

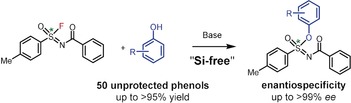
Abstract
SuFEx reactions, in which an S−F moiety reacts with a silyl‐protected phenol, have been developed as powerful click reactions. In the current paper we open up the potential of SuFEx reactions as enantioselective reactions, analyze the role of Si and outline the mechanism of this reaction. As a result, fast, high‐yielding, “Si‐free” and enantiospecific SuFEx reactions of sulfonimidoyl fluorides have been developed, and their mechanism shown, by both experimental and theoretical methods, to yield chiral products.
Keywords: enantioselectivity, kinetics, reaction mechanisms, SuFEx
The SuFEx reaction is shown to proceed quantitatively and fast with wide scope without silyl protection of the phenol. Mechanistic studies reveal a stereospecific bimolecular nucleophilic substitution.
Introduction
Click chemistry1 has been widely used to construct functional molecular skeletons concisely and efficiently.2 A variety of new click reactions has been developed, such as CuI‐catalyzed azide–alkyne cycloadditions (CuAAC)3 as well as metal‐free click reactions4 such as thiol–ene,5a oxime ligation,5b (thio‐)Michael,5c Diels–Alder,5d strain‐promoted cycloadditions between azide–alkyne (SPAAC),5e cyclo‐octyne‐o‐quinone (SPOCQ),5f, 5g and cyclopropene‐o‐quinone5h reactions. More recently, the pioneering work of Gembus6 and Sharpless7 revived the sulfur(VI) fluoride exchange (SuFEx) reaction from “old‐school chemistry” to the latest generation of click reactions. In this reaction, the S−F bond is typically stable enough to be unreactive in a wide variety of reaction conditions, but in the presence of silyl‐protected phenols, fluorosulfates readily react to form sulfates requiring only small amounts of catalyst (base or HF2 −). In this process, Si‐protection is useful, as Si and F form the strongest single bond in nature, allowing the rapid formation of SO2−O bonds from stable silyl ether precursors.8 This synergy of the click reaction with the formation of the thermodynamically favorable Si−F bond (BDE=135 kcal mol−1) results in highly reliable SuFEx reactions under metal‐free conditions.
As a result, research into the SuFEx reaction has surged in the last 5 years.9 To expand the range of useful SuFEx connectors, several SuFEx hubs and fluorosulfuryl transfer reagents have been developed, such as ethenesulfonyl fluoride,10 SO2F2,7 SOF4,11 fluorosulfuryl imidazolium salt,12 and others.13 Hereby, a wide variety of SuFEx‐able substrates with excellent functional group tolerance can be obtained, affording almost quantitative yields and/or polymers with high molecular weights and narrow polydispersity. For example, SuFEx has been used in modifications of peptides and proteins,14 syntheses of functional polymers,15 macrocycles,16 MOFs17 and functionalized surfaces,18 preparations of ionic liquids,19 and late‐stage drug functionalization.20 In addition, SuFEx products have been used as important substrates and key intermediates in various reactions.21
Although the SuFEx reaction has become a very practical click reaction, three key features are as of yet unclear: its mechanism, an evaluation of the necessity and influence of Si‐based protection of the phenols, and the potential of the SuFEx reaction as source of chiral molecules. To address all three facets in one study, we here report on the investigation of the SuFEx reaction of sulfonimidoyl fluorides. Sulfonimidoyl fluorides22 bear a stable chiral sulfur‐centered moiety, and form an ideal platform for mechanistic and enantioselectivity studies, whereby SN1 versus SN2 and addition/elimination pathways can be identified via the chirality of the substrates and products. Although some examples of reactions of unprotected phenols with SO2F2 7, 20, 23 and R‐SO2F24 have been reported, formation of Si−F covalent bonds (as in Figure 1 c) has generally been considered to be a, or even: the, main driving force for the reaction in aprotic solvents,7 but its role has remained under‐investigated. Such studies might not only lead to mechanistic insight, but may also prevent an extra silanization step in cases where the SuFEx reaction might not need it. This therefore inspired us to explore silicon‐free SuFEx reactions. Herein, we thus report our detailed studies on the silicon‐free SuFEx chemistry of sulfonimidoyl fluorides, exploring the scope, kinetics and reaction mechanism by experimental and theoretical (wB97XD/6‐311+G(d,p) density functional theory) studies (Figure 1 d).
Figure 1.
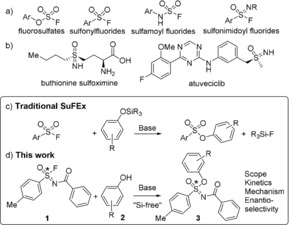
a) Common fluoride substrates in SuFEx reaction; b) sulfoximines in anticancer drugs. c/d) Goal of present study: “Silicon‐free” SuFEx reactions.
While many sulfur‐fluoride reagents with different valency have been studied (Figure 1 a), sulfonimidoyl fluorides have rarely been studied at all, let alone as SuFEx reagents, although they have already been labelled as useful connectors from the start of recent SuFEx literature.7 Despite this lack of exploration, the SuFEx reactivity of sulfonimidoyl fluorides has been shown to be comparable to sulfonyl fluorides,7 with further regulation possible by modification of substituents on the nitrogen atom, making them ideal tunable SuFEx substrates. Further motive is provided by the usefulness of the product, as the resulting sulfoximine group, and structural variations thereof, can express significant medicinal activity.25 For example, buthionine sulfoximine is a potential adjunct with chemotherapy in the treatment of metastatic melanoma,26 while atuveciclib is the first potent and highly selective PTEFb/CDK9 inhibitor to enter clinical trials for the treatment of cancer (Figure 1 b).27
As first part of our studies, the comparison between the traditional and Si‐free SuFEx reaction was investigated. By mixing Si‐protected (short for: tert‐butyl‐dimethylsilyl (TBDMS)‐protected) phenol with unprotected p‐cresol and performing the SuFEx reaction with N‐benzoyl‐4‐methylbenzenesulfonimidoyl fluoride 1 (ratios: 10:10:1, to obtain pseudo first‐order conditions), the relative reactivity of the respective phenol derivative (free or Si‐protected) can be observed from the composition of the final product mixture. This yielded a ratio for Ph‐O‐Si/CH3‐Ph‐OH of 5:95. The analogous experiment with Si‐protected cresol and unprotected phenol yielded a ratio for CH3‐Ph‐O‐Si/Ph‐OH of 9:91 (ratios obtained from 1H NMR of products upon completion of the reaction, typically <2 min). This shows that a roughly 10:1 ratio was found for the reaction rate of unprotected phenols and Si‐protected phenols (see the Supporting Information (SI) for details). During this reaction time, exchange of the TBDMS‐group from the cresol to the phenol or vice versa was negligible. While the formation of the Si−F covalent bond can still be of relevance, it cannot be characterized as the driving force behind the SuFEx reaction.
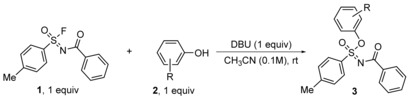
Since the unprotected phenol derivative was clearly more reactive, we set out to outline the scope of Si‐free SuFEx reactions of 1, prepared according to literature,28 via a systematic variation of the nature of the phenol. The results are displayed in Table 1.
Table 1.
Scope of Si‐free SuFEx reactions of sulfonimidoyl fluoride 1 with phenolic derivatives.[a]
|
|
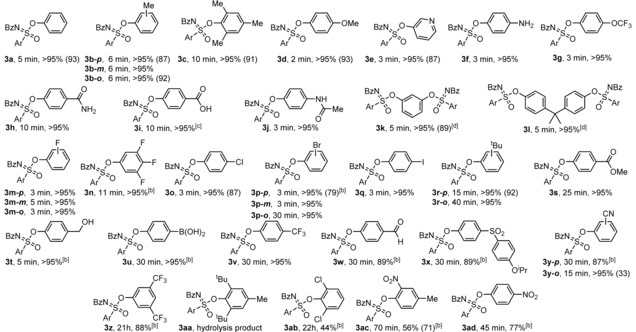
[a] Conversion was determined by 1H NMR measurements. Isolated yield in brackets. Reaction conditions for 1H NMR conversion: 1 (0.05 mmol), phenolic derivative 2 (1.05 equiv) and DBU (1.0 equiv) in CD3CN (0.55 mL), rt. For isolated yield: Fluoride 1 (0.5 mmol), phenolic derivatives 2 (0.5 mmol), DBU (1.5 mmol) in anhydrous CH3CN (1 mL), rt. [b] Hydrolysis by‐product was formed (see SI). [c] 2 equiv of DBU were used. [d] 2 equiv of 1 were used.
In the presence of 1 equivalent 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU), fluoride 1 reacted smoothly with a variety of phenols 2 and 3‐pyridinol to form the product sulfoximine in good to excellent yields under ambient conditions. Unlike in the normal SuFEx reaction, DBU acted both as a catalyst and an acid scavenger due to the absence of a silane group to stabilize the leaving fluoride anions in the Si‐free SuFEx. Most of the reactions actually reached completion within 10 min, providing one product, the sulfoximine, in almost quantitative NMR yield without optimization of the reaction conditions. The reaction exhibited an excellent functional group tolerance, as phenol derivatives containing alkyl (2 b,c), ether (2 d,g), pyridinyl (2 e), amino (2 f), amide (2 h), carboxyl (2 i), halogen (2 m–q,v), ester (2 s), hydroxymethyl (2 t), boronic acid (2 u) and cyano (2 y) functional groups were successfully used as substrates. Similar to the normal SuFEx reaction, alkyl alcohols, anilines and amides, which are weaker nucleophiles compared to phenolates generated from deprotonations by DBU, did not react in Si‐free SuFEx either. Moreover, screening revealed the Si‐free reaction was not very sensitive to steric hindrance although reactions took longer, as target products were obtained successfully for sterically hindered phenols, bearing bulky ortho substituents, such as 2‐bromophenol (2 p‐o), 2‐(tert‐butyl)phenol (2 r‐o) and mesitol (2 c). Unsurprisingly, meta‐substituted phenol derivatives (2 b‐m, 2 m‐m, 2 p‐m,) were found to be excellent substrates. Di‐substituted phenol derivatives resorcinol (2 k) and bisphenol A (BPA) (2 l) also partook in the reaction, albeit with 2 equivalents of the sulfonimidoyl fluoride 1. Unfortunately, the resulting diastereomers (3 k,l) could not be separated by flash column chromatography (see SI). Substrates containing strong electron‐withdrawing substituents in the ortho‐ or para‐position, such as formyl (2 w), cyano (2 y), trifluoromethyl (2 z) or nitro (2 ac, 2 ad) or bearing two large groups (such as 2,6‐di‐tert‐butyl, 2 aa) yielded hydrolysis by‐products at a loss of the target SuFEx products (see SI) at varying degree, and only provide the SuFEx product very slowly under rigorously dry conditions.
To further demonstrate the scope of this reaction, several natural phenolic derivatives were evaluated (Table 2). Eugenol (4 a), vanillic acid (4 b), raspberry ketone (4 c), tyrosol (4 d) sesamol (4 e), ferulic acid (4 f), and vic‐thymol (4 g), salicyl aldehyde (4 h), and vanillin (4 i) all reacted smoothly with fluoride 1 to form the corresponding sulfoximine in good to excellent yields under ambient conditions.
Table 2.
Scope of Si‐free SuFEx reactions of sulfonimidoyl fluoride 1 with naturally occurring phenols.[a]
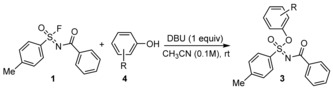
|
|
[a] Conversion was determined by 1H NMR measurements. Reaction conditions for 1H NMR yield: 1 (0.05 mmol) phenolic derivative 2 (1.05 equiv) and DBU (1.0 equiv) in CD3CN (0.55 mL), rt. [b] Hydrolysis by‐product was formed (see SI). [c] 2 equiv of DBU were used.
After having established that the Si‐free SuFEx reaction proceeds smoothly and efficiently for a wide variety of phenol substrates, we aimed for further insight, by studying other catalysts and the reaction kinetics. Regarding the catalysts, HF2 anion has been introduced as a superior rate‐enhancing agent.29 Therefore we investigated the effects of this anion on a range of reactions. When 1 was reacted with Si‐protected 2 b‐p, then this quantitatively yielded product 3 b‐p at rt in 1 h with one equivalent of catalyst. When using catalytic amounts of HF2 anion (0.2 equiv), the same reaction reached completion, albeit at a much slower pace (30 h). When the unprotected phenol was used with this catalyst (even with 1 equiv), then only trace amounts of product were observed. The likely role the HF2 anion plays in these reactions is clarified if it is added to Si‐protected 2 b‐p in the absence of any 1. Within 10 min, full deprotection of the TBDMS group is then achieved. Therefore, when used in a standard SuFEx reaction, HF2 anion might act as follows: it effects the rapid deprotection of Si‐protected phenols. In aprotic solvents, this then yields a highly reactive phenolate, which—upon reaction—frees up a fluoride anion, allowing the reformation of HF2 anion, or fluoride by itself might affect the deprotection of a subsequent Si‐protected phenol. The high rate of both the deprotection and of the SuFEx reaction of the phenolate would then allow the use of only small amounts of catalysts, as has been observed experimentally. In line with this, for unprotected phenols HF2 − is a poor catalyst: since fluoride is only a poor base, it will—unlike DBU—not induce the formation of the reactive phenolates; as a result, nothing happens.
As a start of our kinetics study, we compared the reaction rate of the Si‐free SuFEx reaction of the sterically hindered 2‐(tert‐butyl)phenol (2 r‐o,) with a traditional SuFEx reaction with the Si‐protected 2‐(tert‐butyl)phenol analogue 5. At room temperature, both these reactions were finished in under two minutes; therefore the kinetics were studied in CD3CN using low‐temperature NMR. Under pseudo first‐order conditions, the second‐order rate constant found for 2 r‐o at −30 °C was (3.0±0.1)×10−3 m −1 s−1. For 5 the kinetics were clearly slower, but interestingly showed an activation phase, with a clear S‐shape appearance of the product over time (see SI). To test whether this could be due to the appearance of unprotected phenol 2 r‐o upon a rate‐determining desilylation of 5, we studied this desilylation under these reaction conditions (−30 °C) with equimolar concentration of 5 and (CH3)4NF (both 0.12 m). This yielded a full desilylation within 2 min, suggesting that for silylated phenols the reaction is initiated by freeing up some F− anion, which can deprotect a Si‐protected phenol, which can subsequently react and free up another F−, etc. Here DBU can play a double role, as it has been reported to catalyze the desilylation,30 while as a base, of course, it also increases the nucleophilicity of the phenol.
Further mechanism investigations were made possible by utilizing the chirality of sulfur atom in the sulfonimidoyl fluoride 1 and its SuFEx products. Previous mechanism‐oriented reports have typically focused on substitution reactions on disulfides, yielding addition–elimination and SN2 as the most widely accepted mechanisms, with the detailed mechanism being sensitive to the strain,31a counter cations,31b leaving groups,31c substituents on sulfur atom,31d nucleophiles,31e solvents,31e–31g and so on (see for an overview32). Analogously, we considered as the three most likely mechanisms those summarized in Figure 2.
Figure 2.
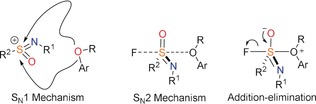
Possible mechanisms of SuFEx reactions.
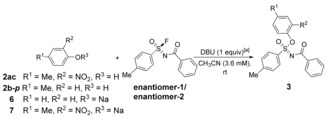
If one enantiomer of 1 is used as the substrate in SuFEx reactions, the SN1 reaction pathway will provide the racemized products. Conversely, stereospecific products will be obtained through the SN2 or addition‐elimination pathway. Firstly, enantiomers of sulfonimidoyl fluoride 1 were shown to be very stable in solution without DBU, but readily racemize when DBU was present. The rate of this racemization reaction, with excess DBU under pseudo first‐order conditions, is (1.9±0.1)×10−3 m −1 s−1 at −30 °C, showing its facile occurrence and competitive character (see SI). Since this would reduce the usefulness of the reaction, further studies were performed.
To this aim, a good and a poor nucleophile—p‐cresol and 4‐methyl‐2‐nitrophenol, respectively—were adopted in SuFEx reactions. With the good nucleophile a good stereoselectivity (ca. 70 % ee) was achieved, whereas with the poor nucleophile only <2 % ee was obtained (Table 3). This provided important mechanistic information, as it shows that SuFEx proceeds through SN2 or addition‐elimination channels rather than via an SN1 mechanism when a good nucleophile was used. The racemization or SN1 mechanism was a possible reason of low stereoselectivity when a poor nucleophile (4‐methyl‐2‐nitrophenol) was used. If the SN2 or addition‐elimination is a competitive reaction with the racemization, we reasoned that the racemization could be inhibited when a large excess of a good nucleophile (10 equiv p‐cresol) was used. In fact, the experimental results showed that under such conditions the %ee was significantly improved (95 % ee). However, the key finding was avoiding DBU altogether, and using sodium phenolate 6 and sodium 4‐methyl‐2‐nitrophenolate 7 as nucleophiles. In this case stereospecific products were obtained, and no racemization of the reactants was observed, even for 7, which takes overnight to get to 80 % conversion. This further demonstrated that the silicon‐free SuFEx reaction mechanism proceeds via an SN2 or addition‐elimination mechanism.
Table 3.
Enantioselective silicon‐free SuFEx reactions.[a]
|
Enantiomer[b] |
Phenol |
ee [c] |
|---|---|---|
|
enantiomer‐1 |
2 b‐p |
73 % (95[d]) |
|
2 ac |
<2 % |
|
|
6 |
>99 % |
|
|
|
7 |
>99 % |
|
|
|
|
|
enantiomer‐2 |
2 b‐p |
71 % (95[d]) |
|
2 ac |
<2 % |
|
|
6 |
>99 % |
|
|
7 |
>99 % |
[a] Reaction conditions: enantiomer‐1/enantiomer‐2 (1 mL, 3.6 mm in CH3CN), phenolic derivative (1 equiv), DBU (1 equiv), rt. When phenolate 6 or 7 was adopted as the substrate, DBU was not added. [b] enantiomer‐1 and enantiomer‐2 refer to two enantiomers with retention times of 16.8 and 18.8 min with chiral HPLC, respectively. [c] ee determined using chiral HPLC and calculated by ee= , [d] 10 equiv 2 b‐p used.
To further distinguish concerted SN2 and addition‐elimination reaction paths, quantum chemical calculations were performed at the wb97xd/6‐311+G(d,p) level of theory, using the SMD solvent model to represent CH3CN as implemented in Gaussian 16.33 We computed the potential energy surfaces (PES) for the SuFEx reaction between fluoride 1 and phenolate 6 (Scheme 1; to slightly simplify the calculation, we replaced the 4‐methylbenzenesulfonimidoyl with a benzenesulfonimidoyl group). Because of the strong S‐F covalent bond (≈90 kcal mol−1), fluoride 1 is very stable, and the leaving of fluoride is generally considered to be one of the rate‐limiting steps. However, for the reaction under study, this does not seem to be the case. Compound 1 undergoes the nucleophilic attack by 6 via TS1, with a rate‐limiting enthalpic barrier of only 2.74 kcal mol−1. This confirms the experimentally observed high reaction rate. However, in TS1 the S‐F bond was still largely unchanged (length increase from 1.64 to 1.72 Å; Wiberg bond order decrease from 0.520 to 0.496), likely due to a relatively early TS (r(O⋅⋅⋅S)=2.33 Å; B.O.=0.151).
Scheme 1.
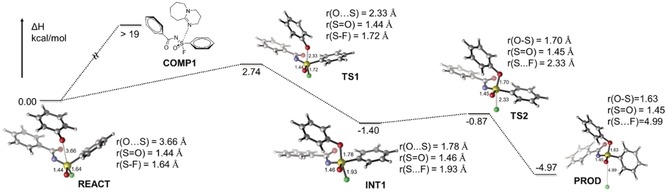
Potential energy surface for SuFEx reaction with phenolate.
The next stationary point, however, was not the set of products, but a five‐coordinated sulfur intermediate, INT1, which was found to be lower in energy than the reactant pair. This intermediate is characterized by an r(O⋅⋅⋅S)=1.78 Å, and a r(S⋅⋅⋅F)=1.93 Å, indicating bonding of the O to S (B.O.=0.544), but also no loss of F yet (B.O.=0.315). Interestingly, the S=O bond does not elongate significantly during the reaction (1.44–1.45 Å, with the bond length of a typical S−O bond=1.64 Å34), nor does the charge on this O atom vary a lot during the reaction.
This intermediate readily loses fluoride via TS2 (ΔH ≠ of this step is only 0.53 kcal mol−1), giving rise to the products. Activation of S−F loss by the base has been one of the most widely accepted mechanism models to date.6, 11, 35 This might be the case for much poorer nucleophiles, including the Si‐protected phenols, but such activation is apparently not needed for phenolate SuFEx reactions. In summary, these DFT calculations thus suggest that SuFEx with a phenolate nucleophile attacking a sulfonimidoyl fluoride takes places with bimolecular kinetics via an addition–elimination mechanism, although a concerted SN2 reaction under experimental conditions cannot be excluded given the very low barrier observed for loss of F−.
This SuFEx reaction has a reaction enthalpy of about −5 kcal mol−1, which is nice to drive a reaction quietly to completion, and better than many other click reactions that are simply too exothermic.36
We also aimed to characterize the nucleophilic attack by DBU. However, no TS could be found, and the energy simply increases upon decreasing the r(N⋅⋅⋅S) distance (to >19 kcal mol−1 for r(N⋅⋅⋅S)=1.7 Å. Apparently, the steric bulk of DBU makes this a really non‐nucleophilic base in this reaction.
Conclusion
In conclusion, we have demonstrated that Si‐free SuFEx reactions with phenolate anions can be readily performed in a stereoselective manner with excellent functional group tolerance and good to quantitative yields in minutes at room temperature. The Si‐free SuFEx is faster than the traditional one, and detailed kinetics of the latter might point to a more complex reaction scheme there. A combination of these experiments with theoretical calculations show that the Si‐free SuFEx reaction is a fast, bimolecular, enantiospecific and slightly exothermic click reaction, making it amenable for a wide range of substrates. These studies contribute to a clearer understanding and continuing development of SuFEx reactions, as well as providing a practical approach for the construction of optically pure sulfoximines.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thanks Tjerk Sminia for synthetic support and Barend van Lagen for technical support. Financial support of the National Science Foundation of China (grant #21871208) and Wageningen University is greatly acknowledged.
D.-D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman, H. Zuilhof, Angew. Chem. Int. Ed. 2020, 59, 7494.
References
- 1. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 2. Moses J. E., Moorhouse A. D., Chem. Soc. Rev. 2007, 36, 1249–1262. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Huisgen R., Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598; [Google Scholar]; Angew. Chem. 1963, 75, 604–637; [Google Scholar]
- 3b. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711. [Google Scholar]
- 4.
- 4a. Becer C. R., Hoogenboom R., Schubert U. S., Angew. Chem. Int. Ed. 2009, 48, 4900–4908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4998–5006; [Google Scholar]
- 4b. Escorihuela J., Marcelis A. T. M., Zuilhof H., Adv. Mater. Interfaces 2015, 2, 1500135. [Google Scholar]
- 5.
- 5a. Hoyle C. E., Bowman C. N., Angew. Chem. Int. Ed. 2010, 49, 1540–1573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1584–1617; [Google Scholar]
- 5b. Kölmel D. K., Kool E. T., Chem. Rev. 2017, 117, 10358–10376; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Nair D. P., Podgórski M., Chatani S., Gong T., Xi W., Fenoli C. R., Bowman C. N., Chem. Mater. 2014, 26, 724–744; [Google Scholar]
- 5d. Tasdelen M. A., Polym. Chem. 2011, 2, 2133–2145; [Google Scholar]
- 5e. Baskin J. M., Prescher J. A., Laughlin S. T., Agard N. J., Chang P. V., Miller I. A., Lo A., Codelli J. A., Bertozzi C. R., Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Borrmann A., Fatunsin O., Dommerholt J., Jonker A. M., Löwik D. W. P. M., van Hest J. C. M., van Delft F. L., Bioconjugate Chem. 2015, 26, 257–261; [DOI] [PubMed] [Google Scholar]
- 5g. Bruins J. J., Albada B., van Delft F., Chem. Eur. J. 2018, 24, 4749–4756; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5h. Gahtory D., Sen R., Kuzmyn A. R., Escorihuela J., Zuilhof H., Angew. Chem. Int. Ed. 2018, 57, 10118–10122; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10275–10279. [Google Scholar]
- 6. Gembus V., Marsais F., Levacher V., Synlett 2008, 1463–1466. [Google Scholar]
- 7. Dong J., Krasnova L., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2014, 53, 9430–9448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9584–9603. [Google Scholar]
- 8. Dong J. J., Sharpless K. B., Kwisnek L., Oakdale J. S., Fokin V. V., Angew. Chem. Int. Ed. 2014, 53, 9466–9470; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9620–9624. [Google Scholar]
- 9. Barrow A. S., Smedley C. J., Zheng Q., Li S., Dong J., Moses J. E., Chem. Soc. Rev. 2019, 48, 4731–4758. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Qin H.-L., Zheng Q., Bare G. A. L., Wu P., Sharpless K. B., Angew. Chem. Int. Ed. 2016, 55, 14155–14158; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14361–14364; [Google Scholar]
- 10b. Xu R., Xu T., Yang M., Cao T., Liao S., Nat. Commun. 2019, 10, 3752–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li S., Wu P., Moses J. E., Sharpless K. B., Angew. Chem. Int. Ed. 2017, 56, 2903–2908; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2949–2954. [Google Scholar]
- 12. Guo T., Meng G., Zhan X., Yang Q., Ma T., Xu L., Sharpless K. B., Dong J., Angew. Chem. Int. Ed. 2018, 57, 2605–2610; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2635–2640. [Google Scholar]
- 13.
- 13a. Tribby A. L., Rodríguez I., Shariffudin S., Ball N. D., J. Org. Chem. 2017, 82, 2294–2299; [DOI] [PubMed] [Google Scholar]
- 13b. Veryser C., Demaerel J., Bieliunas V., Gilles P., De Borggraeve W. M., Org. Lett. 2017, 19, 5244–5247; [DOI] [PubMed] [Google Scholar]
- 13c. Zhou H., Mukherjee P., Liu R., Evrard E., Wang D., Humphrey J. M., Butler T. W., Hoth L. R., Sperry J. B., Sakata S. K., Helal C. J., am Ende C. W., Org. Lett. 2018, 20, 812–815; [DOI] [PubMed] [Google Scholar]
- 13d. Laudadio G., Bartolomeu A. d. A., Verwijlen L. M. H. M., Cao Y., de Oliveira K. T., Noël T., J. Am. Chem. Soc. 2019, 141, 11832–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Chen W., Dong J., Li S., Liu Y., Wang Y., Yoon L., Wu P., Sharpless K. B., Kelly J. W., Angew. Chem. Int. Ed. 2016, 55, 1835–1838; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1867–1870; [Google Scholar]
- 14b. Chen W., Dong J., Plate L., Mortenson D. E., Brighty G. J., Li S., Liu Y., Galmozzi A., Lee P. S., Hulce J. J., Cravatt B. F., Saez E., Powers E. T., Wilson I. A., Sharpless K. B., Kelly J. W., J. Am. Chem. Soc. 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang C., Flynn J. P., Niu J., Angew. Chem. Int. Ed. 2018, 57, 16194–16199; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16426–16431. [Google Scholar]
- 16. Demay-Drouhard P., Du K., Samanta K., Wan X., Yang W., Srinivasan R., Sue A. C. H., Zuilhof H., Org. Lett. 2019, 21, 3976–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S., Song H., Ko N., Kim C., Kim K., Lee E., ACS Appl. Mater. Interfaces 2018, 10, 33785–33789. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Gahtory D., Sen R., Pujari S., Li S., Zheng Q., Moses J. E., Sharpless K. B., Zuilhof H., Chem. Eur. J. 2018, 24, 10550–10556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Yatvin J., Brooks K., Locklin J., Angew. Chem. Int. Ed. 2015, 54, 13370–13373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13568–13571. [Google Scholar]
- 19. Hmissa T., Zhang X., Dhumal N. R., McManus G. J., Zhou X., Nulwala H. B., Mirjafari A., Angew. Chem. Int. Ed. 2018, 57, 16005–16009; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16237–16241. [Google Scholar]
- 20. Liu Z., Li J., Li S., Li G., Sharpless K. B., Wu P., J. Am. Chem. Soc. 2018, 140, 2919–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Smedley C. J., Barrow A. S., Spiteri C., Giel M.-C., Sharma P., Moses J. E., Chem. Eur. J. 2017, 23, 9990–9995; [DOI] [PubMed] [Google Scholar]
- 21b. Zhang G., Zhao Y., Xuan L., Ding C., Eur. J. Org. Chem. 2019, 4911–4915; [Google Scholar]
- 21c. Zhao Y., Mei G., Wang H., Zhang G., Ding C., Synlett 2019, 30, 1484–1488. [Google Scholar]
- 22.
- 22a. Johnson C. R., Bis K. G., Cantillo J. H., Meanwell N. A., Reinhard M. F. D., Zeller J. R., Vonk G. P., J. Org. Chem. 1983, 48, 1–3; [Google Scholar]
- 22b. van Leusen D., van Leusen A. M., Recl. Trav. Chim. Pays-Bas 1984, 103, 41–45; [Google Scholar]
- 22c. Kowalczyk R., Edmunds A. J. F., Hall R. G., Bolm C., Org. Lett. 2011, 13, 768–771; [DOI] [PubMed] [Google Scholar]
- 22d. Kolesnik N. P., Rozhenko A. B., Kinzhybalo V., Lis T., Shermolovich Y. G., J. Fluorine Chem. 2014, 160, 16–19; [Google Scholar]
- 22e. Gao B., Li S., Wu P., Moses J. E., Sharpless K. B., Angew. Chem. Int. Ed. 2018, 57, 1939–1943; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1957–1961; [Google Scholar]
- 22f. Wright M., Martínez-Lamenca C., Leenaerts J. E., Brennan P. E., Trabanco A. A., Oehlrich D., J. Org. Chem. 2018, 83, 9510–9516. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Wan H., Zhou S., Gu P., Zhou F., Lyu D., Xu Q., Wang A., Shi H., Xu Q., Lu J., Polym. Chem. 2020, 11, 1033–1042; [Google Scholar]
- 23b. Park S., Kim S. Y., Cho J., Jung D., Seo D., Lee J., Lee S., Yun S., Lee H., Park O., et al., Bioconjugate Chem. 2019, 30, 1957–1968; [DOI] [PubMed] [Google Scholar]
- 23c. Park S., Kim S. Y., Cho J., Jung D., Seo D., Lee J., Lee S., Yun S., Lee H., Park O., et al., Bioconjugate Chem. 2019, 30, 1969–1978; [DOI] [PubMed] [Google Scholar]
- 23d. Zheng Q., Woehl J. L., Kitamura S., Santos-Martins D., Smedley C. J., Li G., Forli S., Moses J. E., Wolan D. W., Sharpless K. B., Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. He D., Guo Y., Chen Q.-Y., Yang H., Lv T., J. Fluorine Chem. 2019, 222–223, 1–7; [Google Scholar]
- 24b. Semenok D., Kletskov A., Dikusar E., Potkin V., Lukin O., Tetrahedron Lett. 2018, 59, 372–374; [Google Scholar]
- 24c. Zha G.-F., Zheng Q., Leng J., Wu P., Qin H.-L., Sharpless K. B., Angew. Chem. Int. Ed. 2017, 56, 4849–4852; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4927–4930 (see page 8 of ESI); [Google Scholar]
- 24d. Liu F., Wang H., Li S., Bare G. A. L., Chen X., Wang C., Moses J. E., Wu P., Sharpless K. B., Angew. Chem. Int. Ed. 2019, 58, 8029–8033; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8113–8117. [Google Scholar]
- 25. Frings M., Bolm C., Blum A., Gnamm C., Eur. J. Med. Chem. 2017, 126, 225–245. [DOI] [PubMed] [Google Scholar]
- 26. Defty C. L., Marsden J. R., Curr. Top. Med. Chem. 2012, 12, 53–60. [DOI] [PubMed] [Google Scholar]
- 27. Lücking U., Scholz A., Lienau P., Siemeister G., Kosemund D., Bohlmann R., Briem H., Terebesi I., Meyer K., Prelle K., Denner K., Bömer U., Schäfer M., Eis K., Valencia R., Ince S., von Nussbaum F., Mumberg D., Ziegelbauer K., Klebl B., Choidas A., Nussbaumer P., Baumann M., Schultz-Fademrecht C., Rühter G., Eickhoff J., Brands M., ChemMedChem 2017, 12, 1776–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Worch C., Atodiresei I., Raabe G., Bolm C., Chem. Eur. J. 2010, 16, 677–683. [DOI] [PubMed] [Google Scholar]
- 29. Gao B., Zhang L., Zheng Q., Zhou F., Klivansky L. M., Lu J., Liu Y., Dong J., Wu P., Sharpless K. B., Nat. Chem. 2017, 9, 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeom C.-E., Kim H. W., Lee S. Y., Kim B. M., Synlett 2007, 0146–0150. [Google Scholar]
- 31.
- 31a. Bachrach S. M., Woody J. T., Mulhearn D. C., J. Org. Chem. 2002, 67, 8983–8990; [DOI] [PubMed] [Google Scholar]
- 31b. Ren Y., Gai J.-G., Xiong Y., Lee K.-H., Chu S.-Y., J. Phys. Chem. A 2007, 111, 6615–6621; [DOI] [PubMed] [Google Scholar]
- 31c. Sung D. D., Kim T. J., Lee I., J. Phys. Chem. A 2009, 113, 7073–7079; [DOI] [PubMed] [Google Scholar]
- 31d. Höckendorf R. F., Hao Q., Sun Z., Fox-Beyer B. S., Cao Y., Balaj O. P., Bondybey V. E., Siu C.-K., Beyer M. K., J. Phys. Chem. A 2012, 116, 3824–3835; [DOI] [PubMed] [Google Scholar]
- 31e. Paranjothy M., Siebert M. R., Hase W. L., Bachrach S. M., J. Phys. Chem. A 2012, 116, 11492–11499; [DOI] [PubMed] [Google Scholar]
- 31f. Neves R. P. P., Fernandes P. A., Varandas A. J. C., Ramos M. J., J. Chem. Theory Comput. 2014, 10, 4842–4856; [DOI] [PubMed] [Google Scholar]
- 31g. Bortoli M., Wolters L. P., Orian L., Bickelhaupt F. M., J. Chem. Theory Comput. 2016, 12, 2752–2761. [DOI] [PubMed] [Google Scholar]
- 32. Hamlin T. A., Swart M., Bickelhaupt F. M., ChemPhysChem 2018, 19, 1315–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R. B. Gaussian 16, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 34. Chivers T., Laitinen R. S., Chem. Soc. Rev. 2017, 46, 5182–5192. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Vorbrüggen H., Synthesis 2008, 1165–1174; [Google Scholar]
- 35b. Yatvin J., Brooks K., Locklin J., Chem. Eur. J. 2016, 22, 16348–16354. [DOI] [PubMed] [Google Scholar]
- 36. Jones G. O., Houk K. N., J. Org. Chem. 2008, 73, 1333–1342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary