

Abstract
Summary
We present genesis, a library for working with phylogenetic data, and gappa, an accompanying command-line tool for conducting typical analyses on such data. The tools target phylogenetic trees and phylogenetic placements, sequences, taxonomies and other relevant data types, offer high-level simplicity as well as low-level customizability, and are computationally efficient, well-tested and field-proven.
Availability and implementation
Both genesis and gappa are written in modern C++11, and are freely available under GPLv3 at http://github.com/lczech/genesis and http://github.com/lczech/gappa.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The necessity of computation in biology, and in (metagenomic) sequence analysis in particular, has long been acknowledged. In phylogenetics, for example, there is a plethora of software for analyzing data, covering tasks, such as sequence alignment (Pervez et al., 2014), phylogenetic tree inference (Zhou et al., 2018) and diverse types of downstream analyses (Washburne et al., 2018). Furthermore, in metagenomics, a key task is the taxonomic identification of sequences obtained from microbial environments. An increasingly popular method for this is phylogenetic (or evolutionary) placement, which can classify large numbers of (meta-)genomic sequences with respect to a given reference phylogeny. Common tools for phylogenetic placement are pplacer (Matsen et al., 2010), RAxML-EPA (Berger et al., 2011) as well as the more recent and more scalable tools EPA-ng (Barbera et al., 2018), APPLES (Balaban et al., 2019) and RAPPAS (Linard et al., 2019). The result of a phylogenetic placement can be understood as a distribution of sequences over the reference tree, which allows to examine the composition of microbial communities, and to derive biological and ecological insights (Czech et al., 2019; Czech and Stamatakis, 2019).
Here, we introduce Genesis, a library for working with phylogenetic data, as well as Gappa, a command-line tool for typical analyses of such data. They focus on phylogenetic trees and phylogenetic placements, but also offer various additional functionality. Combined, they allow to analyze as well as visualize phylogenetic (placement) data with existing methods and to experiment with and develop novel ideas.
To maximize usability of our tools, our implementation is guided by the following design objectives: (i) most users require a fast and simple application for analyzing their data, (ii) some power users desire customization, for example, via scripting, (iii) developers require a flexible toolkit for rapid prototyping and (iv) with the on-going data growth, the implementation needs to be scalable and efficient with respect to memory and execution times. To this end, Genesis and Gappa are written in C++11, relying on a modern, modular and function-centric software design.
We evaluate the code quality, the runtime behavior and the memory requirements for conducting typical tasks, such as file parsing and data processing in the Supplementary Material. An exemplary benchmark for reading Newick files is shown in Figure 1. We find that Genesis has the overall best code quality score compared to other scientific codes written in C or C++, using softwipe for the comparison (https://github.com/adrianzap/softwipe). It is also consistently faster than all evaluated Python and R libraries in our tests. Furthermore, Gappa is faster and more memory efficient than its main competitor Guppy in almost all tests and, most importantly, it scales better on larger datasets in all benchmarks.
Fig. 1.
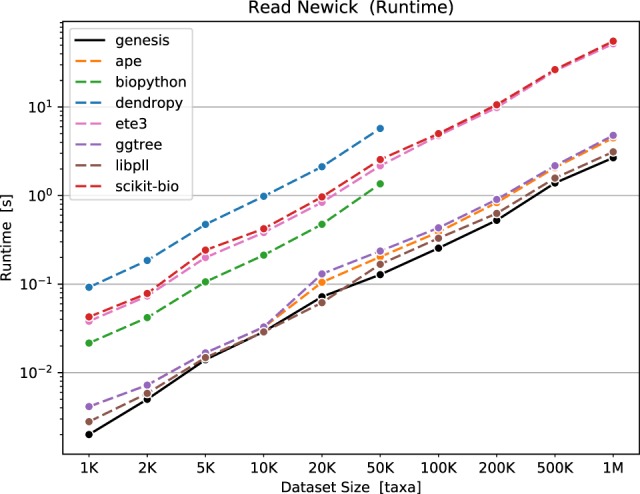
Runtimes for reading Newick files with 1 K–1 M taxa (tip/leaf nodes) and a randomly generated topology, using a variety of different libraries and tools. See the Supplementary Material for details
2 Features of Genesis
Genesis is a highly flexible library for reading, manipulating and evaluating phylogenetic data with a simple and straightforward application programming interface (API). Typical tasks, such as parsing and writing files, iterating over the elements of a data structure, and other frequently used functions are mostly one-liners that integrate well with modern C++. The library is multi-threaded, allowing for fully leveraging multi-core systems for scalable processing of large datasets. The functionality is divided into loosely coupled modules, which are organized in C++ namespaces. We briefly describe them in the following.
2.1 Phylogenetic trees
Phylogenetic trees are implemented via a pointer-based data structure that enables fast and flexible operations, and allows to store arbitrary data at the nodes and at the edges. The trees may contain multifurcations and may have a designated root node. Trees can be parsed from Newick files and be written to Newick, phyloxml and nexus files, again including support for arbitrary edge and node annotations. Traversing the tree starting from an arbitrary node in, for example, post-order, pre-order, or level-order can be accomplished via simple for loops:
// Read a tree from a Newick file.
Tree tree = CommonTreeNewickReader().read(
from_file (“path/to/tree.newick”)
);
// Traverse tree in preorder, print node names.
for (auto it: preorder (tree) ) {
auto& data = it.node().data<CommonNodeData>();
std::cout ≪ data.name ≪ “\n”;
}
Functionality for manipulating trees, finding lowest common ancestors of nodes [e. g. using the Euler tour technique of Berkman and Vishkin (1993)] or paths between nodes, calculating distances between nodes, testing monophyly of clades, obtaining a bitset representation of the bipartitions/splits of the tree and many other standard tasks are provided. Furthermore, functions for drawing rectangular and circular phylograms or cladograms to svg files, using individual custom edge colors and node shapes, are provided for creating publication quality figures.
2.2 Phylogenetic placements
Handling phylogenetic placement data constitutes a primary focus of Genesis. Placement data are usually stored in so-called jplace files (Matsen et al., 2012). Our implementation offers low-level functions for reading, writing, filtering, merging and otherwise manipulating these data, as well as high-level functions for distance calculations (Evans and Matsen, 2012), edge PCA and squash clustering (Matsen and Evans, 2011) and phylogenetic k-means clustering (Czech and Stamatakis, 2019), among others. Advanced functions for analyzing and visualizing the data are implemented as well, for instance, our adaptation of phylofactorization to phylogenetic placement data (Czech and Stamatakis, 2019; Washburne et al., 2017). Lastly, we offer a simple simulator for generating random placement data (e.g. for testing).
To the best of our knowledge, competing software that can parse placement data in form of jplace files [BoSSA (Lefeuvre, 2018), ggtree (Yu et al., 2017) or iTOL (Letunic and Bork, 2016)] merely offers some very basic analyses and visualizations, such as displaying the distribution of placed sequences on the reference phylogeny, but does not offer the wide functionality range of Genesis.
2.3 Other features
Sequences and alignments can be efficiently read from and written to fasta and phylip files; high-level functions for managing sequences include several methods for calculating consensus sequences, the entropy of sequence sets and sequence re-labeling via hashes. Taxonomies and taxonomic paths (e.g. ‘Eukaryota; Alveolata; Apicomplexa’) can be parsed from databases, such as Silva (Quast et al., 2013; Yilmaz et al., 2014) or NCBI (Benson et al., 2009; Sayers et al., 2009), and stored in a hierarchical taxonomic data structure, again with the ability to store arbitrary meta-data at each taxon, and to traverse the taxonomy.
Furthermore, Genesis supports several standard file formats, such as json, csv and svg. All input methods automatically and transparently handle gzip-compressed files. Moreover, a multitude of auxiliary functions and classes is provided: matrices and dataframes to store data, statistical functions and histogram generation to examine such data, regression via the generalized linear model, multi-dimensional scaling, k-means clustering, an efficient bitvector implementation (e. g. used for the bitset representation of phylogenetic trees mentioned above), color support for handling gradients in plots, etc. The full list of functionality is available via the online documentation.
Lastly, Genesis offers a simple architecture for scripting-like development, intended for rapid prototyping or small custom programs (e.g. convert some files or examine some data for a particular experiment).
3 Features of Gappa
The flexibility of a library, such as Genesis is primarily useful for method developers. For most users, it is, however, more convenient to offer a simple interface for typical, frequent tasks. To this end, we have developed the command line program Gappa.
Gappa implements and makes available the methods we presented in Czech et al. (2019) and Czech and Stamatakis (2019), such as: automatically obtaining a set of reference sequences from large databases, which can be used to infer a reference tree for phylogenetic placement; visualization tools to display the distribution of placements on the tree or to visualize per-branch correlations with meta-data features of environmental samples; analysis methods, such as phylogenetic k-means and placement-factorization for environmental samples.
Gappa also contains re-implementations of a few prominent methods of Guppy (Matsen et al., 2010), as well as commands for sanitizing, filtering, and manipulating files in formats, such as jplace, Newick or fasta, and a command for conducting a taxonomic assignment of phylogenetic placements (Kozlov et al., 2016).
As Gappa internally relies on Genesis, it is also efficient and scalable. Hence, Gappa can also be considered as a collection of demo programs for using Genesis, which might be helpful as a starting point for developers who intend to use our library. In comparison to Guppy, we have observed speedups of several orders of magnitude and significantly lower memory requirements in general when processing large data volumes; see the Supplementary Material and Czech et al. (2019) for details.
4 Conclusion
We presented Genesis, a library for working with phylogenetic (placement) data and related data types, as well as Gappa, a command line interface for analysis methods and common tasks related to phylogenetic placements. Genesis and Gappa already formed an integral part in several of our previous publications and programs (Barbera et al., 2018; Czech et al., 2019; Czech and Stamatakis, 2019; Mahé et al., 2017; Zhou et al., 2017), proving their flexibility and utility.
In future Genesis releases, we intend to offer API bindings to Python, thus making the library more accessible to developers. In Gappa, we will implement additional commands, in particular for working with phylogenetic placements, as well as re-implement the remaining commands of Guppy, in order to facilitate analysis of larger datasets.
Both Genesis and Gappa are freely available under GPLv3 at http://github.com/lczech/genesis and http://github.com/lczech/gappa.
Funding
This work was financially supported by the Klaus Tschira Stiftung gGmbH in Heidelberg, Germany.
Conflict of Interest: none declared.
Supplementary Material
References
- Balaban M. et al. (2019) APPLES: scalable distance-based phylogenetic placement with or without alignments. Syst. Biol. doi: 10.1093/sysbio/syz063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera P. et al. (2018) EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol., 68, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A. et al. (2009) GenBank. Nucleic Acids Res., 37, D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S. et al. (2011) Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol., 60, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkman O., Vishkin U. (1993) Recursive star-tree parallel data structure. SIAM J. Comput., 22, 221–242. [Google Scholar]
- Czech L., Stamatakis A. (2019) Scalable methods for analyzing and visualizing phylogenetic placement of metagenomic samples. PLoS One, 14, e0217050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech L. et al. (2019) Methods for automatic reference trees and multilevel phylogenetic placement. Bioinformatics, 35, 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S.N., Matsen F.A. (2012) The phylogenetic Kantorovich-Rubinstein metric for environmental sequence samples. J. R. Stat. Soc. Series B Stat. Methodol., 74, 569–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.M. et al. (2016) Phylogeny-aware identification and correction of taxonomically mislabeled sequences. Nucleic Acids Res., 44, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P. (2018) BoSSA: A Bunch of Structure and Sequence Analysis R package version 3.6. https://rdrr.io/cran/BoSSA/ (22 January 2020, date last accessed).
- Letunic I., Bork P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res., 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linard B. et al. (2019) Rapid alignment-free phylogenetic identification of metagenomic sequences. Bioinformatics, 35, 3303–3312. [DOI] [PubMed] [Google Scholar]
- Mahé F. et al. (2017) Parasites dominate hyperdiverse soil protist communities in Neotropical rainforests. Nat. Ecol. Evol., 1, 0091. [DOI] [PubMed] [Google Scholar]
- Matsen F.A., Evans S.N. (2011) Edge principal components and squash clustering: using the special structure of phylogenetic placement data for sample comparison. PLoS One, 8, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen F.A. et al. (2010) pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen F.A. et al. (2012) A format for phylogenetic placements. PLoS One, 7, e31009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervez M.T. et al. (2014) Evaluating the accuracy and efficiency of multiple sequence alignment methods. Evol. Bioinform. Online, 10, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C. et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res., 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E.W. et al. (2009) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 37, D5–D15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburne A.D. et al. (2017) Phylogenetic factorization of compositional data yields lineage-level associations in microbiome datasets. PeerJ, 5, e2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburne A.D. et al. (2018) Methods for phylogenetic analysis of microbiome data. Nat. Microbiol., 3, 652–661. [DOI] [PubMed] [Google Scholar]
- Yilmaz P. et al. (2014) The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res., 42, D643–D648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol., 8, 28–36. [Google Scholar]
- Zhou X. et al. (2017) Quartet-based computations of internode certainty provide accurate and robust measures of phylogenetic incongruence. Systematic Biology, syz058, doi: 10.1093/sysbio/syz058. [DOI] [PubMed] [Google Scholar]
- Zhou X. et al. (2018) Evaluating fast maximum likelihood-based phylogenetic programs using empirical phylogenomic data sets. Mol. Biol. Evol., 35, 486–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.