promote channel opening (Figure 5B). This coupling mechanism is intrinsically weak for KCNQ1, requiring modulation by auxiliary molecules (see below). Recent work has further elucidated the molecular details of S4–S5 linker relocation, revealing a two-stage mechanism involving alternative binding modes of the S4–S5 linker to the PD (Hou et al., 2020). This two-stage mechanism may apply to the majority of domain-swapped Kv channels.
Figure 5.
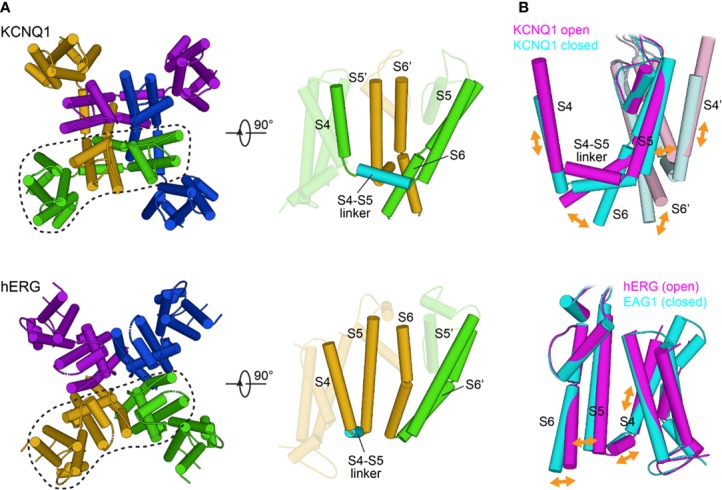
Structural features of electromechanical coupling in the KCNQ1 and hERG channels. (A) Left: Extracellular view of KCNQ1 and hERG transmembrane segments S0 to S6. Individual subunits are drawn with different colors. KCNQ1 (PDB: 6UZZ) (Sun and Mackinnon, 2020) has a domain-swapped architecture whereas hERG (PDB: 5VA1) (Wang and Mackinnon, 2017) channels are non-domain-swapped. Right: Cartoon representation of a single subunit and its neighboring pore segments (S5 to S6). In KCNQ1, the VSD and PD are bridged by an extended α-helical S4–S5 linker, whereas in hERG, S4 and S5 are connected by only a short helix. (B) Implications for the direction of coupled motions between the VSD and PD based on molecular modeling of KCNQ1 in open and closed conformations (top) and comparison of the open state structure of hERG with the closed state structure of EAG1 (PDB: 5K7L) (Whicher and Mackinnon, 2016) (bottom). In the domain-swapped KCNQ1 channel, the S4 movement is transmitted to the gate through the S4–S5 linker. In hERG and EAG1, the S4 movement is proposed to exert a direct force on the S5–S6 interface to compress or open the channel gate.
hERG
The relative positioning of the VSD and PD in hERG (Wang and Mackinnon, 2017) (Figure 5A) and the EAG1 channel (Whicher and Mackinnon, 2016) suggests that the mechanism by which movements in the VSD are transduced to pore opening is different from other KV channels (Toombes and Swartz, 2016; Barros et al., 2019). This notion is supported by the finding that cutting the S4–S5 linker in hERG through separate expression of the VSD and PD fails to significantly perturb activation kinetics (Lorinczi et al., 2015). In contrast to the lever mechanism proposed for KCNQ1, lateral S4 movement in hERG toward the pore could both alter S4–S5 linker/S6 interactions and exert force through the S4–S5 linker directly onto S5. Displacement of S5 may be transmitted through the S5–S6 interface for opening or closing of the cytosolic gate (Wang and Mackinnon, 2017) (Figure 5B). A structure of hERG in the resting state may provide further insight into the molecular features of this distinct coupling mechanism.
SCN5A
Much of our knowledge about the coupling in SCN5A originates from studies of ancestral NaV channels. Comparison of locked resting state structures of the bacterial sodium channel NaVAb (Wisedchaisri et al., 2019) and of the chimeric human NaV1.7–VSDII–NaVAb channel (Xu et al., 2019) with activated state structures (Yan et al., 2017b; Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020) provide new insight into the mechanism of electromechanical coupling in NaV channels. The S4–S5 linker appears to undergo movement similar to that of KCNQ1. The linker constrains the S5 and S6 helices in the resting/closed state, with looser interactions in the activated/open state. Coupling between the VSD and PD during pore opening must involve loosening of linker/PD interactions as S4 moves outward as in KCNQ1, albeit with tighter coupling of S4 and S4–S5 linker movement. The direct connection between S4 movement and pore opening may be crucial for rapid activation of sodium channels.
Comparison of the cockroach NaVPaS channel structure (Shen et al., 2017), featuring a closed pore and VSDs in distinct activation states, to other eukaryotic NaV channel structures suggests that additional structural shifts may be at work in eukaryotic NaV channels to couple VSD to PD movement. Moreover, the distinct sequence of repeats I to IV (including that of the four S4–S5 linkers) and asynchronous voltage sensor movement in eukaryotic NaV channels (Chanda and Bezanilla, 2002) suggest that distinct interactions couple the VSD of each repeat to the PD. Electromechanical coupling mechanisms may thus be more complex in SCN5A.
Inactivation
KCNQ1
Inactivation in KCNQ1 follows a mechanism distinct from canonical mechanisms (Hou et al., 2017). In the absence of KCNE1, KCNQ1 only partially inactivates, in a manner dependent on the IO and AO open states (Pusch et al., 1998). Differences in VSD-PD coupling between these two states appear to contribute to inactivation. The AO state has a lower coupling efficiency than IO, producing a lower open probability. Transition from IO to AO thus results in partial inactivation because the channel is open but less conductive (Hou et al., 2017). However, association with the KCNE1 accessory β subunit removes KCNQ1 inactivation (Pusch et al., 1998; Seebohm et al., 2003c), making inactivation irrelevant to cardiac KCNQ1 function.
hERG
Inactivation plays a critical role in hERG activity in the action potential. Entry into and out of inactivation is both fast and voltage-dependent, properties that maintain the plateau of the action potential (Perry et al., 2015). Additionally, the voltage dependence of inactivation appears to be independent of that of activation (Vandenberg et al., 2006; Cheng and Claydon, 2012), indicating that activation and inactivation may operate through distinct mechanisms. Inactivation in hERG is C-type (Smith et al., 1996), occurring through structural changes in the SF (Herzberg et al., 1998 14243; Hoshi and Armstrong, 2013). While the structural basis of C-type inactivation is not completely understood, work on hERG and other K+ channels has provided useful insight. Several residues in the KcsA SF form stabilizing hydrogen bond networks that suppress inactivation (Doyle et al., 1998; Bhate et al., 2010; Cuello et al., 2010; Vandenberg et al., 2012). While these residues are conserved in many Kv channels (Whicher and Mackinnon, 2016), they are not present in hERG. This implies that the hERG SF is more liable to collapse, leading to inactivation (Fan et al., 1999; Vandenberg et al., 2012). Indeed, in molecular dynamics simulations, several residues in the hERG SF shift in and out of the pore axis, particularly F627 in the GFG motif (Stansfeld et al., 2008). In the hERG cryo-EM structure, the orientation of F627 is offset compared to other Kv channels (Whicher and Mackinnon, 2016), and a hERG S631A mutant, which has an F627 sidechain orientation similar to other structures, does not inactivate (Wang and Mackinnon, 2017). Additionally, mutation of T432 and A443 (corresponding to S620 and S631 in hERG) in non-inactivating EAG1 to serine was sufficient to impart inactivation behavior (Ficker et al., 2001), perhaps due to reorientation of the SF to match that of hERG. These results indicate that the unique positioning of F627 is critical for hERG fast inactivation behavior. C-type inactivation may involve other rearrangements (Loots and Isacoff, 1998), including coupling the SF to motions in S1, S5, and S6 (Ferrer et al., 2011; Wang et al., 2011; Perry et al., 2013a; Perry et al., 2013b).
SCN5A
Fast inactivation in SCN5A halts inward Na+ current triggered by cardiac depolarization, permitting subsequent outward currents (e.g. IKs, IKr) to repolarize the cell in preparation for the next action potential (Ghovanloo et al., 2016) (Figure 6A). The S4 segment in VSDIV (S4IV) is critical for fast inactivation, along with the IFM motif in the III/IV linker (Ahern et al., 2016; Ghovanloo et al., 2016) (Figures 6B, C). Roles of individual basic sites of S4IV in Nav inactivation have been elucidated, with mutations in R1 and R2 delaying inactivation onset and mutations in R3 and R4 delaying recovery from inactivation (Nakajima et al., 2019). Furthermore, structures of an engineered human NaV1.7–NaVPaS channel with VSDIV trapped in a resting state (Clairfeuille et al., 2019) along with the structures of NavPaS (Shen et al., 2017), electric eel (Yan et al., 2017b) and human Nav1.4 channels (Pan et al., 2018) have provided new structural insight into the mechanism of fast inactivation in NaV channels (Figure 6D). In the resting state R5 on S4IV forms an electrostatic bridge with the α1 helix of the C-terminal cytoplasmic domain (CTD), together with K7 and R8 on the S4–S5 linker. The CTD in turn binds the III/IV linker, sequestering the IFM motif (Figure 6D, left plot). VSDIV activation releases the connection between the CTD and the III/IV linker (middle and right plots in Figure 6D), permitting the IFM motif to bind to a hydrophobic pocket formed by the S4–S5 linkers and S6 helices of repeats III and IV, along with S5IV (Figures 6C, E). The insertion of the IFM motif into this pocket causes a twisting in the S6 helices that closes the gate (Yan et al., 2017b; Pan et al., 2018).
Figure 6.

Structural mechanisms of NaV channel inactivation. (A) NaV channels transition from an activated open state to a non-conducting inactivated state after depolarization. Inactivation is induced by binding of a C-terminal motif (yellow) to the cytosolic side of the channel leading to pore closure. The cell membrane is indicated with a gray rectangle, with the extracellular (EXT) and intracellular space (INT) labeled. (B) Closed state model of SCN5A (Kroncke et al., 2019) highlighting structural elements involved in fast inactivation. The III-IV linker (cyan) connecting S6III (blue) with VSDIV (green) contains the IFM motif (yellow spheres) which is the key structural element responsible for inactivation. The position of the III-IV linker is constrained by the CTD following S6IV. (C) Comparison of the SCN5A model in (B) with a cryo-EM structural model of rat SCN5A (PDB: 6UZ3)(Jiang et al., 2020) in a putative inactivated state viewed from the intracellular side. The III-IV linker undergoes a large shift in the SCN5A structure and the IFM motif is docked into a pocket surrounded by the S4–S5 linkers and S6 helices of repeats III and IV. (D) Structural states and transitions proposed to be involved in fast inactivation in NaV channels. Left: Structure of a chimeric NaV1.7–NaVPaS channel (Clairfeuille et al., 2019) (PDB: 6NT3) with S4IV in a “down” position and an IFM-like motif unbound. Basic sites R5–R8 bridge to conserved acidic residues on the CTD which facilitates binding of the III-IV linker to S6IV. Middle: Structure of a chimeric NaV1.7–NaVPaS channel (Clairfeuille et al., 2019) (PDB: 6NT4) with S4IV in a ‘up’ position and an IFM-like motif unbound. The electrostatic bridge between S4IV and the CTD is broken possibly increasing the positional dynamics of the CTD and III-IV linker. Right: Structure of SCN5A (PDB: 6UZ3) (Jiang et al., 2020) with S4IV in an ‘up’ position and the IFM motif plugged into the cytosolic cavity between repeats III and IV. Note, that the CTD is missing in the cryo-EM structure. (E) IFM binding pocket residues on S4–S5III, S4–S5IV, S6III, and S6IV of SCN5A (PDB: 6UZ3) (Jiang et al., 2020). The S6IV helix backbone in the front is not shown for clarity. Residues that are important for fast inactivation and those found in various types of myotonia (Pan et al., 2018) are labeled magenta and orange, respectively.
Regulation of Channel Gating by Intracellular Domains And Auxiliary Molecules
The function of these cardiac channels is tightly regulated to produce currents that faithfully give rise to the cardiac action potential under a host of physiological conditions and prevent early or delayed contractility. We focus here on regulation through the channel cytoplasmic domains as well as regulation by auxiliary (beta) subunits and lipids. Channels are regulated through other mechanisms such as phosphorylation, but are not covered in this review.
Regulation Involving Cytoplasmic Domains
KCNQ1
The C-terminal cytoplasmic domain (CTD) of KCNQ1 contains four alpha helices in lieu of the T1 tetramerization domain common to Kv channels outside the KCNQ family (Haitin and Attali, 2008). The proximal HA and HB helices form an antiparallel bundle with an IQ motif in HA and a 1-5-10 motif in HB that together enable calmodulin (CaM) binding (Yus-Najera et al., 2002). When bound, CaM prevents channel inactivation in KCNQ1, but inhibits the opening of other KCNQ family members (KCNQ2–KCNQ5) (Chang et al., 2018). While the N-lobe of CaM may be constitutively bound to both calcium and the proximal HA and HB helices (Ghosh et al., 2006; Bernardo-Seisdedos et al., 2018), the C-lobe of CaM binds calcium only at higher concentrations (Bernardo-Seisdedos et al., 2018). Calcium binding by the C-lobe induces a conformational change in CaM, facilitating KCNQ1 opening (Bernardo-Seisdedos et al., 2018; Chang et al., 2018). The C-lobe interacts with HA and with a loop in the S2–S3 linker (Sun and Mackinnon, 2017). This feature may contribute to the unique mode of CaM modulation of KCNQ1.
The distal helices HC and HD of KCNQ1 form a self-assembling intersubunit coiled-coil motif that promotes channel tetramerization (Yus-Najera et al., 2002; Howard et al., 2007; Sun and Mackinnon, 2017). The distal coiled-coil domain is not only essential for channel tetramerization, but also for subunit specificity, permitting the exclusive formation of KCNQ1 homotetramers (Schwake et al., 2003; Haitin and Attali, 2008; Sachyani et al., 2014). In contrast, other KCNQ family proteins form heteromers (Schwake et al., 2003). Swapping the coiled-coil domain of KCNQ1 with that of KCNQ3 allows the resulting chimera to co-assemble with other KCNQ isoforms and generate channels with altered gating properties (Schwake et al., 2003; Schwake et al., 2006). Residues conferring specific KCNQ1 homotetramer formation have been identified in the HD helix (Schwake et al., 2006; Wiener et al., 2008). Key interactions involve both the hydrophobic core of the assembly and exterior electrostatic interactions, with a number of the residues involved subject to LQTS-associated mutations (Howard et al., 2007; Wiener et al., 2008).
hERG
hERG channels contain an N-terminal Per-ARNT-Sim (PAS) domain, which is also present in the N-terminus of many signaling proteins (Henry and Crosson, 2011). An additional C-terminal cyclic nucleotide-binding homology domain (CNBHD) similar to cytoplasmic domains in hyperpolarization-sensitive cyclic nucleotide-gated channels is present, but without ligand-binding properties (Codding and Trudeau, 2019). The PAS and the CNBHD appear to be critical for modulating the kinetics of slow deactivation in hERG (Gustina and Trudeau, 2011). Furthermore, slow deactivation is dependent on the direct interaction of these domains, particularly between R56 of the PAS and D803 of the CNBHD and between N12 of the N-terminal Cap (N-Cap) and E788 of the CNBHD (Ng et al., 2014; Kume et al., 2018). Interactions between the N-Cap and PAS domains with the C-linker also appear to be important for deactivation (Gustina and Trudeau, 2011; Ng et al., 2014), and the extreme N-terminus is likely essential, possibly interacting with the VSD through a patch of positively-charged residues in the N-Cap tail (Muskett et al., 2011). Indeed, the hERG cryo-EM structure corroborates the positioning of the N-Cap tail relative to the VSD (Wang and Mackinnon, 2017). The N-Cap tail may even be constitutively bound to the VSD (Morais Cabral et al., 1998; De La Pena et al., 2011), implying that slow deactivation could involve movement of the PAS toward the plasma membrane to alter this interaction (Barros et al., 2018).
SCN5A
Multiple structures of the SCN5A CTD in complex with regulatory factors (Wang et al., 2012; Gabelli et al., 2014; Gardill et al., 2019) have been determined, helping to elucidate how CaM (Johnson et al., 2018) and fibroblast growth factor-homologous factors (FHFs), specifically FGF13, modulate inactivation (Liu et al., 2003; Wang et al., 2012; Musa et al., 2015; Yang et al., 2016). CaM binds the CTD in a calcium-dependent manner (Kim et al., 2004; Hovey et al., 2017; Johnson et al., 2018; Urrutia et al., 2019), altering inactivation kinetics (Yan et al., 2017a; Johnson et al., 2018) and promoting recovery from inactivation (Johnson et al., 2018). In the absence of FHF, the C-lobe of apo-CaM binds to the IQ motif located in an extended helix on the CTD, while the N-lobe binds to the preceding EF hand-like domain (EFL) (Gabelli et al., 2014). With increasing intracellular calcium, holo-CaM has stronger affinity for the SCN5A III/IV linker (Johnson et al., 2018) and the N-lobe disassociates from the EFL (Gardill et al., 2019). These calcium-dependent interactions with the III/IV linker may facilitate CaM modulation of inactivation (Hovey et al., 2017; Johnson et al., 2018). FGF13 slows both inactivation onset and inactivation recovery in SCN5A (Yang et al., 2016), opposing CaM modulation. Importantly, binding of FGF13 to the SCN5A EFL (Wang et al., 2012) prevents binding of the apo-CaM N-lobe to the same domain (Wang et al., 2012). The EFL itself also adopts different orientations when bound to FGF13 (Wang et al., 2012) or CaM (Gabelli et al., 2014). Because the EFL also binds to the III/IV linker containing the IFM inactivation motif (Shen et al., 2017; Clairfeuille et al., 2019), EFL conformational changes upon binding of CaM or FGF13 may contribute to inactivation modulation by these proteins.
SCN5A also contains two cytoplasmic loops linking repeats I and II and repeats II and III, respectively. Unlike the III/IV linker, these loops are around 200 residues long (Pan et al., 2018) and likely disordered, based on the absence of these loops in cryo-EM structures. However, important regulatory events such as phosphorylation (Marionneau et al., 2012; Iqbal et al., 2018) and cofactor binding (Wu et al., 2008) that modulate channel properties have been identified in these loops.
Auxiliary Beta Subunits
KCNQ1
KCNQ1 co-assembles with a family of single span membrane proteins (KCNE1–5) (Abbott, 2014), and this interaction appears to be tissue–specific. In the heart, KCNQ1 is complexed with KCNE1, and the KCNQ1-KCNE1 channel exhibits greater single-channel conductance, opening at more positive potentials, and delayed activation compared to KCNQ1 alone (Barhanin et al., 1996; Sanguinetti et al., 1996). This heteromultimeric channel also exhibits altered Rb+/K+ selectivity (Pusch et al., 2000) and loss of inactivation (Pusch et al., 1998; Pusch et al., 2000). These properties are essential for the generation of the slow delayed rectifier current (IKs) in repolarizing cardiomyocytes. In gastric parietal cells, KCNQ1 co-assembles with KCNE2 to create constitutive K+ currents essential for gastric acid secretion (Heitzmann et al., 2004; Roepke et al., 2006). In intestinal epithelial cells KCNQ1 complexes with KCNE3 to allow K+ recycling for trans-epithelial chloride ion secretion (Preston et al., 2010). In contrast to KCNE1, KCNE2 and KCNE3 render KCNQ1 constitutively open (Schroeder et al., 2000; Tinel et al., 2000).
Due to the critical role of the KCNQ1-KCNE1 channel in the cardiac action potential, many research groups have probed the structural interaction between these proteins (Tapper and George, 2001; Xu et al., 2008; Chung et al., 2009; Strutz-Seebohm et al., 2011; Wang et al., 2011; Chan et al., 2012; Li et al., 2014). Combined results indicate that the transmembrane helix of KCNE1 binds in the cleft between neighboring KCNQ1 subunits and interacts with both the VSD and PD. These binding sites overlap the binding cleft of KCNE3 (Sun and Mackinnon, 2020), indicating that KCNE1 may bind to the same site (Figure 7A). In the human KCNQ1-KCNE3 cryo-EM structure (Sun and Mackinnon, 2020) and an earlier Rosetta model of the complex (Kroncke et al., 2016), this cleft is formed by three KCNQ1 subunits (Figure 7B). KCNE3 contacts the cytoplasmic half of S5 in one subunit, the extracellular side of S6 in a second one, and along the entire length of S1 and the cytoplasmic side of S4 in a third subunit. Importantly, KCNE1 and KCNE3 produce KCNQ1 currents with different voltage dependencies and gating kinetics (Figure 7C). Given the possibility of a common binding cleft, differential modulation of KCNQ1 by KCNE1 and KCNE3 may involve different sidechain interactions within this cleft.
Figure 7.
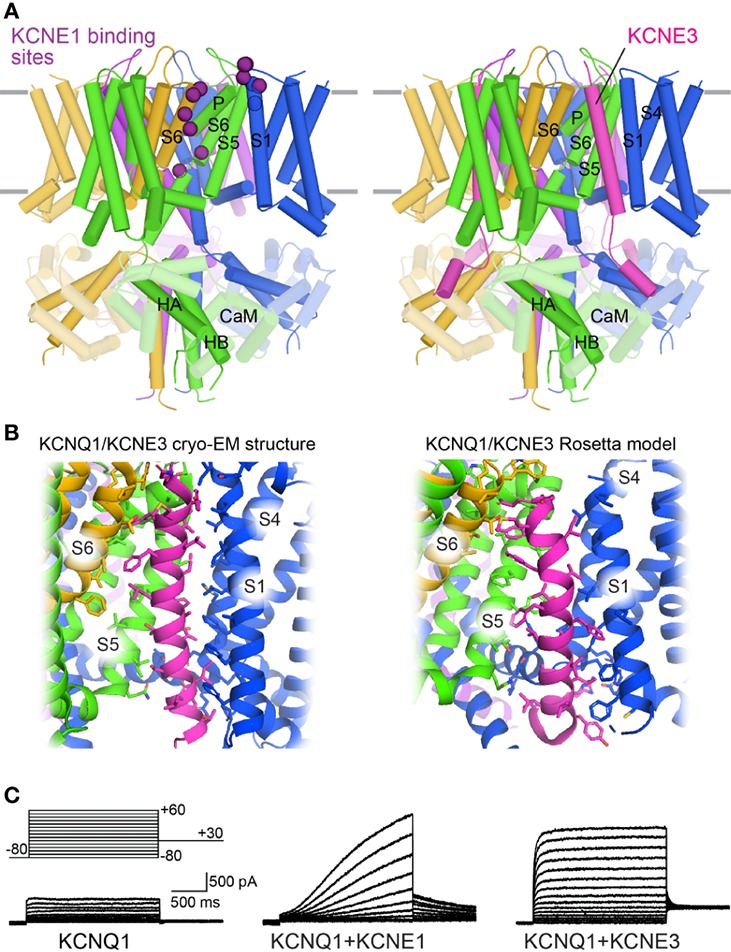
Binding sites of KCNE proteins on the KCNQ1 channel. (A) Left: Mapping of putative KCNE binding sites onto the human KCNQ1 structure (PDB: 6V00) (Sun and Mackinnon, 2020). The position of residues identified to interact with KCNE1 (purple) are indicated by spheres. Right: Cryo-EM structure of human KCNQ1 with CaM and KCNE3 (PDB: 6v00)(Sun and Mackinnon, 2020). (B) KCNQ1/KCNE3 interaction sites. Left: 6V00. Right: KCNQ1/KCNE3 Rosetta model (Kroncke et al., 2016). Interactions sites were calculated with the InterResidues Python script in PYMOL (The PyMOL Molecular Graphics System, Version 2.2 Schrödinger, LLC.). Interaction positions indicated with sticks. (C) Example current traces of KCNQ1 alone or with KCNE proteins. Adapted with permission from (Van Horn et al., 2011).
Mutations in the KCNE1 transmembrane helix, particularly at F57, T58, and L59 (Melman et al., 2001; Melman et al., 2002), alter the ability of this subunit to modulate the function of KCNQ1. The effect of this ‘activation’ triplet on KCNQ1 gating is altered by mutations at residues S338, F339, F340, and A341 in S6 (Melman et al., 2004; Panaghie et al., 2006; Strutz-Seebohm et al., 2011; Li et al., 2014), suggesting a functional link between these residues. However, modeling studies fail to indicate direct contact between the activation triplet on KCNE1 and these S6 sites (Kang et al., 2008; Gofman et al., 2012; Xu et al., 2013), suggesting that the functional coupling may be mediated allosterically. Interestingly, KCNE1 also affects S4 movement (Rocheleau and Kobertz, 2008; Wu et al., 2010a; Nakajo and Kubo, 2014) and shifts VSD voltage-dependence of activation to more negative voltages (Osteen et al., 2010; Ruscic et al., 2013; Barro-Soria et al., 2014). A model of KCNE1 regulation has recently been proposed, in which KCNE1 alters VSD-PD coupling interactions to suppress the IO state and modulate the AO state (Zaydman et al., 2014). This model accounts for many of the observed effects of KCNE1 on KCNQ1, including KCNE1-induced inhibition of inactivation. However, this model is not structurally elaborated.
hERG
Previous studies indicate that KCNE1 (Mcdonald et al., 1997) and KCNE2 (Abbott et al., 1999) can interact with and modulate hERG function. However, whether this interaction occurs under physiological conditions is debated (Weerapura et al., 2002; Anantharam and Abbott, 2005; Abbott et al., 2007). Although KCNE1 and KCNE2 alter hERG gating kinetics in vivo (Mcdonald et al., 1997; Mazhari et al., 2001), co-expression of KCNE2 with hERG in vitro does not reproduce the native IKr current (Weerapura et al., 2002). This suggests that KCNE1 and KCNE2 may not be essential for hERG channel function or that additional factors are required for KCNE-hERG interaction in cardiac cells. The latter possibility is supported by the observation that mutations in KCNE2 may predispose patients to drug-induced LQTS (Abbott et al., 1999; Sesti et al., 2000). As hERG is particularly drug-sensitive, this finding suggests that hERG may, in fact, be modulated by KCNEs in native tissue. Additionally, a T10M mutation in KCNE2 causes arrhythmia induced by auditory stimulation, a known trigger of LQT2, (Gordon et al., 2008). Further studies are needed to resolve these conflicting results and clarify the role of KCNE proteins in regulation of hERG function.
SCN5A
In humans there are five NaV-β-subunit protein isoforms encoded by four genes, SCN1B-SCN4B (β1 to β4) (Detta et al., 2015; O’malley and Isom, 2015). All are expressed in the heart and have been shown to associate with SCN5A in heterologous conditions (Makita et al., 1996; Dhar Malhotra et al., 2001; Malhotra et al., 2004; Medeiros-Domingo et al., 2007; Watanabe et al., 2009; Valdivia et al., 2010). β1 to β4 are single-span transmembrane proteins containing an extracellular N-terminal immunoglobulin (Ig) domain, while β1B, a splice variant of SCN1B, lacks the transmembrane domain (Brackenbury and Isom, 2011). Generally, β-subunits modulate the biophysical properties and cell surface expression of NaV channels in heterologous cells (Calhoun and Isom, 2014). β1 and β3 interact non-covalently with NaV channels (Meadows et al., 2001), while β2 and β4 are covalently bound through cysteine bonds between the extracellular Ig domain and channel pore loops. However, modulation of SCN5A by β-subunits has been difficult to assess. Varying effects of β1 (Qu et al., 1995; Dhar Malhotra et al., 2001; Baroni et al., 2014; Zhu et al., 2017) as well as β3 (Hu et al., 2009; Valdivia et al., 2010; Wang et al., 2010) on SCN5A have been reported. The rat SCN5A structure provides a potential explanation for this difficulty, as SCN5A is missing a cysteine residue critical for covalent interaction with β2, and contains a glycosylation site that may sterically occlude β1 interaction (Jiang et al., 2020). SCN5A may not bind tightly with any β-subunit. Nevertheless, the potential importance of β-subunits in SCN5A modulation has been suggested by arrhythmia-associated mutations in all four β-subunit genes, including mutations causing Brugada syndrome (Watanabe et al., 2008; Hu et al., 2009; Hu et al., 2012), LQT3 (Medeiros-Domingo et al., 2007; Riuro et al., 2014), and atrial fibrillation (Watanabe et al., 2009; Olesen et al., 2011). However, a recent review of genetic evidence supporting these associations has disputed the clinical validity of β subunits as monogenic causes of arrhythmia syndromes (Hosseini et al., 2018; Adler et al., 2020).
SCN5A subcellular localization also contributes importantly to channel regulation. SCN5A channels in cardiac tissue are localized at the lateral membranes and at the anchoring junction between cardiomyocytes (the intercalated disc). These expression patterns give rise to distinct sets of protein-protein interactions and biophysical properties (Lin et al., 2011; Shy et al., 2013). At the lateral membrane SCN5A interacts with the dystrophin/syntrophin multicomplex, but at the intercalated disc SCN5A interacts with ankyrin-G (Lemaillet et al., 2003; Mohler et al., 2004), which links the channel to cytoskeletal proteins such as actin and the desmosomal protein plakophilin-2 (Makara et al., 2014). Intriguingly, the functional properties of SCN5A channels between these two pools also differ: SCN5A at the lateral membrane has smaller current amplitude, distinct voltage-dependence, and slower recovery from inactivation as compared to channels in the intercalated disc (Lin et al., 2011). The functional implications of these differences are not yet clear and remain an active area of study.
Lipid Molecules
KCNQ1
KCNQ1 and other KCNQ channels are dependent on phosphatidyl-4,5-bisphosphate (PIP2) for function (Loussouarn et al., 2003; Zaydman et al., 2013; Taylor and Sanders, 2017), and are inhibited upon stimulation of Gq- and G11-protein coupled receptors, which trigger phospholipase C-catalyzed PIP2 hydrolysis (Selyanko et al., 2000; Loussouarn et al., 2003; Zhang et al., 2003) (Figure 8B). Growing evidence suggests that PIP2 acts as a coupling element for KCNQ1, enhancing weak allosteric interactions between the VSD and PD (Vardanyan and Pongs, 2012; Zaydman et al., 2013; Kasimova et al., 2015; Cui, 2016). Structure-function studies, confirmed by the human KCNQ1/PIP2 structure, localize PIP2 binding to the cleft between neighboring channel subunits, with interactions involving mostly positively-charged residues in the S2–S3 linker, S4–S5 linker, and S6C (Figure 8A) (Thomas et al., 2011; Zaydman et al., 2013; Eckey et al., 2014; Chen et al., 2015; Sun and Mackinnon, 2020). This binding site seems well-suited to modulate coupling of VSD movement to the activation gate. Questions remain regarding the mechanism of PIP2 regulation of VSD-PD coupling, PIP2:KCNQ1 stoichiometry, and binding site differences between the activated and resting states.
Figure 8.
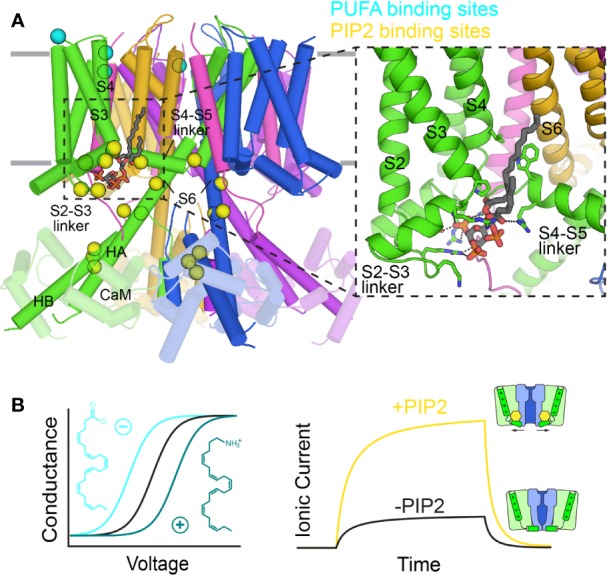
Binding sites of lipids on the KCNQ1 channel. (A) Mapping of positions that have been implicated in the regulation of KCNQ1 by PUFAs (cyan) and PIP2 (yellow) onto the PIP2-bound KCNQ1 structure (6V01) (Sun and Mackinnon, 2020). Inset: the PIP2 cryo-EM binding site. (B) Left: Effect of the PUFA head group charge on the voltage dependence of KCNQ1 conductance (inspired from results in Liin et al., 2015). Right: PIP2 depletion reduces KCNQ1 ionic currents due to decreased VSD-PD coupling.
PIP2 also binds to the CTD at a site shared with CaM (Tobelaim et al., 2017b). These two regulators may competitively regulate KCNQ1 at this site (Tobelaim et al., 2017a). Indeed, in the human KCNQ1 cryo-EM structure, CaM exhibits a nearly 180° rotation when PIP2 is bound, losing contact with the S2–S3 linker (Sun and Mackinnon, 2020). Additionally, S6 and HA in KCNQ1 form a single helix in the open channel. These structural shifts point to an interplay between PIP2 and CaM, but does not clarify the nature of this interaction, as no PIP2 density was seen at the CTD binding site in the cryo-EM structure. The details of the coordination between CaM and PIP2, and its role in channel function, remain to be elucidated.
Polyunsaturated fatty acids (PUFAs) also modulate KCNQ1 function (Taylor and Sanders, 2017). While PUFAs generally inhibit ion channel current (Boland and Drzewiecki, 2008), the KCNQ1-KCNE1 channel is a notable exception. The IKs current is enhanced by docosahexaenoic acid (DHA) and, to a lesser extent, oleic acid (Doolan et al., 2002) by shifting the conductance-vs-voltage curve (GV) to more negative voltages. Interestingly, the charge of the head group determines the direction of the (GV) shift (Figure 8B): negatively-charged DHA causes a negative shift, a neutral head group has no effect, and a positively-charged one shifts the (GV) curve to positive potentials, reducing channel function (Liin et al., 2015). Negative head group charge and a polyunsaturated acyl chain appear to be required for channel activation. The PUFA binding site in KCNQ1 appears to involve residues in the extracellular S3–S4 loop, R1 and R2 on S4, and the PD (Figure 8A), as mutations in these regions either reduce the PUFA effect or completely abolish the (GV) shift (Liin et al., 2015; Liin et al., 2018).
hERG
As with KCNQ1, the function of hERG is upregulated by PIP2 by means of increased current amplitude, a hyperpolarizing shift in voltage-dependence of activation, as well as faster activation and slower inactivation rates (Bian et al., 2001). Stimulation of Gαq-protein coupled receptors also suppresses the IKr current through PIP2 depletion (Bian et al., 2001; Bian et al., 2004). Binding of PIP2 to hERG likely localizes to a cluster of basic residues (R883–Q900) C-terminal to the CNBHD, as substitution of these residues to neutral or negatively-charged amino acids prevented PIP2 effects on hERG function and abolished PIP2 binding (Bian et al., 2004). Unfortunately, these residues are not resolved in the available hERG structure (Wang and Mackinnon, 2017).
SCN5A
In contrast to KCNQ1, PUFAs such as eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), linoleic acid (LA), and α-linolenic acid (ALA) suppress INa current in a concentration-dependent manner by shifting the voltage dependence of INa inactivation to more hyperpolarized potentials (Kang et al., 1995; Kang et al., 1997; Leifert et al., 1999). EPA also accelerates the transition from the resting state to the inactivated state and slows recovery from inactivation (Xiao et al., 1998; Isbilen et al., 2006). However, the effect was reduced by β1 subunit expression (Xiao et al, 2000). Furthermore, N406K renders SCN5A less sensitive to inhibition by EPA, an effect strengthened by β1 subunit expression (Xiao et al., 2001). The structural basis for PUFA binding and modulation is not well understood.
Channel Dysfunction in Congenital Long QT Syndrome
Alterations in the action potential disturb impulse propagation and cause reentry (Kleber Ag, 2004), whereby the impulse re-stimulates the heart tissue that generated it. Reentry promotes cardiac arrhythmias and predisposes to sudden unexplained death (Skinner et al., 2019). LQTS is a prevalent cause of such events, with an estimated population prevalence of approximately 1:2500 (Schwartz et al., 2009).
LQTS is characterized by a prolonged rate-corrected QT interval in patient electrocardiograms (ECGs), indicative of impaired repolarization (Schwartz et al., 2012). In a large proportion of cases, QT interval prolongation is the result of either a loss-of-function in KCNQ1 or hERG, or a gain-of-function in the SCN5A channel (Moss and Kass, 2005; Schwartz et al., 2012; Earle et al., 2013; Skinner et al., 2019). Mutations in these genes confer distinct subtypes of LQTS (denoted LQT1–3, respectively), each with unique ECG features, risk factors, arrhythmia triggers, and responsiveness to β-adrenergic receptor blockers, the most common LQTS treatment (Ackerman, 2005; Moss and Kass, 2005; Skinner et al., 2019; Wallace et al., 2019). Below, the function of each ion channel in the cardiac action potential is discussed, along with how each can cause action potential dysfunction in LQTS.
Channel Function in the Cardiac Action Potential and Dysfunction in LQTS
KCNQ1
In complex with KCNE1, KCNQ1 conducts IKs, which helps shape the plateau and repolarization phases of the action potential, in part by counteracting calcium influx (Skinner et al., 2019). Due to slow activation, the KCNQ1-KCNE1 channel is only slightly open during the plateau phase, with IKs increasing slowly in a nearly linear fashion (Figure 7C) until the repolarization phase is reached and the channel becomes fully activated (Figures 1B, C) (Moss and Kass, 2005). KCNQ1-KCNE1 channels do not inactivate (Pusch et al., 1998), conducting current throughout activation and deactivation. The channel slowly deactivates, shaping the tail of the repolarization phase and mediating return to the resting potential (Moss and Kass, 2005; George, 2013; Skinner et al., 2019). Thus, loss-of-function in KCNQ1 prolongs repolarization primarily by preventing completion of the repolarization phase, broadening the tail of the T-wave (Figure 1A) and causing type 1 LQTS (LQT1). (Ackerman, 2005; Tester and Ackerman, 2014; Skinner et al., 2019; Wallace et al., 2019). The effects of a KCNQ1 mutation are exacerbated upon sympathetic activation of β-adrenergic receptors due to IKs potentiation, while resting heart rates show few perturbations (Shimizu and Antzelevitch, 1998). The QT interval is unable to shorten in response to this stimulation, making physical exertion accompanied by heightened sympathetic nervous system activity a prevalent LQT1 trigger (Schwartz et al., 2012; Bohnen et al., 2017).
hERG
hERG makes up the other primary inward rectifier current IKr, which acts in both the plateau and the repolarization phase (Figures 1B, C) (Moss and Kass, 2005; Skinner et al., 2019). As the channel activates, rapid entry into and out of inactivation (due to SF fluctuations) creates a persistent current through the plateau phase until slow deactivation, regulated by the PAS and CNBHD domains, closes the channel during the repolarization phase. While both hERG and KCNQ1 are conductive during the plateau and repolarization phases, hERG has a higher unitary conductance than KCNQ1 in both phases (Moss and Kass, 2005; George, 2013). The resulting IKr current thus supplies a greater proportion of the overall IK current (Cheng and Kodama, 2004), with loss-of-function in hERG (LQT2) correlating with a lower T-wave amplitude in contrast to the normal T-wave amplitude associated with LQT1. Additionally, LQT2 is often triggered at rest (Skinner et al., 2019), consistent with the greater hERG contribution to IK. However, while IKr is usually more prominent (Cheng and Kodama, 2004), the relative density of IKr and IKs vary by ventricular cell type (Liu and Antzelevitch, 1995; Viswanathan et al., 1999).
SCN5A
SCN5A produces the INa current that shapes the initial upstroke of the cardiac action potential (Skinner et al., 2019), initiating depolarization. SCN5A is rapidly activated through asynchronous motion of the VSD’s in repeats I-III coupled to pore opening, then quickly inactivates through interaction of the IFM motif with the channel, resulting in a brief inward spike of sodium current that depolarizes the cell (Ghovanloo et al., 2016) (Figures 1B, C). Unlike KCNQ1 and hERG, LQT3 is conferred by SCN5A gain-of-function mutations that cause a “leaky” inward sodium current (Wilde and Amin, 2018; Skinner et al., 2019). The functional IKr and IKs outward currents are unable to compensate for the persistent inward INa current, prolonging entry into and completion of the repolarization phase and giving rise to LQT3. As in LQT2, LQT3-related arrhythmias occur most often at rest, rather than in response to stress or exercise (Schwartz et al., 2012). It should be noted that while loss-of-function mutations for SCN5A are not relevant to LQTS, they are prevalent in other channelopathies, particularly Brugada syndrome (Wilde and Amin, 2018; Skinner et al., 2019).
Molecular Mechanisms of LQTS
Mutations in KCNQ1, hERG, and SCN5A can lead to LQTS by perturbing channel function through distinct molecular mechanisms. Structural studies of these channels can help tease apart these molecular mechanisms for a more detailed understanding of LQTS. We have mapped LQTS mutations identified as pathogenic according to ClinVar (Harrison et al., 2016) and HGMD (Stenson et al., 2012) databases onto the human KCNQ1 (Sun and Mackinnon, 2020) and hERG structures (Wang and Mackinnon, 2017), as well as a recent model of SCN5A (Kroncke et al., 2019) and the rat SCN5A homolog (Jiang et al., 2020) (Figures 9–11). The list of curated mutations is found in Table 1: 261 mutations in KCNQ1, 320 mutations in hERG, and 122 mutations in SCN5A. Examination of these mutation sites provides some insight into the channel mechanisms that may be commonly disrupted in LQT1–3.
Figure 9.
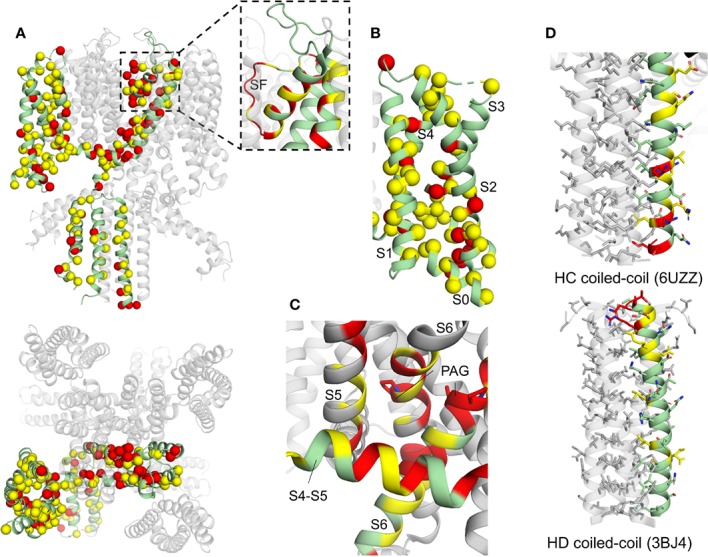
Human KCNQ1 structure with deleterious mutation sites indicated. (A) Full KCNQ1 structure (PDB: 6UZZ) (Sun and Mackinnon, 2020). Yellow sites denote locations of a single deleterious mutation, while red indicates sites with multiple mutations identified. Spheres indicate C-alpha positions of mutation sites. Subunit with LQT1 mutations sites mapped is colored green. Inset: of SF and pore helix. (B) KCNQ1 VSD. (C) S4–S5 linker (S4–S5) interaction site close-up. One subunit is colored green and the others in gray, for clarity. The PAG motif (PAG) is indicated with sticks. (D) Top: HC coiled-coil domain. Bottom: HD coiled-coil domain (PDB: 3BJ4) (Wiener et al., 2008). Residue side chains are indicated with sticks.
Figure 11.

Rat SCN5A structure with deleterious mutation sites indicated. (A) Full SCN5A structure (PDB: 6UZ3) (Jiang et al., 2020). Yellow sites denote locations of a single deleterious mutation, while red indicates sites with multiple mutations identified. Spheres indicate C-alpha positions of mutation sites. Side views of individual repeats are shown with 90° rotations between panels. In each panel, one repeat is colored green and LQT3 mutation sites for that repeat are mapped. (B) C-terminal domain with bound III/IV linker (human SCN5A model: [(Kroncke Bm, 2019)]. (C) Putative inactivation gate. IFM motif is indicated with sticks. (D) SCN5A constriction site. S6 helices are shown at full opacity, with the IFM motif indicated with sticks.
Table 1.
LQTS mutations in KCNQ1, hERG, and SCN5A.
| KCNQ1 | hERG | SCN5A | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutant | Database | Mutant | Database | Mutant | Database | Mutant | Database | Mutant | Database | Mutant | Database |
| A58P* | HGMD | T311I | HGMD | G6R | HGMD | G572R | ClinVar, HGMD | G9V* | HGMD | G1329S | HGMD |
| S66F* | HGMD | T312S | HGMD | G6V | HGMD | G572S | HGMD | R18W* | HGMD | A1330P | HGMD |
| T96R* | HGMD | T312I | ClinVar, HGMD | T13N | HGMD | G572V | HGMD | A29V* | HGMD | A1330T | HGMD |
| T104I | HGMD | I313M | HGMD | D16A | HGMD | M574V | HGMD | E30G* | HGMD | P1332L | HGMD |
| Q107H | HGMD | G314A | HGMD | R20G | HGMD | E575G | HGMD | P52S* | HGMD | S1333Y | HGMD |
| R109L | HGMD | G314D | HGMD | F22S | HGMD | E575K | HGMD | R53Q* | HGMD | L1338V | HGMD |
| Y111C | ClinVar, HGMD | G314C | HGMD | F22Y | HGMD | R582C* | ClinVar | R104G* | HGMD | A1357V | HGMD |
| L114P | HGMD | G314R | ClinVar, HGMD | S26I | HGMD | G584S | HGMD | A110T* | HGMD | G1391R | HGMD |
| E115G | HGMD | G314S | ClinVar, HGMD | R27P | HGMD | G584V | HGMD | V113I* | HGMD | A1428S | HGMD |
| P117L | ClinVar, HGMD | Y315N | HGMD | K28E | HGMD | W585C | HGMD | S115G* | HGMD | S1458Y | ClinVar, HGMD |
| C122Y | ClinVar, HGMD | Y315C | HGMD | F29L | HGMD | L586M | HGMD | I176M | HGMD | N1472S | HGMD |
| Y125D | HGMD | Y315H | HGMD | F29S | HGMD | N588D | HGMD | A185T | HGMD | F1473C | HGMD |
| F127L | HGMD | Y315F | HGMD | I31S | HGMD | G590V | HGMD | I239V | HGMD | F1473S | HGMD |
| L131P | HGMD | Y315S | HGMD | I31T | ClinVar, HGMD | I593T | ClinVar | V240M | HGMD | Q1476R | HGMD |
| I132L | HGMD | G316E | HGMD | A32T | HGMD | I593K | ClinVar, HGMD | Q245K | HGMD | G1481E | HGMD |
| V133I | HGMD | G316V | HGMD | N33T | HGMD | I593R | ClinVar, HGMD | V258A | HGMD | T1488R | HGMD |
| L134P | ClinVar, HGMD | G316R | ClinVar, HGMD | R35W | HGMD | I593V | HGMD | R340Q | HGMD | Y1495S | HGMD |
| C136F | HGMD | D317N | ClinVar, HGMD | V41F | HGMD | G594D | HGMD | A385T | HGMD | K1505N | HGMD |
| L137F | ClinVar, HGMD | D317G | HGMD | V41A | HGMD | K595N | HGMD | I397T | HGMD | T1544P | HGMD |
| S140R | HGMD | D317Y | HGMD | I42N | HGMD | K595E | HGMD | L404Q | HGMD | L1560F | HGMD |
| E146G | HGMD | K318N | ClinVar, HGMD | Y43C | HGMD | P596H | ClinVar, HGMD | N406K | HGMD | I1593M | HGMD |
| E146K | HGMD | P320A | HGMD | Y43D | HGMD | P596A | ClinVar, HGMD | L409P | HGMD | F1594S | HGMD |
| A150G | HGMD | T322K | HGMD | C44F | HGMD | P596R | HGMD | L409V | HGMD | V1597M | HGMD |
| T153M | HGMD | T322A | ClinVar, HGMD | C44W | HGMD | P596L | HGMD | V411M | ClinVar, HGMD | S1609W | HGMD |
| F157C | HGMD | T322M | ClinVar, HGMD | C44Y | HGMD | P596S | HGMD | A413E | HGMD | R1623Q | ClinVar, HGMD |
| E160K | HGMD | G325R | ClinVar, HGMD | N45D | HGMD | P596T | HGMD | R504T* | HGMD | R1623L | HGMD |
| E160V | HGMD | G325E | HGMD | N45S | HGMD | Y597C | HGMD | M506K* | HGMD | R1626P | HGMD |
| G168R | ClinVar, HGMD | G325W | HGMD | N45Y | HGMD | G604D | HGMD | F530V* | HGMD | R1644H | ClinVar, HGMD |
| T169R | HGMD | S338F | HGMD | D46Y | HGMD | G604S | ClinVar, HGMD | D536H* | HGMD | L1646R | HGMD |
| T169K | HGMD | F339S | HGMD | G47D | HGMD | P605S | HGMD | R569W* | HGMD | L1650F | HGMD |
| E170G | HGMD | F339V | HGMD | G47V | HGMD | S606F | HGMD | Q573E* | HGMD | M1652R | HGMD |
| V173D | HGMD | F340L | HGMD | C49Y | HGMD | S606P | HGMD | G579R* | HGMD | M1652T | HGMD |
| R174C | ClinVar, HGMD | A341G | HGMD | G53R | HGMD | D609G | HGMD | P637L* | HGMD | P1725L | HGMD |
| R174H | ClinVar, HGMD | A341V | ClinVar, HGMD | G53D | HGMD | D609H | HGMD | E654K* | HGMD | A1746T | HGMD |
| W176R | HGMD | A341E | ClinVar, HGMD | G53S | HGMD | D609N | ClinVar, HGMD | A665S* | HGMD | I1758V | HGMD |
| A178T | HGMD | L342F | HGMD | G53V | HGMD | D609Y | HGMD | R689C* | HGMD | L1761H | HGMD |
| A178P | ClinVar, HGMD | P343R | HGMD | Y54H | HGMD | K610N | HGMD | G709V* | HGMD | L1761F | HGMD |
| K183R | HGMD | P343L | HGMD | S55L | ClinVar, HGMD | Y611D | HGMD | T731I | HGMD | V1763L | HGMD |
| K183M | HGMD | P343S | HGMD | R56Q | HGMD | Y611H | ClinVar, HGMD | Q750R | HGMD | V1763M | ClinVar, HGMD |
| Y184C | HGMD | A344E | HGMD | A57P | HGMD | V612L | HGMD | Q779K | HGMD | M1766L | ClinVar, HGMD |
| Y184H | HGMD | A344V | ClinVar, HGMD | E58A | HGMD | V612M | HGMD | R800L* | HGMD | M1766V | HGMD |
| Y184S | HGMD | G345R | ClinVar, HGMD | E58D | HGMD | T613A | HGMD | R808P | HGMD | Y1767C | HGMD |
| G186R | HGMD | G345E | ClinVar, HGMD | E58G | ClinVar, HGMD | T613K | HGMD | F816Y | HGMD | I1768V | ClinVar, HGMD |
| G189A | HGMD | G345A | HGMD | E58K | HGMD | T613M | ClinVar, HGMD | L828V | HGMD | L1772V | HGMD |
| G189E | HGMD | G345V | HGMD | Q61R | HGMD | A614V | ClinVar, HGMD | N834D | HGMD | N1774D | HGMD |
| G189R | ClinVar, HGMD | L347P | HGMD | R62Q | HGMD | L615F | HGMD | G840R | HGMD | E1781G | HGMD |
| R190Q | ClinVar, HGMD | S349W | ClinVar, HGMD | C64W | HGMD | Y616C | HGMD | T843A | ClinVar, HGMD | E1784K | HGMD |
| R190L | HGMD | S349P | HGMD | C64Y | HGMD | F617L | HGMD | Q912R | HGMD | D1790G | HGMD |
| R190W | HGMD | G350R | HGMD | T65P | ClinVar, HGMD | F617V | HGMD | S941N | ClinVar | Y1795C | ClinVar, HGMD |
| L191P | HGMD | G350V | HGMD | C66G | HGMD | T618S | HGMD | Q960K* | HGMD | P1824A | HGMD |
| R192P | HGMD | F351L | ClinVar | F68L | HGMD | S620N | HGMD | R975W* | HGMD | D1839G | HGMD |
| F193L | HGMD | F351S | HGMD | L69P | HGMD | S620G | HGMD | C981F* | HGMD | R1860S | HGMD |
| R195P | HGMD | L353P | ClinVar, HGMD | H70R | HGMD | S621R | HGMD | P1021S* | HGMD | A1870T | HGMD |
| K196T | HGMD | K354R | HGMD | H70N | HGMD | S621N | HGMD | D1166N* | HGMD | R1897W* | HGMD |
| P197S | HGMD | Q357R | HGMD | G71E | HGMD | T623I | HGMD | R1175C* | HGMD | E1901Q* | HGMD |
| I198V | ClinVar, HGMD | R360M | HGMD | G71R | ClinVar, HGMD | V625A | HGMD | P1177L* | HGMD | A1949S* | HGMD |
| S199A | HGMD | R360T | HGMD | G71W | HGMD | V625E | HGMD | Y1199S* | HGMD | E1954K* | HGMD |
| I200N | HGMD | K362R | HGMD | P72L | ClinVar, HGMD | G626A | HGMD | Y1241S | HGMD | Y1977N* | HGMD |
| D202G | ClinVar | H363N | ClinVar, HGMD | P72T | HGMD | G626D | HGMD | I1278N | HGMD | L1988R* | HGMD |
| D202H | HGMD | N365H | HGMD | T74P | HGMD | G626S | HGMD | N1325S | ClinVar, HGMD | R1991Q* | HGMD |
| L203P | HGMD | R366Q | ClinVar | T74R | ClinVar, HGMD | G626V | HGMD | A1326S | ClinVar, HGMD | F2004V* | HGMD |
| I204M | HGMD | R366P | HGMD | A78V | HGMD | F627I | HGMD | ||||
| I204F | HGMD | R366W | HGMD | A78T | HGMD | F627L | HGMD | ||||
| V205M | ClinVar, HGMD | Q367H | HGMD | A80P | HGMD | G628R | HGMD | ||||
| S209F | HGMD | A371T | HGMD | A85P | HGMD | G628D | HGMD | ||||
| K218E | HGMD | A372D | HGMD | A85V | HGMD | G628S | ClinVar, HGMD | ||||
| T224M | HGMD | S373P | HGMD | L86R | HGMD | G628V | HGMD | ||||
| S225L | HGMD | W379G | HGMD | L86P | HGMD | G628A | HGMD | ||||
| A226V | HGMD | W379S | HGMD | A89V | HGMD | N629D | ClinVar, HGMD | ||||
| I227L | HGMD | R380G | HGMD | E90K | HGMD | N629I | HGMD | ||||
| G229D | HGMD | R380S | HGMD | V94M | HGMD | N629K | HGMD | ||||
| R231C | HGMD | E385K | HGMD | E95G | HGMD | N629S | ClinVar, HGMD | ||||
| R231H | ClinVar, HGMD | S389P | HGMD | I96T | HGMD | N629T | HGMD | ||||
| I235N | ClinVar, HGMD | S389Y | HGMD | F98S | HGMD | V630A | HGMD | ||||
| L236R | HGMD | T391I | HGMD | Y99S | HGMD | V630L | HGMD | ||||
| L236P | HGMD | W392R | HGMD | R100Q | ClinVar, HGMD | S631A | HGMD | ||||
| L239P | HGMD | Y395S | ClinVar | R100W | HGMD | P632A | HGMD | ||||
| V241G | HGMD | K422T* | HGMD | K101E | HGMD | P632S | HGMD | ||||
| D242N | HGMD | T444M* | HGMD | D102A | HGMD | N633D | HGMD | ||||
| D242Y | HGMD | D446E* | HGMD | D102H | HGMD | N633I | HGMD | ||||
| R243C | ClinVar, HGMD | H455Y* | HGMD | D102V | HGMD | N633K | HGMD | ||||
| R243P | ClinVar, HGMD | R511W | HGMD | F106L | HGMD | N633S | HGMD | ||||
| G245V | HGMD | T513S | HGMD | F106Y | HGMD | T634A | HGMD | ||||
| W248C | HGMD | I517T | HGMD | C108R | HGMD | T634I | HGMD | ||||
| L250H | HGMD | M520R | HGMD | C108Y | HGMD | N635D | HGMD | ||||
| L250P | HGMD | Y522S | HGMD | L109R | HGMD | N635I | HGMD | ||||
| L251Q | HGMD | V524G | HGMD | L109P | HGMD | N635K | HGMD | ||||
| L251P | ClinVar, HGMD | A525T | HGMD | D111V | HGMD | E637D | HGMD | ||||
| G252D | HGMD | A525V | HGMD | P114S | HGMD | E637G | HGMD | ||||
| S253C | ClinVar | R539W | ClinVar, HGMD | M124R | HGMD | E637K | ClinVar, HGMD | ||||
| V254M | ClinVar, HGMD | E543K | HGMD | M124T | HGMD | K638N | HGMD | ||||
| V254L | HGMD | S546L | HGMD | F129I | HGMD | K638E | HGMD | ||||
| H258N | HGMD | Q547R | HGMD | D219V* | ClinVar | F640L | ClinVar, HGMD | ||||
| H258P | HGMD | G548D | HGMD | P334L* | ClinVar | F640V | HGMD | ||||
| H258R | ClinVar, HGMD | V554A | HGMD | I400N | HGMD | V644L | HGMD | ||||
| R259C | ClinVar, HGMD | R555C | ClinVar, HGMD | W410S | HGMD | V644F | HGMD | ||||
| R259L | ClinVar, HGMD | R555H | HGMD | L413P | HGMD | M645L | HGMD | ||||
| R259H | HGMD | R555S | HGMD | Y420C | HGMD | M645V | ClinVar, HGMD | ||||
| E261Q | HGMD | K557E | HGMD | A422D | HGMD | G648S | HGMD | ||||
| E261K | HGMD | R561G | HGMD | A422T | HGMD | S649P | HGMD | ||||
| E261V | HGMD | R562M | HGMD | P426H | HGMD | M651R | HGMD | ||||
| L262V | HGMD | R562S | HGMD | Y427C | HGMD | S654G | HGMD | ||||
| T265I | ClinVar, HGMD | L563P | HGMD | Y427H | HGMD | F656C | HGMD | ||||
| L266P | HGMD | S566F | HGMD | Y427S | HGMD | F656L | HGMD | ||||
| G269S | ClinVar, HGMD | S566P | HGMD | S428L | HGMD | G657R | HGMD | ||||
| G269D | ClinVar, HGMD | I567F | HGMD | S428P | HGMD | G657C | HGMD | ||||
| G272D | ClinVar | I567S | HGMD | A429P | HGMD | G657S | ClinVar, HGMD | ||||
| G272V | HGMD | I567T | HGMD | P451L | HGMD | I662T | HGMD | ||||
| L273F | ClinVar, HGMD | G568A | ClinVar, HGMD | D456Y | HGMD | R685H | HGMD | ||||
| L273R | HGMD | G568R | HGMD | L457P | HGMD | H687Y | HGMD | ||||
| F275S | HGMD | K569E* | HGMD | D460Y | HGMD | R694H | HGMD | ||||
| S277L | ClinVar, HGMD | S571L* | HGMD | F463L | HGMD | R696P | HGMD | ||||
| S277P | HGMD | F573L* | HGMD | D466Y | HGMD | S706C | HGMD | ||||
| S277W | HGMD | R583C* | HGMD | N470D | ClinVar, HGMD | A715V | HGMD | ||||
| Y278H | HGMD | R583G* | HGMD | T473N | HGMD | P721L | HGMD | ||||
| Y281C | HGMD | N586D | HGMD | T473P | HGMD | I728F | HGMD | ||||
| L282P | HGMD | N586S | HGMD | T474I | ClinVar, HGMD | R744P | HGMD | ||||
| A283T | HGMD | T587M | ClinVar, HGMD | Y475C | HGMD | R752Q | HGMD | ||||
| A302T | HGMD | G589D | ClinVar, HGMD | E480V | HGMD | R752W | HGMD | ||||
| A302V | HGMD | T587R | HGMD | I489F | HGMD | A753S | HGMD | ||||
| L303P | HGMD | A590T | HGMD | A490P | HGMD | K757N | HGMD | ||||
| W304R | HGMD | R591C | HGMD | A490T | ClinVar, HGMD | D767Y | HGMD | ||||
| W305R | HGMD | R591H | ClinVar, HGMD | H492Y | HGMD | V770A | HGMD | ||||
| W305L | ClinVar, HGMD | R591L | HGMD | Y493C | HGMD | D774Y | HGMD | ||||
| G306R | ClinVar, HGMD | R594Q | HGMD | Y493F | HGMD | G785A | HGMD | ||||
| G306V | HGMD | E596K | HGMD | Y493S | HGMD | G785V | HGMD | ||||
| V308D | HGMD | L602P | HGMD | W497L | HGMD | G785D | HGMD | ||||
| T309R | HGMD | I609N | HGMD | D501N | HGMD | E788D | HGMD | ||||
| T309I | HGMD | D611Y | HGMD | D501G | ClinVar, HGMD | E788K | HGMD | ||||
| V310I | HGMD | G635R* | HGMD | D501H | HGMD | V795I | HGMD | ||||
| T311A | HGMD | G522R | HGMD | G800A | ClinVar | ||||||
| K525N | HGMD | G800E | HGMD | ||||||||
| R528P | HGMD | G800W | HGMD | ||||||||
| R531Q | HGMD | M801I | ClinVar | ||||||||
| R534C | ClinVar, HGMD | D803Y | HGMD | ||||||||
| R534L | HGMD | F805C | HGMD | ||||||||
| R537W | HGMD | F805S | ClinVar, HGMD | ||||||||
| L552S | ClinVar, HGMD | G806E | ClinVar, HGMD | ||||||||
| E544A | HGMD | P815L | HGMD | ||||||||
| A558E | HGMD | G816V | HGMD | ||||||||
| A558P | ClinVar, HGMD | S818L | HGMD | ||||||||
| L559H | HGMD | S818P | HGMD | ||||||||
| A561P | ClinVar, HGMD | S818W | HGMD | ||||||||
| A561T | ClinVar, HGMD | G820E | HGMD | ||||||||
| A561V | HGMD | G820R | ClinVar, HGMD | ||||||||
| H562R | HGMD | V822I | ClinVar | ||||||||
| W563C | HGMD | V822L | HGMD | ||||||||
| W563G | HGMD | V822M | HGMD | ||||||||
| L564P | HGMD | R823W | HGMD | ||||||||
| A565T | HGMD | T826I | ClinVar, HGMD | ||||||||
| C566F | HGMD | R835W | HGMD | ||||||||
| C566S | HGMD | D837N | HGMD | ||||||||
| W568R | HGMD | D837G | HGMD | ||||||||
| W568C | HGMD | D837H | HGMD | ||||||||
| Y569C | HGMD | D837Y | HGMD | ||||||||
| Y569H | HGMD | V841L | HGMD | ||||||||
| I571L | HGMD | P846T | HGMD | ||||||||
| I571M | HGMD | I858T | HGMD | ||||||||
| G572D | HGMD | N861H | HGMD | ||||||||
| G572C | HGMD | N861I | ClinVar, HGMD | ||||||||
Curated mutations classified as pathogenic in the ClinVar (Harrison et al., 2016) and HGMD (Stenson et al., 2012) databases. Mutations were cross-referenced to remove any mutations with conflicting classifications. Asterisked mutations are not mapped onto the channel structures presented in Figures 9–11 because those positions are not resolved in the final models. The Excel file for this table can be found as “Table 1” in the Supplementary Material.
KCNQ1
KCNQ1 mutations studied to date are highly varied in molecular effects, with the potential to induce defects in channel stability, trafficking, electrophysiology, or all three (Chen et al., 2011; Heijman et al., 2012; Wu et al., 2016; Bohnen et al., 2017). So far, a strong disposition for either expression or functional defects has not been elucidated. However, analysis of mutation sites points to regions of increased pathological risk and provides hints regarding prevalent mechanisms.
LQT1-associated mutations are found in every domain of the protein (Figure 9A), particularly in the transmembrane channel domain (Shimizu et al., 2004; Moss et al., 2007; Kapa et al., 2009). Mutations in the SF, (Figure 9A, inset), VSD (Figure 9B), and the S4–S5 linker (Figure 9C) are especially prominent.
Mutations in and around the SF (Figure 9A, inset) point to a potential ion permeation defect, as conformational changes may prevent potassium ion permeation and thus decrease the effective IKs current (Choveau and Shapiro, 2012), as noted in several SF mutations (Ikrar et al., 2008; Thomas et al., 2010; Burgess et al., 2012; Chen et al., 2019). Molecular dynamics simulations of analogous mutations in KcsA indicated that mutations in the SF-interacting residues (including T322M, T322A, or G325R) may disrupt the organization of backbone atoms in the SF, abolishing K+ ion coordination and producing a dominant-negative effect on channel current (Burgess et al., 2012).
VSD mutations are more common in KCNQ1 compared to hERG and SCN5A (see Figures 9B, 10B, and 11A), indicating that the VSD is more important in LQT1. Analysis of LQTS mutations located in the VSD (Huang et al., 2018; Vanoye et al., 2018) indicate that loss of channel function was most commonly a consequence of mutation-induced destabilization of the channel, resulting in mistrafficking and lower surface expression. These results strongly correlated with Rosetta energy calculations of VSD mutants (Kuenze et al., 2019), supporting the importance of VSD destabilization as a LQT1 mechanism. A number of the deleterious mutations were found in the S0 helix and S0-contacting regions of the VSD. Among the 50 VSD mutations studied, the S0 mutations had the largest energetic destabilizations consistent with the large number of contacts formed between S0 and other regions of the VSD. (Huang et al., 2018; Kuenze et al., 2019). However, these studies also identified variants that did not exhibit folding and trafficking defects, but were either non-functional (low conductance) or dysfunctional, with modified gating, again pointing to a complex spectrum of LQT1 disease mechanisms.
Figure 10.

hERG structure with deleterious mutation sites indicated. (A) Full hERG structure (PDB: 5VA1) (Wang and Mackinnon, 2017). Yellow sites denote locations of a single deleterious mutation, while red indicates sites with multiple mutations identified. Spheres indicate C-alpha positions of mutation sites. Subunit with LQT2 mutations sites mapped is colored green. Inset: close-up of SF and pore helix. (B) hERG VSD. (C). Close-up of the S1/S5 interface. Residues at the interface are indicated with sticks (D) N-terminal PAS (dark gray) and C-terminal CNBHD (light gray) domain interface, cartoon representation with surface representation overlay. One subunit is green and the other gray, for clarity.
Mutations in the KCNQ1 S4–S5 linker and its contacting regions (Figure 9C) have the potential to perturb VSD-PD coupling and channel gating. Such effects have been demonstrated for a number of LQTS-associated mutations in the S4–S5 linker and the cytoplasmic end of S6, particularly for R243C, W248F/R, and Q357R, producing positive shifts in the voltage dependence of activation or inactivation and and/or decreased current amplitude (Franqueza et al., 1999; Boulet et al., 2006; Mousavi Nik et al., 2015). Because the S4–S5 linker is involved in both VSD/PD interactions and in binding of PIP2 (Eckey et al., 2014; Sun and Mackinnon, 2017) and KCNE1 (Kang et al., 2008; Xu et al., 2013; Cui, 2016), mutations in this region may affect gating either by weakening VSD-PD coupling or by hindering modulation by auxiliary molecules. Some LQT1 variant-induced changes in PIP2 binding at the S4–S5 linker have been noted (including R243H, R539W, and R555C) (Park et al., 2005). Due to the complex nature of electromechanical coupling in KCNQ1 and the requirement for auxiliary subunits, a detailed structural understanding of mutation effects in this region is particularly challenging. However, the human KCNQ1, KCNQ1-KCNE3, and KCNQ1-KCNE3/PIP2 channel structures (Sun and Mackinnon, 2020) may provide guidance in experimental design to elucidate LQT1-associated coupling defects in this region.
While the majority of LQT1 mutations map to the transmembrane domain, mutations are also found in the CTD, both in the HA/HB helix bundle, where CaM binds, and in the HC and HD coiled-coil domains (Figure 9A). Mutations in HC and HD are particularly prevalent in sites of hydrophobic and electrostatic interactions that stabilize the coil, hinting at channel assembly defects (Figure 9D). Defects in CaM binding and tetramerization cause some forms of LQT1, as demonstrated for several mutations (Kanki et al., 2004; Ghosh S, 2006; Schwake et al., 2006; Wiener et al., 2008).
hERG
Loss-of-function mutations in hERG may lead to folding/trafficking defects, gating defects, or both (Anderson et al., 2014; Smith et al., 2016; Bohnen et al., 2017). However, as many as 90% of hERG loss-of-function mutations appear to cause trafficking defects, independent of which domain is mutated (Smith et al., 2016). As in KCNQ1, pore domain mutations in hERG demonstrated stronger dominant-negative effects in hERG than other regions, hinting that mutations in this region are more severe (Anderson et al., 2014).
Although hERG mutations span the entire protein, they are prominent in the SF (Figure 10A, inset), the extracellular VSD/PD interface between S1 and S5 (Figure 10C), and in the PAS and CNBHD cytoplasmic domains (Figure 10D). Given the inherent instability of the hERG SF (Fan et al., 1999; Stansfeld et al., 2008; Vandenberg et al., 2012) and the functional necessity of the SF for fast inactivation (Herzberg et al., 1998; Hoshi and Armstrong, 2013), mutations may act by both affecting channel stability and inactivation. Inactivation defects due to LQT2-associated pore helix mutations have been identified (Zhao et al., 2009; Poulsen et al., 2015). However, mutations in LQT2 more commonly cause misfolding and/or trafficking defects. Potassium ions appear to stabilize the mature hERG channel (possibly by interacting with the SF) to promote trafficking (Wang et al., 2009; Apaja et al., 2013), with intracellular K+ depletion resulting in ER retention. G601S mimicked this phenotype (Apaja et al., 2013). G601S, a mutation in the pore loop near the SF, mimicked also impedes trafficking through enhanced chaperone association in the ER (Ficker et al., 2003), supporting the idea of SF destabilization as a cause for channel mistrafficking. A detailed understanding of mutations in the SF region requires both intensive structural and cellular analysis to uncover not only protein conformational changes upon mutation, but also cellular quality control responses to these changes.
The VSD of hERG has fewer pathogenic mutations than that of KCNQ1 (Figure 10B), perhaps an indication that the hERG VSD has a higher folding stability. Indeed, when a set of hERG VSD mutations were assessed for membrane insertion efficiency, the majority of mutations were predicted to have no change in the free energy of insertion (Anderson et al., 2014). However, there are a surprising number of disease mutations at the extracellular interface between S1 and S5, specifically at the A422, P426, Y427, S428, H562, W563, C566, and Y569 positions (Figures 10B, C). This interface has not been studied extensively. However, we speculate that it may be important to channel stability by providing an additional contact surface between the VSD and the PD distinct from the S4–S5 linker. Destabilization of this interface may therefore lead to mistrafficking. Mutation Indeed, A422T results in ER retention but was rescued with a pharmacological chaperone (Guo et al., 2012). Other mutations in S1 alter gating properties in addition to trafficking (Anderson et al., 2014; Phan et al., 2017), indicating that gating properties may also be affected. Further studies are needed to determine the impact of mutations in this interface on folding, trafficking, and gating properties of hERG.
A number of mutations are found in the PAS and CNBHD, particularly in the PAS and at the interface between the two domains (Figures 10A, D). The structural stability of both domains impacts trafficking (Akhavan et al., 2005; Harley et al., 2012; Ke et al., 2013; Ke et al., 2014), and several trafficking-defective mutants have been characterized at the PAS/CNBHD interface (F29L, I31S, I42N, Y43C, R56Q, M124R), suggesting that occlusion of a PAS hydrophobic patch from the cytoplasm through interaction with the CNBHD is critical for proper trafficking (Ke et al., 2013). Trafficking defects due to destabilization of the PAS, the CNBHD, or their interaction may thus be a prominent mechanism in LQT2. The direct interaction of these two domains has also been implicated in slow deactivation of the channel (Ng et al., 2014; Kume et al., 2018), as deactivation defects have been noted in channels with mutations at this interface (Anderson et al., 2014). Deactivation defects may be a secondary source of LQT2. Detailed structural analysis of mutation-induced destabilization of the PAS and the CNBHD as well as studies of cellular responses to PAS/CNBHD destabilization would be highly informative to our understanding of LQT2. Indeed, similar studies have already revealed differential cellular responses to LQT2 PAS mutations, depending on the severity of PAS destabilization (Foo et al., 2019).
SCN5A
LQT3 mutations induce channel gain-of-function in SCN5A (Wilde and Amin, 2018; Skinner et al., 2019). Accordingly, mutations that interfere with SCN5A folding and trafficking do not cause LQT3 (Bohnen et al., 2017; Wilde and Amin, 2018). The primary mechanism leading to LQT3 is rather impairment of fast inactivation (Bohnen et al., 2017; Wilde and Amin, 2018). The results of our analysis are consistent with this notion. The site where the S5 and S6 helices of repeats III and IV cross, in the binding pocket for the IFM motif, contains the majority of the mutations curated in this study (Figures 11A, C). These mutations may impact interaction of the IFM motif with other channel structures, hindering fast inactivation and generating a persistent sodium current. For instance, A1326 (A1328 in rSCN5A) and G1329 (G1331 in rSCN5A) make critical backbone interactions to help form a tight hydrophobic pocket for IFM interactions (Jiang et al., 2020). Mutations A1326P and G1329S (Table 1) may thus compromise the stability of this pocket, hindering inactivation. Additionally, S6 mutations surrounding this putative inactivation gate also map to the constriction site of the channel pore, with additional mutations in S6I and S6II (Figure 11D). It has been proposed that binding of the IFM motif causes a rotation in the S6 helices that closes the pore (Yan et al., 2017b). Therefore mutations in S6 may also alter inactivation by preventing helix rotation. Indeed, residues important to defining the constriction site of the Nav1.7 pore cause LQTS upon mutation in SCN5A (L398 and Y1755) (Shen et al., 2019). A few mutations also map to the CTD of SCN5A (Figure 11B), which could alter interactions with the III/IV linker and hinder fast inactivation.
The recent structure of the rat homolog of SCN5A (Jiang et al., 2020) suggests another gain-of-function mechanism, namely that VSD mutant R225P could promote current leak through the VSD (gating pore current). Experimental evidence supporting this mechanism exists for other SCN5A (Moreau et al., 2015) and NaV1.4 (Sokolov et al., 2007) VSD mutations.
Structural Basis for Channel Pharmacology
Pharmacological management of LQTS aims to minimize symptomatic arrhythmia and prevent life-threatening cardiac events. According to consensus guidelines (Zipes et al., 2006; Priori et al., 2013), treatment with β-blockers is beneficial and effective in the case of LQTS diagnosis, but should be considered only after additional diagnostics for carriers of LQTS mutations. β-blockers work by inhibiting β-adrenergic receptors, consequently slowing heart rate and preventing arrhythmias triggered by activation of the sympathetic nervous system (Bohnen et al., 2017). However, the efficacy of β-blockers is strongly genotype- and trigger-specific (Moss et al., 2000; Schwartz et al., 2001; Bohnen et al., 2017). β-blockers reduce the risk of exercise-induced arrhythmia by 78% and 71% in patients with LQT1 and LQT2, respectively, but neither are protective against cardiac events triggered by emotional arousal or during rest (Kim et al., 2010; Goldenberg et al., 2012). Patients with LQT3 tend to experience the highest breakthrough rate for cardiac events (10–15%) with β-blocker treatment (Giudicessi and Ackerman, 2013). Use of local anesthetic-type anti-arrhythmic agents (e.g. mexiletine) for clinical treatment of LQT3, however, can be accompanied by undesirable side effects (Moreno et al., 2011). The need for improved LQTS pharmacotherapy is further supported by the difficulties surrounding hERG modulation, as several clinically-used drugs can cause drug-induced LQTS by blocking hERG and suppressing IKr (Vandenberg et al., 2012; Kalyaanamoorthy and Barakat, 2018). This section summarizes some known pharmacological agents and the modes of interaction with KCNQ1, hERG, and SCN5A. Special focus is dedicated toward unique structural features of hERG that have been implicated in the promiscuity of this channel for pro-arrhythmic drugs.
KCNQ1
KCNQ1 channels are targeted by several non-selective anti-arrhythmic drugs such as quinidine (Balser et al., 1991), amiodarone (Balser et al., 1991; Kamiya et al., 2001), azimilide (Busch et al., 1997), and clofilium (Yang et al., 1997). More specific KCNQ1 blockers include chromanol 293B (Bosch et al., 1998), and the benzodiazepine L-735821 (Lengyel et al., 2001). The binding site of L-735821 has been located to the SF and S6 (Seebohm et al., 2003a).
Benzodiazepine R-L3 was the first KCNQ1 activator to be described. The compound acts to slow the deactivation rate and causes a hyperpolarizing shift in the voltage-dependence of channel activation (Salata et al., 1998). R-L3 appears to bind residues on S5 and S6 lining the membrane-exposed surface of the PD (Seebohm et al., 2003b) rather than to the inner channel cavity. Drug binding outside of the PD cavity was also reported for quinidine (Yang et al., 2013), which occupies a pocket between S6 and the S4–S5 linker and is postulated to elicit an allosteric channel blocking mechanism. More recently, ML277 was identified as a potent KCNQ1 agonist (Mattmann et al., 2012). ML277 has been proposed to bind to a side pocket surrounded by the S2–S3 and S4–S5 linkers on the intracellular side and helices S4 and S6 on the intramembrane lateral side (Xu et al., 2015). Binding of ML277 may selectively alter VSD/PD coupling to stabilize the AO state relative to the IO state (Hou et al., 2019).
hERG
hERG channels can be blocked by a wide spectrum of compounds, leading to drug-induced LQTS (Cubeddu, 2016). The list of drugs that inhibit hERG includes, among others, antibiotics (grepafloxacin (Bischoff et al., 2000)), antihistamines (astemizole (Zhou et al., 1999)), anti-arrhythmics (quinidine (Roden et al., 1986), dofetilide (Jurkiewicz and Sanguinetti, 1993)), antipsychotics (sertindole (Rampe et al., 1998)), and gastroprokinetic agents (cisapride (Vitola et al., 1998)) (Figure 12A). Several prescription drugs have been withdrawn from the market because of cardiotoxic side effects triggered by off-target interaction with hERG (Kalyaanamoorthy and Barakat, 2018). In fact, an estimated 15% of drugs still on the market may cause QT phase prolongation, and 60% of drugs in development show hERG liability (Vandenberg et al., 2017). It is now required by the US FDA that all compounds considered for advancement to clinical trials be tested for their impact on hERG channel function as part of preclinical toxicity assessments (Center for Drug Evaluation and Research, 2005a; Center for Drug Evaluation and Research, I.C.O.H., 2005b).
Figure 12.
Structural features of the drug-binding cavity of the hERG channel pore. (A) Examples of drugs discontinued by the US FDA because of side effects related to hERG inhibition (adapted from Table 1 in (Kalyaanamoorthy and Barakat, 2018)). (B) Internal molecular surface of the central cavity below the SF in hERG (PDB: 5VA1)(Wang and Mackinnon, 2017) (top) and the KV1.2–2.1 chimeric channel (PDB: 2R9R)(Long et al., 2007) (bottom). The surface is colored by the electrostatic potential calculated with the APBS tool in PyMOL (The PyMOL Molecular Graphics System, Version 2.2 Schrödinger, LLC.). Residues related to drug binding by hERG and the corresponding residues in the KV1.2–2.1 structure are shown as sticks. This panel is inspired by Figure 5 in (Wang and Mackinnon, 2017).
Considerable progress has been made in understanding the basic features of drug binding to hERG, and a plausible mechanism for promiscuity of drug binding has emerged. Pro-arrhythmic drugs commonly bind to the central inner cavity of the PD in hERG. Two pore-lining aromatic residues in S6 (Y652 and F656) are the most crucial for drug binding (Mitcheson et al., 2000), although additional residues at the end of the pore helix (T623, S624, and V625) contribute to binding for some drugs (Mitcheson et al., 2000), and mutations at F557 in the S5 helix can also affect binding (Saxena et al., 2016). The side chains of the S6 and pore helix residues point towards a putative drug binding site located in the central cavity below the SF. This cavity is slightly narrower in hERG than in the Shaker family KV1.2–2.1 chimeric channel (Long et al., 2007). As a consequence, there is a greater negative electrostatic potential in this region (Wang and Mackinnon, 2017), which could enhance drug binding capabilities (Figure 12B). In addition, four lateral hydrophobic pockets, which are not observed in the KV1.2–2.1 structure, extend outwards from that central constriction site, generating additional space to trap chemical moieties (Wang and Mackinnon, 2017).
Quantitative structure-activity relationship studies have identified a pharmacophore for drugs that bind to hERG (Cavalli et al., 2002). The pharmacophore consists of three centers of mass (usually aromatic rings) and an amino group that together form a flattened tetrahedron. It has been suggested that the aromatic rings could fit into one or more lateral hydrophobic pockets in hERG and/or bind by pi-pi stacking or hydrophobic interactions with Y652 and F656 (Chen et al., 2002). Likewise, the greater negative electrostatic potential in the central cavity could explain why many drugs that bind hERG, including off-targets, contain a positive charge (Fernandez et al., 2004). Additionally, a number of docking models for drug-bound hERG, with and without this pharmacophore, are being generated (Wacker et al., 2017; Helliwell et al., 2018; Negami et al., 2019), making possible further delineation of drug binding modes within this pocket and the structural basis of their affinity for hERG.
The majority of high-affinity drugs that bind hERG off-target preferentially bind to the inactivated state rather the open state, with affinity differences ranging from 2- to 100-fold (Perrin et al., 2008). Interestingly, the related EAG1 channel (Whicher and Mackinnon, 2016) is less sensitive to inhibition by most of hERG-blocking drugs even though they share the same pore-lining aromatic residues. One major difference between hERG and EAG1 is that EAG1 channels undergo minimal inactivation (Garg et al., 2012) whereas hERG channels undergo rapid and complete inactivation (Vandenberg et al., 2012). The contribution of inactivation to high-affinity binding is further substantiated by the observation that inactivating EAG1–hERG chimeras, containing the upper half of the hERG PD, can bind drugs with almost hERG-like affinity (Herzberg et al., 1998). While cryo-EM structures have been determined for both hERG and EAG1 (Whicher and Mackinnon, 2016; Wang and Mackinnon, 2017), the structural basis of the functional and pharmacological differences requires higher resolution (Wang and Mackinnon, 2017).
SCN5A
Limited progress has been made in the development of SCN5A-targeted treatment strategies. Classical non-selective sodium channel blockers like the Vaughan-Williams class I anti-arrhythmic drugs quinidine, lidocaine, and propafenone have only limited clinical applicability. Ranolazine is a more selective SCN5A blocker (Belardinelli et al., 2006; Zaza et al., 2008) and has been shown to attenuate action potential duration and reduce or prevent intracellular calcium overload in LQT3 models (Wu et al., 2004; Lindegger et al., 2009). Mexiletine is a potent inhibitor of SCN5A and may be beneficial for treatment of LQT3 (Mazzanti et al., 2016), but effectiveness may depend on the baseline QT prolongation of the mutant channel (Li and Zhang, 2018).
Despite the clinical relevance of SCN5A-targeting drugs, the molecular mechanisms of promising and potentially problematic drugs are not well understood at a structural level, hindering drug design. However, key residues for apparent drug binding have been suggested within the sodium channel pore domain. Specifically, mutations of two conserved aromatic residues in S6IV, F1760 and Y1767, (Ragsdale et al., 1994; Ragsdale et al., 1996) or other S6 residues in repeats I and III (Yarov-Yarovoy et al., 2001; Yarov-Yarovoy et al., 2002) may affect drug binding. Using fluorinated phenylalanine derivatives incorporated at F1760 (Pless et al., 2011a), a strong cation-pi interaction between F1760 and the protonated amine group in class Ib anti-arrhythmic drugs was observed, defining the molecular basis for inhibition of SCN5A by this group of drugs. Additionally, mutations within the SF region can affect apparent drug binding either by enhancing slow inactivation or forming an alternative access pathway (Sunami et al., 1997; Lee et al., 2001; Tsang et al., 2005). Interactions of anti-arrhythmic and local anesthetic drugs (lidocaine, QX-314, etidocaine, flecainide, and ranolazine) docked into an inactivated state model of SCN5A (Nguyen et al., 2019) revealed several key drug binding sites in the inner pore lumen that can simultaneously accommodate up to two drug molecules. Subsequent MD simulations suggested alternative access mechanisms for drugs into the SCN5A lumen – a hydrophilic pathway through the intracellular gate and a hydrophobic pathway through a membrane-exposed fenestration between repeats III and IV.
The recent structure of rat Nav1.5 (Jiang et al., 2020) has provided insight into the binding mode of the class Ic anti-arrhythmic drug flecainide, which has higher affinity for the open conformation of SCN5A (Ramos and O’leary M, 2004). Flecainide binds in the central cavity on the intracellular side of the SF. The piperidine ring of flecainide lies across the top of the central cavity with the positively-charged nitrogen pointing towards the exit of the SF and its hydrophobic edge extending towards the phenyl ring of F1762 (F1760 in human Nav1.5). The trifluoroethoxy tails of flecainide extend into fenestrations formed between the pore helices of repeats I and II as well as repeats II and III, through which the central cavity is accessible from the lipid bilayer. These fenestrations represent possible entry points for flecainide and similar class Ic drugs.
Future Directions
The growing number of structural and functional studies of KCNQ1, hERG, and SCN5A have provided new insight into channel molecular physiology and channel dysfunction in congenital LQTS. This further motivates the study of fundamental biophysical mechanisms in ion channels. One of these mechanisms, electromechanical coupling, remains enigmatic due to a dearth of experimental resting state structures and robust methods to identify allosteric pathways, which cannot easily be discerned from ion channel structures resolved in a single conformation. However, interaction energy and MD dynamical network analysis in the Shaker K+ channel (Fernandez-Marino et al., 2018) has suggested that movements in the VSD and PD are linked by two distinct pathways – a canonical pathway through the S4–S5 linker and a hitherto unknown non-canonical pathway involving contacts at the interface between S4 and S5. Such work may provide a useful alternative approach to explore electromechanical coupling in detail.
Another major effort involves the experimental characterization of hundreds of previously uncharacterized variants of unknown significance (VUS) and LQTS mutations in KCNQ1, hERG, and SCN5A. Widespread use of whole genome and exome sequencing has provided a nearly complete catalog of common sequence variations in protein-coding genes (Lek et al., 2016), although rare sequence variants continue to be discovered. For many LQTS mutations, the molecular mechanisms responsible for impaired channel function remain poorly understood. Such mutations may cause potassium channel loss-of-function or dysfunction by promoting misfolding, mistrafficking, aggregation, or improper gating of the channel protein, as discussed above. Continued experimental testing will be required to complete functional annotation of KCNQ1, hERG, and SCN5A variants and to elucidate how these mutations lead to channel dysfunction. In this regard, new techniques such as high-throughput patch clamp recording (Vanoye et al., 2018; Toh et al., 2020), which allows rapid screening of ion channel variants found in patients, and the use of induced-pluripotent stem cell-derived cardiomyocytes (Moretti et al., 2010; Ma et al., 2013; Terrenoire et al., 2013; Jouni et al., 2015) developed from cells of healthy or mutation-carrying patients, represent helpful tools for large-scale examination of ion channel variants.
Finally, experimentally-informed bioinformatics and modeling approaches are being developed to predict variant pathogenicity (Kroncke et al., 2015; Li et al., 2017; Kroncke et al., 2019). These computational algorithms can help to decrypt newly discovered VUS for which there is not enough data to determine pathogenicity. Decrypting VUS can inform medical practice and has the potential to improve LQTS therapy, both for the prevention of cardiac events in susceptible patients and as a basis for avoiding unneeded treatment in healthy patients. With improved understanding of the molecular pathophysiology and the role of mutations associated with distinct LQTS subtypes, the ultimate hope is that identifying a patient’s genotype will lead to more specific treatment considerations and the delivery of optimal care.
Author Contributions
KB and GK wrote the primary draft of this article, with edits by KB, GK, JM, CV, AG, and CS. KB and GK contributed equally to this manuscript.
Funding
This work was supported by US NIH grant RO1 HL122010. GK was supported by a postdoctoral fellowship from the American Heart Association (18POST34080422) and KB was supported by NIH training grant T32 GM008320.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00550/full#supplementary-material
LQTS mutations in KCNQ1, hERG, and SCN5A.
References
- Abbott G. W., Sesti F., Splawski I., Buck M. E., Lehmann M. H., Timothy K. W., et al. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187. 10.1016/S0092-8674(00)80728-X [DOI] [PubMed] [Google Scholar]
- Abbott G. W., Xu X., Roepke T. K. (2007). Impact of ancillary subunits on ventricular repolarization. J. Electrocardiol. 40, S42–S46. 10.1016/j.jelectrocard.2007.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott G. W. (2014). Biology of the KCNQ1 Potassium Channel. New J. Sci. 2014, 26. 10.1155/2014/237431 [DOI] [Google Scholar]
- Ackerman M. J. (2005). Genotype-phenotype relationships in congenital long QT syndrome. J. Electrocardiol. 38, 64–68. 10.1016/j.jelectrocard.2005.06.018 [DOI] [PubMed] [Google Scholar]
- Adler A., Novelli V., Amin A. S., Abiusi E., Care M., Nannenberg E. A., et al. (2020). An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 141, 418–428. 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S. K., Mackinnon R. (1996). Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 16, 1169–1177. 10.1016/S0896-6273(00)80143-9 [DOI] [PubMed] [Google Scholar]
- Ahern C. A., Payandeh J., Bosmans F., Chanda B. (2016). The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24. 10.1085/jgp.201511492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhavan A., Atanasiu R., Noguchi T., Han W., Holder N., Shrier A. (2005). Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J. Cell Sci. 118, 2803–2812. 10.1242/jcs.02423 [DOI] [PubMed] [Google Scholar]
- Anantharam A., Abbott G. W. (2005). Does hERG coassemble with a beta subunit? Evidence for roles of MinK and MiRP1. Novartis Found Symp 266, 100–112discussion 112–107, 155-108. 10.1002/047002142X.ch9 [DOI] [PubMed] [Google Scholar]
- Anderson C. L., Kuzmicki C. E., Childs R. R., Hintz C. J., Delisle B. P., January C. T. (2014). Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 5, 5535. 10.1038/ncomms6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaja P. M., Foo B., Okiyoneda T., Valinsky W. C., Barriere H., Atanasiu R., et al. (2013). Ubiquitination-dependent quality control of hERG K+ channel with acquired and inherited conformational defect at the plasma membrane. Mol. Biol. Cell 24, 3787–3804. 10.1091/mbc.e13-07-0417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser J. R., Bennett P. B., Hondeghem L. M., Roden D. M. (1991). Suppression of Time-Dependent Outward Current in Guinea-Pig Ventricular Myocytes - Actions of Quinidine and Amiodarone. Circ. Res. 69, 519–529. 10.1161/01.RES.69.2.519 [DOI] [PubMed] [Google Scholar]
- Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996). K(v)LQT1 and IsK (minK) proteins associate to form the I-Ks cardiac potassium current. Nature 384, 78–80. 10.1038/384078a0 [DOI] [PubMed] [Google Scholar]
- Baroni D., Picco C., Barbieri R., Moran O. (2014). Antisense-mediated post-transcriptional silencing of SCN1B gene modulates sodium channel functional expression. Biol. Cell 106, 13–29. 10.1111/boc.201300040 [DOI] [PubMed] [Google Scholar]
- Barros F., Dominguez P., De La Pena P. (2018). Relative positioning of Kv11.1 (hERG) K(+) channel cytoplasmic domain-located fluorescent tags toward the plasma membrane. Sci. Rep. 8, 15494. 10.1038/s41598-018-33492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros F., Pardo L. A., Dominguez P., Sierra L. M., De La Pena P. (2019). New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 20, pii: E248. 10.3390/ijms20020248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro-Soria R., Rebolledo S., Liin S. I., Perez M. E., Sampson K. J., Kass R. S., et al. (2014). KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat. Commun. 5, 3750. 10.1038/ncomms4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro-Soria R., Ramentol R., Liin S. I., Perez M. E., Kass R. S., Larsson H. P. (2017). KCNE1 and KCNE3 modulate KCNQ1 channels by affecting different gating transitions. Proc. Natl. Acad. Sci. U. S. A 114, E7367–E7376. 10.1073/pnas.1710335114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L., Shryock J. C., Fraser H. (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92 Suppl 4, iv6–iv14. 10.1136/hrt.2005.078790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo-Seisdedos G., Nunez E., Gomis-Perez C., Malo C., Villarroel A., Millet O. (2018). Structural basis and energy landscape for the Ca(2+) gating and calmodulation of the Kv7.2 K(+) channel. Proc. Natl. Acad. Sci. U. S. A 115, 2395–2400. 10.1073/pnas.1800235115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. (2005). Voltage-gated ion channels. IEEE Trans. Nanobiosci. 4, 34–48. 10.1109/TNB.2004.842463 [DOI] [PubMed] [Google Scholar]
- Bhate M. P., Wylie B. J., Tian L., Mcdermott A. E. (2010). Conformational dynamics in the selectivity filter of KcsA in response to potassium ion concentration. J. Mol. Biol. 401, 155–166. 10.1016/j.jmb.2010.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J., Cui J., Mcdonald T. V. (2001). HERG K(+) channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ. Res. 89, 1168–1176. 10.1161/hh2401.101375 [DOI] [PubMed] [Google Scholar]
- Bian J. S., Kagan A., Mcdonald T. V. (2004). Molecular analysis of PIP2 regulation of HERG and IKr. Am. J. Physiol. Heart Circ. Physiol. 287, H2154–H2163. 10.1152/ajpheart.00120.2004 [DOI] [PubMed] [Google Scholar]
- Bidaud I., Lory P. (2011). Hallmarks of the channelopathies associated with L-type calcium channels: a focus on the Timothy mutations in Ca(v)1.2 channels. Biochimie 93, 2080–2086. 10.1016/j.biochi.2011.05.015 [DOI] [PubMed] [Google Scholar]
- Bischoff U., Schmidt C., Netzer R., Pongs O. (2000). Effects of fluoroquinolones on HERG currents. Eur. J. Pharmacol. 406, 341–343. 10.1016/S0014-2999(00)00693-2 [DOI] [PubMed] [Google Scholar]
- Bohnen M. S., Peng G., Robey S. H., Terrenoire C., Iyer V., Sampson K. J., et al. (2017). Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 97, 89–134. 10.1152/physrev.00008.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland L. M., Drzewiecki M. M. (2008). Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem. Biophys. 52, 59–84. 10.1007/s12013-008-9027-2 [DOI] [PubMed] [Google Scholar]
- Bosch R. F., Gaspo R., Busch A. E., Lang H. J., Li G. R., Nattel S. (1998). Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc. Res. 38, 441–450. 10.1016/S0008-6363(98)00021-2 [DOI] [PubMed] [Google Scholar]
- Boulet I. R., Raes A. L., Ottschytsch N., Snyders D. J. (2006). Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc. Res. 70, 466–474. 10.1016/j.cardiores.2006.02.006 [DOI] [PubMed] [Google Scholar]
- Boulet I. R., Labro A. J., Raes A. L., Snyders D. J. (2007). Role of the S6 C-terminus in KCNQ1 channel gating. J. Physiol. 585, 325–337. 10.1113/jphysiol.2007.145813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W. J., Isom L. L. (2011). Na Channel beta Subunits: Overachievers of the Ion Channel Family. Front. Pharmacol. 2, 53. 10.3389/fphar.2011.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess D. E., Bartos D. C., Reloj A. R., Campbell K. S., Johnson J. N., Tester D. J., et al. (2012). High-risk long QT syndrome mutations in the Kv7.1 (KCNQ1) pore disrupt the molecular basis for rapid K(+) permeation. Biochemistry 51, 9076–9085. 10.1021/bi3009449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A. E., Busch G. L., Ford E., Suessbrich H., Lang H. J., Greger R., et al. (1997). The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br. J. Pharmacol. 122, 187–189. 10.1038/sj.bjp.0701434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun J. D., Isom L. L. (2014). The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb. Exp. Pharmacol. 221, 51–89. 10.1007/978-3-642-41588-3_4 [DOI] [PubMed] [Google Scholar]
- Capes D. L., Goldschen-Ohm M. P., Arcisio-Miranda M., Bezanilla F., Chanda B. (2013). Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 142, 101–112. 10.1085/jgp.201310998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. (1986). Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55, 953–985. 10.1146/annurev.bi.55.070186.004513 [DOI] [PubMed] [Google Scholar]
- Cavalli A., Poluzzi E., De Ponti F., Recanatini M. (2002). Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 45, 3844–3853. 10.1021/jm0208875 [DOI] [PubMed] [Google Scholar]
- Chakrabarti N., Ing C., Payandeh J., Zheng N., Catterall W. A., Pomes R. (2013). Catalysis of Na+ permeation in the bacterial sodium channel Na(V)Ab. Proc. Natl. Acad. Sci. U. S. A 110, 11331–11336. 10.1073/pnas.1309452110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P. J., Osteen J. D., Xiong D., Bohnen M. S., Doshi D., Sampson K. J., et al. (2012). Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J. Gen. Physiol. 139, 135–144. 10.1085/jgp.201110672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B., Bezanilla F. (2002). Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120, 629–645. 10.1085/jgp.20028679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A., Abderemane-Ali F., Hura G. L., Rossen N. D., Gate R. E., Minor D. L., Jr. (2018). A Calmodulin C-Lobe Ca(2+)-Dependent Switch Governs Kv7 Channel Function. Neuron 97, 836–852 e836. 10.1016/j.neuron.2018.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Seebohm G., Sanguinetti M. C. (2002). Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. U. S. A 99, 12461–12466. 10.1073/pnas.192367299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Weber M., Um S. Y., Walsh C. A., Tang Y., Mcdonald T. V. (2011). A dual mechanism for I(Ks) current reduction by the pathogenic mutation KCNQ1-S277L. Pacing Clin. Electrophysiol. 34, 1652–1664. 10.1111/j.1540-8159.2011.03190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zhang Q., Qiu Y., Li Z., Chen Z., Jiang H., et al. (2015). Migration of PIP2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep. 5, 15079. 10.1038/srep15079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. M., Guo K., Li H., Lu Q. F., Yang C., Yu Y., et al. (2019). A novel mutation KCNQ1p.Thr312del is responsible for long QT syndrome type 1. Heart Vessels 34, 177–188. 10.1007/s00380-018-1223-4 [DOI] [PubMed] [Google Scholar]
- Cheng Y. M., Claydon T. W. (2012). Voltage-dependent gating of HERG potassium channels. Front. Pharmacol. 3, 83. 10.3389/fphar.2012.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. H., Kodama I. (2004). Two components of delayed rectifier K+ current in heart: molecular basis, functional diversity, and contribution to repolarization. Acta Pharmacol. Sin. 25, 137–145. [PubMed] [Google Scholar]
- Choveau F. S., Shapiro M. S. (2012). Regions of KCNQ K(+) channels controlling functional expression. Front. Physiol. 3, 397. 10.3389/fphys.2012.00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D. Y., Chan P. J., Bankston J. R., Yang L., Liu G., Marx S. O., et al. (2009). Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. U. S. A 106, 743–748. 10.1073/pnas.0811897106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairfeuille T., Cloake A., Infield D. T., Llongueras J. P., Arthur C. P., Li Z. R., et al. (2019). Structural basis of alpha-scorpion toxin action on Nav channels. Science 363, pii: eaav8573. 10.1126/science.aav8573 [DOI] [PubMed] [Google Scholar]
- Codding S. J., Trudeau M. C. (2019). The hERG potassium channel intrinsic ligand regulates N- and C-terminal interactions and channel closure. J. Gen. Physiol. 151, 478–488. 10.1085/jgp.201812129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubeddu L. X. (2016). Drug-induced Inhibition and Trafficking Disruption of ion Channels: Pathogenesis of QT Abnormalities and Drug-induced Fatal Arrhythmias. Curr. Cardiol. Rev. 12, 141–154. 10.2174/1573403X12666160301120217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello L. G., Jogini V., Cortes D. M., Perozo E. (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature 466, 203–208. 10.1038/nature09153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. (2016). Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys. J. 110, 14–25. 10.1016/j.bpj.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Pena P., Alonso-Ron C., Machin A., Fernandez-Trillo J., Carretero L., Dominguez P., et al. (2011). Demonstration of physical proximity between the N terminus and the S4-S5 linker of the human ether-a-go-go-related gene (hERG) potassium channel. J. Biol. Chem. 286, 19065–19075. 10.1074/jbc.M111.238899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani-Samani A., Madreseh-Ghahfarokhi S., Dehghani-Samani A. (2019). Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias. Acta Cardiol. Sin. 35, 99–110. 10.6515/ACS.201903_35(2).20181028A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detta N., Frisso G., Salvatore F. (2015). The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 1854, 1502–1509. 10.1016/j.bbapap.2015.07.009 [DOI] [PubMed] [Google Scholar]
- Dhar Malhotra J., Chen C., Rivolta I., Abriel H., Malhotra R., Mattei L. N., et al. (2001). Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103, 1303–1310. 10.1161/01.CIR.103.9.1303 [DOI] [PubMed] [Google Scholar]
- Doolan G. K., Panchal R. G., Fonnes E. L., Clarke A. L., Williams D. A., Petrou S. (2002). Fatty acid augmentation of the cardiac slowly activating delayed rectifier current (IKs) is conferred by hminK. FASEB J. 16, 1662–1664. 10.1096/fj.02-0084fje [DOI] [PubMed] [Google Scholar]
- Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. 10.1126/science.280.5360.6923 [DOI] [PubMed] [Google Scholar]
- Earle N., Crawford J., Smith W., Hayes I., Shelling A., Hood M., et al. (2013). Community detection of long QT syndrome with a clinical registry: an alternative to ECG screening programs? Heart Rhythm. 10, 233–238. 10.1016/j.hrthm.2012.10.043 [DOI] [PubMed] [Google Scholar]
- Eckey K., Wrobel E., Strutz-Seebohm N., Pott L., Schmitt N., Seebohm G. (2014). Novel Kv7.1-phosphatidylinositol 4,5-bisphosphate interaction sites uncovered by charge neutralization scanning. J. Biol. Chem. 289, 22749–22758. 10.1074/jbc.M114.589796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J. S., Jiang M., Dun W., Mcdonald T. V., Tseng G. N. (1999). Effects of outer mouth mutations on hERG channel function: a comparison with similar mutations in the Shaker channel. Biophys. J. 76, 3128–3140. 10.1016/S0006-3495(99)77464-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre I., Moczydlowski E., Schild L. (1996). On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys. J. 71, 3110–3125. 10.1016/S0006-3495(96)79505-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T., Cordero-Morales J. F., Arias M., Ficker E., Medovoy D., Perozo E., et al. (2011). Molecular coupling in the human ether-a-go-go-related gene-1 (hERG1) K+ channel inactivation pathway. J. Biol. Chem. 286 (45), 39091–39099. 10.1074/jbc.M111.292060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D., Ghanta A., Kauffman G. W., Sanguinetti M. C. (2004). Physicochemical features of the HERG channel drug binding site. J. Biol. Chem. 279, 10120–10127. 10.1074/jbc.M310683200 [DOI] [PubMed] [Google Scholar]
- Fernandez-Marino A. I., Harpole T. J., Oelstrom K., Delemotte L., Chanda B. (2018). Gating interaction maps reveal a noncanonical electromechanical coupling mode in the Shaker K(+) channel. Nat. Struct. Mol. Biol. 25, 320–326. 10.1038/s41594-018-0047-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E., Jarolimek W., Brown A. M. (2001). Molecular determinants of inactivation and dofetilide block in ether a-go-go (EAG) channels and EAG-related K(+) channels. Mol. Pharmacol. 60, 1343–1348. 10.1124/mol.60.6.1343 [DOI] [PubMed] [Google Scholar]
- Ficker E., Dennis A. T., Wang L., Brown A. M. (2003). Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 92, e87–100. 10.1161/01.RES.0000079028.31393.15 [DOI] [PubMed] [Google Scholar]
- Foo B., Barbier C., Guo K., Vasantharuban J., Lukacs G. L., Shrier A. (2019). Mutation-specific peripheral and ER quality control of hERG channel cell-surface expression. Sci. Rep. 9, 6066. 10.1038/s41598-019-42331-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L., Lin M., Shen J., Splawski I., Keating M. T., Sanguinetti M. C. (1999). Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274, 21063–21070. 10.1074/jbc.274.30.21063 [DOI] [PubMed] [Google Scholar]
- Gabelli S. B., Boto A., Kuhns V. H., Bianchet M. A., Farinelli F., Aripirala S., et al. (2014). Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat. Commun. 5, 5126. 10.1038/ncomms6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardill B. R., Rivera-Acevedo R. E., Tung C. C., Van Petegem F. (2019). Crystal structures of Ca(2+)-calmodulin bound to NaV C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc. Natl. Acad. Sci. U. S. A 116, 10763–10772. 10.1073/pnas.1818618116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V., Sachse F. B., Sanguinetti M. C. (2012). Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule. J. Gen. Physiol. 140, 307–324. 10.1085/jgp.201210826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A. L., Jr. (2013). Molecular and genetic basis of sudden cardiac death. J. Clin. Invest. 123, 75–83. 10.1172/JCI62928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Nunziato D. A., Pitt G. S. (2006). KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 98, 1048–1054. 10.1161/01.RES.0000218863.44140.f2 [DOI] [PubMed] [Google Scholar]
- Ghovanloo M. R., Aimar K., Ghadiry-Tavi R., Yu A., Ruben P. C. (2016). Physiology and Pathophysiology of Sodium Channel Inactivation. Curr. Top. Membr. 78, 479–509. 10.1016/bs.ctm.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Giudicessi J. R., Ackerman M. J. (2013). Genotype- and phenotype-guided management of congenital long QT syndrome. Curr. Probl. Cardiol. 38, 417–455. 10.1016/j.cpcardiol.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gofman Y., Shats S., Attali B., Haliloglu T., Ben-Tal N. (2012). How does KCNE1 regulate the Kv7.1 potassium channel? Model-structure, mutations, and dynamics of the Kv7.1-KCNE1 complex. Structure 20, 1343–1352. 10.1016/j.str.2012.05.016 [DOI] [PubMed] [Google Scholar]
- Goldenberg I., Thottathil P., Lopes C. M., Moss A. J., Mcnitt S., O-Uchi J., et al. (2012). Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm. 9, 49–56. 10.1016/j.hrthm.2011.08.020 [DOI] [PubMed] [Google Scholar]
- Goldschen-Ohm M. P., Capes D. L., Oelstrom K. M., Chanda B. (2013). Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat. Commun. 4, 1350. 10.1038/ncomms2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon E., Panaghie G., Deng L., Bee K. J., Roepke T. K., Krogh-Madsen T., et al. (2008). A KCNE2 mutation in a patient with cardiac arrhythmia induced by auditory stimuli and serum electrolyte imbalance. Cardiovasc. Res. 77, 98–106. 10.1093/cvr/cvm030 [DOI] [PubMed] [Google Scholar]
- Grant A. O. (2009). Cardiac ion channels. Circ. Arrhythm Electrophysiol. 2, 185–194. 10.1161/CIRCEP.108.789081 [DOI] [PubMed] [Google Scholar]
- Guo J., Zhang X., Hu Z., Zhuang Z., Zhu Z., Chen Z., et al. (2012). A422T mutation in HERG potassium channel retained in ER is rescurable by pharmacologic or molecular chaperones. Biochem. Biophys. Res. Commun. 422, 305–310. 10.1016/j.bbrc.2012.04.153 [DOI] [PubMed] [Google Scholar]
- Gustina A. S., Trudeau M. C. (2011). hERG potassium channel gating is mediated by N- and C-terminal region interactions. J. Gen. Physiol. 137, 315–325. 10.1085/jgp.201010582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitin Y., Attali B. (2008). The C-terminus of Kv7 channels: a multifunctional module. J. Physiol. 586, 1803–1810. 10.1113/jphysiol.2007.149187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox J. C., Whittaker D. G., Du C., Stuart A. G., Zhang H. (2018). Emerging therapeutic targets in the short QT syndrome. Expert Opin. Ther. Targets 22, 439–451. 10.1080/14728222.2018.1470621 [DOI] [PubMed] [Google Scholar]
- Harley C. A., Jesus C. S., Carvalho R., Brito R. M., Morais-Cabral J. H. (2012). Changes in channel trafficking and protein stability caused by LQT2 mutations in the PAS domain of the HERG channel. PloS One 7, e32654. 10.1371/journal.pone.0032654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S. M., Riggs E. R., Maglott D. R., Lee J. M., Azzariti D. R., Niehaus A., et al. (2016). Using ClinVar as a Resource to Support Variant Interpretation. Curr. Protoc. Hum. Genet. 89 8 16, 11–18 16 23. 10.1002/0471142905.hg0816s89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J., Spatjens R. L., Seyen S. R., Lentink V., Kuijpers H. J., Boulet I. R., et al. (2012). Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ. Res. 110, 211–219. 10.1161/CIRCRESAHA.111.249482 [DOI] [PubMed] [Google Scholar]
- Heitzmann D., Grahammer F., Von Hahn T., Schmitt-Graff A., Romeo E., Nitschke R., et al. (2004). Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. 561, 547–557. 10.1113/jphysiol.2004.075168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell M. V., Zhang Y., El Harchi A., Du C., Hancox J. C., Dempsey C. E. (2018). Structural implications of hERG K(+) channel block by a high-affinity minimally structured blocker. J. Biol. Chem. 293, 7040–7057. 10.1074/jbc.RA117.000363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry J. T., Crosson S. (2011). Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 65, 261–286. 10.1146/annurev-micro-121809-151631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg I. M., Trudeau M. C., Robertson G. A. (1998). Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J. Physiol. 511 ( Pt 1), 3–14. 10.1111/j.1469-7793.1998.003bi.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzik M. A., Jr., Fraser J. S., Lander G. C. (2019). A Multi-model Approach to Assessing Local and Global Cryo-EM Map Quality. Structure 27 344-358, e343. 10.1016/j.str.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. (1971). The hydration of sodium ions crossing the nerve membrane. Proc. Natl. Acad. Sci. U. S. A 68, 280–282. 10.1073/pnas.68.2.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoosien M., Ahearn M. E., Myerburg R. J., Pham T. V., Miller T. E., Smets M. J., et al. (2013). Dysfunctional potassium channel subunit interaction as a novel mechanism of long QT syndrome. Heart Rhythm. 10, 728–737. 10.1016/j.hrthm.2012.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Armstrong C. M. (2013). C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J. Gen. Physiol. 141, 151–160. 10.1085/jgp.201210888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini S. M., Kim R., Udupa S., Costain G., Jobling R., Liston E., et al. (2018). Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 138, 1195–1205. 10.1161/CIRCULATIONAHA.118.035070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini S. M. (2018). A useful hand mnemonic to demonstrate ion channel gating. Adv. Physiol. Educ. 42, 321–323. 10.1152/advan.00080.2017 [DOI] [PubMed] [Google Scholar]
- Hou P., Eldstrom J., Shi J., Zhong L., Mcfarland K., Gao Y., et al. (2017). Inactivation of KCNQ1 potassium channels reveals dynamic coupling between voltage sensing and pore opening. Nat. Commun. 8, 1730. 10.1038/s41467-017-01911-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P., Shi J., White K. M., Gao Y., Cui J. (2019). ML277 specifically enhances the fully activated open state of KCNQ1 by modulating VSD-pore coupling. Elife 8, e48576. 10.7554/eLife.48576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P., Kang P. W., Kongmeneck A. D., Yang N. D., Liu Y., Shi J., et al. (2020). Two-stage electro-mechanical coupling of a KV channel in voltage-dependent activation. Nat. Commun. 11, 676. 10.1038/s41467-020-14406-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovey L., Fowler C. A., Mahling R., Lin Z., Miller M. S., Marx D. C., et al. (2017). Calcium triggers reversal of calmodulin on nested anti-parallel sites in the IQ motif of the neuronal voltage-dependent sodium channel NaV1.2. Biophys. Chem. 224, 1–19. 10.1016/j.bpc.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R. J., Clark K. A., Holton J. M., Minor D. L., Jr. (2007). Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53, 663–675. 10.1016/j.neuron.2007.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Barajas-Martinez H., Burashnikov E., Springer M., Wu Y., Varro A., et al. (2009). A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278. 10.1161/CIRCGENETICS.108.829192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Barajas-Martinez H., Medeiros-Domingo A., Crotti L., Veltmann C., Schimpf R., et al. (2012). A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. 9, 760–769. 10.1016/j.hrthm.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Kuenze G., Smith J. A., Taylor K. C., Duran A. M., Hadziselimovic A., et al. (2018). Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 4, eaar2631. 10.1126/sciadv.aar2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research (2005. a). “E14: clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-arrhythmic drugs”, (ed.) U.S.F.a.D. Administration.).
- Center for Drug Evaluation and Research (2005. b). “S7B: The non-clinical evaluation of the potential for delayed ventricular depolarization (QT interval prolongation) by human pharmaceruticals”, (ed.) U.S.F.a.D. Administration.)
- Ikrar T., Hanawa H., Watanabe H., Okada S., Aizawa Y., Ramadan M. M., et al. (2008). A double-point mutation in the selectivity filter site of the KCNQ1 potassium channel results in a severe phenotype, LQT1, of long QT syndrome. J. Cardiovasc. Electrophysiol. 19, 541–549. 10.1111/j.1540-8167.2007.01076.x [DOI] [PubMed] [Google Scholar]
- Iqbal S. M., Aufy M., Shabbir W., Lemmens-Gruber R. (2018). Identification of phosphorylation sites and binding pockets for modulation of NaV 1.5 channel by Fyn tyrosine kinase. FEBS J. 285, 2520–2530. 10.1111/febs.14496 [DOI] [PubMed] [Google Scholar]
- Isbilen B., Fraser S. P., Djamgoz M. B. (2006). Docosahexaenoic acid (omega-3) blocks voltage-gated sodium channel activity and migration of MDA-MB-231 human breast cancer cells. Int. J. Biochem. Cell Biol. 38, 2173–2182. 10.1016/j.biocel.2006.06.014 [DOI] [PubMed] [Google Scholar]
- Jiang Y., Ruta V., Chen J., Lee A., Mackinnon R. (2003). The principle of gating charge movement in a voltage-dependent K+ channel. Nature 423, 42–48. 10.1038/nature01581 [DOI] [PubMed] [Google Scholar]
- Jiang D., Shi H., Tonggu L., Gamal El-Din T. M., Lenaeus M. J., Zhao Y., et al. (2020). Structure of the Cardiac Sodium Channel. Cell 180, 122–134 e110. 10.1016/j.cell.2019.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. N., Potet F., Thompson M. K., Kroncke B. M., Glazer A. M., Voehler M. W., et al. (2018). A Mechanism of Calmodulin Modulation of the Human Cardiac Sodium Channel. Structure 26, 683–694 e683. 10.1016/j.str.2018.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouni M., Si-Tayeb K., Es-Salah-Lamoureux Z., Latypova X., Champon B., Caillaud A., et al. (2015). Toward Personalized Medicine: Using Cardiomyocytes Differentiated From Urine-Derived Pluripotent Stem Cells to Recapitulate Electrophysiological Characteristics of Type 2 Long QT Syndrome. J. Am. Heart Assoc. 4, e002159. 10.1161/JAHA.115.002159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkiewicz N. K., Sanguinetti M. C. (1993). Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 72, 75–83. 10.1161/01.RES.72.1.75 [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S., Barakat K. H. (2018). Development of Safe Drugs: The hERG Challenge. Med. Res. Rev. 38, 525–555. 10.1002/med.21445 [DOI] [PubMed] [Google Scholar]
- Kamiya K., Nishiyama A., Yasui K., Hojo M., Sanguinetti M. C., Kodama I. (2001). Short- and long-term effects of amiodarone on the two components of cardiac delayed rectifier K(+) current. Circulation 103, 1317–1324. 10.1161/01.CIR.103.9.1317 [DOI] [PubMed] [Google Scholar]
- Kang J. X., Xiao Y. F., Leaf A. (1995). ). Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A 92, 3997–4001. 10.1073/pnas.92.9.3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. X., Li Y., Leaf A. (1997). Regulation of sodium channel gene expression by class I antiarrhythmic drugs and n - 3 polyunsaturated fatty acids in cultured neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A 94, 2724–2728. 10.1073/pnas.94.6.2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C., Tian C., Sonnichsen F. D., Smith J. A., Meiler J., George A. L., Jr., et al. (2008). Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 47, 7999–8006. 10.1021/bi800875q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki H., Kupershmidt S., Yang T., Wells S., Roden D. M. (2004). A structural requirement for processing the cardiac K+ channel KCNQ1. J. Biol. Chem. 279, 33976–33983. 10.1074/jbc.M404539200 [DOI] [PubMed] [Google Scholar]
- Kapa S., Tester D. J., Salisbury B. A., Harris-Kerr C., Pungliya M. S., Alders M., et al. (2009). Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 120, 1752–1760. 10.1161/CIRCULATIONAHA.109.863076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimova M. A., Zaydman M. A., Cui J., Tarek M. (2015). PIP2-dependent coupling is prominent in Kv7.1 due to weakened interactions between S4-S5 and S6. Sci. Rep. 5, 7474–7474. 10.1038/srep07474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y., Ng C. A., Hunter M. J., Mann S. A., Heide J., Hill A. P., et al. (2013). Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem. J. 454, 69–77. 10.1042/BJ20130328 [DOI] [PubMed] [Google Scholar]
- Ke Y., Hunter M. J., Ng C. A., Perry M. D., Vandenberg J. I. (2014). Role of the cytoplasmic N-terminal Cap and Per-Arnt-Sim (PAS) domain in trafficking and stabilization of Kv11.1 channels. J. Biol. Chem. 289, 13782–13791. 10.1074/jbc.M113.531277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H. M., Tieleman P. D., Noskov S. Y. (2020). “Refinement of High-resolution Cryo-EM Structure of hERG: What Can We Expect?”, in The Biophysical Society 64th Annual Meeting (San Diego: Cell Press; ). [Google Scholar]
- Kim J., Ghosh S., Liu H., Tateyama M., Kass R. S., Pitt G. S. (2004). Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279, 45004–45012. 10.1074/jbc.M407286200 [DOI] [PubMed] [Google Scholar]
- Kim J. A., Lopes C. M., Moss A. J., Mcnitt S., Barsheshet A., Robinson J. L., et al. (2010). Trigger-specific risk factors and response to therapy in long QT syndrome type 2. Heart Rhythm. 7, 1797–1805. 10.1016/j.hrthm.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber Ag R. Y. (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 84, 431–488. 10.1152/physrev.00025.2003 [DOI] [PubMed] [Google Scholar]
- Kroncke Bm E. A. (2019). Protein structure aids in predicting functional perturbation of missense variants of SCN5A and KCNQ1. Comput. Struct. Biotechnol. J. 17,206–214. 10.1016/j.csbj.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke B. M., Vanoye C. G., Meiler J., George A. L., Jr., Sanders C. R. (2015). Personalized biochemistry and biophysics. Biochemistry 54, 2551–2559. 10.1021/acs.biochem.5b00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke B. M., Van Horn W. D., Smith J., Kang C., Welch R. C., Song Y., et al. (2016). Structural basis for KCNE3 modulation of potassium recycling in epithelia. Sci. Adv. 2, e1501228. 10.1126/sciadv.1501228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke B. M., Mendenhall J., Smith D. K., Sanders C. R., Capra J. A., George A. L., et al. (2019). Protein structure aids predicting functional perturbation of missense variants in SCN5A and KCNQ1. Comput. Struct. Biotechnol. J. 17, 206–214. 10.1016/j.csbj.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenze G., Duran A. M., Woods H., Brewer K. R., Mcdonald E. F., Vanoye C. G., et al. (2019). Upgraded molecular models of the human KCNQ1 potassium channel. PloS One 14, e0220415. 10.1371/journal.pone.0220415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume S., Shimomura T., Tateyama M., Kubo Y. (2018). Two mutations at different positions in the CNBH domain of the hERG channel accelerate deactivation and impair the interaction with the EAG domain. J. Physiol. 596, 4629–4650. 10.1113/JP276208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro A. J., Snyders D. J. (2012). Being flexible: the voltage-controllable activation gate of kv channels. Front. Pharmacol. 3, 168. 10.3389/fphar.2012.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro A. J., Boulet I. R., Choveau F. S., Mayeur E., Bruyns T., Loussouarn G., et al. (2011). The S4-S5 linker of KCNQ1 channels forms a structural scaffold with the S6 segment controlling gate closure. J. Biol. Chem. 286, 717–725. 10.1074/jbc.M110.146977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P. J., Sunami A., Fozzard H. A. (2001). Cardiac-specific external paths for lidocaine, defined by isoform-specific residues, accelerate recovery from use-dependent block. Circ. Res. 89, 1014–1021. 10.1161/hh2301.100002 [DOI] [PubMed] [Google Scholar]
- Leifert W. R., Mcmurchie E. J., Saint D. A. (1999). Inhibition of cardiac sodium currents in adult rat myocytes by n-3 polyunsaturated fatty acids. J. Physiol. 520 (Pt 3), 671–679. 10.1111/j.1469-7793.1999.00671.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaillet G., Walker B., Lambert S. (2003). Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 278, 27333–27339. 10.1074/jbc.M303327200 [DOI] [PubMed] [Google Scholar]
- Lengyel C., Iost N., Virag L., Varro A., Lathrop D. A., Papp J. G. (2001). Pharmacological block of the slow component of the outward delayed rectifier current (I(Ks)) fails to lengthen rabbit ventricular muscle QT(c) and action potential duration. Br. J. Pharmacol. 132, 101–110. 10.1038/sj.bjp.0703777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Zhang L. (2018). The role of mexiletine in the management of long QT syndrome. J. Electrocardiol. 51, 1061–1065. 10.1016/j.jelectrocard.2018.08.035 [DOI] [PubMed] [Google Scholar]
- Li P., Liu H., Lai C., Sun P., Zeng W., Wu F., et al. (2014). Differential modulations of KCNQ1 by auxiliary proteins KCNE1 and KCNE2. Sci. Rep. 4, 4973. 10.1038/srep04973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Mendenhall J. L., Kroncke B. M., Taylor K. C., Huang H., Smith D. K., et al. (2017). Predicting the Functional Impact of KCNQ1 Variants of Unknown Significance. Circ. Cardiovasc. Genet. 10, pii: e001754. 10.1161/CIRCGENETICS.117.001754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. C. H., O’brien T. J., Todaro M., Powell K. L. (2019). Acquired cardiac channelopathies in epilepsy: Evidence, mechanisms, and clinical significance. Epilepsia 60, 1753–1767. 10.1111/epi.16301 [DOI] [PubMed] [Google Scholar]
- Liin S. I., Silvera Ejneby M., Barro-Soria R., Skarsfeldt M. A., Larsson J. E., Starck Harlin F., et al. (2015). Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. U. S. A 112, 5714–5719. 10.1073/pnas.1503488112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S. I., Yazdi S., Ramentol R., Barro-Soria R., Larsson H. P. (2018). Mechanisms Underlying the Dual Effect of Polyunsaturated Fatty Acid Analogs on Kv7.1. Cell Rep. 24, 2908–2918. 10.1016/j.celrep.2018.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Liu N., Lu J., Zhang J., Anumonwo J. M., Isom L. L., et al. (2011). Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 8, 1923–1930. 10.1016/j.hrthm.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegger N., Hagen B. M., Marks A. R., Lederer W. J., Kass R. S. (2009). Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J. Mol. Cell Cardiol. 47, 326–334. 10.1016/j.yjmcc.2009.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. W., Antzelevitch C. (1995). Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ. Res. 76, 351–365. 10.1161/01.RES.76.3.351 [DOI] [PubMed] [Google Scholar]
- Liu C. J., Dib-Hajj S. D., Renganathan M., Cummins T. R., Waxman S. G. (2003). Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278, 1029–1036. 10.1074/jbc.M207074200 [DOI] [PubMed] [Google Scholar]
- Long S. B., Campbell E. B., Mackinnon R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903. 10.1126/science.1116269 [DOI] [PubMed] [Google Scholar]
- Long S. B., Tao X., Campbell E. B., Mackinnon R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. 10.1038/nature06265 [DOI] [PubMed] [Google Scholar]
- Loots E., Isacoff E. Y. (1998). Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Physiol. 112, 377–389. 10.1085/jgp.112.4.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorinczi E., Gomez-Posada J. C., De La Pena P., Tomczak A. P., Fernandez-Trillo J., Leipscher U., et al. (2015). Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat. Commun. 6, 6672. 10.1038/ncomms7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G., Park K. H., Bellocq C., Baro I., Charpentier F., Escande D. (2003). Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421. 10.1093/emboj/cdg526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Klem A. M., Ramu Y. (2001). Ion conduction pore is conserved among potassium channels. Nature 413 (6858), 809–813. 10.1038/35101535 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A. M., Ramu Y. (2002). Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120 (5), 663–676. 10.1085/jgp.20028696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L. J., Ohmert I., Vardanyan V. (2011). Allosteric features of KCNQ1 gating revealed by alanine scanning mutagenesis. Biophys. J. 100, 885–894. 10.1016/j.bpj.2010.12.3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D., Wei H., Zhao Y., Lu J., Li G., Sahib N. B., et al. (2013). Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 168, 5277–5286. 10.1016/j.ijcard.2013.08.015 [DOI] [PubMed] [Google Scholar]
- Makara M. A., Curran J., Little S. C., Musa H., Polina I., Smith S. A., et al. (2014). Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 115, 929–938. 10.1161/CIRCRESAHA.115.305154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita N., Bennett P. B., George A. L., Jr. (1996). Molecular determinants of beta 1 subunit-induced gating modulation in voltage-dependent Na+ channels. J. Neurosci. 16, 7117–7127. 10.1523/JNEUROSCI.16-22-07117.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J. D., Thyagarajan V., Chen C., Isom L. L. (2004). Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J. Biol. Chem. 279, 40748–40754. 10.1074/jbc.M407243200 [DOI] [PubMed] [Google Scholar]
- Marionneau C., Lichti C. F., Lindenbaum P., Charpentier F., Nerbonne J. M., Townsend R. R., et al. (2012). Mass spectrometry-based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J. Proteome Res. 11, 5994–6007. 10.1021/pr300702c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattmann M. E., Yu H., Lin Z., Xu K., Huang X., Long S., et al. (2012). Identification of (R)-N-(4-(4-methoxyphenyl)thiazol-2-yl)-1-tosylpiperidine-2-carboxamide, ML277, as a novel, potent and selective K(v)7.1 (KCNQ1) potassium channel activator. Bioorg. Med. Chem. Lett. 22, 5936–5941. 10.1016/j.bmcl.2012.07.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari R., Greenstein J. L., Winslow R. L., Marban E., Nuss H. B. (2001). Molecular interactions between two long-QT syndrome gene products, HERG and KCNE2, rationalized by in vitro and in silico analysis. Circ. Res. 89, 33–38. 10.1161/hh1301.093633 [DOI] [PubMed] [Google Scholar]
- Mazzanti A., Maragna R., Faragli A., Monteforte N., Bloise R., Memmi M., et al. (2016). Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 67, 1053–1058. 10.1016/j.jacc.2015.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald T. V., Yu Z., Ming Z., Palma E., Meyers M. B., Wang K. W., et al. (1997). A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388, 289–292. 10.1038/40882 [DOI] [PubMed] [Google Scholar]
- Meadows L., Malhotra J. D., Stetzer A., Isom L. L., Ragsdale D. S. (2001). The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J. Neurochem. 76, 1871–1878. 10.1046/j.1471-4159.2001.00192.x [DOI] [PubMed] [Google Scholar]
- Medeiros-Domingo A., Kaku T., Tester D. J., Iturralde-Torres P., Itty A., Ye B., et al. (2007). SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142. 10.1161/CIRCULATIONAHA.106.659086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman Y. F., Domenech A., De La Luna S., Mcdonald T. V. (2001). Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem. 276, 6439–6444. 10.1074/jbc.M010713200 [DOI] [PubMed] [Google Scholar]
- Melman Y. F., Krumerman A., Mcdonald T. V. (2002). A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J. Biol. Chem. 277, 25187–25194. 10.1074/jbc.M200564200 [DOI] [PubMed] [Google Scholar]
- Melman Y. F., Um S. Y., Krumerman A., Kagan A., Mcdonald T. V. (2004). KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 42, 927–937. 10.1016/j.neuron.2004.06.001 [DOI] [PubMed] [Google Scholar]
- Meza U., Beqollari D., Bannister R. A. (2018). Molecular mechanisms and physiological relevance of RGK proteins in the heart. Acta Physiol. (Oxf) 222, e13016. 10.1111/apha.13016 [DOI] [PubMed] [Google Scholar]
- Mitcheson J. S., Chen J., Lin M., Culberson C., Sanguinetti M. C. (2000). A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U. S. A 97, 12329–12333. 10.1073/pnas.210244497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler P. J., Rivolta I., Napolitano C., Lemaillet G., Lambert S., Priori S. G., et al. (2004). Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A 101, 17533–17538. 10.1073/pnas.0403711101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., Mackinnon R. (1998). Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95, 649–655. 10.1016/S0092-8674(00)81635-9 [DOI] [PubMed] [Google Scholar]
- Morais-Cabral J. H., Zhou Y., Mackinnon R. (2001). Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 414, 37–42. 10.1038/35102000 [DOI] [PubMed] [Google Scholar]
- Moreau A., Gosselin-Badaroudine P., Delemotte L., Klein M. L., Chahine M. (2015). Gating pore currents are defects in common with two Nav1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J. Gen. Physiol. 145, 93–106. 10.1085/jgp.201411304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno J. D., Zhu Z. I., Yang P. C., Bankston J. R., Jeng M. T., Kang C., et al. (2011). A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci. Transl. Med. 3, 98ra83. 10.1126/scitranslmed.3002588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A., Bellin M., Welling A., Jung C. B., Lam J. T., Bott-Flugel L., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409. 10.1056/NEJMoa0908679 [DOI] [PubMed] [Google Scholar]
- Moss A. J., Kass R. S. (2005). Long QT syndrome: from channels to cardiac arrhythmias. J. Clin. Invest. 115, 2018–2024. 10.1172/JCI25537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss A. J., Zareba W., Hall W. J., Schwartz P. J., Crampton R. S., Benhorin J., et al. (2000). Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 101, 616–623. 10.1161/01.CIR.101.6.616 [DOI] [PubMed] [Google Scholar]
- Moss A. J., Shimizu W., Wilde A. A., Towbin J. A., Zareba W., Robinson J. L., et al. (2007). Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 115, 2481–2489. 10.1161/CIRCULATIONAHA.106.665406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi Nik A., Gharaie S., Jeong Kim H. (2015). Cellular mechanisms of mutations in Kv7.1: auditory functions in Jervell and Lange-Nielsen syndrome vs. Romano-Ward syndrome. Front. Cell Neurosci. 9, 32. 10.3389/fncel.2015.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa H., Kline C. F., Sturm A. C., Murphy N., Adelman S., Wang C., et al. (2015). SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc. Natl. Acad. Sci. U. S. A. 112, 12528–12533. 10.1073/pnas.1516430112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskett F. W., Thouta S., Thomson S. J., Bowen A., Stansfeld P. J., Mitcheson J. S. (2011). Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 286, 6184–6191. 10.1074/jbc.M110.199364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T., Kaneko Y., Dharmawan T., Kurabayashi M. (2019). Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: The implication of closed-state inactivation. Channels (Austin) 13, 331–343. 10.1080/19336950.2019.1649521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K., Kubo Y. (2007). KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 130, 269–281. 10.1085/jgp.200709805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K., Kubo Y. (2014). Steric hindrance between S4 and S5 of the KCNQ1/KCNE1 channel hampers pore opening. Nat. Commun. 5, 4100–4100. 10.1038/ncomms5100 [DOI] [PubMed] [Google Scholar]
- Naylor C. E., Bagneris C., Decaen P. G., Sula A., Scaglione A., Clapham D. E., et al. (2016). Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J. 35, 820–830. 10.15252/embj.201593285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negami T., Araki M., Okuno Y., Terada T. (2019). Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci. Rep. 9, 16586. 10.1038/s41598-019-53120-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne J. M., Kass R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. 10.1152/physrev.00002.2005 [DOI] [PubMed] [Google Scholar]
- Ng C. A., Phan K., Hill A. P., Vandenberg J. I., Perry M. D. (2014). Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J. Biol. Chem. 289, 25822–25832. 10.1074/jbc.M114.558379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. T., Demarco K. R., Vorobyov I., Clancy C. E., Yarov-Yarovoy V. (2019). Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl. Acad. Sci. U. S. A 116, 2945–2954. 10.1073/pnas.1817446116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’malley H. A., Isom L. L. (2015). Sodium channel beta subunits: emerging targets in channelopathies. Annu. Rev. Physiol. 77, 481–504. 10.1146/annurev-physiol-021014-071846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen M. S., Jespersen T., Nielsen J. B., Liang B., Moller D. V., Hedley P., et al. (2011). Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793. 10.1093/cvr/cvq348 [DOI] [PubMed] [Google Scholar]
- Osteen J. D., Gonzalez C., Sampson K. J., Iyer V., Rebolledo S., Larsson H. P., et al. (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U. S. A 107, 22710–22715. 10.1073/pnas.1016300108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen J. D., Barro-Soria R., Robey S., Sampson K. J., Kass R. S., Larsson H. P. (2012). Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc. Natl. Acad. Sci. U. S. A 109, 7103–7108. 10.1073/pnas.1201582109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Li Z., Zhou Q., Shen H., Wu K., Huang X., et al. (2018). Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 362, pii: eaau2486. 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Pan X., Li Z., Huang X., Huang G., Gao S., Shen H., et al. (2019). Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA. Science 363, 1309–1313. 10.1126/science.aaw2999 [DOI] [PubMed] [Google Scholar]
- Panaghie G., Abbott G. W. (2007). The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 129, 121–133. 10.1085/jgp.200609612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G., Tai K.-K., Abbott G. W. (2006). Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J. Physiol. 570, 455–467. 10.1113/jphysiol.2005.100644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. H., Piron J., Dahimene S., Merot J., Baro I., Escande D., et al. (2005). Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96, 730–739. 10.1161/01.RES.0000161451.04649.a8 [DOI] [PubMed] [Google Scholar]
- Perrin M. J., Kuchel P. W., Campbell T. J., Vandenberg J. I. (2008). Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels. Mol. Pharmacol. 74, 1443–1452. 10.1124/mol.108.049056 [DOI] [PubMed] [Google Scholar]
- Perry M. D., Wong S., Ng C. A., Vandenberg J. I. (2013. a). Hydrophobic interactions between the voltage sensor and pore mediate inactivation in Kv11.1 channels. J. Gen. Physiol. 142 (3), 275–288. 10.1085/jgp.201310975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M. D., Wong S., Ng C. A., Vandenberg J. I. (2013. b). Pore helices play a dynamic role as integrators of domain motion during Kv11.1 channel inactivation gating. J. Biol. Chem. 288 (16), 11482–11491. 10.1074/jbc.M113.461442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M. D., Ng C. A., Mann S. A., Sadrieh A., Imtiaz M., Hill A. P., et al. (2015). Getting to the heart of hERG K(+) channel gating. J. Physiol. 593, 2575–2585. 10.1113/JP270095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan K., Ng C. A., David E., Shishmarev D., Kuchel P. W., Vandenberg J. I., et al. (2017). The S1 helix critically regulates the finely tuned gating of Kv11.1 channels. J. Biol. Chem. 292, 7688–7705. 10.1074/jbc.M117.779298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless S. A., Galpin J. D., Frankel A., Ahern C. A. (2011. a). Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat. Commun. 2, 351. 10.1038/ncomms1351 [DOI] [PubMed] [Google Scholar]
- Pless S. A., Galpin J. D., Niciforovic A. P., Ahern C. A. (2011. b). Contributions of counter-charge in a potassium channel voltage-sensor domain. Nat. Chem. Biol. 7, 617–623. 10.1038/nchembio.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen K. L., Hotait M., Calloe K., Klaerke D. A., Rebeiz A., Nemer G., et al. (2015). The Mutation P.T613a in the Pore Helix of the Kv 11.1 Potassium Channel is Associated with Long QT Syndrome. Pacing Clin. Electrophysiol. 38, 1304–1309. 10.1111/pace.12693 [DOI] [PubMed] [Google Scholar]
- Preston P., Wartosch L., Gunzel D., Fromm M., Kongsuphol P., Ousingsawat J., et al. (2010). Disruption of the K+ channel beta-subunit KCNE3 reveals an important role in intestinal and tracheal Cl- transport. J. Biol. Chem. 285, 7165–7175. 10.1074/jbc.M109.047829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori S. G., Wilde A. A., Horie M., Cho Y., Behr E. R., Berul C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963. 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- Pusch M., Magrassi R., Wollnik B., Conti F. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75, 785–792. 10.1016/S0006-3495(98)77568-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M., Bertorello L., Conti F. (2000). Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/KCNE1 potassium channels. Biophys. J. 78, 211–226. 10.1016/S0006-3495(00)76586-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Isom L. L., Westenbroek R. E., Rogers J. C., Tanada T. N., Mccormick K. A., et al. (1995). Modulation of cardiac Na+ channel expression in Xenopus oocytes by beta 1 subunits. J. Biol. Chem. 270, 25696–25701. 10.1074/jbc.270.43.25696 [DOI] [PubMed] [Google Scholar]
- Ragsdale D. S., Mcphee J. C., Scheuer T., Catterall W. A. (1994). Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265, 1724–1728. 10.1126/science.8085162 [DOI] [PubMed] [Google Scholar]
- Ragsdale D. S., Mcphee J. C., Scheuer T., Catterall W. A. (1996). Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U. S. A 93, 9270–9275. 10.1073/pnas.93.17.9270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos E., O’leary M E. (2004). State-dependent trapping of flecainide in the cardiac sodium channel. J. Physiol. 560, 37–49. 10.1113/jphysiol.2004.065003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampe D., Murawsky M. K., Grau J., Lewis E. W. (1998). The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J. Pharmacol. Exp. Ther. 286, 788–793. [PubMed] [Google Scholar]
- Riuro H., Campuzano O., Arbelo E., Iglesias A., Batlle M., Perez-Villa F., et al. (2014). A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm. 11, 1202–1209. 10.1016/j.hrthm.2014.03.044 [DOI] [PubMed] [Google Scholar]
- Rocheleau J. M., Kobertz W. R. (2008). KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J. Gen. Physiol. 131, 59–68. 10.1085/jgp.200709816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden D. M., Woosley R. L., Primm R. K. (1986). Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am. Heart J. 111, 1088–1093. 10.1016/0002-8703(86)90010-4 [DOI] [PubMed] [Google Scholar]
- Roepke T. K., Anantharam A., Kirchhoff P., Busque S. M., Young J. B., Geibel J. P., et al. (2006). The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J. Biol. Chem. 281, 23740–23747. 10.1074/jbc.M604155200 [DOI] [PubMed] [Google Scholar]
- Ruscic K. J., Miceli F., Villalba-Galea C. A., Dai H., Mishina Y., Bezanilla F., et al. (2013). IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc. Natl. Acad. Sci. U. S. A 110, E559–E566. 10.1073/pnas.1222616110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachyani D., Dvir M., Strulovich R., Tria G., Tobelaim W., Peretz A., et al. (2014). Structural basis of a Kv7.1 potassium channel gating module: studies of the intracellular c-terminal domain in complex with calmodulin. Structure 22, 1582–1594. 10.1016/j.str.2014.07.016 [DOI] [PubMed] [Google Scholar]
- Salata J. J., Jurkiewicz N. K., Wang J., Evans B. E., Orme H. T., Sanguinetti M. C. (1998). A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol. Pharmacol. 54, 220–230. 10.1124/mol.54.1.220 [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., et al. (1996). Coassembly of K(v)LQT1 and minK (IsK) proteins to form cardiac I-Ks potassium channel. Nature 384, 80–83. 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- Saxena P., Zangerl-Plessl E. M., Linder T., Windisch A., Hohaus A., Timin E., et al. (2016). New potential binding determinant for hERG channel inhibitors. Sci. Rep. 6, 24182. 10.1038/srep24182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa N. E., Mccormack K., Tanouye M. A., Sigworth F. J. (1992). The size of gating charge in wild-type and mutant Shaker potassium channels. Science 255, 1712–1715. 10.1126/science.1553560 [DOI] [PubMed] [Google Scholar]
- Schroeder B. C., Waldegger S., Fehr S., Bleich M., Warth R., Greger R., et al. (2000). A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199. 10.1038/35003200 [DOI] [PubMed] [Google Scholar]
- Schwake M., Jentsch T. J., Friedrich T. (2003). A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 4, 76–81. 10.1038/sj.embor.embor715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M., Athanasiadu D., Beimgraben C., Blanz J., Beck C., Jentsch T. J., et al. (2006). Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J. Neurosci. 26, 3757–3766. 10.1523/JNEUROSCI.5017-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. J., Priori S. G., Spazzolini C., Moss A. J., Vincent G. M., Napolitano C., et al. (2001). Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95. 10.1161/01.CIR.103.1.89 [DOI] [PubMed] [Google Scholar]
- Schwartz P. J., Stramba-Badiale M., Crotti L., Pedrazzini M., Besana A., Bosi G., et al. (2009). Prevalence of the congenital long-QT syndrome. Circulation 120, 1761–1767. 10.1161/CIRCULATIONAHA.109.863209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. J., Crotti L., Insolia R. (2012). Long-QT syndrome: from genetics to management. Circ. Arrhythm Electrophysiol. 5, 868–877. 10.1161/CIRCEP.111.962019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G., Chen J., Strutz N., Culberson C., Lerche C., Sanguinetti M. C. (2003. a). Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol. Pharmacol. 64, 70–77. 10.1124/mol.64.1.70 [DOI] [PubMed] [Google Scholar]
- Seebohm G., Pusch M., Chen J., Sanguinetti M. C. (2003. b). Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circ. Res. 93, 941–947. 10.1161/01.RES.0000102866.67863.2B [DOI] [PubMed] [Google Scholar]
- Seebohm G., Sanguinetti M. C., Pusch M. (2003. c). Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. London 552, 369–378. 10.1113/jphysiol.2003.046490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G., Westenskow P., Lang F., Sanguinetti M. C. (2005). Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. 563, 359–368. 10.1113/jphysiol.2004.080887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G., Strutz-Seebohm N., Ureche O. N., Baltaev R., Lampert A., Kornichuk G., et al. (2006). Differential roles of S6 domain hinges in the gating of KCNQ potassium channels. Biophys. J. 90, 2235–2244. 10.1529/biophysj.105.067165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko A. A., Hadley J. K., Wood I. C., Abogadie F. C., Jentsch T. J., Brown D. A. (2000). Inhibition of KCNQ1-4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J. Physiol. 522 (Pt 3), 349–355. 10.1111/j.1469-7793.2000.t01-2-00349.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoh S. A., Sigg D., Papazian D. M., Bezanilla F. (1996). Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 16, 1159–1167. 10.1016/S0896-6273(00)80142-7 [DOI] [PubMed] [Google Scholar]
- Sesti F., Abbott G. W., Wei J., Murray K. T., Saksena S., Schwartz P. J., et al. (2000). A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc. Natl. Acad. Sci. U. States America 97, 10613–10618. 10.1073/pnas.180223197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Zhou Q., Pan X., Li Z., Wu J., Yan N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, pii: eaal4326. 10.1126/science.aal4326 [DOI] [PubMed] [Google Scholar]
- Shen H., Liu D., Wu K., Lei J., Yan N. (2019). Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308. 10.1126/science.aaw2493 [DOI] [PubMed] [Google Scholar]
- Shimizu W., Antzelevitch C. (1998). Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation 98, 2314–2322. 10.1161/01.CIR.98.21.2314 [DOI] [PubMed] [Google Scholar]
- Shimizu W., Horie M., Ohno S., Takenaka K., Yamaguchi M., Shimizu M., et al. (2004). Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J. Am. Coll. Cardiol. 44, 117–125. 10.1016/j.jacc.2004.03.043 [DOI] [PubMed] [Google Scholar]
- Shy D., Gillet L., Abriel H. (2013). Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim. Biophys. Acta 1833, 886–894. 10.1016/j.bbamcr.2012.10.026 [DOI] [PubMed] [Google Scholar]
- Skinner J. R., Winbo A., Abrams D., Vohra J., Wilde A. A. (2019). Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ. 28, 22–30. 10.1016/j.hlc.2018.09.007 [DOI] [PubMed] [Google Scholar]
- Smith P. L., Baukrowitz T., Yellen G. (1996). The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836. 10.1038/379833a0 [DOI] [PubMed] [Google Scholar]
- Smith J. L., Anderson C. L., Burgess D. E., Elayi C. S., January C. T., Delisle B. P. (2016). Molecular pathogenesis of long QT syndrome type 2. J. Arrhythm 32, 373–380. 10.1016/j.joa.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S., Scheuer T., Catterall W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78. 10.1038/nature05598 [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J., Grottesi A., Sands Z. A., Sansom M. S., Gedeck P., Gosling M., et al. (2008). Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry 47, 7414–7422. 10.1021/bi800475j [DOI] [PubMed] [Google Scholar]
- Stenson P. D., Ball E. V., Mort M., Phillips A. D., Shaw K., Cooper D. N. (2012). The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinf. Chapter 1, Unit1 13. 1.13.1–1.13.20 10.1002/0471250953.bi0113s39 [DOI] [PubMed] [Google Scholar]
- Strutz-Seebohm N., Pusch M., Wolf S., Stoll R., Tapken D., Gerwert K., et al. (2011). Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell Physiol. Biochem. 27, 443–452. 10.1159/000329965 [DOI] [PubMed] [Google Scholar]
- Sun J., Mackinnon R. (2017). Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 169, 1042–1050.e1049. 10.1016/j.cell.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Mackinnon R. (2020). Structural Basis of Human KCNQ1 Modulation and Gating. Cell 180, 340–347 e349. 10.1016/j.cell.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami A., Dudley S. C., Jr., Fozzard H. A. (1997). Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. U. S. A 94, 14126–14131. 10.1073/pnas.94.25.14126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X., Lee A., Limapichat W., Dougherty D. A., Mackinnon R. (2010). A gating charge transfer center in voltage sensors. Science 328, 67–73. 10.1126/science.1185954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper A. R., George A. L. (2001). Location and orientation of minK within the I-Ks potassium channel complex. J. Biol. Chem. 276, 38249–38254. 10.1074/jbc.M103956200 [DOI] [PubMed] [Google Scholar]
- Taylor K. C., Sanders C. R. (2017). Regulation of KCNQ/Kv7 family voltage-gated K(+) channels by lipids. Biochim. Biophys. Acta Biomembr 1859, 586–597. 10.1016/j.bbamem.2016.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. C., Kang P. W., Hou P., Yang N.-D., Kuenze G., Smith J. A., et al. (2020). Structure and Physiological Function of the Human KCNQ1 Channel Voltage Sensor Intermediate State. eLife 9, e53901. 10.7554/eLife.53901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C., Wang K., Tung K. W., Chung W. K., Pass R. H., Lu J. T., et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J. Gen. Physiol. 141, 61–72. 10.1085/jgp.201210899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester D. J., Ackerman M. J. (2014). Genetics of long QT syndrome. Methodist Debakey Cardiovasc. J. 10, 29–33. 10.14797/mdcj-10-1-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D., Khalil M., Alter M., Schweizer P. A., Karle C. A., Wimmer A. B., et al. (2010). Biophysical characterization of KCNQ1 P320 mutations linked to long QT syndrome 1. J. Mol. Cell Cardiol. 48, 230–237. 10.1016/j.yjmcc.2009.06.009 [DOI] [PubMed] [Google Scholar]
- Thomas A. M., Harmer S. C., Khambra T., Tinker A. (2011). Characterization of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem. 286, 2088–2100. 10.1074/jbc.M110.153551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel N., Diochot S., Borsotto M., Lazdunski M., Barhanin J. (2000). KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330. 10.1093/emboj/19.23.6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobelaim W. S., Dvir M., Lebel G., Cui M., Buki T., Peretz A., et al. (2017. a). Ca(2+)-Calmodulin and PIP2 interactions at the proximal C-terminus of Kv7 channels. Channels (Austin) 11, 686–695. 10.1080/19336950.2017.1388478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobelaim W. S., Dvir M., Lebel G., Cui M., Buki T., Peretz A., et al. (2017. b). Competition of calcified calmodulin N lobe and PIP2 to an LQT mutation site in Kv7.1 channel. Proc. Natl. Acad. Sci. U. S. A 114, E869–E878. 10.1073/pnas.1612622114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh M. F., Brooks J. M., Strassmaier T., Haedo R. J., Puryear C. B., Roth B. L., et al. (2020). Application of High-Throughput Automated Patch-Clamp Electrophysiology to Study Voltage-Gated Ion Channel Function in Primary Cortical Cultures. SLAS Discovery, 2472555220902388. 10.1177/2472555220902388 [DOI] [PubMed]
- Toombes G. E., Swartz K. J. (2016). STRUCTURAL BIOLOGY. Twists and turns in gating ion channels with voltage. Science 353, 646–647. 10.1126/science.aah4194 [DOI] [PubMed] [Google Scholar]
- Tsang S. Y., Tsushima R. G., Tomaselli G. F., Li R. A., Backx P. H. (2005). A multifunctional aromatic residue in the external pore vestibule of Na+ channels contributes to the local anesthetic receptor. Mol. Pharmacol. 67, 424–434. 10.1124/mol.67.2.424 [DOI] [PubMed] [Google Scholar]
- Ulmschneider M. B., Bagneris C., Mccusker E. C., Decaen P. G., Delling M., Clapham D. E., et al. (2013). Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A 110, 6364–6369. 10.1073/pnas.1214667110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia J., Aguado A., Muguruza-Montero A., Nunez E., Malo C., Casis O., et al. (2019). The Crossroad of Ion Channels and Calmodulin in Disease. Int. J. Mol. Sci. 20, pii: E400. 10.3390/ijms20020400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R., Reilly L., Eckhardt L. L. (2018). Caveolin-3 Microdomain: Arrhythmia Implications for Potassium Inward Rectifier and Cardiac Sodium Channel. Front. Physiol. 9, 1548. 10.3389/fphys.2018.01548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia C. R., Medeiros-Domingo A., Ye B., Shen W. K., Algiers T. J., Ackerman M. J., et al. (2010). Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc. Res. 86, 392–400. 10.1093/cvr/cvp417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn W. D., Vanoye C. G., Sanders C. R. (2011). Working model for the structural basis for KCNE1 modulation of the KCNQ1 potassium channel. Curr. Opin. Struct. Biol. 21, 283–291. 10.1016/j.sbi.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg J. I., Varghese A., Lu Y., Bursill J. A., Mahaut-Smith M. P., Huang C. L. (2006). Temperature dependence of human ether-a-go-go-related gene K+ currents. Am. J. Physiol. Cell Physiol. 291, C165–C175. 10.1152/ajpcell.00596.2005 [DOI] [PubMed] [Google Scholar]
- Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478. 10.1152/physrev.00036.2011 [DOI] [PubMed] [Google Scholar]
- Vandenberg J. I., Perozo E., Allen T. W. (2017). Towards a Structural View of Drug Binding to hERG K(+) Channels. Trends Pharmacol. Sci. 38, 899–907. 10.1016/j.tips.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye C. G., Desai R. R., Fabre K. L., Gallagher S. L., Potet F., Dekeyser J. M., et al. (2018). High-Throughput Functional Evaluation of KCNQ1 Decrypts Variants of Unknown Significance. Circ. Genom. Precis Med. 11, e002345. 10.1161/CIRCGEN.118.002345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardanyan V., Pongs O. (2012). Coupling of voltage-sensors to the channel pore: a comparative view. Front. Pharmacol. 3, 145. 10.3389/fphar.2012.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan P. C., Shaw R. M., Rudy Y. (1999). Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation 99, 2466–2474. 10.1161/01.CIR.99.18.2466 [DOI] [PubMed] [Google Scholar]
- Vitola J., Vukanovic J., Roden D. M. (1998). Cisapride-induced torsades de pointes. J. Cardiovasc. Electrophysiol. 9, 1109–1113. 10.1111/j.1540-8167.1998.tb00888.x [DOI] [PubMed] [Google Scholar]
- Wacker S., Noskov S. Y., Perissinotti L. L. (2017). Computational Models for Understanding of Structure, Function and Pharmacology of the Cardiac Potassium Channel Kv11.1 (hERG). Curr. Top. Med. Chem. 17, 2681–2702. 10.2174/1568026617666170414143430 [DOI] [PubMed] [Google Scholar]
- Wallace E., Howard L., Liu M., O’brien T., Ward D., Shen S., et al. (2019). Long QT Syndrome: Genetics and Future Perspective. Pediatr. Cardiol. 40, 1419–1430. 10.1007/s00246-019-02151-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Mackinnon R. (2017). Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 169, 422–430 e410. 10.1016/j.cell.2017.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Tristani-Firouzi M., Xu Q., Lin M., Keating M. T., Sanguinetti M. C. (1999). Functional effects of mutations in KvLQT1 that cause long QT syndrome. J. Cardiovasc. Electrophysiol. 10, 817–826. 10.1111/j.1540-8167.1999.tb00262.x [DOI] [PubMed] [Google Scholar]
- Wang L., Dennis A. T., Trieu P., Charron F., Ethier N., Hebert T. E., et al. (2009). Intracellular potassium stabilizes human ether-a-go-go-related gene channels for export from endoplasmic reticulum. Mol. Pharmacol. 75, 927–937. 10.1124/mol.108.053793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Yang Q., Wu X., Yang Y., Shi L., Wang C., et al. (2010). Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem. Biophys. Res. Commun. 398, 98–104. 10.1016/j.bbrc.2010.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. H., Jiang M., Xu X. L., Hsu K.-L., Zhang M., Tseng G.-N. (2011). Gating-related molecular motions in the extracellular domain of the IKs channel: implications for IKs channelopathy. J. Membr. Biol. 239, 137–156. 10.1007/s00232-010-9333-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Chung B. C., Yan H., Lee S. Y., Pitt G. S. (2012). Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176. 10.1016/j.str.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Koopmann T. T., Le Scouarnec S., Yang T., Ingram C. R., Schott J. J., et al. (2008). Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268. 10.1172/JCI33891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Darbar D., Kaiser D. W., Jiramongkolchai K., Chopra S., Donahue B. S., et al. (2009). Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm Electrophysiol. 2, 268–275. 10.1161/CIRCEP.108.779181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapura M., Nattel S., Chartier D., Caballero R., Hebert T. E. (2002). A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J. Physiol. 540, 15–27. 10.1113/jphysiol.2001.013296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher J. R., Mackinnon R. (2016). Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669. 10.1126/science.aaf8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R., Haitin Y., Shamgar L., Fernandez-Alonso M. C., Martos A., Chomsky-Hecht O., et al. (2008). The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J. Biol. Chem. 283, 5815–5830. 10.1074/jbc.M707541200 [DOI] [PubMed] [Google Scholar]
- Wilde A., Amin A. S. (2018). Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 4, 569–579. 10.1016/j.jacep.2018.03.006 [DOI] [PubMed] [Google Scholar]
- Wisedchaisri G., Tonggu L., Mccord E., Gamal El-Din T. M., Wang L., Zheng N., et al. (2019). Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 178, 993–1003 e1012. 10.1016/j.cell.2019.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Shryock J. C., Song Y., Li Y., Antzelevitch C., Belardinelli L. (2004). Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J. Pharmacol. Exp. Ther. 310, 599–605. 10.1124/jpet.104.066100 [DOI] [PubMed] [Google Scholar]
- Wu L., Yong S. L., Fan C., Ni Y., Yoo S., Zhang T., et al. (2008). Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. J. Biol. Chem. 283, 6968–6978. 10.1074/jbc.M709721200 [DOI] [PubMed] [Google Scholar]
- Wu D., Delaloye K., Zaydman M. A., Nekouzadeh A., Rudy Y., Cui J. (2010. a). State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 135, 595–606. 10.1085/jgp.201010408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Pan H., Delaloye K., Cui J. (2010. b). KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys. J. 99, 3599–3608. 10.1016/j.bpj.2010.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Ding W. G., Horie M. (2016). Molecular pathogenesis of long QT syndrome type 1. J. Arrhythm 32, 381–388. 10.1016/j.joa.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. F., Wright S. N., Wang G. K., Morgan J. P., Leaf A. (1998). Fatty acids suppress voltage-gated Na+ currents in HEK293t cells transfected with the alpha-subunit of the human cardiac Na+ channel. Proc. Natl. Acad. Sci. U. S. A 95, 2680–2685. 10.1073/pnas.95.5.2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. F., Ke Q., Wang S. Y., Auktor K., Yang Y., Wang G. K., et al. (2001). Single point mutations affect fatty acid block of human myocardial sodium channel alpha subunit Na+ channels. Proc. Natl. Acad. Sci. U. S. A 98, 3606–3611. 10.1073/pnas.061003798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Jiang M., Hsu K.-L., Zhang M., Tseng G.-N. (2008). KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol. 131, 589–603. 10.1085/jgp.200809976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Wang Y., Meng X.-Y., Zhang M., Jiang M., Cui M., et al. (2013). Building KCNQ1/KCNE1 channel models and probing their interactions by molecular-dynamics simulations. Biophys. J. 105, 2461–2473. 10.1016/j.bpj.2013.09.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Wang Y., Zhang M., Jiang M., Rosenhouse-Dantsker A., Wassenaar T., et al. (2015). Probing binding sites and mechanisms of action of an I(Ks) activator by computations and experiments. Biophys. J. 108, 62–75. 10.1016/j.bpj.2014.10.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Li T., Rohou A., Arthur C. P., Tzakoniati F., Wong E., et al. (2019). Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 176 702-715, e714. 10.1016/j.cell.2018.12.018 [DOI] [PubMed] [Google Scholar]
- Yan H., Wang C., Marx S. O., Pitt G. S. (2017. a). Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 149, 277–293. 10.1085/jgp.201611721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Zhou Q., Wang L., Wu J., Zhao Y., Huang G., et al. (2017. b). Structure of the Nav1.4-beta1 Complex from Electric Eel. Cell 170, 470–482 e411. 10.1016/j.cell.2017.06.039 [DOI] [PubMed] [Google Scholar]
- Yang W. P., Levesque P. C., Little W. A., Conder M. L., Shalaby F. Y., Blanar M. A. (1997). KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc. Natl. Acad. Sci. U. S. A 94, 4017–4021. 10.1073/pnas.94.8.4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Smith J. A., Leake B. F., Sanders C. R., Meiler J., Roden D. M. (2013). An allosteric mechanism for drug block of the human cardiac potassium channel KCNQ1. Mol. Pharmacol. 83, 481–489. 10.1124/mol.112.081513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Wang Z., Sinden D. S., Wang X., Shan B., Yu X., et al. (2016). FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific manner. Channels (Austin) 10, 410–420. 10.1080/19336950.2016.1190055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., Brown J., Sharp E. M., Clare J. J., Scheuer T., Catterall W. A. (2001). Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J. Biol. Chem. 276, 20–27. 10.1074/jbc.M006992200 [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., Mcphee J. C., Idsvoog D., Pate C., Scheuer T., Catterall W. A. (2002). Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277, 35393–35401. 10.1074/jbc.M206126200 [DOI] [PubMed] [Google Scholar]
- Yus-Najera E., Santana-Castro I., Villarroel A. (2002). The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 277, 28545–28553. 10.1074/jbc.M204130200 [DOI] [PubMed] [Google Scholar]
- Zaydman M. A., Silva J. R., Delaloye K., Li Y., Liang H., Larsson H. P., et al. (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. U. S. A 110, 13180–13185. 10.1073/pnas.1305167110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman M. A., Kasimova M. A., Mcfarland K., Beller Z., Hou P., Kinser H. E., et al. (2014). Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 3, e03606. 10.7554/eLife.03606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaza A., Belardinelli L., Shryock J. C. (2008). Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol. Ther. 119, 326–339. 10.1016/j.pharmthera.2008.06.001 [DOI] [PubMed] [Google Scholar]
- Zhang H., Craciun L. C., Mirshahi T., Rohacs T., Lopes C. M., Jin T., et al. (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. 10.1016/S0896-6273(03)00125-9 [DOI] [PubMed] [Google Scholar]
- Zhang M., Liu J., Tseng G. N. (2004). Gating charges in the activation and inactivation processes of the HERG channel. J. Gen. Physiol. 124, 703–718. 10.1085/jgp.200409119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. C., Yang H., Liu Z., Sun F. (2018). Thermodynamics of voltage-gated ion channels. Biophys. Rep. 4, 300–319. 10.1007/s41048-018-0074-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. T., Hill A. P., Varghese A., Cooper A. A., Swan H., Laitinen-Forsblom P. J., et al. (2009). Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J. Cardiovasc. Electrophysiol. 20, 923–930. 10.1111/j.1540-8167.2009.01468.x [DOI] [PubMed] [Google Scholar]
- Zhou Z., Vorperian V. R., Gong Q., Zhang S., January C. T. (1999). Block of HERG potassium channels by the antihistamine astemizole and its metabolites desmethylastemizole and norastemizole. J. Cardiovasc. Electrophysiol. 10, 836–843. 10.1111/j.1540-8167.1999.tb00264.x [DOI] [PubMed] [Google Scholar]
- Zhou Y., Morais-Cabral J. H., Kaufman A., Mackinnon R. (2001). Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414, 43–48. 10.1038/35102009 [DOI] [PubMed] [Google Scholar]
- Zhu W., Voelker T. L., Varga Z., Schubert A. R., Nerbonne J. M., Silva J. R. (2017). Mechanisms of noncovalent beta subunit regulation of NaV channel gating. J. Gen. Physiol. 149 (8), 813–831. 10.1085/jgp.201711802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipes D. P., Camm A. J., Borggrefe M., Buxton A. E., Chaitman B., Fromer M., et al. (2006). ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 114, e385–e484. 10.1161/CIRCULATIONAHA.106.178233 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LQTS mutations in KCNQ1, hERG, and SCN5A.