

Abstract
Saturated azacycles are commonly encountered in bioactive compounds and approved therapeutic agents. The development of methods for functionalization of the α-methylene C‒H bonds of these highly privileged building blocks is of great importance, especially in drug discovery. While much effort has been dedicated towards this goal by using a directed C‒H activation approach, the development of directing groups that are both general, as well as practical, remains a significant challenge. Herein, the design and development of novel amidoxime directing groups is described for Ir(I)-catalyzed α-C(sp3)‒H alkylation of saturated azacycles using readily available olefins as coupling partners. This protocol extends the scope of saturated azacycles to piperidines, azepane, and tetrahydroisoquinoline that are incompatible with our previously reported directing group. A variety of olefin coupling partners, including previously unreactive di-substituted terminal olefins and internal olefins, are compatible with this transformation. The selectivity for a branched α-C(sp3)-alkylation product is also observed for the first time when acrylate is used as the reaction partner. The development of practical, one-step installation and removal protocols further add to the utility of amidoxime directing groups.
Graphical Abstract
INTRODUCTION
Saturated azacycles constitute a prevalent structural motif in bioactive natural products and pharmaceutical compounds (Figure 1).1 The development of methods that enable rapid synthesis and late-stage diversification of these heterocycles is appealing from the standpoint of drug discovery.2 Not surprisingly, a variety of methods have been developed for the functionalization of C(sp3)‒H bonds adjacent to nitrogen in saturated azacycles.3 Important progress has been made in the direct functionalization of these heterocycles through iminium ion,4 α-amino carbanion,5 αamino radical,6 carbene insertion7, and other innovative pathways.8 Functionalizations proceeding via transition-metal-catalyzed α-C(sp3)‒H bond activation have also been developed.9–16 These transformations usually entail the installation of a directing group on the azacycle nitrogen for recruitment of the transition-metal catalyst near to the α-C(sp3)‒H bond of interest.17 Although additional steps are required for the installation and eventual removal of the directing group, a directed C‒H activation approach offers important advantages over other methods.4–8 First, regioselectivity can be controlled in the presence of multiple equally reactive C‒H bonds; for example, when more than one amino-alkyl groups are present within the substrate. Second, the formation of a discrete carbon‒metal bond in the intermediate allows for diverse transformations that may not be possible with other approaches.4–8 Third, the directing group can be used as a functional handle for modulating the reactivity and selectivity of a transformation, thus allowing access to different isomers of the product.
Figure 1.
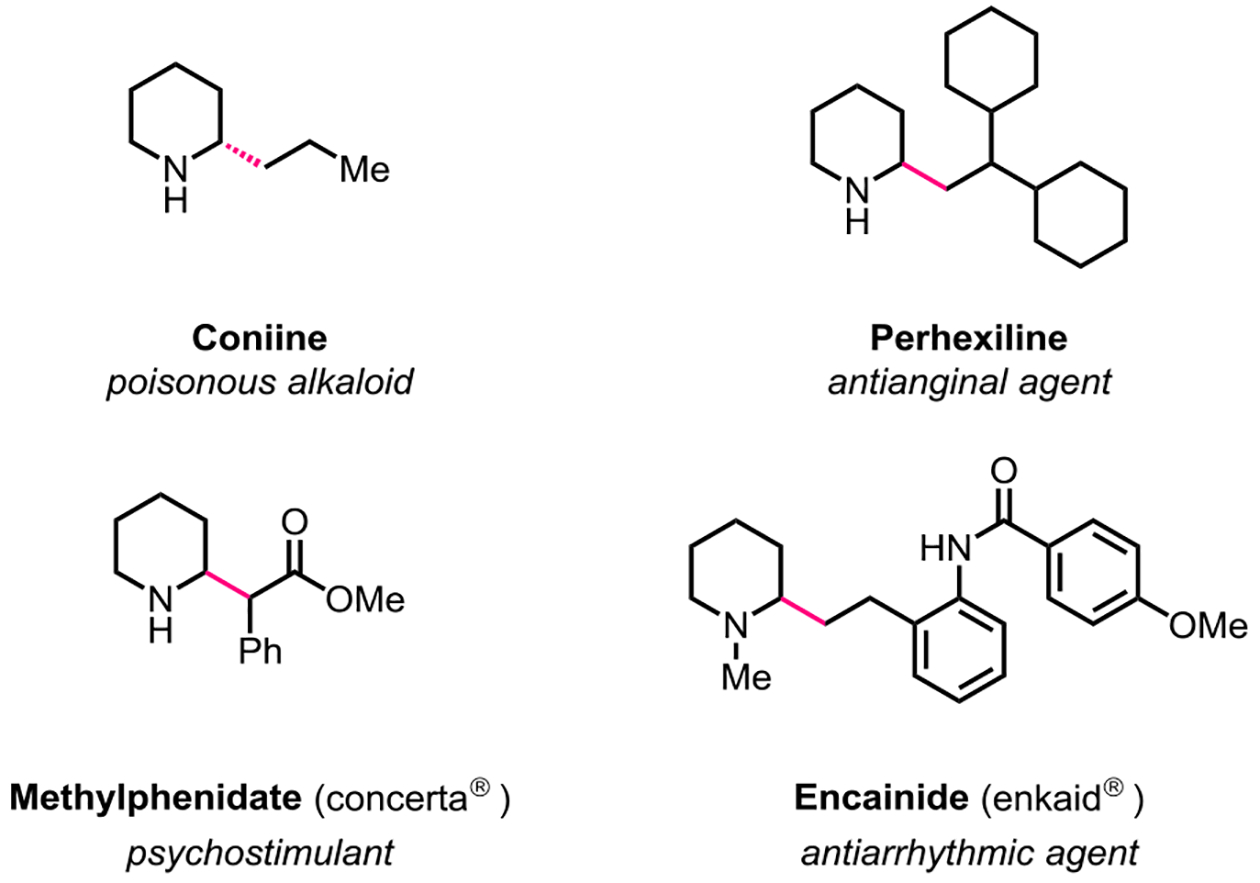
Some biologically significant compounds containing α-alkylated saturated azacycles.
In the realm of transition-metal-catalyzed directed α-C(sp3)‒H activation of saturated azacycles, several arylation transformations have been developed.9 In 2014, our group reported the first example of a palladium(II)-catalyzed directed α-arylation of saturated azacycles using aryl boronic acids as coupling partners (Scheme 1A).18 Our efforts towards developing an alkylation transformation using alkyl boronic acids as coupling partners have met with limited success;19 possibly due to a challenging sp3‒sp3 reductive elimination and competing β-hydride elimination side reactions from the corresponding metal-alkyl intermediate. In contrast, iridium(I)-catalyzed α-alkylation using olefins as coupling partners is a promising approach.20 Such alkylation reactions proceed via the intermediacy of a metal-hydride species, which reacts with olefin coupling partners to affect a net alkylation transformation (Scheme 1B).
Scheme 1.
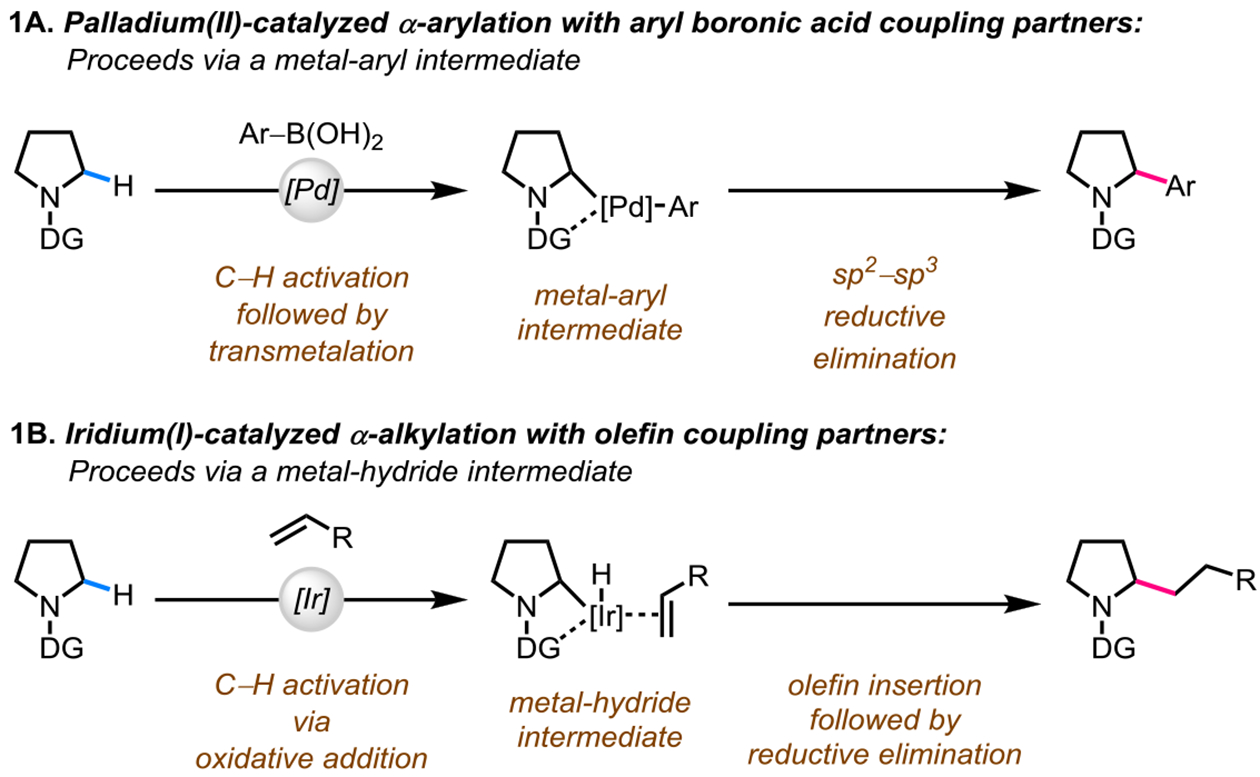
Palladium(II)-and Iridium(I)-catalyzed Directed α-C(sp3)‒H Activation Reactions of Saturated Azacycles. DG = Directing Group.
In 1998, Jun et al. reported the first example of a directed α-C(sp3)–H alkylation of benzylamines using a ruthenium(0) catalyst.10a Later in 2001, a ruthenium-catalyzed directed αC(sp3)–H alkylation of saturated azacycles was reported by Murai and co-workers.10b In 2011, the first example of an iridium-catalyzed directed α-C(sp3)–H alkylation of aliphatic amines was developed by Shibata and co-workers.11a Since these pioneering studies, several groups have developed directed approaches for α-C(sp3)–H alkylation of saturated azacycles via ruthenium10c–e and iridium catalysis11b–g (Scheme 2A). However, the utility of these approaches is limited due to the use of heterocyclic directing groups which require multiple steps and harsh reducing reaction conditions for their removal. Additionally, over-alkylation has been a frequently encountered problem.
Scheme 2.
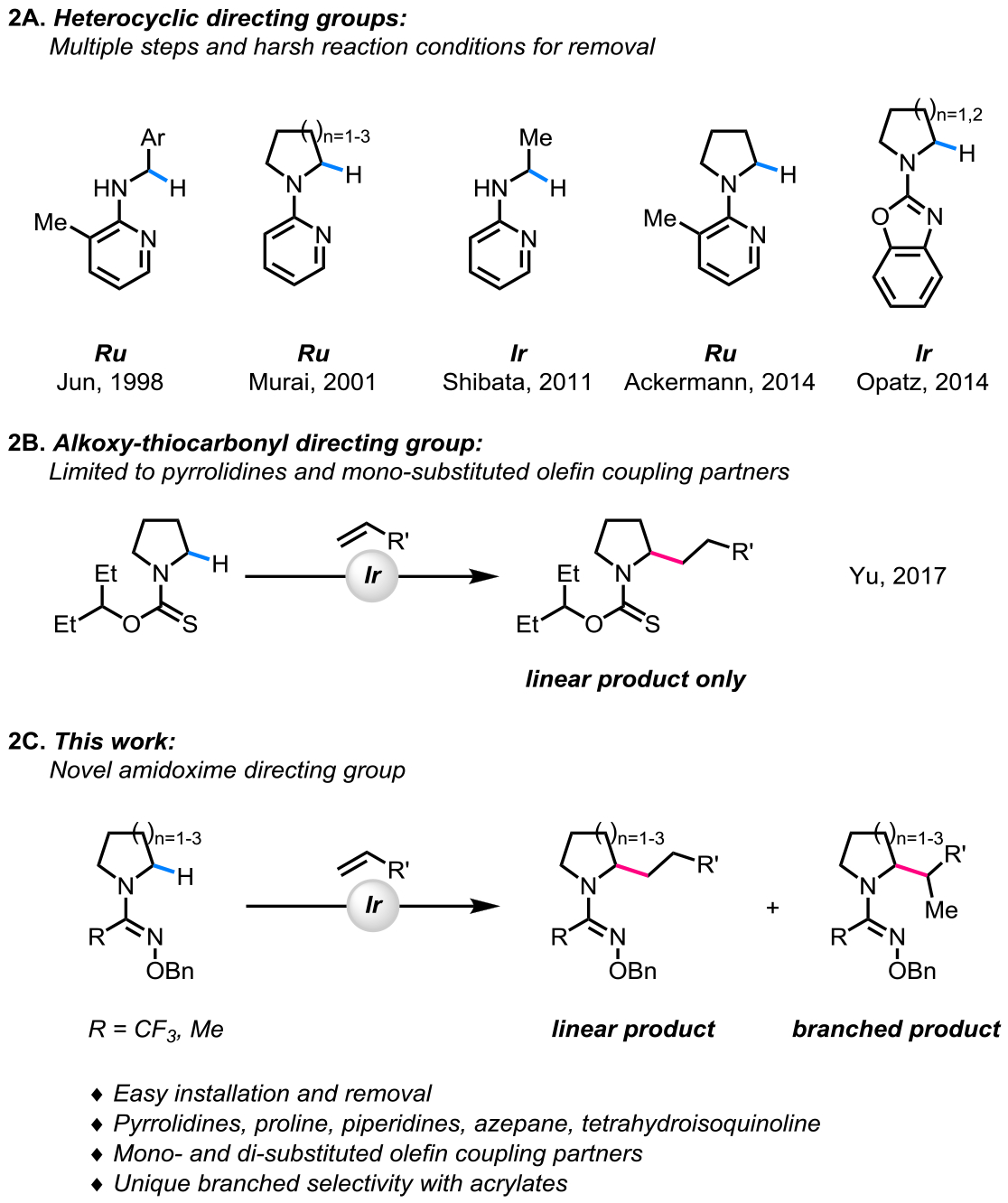
Evolution of Directing Groups for Transition-metal-catalyzed α-C(sp3)‒H Alkylation of Amines with Olefin Coupling Partners.
Recently, our group reported an alkoxy-thiocarbonyl directing group for α-C(sp3)–H alkylation of saturated azacycles using a cationic iridium(I) catalyst (Scheme 2B).21 Although the one-step installation and removal of this directing group was advantageous over previous reports employing heterocyclic directing groups, the synthetic utility of the transformation was limited for the following reasons. First, while pyrrolidines could be alkylated in moderate yields, the alkylation of other azacycles proved to be challenging. Second, the olefin coupling partner scope was limited to only mono-substituted olefins. Third, the alkoxy-thiocarbonyl directing group promoted over-alkylation.
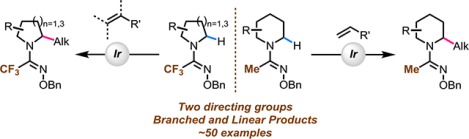
These limitations highlight the challenge associated with designing a practical, as well as a general directing group. Herein, we disclose the design and discovery of novel amidoxime directing groups for an iridium(I)-catalyzed α-C(sp3)–H alkylation of saturated azacycles (Scheme 2C). The amidoxime directing groups are compatible with a wide range of saturated azacycles and olefin coupling partners, while also being easy to install and remove. During our study, we observed an unprecedented selectivity for branched α-C(sp3)-alkylation products with ethyl acrylate as the olefin coupling partner.22 This observation showcases one of the advantages of using a directed C‒H activation approach, wherein a new directing group may impact the reactivity of the catalytic intermediate, thus enabling access to new mechanistic pathways and products.
RESULTS AND DISCUSSION
We designed the amidoxime directing groups (Scheme 3) based on the following considerations:1) an imine moiety to direct the metal insertion, inspired by the high reactivity afforded by heterocyclic directing groups in Scheme 2A, 2) amidoxime moiety to allow easy removal under mild conditions (in comparison with previous reports of amidine directing groups5a,9a), and 3) a modular α-substituent which can be tuned to improve the reactivity and selectivity.
Scheme 3.
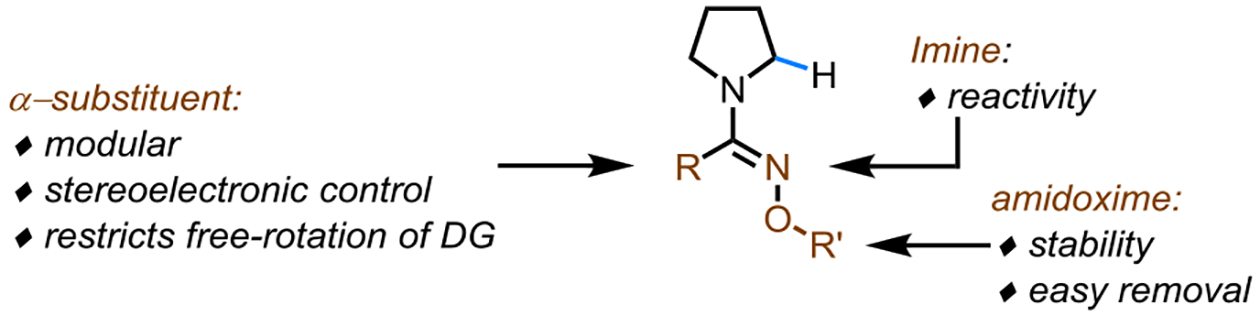
Design Principles of Amidoxime Directing Groups.
Borrowing reaction conditions from our previous work on iridium(I)-catalyzed α-alkylation of pyrrolidines with an alkoxy-thiocarbamate directing group,21 we evaluated various amidoxime directing groups using pyrrolidine as the model substrate and ethyl acrylate as the olefin coupling partner (Table 1). We began our studies with an α-methyl substituted O-benzyl amidoxime directing group and obtained the α-alkylated product in a total yield of 29% (2a-1) as a mixture of branched (B) and linear (L) regioisomers. Keeping the oxime moiety constant, we changed the αsubstituent of the directing group to a bulkier tert-butyl group and observed an increase in the total yield to 67% (2a-2). The introduction of an electron-withdrawing trifluoromethyl group as the αsubstituent further increased the total yield to 86% (2a-3). On the other hand, using a perfluoroethyl group as the α-substituent lowered the total yield to 60% (2a-4). Next, we varied the oxime moiety while fixing the α-substituent as a trifluoromethyl group. Changing the benzyl oxime to a sterically bulky trityl oxime lowered the total yield to 60% (2a-5). Next, we altered the electronics of the oxime moiety by using a para-nitro benzyl oxime which gave a slightly lower total yield of 81% (2a-6) as compared to the simple benzyl oxime. Increasing the electron-withdrawing nature of the oxime moiety further by using a perfluorobenzyl oxime reduced the total yield to 48% (2a-7). In order to test the importance of a pendant benzyl oxime unit, we tied it into a benzoxazole heterocycle and observed a drop in the total yield to 40% (2a-8). Increasing the coordination ability of the pendant oxime unit with a weakly coordinating ester afforded product in 70% total yield (2a-9), while a strongly coordinating pyridine completely shut down the reaction (2a-10). Next, reaction using an O-phenyl amidoxime directing group suffered from poor mass balance and no desired product was observed (2a-11). Employing a tert-butanesulfinyl imine as the directing group did not give any product (2a-12). We next screened the reaction conditions with trifluoromethyl O-benzyl amidoxime as the optimal directing group, and were able to lower the catalyst loading to 5 mol% and the ethyl acrylate loading to 4 equivalents to afford a total yield of 83% (2a-3’) of α-alkylated pyrrolidine products. As seen from Table 1, the regioisomeric product ratio was found to be dependent on the structure of the directing group. Attempts at improving the regioisomeric product ratio by further screening of the reaction conditions were unsuccessful. Moreover, the regioisomeric ratio was affected by the identity of solvent, the catalyst counteranion, and the diene ligand on iridium (see the Supporting Information for details).
Table 1.
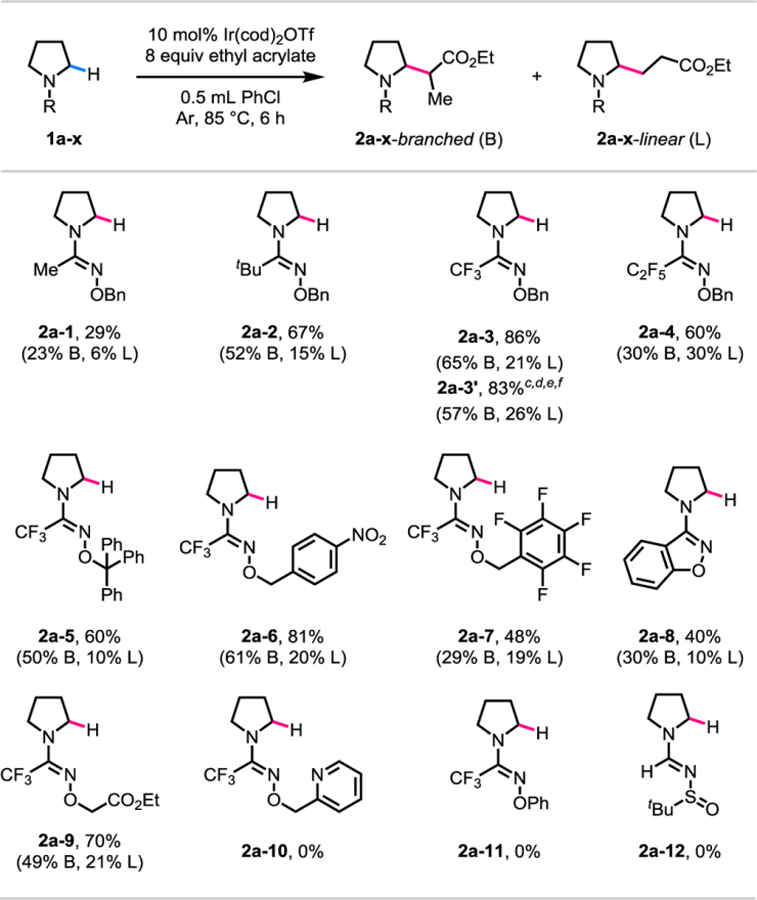 |
Reaction conditions: 1a-1 to 1a-11 (0.1 mmol, 1.0 equiv), Ir(cod)2OTf (0.01 mmol, 0.1 equiv), ethyl acrylate (0.8 mmol, 8.0 equiv), degassed PhCl (0.5 mL), 85 °C, under Ar, 6 h.
Yields were determined by 1H NMR analysis of the crude products using mesitylene as the internal standard.
0.05 equiv of Ir(cod)2OTf (0.005 mmol).
4.0 equiv of ethyl acrylate (0.4 mmol).
0.1 mL of degassed PhCl.
Yield after isolation by chromatography is shown.
We then tested the trifluoromethyl O-benzyl amidoxime directing group against a variety of substituted pyrrolidine substrates (Table 2). When ethyl acrylate was used as the olefin coupling partner, separable mixtures of branched and linearly α-alkylated products were obtained (2b-2i), the ratios of which were dependent on the nature of substituents on the pyrrolidine substrates. On the other hand, when 1-hexene was used as the olefin coupling partner, only linearly α-alkylated products were obtained (2j-2m). With ethyl acrylate as the olefin coupling partner, 2-methyl and 2-phenyl substituted pyrrolidine substrates afforded products in total yields of 80% (2b) and 83% (2c), respectively. A benzyl protected proline substrate was also compatible and afforded product in a total yield of 68% (2d). Spirocyclic and bicyclic pyrrolidine substrates, which are relevant to various medicinal chemistry campaigns,23 reacted in good yields of 98% (2e) and 80% (2f), respectively. Pyrrolidine substrates with 3-alkyl substituents also reacted in good yields (2g, 2h). On the other hand, an electron deficient 3,3-difluoropyrrolidine substrate gave a low yield of 36% (2i). With 1-hexene as the olefin coupling partner, 3-phenyl, 3-alkyl, 3-methoxy, and 3-amino substituted pyrrolidine substrates afforded products in 73% (2j), 86% (2k), 66% (2l), and 77% (2m) yields, respectively. We were pleased to find that the use of trifluoromethyl O-benzyl amidoxime directing group (with adequate substrate-specific tuning of the reaction conditions) prevented over-alkylation of a variety of substituted pyrrolidine substrates, thus addressing one of the major limitations with previous directing group designs.9,10
Table 2.
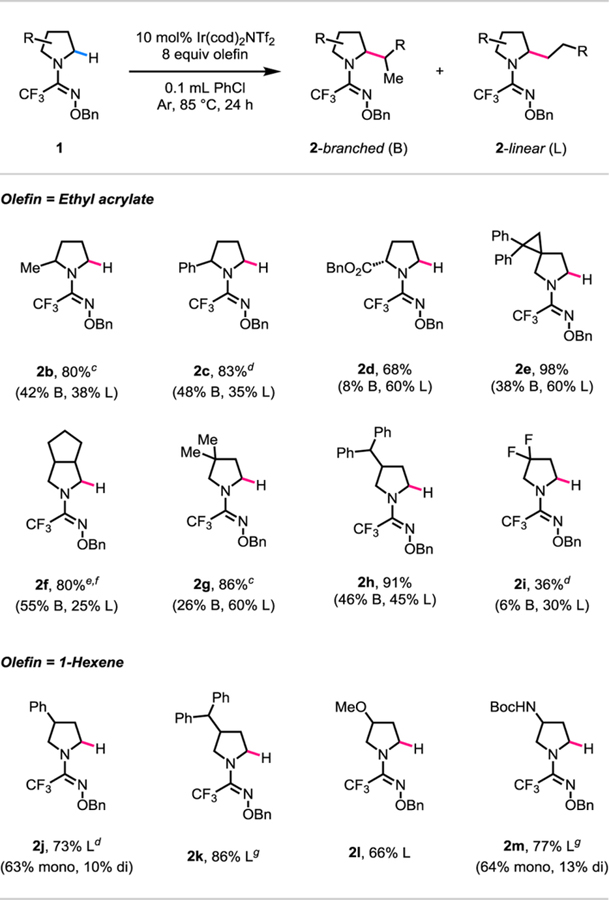 |
Reaction conditions: 1b to 1m (0.1 mmol, 1.0 equiv), Ir(cod)2NTf2 (0.01 mmol, 0.1 equiv), ethyl acrylate (0.8 mmol, 8.0 equiv), degassed PhCl (0.1 mL), 85 °C, under Ar, 24 h.
Yields after isolation by chromatography are shown.
Ir(cod)2OTf instead of Ir(cod)2NTf2.
0.1 equiv of HBF4.Et2O (0.01 mmol) as additive.
0.05 equiv of Ir(cod)2OTf (0.005 mmol).
12 h instead of 24 h.
48 h instead of 24 h.
Having established the scope of pyrrolidine substrates with the trifluoromethyl O-benzyl amidoxime directing group, we next investigated the scope of olefin coupling partners using 3,3dimethyl pyrrolidine (1g) as the standard substrate (Table 3). Olefins with a wide variety of steric and electronic properties proved to be efficient coupling partners. A wide range of styrene analogues, including 4-bromostyrene, afforded the respective linearly α-alkylated products in good yields of 66%−81% (3a-3i). Electron deficient olefins such as vinyl phthalimide and ethyl vinyl ketone also proceeded to give linearly α-alkylated products in 78% (3j) and 65% (3k) yields, respectively. Alternate ester protecting groups on the acrylate were well tolerated (3l) to give a mixture of branched and linearly α-alkylated regioisomeric products. However, vinyl acetate reacted in a low yield of 32% (3m) to give linearly α-alkylated product. Electron neutral olefins all gave their corresponding linearly α-alkylated products selectively. While allylbenzene (3n) reacted in a moderate yield of 51%, allyl silane, vinyl silane, and 1-hexene reacted in good yields of 71% (3o), 74% (3p), and 73% (3q), respectively. When vinyl norbornene was used as the olefin coupling partner, the reaction occurred regioselectively at the vinyl position in 73% yield (3r). Moreover, a selectivity of 6.1:1 was observed in favor of the endo-isomer over the exo-isomer of vinyl norbornene. In contrast, in the absence of a vinyl group, simple norbornene reacted in 78% yield (3s). The reaction also tolerated di-substituted terminal olefins in moderate yields of 55% (3t) and 50% (3u), respectively. When cis-2-hexene was used as the olefin coupling partner, an isomerized linear product was obtained in a moderate yield of 51% (3v). We also observed isomerization of methyl crotonate to give a linear product, albeit in a low yield of 35% (3w).24 A sterically hindered maleate ester also reacted in 23% yield (3x). To the best of our knowledge, this is the first example of an iridium(I)-catalyzed α-C(sp3)‒H alkylation reaction which can utilize di-substituted terminal olefins and internal olefins as effective coupling partners.
Table 3.
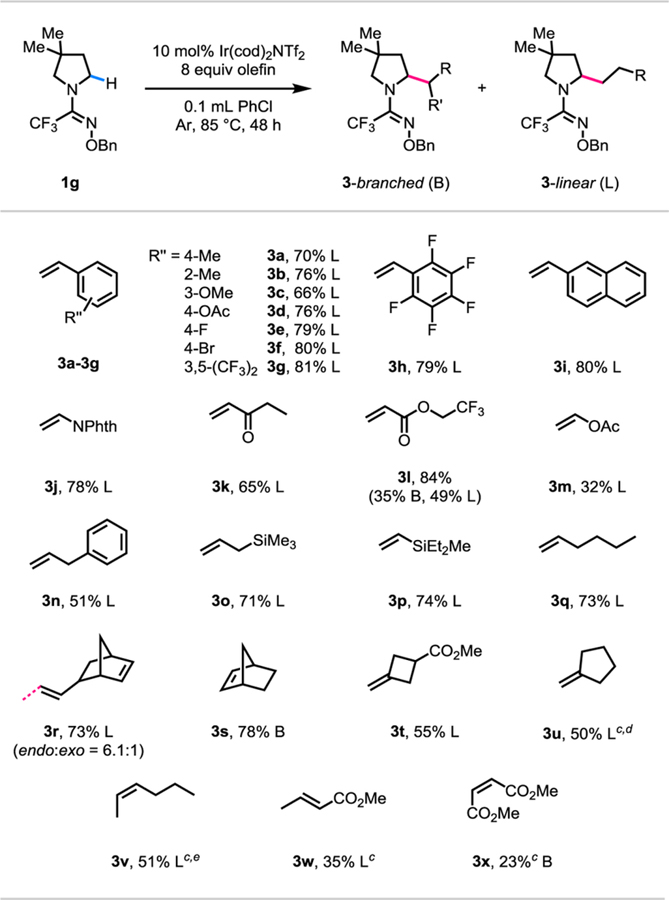 |
Reaction conditions: 1g (0.1 mmol, 1.0 equiv), Ir(cod)2NTf2 (0.01 mmol, 0.1 equiv), olefin coupling partner (0.8 mmol, 8.0 equiv), degassed PhCl (0.1 mL), 85 °C, under Ar, 48 h.
Yields after isolation by chromatography are shown.
1a instead of 1g.
mono:di = 4:1.
mono:di = 7.5:1.
After establishing the performance of the trifluoromethyl O-benzyl amidoxime directing group with a variety of substituted pyrrolidine substrates and olefin coupling partners, we next tested the efficacy of this directing group for azacycles of other ring sizes (Scheme 4). When we subjected the azetidine substrate (1n) to the optimized reaction conditions with ethyl acrylate as the olefin coupling partner, no product was observed. The piperidine substrate afforded a linearly α-alkylated product in a low yield of 25% (2o). In contrast, the azepane substrate reacted in a good yield of 70% (2p) and afforded a mixture of separable branched and linearly α-alkylated regioisomers. A similar reactivity trend has been observed in previous reports where piperidines were found to be a more challenging class of substrates than pyrrolidines and azepanes.
Scheme 4.
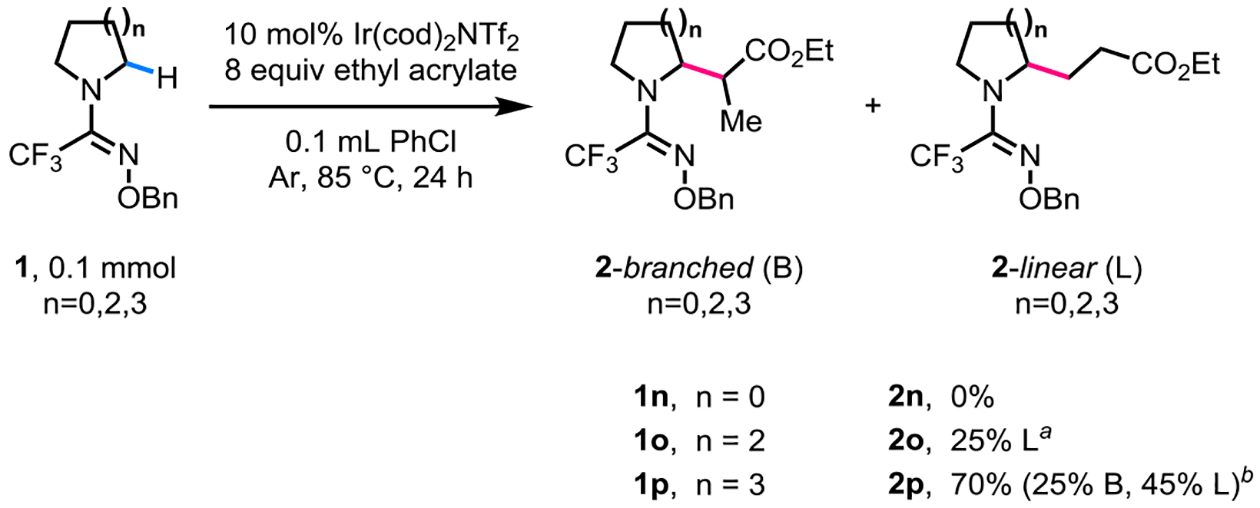
Evaluation of the Trifluoromethyl O-Benzyl Amidoxime Directing Group for α-C(sp3)‒H Alkylation of Azetidine, Piperidine, and Azepane. aYield was determined by 1H NMR analysis of the crude product using mesitylene as the internal standard. bYield after isolation by chromatography is shown.
We reasoned that a lower reactivity for the piperidine substrate (2o) relative to the pyrrolidine and azepane substrates might be associated with an unfavorable conformation of the six-membered saturated azacycle. This led us to consider that a different directing group might be required for the piperidine substrate. Leveraging the modular nature of the amidoxime directing groups, we reevaluated directing groups for the piperidine substrate as shown in Table 4. Under the optimized reaction conditions, with ethyl acrylate as the olefin coupling partner, changing the α-substituent to a perfluoroethyl group had deleterious effect on reactivity (4a-1). On the other hand, a variety of alkyl groups such as, methyl (4a-2), ethyl (4a-3), and isobutyl (4a-4), all worked as efficient αsubstituents and gave a mixture of branched and linearly α-alkylated regioisomers in moderate yields. A bulkier alkyl α-substituent in the directing group led to a higher yield of the linearly αalkylated regioisomer. We selected methyl O-benzyl amidoxime as the optimal directing group for piperidines because we anticipated that a smaller directing group would have an easier removal protocol.
Table 4.
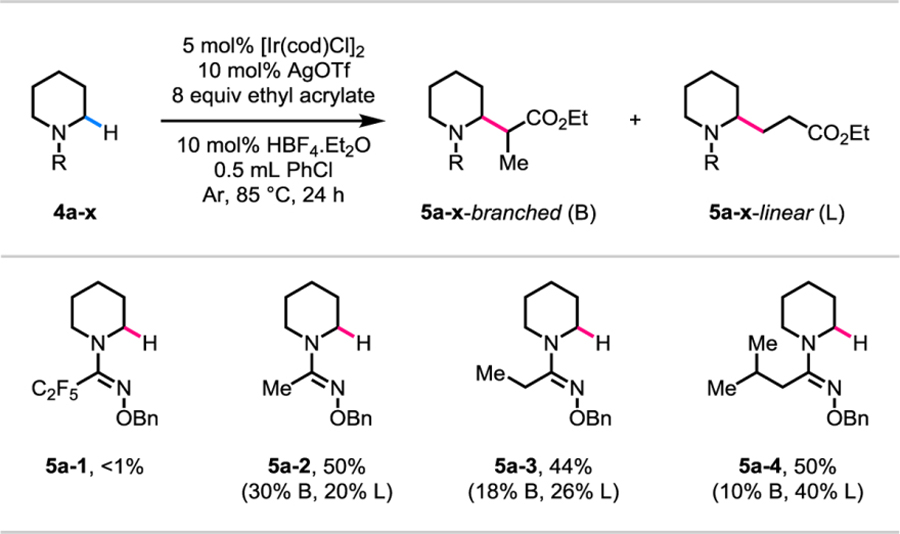 |
Reaction conditions: 4a-1 to 4a-4 (0.1 mmol, 1.0 equiv), [Ir(cod)Cl]2 (0.005 mmol, 0.05 equiv), AgOTf (0.01 mmol, 0.1 equiv), ethyl acrylate (0.8 mmol, 8.0 equiv), HBF4.Et2O (0.01 mmol, 0.1 equiv), degassed PhCl (0.5 mL), 85 °C, under Ar, 24 h.
Yields were determined by 1H NMR analysis of the crude products using mesitylene as the internal standard.
With the optimal directing group for piperidine in hand, we further optimized the reaction conditions using 1-hexene as the olefin coupling partner (see the Supporting Information for details). As shown in Table 5, simple piperidine reacted to give a separable mixture of mono-and di-alkylated products in a total yield of 65% (5b). Next, we tested the methyl O-benzyl amidoxime directing group against a variety of substituted piperidine substrates (5c-5j). 3-Methyl piperidine substrate reacted to give mono-alkylated product in 72% yield (5c). 4-Methyl and 4-phenyl substituted piperidine substrates also reacted in 82% (5d) and 62% (5e) yields, respectively, giving a mixture of separable mono-and di-alkylated products. Piperidine substrates containing various functional groups such as 4-methoxy (5f), 4-amino (5g), 3-trifluoromethyl (5h), and 3-ester (5i) were also compatible and reacted in moderate to good yields. A spirocyclic piperidine substrate yielded a separable mixture of mono-and di-alkylated products in a total yield of 58% (5j). Tetrahydroisoquinoline was also a compatible substrate and reacted to give di-alkylated product in 53% yield (5k). 3-Methyl morpholine was a challenging substrate and gave the corresponding mono-alkylated product in a low yield of 20% (5l). However, complete site-selectivity was achieved for the α-C(sp3)‒H bonds next to the nitrogen atom in the presence of the α-C(sp3)‒H next to an oxygen atom. Finally, the di-alkylation favored anti-stereochemistry; where 5b-di, 5d-di, and 5k were isolated as single diastereomers (see the Supporting Information for details on diastereoselectivity).
Table 5.
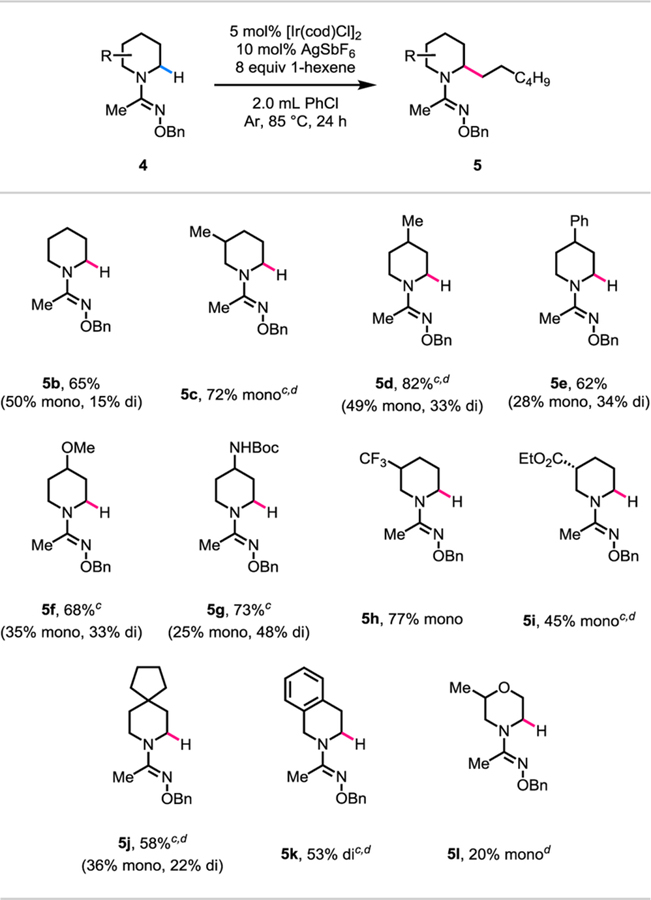 |
Reaction conditions: 4b to 4l (0.1 mmol, 1.0 equiv), [Ir(cod)Cl]2 (0.005 mmol, 0.05 equiv), AgSbF6 (0.01 mmol, 0.1 equiv), 1-hexene (0.8 mmol, 8.0 equiv), degassed PhCl (2.0 mL), 85 °C, under Ar, 24 h.
Yields after isolation by chromatography are shown.
0.1 equiv of Ir(cod)2NTf2 (0.01 mmol) instead of [Ir(cod)Cl]2 and AgSbF6.
48 h instead of 24 h.
Scheme 5A shows the one-step installation protocols developed for both the trifluoromethyl O-benzyl amidoxime and the methyl O-benzyl amidoxime directing groups starting from precursors 6 and 7 (see the Supporting Information for details). These precursors were used for the synthesis of the azacycle substrates (1a-3, 1b-1p, 4a-2, 4b-4l) in a divergent manner. Scheme 5B shows the removal protocols for two representative products, 2k and 5h. Both the directing groups were cleaved effectively under DIBAL-H reduction at 0 °C in a single step.
Scheme 5.
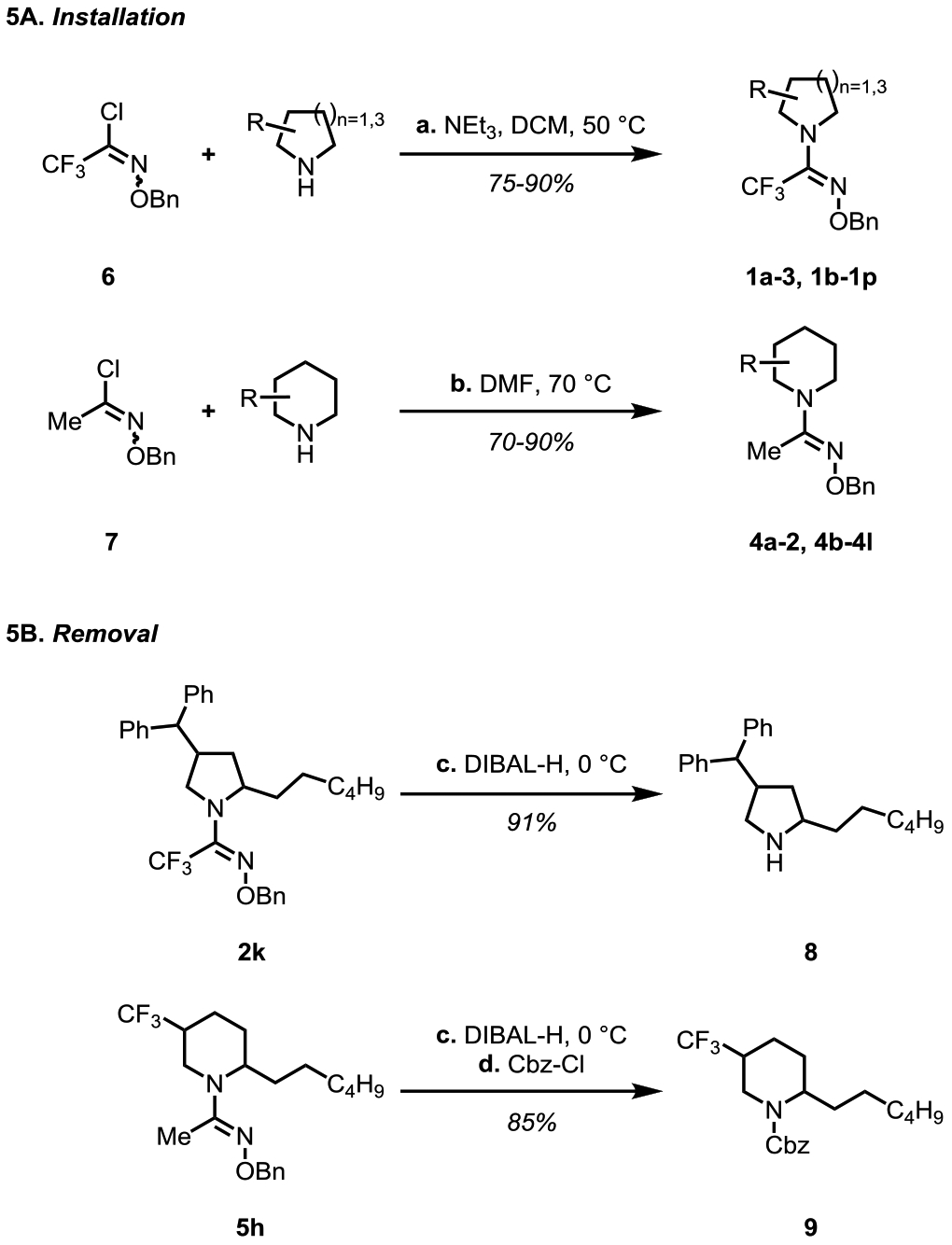
Installation and Removal of Amidoxime Directing Groups.a aReaction conditions: (a) Azacycle (1.0 equiv), 6 (1.2 equiv), triethylamine (1.2 equiv), DCM, 50 °C, under air, 12 h. (b) Azacycle (1.0 equiv), 7 (1.2 equiv), DMF, 70 °C, under air, 12 h. (c) 2k or 5h (0.1 mmol, 1.0 equiv), DIBAL-H (0.5 mmol, 5.0 equiv), toluene (0.5 mL), 0 °C, under N2, 30 min. (d) CbzCl (0.3 mmol, 3.0 equiv), Et3N (0.3 mmol, 3.0 equiv), DCM (1.0 mL), rt, under N2, 12 h.
Due to the observation of unconventional branch-selective α-alkylation with acrylates, we performed deuterium labelling experiments (Scheme 6). When substrate 1a-3 was subjected to the reaction conditions in the presence of D2O and in the absence of an olefin coupling partner, deuterium incorporation at the α-and α’-positions of the pyrrolidine substrate was observed (Scheme 6A). This result implies that the α-C(sp3)‒H bond of 1a-3 is cleaved under the present reaction conditions, without the involvement of an olefin coupling partner. Next, the reaction of substrate 1a-3 with deuterated benzyl acrylate was examined (Scheme 6B). Incorporation of deuterium atoms was detected at the α-and α’-positions of both the branched and linearly alkylated products (3y-B, 3y-L). Moreover, the recovered benzyl acrylate coupling partner was found to have a reduced deuterium content. These observations indicate that the catalytic cycle consists of reversible C‒H bond activation and olefin insertion steps. The experimentally observed isomerization of internal olefins to give linearly α-alkylated products (3v, 3w) lends further support for a reversible olefin insertion step.
Scheme 6.
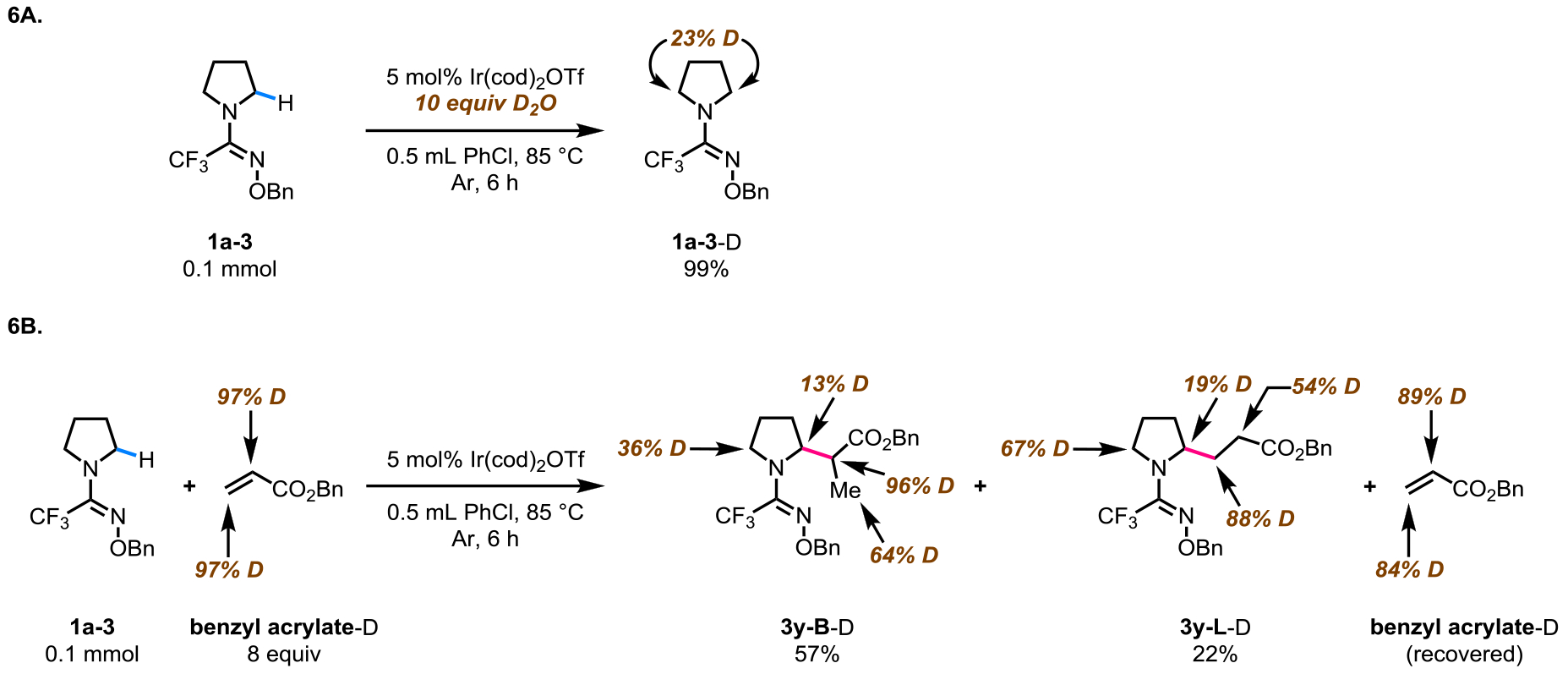
Deuterium labelling experiments.
On the basis of the above labelling experiments, the proposed mechanism for the amidoxime-directed Ir(I)-catalyzed α-C(sp3)‒H alkylation reaction of pyrrolidine with an acrylate coupling partner is shown in Scheme 7. Since the reaction yield and selectivity were found to be dependent on the diene ligand (see the Supporting Information), it is likely that one unit of the diene ligand remains coordinated to iridium.25 The first step involves the directed α-C‒H activation of the pyrrolidine substrate via an oxidative addition mechanism, leading to the formation of a cationic Ir(III) intermediate. Next, a molecule of acrylate may react reversibly with this iridium-hydride species in two different ways - thus giving rise to Pathways L and B. In Pathway L, acrylate undergoes a sterically-controlled 1,2-migratory insertion into the Ir‒H bond, leading to a linear Ir-alkyl species, which upon a C‒C reductive elimination gives the linearly α-alkylated product. In Pathway B, acrylate undergoes an electronically-controlled 2,1-migratory insertion into the Ir‒H bond leading to a branched Ir-alkyl species, which upon a C‒C reductive elimination gives the branched α-alkylated product.
Scheme 7.
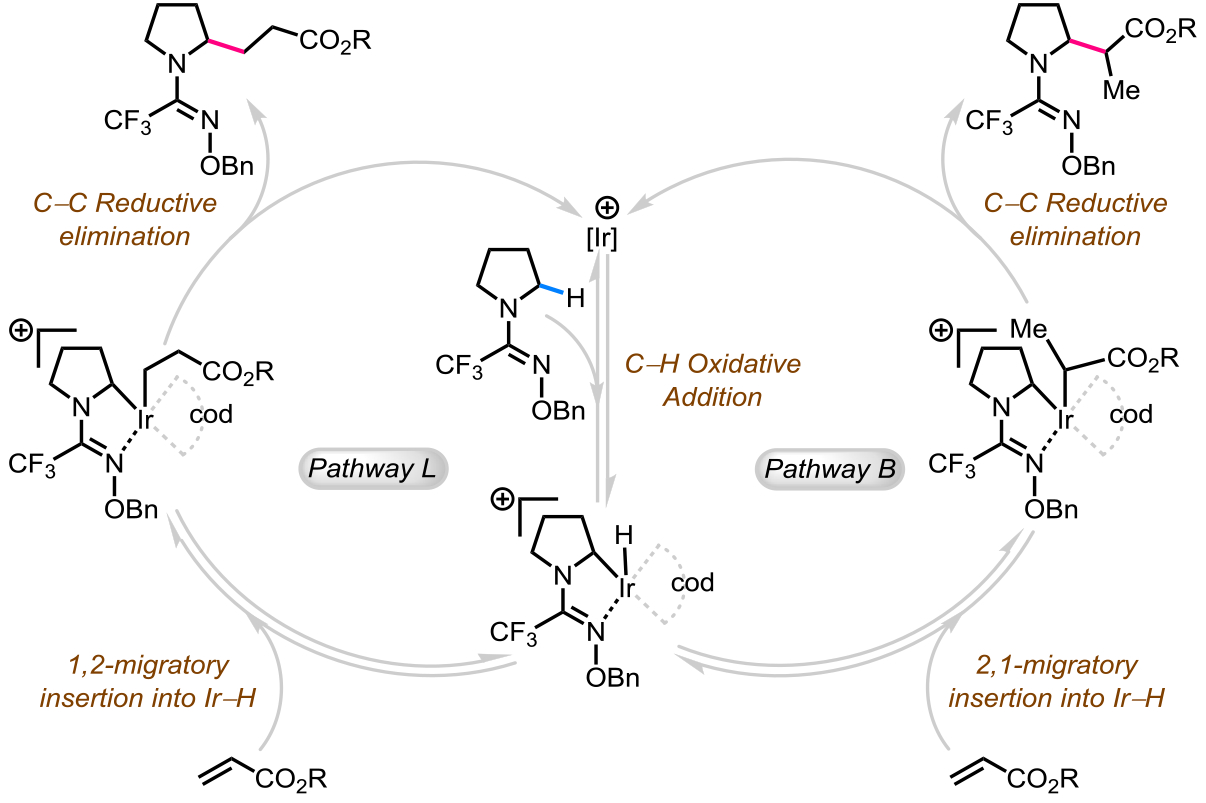
Proposed Mechanistic Pathways.
CONCLUSION
In summary, we report the design and discovery of novel amidoxime directing groups for the iridium(I)-catalyzed α-C(sp3)‒H alkylation of saturated azacycles using readily available olefins as coupling partners. A trifluoromethyl O-benzyl amidoxime directing group was developed for substituted pyrrolidines, proline, and azepane substrates. This transformation is applicable on wide arrays of olefin coupling partners with diverse steric and electronic properties, including previously unreactive di-substituted terminal olefins and internal olefins. For more challenging substrates, such as substituted piperidines and tetrahydroisoquinoline, a methyl O-benzyl amidoxime directing group was developed. The selectivity for a branched α-C(sp3)‒H alkylation product is observed for the first time when acrylate was used as the reaction partner. New protocols enabling practical, one-step installation and removal of these directing groups were also developed. Future work from our group will focus on developing enantioselective α-C(sp3)‒ H alkylation of saturated azacycles and on exploiting the potential of amidoxime directing groups for other interesting substrates and transformations.
EXPERIMENTAL SECTION
A 2-dram vial was charged with the substrate (0.1 mmol, 1.0 equiv) and taken inside an argon glovebox. Ir(cod)2NTf2 (6.9 mg, 0.01 mmol, 0.1 equiv, unless otherwise noted) was added followed by a magnetic stir bar. The vial was sealed with a PTFE septum and taken out of the glovebox. Degassed PhCl (0.1 mL, unless otherwise noted) and olefin coupling partner (0.8 mmol, 8.0 equiv, unless otherwise noted) were added to the vial. The solution was stirred at 85 °C for 24 hours (unless otherwise noted). Upon completion, the reaction mixture was cooled to rt and diluted with 2 mL EtOAc. The mixture was filtered through a pad of celite. The celite was washed thoroughly with EtOAc and the combined organics were concentrated in vacuo. The crude reside was purified by preparative TLC to provide the alkylated product(s).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS, R01 GM084019), and Bristol-Myers Squibb for financial support. We thank Dr. Jason Chen, Ms. Brittany Sanchez, and Ms. Emily Sturgell from Automated Synthesis Facility, The Scripps Research Institute, for their assistance with HRMS analysis and compound purification. Helpful advice on NMR spectroscopy from Dr. Laura Pasternack (TSRI), Dr. Dee-Hua Huang (TSRI), and Dr. Gerard J. Kroon is acknowledged. P.V. thanks Mr. Hojoon Park (TSRI) for helpful discussions.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Detailed experimental procedures and characterization of new compounds (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752. [DOI] [PubMed] [Google Scholar]; (b) Murray CW; Rees DC Opportunity Knocks: Organic Chemistry for Fragment-Based Drug Discovery (FBDD). Angew. Chem. Int. Ed 2016, 55, 488. [DOI] [PubMed] [Google Scholar]; (c) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10, 383. [DOI] [PubMed] [Google Scholar]
- 3.(a) Campos KR Direct sp3 C–H Bond Activation Adjacent to Nitrogen in Heterocycles. Chem. Soc. Rev 2007, 36, 1069. [DOI] [PubMed] [Google Scholar]; (b) Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW Direct α-Functionalization of Saturated Cyclic Amines. Chem. Eur. J 2012, 18, 10092. [DOI] [PubMed] [Google Scholar]
- 4.(a) Jovel I; Prateeptongkum S; Jackstell R; Vogl N; Weckbecker C; Beller M α-Functionalization of Non-activated Aliphatic Amines: Ruthenium-catalyzed Alkynylations and Alkylations. Chem. Commun 2010, 46, 1956. [DOI] [PubMed] [Google Scholar]; (b) Dai C; Meschini F; Narayanam JMR; Stephenson CRJ Friedel–Crafts Amidoalkylation via Thermolysis and Oxidative Photocatalysis. J. Org. Chem 2012, 77, 4425. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Girard SA; Knauber T; Li C-J The Cross-Dehydrogenative Coupling of C–H Bonds: A Versatile Strategy for C–C Bond Formations. Angew. Chem. Int. Ed 2014, 53, 74. [DOI] [PubMed] [Google Scholar]; (d) Seidel D The Azomethine Ylide Route to Amine C–H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc. Chem. Res 2015, 48, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa M C–H Functionalization of Amines via Alkene-Derived Nucleophiles through Cooperative Action of Chiral and Achiral Lewis Acid Catalysts: Applications in Enantioselective Synthesis. J. Am. Chem. Soc 2018, 140, 10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Meyers AI; Edwards PD; Rieker WF; Bailey TR Alpha-amino Carbanions via Formamidines. Alkylation of Pyrrolidines, Piperidines, and Related Heterocycles. J. Am. Chem. Soc 1984, 106, 3270. [Google Scholar]; (b) Beak P; Kerrick ST; Wu S; Chu J Complex Induced Proximity Effects: Enantioselective Syntheses Based on Asymmetric Deprotonations of N-Boc-pyrrolidines. J. Am. Chem. Soc 1994, 116, 3231. [Google Scholar]; (c) Campos KR; Klapars A; Waldman JH; Dormer PG; Chen C-Y Enantioselective, Palladium-Catalyzed α-Arylation of N-Bocpyrrolidine. J. Am. Chem. Soc 2006, 128, 3538. [DOI] [PubMed] [Google Scholar]; (d) Beng TK; Gawley RE Highly Enantioselective Catalytic Dynamic Resolution of N-Boc-2-lithiopiperidine: Synthesis of (R)-(+)-N-Boc-Pipecolic Acid, (S)-(−)-Coniine, (S)-(+)-Pelletierine, (+)-β-Conhydrine, and (S)-(−)Ropivacaine and Formal Synthesis of (−)-Lasubine II and (+)-Cermizine C. J. Am. Chem. Soc 2010, 132, 12216. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hodgson DM; Kloesges J Lithiation–Electrophilic Substitution of NThiopivaloylazetidine. Angew. Chem. Int. Ed 2010, 49, 2900. [DOI] [PubMed] [Google Scholar]; (f) Seel S; Thaler T; Takatsu K; Zhang C; Zipse H; Straub BF; Mayer P; Knochel P Highly Diastereoselective Arylations of Substituted Piperidines. J. Am. Chem. Soc 2011, 133, 4774. [DOI] [PubMed] [Google Scholar]; (g) Cordier CJ; Lundgren RJ; Fu GC Enantioconvergent Cross-Couplings of Racemic Alkylmetal Reagents with Unactivated Secondary Alkyl Electrophiles: Catalytic Asymmetric Negishi α-Alkylations of N-Boc-pyrrolidine. J. Am. Chem. Soc 2013, 135, 10946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liniger M; Estermann K; Altmann K-H Total Synthesis of Hygrolines and Pseudohygrolines. J. Org. Chem 2013, 78, 11066. [DOI] [PubMed] [Google Scholar]; (i) Firth JD; O’Brien P; Ferris L Synthesis of Enantiopure Piperazines via Asymmetric Lithiation–Trapping of N-Boc Piperazines: Unexpected Role of the Electrophile and Distal N-Substituent. J. Am. Chem. Soc 2016, 138, 651. [DOI] [PubMed] [Google Scholar]; (j) Lin W; Zhang K-F; Baudoin O Regiodivergent enantioselective C–H functionalization of Boc-1,3-oxazinanes for the synthesis of β2-and β3amino acids. Nat. Catal 2019, 2, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Booth SE; Benneche T; Undheim K Samarium Diiodide Mediated Alkylation of Saturated Heterocycles Alpha to Nitrogen. Tetrahedron 1995, 51, 3665. [Google Scholar]; (b) Bertrand S; Glapski C; Hoffmann N; Pete J-P Highly Efficient Photochemical Addition of Tertiary Amines to Electron Deficient Alkenes. Diastereoselective Addition to (5R)-5-menthyloxy-2[5H]-furanone. Tetrahedron Lett. 1999, 40, 3169. [Google Scholar]; (c) Catino AJ; Nichols JM; Nettles BJ; Doyle MP The Oxidative Mannich Reaction Catalyzed by Dirhodium Caprolactamate. J. Am. Chem. Soc 2006, 128, 5648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yoshikai N; Mieczkowski A; Matsumoto A; Ilies L; Nakamura E Iron-Catalyzed C–C Bond Formation at α-Position of Aliphatic Amines via C–H Bond Activation through 1,5-Hydrogen Transfer. J. Am. Chem. Soc 2010, 132, 5568. [DOI] [PubMed] [Google Scholar]; (e) McNally A; Prier CK; MacMillan DWC Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-catalysed sp3–sp3 Cross-coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general Alkyl-alkyl Cross-coupling Enabled by Redox-active Esters and Alkylzinc Reagents. Science 2016, 352, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Xie J; Yu J; Rudolph M; Rominger F; Hashmi ASK Monofluoroalkenylation of Dimethylamino Compounds through Radical–Radical Cross-Coupling. Angew. Chem. Int. Ed 2016, 55, 9416. [DOI] [PubMed] [Google Scholar]; (i) Thullen SM; Rovis T A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System. J. Am. Chem. Soc 2017, 139, 15504. [DOI] [PubMed] [Google Scholar]; (j) McManus JB; Onuska NPR; Nicewicz DA Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 9056. [DOI] [PubMed] [Google Scholar]
- 7.Davies HML; Hansen T; Hopper DW; Panaro SA Highly Regio-, Diastereo-, and Enantioselective C–H Insertions of Methyl Aryldiazoacetates into Cyclic N-Boc-Protected Amines. Asymmetric Synthesis of Novel C2-Symmetric Amines and threo-Methylphenidate. J. Am. Chem. Soc 1999, 121, 6509. [Google Scholar]
- 8.(a) Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Oxidative Diversification of Amino Acids and Peptides by Small-molecule Iron Catalysis. Nature 2016, 537, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen W; Ma L; Paul A; Seidel D Direct α-C–H Bond Functionalization of Unprotected Cyclic Amines. Nat. Chem 2017, 10, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Paul A; Seidel D α-Functionalization of Cyclic Secondary Amines: Lewis Acid Promoted Addition of Organometallics to Transient Imines. J. Am. Chem. Soc 2019, 141, 8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For arylation, see:; (a) Pastine SJ; Gribkov DV; Sames D sp3 C–H Bond Arylation Directed by Amidine Protecting Group: α-Arylation of Pyrrolidines and Piperidines. J. Am. Chem. Soc 2006, 128, 14220. [DOI] [PubMed] [Google Scholar]; (b) Prokopcová H; Bergman SD; Aelvoet K; Smout V; Herrebout W; Van der Veken B; Meerpoel L; Maes BUW C-2 Arylation of Piperidines through Directed Transition-Metal-Catalyzed sp3 C–H Activation. Chem. Eur. J 2010, 16, 13063. [DOI] [PubMed] [Google Scholar]; (c) Dastbaravardeh N; Schnürch M; Mihovilovic MD Ruthenium(0)-Catalyzed sp3 C–H Bond Arylation of Benzylic Amines Using Arylboronates. Org. Lett 2012, 14, 1930. [DOI] [PubMed] [Google Scholar]; (d) Kumar NYP; Jeyachandran R; Ackermann L C(sp3)–H Bond Arylations Catalyzed by Well-Defined [Ru(O2CMes)2(p-cymene)]. J. Org. Chem 2013, 78, 4145. [DOI] [PubMed] [Google Scholar]; (e) Greßies S; Klauck FJR; Kim JH; Daniliuc CG; Glorius F Ligand-Enabled Enantioselective C–H Activation of Tetrahydroquinolines and Saturated Aza-Heterocycles by RhI. Angew. Chem. Int. Ed 2018, 57, 9950. [DOI] [PubMed] [Google Scholar]; (f) Jiang H-J; Zhong X-M; Yu J; Zhang Y; Zhang X; Wu Y-D; Gong L-Z Assembling a Hybrid Pd Catalyst from a Chiral Anionic CoIII Complex and Ligand for Asymmetric C(sp3)–H Functionalization. Angew. Chem. Int. Ed 2019, 58, 1803. [DOI] [PubMed] [Google Scholar]
- 10.For Ru-catalyzed alkylation, see:; (a) Jun C-H Chelation-assisted Alkylation of Benzylamine Derivatives by Ru0 Catalyst. Chem. Commun 1998, 1405. [Google Scholar]; (b) Chatani N; Asaumi T; Yorimitsu S; Ikeda T; Kakiuchi F; Murai S Ru3(CO)12-Catalyzed Coupling Reaction of sp3 C–H Bonds Adjacent to a Nitrogen Atom in Alkylamines with Alkenes. J. Am. Chem. Soc 2001, 123, 10935. [DOI] [PubMed] [Google Scholar]; (c) Bergman SD; Storr TE; Prokopcová H; Aelvoet K; Diels G; Meerpoel L; Maes BUW The Role of the Alcohol and Carboxylic Acid in Directed Ruthenium-Catalyzed C(sp3)–H α-Alkylation of Cyclic Amines. Chem. Eur. J 2012, 18, 10393. [DOI] [PubMed] [Google Scholar]; (d) Kulago AA; Van Steijvoort BF; Mitchell EA; Meerpoel L; Maes BUW Directed Ruthenium-Catalyzed C(sp3)–H α-Alkylation of Cyclic Amines Using Dioxolane-Protected Alkenones. Adv. Synth. Catal 2014, 356, 1610. [Google Scholar]; (e) Schinkel M; Wang L; Bielefeld K; Ackermann L Ruthenium(II)-Catalyzed C(sp3)–H α-Alkylation of Pyrrolidines. Org. Lett 2014, 16, 1876. [DOI] [PubMed] [Google Scholar]
- 11.For Ir-catalyzed alkylation, see:; (a) Pan S; Endo K; Shibata T Ir(I)-Catalyzed Enantioselective Secondary sp3 C–H Bond Activation of 2-(Alkylamino)pyridines with Alkenes. Org. Lett 2011, 13, 4692. [DOI] [PubMed] [Google Scholar]; (b) Lahm G; Opatz T Unique Regioselectivity in the C(sp3)–H α-Alkylation of Amines: The Benzoxazole Moiety as a Removable Directing Group. Org. Lett 2014, 16, 4201. [DOI] [PubMed] [Google Scholar]; (c) Tahara Y-K; Michino M; Ito M; Kanyiva KS; Shibata T Enantioselective sp3 C–H alkylation of γ-butyrolactam by a chiral Ir(I) catalyst for the synthesis of 4-substituted γ-amino acids. Chem. Commun 2015, 51, 16660. [DOI] [PubMed] [Google Scholar]; (d) Nagai M; Nagamoto M; Nishimura T; Yorimitsu H Iridium-catalyzed sp3 C–H Alkylation of 3-Carbonyl-2-(alkylamino)pyridines with Alkenes. Chem. Lett 2017, 46, 1176. [Google Scholar]; (e) Yamauchi D; Nishimura T; Yorimitsu H Hydroxoiridium-Catalyzed Hydroalkylation of Terminal Alkenes with Ureas by C(sp3)−H Bond Activation. Angew. Chem. Int. Ed 2017, 56, 7200. [DOI] [PubMed] [Google Scholar]; (f) Hattori H; Nishimura T Iridium-Catalyzed Sequential sp3 C–H Alkylation of an N-Methyl Group with Alkenes Towards the Synthesis of α-Substituted Amines. Adv. Synth. Catal 2018, 360, 4827. [Google Scholar]; (g) Nakamura I; Yamauchi D; Nishimura T Hydroxoiridium-Catalyzed sp3 C–H Alkylation of Indoline Derivatives with Terminal Alkenes. Asian J. Org. Chem 2018, 7, 1347. [Google Scholar]
- 12.For early transition metal catalyzed hydroaminoalkylation, see:; (a) Herzon SB; Hartwig JF Direct, Catalytic Hydroaminoalkylation of Unactivated Olefins with N-Alkyl Arylamines. J. Am. Chem. Soc 2007, 129, 6690. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Eisenberger P; Ayinla RO; Lauzon JMP; Schafer LL Tantalum–Amidate Complexes for the Hydroaminoalkylation of Secondary Amines: Enhanced Substrate Scope and Enantioselective Chiral Amine Synthesis. Angew. Chem. Int. Ed 2009, 48, 8361. [DOI] [PubMed] [Google Scholar]; (c) Kubiak R; Prochnow I; Doye S Titanium-Catalyzed Hydroaminoalkylation of Alkenes by C–H Bond Activation at sp3 Centers in the α-Position to a Nitrogen Atom. Angew. Chem. Int. Ed 2009, 48, 1153. [DOI] [PubMed] [Google Scholar]; (d) Nako AE; Oyamada J; Nishiura M; Hou Z Scandium-catalysed Intermolecular Hydroaminoalkylation of Olefins with Aliphatic Tertiary Amines. Chem. Sci 2016, 7, 6429. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Edwards PM; Schafer LL Early Transition Metal-catalyzed C–H Alkylation: Hydroaminoalkylation for Csp3–Csp3 Bond Formation in the Synthesis of Selectively Substituted Amines. Chem. Commun 2018, 54, 12543. [DOI] [PubMed] [Google Scholar]
- 13.For alkenylation, see:; (a) Tsuchikama K; Kasagawa M; Endo K; Shibata T Cationic Ir(I)-Catalyzed sp3 C–H Bond Alkenylation of Amides with Alkynes. Org. Lett 2009, 11, 1821. [DOI] [PubMed] [Google Scholar]; (b) Pan S; Matsuo Y; Endo K; Shibata T Cationic Iridium-catalyzed Enantioselective Activation of Secondary sp3 C–H Bond Adjacent to Nitrogen Atom. Tetrahedron 2012, 68, 9009. [Google Scholar]
- 14.For borylation, see:; (a) Kawamorita S; Miyazaki T; Iwai T; Ohmiya H; Sawamura M Rh-Catalyzed Borylation of N-Adjacent C(sp3)–H Bonds with a Silica-Supported Triarylphosphine Ligand. J. Am. Chem. Soc 2012, 134, 12924. [DOI] [PubMed] [Google Scholar]; (b) Li Q; Liskey CW; Hartwig JF Regioselective Borylation of the C–H Bonds in Alkylamines and Alkyl Ethers. Observation and Origin of High Reactivity of Primary C–H Bonds Beta to Nitrogen and Oxygen. J. Am. Chem. Soc 2014, 136, 8755. [DOI] [PubMed] [Google Scholar]
- 15.For carbonylation, see:; (a) Ishii Y; Chatani N; Kakiuchi F; Murai S Rhodium-Catalyzed Reaction of N-(2-Pyridinyl)piperazines with CO and Ethylene. A Novel Carbonylation at a C–H Bond in the Piperazine Ring. Organometallics 1997, 16, 3615. [Google Scholar]; (b) Chatani N; Asaumi T; Ikeda T; Yorimitsu S; Ishii Y; Kakiuchi F; Murai S Carbonylation at sp3 C–H Bonds Adjacent to a Nitrogen Atom in Alkylamines Catalyzed by Rhodium Complexes. J. Am. Chem. Soc 2000, 122, 12882. [DOI] [PubMed] [Google Scholar]
- 16.For acetoxylation, see:; Wang D-H; Hao X-S; Wu D-F; Yu J-Q Palladium-Catalyzed Oxidation of Boc-Protected N-Methylamines with IOAc as the Oxidant: A BocDirected sp3 C–H Bond Activation. Org. Lett 2006, 8, 3387. [DOI] [PubMed] [Google Scholar]
- 17. Early transition metal-catalyzed hydroaminoalkylation reactions (ref 11) are exceptions. However, these reactions have limited compatibility with saturated azacycles and a low functional group tolerance.
- 18.Spangler JE; Kobayashi Y; Verma P; Wang D-H; Yu J-Q α-Arylation of Saturated Azacycles and N-Methylamines via Palladium(II)-Catalyzed C(sp3)–H Coupling. J. Am. Chem. Soc 2015, 137, 11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For Pd(II)-catalyzed α-C(sp3)‒H methylation:; Jain P; Verma P; Xia G; Yu J-Q Enantioselective Amine α-Functionalization via Palladium-catalysed C–H Arylation of Thioamides. Nat. Chem 2016, 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan S; Shibata T Recent Advances in Iridium-Catalyzed Alkylation of C–H and N–H Bonds. ACS Catal 2013, 3, 704. [Google Scholar]
- 21.Tran AT; Yu J-Q Practical Alkoxythiocarbonyl Auxiliaries for Iridium(I)-Catalyzed C–H Alkylation of Azacycles. Angew. Chem. Int. Ed 2017, 56, 10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rare examples of transition-metal-catalyzed branch-selective C(sp2)‒H alkylation with acrylates:; (a) Wucher P; Caporaso L; Roesle P; Ragone F; Cavallo L; Mecking S; GöttkerSchnetmann I Breaking the Regioselectivity Rule for Acrylate Insertion in the Mizoroki-Heck Reaction. Proc. Natl. Acad. Sci. U S A 2011, 108, 8955. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kommagalla Y; Srinivas K; Ramana CV Ru-Catalyzed Branched versus Linear Selective C3-Alkylation of 2-Aroylbenzofurans with Acrylates via C‒H Activation. Chem. Eur. J 2014, 20, 7884. [DOI] [PubMed] [Google Scholar]; (c) Srinivas K; Dangat Y; Kommagalla Y; Vanka K; Ramana CV Electronic Control on Linear versus Branched Alkylation of 2-/3-Aroylbenzofurans with Acrylates: Combined DFT and Synthetic Studies. Chem. Eur. J 2017, 23, 7570. [DOI] [PubMed] [Google Scholar]
- 23.(a) Grygorenko OO; Radchenko DS; Volochnyuk DM; Tolmachev AA; Komarov IV Bicyclic Conformationally Restricted Diamines. Chem. Rev 2011, 111, 5506. [DOI] [PubMed] [Google Scholar]; (b) Zheng Y; Tice CM; Singh SB The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg. Med. Chem. Lett 2014, 24, 3673. [DOI] [PubMed] [Google Scholar]
- 24.Borah AJ; Shi Z Rhodium-Catalyzed, Remote Terminal Hydroarylation of Activated Olefins through a Long-Range Deconjugative Isomerization. J. Am. Chem. Soc 2018, 140, 6062. [DOI] [PubMed] [Google Scholar]
- 25.Takebayashi S; Shibata T [Ir(cod)2]BARF-Catalyzed C–H Bond Alkenylation and Alkylation of Ferroecenes. Organometallics 2012, 31, 4114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.