

SUMMARY
In budding yeast, a single DNA double-strand break (DSB) triggers the activation of Mec1ATR-dependent DNA damage checkpoint. After about 12h, cells turn off the checkpoint signaling and adapt despite persistence of the DSB. We report that adaptation involves the autophosphorylation of Mec1 at site S1964. A non-phosphorylatable mec1-S1964A mutant causes cells to arrest permanently in response to a single DSB without affecting the initial kinase activity of Mec1. Autophosphorylation of S1964 is dependent on Ddc1Rad9 and Dpb11TopBP1, and it correlates with the timing of adaptation. We also report that Mec1’s binding partner, Ddc2ATRIP, is an inherently stable protein that is degraded specifically upon DNA damage. Ddc2 is regulated extensively through phosphorylation, which in turn regulates the localization of Mec1-Ddc2 complex to DNA lesions. Taken together, these results suggest that checkpoint response is regulated through the autophosphorylation of Mec1 kinase and through the changes in Ddc2 abundance and phosphorylation.
Keywords: DNA damage response, Mec1/ATR, Ddc2/ATRIP, PIKK, Rad53, Rad9, S. cerevisiae, cell cycle, checkpoint
INTRODUCTION
In response to DNA damage, cells activate the DNA damage response (DDR) (Gobbini et al. 2013, Harrison and Haber 2006). In eukaryotic cells, the DDR is orchestrated by phosphoinositide-3-kinase related protein kinases (PIKKs), which are serine/threonine kinases. The PIKK family of proteins include ATM, ATR and DNA-PKcs in mammals (Lovejoy and Cortez 2009). In the budding yeast Saccharomyces cerevisiae, the major PIKK that is responsible for the activation of DDR is the ATR homolog, Mec1, while the nonessential ATM homolog Tel1 is largely dispensable in response to double strand breaks (DSBs) (Mallory and Petes 2000, Mantiero et al. 2007). Unlike Tel1, Mec1 is essential for survival; its primary role is to stabilize stalled replication forks (Tercero and Diffley 2001). The lethality of mec1Δ can be rescued by upregulation of the dNTP metabolism, for instance, by deleting the ribonucleotide reductase inhibitor SML1 (Zhao et al. 1998).
Under basal conditions, the majority of Mec1 exists as a homodimer of a heterodimer with its binding partner, Ddc2ATRIP (Ball and Cortez 2005, Sawicka et al. 2016). Upon induction of a DSB, the dsDNA ends are resected and coated with the ssDNA binding protein RPA (Alani et al. 1992, Zou and Elledge 2003). Mec1 is recruited to the break site through Ddc2-RPA interactions (Nakada et al. 2005), leading to its activation (Rouse and Jackson 2002). Ddc2 is also phosphorylated in a Mec1-dependent manner in response to both DNA damage and replication stress (Paciotti et al. 2000).
After its recruitment to the DSB site, Mec1 initiates DDR by phosphorylating consensus SQ/TQ sites in its downstream target proteins (Kim et al. 1999). One such early event is the phosphorylation of yeast histone H2A (referred to as γ-H2AX) (Rogakou et al. 1999, Shroff et al. 2004), a modification that spreads extensively around the break site (Lee et al. 2014). γ-H2AX stimulates the recruitment of the scaffold protein, Rad953BP1, which is also phosphorylated in a Mec1-dependent manner (Emili 1998). Rad9 aids the recruitment of the effector kinase Rad53CHEK2 (Sun et al. 1998). Rad53 is initially phosphorylated by Mec1, but it is fully activated through autophosphorylation in the presence of Rad9 (Pellicioli et al. 2001, Sanchez et al. 1996). Another signaling kinase, Chk1, is activated in a similar manner after damage (Chen and Sanchez 2004). Activation of these kinases results in cell cycle arrest by blocking anaphase progression (Yamamoto et al. 1996).
Mec1’s activity is substantially stimulated after its recruitment to DNA lesions by several co-activators (Navadgi-Patil and Burgers 2011). These include the replication protein Dpb11TopBP1, the sliding 9-1-1 clamp subunit Ddc1Rad9, and the endonuclease Dna2. Ddc1 has been implicated in activating Mec1 in G1 and G2/M phases of the cell cycle (Navadgi-Patil and Burgers 2009). During S phase, Mec1’s kinase activity is mainly stimulated by Dna2 (Kumar and Burgers 2013).
A single endonuclease-induced DSB in budding yeast is sufficient to activate DNA damage checkpoint and cause cell cycle arrest prior to anaphase (Lee et al. 1998). This arrest is entirely Mec1-dependent, however Tel1 contributes to the prolongation of arrest (Clemenson and Marsolier-Kergoat 2009). If the DSB is repaired, the cells turn off the checkpoint through a process termed recovery (Lee et al. 2000, Vaze et al. 2002). If the DSB persists, the cells remain arrested at the G2/M phase for 12 to 15h (equivalent to 5–6 normal cell cycles), but eventually turn off the checkpoint through a phenomenon termed adaptation (Lee et al. 1998, Lee et al. 2000, Toczyski et al. 1997). The genetic requirements for adaptation and recovery are overlapping but distinct; in both cases, initial activation of the DDR is Mec1-dependent (Harrison and Haber 2006). Among several factors that are necessary both for recovery and adaptation are the PP2C phosphatases Ptc2 and Ptc3 (Guillemain et al. 2007, Leroy et al. 2003), which dephosphorylate Rad53 and possibly other proteins to terminate the checkpoint signaling. Additionally, the component of Golgi-associated retrograde protein Vps51 is required both for adaptation and recovery (Dotiwala et al. 2013). In contrast, the recombination protein Rdh54/Tid1 (Lee et al. 2001, Vaze et al. 2002) is required specifically for adaptation. Despite the characterization of many proteins that play a role in turning off the checkpoint, how Mec1 signaling is extinguished at the time of adaptation and recovery remains elusive.
Several studies suggest that Mec1 is regulated by phosphorylation in response to DNA damage. Mec1 has been shown to be phosphorylated during replication stress on sites S38 (Bastos de Oliveira et al. 2015) and S1991 (Hustedt et al. 2015). S38 is a Mec1 consensus SQ site, and its phosphorylation is Mec1-dependent, whereas S1991 is not a Mec1 consensus site and is indirectly Mec1-dependent. In mammals, the Mec1 homolog ATR is activated through autophosphorylation of T1989 (Liu et al. 2011, Nam et al. 2011).
We performed a mutational screen to understand if Mec1 autoregulation is important for DDR and for adaptation. We individually mutated each of the nine SQ/TQ sites in Mec1 either to alanine to render these sites non-phosphorylatable, or to glutamic acid to mimic phosphorylation. We find that two previously uncharacterized SQ/TQ sites on Mec1, T1902 and S1964, are critical for the regulation of the DDR and for adaptation. S1964 is phosphorylated only after DNA damage, in a Mec1-dependent manner. An S1964A substitution causes the cells to arrest permanently in response to a single irreparable DSB. In contrast, phosphomimetic substitutions of site T1902 markedly shortens the duration of cell cycle arrest.
In addition to Mec1, we find that Ddc2 is regulated extensively by phosphorylation in response to DNA damage. Two non-SQ sites on Ddc2, S173 and S182, are particularly important. A non-phosphorylatable ddc2-S173A,S182A mutant permanently arrests in response to a single DSB. Abolishing all three SQ/TQ sites on Ddc2 to does not alter cell cycle arrest or adaptation; however, this triple alanine mutant eliminates the DNA damage-dependent change in electrophoretic mobility of Ddc2. Finally, we report that Ddc2 is a stable protein that is specifically degraded upon DNA-damage; this degradation is partially influenced by DDR. Taken together, we show that the regulation of Mec1 kinase in response to DNA damage is multifaceted; its activity is controlled through autophosphorylation and its recruitment to DNA lesions is modulated by the changes in Ddc2 abundance and phosphorylation.
RESULTS
Mutational analysis of SQ/TQ sites in Mec1
We mutated all 9 Mec1 consensus SQ/TQ sites to alanine (AQ) or glutamic acid (EQ) [Figure 1A] at the genomic MEC1 locus individually by using CRISPR/Cas9 as described previously (Anand et al. 2017) to investigate whether these sites are regulated by autophosphorylation. In contrast to the kinase-dead mec1-D2224A mutant (Paciotti et al. 2001), which is only viable in a sml1Δ strain, none of the Mec1 AQ/EQ mutants cause lethality or growth defects in WT [Figure S1A, Figure 2B] or in sml1Δ background (data not shown). While the kinase-dead mec1-D2224A allele conferred sensitivity to the DNA damaging reagents such as hydroxyurea (HU), phleomycin and UV; none of the Mec1 AQ/EQ mutants tested were sensitive to these reagents, with the exception of mec1-T336E’s sensitivity to phleomycin [Figure S1A, Figure 2B].
Figure 1. Mec1 T1902 and S1964 sites regulate Mec1 function.
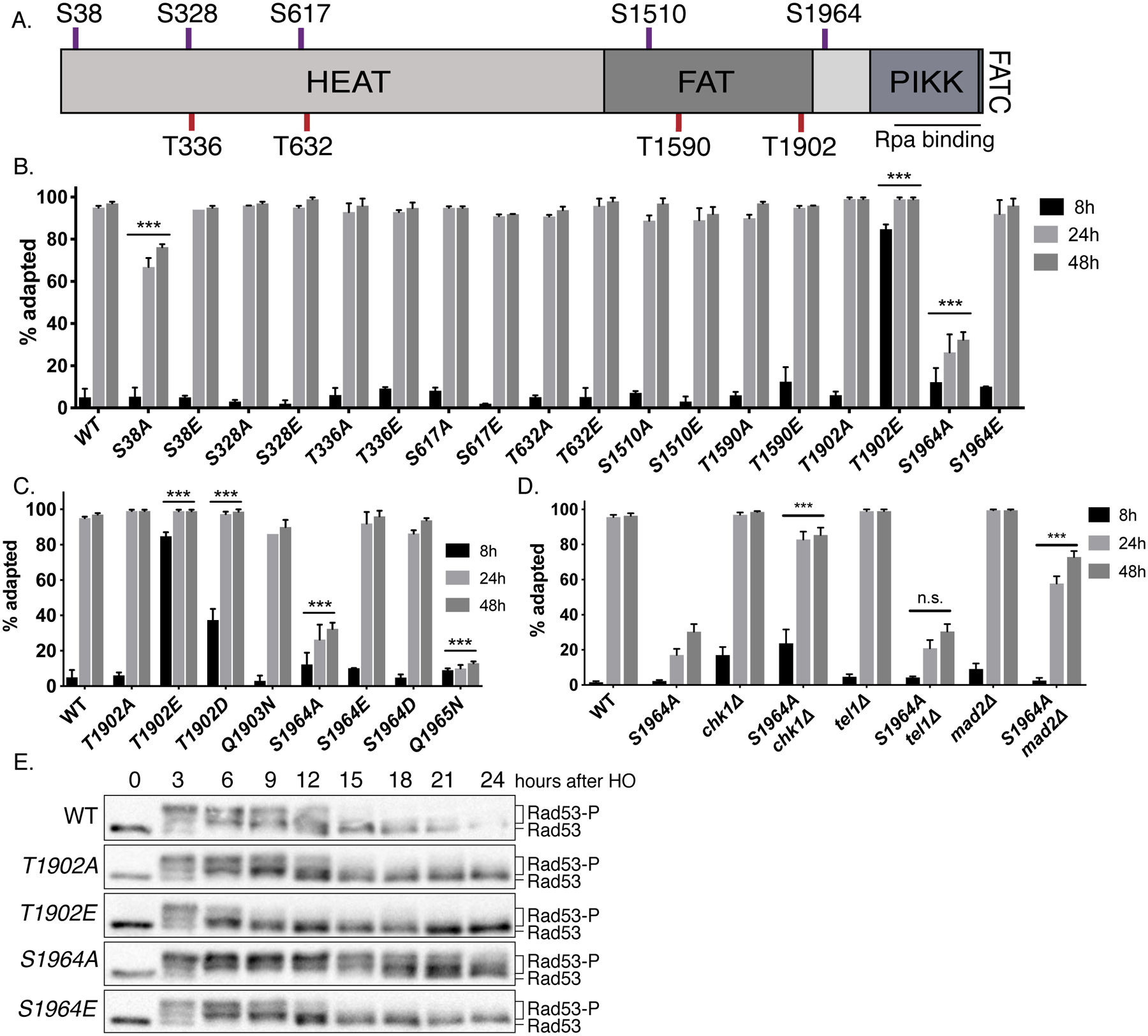
(A) The domain structure of Mec1, including the N-terminus HEAT repeat domain, FAT domain, PIKK domain and the C-terminus FAT domain and the kinase domain. The approximate locations of SQ sites (red) and TQ sites (purple) are indicated.
(B) Cell cycle arrest and adaptation in Mec1 mutants (See Methods). ***p<0.001, compared to WT. Error bars represent SEM.
(C) Cell cycle arrest and adaptation of Mec1 T1902 and S1964 mutants. ***p<0.001, compared to WT. Error bars represent SEM.
(D) Cell cycle arrest and adaptation in mec1-S1964A chk1Δ, mec1-S1964A tel1Δ and mec1-S1964A mad2Δ double mutants. ***p<0.001, compared to mec1-S1964A. Error bars represent SEM.
(E) Rad53 phosphorylation in Mec1 T1902 and S1964 mutants following the induction of an irreparable DSB.
Figure 2. Mec1 S1964 and T1902 mutants do not alter intrinsic Mec1 activity and DNA repair.
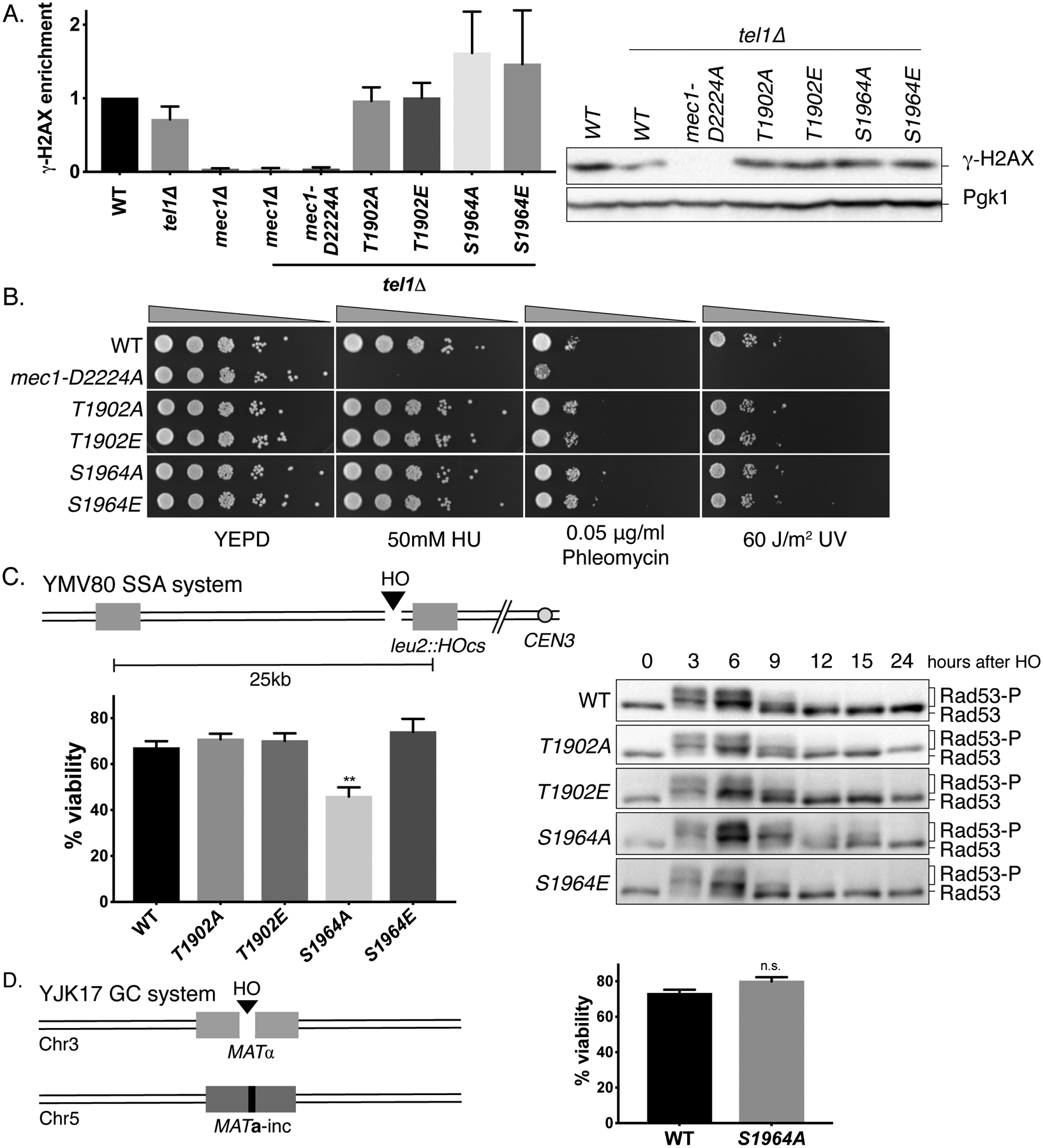
(A) γ-H2AX levels in Mec1 point mutants in a tel1Δ background 3h after the induction of an irreparable DSB, normalized to WT. Error bars represent SEM.
(B) Sensitivity of Mec1 T1902 and S1964 mutants to DNA damaging reagents HU, phleomycin and UV.
(C) The schematic of YMV80 recovery system. The viability (left) and Rad53 hyperphosphorylation (right) of Mec1 T1902 and S1964 mutants after recovery in YMV80. **p=0.0078, compared to WT. Error bars represent SEM.
(D) Schematic of the gene conversion YJK17 system, and the viability of mec1-S1964A mutant in YJK17. p=0.059. Error bars represent SEM.
Mec1 sites T1902 and S1964 are important for its regulation
The site-specific HO endonuclease under the control of a galactose-inducible promoter (GAL::HO) has been used to create an irreparable DSB at the MATα locus (Lee et al. 1998, Lee et al. 2000). This irreparable DSB causes the activation of the DDR and leads to prolonged cell cycle arrest (Lee et al. 1998, Lee et al. 2000). As shown previously (Lee et al. 1998), after the induction of a single DSB, WT cells were mostly arrested prior to anaphase at 8h and were fully adapted by 24h [Figure 1B]. Most Mec1 AQ/EQ mutants behaved similar to WT with two exceptions: mec1-T1902E phosphomimetic substitution led to premature cell cycle progression by 8h, while mec1-S1964A remained arrested up to 24h [Figure 1B, Figure 1C]. Moreover, a mec1-Q1965N substitution, which also disrupts the Mec1 SQ consensus motif, was as adaptation-defective as the mec1-S1964A [Figure 1C].
We created double mutant combinations of sites T1902 and S1964 to examine the possible genetic interactions between these two sites. The double mutant mec1-T1902A,S1964A was as adaptation-defective as the mec1-S1964A single mutant, whereas the phosphomimetic double mutant mec1-T1902E,S1964E was indistinguishable from mec1-T1902E and rapidly turned off the checkpoint signaling [Figure S1C]. Neither of these double mutants were sensitive to the DNA damaging reagents tested [Figure S1B]. The rapid adaptation of mec1-T1902E was partially suppressed by mec1-S1964A, while the mec1-T1902A,S1964E double mutant showed roughly WT behavior [Figure S1C]. These results suggest that sites T1902 and S1964 affect Mec1 activity independently.
Deletion of CHK1 or TEL1 have been shown to modestly shorten, but not eliminate, the cell cycle arrest (Clerici et al. 2014, Dotiwala et al. 2010). Deleting the spindle assembly checkpoint protein Mad2 also suppresses the permanent arrest of several adaptation-defective mutants (Dotiwala et al. 2010). We found that the deletion of MAD2 and CHK1 rescued the adaptation defect of mec1-S1964A [Figure 1D], in agreement with the previously published results with other adaptation-defective mutants (Dotiwala et al. 2010). Deletion of TEL1 had no effect on the cell cycle arrest of mec1-S1964A, implying that the adaptation defect of this mutant is due to the persistence or hyperactivation of the Mec1 branch of the checkpoint pathway.
We monitored the DNA damage response of Mec1 mutants by assaying the phosphorylation of the effector kinase Rad53, the adaptor protein Rad9, and Mec1’s binding partner Ddc2 following the induction of a single irreparable DSB. In a WT strain, Rad53, Rad9 and Ddc2 are each phosphorylated in a Mec1-dependent manner soon after damage and these phosphorylations diminish 12 to 15h after inducing a DSB, as the cells adapt and turn off the checkpoint [Figure 1E and Figure S2] (Emili 1998, Pellicioli et al. 2001). In the adaptation-defective mec1-S1964A mutant, Rad53 and Rad9 phosphorylations were prolonged, whereas mec1-T1902E cells turned off Rad53 and Rad9 signaling by about 9h [Figure 1E and Figure S2A]. Consistent with the persistent cell cycle arrest phenotype of mec1-S1964A, in this mutant, hyperphosphorylated Ddc2 was detectible up to 24h [Figure S2B]. Overall, these data demonstrate that the Mec1 sites T1902 and S1964 are important for regulation of checkpoint response.
Mutating Mec1 sites T1902 and S1964 does not alter the initial kinase activity of Mec1
The checkpoint phenotypes of the T1902 and S1964 mutations could be explained by alterations in Mec1’s kinase activity. As readout of initial kinase activity of Mec1, we measured the levels of phosphorylated histone H2A (γ-H2AX) in Mec1 T1902 and S1964 mutants 3h after DSB induction by western blot in a tel1Δ background, so that the only active kinase available to phosphorylate H2A is Mec1. While γ-H2AX was essentially absent in the mec1-D2224A tel1Δ as well as in the mec1Δ tel1Δ double mutants, Mec1 T1902 and S1964 mutants displayed γ-H2AX levels comparable to WT [Figure 2A]. Although the mec1-T1902E mutant has shortened cell cycle arrest, it retains normal kinase activity towards a substrate that is modified within an hour after the induction of damage (Lee et al. 2014, Shroff et al. 2004). Similarly, the adaptation defect of mec1-S1964A does not appear to reflect an intrinsically elevated kinase activity. These findings are supported by the comparable levels of Rad9 phosphorylation 3h after the induction of a DSB [Figure S2A].
The mec1-S1964A mutant is defective in recovery after repair
We then asked if the altered cell cycle arrest duration we observe in Mec1 T1902 and S1964 mutants would affect recovery after repair. We used strain YMV80, which contains an HO cut site located between two widely-separated homologous sequences [Figure 2C]. In this strain, the HO- induced DSB is repaired through single-strand annealing (SSA) after extensive 5’ to 3’ resection (Vaze et al. 2002), during which time cells fully activate the checkpoint signaling. If the cells fail to turn off the checkpoint after repair, they cannot resume mitosis and they eventually die. In the adaptation-defective mec1-S1964A mutant, we observed a statistically significant decrease in viability, suggesting impaired recovery [Figure 2C]. Consistent with this, mec1-S1964A mutant exhibited prolonged Rad53 hyperphosphorylation after the induction of the DSB [Figure 2D]. On the other hand, the viability of mec1-S1964A in YJK17 strain, which can repair the DSB in approximately 6h through gene conversion (GC) and does not lead to a pronounced cell cycle arrest was comparable to WT [Figure 2D], suggesting mec1-S1964A substitution does not affect repair by homologous recombination. Because mec1-S1964A does not confer sensitivity to DNA damaging reagents and does not lead to repair defects, we conclude that the recovery defect of this strain is due to exacerbated checkpoint signaling downstream of Mec1. We note, however, that this recovery defect is much less profound than that seen by deleting PP2C phosphatases Ptc2 and Ptc3 (Leroy et al. 2003); suggesting that mec1-S1964A may principally affect DNA damage signaling when the DSB is unrepaired.
Mec1 site S1964 is auto-phosphorylated after DNA damage
We performed mass spectrometry on immunoprecipitates of epitope-tagged Ddc2 (see Methods), to see if the Mec1 S1964 and T1902 sites are phosphorylated. The phosphorylation of Mec1-S1964 was detectible 6h after the induction of the DSB, together with the phosphorylation of Rfa1-S178 (Kim and Brill 2003) and Mec1-S38 (Bastos de Oliveira et al. 2015), both of which are shown to be Mec1-dependent [Figure 3A and SI Table 4]. S1964 phosphorylation was not detected prior to the induction of DNA damage, or in an isogenic strain that did not induce damage [SI Table 4], suggesting that Mec1-S1964 phosphorylation is DNA damage-dependent. We were also unable to detect a peptide containing phosphorylation at T1902, as the predicted T1902-containing tryptic peptide is too large for MS analysis. Attempts to detect phospho-T1902 using double protease digestion using trypsin and GluC were also unsuccessful [SI Table 5]. Based on these, we conclude that T1902 is likely not a phosphorylation site.
Figure 3. Mec1 S1964 is autophosphorylated after DNA damage.
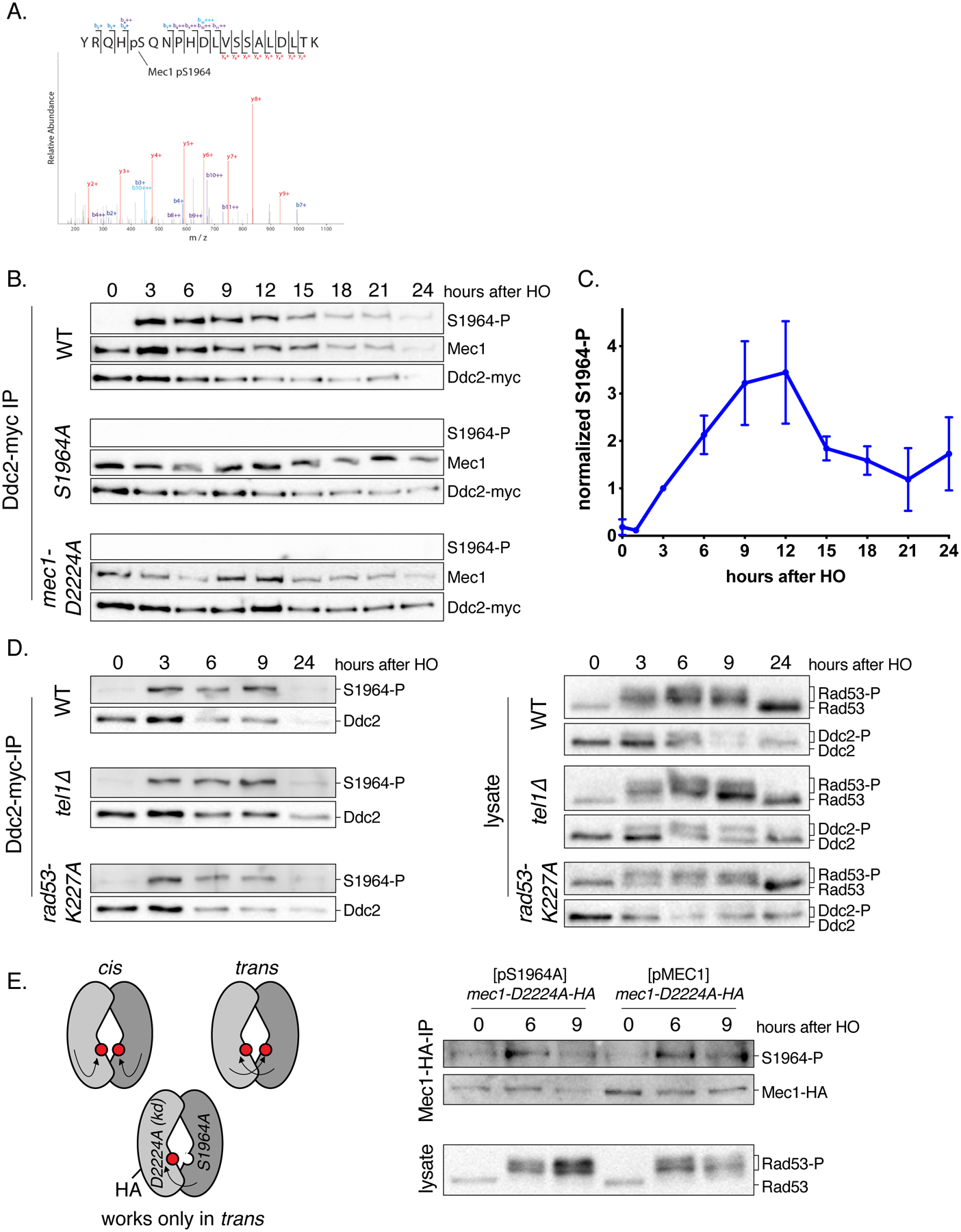
(A) MS2 spectra of a tryptic peptide containing S1964 phosphorylation. Phosphopeptides were enriched from Ddc2-myc immunoprecipitates 6h after the induction of the DSB. For the full list of the phosphopeptides, see SI Table 4.
(B) Mec1-S1964 phosphorylation in WT, mec1-S1964A and mec1-D2224A kinase-dead strains containing Ddc2-myc after the induction of an irreparable DSB. Samples were blotted with anti-myc antibody for Ddc2 detection, anti-Mec1, and a phospho-specific antibody specific to Mec1-S1964 phosphorylation following Ddc2-myc immunoprecipitation.
(C) Mec1-S1964 phosphorylation in a WT strain following the induction of an irreparable DSB, quantified as a ratio of S1964 phosphorylation signal to Ddc2 signal and normalized to the signal at 3h. Error bars represent SEM.
(D) Mec1-S1964 phosphorylation in tel1Δ and rad53-K227A strains following Ddc2-myc immunoprecipitation.
(E) The schematic representation of the strain used for the cis vs trans experiment (left). The “trans-only” strain contains the mec1-D2224A kinase-dead allele tagged with HA integrated in the genome, and a centromeric plasmid expressing MEC1-S1964A. Mec1-S1964 phosphorylation was assayed after Mec1-HA coimmunoprecipitation.
Overexpression of Ddc2 leads to the hyperactivation of Mec1 signaling in WT cells (Clerici et al. 2001). We found that overexpression of Ddc2 suppresses mec1-T1902E and prevents adaptation, whereas Ddc2 overexpression does not rescue the complete lack of cell cycle arrest in kinase-dead mec1-D2224A mutant [Figure S3A]. Moreover, a mec1-T1902E substitution suppresses the adaptation defects of three mutants: ptc2Δ (Leroy et al. 2003), tid1Δ (Lee et al. 2001) and vps51Δ (Dotiwala et al. 2013) [Figure S3B]. Taken together with the cell cycle arrest and adaptation phenotypes of mec1-T1902E and mec1-T1902D, these findings suggest that phosphomimetic substitutions of Mec1-T1902 alter the persistence of Mec1 activity without markedly reducing its intrinsic activity. Recent structural data indicates that T1902 interacts with the kα3 helix of the N lobe of Mec1 catalytic domain (Wang et al. 2017), signifying a regulatory role for the site T1902. To further validate Mec1-S1964 phosphorylation, we generated a phospho-specific antibody against this site. We immunoprecipitated epitope-tagged Ddc2 and probed the samples for Mec1 (Hustedt et al. 2015) and phosphorylated S1964. Phosphorylation of S1964 was undetectable with the phospho-specific antibody in mec1-S1964A mutant, demonstrating the specificity of the antibody [Figure 3B]. Additionally, we could not detect a signal with the phospho-specific antibody in a strain that carries the kinase-dead mec1-D2224A allele. The phosphorylation of S1964 was not apparent prior to DNA damage [Figure 3B and Figure S3C]; thus, this modification only happens upon Mec1 activation. S1964 phosphorylation peaks between 9 and 12h after the induction of a DSB [Figure 3C and Figure S3D], suggesting that it correlates with the timing of adaptation. To determine whether Mec1’s role in S1964 phosphorylation is indirect via Rad53 or Tel1, we examined the S1964 phosphorylation in rad53-K227A kinase-dead and in tel1Δ mutants. Phosphorylation at site S1964 was detectible in the absence of Rad53 kinase activity, as well as in the absence of Tel1 [Figure 3D]. Since the phosphorylation of S1964 is DNA damage and Mec1-dependent and Rad53 and Tel1-independent, we conclude that S1964 is a Mec1 autophosphorylation site.
Two PP2C type phosphatases, Ptc2 and Ptc3, have been shown to promote adaptation through dephosphorylation of Rad53 (Leroy et al. 2003). We asked if Ptc2 and Ptc3 also target Mec1-S1964 phosphorylation. In a ptc2Δ ptc3Δ double mutant, Mec1-S1964 phosphorylation was enriched between 9 and 15 h after DSB induction, but eventually was diminished [Figure S3E], suggesting that Ptc2 and Ptc3 are not likely to play a role in Mec1 dephosphorylation. Other phosphatases, such as Pph3, could be involved in S1964 dephosphorylation (Hustedt et al. 2015).
Mec1 autophosphorylation at site S1964 can occur in trans
Recent structural studies have suggested that budding yeast Mec1-Ddc2 exists as a homodimer of a heterodimer (Sawicka et al. 2016, Wang et al. 2017). Autophosphorylation of Mec1 could therefore happen in cis or in trans. To distinguish between these two possibilities, we created a strain that carries a genomic mec1-D2224A allele that is epitope-tagged with HA, and a centromeric plasmid that bears the kinase-proficient mec1-S1964A allele; hence, Mec1-S1964 autophosphorylation can only be detected if it happens in trans [Figure 3E]. Mec1-S1964 phosphorylation was detectible in this trans configuration upon DNA damage induction [Figure 3E]. We confirmed that the checkpoint was activated properly in the mec1-D2224A strain carrying kinase-proficient mec1-S1964A from a plasmid, as Rad53 was hyperphosphorylated in this strain after DSB induction [Figure 3E]. Given that the mec1-S1964A allele cannot itself be phosphorylated in this strain, we conclude that the Mec1 phosphorylation must have occurred on the kinase-dead monomer, in trans.
Ddc1 and Dpb11 are activators of Mec1 kinase activity for S1964 autophosphorylation
Mec1-S1964 autophosphorylation happens in a DNA damage-dependent manner; implying that Mec1’s basal kinase activity is not sufficient for this autophosphorylation. At least three Mec1 co-activating factors, Ddc1, Dpb11 and Dna2, are shown to further stimulate Mec1’s kinase activity in different contexts (Lanz et al. 2018, Navadgi-Patil and Burgers 2011). We tagged these Mec1 coactivators with auxin-inducible degrons (AID) at their C-termini (Morawska and Ulrich 2013), and depleted them 3h prior to the induction of DSB. Degradation of either Ddc1 or Dpb11 before checkpoint activation resulted in substantially reduced levels of phosphorylated Rad53 after the induction of DSB, whereas the degradation of Dna2 had no effect [Figure 4A]. Furthermore, the degradation of Dpb11 or Ddc1 prior to DNA damage induction caused reduced Ddc2 phosphorylation [Figure 4B].
Figure 4. Ddc1 and Dpb11 regulate the enrichment of Mec1 S1964 autophosphorylation.
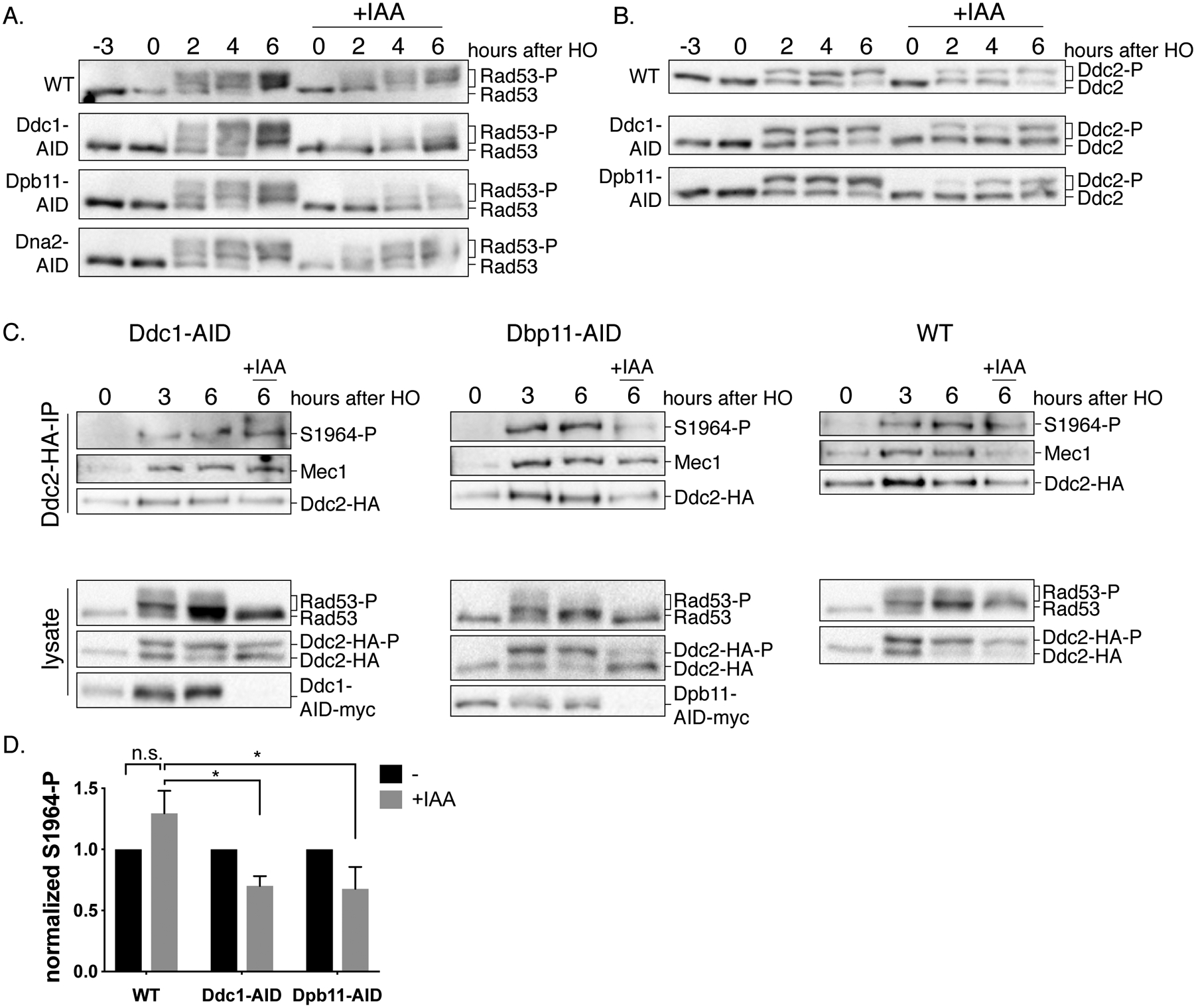
(A) Checkpoint activity after the conditional depletion of Ddc1, Dpb11 or Dna2. Cultures of Ddc1-AID, Dpb11-AID and Dna2-AID strains together with a WT control were grown and split into two. One half was treated with IAA for 3h prior to induction of a DSB to allow the degradation of the AID-tagged proteins (+IAA). Samples were collected and probed for Rad53 and (B) for Ddc2-HA. (C) Strains that contain Ddc2-HA together with either Dpb11-AID-myc or Ddc1-AID-myc, and a WT control strain were treated with galactose for 3h, then cultures were split into two, one to be treated with IAA for 3h. Following Ddc2-HA immunoprecipitation, samples were probed for Mec1, phospho-Mec1-S1964 and HA, for detection of Ddc2 (upper panel). Samples collected from the lysates (bottom panel) were blotted for HA (for detection of Ddc2), myc (for detection of Ddc1 or Dpb11) and Rad53.
(D) Quantification of Mec1-S1964 phosphorylation after the depletion of Ddc1 or Dpb11. *p=0.02. Error bars represent SEM.
Next, we asked if one or more of these co-activators are required for the maintenance of cell cycle arrest in response to an irreparable DSB. We induced the DSB for 6h to allow checkpoint activation, then depleted the AID-tagged proteins. The depletion of Dpb11 and Ddc1 led cells to turn off the Rad53 signaling much earlier compared to WT cells, but degradation of Dna2 did not have any effect [Figure S4]. Because the phosphorylation of Rad53 in this assay is entirely Mec1-dependent [Figure S3C], these findings suggest that Dpb11 and Ddc1 are the major regulators of Mec1 in response to a single DSB.
Finally, we examined the Mec1-S1964 autophosphorylation after degradation of Dbp11 or Ddc1. We induced a DSB for 3h and then treated the cells with IAA for an additional 3h to deplete Ddc1 or Dpb11. We coimmunoprecipitated Mec1 together with Ddc2-HA and assayed Mec1-S1964 autophosphorylation. The degradation of Dpb11 or Ddc1 caused a statistically significant decrease in Mec1-S1964 autophosphorylation at 6h compared to the WT control [Figure 4C and Figure 4D]. Degradation of either Ddc1 or Dpb11 also led to a reduction of Rad53 and Ddc2 hyperphosphorylation [Figure 4C]. Based on these results and previously published studies, we conclude that Dpb11 stimulates Mec1 through Ddc1 in response to a single irreparable DSB, and this stimulation leads to Mec1-S1964 autophosphorylation (Navadgi-Patil and Burgers 2009).
The Ddc2-Mec1 complex persists longer at the break site in mec1-S1964A mutant
If the initial kinase activity of the mec1-S1964A mutant is not altered, then why does it lead to a prolonged checkpoint arrest? We postulated that mec1-S1964A might persist longer around the DSB site which will allow Mec1 kinase to function for an extended period of time. Fluorescently-tagged Ddc2 forms a single focus at the break site; this focus can be used as a proxy for Mec1-Ddc2 localization (Melo et al. 2001). In agreement with previous studies, Ddc2-GFP fluorescent focus intensity decreased after checkpoint adaptation, as seen by comparing focus intensities at 8h and 24h [Figure 5A]. Ddc2 focus intensity was significantly higher at 8h after the induction of a DSB compared to 4h in WT. Moreover, at 4h, fewer cells were able to form Ddc2-GFP focus [Figure S5], implying that continuous checkpoint signaling positively regulates Ddc2 recruitment.
Figure 5. Ddc2 phosphorylation and recruitment is regulated by Mec1-dependent and - independent pathways.
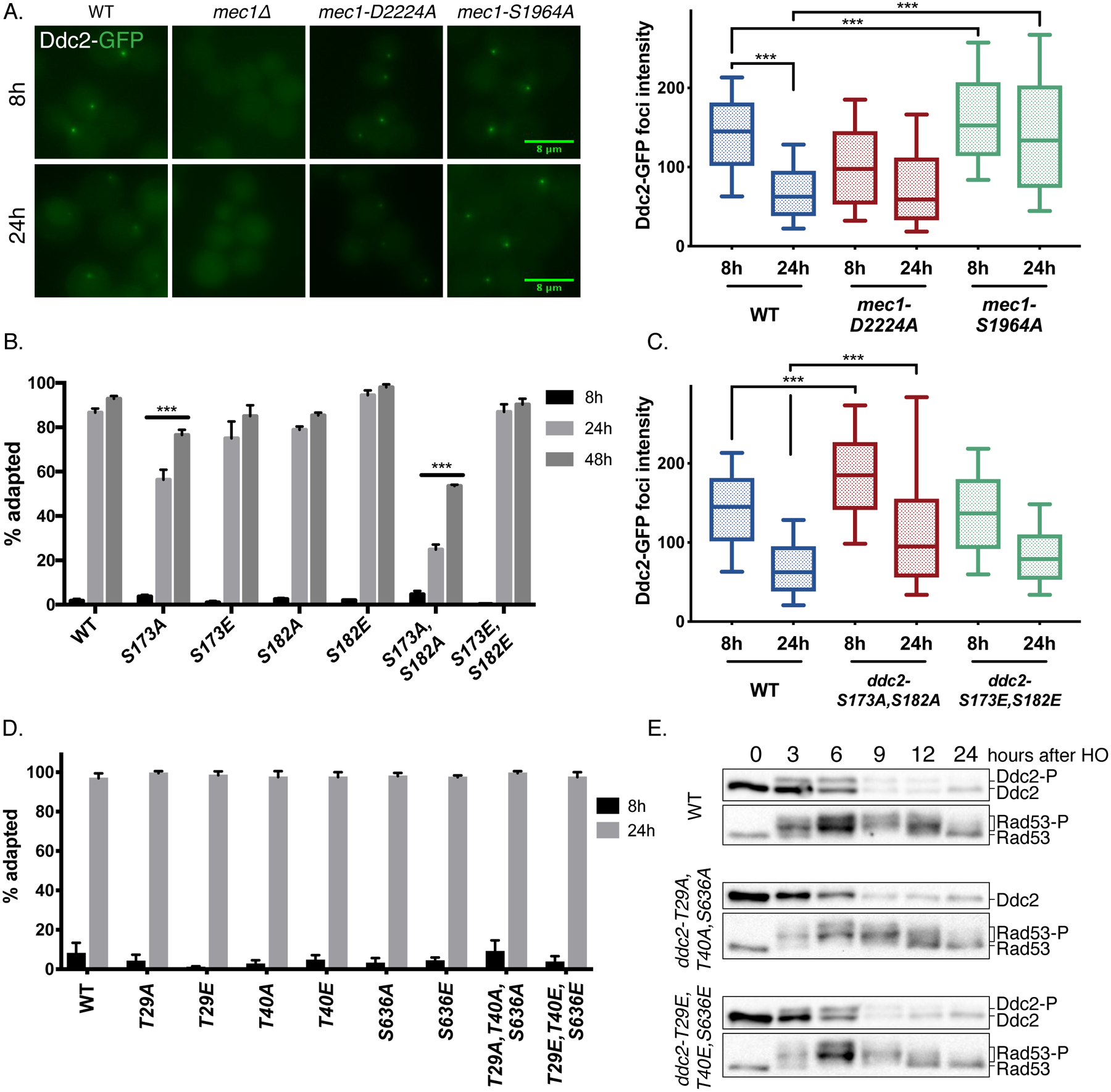
(A) Ddc2-GFP localization and quantification of foci intensity in WT, mec1Δ, mec1-D2224A and mec1-S1964A strains 8h and 24h after induction of an irreparable DSB. *** p<0.001. Error bars represent 10–90% of the data sets.
(B) Cell cycle arrest and adaptation in Ddc2 S173 and S182 mutants. ***p<0.001. Error bars represent SEM.
(C) Ddc2-GFP fluorescent foci intensity in ddc2-S173A,S182A and ddc2-S173E,S182E mutants quantified and plotted as described. ***p<0.001. Error bars represent 10–90% of the data sets.
(D) Cell cycle arrest and adaptation in Ddc2 T29, T40 and S636 mutants by adaptation assay. Error bars represent SEM.
(E) Ddc2 and Rad53 hyperphosphorylation in ddc2-T29A,T40A,S636A and ddc2-T29E,T40E,S636E triple mutants.
Ddc2-GFP focus formation was entirely dependent on the presence of Mec1 [Figure 5A] (Melo et al. 2001). In contrast, the kinase-dead mec1-D2224A mutant was competent for Ddc2 focus formation (Dubrana et al. 2007); but the average Ddc2-GFP focus intensity at 8h in the kinase-dead mutant was significantly reduced compared to WT [Figure 5A]. In the adaptation-defective mutant mec1-S1964A, Ddc2 focus intensities at 8h and 24h after the induction of a DNA break were significantly higher compared to WT, indicating that this mutant is able to stay associated with the DSB for an extended time. Hence, the persistent checkpoint arrest of mec1-S1964A could be due to its prolonged association with the DSB site compared to WT, especially at the later stages of cell cycle arrest.
Ddc2 is regulated by Mec1-dependent and -independent phosphorylation events
Ddc2 is phosphorylated during replication and upon DNA damage (Paciotti et al. 2000), but the effect of Ddc2 phosphorylation on DNA damage response has not been studied in depth. Although the damage-dependent gel migration pattern of Ddc2 is dependent on Mec1’s kinase activity [Figure S3C], we did not detect any phosphorylated SQ/TQ phosphopeptides of Ddc2 in our mass spectrometry analyses [SI Table 4 and 5]. Instead, we found that Ddc2 was phosphorylated at residues S173 and S182, which are positioned near the Mec1-Ddc2 binding interface (Wang et al. 2017) and are not Mec1 SQ/TQ consensus sites. Mutating both of these residues to alanines (ddc2-S173A,S182A) caused the cells to permanently arrest in response to a single DSB, while the phosphomimetic ddc2-S173E,S182E mutant exhibited WT-like behavior [Figure 5B]. Consistent with its hyperactive checkpoint phenotype, ddc2-S173A,S182A displayed prolonged Rad53 and Ddc2 phosphorylation in response to a single DSB [Figure S6A]. Moreover, Ddc2-GFP focus intensities at 8h and 24h in ddc2-S173A,S182A mutant were significantly elevated compared to WT [Figure 5C]. Stable isotope labeling of amino acids in culture (SILAC) assay showed that the phosphorylation of S182 was 3 times more abundant in WT cycling cells compared to cells that suffered a DSB and subsequently arrested at G2/M phase [Figure S6B, SI Table 6]. These data indicate that Ddc2 is constitutively phosphorylated at S173 and S182 and these phosphorylations are important for regulating the DDR.
We find that phosphorylation of S173 and S182 is constitutive; yet, Ddc2 mobility shift is detected specifically after DNA damage. Additionally, alanine substitution of these two sites does not alter the gel migration pattern of Ddc2 [Figure S6A], indicating that Ddc2 is phosphorylated on additional residues in a DNA damage-dependent manner. To understand how Ddc2 is regulated after DNA damage by Mec1, we mutated all 3 Mec1 consensus SQ/TQ sites on Ddc2 to alanine or glutamic acid residues with CRISPR/Cas9. The single mutants of the sites T29, T40 and S636 as well as the triple mutants arrested and adapted like WT after the induction of a single irreparable DSB [Figure 5D]. Although mutating these three sites to alanine residues abolished the DNA damage-dependent mobility shift of Ddc2, this shift was surprisingly still detectible in ddc2-T29E,T40E,S636E triple mutant [Figure 5D]. Mec1-dependent phosphorylation of Ddc2 on these SQ/TQ sites could prime Ddc2 for subsequent phosphorylations at yet unknown sites. It is also possible that the S/T to A substitution by itself eliminates the mobility shift of Ddc2, due to the loss of negatively charged residues.
Ddc2 is an inherently stable protein which is degraded upon the activation of DNA damage checkpoint
While analyzing the phosphorylation of Ddc2 with western blots, we noticed a reduction in Ddc2 protein abundance after the induction of DNA damage in WT cells. We treated the cultures with protein synthesis inhibitor cycloheximide (CHX) and monitored the abundance of Ddc2 to evaluate the inherent stability of Ddc2 in cycling cells, without DNA damage. Ddc2 abundance did not change significantly even after 12h CHX treatment, while DNA damage alone caused a 4-fold reduction in Ddc2 levels by 12h [Figure 6A]. Ddc2 protein levels also diminished with time when the cells were treated with CHX 3h after the induction of DNA damage [Figure 6A]. Hence, we conclude that Ddc2 is targeted for degradation specifically after the induction of DNA damage.
Figure 6. Ddc2 is targeted for degradation after checkpoint induction.
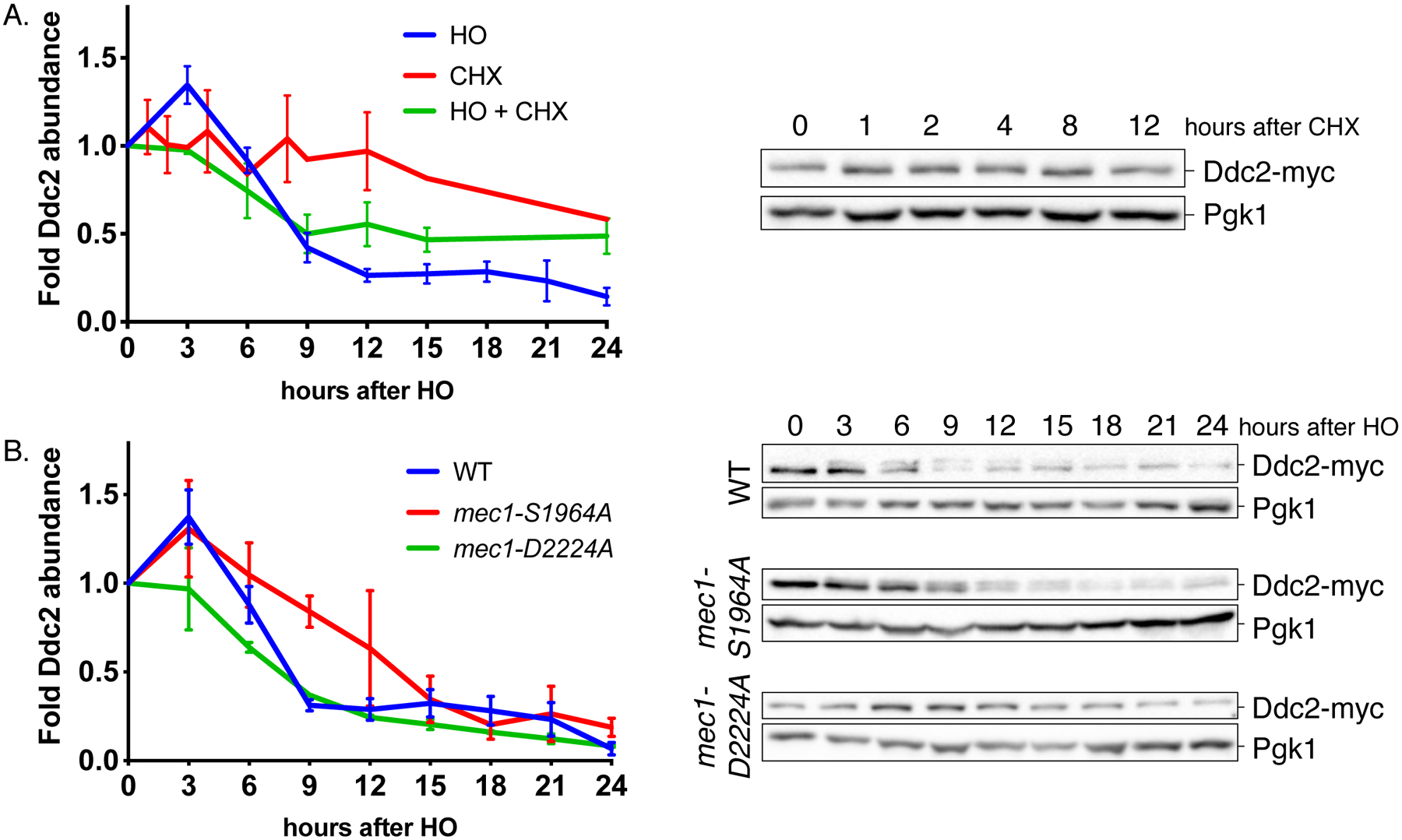
(A) Ddc2 stability following a DSB and cycloheximide (CHX) treatment. Ddc2 signal was normalized to Pgk1 loading control, then to the signal at 0h. Error bars represent SEM.
(B) Ddc2 stability in WT, mec1-S1964A and mec1-D2224A after DNA damage. Error bars represent SEM.
The proteasome-ubiquitin pathway (Finley et al. 2012) and autophagy (Reggiori and Klionsky 2013) are two pathways responsible for protein degradation. The central autophagy kinase ATG1, is essential for proper autophagy function (Matsuura et al. 1997). In an atg1Δ mutant, DNA damage induction led to degradation of Ddc2 with WT kinetics, indicating that the degradation of Ddc2 is autophagy-independent [Figure S6C]. Next, we asked if the Ddc2 stability is correlated with checkpoint signaling. Ddc2 levels were reduced with time after the induction of DNA damage in a mec1-D2224A kinase-dead mutant; albeit with slower kinetics. The DNA-damage dependent increase in Ddc2 abundance seen in WT cells around 3h after damage was also absent in mec1-D2224A mutant [Figure 6B]. On the other hand, in mec1-S1964A mutant which causes the permanent checkpoint arrest, the Ddc2 degradation was slower than in WT [Figure 6B]. Similarly, we detect an upregulation of Ddc2 abundance in adaptation-defective ddc2-S173A,S182A mutant especially at 6h after the induction of DNA damage [Figure S6D]. Based on these findings, we propose that there is a positive feedback loop between Ddc2 stability and checkpoint signaling.
DISCUSSION
How the DNA damage checkpoint is turned off in adaptation or recovery has remained an important question. Previous studies have suggested that maintenance of the checkpoint signaling depends on continuing Mec1 activity (Pellicioli et al. 2001), but how Mec1’s activity would be extinguished during adaptation or recovery has not been well understood. Here, we implicate autoregulation of Mec1 kinase. Abolishing the phosphorylation of the SQ consensus site S1964 caused prolonged checkpoint activation in response to an irreparable DSB. The permanent cell cycle arrest of this mutant in response to a single DSB is rescued by weakening the Mec1 branch of the checkpoint pathway, i.e. by deleting Chk1 or Mad2. Moreover, the prolonged cell cycle arrest observed in the mec1-S1964A mutant impairs recovery after repair is complete, without altering Mec1’s initial kinase activity or DNA repair.
By using a phospho-specific antibody and mass spectrometry, we demonstrated that Mec1 is phosphorylated at site S1964 in a DNA-damage dependent manner. S1964 phosphorylation is dependent on Mec1’s kinase activity but is independent of Tel1 and Rad53. Since Tel1’s contribution to the cell cycle arrest in response to a DSB is minimal and Rad53 activation in response to a DSB is entirely Mec1-dependent, we conclude that the Mec1-S1964 is an autophophosphorylation site.
To understand how Mec1’s kinase activity is stimulated after damage in response to a single DSB, we looked at the three possible Mec1 coactivators: Dpb11, Ddc1 and Dna2. We observe that Dbp11 and Ddc1 are critical regulators of Mec1 during the initiation and maintenance of cell cycle arrest in G2/M phase, agreeing with the previous findings (Navadgi-Patil and Burgers 2009). Conditional depletion of Ddc1 or Dpb11 also interferes with Mec1-S1964 autophosphorylation.
Autophosphorylation of PIKKs is a common regulatory mechanism. Both ATM (Kozlov et al. 2011) and ATR (Liu et al. 2011, Nam et al. 2011) go through autophosphorylation upon activation. Another member of the PIKK family, DNA-dependent protein kinase (DNA-PKcs) is also regulated by an inhibitory autophosphorylation (Chan and Lees-Miller 1996). While the autophosphorylation of Mec1 is Dbp11 and Ddc1-dependent, mammalian ATR autophosphorylation at site T1989 is independent of TopBP1, the mammalian homolog of Dpb11 (Liu et al. 2011). However, ATR-T1989 phosphorylation facilitates TopBP1-ATR binding, which is required for the stimulation of ATR’s kinase activity (Liu et al. 2011). Although the molecular mechanisms are different, Mec1 autophosphorylation of S1964 can also happen in trans, as in the case of ATR autophosphorylation at site T1989 (Liu et al. 2011). Recent studies identified Mec1 S38 and S1991 as phospho-sites, specifically in response to replication stress (Bastos de Oliveira et al. 2015, Hustedt et al. 2015). These sites might also play a role in the regulation of DSB response in addition to the sites we have identified here. In fact, we find that mutating S38 to either alanine or glutamic acid caused the cells to have a slight defect in adaptation in response to an irreparable DSB [Figure 1B].
Given that Mec1 phosphorylates many of its downstream targets within an hour, it is not clear how Mec1 is prevented from autophosphorylating S1964 until much later. Our data indicate that Ddc1 and Dpb11 are needed for fully stimulating Mec1’s kinase activity at the early stages of the cell cycle arrest in response to a single irreparable DSB; however, Mec1 phosphorylation peaks at about 9 to 12h after the induction of a DSB. Possibly one of Mec1’s co-activators, or an as-yet-unidentified protein, blocks access to the site. It is also possible that the reported different conformational states of mammalian ATM (Baretić et al. 2017) are relevant in understanding this regulation.
By using a fluorescently-tagged Ddc2, we show that the adaptation-defective mec1-S1964A persists longer at the DSB site compared to WT. Thus, the prolonged checkpoint arrest seen in the mec1-S1964A mutant can be explained by the prolonged binding of Mec1-Ddc2 complex to the DSB site and subsequent upregulation of the downstream targets. Mec1’s association with DSB appears to depend principally on Ddc2’s affinity for the break; as overexpressing Ddc2 but not Mec1 is shown to enhance the cell cycle arrest (Clerici et al. 2001). Here, we show that Ddc2 overexpression prevents adaptation to a single DSB in WT, and also suppresses the rapid cell cycle exit in mec1-T1902E. These data are consistent with the experiments showing that the artificial recruitment of Mec1-Ddc2 complex to DNA together with the 9-1-1 clamp is sufficient to initiate the DDR and that the strength of the DDR is correlated with the amount of checkpoint proteins recruited to DNA (Bonilla et al. 2008). Moreover, Ddc2 enrichment at the DSB site is reduced after adaptation (Melo et al. 2001), as well as in a mec1-D2224A kinase-dead mutant.
These findings hint a feedback loop between the upstream DDR signaling components such as Mec1, and the downstream targets in DDR pathway to maintain the cell cycle arrest in the presence of a persistent DSB. Therefore, the minimal Ddc2 recruitment seen in the mec1-D2224A cells could be attributed to the lack of proper cell cycle arrest.
Ddc2 has been shown to be regulated by Mec1-dependent phosphorylation; however, the mechanism of Ddc2 regulation still remains unclear (Paciotti et al. 2000). Our initial mass spectrometry analysis identified two previously unknown phospho-sites on Ddc2, S173 and S182. The double alanine substitution mutant ddc2-S173A,S182A leads to prolonged cell cycle arrest and elevated Rad53 hyperphosphorylation in response to a DSB. Further MS2 analysis showed that S182 phosphorylation is more abundant in cycling cells compared to cells that suffer from a DSB. Based on this, we conjecture that Ddc2 is constitutively phosphorylated at these sites, to fine-tune the Mec1-Ddc2 binding under basal conditions, and dephosphorylation of these sites allow stronger Mec1-Ddc2 binding which is mediated by a basic amino acid stretch of Ddc2 (Wang et al. 2017). This mode of Ddc2 regulation could especially be useful during replication, during which there is exposed ssDNA and cell cycle arrest could be deleterious.
Ddc2 is an exceptionally stable protein under basal conditions and is targeted for degradation upon DNA damage induction. We observe that Ddc2 stability is further impaired in the absence of Mec1’s kinase activity, following DNA damage. On the other hand, Ddc2 is more stable in mec1-S1964A and ddc2-S173A,S182A mutants which cannot turn off the checkpoint activity. Based on this and on our Ddc2-GFP localization data, we postulate that the degradation of Ddc2 pool which is not localized to chromatin could be used to limit continuous Mec1-Ddc2 recruitment to the break site in the presence of a persistent DNA lesion. Conditional depletion of Ddc2 by using an auxin-inducible degron system causes rapid Rad53 dephosphorylation (Tsabar et al. 2016). Additionally, the overexpression of Ddc2, but not Mec1, is shown to cause a hyperactivation of checkpoint activity (Clerici et al. 2001). Taken together, these data support the idea that Ddc2 is the limiting factor for Mec1 recruitment to the DSB site.
We propose that adaptation is achieved through the negative regulation of the Mec1-Ddc2 kinase in several ways. The autophosphorylation of Mec1 at site S1964 at the time of adaptation is important for the delocalization of the Mec1-Ddc2 complex from the break site. Moreover, the DNA damage-dependent decrease in Ddc2 abundance attenuates Mec1’s ability to signal to its downstream targets. We also suggestthat the DDR is fine-tuned by at least two Ddc2 phospho-sites, at S173 and S182, which might play a role in Mec1-Ddc2 binding as well as in the positive feedback loop to keep the Ddc2 localized at the break site.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author, Dr. James E. Haber (haber@brandeis.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and plasmids:
Yeast strains were freshly thawed from frozen stocks and grown at 30°C using standard practices. Plasmids and yeast strains used in this study are derivatives of either JKM179 or YMV80; and are listed in SI Table 1 and Table 3 respectively. Deletions of ORFs are introduced through the one step PCR homology cassette amplification and standard yeast transformation method (Wach et al. 1994). Mec1, Rad53, Ddc2 point mutants, and the deletion of vps51Δ were created by using CRISPR/Cas9 as explained previously (Anand et al. 2017), and the reagents used are indicated in SI Table 1 and 2. Strains that contain Ddc2-myc, Ddc2-HA, Ddc2-GFP and Mec1-HA C-terminus epitope tags made by transforming the cells with PCR-amplified cassettes that carry homologies to the C-terminus of the ORFs as explained previously (Longtine et al. 1998). The AID-tagging was performed the same way, and as shown previously (Morawska and Ulrich 2013, Nishimura et al. 2009). Other primer sequences are available upon request.
METHOD DETAILS
Yeast media:
Yeast strains were grown in YEPD (1% yeast extract, 2% peptone, 2% dextrose for normal growth and for spot assays. For adaptation assay, viability assay and HO-induction time courses, the cells were inoculated in YEP containing 3% lactic acid (YEP-lactate) and treated with a final concentration of 2% galactose. For the inducible degradation of the proteins, the cultures were treated with 100μg/ml indole-3-acetic acid (IAA) (Sigma-Aldrich). For protein synthesis inhibition the cells were treated with a final concentration of 100 μg/ml cycloheximide (Sigma-Aldrich).
Spot assay:
Cells were grown in YEPD overnight to saturation. Then, they were diluted to a stock concentration of 107cells/ml. 10-fold serial dilutions of this stock were prepared, and 2μl of each dilution was spotted on the plates that containing 50mM HU (Sigma-Aldrich) or 0.05 μg/ml phleomycin (InvivoGen). For phleomycin-containing plates, the pH of the media was brought to 7.0 with 1M HEPES, pH 8.0. For UV sensitivity, the cells were spotted on YEPD plates, and subjected to a dose of 60J/m2. After the plates were grown at 300C, the photos of the plates were taken by BioRad Gel Doc™ XR+ gel documentation system and manipulated by the Image Lab software (BioRad).
Adaptation assay:
The cells were grown overnight in YEP-lactate and plated on YEP-galactose plates. 50 individual G1 cells were micromanipulated on these plates, and the percentage of adapted cells were calculated as a ratio of cells that are past the 2-cell stage to total number of cells divided (Lee et al. 1998).
Viability assay:
The cells were grown overnight in YEP-lactate, counted, diluted and plated onto YEP-dextrose and YEP-galactose plates. The viability was calculated as a ratio of colony forming units (CFUs) on galactose plates to CFUs on dextrose plates.
HO-induction time course:
Cells were grown in YEP-lactate and grown to early exponential phase (1–10×106 cells/ml). DNA damage was induced by 2% galactose addition to the media. To obtain cell lysate samples for western blots, 50ml of the culture was harvested, and the cells were washed with 1ml of 20% TCA and snap-frozen on dry ice. For coimmunoprecipitation experiments, approximately 500ml of cultures were harvested, and washed with ice cold 1ml 50mM Tris, 5mM EDTA solution. The samples were kept on dry ice.
Sample preparation for western blotting:
Whole cell lysate samples were prepared as previously described (Pellicioli et al. 2001). Frozen cell pellets were resuspended in 200 μl 20% TCA. After the addition of acid-washed glass beads, the samples were vortexed for 2 minutes. The beads were washed with 200 μl of 5% TCA twice, and the extract was collected in a new tube. The crude extract was precipitated by centrifugation at 3000 rpm for 10 minutes. TCA was discarded, and the samples were resuspended in 200 μl 6X Laemmli buffer (60mM Tris, pH6.8, 2% SDS, 10% glycerol, 100mM DTT, 0.2% bromophenol blue) containing 0.9% B-mercaptoethanol and 200 μl 1M Tris (pH8.0). Prior to loading, samples were boiled at 95°C and centrifuged at maximum speed for 2 minutes. Supernatant containing the solubilized proteins were used to run SDS-PAGE gels.
Coimmunoprecipitation:
For coimmunoprecipitation, the cells were resuspended in lysis buffer (50mM HEPES pH7.5, 150mM NaCl, 2mM EDTA, 0.5% NP-40), supplemented with protease inhibitors (Halt Protease Inhibitor Mix, Thermo). An equal volume of acid-washed glass beads was added to the mixture, and samples were vortexed 5 for 2 min, then kept on ice for 5 mins in between. The crude extract was collected and cleared by centrifugation at maximum speed twice for 15 minutes at 4°C. An equal volume of the cleared lysate was mixed with Protein A or G-agarose beads depending on the primary antibody (Roche) pre-incubated with the antibody of interest. After the overnight incubation, the beads were washed twice with 1ml lysis buffer containing protease inhibitors, and the samples were eluted in 100 μl 6X Laemmli buffer containing 0.9% B-mercaptoethanol by boiling at 95°C for 5 minutes. Then, the samples were centrifuged to precipitate the beads, and the soluble part was used to run SDS-PAGE gels.
Western blotting:
Hand-casted 6% gels (for Rad53, Rad9, Mec1, Ddc2), 10% gels (for Pgk1, TIR1) and 12% gels (for γ-H2AX) were run in 1X running buffer (for 10X running buffer, 30g Tris base, 144g glycine, 50ml 20% SDS in 1000ml water) by using BioRad Mini-PROTEAN tetra cell system. Wet transfer was carried out in 1X transfer buffer containing 100ml methanol (for 10X transfer buffer, 30g Tris, 144g glycine in 1000ml water) at 4°C by using BioRad Mini Trans-Blot Cell and BioRad Immun-Blot PVDF membrane. After transfer, the membranes were blocked in 1X TBS (pH 7.25) containing 1ml Tween-20 and 5% dry milk (for 10X TBS, 24g Tris Base, 87g NaCl in 1000ml, pH with concentrated HCl). After blocking and antibody incubation, the membranes were washed 3X with 1X TBS-T for 10 mins, incubated in secondary antibodies and washed again. Then, membranes were developed by using Amersham ECL Prime Western Blotting Detection Reagent and visualized by using by BioRad Gel Doc™ XR+ gel documentation system. BioRad ImageLab software was used to manipulate and quantify the images. Anti-Mec1 antibody was a gift from Dr. Susan Gasser (Hustedt et al. 2015). Anti-Rad9 antibody was a gift from Dr. John Petrini (Usui et al. 2009). Phospho-specific Mec1 antibody was raised by 21st Century Biochemicals upon request. AID plasmids were a gift from Dr. Helle Ulrich’s lab. GAL-DDC2 overexpression plasmids were a gift from Dr. Maria Pia Longhese’s lab. Anti-Pgk1 antibody (Abcam Cat# ab113687, RRID:AB_10861977), anti-myc antibody (Abcam Cat# ab32, RRID:AB_303599), anti-Rad53 antibody (Abcam Cat # ab166859, RRID:AB_2801547), anti-HA antibody (Abcam Cat# ab16918, RRID:AB_302562) and anti phospo-H2A-S129 (Abcam Cat# ab15083, RRID:AB_301630) were purchased from AbCam. For primary antibodies raised in mouse, a secondary mouse IgG HRP-linked antibody (GE Healthcare Cat# NXA931, RRID:AB_772209) was used. For primary antibodies raised in rabbit, a secondary rabbit IgG was used (Sigma-Aldrich Cat# A6154, RRID:AB_258284).
Fluorescence Microscopy:
For live imaging, the cells were grown in YEP-lactate, and cells were treated with 2% galactose at 0h, as mentioned previously. Samples were collected at various time points, and cells were harvested by centrifugation at 3000 rpm for 1 minute. Then, the cells were washed with 1ml of complete synthetic media containing 2 % galactose twice, resuspended in 100μl of synthetic media containing galactose for live imaging. The images were taken with Nikon Eclipse E600 microscope as z-stacks and processed with Fiji software (Fiji, RRID:SCR_002285) (Schindelin et al. 2012) for average intensity. The intensity of GFP foci was calculated manually by using FiJi software, and the intensities were normalized to signal measured from the random portions of cytoplasm as background for at least 300 cells per strain per condition.
Mass spectrometry:
Phosphopeptides were enriched from Ddc2–13xmyc immunoprecipitates via IP-MAC MS, as described previously (Bastos de Oliveira et al. 2015). SILAC analysis was performed as described (Bastos de Oliveira et al. 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed by using Prism7, 2-way ANOVA tests and Tukey correction for multiple comparisons, with a confidence interval of 95%. For the viability assays and the Ddc2-GFP foci quantification, statistical differences were calculated with one-way ANOVA on Prism7 and P-values were corrected with Tukey for multiple comparisons. For adaptation assays, viability assays, γ-H2AX quantification, Ddc2 abundance quantifications and Mec1-S1964 phosphorylation quantification, error bars represent standard error. For Ddc2-GFP foci quantification, the bars represent 10–90% of the data set. For the quantification of γ-H2AX and Ddc2, the signal intensities were normalized to a Pgk1 loading control, and then to a WT control treated identically. For the quantification of Mec1 phosphorylation at S1964 in WT strain, the signal intensities were normalized to 3h, as there is no S1964-P signal at 0h. For the quantification of S1964 phosphorylation after Ddc1 or Dpb11 degradation, the S1964 signal was normalized to anti-Mec1, and then to the untreated control for every strain. P values are indicated in figure legends.
DATA AND SOFTWARE AVAILABILITY
The full list of phospho-peptides to the mass spectrometry and SILAC experiments performed in this study can be found in SI Table 3, SI Table 4 and SI Table 5. All data available from the corresponding author on request.
Supplementary Material
SI Table 4. Full list of phospho-peptides detected with mass spectrometry following trypsin digestion. Related to Figure 3 and 4.
SI Table 5. Full list of phospho-peptides detected with mass spectrometry following trypsin and GluC digestion. Related to Figure 3.
SI Table 3. List of yeast strains used in this study. Related to Star Methods section.
SI Table 6. Full list of phospho-peptides detected with SILAC. Related to Figure 4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Rad53 | AbCam | EL7.E1 |
| Mouse monoclonal anti-HA | AbCam | 12CA5 |
| Mouse monoclonal anti-myc | AbCam | 9E10 |
| Mouse monoclonal anti-Pgk1 | AbCam | 22C5D8 |
| Anti-phospho-H2-S129 | AbCam | ab15083 |
| Rabbit polyclonal anti-Rad9 | (Usui et al. 2009) | N/A |
| Rabbit polyclonal anti-Mec1 | (Hustedt et al. 2015) | N/A |
| Rabbit polyclonal anti-phosho-Mec1-S1964 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hydroxyurea (HU) | Sigma-Aldrich | H8627 |
| Phleomycin | InvivoGen | ant-ph-1 |
| Cycloheximide | Sigma-Aldrich | C7698-5G |
| Indole-3-acetic acid | Sigma-Aldrich | I3750-25G-A |
| Halt Protease Inhibitor | ThermoFisher | PI78430 |
| Protein-A agarose beads | Sigma-Aldrich | 1719408 |
| Protein-G agarose beads | Sigma-Aldrich | 11719416001 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background S228c. Strains are listed in SI Table 3. | This paper | N/A |
| Oligonucleotides | ||
| Oligonucleotides used as repair templates for Cas9-directed mutagenesis are listed in SI Table 2. | This paper | N/A |
| Recombinant DNA | ||
| Plasmids used in this study are listed in SI Table 1. | This paper | N/A |
| Software and Algorithms | ||
| Prism 7.00 | GraphPad Software, Inc. | |
| Image Lab | Bio-Rad | |
| FiJi | ||
ACKNOWLEDGEMENTS
We are grateful to Susan Gasser, John Petrini, Maria Pia Longhese and Helle Ulrich for gifts of reagents. We thank Syed Aleem and Gang Cai for useful discussions. Research was supported by NIH grants GM61766 and GM127029 to J.E.H. and GM097272 to M.B.S.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alani E, Thresher R, Griffith JD and Kolodner RD (1992) Characterization of DNA-binding and strand-exchange stimulation properties of y-RPA, a yeast single-strand-DNA-binding protein. J Mol Biol, 227(1), pp. 54–71. [DOI] [PubMed] [Google Scholar]
- Anand R, Memisoglu G and Haber J (2017) Cas9-mediated gene editing in Saccharomyces cerevisiae. [Google Scholar]
- Ball HL and Cortez D (2005) ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem, 280(36), pp. 31390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretić D, Pollard HK, Fisher DI, Johnson CM, Santhanam B, Truman CM, Kouba T, Fersht AR, Phillips C and Williams RL (2017) Structures of closed and open conformations of dimeric human ATM. Science Advances, 3(5), pp. e1700933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H and Smolka MB (2015) Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell, 57(6), pp. 1124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla CY, Melo JA and Toczyski DP (2008) Colocalization of Sensors Is Sufficient to Activate the DNA Damage Checkpoint in the Absence of Damage. Molecular cell, 30(3), pp. 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW and Lees-Miller SP (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem, 271(15), pp. 8936–41. [DOI] [PubMed] [Google Scholar]
- Chen Y and Sanchez Y (2004) Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair (Amst), 3(8–9), pp. 1025–32. [DOI] [PubMed] [Google Scholar]
- Clemenson C and Marsolier-Kergoat MC (2009) DNA damage checkpoint inactivation: adaptation and recovery. DNA Repair (Amst), 8(9), pp. 1101–9. [DOI] [PubMed] [Google Scholar]
- Clerici M, Paciotti V, Baldo V, Romano M, Lucchini G and Longhese MP (2001) Hyperactivation of the yeast DNA damage checkpoint by TEL1 and DDC2 overexpression. EMBO J, 20(22), pp. 6485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Trovesi C, Galbiati A, Lucchini G and Longhese MP (2014) Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J, 33(3), pp. 198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, Yoshida S and Haber JE (2013) DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc Natl Acad Sci U S A, 110(1), pp. E41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotiwala F, Harrison JC, Jain S, Sugawara N and Haber JE (2010) Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr Biol, 20(4), pp. 328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrana K, van Attikum H, Hediger F and Gasser SM (2007) The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J Cell Sci, 120(Pt 23), pp. 4209–20. [DOI] [PubMed] [Google Scholar]
- Emili A (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell, 2(2), pp. 183–9. [DOI] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T and Kaiser P (2012) The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics, 192(2), pp. 319–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbini E, Cesena D, Galbiati A, Lockhart A and Longhese MP (2013) Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst), 12(10), pp. 791–9. [DOI] [PubMed] [Google Scholar]
- Guillemain G, Ma E, Mauger S, Miron S, Thai R, Guerois R, Ochsenbein F and Marsolier-Kergoat MC (2007) Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol Cell Biol, 27(9), pp. 3378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JC and Haber JE (2006) Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet, 40, pp. 209–35. [DOI] [PubMed] [Google Scholar]
- Hustedt N, Seeber A, Sack R, Tsai-Pflugfelder M, Bhullar B, Vlaming H, van Leeuwen F, Guenole A, van Attikum H, Srivas R, Ideker T, Shimada K and Gasser SM (2015) Yeast PP4 interacts with ATR homolog Ddc2-Mec1 and regulates checkpoint signaling. Mol Cell, 57(2), pp. 273–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS and Brill SJ (2003) MEC1-dependent phosphorylation of yeast RPA1 in vitro. DNA Repair (Amst), 2(12), pp. 1321–35. [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE and Kastan MB (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem, 274(53), pp. 37538–43. [DOI] [PubMed] [Google Scholar]
- Kozlov SV, Graham ME, Jakob B, Tobias F, Kijas AW, Tanuji M, Chen P, Robinson PJ, Taucher-Scholz G, Suzuki K, So S, Chen D and Lavin MF (2011) Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem, 286(11), pp. 9107–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S and Burgers PM (2013) Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev, 27(3), pp. 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Oberly S, Sanford EJ, Sharma S, Chabes A and Smolka MB (2018) Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev, 32(11–12), pp. 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Lee K, Legube G and Haber JE (2014) Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol, 21(1), pp. 103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD and Haber JE (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell, 94(3), pp. 399–409. [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Demeter J, Vaze MP, Gasch AP, Malkova A, Brown PO, Botstein D, Stearns T, Foiani M and Haber JE (2000) Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol, 65, pp. 303–14. [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Malkova A, Foiani M and Haber JE (2001) The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr Biol, 11(13), pp. 1053–7. [DOI] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbein F, Guerois R, Haber JE and Marsolier-Kergoat MC (2003) PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell, 11(3), pp. 827–35. [DOI] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH and Zou L (2011) ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell, 43(2), pp. 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P and Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast, 14(10), pp. 953–61. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA and Cortez D (2009) Common mechanisms of PIKK regulation. DNA Repair (Amst), 8(9), pp. 1004–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory JC and Petes TD (2000) Protein kinase activity of Tel1p and Mec1p, two Saccharomyces cerevisiae proteins related to the human ATM protein kinase. Proc Natl Acad Sci U S A, 97(25), pp. 13749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Clerici M, Lucchini G and Longhese MP (2007) Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep, 8(4), pp. 380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Tsukada M, Wada Y and Ohsumi Y (1997) Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene, 192(2), pp. 245–50. [DOI] [PubMed] [Google Scholar]
- Melo JA, Cohen J and Toczyski DP (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev, 15(21), pp. 2809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M and Ulrich HD (2013) An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast, 30(9), pp. 341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Tanaka Y and Sugimoto K (2005) Role of the C terminus of Mec1 checkpoint kinase in its localization to sites of DNA damage. Mol Biol Cell, 16(11), pp. 5227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB and Cortez D (2011) Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem, 286(33), pp. 28707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM and Burgers PM (2009) The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell, 36(5), pp. 743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM and Burgers PM (2011) Cell-cycle-specific activators of the Mec1/ATR checkpoint kinase. Biochem Soc Trans, 39(2), pp. 600–5. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T and Kanemaki M (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods, 6(12), pp. 917–22. [DOI] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Lucchini G and Longhese MP (2000) The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev, 14(16), pp. 2046–59. [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Scotti M, Lucchini G and Longhese MP (2001) Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol Cell Biol, 21(12), pp. 3913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M and Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell, 7(2), pp. 293–300. [DOI] [PubMed] [Google Scholar]
- Reggiori F and Klionsky DJ (2013) Autophagic processes in yeast: mechanism, machinery and regulation. Genetics, 194(2), pp. 341–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C and Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol, 146(5), pp. 905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J and Jackson SP (2002) Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol Cell, 9(4), pp. 857–69. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B and Elledge SJ (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science, 271(5247), pp. 357–60. [DOI] [PubMed] [Google Scholar]
- Sawicka M, Wanrooij PH, Darbari VC, Tannous E, Hailemariam S, Bose D, Makarova AV, Burgers PM and Zhang X (2016) The Dimeric Architecture of Checkpoint Kinases Mec1ATR and Tel1ATM Reveal a Common Structural Organization. J Biol Chem, 291(26), pp. 13436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods, 9(7), pp. 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE and Lichten M (2004) Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol, 14(19), pp. 1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS and Stern DF (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science, 281(5374), pp. 272–4. [DOI] [PubMed] [Google Scholar]
- Tercero JA and Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature, 412(6846), pp. 553–7. [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ and Hartwell LH (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell, 90(6), pp. 1097–106. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Waterman DP, Aguilar F, Katsnelson L, Eapen VV, Memisoglu G and Haber JE (2016) Asf1 facilitates dephosphorylation of Rad53 after DNA double-strand break repair. Genes Dev, 30(10), pp. 1211–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Foster SS and Petrini JH (2009) Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol Cell, 33(2), pp. 147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M and Haber JE (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell, 10(2), pp. 373–85. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R and Philippsen P (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10(13), pp. 1793–808. [DOI] [PubMed] [Google Scholar]
- Wang X, Ran T, Zhang X, Xin J, Zhang Z, Wu T, Wang W and Cai G (2017) 3.9 A structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science, 358(6367), pp. 1206–1209. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V and Koshland D (1996) Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J Cell Biol, 133(1), pp. 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG and Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell, 2(3), pp. 329–40. [DOI] [PubMed] [Google Scholar]
- Zou L and Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 300(5625), pp. 1542–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI Table 4. Full list of phospho-peptides detected with mass spectrometry following trypsin digestion. Related to Figure 3 and 4.
SI Table 5. Full list of phospho-peptides detected with mass spectrometry following trypsin and GluC digestion. Related to Figure 3.
SI Table 3. List of yeast strains used in this study. Related to Star Methods section.
SI Table 6. Full list of phospho-peptides detected with SILAC. Related to Figure 4.
Data Availability Statement
The full list of phospho-peptides to the mass spectrometry and SILAC experiments performed in this study can be found in SI Table 3, SI Table 4 and SI Table 5. All data available from the corresponding author on request.