

SUMMARY
The cyclin-dependent kinase 1 (Cdk1) drives cell division. To uncover additional functions of Cdk1, we generated knock-in mice expressing an analog-sensitive version of Cdk1 in place of wild-type Cdk1. In our study we focused on embryonic stem (ES) cells, as this cell type displays particularly high Cdk1 activity. We found that in ES cells a large fraction of Cdk1 substrates is localized on chromatin. Cdk1 phosphorylates many proteins involved in epigenetic regulation, including writers and erasers of all major histone marks. Consistent with these findings, inhibition of Cdk1 altered histone-modification status of ES cells. High levels of Cdk1 in ES cells phosphorylate and partially inactivate Dot1l, the H3K79 methyltransferase responsible for placing activating marks on gene bodies. Decrease of Cdk1 activity during ES cell differentiation de-represses Dot1l, thereby allowing coordinated expression of differentiation genes. These analyses indicate that Cdk1 functions to maintain the epigenetic identity of ES cells.
eTOC Blurb
Cdk1 drives mitotic entry and progression. Michowski et al. generated mice expressing analog-sensitive Cdk1. The authors found that in embryonic stem cells Cdk1 phosphorylates a large number of epigenetic regulators and controls the global epigenic ladscape. Decreased Cdk1 avtivity during differentiation allows coordinated expression of differentiation genes.
Graphical Abstract
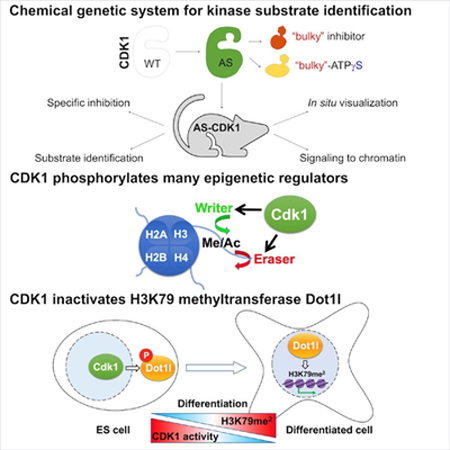
INTRODUCTION
Progression of cells through the cell cycle is driven by protein complexes composed of the regulatory subunits, cyclins, and their catalytic partners, the cyclin-dependent kinases (Malumbres and Barbacid, 2005; Satyanarayana and Kaldis, 2009). In yeast, a single cyclin-dependent kinase, Cdk1 (CDC28 protein in Saccaromyces cerevisiae and Cdc2 in Saccaromyces pombe) is activated at different stages of the cell cycle by different cyclin proteins, and drives progression of cells through the entire cell cycle (Bloom and Cross, 2007; Morgan, 2007). In contrast, mammalian cells express several different cyclin-dependent kinases in addition to Cdk1, such as Cdk2, Cdk3, Cdk4 and Cdk6. These Cdks are activated by different cyclin subunits belonging to the D-, E-, A- and B-type families, and phosphorylate cellular proteins, thereby driving cell cycle progression (Malumbres and Barbacid, 2005; Satyanarayana and Kaldis, 2009).
Genetic experiments revealed that nearly all mammalian Cdks are dispensable for cell proliferation in vivo, and are only required for cell division in a narrow range of specific compartments (Berthet et al., 2003; Malumbres et al., 2004; Ortega et al., 2003; Rane et al., 1999; Tsutsui et al., 1999; Ye et al., 2001). In contrast, Cdk1 is absolutely essential for cell proliferation in all cell types studied. Genetic ablation of Cdk1 resulted in an early embryonic lethality, while conditional shutdown of Cdk1 in postnatal mice halted cell proliferation (Diril et al., 2012; Santamaria et al., 2007). Hence, Cdk1 represents the master regulator of cell cycle progression, which has been conserved from yeast to humans (Morgan, 2007).
Numerous studies have documented the molecular functions of Cdk1 in driving cell division. Mammalian Cdk1 is activated by the B-type cyclins and phosphorylates a large number of proteins that regulate a wide array of events associated with mitotic progression and cell division, such as reorganization of the cytoskeleton, nuclear envelope breakdown, chromosome condensation, mitotic spindle assembly and function, chromosome segregation and cytokinesis (Gavet and Pines, 2010; Kotak et al., 2013; Mishima et al., 2004; Seibert et al., 2019). Cdk1 is also activated by the A-type cyclins; cyclin A-Cdk1 complexes phosphorylate proteins involved in DNA replication, G2 phase progression and entry of cells into mitosis (Furuno et al., 1999; Katsuno et al., 2009; Pagliuca et al., 2011; Vigneron et al., 2018).
While these cell cycle functions of Cdk1 have been extensively studied, little is known whether this kinase performs additional, cell type-specific or tissue-specific, cell cycle-independent functions. To address this question, in this study we developed knock-in mice expressing an analog-sensitive version of Cdk1 in place of the endogenous Cdk1. These mice, and cells derived from them, allow one to identify Cdk1 substrates in essentially any organ or cell type, and at any stage of development.
In our study, we focused on the function of Cdk1 in embryonic stem (ES) cells. These cells display very high activity of Cdk1 (and of closely related Cdk2) that greatly exceed those seen in other proliferating cell types (Fluckiger et al., 2006; Fujii-Yamamoto et al., 2005; Liu et al., 2019; Stead et al., 2002; Wang et al., 2017; White et al., 2005). The molecular function of such high levels of Cdk1 kinase in ES cells has been unknown. Using our system, we uncovered an unexpected function of Cdk1 in regulating the global epigenetic landscape of pluripotent stem cells.
RESULTS
Mice Expressing Analog-Sensitive Cdk1 Allow Identification of Cdk1 Substrates
To generate an ‘analog-sensitive’ version of Cdk1 (further referred to as AS-Cdk1), we mutated the conserved bulky ‘gatekeeper’ residue in Cdk1’s ATP-binding pocket from phenylalanine 80 to a smaller amino acid, glycine (Figure S1A). Such amino acid substitution creates an enlarged ATP-binding pocket not found in any wild-type kinase (Bishop et al., 2000) (Figure 1A). In addition, methionine 32 in the third exon of Cdk1 has been changed to valine. This additional substitution, called ‘suppressor of glycine gatekeeper’ is often needed to maintain normal kinase activity of analog-sensitive kinases (Figure S1B) (Zhang et al., 2005). Several studies documented that such substitutions do not alter the kinase specificity (Blethrow et al., 2008; Shah et al., 1997; Witucki et al., 2002). However, the substituted kinase can be selectively inhibited by ‘bulky’ chemical compounds such as 1-NM-PP1 or 3-MB-PP1, which can occupy the enlarged pocket (Figure 1A). Importantly, these compounds do not inhibit any other kinase in the mammalian kinome (Bishop et al., 2000). Moreover, the analog-sensitive kinases can accept the N6-substituted bulky ATP analogs, while wild-type kinases cannot utilize these compounds, due to steric hindrance of the gatekeeper residue (Hertz et al., 2010) (Figure 1A). Hence, by providing cells expressing an analog-sensitive kinase with bulky ATP analogs in which the gamma phosphate has been replaced with a thiophosphate moiety, one can uniquely label direct kinase substrates with thiophosphate (Hertz et al., 2010). This thiophosphate tag can be then used to purify and identify target proteins (Hertz et al., 2010).
Figure 1. Analyses of Embryonic Stem Cells and Organs Expressing Analog-Sensitive Cdk1.

(A) Diagram illustrating different experimental approaches using analog-sensitive kinases.
(B) Endogenous Cdk1 was immunoprecipitated from Cdk1+/+ or Cdk1AS/AS ES cells and subjected to in vitro kinase reactions with histone H1 as a substrate. For control, immunoprecipitates obtained with normal IgG (IgG) were used for kinase reactions. Lower panel, immunoblots were probed with antibodies against Cdk1 (arrowhead points to the band corresponding to Cdk1). +3-MB-PP1 denotes addition of 3-MB-PP1.
(C) Cdk1+/+ or Cdk1AS/AS ES cells were cultured for 18 h in the presence of vehicle (DMSO), or 3-MB-PP1, stained with propidium iodide and analyzed by flow cytometry.
(D) Cdk1+/+ or Cdk1AS/AS ES cells were cultured for 4 d in the presence of the indicated concentrations of 3-MB-PP1, and stained with crystal violet. Vertical lines mark places where the image was combined from non-adjacent wells.
(E) Experimental outline of using Cdk1AS/AS cells for identification of substrates. PNBM, p-nitrobenzyl mesylate used to alkylate proteins in order to generate epitopes for an anti-thiophosphate ester antibody.
(F) In vitro cultured Cdk1AS/AS or Cdk1+/+ ES cells were permeabilized by treatment with digitonin, and supplemented with N6-substituted bulky ATP analog, N6-furfuryl-ATPγS for 20 min. Cells were then alkylated with PNBM, and thio-labeled substrates visualized by immunostaining with an anti-thiophosphate ester antibody. Scale bar, 50 μm.
(G) Detection of Cdk1 substrates in in vitro cultured Cdk1AS/AS ES cells or in frozen sections prepared from embryonic day 14.5 fetal livers or postnatal day 21 thymuses isolated from Cdk1AS/+ (AS/+) or control Cdk1+/+ mice (+/+). Staining for phospho-histone H3 (pH3) was used to detect mitotic cells. Scale bar, 20 μm
(H)In vitro cultured ES cells were permeabilized and supplemented with one of N6-substituted bulky ATPγS analogs, N6-benzyl-ATPγS (Bn), N6-phenylethyl-ATPγS (Ph), or N6-furfuryl-ATPγS (Fu) for 20 min. Proteins were then analyzed by western blotting with an anti-thiophosphate ester antibody to detect thio-phosphorylated proteins (representing Cdk1 phosphorylation substrates).
(I) Protein lysates were prepared from the indicated organs of Cdk1AS/+ mice and supplemented for 20 min with N6-furfuryl-ATPγS (to allow labeling of direct Cdk1 substrates). Lysates were then immunoblotted using an anti-thiophosphate ester antibody to detect thio-phosphorylated proteins.
See also Figure S1.
We first verified that AS-Cdk1 can be potently and reversibly inhibited by 3-MB-PP1 (Figure S1B). We then used gene-targeting and knocked-in the analog-sensitive substitutions into the endogenous Cdk1 locus in embryonic stem (ES) cells, thereby generating homozygous Cdk1AS/AS ES cells expressing inhibitable Cdk1 (Figures 1B and S1C–S1G). As expected, when cultured in the absence of inhibitors of analog-sensitive kinases, Cdk1AS/AS cells proliferated normally and displayed unperturbed cell cycle profiles (Figure S1H). Addition of analog-sensitive inhibitors 1-NM-PP1 or 3-MB-PP1 to the culture medium blocked proliferation of Cdk1AS/AS cells and arrested their cell cycle progression in G2/M, consistent with the rate-limiting role of Cdk1 in mitotic entry (Diril et al., 2012) (Figures 1C, 1D and S1H). As expected, 3-MB-PP1 has no effect on proliferation of Cdk1+/+ cells (Figures 1C, 1D and S1H).
To label direct Cdk1 substrates, we cultured Cdk1AS/AS cells in the presence of N6-substituted ATPγS analogs bearing a transferable thiophosphate group (Figure 1E). Subsequently, thiophosphorylated proteins were intracellularly visualized by immunostaining of cells with an anti-thiophosphate ester antibody. Indeed, we detected the presence of abundant Cdk1 substrates in Cdk1AS/AS cells (Figures 1F and 1G, top row). As expected, Cdk1+/+ cells were unable to utilize bulky ATPγS analogs, and hence displayed no staining (Figure 1F). In an alternative approach to detect Cdk1 substrates, we cultured Cdk1AS/AS cells with three chemically distinct types of N6-substituted ATPγS as above, prepared protein lysates and immunoblotted them with an anti-thiophosphate ester antibody. Again, we observed thiolabeling of a large number of Cdk1 substrates in Cdk1AS/AS , but not in CDK1+/+ cells (Figure 1H).
We next injected Cdk1AS/AS cells into mouse blastocysts, and generated Cdk1AS/+ animals using standard methods. These mice were viable and displayed no overt abnormalities. We collected different organs from Cdk1AS/+ animals and incubated protein lysates in the presence of bulky ATPγS analogs. As it was the case in ES cells, this led to thiolabeling of Cdk1 substrates, which were visualized by probing the immunoblots with an anti-thioester antibody (Figure 1I). Moreover, by supplementing histologic sections of organs from Cdk1AS/+ mice with bulky ATPγS, we were able to intracellularly label Cdk1 substrates, which were detected by immunostaining with an anti-thioester antibody (Figure 1G, 2nd to 4th rows). Hence, our approach allows to visualize Cdk1 substrates at a single-cell resolution.
Identification of Cdk1 Substrates in Pluripotent Stem Cells
In our study we decided to focus on ES cells, as this cell type expresses high activity of Cdk1 (and of closely related Cdk2), which by far exceeds that seen in other proliferating compartments (see Figure 6A) (Fluckiger et al., 2006; Fujii-Yamamoto et al., 2005; Liu et al., 2019; Stead et al., 2002; Wang et al., 2017; White et al., 2005). The molecular role of the very high Cdk1/2 levels in ES cells remains largely unknown.
Figure 6. Regulation of H3K79me2 marks by Cdk1 During ES Cell Differentiation.

(A) Cdk1 or Cdk2 were immunoprecipitated from in vitro cultured 2 different ES cell lines, or mouse embryonic fibroblasts (MEFs) derived from 3 different embryos, or 2 independent lines of induced pluripotent stem cells (iPSC; line iPSC-1 was derived from MEF-3 cells, by reprogramming), and used for in vitro kinase reactions with histone H1 as a substrate. For control, immunoprecipitates from ES-J1 with normal IgG (IgG) were used.
(B) Wild-type ES cells and MEFs were transduced with a lentivirus encoding Flag-tagged wild-type Dot1l. Undifferentiated ES cells were maintained by culturing in a stem cell medium containing serum and LIF (SL), or were induced to differentiate with retinoic acid (RA) or Wnt3a and activin A (Wnt/Act). The localization of Dot1l was assessed by immunostaining with an anti-Flag antibody. DAPI was used to visualize cell nuclei. Scale bar, 20 μm.
(C) Densitometric quantification of H3K79me2 marks in 3 ES cell lines and MEFs derived from 3 independent mouse embryos, normalized against total histone H3 levels. Below, immunoblotting of chromatin fractions for H3K79me2 and total histone H3.
(D) Similar analysis as in C, using in vitro cultured MEFs derived from 3 different mice and 3 iPSC lines (iPSC-1 cells were derived from MEF-3 cells, by reprogramming).
(E) Cdk1AS/AS ES cells were treated with the increasing concentrations of 3-MB-PP1. Total lysates were probed with antibodies against H3K79me2 or histone H3. Shown is densitometric quantification of H3K79me2 marks, normalized against total histone H3 levels (mean values ±SD from 2 independent experiments). *, p<0.05 (one-way Anova with Tukey’s multiple comparison test). Identical results were obtained when chromatin fraction was analyzed this way.
(F) Wild-type ES cells were cultured in a stem cell medium (SL), or switched to differentiation medium containing retinoic acid (RA) or Wnt3a and activin (Wnt/Act) for 4 or 7 days. Cdk1 or Cdk2 were immunoprecipitated and subjected to in vitro kinase assays using histone H1 as a substrate. For control, immunoprecipitates from pluripotent ES cells with normal IgG (IgG) were used
(G) Densitometric quantification of H3K79me2 marks in chromatin fractions of Cdk1AS/AS ES cells cultured in stem cell medium (SL), or switched to differentiation medium containing Wnt3a and activin (Wnt/Act) for 4 and 7 days.
Mean values ±SD from 2 replicates. **, p<0.05 (paired t-test).
First, we took advantage of our system to investigate the subcellular localization of Cdk1 substrates in ES cells. To this end, we labeled Cdk1 substrates (by supplementing in vitro cultured Cdk1AS/AS ES cells with bulky ATPγS analogs), performed subcellular fractionation, and detected thiophosphorylated proteins in different fractions by immunoblotting. Very surprisingly, we found that a large fraction of Cdk1 substrates was present in the chromatin fraction, suggesting that many of Cdk1 substrates in ES cells may be DNA-bound (Figure 2A).
Figure 2. Localization of Cdk1 Substrates on Chromatin.

(A) In vitro cultured Cdk1AS/AS or Cdk1+/+ ES cells were supplemented with 6-Fu-ATP-γ-S for 20 min to thiolabel direct Cdk1 substrates. The indicated fractions were prepared, and analyzed by western blotting with an anti-thiophosphate ester antibody (to detect thio-phosphorylated proteins), or with an anti-Cdk1 antibody. Immunoblots were also probed with antibodies against α-tubulin (marker of the cytoplasmic fraction), calreticulin (membranes), topoisomerase I (topo I, soluble nuclear and chromatin), histone H3 (chromatin) and vimentin (cytoskeleton).
(B) The outline of ‘thio-ChIP-sequencing’ approach. PNBM, p-nitrobenzyl mesylate used to alkylate proteins in order to generate epitopes for an anti-thiophosphate ester antibody.
(C) Distribution of thiopeaks relative to transcription start sites (TSS) of annotated genes.
(D) A pie-chart depicting the fraction of genes with thiopeaks among active, bivalent and silent (containing no H3K4me3 and no H3K79me2 marks) genes.
(E) Examples of thio-ChIP-seq peaks (representing DNA-bound Cdk1 substrates) and histone modification profiles for actively transcribed (Ctnnb1) and bivalent (Irf8) genes. Shown are genome browser tracks for thio-ChIP-seq as well as publicly available ChIP-seq data for the indicated histone marks in mouse ES cells.
(F) Boxplots illustrating expression levels of ES cell genes containing thiopeaks in their promoter regions (Thio-genes) versus of all other genes (Other genes). p < 2.2 × 10−16 (Mann-Whitney test).
See also Table S1.
To further investigate this possibility, we developed the ‘thio-ChIP-sequencing’ method. Specifically, we cultured Cdk1AS/AS cells in the presence of bulky ATPγS analogs (to label direct Cdk1 substrates), crosslinked proteins to DNA, immunoprecipitated thiophosphorylated proteins with an anti-thiophosphate ester antibody, and subjected the immunoprecipitates to DNA sequencing (Figure 2B). These analyses revealed 7924 specific enrichment regions (‘thiopeaks’) corresponding to DNA-bound Cdk1 substrates, which were assigned to 8352 genes (Table S1). 66.4% of these peaks were located within 5 kb of transcription start sites (Figure 2C). By overlaying the thiopeaks with available ChIPsequencing data of histone modifications in ES cells, we found that these peaks occupy promoters of active (marked by H3K4me3 and H3K79me2) or bivalent (marked by H3K4me3 and H3K27me3) genes (Figures 2D and 2E). Also intersection of thio-ChIP-sequencing with our RNA-sequencing data (Table S1) revealed that genes containing Cdk1 substrates on their promoters are generally expressed at higher levels than the remaining genes (Figure 2F).
To determine the identity of Cdk1 substrates in ES cells, we captured thiophosphorylated proteins from whole cell lysates and analyzed them by mass spectrometry (Figure 3A). In total, we detected 1181 unique peptides that were labeled by thiophosphate moieties (Table S2). 64% of these conformed to the consensus serine/threonine-proline (S/T-P) Cdk1 phosphorylation sequence (Figure 3B), and we considered proteins containing these residues (417 in total) as putative direct Cdk1 phosphorylation substrates (Table S2).
Figure 3. Identification and Analyses of Cdk1 Substrates in ES Cells.

(A) Schematic representation of the direct labeling and inhibition approaches.
(B) Summary of the direct labeling approach. A total of 1181 unique thiophosphorylated peptides were detected; 92% of these were ‘Cdk1-specific’, since they were never detected in Cdk1+/+ cells. Only ‘Cdk1-specific’ peptides were used for subsequent analyses. 64% of ‘Cdk1-specific’ peptides contained thiophosphorylated serine or threonine residue followed by a proline.
(C) Summary of the inhibition approach. In total, we quantified 11842 unique phosphopeptides. 19% of them showed altered phosphorylation status (p<0.05 using the t-test with Benjamini-Hochberg correction) in Cdk1-inhibited cells. 97% of these peptides displayed reduction in phosphorylation upon Cdk1 inhibition. In 53% of peptides showing decreased phosphorylation upon Cdk1 inhibition, the residue displaying decreased phosphorylation corresponded to serine or threonine residue followed by a proline.
(D)Waterfall plot depicting changes in phosphorylation of peptides upon Cdk1 inhibition. Shown are log2 values of the ratio between abundance of each phosphopeptide in 3-MB-PP1- versus in vehicle-treated cells. Negative values correspond to decreased phosphorylation, positive to increased phosphorylation.
(E) Venn diagram summarizing results of the inhibition approach. The blue square depicts all phosphoproteins quantified by mass spectrometry (the number of unique phosphopeptides is in brackets). The yellow rectangle corresponds to quantified proteins (peptides) containing S/T-P sequence. The red rectangle depicts proteins (peptides) showing decreased phosphorylation upon Cdk1-inhibition (Benjamini-Hochberg p-value < 0.05). We considered the overlap between the yellow and red rectangles as putative Cdk1 substrates (622 proteins, 1177 peptides).
(F) Venn diagram illustrating the overlap between proteins identified by us as Cdk1 substrates in the direct labeling (green circle) and inhibition (orange circle) approaches. Proteins were considered as substrates only if the phosphorylation site matched the consensus S/T-P sequence. The number (and percentage) of known Cdk1 substrates (as reported by PhosphoSite Plus substrate page for Cdk1) identified using each approach, and by both approaches (intersection) is shown.
(G) Graphic representation of the top ten significantly enriched GO categories among Cdk1 substrates identified using the inhibition approach. p-values were adjusted using the Benjamini-Hochberg procedure.
See also Figures S2, S3, Tables S2, S3, S5.
We next used an orthogonal approach to identify proteins that depend on Cdk1 for their phosphorylation in ES cells. We acutely inhibited Cdk1 by treating Cdk1AS/AS ES cells with 3-MB-PP1 for 30 min, and verified that such short treatment did not significantly alter the cell cycle profile of cells (Figure S1I). Subsequently, we prepared lysates from Cdk1-inhibited and control (vehicle-treated) ES cells, enriched the samples for phosphopeptides, labeled them with different isobaric tandem mass tags (TMT) (McAlister et al., 2012; Wuhr et al., 2012), and quantified phosphopeptides by mass spectrometry (Figure 3A). Inhibition of Cdk1 resulted in decreased phosphorylation (Benjamini-Hochberg corrected p-value <0.05) of 2228 unique peptides (Table S2). 52.8% of these peptides contained phosphoresidues that conformed to the consensus Cdk1 phosphorylation S/T-P sequence, and we considered proteins carrying these residues (662 in total) as potential Cdk1 substrates (Figures 3C–3E; Table S2). Importantly, we observed an over 50% overlap between Cdk1 substrates identified by the two approaches (direct labeling vs. acute inhibition, Figure 3F; p<1.6 × 10−115). Among proteins identified in each of these approaches we detected several known Cdk1 substrates, with the highest number present among proteins commonly identified by the two methods (Figure 3F).
Computational Analyses of Cdk1 Substrates
We observed that proteins identified by us as Cdk1 substrates displayed higher number of Cdk1-dependent phosphoresidues than that predicted by a random chance (Figure S2A); moreover these residues non-randomly clustered along the amino acid sequence of target proteins (Figures S2B–S2D). We also observed that most of Cdk1 phosphorylation sites (72.7%) were located in disordered domains of target proteins. Analysis of the amino acid sequences surrounding the Cdk1-dependent residues recovered a consensus sequence S/T-P-X-K-K-KK-K (where X represents any amino acid); almost exactly the same motif was identified in the labeling and in the inhibition approaches (Figures S3A–S3D). These analyses indicate that the Cdk1 consensus sequence displays a stronger preference for basic residues following the +2 position than currently appreciated.
Intriguingly, in the direct labeling approach, we observed that in several instances AS-Cdk1 phosphorylated serines or threonines that were not followed by the proline residue. Analysis of these non-canonical targets recovered a consensus S/T-X-K/X-K-K-K-K-K sequence (Figures S3E and S3F). These findings are consistent with the observations that in vitro Cdk1 can phosphorylate non-S/T-P sites (Harvey et al., 2005; McCusker et al., 2007; Suzuki et al., 2015; Brown et al., 2015), and suggest that Cdk1 may phosphorylate in vivo a broader range of substrates than anticipated. For the purpose of this study, however, we only focused on proteins phosphorylated by Cdk1 within the canonical S/T-P sequence.
We next analyzed putative Cdk1 substrates for enrichment in Gene Ontology (GO) categories. As expected, we observed enrichment for functions involved in cell division. Strikingly, the GO term ‘covalent chromatin modification’ was significantly enriched among substrates identified by the direct labeling, in the inhibition approach, as well as by both methods (Figures 3G, S2E and S2F; Table S3). In total, we enumerated 76 epigenetic regulators among Cdk1 substrates, representing writers (such as Mll2, G9a, Prdm2, Setdb1, Setd8, Suv39h2, Ezh2, Nsd1, Nsd2, Setd2, Dot1l, Ep300) and erasers (Lsd1/Kdm1a, Phf2, Phf8, Jmjd1c, Kdm2a, Kdm3b, Kdm4b, Kdm4c, Kdm5a, Kdm6b, Hdac6) of essentially all major histone modifications: H3K4me, H3K9me, H3K27me, H3K36me, H3K79me, H4K20me, H3K9Ac and H3K27Ac, as well as several epigenetic readers (such as Brd4 and Ledgf/Psip1) (Figure 4A; Table S4).
Figure 4. Phosphorylation of Epigenetic Regulators by Cdk1.

(A) A table listing chromatin modifying enzymes (writers and erasers) identified by us as Cdk1 substrates using the direct labeling and/or inhibition approaches.
(B) Examples of murine epigenetic regulators identified by us as Cdk1 substrates, which are phosphorylated by Cdk1 within the domain that is absent in the corresponding ortholog from S. cerevisiae. Shown is alignment of the murine and the corresponding S. cerevisiae protein. Residues of mouse proteins found to be phosphorylated by Cdk1 in inhibition and/or labeling approaches are marked in red.
(C) In vitro kinase reactions with the indicated wild-type cyclin-Cdk kinases, using the indicated epigenetic regulators as substrates. In vitro kinase reactions with the canonical Cdk1 and Cdk2 substrate, histone H1, were used to ensure the comparable specific activity of all cyclin-Cdk kinases used (Hist H1, lowest panel). Lanes marked ‘−’ indicate incubation of substrates with [γ32P]-ATP without any added kinase.
(D) 293T cells were transfected with constructs encoding cyclin E, analog-sensitive Cdk2 (AS), and Flag-tagged versions of the indicated epigenetic regulators. Cells were permeabilized with digitonin and supplemented with N6-furfuryl-ATPγS. Subsequently, epigenetic regulators were immunoprecipitated with an anti-Flag antibody, and immunoblots probed with an anti-thiophosphate ester antibody. As a negative control, Flag-tagged version of Hsp90 (not phosphorylated by cyclin E-Cdk2) was expressed in cells together with cyclin E and analog-sensitive Cdk2. For another negative control, epigenetic regulators were expressed in cells with cyclin E and wild-type Cdk2 (WT). Lower panel, whole cell lysates (WCL) were probed with an anti-Flag antibody. 3xAla and T60E, mutants of G9a and Lsd1, respectively.
(E) Cdk1AS/AS ES cells were left untreated (lanes 1 and 4), or treated with 3-MB-PP1 for 3 (lane 2) or 4 days (lane 3). Chromatin fractions were probed with antibodies against the indicated histone modifications, and against histone H3 (Total H3, loading control). Right, densitometric quantification of the indicated marks, normalized against total histone H3 levels. Mean values ±SD of 2 replicates.
See also Figures S4, S5, Table S4.
Comparison with Yeast Cdk1 Substrates
To extend these findings, we took advantage of a study which identified yeast Cdk1 substrates using the inhibition approach in Saccharomyces cerevisiae expressing analog-sensitive Cdk1 (CDC28) (Holt et al., 2009). We observed a significant overlap (p=3.34 × 10−10) between Cdk1 substrates identified by the inhibition approach in yeast versus in mouse ES cells, including several well-established targets of Cdk1 such as Cdc20, Bub1, Cdc6, Cdc25b and Plk1 (Figure S2G; Table S5). However, yeast Cdk1 substrates did not show GO enrichment for ‘chromatin modification’ category, indicating that this function of Cdk1 is not evolutionarily conserved (Figure S2H; Table S3). Indeed, analysis of murine epigenetic regulators identified by us as Cdk1 substrates revealed that 24 of them do not have yeast orthologs (Table S4). In case of additional 27 murine epigenetic regulators where yeast orthologs are present, the Cdk1-dependent phosphorylation events occur on domains that are absent in the yeast counterparts (Figures 4B and S4; Table S4).
The Function of Cdk1 in Epigenetic Regulation
To further probe a possible role of mammalian Cdk1 in epigenetic regulation, we tested phosphorylation of 19 epigenetic regulators, selected from proteins identified by us as potential Cdk1 substrates, using three approaches: (a) We performed in vitro kinase reactions with wild-type Cdk1 together with cyclins A or B (Figure 4C). (b) Since Cdk1 shares many of its substrates with a closely related cyclin-dependent kinase Cdk2, which is also highly active in ES cells (Liu et al., 2019; Stead et al., 2002; White et al., 2005), we performed in vitro kinase reactions with Cdk2 (Figure 4C). (c) To test if epigenetic regulators are phosphorylated by Cdk2 in vivo, we ectopically co-expressed epigenetic regulators together analog-sensitive Cdk2. Bulky ATPγS was added to cells, resulting in thiolabeling of direct Cdk2 substrates. Epigenetic regulators were then immunoprecipitated, and immunoblots probed with an anti-thiophosphate ester antibody to detect thiophosphorylation by AS-Cdk2 (Figure 4D). Using these approaches we verified 18/19 tested epigenetic regulators as direct Cdk1 and Cdk2 substrates. Intriguingly, we observed some specificity in the ability of different cyclin-Cdk combinations to phosphorylate different epigenetic regulators. For example, Phf8, G9a and Arid4b were efficiently phosphorylated by cyclin A-Cdk1, B-Cdk1 and E-Cdk2, Lsd1 and Jarid2 by cyclin A-Cdk1 and E-Cdk2, while Kdm6b, Kdm4c and Kdm2a mainly by cyclin A-Cdk1). These observations raise a possibility that different cyclin-Cdks may play distinct roles in the epigenetic regulation.
To further verify the significance of phosphorylation of epigenetic regulators by Cdk1, we inserted phosphomimetic or phosphoinactivating substitutions into Cdk1-dependent phosphoresidues of the H3K9 methyltransferase G9a. Mutation of these residues to alanines significantly increased the levels of G9a protein in ES cells, while the phosphomimetic G9a mutant was expressed at strongly reduced levels (Figure S5A). These observations suggest that phosphorylation of G9a by Cdk1 may destabilize G9a. Consistent with this notion, we observed that inhibition of Cdk1 kinase activity in ES cells resulted in a strong upregulation of G9a protein levels, which was followed by significantly increased global H3K9me3 mark (Figure S5B).
Collectively, these observations raised a possibility that Cdk1 might regulate the epigenetic landscape in ES cells by directly phosphorylating a large number of epigenetic regulators. To test this, we treated Cdk1AS/AS cells with 3-MB-PP1, isolated the chromatin fraction and probed immunoblots with antibodies against nine different histone modifications. We observed that the levels of all nine modifications were significantly altered upon Cdk1 inhibition (Figure 4E).
Regulation of Dot1l by Cdk1 in ES Cells
To start dissecting the role of Cdk1 in epigenetic regulation of ES cells, we focused on Dot1l, an enzyme catalyzing transfer of methyl groups to lysine 79 of histone H3 (Nguyen and Zhang, 2011). H3K79 methylation marks are localized within the bodies of transcribed genes, and the amount of the modification correlates with gene expression levels (Nguyen and Zhang, 2011; Schubeler et al., 2004; Steger et al., 2008; Vakoc et al., 2006). The rationale for focusing on Dot1l was as follows: (a) We identified Dot1l as a Cdk1 substrate both by the labeling and the inhibition approaches. (b) Dot1l is the only enzyme responsible for placing the H3K79 methyl marks (Nguyen and Zhang, 2011), thereby rendering mechanistic analyses more feasible. This is in contrast to many other histone modifications, where multiple different enzymes (several of which were identified by us as Cdk1 substrates) can redundantly perform the same task (Kouzarides, 2007). (c) No H3K79 demethylase has been identified, and Dot1l is considered to be the sole enzyme regulating this mark (Nguyen and Zhang, 2011).
We first verified using in vitro kinase reactions with recombinant proteins that Dot1l represents a direct substrate of wild-type Cdk1 and Cdk2 (Figure 5A). Mass spectrometric analysis of the phosphorylation status of the endogenous Dot1l upon an acute (30 min) inhibition of Cdk1 in ES cells revealed that the residue most strongly decreased in phosphorylation, serine 1105, is located within Dot1l’s predicted bipartite nuclear localization signal (NLS) (Figure 5B; Table S2). The very same residue (serine 1105) has been identified as a direct Cdk1 phosphorylation site in our thiolabeling approach (Table S2). These findings suggested that phosphorylation of Dot1l by Cdk1 might disrupt the NLS, thereby preventing Dot1l nuclear localization. To test this possibility, we first verified, using recombinant proteins and mass spectrometry, that wild-type Cdk1 and Cdk2 can directly phosphorylate serine 1105 residue of Dot1l (Table S6). We next expressed wild type Dot1l, or phosphomimetic serine 1105 Dot1l mutant in human breast cancer MCF7 cells, which display much lower levels of Cdk1 kinase than those seen in ES cells (Figure 5C). We observed that whereas wild-type Dot1l was present predominantly in the nucleus, the phosphomimetic mutant was almost exclusively cytoplasmic (Figure 5D). We also examined localization of Dot1l in asynchronously growing ES cells (displaying very high Cdk1 activity, Figures 5C and 6A). We observed that in ES cells Dot1l was present both in the cytoplasm and in the nucleus, with predominant localization in the cytoplasm. Strikingly, an acute inhibition of Cdk1 triggered translocation of Dot1l to the nucleus (Figures 5E and S5C–S5F, see also Figure 6B, upper row). Consistent with our hypothesis that phosphorylation of Dot1l inhibits its nuclear import, phosphomimetic Dot1l mutant was predominantly cytoplasmic in ES cells, and failed to undergo significant re-localization to the nucleus upon Cdk1 inhibition (Figures 5F and S5G).
Figure 5. Negative Regulation of Dot1l by Cdk1.

(A) In vitro kinase reactions with the indicated wild-type cyclin-Cdk kinases using Dot1l as a substrate. Lower panel, kinase reactions with histone H1 were used to ensure the comparable specific activity of all cyclin-Cdk kinases used. Lanes marked ‘−’ indicate incubation of Dot1l or histone H1 with [γ32P]-ATP without any added kinase. In the upper right panel, the middle lanes were spliced out (indicated by a vertical line).
(B) Amino acid sequence of Dot1l containing the bipartite predicted nuclear localization signal (in bold). Cdk1-dependent phosphorylation site (S1105) is in red. Below, changes in the phosphorylation status of the indicated Dot1l phosphopeptides upon an acute 30 min Cdk1 inhibition. Note that the most affected peptide (>40% decrease in phosphorylation), contains the S1105 phosphosite.
(C) Cdk1 or Cdk2 were immunoprecipitated from human breast cancer MCF7 or wild-type ES (ES-V6.5) cells and used for in vitro kinase reactions with histone H1 as a substrate. For control, immunoprecipitates from ES cells with normal IgG (IgG) were used.
(D) MCF7 cells were transfected with constructs encoding Flag-tagged wild-type Dot1l or a phosphomimetic S1105 Dot1l mutant. The localization of Dot1l was assessed by immunostaining with an anti-Flag antibody. DAPI was used to visualize cell nuclei. Scale bar, 10 μm.
(E and F) Cdk1AS/AS ES cells were transduced with lentiviruses encoding Flag-tagged wild-type Dot1l (E), or phosphomimetic S1105 Dot1l mutant (F). Cells were treated for 6 h with vehicle (DMSO), or 3-MB-PP1, and Dot1l localization was assessed by immunostaining with an anti-Flag antibody. DAPI was used to visualize cell nuclei. Scale bar, 5 μm.
(G) Cdk1AS/AS ES cells were cultured in the presence of vehicle (DMSO) or 3-MB-PP1, together with retinoic acid, and immunostained with an antibody against H3K79me2; DAPI was used to visualize cell nuclei. Scale bar, 10 μm.
(H) Distribution of H3K79me2 peak widths in Cdk1AS/AS ES cells threated with vehicle (DMSO), or 3-MB-PP1.
(I) H3K79me2 ChIP-sequencing signal height and position relative to transcription start sites (TSS) for all genes containing this mark in Cdk1AS/AS ES cells treated with DMSO or 3-MB-PP1.
(J) ChIP-sequencing density heatmap of H3K79me2 enrichment in DMSO- and 3-MB-PP1-treated Cdk1AS/AS ES cells, within 20 kb around the transcription start site. Gene order was arranged from highest to lowest density.
(K) Examples of H3K79me2 modification profiles in Cdk1AS/AS ES cells treated with DMSO or with 3-MB-PP1. Shown are genome browser tracks for H3K79me2 ChIP-sequencing.
We next synchronized wild-type ES cells and monitored the activity of Cdk1 and Cdk2 as well as localization of Dot1l throughout cell cycle progression. We found that both kinases were active across the ES cell cycle, as previously reported (Liu et al., 2019); the kinase activity associated with cyclin A was constant, while the activity of cyclin B peaked in G2/M phase (Figures S5H and S5I). Dot1l remained predominantly cytoplasmic in all cell cycle phases, suggesting that its phosphorylation is maintained across the cell cycle (Figures S5J and S5K). Consistent with this notion, the H3K79me2 mark also remained unchanged throughout the cell cycle (Figure S5L).
Collectively, these results indicate that in ES cells high activity of Cdk1 inhibits Dot1l throughout the cell cycle by impeding its nuclear localization. Inhibition of Cdk1 triggers translocation of Dot1l to the nucleus, where it can perform its histone modifying function. Consistent with this model, we observed that inhibition of Cdk1 in ES cells significantly increased H3K79me2 modification, as judged by immunoblotting (Figure 4E) or immunostaining with anti-H3K79me2 antibodies (Figure 5G). Also ChIP-sequencing revealed that inhibition of Cdk1 increased H3K79me2 marks on gene bodies (Figures 5H–5K; Table S7).
We next compared Dot1l localization and H3K79me2 marks between murine ES cells versus mouse fibroblasts (MEF), a cell type expressing much lower levels of Cdk1/2 kinase (Figure 6A). ES cells displayed very high Cdk1/2 activity (Figure 6A), Dot1l was mostly cytoplasmic (Figure 6B, top row, ‘SL’) and the level of the H3K79me2 mark was relatively low (Figure 6C). Conversely, in MEFs Cdk1/2 activity was low (Figure 6A), Dot1l was mostly nuclear (Figure 6B, bottom row, MEFs), and the level of the H3K79me2 mark was substantially higher than that in ES cells (Figure 6C).
The cell cycle organization of induced pluripotent stem cells (iPSC) resembles that of ES cells (Ghule et al., 2011; Ruiz et al., 2011). Therefore, we compared the levels of Cdk1/2 and of H3K79me2 marks between iPSC versus fibroblasts from which these iPSC were derived through cell reprogramming. Again, we observed an inverse correlation between Cdk1/2 activity and H3K79me2 marks. Thus, in iPSC the Cdk1/2 activity was very high (Figure 6A) and H3K79me2 marks were low (Figure 6D), exactly as seen in ES cells, while the opposite was true in parental fibroblasts from which these iPSC were made (see MEF-3 in Figures 6A and 6D). Collectively, these analyses suggest that high levels of Cdk1/2 in ES cells and in iPSC partially inactivate Dot1l, while decreased Cdk1/2 activity promotes Dot1l chromatin-modifying function.
Regulation of Dot1l by Cdk1 During Differentiation
Since the dissolution of pluripotency and the onset of differentiation is accompanied by strongly decreased Cdk1/2 kinase activity (Fluckiger et al., 2006; Fujii-Yamamoto et al., 2005; Liu et al., 2019; Wang et al., 2017; White et al., 2005), we hypothesized that Dot1l may become activated during this process. To test this, we first progressively inhibited Cdk1 by culturing Cdk1AS/AS ES cells in a medium containing increasing 3-MB-PP1 concentrations. We observed that this resulted in a progressive increase of the H3K79me2 marks (Figure 6E). We next subjected wild-type ES cells to differentiation induced by retinoic acid or Wnt3a/Activin A. As expected, Cdk1/2 kinase activity progressively declined during this process (Figure 6F). This was accompanied by translocation of Dot1l from the cytoplasm to the nucleus (Figure 6B, second and third rows), and by the increase in H3K79me2 marks (Figure 6G). Importantly, Dot1l phosphomimetic mutant failed to undergo nuclear localization upon differentiation of ES cells, and remained largely cytoplasmic (Figure S6A). Collectively, these observations suggest that during differentiation, decreasing levels of Cdk1/2 kinase liberate Dot1l, which then translocates to the nucleus to perform its histone-modifying function.
We noticed that further inhibition of Cdk1 during differentiation (by addition of 3-MB-PP1 to the culture medium) synergized with the retinoic acid and Wnt3a/Activin in inducing the expression of endodermal genes, such as Gata6, Sox17 and Foxa2 (Figure 7A). Targeted ChIP revealed that inhibition of Cdk1 resulted in increased H3K79me2 marks on gene bodies of Gata6, Sox17 and Foxa2 (Figure 7B). Consistent with these findings, we observed that inhibition of Cdk1 cooperated with weak differentiation signals to induce the endodermal fate (Figure 7C). We extended these observations using embryoid bodies derived from Cdk1AS/AS or Cdk1+/+ ES cells. Again, treatment of Cdk1AS/AS embryoid bodies with 3-MB-PP1, or treatment of Cdk1 wild-type embryoid bodies with a Cdk1 inhibitor RO-3306 strongly increased transcript and protein levels of the endodermal markers (Figures 7D–7G).
Figure 7. Inhibition of Cdk1 Promotes Endodermal Differentiation.

(A) RT-qPCR analysis of the levels of the indicated transcripts in Cdk1AS/AS ES cells cultured in stem cell medium (SL) or switched to differentiation medium containing Wnt3a and activin A or retinoic acid (RA), with vehicle (DMSO) or 3-MB-PP1. T represents a mesodermal marker, Sox1 and Pax6 markers of neural ectoderm. Mean values ±SD from 3 independent experiments. *, p<0.05; **, p<0.01 (unpaired t-test).
(B) Targeted ChIP – qPCR analysis to assess enrichment of H3K79me2 marks on the promoter and gene body regions of the Gata6, Sox17 and Foxa2 genes. Cdk1AS/AS ES cells were cultured in stem cell medium (SL), or switched to differentiation medium containing retinoic acid (RA), or retinoic acid plus 3-MB-PP1, and subjected to ChIP with an anti-H3K79me2 antibody, followed by PCR amplification of the indicated gene segments. Mean values ±SD from 2 independent experiments. *, p<0.05; **, p<0.01 (multiple t-test with Benjamini’s correction).
(C) Cdk1AS/AS ES cells were cultured for 2 days in a serum- and LIF-free medium to dissolve pluripotency, followed by 24 h culture in the presence of the indicated concentrations of retinoic acid with DMSO or 3-MB-PP1. Expression of transcripts encoding endodermal (Gata6, Foxa2 and Sox17) and ectodermal (Sox1, Pax6) markers was gauged by RT-qPCR. Shown is fold induction relative to expression levels in cells growing in stem cell SL medium.
(D) Cdk1AS/ASES cells were allowed to form embryoid bodies for 2 days and then cultured in the presence of retinoic acid (RA, upper row) or with retinoic acid plus 3-MB-PP1 (lower row) for 24 h. Embryoid bodies were then cryosectioned and stained with antibodies against Gata6 and Sox17. DAPI was used to visualize cell nuclei. Scale bar, 20 μm.
(E) Bright field images of embryoid bodies cultured as above. Scale bar, 100 μm. Note that treatment with 3-MB-PP1 did not have any gross effects on morphology of embryoid bodies.
(F) Transcript levels of the indicated markers in embryoid bodies grown and treated as in D.
(G) Embryoid bodies derived from wild-type ES cells were generated as in D and F and supplemented with retinoic acid alone, or retinoic acid plus a chemical inhibitor of Cdk1, RO-3306. The expression of the indicated markers was analyzed by RT-qPCR. In (F) and (G), mean values ±SD from 3 biological replicates. **, p<0.01; **, p<0.001 (multiple t-test with Benjamini’s correction).
To further test the biological significance of Dot1l activation during the differentiation process, we used CRISPR/Cas9 to knock out Dot1l in ES cells (Figures S6B–S6F). RNA-sequencing revealed that expression of only a small number of genes was deregulated (25 downregulated, 228 upregulated over 2-fold, Benjamini-Hochberg corrected p-value <0.05, Table S8), consistent with our observation that in undifferentiated ES cells Dot1l is largely cytoplasmic, and hence partially inactivated. In contrast, upon switching to differentiation-inducing medium, the expression profile of Dot1l-deficient cells became strongly deregulated, as compared to wild-type cells (505 downregulated, 961 upregulated over 2-fold, p<0.05, Table S8). While differentiating wild-type cells became enlarged and flattened, Dot1l KO colonies remained rounded and maintained compact appearance conforming more closely to undifferentiated ES cell phenotype (Figure S6G). These observations suggest that Dot1l becomes rate-limiting for gene expression during differentiation, when it becomes de-repressed by declining Cdk1 kinase activity.
DISCUSSION
One of the main limitations in studying the in vivo functions of protein kinases is the lack of tools to identify tissue-specific substrates of the kinase of interest. In this study we developed a novel approach to overcome this limitation, which utilizes a knock-in strain of mice expressing an analog-sensitive version of a kinase in place of the wild-type protein. These mice express the kinase of interest at physiological levels, maintain its tissue-specific expression, and allow one to ‘tag’ direct substrates of the kinase in any organ or cell type using ‘bulky’, thio-substituted ATP analogs. In addition, one can acutely inhibit the analog-sensitive kinase for a very short period, and then use quantitative phosphoproteome profiling to identify proteins displaying decreased phosphorylation. These two orthogonal approaches allow one to identify with high confidence proteins that represent direct substrates of the kinase (labeling approach) and which critically depend on this kinase for their phosphorylation (inhibition approach). We also developed methods that allow to visualize direct substrates of the kinase in tissue sections and to determine the subcellular localization of the substrates in a cell type of interest. In addition, we developed a ‘thio-ChIP-sequencing’ method that allows to reveal the localization of kinase substrates on specific DNA sequences. These approaches can be used to study the function of the endogenous kinase in vivo at any stage of development, or in any disease state, for example during cancer initiation, progression, and in the metastatic spread.
We applied this approach to Cdk1, an ancient cell cycle kinase that has been conserved throughout the evolution from yeast to humans. One of the major challenges in studying the functions of Cdk1 is the fact that its inhibition or genetic ablation halts cell proliferation. Since the cell cycle effect is so dominant, it has been difficult to dissect whether Cdk1 plays other functions that are unrelated to cell cycle progression. We circumvented this limitation by identifying direct Cdk1 substrates in exponentially growing cells (labeling approach), or by identifying proteins that depend on Cdk1 for phosphorylation using a transient inhibition of Cdk1 activity, which did not affect cell cycle progression. In contrast, most cellular assays require longer Cdk1 inhibition, and the resulting cell cycle arrest may confound the interpretation of the results. For instance, we observed that inhibition of Cdk1 in ES cells altered global histone modification status, and it could be argued that this phenomenon represents an indirect effect. However, our mechanistic experiments indicate that this is not the case. The direct link between Cdk1 activity and the levels H3K9 methyl marks was provided by our observation that Cdk1 directly phosphorylates H3K9 methyltransferase G9a, resulting in G9a protein destabilization. Likewise, we demonstrated that Cdk1 directly phosphorylates and inhibits Dot1l by impeding its nuclear localization. Similar mechanistic analyses of other epigenetic regulators, identified by us as direct Cdk1 substrates will allow to elucidate how Cdk1 modulates their enzymatic activity, localization, stability or interaction with other proteins.
We propose that in ES cells, high Cdk1 levels serve to globally regulate the epigenetic landscape by phosphorylating epigenetic regulators, thereby helping to maintain the undifferentiated and pluripotent state. The epigenetic state of ES cells is characterized by low levels of H3K9me3, H3K27me3 and H3K79me2 marks; these marks increase during differentiation, and are largely reverted to the ES cell-like levels during somatic reprogramming (Hawkins et al., 2010; Sridharan et al., 2013). We propose that high Cdk1 activity helps to maintain the hypomethylated state of these marks in ES cells and in iPSC. Consistent with this model, we observed that inhibition of Cdk1 activity in ES cells increased methylation of H3K9, H3K27 and H3K79.
Cdk1 was previously shown to play an important role in enforcing the undifferentiated state of ES cells, as inhibition of Cdk1 resulted in the loss of pluripotency and triggered ES cell differentiation (Neganova et al., 2014; Wang et al., 2017). While in the current study we mechanistically focused on regulation of Dot1l, several other Cdk1 substrates identified in our study were shown to play key roles in maintaining ES cell identity. For example, H3K9/H3K36 histone demethylases Kdm4a, Kdm4b and Kdm4c acting on gene promoters were shown to be essential in ensuring transcriptional competence and enforcing the identity of pluripotent ES cells (Das et al., 2014; Pedersen et al., 2016). Lsd1 regulates decommissioning of enhancers during dissolution of pluripotency (Whyte et al., 2012), while H3K4 and H3K27 methyltransferases Mll and Ezh2, respectively, control ‘bivalent’, developmentally regulated genes (Bernstein et al., 2006). Since we found that all these enzymes represent direct Cdk1 substrates, it is likely that their activity is regulated by Cdk1 in ES cells. Consistent with our findings, it has been reported that Cdk1 can phosphorylate Ezh2, with different studies ascribing activating or inhibitory impact on Ezh2’s enzymatic activity (Chen et al., 2010; Kaneko et al., 2010; Wei et al., 2011). Collectively, these observations indicate that the function of Cdk1 in ES cells extends well beyond its well-established role as the regulator of cell cycle progression and cell division, and that Cdk1 plays a central role in maintaining ES cell identity.
The unique role of Cdk1 likely extends to iPSC cells, a cell type with very high levels of Cdk1/2 kinase that resemble those seen in ES cells. We speculate that re-activation of high Cdk1 levels during somatic reprogramming helps to re-set the epigenetic landscape to the ES cell-like one. Indeed, the epigenetic state of iPSC is highly similar to that of ES cells (Hawkins et al., 2010; Mikkelsen et al., 2008; Sridharan et al., 2013). Like in ES cells, depletion of Cdk1 in reprogrammed iPSC cells attenuated the pluripotent state, while ectopic expression of cyclin B-Cdk1 enhanced the reprogramming efficiency of human fibroblasts (Neganova et al., 2014; Wang et al., 2017). Lastly, while we observed that high Cdk1 activity in ES cells maintains low levels of H3K79 or H3K9 methyl marks, inhibition of H3K79 or H3K9 methyltransferases in somatic cells was shown to augment reprogramming (Chen et al., 2013; Onder et al., 2012; Sridharan et al., 2013). These observations indicate that Cdk1 is likely to play a major role in maintaining the epigenetic state of reprogrammed stem cells. Hence, elucidation of the full range of Cdk1 epigenetic functions may be of interest also for regenerative medicine.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for all unique reagents generated in this study should be directed to and will be fulfilled with a completed Materials Transfer Agreement by the Lead Contact Piotr Sicinski (peter_sicinski@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of Cdk1AS knock-in embryonic stem cells and mice
All mouse procedures have been described in the protocol, which has been approved by the Dana-Farber Cancer Institute Institutional Animal Care and Use Committee. The targeting construct was generated by cloning three fragments of the Cdk1 gene from V6.5 mouse embryonic stem cells (mixed C57BL/6 and 129/Sv background) into a pSL301 plasmid. First, a 3 kb-long fragment constituting the left arm of homology was inserted upstream of the puromycin resistance cassette, which was flanked by two loxP sites. Second 2.1 kb fragment encompassing exons 3 and 4 was mutagenized to introduce M32V (suppressor of glycine gatekeeper) and F80G (analog-sensitive) substitutions (all cells and mice designated as Cdk1AS contained both mutations). Third 3.85 kb fragment served as the right arm of homology. The targeting construct was linearized and electroporated into V6.5 mES cells. Cdk1puro/+ clones were identified by long-range PCR; Southern blotting was used to confirm the correct integration. Subsequently, cells were electroporated with plasmid encoding Cre recombinase giving rise to Cdk1AS/+ cells. Cdk1AS/+ cells were subjected to a second round of targeting of the remaining Cdk1+ allele to obtain Cdk1AS/puro cells. Subsequent electroporation with Cre produced Cdk1AS/AS ES cells. To generate mice, Cdk1AS/+ ES cells were injected into mouse blastocysts, and Cdk1 AS/+ animals were generated as before (Geng et al., 1999). Intercrossing of Cdk1AS/+ heterozygotes revealed that homozygous Cdk1AS/AS embryos die during mid-gestation (around embryonic day 10.5) and display relatively normal development of all internal organs. This is in stark contrast to Cdk1−/− embryos which die prior to 2–4 cell stage (Santamaria et al., 2007). Hence, the analog-sensitive Cdk1 allele, despite its normal biochemical function in ES cells, is hypomorphic in vivo. The livers used for in situ staining of CDK1 substrates were collected from embryonic day 14.5 (E14.5) (Figure 1G) or E15.5 (Figure 1I) animals.
METHODS DETAILS
ES and iPS cell culture, differentiation and thiolabeling of substrates
Mouse embryonic stem cells V6.5 (Rideout et al., 2000) and J1 (Li et al., 1992) were maintained on a monolayer of mitotically inactivated mouse embryonic fibroblasts in SL medium consisting of Knockout DMEM medium (Invitrogen), 15% HyClone fetal bovine serum (Invitrogen), 1000 U/ml leukemia inhibitory factor (LIF) (Millipore), 1% MEM non-essential amino acids, 1% penicillin/streptomycin, 1% glutamine and 0.1 mM 2-mercaptoethanol. ES-2 is an ES cell line derived by us from embryonic day 3.5 blastocyst collected from a mouse of the mixed C57BL/6 and 129/Sv background and was cultured as above. iPS cells were generated as in (Wernig et al., 2007) and cultured identically to ES cells. Differentiation was induced as in (Thomson et al., 2011) with modifications. ES cells were trypsinized and plated at 3 × 104/cm2 on gelatin-coated dishes in N2B27 medium (Knockout DMEM, 1% N2 and 2% B27 without vitamin A (Invitrogen), 1% MEM non-essential amino acids, 1% penicillin/streptomycin, 1% glutamine, 0.1 mM 2-mercaptoethanol and 10 μM Y27632 (Selleckchem). Two days later medium was changed to N2B27 with 500 nM retinoic acid (Sigma R2625) or 50 ng/ml Wnt3a (R&D Systems, 5036-WN) and 10 ng/ml Activin A (R&D Systems, 330-AC). Cells were usually harvested after 24 h for gene expression and Dot1l localization analyses, or allowed to differentiate for 4 and 7 days to follow changes in Cdk1/2 activity and H3K79me2 mark.
To thiolabel Cdk1 substrates (Figure 1H), in vitro cultured Cdk1AS/AS ES cells were permeabilized with 45 μg/ml digitonin and supplemented with N6substituted bulky ATPγS analogs, N6-benzyl-ATPγS, N6-phenylethyl-ATPγS, or N6-furfuryl-ATPγS for 20 min. Specifically, Cdk1AS/AS ES cells were incubated with the labeling buffer modified from (Banko et al., 2011) (20 mM HEPES pH 7.5, 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 0.5 mM DTT, 45 μg/ml digitonin, 5 mM GTP, 0.2 mM ATP, 0.1 mM ATP analog, Roche protease inhibitors cocktail) for 20 min. Subsequently, lysates were supplemented with 20 mM EDTA and 1.5 mM p-nitrobenzyl mesylate (PNBM), to alkylate proteins in order to generate epitopes for an anti-thiophosphate ester antibody. Lysates were separated on SDS-PAGE gels, transferred to membranes which were probed with an anti-thiophosphate ester antibody.
To label Cdk1 substrates in embryonic livers and postnatal thymuses (Figure 1I), freshly harvested organs were pulverized in a mortar with liquid nitrogen, and lysed in a buffer containing 50 mM HEPES-KOH pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.2% NP-40 and Roche protease inhibitor cocktail. Cleared lysates were then supplemented with 0.1 mM N6-furfuryl ATPγS, 0.2 mM ATP, 3 mM GTP and 2 mM TCEP, and labeling reaction was carried out at 30°C for 20 min. Samples were next alkylated and analyzed by western blotting as described above.
In situ staining of Cdk1 substrates
Freshly harvested embryonic day 14.5 (E14.5) livers or postnatal day 21 (P21) thymi from Cdk1AS/+ or wild-type mice were frozen in isopentane/dry ice bath in OCT medium. The organs were cryosectioned (15 μm-thick slices) directly onto poly-L-lysine coated glass slides (Sigma), and fixed for 4 min in 4% PFA. The sections were overlaid with 200 μl of labeling buffer containing 20 mM HEPES pH 7.5, 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 5 mM MnCl2, 5 mM TCEP, 5 mM GTP, 0.2 mM ATP, 0.1 mM N6-furfuryl ATPγS (Biolog, Germany) and 45 μg/ml digitonin. The kinase reaction was carried out for 30 min, and arrested by submerging the slides in 4% PFA, 20 mM EDTA in PBS for 5 min. The specimens were then alkylated for 15 min with 1 mM PNBM, 45 μg/ml digitonin, pH 4.5 (adjusted with formic acid). The alkylation reaction was quenched with 10 mM DTT in TBS, and tissue sections blocked with 5% BSA in TBS containing 0.1% Triton X-100 (TBS-T) for 30 min. Immunostaining was performed with a mix of anti-thiophosphate ester rabbit antibody (Abcam, ab92570) and anti-phospho-histone H3 (Ser10) mouse antibody (CST, 9706), both at 1:1500 dilution in 2% BSA/TBS-T, overnight at 4° C. Specimens were then incubated for 30 min with 1:500 dilution of donkey anti-rabbit Alexa Fluor 594 and donkey anti-mouse Alexa Fluor 488 secondary antibodies in TBS-T with Hoechst DNA stain. For visualization of Cdk1 substrates in ES cells (Figure 1F), in vitro cultured Cdk1AS/AS or wild-type ES cells were fixed, permeabilized, labeled, alkylated and immunostained as described above.
Cell lysis, cell fractionation and western blotting
Cell lysates were prepared in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.1% SDS, 10 mM EDTA) with Roche protease inhibitors and cleared by centrifugation at 16000 rpm, 4°C. Protein content was quantified using BCA Protein Assay Kit (Pierce). Twenty to fifty micrograms of total proteins were mixed with reducing SDS-PAGE sample buffer, heated to 95°C for 5 min, and resolved on SDS-PAGE. Proteins were transferred to nitrocellulose membranes in Towbin buffer (1 h, 450 mA) The membranes were incubated with primary antibodies in 5% milk, TBS plus 0.1% Tween overnight at 4°C, and then with a 1:10000 dilution of HRP-conjugated anti-mouse or anti-rabbit secondary antibodies. The signal was detected using Pierce ECL Western Blotting substrate (Thermo Fisher). The following primary antibodies were used: anti-Cdk1 1:2000 (Abcam, ab71939), anti-thiophosphate ester 1:4000 (Abcam, ab92570), anti-Dot1l 1:500 (Bethyl, A300–954A), anti-G9a 1:1000 (Abcam, ab185050), anti-Flag 1:10000 (Sigma, F1804), anti-Hsp90 1:1000 (CST, 4877), anti-α-tubulin 1:5000 (Sigma, T5168), anti-calreticulin 1:5000 (Abcam, ab92516), anti-topoisomerase I 1:500 (BD, 556597), anti-histone H3 1:2000 (CST, D1H2), and anti-vimentin 1:1000 (CST, D21H3).
In order to determine subcellular localization of Cdk1 substrates, ES cells were fractionated using Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific, 78840) according to manufacturer’s manual, with modifications to accommodate for thio-labeling. Specifically, one 80% confluent 10 cm plate of Cdk1AS/AS or wild-type V6.5 cells was washed twice with PBS and incubated with 2.5 ml of the labeling buffer modified from (Banko et al., 2011) (20 mM HEPES pH 7.5, 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 0.5 mM DTT, 45 μg/ml digitonin, 5 mM GTP, 0.2 mM ATP, 0.1 mM N6-furfuryl ATPγS (Biolog), Roche protease inhibitors cocktail) at 30°C for 15 min. The cells were next scraped off, supplemented with 20 mM EDTA and 1.5 mM p-nitrobenzyl mesylate, and incubated for 15 min at 4° C with rotation. The samples were then spun at 500 × g, 4°C, 7 min, and cell pellets were resuspended in the kit’s CEB buffer. The kit manufacturer’s protocol was then followed. The subcellular fractions were analyzed by western blotting for the presence of thio-labeled Cdk1 substrates.
Identification of Cdk1 substrates in ES cells by direct labeling
Cdk1 substrates reported in this study were identified using two methods of thiophosphate capturing. The first method was through covalent bond formation between thiophosphorylated peptides and iodoacetyl groups of SulfoLink resin (Thermo Scientific). The second method involved alkylation of thiophosphate moieties with p-nitrobenzyl mesylate (PNBM), followed by immunoprecipitation of modified peptides with anti-thiophosphate ester antibody. Both methods produced comparable results.
Capturing of thiophosphorylated peptides on SulfoLink resin
Cdk1AS/AS or wild-type ES cells were harvested and resuspended in a buffer: 50 mM HEPES-KOH pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.2% NP-40, Roche protease inhibitor cocktail, 1 mM PMSF. Lysates containing 20 mg protein were supplemented with 0.1 mM N6-furfuryl ATPγS (Biolog, Germany), 0.2 mM ATP, 3 mM GTP and 2 mM TCEP in a final volume of 10 ml. Labeling reaction was carried out at 30° C for 30 min and stopped with 20 mM EDTA. The reaction was divided into two 50 ml tubes and the proteins were precipitated. Briefly, 5 volumes (25 ml) of ice-cold chloroform/methanol mix (1:4) was added to the labeling reactions, followed by addition of 15 ml of cold H2O and 10 min incubation on ice. Samples were next centrifuged for 20 min at 1000 × g, pellets washed in 25 ml of ice-cold methanol, the samples were re-centrifuged and pellets dissolved in 5 ml of 8 M urea in 0.2 M Tris pH 8.8. The urea was next diluted to 1 M with water, and 10 mM TCEP and Tris pH 8.8 (to bring the pH to 8.0) were added. The proteins were digested with 200 μg LysC (Wako) (1:50 m/m) overnight at RT. The digest was acidified to pH 2.5 with trifluoroacetic acid (TFA) and centrifuged at 1000 × g. The supernatant was desalted using C-18 Sep Pak Plus columns, the peptides were eluted with 5 ml 80% acetonitrile/0.5% acetic acid solution and lyophilized overnight. Dry peptides were dissolved in 1.5 ml of 4 M urea, 0.1 M Tris pH 8.8, 10 mM TCEP, acidified to pH 5 with formic acid, applied to SulfoLink beads suspension, and rotated overnight. The beads were then spun at 1000 × g and washed sequentially with 10 ml of a) 4M urea in 20 mM HEPES pH 7.0; b) ddH2O; c) 5 M NaCl; and d) 50% acetonitrile in ddH2O. The beads were spun at 2400 × g, washed with 5% formic acid, resuspended in 1 ml of 10 mM DTT, spun again and the phosphorylated peptides were eluted with 750 μl of 2 mg/ml Oxone (Sigma) for 5 min. The elution was repeated 2 more times, the samples were pooled and frozen before being analyzed by tandem mass spectrometry.
Immunopurification of thiophosphorylated peptides
The labeling was performed in the same way as in the SulfoLink method. The reaction was stopped with 20 mM EDTA and the cells were further lysed by adding SDS to 0.1%. The samples were sonicated and cleared by centrifugation at 16000 × g. Labeled lysates were acidified to pH 5.0 with formic acid and alkylated with 1.5 mM PNBM for 1 h. The proteins were then precipitated, dissolved in urea, digested with LysC protease, desalted and lyophilized, as above. Peptides where dissolved in 2 ml of PTMScan IAP Buffer (Cell Signaling Technology), and incubated for 2 h with an anti-thiophosphate ester antibody (Abcam, ab133473) immobilized on protein A/G resin (Pierce). The beads were washed 4 times with IAP buffer and 2 times with PBS. Thiophosphorylated peptides were seluted with 150 μl of 100 mM formic acid. Purified peptides were frozen until ready for identification by tandem mass spectrometry. The thiolabeled and PNBM-modified peptides were identified as described (Allen et al., 2007).
Identification of substrates using the inhibition approach
Cdk1AS/AS ES cells were cultured in SL medium without feeders. Four plates were cultured for 30 min with 1 μM 3-MB-PP1 and the five were treated with equal volume of DMSO. The cells were washed with ice-cold PBS and cells from each plate were harvested separately in 5 ml of 2% SDS in 50 mM Tris pH 8.5 with Halt protease and phosphatase inhibitors (Pierce). The lysates were flash frozen in liquid nitrogen and stored frozen until analysis by tandem mass spectrometry.
Phosphopeptide enrichment, Tandem Mass Tag labeling, and fractionation of phosphopeptides obtained from the inhibition approach
Phosphopeptide enrichment.
Digested peptides (~10 mg per TMT channel) were resuspended in 1 ml of 2 M lactic acid/50% acetonitrile (ACN) and centrifuged at 15 000g for 20 min. Supernatants were removed, placed in an Eppendorf tube containing 15 mg of titanium dioxide beads (GL Sciences, Japan), and vortexed for 1 h at room temperature. Beads were washed twice with 2 M lactic acid/50% ACN and once with 0.1% TFA in 50% ACN. Phosphopeptides were eluted twice with 150 μl of 50 mM K2HPO4, pH 10, acidified with 40 μl of 20% formic acid, and subjected to C18 StageTip desalting (3M Empore, South Eagan, MN).
TMT labeling.
Isobaric labeling of the enriched phosphopeptides was performed using 10-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Rockford, IL). TMT reagents (5 mg) were dissolved in 250 μl of dry acetonitrile (ACN), and 10 μl was added to 100 μg (quantified by Micro BCA, Thermo Scientific, Rockford, IL) of phosphopeptides dissolved in 100 μl of 200 mM HEPES, pH 8.5. After 1 h, the reaction was quenched by adding 8 μl of 5% hydroxylamine. Labeled peptides were combined, acidified with 20 μl of 20% FA (pH ~2), and concentrated via C18 SPE on Sep-Pak cartridges (50 mg bed volume).
Basic pH reverse phase liquid chromatography.
TMT labeled phosphopeptides were subjected to orthogonal basic-pH reverse phase (bpHrp) fractionation. Labeled phosphopeptides were solubilized in buffer A (5% ACN, 10 mM ammonium bicarbonate, pH 8.0) and separated on an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm i.d. and 220 mm in length). Using an Agilent 1100 binary pump equipped with a degasser and a photodiode array (PDA) detector (Thermo Scientific, San Jose, CA), a 45 min linear gradient from 8% to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 (flow rate of 0.8 ml/min) separated the peptide mixtures into a total of 96 fractions. The 96 fractions were consolidated into 24 samples in a checkerboard manner, acidified with 10 μl of 20% formic acid and vacuum-dried. Each sample was redissolved in 5% formic acid, desalted via StageTip, dried via vacuum centrifugation, and reconstituted for LC–MS/MS analysis.
Liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) and data analysis
Identification of thiolabeled peptides.
Peptides were fractionated as described above and samples were analysed using an Orbitrap Fusion Tribrid-MS (Thermo Scientific, USA) equipped with an ultra-high pressure liquid chromatography unit (Proxeon). Peptides were separated on a 3 h RP gradient of 6–30% acetonitrile in 0.125% formic acid at a flow rate of ~400 nl/min. In each data collection cycle, one full MS scan (400–1400 m/z) was acquired in the Orbitrap (1.2 × 105 resolutions and an AGC of 2 × 105 ions). Top 10 most abundant ions were selected for isolation (0.5 Da), fragmentation (CID with 30% collision energy), and subsequent MS2 scans were acquired in the Orbitrap (15,000 resolution, AGC of 1 × 105 ions). Ions were dynamically excluded for 60 seconds and had a maximum ion accumulation time of 150 ms.
2D phosphopeptide profiling of TMT labeled peptides from the inhibition approach.
Using the same LC conditions as the direct labeling experiments above, each data collection cycle, one full MS scan (400–1400 m/z) was acquired in the Orbitrap (1.2 × 105 resolutions and an AGC of 2 × 105 ions). The subsequent MS2-MS3 analysis was conducted for top 10 most intense ions and fragmented by CID with following settings: collision energy of 35%, AGC 4×103, isolation window 0.5 Da, maximum ion accumulation time 150 ms with 40 seconds of dynamic exclusion. Following the MS2 scan, for the MS3 analyses, precursor isolation was performed using a 2.5 Da window and fragmented in the ion trap using CID using a collision energy of 35%, AGC targeted of 8,000 ions and a maximum ion injection time of 150 ms. Multiple fragment ions (SPS ions) were co-isolated and further fragmented by HCD. Selection of fragment ions was based on the previous MS2 scan and the MS2-MS3 was conducted using recently described sequential precursor selection (SPS) methodology (McAlister et al., 2014). MS3 was performed using HCD with 55% collision energy and reporter ion detection in the Orbitrap with an AGC of 150,000 ions, a resolution of 60,000 and a maximum ion accumulation time of 150 ms.
Database searching and reporter ion quantitation in-house software tools were used to convert RAW file to the .mzxml format (Huttlin et al., 2010). Correction of erroneous charge state and monoisotopic m/z values were performed using method detailed in (Huttlin et al., 2010). Assignment of MS/MS spectra were made with the Sequest algorithm (Eng et al., 1994) using an indexed Uniprot mouse database (compiled 2016) prepared with forward and reversed sequences concatenated as per the target-decoy strategy (Elias and Gygi, 2007). Database searches were conducted using cysteine alkylation and TMT on the peptide N-termini and lysines residues as static modification, oxidation of methionine as a dynamic modification, precursor ion tolerance of 20 ppm and a fragment ion tolerance of 0.8 Da (for CID). Sequest matches were filtered using linear discriminant analysis as previously reported (Elias and Gygi, 2007) first to a dataset level error of 1% at the peptide level based on matches to reversed sequences. Protein rankings were generated by multiplying peptide probabilities and the dataset was finally filtered to 1% FDR at the protein level. The final peptide-level FDR fell well below 1% (~0.2% peptide level). A reductionist model was used for assignment of peptides to protein matches, where all peptides were explained using the least number of proteins.
Quantitation of peptides using TMT reporter ions was performed as previously published (McAlister et al., 2014; Ting et al., 2011). Briefly, a 0.003 Th window centered on the theoretical m/z value of each reporter ion was recorded for each of the 6 reporter ions, and the intensity of the signal closest to the theoretical m/z value was recorded. TMT signals were also corrected for isotope impurities as per manufacturer’s instructions. Peptides were only considered quantifiable if the total signal-to-noise for all channels was >200 and an isolation specificity of >0.75. Within each TMT experiment, peptide quantitation was normalized by summing the values across each channel and then each channel was corrected so that each channel had the same summed value. Protein quantitation was performed by summing the signal-to-noise for all peptides for a given protein.
Statistical analyses and dissemination of proteomic data
Phosphopeptide site localisation.
We used a modified version of the Ascore algorithm to quantify the confidence with which phosphorylation site could be assigned to a particular residue. Phosphorylation sites with Ascore values >13 (P ≤ 0.05) were considered confidently localized to a particular residue (Huttlin et al., 2010).
Statistical analyses for phosphopeptide profiling.
Student’s t test was used to assign confidence in changes in phosphopeptide abundance. Multiple test correction was performed by adjusting the calculated p-values according to Benjamini–Hochberg. All proteomic data have been deposited into the proteome exchange repository - www.proteomexchange.org/ with the following dataset ID PXD015173.
Analysis of the direct labeling and inhibition approaches and comparison between mouse and yeast Cdk1 substrates
Directly labeled (thiophosphorylated) proteins were considered Cdk1 substrates if the thiophosphorylated serine or threonine residue was followed by a proline. For the inhibition screen, we identified Cdk1 substrates using the criteria: log2 control/inhibited > 0, p-value < 0.05 (t-test with Benjamini-Hochberg correction), phosphorylated residue followed by a proline. To compare mouse Cdk1 substrates identified by us in the inhibition approach versus the yeast Cdk1 substrates (Holt et al., 2009) (Figure S2G), we used DIOPT software v7.1 (Hu et al., 2011) (default parameter) to translate S. cerevisiae genes to mouse orthologs. In cases when a certain S. cerevisiae gene was translated to multiple mouse orthologs, all possible mouse orthologs were kept for subsequent analyses. S. cerevisiae genes were downloaded from the Ensembl database (Zerbino et al., 2018). For analyses shown in Figures 4B and S4, domain information for each substrate protein was retrieved from Uniprot and DIOPT databases. The domain organization of the ortholog proteins was plotted using DOG v2.0.1 (Ren et al., 2009). We aligned the mouse and yeast orthologs using the DIOPT software. If an S#P/T#P residue on a given mouse epigenetic regulator has been determined by us to represent Cdk1 phosphorylation substrate in ES cells, there can be four possibilities: (1) There is no yeast ortholog of this protein; (2) The corresponding site on the yeast ortholog is also S#P/T#P; (3) The corresponding site on the yeast ortholog is not S#P/T#P; (4) The sequence containing mouse S#P/T#P phosphosite does not have a corresponding sequence in yeast (no alignment). Phosphosites fulfilling criterion #4 were considered to reside on a domain that are absent in the yeast counterpart.
Analyses of the distribution of phosphorylation sites
To compare the actual number of Cdk1 phosphorylation sites per protein versus the expected number (Figure S2A), we performed 1000 simulations in which the same number of phosphorylation sites was randomly distributed to mouse ES cell proteome with probability proportional to the number of minimal Cdk1 consensus sites (S/T-P) in the protein. The two distributions were then compared using Mann Whitney test. For analyses of the inter-phosphosite distances (Figures S2B–S2D), distances between adjacent phosphorylation sites in actual and simulated cases were calculated and the two distributions were then compared using Mann Whitney test. In the 1000 simulated cases, the same number of phosphorylation sites was randomly distributed to minimal Cdk1 consensus sites in each protein.
Analyses of consensus Cdk1 phosphorylation motif
For analyses shown in Figure S3, the enrichment scores represent log10 p-values for the frequency of each amino acid at positions −7 to +7 relative to Cdk1-dependent phosphorylated serine or threonine. p-values were calculated relative to the mouse ES cell phosphoproteome under normal approximation of binominal distribution. Positive signs were assigned to the positions where the actual frequency was greater than expected. Sequence logos were generated using the sequence logo analysis tool in PhosphoSitePlus (https://www.phosphosite.org/).
Gene Ontology analyses
Gene Ontology (GO) analyses for enriched ‘biological process’ terms were performed using the web tool DAVID Bioinformatics Database v6.8 (https://david.ncifcrf.gov/tools.jsp) (Huang da et al., 2009a, b) for the ‘GOTERM_BP_DIRECT’ category. The complete set of all RefSeq genes was used as a background. The DAVID outputs are shown in Table S3. For plotting purposes, the enriched GO terms were further subjected to the web tool REVIGO (Supek et al., 2011) with the default parameter to filter redundant terms.
In vitro kinase assays
Kinase assays were performed in a final volume of 30 μl of the kinase buffer: 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM DTT, 1 mM EGTA, 0.1 mM NaF, containing 10 μM ATP and 0.4 μCi [32P]γATP (Perkin Elmer). To assess the endogenous kinase activity (Figures 1B, 5C, 6A, 6F, S1F and S5I), Cdk1, Cdk2, cyclin A2 or cyclin B1 were immunoprecipitated from whole cell lysates using anti-CDK1 (sc-54), anti-CDK2 (sc-163), anti-cyclin A2 (sc-751AC), or anti-cyclin B1 (sc-245AC) antibodies from Santa Cruz. For control, immunoprecipitates obtained with normal rabbit IgG (sc-2345) or normal mouse IgG (sc-2343) from Santa Cruz were used for kinase reactions. Histone H1 (EMD Millipore, 14–155) was used as kinase substrate, 1 μg per reaction. To verify selected epigenetic regulators as Cdk1/2 substrates (Figures 4C and 5A), Flag-tagged proteins were immunoprecipitated from transiently transfected 293 cells using anti-Flag M2 affinity gel (Sigma). The beads-bound proteins were subjected to in vitro kinase reactions with recombinant Cdk1/cyclin A2, Cdk1/cyclin B1, Cdk2/cyclin A2 or Cdk2/cyclin E1 complexes (Proqinase) in the kinase buffer. Histone H1 was used to compare the specific activity of the these Cdk/cyclin complexes. Proteins were then resolved by SDS-PAGE, transferred to nitrocellulose membranes and radiolabeled proteins detected by autoradiography. For mass spectrometric identification of phosphorylated residues of Dot1l, in vitro kinase reactions were performed as above, using immunoprecipitated Dot1l as a substrate, without radioactive ATP.
In-cell substrate labeling by analog-sensitive Cdk2
For analyses shown in Figure 4D, 293 cells were co-transfected using Polyfect (Qiagen) with 0.4 μg of plasmids encoding Flag-tagged versions of epigenetic regulators representing candidate Cdk1 substrates, together with 1 μg of AS-Cdk2 (F80G) and 1 μg of cyclin E1 encoding plasmids. Next day, cells were harvested in 200 μl of the labeling buffer modified from (Banko et al., 2011) (20 mM HEPES pH 7.5, 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 0.5 mM DTT, 45 μg/ml digitonin, 5 mM GTP, 0.2 mM ATP, 0.1 mM N6-furfuryl ATPγS (Biolog), Roche protease inhibitors cocktail) and incubated for 20 min at 30° C. Equal volume of 2x RIPA buffer wa s added (100 mM Tris pH 8.0, 300 mM NaCl, 2% NP-40, 0.2% SDS, 20 mM EDTA) and lysates were cleared by centrifugation at 16000 × g. Flag-tagged proteins were immunoprecipitated from thiolabeled extracts for 2 h at 4° C using 10 μl of an anti-Flag M2 antibody-coupled resin (Sigma), the beads were washed 3 times in 1x RIPA buffer, resuspended in 20 μl of 1.5 mM p-nitrobenzyl mesylate (Abcam), pH 4.5 adjusted with formic acid, and incubated for 15 min. Proteins were resolved by SDS-PAGE and immunoblots probed with an anti-thiophosphate ester antibody.
Analysis of endogenous Dot1l phosphorylation with anti-phospho-Ser/Thr-Pro antibody.
Wild-type or Flag-Dot1l V6.5 cells were treated for 2 h with 10 μM RO-3306 or with 20 μM CVT-313 or DMSO. Cells were next harvested in RIPA buffer containing protease and phosphatase inhibitors and Dot1l was immunoprecipitated with anti-Dot1l or anti-Flag antibody. The immunoprecipitates were analyzed by western blot using anti-phospho-Ser/Thr-Pro MPM-2 antibody (05–368MG Millipore) and anti-Dot1l antibody (A300–954A, Bethyl). Normal IgG (IgG) was used as an immunoprecipitation control.
Mass spectrometric analysis of Dot1l phosphorylation by Cdk1 and Cdk2
In vitro kinase assay products were processed and digested using the in-gel digestion method as published previously (Paulo, 2016). Five μl (100 μg) TMT reagents (127C, 128C, 129C, 130C) in ACN were used to label each sample. Then, 0.05% hydroxylamine was used to quench the reaction. Labeled peptides were mixed and dried in a vacuum centrifuge. Mass spectrometric analysis was conducted as described previously (Navarrete-Perea et al., 2018). Following data acquisition, database searching was performed against the Dot1l-only database using a SEQUEST-based suite. The ratio of the Dot1l protein level in each channel was 1.0:0.93:1.30:1.68. Phosphorylation site quantification output was normalized using the Dot1L protein levels, and scaled based on intensities in no kinase control sample. Data are organized and listed in Table S6.
Thio-ChIP-sequencing
Cdk1AS/AS ES cells were plated in N2B27 medium. Two days later, cells were co-treated for 16 h with 500 nM retinoic acid and 1 μM 3-MB-PP1 to obtain cells synchronized at G2/M phase with peak Cdk1 activity. The inhibitor was then washed off with PBS and 2 ml of the labeling buffer, modified from (Banko et al., 2011) (20 mM HEPES pH 7.5, 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 0.5 mM DTT, 45 μg/ml digitonin, 5 mM GTP, 0.2 mM ATP, 0.1 mM N6-furfuryl ATPγS (Biolog), Roche protease inhibitors cocktail) were added for 25 min. Control cells were treated the same way except that N6furfuryl ATPγS was omitted. Formaldehyde was then added to 0.5% final concentration and plates were incubated for 15 min, followed by quenching with 150 mM glycine. Cells were scraped and collected by centrifugation at 1350 rpm for 5 min at 4° C, resuspended in 6 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.1, Halt phosphatase and protease inhibitors) and sonicated for 10 cycles 30 s ON and 30 s OFF, using Diagenode Bioruptor Pico. The lysates were then cleared by centrifugation at 16000 × g for 5 min, the pH was adjusted to 6.0 with formic acid, and p-nitrobenzyl mesylate was added to 1.5 mM for 30 min. Two percent of the labeled and control alkylated lysates volume were saved as input controls and the rest was incubated with 10 μl of anti-thiophosphate ester antibody overnight at 4° C. Fifty μl of protein G Dybabeads (Invitrogen) were then added and the reactions incubated for another 5 h. Beads were then washed twice with RIPA buffer (0.1% SDS, 1% Triton X-100, 10 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.1% sodium deoxycholate), twice with RIPA buffer with 0.3 M NaCl, twice with LiCl buffer (250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1) and two times with TE buffer (1 mM EDTA, 10 mM Tris-HCl pH 8.0, 0.1% Triton X-100). The beads and the previously saved input controls were then rotated with 300 μl elution buffer (1% SDS, 0.1 M NaHCO3) overnight, 65° C. Next day, the beads were discarded and DNA recovered by adding 1.5 μl RNase A (10 mg/ml) and incubated for 30 min at 37° C. Next, 2.4 μl EDTA (0.5 M), 4.8 μl Tris-HCl (1 M), 2 μl glycogen (20 mg/ml) and 3 μl proteinase K (20 mg/ml) were added, followed by 2 h incubation at 62° C. DNA was then extracted using phenol/chloroform, washed with 75% ethanol, air dried and reconstituted in 24 μl TE buffer. The samples were submitted to Next Generation sequencing by the Whitehead Institute Genome Technology Core. Thio-ChIP-seq reads were aligned to the non-random chromosomes of the mm9 using Bowtie v1.2 (Langmead et al., 2009) (default parameter, except: -k 1 -m 1 --best). The thiopeaks were called using MACS v1.4.2 (Zhang et al., 2008) with the corresponding input control (default parameter, except: -p 1e-9). The GREAT software (McLean et al., 2010) (default parameter) was used to assign peaks to genes of the mouse mm9 assembly.
Analyses of genes associated with Cdk1 substrate peaks (‘thio-genes’)
Genes which were associated with thiopeaks (thio-genes) were classified into active, bivalent or silent groups (Figure 2D) based on the presence of H3K4me3, H3K79me2 and H3K27me3 histone marks, as in (Whyte et al., 2012). The ChIP-seq profiles of histone modifications from mouse ES cells were obtained from the GEO database (accession numbers are listed below).
| ChIP-seq, V6.5 cells | Source |
|---|---|
| H3K4me3 | GSM307146, GSM307147, GSM307148, GSM307149 (Marson et al., 2008) |
| H3K79me2 | GSM307150, GSM307151 (Marson et al., 2008) |
| H3K27me3 | GSM307619 (Mikkelsen et al., 2007) |
| WCE mES (Input control) |
GSM307154, GSM307155 (Marson et al., 2008) |
Sequence reads from multiple biological replicates were combined. ChIP-seq reads were aligned to the non-random chromosomes of the mouse (mm9 assembly) genome using Bowtie 2 v2.2.9 (Langmead et al., 2009) (default parameter, except: --local). Peak calling was performed using MACS2 v2.1.1 (Zhang et al., 2008) with corresponding input control (default parameter). H3K79me2 and H3K27me3 ChIP-seq peaks were called using the additional parameter ‘--broad’. The GREAT software (McLean et al., 2010) (default parameter) was used to assign peaks to genes based on the classification rules listed below (Whyte et al., 2012).
| Classification rules | ||||
|---|---|---|---|---|
| H3K4me3 within +/− 2 kb of the TSS | H3K79me2 within first 5 kb of gene body | H3K27me3 within +/− 5 kb of the TSS | Number genes of genes | |
| Active | Required | Required | - | 7193 |
| Bivalent | Required | - | Required | 3174 |
| Silent | Absent | Absent | - | 7140 |
p-values for enrichment of the thio-genes in each of these classes were determined using the hypergeometric test.
Histone H3K79me2 ChIP-sequencing
Cdk1AS/AS ES cells were grown without feeders and treated with 1 μM 3-MB-PP1 or DMSO for 4 days. Cells were crosslinked with 1% formaldehyde for 20 min, glycine (0.125 M final concentration) was added, cells were scraped, collected by centrifugation at 1350 × g, 10 min, 4° C, flash frozen in liquid nitrogen and stored at −70° C. Pellets were thawed into Nuclei EZ lysis buffer (Sigma) with protease inhibitor cocktail (Roche), incubated for 30 min on ice, centrifuged as above, and the nuclei resuspended in 3 ml of sonication buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitors). Nuclei were then sonicated 10× 30 s ON (output level 3, 24W) / 60 s OFF cycles with probe sonicator, on ice. Sonicated nuclear extracts were then cleared by centrifugation at 16000 × g, 15 min, 4° C. Twenty five microliters of the nuclear extracts were saved as input material. Chromatin was immunoprecipitated from nuclear extracts with antihistone H3 Lys79me2 antibody bound to beads (obtained by binding ab3594 antibody, Abcam, to protein G Dynabeads, Novex), overnight at 4° C. The beads were collected using magnetic separator, washed, and chromatin was eluted for 30 min at 65° C with 200 μl of buffer containing 50 mM Tris-HCl, pH 8.0, 10 mM EDTA and 1% SDS. Crosslinking of ChIP and input samples was reversed for 16 h at 65° C. Samples were treated with RNase A at 0.5 mg/ml for 2 h at 37° C, supplemented with 5 mM CaCl2 and 0.3 mg/ml Proteinase K and digested for 30 min at 55° C. DNA was next purified with phenol-chloroform-isoamyl alcohol mix (Qiagen) and 2 ml MaXtract gel tubes (Qiagen). After 10 min centrifugation at 16000 × g, DNA was precipitated by adding 16 μl of 5 M NaCl, 1 μl of 20 mg/ml glycogen (Fermentas) and 900 μl ethanol, centrifuged at 16000 × g, 4° C, and dissolved in 25 μl of TE buffer. The ChIP and input samples were submitted to the Whitehead Institute Genome Technology Core for library preparation and DNA sequencing. ChIP-seq reads were aligned to the non-random chromosomes of the mouse (mm9 assembly) genome using Bowtie 2 v2.2.9 (Langmead and Salzberg, 2012) (default parameter). Peak calling was performed using MACS2 v2.1.1 (Zhang et al., 2008) with corresponding input controls (default parameter, except: --broad). Selected enriched regions were aligned with each other according to the position of TSS. For each experiment, the ChIP-seq density profiles were normalized to the density per million total reads. The metagene plot (Figure 5I) and heatmaps (Figure 5J) were generated using ngsplot (Shen et al., 2014) with color saturation as indicated.
Targeted histone H3K79me2 chromatin immunoprecipitation (ChIP-qPCR)
Cdk1AS/AS ES cells were grown without feeders in SL medium or were differentiated with 500 nM retinoic acid plus 1 μM 3-MB-PP1 or DMSO for 24 h. Cells were fixed with formaldehyde and harvested as described in the ChIPsequencing section. Cell pellets were lysed in a buffer containing 50 mM TrisHCl pH 8.1, 10 mM EDTA, 1% SDS, protease and phosphatase inhibitors and sonicated as described in the Thio-ChIP-sequencing section. The samples were cleared by centrifugation at 16000 × g, 4°C, and 10% of the supernatant was saved as input control. The remaining extracts were diluted 4 times with a buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 and protease and phosphatase inhibitors, and 3 μg of anti-histone H3 Lys79me2 antibody (Abcam, ab3594) or rabbit IgG (Santa Cruz Biotechnology, sc-2027) were added, followed by overnight incubation at 4° C. Thirty microliters of protein G Dynabeads (Invitrogen) were added and ChIP samples were agitated for additional 4 h at 4° C. The beads were recovered using magnetic separator, washed 2 times, and 300 μl of the elution buffer (1% SDS, 100 mM NaHCO3) was added to the beads and input samples. DNA was recovered as in ChIPsequencing protocol and dissolved in 50 μl of 10 mM Tris-HCl pH 8.0. Half microliter of ChIP or input DNA was used in 20 μl qPCR reaction with 200 nM of each primer and AzuraView GreenFast qPCR Blue Mix (Azura Genomics). Enrichment of histone H3K79me2 mark over input DNA on different regions of the endodermal genes was determined by ΔΔCt method using primers designed with help of Primer-BLAST (NCBI). Quantitative PCR performed on DNA recovered with normal IgG yielded no enrichment over input and these data are not presented.
| Gene | Region | Forward | Reverse |
|---|---|---|---|
| Gata6 | promoter | TAGAGAGCAGTTCCGACCCA | GCTAGTCGCTGCTGGTGAAT |
| intron 1 | GTGTCCGGTCCTTCGCTTTA | GCGCTGTGATAACTCGGAGA | |
| intron 1 | ATTTCCACGTGCGGAACTCT | AACTCTGAGCCGGTGACAAG | |
| exon 2 | GCCCCAGCACAGGTAAAGAT | GCCTGCCACGTAGAAAGTCT | |
| Sox17 | promoter | TCTACAGCGAGCCTCAGAGT | ACCACGGATCTGCTTGAGTG |
| intron 3 | TCGGAAGCTAGCCTGGAGAT | GCGGATTAGGCGAAGGGTT | |
| Foxa2 | promoter | GCCTGGAGAGACCCGTTTAG | ATGCTTTGGGCACCTTGGAT |
| intron 3 | TTCATGCCATTCATCCCCAGG | CAGCCTAGGTGCTAACCTGTC |
Gene expression analysis by RNA-sequencing
To compare the results of thio-ChIP-sequencing with gene expression levels (Figure 2F), ES cells were grown in SL medium. To compare gene expression between wild-type and Dot1l KO ES cells (Table S8), V6.5 wild-type and Dot1l-knockout ES cells were grown in SL medium, or were induced to differentiate with Wnt3a and activin A for 24 h. Cell pellets were flash frozen in liquid nitrogen and stored at −70°C. Total RNA was purified using RNeasy Mini Kit (Qiagen) and submitted for Kapa mRNA library preparation and Illumina NS500 Single-End 75bp sequencing by Molecular Biology Core Facility at Dana-Farber Cancer Institute. RNA-sequencing data was aligned to GRCm38 using STAR v2.5.4a (Dobin et al., 2013) (default parameter, except: --outSAMstrandField intronMotif, --outFilterIntronMotifs RemoveNoncanonical). Differential gene expression analysis was performed using the Cuffdiff function in the Cufflinks v2.2.1 (Trapnell et al., 2012). Analysis was performed on the gene level. A significant change was defined by the q-value (adjusted p-value using the Benjamini-Hochberg procedure) < 0.05.
Reverse transcription and quantitative PCR
Five hundred nanograms of RNA, extracted from cells using RNeasy Mini Kit (Qiagen), were used to generate cDNA with High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems). Quantitative PCR reactions were performed using Power SYBR Green PCR Master Mix with 10 ng cDNA and the final 150 nM concentration of each primer in a total volume of 20 μl. Transcript levels of genes of interest were expressed as percent of GAPDH by ΔΔ Ct method. Sequences of gene-specific primers were obtained from PrimerBank Massachusetts General Hospital (Wang et al., 2012).
| Gene | Forward | Reverse |
|---|---|---|
| Gapdh | TCAAGCTCATTTCCTGGTATGAC | CTTGCTCAGTGTCCTTGCTG |
| Pax6 | TACCAGTGTCTACCAGCCAAT | TGCACGAGTATGAGGAGGTCT |
| Gata6 | TTGCTCCGGTAACAGCAGTG | GTGGTCGCTTGTGTAGAAGGA |
| Sox17 | CACAACGCAGAGCTAAGCAA | CGCTTCTCTGCCAAGGTC |
| Foxa2 | CCCTACGCCAACATGAACTCG | GTTCTGCCGGTAGAAAGGGA |
| T | GCTTCAAGGAGCTAACTAACGAG | CGTCACGAAGTCCAGCAAGA |
| Sox1 | GCAGCGTTTCCGTGACTTTAT | GGCAGAACCACAGGAAAGAAA |
Molecular cloning
Mouse Cdk1 was cloned into pCMV10 (no tag) and p3xFLAG-CMV10 by RT-PCR from mouse ES cell total RNA using forward (GCGAATTCGATGGAAGACTATATCAAAATAGAG) and reverse (ACGGATCCCTACATCTTCTTAATCTGATTGTC) primers containing EcoRI and BamHI restriction enzyme sites, respectively. AS-Cdk1 mutant was generated using Stratagene Site-Directed mutagenesis kit according to the manufacturer’s instructions with mutagenesis primers
GACTCCAGGCTGTATCTCATCGGGGAATTCCTGTCCATGGACCTC (Phe80Gly) and CACTGGCCAGATAGTGGCCGTGAAGAAGATCAGACTTG (Met32Val).
Epigenetic regulators were cloned using Gateway system by RT-PCR from mouse ES cell total RNA using primers listed in the table below. The ORFs were transferred by LR reactions or using NEB builder into destination plasmids: p3xFLAG-CMV10, pLenti-FUGW-3xFLAG-ires-Neo (modified from Addgene plasmid # 14883), or pLenti-EF1a-3xFLAG. The cDNA encoding first 84 amino acids of Lsd1 contained a long region of GCG repeats. To enable cloning, this fragment was codon optimized, synthesized (Genewiz) and assembled together with the remaining part of the cDNA in a plasmid.
| Protein | Plasmid | Forward | Reverse |
|---|---|---|---|
| Dot1l | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGGCGAGAAGCTGGAGCTGAGGCTCAAG | GGGGACCACTTTGTACAAGAAAGCTGGGTCTTACTAATTACCTCCAACGGTTCCGCCGGC |
| Kdm6b | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGCTCCAAAGAAGAAGCGTAAGGTAAATCG | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAGTCTCACTTGTCATCGTCGTCCTTGTAGTC |
| Kdm4a | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGCTTCTGAGTCTGAAACTCTGAATCCCAG | GGGGACCACTTTGTACAAGAAA GCTGGGTCCTAGTCCTACTCCATGATGGCCCGGTATAGTG |
| Kdm4c | p3xFLAG-CMV10 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAGGTGGCCGAGGTGGAAAGTCC | GGGGACCACTTTGTACAAGAAA GCTGGGTCCTAGTCCTACTGTCTCTTCTGGCACTTCTTCTGGAAAG |
| Kdm2a | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAACCTGAAGAAGAAAGGATTCGGTACAGC | GGGGACCACTTTGTACAAGAAAGCTGGGTCCCTGTTCAGTCTGGGCTGAGCATG |
| Phf8 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGCCTCGGTGCCTGTGTATTGCCTCTG | GGGGACCACTTTGTACAAGAAA GCTGGGTCTCACAGAAGTAACTTGCCATTTCTGTGGATTTTCAGG |
| Ehmt2 (G9a) | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGGCGGCGGCGGCGGGAGC | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAGTCTCAGGTGTTGATGGGGGGCAGGGAG |
| Nsd1 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCCCTCCGAAGACAAGGACAGCCCTTTC | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAGTCTTATTTCTTTTCTGAATCTGCACATTTGCGACTAG |
| Nsd2 | FUGW-3xFLAG | TTGTACAAAAAAGCAGGCTTCGAATTTAGCATCAGAAAAAGTCCTCTTTCTGTTCAG | TTTGTACAAGAAAGCTGGGTCCTATTTGCCATCTGTGACCCTTCGCC |
| Ncoa3 | pEXPEF1a3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGAGTGGACTAGGCGAAAGCTCTTTGGATCCGCTGGCC | GGGGACCACTTTGTACAAGAAAGCTGGGTCTTATCAGCAGTATTTCTGATCGGGGCCCATGGGCATGCCAG |
| Hdac6 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCTCAACCGGCCAGGATTCCAC | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAGTCTTACTTGTCATCGTCGTCCTTGTAGTCTCC |
| Mta1 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGGCCGCCAACATGTACAGGGTC | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAGTCCTCAATAACAATGGGCTCATC |
| Phf17 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGAAACGAGGTCGCCTTCCCAGCAGC | GGGGACCACTTTGTACAAGAAAGCTGGGTCTTATCAAGAGGCCAAGATGCTTCGCCTGATG |
| Jarid2 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCAGCAAGGAAAGACCCAAGAGGAATATC | GGGGACCACTTTGTACAAGAAAGCTGGGTCTCATGAGGATGGGAGCCGAGATG |
| Psip1 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGACTCGCGATTTCAAACCTGGAGACCTC | GGGGACCACTTTGTACAAGAAAGCTGGGTCTTACTAGTTATCTAGTGTAGACTCTTTCAGAGATATTTCAGC |
| Arid4b | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTC AAGGCCCTTGATGAACCTCCTTATTTG |
GGGGACCACTTTGTACAAGAAAGCTGGGTC TCACCTGCACTCAACTGACATTCC |
| Setdb1 | FUGW-3xFLAG | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCTCCTCCCTCCCTGGGTGCATGAGTTTG | GGGGACCACTTTGTACAAGAAAGCTGGGTCCTAAAGAAGTCTCCCTCTGCATTCAATGGC |
| Hsp90 | FUGW3xFLAG-2xGFP | CCTGAGGAAGTGCACCATGGAGAGGAG | ATCCACCTCTTCCATGCGCGAGGC |
The phosphorylation sites substitutions were introduced into plasmids using Multisite Directed Mutagenesus kit from Stratagene and primers listed below.
| Protein | Substitution | Mutagenesis primer (substituted nucleotides) |
|---|---|---|
| Dot1l | S1105D,P1106D | CTCAGCACGGGTGTGGACGACAAGCGCCGGGCTC |
| LSD1 | T60E | GGAGCAGCTGGAGAGCGAGAGCCTCGAAAGAAGGAGCC |
| G9a | S115A | AAGTCGTTCCCCTCTGCCCCCAGCAAGGG |
| G9a | S346A | AAATGGCGGAAAGACGCCCCGTGGGTGAAGCC |
| G9a | T853A | GGCATGGGATCTGGCCCCAGAGCGCTC |
| G9a | S115D | AAGTCGTTCCCCTCTGACCCCAGCAAGGGGGG |
| G9a | S346D | AAATGGCGGAAAGACGACCCGTGGGTGAAGCC |
| G9a | T853E | GACACGGCATGGGATCTGGAACCAGAGCGCTCTGA |
Generation of knockout ES cell lines
Guide RNA (CTGCAAACATCACTACGGAG) to generate Dot1l KO A and to target G9a (CCTGCTGCACTCACCTCTCT) were designed using CRISPOR (Haeussler et al., 2016) and cloned into lentivCRISPR v2 plasmid which was a gift from Dr. Feng Zhang (Addgene plasmid # 52961; http://n2t.net/addgene:52961 ; RRID:Addgene_52961) (Sanjana et al., 2014). A non-targeting gRNA (ACGGAGGCTAAGCGTCGCTT) from GeCKO v2 libraries was cloned as well. Dot1l KO B (GATGACCTGTTTGTCGACCT) and C (GCTGTGTGACAAATACAACC) were generated with Edit-R predesigned all-inone lentiviral sgRNAs from Dharmacon. Wild-type V6.5 and Cdk1AS/AS ES cells were transduced with lentiviral particles carrying Dot1l or G9a gRNAs, as well as non-targeting gRNA, respectively. The cells were selected with 2 μg/ml puromycin and analyzed by western blotting for knockout efficiency. In case of G9a, single clones were picked, expanded and pooled to obtain cell population without detectable G9a protein.
It was reported that prolonged inhibition of Cdk1 causes apoptosis of ES cells (Huskey et al., 2015). It was also reported that inactivation of the intrinsic apoptotic pathway, by ablating Bak and Bax function, prevented the death of ES cells upon Cdk1 inhibition, without affecting expression of pluripotency markers (Huskey et al., 2015). However, we observed that ES cell death upon prolonged Cdk1 inhibition could be prevented by culturing the cells without feeders for 2–3 passages, and this allowed us to study the consequences of Cdk1 inhibition for up to 4 days, without encountering any apoptosis. Nevertheless, to ensure that the observed effects of Cdk1-inhibition are not due to the impending apoptosis, we repeated our analyses using Bak- and Bax-deficient ES cells. Guide RNAs targeting Bak (TCATCGCAGCCCACCTTCGG) and Bax (AGCGAGTGTCTCCGGCGAAT) were selected with help of CRISPOR (Haeussler et al., 2016), cloned into pX330-Cas9 vector (a gift from Dr. Feng Zhang, Addgene plasmid # 42230 (Cong et al., 2013)) and co-electroporated into Cdk1AS/AS cells. Five independent clones with homozygous insertions or deletions in Bak and Bax genes were identified by PCR – DNA sequencing. These cells showed no signs of cell death after Cdk1 inhibition. The results obtained using Cdk1AS/AS Bak −/− Bax −/− cells were identical to the results with Cdk1AS/AS cells.
Expression of proteins in ES cells, MEFs and MCF7 cells
To generate Ehmt2 (G9a) mutants (Figure S5A), we inserted phosphomimetic (aspartic acid) or phosphoinactivating (alanine) substitutions in place of serine 115 of G9a, the residue identified as Cdk1 target in our inhibition approach (Table S2), together with similar substitutions (Ser346Asp, Thr853Glu or Ser346Ala, Thr853Ala) in serine 346 and threonine 853. Although the latter two phosphoresidues were not identified in our approach, we included them into our mutational analysis as they are a part of a full Cdk1 consensus sequence (S/TP(X)1–2R/K), and are hence likely phosphorylated by Cdk1. Note that mutation of these three residues to alanines abolished phosphorylation of G9a by Cdk1 – See Figure 4D, lane “3xAla”. To generate the Dot1l phosphomimetic mutant (Figures 5D, 5F, S5G and S6A), we substituted serine 1105 of Dot1l (identified as a Cdk1 phosphorylation target both in the direct labeling and the inhibition approaches, see Table S2) with aspartic acid. To better mimic phosphorylation (by providing the second negative charge), the mutant contained an additional aspartic acid substitution in the adjacent proline 1106. Such an approach has been shown to better mimic the charge introduced by Cdk-driven phosphorylation of a serine or threonine residue (Strickfaden et al., 2007). Transfer plasmids encoding wild-type or phospho-site mutants of Flag-Dot1l or G9a were co-transfected with lentiviral envelope (VSVG) and packaging (d8.9) plasmids into 293FT cells. Virus containing medium was collected after 2 days and passed through a 0.45 μm syringe filter, followed by virus concentration on Amicon Ultra-15 100kDa centrifugal columns (Millipore). Concentrated virus was added to freshly trypsinized ES cells supplemented with 10 μg/ml polybrene in a gelatinized well. Next day, medium was exchanged, and 2 days later, ES cells were passaged onto a monolayer of mitotically inactivated, neomycin-resistant feeder cells and selected with 400 μg/ml G418. Primary E13.5 mouse embryonic fibroblasts at passage three were transduced as described for ES cells with lentiviral particles encoding Flag-Dot1l. MCF7 cells were transiently transfected with plasmids encoding WT or phosphomimetic Flag-Dot1l using Lipofectamine 2000.
Quantification of histone modifications
ES cells, iPSC or MEFs were dissociated with trypsin and counted. Cells were lysed in 250 μl of 1x SDS-PAGE reducing sample buffer and sonicated to fragment genomic DNA. The lysates were then diluted ten fold, 20 μl were resolved on 15% SDS-PAGE and transferred to 0.2 μm pore-size nitrocellulose membrane by wet transfer (1 h, 450 mA). The membranes were probed with a mix of a histone mark-specific antibody from rabbit and an anti-histone H3 antibody from mouse (Millipore Sigma, 05–499), both diluted 1:1000 in 5% BSA/TBS-T, overnight at 4° C. Membranes were probed with IRDye 680 goat anti-rabbit and IRDye 800 Goat anti-mouse antibodies (Licor) diluted 1:10000 in BSA/TBS-T, and visualized using Odyssey scanner. The level of each histone modification was expressed as the ratio of fluorescence signal from the histone mark antibody to the signal from total histone H3 channel. The following histone mark antibodies from Cell Signaling Technology were used: acetyl-histone H3K9 (#9649), acetyl-histone H3K27 (#8173), tri-methyl-histone H3K4 (#9751), trimethyl-histone H3K9 (#13969), tri-methyl-histone H3K27 (#9733), tri-methylhistone H3K36 (#4909), mono-methyl-histone H3K79 (#12522), di-methyl-histone H3K79 (#5427), mono-methyl-histone H3K4 (#5326), tri-methyl-histone H4K20 (#5737). Identical results were obtained when chromatin fraction was analyzed this way.
Crystal violet staining
For analyses shown in Figure 1D, Cdk1AS/AS or wild-type V6.5 ES cells were seeded in a 24-well plate. Cells were treated with increasing concentrations of 3MB-PP1 or DMSO. After 4 days, cells were fixed with 10% buffered formalin and stained with crystal violet solution for 5 min.
Alkaline phosphatase staining
Wild-type V6.5, Cdk1AS/AS or Dot1l-knockout ES cells were cultured in SL medium, fixed with 4% paraformaldehyde (PFA) in PBS for 2 min, rinsed with TBS-T, and incubated for 20 min with Alkaline Phosphatase Substrate (Vector Red, SK-5100). Uniformly red staining is indicative of pluripotent, undifferentiated state of ES cells.
Analyses of embryoid bodies
Cdk1AS/AS or Cdk1+/+ ES cells were trypsinized and depleted of feeders by incubation on gelatin-coated plates 2 × 20 min. Cells were resuspended in SL medium without LIF and counted. Droplets of 20 μl, each containing 2000 cells, were pipetted onto the inner surface of a 150 mm plate lid in a 2D array. The lid was then inverted (‘hanging drops’) to cover the dish containing 20 ml PBS. After 24 h, early embryoid bodies (EB) were washed off the lids, allowed to sediment, re-suspended in N2B27 medium, and cultured for 1 day in ultra-low attachment 6-well plates (Corning, CLS3471). Next day, medium was changed to N2B27 with 1 μM retinoic acid in the absence or presence of 1 μM 3MB-PP1 (Cdk1AS/AS cells) or 10 μM RO-3306 (Cdk1+/+ cells). The EB were harvested for RNA isolation or immunostaining after 24 h. To obtain sections for staining, EBs were collected by sedimentation in silicon-coated 1.5 ml tubes, fixed in 4% PFA for 20 min and equilibrated in 30% sucrose at 30°C for 20 min. Pellets of embryoid bodies were frozen in O.C.T. freezing medium (VWR, 25608–930), and stored at −80°C before cryosectioning onto poly-lysine coated slides.
Immunostaining
Cells were fixed with 4% PFA in PBS for 15 min and incubated in the blocking buffer (5% BSA in PBS, 0.1% Triton X-100) for 30 min. Slides were next incubated overnight at 4°C with primary antibodies diluted in the blocking buffer, washed with PBS, 0.1% Triton X-100, and incubated with fluorophore-conjugated secondary antibodies and Hoechst stain diluted in PBS, 0.1% Triton X-100.
For staining of embryoid bodies, slides with cryosections were fixed with 4% PFA for 7 min and incubated for 30 min in PBS containing 0.25% Triton X-100 and 3% BSA. Slides were then incubated with primary antibodies diluted in PBS 0.25% Triton X-100 for 2h, washed and incubated with secondary antibodies in PBS 0.1% Triton X-100. Slides with stained cells or EBs were mounted with coverslips using Fluoromount G medium (Southern Biotech). The following antibodies were used for immunostaining: anti-Flag, 1:2000 (Sigma, F1804), anti-histone H3 Lys79me2 1:500 (Abcam, ab3594), anti-Gata6 1:1500 (CST, D61E4); anti-Sox17 1:50 (R&D, AF1924) anti-Foxa2 1:50 (Santa Cruz, sc-6554), and 1:500 dilutions of secondary donkey anti-mouse Alexa Fluor 594 and Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 488, and donkey anti-goat Alexa Fluor 568.
ES cell cycle synchronization and propidium iodide cell cycle analysis
For analyses shown in Figures S5H and S5J, cells were plated on gelatin-coated plastic 24 hours prior to nocodazole treatment. The cells were then blocked with nocodazole at 50 ng/ml for 12 hours. Next, nocodazole-containing medium was removed, the cells washed with PBS and supplemented with fresh SL medium. For data shown in Figures S5H, S5I and S5K, cells were collected and fixed in 90% EtOH immediately (G2/M) or after 6 h (G1/S), 10 h (early S) and 12 h (late S). For data shown in Figures S5J and S5K, cells were collected right away (G2/M) or after 6 h (G1/S) and 8 h (S-phase). Subsequently, the cells were stained with 40 μg/ml propidium iodide with 0.1 mg/ml RNase A and analyzed by flow cytometry using the BD LSR Fortessa.
QUANTIFICATION AND STATISTICAL ANALYSES
For Figures 2D, 2F, 3F (significance of overlap), the p-values were determined by the hypergeometric test; for Figure 3G and Figures S2E, S2F, S2H, by the t-test with Benjamini-Hochberg correction; for Figure 5H, by the Mann-Whitney test; for Figure 6E, by one-way Anova with Tukey’s multiple comparisons test; for Figure 6G by paired two-tailed t-test; for Figure 7A, using the unpaired two-tailed t-test; for Figures 7B, 7F and 7G by multiple t-test with Benjamini’s correction; for Figures S2A–S2D, by the Mann-Whitney test, for Figure S2G, by the hypergeometric test.
DATA AND CODE AVAILABILITY
Data resources
All proteomic data have been deposited into the proteome exchange repository – www.proteomexchange.org/ with the accession number PXD015173.
The next generation sequencing (NGS) data generated in this study have been submitted to the Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE134645.
Supplementary Material
Table S1. Results of ‘Thio-ChIP-Sequencing’ and RNA-Sequencing of ES Cells, Related to Figure 2
Sheet 1: Listed are thiopeaks and genes associated with them. Numbers in parentheses denote distance (bp) of a thiopeak from the gene’s transcription start site (TSS). We considered a thiopeak to be associated with a gene, if it overlapped with gene’s proximal regulatory region (5 kb upstream to 1 kb downstream of TSS), regardless of other nearby genes, or with its distal 59 regulatory region (proximal regulatory region extended in both directions, up to another gene’s proximal regulatory region, but not further than 1000 kb).
Sheet 2: Listed are the transcriptomic data of ES cells. Gene expression levels from three replicates are shown in FPKM (Fragments Per Kilobase Million) values.
Table S2. Results of the Labeling and Inhibition Approaches, Related to Figure 3 (including the list of 417 direct and 662 inhibition substrates).
Listed are phosphopeptides identified by the labeling approach (sheet 1) and by the inhibition approach (sheet 3), as well as 417 substrates identified by the labeling approach (sheet 2) and 662 substrates identified by the inhibition approach (sheet 4).
Table S3. Results of Gene Ontology Analyses of Cdk1 Substrates, Related to Figure 3
Listed are enriched ‘biological process’ terms of 308 yeast substrates (sheet 1), 662 substrates identified by the inhibition approach (sheet 2), 417 substrates identified by the labeling approach (sheet 3), and 210 overlapping substrates identified by both approaches (sheet 4).
Table S4. Epigenetic Regulators Identified as Cdk1 Substrates in ES Cells, Related to Figure 4
Shown are: the symbol of the protein, the approach which led to identification of a given protein as a Cdk1 substrate in mouse ES cells (I, inhibition; L, labeling; I/L identified by both approaches), the name of S. cerevisiae ortholog (if present), the information whether the ortholog was found to be phosphorylated by Cdk1 in S. cerevisiae CDC28 (Cdk1) inhibition approach (Holt et al., 2009), and additional information regarding the localization of the phosphorylated residues relative to the yeast ortholog.
Table S5. Comparison of S. cerevisiae and Mouse Cdk1 Substrates Identified by Yeast CDC28 Inhibition or Mouse Cdk1 Inhibition Approaches, Related to Figure 3
Sheet 1: Listed is the output of DIOPT translation of yeast substrates to their mouse orthologs. Sheet 2: Listed are the pairs of common yeast and mouse Cdk1 substrates.
Table S6. In vitro Phosphorylation of Dot1l by Cdk1 and Cdk2, Related to Figure 5
Shown is mass spectrometric quantification of in vitro phosphorylation of Dot1l at different sites by Cdk1 and Cdk2. Dotl1 was phosphorylated in vitro with Cdk1/cyclin A1, Cdk1/ cyclin B1, Cdk2/ cyclin E1, or incubated without any added kinase. The products of kinase reactions were labeled using TMT reagents 127C, 128C, 129C, or 130C. Shown are raw intensities (Phosphorylation sites quantification), intensities normalized by Dot1l total intensity in each channel (Normalization using Dot1l protein level) and scaled intensities after normalization against no kinase control sample (Scaled based on no kinase control sample). U denotes unique peptides.
Table S7. Results of H3K79me2 ChIP-Sequencing, Related to Figure 5
Listed are H3K79me2 ChIP-sequencing peaks called by MACS2 in cells cultured in DMSO (sheet 1) or 3-MB-PP1 (sheet 2). ‘chrom’: chromosome name; ‘chromStart’: chromosome start position; ‘chromEnd’: chromosome end position; ‘score’: integer of ‘-10*log10(q-value)’.
Table S8. Results of RNA Sequencing of Dot1l-Knockout ES Cells, Related to Figure 6
Listed are the results of Cuffdiff differential gene expression analysis of wild-type and Dot1l KO ES cells grown in SL medium (sheet 1), or induced to differentiate with Wnt3a and activin A (sheet 2). Gene expression levels from three replicates are shown in FPKM (Fragments Per Kilobase Million) values.
Highlights.
Mice expressing analog-sensitive Cdk1 allow identification of Cdk1 substrates
Many Cdk1 substrates in ES cells are chromatin-bound at poised or transcribed genes
Cdk1 phosphorylates a large number of epigenetic regulators
Cdk1 phosphorylates and inactivates H3K79 methyltransferase Dot1l
ACKNOWLEDGEMENTS
W.M. was supported by a postdoctoral fellowship from Foundation for Polish Science, Ruth L. Kirschstein Institutional National Research Service Award from the NIH, and Claudia Adams Barr Program for Innovative Basic Cancer Research Award. This work was supported by grants R01CA202634 and R01CA190509 (to P.S.) and R01 GM123511 (to R.A.Y.). We thank Drs. J. Spangle and H. Gardner for help.
DECLARATION OF INTERESTS
W.M. is currently an employee of Cedilla Therapeutics. BJA is a shareholder of Syros Pharmaceuticals. RAY is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics and Dewpoint Therapeutics. P.S. has been a consultant at Novartis, Genovis, Guidepoint, The Planning Shop, ORIC Pharmaceuticals and Exo Therapeutics; his laboratory receives research funding from Novartis. In situ visualization of kinase substrates has been patented (P. Sicinski and W. Michowski, WO2018/035296A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hubner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, et al. (2007). A semisynthetic epitope for kinase substrates. Nature Methods 4, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, et al. (2011). Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Molecular Cell 44, 878–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, and Kaldis P. (2003). Cdk2 knockout mice are viable. Current biology 13, 1775–1785. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. (2000). A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401. [DOI] [PubMed] [Google Scholar]
- Blethrow JD, Glavy JS, Morgan DO, and Shokat KM (2008). Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proceedings of the National Academy of Sciences of the United States of America 105, 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J, and Cross FR (2007). Multiple levels of cyclin specificity in cell-cycle control. Nature Reviews Molecular Cell Biology 8, 149–160. [DOI] [PubMed] [Google Scholar]
- Brooks EE, Gray NS, Joly A, Kerwar SS, Lum R, Mackman RL, Norman TC, Rosete J, Rowe M, Schow SR, et al. (1997). CVT-313, a specific and potent inhibitor of CDK2 that prevents neointimal proliferation. Journal of Biological Chemistry 272, 29207–29211. [DOI] [PubMed] [Google Scholar]
- Brown NR, Korolchuk S, Martin MP, Stanley WA, Moukhametzianov R, Noble MEM, and Endicott JA (2015). CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat Commun 6, 6769 10.1038/ncomms7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Chen J, Wu Y, et al. (2013). H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nature Genetics 45, 34–42. [DOI] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, and Huang H. (2010). Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nature Cell Biology 12, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, O’Brien K, Atsma JM, Fujiwara Y, Nguyen M, et al. (2014). Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Molecular Cell 53, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, and Kaldis P. (2012). Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proceedings of the National Academy of Sciences of the United States of America 109, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fluckiger AC, Marcy G, Marchand M, Negre D, Cosset FL, Mitalipov S, Wolf D, Savatier P, and Dehay C. (2006). Cell cycle features of primate embryonic stem cells. Stem Cells 24, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii-Yamamoto H, Kim JM, Arai K, and Masai H. (2005). Cell cycle and developmental regulations of replication factors in mouse embryonic stem cells. The Journal of Biological Chemistry 280, 12976–12987. [DOI] [PubMed] [Google Scholar]
- Furuno N, den Elzen N, and Pines J. (1999). Human cyclin A is required for mitosis until mid prophase. The Journal of Cell Biology 147, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, and Pines J. (2010). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Developmental Cell 18, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, and Sicinski P. (1999). Rescue of cyclin D1 deficiency by knockin cyclin E. Cell 97, 767–777. [DOI] [PubMed] [Google Scholar]
- Ghule PN, Medina R, Lengner CJ, Mandeville M, Qiao M, Dominski Z, Lian JB, Stein JL, van Wijnen AJ, and Stein GS (2011). Reprogramming the pluripotent cell cycle: restoration of an abbreviated G1 phase in human induced pluripotent stem (iPS) cells. Journal of Cellular Physiology 226, 11491156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SL, Charlet A, Haas W, Gygi SP, and Kellogg DR (2005). Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122, 407–420. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. (2010). Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Wang BT, Allen JJ, Zhang C, Dar AC, Burlingame AL, and Shokat KM (2010). Chemical Genetic Approach for Kinase-Substrate Mapping by Covalent Capture of Thiophosphopeptides and Analysis by Mass Spectrometry. Curr Prot Chem Bio, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, and Morgan DO (2009). Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huskey NE, Guo T, Evason KJ, Momcilovic O, Pardo D, Creasman KJ, Judson RL, Blelloch R, Oakes SA, Hebrok M, et al. (2015). CDK1 inhibition targets the p53-NOXA-MCL1 axis, selectively kills embryonic stem cells, and prevents teratoma formation. Stem Cell Reports 4, 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, and Reinberg D. (2010). Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes & Development 24, 2615–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno Y, Suzuki A, Sugimura K, Okumura K, Zineldeen DH, Shimada M, Niida H, Mizuno T, Hanaoka F, and Nakanishi M. (2009). Cyclin A-Cdk1 regulates the origin firing program in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 106, 3184–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak S, Busso C, and Gonczy P. (2013). NuMA phosphorylation by CDK1 couples mitotic progression with cortical dynein function. The EMBO Journal 32, 2517–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Liu L, Michowski W, Kolodziejczyk A, and Sicinski P. (2019). The cell cycle in stem cell proliferation, pluripotency and differentiation. Nature Cell Biology 21, 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M. (2005). Mammalian cyclin-dependent kinases. Trends in Biochemical Sciences 30, 630–641. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, and Barbacid M. (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases CDK4 and CDK6. Cell 118. [DOI] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. (2008). Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker D, Denison C, Anderson S, Egelhofer TA, Yates JR 3rd, Gygi SP, and Kellogg DR (2007). Cdk1 coordinates cell-surface growth with the cell cycle. Nature Cell Biology 9, 506–515. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G. (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, and Meissner A. (2008). Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima M, Pavicic V, Gruneberg U, Nigg EA, and Glotzer M. (2004). Cell cycle regulation of central spindle assembly. Nature 430, 908–913. [DOI] [PubMed] [Google Scholar]
- Morgan DO. (2007). The Cell Cycle: Principles of Control. New Science Press Ltd. [Google Scholar]
- Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neganova I, Tilgner K, Buskin A, Paraskevopoulou I, Atkinson SP, Peberdy D, Passos JF, and Lako M. (2014). CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death & Disease 5, e1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, and Zhang Y. (2011). The diverse functions of Dot1 and H3K79 methylation. Genes & Development 25, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. (2012). Chromatin-modifying enzymes as modulators of reprogramming. Nature 483, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, and Barbacid M. (2003). Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nature Genetics 35, 25–31. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Collins MO, Lichawska A, Zegerman P, Choudhary JS, and Pines J. (2011). Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Molecular Cell 43, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA (2016). Sample preparation for proteomic analysis using a GeLC-MS/MS strategy. J Biol Methods 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen MT, Kooistra SM, Radzisheuskaya A, Laugesen A, Johansen JV, Hayward DG, Nilsson J, Agger K, and Helin K. (2016). Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. The EMBO Journal 35, 1550–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, and Barbacid M. (1999). Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nature Genetics 22, p44–52. [DOI] [PubMed] [Google Scholar]
- Ren J, Wen L, Gao X, Jin C, Xue Y, and Yao X. (2009). DOG 1.0: illustrator of protein domain structures. Cell Res 19, 271–273. [DOI] [PubMed] [Google Scholar]
- Rideout WM, Wakayama T, Wutz A, Eggan K, Jackson-Grusby L, Dausman J, Yanagimachi R, Jaenisch R, (2000). Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet. 24, 109–110 [DOI] [PubMed] [Google Scholar]
- Ruiz S, Panopoulos AD, Herrerias A, Bissig KD, Lutz M, Berggren WT, Verma IM, and Izpisua Belmonte JC (2011). A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Current Biology 21, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, and Barbacid M. (2007). Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, and Kaldis P. (2009). Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28, 2925–2939. [DOI] [PubMed] [Google Scholar]
- Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O’Neill LP, Turner BM, Delrow J, et al. (2004). The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes & Development 18, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert M, Kruger M, Watson NA, Sen O, Daum JR, Slotman JA, Braun T, Houtsmuller AB, Gorbsky GJ, Jacob R, et al. (2019). CDK1-mediated phosphorylation at H2B serine 6 is required for mitotic chromosome segregation. The Journal of Cell Biology 218, 1164–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Liu Y, Deirmengian C, and Shokat KM (1997). Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proceedings of the National Academy of Sciences of the United States of America 94, 3565–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E. (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, Patel S, Lopez D, Mishra N, Pellegrini M, et al. (2013). Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nature Cell Biology 15, 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, and Dalton S. (2002). Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 21, 8320–8333. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. (2008). DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Molecular and Cellular Biology 28, 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, and Pryciak PM (2007). A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell 128, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PloS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sako K, Akiyama K, Isoda M, Senoo C, Nakajo N, and Sagata N. (2015). Identification of non-Ser/Thr-Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Scientific Reports 5, 7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Berge D, Koole W, Fuerer C, Fish M, Eroglu E, and Nusse R. (2008). Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 3, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, and Ramanathan S. (2011). Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W. (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, and Kiyokawa H. (1999). Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Molecular and Cellular Biology 19, 7011–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Sachdeva MM, Wang H, and Blobel GA (2006). Profile of histone lysine methylation across transcribed mammalian chromatin. Molecular and Cellular Biology 26, 9185–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar K, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, and Chen L. (2006). Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proceedings of the National Academy of Sciences of the United States of America 103, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Sundermann L, Labbe JC, Pintard L, Radulescu O, Castro A, and Lorca T. (2018). Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Developmental Cell 45, 637–650 e637. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, and Seed B. (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Lo CM, Chen L, Ngan ES, Xu A, and Poon RY (2017). CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death and Differentiation 24, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, et al. (2011). CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nature Cell Biology 13, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324. [DOI] [PubMed] [Google Scholar]
- White J, Stead E, Faast R, Conn S, Cartwright P, and Dalton S. (2005). Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Molecular Biology of the Cell 16, 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, and Young RA (2012). Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witucki LA, Huang X, Shah K, Liu Y, Kyin S, Eck MJ, and Shokat KM (2002). Mutant tyrosine kinases with unnatural nucleotide specificity retain the structure and phospho-acceptor specificity of the wild-type enzyme. Chem Biol 9, 25–33. [DOI] [PubMed] [Google Scholar]
- Wuhr M, Haas W, McAlister GC, Peshkin L, Rad R, Kirschner MW, and Gygi SP (2012). Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal Chem 84, 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zhu C, and Harper JW (2001). A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proceedings of the National Academy of Sciences of the United States of America 98, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokochi T, Poduch K, Ryba T, Lu J, Hiratani I, Tachibana M, Shinkai Y, and Gilbert DM (2009). G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proceedings of the National Academy of Sciences of the United States of America 106, 19363–19368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, et al. (2018). Ensembl 2018. Nucleic Acids Res 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, Templeton DJ, and Shokat KM (2005). A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nature Methods 2, 435–441. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137.psf. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of ‘Thio-ChIP-Sequencing’ and RNA-Sequencing of ES Cells, Related to Figure 2
Sheet 1: Listed are thiopeaks and genes associated with them. Numbers in parentheses denote distance (bp) of a thiopeak from the gene’s transcription start site (TSS). We considered a thiopeak to be associated with a gene, if it overlapped with gene’s proximal regulatory region (5 kb upstream to 1 kb downstream of TSS), regardless of other nearby genes, or with its distal 59 regulatory region (proximal regulatory region extended in both directions, up to another gene’s proximal regulatory region, but not further than 1000 kb).
Sheet 2: Listed are the transcriptomic data of ES cells. Gene expression levels from three replicates are shown in FPKM (Fragments Per Kilobase Million) values.
Table S2. Results of the Labeling and Inhibition Approaches, Related to Figure 3 (including the list of 417 direct and 662 inhibition substrates).
Listed are phosphopeptides identified by the labeling approach (sheet 1) and by the inhibition approach (sheet 3), as well as 417 substrates identified by the labeling approach (sheet 2) and 662 substrates identified by the inhibition approach (sheet 4).
Table S3. Results of Gene Ontology Analyses of Cdk1 Substrates, Related to Figure 3
Listed are enriched ‘biological process’ terms of 308 yeast substrates (sheet 1), 662 substrates identified by the inhibition approach (sheet 2), 417 substrates identified by the labeling approach (sheet 3), and 210 overlapping substrates identified by both approaches (sheet 4).
Table S4. Epigenetic Regulators Identified as Cdk1 Substrates in ES Cells, Related to Figure 4
Shown are: the symbol of the protein, the approach which led to identification of a given protein as a Cdk1 substrate in mouse ES cells (I, inhibition; L, labeling; I/L identified by both approaches), the name of S. cerevisiae ortholog (if present), the information whether the ortholog was found to be phosphorylated by Cdk1 in S. cerevisiae CDC28 (Cdk1) inhibition approach (Holt et al., 2009), and additional information regarding the localization of the phosphorylated residues relative to the yeast ortholog.
Table S5. Comparison of S. cerevisiae and Mouse Cdk1 Substrates Identified by Yeast CDC28 Inhibition or Mouse Cdk1 Inhibition Approaches, Related to Figure 3
Sheet 1: Listed is the output of DIOPT translation of yeast substrates to their mouse orthologs. Sheet 2: Listed are the pairs of common yeast and mouse Cdk1 substrates.
Table S6. In vitro Phosphorylation of Dot1l by Cdk1 and Cdk2, Related to Figure 5
Shown is mass spectrometric quantification of in vitro phosphorylation of Dot1l at different sites by Cdk1 and Cdk2. Dotl1 was phosphorylated in vitro with Cdk1/cyclin A1, Cdk1/ cyclin B1, Cdk2/ cyclin E1, or incubated without any added kinase. The products of kinase reactions were labeled using TMT reagents 127C, 128C, 129C, or 130C. Shown are raw intensities (Phosphorylation sites quantification), intensities normalized by Dot1l total intensity in each channel (Normalization using Dot1l protein level) and scaled intensities after normalization against no kinase control sample (Scaled based on no kinase control sample). U denotes unique peptides.
Table S7. Results of H3K79me2 ChIP-Sequencing, Related to Figure 5
Listed are H3K79me2 ChIP-sequencing peaks called by MACS2 in cells cultured in DMSO (sheet 1) or 3-MB-PP1 (sheet 2). ‘chrom’: chromosome name; ‘chromStart’: chromosome start position; ‘chromEnd’: chromosome end position; ‘score’: integer of ‘-10*log10(q-value)’.
Table S8. Results of RNA Sequencing of Dot1l-Knockout ES Cells, Related to Figure 6
Listed are the results of Cuffdiff differential gene expression analysis of wild-type and Dot1l KO ES cells grown in SL medium (sheet 1), or induced to differentiate with Wnt3a and activin A (sheet 2). Gene expression levels from three replicates are shown in FPKM (Fragments Per Kilobase Million) values.
Data Availability Statement
Data resources
All proteomic data have been deposited into the proteome exchange repository – www.proteomexchange.org/ with the accession number PXD015173.
The next generation sequencing (NGS) data generated in this study have been submitted to the Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE134645.