

Abstract
Hyperinsulinemia and insulin resistance were proposed more than 30 years ago to be important contributors to elevated blood pressure (BP) associated with obesity and the metabolic syndrome, also called syndrome X. Support for this concept initially came from clinical and population studies showing correlations among hyperinsulinemia, insulin resistance and elevated BP in individuals with metabolic syndrome. Short-term studies in experimental animals and in humans provided additional evidence that hyperinsulinemia may evoke increases in sympathetic nervous system (SNS) activity and renal sodium retention that, if sustained, could increase BP. Although insulin infusions may increase SNS activity and modestly raise BP in rodents, chronic insulin administration does not significantly increase BP in lean or obese insulin resistant rabbits, dogs, horses, or humans. Multiple studies in humans and experimental animals have also shown that severe insulin resistance and hyperinsulinemia may occur in the absence of elevated BP. These observations question whether insulin resistance and hyperinsulinemia are major factors linking obesity/metabolic syndrome with hypertension. Other mechanisms, such as physical compression of the kidneys, activation of the renin-angiotensin-aldosterone system, hyperleptinemia, stimulation of the brain melanocortin system, and SNS activation appear to play a more critical role in initiating hypertension in obese subjects with metabolic syndrome. However, the metabolic effects of insulin resistance, including hyperglycemia and dyslipidemia, appear to interact synergistically with increased BP to cause vascular and kidney injury which can exacerbate the hypertension and associated injury to the kidneys and cardiovascular system.
Keywords: Blood pressure, kidney, leptin, melanocortins, sympathetic activity, renin-angiotensin-aldosterone system, obesity
BRIEF SUMMARY
More than three decades ago hyperinsulinemia and insulin resistance were proposed as important contributors to elevated blood pressure (BP) in syndrome X/metabolic syndrome. Although insulin resistance and hyperinsulinemia are correlated with high BP, extensive studies in experimental animals and humans have provided little evidence that elevated insulin or insulin resistance directly mediate increased BP in metabolic syndrome, although the metabolic effects of insulin resistance may contribute to target organ injury and exacerbate hypertension.
INTRODUCTION
Cardiovascular (CV) diseases continue to be the main cause of mortality and morbidity worldwide. Despite major advances in diagnosis and treatment in the past 30–40 years, the prevalence of CV disease continues to increase in parallel with aging populations and ever growing prevalence of obesity and associated metabolic derangements1. Elevated blood pressure (BP), even when not reaching defined cutoff levels that are considered to be diagnostic of hypertension, is a major risk factor for CV diseases including vascular injury, stroke, myocardial infarction, and heart failure2, 3.
Although the causes of primary (essential) hypertension are not completely understood, excess adiposity appears to be a major culprit. Risk estimates from studies in multiple populations indicate that as much as 65–75% of the risk for primary hypertension can be attributed to excess weight gain and obesity4. However, the distribution of adipose tissue also appears to be important in determining the impact of obesity on BP and metabolic abnormalities such as dyslipidemia, insulin resistance, hyperinsulinemia, and diabetes mellitus5, 6 Experimental and clinical studies have provided strong evidence that excess visceral fat conveys much higher risk for CV disease and metabolic disorders compared to excess subcutaneous fat6.
The concept that CV disease is closely associated with a cluster of metabolic disorders was recognized more than 80 years ago when Banting and Best discovered insulin7. The interdependence of metabolic abnormalities, hypertension, and CV diseases was also described by physician scientists in the early and mid-1900’s and the term “metabolic syndrome” was introduced in the 1970’s8, 9. In 1988, Gerald Reaven hypothesized that insulin resistance was the key factor underlying a group of metabolic disorders which included impaired glucose tolerance (IGT), hyperinsulinemia, high levels of very low-density lipoprotein (VLDL) triglycerides, low levels of high-density lipoprotein (HDL) cholesterol, and hypertension10; Reaven coined the term “Syndrome X” to emphasize the unknown features of this group of disorders10, 11. Norman Kaplan added central adiposity as a key driver of CV disease and named this cluster of disorders as the “deadly quartet”, consisting of visceral obesity, IGT/insulin resistance, hypertriglyceridemia, and hypertension12. Other investigators used the term “insulin resistance syndrome” to emphasize what they believed to be the primary initiator of these cardiometabolic disorders13, 14.
In this brief review we provide an update of the hypothesis that insulin resistance and compensatory hyperinsulinemia are primary mediators of elevated BP in metabolic syndrome and obesity, and then focus on other factors that may exert even greater impact on BP regulation in obesity/metabolic syndrome.
ROLE OF HYPERINSULINEMIA AND INSULIN RESISTANCE IN HYPERTENSION
Epidemiological Evidence Associating Insulin Resistance and Hyperinsulinemia with Hypertension
More than 30 years ago investigators observed that people with high plasma insulin concentration and insulin resistance often had higher BP compared to those with normal insulin levels12–17. The majority of people with insulin resistance and hyperinsulinemia also exhibited a cluster of other metabolic abnormalities, including elevated serum triglycerides, dyslipidemia with low HDL and high low-density lipoproteins (LDL), among other factors. This “metabolic syndrome” included hypertension as a hallmark feature18–20 (Table 1).
Table 1 -.
Clinical Definition of the Metabolic Syndrome. The criteria for an individual to be classified as having the metabolic syndrome include the presence of 3 or more of the following characteristics18–20:
| Measurement | Cutoff points |
|---|---|
| Increased waist circumference | Population- and country-specific definitions |
| Increased TG (drug treatment for elevated TG can be used as alternative indicator) | ≥150 mg/dL (1.7 mmol/L) |
| Reduced HDL cholesterol (drug treatment for reduced HDL cholesterol can be used as alternative indicator) | ≤40 mg/dL (1.0 mmol/L) in men; ≤50 mg/dL (1.3 mmol/L) in women |
| Increased BP (use of antihypertensive medication can be used as alternative indicator) | Systolic BP ≥130 mmHg and/or diastolic BP ≥85 mmHg |
| Increased fasting plasma glucose (drug treatment for increased plasma glucose levels can be used as alternative indicator) | >100 mg/dL (5.5 mmol/L) |
BP, blood pressure; HDL, high density lipoprotein; TG, triglycerides.
Support for the concept that insulin resistance and hyperinsulinemia may mediate hypertension came from observations that these metabolic disorders were correlated with increased BP in non-obese as well as obese people. Ferrannini et al.21 reported that hyperinsulinemia was correlated with BP in non-obese subjects; however, the slope of the relationship between BP and plasma insulin concentration in 2,241 normotensive, non-diabetic subjects predicted that a 200 μU/mL increase in plasma insulin concentration could account for only a 1 mm Hg rise in BP21. These findings suggested minimal effects of insulin on BP in non-obese subjects. Other investigators found that plasma insulin concentrations are similar in non-diabetic normotensive and hypertensive people and that there was a tendency toward a negative correlation between insulin and BP22. After stratifying for obesity, the relationship between plasma insulin and BP is weak or nonexistent23. Chen and colleagues24, for example, found that, despite similar body mass index (BMI) and subcutaneous fat, obese insulin sensitive individuals exhibited lower systolic and diastolic BPs, higher HDL and lower triglycerides than their obese resistant counterparts. However, obese insulin sensitive individuals also had lower visceral and liver fat compared to their obese insulin resistant counterparts24. Also, in most observational studies, including those in which insulin resistance was reported in non-obese people with hypertension, visceral adiposity was not assessed. Many people with hypertension and normal BMI may have increased visceral adiposity which can contribute to increased BP as well as to insulin resistance6. Therefore, despite the fact several population studies have reported an association between plasma insulin concentration, insulin resistance, BP and risk for developing hypertension, obesity (especially when associated with excess visceral fat) is often a confounding factor. Moreover, regardless of whether insulin and BP are correlated in obese or non-obese subjects, the quantitative importance of hyperinsulinemia and insulin resistance in causing hypertension cannot be established solely from cross sectional, or even longitudinal, correlational studies.
Some investigators have suggested that insulin resistance and compensatory hyperinsulinemia may be secondary to hypertension due to vascular rarefaction and increased peripheral vascular resistance which could reduce delivery of glucose and insulin to skeletal muscle thereby impairing glucose uptake25. However, in many forms of secondary hypertension not associated with obesity, such as renovascular or mineralocorticoid hypertension, there is no evidence of insulin resistance26. Moreover, most obese subjects with insulin resistance have normal or elevated skeletal muscle blood flow and insulin resistance appears to be related mainly to lipid accumulation and post-receptor signaling abnormalities26–28.
Impact of Acute Hyperinsulinemia on Renal Sodium Excretion, SNS Activity and BP
Compensatory increases in plasma insulin levels in response to worsening insulin resistance have been proposed to trigger mechanisms that directly or indirectly increase renal sodium reabsorption29, 30 and sympathetic nervous system (SNS) activity31, 32. If persistent, these changes could raise BP and eventually lead to hypertension in patients with syndrome X/metabolic syndrome.
Acute antinatriuretic effects of insulin.
Support for the concept that hyperinsulinemia may contribute to hypertension came from acute studies in which insulin was shown to have antinatriuretic effects. For example, acute insulin administration in humans can promote antinatriuresis33, 34, while stopping insulin therapy in diabetic patients was associated with acute increases in sodium excretion34. The antinatriuretric effect of acute hyperinsulinemia was also observed in isolated kidneys35. These studies support the hypothesis that acute hyperinsulinemia can directly stimulate renal tubular sodium reabsorption, an effect that if sustained could translate into increased BP.
Acute effects of insulin to increase SNS activity.
Another mechanism proposed to mediate the effects of hyperinsulinemia on BP regulation is activation of the SNS. Excess caloric intake, commonly associated with development of insulin resistance and hyperinsulinemia, raises SNS activity31, 32, 36, and acute insulin administration in humans to increase plasma insulin levels to those found in morbid obesity also increases skeletal muscle SNS activity37–40. If this increase in muscle SNS activity is paralleled by increased SNS to CV relevant tissues, including the kidneys and vasculature, then insulin-mediated SNS activation may raise BP.
Although the mechanisms by which acute elevations in plasma insulin concentration promote sympathetic activation are not completely understood, insulin appears to have direct central nervous system (CNS) effects41. Insulin crosses the blood brain barrier, and acute injections of insulin into the cerebral ventricles or in specific brain nuclei increase SNS activity in rodents42–45. Elevations in plasma insulin concentration during euglycemic-hyperinsulinemic clamp increase lumbar SNS activity46, 47 and this effect appears to be mediated by stimulation of proopiomelanocortin (POMC) neurons in the arcuate nucleus (ARC) of the hypothalamus and activation of glutamatergic neurons and melanocortin 4 receptor (MC4R) expressing neurons of the paraventricular nucleus (PVN)43, 47, 48. Some of these direct CNS effects of insulin, however, may be influenced by sex hormones as males appear to be more sensitive than females, particularly in obese rodents49.
Additional mechanisms have been proposed to contribute to insulin’s effects on BP regulation including insulin-mediated reduction in natriuretic peptides which appears to be accentuated in obesity50 and loss of insulin-induced vasodilation which could potentiate the actions of insulin on BP51–53. However, as discussed below, the acute actions of hyperinsulinemia on renal sodium handling and SNS activity do not appear to translate into elevations in BP.
Acute hyperinsulinemia does not raise BP.
Although acute increases in plasma or CNS insulin levels may promote sodium retention and increased SNS activity, almost invariably these acute studies have not demonstrated an effect of insulin in elevating BP. For example, acute hyperinsulinemia in healthy people increased forearm blood flow and reduced vascular resistance but did not alter BP when compared to baseline values before insulin infusion, despite evoking increases in skeletal muscle SNS activity37. Even borderline hypertensive individuals who may be more sensitive to the pressor effects of insulin did not show elevations in BP during acute hyperinsulinemia despite insulin-mediated sympathoexcitation38. Similar findings were observed in lean healthy rabbits, dogs and horses in which acute hyperinsulinemia during euglycemic-hyperinsulinemic clamp also failed to increase BP54–56. In rodents, no significant elevation in BP was observed despite increased SNS activity during acute increases in plasma or CNS insulin concentrations41–49.
It could be speculated that lean healthy humans and experimental animals do not resemble the milieu most commonly associated with hyperinsulinemia/insulin resistance including dyslipidemia, hyperglycemia, and other abnormalities caused by obesity such as impaired insulin-mediated vasodilation, reduced vascular nitric oxide (NO) availability, and impaired baroreflexes, which could amplify the effects of insulin to raise BP. The evidence, however, does not support this speculation. For instance, acute insulin administration in older adults who lacked insulin-mediated forearm vasodilation did not raise their BP52. Acute insulin administration in obese horses with metabolic syndrome also failed to evoke increases in BP57, and in obese rats injected centrally with insulin, BP remained unaltered despite greater increases in lumbar SNS activity than in lean controls49. Acute hyperinsulinemia in baroreceptor denervated or intact dogs caused no significant changes in BP56. Thus, short-term hyperinsulinemia, due to acute systemic or CNS insulin administration, does not raise BP even in the presence of metabolic abnormalities associated with insulin resistance/metabolic syndrome in these animal models.
Impact of Chronic Hyperinsulinemia on Renal Sodium Excretion and BP Regulation
Although acute increases in plasma insulin concentration or direct intrarenal insulin infusion can elicit sodium retention and CNS injections of insulin can promote SNS activation, these effects do not appear to be sufficient to cause elevations in BP. It is possible, however, that chronic hyperinsulinemia associated with obesity/metabolic syndrome could contribute to hypertension if these effects on SNS activation and renal function were sustained.
Chronic hyperinsulinemia increases BP in rodents.
Chronic intravenous infusion of insulin in lean rats causes modest, but significant, increases in BP58, 59. This increase in BP requires an intact renin-angiotensin-aldosterone system (RAAS)60 and can be significantly attenuated by inhibiting thromboxane synthesis61, but is independent of adrenergic receptor activation62. Irsik et al.63 also showed that chronic intrarenal infusion of insulin, at a dose that did not significantly alter circulating insulin levels, caused increases in BP similar to those observed with larger doses of systemically infused insulin, and that this pressor effect was not associated with activation of the RAAS. Other investigators also reported that chronic systemic insulin infusions in lean rats is associated with increased sensitivity to adrenergic stimulation and impaired reflex inhibition of splanchnic nerve activity64. These observations suggest that in rats hyperinsulinemia may elicit modest, sustained increases in BP. However, as discussed below, results from studies in larger experimental animals and humans do not corroborate these observations in rodents.
Evidence against a major role for hyperinsulinemia/insulin resistance in initiating hypertension.
Although previous studies demonstrated that chronic insulin infusion in rats can increase BP, studies in other experimental animals as well as in humans show that chronic hyperinsulinemia, even in the presence of metabolic syndrome/insulin resistance, does not cause hypertension. In humans, the correlation between plasma insulin levels and BP is usually confounded by visceral obesity that often precedes insulin resistance and hyperinsulinemia. For instance, the higher BP observed in insulin resistant individuals compared to insulin sensitive subjects with similar BMI is confounded by reduced visceral adiposity and liver fat infiltration in the insulin sensitive group24. The positive correlations between changes in fasting insulin, insulin sensitivity, and risk of developing hypertension are lost after adjusting for BMI or waist circumference65; and when a positive correlation between insulin levels and BP still remains after adjusting for obesity it is often very modest (only 1 mmHg BP for each 200 μU/mL increment in insulin level13).
Stronger evidence that insulin may not be a major hypertensive factor in humans comes from studies in insulinoma patients who are not hypertensive despite severe hyperinsulinemia66, 67, even when they become resistant to the metabolic/vasodilatory effects of insulin68, 69. Also, patients with type 1 diabetes exhibited reduced rather than increased BP during a 15-month follow-up treatment with continuous subcutaneous insulin infusion70.
Contrary to what has been demonstrated in rodents, insulin does not appear to play a critical role in long-term BP regulation in other experimental animals. Administration of an insulin antagonist in hypertensive and hyperinsulinemic obese rabbits resulted in only modest attenuation of hypertension71. In addition to the lack of a pressor effect of acute hyperinsulinemia to elevate BP in lean or obese dogs and horses discussed previously, intravenous insulin infusions for several days or even weeks in lean healthy dogs did not raise BP72–75. When insulin was chronically infused directly into the renal arteries of conscious dogs, no increase in BP was observed and the dogs exhibited only mild transient sodium retention76. Chronic insulin infusions directly into the CNS via the carotid or vertebral arteries of conscious dogs also failed to increase BP77.
Although insulin infusion has been reported to increase SNS activity without raising BP in several species, including humans, at least part of the sympathetic activation may be a compensatory response to the vasodilator effects of insulin. Insulin infusion in humans causes vasodilation of skeletal muscle37 and chronic intravenous insulin administration in dogs decreased total peripheral vascular resistance, increased cardiac output, and increased HR, which may suggest compensatory activation of the SNS75, 78. Thus, it is possible that insulin’s action to cause systemic vasodilation may trigger reflex activation of the SNS to buffer a potential fall in BP caused by its vasodilatory effects. In fact, patients with autonomic insufficiency and/or impaired baroreflex show greater vasodilation in response to insulin and more pronounced reductions in BP79, 80. These studies suggest that insulin may not cause sustained increases in SNS activity to CV relevant tissues via direct actions in the CNS and that its hypertensive effects in rodents are largely mediated by action on peripheral tissues, such as the kidneys63, 81.
In obesity/metabolic syndrome the vasodilator effects of insulin are attenuated82 and it has been suggested that vascular insulin resistance and impaired vasodilation may be required for hyperinsulinemia to cause hypertension. However, obese insulin resistant dogs exhibited no significant increases in BP during chronic insulin infusions that raised their already high baseline insulin levels83. This failure of insulin to raise BP occurred despite impaired insulin-mediated vasodilation83. Moreover, hypertensive dogs infused chronically with angiotensin II (Ang II) did not show potentiated BP responses to insulin when compared to Ang II infusion alone73. This suggests that obesity-induced activation of the RAAS does not interact additively or synergistically with insulin to increase BP. Similar findings were obtained in dogs infused chronically with norepinephrine, to mimic the increased adrenergic activity that is also commonly observed in obese/metabolic syndrome patients74. Chronic insulin infusion in dogs with 70% reduction in kidney mass and fed high sodium diet, to increase their susceptibility to hypertensive stimuli, also did not raise BP73. Therefore, even with a background of insulin resistance, dyslipidemia, increased Ang II, increased adrenergic activity, and cardiorenal dysfunction, chronic hyperinsulinemia failed to significantly increase BP in obese dogs.
Overall, as summarized in Table 2 and Figure 1, most previous studies suggest that insulin resistance and hyperinsulinemia do not play a major role in initiating hypertension associated with obesity/metabolic syndrome in humans or large experimental animals such as dogs and horses. However, chronic insulin administration may cause modest increases in BP in rodents. Yet, as discussed later, several studies have also shown that severe insulin resistance and hyperinsulinemia may not lead to hypertension even in rodents when there is morbid obesity due to deficiency of the leptin-POMC-MC4R pathway.
Table 2 -.
Differential impact of insulin infusion on blood pressure regulation in humans and experimental animal models.
| Model | Effects |
|---|---|
| Humans | |
| Lean |
|
| Obese |
|
| Horses | |
| Lean |
|
| Obese |
|
| Dogs | |
| Lean |
|
| Obese |
|
| Rabbits | |
| Lean |
|
| Obese |
|
| Rats | |
| Lean |
|
| Obese |
|
BP, blood pressure; Na+, sodium; SNS, sympathetic nervous system.
Figure 1 -. Main actions of insulin on BP regulation.
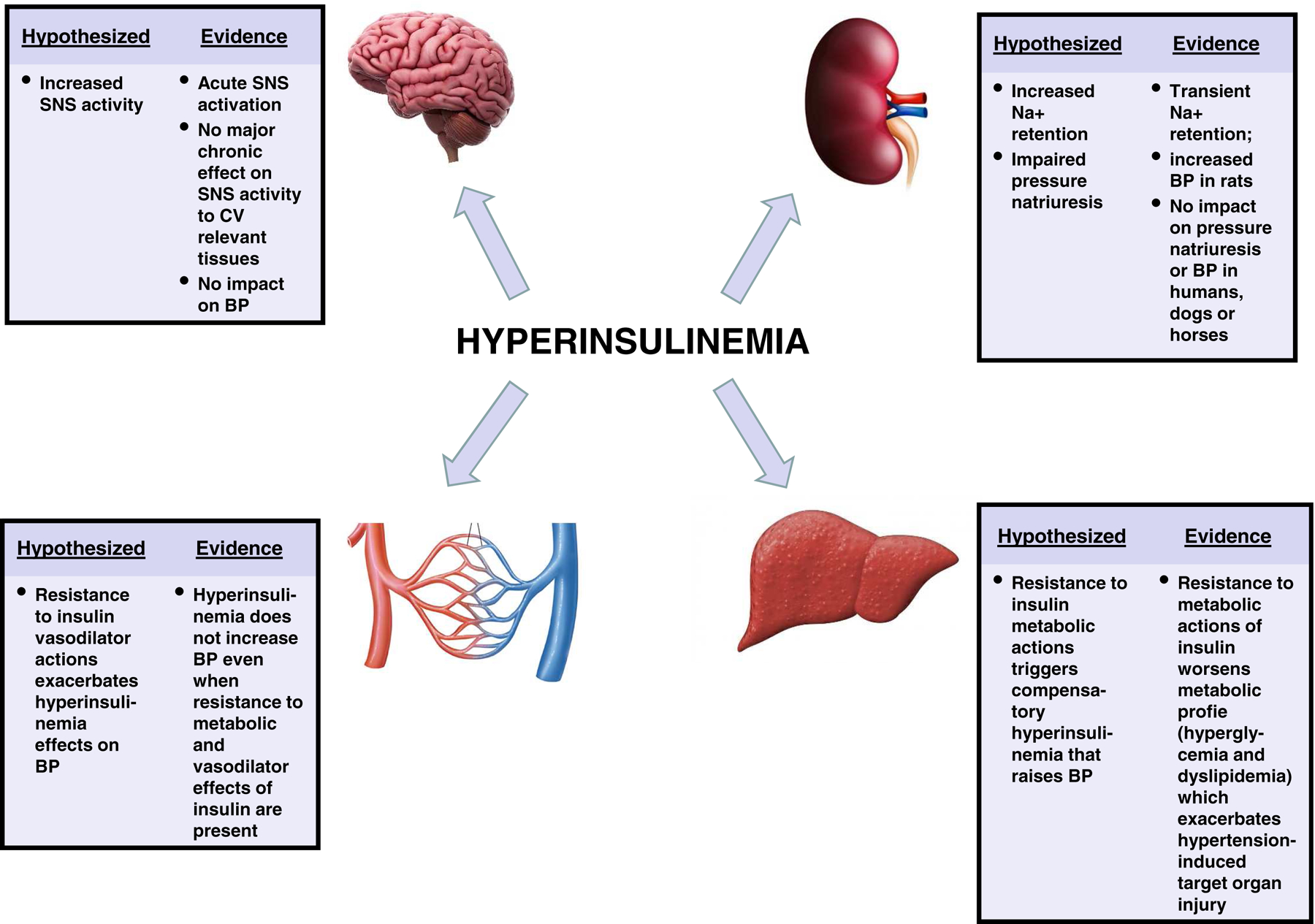
This figure summarizes the main effects of hyperinsulinemia on blood pressure regulation that have been proposed and the evidence from studies in humans and several experimental animal models supporting or refuting these hypothesized effects. BP, blood pressure; Na+, sodium; SNS, sympathetic nervous system.
MECHANISMS THAT INITIATE HYPERTENSION IN OBESITY AND METABOLIC SYNDROME
The list of additional factors, besides insulin resistance and hyperinsulinemia, that have been postulated to mediate hypertension on obesity/metabolic syndrome is extensive and includes various adipokines from adipose tissue, abnormal gut microbiota, SNS activation, excess antinatriuretic hormones, deficiency of natriuretic hormones, vascular and kidney dysfunction, and others mechanisms that have been previously reviewed84–87. For many of these factors, however, clear cause and effect relationships with increased BP have not been established.
Studies in experimental animals and in humans indicate that hypertension associated with obesity/metabolic syndrome is initiated by multiple factors that increase renal sodium reabsorption and cause expansion of extracellular fluid volume84, 88. Three mechanisms appear to be especially important in initiating these renal changes and hypertension associated with visceral obesity: 1) physical compression of the kidneys by fat in and around the kidneys, 2) activation of the RAAS, and 3) increased SNS activity. With prolonged obesity over several years, elevated BP interacts synergistically with metabolic abnormalities, especially hyperglycemia and hyperlipidemia, to cause kidney and cardiovascular injury which exacerbates hypertension and injury to the CV system and kidneys89, 90. We have previously discussed the importance of these mechanisms84, 91, 92 and therefore briefly outline them in this review.
Compression of the Kidneys by Visceral, Perirenal and Renal Sinus Fat
In obese dogs, rabbits and humans, but not in rodents, perirenal fat often encapsulates the kidneys, adheres tightly to the renal capsule, and invades the renal sinuses, causing kidney compression and increased intrarenal pressures which, in turn, raises BP91. In patients with visceral obesity, intra-abdominal pressures also rise in proportion to sagittal abdominal diameter, further compressing the kidneys93. Population studies indicate that visceral obesity and especially retroperitoneal and renal sinus fat are uniquely correlated with incident hypertension94–96. Also, fatty kidneys are associated with increased risk for chronic kidney disease (CKD) even after adjustment for BMI and visceral adiposity95.
Compression of the kidneys by fat around the kidneys and in the renal sinuses raises intrarenal pressure to as high as 19 mmHg in obese dogs84 and perhaps even higher in people with severe abdominal obesity93. High intrarenal pressures compress the vasa recta capillaries and thin loops of Henle, reducing blood flow in the renal medulla, increasing sodium reabsorption in the loop of Henle, and contributing to volume expansion and hypertension. Increased sodium reabsorption in the loop of Henle sodium would also tend to reduce sodium chloride delivery to the macula densa, causing feedback mediated renal vasodilation, increased glomerular filtration rate (hyperfiltration), and increased renin secretion – phenotypes that are characteristic of obesity-associated hypertension prior to renal injury and eventual loss of nephrons84
Although excess fat in and around the kidneys cannot explain rapid increases in BP that occur shortly after increased caloric intake and weight gain, kidney compression and “lipotoxic” effects of kidney fat may help explain why visceral adiposity is much better correlated with hypertension than is subcutaneous adiposity5, 6, 84.
RAAS Activation in Obesity/Metabolic Syndrome
The RAAS is the most powerful hormonal system for regulating renal sodium excretion and plays a critical role in BP regulation97. Excess weight gain, especially when associated with increased visceral adiposity, causes mild to moderate increases in several components of the RAAS, including Ang II and aldosterone91, 98. Even with only modest activation of the RAAS, blockade of Ang II receptors or angiotensin converting enzyme (ACE), or mineralocorticoid receptor (MR) antagonism attenuates sodium retention, volume expansion, and increased BP in obesity84, 91, 99. Thus, obesity appears to increase BP sensitivity to RAAS activation100.
MR antagonism may also reduce BP and protect the kidneys from injury through mechanisms that are at least partly independent of aldosterone. For example, administration of the MR antagonist spironolactone in obese patients and patients with treatment-resistant hypertension caused reductions in BP that did not correlate with circulating aldosterone levels101–103. In obese hypertensive patients treated with ACE inhibitors, addition of spironolactone to their treatment regimen further reduced BP indicating that MR antagonism reduces BP in obesity despite prior blockade of Ang II formation101.
Although the mechanisms responsible for aldosterone-independent MR activation in obesity are still unclear there is evidence that obesity and its associated metabolic disorders may increase Rac1, a GTP-binding protein that stimulates MR signaling104. Other studies suggest that oxidative stress and cortisol may activate MR in obesity due to downregulation of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2)91, 105, 106; this enzyme normally converts cortisol, which can activate the MR, to inactive cortisone in the renal tubular cells106. Although the mechanisms by which MR is activated in obesity are not fully understood, MR antagonism is clearly an effective treatment for obese patients with hypertension that are resistant to antihypertensive therapy101–103, 107, 108.
Sympathetic Nervous System Activation in Obesity/Metabolic Syndrome
The importance of SNS activation in contributing to hypertension in obesity/metabolic syndrome has been clearly documented109, 110. Increases in sympathetic nerve activity in obese experimental animals and in humans are often modest and do not reduce tissue blood flow. However, increased renal sympathetic activity is sufficient to increase sodium reabsorption and renin secretion. Moreover, renal denervation greatly attenuates sodium retention and hypertension in obese experimental animals as well as obese humans with metabolic syndrome111–113.
Multiple factors have been proposed to stimulate SNS activity in obesity. The roles of some of these factors, such as insulin resistance, hyperinsulinemia, fatty acids and Ang II, are beyond the scope of this review and have previously been discussed5, 109, 114. Some of the more important factors that may contribute to SNS activation in obesity include impaired baroreceptor reflexes, activation of chemoreceptors, especially in patients with obstructive sleep apnea and hypoxemia, and hyperleptinemia with activation of the CNS POMC-MC4R pathway84, 109, 115, 116.
Hyperleptinemia and activation of CNS melanocortin system in obesity/metabolic syndrome.
Leptin, the product of the ob/ob gene, is secreted by adipocytes in proportion to the degree of adiposity and obese individuals exhibit higher plasma leptin concentrations than lean people117. There is a positive correlation between plasma leptin concentration, skeletal muscle SNS activity, and BP in humans118. Moreover, acute leptin administration increases SNS activity to various tissues including the kidneys, brown adipose tissue, skeletal muscle, and adrenal glands in rodents119, 120. Leptin also increases skeletal muscle SNS activity in humans121. Acute leptin infusions, however, often have minimal effect on BP in rodents or humans despite increasing SNS activity, likely due to counterbalancing vasodilator effects of NO, also stimulated by leptin122, and the fact the hypertensive effects of leptin are slow in onset, requiring several days to occur123.
In lean rodents, chronic leptin administration to raise leptin concentration to that observed in severe obesity causes sustained, albeit modest, increases in BP122–124 which can be blocked by adrenergic receptor antagonism in male rodents and markedly attenuated by MR antagonism in females125–128. The hypertensive effects of leptin are exacerbated in animals with reduced NO availability122 as often occurs in obese patients with endothelial injury and atherosclerosis. Further support for a role of hyperleptinemia in contributing to elevated BP in obesity also comes from studies showing significant attenuation of the hypertension in obese rabbits treated with leptin receptor antagonist71.
Although studies of the chronic BP effects of leptin in humans are limited, people with leptin gene or leptin receptor mutations are extremely obese with many characteristics of the metabolic syndrome, including marked hyperinsulinemia and severe insulin resistance, but they are not hypertensive129. Despite severe obesity, these patients also appear to have sympathetic hypofunction, postural hypotension, attenuated RAAS responses to upright posture, and reduced BP responses to cold pressor tests129. Similar findings have been reported in male leptin deficient (ob/ob) mice which have severe obesity, insulin resistance, hyperinsulinemia, and dyslipidemia but lower BP and reduced SNS activity compared to lean mice128, 130; and leptin infusion in ob/ob mice increased BP despite reducing body weight131. These observations collectively support a role for leptin as an important link between obesity/metabolic syndrome, increased SNS activity and elevated BP.
One mechanism by which leptin raises BP is by stimulating the brain POMC neuron-MC4R pathway92, 132. The CNS POMC-MC4R pathway regulates appetite, energy expenditure and body weight133. POMC-expressing neurons, located in hypothalamus and brainstem, send projections to second order neurons where they release α-melanocyte stimulating hormone (α-MSH), an agonist for MC4R133. MC4R are located in several regions of the CNS but are particularly abundant in areas that participate in cardiovascular regulation including the hypothalamus, brainstem, and spinal cord92, 134, 135.
Genetic deletion of leptin receptors on POMC neurons, MC4R deficiency, or pharmacological blockade of MC4R all completely abolish the rise in BP observed during chronic leptin infusion124, 132, 136. Also, MC4R activation using synthetic or natural agonists injected into the CNS raises SNS to various tissues including the kidneys137, 138 and chronic activation of brain MC4R evokes sustained elevations in BP in rodents139, 140. MC4R-induced elevations in BP are also observed in lean and obese humans treated with MC4R agonists134, 135, 141, 142. Perhaps the most compelling evidence for a role of MC4R in obesity/metabolic syndrome-induced hypertension is the finding that patients with loss-of-function mutations of the POMC gene, genes involved in POMC processing, or MC4R gene have severe obesity and most features of the metabolic syndrome but are less likely to develop hypertension than less obese individuals with normal POMC-MC4R genotypes141, 142. Humans with MC4R mutations also have attenuated SNS responses to stimuli that increase SNS activity such as inspiratory hypoxia143.
The attenuated SNS response and lower BP in humans with POMC/MC4R mutations despite severe insulin resistance, impaired IGT, hyperinsulinemia and dyslipidemia also suggest that insulin resistance and hyperinsulinemia are not the primary drivers of hypertension in obesity/metabolic syndrome.
CONCLUSIONS
Obesity/metabolic syndrome is a major risk factor for multiple chronic diseases including CV diseases. Many hypotheses have been proposed to explain how excess adiposity increases SNS activity, impairs kidney function, and elevates BP. One hypothesis that gained traction more than 30 years ago is that hyperinsulinemia and insulin resistance are major contributors to hypertension in people with obesity/metabolic syndrome. This hypothesis is mainly supported by epidemiological studies showing positive correlations among plasma insulin concentration, insulin resistance, and BP and from studies in rodents. Although hyperinsulinemia and insulin resistance are major players in other disturbances that occur in metabolic syndrome (e.g., dyslipidemia and dysglycemia) and may increase the risk for CV disease, there is strong evidence from studies in humans and experimental animals that hyperinsulinemia, with or without insulin resistance, does not play a major role in initiating hypertension in obesity/metabolic syndrome. As summarized in Figure 2, other factors such as physical compression of the kidneys, RAAS activation, SNS activation, hyperleptinemia, and activation of brain MC4R have been demonstrated to be critical in linking excess visceral adiposity with increased BP associated with obesity/metabolic syndrome. Although hyperinsulinemia may not initiate obesity hypertension, hyperglycemia and dyslipidemia associated with insulin resistance likely contribute to progressive vascular and kidney injury which, over the long-term, can exacerbate hypertension and lead to further target organ injury. The precise mechanisms by which the metabolic syndrome contributes to hypertension and progressive target organ injury are not completely understood and remain an important area for research, especially considering the ever growing prevalence of obesity and the limited effectiveness of current therapies for many of the associated metabolic disorders.
Figure 2 -. Potential mechanisms of hypertension and cardiovascular and renal injury in obesity.
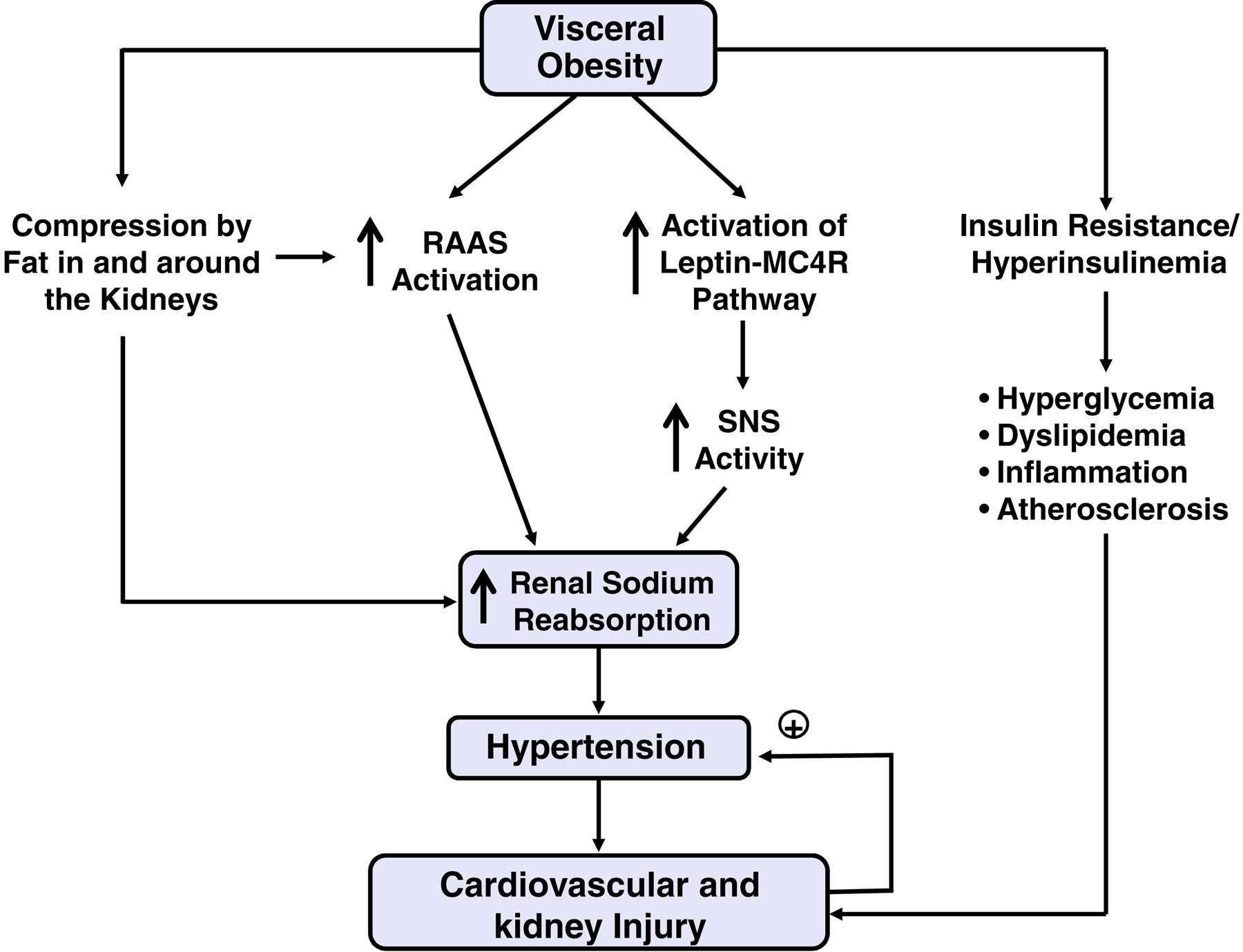
Obesity, particularly visceral obesity, triggers activation of the renin-angiotensin-aldosterone system (RAAS) which together with increased sympathetic nervous system (SNS) activity increases renal sodium reabsorption and blood pressure. Physical compression of the kidneys caused by increased fat in and around the kidneys may also stimulate renal sodium reabsorption and contribute to RAAS activation in obesity. When obesity is sustained, progressive metabolic impairments including insulin resistance, compensatory hyperinsulinemia, and accompanying hyperglycemia and dyslipidemia may contribute to inflammation and atherosclerosis and interact with hypertension to cause further injury to the kidneys and cardiovascular system that worsens the hypertension and creates a vicious cycle.
FUNDING SOURCES:
The authors’ research was supported by National Heart, Lung, and Blood Institute (P01 HL51971), National Institute of General Medical Sciences (P20 GM104357 and U54 GM115428), National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK121411 and R00 DK113280), and the American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES:
The authors declare no conflict of interest relevant to this manuscript.
REFERENCES:
- 1. https://www.who.int/cardiovascular_diseases/world-heart-day/en/.
- 2.Dai S, Huang B, Zou Y, Liu Y. Associations of dipping and non-dipping hypertension with cardiovascular diseases in patients with dyslipidemia. Arch Med Sci. 2019;15:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. [DOI] [PubMed] [Google Scholar]
- 4.Garrison RJ, Kannel WB, Stokes J 3rd, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med. 1987;16:235–251. [DOI] [PubMed] [Google Scholar]
- 5.Davy KP, Hall JE. Obesity and hypertension: two epidemics or one? Am J Physiol Regul Integr Comp Physiol. 2004;286:R803–813. [DOI] [PubMed] [Google Scholar]
- 6.Neeland IJ, Poirier P, Despres JP. Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation. 2018;137:1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kylin E [Studies of the hypertension-hyperglycemia-hyperuricemia syndrome] (German). Zentralbl Inn Med. 1923;44:105–127. [Google Scholar]
- 8.Haller H [Epidermiology and associated risk factors of hyperlipoproteinemia]. Z Gesamte Inn Med. 1977;32:124–128. [PubMed] [Google Scholar]
- 9.Singer P [Diagnosis of primary hyperlipoproteinemias]. Z Gesamte Inn Med. 1977;32:128 contd. [PubMed] [Google Scholar]
- 10.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. [DOI] [PubMed] [Google Scholar]
- 11.Reaven GM. Syndrome X: 6 years later. J Intern Med Suppl. 1994;736:13–22. [PubMed] [Google Scholar]
- 12.Kaplan NM. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Arch Intern Med. 1989;149:1514–1520. [DOI] [PubMed] [Google Scholar]
- 13.Ferrannini E, Haffner SM, Stern MP. Essential hypertension: an insulin-resistant state. J Cardiovasc Pharmacol. 1990;15 Suppl 5:S18–25. [PubMed] [Google Scholar]
- 14.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. [DOI] [PubMed] [Google Scholar]
- 15.Reaven GM. Syndrome X. Blood Press Suppl. 1992;4:13–16. [PubMed] [Google Scholar]
- 16.Christlieb AR, Krolewski AS, Warram JH, Soeldner JS. Is insulin the link between hypertension and obesity? Hypertension. 1985;7:II54–57. [DOI] [PubMed] [Google Scholar]
- 17.Modan M, Halkin H, Almog S, et al. Hyperinsulinemia. A link between hypertension obesity and glucose intolerance. J Clin Invest. 1985;75:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech 2009;2:231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. [DOI] [PubMed] [Google Scholar]
- 20.Eckel RH, Alberti KG, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2010;375:181–183. [DOI] [PubMed] [Google Scholar]
- 21.Ferrannini E, Buzzigoli G, Bonadonna R, et al. Insulin resistance in essential hypertension. N Engl J Med. 1987;317:350–357. [DOI] [PubMed] [Google Scholar]
- 22.Mbanya JC, Thomas TH, Wilkinson R, Alberti KG, Taylor R. Hypertension and hyperinsulinaemia: a relation in diabetes but not essential hypertension. Lancet. 1988;1:733–734. [DOI] [PubMed] [Google Scholar]
- 23.Hall JE. Hyperinsulinemia: a link between obesity and hypertension? Kidney Int. 1993;43:1402–1417. [DOI] [PubMed] [Google Scholar]
- 24.Chen DL, Liess C, Poljak A, et al. Phenotypic Characterization of Insulin-Resistant and Insulin-Sensitive Obesity. J Clin Endocrinol Metab. 2015;100:4082–4091. [DOI] [PubMed] [Google Scholar]
- 25.Julius S, Gudbrandsson T, Jamerson K, Tariq Shahab S, Andersson O. The hemodynamic link between insulin resistance and hypertension. J Hypertens. 1991;9:983–986. [DOI] [PubMed] [Google Scholar]
- 26.Hall JE, Summers RL, Brands MW, Keen H, Alonso-Galicia M. Resistance to metabolic actions of insulin and its role in hypertension. Am J Hypertens. 1994;7:772–788. [DOI] [PubMed] [Google Scholar]
- 27.Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98:2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019;576:51–60. [DOI] [PubMed] [Google Scholar]
- 29.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeFronzo RA. Insulin and renal sodium handling: clinical implications. Int J Obes. 1981;5 suppl 1:93–104. [PubMed] [Google Scholar]
- 31.Landsberg L, Krieger DR. Obesity, metabolism, and the sympathetic nervous system. Am J Hypertens. 1989;2:125S–132S. [DOI] [PubMed] [Google Scholar]
- 32.Tuck ML. Obesity, the sympathetic nervous system, and essential hypertension. Hypertension. 1992;19:I67–77. [DOI] [PubMed] [Google Scholar]
- 33.Miller JH, Bogdonoff MD. Antidiuresis associated with administration of insulin. J Appl Physiol. 1954;6:509–512. [DOI] [PubMed] [Google Scholar]
- 34.Atchley DW, Loeb RF, Richards DW, Benedict EM, Driscoll ME. ON DIABETIC ACIDOSIS: A Detailed Study of Electrolyte Balances Following the Withdrawal and Reestablishment of Insulin Therapy. J Clin Invest. 1933;12:297–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nizet A, Lefebvre P, Crabbe J. Control by insulin of sodium potassium and water excretion by the isolated dog kidney. Pflugers Arch. 1971;323:11–20. [DOI] [PubMed] [Google Scholar]
- 36.Ohtani N, Okamoto Y, Tateishi K, Uchiyama H, Ohta M. Increased Feeding Speed Is Associated with Higher Subsequent Sympathetic Activity in Dogs. PLoS One. 2015;10:e0142899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest. 1991;87:2246–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson EA, Balon TW, Hoffman RP, Sinkey CA, Mark AL. Insulin increases sympathetic activity but not blood pressure in borderline hypertensive humans. Hypertension. 1992;19:621–627. [DOI] [PubMed] [Google Scholar]
- 39.Berne C, Fagius J, Pollare T, Hjemdahl P. The sympathetic response to euglycaemic hyperinsulinaemia. Evidence from microelectrode nerve recordings in healthy subjects. Diabetologia. 1992;35:873–879. [DOI] [PubMed] [Google Scholar]
- 40.Vollenweider P, Randin D, Tappy L, Jequier E, Nicod P, Scherrer U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest. 1994;93:2365–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muntzel MS, Anderson EA, Johnson AK, Mark AL. Mechanisms of insulin action on sympathetic nerve activity. Clin Exp Hypertens. 1995;17:39–50. [DOI] [PubMed] [Google Scholar]
- 42.Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol. 1994;267:R1350–1355. [DOI] [PubMed] [Google Scholar]
- 43.Stocker SD, Gordon KW. Glutamate receptors in the hypothalamic paraventricular nucleus contribute to insulin-induced sympathoexcitation. J Neurophysiol. 2015;113:1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol. 2013;304:H1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol. 2011;589:1643–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension. 2010;55:284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension. 2011;57:435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci. 2003;23:5998–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Z, Cassaglia PA, Pelletier NE, Brooks VL. Sex differences in the sympathoexcitatory response to insulin in obese rats: role of neuropeptide Y. J Physiol. 2019;597:1757–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachmann KN, Deger SM, Alsouqi A, et al. Acute effects of insulin on circulating natriuretic peptide levels in humans. PLoS One. 2018;13:e0196869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brands MW, Hall JE. Insulin resistance, hyperinsulinemia, and obesity-associated hypertension. J Am Soc Nephrol. 1992;3:1064–1077. [DOI] [PubMed] [Google Scholar]
- 52.Hausberg M, Hoffman RP, Somers VK, Sinkey CA, Mark AL, Anderson EA. Contrasting autonomic and hemodynamic effects of insulin in healthy elderly versus young subjects. Hypertension. 1997;29:700–705. [DOI] [PubMed] [Google Scholar]
- 53.Hall JE, Brands MW, Zappe DH, Alonso Galicia M. Insulin resistance, hyperinsulinemia, and hypertension: causes, consequences, or merely correlations? Proc Soc Exp Biol Med. 1995;208:317–329. [DOI] [PubMed] [Google Scholar]
- 54.Nostell KE, Lindase SS, Brojer JT. Blood pressure in Warmblood horses before and during a euglycemic-hyperinsulinemic clamp. Acta Vet Scand. 2016;58:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takishita S, Takata Y, Abe I, et al. Lack of sympathetic augmentation in response to intravenous load of glucose in rabbits. Clin Exp Pharmacol Physiol. 1991;18:525–531. [DOI] [PubMed] [Google Scholar]
- 56.Richey JM, Poulin RA, Buchanan TA, Galperin E, Moore DM, Bergman RN. Failure of acute hyperinsulinemia to alter blood pressure is not due to baroreceptor feedback. Am J Hypertens. 1999;12:405–413. [DOI] [PubMed] [Google Scholar]
- 57.Nostell K, Lindase S, Edberg H, Brojer J. The effect of insulin infusion on heart rate and systemic blood pressure in horses with equine metabolic syndrome. Equine Vet J. 2019;51:733–737. [DOI] [PubMed] [Google Scholar]
- 58.Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Sustained hyperinsulinemia increases arterial pressure in conscious rats. Am J Physiol. 1991;260:R764–768. [DOI] [PubMed] [Google Scholar]
- 59.Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Hypertension during chronic hyperinsulinemia in rats is not salt-sensitive. Hypertension. 1992;19:I83–89. [DOI] [PubMed] [Google Scholar]
- 60.Brands MW, Harrison DL, Keen HL, Gardner A, Shek EW, Hall JE. Insulin-induced hypertension in rats depends on an intact renin-angiotensin system. Hypertension. 1997;29:1014–1019. [DOI] [PubMed] [Google Scholar]
- 61.Keen HL, Brands MW, Smith MJ Jr., Shek EW, Hall JE. Inhibition of thromboxane synthesis attenuates insulin hypertension in rats. Am J Hypertens. 1997;10:1125–1131. [DOI] [PubMed] [Google Scholar]
- 62.Keen HL, Brands MW, Alonso-Galicia M, Hall JE. Chronic adrenergic receptor blockade does not prevent hyperinsulinemia-induced hypertension in rats. Am J Hypertens. 1996;9:1192–1199. [DOI] [PubMed] [Google Scholar]
- 63.Irsik DL, Chen JK, Brands MW. Chronic renal artery insulin infusion increases mean arterial pressure in male Sprague-Dawley rats. Am J Physiol Renal Physiol. 2018;314:F81–F88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bunag RD, Krizsan-Agbas D, Itoh H. Sympathetic activation by chronic insulin treatment in conscious rats. J Pharmacol Exp Ther. 1991;259:131–138. [PubMed] [Google Scholar]
- 65.Arshi B, Tohidi M, Derakhshan A, Asgari S, Azizi F, Hadaegh F. Sex-specific relations between fasting insulin, insulin resistance and incident hypertension: 8.9 years follow-up in a Middle-Eastern population. J Hum Hypertens. 2015;29:260–267. [DOI] [PubMed] [Google Scholar]
- 66.Tsutsu N, Nunoi K, Kodama T, Nomiyama R, Iwase M, Fujishima M. Lack of association between blood pressure and insulin in patients with insulinoma. J Hypertens. 1990;8:479–482. [DOI] [PubMed] [Google Scholar]
- 67.Sawicki PT, Heinemann L, Starke A, Berger M. Hyperinsulinaemia is not linked with blood pressure elevation in patients with insulinoma. Diabetologia. 1992;35:649–652. [DOI] [PubMed] [Google Scholar]
- 68.Pontiroli AE, Alberetto M, Pozza G. Patients with insulinoma show insulin resistance in the absence of arterial hypertension. Diabetologia. 1992;35:294–295. [DOI] [PubMed] [Google Scholar]
- 69.Sawicki PT, Baba T, Berger M, Starke A. Normal blood pressure in patients with insulinoma despite hyperinsulinemia and insulin resistance. J Am Soc Nephrol. 1992;3:S64–68. [DOI] [PubMed] [Google Scholar]
- 70.Markakis K, Alam T, Jinadev P, et al. Continuous Subcutaneous Insulin Infusion Initiation Is Associated With Blood Pressure Reduction in Adults With Type 1 Diabetes. J Diabetes Sci Technol 2019;13:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim K, Burke SL, Head GA. Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension. 2013;61:628–634. [DOI] [PubMed] [Google Scholar]
- 72.Hall JE, Coleman TG, Mizelle HL. Does chronic hyperinsulinemia cause hypertension? Am J Hypertens. 1989;2:171–173. [DOI] [PubMed] [Google Scholar]
- 73.Hall JE, Coleman TG, Mizelle HL, Smith MJ Jr. Chronic hyperinsulinemia and blood pressure regulation. Am J Physiol. 1990;258:F722–731. [DOI] [PubMed] [Google Scholar]
- 74.Hall JE, Brands MW, Kivlighn SD, Mizelle HL, Hildebrandt DA, Gaillard CA. Chronic hyperinsulinemia and blood pressure. Interaction with catecholamines? Hypertension. 1990;15:519–527. [DOI] [PubMed] [Google Scholar]
- 75.Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA, Hall JE. The hemodynamic response to chronic hyperinsulinemia in conscious dogs. Am J Hypertens. 1991;4:164–168. [DOI] [PubMed] [Google Scholar]
- 76.Hall JE, Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA. Chronic intrarenal hyperinsulinemia does not cause hypertension. Am J Physiol. 1991;260:F663–669. [DOI] [PubMed] [Google Scholar]
- 77.Hildebrandt DA, Smith MJ Jr., Hall JE. Cardiovascular regulation during insulin infusion into the carotid or vertebral artery in dogs. J Hypertens. 1999;17:251–260. [DOI] [PubMed] [Google Scholar]
- 78.Hall JE, Brands MW, Zappe DH, Alonso-Galicia M. Cardiovascular actions of insulin: are they important in long-term blood pressure regulation? Clin Exp Pharmacol Physiol. 1995;22:689–700. [DOI] [PubMed] [Google Scholar]
- 79.Mathias CJ, da Costa DF, Fosbraey P, Christensen NJ, Bannister R. Hypotensive and sedative effects of insulin in autonomic failure. Br Med J (Clin Res Ed). 1987;295:161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown RT, Polinsky RJ, Baucom CE. Euglycemic insulin-induced hypotension in autonomic failure. Clin Neuropharmacol. 1989;12:227–231. [DOI] [PubMed] [Google Scholar]
- 81.Liu J, da Silva AA, Tallam LS, Hall JE. Chronic central nervous system hyperinsulinemia and regulation of arterial pressure and food intake. J Hypertens. 2006;24:1391–1395. [DOI] [PubMed] [Google Scholar]
- 82.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hall JE, Brands MW, Zappe DH, et al. Hemodynamic and renal responses to chronic hyperinsulinemia in obese, insulin-resistant dogs. Hypertension. 1995;25:994–1002. [DOI] [PubMed] [Google Scholar]
- 84.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity, kidney dysfunction and hypertension: mechanistic links. Nat Rev Nephrol. 2019;15:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. 2018;14:442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10:364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jordan J, Birkenfeld AL, Melander O, Moro C. Natriuretic Peptides in Cardiovascular and Metabolic Crosstalk: Implications for Hypertension Management. Hypertension. 2018;72:270–276. [DOI] [PubMed] [Google Scholar]
- 88.Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41:625–633. [DOI] [PubMed] [Google Scholar]
- 89.Hall JE, Henegar JR, Dwyer TM, et al. Is obesity a major cause of chronic kidney disease? Adv Ren Replace Ther. 2004;11:41–54. [DOI] [PubMed] [Google Scholar]
- 90.Hall ME, do Carmo JM, da Silva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovasc Dis. 2014;7:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.da Silva AA, do Carmo JM, Wang Z, Hall JE. Melanocortin-4 Receptors and Sympathetic Nervous System Activation in Hypertension. Curr Hypertens Rep. 2019;21:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sugerman H, Windsor A, Bessos M, Wolfe L. Intra-abdominal pressure, sagittal abdominal diameter and obesity comorbidity. J Intern Med. 1997;241:71–79. [DOI] [PubMed] [Google Scholar]
- 94.Chughtai HL, Morgan TM, Rocco M, et al. Renal sinus fat and poor blood pressure control in middle-aged and elderly individuals at risk for cardiovascular events. Hypertension. 2010;56:901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foster MC, Hwang SJ, Porter SA, Massaro JM, Hoffmann U, Fox CS. Fatty kidney, hypertension, and chronic kidney disease: the Framingham Heart Study. Hypertension. 2011;58:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandra A, Neeland IJ, Berry JD, et al. The relationship of body mass and fat distribution with incident hypertension: observations from the Dallas Heart Study. J Am Coll Cardiol. 2014;64:997–1002. [DOI] [PubMed] [Google Scholar]
- 97.Hall JE, Granger JP, do Carmo JM, et al. Hypertension: physiology and pathophysiology. Compr Physiol. 2012;2:2393–2442. [DOI] [PubMed] [Google Scholar]
- 98.Engeli S, Sharma AM. The renin-angiotensin system and natriuretic peptides in obesity-associated hypertension. J Mol Med (Berl). 2001;79:21–29. [DOI] [PubMed] [Google Scholar]
- 99.de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43:41–47. [DOI] [PubMed] [Google Scholar]
- 100.Alonso-Galicia M, Brands MW, Zappe DH, Hall JE. Hypertension in obese Zucker rats. Role of angiotensin II and adrenergic activity. Hypertension. 1996;28:1047–1054. [DOI] [PubMed] [Google Scholar]
- 101.Bomback AS, Muskala P, Bald E, Chwatko G, Nowicki M. Low-dose spironolactone, added to long-term ACE inhibitor therapy, reduces blood pressure and urinary albumin excretion in obese patients with hypertensive target organ damage. Clin Nephrol. 2009;72:449–456. [DOI] [PubMed] [Google Scholar]
- 102.Dudenbostel T, Calhoun DA. Use of Aldosterone Antagonists for Treatment of Uncontrolled Resistant Hypertension. Am J Hypertens. 2017;30:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Souza F, Muxfeldt E, Fiszman R, Salles G. Efficacy of spironolactone therapy in patients with true resistant hypertension. Hypertension. 2010;55:147–152. [DOI] [PubMed] [Google Scholar]
- 104.Fujita T Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol. 2014;25:1148–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Funder JW. Apparent mineralocorticoid excess. J Steroid Biochem Mol Biol. 2017;165:151–153. [DOI] [PubMed] [Google Scholar]
- 107.Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Williams B, MacDonald TM, Morant SV, et al. Endocrine and haemodynamic changes in resistant hypertension, and blood pressure responses to spironolactone or amiloride: the PATHWAY-2 mechanisms substudies. Lancet Diabetes Endocrinol. 2018;6:464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res. 2015;116:976–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hall JE, da Silva AA, do Carmo JM, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–17276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Henegar JR, Zhang Y, De Rama R, Hata C, Hall ME, Hall JE. Catheter-based radiorefrequency renal denervation lowers blood pressure in obese hypertensive dogs. Am J Hypertens. 2014;27:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lohmeier TE, Hall JE. Device-Based Neuromodulation for Resistant Hypertension Therapy. Circ Res. 2019;124:1071–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsioufis C, Ziakas A, Dimitriadis K, et al. Blood pressure response to catheter-based renal sympathetic denervation in severe resistant hypertension: data from the Greek Renal Denervation Registry. Clin Res Cardiol. 2017;106:322–330. [DOI] [PubMed] [Google Scholar]
- 114.Esler M, Lambert G, Schlaich M, Dixon J, Sari CI, Lambert E. Obesity Paradox in Hypertension: Is This Because Sympathetic Activation in Obesity-Hypertension Takes a Benign Form? Hypertension. 2018;71:22–33. [DOI] [PubMed] [Google Scholar]
- 115.da Silva AA, do Carmo JM, Wang Z, Hall JE. The brain melanocortin system, sympathetic control, and obesity hypertension. Physiology (Bethesda). 2014;29:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Floras JS. Sleep Apnea and Cardiovascular Disease: An Enigmatic Risk Factor. Circ Res. 2018;122:1741–1764. [DOI] [PubMed] [Google Scholar]
- 117.Friedman J The long road to leptin. J Clin Invest. 2016;126:4727–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Snitker S, Pratley RE, Nicolson M, Tataranni PA, Ravussin E. Relationship between muscle sympathetic nerve activity and plasma leptin concentration. Obes Res. 1997;5:338–340. [DOI] [PubMed] [Google Scholar]
- 119.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Haynes WG, Sivitz WI, Morgan DA, Walsh SA, Mark AL. Sympathetic and cardiorenal actions of leptin. Hypertension. 1997;30:619–623. [DOI] [PubMed] [Google Scholar]
- 121.Machleidt F, Simon P, Krapalis AF, Hallschmid M, Lehnert H, Sayk F. Experimental hyperleptinemia acutely increases vasoconstrictory sympathetic nerve activity in healthy humans. J Clin Endocrinol Metab. 2013;98:E491–496. [DOI] [PubMed] [Google Scholar]
- 122.Kuo JJ, Jones OB, Hall JE. Inhibition of NO synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension. 2001;37:670–676. [DOI] [PubMed] [Google Scholar]
- 123.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31:409–414. [DOI] [PubMed] [Google Scholar]
- 124.Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension. 2005;46:326–332. [DOI] [PubMed] [Google Scholar]
- 125.Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. 2002;39:496–501. [DOI] [PubMed] [Google Scholar]
- 126.Faulkner JL, Belin de Chantemele EJ. Sex Differences in Mechanisms of Hypertension Associated With Obesity. Hypertension. 2018;71:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Faulkner JL, Belin de Chantemele EJ. Leptin and Aldosterone. Vitam Horm. 2019;109:265–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.do Carmo JM, da Silva AA, Gava FN, Moak SP, Dai X, Hall JE. Impact of leptin deficiency compared with neuronal-specific leptin receptor deletion on cardiometabolic regulation. Am J Physiol Regul Integr Comp Physiol. 2019;317:R552–R562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84:3686–3695. [DOI] [PubMed] [Google Scholar]
- 130.Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–1953. [DOI] [PubMed] [Google Scholar]
- 131.Aizawa-Abe M, Ogawa Y, Masuzaki H, et al. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105:1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.da Silva AA, Kuo JJ, Hall JE. Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension. 2004;43:1312–1317. [DOI] [PubMed] [Google Scholar]
- 133.Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31:506–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kuhnen P, Krude H, Biebermann H. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol Med. 2019;25:136–148. [DOI] [PubMed] [Google Scholar]
- 135.Baldini G, Phelan KD. The melanocortin pathway and control of appetite-progress and therapeutic implications. J Endocrinol. 2019;241:R1–R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dunbar JC, Lu H. Leptin-induced increase in sympathetic nervous and cardiovascular tone is mediated by proopiomelanocortin (POMC) products. Brain Res Bull. 1999;50:215–221. [DOI] [PubMed] [Google Scholar]
- 138.Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33:542–547. [DOI] [PubMed] [Google Scholar]
- 139.Kuo JJ, Silva AA, Hall JE. Hypothalamic melanocortin receptors and chronic regulation of arterial pressure and renal function. Hypertension. 2003;41:768–774. [DOI] [PubMed] [Google Scholar]
- 140.Kuo JJ, da Silva AA, Tallam LS, Hall JE. Role of adrenergic activity in pressor responses to chronic melanocortin receptor activation. Hypertension. 2004;43:370–375. [DOI] [PubMed] [Google Scholar]
- 141.Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol. 2011;23:186–193. [DOI] [PubMed] [Google Scholar]
- 142.Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52. [DOI] [PubMed] [Google Scholar]
- 143.Sayk F, Heutling D, Dodt C, et al. Sympathetic function in human carriers of melanocortin-4 receptor gene mutations. J Clin Endocrinol Metab. 2010;95:1998–2002. [DOI] [PubMed] [Google Scholar]