

Abstract
Over recent decades, gene therapy, which has enabled the treatment of several incurable diseases, has undergone a veritable revolution. Cell therapy has also seen major advances in the treatment of various diseases, particularly through the use of adult stem cells (ASCs). The combination of gene and cell therapy (GCT) has opened up new opportunities to improve advanced therapy medicinal products for the treatment of several diseases. Despite the considerable potential of GCT, the use of retroviral vectors has major limitations with regard to oncogene transactivation and the lack of physiological expression. Recently, gene therapists have focused on genome editing (GE) technologies as an alternative strategy. In this review, we discuss the potential benefits of using GE technologies to improve GCT approaches based on ASCs. We will begin with a brief summary of different GE platforms and techniques and will then focus on key therapeutic approaches that have been successfully used to treat diseases in animal models. Finally, we discuss whether ASC GE could become a real alternative to retroviral vectors in a GCT setting.
Keywords: adult stem cells, CRISPR, electroporation, gene delivery systems in vivo or in vitro, gene therapy, hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), pluripotent hemopoietic stem cells
Diagram showing potential clinical HPSCs genome editing applications using either NHEJ‐ or HR‐based approaches (blue and red arrows, respectively). The target conditions are indicated in gray boxes, and each arrow points to the locus targeted in each case
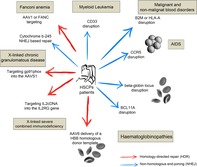
Significance statement.
Recent advances in adult stem cells and genome editing techniques have enabled scientists to envisage the generation of efficient and safe advanced therapy medicinal products for the treatment of untreatable diseases. Hematopoietic progenitor stem cells are now clearly regarded as the cell type of reference. Promising results have been achieved in controlling AIDS and hemoglobinopathies, resulting in several clinical trials. Promising results have also been obtained in the treatment of monogenic diseases, including X‐SCID, SCID‐ADA, X‐CGD, and Fanconi anemia, indicating that further clinical trials will be approved in the near future.
1. REASONS FOR USING ADULT STEM CELLS
Gene and cell therapy (GCT) strategies utilize multiple cell types for the treatment of different diseases. 1 The most common approaches use adult stem cells (ASCs), also known as somatic stem cells, as well as T cells. However, other differentiated cell types, such as B, NK and macrophage cells, as well as pluripotent stem cells (PSCs), have also been used.2, 3 In particular, PSCs, which are essentially embryonic stem cells (ESCs), and induced PSCs (iPSCs), have been proposed as potent therapeutic tools due to their ability to produce all types of mature cells in the human body. However, although PSCs are widely used in basic research, very few studies have been carried out on their clinical applications. PSCs are restricted to a small number of applications in clinical trials according to recent data published on the U.S. National Institute of Health's web page regarding clinical trials (https://clinicaltrials.gov). The possibility of PSC‐derived products being contaminated by potentially tumorigenic undifferentiated cells, as well as the lack of clear regulatory guidelines, has delayed their clinical application. In addition, genetic alterations can be accumulated during PSC passage and/or differentiation.4, 5 Of the 25 clinical studies using PSCs, 21 use hESCs, 4 are based on iPSCs, while none use genetically modified PSCs.
Unlike PSCs, multipotent, undifferentiated ASCs, which are found in all organs of living organisms, are involved in physiological tissue regeneration. 6 ASCs, which have a self‐renewal capacity, can give rise to some or all of the differentiated cells of the tissue in which they reside. They have been widely used in clinic due to their ability to regenerate tissues, such as blood and skin, and to dampen immune responses. Most ASCs used in clinical trials are hematopoietic progenitor stem cells (HPSCs) and mesenchymal stem cells (MSCs), with over 3000 clinical trials carried out so far (http://clinicaltrials.gov 2019). A major reason for the success of ASC transplants is their safety. However, in several applications, genetic modification of ASCs is necessary in order to achieve the desired therapeutic benefits. 1 Genetically modified ASCs have been successfully employed in the treatment of several disorders through the use of integrative viral vectors. 7 These ASCs include HSPCs which are chosen due to their capacity to be grafted in bone marrow and give rise to all hematopoietic lineages. Over 120 clinical trials involving genetically modified HSPCs are on‐going or have been completed worldwide, 7 of which are now in Phase III or IV, with one medicinal treatment (Strimvelis) already approved by the Food and Drug Administration (FDA) and European Medicines agency (EMA). In addition to HSPCs, other gene‐modified ASCs have also reached Phase I/II clinical trials, including MSCs, T stem cell memory (TSCM) cells, epidermal stem cells (EpSCs), endothelial stem cells (EnSCs), and neural stem cells (NSCs) (data obtained from https://clinicaltrials.gov and http://www.abedia.com/wiley/). Most of the clinical trials mentioned above rely on semi‐random integration of one or more copies of the therapeutic gene introduced into the host genome using γ‐retroviral or lentiviral vectors. However, this type of genetic integration has generated concerns regarding the possibility of cellular transformation and expression variability. 8 In this review, we discuss the potential role of genome editing (GE) technologies in overcoming the limitations of retroviral vectors. We will focus on ex vivo strategies using ASC GE in clinical and/or preclinical settings.
2. GE STRATEGIES
GE involves a group of technologies that enable the cellular genome to be modified. However, for its successful in‐clinic application, GE needs to be used efficiently either in vitro or in vivo without affecting the normal physiology of targeted human cells. Nuclease‐independent9, 10 technologies, as well as those based on the use of specific endonucleases (SENs), are used to carry out GE. 11 The nuclease‐independent strategy facilitates GE without generating double strand breaks (DSBs) by using systems that improve homologous directed recombination (HDR) such as adeno‐associated virus (AAV) vectors 10 or that introduce distortions in the target DNA that triggers repair mechanisms, such as triplex‐forming oligonucleotides (TFOs) 9 (Figure 1).
Figure 1.
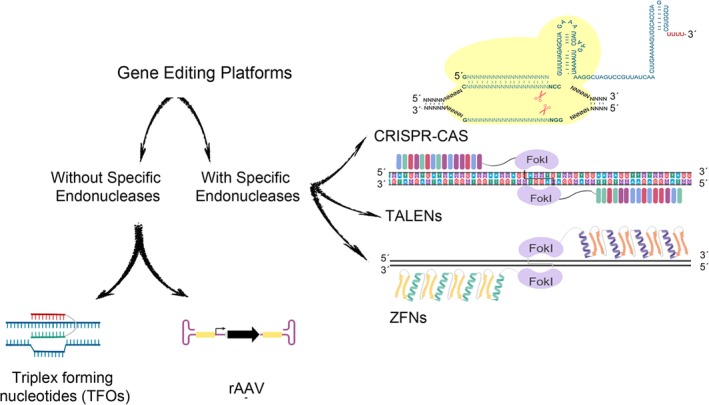
Current genome editing technology platforms can be divided into two main groups: specific endonuclease (SEN)‐based (right) and nuclease‐independent (left) platforms. The three main types of SEN‐based genome editing platforms are the transcription activator‐like effector nuclease (TALEN), zinc finger nuclease (ZFN), and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR‐associated protein 9 (Cas9) systems. The principal SEN‐free gene editing platforms use recombinant adeno‐associated virus (rAAV) vectors and triplex‐forming oligonucleotides (TFOs)
Whether GE systems generate DNA breaks or distortions, the target cell triggers different DNA repair mechanisms, mainly through non‐homologous end joining (NHEJ) or homology‐directed repair (HDR). NHEJ is a type of double‐stranded break repair mechanism that does not require a DNA donor. The targeted sequences are rapidly processed by cellular machinery which generates small insertions or deletions (indels). Although a less efficient DNA repair mechanism, HDR is more precise than NHEJ. When a compatible donor DNA template is delivered to the cell, these DNA molecules are incorporated into the endogenous locus, thus enabling precise modifications to be carried out (Figure 2). The most advanced strategy in terms of preclinical and clinical applications is NHEJ‐mediated GE which is highly efficient. NHEJ‐based GE strategies using SENs and ASCs have already reached the clinical stage for the treatment of sickle cell disease (SCD), B‐thalassemia, AIDS, and acute lymphoblastic leukemia.
Figure 2.
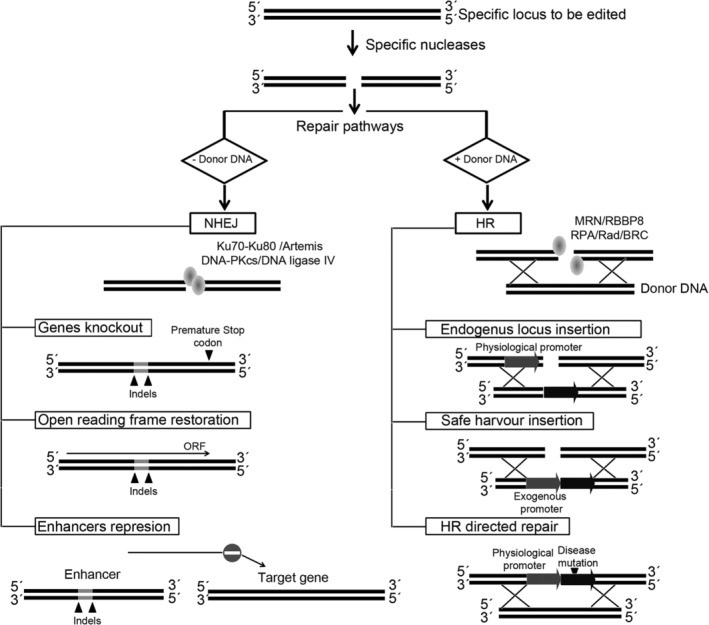
Genome editing strategies based on the activity of specific nucleases (SENs). Once SENs generate a double strand break in the target locus (top), the cell triggers two main repair pathways depending on the availability of homologous DNA and cell type. Non‐homologous end joining (NHEJ, left) preferentially occurs on G1 phase and quiescence cells, whereas homologous recombination (HR, right) generally requires cell division and takes place in the S phase of the cell cycle. In the NHEJ pathway, the donor or template DNA are not available and, after SEN cleavage, the binding of the proteins Ku70‐Ku80 protects the DNA ends against excessive resection and promotes DNA repair by recruiting the Artemis, DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) and DNA ligase IV complex. This repair pathway introduces short DNA insertions or deletions (indels) into the target site and facilitates different GE approaches: (a) generation of knockout genes by eliminating the ATG or by changing the open reading frame, thus generating premature stop codons, (b) repair of the correct open reading frame on mutated genes, or (c) elimination/alteration of enhancers/promoter regions. In contrast to NHEJ, in the HR pathway, the cell uses a donor DNA to fix DNA breaks introduced by SENs. HR repair is initiated by the combined action of the MRE11‐RAD50‐NBS1 (MRN) complex and RBBP8 generating single strand DNA where replication protein A (RPA), in association with Rad and BRC proteins bind and promote HR by invading the homologous template. By providing abundant homologous DNA donors, the HR pathway can also be used for different GE strategies: (a) insertion of a cDNA sequence under the regulation of a specific locus in order to provide locus‐specific, physiological expression of the particular cDNA. (b) Insertion of an expression cassette (promoter and cDNA) into a safe location (harbor). (c) Alternatively, HR can be used for HR‐directed repair (HRDR) of disease‐causing mutations (precise gene editing) by providing DNA donor harboring the corrected DNA sequences
There are four main types of SEN: mega nucleases (MGNs), transcription activator‐like effectors nucleases (TALENs), zinc finger nucleases (ZFNs), and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR‐associated system 9 (CRISPR‐Cas9) systems. 11 The success of GE approaches greatly depends on the type of gene editing tools used and on how these tools are delivered to the cells and tissues. Another important feature is safety, which can be measured by the levels of unwanted off‐target modifications outside the target locus. As both NHEJ and HDR strategies are capable of introducing undesirable modifications into the host genome, it is crucial to accurately determine the safest system to be used (a combination of the appropriate GE tool, delivery system, and strategy).12, 13, 14, 15, 16
In order to obtain GE‐ASCs, cells must first be isolated from their original tissue and then edited ex vivo. As explained above, how GE tools (SENs and/or donor) are delivered to the ASCs is crucial for the success of the strategy. These delivery methods can be viral,16, 17, 18, 19 nonviral,20, 21, 22 or a combination of both, 13 and should be transient, highly efficient, and nontoxic. Recently, a hybrid method based on murine leukemia virus particles has produced interesting results.23, 24 A more detailed review of delivery systems for GE can be found elsewhere. 25
The most effective platforms for NHEJ‐GE of ASCs are mRNA nucleofections for ZFNs and the ribonucleoprotein complex for CRISPR. Nucleofection, a type of electroporation system, is probably the most successful physical non‐viral‐based method for delivering macromolecules to target cell nuclei. It is important to note that nucleofection produces a spike in SEN expression, thus reducing toxicity and increasing GE specificity.7, 26 Clinical‐grade electroporators, which can be used in clinical trials for the treatment of AIDS and blood disorders, have been developed. In addition to nucleofection, adenoviral (AdV), AAV, and integration‐deficient lentiviral vectors (IDLVs)16, 27 are often used in HDR‐GE strategies. Although capable of efficiently delivering large donor DNAs, these viral vectors can also be used to deliver specific nucleases.
A major concern with using GE technologies as a treatment option arises from the possibility of introducing off‐target unwanted alterations into the modified genome.12, 28 However, none of the methods for detecting the distribution and frequency of off‐targets introduced by SENs are regarded as sufficiently robust to be implemented in clinical trials. 14 Nevertheless, it is very useful to compare different off‐target SEN activities in order to develop more effective and safer strategies. Some research groups have focused on CRISPR‐based systems which have no endonuclease activity but maintain the capacity to bind to the site indicated by the gRNA. New CRISPR/Cas9, such as cytosine base editors and adenine base editors,29, 30 have been developed. These editors combine a catalytically dead Cas9 (dCas9) with a cytosine or adenosine deaminase domain in order to facilitate direct single‐base pair substitutions (C:G to T:A and A:T to G:C) without generating DSBs. More recently, the group led by Dr. Liu has developed “prime editing” technology by combining a dCas9, a reverse transcriptase and a prime editing guide RNA (pegRNA). This technique enables DNA to be edited with unprecedented precision, with fewer errors being introduced than previous gene‐editing technologies. 31
Although technical issues still need to be addressed, 23 GE clinical trials using ZFNs (14 clinical trials), CRISPR/Cas9 (16 clinical trials), and TALENs (3 clinical trials) for the treatment of infectious diseases (HIV‐1 and HPV), cancer, as well as blood and metabolic disorders, are currently on‐going (http://clinicaltrials.gov Dec 2019). Six of these clinical trials use ex vivo ASC techniques to treat AIDS and blood disorders.
3. GE of ASCs
As mentioned above, the vast majority of clinical trials involving gene‐edited stem cells use ASCs. Although those using HPSCs are by far the most successful, 1 EpSCs, MSCs, TSCM cells, and NSCs have also produced promising results.
3.1. Genome‐edited HSPCs
The engraftment of HSPCs in a recipient's bone marrow, which gives rise to all types of hematopoietic cells, provides a wide range of intervention opportunities for a large number of disorders. 7 Despite being some of the most desirable target cells for stem cell‐based therapies, HSPCs are highly resistant to gene modification and preferentially use NHEJ rather than HR pathways to repair damage to DNA. 11 This preference explains why the vast majority of HSPC GE studies and clinical trials use NHEJ‐based strategies rather than HR‐based approaches (Figure 3). Table 1 summarizes important preclinical studies of HSPC gene therapies (GTs).
Figure 3.
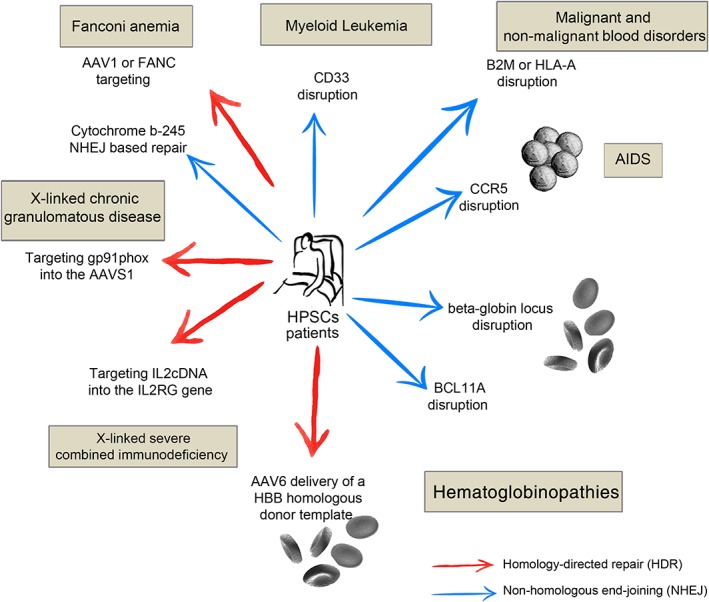
Diagram showing potential clinical HPSCs genome editing applications using either NHEJ‐ or HR‐based approaches (blue and red arrows, respectively). The target conditions are indicated in gray boxes, and each arrow points to the locus targeted in each case
Table 1.
Examples of successful preclinical studies, combining different type of ASCs and genome editing tools, for the treatment of different genetic and infectious diseases
| Cells type | Diseases | GE strategies | Tools | Ref | |
|---|---|---|---|---|---|
| Hematopoietic progenitor stem cells | Primary immune deficiency |
HR ‐ insertion in safe harbor HR ‐ insertion in affected locus |
CRISPR‐Cas9/ZFNs | 32, 33 | |
| ZFNs | 27 | ||||
| Hematoglobinopathies | HR ‐ gene repair | CRISPR‐Cas9 | 34 | ||
| ZFNs | 35 | ||||
| NHEJ ‐ therapeutic mutation | TALENs | 36 | |||
| CRISPR‐Cas9 | 37 | ||||
| ZFNs | 38 | ||||
| Fanconi anemia | HR ‐ insertion ‐ safe harbor | ZFNs | 39 | ||
| Cancer |
HR ‐ therapeutic mutation NHEJ ‐ exon deletion |
CRISPR‐Cas9 | 40, 41 | ||
| Infectious diseases | NHEJ ‐ gene disruption | CRISPR‐Cas9 | 42 | ||
| ZFNs | 43, 44, 45 | ||||
| Other stem cells | Mesenchymal stem cells | Liver fibrosis | HR ‐ insertion in safe harbor | TALENs | 46 |
| Parkinson's disease | 47 | ||||
| Epidermal stem cells | Dystrophic epidermolysis bullosa | NHEJ ‐ therapeutic mutation | CRISPR‐Cas9 | 48, 49 | |
| TALENs | 50 | ||||
| Junctional epidermolysis bullosa | HR ‐ insertion in affected locus | CRISPR‐Cas9 | 51 | ||
| Neural stem cells | Krabbe disease | HR ‐ insertion in safe harbour | CRISPR‐Cas9/TALENs | 52 | |
| Muscle stem cells | Duchenne muscular dystrophy | HR ‐ gene repair | CRISPR‐Cas9 | 53 | |
3.1.1. HSPC GT for infectious diseases (HIV‐1)
Although several strategies based on GE technology have been designed to fight different infectious agents, only human immunodeficiency virus type I (HIV‐1) has been targeted using HSPC GE. Initial studies have demonstrated that long‐term HSCP‐CCR5‐KO repopulation can be achieved using ZFN43, 44 and CRISPR/CAS942, 54 strategies, both of which provide protection for HIV‐1 in humanized NOD/SCID/IL2R gamma mice. Paterson et al. were the first to use nonhuman primates (NHPs) to demonstrate multilineage repopulation of genome‐edited HSPCs. 45 These preclinical studies led to the first two clinical trials to evaluate the feasibility, safety, and engraftment of allogeneic and autologous HSCP‐CCR5‐KO in China (NCT03164135) and the United States (NCT02500849), respectively (Figure 4). The clinical trial being carried out in the United States using autologous HSPCs (CCR5‐KO) and ZFNs, for which patients were recruited in September 2019, is sponsored by the City of Hope Medical Center in collaboration with Sangamo Therapeutics. These patients were placed on either a 2‐ or 3‐day course of busulfan prior to product infusion in order to enhance HSPC engraftment. The clinical trial in China has already produced an initial report on HIV‐1‐infected patients treated with genome‐edited HSPCs. 55 Paterson et al. treated HIV‐infected patients with acute lymphocyte leukemia (ALL) at the Academy of Military Medical Sciences in China using CRISPR/CAS9 to edit the CCR5 locus of allogeneic HSPCs. The patients with ALL achieved complete remission, with CD4+ cells in CCR5KO mice found to increase following a pause in antiretroviral therapy. However, the percentage (5%) of CCR5 disrupted was relatively low, indicating the need for further improvement.
Figure 4.
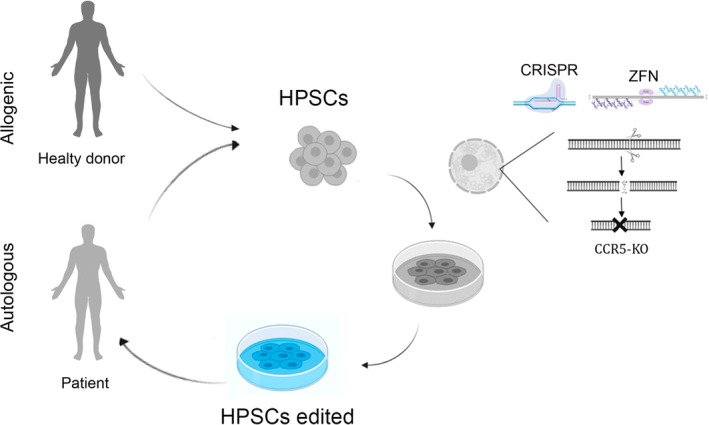
Diagram showing the principal steps in a clinical trial using autologous as compared to allogenic HSPCs. HPSCs were harvested from patients and healthy donors and cultivated in vitro. Once an optimal number of cells with the appropriate phenotype were obtained, they were subjected to gene editing and then infused back into the patient who was treated with the appropriate conditioning regimen
3.1.2. GE‐HPSCs for monogenetic diseases
Monogenetic diseases are a series of inherited pathologies associated with alterations in a single gene that can be point mutations, indels, or large deletions. These diseases include hemoglobinopathy, X‐linked severe combined immunodeficiency (X‐SCID), and Fanconi anaemia (FA), which were initially considered to be targets for treatment with gene‐corrected HPSCs.
Hemoglobinopathies
Hemoglobinopathies, characterized by defective hemoglobin synthesis, include SCD, and β‐thalassemia. Lentiviral vector (LV)‐based GT has been highly successful in integrating the normal β‐globin gene into HPSCs, a strategy which has reached clinical trial phase III using LentiGlobin (NCT03207009). Despite their considerable success, LVs, which are integrated into the transcriptionally active locus, represent a potential risk of cellular transformation. 8 GE appears to be a potentially safer alternative for restoring normal β‐globin expression either through insertion of the healthy β‐globin gene via the HR pathway or through reactivation of the fetal γ‐globin gene34, 35, 36, 37, 38, 56, 57, 58 (Figure 5). The most successful strategies are aimed at reactivating fetal γ‐globin gene expression by disrupting the negative regulatory region of the γ‐globin gene.59, 60 These were the first human GE strategies to be investigated in clinical trials using CRISPR/CAS9 (CTX001, NCT03745287) and later ZFNs (ST‐400; NCT03432364) for the treatment of β‐thalassemia and SCD. New clinical trials have been approved for these diseases using CRISPR (iHSCs; NCT03728322) and ZFNs (PRECIZN‐1; NCT03653247). Recently, CRISPR Therapeutics and Vertex published the initial results of monitoring the CTX001 trial at month 9 for a patient with β‐thalasemia and at month 4 for a patient with SCD (http://www.crisprtx.com). The β‐thalasemia patient in the transfusion‐independent clinical trial had total hemoglobin levels of 11.9 g/dL, fetal hemoglobin of 10.1 g/dL, and erythrocyte‐expressing fetal hemoglobin of 99.8%, with no severe side effects. The SCD patient also underwent significant improvement, with the occurrence of novaso‐occlusive crisis (VOC) episodes (total hemoglobin levels of 11.3 g/dL and 94.7% of erythrocytes expressing fetal hemoglobin).
Figure 5.
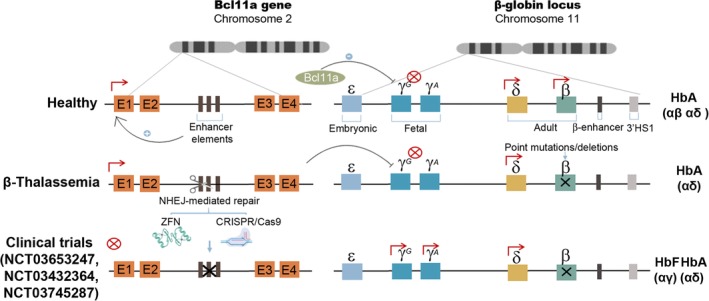
NHEJ GE strategies for treating β‐thalassemic and sickle cell disease (SCD) patients. Schematic representation of the β‐globin cluster in healthy individuals (top drawing). Only adult globins, δ‐globin and β‐globin, are expressed in healthy adult individuals. The Bcl11a gene is expressed thanks to the erythroid‐specific enhancer and blocks fetal globin (γG and γA) expression. In β‐thalassemia and SCD patients (middle drawing), mutations in the β‐globin gene abrogate its normal expression, preventing the generation of the predominant adult hemoglobin form (αβ). Three different clinical trials are currently on‐going to investigate the feasibility of eliminating the erythroid‐specific enhancer of the Bcl11a gene on HSPCs (Bottom drawing) using ZFNs (NCT03432364 and NCT03745287) or CRISPR/Cas9 (NCT03653247). By disrupting Bcl11a gene expression in erythroid cells, the fetal γ‐globin will be expressed in adults forming fetal hemoglobin (αγ) which should restore normal function
Fanconi anemia (FA)
FA, characterized by congenital malformation and cancer susceptibility, with defective repair of DNA inter‐strand crosslinks (ICLs), is a rare disease associated with genetic mutations in one or more of the 22 FANC genes. FA is an excellent target for genetic correction, as corrected stem cells and their progeny have a strong selective in vivo advantage. 61 Five on‐going GT trials, including one at the phase II stage (NCT02931071), are investigating the efficacy and safety of HSPCs expressing the correct version of the FANCA gene through the use of LVs (http://clinicaltrials.gov Sept 2019). Despite the positive outcome of the latest clinical trial using LVs, 61 GE is also being studied as a potential alternative to FA GT. However, the intrinsic problems of repairing ICL lesions in the cells of FA patients have made gene therapists cautious about using gene editing to treat these patients. Nevertheless, several studies have demonstrated the feasibility of the GE of fibroblasts from FA patients harboring mutations. 62 Diez et al., who have demonstrated the feasibility of phenotypic correction of FA HPSCs by inserting a FANCA donor in an AAVS1 locus via HR, have also shown that GE can be effective in HPSCs when ZFNs are used. 39
Primary immunodeficiencies (PIDs)
Several PIDs, including severe combined immune deficiencies (SCID‐X1, SCID‐ADA), Wiskott‐Aldrich syndrome (WAS), chronic granulomatous disease (CGD), and X‐linked hyper‐IgM (X‐HIM), have been successfully treated using GT‐based approaches involving both γ‐retroviral and LVs. 63 Given the mutagenic nature of first‐generation γ‐retrovirus‐based vectors, some patients developed leukemia. However, the latest generation of these vectors has produced better results, with physiological promoter‐driven, self‐inactivating γ‐retrovirus 64 and lentivirus65, 66 vectors, in particular, found to be safer and more effective in clinical settings. 1 As a result, a new advanced therapy medicinal product (ATMP), named Strimvelis, has been approved for the treatment of SCID‐ADA, with several others on the way. GE has opened up the possibility of further improvements in GT strategies by expressing the therapeutic transgene in a more physiological manner through the use of endogenous regulatory sequences and/or reductions in genotoxicity caused by semi‐random integration of retroviral vectors. Different cell models have been used to test the feasibility of using GE to correct genetic mutations causing PIDs. 67 However, HPSC GE is problematic due to poor permissiveness to gene transfer and limited HD DNA repair pathways in these cells. In 2014, Genovese et al. demonstrated, for the first time, successful correction of target genes in human HPSCs. 27 Using a HR‐based approach based on ZFN mRNA nucleofection and IDLV‐DNA donors, they repaired the mutated IL2RG gene of HPSCs from a patient with SCID‐X1 and successfully engrafted genome‐edited HPSCs, giving rise to functioning hematopoietic cells. In recent years, using gene‐editing tools, other research groups, who have produced new studies on SCID‐X132, 33 using AAV6 for donor delivery and ZFN mRNA, 32 as well as CRISPR/Cas9 RNP 33 nucleofection, have confirmed the feasibility of correcting HPSCs from different PIDs.32, 33, 68, 69 Kuo et al. have shown that AAV6 harboring the donor DNA could also be combined with TALEN mRNA or CRISPR/Cas9 RNP to restore X‐linked hyper‐IgM syndrome. 68 Using a different platform (Cas9 mRNA, gRNA and ssODN), De Ravin et al. 69 repaired up to 20% of HSPCs from patients with X‐linked chronic granulomatous disease (X‐CGD). See Table 1 for further information on GE for PID.
HLA gene editing enables the generation of HSPC universal donor cells
In recent decades, unmodified allogeneic HLA‐matched HSPCs have been used to treat malignant and non‐malignant blood disorders. However, the success of transplants depends on the existence of compatible donors, whereas the risk of graft rejection is still a major concern. A definitive approach could be developed through the genetic elimination of HLA mediated by GE. Torikai et al. have used ZFNs to fully disrupt HLA‐A in T cells. 70 Recognition of GE‐T cells by natural killers was circumvented by the expression of non‐classical HLA molecules. This research group later demonstrated the feasibility of their approach with HSCs which maintains the engraftment of the engineered cells and reconstitutes hematopoiesis in immunocompromised mice. 71 Other strategies could benefit from using universal HLA−HSPCs as ATMP cells by, for example, enabling the manufacture of erythrocytes and/or platelets from universal HLA−HSPCs.
3.1.3. HSPC GE for cancer GT
Genetically modified T cells expressing a chimeric antigen receptor (CAR) are a powerful new class of advanced therapy medicinal product. 72 CD19‐targeting CAR‐T cells were recently approved by the FDA for the treatment of refractory type B leukemia and lymphomas. This approach does not discriminate between normal and malignant B cells, although patients can live almost normal lives without B cells if treated with immunoglobulins. CD33 is an interesting target for acute myeloid leukemia (AML). 73 Unfortunately, CD33 is expressed in both malignant and in normal myeloid cells (including progenitors), which are destroyed by CD33‐CAR‐T cells. In order to overcome this limitation, several research groups have targeted CD33 from normal HSPCs using CRISPR/Cas9 in order to generate functional myeloid cells resistant to CAR‐T αCD33.40, 41, 74 Gene‐edited HSPCs showed normal myeloid function and resistance to CD33 therapy mediated by T cells engineered to express CAR‐targeting CD33.40, 41, 74 It is important to note that multilineage reconstitution has been demonstrated in both mouse and NHP models.40, 41, 74
3.2. Other stem cells
GE of other types of cells, such as EpSCs, MSCs, as well as muscle and neural stem cells, has also produced interesting results in the treatment of several diseases. However, except for EpSCs, the results have been insufficiently conclusive to test these strategies in clinical trials. The most important studies in the field are summarized below.
3.2.1. Epidermal stem cells
The genetic modification of EpSCs has important applications in the treatment of diseases such as recessive dystrophic epidermolysis bullosa (RDEB) and junctional epidermolysis bullosa (JEB). Major advances have been made using gamma‐retroviral and LV vectors to generate autologous artificial skin expressing Col7a1 for RDEB and LAMB3 for JEB; JEB can be genetically corrected by transplanting genetically modified EpSCs.75, 76 As discussed above, EPSC GE could be a good alternative to retroviral vectors, as several research groups have managed to restore Col7Aa148, 49, 50 and LAMB3 51 expression by genetically editing EpSCs from RDEB and JEB patients, respectively. In both these pathologies, artificial skin generated using genetically modified EpSCs has enabled long‐term engraftment of phenotypically normal skin. This provides strong support for future ex vivo GE clinical trials for the treatment of RDEB and JEB patients.
3.2.2. Mesenchymal stem cells
Due to their regenerative potential and anti‐inflammatory properties, MSCs have been widely used in clinical trials for multiple diseases. However, despite the major successes of MSCs in some disorders, which have led to the approval of MSC therapies,77, 78, 79 they have had limited therapeutic benefits in other applications. To overcome these limitations, several research groups are investigating the feasibility of genetically modifying MSCs to express different genes that enhance their therapeutic efficacy. 80 As previously described with respect to other cell types, GE has also become an alternative to GT vectors. GE‐MSCs, which are being studied as a platform for the delivery of proteins into the blood stream, 81 are an interesting tool in the treatment of blood disorders caused by the absence of plasma proteins. In this setting, GE has been used to insert hFIX and hFVIII into the AAVS1 locus of MSCs through homologous recombination (HR) in order to treat hemophilia A and B.82, 83 GE‐MSCS are also considered a potential alternative treatment for neurodegenerative diseases such as Parkinson's disease (PD). Using this approach, MSCs have been engineered to secrete soluble receptors of advanced glycation end products from AAVS1 loci. 47 The aim is to reduce advanced glycation end product concentrations involved in PD and Alzheimer's disease. GE‐MSCs have also been used in the treatment of ischemia in an animal infarct model. Meng et al. have used TALENs to integrate the IL‐10 gene into the AAVS1 safe harbor locus of MSCs and have performed an intra‐myocardial transplant in the infracted myocardium, which reduced pro‐inflammatory factor expression and increased vascular density.84, 85 The same approach was used to ameliorate liver fibrosis. 46 All these data indicate that GE is becoming a real alternative to viral and nonviral vectors in generating genetically modified MSCs.
3.2.3. Neural stem cells
Neural stem cells (NSCs), whose regenerative capacity, as with MSCs, can be improved by GE, have promising, although not immediate, clinical potential as a cellular treatment for neurological diseases. Recently, Dever et al. demonstrated that NSCs can be modified genetically at multiple loci using Cas9 mRNA and DNA donors. They showed that, upon transplantation, GE‐NSCs can migrate and differentiate into astrocytes, neurons and myelin‐producing oligodendrocytes. They also highlighted the therapeutic potential of GE‐NSCs by generating NSCs overexpressing the GALC enzyme which can cross‐correct the GALC enzyme activity of fibroblasts obtained from Krabbe disease patients. 52 These findings highlight the therapeutic potential of GE‐NSCs, not only for the regeneration of neural cells but also as a Trojan horse to deliver proteins to the central nervous system.
3.2.4. Muscle stem cells
Muscle stem cells are undifferentiated cells capable of producing new muscle tissue and of fusing with pre‐existing myofibers in order to repair damaged myofibers. Muscle satellite cells (MuSCs) are probably the most studied muscle stem cells, a population of cells that are capable of self‐renewal and differentiation into muscle fibers which represent an ideal target for therapeutic GE. 86 Zhu et al. have developed a fibrin gel culture system to selectively expand MuSCs from an mdx mouse model for Duchenne muscular dystrophy (DMD) research. These cells were successfully corrected using CRISP/Cas9‐based GE and, following transplantation to mdx mice, restored dystrophin expression in skeletal muscle. 53 This demonstrates the feasibility of using ex vivo GE‐MuSCs to target and correct DMD.
3.2.5. T stem cell memory cells
TSCM cells, which constitute a recently described 2% to 3% circulating T‐cell subpopulation, 87 have a naive T‐cell phenotype, express a CD62L memory marker, proliferate, self‐renew, and generate effector/memory T cells. TSCM cells have emerged as a highly interesting population for adoptive T‐cell therapies for cancer 88 and inherited immunodeficiency. 89 As with other ASCs, GE has also been applied to TSCM cells to enhance the potency of CAR T cells for the treatment of hematological malignancies and solid tumors. Eyquem et al. 90 inserted a α‐CD19 CAR into the T‐cell receptor A constant (TRAC) locus using Cas9 mRNA and gRNA electroporation followed by transduction with AAV6 harboring the donor DNA containing the CAR. This strategy generated off‐the‐shelf CAR‐T cells (without TCR) which expressed the CAR physiologically through the endogenous TCR promoter. This approach has also been used to improve efficiency thanks to the maintenance of the TSCM phenotype following repeated exposure to the antigen. Recently Sachdeva et al. used a similar approach, involving TALENs instead of CRISPR/Cas9, to insert CAR cDNAs into the TRAC locus and to insert the proinflammatory cytokine IL12 into the CD25 or PD1 loci. 91 This resulted in the physiological expression of the CAR and a transient secretion of IL12 which depends on tumor engagement (following the expression patterns of CD25 or PD1 locus). In addition, the targeted integration of IL12 at the PD1 locus inactivated PD1, a major T‐cell immune checkpoint, and increased the cell surface of CD62L, a marker of TSCM cells. This strategy resulted in increased CAR‐T cell cytotoxicity and extended survival of mice engrafted with solid tumors. A further potential improvement in generating universal off‐the‐shelf CAR‐T cells involves simultaneously deleting TCR and beta‐2 microglobulin (B2M) genes to reduce graft‐vs‐host disease (GVHD) and CAR‐T‐cell rejection. Recently, Choi et al. 92 generated αEGFRvIII CAR‐T cells lacking the expression of the TCR, B2M, and PD1 electroporating RNP (Cas9 protein and sgRNAs targeting TRAC, B2M, and PD1 loci) and transducing with AAV6 vectors containing donor DNAs for insertion of the EGFRvIII CAR construct at the different loci. The authors showed increased survival of mice in mouse models of glioma, which correlated with the increased presence of CAR‐T cells with a TSCM phenotype. 92
4. CONCLUSION
In recent decades, genetic modification of ASCs using traditional GT vectors has opened up new opportunities to improve ATMPs for the treatment of several diseases. Most approaches use retroviral vectors to achieve stable transgene expression in ASCs upon expansion and differentiation. However, the use of retroviral vectors has several drawbacks associated with oncogene activation and the lack of physiological transgene expression. Recent advances in GE technologies have enabled researchers to design next‐generation GM‐ATMPs based on ASCs. Using cellular HR‐ and NHEJ‐based repair pathways, GE achieves precise, and safe ASC genetic modifications such as gene disruption, addition and repair, as well as the generation of therapeutic mutations. Six ongoing phase I and II clinical trials are currently being carried out to study the safety and effectiveness of GE‐ASCs in the treatment of AIDS, SCD, and β‐thalassemia. The trials are based on GE‐HSPCs and NHEJ disruption of CCR5 for the treatment of AIDS and on the inactivation of regulatory regions controlling the fetal γ‐globin gene repression for the treatment of SCD and β‐thalassemia. GE‐HSPCs have also produced promising results in preclinical models for monogenetic diseases such as severe X‐SCID, SCID‐ADA, X‐CGD, and FA, as well as for cancer and transplantation. In addition to HSPCs, several other ASCs have been studied as GE targets in therapeutic applications. GE of EpSCs, MSCs, NPCs, and MuSCs has also produced interesting results in animal models of RDEB, JEB, hemophilia, PD, and Krabbe disease.
5. FUTURE PERSPECTIVES
It is still too early to speculate whether GE‐ASC clinical trials for the treatment of AIDS, SCD, and β‐thalassemia will lead to the approval of ATMPs for clinical use. For that to happen, these strategies need to demonstrate improved therapeutic effectiveness as compared to other GT approaches based on retroviral vectors already in phase III. Some of these GT techniques are about to be authorized as ATMPs. 7 For example, Lentiglobin, a HSPC‐LV‐based ATMP, has demonstrated excellent, long‐term therapeutic benefits in β‐thalassemia patients, with no severe secondary effects. Therefore, provided GE‐ASC clinical trials demonstrate a similar level of therapeutic efficiency and reduced genome alterations as compared to RV‐based GT, GE‐ASCs will be approved as ATMPs. Regardless of the results achieved in ongoing clinical trials, we believe that, in the near future, next‐generation ATMPs will incorporate GE‐ASCs in their arsenal. The field of GE is advancing at an unprecedented pace, with new, more efficient and safer tools being developed each year. Advances in the specificity and versatility of SENs,28, 93, 94, 95 in strategies to improve HR repair96, 97 and in delivery methods13, 16, 98, 99, 100 have been and will remain crucial. In addition, the field of ASCs is evolving exponentially due to their safety and potential applications in regenerative medicine.26, 101, 102, 103, 104, 105 New developments in both GE and ASCs are bound to provide opportunities to improve the safety and efficacy of GE‐ASCs in order to achieve the final objective: the approval by official medical authorities of GE‐ASCs as ATMPs.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
S.S.H., A.A.G., N.M.P., A.G.G., M.T.M, M.C.G., I.R.H.: manuscript writing and illustrations; P.G.M., C.H.: manuscript review; K.B., F.M.: manuscript writing, figure artwork, final approval of manuscript.
ACKNOWLEDGMENTS
This study was funded by the Spanish ISCIII Health Research Fund and the European Regional Development Fund (FEDER) through research grants PI12/01097 and PI18/00337 (F.M.), PI18/00330 (K.B.), and PI18/01610 (C.H.). The CECEyU and CSyF of the Junta de Andalucía FEDER/European Cohesion Fund (FSE) for Andalusia provided the following research grants: 2016000073391‐TRA, 2016000073332‐TRA, PI‐57069, and PAIDI‐Bio326 (F.M.) and PI‐0160/2012 and PI‐0014‐2016 (K.B.); K.B. also held a Nicolas Monardes regional Ministry of Health contract (0006/2018). M.T.‐M, A.F.‐A, and M.P.‐N were awarded FPU fellowships (FPU16/05467, FPU17/04327, and FPU1702268, respectively), whereas S.S.‐H was employed on a PEJ contract basis (PEJ‐2014‐A‐17105). Finally, we wish to thank Michael O'Shea for proofreading the manuscript.
Benabdellah K, Sánchez‐Hernández S, Aguilar‐González A, et al. Genome‐edited adult stem cells: Next‐generation advanced therapy medicinal products. STEM CELLS Transl Med. 2020;9:674–685. 10.1002/sctm.19-0338
Karim Benabdellah and Francisco Martin are senior authors.
Funding information European Regional Development Fund (FEDER), Grant/Award Numbers: PI18/01610, PI18/00330, PI18/00337, grants PI12/01097; Spanish ISCIII Health Research Fund
Contributor Information
Karim Benabdellah, Email: karim.benabdel@genyo.es.
Francisco Martin, Email: francisco.martin@genyo.es.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Naldini L. Ex vivo gene transfer and correction for cell‐based therapies. Nat Rev Genet. 2011;12(5):301‐315. [DOI] [PubMed] [Google Scholar]
- 2. Doss MX, Sachinidis A. Current challenges of iPSC‐based disease modeling and therapeutic implications. Cell. 2019;8(5):E403 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montel‐Hagen A, Crooks GM. From pluripotent stem cells to T cells. Exp Hematol. 2019;71:24‐31. [DOI] [PubMed] [Google Scholar]
- 4. Haake K, Ackermann M, Lachmann N. Concise review: towards the clinical translation of induced pluripotent stem cell‐derived blood cells‐ready for take‐off. Stem Cells Translational Medicine. 2019;8(4):332‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clevers H, Watt FM. Defining adult stem cells by function, not by phenotype. Annu Rev Biochem. 2018;87:1015‐1027. [DOI] [PubMed] [Google Scholar]
- 7. Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med. 2019;11(3):e9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montini E, Cesana D. Genotoxicity assay for gene therapy vectors in tumor prone Cdkn2a(−)/(−) mice. Methods Enzymol. 2012;507:171‐185. [DOI] [PubMed] [Google Scholar]
- 9. Ricciardi AS et al. Peptide nucleic acids as a tool for site‐specific gene editing. Molecules. 2018;23(3):632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gaj T, Epstein BE, Schaffer DV. Genome engineering using adeno‐associated virus: basic and clinical research applications. Mol Ther. 2016;24(3):458‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porteus MH. A new class of medicines through DNA editing. N Engl J Med. 2019;380(10):947‐959. [DOI] [PubMed] [Google Scholar]
- 12. Tsai SQ, Joung JK. Defining and improving the genome‐wide specificities of CRISPR‐Cas9 nucleases. Nat Rev Genet. 2016;17(5):300‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Exline CM, DeClercq JJ, et al. Homology‐driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33(12):1256‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin F, Sánchez‐Hernández S, Gutiérrez‐Guerrero A, Pinedo‐Gomez J, Benabdellah K. Biased and unbiased methods for the detection of off‐target cleavage by CRISPR/Cas9: an overview. Int J Mol Sci. 2016;17(9):E1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gutierrez‐Guerrero A, Sanchez‐Hernandez S, Galvani G, et al. Comparison of zinc finger nucleases versus CRISPR‐specific nucleases for genome editing of the Wiskott‐Aldrich syndrome locus. Hum Gene Ther. 2017;29(3):366‐380. [DOI] [PubMed] [Google Scholar]
- 16. Sanchez‐Hernandez S et al. The IS2 element improves transcription efficiency of integration‐deficient lentiviral vector episomes. Mol Ther Nucleic Acids. 2018;13:16‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi JG, Dang Y, Abraham S, et al. Lentivirus pre‐packed with Cas9 protein for safer gene editing. Gene Ther. 2016;23(7):627‐633. [DOI] [PubMed] [Google Scholar]
- 18. Gwiazda KS et al. High efficiency CRISPR/Cas9‐mediated gene editing in primary human T‐cells using mutant adenoviral E4orf6/E1b55k "helper" proteins. Mol Ther. 24(9):1570‐1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai Y, Bak RO, Mikkelsen JG. Targeted genome editing by lentiviral protein transduction of zinc‐finger and TAL‐effector nucleases. Elife. 2014;3:e01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Osman G, Rodriguez J, Chan SY, et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J Control Release. 2018;285:35‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Finn JD, Smith AR, Patel MC, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22(9):2227‐2235. [DOI] [PubMed] [Google Scholar]
- 22. Mastorakos P, da Silva AL, Chisholm J, et al. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc Natl Acad Sci USA. 2015;112(28):8720‐8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mangeot PE, Risson V, Fusil F, et al. Genome editing in primary cells and in vivo using viral‐derived Nanoblades loaded with Cas9‐sgRNA ribonucleoproteins. Nat Commun. 2019;10(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mangeot PE. Nanoblades: pseudoviral shuttles for CRISPR‐CAS9 delivery. Virologie. 2019;23(1):3‐6. [DOI] [PubMed] [Google Scholar]
- 25. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16(6):387‐399. [DOI] [PubMed] [Google Scholar]
- 26. Schiroli G et al. Precise gene editing preserves hematopoietic stem cell function following transient p53‐mediated DNA damage response. Cell Stem Cell. 2019;24(4):551‐565.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Genovese P, Schiroli G, Escobar G, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paschon DE, Lussier S, Wangzor T, et al. Diversifying the structure of zinc finger nucleases for high‐precision genome editing. Nat Commun. 2019;10(1):1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim HS, Jeong YK, Hur JK, Kim JS, Bae S. Adenine base editors catalyze cytosine conversions in human cells. Nat Biotechnol. 2019;37(10):1145‐1148. [DOI] [PubMed] [Google Scholar]
- 30. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature. 2016;533(7603):420‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anzalone AV et al. Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature. 2019;576(7785):149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schiroli G, Ferrari S, Conway A, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID‐X1. Sci Transl Med. 2017;9(411):eaan0820. [DOI] [PubMed] [Google Scholar]
- 33. Pavel‐Dinu M, Wiebking V, Dejene BT, et al. Gene correction for SCID‐X1 in long‐term hematopoietic stem cells. Nat Commun. 2019;10(1):1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 beta‐globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125(17):2597‐2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lux CT, Pattabhi S, Berger M, et al. TALEN‐mediated gene editing of HBG in human hematopoietic stem cells leads to therapeutic fetal hemoglobin induction. Mol Ther Methods Clin Dev. 2019;12:175‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang KH, Smith SE, Sullivan T, et al. Long‐term engraftment and fetal globin induction upon BCL11A gene editing in bone‐marrow‐derived CD34(+) hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev. 2017;4:137‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Psatha N, Reik A, Phelps S, et al. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with beta‐thalassemia major. Mol Ther Methods Clin Dev. 2018;10:313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Diez B, Genovese P, Roman‐Rodriguez FJ, et al. Therapeutic gene editing in CD34(+) hematopoietic progenitors from Fanconi anemia patients. EMBO Mol Med. 2017;9(11):1574‐1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim MY et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173(6):1439‐1453.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Humbert O, Laszlo GS, Sichel S, et al. Engineering resistance to CD33‐targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9‐deletion of CD33 exon 2. Leukemia. 2019;33(3):762‐808. [DOI] [PubMed] [Google Scholar]
- 42. Xu L, Yang H, Gao Y, et al. CRISPR/Cas9‐mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV‐1 resistance in vivo. Mol Ther. 2017;25(8):1782‐1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holt N, Wang J, Kim K, et al. Human hematopoietic stem/progenitor cells modified by zinc‐finger nucleases targeted to CCR5 control HIV‐1 in vivo. Nat Biotechnol. 2010;28(8):839‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li L, Krymskaya L, Wang J, et al. Genomic editing of the HIV‐1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol Ther. 2013;21(6):1259‐1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peterson CW, Wang J, Norman KK, et al. Long‐term multilineage engraftment of autologous genome‐edited hematopoietic stem cells in nonhuman primates. Blood. 2016;127(20):2416‐2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi JS, Jeong IS, Han JH, Cheon SH, Kim SW. IL‐10‐secreting human MSCs generated by TALEN gene editing ameliorate liver fibrosis through enhanced anti‐fibrotic activity. Biomater Sci. 2019;7(3):1078‐1087. [DOI] [PubMed] [Google Scholar]
- 47. Lee J, Bayarsaikhan D, Arivazhagan R, et al. CRISPR/Cas9 edited sRAGE‐MSCs protect neuronal death in Parkinson's disease model. Int J Stem Cells. 2019;12:114‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bonafont J, Mencía Á, García M, et al. Clinically relevant correction of Recessive Dystrophic Epidermolysis Bullosa by dual sgRNA CRISPR/Cas9‐mediated gene editing. Mol Ther. 2019;27(5):986‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hainzl S, Peking P, Kocher T, et al. COL7A1 editing via CRISPR/Cas9 in recessive dystrophic Epidermolysis Bullosa. Mol Ther. 2017;25(11):2573‐2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mencia A et al. Deletion of a pathogenic mutation‐containing exon of COL7A1 allows clonal gene editing correction of RDEB patient epidermal stem cells. Mol Ther Nucleic Acids. 2018;11:68‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benati D, Miselli F, Cocchiarella F, et al. CRISPR/Cas9‐mediated in situ correction of LAMB3 gene in keratinocytes derived from a Junctional Epidermolysis Bullosa patient. Mol Ther. 2018;26(11):2592‐2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dever DP, Scharenberg SG, Camarena J, et al. CRISPR/Cas9 genome engineering in engraftable human brain‐derived neural stem cells. iScience. 2019;15:524‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu P, Wu F, Mosenson J, Zhang H, He TC, Wu WS. CRISPR/Cas9‐mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol Ther Nucleic Acids. 2017;7:31‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mandal PK, Ferreira LMR, Collins R, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15(5):643‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu L, Wang J, Liu Y, et al. CRISPR‐edited stem cells in a patient with HIV and acute lymphocytic leukemia N Engl J Med. 2019;381:1240‐1247. [DOI] [PubMed] [Google Scholar]
- 56. Bjurstrom CF et al. Reactivating fetal hemoglobin expression in human adult erythroblasts through BCL11A knockdown using targeted endonucleases. Mol Ther Nucleic Acids. 2017;5:e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mettananda S, Fisher CA, Hay D, et al. Editing an alpha‐globin enhancer in primary human hematopoietic stem cells as a treatment for beta‐thalassemia. Nat Commun. 2017;8(1):424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ye L, Wang J, Tan Y, et al. Genome editing using CRISPR‐Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and beta‐thalassemia. Proc Natl Acad Sci USA. 2016;113(38):10661‐10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis R, Gurumurthy A, Hossain MA, Gunn EM, Bungert J. Engineering globin gene expression. Mol Ther Methods Clin Dev. 2019;12:102‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cavazzana M, Mavilio F. Gene therapy for hemoglobinopathies. Hum Gene Ther. 2018;29(10):1106‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rio P et al. Successful engraftment of gene‐corrected hematopoietic stem cells in non‐conditioned patients with Fanconi anemia. Nat Med. 2019;25(9):1396‐1401. [DOI] [PubMed] [Google Scholar]
- 62. Osborn MJ et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther. 2014;26(2):114‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Booth C, Gaspar HB, Thrasher AJ. Treating immunodeficiency through HSC gene therapy. Trends Mol Med. 2016;22(4):317‐327. [DOI] [PubMed] [Google Scholar]
- 64. Cavazza A, Cocchiarella F, Bartholomae C, et al. Self‐inactivating MLV vectors have a reduced genotoxic profile in human epidermal keratinocytes. Gene Ther. 2013;20(9):949‐957. [DOI] [PubMed] [Google Scholar]
- 65. Martin F et al. Lentiviral vectors transcriptionally targeted to hematopoietic cells by WASP gene proximal promoter sequences. Gene Ther. 2005;12(8):715‐723. [DOI] [PubMed] [Google Scholar]
- 66. Dupre L et al. Lentiviral vector‐mediated gene transfer in T cells from Wiskott‐Aldrich syndrome patients leads to functional correction. Mol Ther. 2004;10(5):903‐915. [DOI] [PubMed] [Google Scholar]
- 67. Kohn DB, Kuo CY. New frontiers in the therapy of primary immunodeficiency: from gene addition to gene editing. J Allergy Clin Immunol. 2017;139(3):726‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuo CY, Long JD, Campo‐Fernandez B, et al. Site‐specific gene editing of human hematopoietic stem cells for X‐linked hyper‐IgM syndrome. Cell Rep. 2018;23(9):2606‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. De Ravin SS et al. CRISPR‐Cas9 gene repair of hematopoietic stem cells from patients with X‐linked chronic granulomatous disease. Sci Transl Med. 2017;9(372):eaah3480. [DOI] [PubMed] [Google Scholar]
- 70. Torikai H, Reik A, Soldner F, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122(8):1341‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Torikai H, Mi T, Gragert L, et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci Rep. 2016;6:21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kenderian SS, Ruella M, Shestova O, et al. CD33‐specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Borot F, Wang H, Ma Y, et al. Gene‐edited stem cells enable CD33‐directed immune therapy for myeloid malignancies. Proc Natl Acad Sci USA. 2019;116(24):11978‐11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hirsch T, Rothoeft T, Teig N, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lwin SM et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(11):e126243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mastrolia I et al. Concise review: challenges in clinical development of Mesenchymal stromal/stem cells. Stem Cells Translational Medicine. 2019;8(11):1135‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cobo M et al. Mesenchymal stem cells expressing vasoactive intestinal peptide ameliorate symptoms in a model of chronic multiple sclerosis. Cell Transplant. 2012;22(5):839‐854. [DOI] [PubMed] [Google Scholar]
- 79. Martin F et al. Stable genetic modification of mesenchymal stromal cells using lentiviral vectors. Methods Mol Biol. 1937;2019:267‐280. [DOI] [PubMed] [Google Scholar]
- 80. Wei W, Huang Y, Li D, Gou HF, Wang W. Improved therapeutic potential of MSCs by genetic modification. Gene Ther. 2018;25(8):538‐547. [DOI] [PubMed] [Google Scholar]
- 81. Benabdallah BF, Allard E, Yao S, et al. Targeted gene addition to human mesenchymal stromal cells as a cell‐based plasma‐soluble protein delivery platform. Cytotherapy. 2010;12(3):394‐399. [DOI] [PubMed] [Google Scholar]
- 82. Li SJ, Luo Y, Zhang LM, Yang W, Zhang GG. Targeted introduction and effective expression of hFIX at the AAVS1 locus in mesenchymal stem cells. Mol Med Rep. 2017;15(3):1313‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lee SS, Sivalingam J, Nirmal AJ, et al. Durable engraftment of genetically modified FVIII‐secreting autologous bone marrow stromal cells in the intramedullary microenvironment. J Cell Mol Med. 2018;22(7):3698‐3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Meng D et al. Interleukin 10‐secreting MSCs via TALEN‐mediated gene editing attenuates left ventricular remodeling after myocardial infarction. Cell Physiol Biochem. 2019;52(4):728‐741. [DOI] [PubMed] [Google Scholar]
- 85. Cho HM, Kim PH, Chang HK, et al. Targeted genome engineering to control VEGF expression in human umbilical cord blood‐derived mesenchymal stem cells: potential implications for the treatment of myocardial infarction. Stem Cells Translational Medicine. 2017;6:1040‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schmidt M, Schüler SC, Hüttner SS, von Eyss B, von Maltzahn J. Adult stem cells at work: regenerating skeletal muscle. Cell Mol Life Sci. 2019;76:2559‐2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lugli E et al. Stem, effector, and hybrid states of memory CD8(+) T cells. Trends Immunol. 2020;41:17‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Biasco L et al. In vivo tracking of T cells in humans unveils decade‐long survival and activity of genetically modified T memory stem cells. Sci Transl Med. 2015;7(273):273ra13. [DOI] [PubMed] [Google Scholar]
- 90. Eyquem J, Mansilla‐Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sachdeva M, Busser BW, Temburni S, et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10(1):5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Choi BD, Yu X, Castano AP, et al. CRISPR‐Cas9 disruption of PD‐1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019;7(1):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol. 2019;37(6):657‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hu JH, Miller SM, Geurts MH, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Iyer S, Suresh S, Guo D, et al. Precise therapeutic gene correction by a simple nuclease‐induced double‐stranded break. Nature. 2019;568(7753):561‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tang XD, Gao F, Liu MJ, Fan QL, Chen DK, Ma WT. Methods for enhancing clustered regularly interspaced short palindromic repeats/Cas9‐mediated homology‐directed repair efficiency. Front Genet. 2019;10:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chu VT, Weber T, Wefers B, et al. Increasing the efficiency of homology‐directed repair for CRISPR‐Cas9‐induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33(5):543‐548. [DOI] [PubMed] [Google Scholar]
- 98. Zuris JA, Thompson DB, Shu Y, et al. Cationic lipid‐mediated delivery of proteins enables efficient protein‐based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang Y, Wang Y, Chang T, Huang H, Yee JK. Integration‐defective lentiviral vector mediates efficient gene editing through homology‐directed repair in human embryonic stem cells. Nucleic Acids Res. 2017;45(5):e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lattanzi A, Meneghini V, Pavani G, et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol Ther. 2019;27(1):137‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chan CKF et al. Identification of the human skeletal stem cell. Cell. 2018;175(1):43‐56.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Baser A, Skabkin M, Kleber S, et al. Onset of differentiation is post‐transcriptionally controlled in adult neural stem cells. Nature. 2019;566(7742):100‐104. [DOI] [PubMed] [Google Scholar]
- 103. Munoz‐Canoves P, Huch M. Definitions for adult stem cells debated. Nature. 2018;563(7731):328‐329. [DOI] [PubMed] [Google Scholar]
- 104. Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553(7689):418‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Leberfinger AN et al. Concise review: bioprinting of stem cells for transplantable tissue fabrication. Stem Cells Translational Medicine. 2017;6(10):1940‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.