

Abstract
Recent data show that anti-CD20 therapy is effective for some autoimmune diseases, including multiple sclerosis (MS). However, the efficacy of anti-CD20 therapy for MS is largely limited because anti-CD20 antibodies target only B cells. In previous studies, we have investigated the function of MS4a4B, a novel CD20 homologue, in T cell proliferation. Here, we found that MS4a4B regulates not only T cell proliferation but also T cell apoptosis. Knockdown of MS4a4B by MS4a4B-siRNA or MS4a4B-shRNA-expressing vector promoted apoptosis in primary T cells and T32 cell line. In contrast, vector-driven over-expression of MS4a4B reduced apoptosis in EL-4 cells. Machinery analysis showed that MS4a4B-mediated T cell survival was associated with decreased activity of caspases 3, 8 and 9. Interestingly, binding of anti-MS4a4B antibodies to T cells induced activated T cells to undergo apoptosis. To test whether anti-MS4a4B antibody interferes with MS4a4B-mediated protection of T cells, we injected anti-MS4a4B antibodies into mice with experimental autoimmune encephalomyelitis (EAE). The results show that anti-MS4a4B treatment ameliorated the severity of EAE, accompanied by decreased Th1 and Th17 cell responses and reduced levels of pro-inflammatory cytokines in the central nervous system, suggesting that MS4a4B may serve as a target of antibody-based therapy for T cell-mediated diseases.
Keywords: MS4A, T cell, Apoptosis, EAE, Antibody therapy
Introduction
Anti-CD20 antibody therapy has been considered as the most successful treatment for B cell lymphoma [1–3]. Recent data show that anti-CD20 antibody therapy is effective for some autoimmune diseases, including multiple sclerosis (MS) [4], a disease of the central nervous system (CNS) [5]. However, the efficacy of anti-CD20 therapy for MS is largely limited because anti-CD20 antibodies target only B cells [6]. Several therapeutic approaches directly targeting T cells have been developed, including CD3-specific antibody therapy. Studies in animal models and clinical trials show that administration of humanized non-FcR-binding anti-CD3 antibody, which largely overcomes the side-effects of traditional anti-CD3 antibody (immunogenicity and mitogenicity), suppresses T cell responses and ameliorates autoimmune diseases by blockading antigenic responsiveness of T cells through induction of TCR internalization and T cell depletion or through induction of T cell apoptosis [7], and for long term immune suppression, through induction of apoptotic T cell-induced regulatory T cells (Treg) [8]. However, administration of CD3-specific antibody, in case of humanized non-FcR binding form, depletes only about 20–30 % of CD3+ T cells in peripheral blood [9]. In addition, the idiotypic epitopes in the hypervariable regions of anti-CD3 antibody are still immunogenic for induction of humoral anti-idiotype responses. The generated anti-idiotype antibodies can neutralize CD3-specific antibody’s effects and promote recovery of TCR-expression and antigenic responses of T cells. Thus, a regimen including combination of antibodies against different T cell markers may compensate the limitation of existing CD3-specific antibody therapy. Therefore, identification of novel proteins in T cells as targets of antibody therapy for inhibition of T cell responses is still a challenge.
In a study for screening novel regulatory proteins during thymocyte development, we cloned a novel CD20 homologue, termed MS4a4B, from mouse thymus [10]. MS4a4B is a member of the MS4A gene family (membrane-spanning 4-domain family, subfamily A, MS4As) [11, 12]. MS4a4B protein contains four membrane-spanning domains, two extracellular domains and two cytoplasmic regions (Fig. S1). MS4a4B protein is highly conserved during evolution, especially its transmembrane domains. There is 41 % homology between MS4a4B and its potential human counterpart, MS4A4A [12]. Relatively, its extracellular domains are more diverse, which provide potential antigenic epitopes for specific antibody recognition. Previous data show that MS4a4B is highly expressed on T cell membrane and is closely related to the regulation of T cell responses [10, 13, 14]. One of our interesting observations is that reduced expression of MS4a4B is associated with apoptotic T cells during T cell activation [15], suggesting that MS4a4B may play a role in regulating apoptosis and survival of activated T cells.
In this report, we investigated the role of MS4a4B in T cell apoptosis by over-expression or knockdown of MS4a4B in a T cell line and primary T cells. We found that MS4a4B not only regulates T cell proliferation [10, 16] but also plays a protective role in T cell apoptosis. Binding of anti-MS4a4B antibodies to T cells abrogated MS4a4B-mediated protection and induced T cells to undergo apoptosis. We tested whether MS4a4B might serve as a target in T cells for antibody-based therapy by administration of anti-MS4a4B antibodies in the mouse experimental autoimmune encephalomyelitis (EAE) model, an animal model of MS [17]. Our data indicate that anti-MS4a4B antibody treatment suppresses T cell-mediated immune responses and reduces severity of EAE, suggesting that the anti-MS4a4B approach may have therapeutic effect for inhibition of T cell-mediated immune responses in addition to CD3-specific antibody therapy.
Materials and methods
Mice and cells
C57BL/6J mice, 8–10 weeks old, female (The Jackson Laboratory), were used for in vivo study and as donors for primary cells. All mice were used in accordance with National Institutes of Health and Thomas Jefferson University Institutional Animal Care and Use Committee guidelines. MS4a4B-expressing lentiviral vector-transduced EL-4 cells and shMS4a4B-expressing lentiviral vector-transduced T cell clone T32 were described previously [15]. All cells were cultured with RPMI 1640 complete medium containing 10 % of fetal calf serum.
Transfection of primary CD4+ T cells by MS4a4B-specific siRNA
CD4+ T cells were isolated from C57BL/6 mice by anti-CD4 magnetic beads (Miltenyi Biotec Inc) and were stimulated by anti-mouse CD3e (145–2C11) and anti-mouse CD28 (37.51) antibodies (BD Biosciences) for 36 h. Activated T cells were transfected with FAM-labeled MS4a4B-specific siRNA (siMS4a4B2) using Lipofect-amine 2000 (Invitrogen) as described previously [15]. FAM-labeled Silencer Negative Control #1 siRNA (Ambion, Applied Biosystems) was used as control.
Flow cytometric analysis
Cells harvested from culture were washed with cold-PBS containing 3 % horse serum and 0.02 % NaN3. To determine GFP expression, washed cells were acquired by using either FACSCalibur, or FACSAria (BD Biosciences). For detection of MS4a4B expression, cells were stained with biotin-labeled anti-MS4a4B antibody as described previously [10]. To detect cell apoptosis, cells were stained with Cy™5.5 Annexin V and propidium iodide (PI) according to manufacturer’s instructions (BD Biosciences). To analyze T cells in peripheral circulation, blood samples were primed with red blood cell lysis buffer and then stained with fluorescently-labeled antibodies against mouse CD3, CD4, CD8 and CD25 (BD Biosciences). Data were analyzed using FlowJo Software (Tree Star).
Spinal cord mononuclear cell (MNC) isolation and intracellular staining
Infiltrating MNCs in CNS were isolated as described previously [18]. Briefly, spinal cords were mechanically dissociated through a cell strainer. Cells were washed with PBS and then were fractionated on a 60/30 % Percoll gradient by centrifugation at 300×g for 20 min. Cell layer at the interface was collected for use. To detect intracellular cytokine, cells were adjusted to 1 × 106/ml in RPMI 1640 complete medium and then stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) (Sigma–Aldrich) and GolgiStop (1 μg/106 cells) (BD Biosciences) for 4 h at a density of 1 × 106/ml in RPMI 1640 complete medium. For determination of intracellular cytokines or FoxP3 protein, cells were first stained with fluorescent antibodies to surface markers and then were fixed and permeabilized using Fix/Perm® cell permeabilization reagents (BD Biosciences), followed by incubation with fluorescently-labeled antibodies against intracellular cytokine or FoxP3. Cells were acquired by using either FACSCalibur, or FACSAria (BD Biosciences).
Cytokine measurement
For cytokine detection, supernatants were collected from culture at 48 h of stimulation. Levels of IL-5, IFN-γ and IL-17 in supernatants were determined by ELISA with Duoset cytokine assay reagents (R&D Systems). Data were read with Multiskan® FC microplate photometer (Thermo Fisher Scientific). To detect IL-4, IL-6, IL-10, IFN-γ, IL-17 and TNF levels in nerve tissues by Cytokine Beads Array (CBA), spinal cords from tested mice were added by five time weight PBS (1 mg tissue + 5 μl PBS) containing 0.002 % Tween-20 and protease inhibitor (Complete Mini, EDTA-free, Roche Diagnostics). Cells were homogenized with homogenizer (Tissue Master 125, OMNI, International). After centrifugation, supernatants were collected for cytokine assay by CBA with CBA Th1/Th2/Th17 assay kit (BD Biosciences) according to the manufacturer’s instructions.
Caspase activity analysis
Bioactivities of caspase-3/7, caspase-8 and caspase-9 in cells were determined by luminescent assay with homogeneous Caspase-Glo® Assay kits (G8090, G8201 and G8210, Promega) according to the manufacturer’s instructions. To detect caspase proteins by western blotting, cells were lysed in lysis buffer (Cell Signaling) supplemented with protease inhibitor (Complete Mini, EDTA-free; Roche Diagnostics). Cell lysates were separated by 10 % SDS-PAGE and transferred onto Immun-Blot PVDF membrane (Bio-Rad Laboratories). Membranes were blotted with primary antibodies followed by incubation with HRP-conjugated secondary antibodies. The blots were developed by ECL reagents and exposed on HyperFilm™ (Amersham). The following antibodies were used for western blotting: antibodies against caspase 3, caspase 8 and caspase 9 (Cell Signaling, #9662, #4927 and #9504); β-Actin (AC-15) (Santa Cruz Biotechnology).
EAE induction and MS4a4B-specific antibody treatment
Mice were immunized subcutaneously on the back with 150 μg of MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) emulsified in CFA (Difco Lab) containing 4 mg/ml Mycobacterium tuberculosis H37Ra (Difco Lab). 200 ng of pertussis toxin (List Biological Lab) was given i.p. on days 0 and 2 post immunization (p.i). Mice were scored daily for appearance of clinical signs of EAE by a scale from 0 to 5 as described previously [18]. For antibody therapy, mice were treated by tail vein injection with antibodies against the second extracellular domain of MS4a4B (Fig. S1), which were purified by antigen-specific affinity chromatography from sera of MS4a4B peptide-immunized rabbits.
Statistical analysis
For clinical scores of EAE, significance between two groups was examined by using the Two-way ANOVA test. For other data, statistical difference between two groups was determined by unpaired or paired, two-tailed Student’s t test as indicated in figure legends. Data are presented as mean ± SE or SD as specified in figure legends. In all cases, a value of p < 0.05 was considered statistically significant.
Results
EL4 cell co-culture reveals a dual role of MS4a4B in regulation of both T cell proliferation and T cell apoptosis
CD20, a well-known member of the MS4A family, has been found to be involved in regulation of B cell proliferation and apoptosis [19, 20]. In our previous studies, activated T cells expressing high levels of MS4a4B displayed less apoptosis, which prompted us to hypothesize that MS4a4B may function as a regulator for T cell apoptosis. To test this hypothesis, we over-expressed MS4a4B protein in EL4 cells by lentiviral vector.
EL4 is a thymoma cell line, which has been widely used as in vitro and in vivo models for studying T cell function and thymoma [21–23]. Our previous studies showed that EL4 cells do not express MS4a4B protein and we generated a MS4a4B-expressing cell line by transfecting EL4 cells with a MS4a4B-lentiviral vector [15]. To explore MS4a4B function in T cell apoptosis, we co-cultured EL-4 cells (MS4a4B−GFP−) with MS4a4B-transduced EL4 cells (MS4a4B+GFP+), or mock lentiviral vector-transfected EL4 cells (LV-EL4) as control, at 1:1 ratio and assessed the growth rates of GFP− and GFP+ EL4 cells by flow cytometry and cell counting. We found that the total number of cells in EL4/MS4a4B-EL4 co-culture was less than that in EL4/LV-EL4 co-culture during the period from days 0 to 8 (Fig. 1a). We further analyzed GFP+ cells and GFP− cells in EL4/MS4a4B-EL4 co-culture and EL4/LV-EL4 co-culture respectively. The results showed that MS4a4B+GFP+ EL4 cells proliferated more slowly than MS4a4B−GFP− EL4 cells during days 0–4. However, the dominant population shifted from MS4a4B−GFP− to MS4a4B+GFP+ cells after day 12 of culture (Fig. 1b). In contrast, there was no significant difference in cell numbers between GFP+ cells and GFP− cells during the early culture period but even less numbers of mock viral vector-transfected cells in EL4/LV-EL4 co-culture after day 8 (Fig. 1c). These results suggest that un-transduced EL4 cells in EL4/MS4a4B-EL4 co-culture may undergo apoptosis after a rapid proliferating period. On the other hand, forced-MS4a4B expression in MS4a4B-transduced cells inhibited EL4 cell proliferation at the early stage of culture. At the later stages of culture, in the case of MS4a4B+ EL4 cells, MS4a4B expression may protect cells from apoptosis. To determine whether MS4a4B plays a protective role in apoptosis, we collected cell samples from EL4/MS4a4B-EL4 and EL4/LV-EL4 co-cultures to analyze the percentage of apoptotic cells. The results showed that there was no significant difference in apoptotic percentages between un-transduced cells and mock LV-transfected cells in EL4/LV-EL4 coculture (Fig. 1d). However, in EL4/MS4a4B-EL4 co-culture, MS4a4B-expressing EL4 cells have a lower apoptotic percentage compared with that of un-transduced EL4 cells (Fig. 1e), suggesting that MS4a4B plays a protective role in cells under stress culture conditions.
Fig. 1.
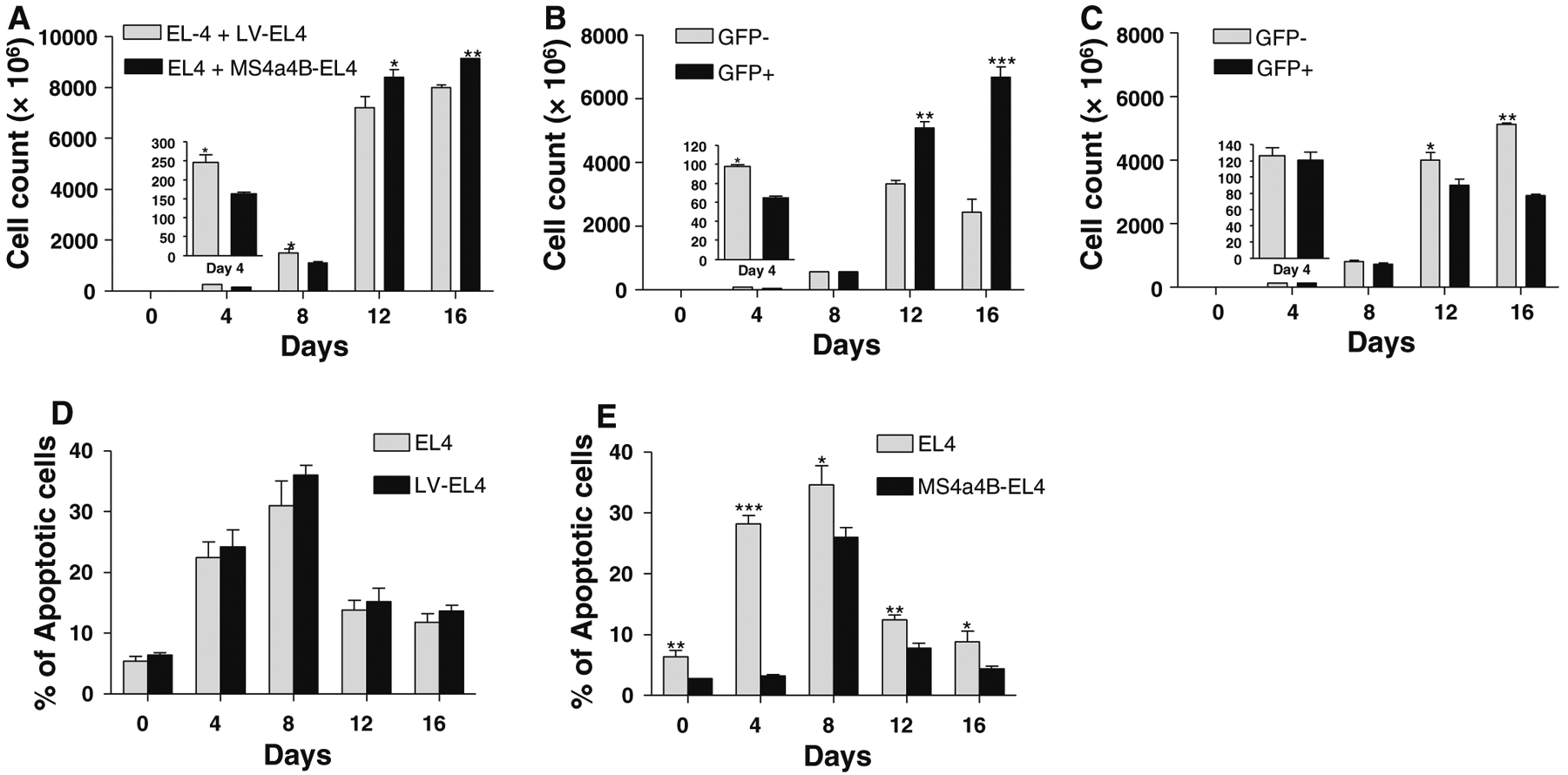
MS4a4B inhibits proliferation and apoptosis of EL4 cells. MS4a4B-transduced EL4 cells (purified GFP+) were co-cultured with an equal number of un-transduced EL4 cells (GFP−). Co-culture of mock lentiviral vector (LV)-infected EL4 cells with un-transduced EL4 cells was parallel performed as control. Cell samples were collected from culture at the time points indicated in the figures for analysis of cell proliferation and apoptosis by flow cytometry. All data are presented as mean ± SE of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). a Total cell number in EL4/LV-EL4 co-culture was compared with that in EL4/MS4a4B-EL4 coculture. b Number of MS4a4B-transduced cells (GFP+) vs. that of un-transduced EL4 cells (GFP−) in EL4/MS4a4B-EL4 co-culture. c Number of LV-infected cells (GFP+) versus that of un-transduced EL4 cells (GFP−) in EL4/LV-EL4 co-culture. d Apoptotic percentages of LV-infected EL4 cells and un-transduced EL4 cells in EL4/LV-EL4 co-culture were determined by Annexin V assay. e Apoptosis of MS4a4B-transduced EL4 cells and un-transduced EL4 cells in EL4/MS4a4B-EL4 co-culture. Apoptotic percentages were determined by Annexin V assay
Manipulation of MS4a4B expression in T cells shows that MS4a4B plays a protective role in T cell apoptosis
To confirm whether MS4a4B has a role in protecting cells from apoptosis, we treated MS4a4B-transduced EL4 cells or MS4a4B shRNA-vector-transduced T32 cells (knock-down) with staurosporine (Sigma–Aldrich), an universal apoptosis inducer [24], and analyzed the apoptotic percentage in comparison with mock-vector-transfected control cells. The results showed that forced MS4a4B expression markedly reduced apoptosis of EL4 cells (Fig. 2a–c). On the other hand, knockdown of MS4a4B increased apoptosis of T32 cells (Fig. 2d–f). These data indicate that MS4a4B may serve as a survival protein against apoptosis of activated T cells.
Fig. 2.
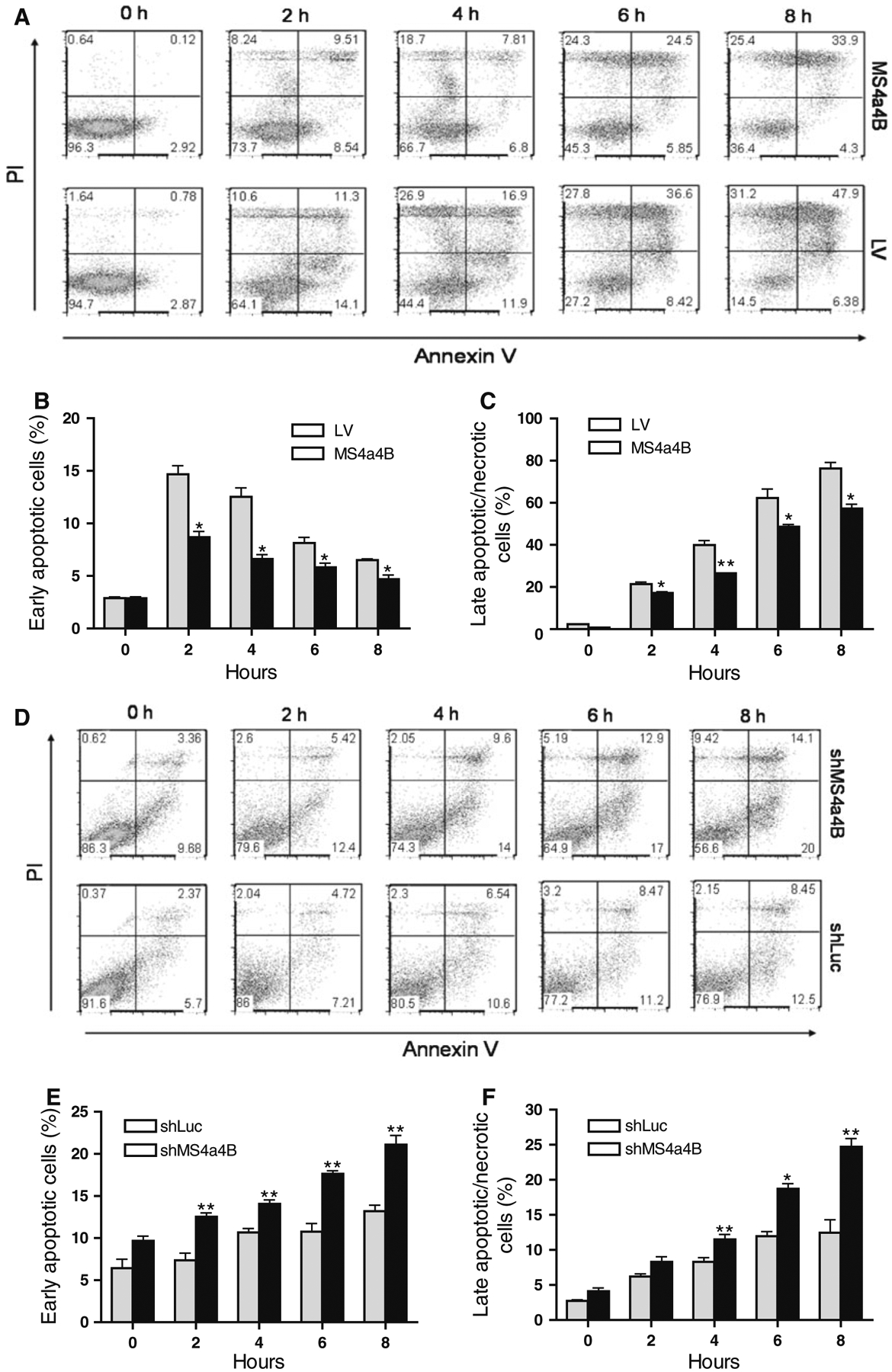
MS4a4B inhibits staurosporine-induced cell apoptosis. MS4a4B-virus-infected EL4 cells (for MS4a4B overexpression) or MS4a4B-shRNA vector (shMS4a4B)-infected T32 cells (for MS4a4B knockdown) were primed in the presence of staurosporine (2 μM) for the period indicated in the figures. Corresponding lentiviral vectors (LV mock lentiviral vector, shLuc lentiviral vector expressing luciferase-specific shRNA) were used as controls. Cell samples were collected from culture for assessment of cell apoptosis by Annexin V assay and PI staining. a Overexpression of MS4a4B reduces apoptosis percentage in EL4 cells. A representative of three experiments is shown as flow cytometric histograms (PI vs. Annexin V in GFP+ gate). Percentages of early apoptotic cells (Annexin V+PI−) (b) and late apoptotic/necrotic cells (PI+ plus Annexin V+ PI+) (c) in MS4a4B-EL4 cells were compared with that in LV-EL4 cells (GFP+ gate). The results in “b” and “c” are shown as mean ± SE of three independent experiments. *p < 0.05, **p < 0.01. d Knockdown of MS4a4B by shMS4a4B vector promotes apoptosis of T32 cells. A representative of three independent experiments is shown as flow cytometric histograms as described in “a”. Early apoptotic (e) and late apoptotic/necrotic (f) percentages of three independent knockdown experiments are presented as mean ± SE. *p < 0.05, **p < 0.01
To further determine whether MS4a4B-mediated protection is relevant to TCR stimulation-induced T cell apoptosis, we knocked down MS4a4B in αCD3/αCD28-preactivated T32 cells and primary T cells by shMS4a4B-vector and synthesized MS4a4B-specific siRNA (siM-S4a4B) respectively. The transduced cells were then primed with CD3-specific antibody alone for induction of apoptosis. T cell apoptosis was detected by flow cytometry with Annexin V assay. Consistent with the observation from EL4 cells, knockdown of MS4a4B in T cells enhanced the apoptotic percentage in both T32 cell line (Fig. 3a–c) and primary CD4+ T cells (Fig. 3d–f). Thus, MS4a4B expression plays a protective role in preventing TCR-activated T cells from activation-induced cell death (ACID). In comparison with primary T cells, activated T32 cells showed a higher background of apoptosis and that increase of apoptosis by knockdown of MS4a4B in these cells was only observed in early apoptotic, but not late/necrotic, populations. Whether it is due to the high apoptotic background of T32 cells, or that these cells have a different time course in response to anti-CD3-induced apoptosis remains to be further investigated.
Fig. 3.
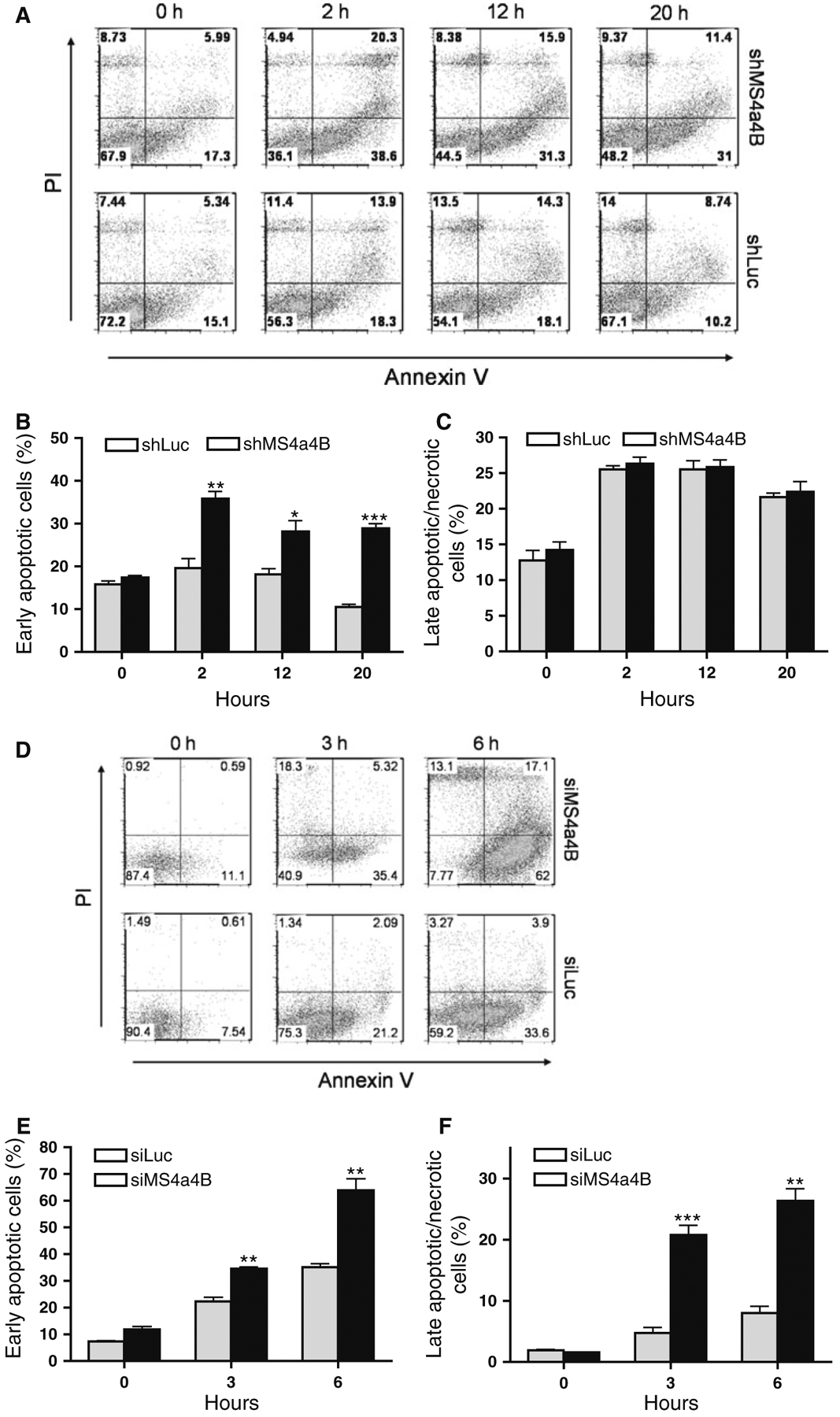
Knockdown of MS4a4B enhances TCR-stimulation-induced apoptosis. a shMS4a4B-vector (or control shLuc vector)-infected T32 cells were pre-activated by anti-CD3/anti-CD28-stimulation. The activated cells were then transferred to 24 well plates and primed with anti-CD3 antibody. Cell samples were collected at serial time points and analyzed for cell apoptosis by flow cytometry with Annexin V assay and PI staining. The results are presented as flow cytometric histograms with apoptotic percentages in GFP+ gate. One of three repeat experiments is shown. b, c Percentages of early apoptotic cells and late apoptotic/necrotic cells in MS4a4B virus-infected (or LV-infected as control) T32 cells. The data are presented as mean ± SE of cell percentages from three experiments. *p < 0.05, **p < 0.01. d Purified mouse primary CD4+ T cells were pre-activated as described in “a”. The activated cells were then transfected with Cy5-labeled MS4a4B-siRNA (siMS4a4B) or luciferase-specific siRNA (siLuc) as control. The siRNA-transduced cells were then stimulated by anti-CD3 antibody. Cell apoptosis was assessed as described in “a”. The results are presented as flow cytometric histograms with apoptotic % in Cy5+ gate. A representative of three independent experiments is shown. e, f. Percentages of early apoptotic cells and late apoptotic/necrotic cells described in “d” are presented as mean ± SE of three experiments. *p < 0.05, **p < 0.01, ***p < 0.001
MS4a4B-mediated protection in T cell apoptosis is associated with inhibition of caspase activities
We analyzed TCR-activation-induced caspase expression and activation in T32 cells and primary T cells. Caspases are essential for cell apoptosis. Caspases 8 and 9 play a critical role in activating apoptotic process. Caspases 3 and 7 are the main executers for apoptosis [25, 26]. To determine if MS4a4B protects cells from apoptosis by regulating caspase activity, we compared anti-CD3-induced caspase activity in MS4a4B-specific shRNA lentiviral vector (shMS4a4B)-infected T32 cells with that in mock vector-infected control T32 cells. We harvested shMS4a4B or control vector-infected T32 cells at 24 h after incubation with anti-CD3 antibody and measured caspase activity by Caspase-Glo® Assay Systems (Promega). The results showed that knockdown of MS4a4B significantly augmented anti-CD3-induced caspase activity, including caspase 3/7, caspase 8 and caspase 9 (Fig. 4a). To further confirm these results, we isolated CD4+ T cells from mouse spleen and knocked down MS4a4B expression by transfection of primary T cells with siMS4a4B or siRNA for luciferase (siLuc) as an irrelevant siRNA control. Cell samples were collected from culture at 24 h after siRNA transfection for determination of MS4a4B protein level by flow cytometry, and the remaining cells were re-stimulated with anti-CD3 antibody alone for detection of caspase activation by western blotting. The results showed that transfection of T cells with siMS4a4B reduced MS4a4B protein expression (Fig. 4b) and increased levels of CD3-specific antibody-induced pro-and cleaved caspases 3, 8 and 9 (Fig. 4c).
Fig. 4.

Knockdown of MS4a4B by shRNA vector enhanced anti-CD3-induced caspase activities. a shMS4a4B-vector (or control shLuc vector)-infected T32 cells were pre-activated by αCD3/αCD28 stimulation, and then were primed with αCD3 antibody alone. Cells were harvested from culture at 24 h of αCD3 stimulation for analyzing activation of caspases by Caspase-Glo@-Assays. Data were presented as mean ± SD of relative light units from duplicate. **p < 0.01. One of two independent experiments is shown. b Primary CD4+ T cells were isolated from C57BL/6 mice. Isolated cells were activated for 36 h with αCD3/αCD28 antibodies and then were transfected with siLuc or siMS4a4B. Cells were harvested at 24 h from culture. MS4a4B protein level was determined by intracellular flow cytometry with anti-MS4a4B antibody staining. Grey peak: isotype control; solid line: anti-MS4a4B antibody. c Harvested cells as described in “b” were re-stimulated with αCD3 antibody alone (5 μg/ml) for an additional 24 h and then were lysed with cell lysis buffer. β-Actin control and pro- and cleaved caspases 3, 8 and 9 were detected by western blotting with corresponding antibodies. A representative of three experiments is shown
MS4a4B-specific antibody binding abrogates MS4a4B-mediated protection in TCR-activation-induced apoptosis, leads to depletion of T cells and enhances levels of Treg cells in vivo
To test whether direct anti-MS4a4B antibody binding can induce T cell apoptosis, we stimulated mouse CD4+ T cells with αCD3/αCD28 antibodies for 2 days. Activated T cells were harvested and incubated with either anti-MS4a4B (extracellular domain) antibody or Ig control. After 48 h, cells were collected for assessment of apoptosis by Annexin V assay. To rule out whether anti-MS4a4B antibodies have pro-apoptotic effects on resting T cells, un-stimulated CD4+ T cells were parallel primed as described for activated T cells. The results showed that anti-MS4a4B-binding markedly enhanced apoptosis of TCR-activated T cells. We noticed that anti-MS4a4B priming also enhanced apoptosis in un-stimulated T cells. However, apoptotic percentage in un-stimulated T cells was much lower than that in activated T cells (Fig. 5a). These data suggest that MS4a4B-specific binding by antibody may interfere with MS4a4B mediated-protection in T cell apoptosis, especially in activated T cells.
Fig. 5.
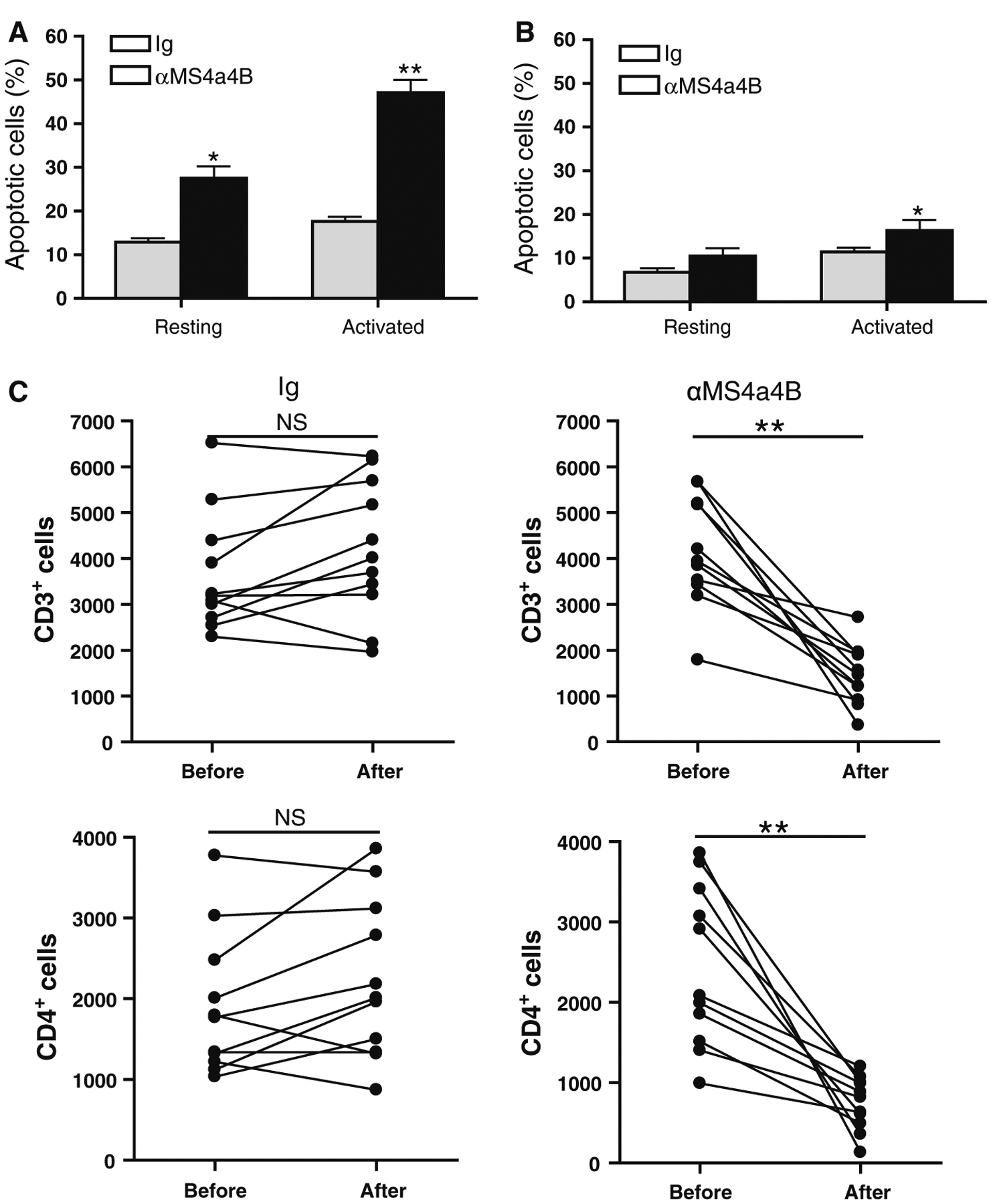
Anti-MS4a4B binding enhances T cell apoptosis and decreases number of CD3+ T cells in vivo. a Mouse spleen CD4+ T cells were isolated from C57BL/6 mice by anti-CD4 magnetic beads. Purified T cells were stimulated with pre-coated αCD3 (5 μg/ml)/αCD28 (2 μg/ml) for 2 days. The activated T cells were harvested from culture and then primed with anti-MS4a4B antibody or normal rabbit IgG control (20 μg/ml) for 48 h. Un-stimulated resting CD4+ T cells were parallel primed as described for activated T cells for comparison. T cell apoptosis was detected by Annexin V and anti-CD3 antibody. The percentages of apoptotic cells are shown as mean ± SE of three independent experiments. Statistical significance was analyzed by unpaired, two-tailed Student’s t test. *p < 0.05, **p < 0.01. b Naturally-occurring Treg cells were isolated from spleens of naїve C57BL/6 mice by CD4+CD25+ regulatory T cells isolation kit (Miltenyi Biotec Inc). Purified Treg cells were activated for 2 days as described in “a” or remained in culture without stimulation. The activated and un-stimulated cells were harvested from culture, and then primed with anti-MS4a4B antibody or normal rabbit IgG control (20 μg/ml) for 48 h. Apoptotic Treg cells were detected by Annexin V assay. The percentages of apoptotic Treg cells were presented as mean ± SE of three experiments. Statistical significance was determined as described in “a” (*p < 0.05). c MOG-immunized C57BL/6 mice were injected with 2 mg/mouse anti-MS4a4B antibody or control IgG on day 9 post immunization. Mouse peripheral blood samples (30 μl/mouse) were collected prior to and day 3 after injection with antibody or Ig control. After red blood cells were lysed, total cell numbers were counted with hemacytometer and apoptotic T cells in cell samples were analyzed with Annexin V assay. Percentage of CD3+ and CD4+ T cells was determined by flow cytometry. Total numbers of CD3+ and CD4+ T cells in 30 μl blood were calculated as total cell number × percentage of CD3+ and CD4+ cells respectively. Data are presented as cell number/μl. Three independent experiments were pooled (N = 11).Statistical significance was determined by paired, two-tailed Student’s t test. NS, not significant; **p < 0.01
To rule out whether anti-MS4a4B-induced apoptosis also affect Treg cells, we isolated naturally-occurring Treg cells from spleens of naїve C57BL/6 mice and primed these cells as described for total CD4+ T cells above. The results showed that binding of anti-MS4a4B antibody also enhanced the percentage of apoptosis in CD3/CD28-prestimulated Treg cells. However, the level of apoptosis in Treg cells is markedly lower than that in total CD4+ T cells (Fig. 5b).
To test whether administration of anti-MS4a4B antibodies can induce apoptosis in T cells and reduce T cell numbers in vivo, we treated mice with anti-MS4a4B antibodies by i.v. injection. Peripheral blood was collected prior to and after treatment. After red blood cells were lysed, apoptotic T cells in MNCs from blood samples were analyzed with Annexin V assay. Consistent with our observations from in vitro studies, we found that anti-MS4a4B antibody treatment markedly enhanced the percentage of apoptotic T cells compared with Ig-injected mice in control group (mean ± SD: 23 ± 3.7 vs. 10.3 ± 0.9). To further analyze whether anti-MS4a4B antibody treatment caused T cell depletion in vivo, total MNC numbers were counted by hemacytometer. Percentages of CD3+ and CD4+ T cells were determined by flow cytometry with anti-CD3 and anti-CD4 staining. The results showed that total numbers of CD3+ and CD4+ T cells were markedly reduced after injection with anti-MS4a4B antibodies (Fig. 5c). However, T cell numbers in the peripheral blood were gradually recovered after day 4 of antibody treatment (data not shown).
It has been documented that administration of CD3-specific antibody depletes effector T cells and subsequently induces an increase in Treg cells in mice [8]. We therefore explored whether anti-MS4a4B-mediated T cell apoptosis was associated with induction of Treg cells in vivo. We harvested peripheral blood samples and spleens on days 2, 4 and 8 post antibody treatment to compare levels of apoptotic T cells and Treg cells between anti-MS4a4B-and Ig control-treated mice. We found that T cell apoptosis mainly occurred during the early period after antibody injection, especially on day 2 post antibody injection. No difference was observed on day 8 post treatment in percentage of apoptotic T cells between anti-MS4a4B antibody-treated and Ig-treated mice (Fig. 6a). We observed a trend in increase of the percentage of blood Treg cells on day 2 post antibody treatment compared with Ig-injected mice. Interestingly, percentages of Treg cells in the peripheral blood (Fig. 6b) and spleen (Fig. 6c) were elevated post day 4 of anti-MS4a4B antibody treatment. These data suggest that anti-MS4a4B antibody-mediated T cell apoptosis may be potentially related to up-regulation of Treg cells.
Fig. 6.
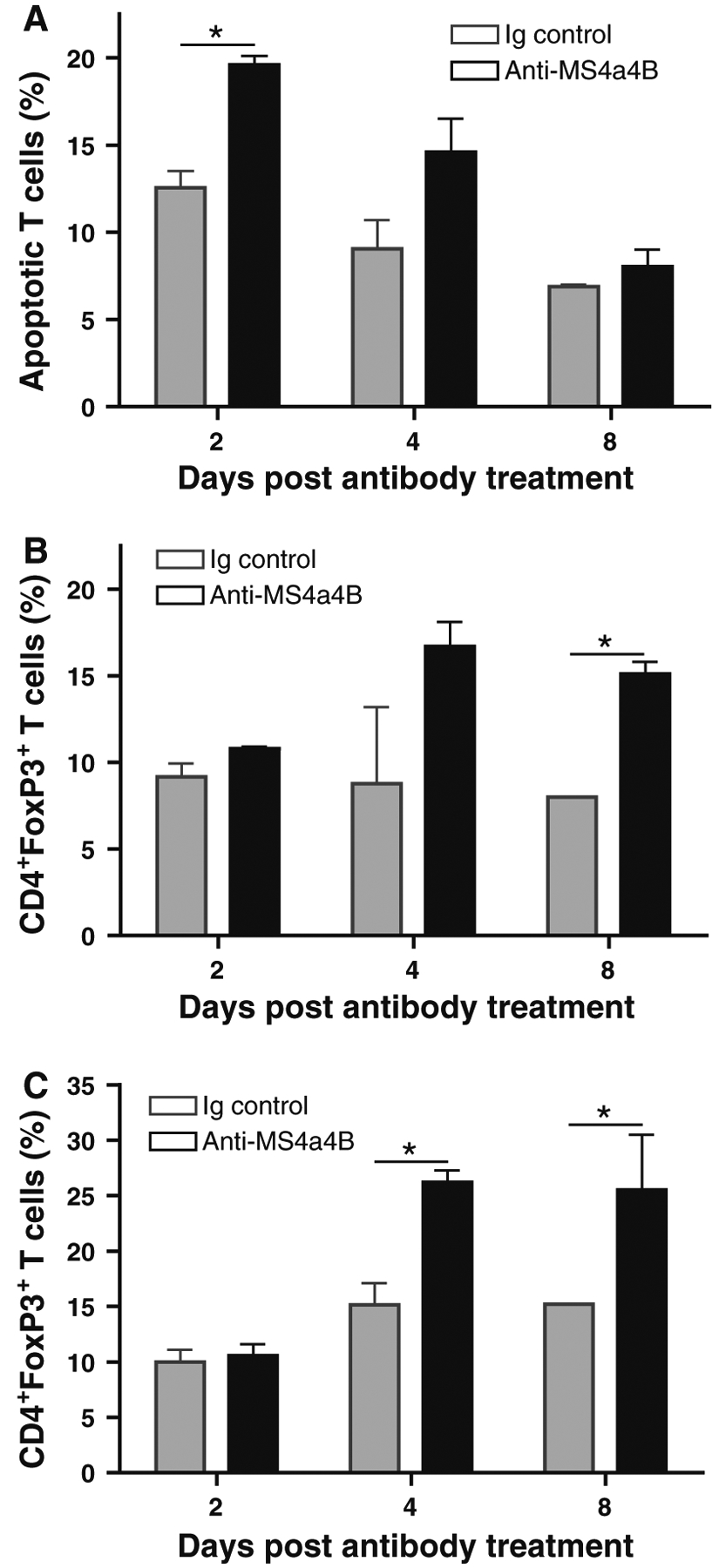
Anti-MS4a4B-mediated T cell apoptosis is accompanied by an elevated level of Treg cells. MOG-immunized C57BL/6 mice were injected with 1 mg/mouse anti-MS4a4B antibody or control rabbit IgG on day 9 post immunization. a Peripheral blood samples were collected from mice on days 2, 4 and 8 of antibody (or Ig) injection. After red blood cells were lysed, apoptotic T cells in samples were analyzed with Annexin V assay. Data are presented as mean ± SD of apoptotic T cell percentage (N = 3). b Treg cells in blood samples described in “a” were detected by flow cytometry with anti-CD4-FITC/anti-FoxP3-PE staining. Data are presented as mean ± SD of percentage of FoxP3+ T cells in CD4+ gate. c Treg cells in spleens from mice described in “a” were analyzed by flow cytometry as described in “b”. The results were presented as mean ± SD of percentage of FoxP3+ T cells in CD4+ gate (N = 3). A representative of two experiments is shown
Administration of MS4a4B antibody suppresses encephalitogenic T cell responses and ameliorates severity of EAE
We next tested the therapeutic effect of anti-MS4a4B antibodies in treatment of EAE. We induced EAE in mice with myelin oligodendrocyte glycoprotein (MOG) [18] and treated mice with anti-MS4a4B antibody by i.v injections. EAE severity was monitored daily as previously described [18, 27, 28]. In earlier experiments, we found that treatment of EAE mice by multiple injections with MS4a4B-specific antibody either during antigen-priming phase (1 mg/mouse on days 3, 6, 9, 12 p.i) or after early clinical onset of disease (2 mg/mouse on day 10; 1 mg/mouse on days 13 and 16 respectively) significantly reduced EAE severity (Fig. S2A, B). We then tested whether single injection with anti-MS4a4B antibody was still effective. The results showed that injection with MS4a4B-specific antibody (0.5 or 1 mg/mouse) at clinical onset (day 11 p.i) significantly ameliorated disease severity (Fig. 7a). Immunological analysis showed that MS4a4B-specific antibody treatment-achieved amelioration of EAE was associated with a decreased percentage of IFN-γ-expressing T cells and IL-17-expressing T cells, and increased levels of Treg cells in spinal cords (Fig. 7b). Moreover, levels of soluble pro-inflammatory cytokines, including IL-6, IFN-γ, IL-17 and TNF, in tissue extract of spinal cords were significantly decreased in MS4a4B-specific antibody-treated mice (Fig. 7c). In addition, MOG-induced T cell proliferation and production of IFN-γ and IL-17 of spleen cells from MS4a4B antibody-treated mice were also reduced (Fig. S3 and S4). Histology showed that spinal cord samples from MS4a4B-antibody-treated mice had less inflammatory cell infiltration (Fig. 7d). These results suggest that anti-MS4a4B antibody treatment effectively inhibits Th1 and Th17 encephalitogenic responses and decreases T cell-mediated inflammation in CNS in the MOG-induced EAE model.
Fig. 7.
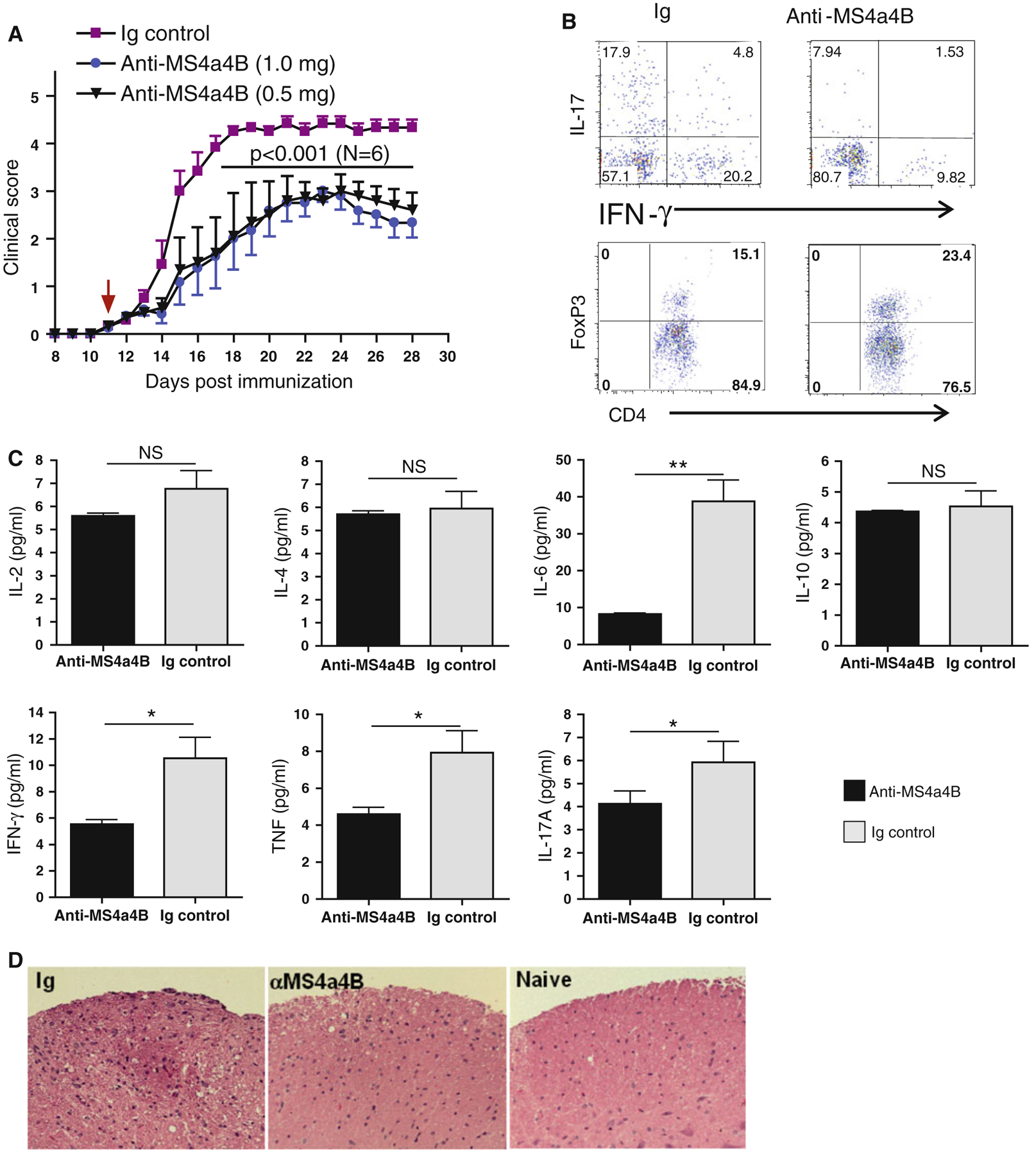
Anti-MS4a4B treatment ameliorates EAE. EAE in C57BL/6 mice was induced with MOG/CFA protocol. Mice were then injected i.v with anti-MS4a4B antibody (1 mg/mouse for test group 1; 0.5 mg/mouse for test group 2) on day 11 post immunization (p.i). Normal Ig (1 mg/mouse) was used for control group. a Anti-MS4a4B therapy reduced clinical severity of EAE. Clinical scores of MOG-immunized mice were monitored daily. Data are shown as mean ± SE of clinical scores. Statistical significance was determined by two-way ANOVA test. One of three independent experiments is shown. b Th1, Th17 and Treg cells in CNS. Mice in “A” were sacrificed on day 28 p.i. Infiltrating lymphocytes in CNS were isolated from spinal cords (pooled for anti-MS4a4B treated group (1 mg/mouse) and Ig control group respectively). Cells were stimulated with PMA/ionomycin/GolgiStop for 4 h. Cells were first stained with anti-CD4 antibody, and then fixed/permeabilized. IFN-γ, IL-17 and FoxP3 expression in cells was detected by intracellular staining with anti-IFN-γ, anti-IL-17 and anti-FoxP3 antibodies respectively. Results for Th1/Th17 are shown as IFN-γ vs. IL-17 in CD4+ gates; levels of Treg cells are shown as percentage of FoxP3+ cells in CD4+ gates. The increase of CD4+FoxP3+ cells in anti-MS4a4B-treated mice was consistent over all three repeated experiments (mean ± SD: Ig vs. anti-MS4a4B = 16.1 ± 2 vs. 22.6 ± 2.1, p < 0.05). c Levels of pro-inflammatory cytokines in CNS tissues. Tissue extracts were prepared from spinal cord samples (pooled for each group) as described in Methods. Levels of pro-inflammatory cytokines were determined by CBA kit. Results are shown as mean ± SD of duplicate. Statistical significance was determined by t test. *p < 0.05, **p < 0.01, NS not significant. d Representative images (×20 magnification) of H&E-stained spinal cord sections from anti-MS4a4B-treated and control mice
Discussion
The importance of MS4A proteins in regulation of cell proliferation and apoptosis has previously been shown in studies on MS4a1 (murine CD20) and MS4A1 (human CD20), and MS4A3 (HTm4) [3, 19, 29]. Although we still know little about the function of the MS4A family, available data show that CD20 and HTm4 play an inhibitory role in cell cycle progression of B cells and human hematopoietic cells respectively [29, 30]. Interestingly, cross-linking of CD20 by anti-CD20 mAb promoted apoptosis of B cells in vitro, indicating that in addition to the effects of CD20 in cell proliferation, it may also regulate B cell apoptosis [31]. We previously reported the inhibitory function of MS4a4B in T cell proliferation [15]. Here, we demonstrate, for the first time, that MS4a4B protein in T cells plays a dual role in T cell proliferation and apoptosis. On the one hand, MS4a4B functions as an inhibitory protein to prevent over-growth of cells; on the other hand, it plays a role in protecting T cells from ACID, suggesting that MS4a4B may serve as a survival protein for maintenance of normal cell function.
The mechanisms underlying MS4a4B-mediated protection in T cell apoptosis is likely related to its effects in regulation of the pro-apoptotic signaling pathway, especially inhibition of caspase cascades. Our data showed that levels of MS4a4B protein are negatively correlated to expression and activation of caspase 3, caspase 8 and caspase 9. At present, we do not know whether MS4a4B affects caspase activation directly or through interaction with a third-party protein. In regulatory T cells, MS4a4B has been found to directly interact with GITR (glucocorticoid induced tumor necrosis factor receptor) for function (13). Responder resting T cells express only basal levels of GITR. However, GITR expression is upregulated upon T cell activation [32]. GITR has been shown to induce pro-apoptotic or anti-apoptotic signals [33, 34]. It remains to be elucidated whether interaction of MS4a4B with GITR will interfere with GITR-mediated pro-apoptotic signals and enhance its anti-apoptotic effects.
One important finding from our studies is that binding of anti-MS4a4B antibodies to T cells can induce activated T cells to undergo apoptosis. This phenomenon has been confirmed by both in vitro and in vivo approaches. Anti-MS4a4B antibody-induced apoptosis in T cells is similar to anti-CD20 antibody-induced apoptosis in B cells observed by others [31]. Interestingly, intravenous injection of anti-MS4a4B antibodies caused T cell depletion, reduced the number of T cells in the peripheral blood, and enhanced levels of Treg cells in vivo. Moreover, administration of anti-MS4a4B antibodies in the mouse EAE model significantly ameliorated clinical severity of EAE and markedly reduced inflammation in CNS, including decreased Th1 and Th17 cell responses and levels of inflammatory cytokines, especially IL-6. The mechanisms underlying the therapeutic effects of anti-MS4a4B antibodies in the EAE model is still not completely clear. Based on the evidences from our studies, at least two potential mechanisms are involved in anti-MS4a4B treatment-mediated immune suppression: (1) anti-MS4a4B antibody-mediated temporary T cell depletion hampers function of T cells and interfere with the process of the cellular immune responses; (2) given that anti-MS4a4B-mediated T cell depletion is followed by an increase in Treg cell percentage, anti-MS4a4B antibody treatment may up-regulate function of Treg cells, that will induce an long-lasting immune tolerance. The mechanism underlying anti-MS4a4B antibody-mediated up-regulation of Treg cells still remains unclear. Based on the data from our in vitro studies (Fig. 5a, b), priming with anti-MS4a4B antibodies induces high levels of apoptosis in activated CD4+ T cells but relatively lower levels of apoptosis in Treg cells, which may change the balance of Treg cells/effector T cells and relatively up-regulates function of Treg cells. Moreover, it is possible that anti-MS4a4B therapy may modulate Treg cell function through some mechanisms similar to that observed in anti-CD3 therapy [8, 35, 36]. Given that administration of anti-MS4a4B antibodies enhanced percentages of apoptotic T cells in vivo, which was accompanied by an increase of Treg cell percentage (Fig. 6), Treg cells seem less sensitive to induction of apoptosis in comparison with activated T cells, which is consistent to the observation in anti-CD3 therapy [36]. We did not analyze absolute numbers of Treg cells in this study. However, our data showed that anti-MS4a4B antibody-mediated T cell depletion mainly occurred in the early period post treatment (day 0–4). The T cell population was gradually recovered after day 4 of treatment, which was accompanied by an increased level of Treg cells (Fig. 6), suggesting that some “new” Treg cells had been generated. Therefore, less sensitivity of Treg cells to anti-MS4a4B antibody-mediated depletion and anti-MS4a4B treatment-induced expansion of Treg cells may contribute to the up-regulation of Treg cell function. We are currently not clear whether anti-MS4a4B-mediated apoptotic T cells contribute to induction of Treg cells as observed in anti-CD3 antibody therapy in EAE model [8] since we did not find a lift of levels of Treg cell skewing cytokine TGF-β in sera of anti-MS4a4B-treated mice. Also, we did not analyze whether the up-regulation of Treg cells observed in anti-MS4a4B-treated mice was limited in some particular Treg clonetypes as reported in anti-CD3 therapy [35]. This possibility is certainly worthy to be examined in future studies. Thus, the deeper interplays in anti-MS4a4B therapy-induced up-regulation of Treg cells remain to be further explored.
In conclusion, we have demonstrated that MS4a4B plays an important role in regulating apoptosis and survival of T cells. Stimulatory signals through TCR can activate T cells and up-regulate expression of MS4a4B in these cells. MS4a4B, as a survival protein in activated T cells, may serve as a modulator in controlling the fate of activated T cells by protecting activated T cells from AICD and prolonging their survival. The protective effect of MS4a4B in T cell apoptosis is likely similar to that of CD20 in B cells, although MS4a4B may function through different mechanisms. Anti-MS4a4B antibody binding to T cells will abrogate MS4a4B-mediated protection and facilitates activated T cells to undergo apoptosis, subsequently leading to expansion of Treg cells and suppression of T cell responses, as we have demonstrated in the EAE model. Thus, MS4a4B may serve as a target of antibody-based therapy for inhibition of T cell responses. Our studies also provide a rationale for investigation of the MS4a4B counterpart in humans, which may be potentially beneficial in development of antibody-based treatment for T cell-mediated diseases.
Supplementary Material
Acknowledgments
We are grateful to Katherine Regan for editorial assistance. We thank Hong Dai and Ke Li for helpful technical assistance. This work was supported by Grants from the American Heart Association (H.X.) and from the National Institutes of Health (A.M.R.).
Footnotes
Conflict of interest The authors declare that they have no competing financial interests.
Electronic supplementary material The online version of this article (doi:10.1007/s10495-013-0870-2) contains supplementary material, which is available to authorized users.
Contributor Information
Yaping Yan, Department of Neurology, Thomas Jefferson University, 900 Walnut Street, JHN 300, Philadelphia, PA 19107, USA.
Zichen Li, Department of Neurology, Thomas Jefferson University, 900 Walnut Street, JHN 300, Philadelphia, PA 19107, USA.
Guang-Xian Zhang, Department of Neurology, Thomas Jefferson University, 900 Walnut Street, JHN 300, Philadelphia, PA 19107, USA.
Mark S. Williams, Department of Microbiology and Immunology, University of Maryland School of Medicine, 800 W Baltimore Street, Baltimore, MD 21201, USA
Gregory B. Carey, Department of Microbiology and Immunology, University of Maryland School of Medicine, 800 W Baltimore Street, Baltimore, MD 21201, USA
Jianke Zhang, Department of Microbiology and Immunology, Thomas Jefferson University, 233 South 10th Street, Philadelphia, PA 19107, USA.
Abdolmohamad Rostami, Department of Neurology, Thomas Jefferson University, 900 Walnut Street, JHN 300, Philadelphia, PA 19107, USA.
Hui Xu, Department of Neurology, Thomas Jefferson University, 900 Walnut Street, JHN 300, Philadelphia, PA 19107, USA; Division of Therapeutic Proteins, Center for Drug Evaluation and Research, Food and Drug Administration, 29 Lincoln Drive, Bethesda, MD 20892, USA.
References
- 1.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC (2005) Monoclonal antibody successes in the clinic. Nat Biotechnol 23:1073–1078 [DOI] [PubMed] [Google Scholar]
- 2.Taylor RP, Lindorfer MA (2008) Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol 20:444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tedder TF, Engel P (1994) CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today 15:450–454 [DOI] [PubMed] [Google Scholar]
- 4.Buttmann M (2010) Treating multiple sclerosis with monoclonal antibodies: a 2010 update. Expert Rev Neurother 10:791–809 [DOI] [PubMed] [Google Scholar]
- 5.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG et al. (2010) T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 16:406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF (2008) Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Investig 118:3420–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatenoud L, Bluestone JA (2007) CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 7:622–632 [DOI] [PubMed] [Google Scholar]
- 8.Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W (2008) CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med 14:528–535 [DOI] [PubMed] [Google Scholar]
- 9.Chatenoud L (2003) CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol 3:123–132 [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Williams MS, Spain LM (2006) Patterns of expression, membrane localization, and effects of ectopic expression suggest a function for MS4a4B, a CD20 homolog in Th1 T cells. Blood 107:2400–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishibashi K, Suzuki M, Sasaki S, Imai M (2001) Identification of a new multigene four-transmembrane family (MS4A) related to CD20, HTm4 and beta subunit of the high-affinity IgE receptor. Gene 264:87–93 [DOI] [PubMed] [Google Scholar]
- 12.Liang Y, Tedder TF (2001) Identification of a CD20-, Fcepsi-lonRIbeta-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics 72:119–127 [DOI] [PubMed] [Google Scholar]
- 13.Howie D, Nolan KF, Daley S, Butterfield E, Adams E, Garcia-Rueda H, Thompson C, Saunders NJ, Cobbold SP, Tone Y et al. (2009) MS4A4B is a GITR-associated membrane adapter, expressed by regulatory T cells, which modulates T cell activation. J Immunol 183:4197–4204 [DOI] [PubMed] [Google Scholar]
- 14.Venkataraman C, Schaefer G, Schindler U (2000) Cutting edge: Chandra, a novel four-transmembrane domain protein differentially expressed in helper type 1 lymphocytes. J Immunol 165:632–636 [DOI] [PubMed] [Google Scholar]
- 15.Xu H, Yan Y, Williams MS, Carey GB, Yang J, Li H, Zhang GX, Rostami AM (2010) MS4a4B, a CD20 homologue in T cells, inhibits T cell propagation by modulation of cell cycle. PLoS ONE 5:e13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan Y, Zhang GX, Williams MS, Carey GB, Li H, Yang J, Rostami A, Xu H (2012) TCR stimulation upregulates MS4a4B expression through induction of AP-1 transcription factor during T cell activation. Mol Immunol 52:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinman L, Zamvil SS (2006) How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol 60:12–21 [DOI] [PubMed] [Google Scholar]
- 18.Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, Cullimore ML, Rostami A, Xu H (2010) IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol 185:5953–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deans JP, Li H, Polyak MJ (2002) CD20-mediated apoptosis: signalling through lipid rafts. Immunology 107:176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmeister JK, Cooney D, Coggeshall KM (2000) Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis 26:133–143 [DOI] [PubMed] [Google Scholar]
- 21.Arens R, Schepers K, Nolte MA, van Oosterwijk MF, van Lier RA, Schumacher TN, van Oers MH (2004) Tumor rejection induced by CD70-mediated quantitative and qualitative effects on effector CD8? T cell formation. J Exp Med 199:1595–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dayde D, Ternant D, Ohresser M, Lerondel S, Pesnel S, Watier H, Le Pape A, Bardos P, Paintaud G, Cartron G (2009) Tumor burden influences exposure and response to rituximab: pharma-cokinetic-pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood 113:3765–3772 [DOI] [PubMed] [Google Scholar]
- 23.Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O (2009) Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol 182:2221–2230 [DOI] [PubMed] [Google Scholar]
- 24.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L et al. (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410:549–554 [DOI] [PubMed] [Google Scholar]
- 25.Fan TJ, Han LH, Cong RS, Liang J (2005) Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 37:719–727 [DOI] [PubMed] [Google Scholar]
- 26.Krammer PH, Arnold R, Lavrik IN (2007) Life and death in peripheral T cells. Nat Rev Immunol 7:532–542 [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Zhang GX, Wysocka M, Wu CY, Trinchieri G, Rostami A (2000) The suppressive effect of TGF-beta on IL-12-mediated immune modulation specific to a peptide Ac1–11 of myelin basic protein (MBP): a mechanism involved in inhibition of both IL-12 receptor beta1 and beta2. J Neuroimmunol 108:53–63 [DOI] [PubMed] [Google Scholar]
- 28.Zhang GX, Xu H, Kishi M, Calida D, Rostami A (2002) The role of IL-12 in the induction of intravenous tolerance in experimental autoimmune encephalomyelitis. J Immunol 168:2501–2507 [DOI] [PubMed] [Google Scholar]
- 29.Donato JL, Ko J, Kutok JL, Cheng T, Shirakawa T, Mao XQ, Beach D, Scadden DT, Sayegh MH, Adra CN (2002) Human HTm4 is a hematopoietic cell cycle regulator. J Clin Investig 109:51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tedder TF, Forsgren A, Boyd AW, Nadler LM, Schlossman SF (1986) Antibodies reactive with the B1 molecule inhibit cell cycle progression but not activation of human B lymphocytes. Eur J Immunol 16:881–887 [DOI] [PubMed] [Google Scholar]
- 31.Janas E, Priest R, Wilde JI, White JH, Malhotra R (2005) Rituxan (anti-CD20 antibody)-induced translocation of CD20 into lipid rafts is crucial for calcium influx and apoptosis. Clin Exp Immunol 139:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhan Y, Funda DP, Every AL, Fundova P, Purton JF, Liddicoat DR, Cole TJ, Godfrey DI, Brady JL, Mannering SI et al. (2004) TCR-mediated activation promotes GITR upregulation in T cells and resistance to glucocorticoid-induced death. Int Immunol 16:1315–1321 [DOI] [PubMed] [Google Scholar]
- 33.Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, Migliorati G, Riccardi C (1997) A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci USA 94:6216–6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spinicelli S, Nocentini G, Ronchetti S, Krausz LT, Bianchini R, Riccardi C (2002) (GITR) interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ 9:1382–1384 [DOI] [PubMed] [Google Scholar]
- 35.Nishio J, Feuerer M, Wong J, Mathis D, Benoist C (2010) Anti-CD3 therapy permits regulatory T cells to surmount T cell receptor-specified peripheral niche constraints. J Exp Med 207:1879–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penaranda C, Tang Q, Bluestone JA (2011) Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol 187:2015–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.