

Abstract
SCN8A is a novel causal gene for early infantile epileptic encephalopathy. It is well accepted that gain-of-function mutations in SCN8A underlie the disorder, but the remarkable heterogeneity of its clinical presentation and poor treatment response demand for better understanding of the disease mechanisms. Here, we characterize a new epilepsy-related SCN8A mutation, R850Q, in human Nav1.6. We show that it is a gain-of-function mutation, with a hyperpolarizing shift in voltage dependence of activation, a 2-fold increase of persistent current and a slowed decay of resurgent current. We systematically compare its biophysics with three other SCN8A epilepsy mutations, T767I, R1617Q and R1872Q, in the human Nav1.6 channel. Although all of these mutations are gain-of-function, the mutations affect different aspects of channel properties. One commonality we discovered is an alteration of resurgent current kinetics, but the mechanisms by which resurgent currents are augmented is not yet clear for all of the mutations. Computational simulations predict increased excitability of neurons carrying these mutations with differential enhancement by open channel block.
Keywords: epilepsy, SCN8A, resurgent current, computational model
1. Introduction
Early infantile epileptic encephalopathy (EIEE) is a heterogeneous group of disorders characterized by early onset epileptic seizures and developmental delays. The majority of EIEE patients are resistant to seizure control treatments. Important causes for EIEEs include mutations in voltage-gated sodium channel (Nav) genes SCN1A, SCN2A and SCN8A. Among them, SCN8A is a relatively novel EIEE gene with the first case reported in 2012 (Veeramah et al., 2012). The number of SCN8A mutations and clinical cases have been growing rapidly with the advances of whole genome / exome sequencing as well as the inclusion of SCN8A in the diagnostic panels for infantile epileptic encephalopathy (Meisler et al., 2016). These patients have seizure onset within 18 months of life (5 months on average), after which development delay or regression is typical. Moderate to severe intellectual disability is common and motor deficits are found in 50% of affected individuals. 10% of the published cases reported sudden unexpected death in epilepsy (SUDEP) (Meisler et al., 2016).
SCN8A encodes Nav1.6, one of the brain isoforms of voltage-gated sodium channel in human, along with Nav1.1 (SCN1A), Nav1.2 (SCN2A) and Nav1.3 (SCN3A). Nav1.6 is ubiquitously expressed in almost all neurons throughout the central nervous system (CNS) and the peripheral nervous system (PNS), contrasting with the region- and cell type-restricted distribution of other Navs. Developmentally, Nav1.6 replaces Nav1.2 as the major isoform at the axon initial segment (AIS) and the nodes of Ranvier in myelinated excitatory neurons (Boiko et al., 2001; Boiko et al., 2003; Van Wart & Matthews, 2006a), where it plays a dominating role in action potential initiation and propagation (Hu et al., 2009). Nav1.6 is also found at the AIS of some inhibitory interneurons (Ogiwara et al., 2007; Lorincz & Nusser, 2008; Makinson et al., 2017), although Nav1.1 is considered to be the primary driving force for action potential initiation, and thus for excitability of most inhibitory neurons. The functional significance of Nav1.6 in inhibitory interneurons is largely unclear. Although some evidence indicates a role for Nav1.6 in establishing synaptic inhibition in the thalamic network (Makinson et al., 2017), conditional expression of the R1872W mutation in mouse inhibitory neurons did not induce seizures but expression in excitatory neurons did (Bunton-Stasyshyn et al., 2019).
Nav1.6 is unique in its biophysical properties. It activates at more hyperpolarized voltages than Nav1.1 (Spampanato et al., 2001) and Nav1.2 (Rush et al., 2005), allowing a lower threshold for action potential generation. It can conduct higher levels of persistent current (Smith et al., 1998; Maurice et al., 2001) and resurgent current (Raman et al., 1997; Jarecki et al., 2010b). Persistent currents often reflect incomplete inactivation. While incomplete inactivation can lead to enhanced resurgent currents, resurgent currents also reflect the activity of an open channel blocker. Substantial evidence indicates that in many neurons an open channel blocker can mimic the normal inactivation process, but if the open channel blocker unbinds during repolarization sodium flux through the channel can resurge, leading to increased action potential firing (Raman & Bean, 2001; Khaliq et al., 2003). The intracellular segment of the Navβ4 subunit has been implicated in resurgent current induction by behaving similar to an open channel blocker and Nav1.6 has been identified as a predominant generator of resurgent currents in neurons (Grieco et al., 2005; Bant & Raman, 2010; Jarecki et al., 2010a; Barbosa et al., 2015; Patel et al., 2015; Xie et al., 2016). Moreover, Nav1.6 is more resistant to use-dependent inactivation during high-frequency firing compared to Nav1.1 and Nav1.2 (Spampanato et al., 2001; Rush et al., 2005; Patel et al., 2016). The combination of these features makes Nav1.6 a favorable isoform to support high frequency, repetitive firing in a wide range of neurons in CNS and PNS (Maurice et al., 2001; Khaliq et al., 2003; Do & Bean, 2004; Van Wart & Matthews, 2006b; Enomoto et al., 2007; Mercer et al., 2007; Royeck et al., 2008).
More than 150 epilepsy-related mutations have been identified in SCN8A (Meisler et al., 2016). Most of these are de novo missense mutations. To date, only about 20 mutations have been functionally characterized. 15 of them are gain-of-function, highlighting Nav1.6 as a potent contributor to neural excitation. Indeed, SCN8A expression was increased in chemical and electrical induced seizure models (Blumenfeld et al., 2009; Hargus et al., 2013; Sun et al., 2013), while reducing SCN8A activity conferred seizure protection (Martin et al., 2007; Makinson et al., 2014; Wong et al., 2018). Importantly, the seizure phenotypes presented in SCN1A epilepsy mouse models can be rescued by an additional introduction of loss-of-function mutations in SCN8A, presumably by tuning down excitation in the inhibition-defected network (Martin et al., 2007; Hawkins et al., 2011). Moreover, the SCN8A-N1768D knockin mouse, carrying the gain-of-function epilepsy mutation, recapitulated the main features in the human disease (Wagnon et al., 2014).
The molecular mechanisms and phenotypic outcomes of SCN8A epilepsy mutations are highly heterogeneous, in part due to the various location of these mutations in the channel. Voltage-gated sodium channels consist of four domains (DI-DIV), with each containing six transmembrane segments (S1-S6) (Fig.1). S1-S4 are the voltage sensors, while the S5-S6 regions form the ion conducting pore. These highly conserved regions are critical for normal channel functions, and thus are the frequent targets of disease mutations.
Figure 1.
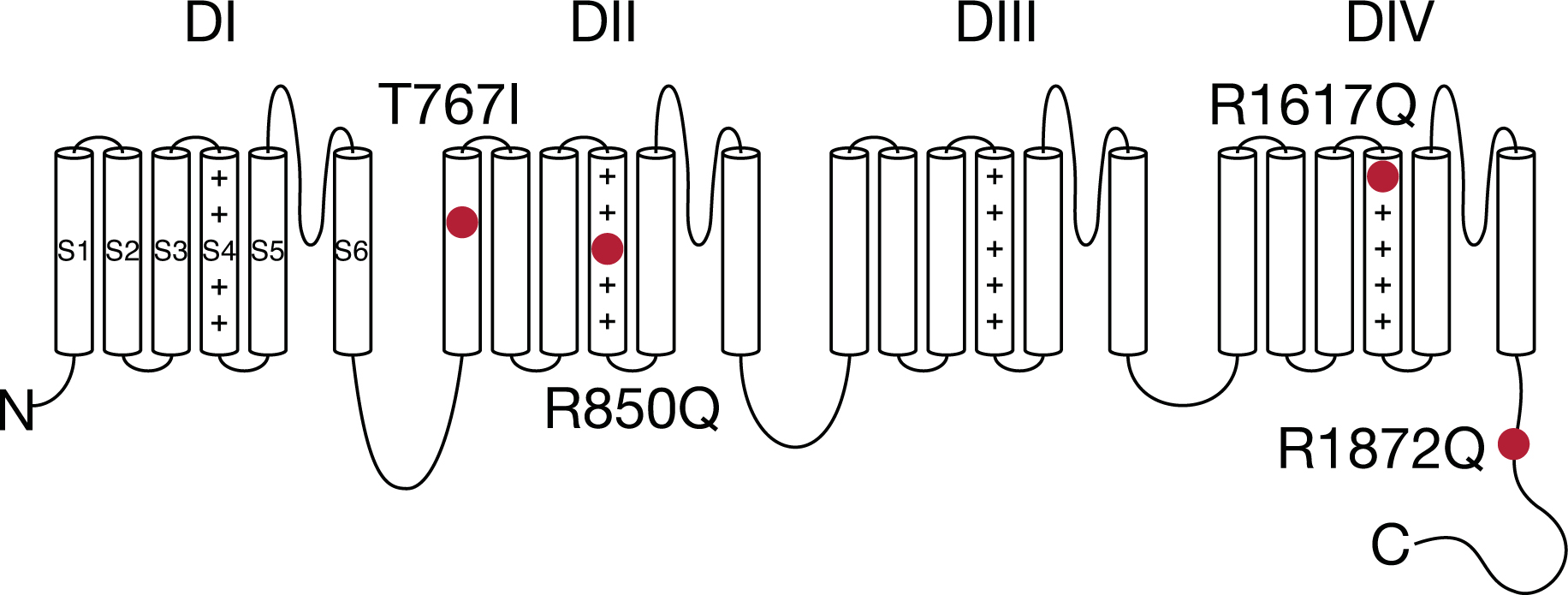
Topology of voltage-gated sodium channel with SCN8A epilepsy mutations.
In this study, we aimed to characterize a novel SCN8A epilepsy mutation R850Q and to identify potential commonalities among different SCN8A epilepsy mutations in the human Nav1.6 (hNav1.6) channel. We systematically compared R850Q to three other SCN8A epilepsy mutants in hNav1.6 and discovered that although all of them were gain-of-function, they affected various aspects of channel functions. Interestingly, all the evaluated mutant channels presented distinct kinetics of resurgent currents compared to the WT channel. Using computational simulations, we demonstrated increased excitability of both modeled Purkinje neurons and modeled cortical pyramidal neurons harboring these mutant channels.
2. Material and methods
2.1. SCN8A channel constructs
Our optimized human cDNA construct for wild-type (WT) Nav1.6 was designed in-house (Patel et al., 2016), encoding for the amino acid sequence corresponding to the accession number NP_055006.1 in the NCBI database. A Y371S amino acid substitution was introduced into the construct, rendering the channel resistant to tetrodotoxin (TTX) block. Mutations R850Q, T767I, R1617Q and R1872Q were introduced into the WT construct using QuikChange® II XL site-directed mutagenesis kit from Agilent Technologies according to the manufacturer’s instructions. Mutant channel constructs were fully sequenced (ACGT, Inc.) to confirm the presence of the correct mutation and the absence of additional mutations.
2.2. Cell culture and transfection
The neuronal cell line ND7/23 was used to express WT and mutant hNav1.6 channels for functional characterization. The cells were grown under standard tissue culture conditions and transiently transfected using Invitrogen Lipofectamine 2000 Transfection Reagent according to the manufacturer’s instructions. Briefly, lipid-DNA mixture [5μg channel construct and 0.5μg enhanced green fluorescent protein (EGFP) construct] in Opti-MEM medium was added to cells for 4h, after which transfected cells were split onto 35mm dish with fresh medium. Transfected cells were incubated at 30°C overnight to increase channel surface expression. 24–32h after transfection, whole-cell voltage-clamp recordings were performed. Transfected cells were identified by EGFP expression under a fluorescent microscope.
2.3. Whole-cell voltage-clamp recordings
All recordings were obtained at room temperature (~22°C) using a HEKA EPC-10 amplifier and the PatchMaster program (v2×73.2, HEKA Electronic). Electrodes were fabricated from 1.7mm capillary glass and fire-polished to a resistance of 0.8–1.0 MΩ using a Sutter P-1000 Micropipette puller (Sutter Instrument Company). The series of recording protocols was started 3 min after break-in for each cell, which controlled for time-dependent shifts in channel properties. For resurgent current recording, the protocol was started 5 min after break-in to allow sufficient time for Navβ4 peptide diffusion. Cells were not considered for analysis if the initial seal resistance was < 1 GΩ or if they had a series resistance > 3 MΩ. Voltage errors were minimized using 80% series resistance compensation and passive leak currents were cancelled by subtraction. The extracellular solution contained (in mM): 140 NaCl, 20 TEA-Cl, 3 KCl, 1 MgCl2, 1 CaCl2, and 10 HEPES, adjusted to a pH of 7.30 with NaOH. 500nM TTX was added to the extracellular solution to block endogenous sodium currents and isolate TTX-resistant current generated by transfected channels. The intracellular solution contained (in mM): 140 CsF, 10 NaCl, 1.1 EGTA, and 10 HEPES, adjusted to a pH of 7.30 with CsOH. Osmolarity of all solutions was adjusted to 300 mOsm. To induce resurgent currents in ND7/23 cells, 200μM Navβ4 peptide (KKLITFILKKTREK-OH) (Biopeptide Co), a peptide that corresponds to part of the C-terminal tail of the full-length Navβ4 subunit, was included in the intracellular solution.
2.4. Voltage protocols and data analysis
2.4.1. Activation protocol.
Transient sodium current (INaT) was measured during a 50ms depolarizing step (–80mV to +45mV; 5mV increment) from a holding potential of –120mV (Fig.10D upper). The current density was calculated by dividing the measured INaT by the capacitance of the cell. Persistent sodium current (INaP) was measured at the last 1ms during the depolarizing steps and presented as a percentage normalized to the maximal INaT of each cell. Sodium current conductance (GNa) was converted from INaT using the equation
where Vrev is the reversal potential of Na+ obtained in FitMaster (v2×73.5, HEKA Electronic) for each cell. Activation curves were generated by plotting normalized GNa against depolarizing potentials and fitting it with the Boltzmann function in the form of
where Gmax is the maximal GNa, V50,act is the potential at which activation is half-maximal, V is the depolarizing potential, and kact is the slope factor. Rate of decay (τNaT) for INaT was obtained in FitMaster by fitting the current traces 1ms into the depolarizing steps with a single exponential function.
Figure 10. R1872Q has altered pattern of persistent current and enhanced resurgent currents.
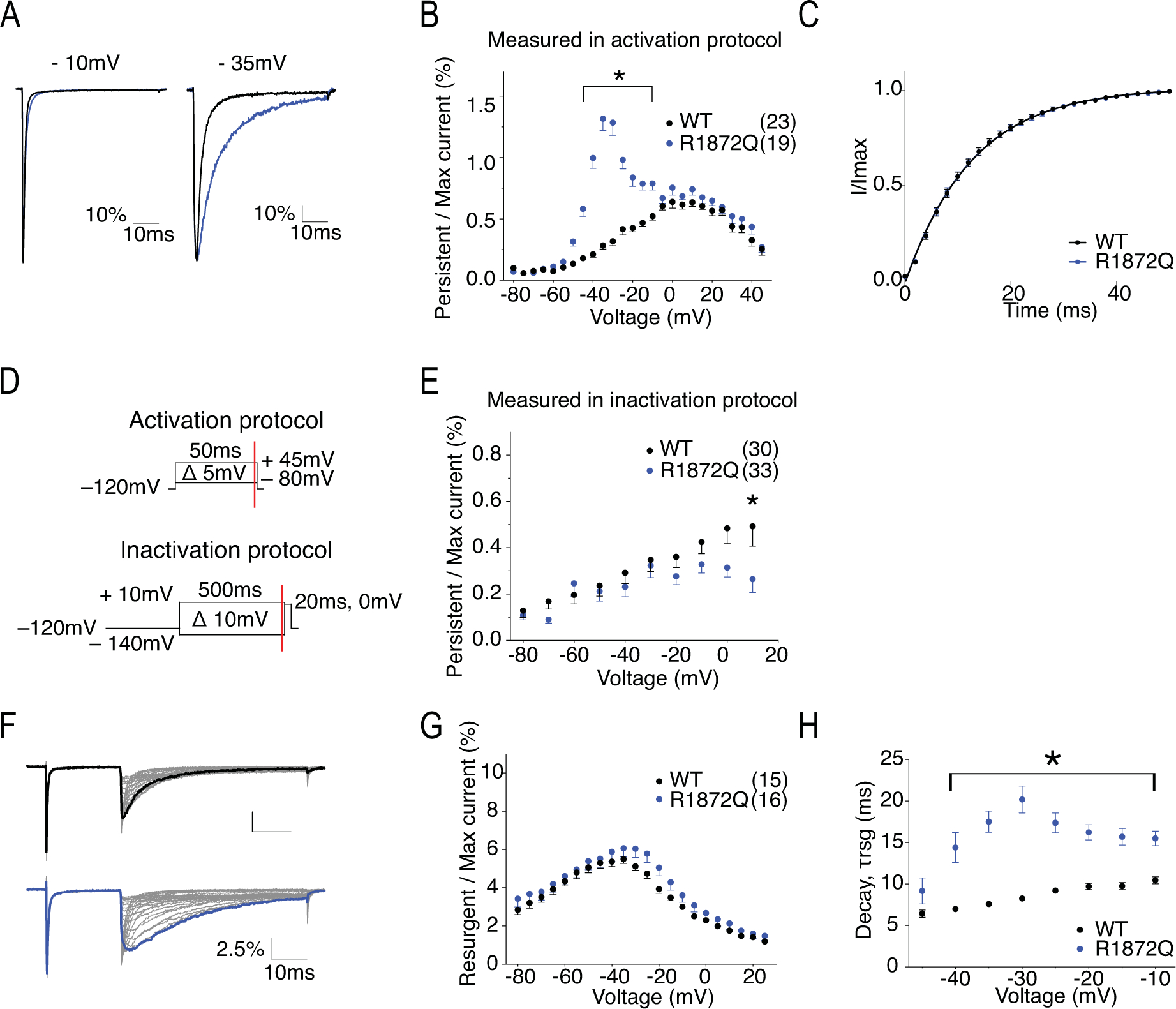
A, representative traces showing persistent current from WT (black) and R1872Q (blue) channels elicited at 0mV (left) and −35mV (right). Traces shown are presented as the percentage amplitude normalized to the maximum peak current during the 50ms activation protocol. B, per cent persistent current-voltage plot with persistent current measured during the last 1ms of the 50ms activation protocol. P < 0.001 at −45mV and −10mV in two-way ANOVA with Sidak multiple comparison test. C, recovery from inactivation with recovery time duration from 0 to 50ms. D, persistent current measurement at 50ms (red line) of the test pulse in the activation protocol (upper) and at 500ms (red line) of the prepulse of the steady-state inactivation protocol. E, per cent persistent current-voltage plot with persistent current measured during the last 1ms of the 500ms prepulse in steady-state inactivation protocol. P < 0.05 at 10mV in two-way ANOVA with Sidak multiple comparison test. F, representative traces showing resurgent current from WT (top) and R1872Q (bottom) channels. G, per cent resurgent current plotted against repolarization voltage. H, Rate of decay for resurgent current (τrsg) at each repolarization voltages. P < 0.0001 from −40mV to −20mV, P < 0.001 at −15mV, P < 0.01 at −10mV in two-way ANOVA with Sidak multiple comparison test. All values and statistical significance are reported in Table1.
2.4.2. Steady-state inactivation.
Availability of sodium channels was measured by the peak sodium current during a 20ms test pulse at 0mV following a 500ms prepulse (–140mV to +10mV; 10mV increment) that allows channels to enter equilibrium states (Fig.10D lower). Steady-state inactivation curves were generated by plotting normalized sodium current against prepulse potentials and fitting it with the Boltzmann function in the form of
where Imax is the maximal sodium current obtained in this protocol, V50,inact is the potential at which half of the sodium channels are available for activation, V is the prepulse potential, and kinact is the slope factor.
2.4.3. Recovery from inactivation.
A 20ms depolarization prepulse at 0mV was applied to allow channel activation and subsequent inactivation, which was followed by a repolarizing step to –80mV for durations ranging from 0ms to 50ms with 2ms increment. The non-inactivated sodium currents were measured during a subsequent 20ms test pulse at 0mV and normalized to the maximum current obtained in this protocol. The normalized non-inactivated sodium current was plotted against the duration of repolarizing step and fitted with a single exponential function.
2.4.4. Resurgent current.
Resurgent currents (INaR) were recorded with 200μM Navβ4 peptide in the intracellular solution and elicited by a protocol (Fig.4D top) with a step depolarization from –120 mV to +60 mV for 20 ms to open channels, allowing them to undergo open-channel block and subsequently repolarizing to a series of potentials (+25mV to –80mV; 5mV decrement) for 50 ms to allow the blocker to unbind, generating the resurgent current. INaR was presented as a percentage normalized to the maximal INaT obtained from the activation protocol and plotted against the repolarization potentials. Rate of decay (τrsg) for resurgent current was obtained in FitMaster by fitting the decay phase of the resurgent current traces, starting at 3ms (or 6.5ms for R1617Q due to its extremely slow resurgent current kinetics) in the repolarizing steps of the resurgent current protocol with a single exponential function.
Figure 4. R850Q has increased persistent current, increased resurgent current, and altered resurgent current kinetics.
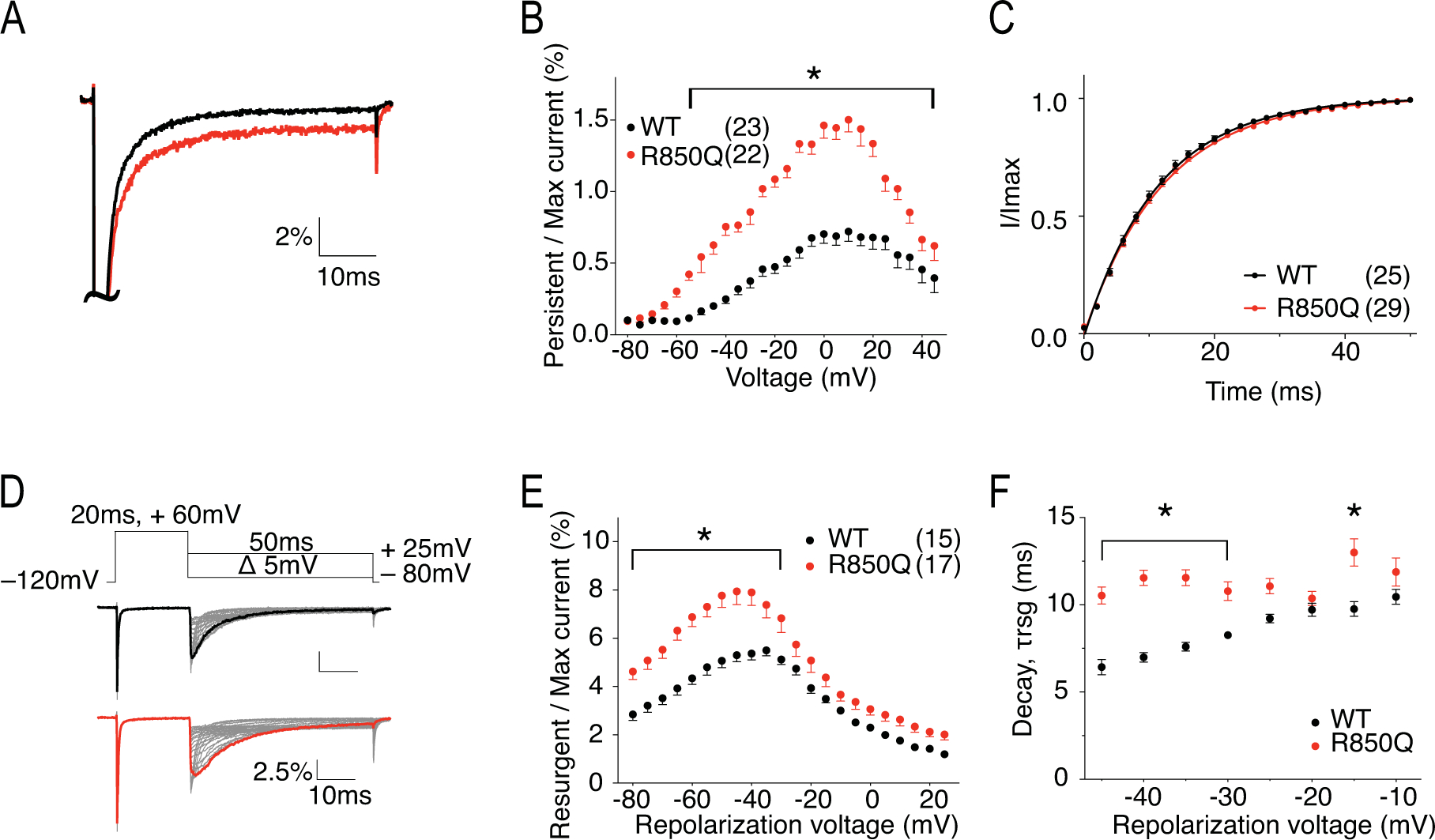
A, representative traces showing persistent current from WT (black) and R850Q (red) channels. Traces shown are elicited at 0mV and are presented as the percentage amplitude normalized to the maximal transient current during the 50ms activation protocol. B, per cent persistent current-voltage plot with persistent current measured during the last 1ms of the 50ms activation protocol. P < 0.01 from −55mV to 45mV in two-way ANOVA with Sidak multiple comparison test. C, recovery from inactivation with recovery duration from 0 to 50ms. D, resurgent current protocol and representative traces showing resurgent current from WT (upper) and R850Q (lower) channels. E, per cent resurgent current plotted against repolarization voltages. P < 0.01 from −80mV to −30mV in two-way ANOVA with Sidak multiple comparison test. F, Rate of decay for resurgent current (τrsg) at each repolarization voltages. P < 0.0001 from −45mV to −35mV and at −15mV in two-way ANOVA with Sidak multiple comparison test. All values and statistical significance are reported in Table1.
2.5. Simulations
Computational modeling of sodium currents and simulation of action potential firing were conducted in NEURON v7.5 (Hines & Carnevale, 1997). The sodium current model was based on a Markov state model described in detail previously (Khaliq et al., 2003) with minor modifications to partially segregate the coupling of voltage dependence of activation and inactivation (Fig.2). To model WT Nav1.6 current, the kinetic parameters remained identical to the original model, except for Oon, the rate constant for the O→I6 transition (INaT decay), was modified from 0.75 to 1.132 to match our experimental result. Mutant Nav1.6 currents were modeled as changes relative to this baseline. Quantitative analysis and curve fitting were performed to reconcile experimental and modelled data. ε, the rate constant for O→OB was not altered for experiments with open channel block (resurgent current) implemented, while it was set to 0 otherwise. All modified parameters were reported in Table 2.
Figure 2. Simple Markov state model of the sodium channel.
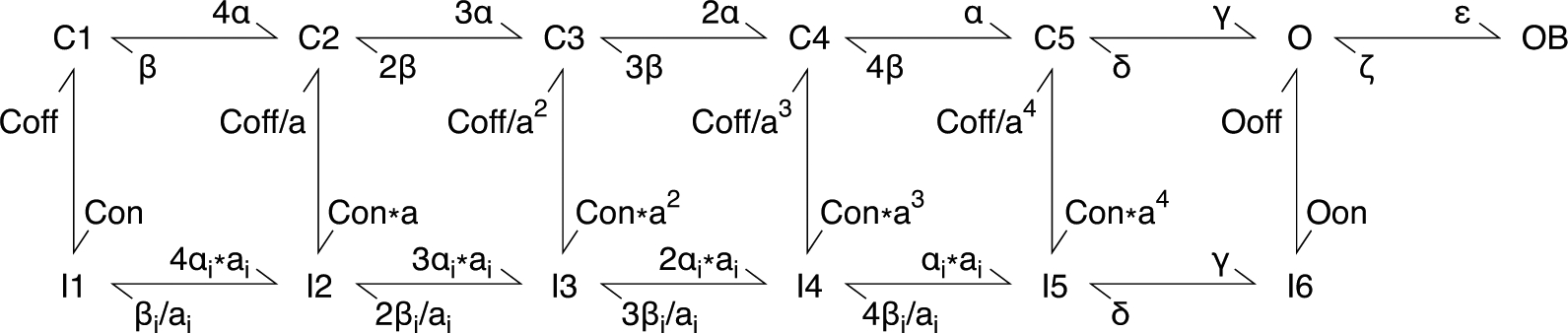
Modified based on the model described by Khaliq et al. (2003). States of the channel include closed (C), inactivated (I), open (O), and open-blocked (OB).
Table 2.
Parameters of modeled sodium channels
| WT | R850Q | T767I | R1617Q | R1872Q | |
|---|---|---|---|---|---|
| α | 150 | 85 | 700 | 150 | 330 |
| β | 3 | 0.9 | 5 | 3 | 4 |
| αi | 150 | 126 | 110 | 650 | 80 |
| βi | 3 | 3.4 | 6 | 3 | 6 |
| γ | 150 | 137 | 94 | 111 | 103 |
| Coff | 0.5 | 0.4 | 0.34 | 5 | 0.35 |
| Oon | 1.132 | 1.018 | 1.213 | 0.2866 | 0.656 |
| Ooff | 0.005 | 0.018 | 0.018 | 0.005 | 0.005 |
Simulations of neuronal activities were performed in a single compartment model of a Purkinje neuron (Khaliq et al., 2003) and a multi-compartment model of a cortical pyramidal neuron (Ben-Shalom et al., 2017). To simulate action potential firing of the Purkinje neuron, sodium conductance in the original model (Khaliq et al., 2003) was replaced by our modeled Nav1.6 current. For the simulation in the cortical pyramidal neuron model (Ben-Shalom et al., 2017), the sodium conductance that represents Nav1.6 current was replaced by our modeled Nav1.6 current. To simulate heterozygous mutant conditions in both models, 50% of the modelled WT Nav1.6 conductance was replaced by the modelled mutant Nav1.6 conductance. Neuronal morphology, channel distributions and kinetics of other channels remained the same as the original models.
2.6. Statistics
GraphPad Prism (v 6.00, GraphPad Software) was used for statistical analysis and curve fitting. The ROUT method in GraphPad Prism was used to identify outliers, which were then excluded from plotting and analysis. D’Agostino & Pearson omnibus normality test was used to confirm normal distribution of the data. Nonlinear least-squares minimization method was used for curve fitting. All data are presented as mean ± standard error of mean (SEM) of the indicated number of cells (n). Electrophysiological data for the WT group was collected along with each mutant group to eliminate potential differences in cell batch and in transfection efficiency. We found some significant difference of INaT current density, but not any other channel properties, between the various WT groups from different batches. Therefore, for comparisons of INaT current density, data from different WT batches was not pooled for comparison to mutants, and thus was presented with different number of cells (n). For comparisons of INaT current density, INaP percentage, INaR percentage, rate of decay for INaT, and rate of decay for INaR, two-way ANOVA with Sidak’s multiple comparisons test was performed between WT and mutants. For comparisons of V50,act, kact, V50,inact, kinact, and recovery rate, Student’s t test was performed on the parameters extrapolated from curve fitting.
3. Results
3.1. R850Q
R850 is the third charged residue in the voltage-sensing fourth transmembrane segment (S4) in the second domain (DII) of Nav1.6 (Fig.1). Substitution of this Arg with an uncharged Gln is predicted to impair channel activation and can potentially generate gating pore current. Fig.3A shows the representative traces of transient current from hNav1.6 WT and R850Q. The peak current density (Fig.3B) and rate of decay for the transient current (Fig.3C) were not significantly changed by the R850Q mutation (Table 1). However, R850Q exhibited altered channel activation as predicted, with a midpoint voltage 4.5mV more negative than the WT channel (V50 = −25.61 ± 0.67 mV in R850Q vs V50 = −21.07 ± 0.79 mV in WT, P < 0.0001) (Fig.3D). This shift indicated premature opening of the mutant channel, while the voltage-dependence of steady-state inactivation remained unchanged (Fig.3E) (Table 1). Collectively, this resulted in larger window currents in the mutant channel as demonstrated in Fig.3F. Fig.4A shows the normalized current traces of WT and R850Q, revealing an increased percentage of persistent current from the mutant channel (1.33 ± 0.06 % in R850Q vs. 0.52 ± 0.03 % in WT, P < 0.0001) (Fig.4B). Compared to WT, R850Q did not have a significant change in its rate of recovery from inactivation (Fig.4C) (Table 1). However, the resurgent current amplitude of R850Q increased (7.40 ± 0.53 % in R850Q vs. 5.50 ± 0.23 % in WT, P < 0.0001) (Fig.4E), and the decay of the resurgent current was slower compared to the WT channel (at −35mV repolarization step, 11.35 ± 0.44 ms in R850Q vs. 7.25 ± 0.09 ms in WT, P < 0.0001) (Fig.4F) (Table 1).
Figure 3. R850Q displayed altered voltage-dependence of activation.
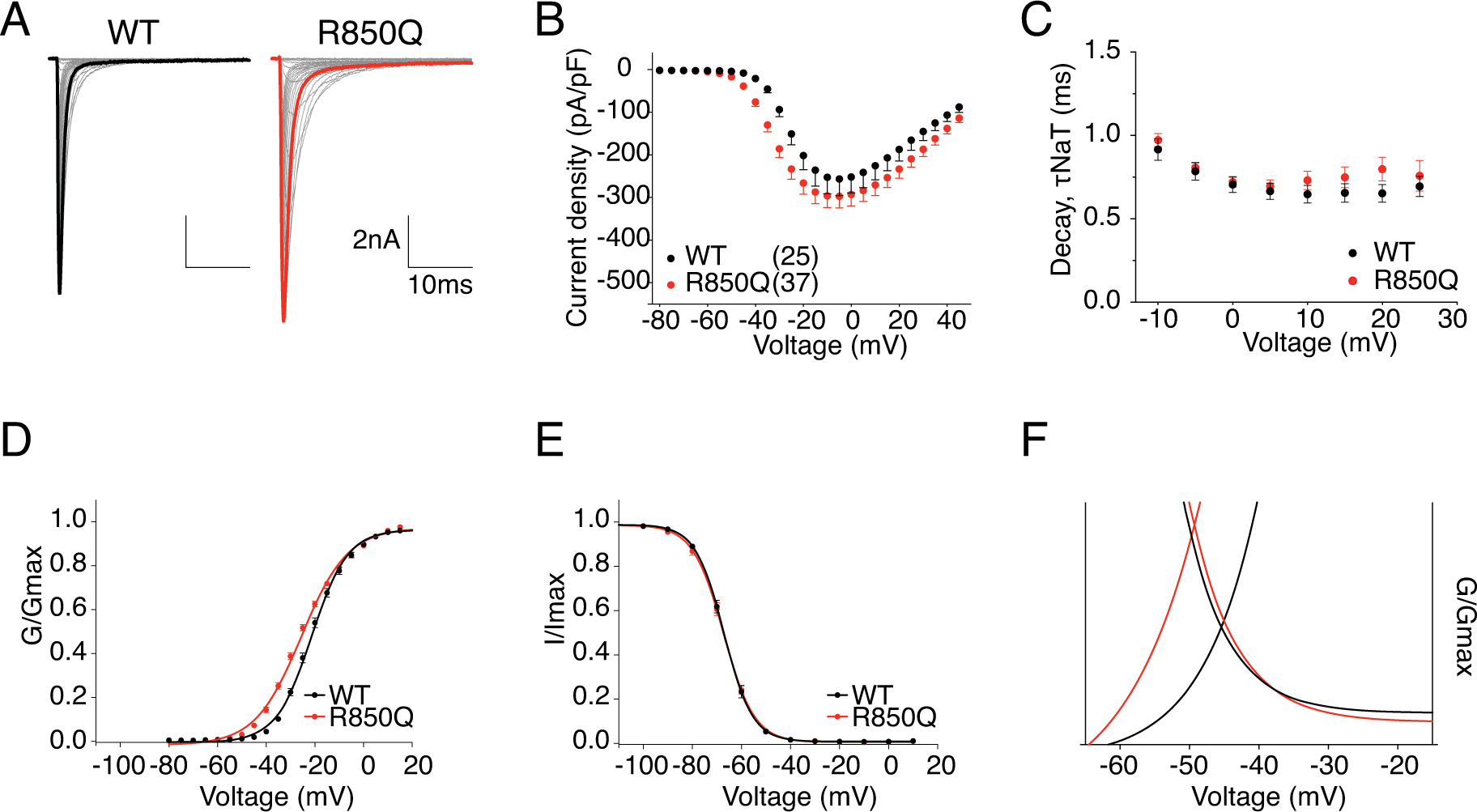
A, representative current trace families from hNav1.6 WT (left) and R850Q (right) channels elicited by an activation protocol. Maximum current traces are highlighted with bolded black and red respectively. B, Current density-voltage plot of hNav1.6 WT and R850Q. C, Rate of decay for transient sodium current (τNaT) at each depolarization voltages. D,E, activation and steady-state inactivation curves fitted with Boltzmann functions. F, window currents shown by Boltzmann fitted curves enlarged from DE combined. All values and statistical significance are reported in Table1.
Table 1.
Biophysical properties of WT and mutant SCN8A channel
| WT | n | R850Q | n | T767I | n | R1617Q | n | R1872Q | n | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximal transient I | (pA/pF) | −258.0 ± 38.81 | 25 | −302.1 ± 27.95 | 37 | ||||||
| −756.8 ± 147.2 | 10 | −552.7 ± 68.49 | 20 | ||||||||
| −351.0 ± 78.56 | 12 | −333.2 ± 58.85 | 15 | ||||||||
| −498.1 ± 95.49 | 21 | −861.00 ± 97.40* | 25 | ||||||||
| Transient I decay (−10mV) | (ms) | 0.89 ± 0.03 | 18 | 0.97 ± 0.04 | 28 | 0.80 ± 0.02* | 19 | 3.49 ± 0.15**** | 14 | 1.52 ± 0.06**** | 24 |
| Activation | V1/2 (mV) | −21.07 ± 0.79 | 21 | −25.61 ± 0.67**** | 31 | −31.68 ± 0.81**** | 21 | −21.25 ± 0.73 | 14 | −25.5 ± 0.83*** | 27 |
| K | 6.89 ± 0.20 | 9.26 ± 0.13**** | 6.29 ± 0.15* | 6.76 ± 0.16 | 5.69 ± 0.20*** | ||||||
| Inactivation | V1/2 (mV) | −67 ± 0.66 | 19 | −66.95 ± 0.64 | 36 | −68.20 ± 0.60 | 20 | −67.64 ± 0.65 | 12 | −61.69 ± 0.61**** | 24 |
| K | 5.30 ± 0.38 | 5.66 ± 0.37** | 5.41 ± 0.49 | 10.07 ± 0.67**** | 5.39 ± 0.36 | ||||||
| Recovery (τ) | (ms) | 13.2 ± 0.72 | 19 | 12.48 ± 0.53 | 29 | 12.39 ± 0.42 | 18 | 5.33 ± 0.20**** | 15 | 12.57 ± 0.71 | 21 |
| Persistent I (−10mV) | (%) | 0.52 ± 0.03 | 23 | 1.33 ± 0.06**** | 22 | 1.21 ± 0.06** | 17 | 2.68 ± 0.20**** | 14 | 0.79 ± 0.05 | 19 |
| Resurgent I (−35mV) | (%) | 5.50 ± 0.23 | 15 | 7.40 ± 0.53** | 17 | 7.91 ± 0.42**** | 13 | 14.13 ± 0.72**** | 18 | 6.07 ± 0.45 | 16 |
| Resurgent I decay (−35mV) | (ms) | 7.25 ± 0.09 | 12 | 11.35 ± 0.44**** | 13 | 11.24 ± 0.29**** | 12 | 8.33 ± 0.37* | 15 | 17.52 ± 1.29**** | 15 |
| Resurgent I decay (−10mV) | (ms) | 10.46 ± 0.43 | 12 | 11.18 ± 0.44 | 13 | 11.98 ± 0.62 | 10 | 25.87 ± 1.26**** | 16 | 15.51 ± 0.89** | 14 |
P < 0.0001,
P < 0.001,
P < 0.01,
P < 0.05 in unpaired t test
3.2. T767I
T767 is located in the first transmembrane segment of DII (Fig.1). The mutation from Thr to Ile has been shown to disrupt channel activation in mouse Nav1.6 (Estacion et al., 2014). Representative current traces of hNav1.6 WT and T767I are shown in Fig.5A. The current density (Fig.5B) and rate of decay for transient current (Fig.5C) of T767I in hNav1.6 were not significantly different from the WT channel (Table 1). In agreement with previous literature, T767I displayed a 10mV hyperpolarizing shift in the voltage-dependence of activation (V50 = −31.68 ± 0.81 mV vs V50 = −21.07 ± 0.79 mV in WT, P < 0.0001) (Fig.5D), indicative of premature channel opening, while it did not alter the steady-state inactivation (Fig.5E) (Table 1). As a result, larger window currents were observed (Fig.5F). The normalized current traces of WT and T767I shown in Fig.6A demonstrates an increased percentage of persistent current in the mutant channel (1.21 ± 0.06 % in T767I vs. 0.52 ± 0.03 % in WT, P = 0.0012) (Fig.6B). Compared to WT, T767I had no significant change in the rate of recovery from inactivation (Fig.6C) (Table 1). Fig.6D shows the representative traces of resurgent current generated by WT and T767I. Similar to R850Q, T767I showed significant increase in resurgent current amplitude (7.91 ± 0.42 % in T767I vs. 5.50 ± 0.23 % in WT, P < 0.0001) (Fig.6E), and the resurgent currents displayed slower decay kinetics (at −35mV repolarization step, 11.24 ± 0.29 ms in T767I vs. 7.25 ± 0.09 ms in WT, P < 0.0001) (Fig.6F) (Table 1).
Figure 5. T767I displayed depolarized voltage-dependence of activation.
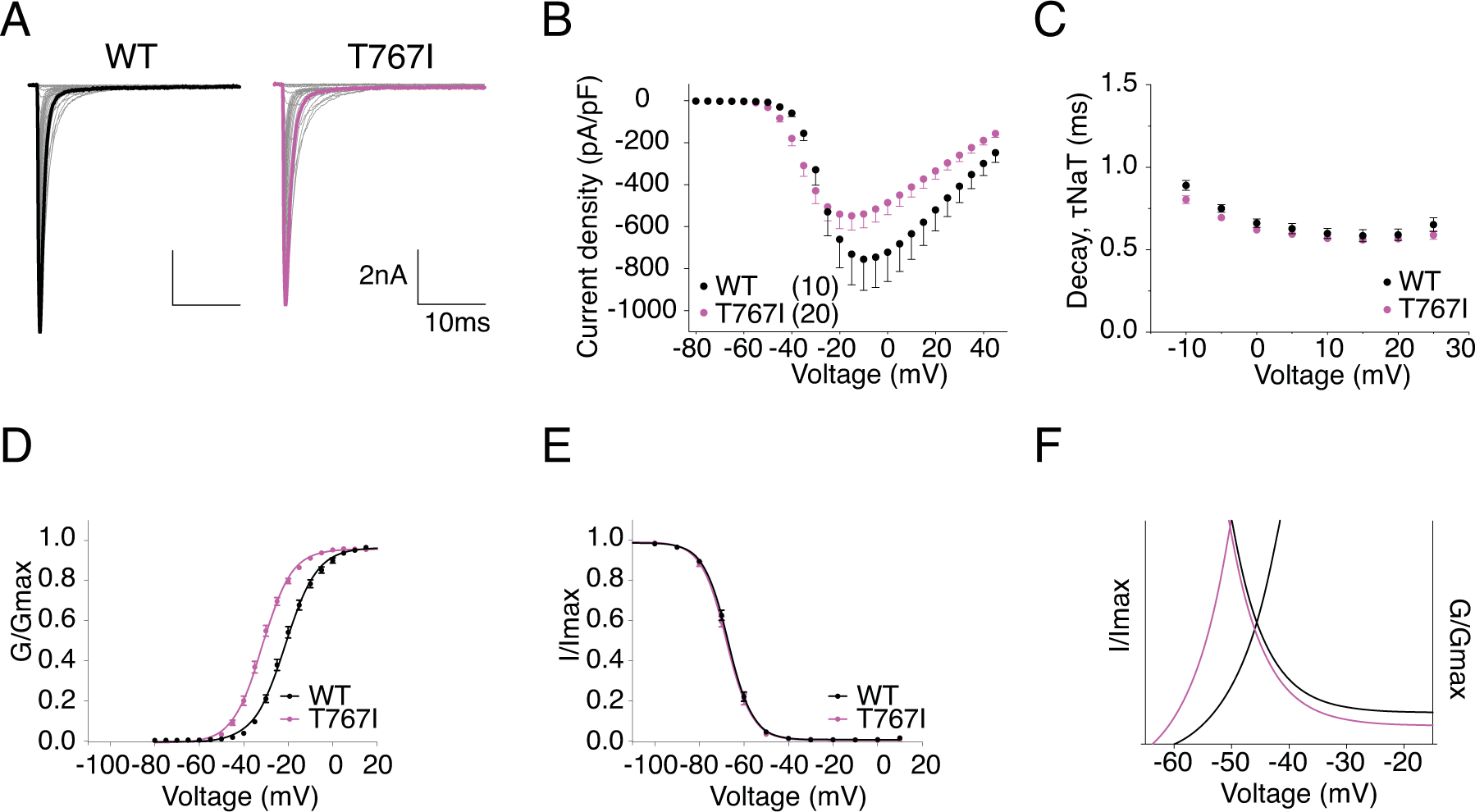
A, representative current trace families from hNav1.6 WT (left) and T767I (right) channels elicited by an activation protocol. Maximum current traces are highlighted with bolded black and pink respectively. B, Current density-voltage plot of hNav1.6 WT and T767I. C, Rate of decay for transient sodium current (τNaT) at each depolarization voltages. D,E, activation and steady-state inactivation curves fitted with Boltzmann functions. F, window currents shown by Boltzmann fitted curves enlarged from DE combined. All values and statistical significance are reported in Table1.
Figure 6. T767I has increased persistent current, increased resurgent current, and altered resurgent current kinetics.
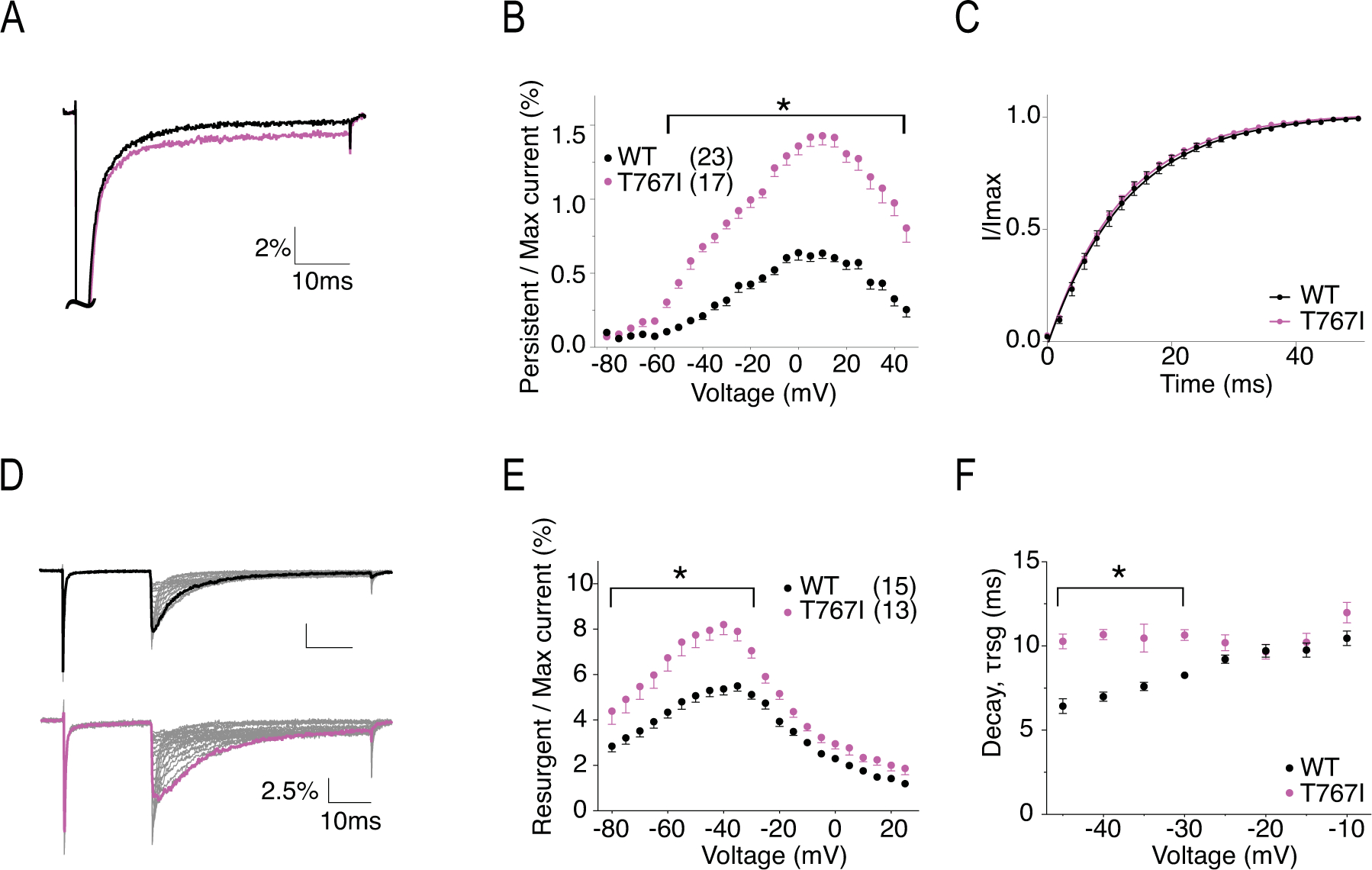
A, representative traces showing persistent current from WT (black) and T767I (pink) channels. Traces shown are elicited at 0mV and presented as the percentage amplitude normalized to the maximum peak current during the 50ms activation protocol. B, per cent persistent current-voltage plot with persistent current measured during the last 1ms of the 50ms activation protocol. P < 0.0001 from −50mV to 45mV in two-way ANOVA with Sidak multiple comparison test. C, recovery from inactivation with recovery duration from 0 to 50ms. D, representative traces showing resurgent current from WT (top) and T767I (bottom) channels. E, per cent resurgent current plotted against repolarization voltage. P < 0.05 from −80mV to −30mV in two-way ANOVA with Sidak multiple comparison test. F, Rate of decay for resurgent current (τrsg) at each repolarization voltages. P < 0.0001 from −45mV to −35mV in two-way ANOVA with Sidak multiple comparison test. All values and statistical significance are reported in Table1.
3.3. R1617Q
R1617 is the first positively charged arginine in DIV S4 (Fig.1). Loss of the positive charge is predicted to and has been shown to affect channel inactivation in mouse Nav1.6 (Wagnon et al., 2016). Representative current traces of hNav1.6 WT and R1617Q are shown in Fig.7A. The transient current of R1617Q was not significantly different from WT in current density (Fig.7B) (Table 1). However, it decayed far slower with a time constant about 3.5 times larger than that of the WT channel (τ = 3.49 ± 0.15 in R1617Q vs. τ = 0.89 ± 0.03 in WT, P < 0.0001) (Fig.7C). Voltage-dependence of activation of R1617Q remained unchanged (Fig.7D), while steady-state inactivation had a shallower slope (k = 10.07 ± 0.67) compared to WT (k = 5.30 ± 0.38) (P < 0.0001), but was unaltered in midpoint voltage (Fig.7E), in contrast to the 9 mV depolarizing shift seen by Wagnon et al. with the mouse orthologue (Table 1). Fig.7F shows the larger window currents of R1617Q due to the change of inactivation time constant. The normalized current traces of WT and R1617Q in Fig.8A demonstrates an increase of persistent current in the mutant channel (2.68 ± 0.20% in R1617Q vs. 0.52 ± 0.03 % in WT, P < 0.0001) (Fig.8B). Compared to WT, R1617Q had faster recovery from inactivation with a time constant of 5.33 ± 0.20 ms compared to 13.2 ± 0.72 ms in WT (P < 0.0001) (Fig.8C). Fig.8D shows the representative resurgent current traces from WT and R1617Q, demonstrating larger resurgent current amplitude generated by R1617Q (14.13 ± 0.72 % in R1617Q vs. 5.50 ± 0.23 % in WT, P < 0.0001) (Fig.8E). In addition, the R1617Q resurgent current also displayed slower decay kinetics (at −10mV repolarization step, 25.87 ± 1.26 ms in R1617Q vs. 10.46 ± 0.43 ms in WT, P < 0.0001) (Fig.8F) (Table 1).
Figure 7. R1617Q has altered voltage-dependence of inactivation and slower rate of decay for transient current.
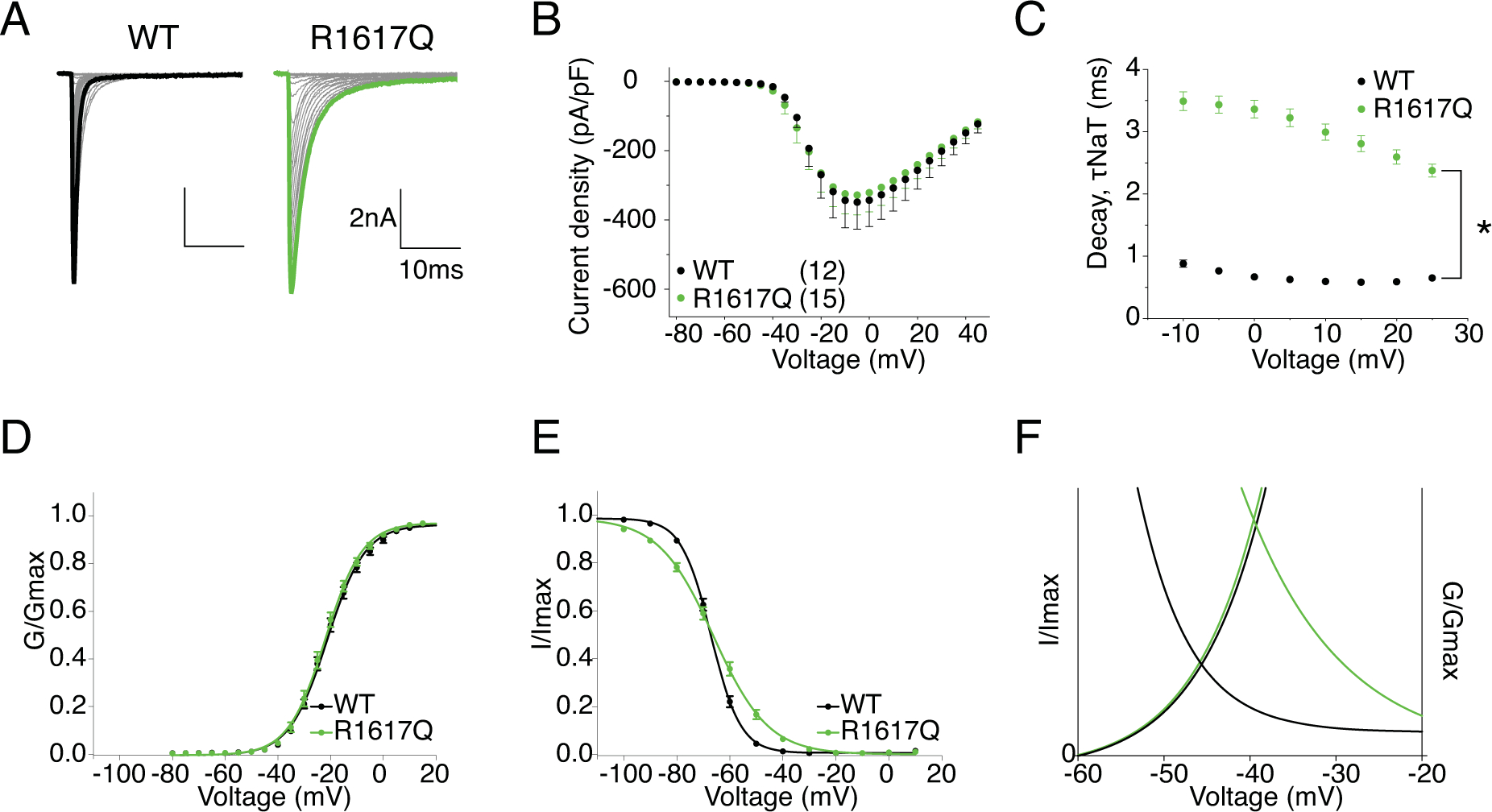
A, representative current trace families from hNav1.6 WT (left) and R1617Q (right) channels elicited by an activation protocol. Maximum current traces are highlighted with bolded black and green respectively. B, Current density-voltage plot of hNav1.6 WT and R1617Q. C, Rate of decay for transient sodium current (τNaT) at each depolarization voltages. P < 0.0001 at all voltages in two-way ANOVA with Sidak multiple comparison test. D,E, activation and steady-state inactivation curves fitted with Boltzmann functions. F, window currents shown by Boltzmann fitted curves enlarged from DE combined. All values and statistical significance are reported in Table1.
Figure 8. R1617Q has increased persistent current, faster recovery from inactivation, increased resurgent current and altered resurgent current kinetics.
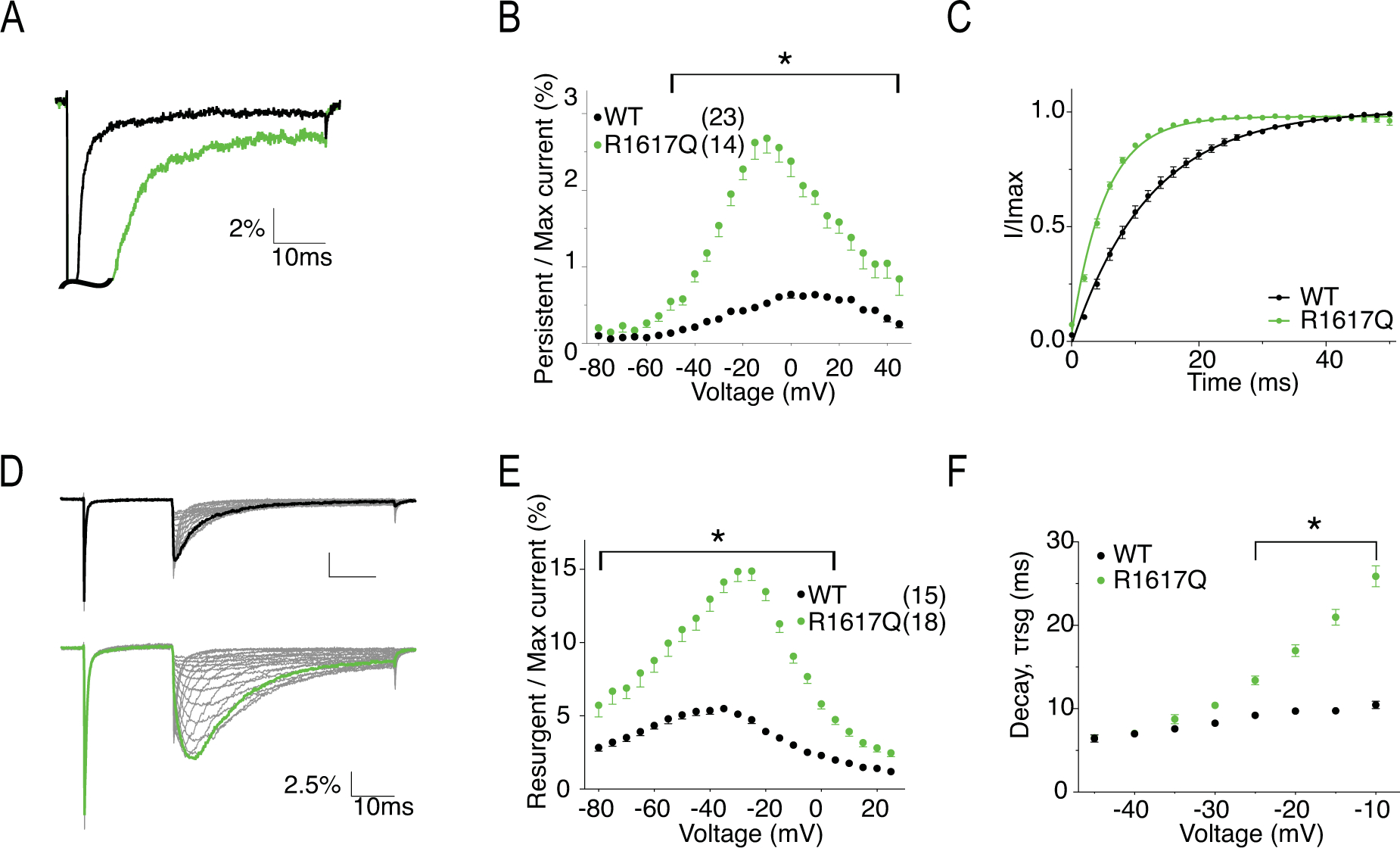
A, representative traces showing persistent current from WT (black) and R1617Q (green) channels. Traces shown are elicited at 0mV and presented as the percentage amplitude normalized to the maximum peak current during the 50ms activation protocol. B, per cent persistent current-voltage plot with persistent current measured during the last 1ms of the 50ms activation protocol. P < 0.0001 from −40mV to 40mV in two-way ANOVA with Sidak multiple comparison test. C, recovery from inactivation with recovery time duration from 0 to 50ms. D, representative traces showing resurgent current from WT (top) and R1617Q (bottom) channels. E, per cent resurgent current plotted against repolarization voltage. P < 0.0001 from −75mV to 0mV in two-way ANOVA with Sidak multiple comparison test. F, Rate of decay for resurgent current (τrsg) at each repolarization voltages. P < 0.0001 from −25mV to −10mV in two-way ANOVA with Sidak multiple comparison test. All values and statistical significance are reported in Table1.
3.4. R1872Q
R1872 locates near the center of the cytoplasmic c-terminal domain (Fig.1). The loss of the positively charged Arg in this region is predicted to interfere with channel inactivation due to its presumed interaction with the channel inactivation gate. Representative traces of hNav1.6 WT and R1872Q are shown in Fig.9A. Fig.9B shows that R1872Q generated larger hNav1.6 current, with a current density of −861.00 ± 97.40 pA/pF compared to −498.10 ± 95.49 pA/pF of WT (P = 0.016). Interestingly, the R1872Q transient current decayed slower, particularly at more negative voltages (1.52 ± 0.06 ms in R1872Q vs. 0.89 ± 0.03 ms in WT, P < 0.0001) (Fig.9C). This is clearly shown in Fig.10A, where the decay of R1872Q was almost indistinguishable from WT at −10mV, but was dramatically slower at −35mV. This voltage-dependent change of current decay likely contributed to a peculiar pattern of the persistent current-voltage plot (Fig.10B), showing significantly larger percentage of persistent current only at depolarizing voltages ranging from −40mV to −10mV. Important to note, this pattern was not observed when persistent current was measure at 500ms of the prepulse depolarization step in the steady-state inactivation protocol (Fig.10DE). We reasoned that the increased persistent current observed with the 50ms stimulation protocol was due to the slow decay at more negative voltages, instead of a non-inactivating component of the transient current. The voltage-dependence of activation and stead-state inactivation of R1872Q were shifted in opposite directions: a hyperpolarizing shift in activation (V50 = −25.5 ± 0.83 mV in R1872Q vs V50 = −21.07 ± 0.79 mV in WT, P = 0.0004) and a depolarizing shift in steady-state inactivation (V50 = −61.69 ± 0.61 mV in R1872Q vs. V50 = −67 ± 0.66 mV in WT, P < 0.0001) (Fig.9DE) (Table 1). As a result, R1872Q displayed larger window currents (Fig.9F). R1872Q had no significant change in recovery from inactivation (Fig.10C) (Table 1). Fig.10F shows the representative resurgent current traces from WT and R1872Q. The resurgent current of R1872Q had no significant change in amplitude (Fig.10G), but exhibited a substantially slower rate of decay (at −35mV repolarization step, 17.52 ± 1.29 ms in R1872Q vs. 7.25 ± 0.09 ms in WT, P < 0.0001) (Fig.10H) (Table 1).
Figure 9. R1872Q has increased current amplitude, altered voltage-dependence of both activation and steady-state inactivation.
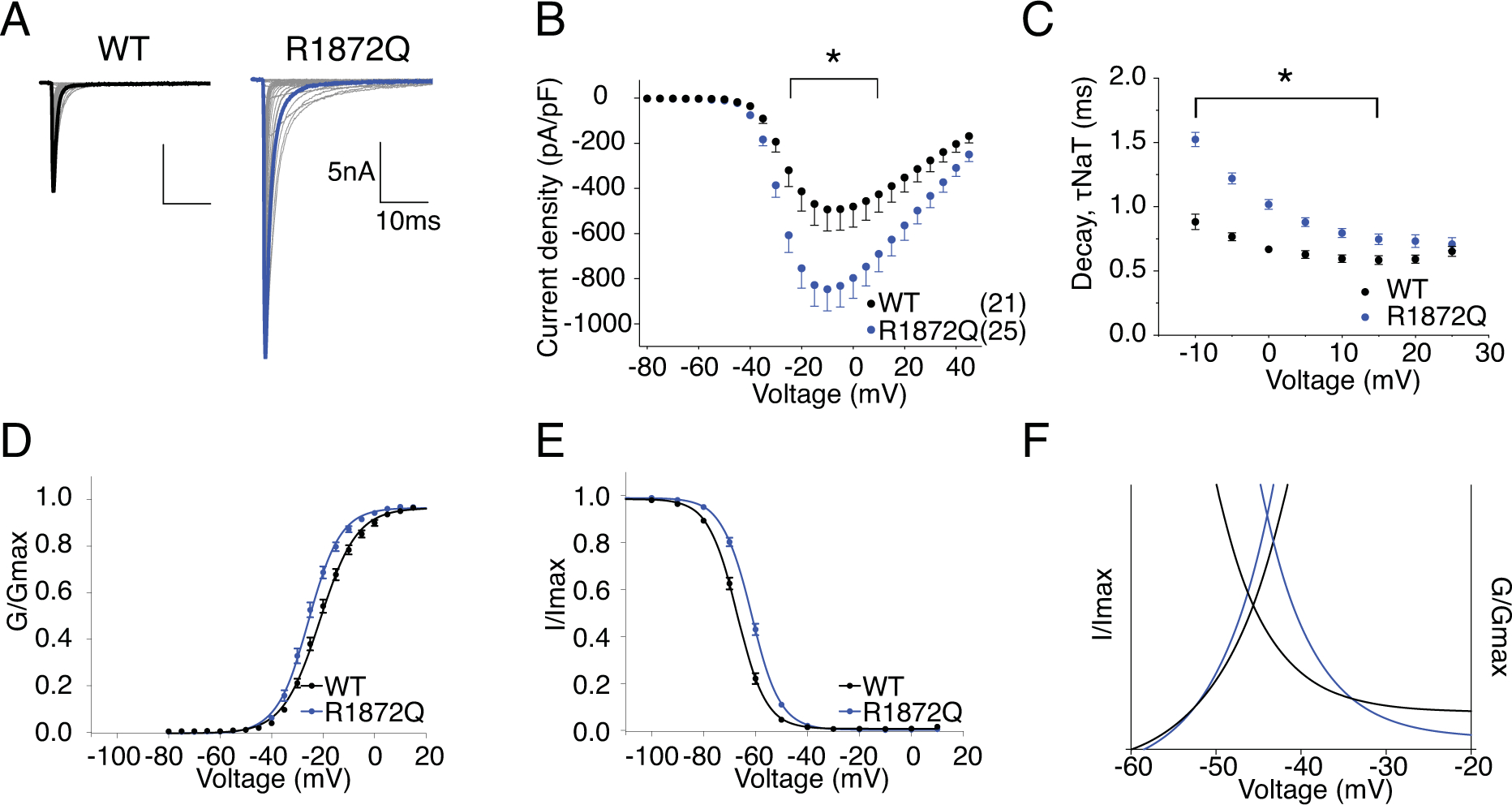
A, representative current trace families from hNav1.6 WT (left) and R1872Q (right) channels elicited by an activation protocol. Maximum current traces are highlighted with bolded black and blue respectively. B, Current density-voltage plot of hNav1.6 WT and R1872Q. P < 0.05 from −25mV to +10mV in two-way ANOVA with Sidak multiple comparison test. C, Rate of decay for transient sodium current (τNaT) at each depolarization voltages. P < 0.0001 from −10mV to 0mV in two-way ANOVA with Sidak multiple comparison test. D,E, activation and steady-state inactivation curves fitted with Boltzmann functions. F, window currents shown by Boltzmann fitted curves enlarged from DE combined. All values and statistical significance are reported in Table1.
3.5. Simulation
To explore how Nav1.6 epilepsy mutations affect neuronal excitability, we simulated action potential firing in a single compartment model of Purkinje neuron (Khaliq et al., 2003) and a multi-compartment model of cortical pyramidal neuron (Ben-Shalom et al., 2017). We began by modeling the WT and mutant Nav1.6 currents based on the Markov state sodium channel model (Khaliq et al., 2003) with minor modifications. Voltage-clamp simulations validated that the modelled R850Q, T767I and R1872Q channels successfully recapitulated all the functional alterations observed in our experimental data (Fig.11). The modelled R1617Q channel, however, under represented the experimental changes of the slope of steady-state inactivation curve and the increase of resurgent current amplitude. A summary of the biophysical properties of modeled channels are reported in Table 3.
Figure 11. Simulation of sodium current generated by modeled WT and mutant Nav1.6.
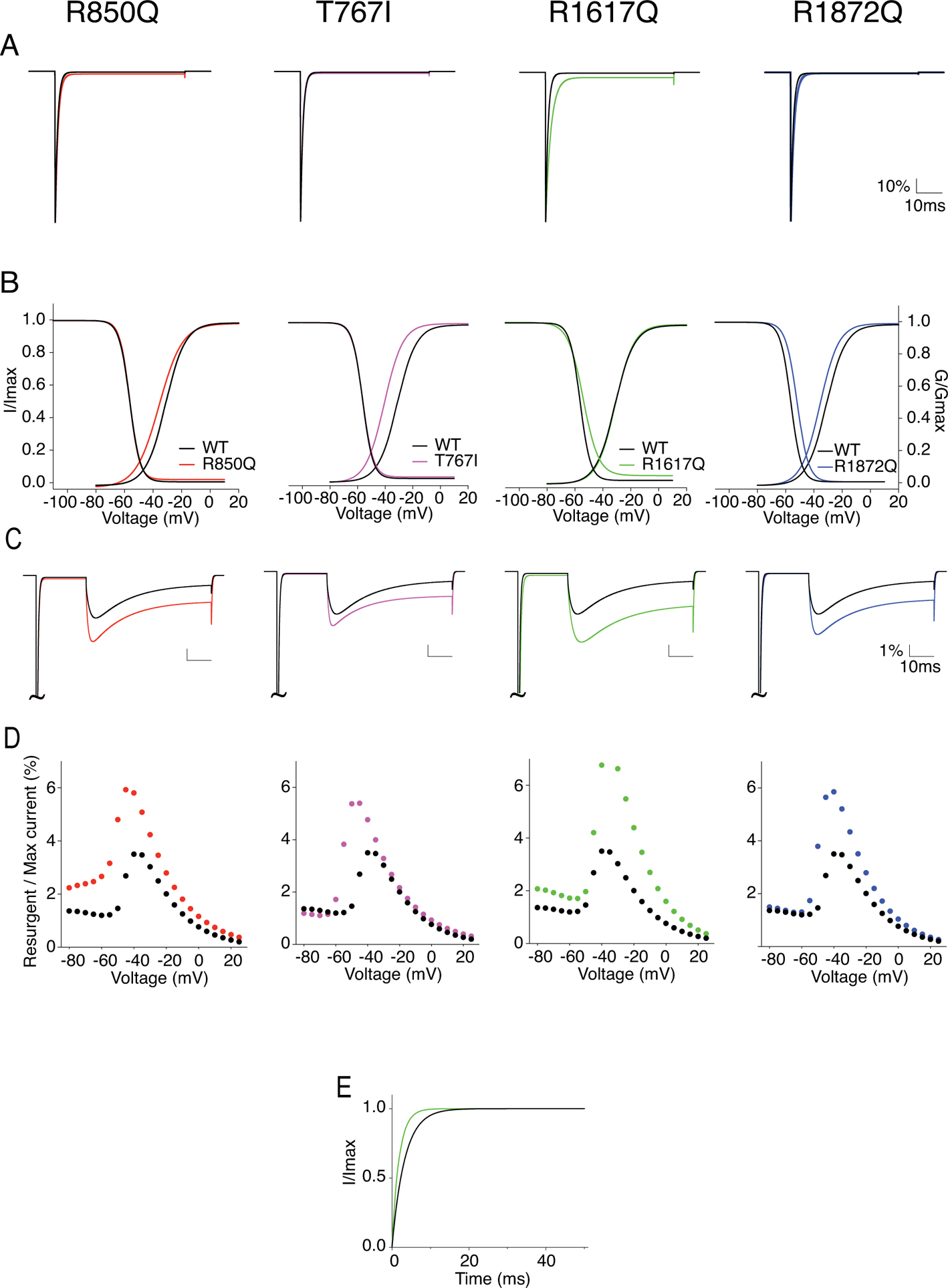
A, example WT (black) and mutant Nav1.6 currents (red: R850Q, pink: T767I, green: R1617Q, blue: R1872Q) generated from modeled neurons without open channel block. B, I-V/G-V plots showing the voltage dependence of activation and steady-state inactivation of the modeled WT and mutant Nav1.6 channels. C, example resurgent currents generated by modeled WT and mutant Nav1.6 channels with open channel block. D, per cent resurgent currents plotted against repolarization voltages from simulated recordings. Peak resurgent currents were measured starting 1.4ms into the repolarization step and normalized to the maximum peak current during a 50ms activation protocol. E, recovery from inactivation plot with recovery time duration from 0 to 50ms for R1617Q. Values are reported in Table 3.
Table 3.
Biophysical properties of modeled WT and mutant SCN8A channel
| WT | R850Q | T767I | R1617Q | R1872Q | ||
|---|---|---|---|---|---|---|
| Transient I | (pA/pF) | −714.6 | −713.2 | −735.3 | −724.5 | −716.7 |
| Transient I decay | 0.92 | 1.06 | 0.85 | 2.01 | 1.30 | |
| Activation | V1/2(mV) | −31.23 | −35.78 | −40.59 | −30.92 | −35.59 |
| K | 6.90 | 8.08 | 6.73 | 6.45 | 6.60 | |
| Inactivation | V1/2(mV) | −55.85 | −56.16 | −56.41 | −53.86 | −51.57 |
| K | 3.84 | 3.74 | 4.00 | 6.15 | 3.96 | |
| Recovery | (ms) | 3.30 | 3.35 | 3.36 | 1.68 | 3.26 |
| Resurgent I | (%) | 3.50 | 5.93 | 5.40 | 7.43 | 5.85 |
To simulate spontaneous and evoked activities of Purkinje neurons, the total sodium conductance of the Khaliq model was replaced by our modeled hNav1.6. To represent heterozygous conditions, 50% of our Nav1.6 sodium conductance was replaced by the modeled mutant channels. Fig.12A shows the spontaneous action potential (AP) firing from WT and mutant Purkinje neurons with open channel block implemented. Modeled neurons incorporated with R850Q, T767I and R1872Q currents demonstrated increased excitability exemplified by a higher frequency of spontaneous (Fig.12AC) as well as evoked APs (Fig.12D) (Table 4). The R1617Q neuron was not more excitable than the WT neuron in response to lower range of stimulation, but its firing frequency continued increasing in the higher stimulation range, showing resistance to depolarization block and reaching a higher maximal AP frequency. This enables the R1617Q neuron to sustain high-frequency, repetitive firing, which is an important characteristic of neurons involved in seizure activities. Our simulations demonstrated a differential pattern of excitability outcome caused by functional alterations in activation versus inactivation. Premature opening, as implemented in R850Q, T767I and R1872Q, has prominent effect on enhancing firing, either spontaneous or in response to low intensity stimulation. On the other hand, altered inactivation that led to increased channel availability, as exhibited mainly with R1617Q and R1872Q, boosts the ability of a neuron to sustain high frequency firing.
Figure 12. Simulation of action potential firing in Purkinje neuron expressing WT or heterozygous mutant Nav1.6 channels.
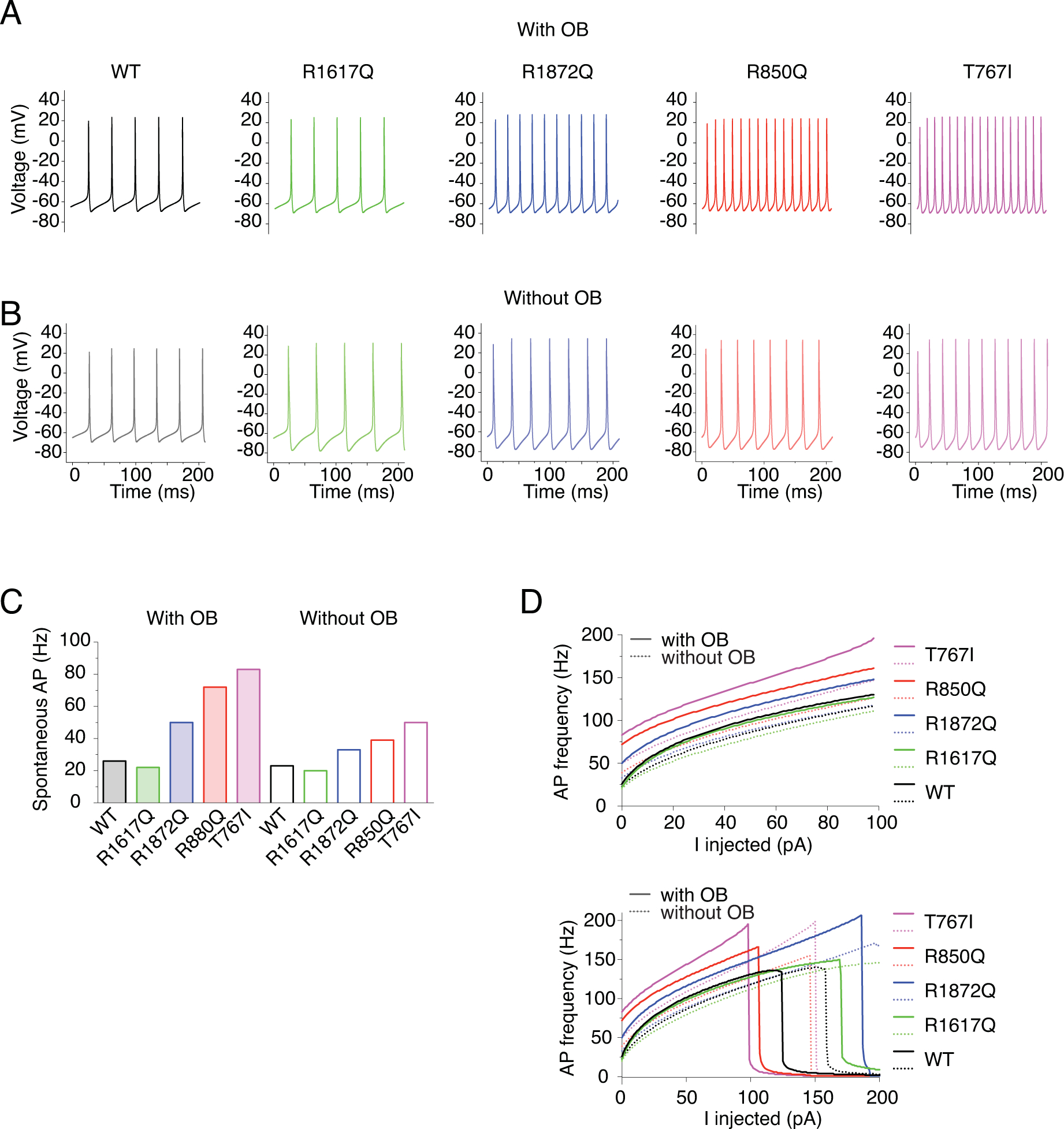
A-B, example traces of spontaneous firing of modeled Purkinje neurons either with 100% WT Nav1.6 current or with 50% WT and 50% mutant Nav1.6 currents (black: WT, green: R1617Q, blue: R1872Q, pink: T767I, red: R850Q) with OB (A) and without OB (B). C, Quantification of the frequency of spontaneous action potentials. Values are reported in Table 4. D, Quantification of evoked action potentials.
Table 4.
Spontaneous action frequency (Hz) of simulated Purkinje neurons
| WT | R1617Q | R1872Q | R850Q | T767I | ||
|---|---|---|---|---|---|---|
| Spontaneous | With OB | 26 | 22 | 50 | 72 | 83 |
| Without OB | 23 | 20 | 33 | 39 | 50 | |
| Maximum | With OB | 136 | 150 | 207 | 166 | 196 |
| Without OB | 140 | 146 | 171 | 155 | 199 |
In addition, simulations without the open channel block mechanism suggested an important role of resurgent current in affecting excitability, especially when mutant channels were involved. As shown in Fig.12B and Table 4, all variants are consistently less excitable without OB, suggesting that resurgent current is an important contributor to high frequency firing associated with epilepsy mutations. Moreover, the WT neuron demonstrated about 12% reduction of spontaneous AP firing, while the R1617Q, R1872Q, R850Q and T767I mutants showed 9%, 34%, 46% and 40% reduction, respectively. This indicated that the excitability of neurons with mutant channels was generally more sensitive to the existence of the open channel block. As a result, the R1872Q, R850Q and T767I mutant neurons with OB implemented presented larger percentage increase of spontaneous APs (92%, 177%, 219%) compared to those without OB implemented (43%, 70%, 117%). Therefore, resurgent currents can have significant impact on the excitability of neurons with mutant Navs, although resurgent currents did not display as remarkable an influence on the excitability of WT neurons in our simulations. Intriguingly, the effect size of OB inclusion in the model did not correlate with the amplitude of resurgent current (Table 3). For example, the excitability of the R850Q-containing model neuron was most affected by the inclusion of OB, although the modeled R850Q channel did not exhibit the most prominent increase of resurgent current (Fig.11CD). On the contrary, the spontaneous activity of R1617Q was unexpectedly reduced by the inclusion of OB, although it had the largest increase of resurgent current. This emphasizes that neuronal excitability is an integrated outcome of different aspects of channel functions, and such complex integration can potentially be extended to the inter-cellular and network levels.
To further investigate the impact of Nav1.6 mutations on the integrated outcome of neuronal excitability, we incorporated our modeled Nav1.6 in a more sophisticated cortical pyramidal neuron model (Ben-Shalom et al., 2017). This model implemented multiple sodium channel isoforms that were differentially distributed in soma, dendrites, axon initial segment, nodes of Ranvier and along the myelinated axon. The sodium conductance that represents Nav1.6 current in the original model was replaced by our modeled hNav1.6 current. Similar to the Purkinje neuron model, heterozygous mutations of Nav1.6 R1872Q, R850Q and T767I in modeled cortical pyramidal neuron lead to marked increase of excitability with or without OB (Fig.13). However, unlike the Purkinje neuron model, the inclusion of the OB mechanism did not consistently increase action potential firing across all variants. In response to lower range of stimulations, WT and mutant neurons fired more action potentials without OB, while in the higher range of stimulations (up to 4nA) the firing rates were eventually surpassed by those with OB implemented. This supports the notion that resurgent current facilitates but does not guarantee high frequency firing (Lewis & Raman, 2014). Therefore, the precise impact of resurgent currents on neuronal excitability also likely depends on cellular background.
Figure 13. Simulation of action potential firing in cortical pyramidal neuron expressing WT or heterozygous mutant Nav1.6 channels.
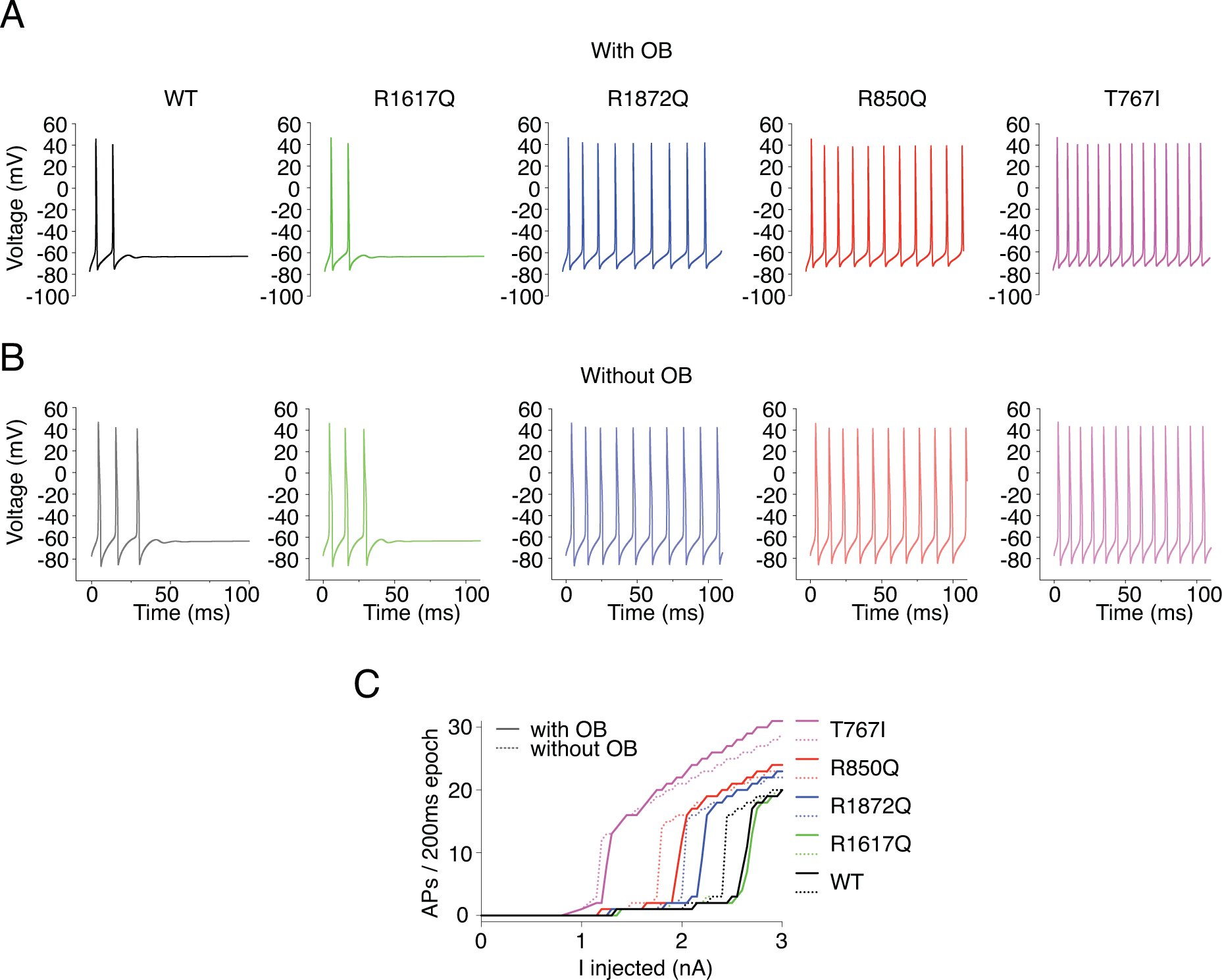
A-B, example traces of action potential firing evoked by 2.3nA current injection in cortical pyramidal neuron models either with 100% WT Nav1.6 current or with 50% WT and 50% mutant Nav1.6 currents (black: WT, green: R1617Q, blue: R1872Q, pink: T767I, red: R850Q) with OB (A) and without OB (B). C, Quantification of evoked action potentials.
4. Discussion
A growing number of SCN8A mutations have been associated with EIEE, but the pathogenic outcomes are hard to predict without functional characterization. In this study, we report that the novel epilepsy mutation R850Q is gain-of-function, consistent with the hypothesis that a majority of SCN8A epilepsy mutations primarily lead to enhanced channel function. R850Q exhibits multiple aberrant properties, including premature opening, impaired inactivation and increased resurgent current. By comparing R850Q with three other SCN8A epilepsy mutations that have been previously characterized in mouse Nav1.6 constructs, we show that the mutations affect different aspects of the human Nav1.6 channel properties despite all being gain-of-function. To our surprise, all the evaluated mutations display enhanced resurgent currents, pointing to a potential commonality among gain-of-function SCN8A epilepsy mutations. Using computational simulations, we demonstrate that modeled neurons bearing these SCN8A epilepsy mutations are hyperexcitable, further supporting the proposed mechanism for SCN8A epilepsy being neuronal over-excitation and the hypothesis that altered resurgent currents in hNav1.6 channels likely contribute to increased seizure susceptibility.
EIEE with a SCN8A mutation is a devastating condition due to its phenotypic severity and resistance to classic anti-epileptic drugs. Additionally, SCN8A cases are highly heterogeneous, displaying variable clinical presentation and treatment response. The disease mechanism is not well understood and requires functional characterization of a wide range of SCN8A mutations for more individualized therapeutic development. However, the majority of previous functional analysis of SCN8A mutations were conducted with the mouse cDNA construct expressed in heterologous systems. Although these results have provided valuable insights for the molecular underpinnings of the disease, the alterations of channel functions they presented may differ from the same mutations in human DNA. Moreover, most of the previous studies on SCN8A epilepsy mutations did not test the involvement of resurgent current, the alteration of which can affect neuronal excitability, as it does in many other sodium channelopathies (Jarecki et al., 2010b; Hargus et al., 2011; Sittl et al., 2012; Hargus et al., 2013; Tan et al., 2014; Patel et al., 2016; Xiao et al., 2019). In our study, we used a human SCN8A cDNA construct (hNav1.6) for functional characterization and systematically compare the outcomes with those from previous studies using the mouse construct. We observed consistent functional changes for the mutation R1872Q, but important functional differences for mutations T767I and R1617Q in the human compared to the mouse channel background. For example, the current amplitude produced by the T767I mutation in hNav1.6 is similar to the amplitude observed with WT hNav1.6 (Fig.5AB), while the T767I mutation was reported to have a 2.5-fold reduction in mouse Nav1.6 (Estacion et al., 2014). On the other hand, the hyperpolarizing shift of voltage dependence of activation and the increase of persistent current are comparable in both backgrounds. As for R1617Q, a similar slowing of current decay and increase of persistent currents are observed with both the human and mouse channel background, but there are differential effects on the voltage dependence of activation and inactivation (Fig.7,8) (Wagnon et al., 2016). In hNav1.6, the R1617Q mutation altered only the slope of steady-state inactivation, while in the mouse Nav1.6, there is a hyperpolarizing shift of activation and a depolarizing shift of inactivation along with a slope change. Such differences may seem subtle, but they suggest molecular and functional distinctions between the human and mouse channels, which in turn could alter how individual mutations impact intrinsic neuronal excitability and even affect their pharmacological responsiveness, and thus could have important impact when it comes to therapeutic development and testing.
Although it is generally accepted that SCN8A epilepsy mutations are primarily gain-of-function, the molecular mechanisms often differ among individual mutations. The heterogeneity of SCN8A epilepsy phenotypes likely stems in part from the various locations and functional consequences of the mutations. Due to the clear functional segregation of channel regions, outcomes of some mutations can be predicted by their location. For example, mutations in DI-DIII voltage sensors are likely to alter channel activation. As shown in this study, missense mutations at R850 and T767, located in DII S4 and S1 respectively, result in premature opening, a functional alteration in activation that is highly predicted to lead to hyperactivity. On the other hand, mutations in DIV voltage sensors usually disrupt channel inactivation. An Arg to Gln substitution at 1617 in DIV S4 exhibits a dramatically slower decay of the transient current, indicating impaired inactivation. Although persistent currents and resurgent currents can be related, persistent currents occur due to incomplete inactivation. Resurgent currents occur via a distinct mechanism in response to an alternative inactivation particle that has been referred to as an open channel blocker (Raman & Bean, 2001; Grieco et al., 2005). Since the open channel block competes with the IFM (Ile-Phe-Met motif) inactivation gate for channel binding after activation (Khaliq et al., 2003; Grieco & Raman, 2004), the slower inactivation kinetics of R1617Q and many other disease mutations (Jarecki et al., 2010b) increase the probability of open channel block, and thus the generation of larger resurgent currents. Indeed, the slowed inactivation induced by R1617Q is associated with a 2-fold increase of resurgent current, the mechanism of which involves an open channel block. Nav1.6 resurgent currents flows following open channel block when channels reopen during repolarizations before undergoing either inactivation or deactivation (Bant & Raman, 2010) . This consequence of impaired inactivation applies to R850Q as well as R1617Q. In addition it is interesting to note that both R850Q and R1617Q neutralize charged residues in S4 segments. While R1617Q may not induce gating pore currents due to its location at the extracellular extreme of the S4 segment, R850Q is predicted to induce gating pore current due to the S4 charge neutralization (Sokolov et al., 2005). However, because gating pore currents are very small and difficult to measure in mammalian expression systems, we were unable to directly determine if either of these mutations induced gating pore currents.
In addition to direct lesion to the molecular components responsible for specific channel function, mutations located in regions involved in intramolecular interactions can also cause predictable functional defects. The C-terminal domain (CTD) has been shown to interact with the IFM inactivation gate via static interaction between charged residues (Shen et al., 2017; Johnson et al., 2018). Removing any of the charges can disrupt the interaction and affect normal channel inactivation (Lee & Goldin, 2008; Nguyen & Goldin, 2010; Johnson et al., 2018). Indeed, we show that an Arg to Gln mutation at 1872, located near the center of CTD, displays abnormal inactivation properties, including shifted voltage dependence of inactivation, slower decay of transient current and higher level of persistent current. Similarly, Wagnon et al. (2016) demonstrated inactivation-related dysfunctions of two other epilepsy mutations at R1872 in mouse Nav1.6, highlighting the functional significance of the IFM-CTD interaction for channel inactivation.
The complicated network of sodium channel regulation is another source of heterogeneity for EIEE13. As shown in this study, the functional consequences of a single residue substitution can impact properties beyond what is predicted based on its location. For example, R850Q and T767I in the domain II voltage-sensor both impair inactivation and R1872Q in the CTS has premature activation. Most interestingly, R850Q, T767I and R1872Q demonstrate slowed resurgent kinetics that are not readily predicted by their location. These alterations may be consequences of the complex nature of subtle intramolecular interactions within the channel as well as intermolecular interactions between the channel and a great variety of regulatory proteins. So far, Nav-β subunits (Hull & Isom, 2018), calmodulin, fibroblast growth factor homologous factors (FHFs) (Wang et al., 2012; Wang et al., 2014) and ankyrinG (ankG) (Shirahata et al., 2006; Gasser et al., 2012) are identified as important interacting partners of Nav channels. They regulate Nav trafficking, expression pattern and gating in isoform- and cell type-specific manners (Deschênes et al., 2002; Herzog et al., 2003; Chichili et al., 2013; Ben-Johny et al., 2014; Barbosa et al., 2017; Yan et al., 2017). Mutations within or near the interacting interface on either protein can alter the interaction and cause channel dysregulation. Moreover, the wide range of post-translational modifications of voltage-gated sodium channels (Pei et al., 2017) and the existence of modifier genes also likely contribute to the heterogeneity of SCN8A epilepsy (Sprunger et al., 1999; Bergren et al., 2005; Rusconi et al., 2007; Bergren et al., 2009; Singh et al., 2009).
Due to the vast number of epilepsy mutations and the remarkable complexity of sodium channel regulation, it may be impractical to extend functional assays to all combinations of mutations and genetic variants appearing in patients. Computational simulation can be of great value in this matter. In our study, we perform excitability simulations using two neuron models and show predictions of the outcome of allelic epilepsy mutations. To model sodium channel kinetics, we adapted a simple Markov state model that included the open channel block mechanism (Khaliq et al., 2003) (Fig.2). This feature enables the simulation of resurgent sodium currents. Extensive evidence has showed that physiological resurgent currents support high-frequency repetitive firing of action potentials by maintaining a relatively constant availability of sodium channels (Raman & Bean, 2001). Pathological alterations of resurgent current kinetics are involved in a wide range of excitability disorders (Jarecki et al., 2010b; Hargus et al., 2011; Sittl et al., 2012; Hargus et al., 2013; Tan et al., 2014; Patel et al., 2016; Xiao et al., 2019). More importantly, we revealed enhanced resurgent current as a commonality among the four Nav1.6 epilepsy mutations in our study. Therefore, abnormal resurgent currents found in epilepsy mutant Nav1.6 are likely to contribute to neuronal hyperexcitability. Indeed, our simulations demonstrated a cell type- and frequency- dependent role of resurgent current in heightening excitability, which in turn can lead to uncontrolled, synchronized firing involved in seizure activities.
The computational simulations we performed can be expanded to accommodate variants in multiple ion channel genes to predict the integrated effect on neuronal as well as network excitability (Klassen et al., 2011). However, such simulations are not without limitations. The original sodium channel model (Khaliq et al., 2003) couples the voltage dependence of inactivation with activation, which does not necessarily reflect the complex behaviors of a voltage-gated sodium channel. As illustrated by the R1872Q mutant channel, where the voltage-dependence of activation and inactivation are shifted in opposite directions. Such channel behavior is impossible to be represented by a model constrained by activation-inactivation coupling. Therefore, we partially segregated the coupling to closely mimic channel behaviors observed in our experiments. The modified model allowed us to accurately simulate the opposite direction shifts in R1872Q, and the depolarizing shifts of activation in R850Q and T767I without immensely affecting the voltage dependence of inactivation. Nonetheless, we were not able to satisfactorily achieve all functional alterations observed in R1617Q. The inability to completely mirror R1617Q channel behavior may account for the mild increase of excitability observed in the simulation. Therefore, there is a clear need for improved channel models to accurately capture the dynamics of ionic flows in excitable cells in order to produce reliable excitability prediction and assist in individualized medicine.
Taken together, our study demonstrates that while many epilepsy-related SCN8A mutations are gain-of-function, they can impact multiple gating properties in distinct combinations. One commonality among them is an alteration in resurgent current, which can play a critical role in determining neuronal excitability and thus seizure activities in patients. We demonstrated that computational simulation is a useful tool to explore the influence of mutations on excitability. Using the human Nav1.6 channel, our data also indicates that the SCN8A mutations can exhibit subtle yet potentially important differences in their biophysical consequences in human versus rodent channels. These important distinctions could influence how specific mutations are targeted therapeutically.
Key points.
Mutations in the SCN8A gene cause early infantile epileptic encephalopathy.
We characterize a new epilepsy-related SCN8A mutation, R850Q, in the human SCN8A channel and present gain-of-function properties of the mutant channel.
Systematic comparison of R850Q with three other SCN8A epilepsy mutations, T761I, R1617Q, R1872Q, identifies one common dysfunction in resurgent current, although these mutations alter distinct properties of the channel.
Computational simulations in two different neuron models predict increased excitability of neurons carrying these mutations, which explains the over-excitation that underlies seizure activities in patients.
These data provide further insights into the mechanism of SCN8A-related epilepsy and reveal subtle but potentially important distinction of functional characterization performed in the human vs. rodent channels.
Acknowledgements:
This work was partially supported by the National Institute of Neurological Disorders and Stroke (NINDS), NIH award U54NS108874–01.
Footnotes
Competing interests
The authors declare that they have no competing interests.
References
- Bant JS & Raman IM. (2010). Control of transient, resurgent, and persistent current by open-channel block by Na channel {beta}4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Tan ZY, Wang R, Xie W, Strong JA, Patel RR, Vasko MR, Zhang JM & Cummins TR. (2015). Navbeta4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol Pain 11, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Xiao Y, Johnson AJ, Xie W, Strong JA, Zhang J-M & Cummins TR. (2017). FHF2 isoforms differentially regulate Nav1. 6-mediated resurgent sodium currents in dorsal root ganglion neurons. Pflügers Archiv-European Journal of Physiology 469, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R & Yue DT. (2014). Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell 157, 1657–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ & Bender KJ. (2017). Opposing effects on NaV1. 2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biological psychiatry 82, 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergren SK, Chen S, Galecki A & Kearney JA. (2005). Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channelScn2a. Mammalian Genome 16, 683–690. [DOI] [PubMed] [Google Scholar]
- Bergren SK, Rutter ED & Kearney JA. (2009). Fine mapping of an epilepsy modifier gene on mouse Chromosome 19. Mammalian genome 20, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, Lampert A, Klein JP, Mission J, Chen MC, Rivera M, Dib-Hajj S, Brennan AR, Hains BC & Waxman SG. (2009). Role of hippocampal sodium channel Nav1. 6 in kindling epileptogenesis. Epilepsia 50, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS & Matthews G. (2001). Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30, 91–104. [DOI] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS & Matthews G. (2003). Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23, 2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RK, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, Bhatia K, Jones JM, Maniaci MR & Parent JM. (2019). Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichili VPR, Xiao Y, Seetharaman J, Cummins TR & Sivaraman J. (2013). Structural basis for the modulation of the neuronal voltage-gated sodium channel Na V 1.6 by calmodulin. Scientific reports 3, 2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes I, Neyroud N, DiSilvestre D, Marbán E, Yue DT & Tomaselli GF. (2002). Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circulation research 90, e49–e57. [DOI] [PubMed] [Google Scholar]
- Do MTH & Bean BP. (2004). Sodium currents in subthalamic nucleus neurons from Nav1. 6-null mice. Journal of neurophysiology 92, 726–733. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao C-F & Chandler SH. (2007). Sodium currents in mesencephalic trigeminal neurons from Nav1. 6 null mice. Journal of neurophysiology 98, 710–719. [DOI] [PubMed] [Google Scholar]
- Estacion M, O’Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD & Meisler MH. (2014). A novel de novo mutation of SCN8A (Nav1. 6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiology of disease 69, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, Ho TS-Y, Cheng X, Chang K-J, Waxman SG, Rasband MN & Dib-Hajj SD. (2012). An ankyrinG-binding motif is necessary and sufficient for targeting Nav1. 6 sodium channels to axon initial segments and nodes of Ranvier. Journal of Neuroscience 32, 7232–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL & Raman IM. (2005). Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45, 233–244. [DOI] [PubMed] [Google Scholar]
- Grieco TM & Raman IM. (2004). Production of resurgent current in NaV1. 6-null Purkinje neurons by slowing sodium channel inactivation with β-pompilidotoxin. Journal of Neuroscience 24, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram III EH & Patel MK. (2011). Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiology of disease 41, 361–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Nigam A, Bertram III EH & Patel MK. (2013). Evidence for a role of Nav1. 6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. Journal of neurophysiology 110, 1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA & Escayg A. (2011). Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiology of disease 41, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Liu C, Waxman SG & Cummins TR. (2003). Calmodulin binds to the C terminus of sodium channels Nav1. 4 and Nav1. 6 and differentially modulates their functional properties. Journal of Neuroscience 23, 8261–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML & Carnevale NT. (1997). The NEURON simulation environment. Neural computation 9, 1179–1209. [DOI] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H & Shu Y. (2009). Distinct contributions of Na v 1.6 and Na v 1.2 in action potential initiation and backpropagation. Nature neuroscience 12, 996. [DOI] [PubMed] [Google Scholar]
- Hull JM & Isom LL. (2018). Voltage-gated sodium channel β subunits: the power outside the pore in brain development and disease. Neuropharmacology 132, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO 2nd & Cummins TR. (2010a). Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. The Journal of clinical investigation 120, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO & Cummins TR. (2010b). Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. The Journal of clinical investigation 120, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CN, Potet F, Thompson MK, Kroncke BM, Glazer AM, Voehler MW, Knollmann BC, George AL & Chazin WJ. (2018). A mechanism of calmodulin modulation of the human cardiac sodium channel. Structure 26, 683–694. e683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW & Raman IM. (2003). The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. Journal of Neuroscience 23, 4899–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L & Villasana D. (2011). Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A & Goldin AL. (2008). Role of the amino and carboxy termini in isoform-specific sodium channel variation. The Journal of physiology 586, 3917–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH & Raman IM. (2014). Resurgent current of voltage-gated Na+ channels. The Journal of physiology 592, 4825–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A & Nusser Z. (2008). Cell-type-dependent molecular composition of the axon initial segment. Journal of Neuroscience 28, 14329–14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Lamar T, Goldin AL & Escayg A. (2014). Role of the hippocampus in Na V 1.6 (Scn8a) mediated seizure resistance. Neurobiology of disease 68, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A & Huguenard JR. (2017). Regulation of thalamic and cortical network synchrony by Scn8a. Neuron 93, 1165–1179. e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA & Escayg A. (2007). The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Human molecular genetics 16, 2892–2899. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK & Surmeier DJ. (2001). D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. Journal of Neuroscience 21, 2268–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Helman G, Hammer MF, Fureman BE, Gaillard WD, Goldin AL, Hirose S, Ishii A, Kroner BL, Lossin C, Mefford HC, Parent JM, Patel M, Schreiber J, Stewart R, Whittemore V, Wilcox K, Wagnon JL, Pearl PL, Vanderver A & Scheffer IE. (2016). SCN8A encephalopathy: Research progress and prospects. Epilepsia 57, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JN, Chan CS, Tkatch T, Held J & Surmeier DJ. (2007). Nav1. 6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. Journal of Neuroscience 27, 13552–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HM & Goldin AL. (2010). Sodium channel carboxyl-terminal residue regulates fast inactivation. Journal of Biological Chemistry 285, 9077–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y & Obata K. (2007). Nav1. 1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. Journal of Neuroscience 27, 5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N & Cummins TR. (2016). Aberrant epilepsy-associated mutant Nav1. 6 sodium channel activity can be targeted with cannabidiol. Brain 139, 2164–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Xiao Y & Cummins TR. (2015). Human Nav1.6 Channels Generate Larger Resurgent Currents than Human Nav1.1 Channels, but the Navbeta4 Peptide Does Not Protect Either Isoform from Use-Dependent Reduction. PloS one 10, e0133485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z, Pan Y & Cummins TR. (2017). Posttranslational modification of sodium channels. [DOI] [PubMed]
- Raman IM & Bean BP. (2001). Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J 80, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH & Bean BP. (1997). Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19, 881–891. [DOI] [PubMed] [Google Scholar]
- Royeck M, Horstmann M-T, Remy S, Reitze M, Yaari Y & Beck H. (2008). Role of axonal NaV1. 6 sodium channels in action potential initiation of CA1 pyramidal neurons. Journal of neurophysiology 100, 2361–2380. [DOI] [PubMed] [Google Scholar]
- Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S, Annesi G, Wanke E & Mantegazza M. (2007). Modulatory proteins can rescue a trafficking defective epileptogenic Nav1. 1 Na+ channel mutant. Journal of Neuroscience 27, 11037–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD & Waxman SG. (2005). Electrophysiological properties of two axonal sodium channels, Nav1. 2 and Nav1. 6, expressed in mouse spinal sensory neurones. The Journal of physiology 564, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J & Yan N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326. [DOI] [PubMed] [Google Scholar]
- Shirahata E, Iwasaki H, Takagi M, Lin C, Bennett V, Okamura Y & Hayasaka K. (2006). Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1. 6. Journal of neurophysiology 96, 1347–1357. [DOI] [PubMed] [Google Scholar]
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C & Peiffer A. (2009). A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS genetics 5, e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P & Carr RW. (2012). Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1. 6-resurgent and persistent current. Proceedings of the National Academy of Sciences 109, 6704–6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH & Goldin AL. (1998). Functional analysis of the mouse Scn8a sodium channel. Journal of Neuroscience 18, 6093–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T & Catterall WA. (2005). Ion permeation through a voltage- sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47, 183–189. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH & Goldin AL. (2001). Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. Journal of neuroscience 21, 7481–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprunger LK, Escayg A, Tallaksen-Greene S, Albin RL & Meisler MH. (1999). Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Human molecular genetics 8, 471–479. [DOI] [PubMed] [Google Scholar]
- Sun W, Wagnon JL, Mahaffey CL, Briese M, Ule J & Frankel WN. (2013). Aberrant sodium channel activity in the complex seizure disorder of Celf4 mutant mice. The Journal of physiology 591, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z-Y, Piekarz AD, Priest BT, Knopp KL, Krajewski JL, McDermott JS, Nisenbaum ES & Cummins TR. (2014). Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. Journal of Neuroscience 34, 7190–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A & Matthews G. (2006a). Expression of sodium channels Nav1.2 and Nav1.6 during postnatal development of the retina. Neurosci Lett 403, 315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A & Matthews G. (2006b). Impaired firing and cell-specific compensation in neurons lacking nav1. 6 sodium channels. Journal of Neuroscience 26, 7172–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP & Hammer MF. (2012). De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK & Meisler MH. (2016). Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Annals of clinical and translational neurology 3, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM & Meisler MH. (2014). Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Human molecular genetics 24, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chung BC, Yan H, Lee S-Y & Pitt GS. (2012). Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chung BC, Yan H, Wang H-G, Lee S-Y & Pitt GS. (2014). Structural analyses of Ca 2+/CaM interaction with Na V channel C-termini reveal mechanisms of calcium-dependent regulation. Nature communications 5, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JC, Makinson CD, Lamar T, Cheng Q, Wingard JC, Terwilliger EF & Escayg A. (2018). Selective targeting of Scn8a prevents seizure development in a mouse model of mesial temporal lobe epilepsy. Scientific reports 8, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Barbosa C, Pei Z, Xie W, Strong JA, Zhang J-M & Cummins TR. (2019). Increased resurgent sodium currents in Nav1. 8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. Journal of Neuroscience 39, 1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Tan ZY, Barbosa C, Strong JA, Cummins TR & Zhang JM. (2016). Upregulation of the sodium channel NaVbeta4 subunit and its contributions to mechanical hypersensitivity and neuronal hyperexcitability in a rat model of radicular pain induced by local dorsal root ganglion inflammation. Pain 157, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wang C, Marx SO & Pitt GS. (2017). Calmodulin limits pathogenic Na+ channel persistent current. The Journal of general physiology, jgp. 201611721. [DOI] [PMC free article] [PubMed] [Google Scholar]