

Abstract
Motivation
The biological effects of human missense variants have been studied experimentally for decades but predicting their effects in clinical molecular diagnostics remains challenging. Available computational tools are usually based on the analysis of sequence conservation and structural properties of the mutant protein. We recently introduced a new machine learning method that demonstrated for the first time the significance of protein dynamics in determining the pathogenicity of missense variants.
Results
Here, we present a new interface (Rhapsody) that enables fully automated assessment of pathogenicity, incorporating both sequence coevolution data and structure- and dynamics-based features. Benchmarked against a dataset of about 20 000 annotated variants, the methodology is shown to outperform well-established and/or advanced prediction tools. We illustrate the utility of Rhapsody by in silico saturation mutagenesis studies of human H-Ras, phosphatase and tensin homolog and thiopurine S-methyltransferase.
Availability and implementation
The new tool is available both as an online webserver at http://rhapsody.csb.pitt.edu and as an open-source Python package (GitHub repository: https://github.com/prody/rhapsody; PyPI package installation: pip install prody-rhapsody). Links to additional resources, tutorials and package documentation are provided in the 'Python package' section of the website.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Single-nucleotide polymorphisms (SNPs) are single DNA base pair changes that are inherited (germline variants) or occur during the organism’s lifetime (somatic variants). A SNP located in a coding region of the DNA may lead to the translation of the gene codon into a different amino acid than the wild-type (non-synonymous SNPs), giving rise to a single amino acid variant (SAV or missense variant). Both synonymous and non-synonymous SNPs can perturb the normal activity of a cell. For example, synonymous SNPs can affect splicing, regulatory mechanisms and gene and/or protein expression levels although they do not affect the encoded protein’s sequence. SAVs can additionally have molecular effects, e.g. by altering a protein’s orthosteric or allosteric sites, its interaction with substrates or its stability.
More than half of the mutations implicated in human inherited diseases are estimated to be associated with SAVs (Stenson et al., 2017). As a result, devising analytical and computational approaches for predicting their effect has been of broad interest, but equally challenging due to complex effects in the cell. In recent years, it became evident that comprehensive approaches integrating multiple perspectives are the only viable solutions to achieve higher accuracy in pathogenicity predictions and to interpret experimental data at the molecular level. In the case of SAVs, this means understanding not only the significance of the mutated amino acid vis-à-vis the biological function of the protein, often captured by sequence-based conservation models, but also its importance for the fold stability and conformational mechanics and interactions, both intra- and intermolecular (Ancien et al., 2018).
Significant progress has been made in tools that focus on protein sequence conservation and residue coevolution, such as context-dependent modeling of sequence evolution (Feinauer and Weigt, 2017; Hopf et al., 2017) in recent years. In contrast, structure-based modeling approaches have been lagging behind compared to sequence-based approaches in evaluating the effect of SAVs, even though the first-generation classifiers that take account of 3D structures have shown considerable success (Adzhubei et al., 2010; Ancien et al., 2018; Capriotti and Altman, 2011). The importance of considering structure, or solvent accessibility, especially when relatively few homologs are available, has been pointed out in early studies (Saunders and Baker, 2002) and in more recent works based on residue network analysis (Brown et al., 2017; Brown and Tastan Bishop, 2017). This class of computations has been limited by two factors: first, they are possible only when the 3D structure of the protein is known, either from experiments or from comparative modeling. Second, even when a structure is available, the traditional methods to investigate the effect of missense variants such as molecular dynamics (MD) simulations require expensive computations which do not lend themselves to genome-scale analyses. While MD studies have shown success in predicting the impact of SAVs (Abdul Samad et al., 2016; Kumar and Purohit, 2014; LaRusch et al., 2014; Parveen et al., 2019; Priya Doss et al., 2014), they are applicable on a case-by-case basis only and are limited by the time and space limitations of MD simulations.
Yet, recent years have seen a rapid growth in the structural characterization of the proteome with advances in structure determination (e.g. cryo-EM) technologies. In parallel, computationally efficient methods such as those based on elastic network models (ENMs) have been developed, which efficiently provide insights into the intrinsic dynamics of proteins uniquely defined by their inter-residue contact topology (Bahar et al., 2010; Li et al., 2017). Many analytical tools have been developed within the framework of ENMs, which focus on different aspects of protein equilibrium dynamics, both on a local (e.g. fluctuations in residue positions) and a global (e.g. coupled domain movements and allosteric switches) scale. ENMs are broadly used for mechanistic studies, but their utility in genome-scale studies of the impact of mutations is becoming clear only in recent studies (Ponzoni and Bahar, 2018; Rodrigues et al., 2018).
The rapidly growing experimental data on the functional impact of SAVs and on protein structures provide a unique opportunity for building upon that first generation of pathogenicity predictors to develop a machine learning approach trained not only on well-established sequence- and structure-dependent properties, but also on intrinsic dynamics, derived from ENMs. A first attempt in that direction (Ponzoni and Bahar, 2018) paved the way to the current development and implementation of Rhapsody, an advanced tool and user-friendly server for Rapid High-Accuracy Prediction of SAV Outcome based on DYnamics, accessible at http://rhapsody.csb.pitt.edu.
The inclusion of dynamics-based features distinguishes Rhapsody from tools broadly used in the field such as PolyPhen-2 (Adzhubei et al., 2010), SIFT (Ng and Henikoff, 2003), CADD (Kircher et al., 2014) and others [see Grimm et al. (2015) for a critical review of some of these methods and Hu et al. (2019) for an updated list of tools]. We presently introduce a ‘full’ version of Rhapsody that incorporates coevolution features extracted from Pfam domains, inspired by the success of recent studies (Feinauer and Weigt, 2017; Hopf et al., 2017). We provide extensive comparisons of Rhapsody against PolyPhen-2 (Adzhubei et al., 2010) and EVmutation (Hopf et al., 2017), utilizing a refined dataset of about 20 000 human SAVs, built from consensual clinical interpretations between multiple databases (DBs). PolyPhen-2 is a broadly used tool for predicting the functional effects of human variants, which relies on a supervised naïve Bayes classifier trained on annotations, conservation scores and structural features that characterize the amino acid substitution. It is chosen here as a representative tool among several other publicly available methods because of its widespread use. EVmutation, on the other hand, emerges as one of the most accessible and powerful tools among the recent wave of tools that leverage coevolution analysis for predicting the fitness of mutants, going beyond the limitations of conservation analyses by taking account of the inter-dependencies between pairs of sequence positions. The change in ‘evolutionary statistical energy’ ΔE incurred upon mutation is directly interpreted as a proxy for the mutant fitness. However, a cutoff energy for binary classification of mutants as deleterious or neutral is not defined.
Rhapsody is implemented as a standalone package, which may be used in conjunction with our ProDy API (Bakan et al., 2011). The server offers the option of using as input customized Protein Data Bank (PDB) structures, such as those stabilized under different conformational and oligomerization states as well as those resolved for orthologues or generated by comparative modeling.
We illustrate the utility of Rhapsody by way of applications to human H-Ras, a highly conserved G-protein belonging to Ras subfamily of small GTPases for which deep mutational scanning data have been recently reported (Bandaru et al., 2017), and to two human proteins featured in a recent Critical Assessment of Genome Interpretation (CAGI) competition (Andreoletti et al., 2019): PIP3 phosphatase, also called phosphatase and tensin homolog (PTEN) and thiopurine S-methyltransferase (TPMT). The new tool provides not only an efficient independent assessment of potential pathogenic effect of mutations, but also mechanistic insights into the molecular basis of the observed and/or predicted effects.
2 Materials and methods
2.1 Development of an upgraded dynamics-based pathogenicity predictor
Three groups of features, sequence-based (SEQ), structure-based (STR) and dynamics-based (DYN), computed for each position along the sequence and/or specific amino acid substitution (e.g. ‘P01112 10 G A’ in UniProt coordinates, indicating variant G10A of GTPase H-Ras), are used for training a random forest classifier, following the approach described in our earlier work (Ponzoni and Bahar, 2018). In the original version of the algorithm, SEQ features were computed by the PolyPhen-2 server (Adzhubei et al., 2010), STR features by using structural data from the PDB and DYN features by the ProDy API (Bakan et al., 2011). This classifier proved to achieve accuracy levels comparable to, if not better, than 11 existing tools (Ponzoni and Bahar, 2018).
In this study, we introduce two upgraded versions, referred to as ‘reduced’ and ‘full’ Rhapsody classifiers. Supplementary Table S1 provides a detailed list of the features used in both versions along with their definition and interpretation. The reduced version includes BLOSUM62 amino acid substitution scores (Henikoff and Henikoff, 1992) as an additional feature and upgraded DYN features calculations (Fig. 1A and B). The full Rhapsody classifier uses as additional features the mutation site entropy and coevolution properties deduced from Pfam domains (El-Gebali et al., 2019).
Fig. 1.

Rhapsody features and prediction accuracy. (A) Random forest features used in Rhapsody classifiers. See Supplementary Table S1 for detailed descriptions. (B) Comparison of the accuracy of three Rhapsody classification schemes of different complexities and coverage (full, reduced and combined with EVmutation, shown by the three sets of bars on the left) with that of two other tools, PolyPhen-2 and EVmutation, measured by area under the ROC plot (AUROC) values (and relative error bars from 10-fold cross-validation) obtained using OPTIDS (Supplementary Table S2). As SAVs from the same residue or protein could be found in both training and testing subsets (gray bars), we repeated the Rhapsody computations for residue- (orange) and protein-stratified (blue) versions of our dataset to ensure unbiased evaluations. Light green bars in the background show the relative size of the datasets of variants (right ordinate) used for 10-fold cross-validations of Rhapsody and for testing the other two methods. See also Supplementary Figures S1 and S2 for further comparison of these methods using additional metrics and for comparisons with outputs from other tools, and Supplementary Materials and Methods for more details. (C) Effect of excluding variants of various confidence levels (based on ClinVar DB review rates/stars) from the training dataset. Light green bars represent the numbers of SAVs (right ordinate) that could be processed by Rhapsody’s full classifier, for different subsets. The leftmost bar refers to the complete IDS (with PDB structures larger than 150 residues); the second bar excludes those with 0-star; the third excludes those with 0- and 1-stars and so on. The blue curve (left ordinate) displays the prediction accuracy levels with error bars computed through cross-validations
We also designed a new interface (http://rhapsody.csb.pitt.edu) that enables efficient use of the algorithm and visualization of its output. A detailed description of random forest features and hyper-parameter optimization, Python package implementation and interface design is presented in Supplementary Materials and Methods.
2.2 Construction of an integrated dataset of annotated human variants
The dataset for training the algorithm has been generated by combining five publicly available datasets [HumVar (Adzhubei et al., 2010), ExoVar (Li et al., 2013), PredictSNP (Bendl et al., 2014), VariBench (Thusberg et al., 2011) and SwissVar (Mottaz et al., 2010)] with the Humsavar DB of all human missense variants annotated in the UniProtKB/Swiss-Prot DB and the ClinVar archive of reports on the level of concordance between human variations and phenotypes (Landrum et al., 2016). Supplementary Table S2 provides information on the content of these datasets and their level of agreement. After filtering out discordant labels, we obtained an ‘Integrated Dataset’ (IDS) of 87 726 SAVs, of which 27 655 could be mapped onto PDB structures, a prerequisite for computing STR/DYN features, and 23 085 had PDB structures with at least 150 residues.
The ClinVar DB provides a reliability level for each variant, with the help of zero (weak) to four (best) ‘review stars’ assigned to each SAV, based on the number of, and consensus between, various sources. Variants with ‘no assertion’ or ‘no assertion criteria provided’ are assigned 0-star; those characterized by ‘single submitter’ or ‘conflicting interpretations’ are assigned 1-star; a 2-star assignment refers to ‘no conflicts and multiple submitters’; 3-star, to ‘reviewed by experts’ and 4-star, to ‘practice guideline’. As will be shown in Section 3, removal of the 0-star cases led to improved prediction accuracy. The final, optimized integrated dataset (OPTIDS) after eliminating these low-confidence cases contains 20 361 SAVs with at least 1 ClinVar review star, mapped onto 2828 unique chains in the PDB, each containing at least 150 residues.
3 Results
3.1 Cross-validation and comparison with other tools
In a preliminary analysis (Fig. 1C), we monitored the average area under the ROC curve (AUROC) attained by the full classifier in a 10-fold cross-validation procedure while gradually excluding from the IDS those SAVs with lower ClinVar rating. The exclusion of SAVs with 0-stars helped improve the accuracy (blue curve in Fig. 1C). This was followed by a plateau or minimal decrease in accuracy when further excluding 1-, 2-, 3- and 4-star SAVs. These additional changes were within the error bars computed from 10 cross-validation iterations, so we opted to exclude SAVs with 0-stars only, which accounted for ∼12% of cases, from our training dataset in all subsequent analyses.
This OPTIDS was used for evaluating the accuracy of the classifier through cross-validation. In Figure 1B, we compare the performances of three variants of Rhapsody against PolyPhen-2 (Adzhubei et al., 2010) and EVmutation (Hopf et al., 2017). The colored bars represent accuracy measurements for each method’s predictions. For the three Rhapsody variants on the left, we calculated the average AUROC and associated SDs from a 10-fold cross-validation on OPTIDS, while for PolyPhen-2 and EVmutation, we plotted the AUROC values over the same dataset of variants. The light green bars in the background indicate the actual number of SAVs that could be evaluated by each approach. The cross-validation for Rhapsody classifiers has been carried out through random partitioning of OPTIDS, stratified by mutation classes to ensure equivalent bias in each fold (gray bars in Fig. 1B). Additional low-redundancy measurements have been performed as more stringent tests, by removing the variants of the same residue (‘residue-stratification’, orange bars) or within the same protein (‘protein-stratification’, blue bars) from the training subsets. Each of these steps resulted in lower estimates of accuracy, by up to ∼0.03.
We notice that the full Rhapsody classifier outperforms both PolyPhen-2 and EVmutation, based on AUROC values. Similar conclusions could be drawn by evaluating the performance of these methods with other metrics, such as the Matthews correlation coefficient (MCC) and F1-score, as presented in Supplementary Figure S1. The latter are known to be less affected by high class imbalance (bias toward deleterious mutations in OPTIDS), and therefore may provide a better estimate of accuracy. Note that about 70% of our training dataset consists of deleterious variants while an opposite composition bias is observed in naturally-occurring human variants (Lek et al., 2016). To mitigate the effect of such imbalances, the random forest models have been trained by assigning to training examples weights inversely proportional to class frequency.
The full Rhapsody classifier is also seen to outperform the reduced version, although within the error margins defined by the metrics’ SD. Further comparison with the original version introduced in 2018 (Ponzoni and Bahar, 2018), presented in Figure 2C, shows the statistically significant improvement achieved in the full version, using two different ENMs, the Gaussian Network Model (GNM) (Li et al., 2016) and the Anisotropic Network Model (ANM) (Eyal et al., 2015), for evaluating DYN properties. However, the introduction of Pfam-derived features in the full classifier comes at the cost of a slight decrease in coverage, since Pfam domains often do not encompass the full span of a protein sequence, but only those portions that are preserved across species. In this regard, PolyPhen-2 has the widest coverage, being able to return a prediction even for variants without a PDB structure.
Fig. 2.

Analysis of the Rhapsody classifier. (A) Weights of features in the Rhapsody classifier integrated with EVmutation. See also Supplementary Figure S4. (B) Spearman’s correlations between all pairs of features. (C) Accuracy of the full Rhapsody algorithm (repeated using either GNM- or ANM-predicted DYN features, with and without environmental effects) on different subsets of the OPTIDS obtained by setting a minimum PDB structure size (i.e. number N of resolved residues). In yellow, we show the performance of the original algorithm (Ponzoni and Bahar, 2018). Error bars represent the SD computed during cross-validations. See also Supplementary Figure S5 for similar results with other accuracy metrics. (D) Training dataset size (SAVs successfully processed by ‘full’ classifier, in blue) and fraction of positive training examples (i.e. deleterious SAVs, in red) as a function of the minimum number of residues used to filter PDB structures based on their size
In addition to the full and reduced versions of Rhapsody, we also considered a third option, designated as ‘Rhapsody + EVmut’, which incorporated the EVmutation ‘epistatic’ score ΔE within the feature set. This variant slightly improved upon the full classifier, but it also further reduced the coverage. Of note, the integration of EVmutation and Rhapsody leads to significantly more accurate predictions than EVmutation used alone.
In the above comparative evaluations, we note that PolyPhen-2’s training dataset partially overlaps with OPTIDS, as discussed earlier (Ponzoni and Bahar, 2018), which may lead to an overestimation of the accuracy of PolyPhen-2 (Grimm et al., 2015). More generally, it is not always possible nor feasible to account for such ‘training biases’, unless a completely novel and independent testing dataset is designed. In order to facilitate future assessments, the output from our algorithm explicitly acknowledges whenever a tested variant is also listed in the training dataset. We presented in Supplementary Figure S2 an additional comparison of the outputs from Rhapsody with those from 27 other tools currently compiled in dbNSFP, a DB of functional predictions and annotations for all potential non-synonymous single-nucleotide variants in the human genome (Liu et al., 2011, 2016). Yet, the same type of training bias may also hold for the precomputed outputs in dbNSFP which may preclude an objective assessment, even though an exhaustive list of metrics has been considered therein. The large discrepancies in the accuracy levels for individual classes [neutral and deleterious SAVs, indicated by suffixes ‘(0)’ and ‘(1)’, respectively] observed for all methods reflects the imbalance of the dataset and the challenges associated with it.
Finally, we carried out an additional benchmarking study against predictions from SNPs3D (Yue et al., 2006). The latter is notable among pathogenicity prediction tools because it evaluates the functional consequences of a SAV by assessing its impact on structural stability, in addition to identifying candidate genes for specific diseases and providing information on the relationships between these candidates. For this comparison, a new classifier was trained. A relatively small subset of variants in our OPTIDS was chosen as a test set, given the availability of precomputed predictions from SNPs3D, and the proteins containing those variants were excluded from the training set. The results presented in Supplementary Figure S3 show equal or better performance of Rhapsody in general over SNPs3D using a broad range of metrics, even on this particularly challenging (imbalanced) test set that included a small proportion of deleterious SAVs, strongly departing from the composition of OPTIDS.
Overall, these results confirm the usefulness of including intrinsic dynamics features in the context of functional assessment of variants, and further demonstrate the power of adopting an integrative approach that incorporates coevolution analysis into supervised learning approaches, thus taking advantage of its superior predictive power compared to single amino acid conservation properties.
3.2 Contribution of selected features
Figure 2A illustrates the relative weights of the features used in the integrated classifier 'Rhapsody + EVmut'. The counterparts for the 'full' and 'reduced' Rhapsody classifiers can be seen in the Supplementary Figure S4. In parallel with previous observations (Ponzoni and Bahar, 2018), sequence-based features (wtPSIC, ΔPSIC and entropy of Pfam domain) rank higher than dynamics-based (ENM-derived) features, since the latter lack residue specificity. Dynamics-based features, in turn, prove to be more informative than a widely used structural property, solvent accessibility.
We note that these features are not necessarily independent. The heat map in Figure 2B provides a quantitative description of their similarities. Yet, their explicit inclusion in the training algorithm assists in increasing prediction accuracies. We note, in this context, the remarkable weight difference between two coevolution properties, the ‘ranked’ mutual information (MI) and EVmutation’s ΔE score. The former was chosen for its simplicity, which makes it orders of magnitude faster to evaluate computationally than EVmutation scores, for which a DB of precomputed values was used in practice (Hopf et al., 2017). For real-time evaluation of coevolution properties, the integration of more efficient coevolution algorithms might be envisioned.
3.3 Higher accuracy achieved with larger structures
Figure 2C illustrates the dependency of pathogenicity prediction accuracy on the minimum size of the PDB structure included in the evaluation of the STR and DYN features. More detailed results with different metrics are presented in the Supplementary Figure S5. A slight improvement in accuracy is observed when excluding structures with fewer than N = 150 residues, and again when limiting the analysis to structures with at least 500 residues. Examination of the dependency of feature weights on protein size illustrated in Supplementary Figure S7 indicated that the observed pattern did not originate from differences in feature weights which remained relatively constant in the range N < 300. The increased accuracy upon exclusion of small (N < 150) structures could be attributed to the fact that sequence/structure data in this range might be incomplete and not representative of the intact protein. Conversely, the relatively high accuracy in the range N > 500 could reflect the more complete inclusion of physical and evolutionary interactions between sequentially distal but spatially close neighbors in the multi-domain or multi-subunit proteins.
The existence of a direct correlation between prediction accuracy and size of PDB structures, if any, is blurred by the concurrent changes in the training dataset size and composition (blue and red curves, respectively, in Figure 2D). The non-monotonic behavior of the AUROC plot in Figure 2C could thus be attributed to the changing imbalance between deleterious and neutral variants in the training dataset at different PDB size cutoffs. Such non-uniform distributions are also viewed in the breakdown of the IDS population and imbalance at various PDB chain length intervals in Supplementary Figure S6. However, the pattern observed in Figure 2C is robustly displayed by other metrics that are less susceptible to dataset imbalance, namely MCC and F1-score (Supplementary Fig. S5). Thus, we deemed it safe to use the SAVs with N > 150 for training purposes.
3.4 Application to H-Ras
3.4.1 Saturation mutagenesis analysis of human H-Ras protein
Kuriyan and coworkers recently presented results from deep mutational scanning of human H-Ras (Bandaru et al., 2017), a highly conserved signaling protein which transduces signals through a nucleotide-dependent switch between active (GTP-bound) and inactive (GDP-bound) conformations. The impact of a single mutation on the protein’s normal activity was experimentally linked to the survival of the hosting bacterial system and quantified by a ‘fitness score’ (ΔE), under different contexts. Here, we focus on the complete (‘regulated Ras’) experimental setup, designed to include regulatory factors that might constrain Ras sequence variability and that are necessary to obtain a realistic assessment of mutants’ fitness.
Figure 3 presents the results from our so-called ‘in silico saturation mutagenesis’ analysis. The results are presented in a 20 x N heat map (Figure 3A) where the entries are color-coded by pathogenicity probabilities (Supplementary Materials and Methods) predicted for all 19 possible substitutions at each of the N = 171 structurally resolved sequence positions of H-Ras (UniProt sequence ID: P01112). The entries corresponding to the wild-type amino acids are in white. The map structure mirrors that of analogous maps of experimental fitness measurements (Bandaru et al., 2017).
Fig. 3.

In silico saturation mutagenesis results for human H-Ras. (A) The predicted pathogenicity probabilities for all possible SAVs in H-Ras computed by Rhapsody are shown as a heatmap with a color code ranging from red (deleterious) to blue (neutral); see Supplementary Materials and Methods for more details on the definition of pathogenicity probability. The corresponding residue-averaged pathogenicity profile is shown in red in the bottom panel, compared to analogous profiles from PolyPhen-2 (blue) and EVmutation (green) and from experimental fitness measures (grey). The two strips along the upper abscissa of the heatmaps display the secondary structure and solvent accessibility (SASA) along the sequence. The Rhapsody results are obtained for the structure in the active state. The counterpart for the inactive state is presented in Supplementary Fig. S8. (B) Residue pathogenicities displayed by color-coded ribbon diagrams for active (top) and inactive (middle) H-Ras. Red and blue colors indicate the regions with high and low propensities for pathogenicity, respectively. The difference is shown in the bottom panel. The respective purple and green regions refer to sites exhibiting increased and decreased pathogenicities in the active form. The purple regions include the two switches involved in activation
The structure-dependent (STR) and dynamics-based (DYN) features required by Rhapsody were computed on the active, GTP-bound conformation of H-Ras (PDB ID: 6Q21, chain A). Computations repeated for the inactive state (PDB ID: 4Q21, chain A) showed that the predictions were very similar (Supplementary Figs. S8 and S9), with the main differences localized at the switches I and II (Fig. 3B). These results are consistent with the robustness of ENM results to structural details, i.e. H-Ras structural dynamics is predominantly defined by its 3D fold, which defines its inter-residue contact topology. The contact topology, in turn, determines the intrinsically accessible spectrum of motions. The impact of SAVs on collective mechanics can thus be inferred from either active or inactive state, provided that the overall fold remains unchanged.
At first glance, the heat maps in Figure 3A show an alternating pattern of blue (neutral) and red (pathogenic) vertical bands that loosely correlate with either secondary structure or surface exposure of residues (top strips). Such a pattern can also be discerned in the bottom panels of Figure 3A. The red curve therein shows the residue-based pathogenicity profile predicted by Rhapsody upon averaging the entries in the corresponding column of the map. Analogous profiles obtained using PolyPhen-2 (blue), EVmutation (green) and experimental fitness scores for ‘regulated-Ras’ (Bandaru et al., 2017) (–ΔE, gray) reveal an overall agreement between computations and experiments.
Rhapsody performs better than EVmutation and PolyPhen-2 when comparing the predicted residue-averaged pathogenicities with experimental data, as can be seen in Supplementary Table S3. The table lists the Spearman’s rank-order correlations, |ρ|, between experimental and (different types of) computational data. For the ‘regulated’ case (Fig. 4A and Supplementary Fig. S10), |ρ| = 0.60 and 0.57 for Rhapsody predictions based on the inactive and active states, respectively, as opposed to |ρ| = 0.52 and 0.51 for EVmutation and PolyPhen-2. Both Rhapsody and EVmutation outperform PolyPhen-2 in predicting individual fitness scores (|ρ| ≈ 0.42 versus 0.36). We also estimated the prediction accuracies using AUROC and AUPRC as metrics. These required a binary labeling of variants (neutral/pathogenic) that cannot be readily deduced from the distribution of experimental ΔE values, see Supplementary Figure S11. We arbitrarily set the median of the distribution as a cutoff, while the 40th and 60th percentiles have been used to compute an uncertainty interval (an alternative labeling scheme and relative metrics calculations are shown in Supplementary Figs. S14 and S15). The resulting ROC curves (Fig. 4B and Supplementary Fig. S12) confirm similar accuracy levels for Rhapsody and EVmutation, with respect to both individual (AUC) and residue-averaged («AUC»res) experimental data, and slightly lower accuracies for PolyPhen-2. Analogous conclusions emerge from the analysis of Precision–Recall curves, presented in Figure 5A and Supplementary Figure S13.
Fig. 4.

Pathogenicity predictions of human Ras protein variants. (A) Scatter plots and Spearman’s ρ correlations between experimental fitness scores from (Bandaru et al., 2017) and predictions from Rhapsody (based on inactive/active conformations), EVmutation and PolyPhen-2. Red circles correspond to residue-averaged values. See also Supplementary Figure S10. (B) ROC curves for substitution-specific (blue) and residue-averaged (red) predictions. The median of experimental ΔE values is used as cutoff to assign binary labels to variants (Supplementary Fig. S11). The 40th and 60th percentiles have also been considered and used to compute uncertainty bands, represented in figure by semi-transparent blue/red shades. See also Supplementary Figure S12
Fig. 5.

Analysis of Ras predictions. (A) Precision-recall plot for individual (blue curve) and residue-averaged (red curve) Rhapsody predictions of experimental fitness values. Corresponding AUCs are 0.72 and 0.80, respectively. Analogous plots for EVmutation and PolyPhen-2 are reported in Supplementary Figure S13. (B) Scatter plot of Rhapsody predicted pathogenicity probabilities versus experimental measurements. See Supplementary Figure S11 for the definition of the vertical boundary separating experimental fitness effects. (C) False positives (green) and False negatives (red) highlighted in panel (B) and displayed on the protein structure (active conformation)
These results show that Rhapsody can be advantageously used for a first assessment of the regions that are sensitive to mutations. Moreover, the consideration of a more diverse set of properties, such as dynamics-based features on top of sequence- and structure-based ones, as in Rhapsody, provides the opportunity of interpreting the observations in the light of the protein’s structural and dynamic features.
A visualization of Rhapsody incorrect predictions on Ras 3D structure (Fig. 5B and C) reveals that most False Negatives are localized on the protein’s surface, while False Positives are generally found in less exposed positions. A possible explanation is that the method is inherently biased toward the identification of residues important for the fold stability or internal dynamics, while locations subjected to other kinds of constraints, e.g. allostery and interactions with other proteins and small molecules, are more difficult to evaluate with the current set of features.
3.4.2 Analysis of H-Ras variants in gnomAD
We tested our predictions on a set of human variants found in healthy individuals, as collected by the gnomAD DB (Karczewski et al., 2019). The assumption is that those substitutions seen in the 140 000 people tested (mostly normal population) are somewhat permissive. We therefore compared the distribution of predictions obtained by Rhapsody on this set of gnomAD SAVs with the corresponding fitness scores from the experimental study considered above (Bandaru et al., 2017).
The results, illustrated in Figure 6, show that the predictions for the gnomAD SAVs are skewed toward ‘neutral’ classification in both distributions, with 49 out of 82 total variants classified as ‘neutral’ or ‘probably neutral’ by our algorithm. Of note, 3 out of 4 ‘high count’ SAVs (i.e. seen in 10 or more people) are interpreted as non-pathogenic by Rhapsody, while 2 out of 4 SAVs have a fitness score ΔE, as measured in the saturation mutagenesis study, significantly lower than the wild-type amino acid (when choosing the median of all values as cutoff).
Fig. 6.
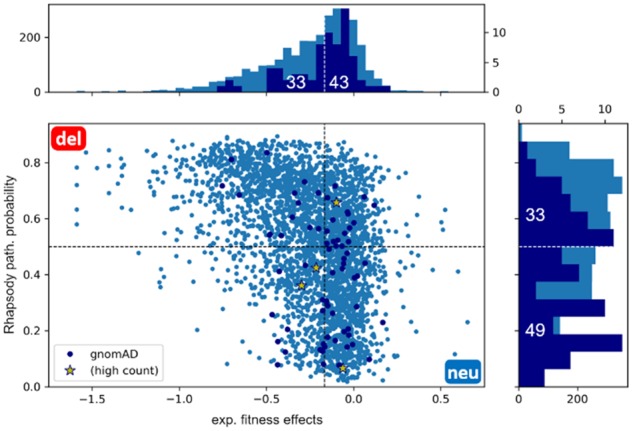
Analysis of H-Ras SAVs from gnomAD DB. H-Ras SAVs (dark blue dots) collected from the gnomAD DB found in healthy population are shown along with the results for all SAVS (light blue dots) on the scatter plot between Rhapsody pathogenicity probabilities and experimental fitness scores (Bandaru et al., 2017). ‘High-count’ SAVs (yellow stars) were seen in at least 10 individuals. The marginal plots show the corresponding distributions computed for all variants (light blue) and gnomAD variants (dark blue)
3.5 Application to PTEN and TPMT variants from CAGI competition
As an additional test, we considered a dataset of over 7000 SAVs for the tumor suppressor protein PTEN and the enzyme TPMT. The pathogenicity of these proteins’ variants has been recently investigated by massively parallel sequencing (VAMP-seq), a functional assay that measures the steady-state abundance of variants in cultured human cells (Matreyek et al., 2018). The results for PTEN/TPMT datasets were featured in the fifth edition of CAGI, a series of competitions that aim to objectively assess computational methods on blind prediction tasks (Andreoletti et al., 2019). We performed a direct comparison of our predictor with other computational methods that, although adapted for the specific challenges proposed in the competition, were tasked with providing blind predictions, without having access to the experimental results (Pejaver et al., 2019). To ensure as much as possible a similar unbiased evaluation, new Rhapsody classifiers were trained by excluding SAVs of PTEN (56 deleterious, 1 neutral) and TPMT (3 deleterious, 4 neutral) from our training dataset, as previously done for H-Ras.
We first evaluated the predictions from Rhapsody, PolyPhen-2 and EVmutation by computing their Spearman’s correlation with experimental ‘protein-abundance’ scores from VAMP-seq data (Supplementary Fig. S16). Low-abundance variants were found to be enriched in pathogenic variants and they correlated with low protein thermodynamic stability (Matreyek et al., 2018), thus abundance score has been used as a proxy for variant impact on proteins (Pejaver et al., 2019). A classification of variants into ‘abundancy’ classes (‘low-abundance’, ‘possibly low-abundance’, ‘possibly WT-like’ and ‘WT-like’) was also provided (Matreyek et al., 2018), thus allowing the use of other class-based accuracy metrics, such as AUROC, MCC and F1 score. Based on these metrics, we see in Figure 7 that Rhapsody and EVmutation are distinguished by their respective higher accuracy levels on TPMT and PTEN variants, and both consistently outperform PolyPhen-2. EVmutation, however, could only provide predictions for a small fraction (∼13%) of PTEN variants.
Fig. 7.

Assessment of pathogenicity predictions applied to PTEN and TPMT variants. The level of agreement between computationally predicted pathogenicity scores and experimental abundance scores is shown, based on four metrics (Spearman’s, AUROC, MCC and F1-score). Results are presented for Rhapsody, PolyPhen-2 and EVmutation, represented by bar plots (see also scatter plots in Supplementary Fig. S16), and for CAGI predictors, represented by violin plots showing the distribution, median and range of values. The ‘low-abundance’ and ‘possibly low-abundance’ classes [as reported in (Matreyek et al., 2018)] were considered as ‘deleterious’, and ‘possibly WT-like’ and the ‘WT-like’ classes, as ‘neutral’. The rightmost bars in each plot represent the number of SAVs successfully predicted by each method (in red, the common value for CAGI predictors)
Prediction accuracies from participants to the CAGI5 challenge, described in (Pejaver et al., 2019) (data available from CAGI website to registered users only), are also shown in aggregated form as violin plots in Figure 7. In both cases, Rhapsody, EVmutation and Polyphen-2 all fall within the range of prediction accuracies measured for CAGI predictors. In the case of TPMT, we notice that Rhapsody consistently ranks between the median of the CAGI methods and the best-performing one.
These results demonstrate the validity of Rhapsody predictions in tasks specifically designed for testing computational methods, and against tools specifically adapted for these tasks. The modest performances demonstrated by all methods, on the other hand, also highlight the need for more effective computational approaches. Systematic assessment campaigns such as CAGI constitute an invaluable platform for evaluating the progress in the field.
4 Discussion
In the present study, we presented a novel machine learning approach for evaluating the functional impact of human SAVs, and illustrated its application to H-Ras, PTEN and TPMT. In a strict sense, Rhapsody, like many other tools in the field, predicts whether a given mutation is neutral or deleterious to protein activity, whereas pathogenicity entails many other factors, including inheritance pattern, penetrance, expressivity and environment. Thus, the outcome from the tool rather indicates a potential to be pathogenic. The newly introduced interface, Rhapsody, integrates dynamical features computed from the ENM-based analyses of protein structures and attains a state-of-the-art accuracy for predicting such a potential with a relatively simple design. We also highlighted how the method can be used not only for hypothesis generation (predictions for variants of unknown significance) but also for hypothesis testing, by providing a unified framework for comparing the predictive power of new as well as more established features. For instance, we demonstrated the utility of including in our machine-learning algorithm the ENM-derived dynamics-based features, in addition to more traditional features such as sequence conservation and structural accessibility, and emphasized the need for a better integration with coevolution analysis that recently showed significant success in evaluating the effect of SAVs.
Through the analysis of saturation mutagenesis studies and other experimental and clinical data, we identified the strengths and limitations of our approach and compared it against other prediction tools. We observed a general robustness of computational predictions, especially in the identification of residue sites that are sensitive to any mutation, regardless of the specific amino acid substitution. This information can be invaluable for the study of the functional mechanisms of proteins, especially when projected on the 3D structures. The use of structure-based properties, in combination with sequence conservation properties (reduced classifier), can be used as an alternative approach to the more sophisticated coevolutionary analysis, whenever the latter cannot be applied due to lack of suitable multiple sequence alignments. The current algorithm has been designed to be easily expandable with new features and functionalities. Structural features such as those used in the FEATURE framework for protein annotation (Halperin et al., 2008) could be incorporated in future versions for possibly enhancing the utility of Rhapsody.
The comparison with clinical and experimental data also revealed a few issues that need to be resolved in order to advance the field. Apart from the obvious shortcomings such as the imbalance of available datasets toward pathogenic variants and the often-contradictory clinical interpretations in different DBs, we reported our difficulties in interpreting data from large-scale experimental studies. These studies provide a unique opportunity for dramatically increasing the size of training datasets. However, there is a need for a systematic definition of what is considered as a ‘pathogenic’ variant, that would account for both loss-of-function and gain-of-function effects in relation to the biological role of the affected protein.
We expect future improvements to our method to address some of these shortcomings. A recent ENM study has demonstrated how the consideration of the intact structures of multimers, complexes or assemblies improves the accuracy of predicted fluctuation spectrum of residues, and predictions from that server (DynOmics) (Li et al., 2017) could be used for evaluating context-dependent structural and dynamic properties. For example, a region that is deemed to be tolerant to mutations by virtue of its solvent-exposure in the PDB resolved structure, may become a buried site in a complex/assembly, and a substitution at that region could alter its binding properties. A recent study has demonstrated how disease-associated SAVs are likely to be located at singlet hot spots at protein–protein interfaces (Ozdemir et al., 2018). Consideration of the involvement of residues in interfacial interactions is expected to improve the prediction accuracy of current algorithms.
Another possible improvement would be the consideration of the signature dynamics of the protein family to which the investigated protein belongs, as opposed to the dynamics of the protein alone (Zhang et al., 2019). In the same way as variations in sequence among family members point to sites that can, or cannot, tolerate mutations, family-based analyses can provide deeper insights into sites whose mechanistic properties are indispensable for function or for differentiation among subfamily members. Finally, a decomposition of the mode spectrum could help extract information on high-energy localization (hot) spots emerging as peaks in high frequency modes, as well as the hinge regions between domains, where substitutions may be detrimental (Dorantes-Gilardi et al., 2018; Rodrigues et al., 2018; Sayılgan et al., 2019).
The Rhapsody algorithm is provided both as an open-source Python package (pip install prody-rhapsody) and a web tool (http://rhapsody.csb.pitt.edu). The latter has been designed as a user-friendly service that requires minimal user input or computing skills, but also allows for some customization, such as selecting or uploading a specific PDB structure. The Rhapsody webserver can be used for both obtaining predictions on a list of human SAVs (batch query) and for visualizing a complete in silico saturation mutagenesis analysis of a human sequence, akin to those presented in Figure 3 for H-Ras. Finally, the site offers tutorials, training data (OPTIDS) and precomputed features needed for reproducing all results presented here, or for analyzing new variants. The documentation also explains how to train a model on a completely different set of features and using a different training dataset, thus providing researchers with a flexible tool for analyzing personalized datasets and testing new predictors with the help of all the functionalities implemented in Rhapsody.
Supplementary Material
Acknowledgements
The authors are grateful to Dr. Reece Hart for constructive comments.
Author contributions
The project was designed by L.P. and I.B. The in silico models were generated by L.P. with input from I.B. The Rhapsody Python package, documentation, tutorials and webserver were implemented by L.P. Clinical interpretations of the variants were contributed by Z.N.O. D.A.P contributed to data retrieval and processing. The manuscript was written by L.P., Z.N.O. and I.B. All authors approved the manuscript.
Funding
This work was supported by National Institutes of Health [P41 GM103712 and P30 DA035778 to I.B.].
Conflict of Interest: none declared.
References
- Abdul Samad F. et al. (2016) A comprehensive in silico analysis on the structural and functional impact of SNPs in the congenital heart defects associated with NKX2-5 gene—a molecular dynamic simulation approach. PLoS One, 11, e0153999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I.A. et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancien F. et al. (2018) Prediction and interpretation of deleterious coding variants in terms of protein structural stability. Sci. Rep., 8, 4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoletti G. et al. (2019) Reports from the fifth edition of CAGI: the critical assessment of genome interpretation. Hum. Mutat., 40, 1197–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I. et al. (2010) Global dynamics of proteins: bridging between structure and function. Annu. Rev. Biophys., 39, 23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A. et al. (2011) ProDy: protein dynamics inferred from theory and experiments. Bioinformatics, 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru P. et al. (2017) Deconstruction of the Ras switching cycle through saturation mutagenesis. Elife, 6, e27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J. et al. (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol., 10, e1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.K. et al. (2017) Structure-based analysis of single nucleotide variants in the renin-angiotensinogen complex. Glob. Heart, 12, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.K. and Tastan Bishop,Ö. (2017) Role of structural bioinformatics in drug discovery by computational SNP analysis: analyzing variation at the protein level. Glob. Heart, 12, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E., Altman R.B. (2011) Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinformatics, 12, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorantes-Gilardi R. et al. (2018) In proteins, the structural responses of a position to mutation rely on the Goldilocks principle: not too many links, not too few. Phys. Chem. Chem. Phys., 20, 25399–25410. [DOI] [PubMed] [Google Scholar]
- El-Gebali S. et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res., 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal E. et al. (2015) The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics, 31, 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinauer C., Weigt M. (2017) Context-aware prediction of pathogenicity of missense mutations involved in human disease. ArXiv, arXiv:1701.07246.
- Grimm D.G. et al. (2015) The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat., 36, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin I. et al. (2008) The FEATURE framework for protein function annotation: modeling new functions, improving performance, and extending to novel applications. BMC Genomics, 9, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S. et al. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf T.A. et al. (2017) Mutation effects predicted from sequence co-variation. Nat. Biotechnol., 35, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z. et al. (2019) VIPdb, a genetic variant impact predictor database. Hum. Mutat., 40, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K.J. et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv, 531210.
- Kircher M. et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Purohit R. (2014) Use of long-term molecular dynamics simulation in predicting cancer associated SNPs. PLoS Comput. Biol., 10, e1003318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRusch J. et al. (2014) Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet., 10, e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2016) iGNM 2.0: the Gaussian network model database for biomolecular structural dynamics. Nucleic Acids Res., 44, D415–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2017) DynOmics: dynamics of structural proteome and beyond. Nucleic Acids Res., 45, W374–W380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.-X. et al. (2013) Predicting Mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet., 9, e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2011) dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat., 32, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat., 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek K.A. et al. (2018) Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet., 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottaz A. et al. (2010) Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics, 26, 851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir E.S. et al. (2018) Analysis of single amino acid variations in singlet hot spots of protein–protein interfaces. Bioinformatics, 34, i795–i801. [DOI] [PubMed] [Google Scholar]
- Parveen A. et al. (2019) A novel pathogenic missense variant in CNNM4 underlying Jalili syndrome: insights from molecular dynamics simulations. Mol. Genet. Genomic Med., 7, e902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejaver V. et al. (2019) Assessment of methods for predicting the effects of PTEN and TPMT protein variants. Hum. Mutat., 40, 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzoni L., Bahar I. (2018) Structural dynamics is a determinant of the functional significance of missense variants. Proc. Natl. Acad. Sci., 115, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya Doss C.G. et al. (2014) Integrating in silico prediction methods, molecular docking, and molecular dynamics simulation to predict the impact of ALK missense mutations in structural perspective. Biomed. Res. Int., 2014, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues C.H. et al. (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res., 46, W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C.T., Baker D. (2002) Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J. Mol. Biol., 322, 891–901. [DOI] [PubMed] [Google Scholar]
- Sayılgan J.F. et al. (2019) Protein dynamics analysis reveals that missense mutations in cancer‐related genes appear frequently on hinge‐neighboring residues. Prot. Struct. Funct. Bioinform., 87, 512–519. [DOI] [PubMed] [Google Scholar]
- Stenson P.D. et al. (2017) The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet., 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J. et al. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat., 32, 358–368. [DOI] [PubMed] [Google Scholar]
- Yue P. et al. (2006) SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics, 7, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. (2019) Shared signature dynamics tempered by local fluctuations enables fold adaptability and specificity. Mol. Biol. Evol., 36, 2053–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.