

Abstract
Given the intersection between diabetes mellitus and cardiovascular disease (CVD), pharmacologic agents used to treat type 2 diabetes mellitus must show cardiovascular safety. Comorbid conditions, including heart failure and chronic kidney disease, are increasingly prevalent in patients with diabetes; therefore, they also play a large role in drug safety. Although biguanides, sulfonylurea, glitazones, and dipeptidyl peptidase 4 inhibitors have variable effects on cardiovascular events, sodium glucose cotransporter 2 inhibitors and glucagon-like peptide 1 receptor agonists have consistently shown safety and reduction in cardiovascular events in patients with established CVD. These medications are becoming essential tools for cardioprotection for patients with diabetes and CVD. They may also have roles in primary prevention and renal protection. This paper will review the cardiovascular impact, adverse effects, and possible mechanisms of action of pharmacologic agents used to treat patients with type 2 diabetes.
Keywords: cardiovascular outcomes trials, diabetes drugs, type 2 diabetes
Type 2 diabetes mellitus (T2DM) is a well established risk factor for cardiovascular disease (CVD), and CVD is the leading cause of death in adults with T2DM. Compared with an individual without T2DM, the life expectancy of a 50- year-old with T2DM is on average 6 years shorter. The lifespan of an individual with T2DM and a prior myocardial infarction (MI) is shortened further still by 12 years. Sixty percent of the difference in survival is attributable to excess CVD mortality (1). As previously characterized (2), heart failure (HF) is also underrecognized among T2DM patients and increases mortality (3). In recent years, the scope of diabetes treatment has broadened to reversal of known pathophysiologic defects and not simply on improving dysglycemia. Glycemic control, a traditional mainstay of T2DM management, overall does not correlate with reduced burden of CVD or mortality, particularly in the near-term (4,5). Insulin resistance in liver and muscle and eventual β-cell failure constitute the core pathophysiologic defects in T2DM. Additional mechanisms of disease include hyperglucagonemia, incretin deficiency or resistance, and maladaptive increases in renal glucose reabsorption. Defects in the fat cells, such as increased lipolysis, and impaired hypothalamic appetite regulation have also been implicated (6). Because of the progressive and multifaceted pathophysiology of type 2 diabetes, pharmacologic agents with distinct yet complementary actions are needed. Obesity, hypoglycemia, and CVD risk are important considerations in the treatment of T2DM, and interventions aimed at reducing chronic micro and macrovascular complications and improving cardiorenal outcomes are of paramount importance (7).
The perceived cardiovascular risk with certain glucose-lowering agents and evidence that hemoglobin a1c (HbA1c) lowering per se did not significantly reduce cardiovascular risk or mortality led to the regulatory requirement for cardiovascular safety trials for new agents beginning in 2009. Before the EMPA-REG OUTCOME trial, antihyperglycemic agents were believed to prevent or delay the development of microvascular complications, but were not able to reduce major adverse cardiovascular events. Since December 2008, the United States Food and Drug Administration (FDA) regulatory guidance for industry mandated cardiovascular outcome trials (CVOTs) for cardiovascular safety of novel antidiabetic agents to ensure their cardiovascular safety. This statute is met by blinding central adjudication of CVD events and inclusion of high-risk subjects such as those with advanced age, advanced CVD, and kidney disease. Medications must also be studied for 2 years or approximately 15,000 patient-years. In this setting, studies that evaluate novel medications for T2DM are well-positioned to evaluate cardiovascular risk, resulting in a surge of data on managing patients with both T2DM and CVD. Since the FDA mandate, 21 CVOT studies are on track to be completed by 2020, first in predominantly high-risk T2DM patients with established CVD (secondary prevention) and later in broader populations with multiple CVD risk factors (primary prevention) (8). After the success of many of these trials, the FDA commissioned additional labels specifically evaluating CVD risk reduction for empagliflozin, canagliflozin, and liraglutide.
The advent of CVOTs has led to a paradigm shift in the clinical practice recommendations for the management of T2DM. Until 2008, the approval of novel antidiabetic agents was based on their glucoselowering potential (9). In 2012, guidelines proposed that HbA1c targets should be individualized according to patient’s risk profile, in the context of potential risks associated with hypoglycemia and other adverse drug effects, disease duration, life expectancy, comorbidities, vascular complications, patient attitude, and expected treatment efforts and resources (10). The strategy for the management of type 2 diabetes was updated in 2018 in response to the abundance of new cardiovascular outcome data from the CVOTs published since 2015, which showed safety, tolerability, and cardiovascular and renoprotection with 2 classes of agents, sodium glucose cotransporter 2 inhibitors (SGLT2i) and glucagon-like peptide receptor agonists (GLP1RA) in patients with established CVD (11). In response to these findings, in 2018 European Association for the Study of Diabetes (EASD) and the American Diabetes Association (ADA) consensus guidance provides a decision cycle for patient-centered management of T2DM, taking into account not only key patient characteristics (age, weight, CVD, and renal history), but also specific factors such as HbA1c lowering effect, hypoglycemic risk, effect on weight, side effects, complexity, costs, and cardiorenal effects. These guidelines integrate these data for recommendations on choice of treatment and a shared decision-making strategy to create a management plan. In this plan, the focus has shifted from a pure glucocentric approach towards a holistic approach, with a preferred use of agents with proven cardiorenal superiority (11).
To synthesize this wealth of new data, we will provide an updated overview of pharmacologic agents for cardiovascular care in T2DM from metformin, sulfonylureas, and glitazones to dipeptidyl peptidase 4 inhibitors (DPP4i), SGLT2i, and GLP1RA, discussing mechanism of action, metabolic and cardiorenal effects, and benefits and limitations of current design. The evidence for cardiovascular benefit of SGLT2i and GLP1RA has rightfully prompted the diabetes and cardiovascular communities to incorporate these new classes of agents into clinical management guidance.
BIGUANIDES
Metformin has remained first-line treatment for T2DM due to its efficacy, safety, duration of evidence, affordability, and limited side-effect profile. The biguanide was developed in the 1920s, before the era of target-specific drug development; therefore, exact cellular mechanisms of metformin remain ill-defined. Metformin has been used Europe since the 1950s whereas phenformin, another biguanide, was primarily used in the United States until metformin was approved in 1990 (12). Metformin lowers blood glucose by increasing peripheral uptake of glucose and decreasing hepatic glucose production, likely via inhibition of mitochondrial enzymes. Metformin’s role in inflammatory pathways may also underpin the non-metabolic benefits of the drug (13). In the last decade, our understanding of metformin’s mechanism has expanded from alterations in liver metabolism leading to improvements glycemic control, to a much more complex picture reflecting its multiple modes of action, including a key role in the gut (13).
Data on the cardiovascular impact of metformin rely heavily on the United Kingdom Prospective Diabetes Study (UKPDS). In 1970s, the study group assigned a total of 1,704 overweight adults with T2DM aged 25 to 65 years to 1 of 5 treatment groups: diet only, diet plus metformin, diet plus sulfonylurea, or diet plus insulin, then followed a number of metabolic, renal and cardiovascular markers for 10 years (14). Compared to diet alone, in the group of 342 newly diagnosed overweight patients with T2DM treated with metformin, MI was reduced by 39%, coronary deaths by 50%, stroke by 41%, and all-cause mortality by 36% after a median 10.7 years (14). These reductions in major CVD events with diet plus metformin were greater than diet with either a sulfonylurea or insulin. Additional follow-up for 8 to 10 years when all patients received intensive therapy found that the reduced risk of MI and mortality with initial metformin therapy persisted over time compared with early treatment using a sulfonylurea or insulin (15). Metformin use was also associated with fewer hypoglycemic episodes and less weight gain.
However, any conclusions drawn from the UKPDS data is tempered by major limitations in study design. For example, the study population was low risk and excluded recent acute coronary syndrome, HF, or microvascular disease events and was performed in the absence of contemporary lipid-lowering therapy with statins. Moreover, compared to recent CVOTs, the UKPDS study population was small, incompletely blinded, and lacked placebo-control. Additional data on the cardiovascular benefits of metformin relative to placebo remain sparse, limited to meta-analyses with wide confidence intervals (CIs) for most cardiovascular endpoints (16). The VA-IMPACT trial is attempting fill this gap by evaluating cardiovascular outcomes in patients with pre-diabetes and established CVD treated with metformin versus placebo (NCT02915198).
Patients with chronic kidney disease (CKD) are also underrepresented in the evidence base of metformin use for CVD risk reduction in T2MD. Per current FDA guidelines, metformin is contraindicated at an estimated glomerular filtration rate (eGFR) less than 30 ml/min/1.73 kg/m2, and initiation is not recommended at an eGFR between 30 and 45 ml/min/1.73 kg/m2 (17). For patients tolerating the drug who experience a decreases in glomerular filtration rate (GFR), new guidelines state reduced renal dosing is a safe option (17,18). This was supported in a post hoc analysis of SAVOR TIMI 53 participants showing that exposure to metformin did not significantly affect cardiovascular outcomes in patients with to severe CKD (19). Metformin’s major adverse effect is a type B lactic acidosis that may develop at the upper therapeutic limit of drug dosing, which current evidence indicates is rare in contemporary practice (20). Withholding metformin during “sick days” may mitigate this risk, but trial evidence to support this approach is lacking. Metformin was background medical therapy for most patients in recent CVOTs, further enshrining its use as first-line therapy for most patients with T2DM. Given the duration of evidence, low cost, favorable safety profile, and background use in recent CVOTs, metformin has, until now, remained first-line therapy onto which additional agents can be considered for cardiovascular risk reduction in T2DM. However, new European Society of Cardiology (ESC)/EASD guidelines recommend initiating SGLT2i or GLP1RA monotherapy in drug-naive patients with T2DM with established or high risk for CVD (21). This recommendation is made despite high prevalence (51% to 83%) of baseline metformin use in these trials (Table 1).
TABLE 1.
SGLT2i Cardiovascular Outcome Trials
| EMPA-REG Empagliflozin (n = 7,020) | CANVAS Program Canagliflozin (n = 10,142) | DECLARE Dapagliflozin (n = 17,160) | CREDENCE Canagliflozin (n = 4,401) | DAPA-HF Dapagliflozin (n = 4744) | |
|---|---|---|---|---|---|
| Median follow-up, yrs | 3.1 | 2.4 | 4.2 | 2.6 | 1.5 |
| Mean age, yrs | 63 | 63 | 64 | 63 | 66 |
| Female, % | 29 | 36 | 37 | 34 | 23 |
| BMI, kg/m2 | 30.6 | 32.0 | 32.1 | 31.3 | 28.2 |
| HbA1c, % | 8.1 | 8.3 | 8.3 | 8.3 | NR |
| Baseline metformin, %* | 73 | 77 | 82 | 66 | 73 |
| Baseline eGFR† | 74 | 77 | 85 | 56 | 65 |
| eGFR† <60 ml/min, % | 26 | 20 | 7 | 59 | 40 |
| Prior CVD, % | 99 | 66 | 41 | 50 | |
| Prior HF, % | 10 | 14 | 10 | 15 | |
| 3P-MACE | 0.86 (0.74–0.99) | 0.86 (0.67–0.91) | 0.93 (0.84–1.03) | 0.80 (0.67–0.95) | 0.74 (0.65–0.85) |
| CV death | 0.62 (0.49–0.77) | 0.87 (0.72–1.06) | 0.98 (0.82–1.17) | 0.78 (0.61–1.00) | 0.75 (0.65–0.85) |
| Nonfatal MI | 0.87 (0.70–1.09) | 0.89 (0.73–1.09) | 0.89 (0.77–1.01) | NR | 0.82 (0.69–0.98) |
| Nonfatal stroke | 1.18 (0.89–1.56) | 0.87 (0.69–1.09) | 1.01 (0.84–1.21) | NR | 0.83 (0.71–0.97) |
| CV death or HHF | 0.66 (0.55–0.79) | 0.78 (0.67–0.91) | 0.83 (0.73–0.95) | 0.69 (0.57–0.83) | |
| All-cause mortality | 0.68 (0.57–0.82) | 0.87 (0.74–1.01) | 0.93 (0.82–1.04) | 0.83 (0.68–1.02) | |
| HHF | 0.65 (0.50–0.85) | 0.67 (0.52–0.87) | 0.73 (0.61–0.88) | 0.61 (0.47–0.80) | 0.70 (0.59–0.83) |
| Renal events‡ | 0.61 (0.53–0.70) | 0.60 (0.47–0.77) | 0.53 (0.43–0.66) | 0.70 (0.59–0.82) | 0.83 (0.44–1.16) |
Values are hazard ratio (confidence interval) unless otherwise indicated.
Average of entire study group (treatment and control).
eGFR units ml/min/1.73 m2.
Definition varied across trials.
CANVAS = ■■■; CREDENCE = ■■■; DAPA-HF = ■■■; DECLARE = ■■■; EMPA-REG = ■■■; SGLT21 = sodium glucose cotransporter 2 inhibitor; other abbreviations as in Table 1.
SULFONYLUREAS
Sulfonylureas have historically been considered second-line treatment for T2DM for patients with uncontrolled hyperglycemia on metformin. In contrast to metformin, sulfonylureas increase blood insulin concentration via stimulation of pancreatic beta cells. Augmented insulin secretion and sensitivity can increase risk of hypoglycemia and lead to weight gain (Figure 1) (22). Although sulfonylureas are associated with slightly greater upfront reductions in glycosylated hemoglobin levels (HbA1c, 1% to 1.25% reduction) relative to metformin (0.5% to 1.25%) (23) in the UKPDS study, after 6 months, reduction in a1c levels were similar between groups on either therapy. Over 6 years, 54% of patients allocated sulfonylureas alone required the addition of insulin to achieve the prespecified target of a fasting glucose under 106 mg/dl (24).
FIGURE 1. Physiologic Mechanisms of Novel Pharmacologic Agents for Diabetes.
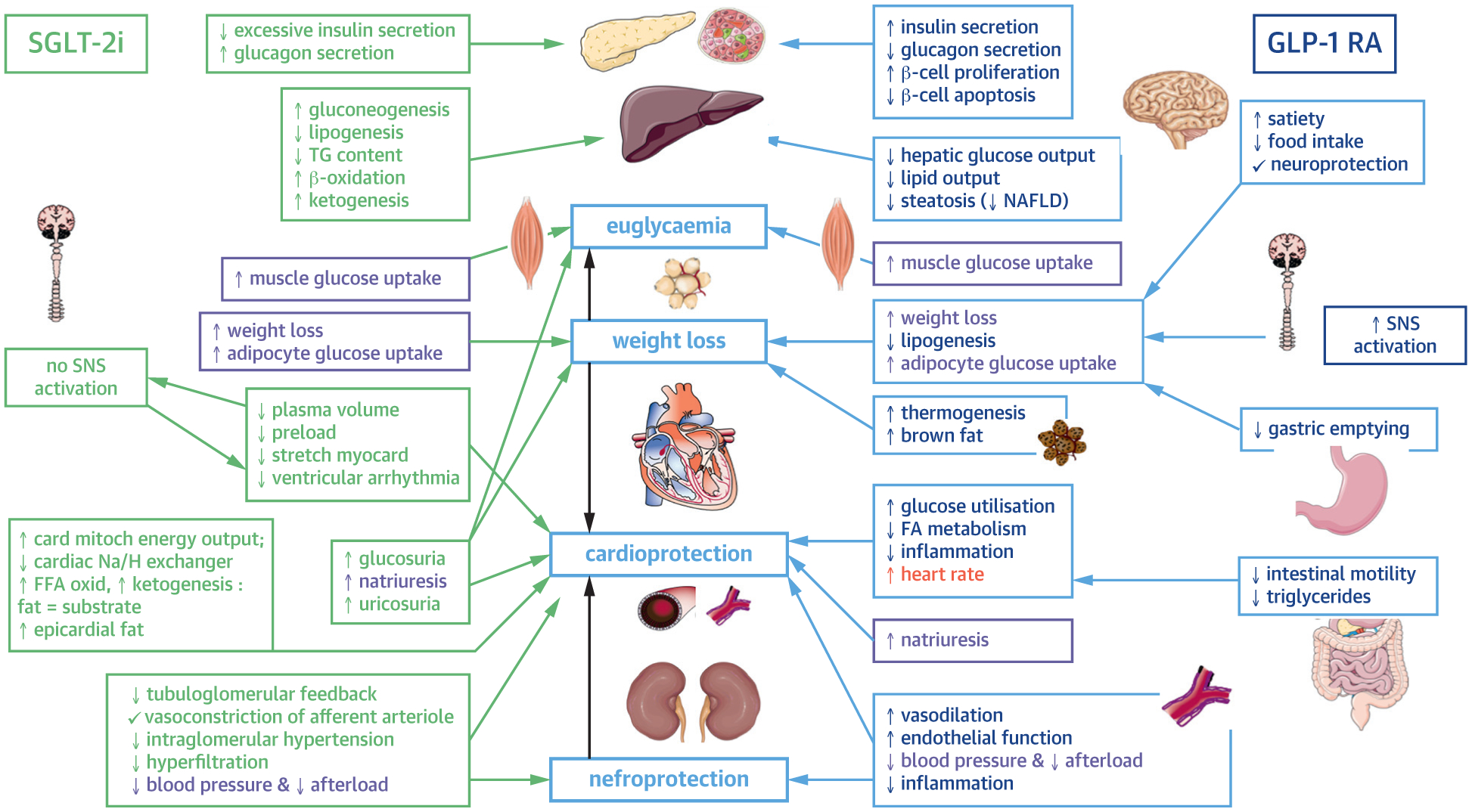
Both SGLT2i and GLPRA have effects on multiple organs in the body, modulating not only glycemic control but lipogenesis, satiety, smooth muscle tone, and renal filtration. From top to bottom, the pancreas, pancreatic islet cells, liver, skeletal muscle adipocytes, heart, vasculature, kidneys, central nervous system, and gastrointestinal track are affected by these molecules. Up and down arrows denote whether a process is increased or decreased by each medication. Directional arrows denote the proposed effects of modulations in these processes. GLPRA = glucagon-like peptide 1 receptor agonist; FA = ■■■; FFA = ■■■; NAFLD = ■■■; Na/H = ■■■; SGLT2i = sodium glucose cotransporter 2 inhibitor; SNS = ■■■; TG = ■■■.
The UKPDS and ADVANCE trials have shown microvascular benefits of sulfonylureas, including a reduction in the incidence or worsening of nephropathy and retinopathy, and no increase in all-cause mortality. However, whether these benefits were due to sulfonylurea therapy versus an overall glucose-lowering effect could not be confirmed (4).
Since the 1960s, sulfonylureas have been implicated with increased risk of adverse cardiovascular outcomes. The University Group Diabetes Program observed increased risk of all cause and cardiovascular mortality in those treated with the first generation sulfonylurea tolbutamide versus placebo (25). The UKPDS study randomized patients to either first (chlorpropamide) or second (glibenclamide) generation sulfonylureas, but did not report differences in outcomes between the two. Subsequent data have been inconclusive, with observational studies corroborating these results while underpowered randomized control trials have not (26). A recent metaanalysis identified selected observational studies comparing only second and third-generation sulfonylureas to metformin, most of which evaluated the outcome of death. After excluding studies for misclassification or other biases, the relative risk of cardiovascular events with sulfonylureas was consistently elevated (relative risk [RR] range: 1.16 to 1.55) compared to metformin, with the exception of one trial comparing sulfonylurea plus metformin to metformin alone, which found no difference in all cause mortality (26). The recently published the CAROLINA trial comparing second-generation sulfonylurea glimepiride to linagliptin (a DPP4i) found no differences for incidence of nonfatal MI, nonfatal stroke, and CVD during a median of 6 years (27). As discussed below, a large body of evidence has supported the cardiovascular safety of the entire class of DPP4i. Taken in this context, the findings of non inferiority of glimepiride in CAROLINA provide reassuring evidence for cardiovascular safety. Overall, the sulfonylurea data indicate that second and third generation agents, such as glimepiride, likely have a reassuring cardiovascular safety profile similar to that of newer glucose-lowering therapies such as linagliptin. Trials have not shown the cardiovascular benefit of sulfonylureas. Given their adverse side effect profile, they should be reserved for a select group of patients in whom medications with cardiovascular benefit, discussed below, are not an option.
Patients older than 75 years are poorly represented in sulfonylurea trials, with a mean age of 57 years in a large meta-analysis of 37,650 patients using sulfonylureas (26). However, because of the known risk of hypoglycemia, most expert consensus opinions, including those issued by the ADA and the Choosing Wisely campaign, recommend against sulfonylurea use in older, frail patients (28,29). Similarly, in patients with advanced CKD stage 3 or higher, first generation sulfonylureas should be avoided due to risk of prolonged hypoglycemia from active metabolites (28).
GLITAZONES
Thiazolidinediones (glitazones) (Figure 1) are nuclear peroxisome proliferator–activated receptor agonists that increase insulin sensitivity in muscle, adipose tissue, and liver (30). Similar to metformin, glitazones may have anti-inflammatory properties and have shown salutary vascular effects in preclinical trials (31). In clinical trials, however, preclinical cardiovascular benefits were not sustained, and increased hospitalizations for HF and fractures were observed. Published in 2005, the PROACTIVE trial prospectively enrolled 5,328 patients with T2DM and established CVD to treatment with pioglitazone versus placebo (Figure 4). Four years later, the RECORD was published, which trial enrolled 4,447 patients with uncontrolled T2DM on maximum tolerated monotherapy for at least 2 months and assigned them to rosiglitazone versus sulfonylurea or metformin. In PROACTIVE, there was no difference in the composite primary outcome of death, non-fatal MI, non-fatal stroke, acute coronary syndrome, endovascular or surgical intervention in the coronary or lower extremity arteries, or amputation above the ankle after a median follow-up of just under 3 years (hazard ratio [HR]: 0.90; 95%; CI: 0.8 to 1.02) (32), with a signal of benefit in the pre-specified secondary composite of death, non-fatal MI and non-fatal stroke (HR: 0.84; 95% CI: 0.72 to 0.98). RECORD had a significantly longer median follow-up of 5.5 years, and similar to PROACTIVE found no difference in the primary outcome of cardiovascular hospitalization or CVD (HR: 0.99; 95% CI: 0.85 to 1.16; p = 0.93) (33). There was also no between-group difference in the secondary outcomes of cardiovascular mortality with or without MI and stroke (mortality HR: 0.84, 95% CI: 0.59 to 1.18; mortality stroke or MI HR: 0.93, 95% CI: 0.74 to 1.15).
FIGURE 4. Timeline of Pharmacologic Agents Used in Type 2 Diabetes.
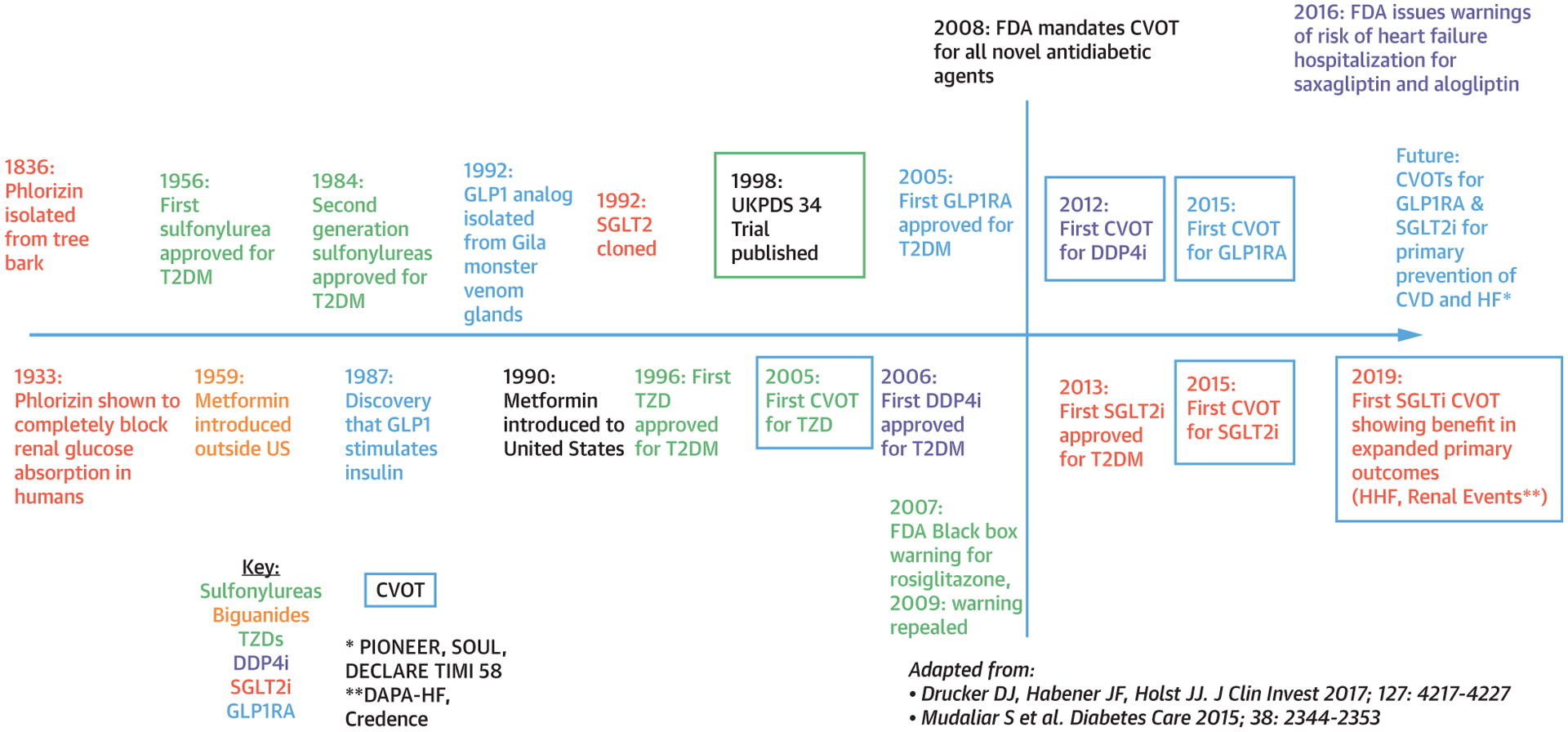
Timeline delineating the dates of discovery, U.S. Food and Drug Administration (FDA) approval and, if applicable, cardiovascular outcome trial publication for biguanides, sulfonylureas, TZDs, DPP4is, GLP1RAs, and SGLTis. Adapted with permission from Mudaliar (72) and Drucker et al. (103). HHF =; T2DM = type 2 diabetes mellitus; UKPDS = United Kingdom Prospective Diabetes Study; other abbreviations as in Figures 1 and 2.
Both trials were concerning for an increase in HF events for patients in the glitazones treatment arms (PROACTIVE 11% vs. 8%, p < 0.0001; RECORD 3.7% vs. 1.9%, p = 0.0003, respectively). Although patients enrolled in both trials were at high cardiovascular risk, patients with symptomatic HF or those receiving medications for HF were excluded, making these findings more concerning. Moreover, treatment with pioglitazone in PROACTIVE trended toward increased risk of lower-extremity revascularization (80 vs. 65 HR: 1.25; 95% CI: 0.9 to 1.73), and weight gain (þ3.6 kg vs. 0.4 kg, p < 0.0001). In response to these and other data, including a meta-analysis of 42 trials suggesting that rosiglitazone was associated with increased risk of MI (34), the FDA briefly issued a black box warning for cardiovascular risk with rosiglitazone in 2007. However, following the results of the RECORD trial published in 2009, this warning was reconsidered and removed (31). Despite this, rosiglitazone is unavailable in the United States and Europe.
Glitazones are potent insulin sensitizers, and there is some evidence that their use is associated with improved cardiometabolic outcomes in patients with insulin resistance, but without established diabetes. The IRIS trial assigned 3,895 non-diabetic participants with insulin resistance and a recent stroke to pioglitazone versus placebo, and found lower rates of recurrent stroke (HR: 0.7; 95% CI: 0.62 to 0.93) and progression of insulin resistance to diagnosed diabetes (HR: 0.48; 95% CI: 0.33 to 0.69) at a median follow-up of nearly 5 years. Adverse events associated with pioglitazone in IRIS included significantly greater weight gain (þ2.6 kg vs. 0.5 kg, p = 0.001) and bone fractures requiring surgery or hospitalization in the pioglitazone group (5% vs. 3%, p = 0.003) (33).
PROACTIVE and IRIS were both secondary prevention trials, and participants in the glitazone CVOTs were predominantly obese with a median HbA1c of approximately 8%. Most participants in these trials were treated with appropriate cardiovascular medications, including beta blockers, angiotensin-converting enzyme (ACE) inhibitors, antiplatelet medications, and statins. Similar to prior trials, the mean age was between 50 and 60 years, so it is challenging to extrapolate these findings to older T2DM patients.
Glitazones are tolerated in patients with kidney diseases; however, studies are equivocal regarding whether they provide benefit in this population. In a PROACTIVE post-hoc analysis of patients with at least stage II CKD, pioglitazone was associated with a mortality benefit (35). However, a retrospective analysis of dialysis patients on rosiglitazone had higher odds of all-cause and cardiovascular mortality compared to patients treated with other oral hypoglycemic agents. Secondary analyses of clinical trials and observational analyses also support reduction in albuminuria associated with glitazone use. However, there is an absence of randomized trial data to evaluate glitazone use in patients with CKD. Overall, in a manner similar to sulfonylureas, the glitazones have not shown a clear cardiovascular or renal benefit, and carry a less-than-ideal side-effect profile than either metformin or the sulfonylureas.
DIPEPTIDYL PEPTIDASE 4 INHIBITORS
DPP4i (Figure 1) modulate the incretin pathway, a hormonal cascade initiated by the gastrointestinal (GI) tract post-prandially. The incretin pathway culminates in the release of glucagon-like peptide 1 (GLP1) which potentiates glucose-dependent insulin release and glucagon suppression. The DPP4 enzyme deactivates GLP1; thus DPP4 inhibition extends the function of endogenous GLP1 (36). Beyond the glucose-lowering effects, DPP4i have neutral to beneficial effects on weight, blood pressure, postprandial lipid status, inflammation, oxidative stress, and endothelial function (37). Unlike sulfonylureas and metformin, DPP4i metabolism is via the portalhepatic circulation and is not affected by impaired renal function. Therefore, the DPP4i appear safe in patients with advanced kidney disease, including those on dialysis (38).
TRIAL DESIGN AND MAJOR OUTCOMES.
Five major clinical trials have examined the cardiovascular outcomes associated with DPP4i: SAVOR-TIMI 53 (sax-agliptin), EXAMINE (alogliptin), TECOS (sitagliptin), CARMELINA (linagliptin), and CAROLINA (linagliptin) (39–45). These trials compared cardiovascular outcomes in large groups of patients (5,000 to 16,000) treated with DPP4i versus placebo over approximately 1.5 to 6 years. In CAROLINA, a sulfonylurea, glimepiride, was chosen as an active comparator. All DPP4i trials enrolled high-risk T2DM patients with either established CVD or multiple cardiovascular risk factors and a baseline T2DM defined as HbA1c greater than 6.5% with either established CVD or multiple cardiovascular risk factors. The DPP4i trials included on average older patients than prior trials, in TECOS, SAVOR, CARMELINA, and CAROLINA the average age was approximately 65 years, and in EXAMINE it was 61 years of age. The primary outcome for all 5 trials was cardiovascular death, nonfatal MI, and nonfatal stroke with the exception of TECOS, which added unstable angina requiring hospitalization. Across all DPP4i trials, there was no difference in cardiovascular outcomes between DPP4i and placebo (Table 2). However, secondary analyses revealed higher rate of HF hospitalizations in participants treated with sax-agliptin in the SAVOR-TIMI 53 (HR: 1.27; 95% CI: 1.07 to 1.15; p = 0.007), with a trend towards increased HF with alogliptin in EXAMINE (HR: 1.19; 95% CI: 0.89 to 1.59). For this reason, the FDA added warnings about HF risk in medicines containing saxagliptin and alogliptin in 2016 (46) (Figure 4). However, this HF signal was not observed in TECOS, CARMELINA, or CAROLINA, suggesting that sitagliptin and linagliptin may have a neutral side effect profile on HF hospitalization risk.
TABLE 2.
DPP4i Cardiovascular Outcome Trials
| SAVOR TIMI 53 Saxaglipitin (n = 16,492) | EXAMINE Alogliptin (n = 5,380) | TECOS Sitagliptin (n = 14,523) | CARMELINA Linagliptin (n = 6,979) | CAROLINA Linagliptin vs. Glimepiride (n = 6,033) | |
|---|---|---|---|---|---|
| Median follow-up, yrs | 2.1 | 1.5 | 3.0 | 2.2 | 5.9 |
| Mean age, yrs | 65 | 61 | 65 | 66 | 64 |
| Female, % | 33 | 32 | 29 | 37 | 40 |
| BMI, kg/m2 | 31.2 | 28.7 | 30.2 | 31.4 | 30.1 |
| HbA1c, % | 8.0 | 8.0 | 7.2 | 8.0 | 7.2 |
| Baseline metformin, % | 69 | NA | 81 | 54 | 83 |
| Baseline eGFR* | 73 | 71 | 75 | 55 | 77 |
| eGFR* <60 ml/min, % | < 50 ml/min: 16 | 29 | NR | 62 | 19 |
| Prior CVD, % | 78 | 100 | 100 | 57 | 42 |
| Prior HF, % | 13 | 28 | 18 | 27 | 4 |
| 3P-MACE | 1.00 (0.89–1.12) | 0.96 (≤1.16) | 0.98 (0.89–1.08) | 1.02 (0.89–1.17) | 0.98 (0.84–1.13) |
| CV death | 1.03 (0.87–1.22) | 0.85 (0.66–1.10) | 1.03 (0.89–1.19) | 0.96 (0.81–1.14) | 1.00 (0.81–1.24) |
| Non-fatal MI | 0.95 (0.80–1.12) | 1.08 (0.88–1.33) | 0.95 (0.81–1.11) | 1.12 (0.90–1.40) | 1.01 (0.80–1.28) |
| Non-fatal stroke | 1.11 (0.88–1.39) | 0.95 (≤ 1.14) | 0.97 (0.79–1.19) | 0.91 (0.67–1.23) | 0.86 (0.66–1.12) |
| CV death or HHF | NR | 1.00 (0.82–1.20) | 1.02 (0.90–1.15) | NR | 1.00 (0.84–1.20) |
| All-cause mortality | 1.11 (0.96–1.27) | 0.88 (0.71–1.09) | 1.01 (0.90–1.14) | 0.98 (0.84–1.13) | 0.91 (0.78–1.06) |
| HHF | 1.27 (1.07–1.51) | 1.07 (0.79–1.46) | 1.00 (0.83–1.20) | 0.90 (0.74–1.08) | 1.21 (0.92–1.59) |
| Renal events† | 1.08 (0.88–1.3) | NR | NR | 1.04 (0.89–1.22) | NR |
Values are hazard ratio (confidence interval) unless otherwise indicated.
eGFR units ml/min/1.73 m2.
Definition varied across trials.
3P-MACE = 3-point major adverse cardiac events; BMI = body mass index; CARMALINA = ■■■; CAROLINA = ■■■; CV = cardiovascular; CVD = cardiovascular disease; DPP4i = dipeptidyl peptidase 4 inhibitor; eGFR = estimated glomerular filtration rate; EXAMINE = ■■■; Hb1Ac = glycolated hemoglobin; HF = heart failure; HHF = ■■■; NR = not reached; SAVOR TIMI 53 = ■■■; TECOS = ■■■.
The CAROLINA trial is the first active comparator CVOT in the DPP4i class and compared linagliptin to glimepiride on cardiovascular safety and non-inferiority with subsequent hierarchical testing for superiority of among adults with T2DM and estab-lished CVD (34.5%) or increased cardiovascular risk factors alone, with a median follow-up of more than 6 years — the longest follow-up of any DPP4i studied to date. The primary endpoint of non-inferiority in time to first occurrence of cardiovascular death, nonfatal MI or nonfatal stroke (the 3-point major adverse car-diac event [3P-MACE]) was met. Linagliptin was also similar to glimepiride on the secondary endpoint of 3P-MACE plus hospitalization for unstable angina. The additional evidence from CAROLINA seems to confirm that although DPP4i are safe for most pa-tients, they are not efficacious in reducing cardio-vascular outcomes.
In addition to cardiovascular safety, there is some evidence for renoprotection for diabetic nephropathy with some DPP4i agents. For example, in SAVOR-TIMI 53, saxagliptin use was associated with slowing in the rate of progression of albuminuria in patients with and without CKD. This benefit was observed regard-less of improvements in HbA1c, suggesting that some of the DPP4i may have renoprotection regardless of glycemic control (41). To date, however, no large DPP4i CVOT has shown independent renoprotective effects in T2MD patients with CKD. Most DPP4i trials were of limited duration (2- to 4-year median follow up, except for CAROLINA) to detect renal or benefit for reductions in CVD from this therapeutic class (27). Because of advanced CVD, prolonged diabetes duration, and relatively short exposure to study drug, the DPP4i trials may have had limited ability to detect renoprotective benefit with these agents.
GLUCAGON-LIKE PEPTIDE 1 RECEPTOR AGONISTS
In the 1930s, intestinal factors called incretins were suggested to be secreted in response to a meal, enhancing glucose lowering (47). The incretin effect describes the observation that glucose oral ingestion elicits a much greater insulin response than an intravenous glucose infusion (48). The incretin effect causes greater than 50% of meal-related insulin secretion in healthy individuals (47). Two gut hormones, GLP1 and gastric inhibitory polypeptide, are responsible for the incretin effect. GLP1 not only potentiates/enhances insulin secretion but also inhibits glucagon secretion, thereby limiting the excessive hepatic glucose output, and reducing appetite leading to weight loss (Figure 1). Although there is a deficiency of GLP1 in T2DM (48,49), its biological potency is largely retained, making GLP1 an attractive target molecule for therapeutics. However, native GLP1 is rapidly inactivated by the ubiquitous enzyme DPP4. Therefore, agents that enhance or mimic the actions of GLP1 are of great clinical interest. To extent the half-life of GLP1, DPP4-resistant GLP1 analogues were in the 1980s and 1990s which were approved for medical use in 2005 (Figure 4) (12). Early trials have proven the efficacy of GLP1 receptor agonists to improve HbA1c without increasing risk of hypoglycemia; lower body weight also showed promising cardiovascular effects.
GLP1 receptors are present in the brain, pancreas, and stomach; thus, agonists prolong the typically rapid effects on centrally mediated satiety and nausea, post-prandial insulin release, gastric motility, and chylomicron formation. Mechanisms of weight loss are multifactorial and independent of diabetes status, as seen in the SCALE Obesity and Prediabetes Study Group trial of GLP1RA in obese patients without diabetes, who lost 5.6 kg more weight than placebo (50). Hypothalamic and GI mechanisms including satiety, appetite suppression and delayed gastric emptying likely contribute additional benefits for weight loss. In addition, GLP1RAs have beneficial effects on inflammation and on blood pressure (via release of atrial natriuretic peptide by cardiomyocytes, improved endothelial function, vasodilatory effects, and natriuresis); thereby reducing afterload (36). They also alter renal natriuresis and diuresis via effects on proximal tubule cells, providing a postulated mechanism for the emerging evidence of renoprotection with this class of drugs (51) (Figure 1).
Previously, while differing in structure and duration of effect, all GLP1RAs approved for T2DM are administered through subcutaneous injection. However, after the results of PIONEER 6, the FDA approved oral semaglutide. Exenatide and lixisenatide are derived from exogenous Gila monster venom and have an exendin-4 backbone, whereas the remaining 4 commercially available GLP1RAs are modifications of endogenous GLP1 (52). In July 2018, the manufacturer removed albiglutide from the market for financial reasons.
TRIAL DESIGN AND MAJOR OUTCOMES.
Six major randomized control trials, ELIXA, LEADER, SUSTAIN-6, EXSCEL, HARMONY, and REWIND have investigated cardiovascular outcomes in patients treated with injectable GLP1 agonists (53–58). They were all large trials of 3,000 to 14,000 patients with similar baseline patient demographics: 30% to 45% were female, average age was between 54 and 66 years, baseline HbA1c was 7.7% to 8.8%, with the exception of the REWIND trial which enrolled a lower-risk population with better controlled T2DM (HbA1c, 7.3%) and a greater proportion of females (46%). Recently, a seventh trial, PIONEER 6 showed noninferiority of oral semaglutide compared with placebo for 3P-MACE among 3,183 T2DM patients, 85% of whom had established CVD, followed for a median of approximately 1.3 years (59). All 7 trials enrolled patients either determined to be at high risk or have established CVD; however, degree of disease varied by trial. In ELIXA and HARMONY all patients had established CVD, whereas ELIXA was the only trial to evaluate patients who had had an acute coronary event within 180 days. EXSCEL and REWIND had a larger percentage of patients without established CVD, (EXSCEL 30%, REWIND 68.5%) lending evidence for extrapolation of use of this class of agents to the primary prevention of CVD in T2DM.
All trials were designed to evaluate the primary outcome of MACE defined as cardiovascular death, MI or stroke, with the exception of ELIXA, which included hospitalization for unstable angina as an additional component of their primary outcome. Based on 2008 FDA guidance, early GLP1RA trials were designed to show non-inferiority for cardiovascular safety compared with placebo; 2 of these non-inferiority trials were also powered for and showed superiority, including LEADER (liraglutide) and HARMONY-OUTCOMES (albiglutide weekly). SUSTAIN-6 showed a 26% reduction in 3P-MACE in a secondary prevention population, prompting the FDA to expand indications for injectable semaglutide to include reducing risk of cardiovascular events in patients with T2DM and established CVD (60). Currently, the FDA has approved oral semaglutide for the indication of glycemic control; the results of PIONEER and the ongoing SOUL trial are building an evidence base for use of semaglutide for CVD prevention (61). REWIND was the first trial of a weekly GLP1RA studied in a majority primary prevention population; it showed established superiority compared with placebo for reduction in 3P-MACE (Table 3). The results of REWIND support consideration of GLP1RA for primary prevention. To date, liraglutide, semaglutide, albiglutide, and dulaglutide (but not exenatide or lixisenatide) have shown superiority compared to placebo for reduction in a 3P-MACE endpoint (62). However, to date, only liraglutide has shown reductions in both cardiovascular and all-cause mortality (54). When all 56,004 participants from all 7 trials were analyzed together, primary and secondary endpoints of death from cardiovascular causes (fatal or nonfatal stroke, fatal or nonfatal MI) remained significant, albeit modest. This meta-analysis found that GLP1RA reduced all-cause mortality by 12%, and adverse renal outcomes by 17% (63). They also report reduction in hospital admissions for HF that was not present in any of the individual trials, and thus should be interpreted with caution given the limitations in statistical design of meta-analyses. Evidence for the atherosclerotic benefits of GLP1RA remains consistently strong, while benefits in HF are not widely accepted.
TABLE 3.
GLP1RA Cardiovascular Outcome Trials
| ELIXA Lixisenatide (n = 6,068) | LEADER Liraglutide (n = 9,340) | SUSTAIN-6 Semaglutide (n = 3,297) | EXSCEL Exenatide Every Week (n = 14,752) | HARMONY Albiglutide Every Week (n = 9,463) | REWIND Dulaglutide Every Week (n = 9,901) | PIONEER Semaglutide Oral (n = 3,182) | |
|---|---|---|---|---|---|---|---|
| Median follow-up, yrs | 2.1 | 3.8 | 2.1 | 3.2 | 1.6 | 5.4 | 1.3 |
| Mean age, yrs | 60 | 64 | 54 | 62 | 64 | 66 | 66 |
| Female, % | 30 | 36 | 39 | 38 | 31 | 46 | 32 |
| BMI, kg/m2 | 30.2 | NR | NR | NR | 32.3 | 32.3 | 32.3 |
| HbA1c, % | 7.7 | 8.7 | 8.7 | 8.1 | 8.8 | 7.3 | 8.2 |
| Baseline metformin, %* | 76 | 73 | 76 | 74 | 81 | 57 | 51 |
| Baseline eGFR† | 76 | 75 | 75 | 76 | 79 | 75 | 74 |
| eGFR† <60, % | 23 | 23 | 28.5 | 18 | 23 | 22 | 27 |
| Prior CVD, % | 100 | 81 | 83 | 73 | 100 | 32 | 85 |
| Prior HF, % | 22 | 18 | 24 | 16 | 20 | 9 | NR |
| 3P-MACE | 1.02 (0.89–1.17) | 0.87 (0.78–0.97) | 0.74 (0.58–0.95) | 0.91 (0.83–1.00) | 0.78 (0.68–0.90) | 0.88 (0.79–0.99) | 0.79 (0.57–1.11) |
| CV death | 0.98 (0.78–1.22) | 0.78 (0.66–0.93) | 0.98 (0.65–1.48) | 0.88 (0.76–1.02) | 0.93 (0.73–1.19) | 0.91 (0.78–1.06) | 0.49 (0.27–0.92) |
| Nonfatal MI | 1.03 (0.87–1.22) | 0.86 (0.73–1.00) | 0.74 (0.51–1.08) | 0.97 (0.85–1.10) | 0.75 (0.61–0.90) | 0.96 (0.79–1.16) | 1.18 (0.73–1.90) |
| Nonfatal stroke | 1.12 (0.79–1.58) | 0.86 (0.71–1.06) | 0.61 (0.38–0.99) | 0.85 (0.70–1.03) | 0.86 (0.66–1.14) | 0.76 (0.61–0.95) | 0.74 (0.35–1.57) |
| All-cause mortality | 0.94 (0.78–1.13) | 0.85 (0.74–0.97) | 1.05 (0.74–1.50) | 0.86 (0.77–0.97) | 0.95 (0.79–1.16) | 0.90 (0.80–1.01) | 0.51 (0.31–0.84) |
| HHF | 0.96 (0.75–1.23) | 0.87 (0.73–1.05) | 1.11 (0.77–1.61) | 0.94 (0.78–1.13) | NR | 0.93 (0.77–1.12) | 0.86 (0.48–1.55) |
| Renal events‡ | 0.81 (0.66–0.99) | 0.78 (0.67–0.92) | 0.64 (0.46–0.88) | 0.85 (0.73–0.98) | NR | 0.85 (0.77–0.93) | NR |
| Weight loss§ | 0.7 (0.9–0.5) | 2.3 (2.5–2.0) | 2.9 (2.3–3.5)/3.6 (3.8–4.9)* | 1.3 (1.1–1.4) | 0.83 (0.6–1.1) at 16 months | 1.5 (1.3–1.7) | 2.9/4.3¶ |
Values are hazard ratio (confidence interval) unless otherwise indicated.
Average of entire study group (treatment and control).
eGFR units ml/min/1.73 m2.
Definition varied across trials.
Kilogram difference from placebo, 95% confidence interval.
0.5 mg dose/1 mg dose.
ELIXA = ■■■; GL1PRA = glucagon-like peptide 1 receptor agonist; HARMONY = ■■■; LEADER = ■■■; PIONEER = ■■■; REWIND = ■■■; SUSTAIN 6 = ■■■; other abbreviations as in Table 1.
ADDITIONAL OUTCOMES.
The cardiovascular benefits shown in these trials are likely independent of glycemic control; all trials showed modest reduction in glycosylated hemoglobin levels in the treatment groups (0.2% to 1%) varying by dose and follow-up period. Although the mechanism of the cardiovascular benefit of the GLP1RA is unknown, it may be related to the positive effects on weight loss, blood pressure, and renal outcomes. Weight was modestly but universally lower in the treatment arm of all of the aforementioned trials. Participants treated with high-dose (1-mg) semaglutide lost the greatest amount of weight; SUSTAIN and PIONEER report 3.6 and 4.3-kg weight loss with injectable and oral formulations, respectively. The remainder of trials report weight loss of 1 to 2 kg relative to placebo (Table 1). Blood pressure reduction was also modest (1 to 2 mm Hg vs. placebo) but universal across these trials. REWIND, which was performed over the longest follow-up period of 5 years, supports the long-term durability of weight loss and blood pressure reductions with weekly injectable dulaglutide (36).
Several trials suggested a reduction in adverse renal outcomes in prespecified secondary analysis. SUSTAIN-6 (semaglutide), LEADER (liraglutide), and REWIND (dulaglutide) all found significantly lower odds of new or worsening nephropathy with GLP1RA compared to placebo (54,55,58) (Table 3). In the AWARD-7 trial (which was not a CVOT) performed in T2DM subjects with moderate-to-severe CKD, onceweekly dulaglutide produced glycemic control similar to that achieved with insulin glargine, but showed evidence of reduced decline in eGFR (64).
Other microvascular outcomes did not improve in tandem with the observed renoprotective effects of GLP1RA use, and retinopathy was observed more frequently with GLP1RA use compared to placebo in some of the CVOT trials to date (see below).
SAFETY AND ADVERSE OUTCOMES.
Adverse GI outcomes are the most consistent adverse effect of GLP1RA use. In REWIND, there was no difference in serious GI events, severe hypoglycemia, cancer, or pancreatitis; however, more patients treated with dulaglutide in REWIND had a reportable, nonsevere GI event (47.4% vs. 34.1%, p < 0.0001). Similarly, of the numerous adverse effects tracked in LEADER, only GI events were significantly higher in those treated with liraglutide (acute gallstone disease, nausea, vomiting, and diarrhea).
Retinopathy is another important potential side effect associated with GLP1RA (62). In SUSTAIN-6, there were significantly higher rates of retinopathy complications (vitreous hemorrhage, blindness, condition requiring intravitreal agent, or photocoagulation [HR: 1.76; 95% CI: 1.11 to 2.78) observed with semaglutide compared to placebo. A nonsignificant increase in retinopathy was also observed in LEADER. A possible explanation for the early worsening in retinopathy in the SUSTAIN-6 trial is the rapid improvement in glycemic control (65). Also, none of the GLP1 trials were specifically designed to assess retinopathy in detail, and GLP1 trials were not designed to provide a sensitive assessment of the progression of clinically evident or subclinical diabetic retinopathy. Moreover, a recent large meta analysis failed to show an increase in retinopathy across GLP1RA trials to date (66). Nevertheless, as with insulin, guidance regarding the potential for early worsening of diabetic retinopathy with GLP1RA may be appropriate (65).
HF was an initial concern with GLP1RA use. This signal, however, was not confirmed in the FIGHT trial which showed nonsignificant increases in death or HF hospitalization with liraglutide compared with placebo for patients with HF with reduced ejection fraction (HR: 1.30; 95% CI: 0.92 to 1.83; p = 0.14) (67). Subsequent GLP1RA trials including LEADER and SUSTAIN-6 had small populations of patients with established HF, therefore limiting any potential generalizations about HF risk with GLP1RA (54,55). Twenty percent of both the treatment and control groups in HARMONY had prevalent HF, and no difference was observed in the composite outcome of death from cardiovascular causes combined with hospitalization for HF (57); however, HF was not evaluated as an outcome individually.
SODIUM GLUCOSE COTRANSPORTER 2 INHIBITORS
The kidneys play an important role in glucose homeostasis via gluconeogenesis (20% to 25% of total glucose production), glucose use, and glucose reabsorption (68). In diabetes, renal gluconeogenesis is increased and the ability to reabsorb glucose in the convoluted segment of the proximal tubule is pathologically increased via upregulation of SGLT2 transporters. Phlorizin, found in the root bark, leaves, shoots, and fruit of the apple tree, is a naturally occurring competitive SGLT2 and SGLT1i. This molecule, first identified to cause glucosuria in the late 1800s, has been explored for various pharmacologic indications, but its use was hampered by intestinal side effects (69,70). Moreover, the drug’s mechanism of action was not discovered until the 1990s when scientists were exploring glucose transport within renal cells. In 1992, the gene encoding the SGLT2 protein was discovered, and was so named for its structural similarly to SGLT1 (Figure 4). This protein was determined to mediate renal glucose resorption and was also found to be sensitive to phlorizin (71,72). Subsequently, the gene was evaluated in patients with familial renal glycosuria, and it was found that multiple mutations in the coding region in affected families (73). Drug development was focused on created phlorizin-based analogs with SGLT2 selectivity. Thus, phlorizin was a starting point for the development of specific inhibitors of SLGT2. The SGLT2i (Table 1) inhibit the high-capacity, low-affinity SGLT2 receptors in the proximal tubule of the nephron in which more than 90% of filtered glucose is reabsorbed (74). Importantly, SGLT2 receptor activity is increased in chronic hyperglycemia, leading to enhanced resorption of glucose and sodium (75). Conversely, the SGTL2 receptor is downregulated at lower glucose concentrations and glucosuria declines with declines in eGFR. This provides an important clinical benefit of minimizing the risk of hypoglycemia with SGLT2i (76), especially at lower levels of glycemia or at lower GFR.
Since the approval of the first SGLT2i in 2013 by the FDA, these agents have become the mainstay in the treatment of T2DM. SGLT2i improve glucose control without increasing the risk of hypoglycemia and their glucosuric effect contributes to weight loss (70 g of carbohydrates accounts for 280 kcal). In addition, they have shown benefits on the cardiovascular system, including protection against HF, and the kidney. Across all SGLT2i CVOTs, because of the quest for glycemic equipoise in the trials, the reduction in % HbA1c during the trial was modest at approximately 0.1% to 0.6%, indicating that glycemic control alone is unlikely to account for the cardiovascular benefit observed with use of this class of agents (62). Although unproven, nonglycemic hemodynamic effects may promote cardiovascular benefits with SGLT2i (Figure 1). For example, SGTL2i can increase uric acid excretion through co-inhibition of glucose and uric acid reabsorption (77), reducing uric acid levels which have been independently associated with cardiovascular and renal events (77). Further evidence of hemodynamic benefit is suggested by a recent mediation analysis from EMPA-REG showing that markers of plasma volume such as hematocrit and hemoglobin were important markers associated with reduction in cardiovascular mortality observed with SGLT2i use (10,78). These observations may be due in part to the improved oxygenation and normalization of erythropoietin production observed with SGLT2i (79) and observed renoprotective effects (80). Mechanisms whereby SGLT2 inhibition might contribute to cardioprotection include improved glycemic control, blood pressure, and weight, and improved endothelial function and reduced arterial stiffness thereby reducing afterload (62). SGLT2i stimulate natriuresis, diuresis, and glucosuria thereby reducing plasma volume and lowering cardiac preload (81,82). Weight loss limits epicardial deposition. Other putative mechanisms include improved cardiac mitochondrial energy output, inhibition of the cardiac Na/hydrogen exchanger leading to lower cytosolic concentrations of sodium and calcium, and increased mitochondrial concentrations of calcium (83,84). Finally, an increase free fatty oxidation and stimulation of ketogenesis has been proposed, thereby shifting cardiac substrate to more energy efficient use of fatty acids and ketones. Postulated renoprotective effects include constriction of afferent glomerular arterioles (via SGLT2i-mediated diuresis, natriuresis, and reduction of natriuretic peptides) leading to suppression of tubuloglomerular feedback along with reduction in renin and angiotensin II production, leading overall to efferent arterioles dilatation and reduction in glomerular hydrostatic pressure. The combined effect reduces intraglomerular hypertension and hyperfiltration. Thus, despite the initial modest decline in GFR (an effect also observed with use of and attenuated by renin-angiotensin-aldosterone system blockers), reductions in hyperfiltration and intraglomerular pressure are renoprotective (82,85).
TRIAL DESIGN AND MAJOR OUTCOMES.
Three large CVOTs have been completed for the SGT2i in patients with T2DM: EMPA-REG OUTCOME (empagliflozin), CANVAS (canagliflozin), and DECLARE-TIMI 58 (dapagliflozin) (Table 1) (78,83–88). All 3 trials showed non-inferiority for cardiovascular safety compared placebo, and in some cases showed impressive reductions in adverse cardiovascular outcomes. Two additional trials, CREDENCE (canagliflozin) and DAPA-HF (dapagliflozin), evaluated alternative primary outcomes of renal events and HF events in patients with CKD stage 2/3 and chronic HF, respectively, and analyzed 3P-MACE as a secondary endpoint. A sixth study, VERTIS-CV, evaluating ertugliflozin, is expected to be completed in December 2019 (89). EMPA-REG OUTCOME, CANVAS, CREDENCE, and DAPA-HF all showed reductions in a 3P-MACE outcome of cardiovascular death, nonfatal MI, or nonfatal stroke (Table 1). DECLARE TIMI 58 had 2 primary endpoints: a 3P-MACE, and a 2-item composite of cardiovascular death or hospitalization for HF. In contrast to EMPA-REG OUTCOME and CANVAS, the 3P-MACE was not met in DECLARE TIMI-58 (HR: 0.93; 95% CI: 0.84 to 1.03; p = 0.17). However, there was a nearly 20% RR reduction in cardiovascular death or hospitalization for HF (HR: 0.83; 95% CI: 0.73 to 0.95; p = 0.005), driven by approximately 30% reduction in hospitalization for HF (HR: 0.73; 95% CI: 0.61 to 0.88). Consistent reductions in hospitalization for HF and renal outcomes were observed in all 4 SGLT2i CVOTs (Table 1), but due to the prespecified hierarchical testing plan in CANVAS, estimates for secondary outcomes are considered nonsignificant and exploratory only.
ADDITIONAL OUTCOMES.
All the SGLT2i trials to date have shown impressive cardiorenal protective effects in patients with T2DM (Table 1). The definition of renal outcomes across these in these trials varied, but included measures such as incident or worsening nephropathy (progression to macroalbuminuria, doubling of the serum creatinine level, initiation of renal replacement therapy, or death from renal disease) and incident albuminuria (86,90). However, in all trials, there have been relatively few participants with clinically significant impairments in renal function at baseline. Participants in EMPA-REG OUTCOME and CANVAS were required to have an eGFR greater than or equal to 30 ml/min/1.73 m2 per the Modification of Diet in Renal Disease formula (MDRD) (86,90). In EMPA-REG, CANVAS, and DECLARE, less the 25% of participants had an eGFR less than 60 ml/min (78,86,87). In contrast, nearly 60% of patients in CREDENCE had an eGFR less than 60 ml/min (88). Although the analyses plan in CANVAS precluded formal testing of renal efficacy, the point estimates suggest a consistency of renal benefit with canagliflozin compared to the other SGLT2i (86). Overall, SGLT2i as a class have shown renoprotection and reduced composite measures of adverse renal effects by 45% (HR: 0.55; 95% CI: 0.48 to 0.64; p < 0.001) (91). As noted, there may be heterogeneity of benefit of SGLT2i among participants with less severe renal dysfunction at baseline (91). Conversely, the reduction in HF with SGLT2i increased with worsening renal failure across trials of SGLT2i to date (91). All 5 CVOTs reported reduction in hospitalization for HF, including DAPA-HF, which evaluated HF hospitalization and cardiovascular death as a composite primary outcome in patients with baseline chronic HF with reduced ejection fraction.
HETEROGENEITY OF BENEFIT ACROSS RISK CATE-GORIES WITH SGLT2i.
Baseline CVD risk among patients with T2DM also varied across the 4 SGLT2i CVOTs to date (Table 1). For example, all participants in EMPA-REG OUTCOME had established atherosclerotic CVD, compared with approximately 65% of participants in CANVAS, 50% in CREDENCE, and approximately 40% in DECLARE TIMI-58. As a result, the MACE event rate varied substantially for participants assigned to placebo, being greatest in EMPA-REG OUTCOME (43.9 per 1,000 patient-years), followed by CREDENCE (38.7 per 1,000 patient-years), CANVAS (31.5 per 1,000 patient-years), and finally DECLARE TIMI-58 (24.2 per 1,000 patient-years). As previously shown (91), there appears to be heterogeneity of benefit for SGLT2i for cardiovascular outcomes based on baseline risk factor profile. In a meta-analysis of participants from EMPA-REG, CANVAS, and DECLARE TIMI 58, SGLT2i reduced MACE by 11% (HR: 0·89; 95% CI: 0·83 to 0·96; p = 0.0014), with benefit only seen in patients with atherosclerotic CVD (HR: 0.86; 95% CI: 0.80 to 0.93) and not in those without (HR: 1.00; 95% CI: 0.87 to 1.16; p for interaction = 0.0501) (91). In comparison, SGLT2i reduced the risk of cardiovascular death or hospitalization for HF by 23% (HR: 0.77; 95% CI: 0.71 to 0.84; p < 0.0001), with a similar benefit in patients with and without atherosclerotic CVD and with and without a history of HF (91). While this recent metaanalysis did not include data from CREDENCE, the HR for CREDENCE participants with established CVD (HR: 0.70; 95% CI: 0.56 to 0.88) compared to without Q18 (HR: 0.69; 95% CI: 0.53 to 0.87; p for interaction = 0.91) suggests a similar magnitude of benefit for the primary outcome in this trial based on presence or absence of comorbid CVD on a background of renal impairment (88).
SIDE EFFECTS.
The SGLT2i were overall welltolerated in the large CVOT with some notable exceptions. A similar proportion of participants randomized to active therapy (20% to 30%) in the 4 SGLT2i CVOT trials discontinued the study drug, compared with 25% to 30% for placebo (78,86–88). Genital infections, definitions of which varied by sex and across trials, were more common with SGLT2i than with placebo; however, these infections infrequently(<1%) resulted in study drug discontinuation (78,86–88). Additionally, there was no difference in the occurrence of complicated urinary tract infections between participants randomized to study drug versus placebo in these trials. Volume depletion has been described as a known side effect with all SGLT2i and appears similar with all 4 SGLT2i agents, although an increased risk of volume depletion reached statistical significance in CANVAS (78,86–88). CANVAS also identified amputations and fractures, captured as adverse events of special interest, as new safety concerns. Additionally, CREDENCE amended their protocol to mandate that participants who developed an active foot lesion were required to interrupt use of the study drug until the lesion resolved (as presented at the American Diabetes Association Scientific Sessions in 2019) (92). This pattern was also observed with use of canagliflozin in a “realworld” observation study in the U.S. Department of Defense Military Health System (93). However, as noted, an increased risk of amputations or fractures was not observed with use of other SGLT2i. Additionally, canagliflozin was not associated with fractures or amputations in the CREDENCE trial (86). Increased fractures or amputations have not been consistently shown with either empagliflozin or dapagliflozin, although monitoring for amputations was not a primary safety concern in EMPA-REG OUTCOME. Euglycemic ketoacidosis has been reported, mainly in insulin-treated T2DM patients. However, no increased risk of ketoacidosis has been reported as a trial outcome in the CVOTs completed to date (94).
DISCUSSION
CVD is the leading cause of death in adults with T2DM. Sixty percent of the difference in survival is attributable to excess CVD mortality (1). Additionally HF is underrecognized among T2DM patients and further increases morbidity and mortality associated with T2DM (3). Despite the overlap between T2DM and CVD, antidiabetic therapeutics were either discovered by chance or designed to target metabolic, rather than cardiovascular, mechanisms. Biguanides were derived from plants used to treat diabetes in antiquity before the era of targeted drug development (12). Sulfonylureas, first under investigation in the 1940s as antibacterial agents, and TZDs, investigated in the 1980s as a lipid lowering therapies, were observed to induce hypoglycemia in animal models, and were then repurposed as antidiabetic drugs (95,96). The remainder of pharmacologic agents emerged from research focused on pathways known to be involved in insulin and energy regulation. For example DPP4is and GLP1RAs both target the incretin pathway, which has been investigated since the 1960s for its involvement in postprandial energy metabolism (97). In the 1990s, SGLT2i precursors were isolated from phlorizin, a compound found primarily in the root bark of apple trees known to cause glycosuria since 1886 (98) (Figure 4). In 2008, the adverse cardiovascular outcomes observed in trials involving tolbutamide and rosiglitazone prompted mandates that all antidiabetic agents meet cardiovascular safety endpoints. Similar guidance was later adopted by the European Medicines Agency (EMA) (99). Thus, it was by happenstance, rather than by design, that the cardiovascular benefits of antidiabetic agents were discovered.
Twenty-one CVOT studies are on track for completion by 2020, first in high-risk T2DM patients with established CVD (secondary prevention) and subsequently among T2MD patients with multiple CVD risk factors (primary prevention). Not every trial was powered or designed to show superiority, and differences in trial design and in participant baseline characteristics make it challenging to compare results across trials. All recent CVOTs to date, except for CAROLINA, have compared active therapy against placebo. Despite limitations of comparisons across trials, some overall themes emerge.
In current clinical practice, metformin remains the first-line pharmacologic treatment due to established evidence of its metabolic benefits, along with long term data on safety, drug affordability, and limited trial data to support its use for reduction in MI. As noted, metformin was background medical therapy for most patients in recent CVOTs, further enshrining its use as first-line therapy for cardiovascular risk reduction in T2DM. However, clinical practice in Europe may shift away from universal first-line use of metformin given new ESC/EASD guidelines recommending initiating SGLT2i or GLP1RA monotherapy with drug-naive patients with T2DM with established or high risk for CVD (21). The 2020 updated ADA guidelines continue to recommend metformin as first-line therapy for all comers, with consideration of concurrent therapy with SGLTi in patients with HF or kidney disease and SGLT2i or GLP1RA in patients with predominantly atherosclerotic CVD (100).
The place of sulfonylureas is controversial as they may induce hypoglycemia, and as suggested by the UGDP study, tolbutamide use may also be associated with an increased risk of cardiovascular mortality. However, subsequent trials including ADVANCE (with gliclazide modified release) and CAROLINA (with glimepiride) have overall shown cardiovascular safety of second- and third-generation sulfonylurea. However, the well-documented weight gain and lack of established cardiovascular outcome benefit with the sulfonylureas and adverse side-effect profile indicates that these agents should be further down the list of treatment options for cardiovascular risk reduction among T2DM patients. Despite promising protective preclinical and metabolic effects of DPP4i, none of the DPP4i CVOTs showed a reduction in cardiovascular events. The lack of cardiovascular benefit to date and the high cost of the DPP4is suggest a limited role in current paradigms to reduce cardiovascular risk for T2DM patients.
In contrast, the CVOTs with GLP1RA and SGLT2i have overall shown beneficial effects on cardiovascular outcome. Liraglutide, subcutaneous semaglutide, albiglutide, and dulaglutide (but not exenatide or lixisenatide) showed superiority compared with placebo for reduction in a 3P-MACE endpoint. Furthermore, all GLP1RA CVOTs showed noninferiority for cardiovascular safety compared with placebo. Similarly, the 4 SLGT2i CVOTs completed to date have also shown non-inferiority for cardiovascular safety compared with placebo, and in some cases showed impressive reductions in adverse cardiovascular outcomes. Empagliflozin and canagliflozin showed reductions in a 3P-MACE outcome of cardiovascular death, nonfatal MI, or nonfatal stroke. Dapagliflozin showed a reduction in a 2-item composite of cardiovascular death or hospitalization for HF. As reviewed, there is good evidence of renoprotection across many of the GLP1RA trials (Table 3) and SGLT2i trials (Table 1), including recent evidence on the use of canagliflozin in the CREDENCE trial focused on patients with T2DM and diabetic nephropathy, and of dulaglutide in the AWARD7 trial. If data continue to support consistent benefit in patients with renal disease, the tide may continue to shift away from first-line use of metformin, and to-ward a strategy that begins with an SGLT2i or GLP1RA.
The EASD/ADA consensus proposes SGLT2i for patients with atherosclerotic CVD (ASCVD), and does not prioritize these agents for those with multiple risk factors, as the magnitude of benefit on MACE appeared greater among patients with established ASCVD (101). However, no single trial has been adequately powered to test heterogeneity of cardiovascular effect for T2DM patients with established CVD compared to patients with T2DM and cardiovascular risk factors only, limiting conclusions of heterogeneity of effect to meta-analyses of CVOT trials completed to date (91). Given the fact that SGLT2i and GLP1RA safely reduce HbA1c without increased risk of hypoglycemia or weight gain and have beneficial cardiorenal effects across a broad range of subjects with T2DM, we believe these agents should be considered regardless of the presence of CVD. Evidence is amassing that the benefits of these agents are not restricted to patients with ASCVD, indicating that the GLP1RA and SGLT2i may be effective therapies for CVD prevention among patients with T2DM. Most recently, the Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF) showed that SGLT2i’s appear to reduce MACE and cardiovascular mortality among patients with HF with reduced ejection fraction, with or without comorbid T2DM (102). Along with prior studies showing that SGLT2i robustly reduce the risk of hospitalization for HF among patients with T2DM with or without established CVD or HF (91), these results suggest adaptations of consensus proposals to prioritize use of SGLT2i in T2DM patients at risk for or with established HF (Figure 3, Central Illustration). Finally, as previously reviewed, there is a need to examine the use of GLP1RA and SGLT2is in patients with disordered glucose metabolism, including pre-diabetes or metabolic syndrome (62). Earlier adoption of these therapies before the diagnostic threshold of T2DM is reached may be a transformative use of these therapies for the primordial prevention of CVD.
FIGURE 3. Algorithm Choosing Pharmacologic Therapy for Type 2 Diabetes.
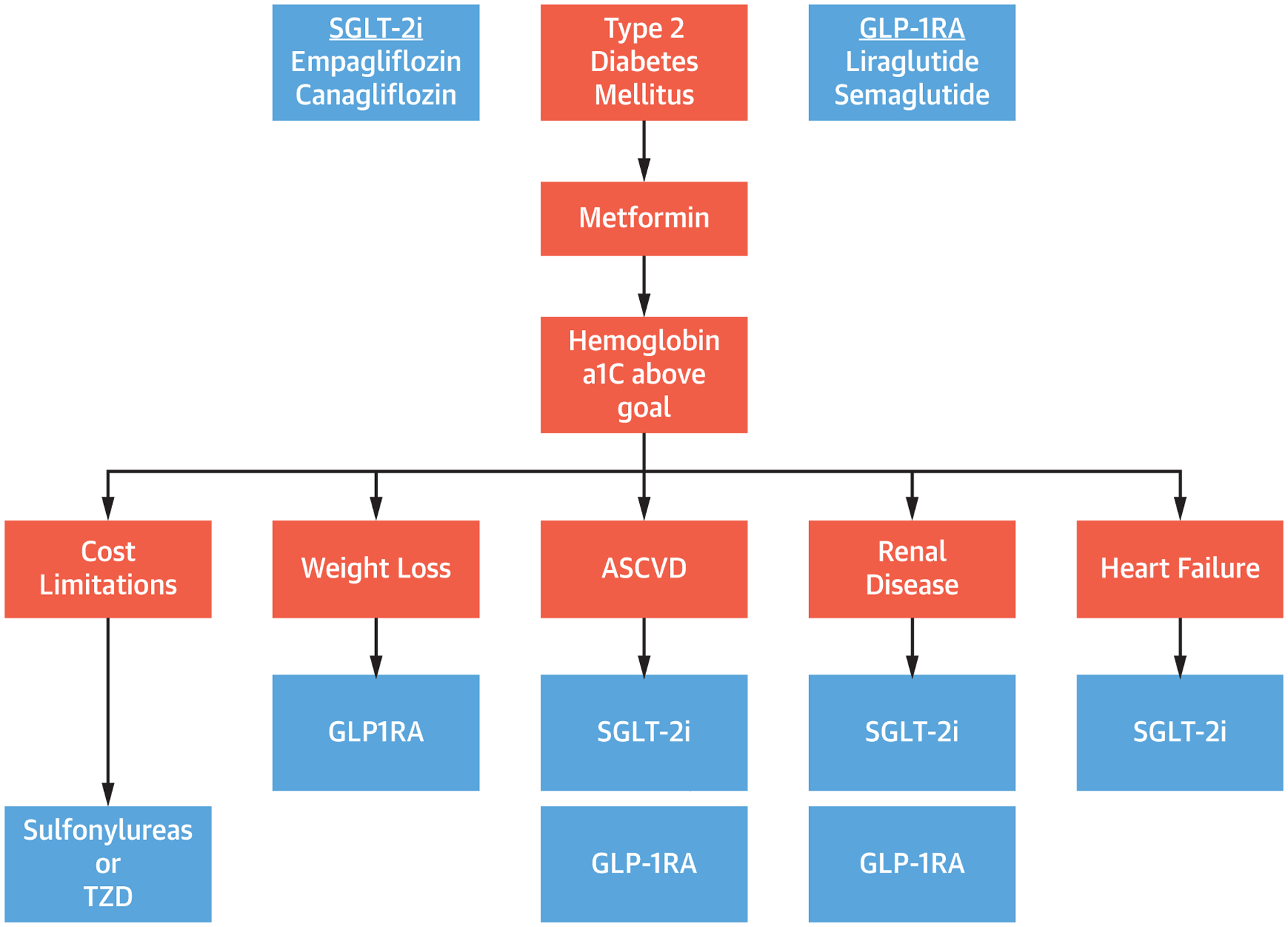
In patients with type 2 diabetes on metformin with hemoglobin a1c above goal, second-line therapy depends on cost limitations, comorbid disease, and desire for weight loss. Abbreviations as in Figures 1 and 2.
CENTRAL ILLUSTRATION. Diversity of Physiologic Effects of SGLT2i and GLP1RA.
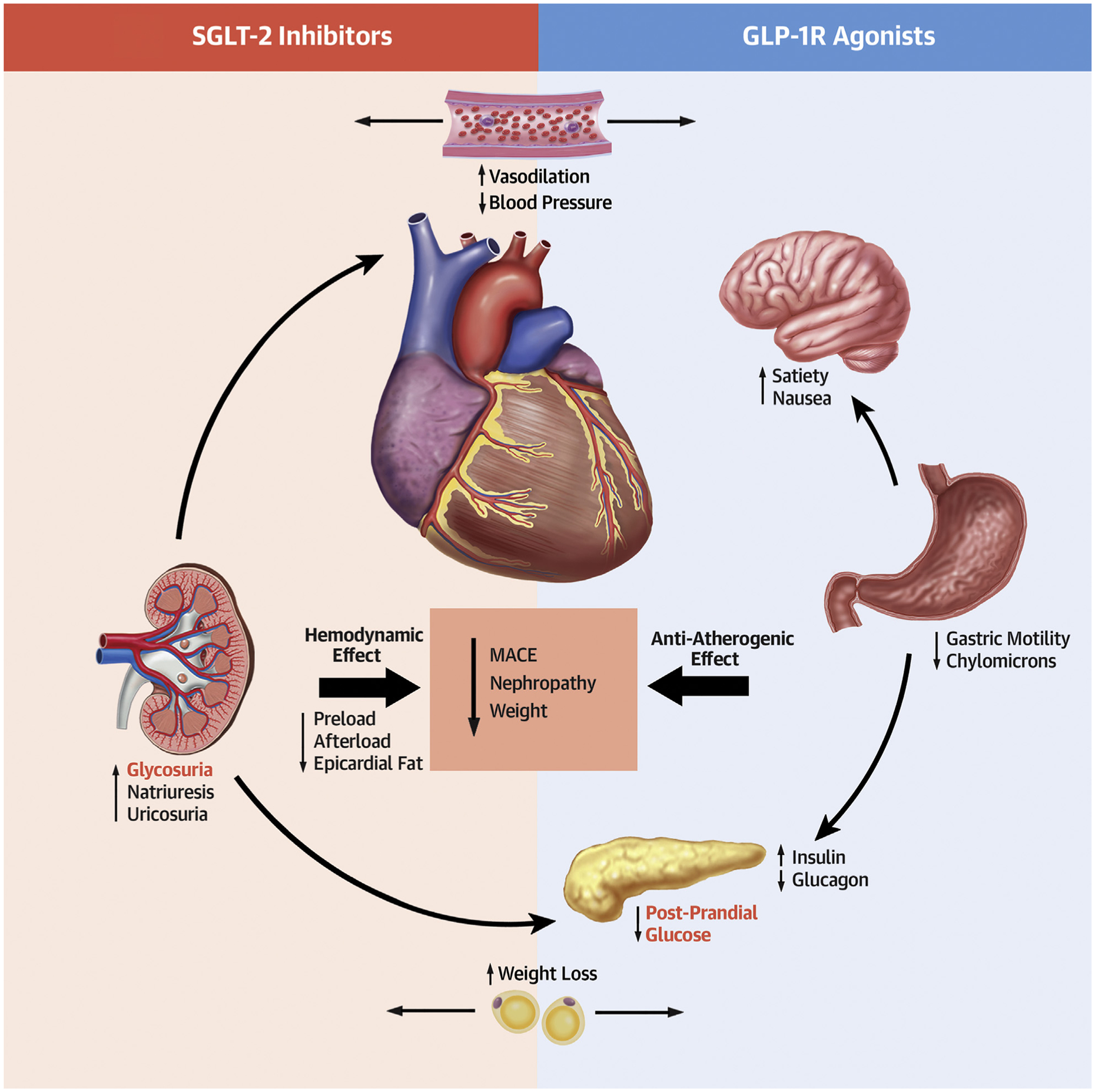
Increasing evidence supports the role of both SGLT2i and GLP1RA in reducing major adverse cardiac events and progression of renal disease while increasing weight loss and reducing blood pressure. SGLT2i accomplish this primarily via hemodynamic effects, whereas GLP1RAs have stronger anti-atherogenic effects.GLP1RA = glucagon-like peptide 1 receptor agonist; MACE = major adverse cardiac events; SGLT21 = sodium glucose cotransporter 2 inhibitor.
FIGURE 2. Selecting Pharmacologic Therapy for Patients With Type 2 Diabetes.
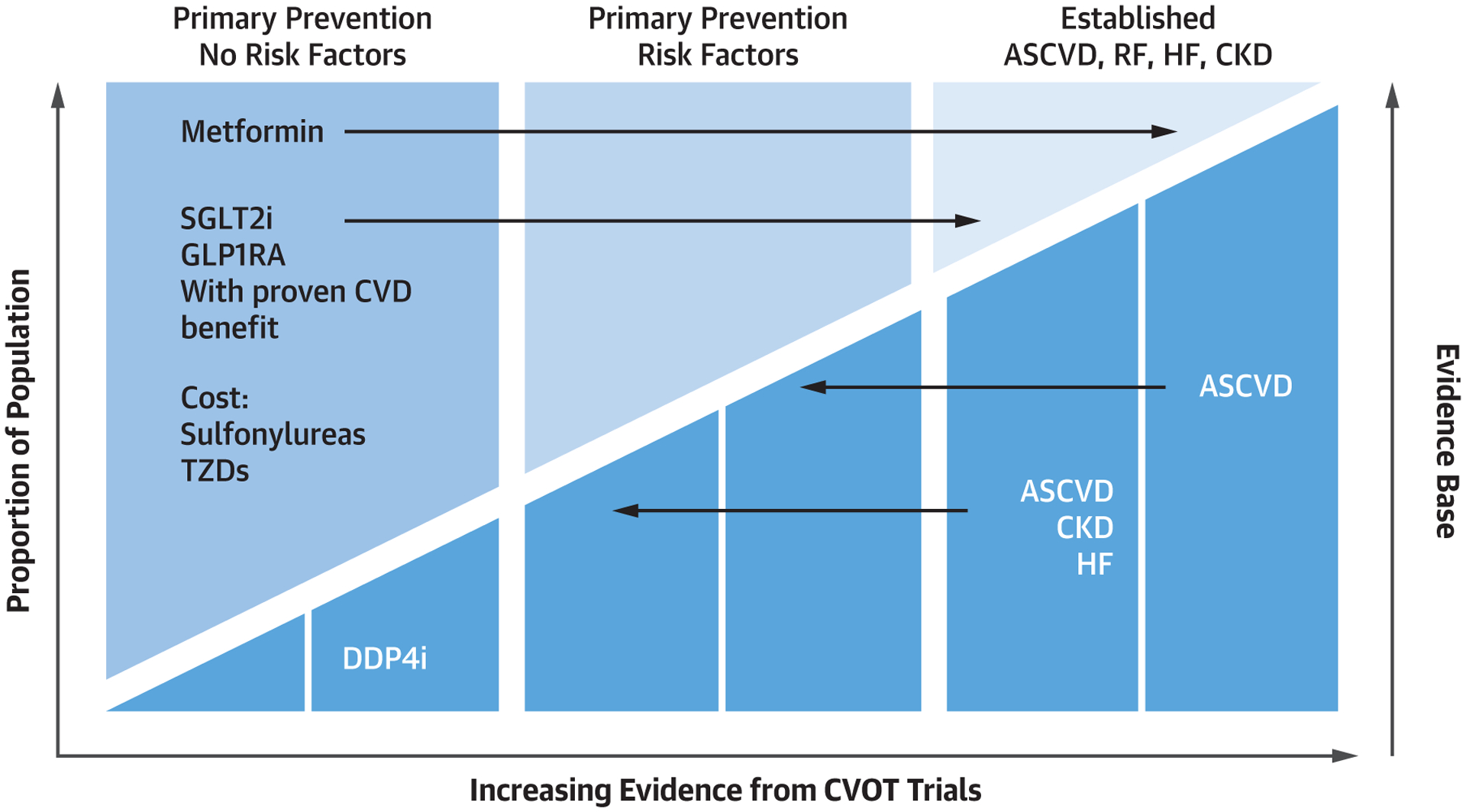
The evidence base for selecting pharmacologic therapy for patients with type 2 diabetes depends on presence of risk factors, comorbid heart failure, atherosclerotic disease, and renal disease. ASCVD = atherosclerotic cardiovascular disease; CKD = chronic kidney disease; CVD = cardiovascular disease; CVOT = cardiovascular outcome trial; DDP4i = dipeptidyl peptidase 4 inhibitor; HF = heart failure; RF = ■■■; TZD = ■■■; other abbreviations as in Figure 1.
HIGHLIGHTS.
Historically, glycemic control was the primary focus in reducing cardiovascular risk in patients with diabetes mellitus.
Although historic agents effectively lower blood sugar, evidence for cardiovascular benefit was lacking.
Newer glucose-lowering medications target numerous novel pathways to reduce cardiovascular and renal events in patients with type 2 diabetes.
These medications should be considered in patients with diabetes and CVD and may play a role in primary prevention of cardiovascular and renal disease.
Acknowledgments
Dr. De Block has received personal fees from AstraZeneca, Boehringer Ingelheim, Johnson & Johnson, Lilly, Merck Sharp & Dohme, Novo Nordisk A/S, and Sanofi. Dr. Schwartzbard has received grants from Merck/Pfizer, Amarin, Sanofi, Novartis, and Amgen; and has served as a consultant to the formulary committee for optum Rx. Dr. Newman has received grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) (K23HL K23HL125991); and has received honoraria from Creative Educational Concepts. Dr. Wilcox has reported that she has no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- CKD
chronic kidney disease
- CVD
cardiovascular disease
- CVOT
cardiovascular outcome trials
- DPP4i
dipeptidyl peptidase 4 inhibitors
- GLP1RA
glucagon-like peptide 1 receptor antagonist
- HbA1c
hemoglobin A1c
- HF
heart failure
- MI
myocardial infarction
- SGLT2i
sodium glucose transporter 2 inhibitor
- T2DM
type 2 diabetes mellitus
- TZD
thiazolidinediones
REFERENCES
- 1.Di Angelantonio E, Kaptoge S, Wormser D, et al. Association of cardiometabolic multimorbidity with mortality. JAMA 2015;314: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet 2015;385:2107–17. [DOI] [PubMed] [Google Scholar]
- 3.McMurray JJ, Packer M, Desai AS, et al. Angiotensin–neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371: 993–1004. [DOI] [PubMed] [Google Scholar]
- 4.ADVANCE Collaborative Group, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–72. [DOI] [PubMed] [Google Scholar]
- 5.Action to Control Cardiovascular Risk in Diabetes Study Group et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeFronzo RA. From the triumvirate to the “ominous octet”: a new paradigm for the treatment of type 2 diabetes mellitus. Clini Diabetol 2009;10:101–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet 2011;378: 182–97. [DOI] [PubMed] [Google Scholar]
- 8.Cefalu WT, Kaul S, Gerstein HC, et al. Cardiovascular outcomes trials in type 2 diabetes: where do we go from here? Reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2018; 41:14–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathan DM, Buse JB, Davidson MB, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009;32:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inzucchi SE, Zinman B, Fitchett D, et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care 2018;41:356–63. [DOI] [PubMed] [Google Scholar]
- 11.Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018;41:2669–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White JR. A brief history of the development of diabetes medications. Diabetes Spectr 2014;27: 82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rena G, Lang CC. Repurposing metformin for cardiovascular disease. Circulation 2018;137: 422–4. [DOI] [PubMed] [Google Scholar]
- 14.Group UPDS. Effect of intensive blood-glucose control with metformin on complications in over weight patients with type 2 diabetes (UKPDS 34). Lancet 1998;352:854–65. [PubMed] [Google Scholar]
- 15.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–89. [DOI] [PubMed] [Google Scholar]
- 16.Selvin E, Bolen S, Yeh H-C, et al. Cardiovascular outcomes in trials of oral diabetes medications: a systematic review. Arch Intern Med 2008; 168:2070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care 2011;34:1431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inzucchi SE, Lipska KJ, Mayo H, Bailey CJ, McGuire DK. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA 2014;312:2668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergmark BA, Bhatt DL, McGuire DK, et al. Metformin use and clinical outcomes among patients with diabetes with or without heart failure or kidney dysfunction: observations from the SAVOR-TIMI 53 trial. Circulation 2019;140: 1004–14. [DOI] [PubMed] [Google Scholar]
- 20.Lalau J-D, Kajbaf F, Bennis Y, Hurtel-Lemaire A-S, Belpaire F, De Broe ME. Metformin treatment in patients with type 2 diabetes and chronic kidney disease stages 3A, 3B, or 4. Diabetes Care 2018;41:547–53. [DOI] [PubMed] [Google Scholar]
- 21.Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the task force for diabetes, prediabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and the European Association for the Study of Diabetes (EASD). Eur Heart J 2019. 10.1093/eurheartj/ehz486 [published Online First: Epub Date]j. [DOI] [Google Scholar]
- 22.Sigal RJ, El-Hashimy M, Martin BC, Soeldner JS, Krolewski AS, Warram JH. Acute postchallenge hyperinsulinemia predicts weight gain: a prospective study. Diabetes 1997;46: 1025–9. [DOI] [PubMed] [Google Scholar]
- 23.Sherifali D, Nerenberg K, Pullenayegum E, Cheng JE, Gerstein HC. The effect of oral antidiabetic agents on A1C levels: a systematic review and meta-analysis. Diabetes Care 2010;33: 1859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright A, Burden AF, Paisey RB, Cull CA, Holman RR. Sulfonylurea inadequacy: efficacy of addition of insulin over 6 years in patients with type 2 diabetes in the UK Prospective Diabetes Study (UKPDS 57). Diabetes Care 2002;25:330–6. [DOI] [PubMed] [Google Scholar]
- 25.Meinert CL, Knatterud GL, Prout TE, Klimt CR. A study of the effects of hypoglycemic agents on vascular complications in patients with adultonset diabetes. II. Mortality results. Diabetes 1970;19 suppl:789–830. [PubMed] [Google Scholar]
- 26.Rados DV, Pinto LC, Remonti LR, Leitão CB, Gross JL. The association between sulfonylurea use and all-cause and cardiovascular mortality: a meta-analysis with trial sequential analysis of randomized clinical trials. PLoS Med 2016;13: e1001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenstock J, Kahn SE, Johansen OE, et al. Effect of linagliptin vs glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes: the CAROLINA randomized clinical trial. JAMA 2019;322:1155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Association AD. Standards of medical care in diabetes—2016 abridged for primary care providers. Clin Diabetes 2016;34:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng C-L, Soroka O, Maney M, Aron DC, Pogach LM. Assessing potential glycemic overtreatment in persons at hypoglycemic risk. JAMA Intern Med 2014;174:259–68. [DOI] [PubMed] [Google Scholar]
- 30.Yki-Jarvinen H Thiazolidinediones. N Engl J Med 2004;351:1106–18. [DOI] [PubMed] [Google Scholar]
- 31.Lathief S, Inzucchi SE. Approach to diabetes management in patients with CVD. Trends Cardiovasc Med 2016;26:165–79. [DOI] [PubMed] [Google Scholar]
- 32.Dormandy JA, Charbonnel B, Eckland DJA, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005;366:1279–89. [DOI] [PubMed] [Google Scholar]
- 33.Kernan WN, Viscoli CM, Furie KL, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 2016;374:1321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356: 2457–71. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Ferrannini E, DeFronzo R, Schernthaner G, Yates J, Erdmann E. Effect of pioglitazone on cardiovascular outcome in diabetes and chronic kidney disease. J Am Soc Nephrol 2008;19:182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 2013;17:819–37. [DOI] [PubMed] [Google Scholar]
- 37.Scheen AJ. Cardiovascular effects of gliptins. Nat Rev Cardiol 2013;10:73. [DOI] [PubMed] [Google Scholar]
- 38.Bloomgarden Z The kidney and cardiovascular outcome trials. J Diabetes 2018;10:88–9. [DOI] [PubMed] [Google Scholar]
- 39.Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;373:232–42. [DOI] [PubMed] [Google Scholar]
- 40.McGuire D, Van de Werf F, Armstrong P, et al. Trial Evaluating Cardiovascular Outcomes With Sitagliptin (TECOS) study group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol 2016;1:126–35. [DOI] [PubMed] [Google Scholar]
- 41.Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317–26. [DOI] [PubMed] [Google Scholar]
- 42.Zannad F, Cannon CP, Cushman WC, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015;385:2067–76. [DOI] [PubMed] [Google Scholar]
- 43.White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013;369: 1327–35. [DOI] [PubMed] [Google Scholar]
- 44.Rosenstock J, Perkovic V, Johansen OE, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA 2019;321:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ingelheim B CAROLINA: cardiovascular outcome study of linagliptin versus glimepiride in patients with type 2 diabetes ClinicalTrials. gov [Internet] Bethesda, MD, National Library of Medicine; Available at, https://clinicaltrials/.gov/show/NCT01243424. NLM Identifier: NCT01243424. Accessed 2017;6. [Google Scholar]
- 46.FDA Drug Safety Communication, https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-adds-warnings-about-heart-failure-risk-labels-type-2-diabetes.
- 47.Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia 2011;54:10–8. [DOI] [PubMed] [Google Scholar]
- 48.Drucker DJ. The biology of incretin hormones. Cell Metab 2006;3:153–65. [DOI] [PubMed] [Google Scholar]
- 49.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006;368:1696–705. [DOI] [PubMed] [Google Scholar]
- 50.Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015;373:11–22. [DOI] [PubMed] [Google Scholar]
- 51.Muskiet MH, Tonneijck L, Smits MM, et al. GLP-1 and the kidney: from physiology to pharmacology and outcomes in diabetes. Nat Rev Nephrol 2017;13:605. [DOI] [PubMed] [Google Scholar]
- 52.Madsbad S, Holst JJ. Treatment with GLP-1 receptor agonists In: Bonora E, DeFronzo R, editors. Diabetes. Epidemiology, Genetics, Pathogenesis, Diagnosis, Prevention, and Treatment Cham, Germany: Springer International Publishing, 2018:1–45. [Google Scholar]
- 53.Holman RR, Bethel MA, Mentz RJ, et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2017; 377:1228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marso SP, Bain SC, Consoli A, et al. Semaglu-tide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016;375:1834–44. [DOI] [PubMed] [Google Scholar]
- 56.Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015;373: 2247–57. [DOI] [PubMed] [Google Scholar]
- 57.Hernandez AF, Green JB, Janmohamed S, et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 2018;392:1519–29. [DOI] [PubMed] [Google Scholar]
- 58.Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019;394:121–30. [DOI] [PubMed] [Google Scholar]
- 59.Husain M, Birkenfeld AL, Donsmark M, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2019; 381:841–51. [DOI] [PubMed] [Google Scholar]
- 60.Medscape. FDA Approves CVD Benefit for Once-Weekly Semaglutide, https://www.medscape.com/viewarticle/923922.
- 61.Heart Disease Study of Semaglutide in Patients With Type 2 Diabetes (SOUL), https://clinicaltrials.gov/ct2/show/NCT03914326.
- 62.Newman JD, Vani AK, Aleman JO, Weintraub HS, Berger JS, Schwartzbard AZ. The changing landscape of diabetes therapy for cardiovascular risk reduction: JACC State-of-the-Art Review. J Am Coll Cardiol 2018;72:1856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kristensen SL, Rorth R, Jhund PS, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol 2019;7:776–85. [DOI] [PubMed] [Google Scholar]
- 64.Tuttle KR, Lakshmanan MC, Rayner B, et al. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open label, randomised trial. Lancet Diabetes Endocrinol 2018;6:605–17. [DOI] [PubMed] [Google Scholar]
- 65.Vilsboll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated haemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab 2018;20:889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monami M, Dicembrini I, Mannucci E. Effects of SGLT-2 inhibitors on mortality and cardiovascular events: a comprehensive meta-analysis of randomized controlled trials. Acta Diabetol 2017; 54:19–36. [DOI] [PubMed] [Google Scholar]
- 67.Margulies KB, Hernandez AF, Redfield MM, et al. Effects of liraglutide on clinical stability among patients with advanced heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 2016;316:500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabetes Med 2010;27:136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018;61:2079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chasis H, Jolliffe N, Smith HW. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J Clin Invest 1933;12:1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Naþ/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest 1994;93:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mudaliar S, Polidori D, Zambrowicz B, Henry RR. Sodium–glucose cotransporter inhibitors: effects on renal and intestinal glucose transport: from bench to bedside. Diabetes Care 2015;38:2344–53. [DOI] [PubMed] [Google Scholar]
- 73.Santer R, Calado J. Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 2010;5:133–41. [DOI] [PubMed] [Google Scholar]
- 74.Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diabetes Vasc Dis Res 2015;12:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non–insulindependent diabetes. Diabetes 2005;54:3427–34. [DOI] [PubMed] [Google Scholar]
- 76.Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016;134:752–72. [DOI] [PubMed] [Google Scholar]
- 77.Borghi C, Rosei EA, Bardin T, et al. Serum uric acid and the risk of cardiovascular and renal disease. J Hypertens 2015;33:1729–41. [DOI] [PubMed] [Google Scholar]
- 78.Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373: 2117–28. [DOI] [PubMed] [Google Scholar]
- 79.Carson JL, Brooks MM, Abbott JD, et al. Liberal versus restrictive transfusion thresholds for patients with symptomatic coronary artery disease. Am Heart J 2013;165:964–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sano M, Takei M, Shiraishi Y, Suzuki Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J Clin Med Res 2016;8:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferrannini G, Rydén L. Sodium-glucose transporter 2 inhibition and cardiovascular events in patients with diabetes: information from clinical trials and observational real-world data. Clin Sci 2018;132:2003–12. [DOI] [PubMed] [Google Scholar]
- 82.Lytvyn Y, Bjornstad P, Udell JA, Lovshin JA, Cherney DZ. Sodium glucose cotransporter-2 inhibition in heart failure: potential mechanisms, clinical applications, and summary of clinical trials. Circulation 2017;136:1643–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scheen AJ. Cardiovascular effects of new oral glucose-lowering agents: DPP-4 and SGLT-2 inhibitors. Circ Res 2018;122:1439–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol 2017;2:1025–9. [DOI] [PubMed] [Google Scholar]
- 85.Heerspink HJ, Kosiborod M, Inzucchi SE, Cherney DZ. Renoprotective effects of sodium glucose cotransporter-2 inhibitors. Kidney Int 2018;94:26–39. [DOI] [PubMed] [Google Scholar]
- 86.Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017;377:644–57. [DOI] [PubMed] [Google Scholar]
- 87.Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2018;380:347–57. [DOI] [PubMed] [Google Scholar]
- 88.Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019;380:2295–306. [DOI] [PubMed] [Google Scholar]
- 89.Effect of High Dose Fish Oil Supplementation After Recent Heart Attack Using Magnetic Resonance Imaging, https://clinicaltrials.gov/ct2/results?cond=&term=NCT00729430&cntry=&state=&city=&dist=.
- 90.Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016;375:323–34. [DOI] [PubMed] [Google Scholar]
- 91.Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019;393: 31–9. [DOI] [PubMed] [Google Scholar]
- 92.CREDENCE: Canagliflozin and Renal Events in Diabetes With Established Nephropathy Clinical Evaluation, http://med.stanford.edu/content/dam/sm/sccr/documents/June%202019ADA%202019_CREDENCE%20symposium_FINAL%20for%20posting.pdf.
- 93.Udell JA, Yuan Z, Rush T, Sicignano NM, Galitz M, Rosenthal N. Cardiovascular outcomes and risks after initiation of a sodium glucose cotransporter 2 inhibitor: results from the EASEL population-based cohort study (Evidence for Cardiovascular Outcomes With Sodium Glucose Cotransporter 2 Inhibitors in the Real World). Circulation 2018;137:1450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care 2015;38:1638–42. [DOI] [PubMed] [Google Scholar]
- 95.Levine R Sulfonylureas: background and development of the field. Diabetes Care 1984;7: 3–7. [PubMed] [Google Scholar]
- 96.Hulin B, McCarthy PA, Gibbs EM. The glitazone family of antidiabetic agents. Curr Pharm Des 1996;2:85–102. [Google Scholar]
- 97.Sebokova E, Christ AD, Boehringer M, Mizrahi J. Dipeptidyl peptidase IV inhibitors: the next generation of new promising therapies for the management of type 2 diabetes. Curr Top Med Chem 2007;7:547–55. [DOI] [PubMed] [Google Scholar]
- 98.Ehrenkranz JR, Lewis NG, Ronald Kahn C, Roth J. Phlorizin: a review. Diabetes Metab Res Rev 2005;21:31–8. [DOI] [PubMed] [Google Scholar]
- 99.American Diabetes Association. 1. Improving care and promoting health in populations: Standards of Medical Care in Diabetes—2018. Diabetes Care 2018;41 suppl 1:S7–12. [DOI] [PubMed] [Google Scholar]
- 100.American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020;43 suppl 1:S98–110. [DOI] [PubMed] [Google Scholar]
- 101.Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018;41: 2669–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McMurray J DAPA-HF trial. Paris, France: European Society of Cardiology Congress, 2019. [Google Scholar]
- 103.Drucker DJ, Habener JF, Hoist JJ. Discovery, characterization, and clinical development of the glucagon-like peptides. J Clin Invest 2017;127: 4217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]