

Abstract
We report on the structures of three unprecedented heteroleptic Sb‐centered radicals [L(Cl)Ga](R)Sb. (2‐R, R=B[N(Dip)CH]2 2‐B, 2,6‐Mes2C6H3 2‐C, N(SiMe3)Dip 2‐N) stabilized by one electropositive metal fragment [L(Cl)Ga] (L=HC[C(Me)N(Dip)]2, Dip=2,6‐i‐Pr2C6H3) and one bulky B‐ (2‐B), C‐ (2‐C), or N‐based (2‐N) substituent. Compounds 2‐R are predominantly metal‐centered radicals. Their electronic properties are largely influenced by the electronic nature of the ligands R, and significant delocalization of unpaired‐spin density onto the ligands was observed in 2‐B and 2‐N. Cyclic voltammetry (CV) studies showed that 2‐B undergoes a quasi‐reversible one‐electron reduction, which was confirmed by the synthesis of [K([2.2.2]crypt)][L(Cl)GaSbB[N(Dip)CH]2] ([K([2.2.2]crypt)][2‐B]) containing the stibanyl anion [2‐B]−, which was shown to possess significant Sb−B multiple‐bonding character.
Keywords: antimony, EPR spectroscopy, main-group elements, radicals, X-ray diffraction
Radical tuning: Three unprecedented heteroleptic stibanyl radicals were synthesized and their electronic structures elucidated by single‐crystal X‐ray diffraction, EPR spectroscopy, UV/Vis spectroscopy, cyclic voltammetry, and DFT calculations. The boryl‐substituted radical undergoes one‐electron reduction to give the corresponding stibanyl anion, which was shown to possess partial Sb−B double‐bond character.
Introduction
Main group element centered radicals1, 2, 3, 4 not only contributed to our current understanding of chemical bonding in open‐shell main group element compounds,5 but also show intriguing reactivities such as the activation of small molecules, that is, H2, CO, CO2, and others.6, 7 They also form important intermediates in various chemical and biological processes. Unfortunately, due to a limited number of energetically accessible valence orbitals, stable main group element centered radicals are still elusive in nature. With respect to Group‐15 elements, stable and persistent neutral,8 anionic,9 and cationic P‐centered radicals10 are known. In remarkable contrast, the chemistry of Sb‐centered radicals is far less explored due to their high tendency toward either Sb−Sb bond formation (dimerization) or disproportionation into elemental Sb and stibanes R3Sb.11 Neutral stibanyl radicals R2Sb. were first postulated by Paneth as intermediates in the reaction of elemental Sb with methyl radicals,12 and later found in chemical vapor deposition (CVD) processes.13 Unfortunately, their electronic stabilization and isolation in the solid state remains challenging, and only a few persistent or stable Sb‐centered radicals have been reported (Figure 1).
Figure 1.
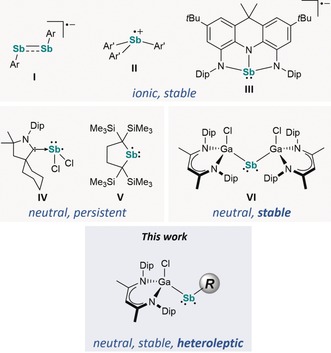
Stable and persistent neutral and charged Sb‐centered radicals.
Ishida et al. spectroscopically characterized a neutral stibanyl radical [H2CC(SiMe3)2]2Sb. (V) in solution, formed by homolytic Sb−Sb bond cleavage of Sb2{[C(SiMe3)2CH2]2}4.14 Bertrand et al. reported on the carbene‐coordinated stibanyl radical (cAAC)SbCl2 (IV) (cAAC=cyclic (alkyl)(amino)carbene),15 whereas the distibene radical anion [ArSbSbAr].− (I) (Ar=2,6‐(CH(SiMe3)2)2‐4‐C(SiMe3)3C6H2)16 and the stibane radical anion [NNNSb].− containing a diamidodihydroacridinide ligand (III)9d as well as triarylstibane radical cations [Ar′3Sb].+ (II) (Ar′=2,6‐i‐Pr2‐4‐OMeC6H2, 2,4,6‐i‐Pr3C6H2)17 were synthesized by one‐electron reduction and oxidation reactions of the corresponding neutral counterparts. Recently, we reported on the isolable neutral, two‐coordinated stibanyl radical [L(Cl)Ga]2Sb. (VI), formed by reaction of [L(Cl)Ga]Sb(Cl)Cp* (VII) (Cp*=C5Me5) with LGa, which occurred through an insertion of LGa into the Sb−Cl bond followed by a homolytic Sb−Cp* bond cleavage.18
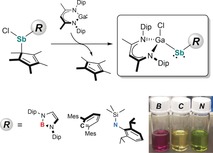
We became interested in extending this novel approach to heteroleptic stibanyl radicals [L(Cl)Ga]SbR in order to elucidate the effect of differing electronic properties of the ligand R on the electronic structure of the radicals and report herein on the synthesis, structure, and reactivity of a series of Sb‐centered radicals [L(Cl)Ga]{B[N(Dip)CH]2}Sb. (2‐B), [L(Cl)Ga](2,6‐Mes2C6H3)Sb. (2‐C), and [L(Cl)Ga][N(SiMe3)Dip]Sb. (2‐N).
Results and Discussion
Replacement of one [L(Cl)Ga] ligand in VI by a boryl, aryl, or amido substituent with comparable size was expected to influence the electronic structure of the radicals due to the differing electronegativity (B<C<N) as well as the availability of a vacant p orbital (B) or a lone pair of electrons in a p orbital (N). Based on the calculated buried volume V bur 19 of [L(Cl)Ga] (35.3 %), ligands B (34.9 %), C (36.8 %), and N (34.5 %) were identified as promising candidates with appropriate steric demand (Figure S1, Supporting Information). Compounds RSb(Cl)Cp* (1‐R) were synthesized in high yields by salt‐metathesis reactions of Cp*SbCl2 with RLi (Scheme 1) and used without further work‐up. The synthesis of 1‐B, a rare borylstibane with Sb−B single bond, is remarkable, since Jones et al. observed reduction of (2,6‐{CHPh2}‐4‐i‐PrC6H2)(Si(i‐Pr3))NSbBr2 upon treatment with BLi(THF)2.20
Scheme 1.
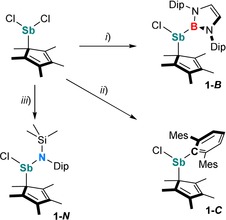
Synthesis of 1‐R (R=B, C, N). Reagents: i) BLi(THF)2, −LiCl, −2 THF; ii) CLi, −LiCl; iii) NLi, −LiCl.
Stibanes 1‐R are thermally stable solids and soluble in common organic solvents. Their 1H and 13C{1H} NMR spectra exhibit the expected resonances of the organic ligands (R, Cp*). The 1H NMR spectrum of 1‐B shows two septets and four doublets for the CHMe2 and CHMe 2 protons of the Dip groups, indicating an asymmetrical environment at the Sb center, whereas no comparable splitting was observed in the 1H NMR spectra of 1‐C and 1‐N. The Cp* methyl protons give single resonances in all cases. 1‐B shows a broad 11B NMR resonance at 38.1 ppm, comparable to that of [DipNC(t‐Bu)NN(Ph)B]SbPh2 (30.9 ppm).21
Compounds 1‐R were then reacted with LGa (Scheme 2). While no reactions occurred at ambient temperature, a color change and formation of decamethyl‐1,1′‐dihydrofulvalene (Cp*2), identified by in‐situ 1H NMR spectroscopy, was observed at elevated temperatures (80–95 °C). After recrystallization from n‐hexane, the stibanyl radicals 2‐R were isolated as red (2‐B), orange (2‐C), and green (2‐N) crystals. Noticeably, the formation of radicals 2‐R was only observed when reacting 1‐R with an equimolar amount of LGa, whereas salt‐elimination reactions of VII with RLi only gave mixtures of so far unidentified species in which none of the radicals 2‐R were observed.
Scheme 2.
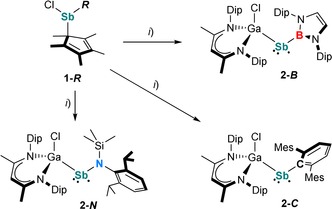
Synthesis of 2‐R (R=B, C, N). Reagents: i) LGa, −0.5 Cp*2.
Radicals 2‐R are stable both in the solid state and in solution at ambient temperature under an argon atmosphere, but immediately decompose upon exposure to air or moisture. Due to their paramagnetic nature, the 1H NMR spectra of 2‐R only exhibit broad resonances, as observed for [L(Cl)Ga]2Sb.18 The paramagnetic character of 2‐R was further confirmed by their effective magnetic moments μ eff of 1.58 μ B (2‐B), 1.61 μ B (2‐C), and 1.56 μ B (2‐N), determined using the Evans method. These values correspond well to the expected value of 1.73 μ B for a single unpaired electron.22 Based on variable‐temperature (VT) 1H NMR studies (Figures S13, S16, and S19), the dimerization of the radicals to diamagnetic distibanes, which was observed for [H2CC(SiMe3)2]2Sb. V,14 can be excluded even at −80 °C. The formation of stibanes [L(Cl)Ga]Sb(H)R containing an Sb−H function was also excluded due to the absence of any absorption bands in the IR spectra of 2‐R between 1600 to 2000 cm−1, the expected region for an Sb−H group ([L(Cl)Ga]Sb(H)Cp*, ν Sb−H=1855 cm−1).18
Single crystals of radicals 2‐R were obtained from solutions in n‐hexane upon storage at ambient temperature.23 The molecular structures of 2‐R (Figure 2) feature two‐coordinate Sb centers. The Ga‐Sb‐E bond angles of 113.40(14)° (2‐B), 114.80(4)° (2‐C), and 111.45(5)° (2‐N) are much larger compared to [L(Cl)Ga]2Sb. VI (104.89(1)°),18 but fairly agree with the Ga‐As‐Ga bond angle in [L(Cl)Ga]2As. (109.43(6)°).24 The Ga−Sb bond lengths (2‐B 2.5959(7) Å, 2‐C 2.635(3) Å, 2‐N 2.6258(3) Å) compare well to those of VI (2.5899(4) Å, 2.5909(3) Å),18 [L(Cl)Ga]2Sb2 (2.58178(19) Å),25 {[L(Cl)Ga]2(μ,η1:1‐Sb4)} (2.6008(13) Å, 2.6044(14) Å),26 and the sum of the computed covalent radii (∑r cov(Ga−Sb)=2.64 Å).27 The Sb−B bond length in 2‐B (2.245(5) Å) is comparable to that of [DipNC(t‐Bu)NN(Ph)B]SbPh2 (2.257(5) Å)21 and the sum of the covalent radii (∑r cov(Sb−B)=2.25 Å),27 while the Sb−C bond length in 2‐C (2.1716(16) Å) is virtually identical with those of LGa=Sb‐C (2.180(3) Å)24 and C‐Sb=Sb‐C (2.169(4) Å).29 The Sb−N distance in 2‐N (2.0502(18) Å) is close to the average value of Sb−N bond lengths observed in aminostibanes (2.036 Å)29 containing a SbN(Ar)(SiR2X) group (Ar=2,4,6‐t‐Bu3C6H2, 2,6‐Mes2C6H3, 2,4,6‐CHPh2‐4‐i‐PrC6H2, Dip; R=Me, i‐Pr; X=Me, i‐Pr, Cl, N3, OSO2CF3, N(H)Dip). The N atom in 2‐N is planar‐coordinated (∑ϕ(N3)=359.9(4)°) and the Ga atom adopts an in‐plane orientation with respect to the Sb‐N‐Si‐C plane (torsion angles: Si1‐N3‐Sb1‐Ga1 9.55(12)°, C33‐N3‐Sb1‐Ga1 168.66(12)°).
Figure 2.
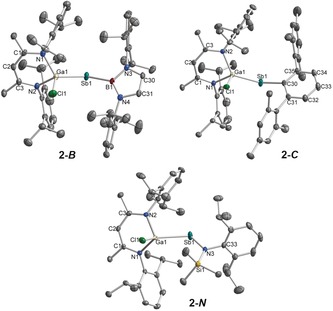
Molecular structures of 2‐R. Hydrogen atoms and the minor component of the disordered i‐Pr group in 2‐C were omitted for clarity. Displacement ellipsoids are drawn at 50 % probability level. Selected bond lengths [Å] and angles [°]: 2‐B: Ga1−Sb1 2.5959(7), Ga1−Cl1 2.2085(13), Sb1−B1 2.245(5), Ga1−Sb1−B1 113.40(14), Sb1−B1−N3 119.5(3), Sb1−B1−N4 133.3(3); 2‐C: Ga1−Sb1 2.6335(3), Ga1−Cl1 2.2178(5), Sb1−C30 2.1716(16), Ga1−Sb1−C30 114.80(4), Sb1−C30−C31 123.17(12), Sb1−C30−C35 117.10(12); 2‐N: Ga1−Sb1 2.6258(3), Ga1−Cl1 2.2213(6), Sb1−N3 2.0502(18), Ga1−Sb1−N3 111.45(5), Sb1−N3−Si1 130.05(10), Sb1−N3−C33 109.17(13).
The continuous‐wave (CW) X‐band (≈9.6 GHz) and Q‐band (≈34 GHz) frozen‐solution EPR spectra of 2‐R exhibit S=1/2 signals over a broad magnetic‐field range (Figure 3). The complex multi‐line EPR spectra observed are similar to that of homoleptic radical VI. The heteroleptic nature of 2‐R offers differing electronic structures and nuclear hyperfine properties from the naturally abundant isotopes of 121Sb (57.2 %, I=5/2), 123Sb (42.8 %, I=7/2), 69Ga (60.1 %, I=3/2), 71Ga (38.9 %, I=3/2), 10B (19.9 %, I=3), 11B (80.1 %, I=3/2), and 14N (99.6 %, I=1). Only a single Ga atom is present in 2‐R, potentially simplifying the observed super‐hyperfine splitting pattern of the EPR spectra compared to VI.18 For the 2‐R series, both the high‐field and low‐field extremes of the X‐band EPR spectrum allow for the refinement of g 3 and associated Sb and Ga A 3 hyperfine couplings.18 The high‐field edges of the X‐ and Q‐band spectra of 2‐C have identical patterns, resolving the g 3, A 3(121Sb), and A 3(69Ga) values (Table 1). The low‐field region of the spectra differs dramatically due to the increased g‐separation at higher microwave frequencies. The Q‐band spectrum of 2‐C exhibits broader, less resolved hyperfine splittings at the low‐field region where g 1 and A 1 hyperfine splittings are observed. This broadening is possibly due to orientation or g‐strain. Refinement of the remaining EPR parameters is possible through the parallel simulation of both the X‐ and Q‐band EPR spectra and evaluation of the integrated EPR spectra and simulations (Figure S25).
Figure 3.
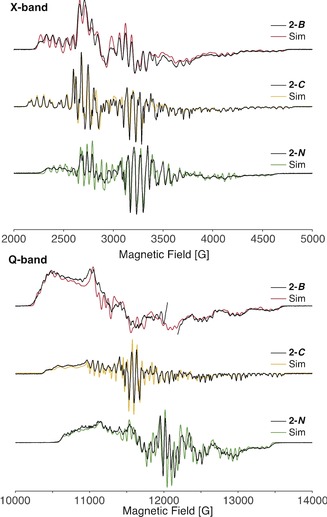
Continuous‐wave X‐band (top) and Q‐band (bottom) of 2‐R (black) with simulations (colored lines). A sharp radical impurity of the Q‐band 2‐B sample at the g≈2 region (≈12 050 G) is omitted. Spectrometer conditions detailed in the Supporting Information. EPR simulation parameters; 2‐B: g=[2.279, 2.071, 1.970]; A(Sb)=[300, 500, 1240] MHz; A(Ga)=[173, 168, 150] MHz; A(B)=[20.8, 23.5, 25.1] MHz, [α, β, γ]=[‐10°, 35°, 0°]; g‐strain=[0.008, 0.008, 0]; X‐band lw (linewidth) (fwhm)=3.0 G; Q‐band lw=4.0 G. 2‐C: g=[2.215, 2.025, 1.972]; A(Sb)=[500, 560, 1330] MHz; A(Ga)=[169, 179, 169] MHz; X‐band lw=2.0 G; Q‐band lw=1.2 G; g‐strain=[0.012, 0, 0] (Q‐band only). 2‐N: g=[2.151, 1.980, 1.972]; A(Sb)=[651, 782, 1140] MHz; A(Ga)=[149, 163, 155] MHz; X‐band lw=2.0 G H‐strain=[30, 10, 80] MHz; Q‐band lw=1.5 G; H‐strain=[160, 50, 120] MHz.
Table 1.
EPR parameters of VI and 2‐R.
|
|
[L(Cl)Ga]2Sb[a] |
2‐B |
2‐C |
2‐N |
|---|---|---|---|---|
|
g 1 |
2.298 |
2.279 |
2.215 |
2.151 |
|
g 2 |
2.114 |
2.071 |
2.025 |
1.977 |
|
g 3 |
1.967 |
1.970 |
1.972 |
1.976 |
|
g iso [b] |
2.126 |
2.107 |
2.071 |
2.035 |
|
%R |
80 % |
49 % |
28 % |
1 % |
|
| ||||
|
121Sb Hyperfine Couplings [MHz][c] | ||||
|
A 1 |
−385 |
−300 |
−500 |
−651 |
|
A 2 |
−496 |
−500 |
−560 |
−763 |
|
A 3 |
1138 |
1240 |
1330 |
1160 |
|
a iso |
86 |
147 |
90 |
−86 |
|
T 1 |
−471 |
−447 |
−590 |
−569 |
|
T 2 |
−582 |
−647 |
−650 |
−677 |
|
T 3 |
1052 |
1093 |
1240 |
1246 |
|
| ||||
|
69Ga hyperfine couplings [MHz][d] | ||||
|
A 1 |
132 |
173 |
169 |
149 |
|
A 2 |
176 |
168 |
179 |
166 |
|
A 3 |
134 |
150 |
169 |
154 |
[a] Data taken from ref. 18. [b] g iso=(g 1+g 2+g 3)/3. [c] Values reported for the 121Sb isotope (gn=1.345), 123Sb hyperfine (gn=0.729) values obtained from the gyromagnetic ratio (A(123Sb)=gn(123Sb)/gn(123Sb)*A(121Sb)), [d] 69Ga isotope (gn=1.344) hyperfine values reported, 71Ga hyperfine value (gn=1.708) obtained from the gyromagnetic ratio.
The fitted EPR parameters for 2‐R are detailed in Table 1. All three complexes exhibit g 3‐values in the range of 1.970–1.976, close to the free‐electron value of ge=2.0023. Clear shifts of the g 1 feature of the Q‐band spectra and giso‐values are observed: 2‐B>2‐C>2‐N. 2‐B also exhibits the most rhombic EPR spectrum of the 2‐R group with a 49 % R‐value, where the rhombicity is given by [Eq. (1)]:
| (1) |
2‐N exhibits the least g‐anisotropy with a low g 1‐value of 2.151 and a nearly axial EPR spectrum (R=1 %). Both 2‐B and 2‐C have g‐tensors of the form g 1>g 2>ge>g 3, whereas the g‐tensor of 2‐N is almost axial and has a slightly different form with g 2<ge.
The solution EPR spectra of 2‐B and 2‐N yield a single broad ≈300 G wide (peak‐to‐peak) feature centered at approximately the g iso‐values determined for the frozen‐solution samples (Figure S26). The solution spectra fail to exhibit any resolved hyperfine splittings in the very broad signal.
For the 2‐R series and the previously published [L(Cl)Ga]2Sb. (VI),18 excellent periodic trends emerge. For instance, the less electronegative the Sb‐coordinating atom in R, the more anisotropic the EPR spectrum is, and the larger the g 1‐value is (Figure 4). The same trend is observed for the relationship of electronegativity of the R substituent and degree of rhombicity (%R) of the EPR spectrum.
Figure 4.
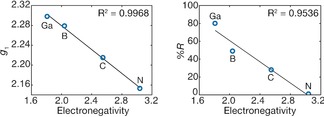
Linear trends of observed g 1‐value (left) and rhombicity (%R, right) vs. Pauling electronegativity value of the ligand.
The hyperfine tensor of the Sb centers, including signs, has been determined as previously published.18, 24, 30 For each, a minimal isotropic hyperfine coupling value, a iso, is observed as expected for minimal s‐orbital spin density.31 The resultant anisotropic Sb hyperfine tensor, T=A−a iso, observed for 2‐R are all nearly axial, [‐t, ‐t, 2t], characteristic of the unpaired electron centered in a p orbital,18, 24 with an estimated p‐orbital density from +0.87 (2‐B) to +0.98 (2‐N).32 The anisotropic tensor of 2‐B exhibits the largest degree of rhombicity, indicating small perturbations to the Sb spin density induced by the π‐accepting character of the boron substituent, which facilitates delocalization of spin density onto the ligand. Each complex clearly exhibits a modest amount of delocalization of spin density onto its [L(Cl)Ga] ligand, as indicated by the comparable Ga hyperfine couplings. Because the Ga hyperfine coupling appears relatively constant throughout the series, it is immediately concluded that the ligands R are dominating the electronic properties of the Sb radical center.
2‐C exhibits the narrowest linewidth and most well‐resolved hyperfine pattern due to the lack of hyperfine broadening of the unlabeled C substituent. 2‐B and 2‐N both exhibit broader lines due to additional possible 10/11B and 14N hyperfine interactions but are not well‐resolved from analysis of the EPR spectra alone.
The B hyperfine interaction of 2‐B was measured by pulsed Q‐band Davies electron–nuclear double‐resonance (ENDOR) spectroscopy (Figure S27), where both the 11B and 10B isotopes are observed. Analysis of the isotropic and local anisotropic hyperfine components reveals a modest unpaired spin density on the boron atom: ρ(s)≈0.009, ρ(p)≈0.031 (see Supporting Information). This is moderately less than the observed spin density observed on the more electropositive Ga ligands of VI 18 and 2‐B, which is consistent with previous observations that the spin density of a heteroatom bound to a radical center decreases as the electronegativity of the heteroatom increases.16 Hence, small 14N hyperfine couplings from the increased electronegativity of the N ligand (see Supporting Information) were anticipated, but no identifiable 14N ENDOR, ESEEM, or HYSCORE responses were observed for 2‐N. A maximum A 3(14N) hyperfine coupling of ≈40 MHz may be used to broaden the simulated EPR spectrum without further splitting the EPR spectrum (Figure S29), but does not reliably estimate the 14N hyperfine coupling.
DFT calculations34 closely reproduce the central parameters of the XRD structures of 2‐R (Δr<0.02 Å, Δϕ<2°) and support the assignment of 2‐R as Sb p‐orbital centered radicals (Figure S25). The singly occupied molecular orbitals (SOMO) in 2‐R consist mainly of the Sb 5p orbital, whereas 2‐B and 2‐N exhibit additional contributions of the diazaborolyl π‐system and the nitrogen lone pair (Figure 5). The calculated SOMO levels of 2‐R (2‐B −5.13 eV, 2‐C −5.05 eV, 2‐N −4.78 eV) decrease in energy from 2‐N to 2‐C and 2‐B, which is in agreement with the UV/Vis and CV results (see below). The charges at the Sb center (2‐B 0.09 e, 2‐C 0.35 e, 2‐N 0.51 e) and bond polarizations of the Sb−R bonds (2‐B 53.4 % B, 46.6 % Sb; 2‐C 68.7 % C, 31.3 % Sb; 2‐N 82.6 % N, 17.4 % Sb) obtained from natural population analysis (NPA) and natural bond orbital (NBO) analysis correlate with the electronegativity of the Sb‐bound atom in the ligands R.
Figure 5.
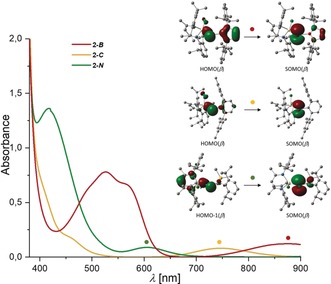
UV/Vis absorption spectra of 2‐R and associated orbital transitions obtained from TD‐DFT calculations.
The ambient‐temperature UV/Vis spectra of stibanyl radicals 2‐R in toluene (Figure 5) feature low‐energy absorptions at λ max=875 (2‐B), 746 (2‐C), and 605 nm (2‐N). Based on time‐dependent (TD) DFT calculations, these absorptions were attributed to electronic transitions from mainly σ(Ga−Sb)‐type orbitals to the SOMOs, thus supporting the observed trend in the SOMO levels. Additional absorption bands at λ max=526 (2‐B), 457 (2‐C), and 417 nm (2‐N), which correspond to ligand‐to‐metal transfer processes, show a similar trend (Figures S26–S28). These findings are consistent with the EPR results. Since the g‐tensor is a measurement of the interaction of the ground‐state electron with low‐lying unoccupied orbitals among several other interactions, the observed g‐shift is inversely proportional to the energy gap between the ground state and the excited state. The trend of the energy gap for the low‐lying excited state and the SOMO matches that observed by UV/Vis spectroscopy, ΔE: 2‐B<2‐C<2‐N.
One‐electron reduction and oxidation reactions of the radicals 2‐R were studied by cyclic voltammetry (CV, Figure 6). The CV traces of 2‐R each feature irreversible one‐electron oxidations at E p,a=0.10 V (2‐B), −0.07 V (2‐C), and −0.14 V (2‐N) against Fc0/+, which are attributed to the formation of the corresponding cationic counterparts [2‐R]+ by removal of the unpaired electron from the SOMO. The increasing oxidation potentials (2‐B>2‐C>2‐N) are consistent with the computed SOMO energies, as the unpaired electron is more easily removed from a high‐lying SOMO. 2‐C and 2‐N further show irreversible one‐electron reductions at E p,c=−1.83 V and −1.84 V, respectively, whereas 2‐B features a quasi‐reversible one‐electron reduction at E 1/2=−1.78 V. These reduction events are attributed to the formation of the corresponding anionic stibanides [2‐R]−, of which [2‐B]− is seemingly stable. In contrast to the oxidation potentials, the reduction potentials of 2‐R are similar.
Figure 6.
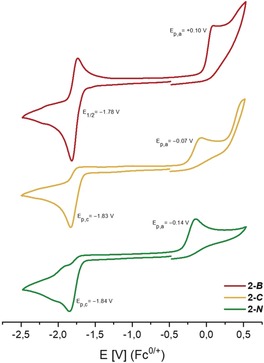
Cyclic voltammograms of 2‐R in THF solution (1 mm) containing [n‐Bu4N][B(3,5‐(CF3)2‐C6H3)4] (50 mm) as electrolyte at 100 mV s−1 scan rate.
DFT calculations, UV/Vis, EPR, and CV studies show decreasing SOMO energies in the order 2‐N>2‐C>2‐B. This is remarkable, because more electronegative substituents (−I effect) typically lead to a stronger stabilization of the frontier orbitals, hence the opposite trend would be expected based on the electronegativities of the Sb‐bound atoms in R (B: 2.04, C: 2.55, N: 3.04 by Pauling's scale).35 However, in 2‐B, the unique π‐acceptor properties (−M effect) of the boryl ligand, which allowed for the stabilization of unusual B−O and B−S double bonds,36 stabilizes the SOMO in 2‐B. The π‐donating amido substituent (+M effect) in 2‐N destabilizes the SOMO level due to an interaction comparable to the α‐effect.37 In 2‐C, an interaction of the SOMO with the π‐system of the central aryl ring of the terphenyl ligand is not observed. Furthermore, it was shown that the π‐acidic nature of the [L(Cl)Ga] ligand, as confirmed by EPR spectroscopy, efficiently stabilizes main group element radical centers.18, 24
According to the promising results of the electrochemical studies, 2‐B was reacted with [K([2.2.2]crypt)][C10H8] (Scheme 3). Dark green crystals of [K([2.2.2]crypt)][L(Cl)GaSb−B] ([K([2.2.2]crypt)][2‐B]) containing the stibanyl anion [2‐B]− were obtained and characterized by multinuclear NMR and IR spectroscopy as well as single‐crystal X‐ray diffraction. The 1H NMR spectrum of [K([2.2.2]crypt)][2‐B] exhibits the expected resonances for the ligands (B, [L(Cl)Ga]) and [2.2.2]cryptand in a 1:1:1 ratio. The 11B NMR spectrum shows a broad resonance at 31.8 ppm, indicating increased electron density at B compared to 1‐B.
Scheme 3.
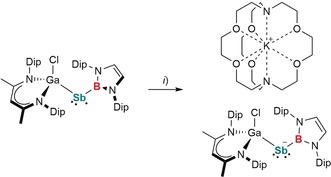
Synthesis of [K([2.2.2]crypt)][2‐B]. Reagents: i) [K([2.2.2]crypt)][C10H8], −C10H8.
Single crystals of [K([2.2.2]crypt)][2‐B] suitable for X‐ray diffraction were obtained from a saturated fluorobenzene solution upon storage at ambient temperature (Figure 7).23
Figure 7.
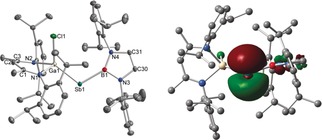
Molecular structure and HOMO of the anion in [K([2.2.2]crypt)][2‐B]. Hydrogen atoms, the cation [K([2.2.2]crypt)]+, and one molecule of fluorobenzene were omitted for clarity. Displacement ellipsoids are drawn at 50 % probability level. Selected bond lengths [Å] and angles [°]: Ga1−Sb1 2.5344(2), Ga1−Cl1 2.2820(4), Sb1−B1 2.2067(13), Ga1‐Sb1‐B1 110.79(4).
The Ga1−Sb1−B1 angle of 110.79(4)° in the anion [2‐B]− is slightly more acute in comparison to the neutral stibanyl radical 2‐B, which is in accordance with VSEPR theory, and caused by the electron lone pair occupying more space than a single unpaired electron. The Ga1−Sb1 (2.5344(2) Å) and Sb1−B1 (2.2067(13) Å) bond lengths in [2‐B]− are significantly shorter compared to those of 2‐B (Ga1−Sb1 2.5959(7), Sb1−B1 2.245(5) Å), indicating back‐bonding from the electron‐rich Sb to the Ga and B atoms. The Ga1−Sb1−B1−N3/N4 torsion angles (163.41(8)°, 23.86(16)°) in [2‐B]− are less twisted compared to 2‐B (123.6(4)°, 76.4(5)°), hence enabling an Sb−B π‐interaction. Compared to 2‐B, the Ga1−Cl1 (2.2820(4) Å) and Ga1−N1/2 (2.0156(10) Å, 2.0395(10) Å) bond lengths in [K([2.2.2]crypt)][2‐B] are elongated, further pointing to a back‐bonding contribution from Sb to the Ga atom. These conclusions are supported by DFT calculations. The optimized geometry of [2‐B]− shows Ga−Sb and Sb−B bond lengths of 2.541 Å and 2.204 Å, respectively, and a Ga‐Sb‐B bond angle of 107.91°, in excellent agreement with the experimental findings. The HOMO of [2‐B]− (Figure 7) is the Sb p‐orbital centered lone pair of electrons. According to NBO and second‐order perturbation theory (SOPT) analyses, the HOMO of [2‐B]− is of pure p‐orbital character (99.8 % p) and exhibits a strong π‐interaction of 50.9 kcal mol−1 to the vacant p‐orbital at B (99.9 % p). An additional interaction of the p‐orbital centered Sb lone pair to the antibonding Ga−N orbitals of 30.3 kcal mol−1 is also observed. Wiberg bond indices (WBI) of 1.25 and 1.19 for the Sb−B and Sb−Ga bonds, respectively, also evidence significant Sb−B and Sb−Ga multiple bonding. Hence, these findings give a first insight into Sb−B double bonding, which hitherto remained unknown, as double‐bonding interactions to B are only known for the lighter Group‐15 homologues N, P, and As.38
Conclusion
A series of stable, neutral, and heteroleptic antimony‐centered radicals 2‐B, 2‐C, and 2‐N were synthesized by a LGa‐induced Sb−Cp* bond‐cleavage reaction. EPR characterization of 2‐R demonstrates an antimony p‐orbital centered radical character and modest delocalization of the unpaired spin onto the ligands. The electronegativity of the ligand significantly perturbs the electronics of the Sb radical and correlates with the EPR parameters. The 2‐R series studied here, with the inclusion of radical VI, sets the foundation for future heavy main group electronic‐structure predictions. The effect of the ligands on the mainly antimony‐centered SOMO was verified by DFT calculations, UV/Vis spectroscopy, and cyclic voltammetry. Contrary to the general understanding, the electropositive boryl ligand in 2‐B causes a stabilization of the SOMO due to its π‐acceptor ability, whereas the π‐donor property of the electronegative amide ligand in 2‐N leads to a destabilization of the SOMO. The unique properties of the boryl ligand further enabled the one‐electron reduction of radical 2‐B, yielding the stibanide anion [2‐B]− with Sb−B multiple bonding character. These novel and uncommon species illustrate the versatility of the metal–carbon bond‐cleavage approach to generate radical species which could be applied for the synthesis of rare main group element centered radicals.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The University of Duisburg‐Essen (S.S.), the German Research Foundation DFG (S.S.; research grant SCHU 1069/23‐1), Evonik Industries (doctoral fellowship C.H.), and the Max‐Planck‐Gesellschaft (G.E.C.) are gratefully acknowledged for financial support.
C. Helling, G. E. Cutsail, H. Weinert, C. Wölper, S. Schulz, Angew. Chem. Int. Ed. 2020, 59, 7561.
In memory of Professor Carsten Schmuck
Contributor Information
Dr. George E. Cutsail, III, Email: george.cutsail@cec.mpg.de.
Prof. Dr. Stephan Schulz, Email: stephan.schulz@uni-due.de.
References
- 1.
- 1a. Power P. P., Chem. Rev. 2003, 103, 789–809; [DOI] [PubMed] [Google Scholar]
- 1b. Tan G., Wang X., Chin. J. Chem. 2018, 36, 573–586. [Google Scholar]
- 2. Hicks R. G., Org. Biomol. Chem. 2007, 5, 1321–1338. [DOI] [PubMed] [Google Scholar]
- 3. Chivers T., Konu J. in Comprehensive Inorganic Chemistry II, Vol. 1 (Eds.: J. Reedijk, K. Poeppelmeier), Elsevier, Amsterdam, 2013, pp. 349–373. [Google Scholar]
- 4. Martin C. D., Soleilhavoup M., Bertrand G., Chem. Sci. 2013, 4, 3020–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boeré R. T. in Electron Paramagnetic Resonance, Vol. 23 (Eds.: B. C. Gilbert, D. M. Murphy, V. Chechik), RSC, London, 2012, pp. 22–57. [Google Scholar]
- 6. Power P. P., Acc. Chem. Res. 2011, 44, 627–637. [DOI] [PubMed] [Google Scholar]
- 7. Yadav S., Saha S., Sen S. S., ChemCatChem 2016, 8, 486–501. [Google Scholar]
- 8.Selected neutral P-centered radicals:
- 8a. Hinchley S. L., Morrison C. A., Rankin D. W. H., Macdonald C. L. B., Wiacek R. J., Cowley A. H., Lappert M. F., Gundersen G., Clyburne J. A. C., Power P. P., Chem. Commun. 2000, 2045–2046; [DOI] [PubMed] [Google Scholar]
- 8b. Hinchley S. L., Morrison C. A., Rankin D. W. H., Macdonald C. L. B., Wiacek R. J., Voigt A., Cowley A. H., Lappert M. F., Gundersen G., Clyburne J. A. C., Power P. P., J. Am. Chem. Soc. 2001, 123, 9045–9053; [DOI] [PubMed] [Google Scholar]
- 8c. Ito S., Kikuchi M., Yoshifuji M., Arduengo A. J., Konovalova T. A., Kispert L. D., Angew. Chem. Int. Ed. 2006, 45, 4341–4345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4447–4451; [Google Scholar]
- 8d. Agarwal P., Piro N. A., Meyer K., Müller P., Cummins C. C., Angew. Chem. Int. Ed. 2007, 46, 3111–3114; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3171–3174; [Google Scholar]
- 8e. Beweries T., Kuzora R., Rosenthal U., Schulz A., Villinger A., Angew. Chem. Int. Ed. 2011, 50, 8974–8978; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9136–9140; [Google Scholar]
- 8f. Ishida S., Hirakawa F., Iwamoto T., J. Am. Chem. Soc. 2011, 133, 12968–12971; [DOI] [PubMed] [Google Scholar]
- 8g. Tondreau A. M., Benko Z., Harmer J. R., Grützmacher H., Chem. Sci. 2014, 5, 1545–1554; [Google Scholar]
- 8h. Hirakawa F., Ichikawa H., Ishida S., Iwamoto T., Organometallics 2015, 34, 2714–2716; [Google Scholar]
- 8i. Guo J.-D., Nagase S., Power P. P., Organometallics 2015, 34, 2028–2033; [Google Scholar]
- 8j. Blum M., Puntigam O., Plebst S., Ehret F., Bender J., Nieger M., Gudat D., Dalton Trans. 2016, 45, 1987–1997; [DOI] [PubMed] [Google Scholar]
- 8k. Fischbach U., Trincado M., Grützmacher H., Dalton Trans. 2017, 46, 3443–3448; [DOI] [PubMed] [Google Scholar]
- 8l. Abbenseth J., Delony D., Neben M. C., Würtele C., de Bruin B., Schneider S., Angew. Chem. Int. Ed. 2019, 58, 6338–6341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6404–6407. [Google Scholar]
- 9.Selected anionic P-centered radicals:
- 9a. Pan X., Wang X., Zhao Y., Sui Y., Wang X., J. Am. Chem. Soc. 2014, 136, 9834–9837; [DOI] [PubMed] [Google Scholar]
- 9b. Tan G., Li S., Chen S., Sui Y., Zhao Y., Wang X., J. Am. Chem. Soc. 2016, 138, 6735–6738; [DOI] [PubMed] [Google Scholar]
- 9c. Asami S. S., Ishida S., Iwamoto T., Suzuki K., Yamashita M., Angew. Chem. Int. Ed. 2017, 56, 1658–1662; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1680–1684; [Google Scholar]
- 9d. Mondal M. K., Zhang L., Feng Z., Tang S., Feng R., Zhao Y., Tan G., Ruan H., Wang X., Angew. Chem. Int. Ed. 2019, 58, 15829–15833; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15976–15980. [Google Scholar]
- 10.Selected cationic P-centered radicals:
- 10a. Back O., Celik M. A., Frenking G., Melaimi M., Donnadieu B., Bertrand G., J. Am. Chem. Soc. 2010, 132, 10262–10263; [DOI] [PubMed] [Google Scholar]
- 10b. Back O., Donnadieu B., Parameswaran P., Frenking G., Bertrand G., Nat. Chem. 2010, 2, 369–373; [DOI] [PubMed] [Google Scholar]
- 10c. Pan X., Chen X., Li T., Li Y., Wang X., J. Am. Chem. Soc. 2013, 135, 3414–3417; [DOI] [PubMed] [Google Scholar]
- 10d. Pan X., Su Y., Chen X., Zhao Y., Li Y., Zuo J., Wang X., J. Am. Chem. Soc. 2013, 135, 5561–5564; [DOI] [PubMed] [Google Scholar]
- 10e. Brückner A., Hinz A., Priebe J. B., Schulz A., Villinger A., Angew. Chem. Int. Ed. 2015, 54, 7426–7430; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7534–7538; [Google Scholar]
- 10f. Wang W., Xu C.-Q., Fang Y., Zhao Y., Li J., Wang X., Angew. Chem. Int. Ed. 2018, 57, 9419–9424; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9563–9568; [Google Scholar]
- 10g. Sharma M. K., Rottschäfer D., Blomeyer S., Neumann B., Stammler H.-G., van Gastel M., Hinz A., Ghadwal R. S., Chem. Commun. 2019, 55, 10408–10411. [DOI] [PubMed] [Google Scholar]
- 11. Balázs L., Breunig H. J., Coord. Chem. Rev. 2004, 248, 603–621. [Google Scholar]
- 12. Paneth F. A., Loleit H., J. Chem. Soc. 1935, 366–371. [Google Scholar]
- 13. Salim S., Lim C. K., Jensen K. F., Chem. Mater. 1995, 7, 507–516. [Google Scholar]
- 14. Ishida S., Hirakawa F., Furukawa K., Yoza K., Iwamoto T., Angew. Chem. Int. Ed. 2014, 53, 11172–11176; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11354–11358. [Google Scholar]
- 15. Kretschmer R., Ruiz D. A., Moore C. E., Rheingold A. L., Bertrand G., Angew. Chem. Int. Ed. 2014, 53, 8176–8179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8315–8318. [Google Scholar]
- 16. Sasamori T., Mieda E., Nagahora N., Sato K., Shiomi D., Takui T., Hosoi Y., Furukawa Y., Takagi N., Nagase S., Tokitoh N., J. Am. Chem. Soc. 2006, 128, 12582–12588. [DOI] [PubMed] [Google Scholar]
- 17. Li T., Wei H., Fang Y., Wang L., Chen S., Zhang Z., Zhao Y., Tan G., Wang X., Angew. Chem. Int. Ed. 2017, 56, 632–636; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 647–651. [Google Scholar]
- 18. Ganesamoorthy C., Helling C., Wölper C., Frank W., Bill E., G. E. Cutsail III , Schulz S., Nat. Commun. 2018, 9, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Falivene L., Credendino R., Poater A., Petta A., Serra L., Oliva R., Scarano V., Cavallo L., Organometallics 2016, 35, 2286–2293. [Google Scholar]
- 20. Dange D., Davey A., Abdalla J. A. B., Aldridge S., Jones C., Chem. Commun. 2015, 51, 7128–7131. [DOI] [PubMed] [Google Scholar]
- 21. Lu W., Hu H., Li Y., Ganguly R., Kinjo R., J. Am. Chem. Soc. 2016, 138, 6650–6661. [DOI] [PubMed] [Google Scholar]
- 22. Evans D. F., J. Chem. Soc. 1959, 2003–2005. [Google Scholar]
- 23.Full crystallographic data of all structurally characterized compounds are given in the Supporting Information.
- 24. Helling C., Wölper C., Schulte Y., G. E. Cutsail III , Schulz S., Inorg. Chem. 2019, 58, 10323–10332. [DOI] [PubMed] [Google Scholar]
- 25. Tuscher L., Helling C., Ganesamoorthy C., Krüger J., Wölper C., Frank W., Nizovtsev A. S., Schulz S., Chem. Eur. J. 2017, 23, 12297–12304. [DOI] [PubMed] [Google Scholar]
- 26. Tuscher L., Ganesamoorthy C., Bläser D., Wölper C., Schulz S., Angew. Chem. Int. Ed. 2015, 54, 10657–10661; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10803–10807. [Google Scholar]
- 27. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 12770–12779. [DOI] [PubMed] [Google Scholar]
- 28. Twamley B., Sofield C. D., Olmstead M. M., Power P. P., J. Am. Chem. Soc. 1999, 121, 3357–3367. [Google Scholar]
- 29.Cambridge Structural Database, Version 5.40, see also: Allen F. H., Acta Crystallogr. Sect. B 2002, 58, 380–388; Search for Sb-N(Ar)(SiR2X) gave 34 hits. The Sb−N bond lengths range from 1.965 Å to 2.104 Å, with a mean value of 2.04(3) Å.12037359 [Google Scholar]
- 30. Weil J., Bolton J. In Electron Paramagnetic Resonance: Elementary Theory and Practical Applications , 2nd ed., Wiley, Hoboken, 2007. [Google Scholar]
- 31.The assigned positive t component yields a negative spin (or opposite) a iso hyperfine sign for 2-N only. While this is unusual, we may still consider the other possible sign assignments. An A-tensor of all positive values for 2-N yields a large a iso value, but only an absolute s-orbital spin density of <0.03. However, the resulting p-orbital spin-density estimate would become 0.24. This is significantly smaller and in major disagreement with previous analysis of analogous Sb radicals. Furthermore, a complete sign reversal of that shown 2-N would yield a majority “negative p-orbital spin density” on the Sb radical center, an assignment that is opposite of accepted sign conventions. The nitrogen ligand of 2-N is an excellent π-donor due to the lone pair of electrons on the nitrogen. These electrons have the potential to perturb the Sb spin density through a spin-polarization mechanism that may reverse the sign of the minimal Sb s-orbital spin density.
- 32.Isotropic coupling constant a 0(121Sb)=35098 MHz; anisotropic coupling constant b 0(121Sb)=629 MHz; a 0(69Ga)=12210 MHz; b 0(69Ga)=204 MHz; a 0(14N)=1811 MHz; b 0(14N)=55.5 MHz; a 0(11B)=2547 MHz; b 0(11B)=63.6 MHz.[30]
- 33. Zhuravleva T. S., J. Struct. Chem. 1967, 7, 489–492. [Google Scholar]
- 34.Full details on the quantum-chemical calculations are given in the Supporting Information.
- 35. Allred A. L., J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar]
- 36. Loh Y. K., Porteous K., Fuentes M. Á., Do D. C. H., Hicks J., Aldridge S., J. Am. Chem. Soc. 2019, 141, 8073–8077. [DOI] [PubMed] [Google Scholar]
- 37. Edwards J. O., Pearson R. G., J. Am. Chem. Soc. 1962, 84, 16–24. [Google Scholar]
- 38. Rivard E., Merrill W. A., Fettinger J. C., Power P. P., Chem. Commun. 2006, 3800–3802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary