

Abstract
Despite recent advances, reactivity and site-selectivity remain significant obstacles for the practical application of C(sp3)−H bond functionalization methods to synthetic chemistry. Here, we describe a system that combines a salicylic-aldehyde-derived L,X-type directing group with an electron-deficient pyridone ligand to enable the β-methylene C(sp3)−H arylation of aliphatic alcohols, which has not been possible previously. Notably, this protocol is compatible with heterocycles embedded in both alcohol substrates and aryl coupling partners. A site-and stereo-specific annulation of dihydrocholesterol and the synthesis of a key intermediate of englitazone illustrate the practicality of this method.
Keywords: aliphatic alcohol, C-H activation, directing group, ligand
Incorporation of directing group and ligand:
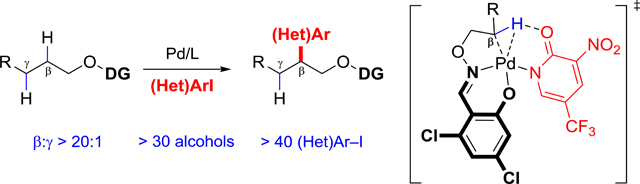
β-C(sp3)−H arylation of aliphatic alcohols was realized by the combination of a new designed L,X-type bidentate directing group and 2-pyridone ligand.
Palladium-catalyzed C(sp3)−H functionalization reactions are uniquely suited for diversity-oriented synthesis and late-stage diversification, owing to the versatile reactivity of the intermediate carbon-palladium bonds for forging a wide range of carbon-carbon and carbon-heteroatom bonds.[1] However, most reported palladium-catalyzed C(sp3) −H arylation reactions present three significant obstacles that limit their practical application: (1) secondary C(sp3) −H bonds are less readily functionalized than the more reactive and less hindered primary C−H bonds;[2] (2) aza-heterocyclic substrates, in which the nitrogen atom may provide an extra coordination site with palladium, can suffer from sluggish C−H activation;[3] (3) aza-heteroaryl iodides remain largely inefficient coupling partners for C(sp3) −H functionalization.[4] Generally, efforts to address these limitations have focused on carboxylic acids[3a–3e] and amines[3c,4a] (or derivatives thereof) as C−H activation substrates. In comparison, fewer methods and practical directing groups exist to achieve the C−H functionalization of aliphatic alcohols, despite the abundance of this motif among therapeutically relevant natural and unnatural products.[5]
Perhaps owing to the weak coordination of transition metals with the hydroxyl moiety, C−H activation reactions on free alcohol substrates are limited to sp2 C−H bonds[6] or photoredox processes where site-selectivity is dictated by 1,5-hydrogen-atom transfer (HAT).[7] Recently, iridium-catalyzed intramolecular C(sp3)−H silylation of alcohols using a tethered silyl hydride have been reported.[8] A common strategy to promote selective C−H metalation consists in preinstalling an external chelating group onto the hydroxyl group, which both accelerates and imparts selectivity to a critical step of cyclometallation. We and others employed various directing groups for palladium-catalyzed C(sp3)−H oxidation,[9] fluorination,[10] amination,[11] carboxylation and olefination[12] of alcohol substrates. Notably, these reactions are largely limited to primary C−H bonds with a few exceptional examples of fluorination and acetoxylation[9a, 10] (Scheme 1a).
Scheme 1.

Versatile C(sp3)-H Functionalizations of Aliphatic Alcohols.
These advances notwithstanding, examples of C−C bond formation reactions based on aliphatic-alcohol C−H bond activation remain limited. Recently, we reported a method for γ-C−H arylation of aliphatic alcohols via the incorporation of directing group and 2-pyridone ligand (Scheme 1b). Although the reaction proceeds with certain secondary C−H bonds (specifically, cycloalkane methylene C−H), it remains incompatible with aza-heterocyclic substrates and heteroaryl iodides.[13] The γ-selectivity in this reaction is thought to arise from the preference, upon C−H for a [5,6]-fused palladacycle intermediate (Scheme 1b). We thus hypothesized that the choice of a directing group that would instead form a [6,5]-bicyclic palladacycle intermediate, would facilitate β-selective C−H metalation (Scheme 1c).
In addition, by introducing an external 2-pyridone ligand, which has been proved to significantly lower the transition state energy of C−H activation step,[14] less reactive C(sp3)−H bonds (e.g., secondary, tertiary) would be activated (Figure 1a). Because the β-C−H arylation of aliphatic alcohols at non-primary sites still remains unexplored, developing such a transformation could effectively offer a positionally diverse C−H bond editing method.
Figure 1.

Ligand Acceleration Effect and directing group installation.
To evaluate the feasibility of our approach, we first attempted the β-arylation of cyclopentanol employing a range of aldoxime-and alkoxamide-based directing groups. A representative example of directing group installation, achieved in a single operation from the corresponding alcohol, is depicted in Figure 1b, and results of the directing group study are summarized in Table 1a. Cyclopentanol had indeed shown limited reactivity at the β-methylene in our previous studies (Table 1a, DG1, β:γ = 2:1);[13] moreover, some β-methyl C−H arylation was also reported using a closely related directing group.[15] Two sets of reaction conditions that had proved effective in our former work13 were selected for the initial directing-group screening. These conditions differ, notably, in the reagent used to facilitate the concerted metalation-deprotonation step: lithium carbonate (2.0 equiv, condition A) or 3-nitro-5-chloro-2-pyridone (L1, 40 mol%, condition B). Directing groups based on pyridine-2-carboxylic acid (DG2) or quinoline-8-carboxylic acid motif (DG3), frequently used in directed C−H activations, failed to afford the desired product (2) under the attempted conditions. To our delight, a salicylic-aldehyde-derived directing group (DG4), inert under condition A, under condition B led to the formation of β-arylation product 2 in 44% yield without evidence of γ-arylation product in the crude reaction mixture (1H NMR). Given this encouraging result, we screened several other salicylic aldehydes with different substituents on the aromatic ring (DG5-DG9): dichloro-derivative DG9 was found to be the most effective directing group, resulting in 56% yield of 2. Notably, none of the examples that employed salicylic-aldehyde-derived directing groups led to observable quantities of γ-arylation product. In addition, methylation of the free hydroxyl group (DG10) abolished reactivity, indicating that L,X-type directing groups are crucial to the success of this reaction.
Table 1.
Directing group and ligand screening.[a]
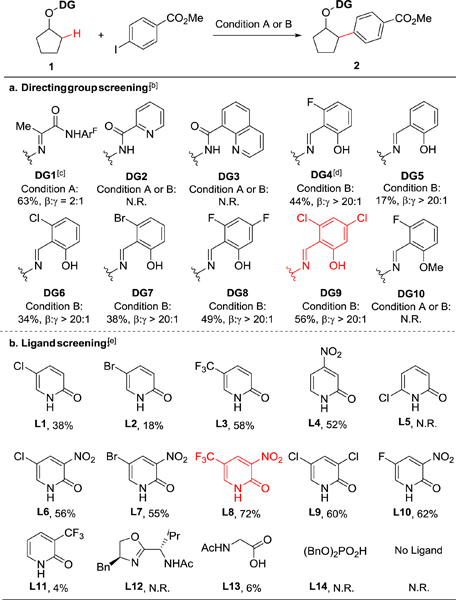 |
Yields were determined by 1H NMR using dibromomethane as internal standard. Condition A: Substrate 1 (0.1 mmol), aryl iodide (0.3 mmol), Pd(OAc)2 (10 mol%), AgOAc (2.5 equiv), Li2CO3 (2.0 equiv) in DCE (1.0 mL), 120 °C, 24 h; Condition B: Substrate 1 (0.1 mmol), aryl iodide (0.3 mmol), Pd(OAc)2 (10 mol%), Ligand (40 mol%), AgTFA (1.5 equiv), in HFIP (1.0 mL), 100 °C, 4 h.
Ligand L6 was used with condition B.
No reaction under condition B.
No reaction under condition A.
Using condition B and DG9 as directing group.
In order to further increase the efficiency of this reaction, we investigated the effect of the 2-pyridone ligand (Table 1b). We first found that no reaction occurred in its absence, both evidencing its crucial role and indicating an opportunity for reaction optimization. Indeed, after screening a series of 2-pyridone ligands, we found that electron-withdrawing groups at C-4 or C-5 position generally promoted the reaction, although with moderate yields (L1-L4); while substitution at C-6 position did not give any product (L5). Electron-withdrawing groups at both C-3 and C-5 resulted in better reactivity; among these, L8 was the most reactive and gave 72% yield. Other types of ligands such as oxazoline-acetamide (L12), acetylglycine (L13) and dibenzyl phosphoric acid (L14) were also evaluated in this reaction, but led virtually to no reactivity (< 10% yield).
With optimized conditions in hand, we examined the scope of this β-C−H arylation protocol (Table 2). As expected, alcohols with primary β-C−H bonds exhibited high reactivity, leading to diarylation product along with monoarylation product in most cases (1a-1c). Lower stoichiometry of aryl iodide was used in the cases of alcohols bearing multiple reactive sites in order to mitigate over-functionalization (1d, 1e). Next, we set out to investigate the reactivity of secondary C−H bonds. We were pleased to find that, with our method, a series of linear primary alcohols exhibited very good reactivity and exclusive selectivity for the β-position (1f-1h, from 72% to 85% yield). Naturally occurring cetyl alcohol (1k) and lauryl alcohol (1l) also underwent arylation at the β-position in 71% and 76% yield, respectively. Benzylic (1i, 61% yield) and homobenzylic (1j, 54% yield) methylene C−H bonds proved less reactive, although affording product with high site-selectivity.
Table 2.
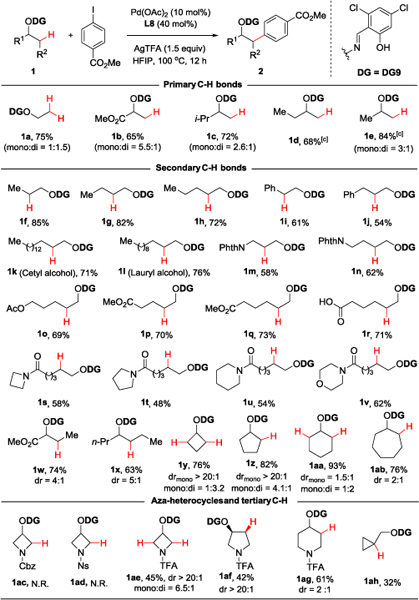 |
Isolated yields.
The diastereomeric ratio was determined by 1H NMR spectroscopy.
1.3 equivalent of aryl iodide was used.
We next evaluated the compatibility of various functional groups with this transformation. Protected 1,3-and 1,4-amino alcohols (1m, 1n), as well as substrates bearing acetoxy or carboxylate groups (1o-1q), led to good product yields (58–73%). Interestingly, a free carboxylic acid was also compatible with the reaction (1r, 71% yield). A series of tertiary amide substrates were tested under the same conditions, all of them giving moderate to good yields (48–62%, 1s-1v). Secondary alcohols with available β-methylene C-H bonds (1w, 1x) also resulted in good yields, but with moderate diastereocontrol (d.r. = 4:1 and 5:1, respectively). Cyclobutanol (1y) and cyclopentanol (1z) afforded the expected products in high yields (76% and 82%, respectively) and high selectivity for the cis-diastereomers. In contrast, medium-sized cycloalkanols such as cyclohexanol (1aa) and cycloheptanol (1ab) gave diastereomeric ratios of 1.5:1 and 2:1, respectively, although exhibiting excellent reactivity.
Having established the high efficiency of this protocol for the β-arylation of aliphatic alcohols, we turned our attention to azaheterocycles, more challenging C−H activation substrates of potential interest in probe and drug development.[16] We found that the choice of protecting group on the nitrogen atom was important for reactivity, with N-Cbz-protected and N-Ns-protected 3-hydroxy azetidine leading to no C−H activation product (1ac, 1ad), while the corresponding N-trifluoroacetyl (N-TFA) resulted in 45% yield (mono and di, 1ae). Similarly, N-TFA 3-hydroxy-pyrrolidine (1af) and N-TFA 4-hydroxypiperidine (1ag) gave the expected arylation products in 42% yield and 61% yield (d.r. = 2:1), respectively. Finally, cyclopropyl methanol (1ah) was able to undergo C−H arylation at the β-site, a rare instance of palladium inserting into a tertiary C−H bond to form a quaternary carbon center.[17]
Next, we examined the scope of aryl iodides as the coupling partner, using the propanol DG9-derivative as model substrate (Table 3). Both electron-donating and electron-withdrawing groups on the para or meta positions of aryl iodides ring resulted in good to excellent yields (3a-3e, 3h-3l, 3n, 3o). Of note, products containing aryl bromide and aryl iodide functionality were readily accessed, (3f, 3g, 3m), offering the possibility for further diversification. In addition, ortho-substituted aryl iodides were also reactive with our method (3p, 3q). Given the ubiquity of heteroaromatics in small-molecule drug discovery, we examined the reactivity of a wide range of aza-heteroaryl iodides. We were delighted to find that 2-substituted iodopyridines behaved well, leading to good to excellent product yields (3v-3aj). Unsubstituted iodopyridines also resulted in arylation product, albeit in moderate yield (3t, 3u) presumably due to competitive coordination to the palladium catalyst. Other heteroaryl iodides such as iodothiophene, -quinoline, -quinoxaline, -indole and -oxindole were also suitable coupling partners (3ak-3ao), providing further support to the generality of this protocol.
Table 3.
Scope of aryl and heteroaryl iodides.[a]
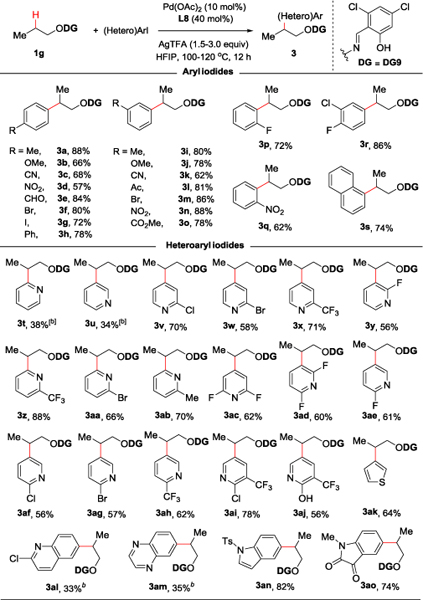 |
Isolated yields.
The reaction was conducted at 120 °C.
To demonstrate the practical application of this method, we first turned to dihydrocholesterol as a substrate for C-H functionalization. Following directing group installation, a short sequence involving C−H arylation with 3-iodo-2-fluoropyridine, directing group removal and SNAr cyclization generated a novel dihydrofuro[2,3-b]pyridine cis-fused structure (5, 36% yield from 4, Scheme 2a). The new C−H functionalization protocol was further showcased in the synthesis of 9, a known precursor of englitazone (of antihyperglycemic activity),[18] via sequential β-and γ-C−H arylations. This route required a total of 7 transformations and proceeded in 23% yield, comparing favorably to the 11 steps and 4.5% overall yield previously reported (Scheme 2b).[19]
Scheme 2.

Practical application. Conditions: a) Substrate 6 (0.5 mmol), iodobenzene (0.6 mmol), Pd(OAc)2 (10 mol%), L8 (40 mol%), AgTFA (1.5 equiv), in HFIP (3.0 mL), 100 °C, 4 h; then Pd(OH)2/C (20 mol%), H2 (1 atm), in EtOH (2.0 mL), r.t., overnight, 65% yield over 2 steps; b) Ph3P (1.2 equiv), NHPI (1.2 equiv), DIAD (1.5 equiv), in THF (2 mL), r.t., 95% yield; c) Hydrazine monohydrate (2.0 equiv), in CH2Cl2/MeOH, 2 h, then pyruvic acid (5 equiv), 1 h, 95% yield; d) Methyl 4-fluoro-3-iodobenzoate (1.5 equiv), Pd(OAc)2 (10 mol%), L8 (40 mol%), AgTFA (1.5 equiv), in HFIP (3.0 mL), 100°C, 4 h; then SOCl2/MeOH, 49% yield; e) Pd(OH)2/C (20 mol%), H2 (1 atm), in EtOH (2.0 mL), r.t., overnight; then NaH (3.0 equiv), in THF, 70 °C, overnight, 81% yield over 2 steps.
In summary, we have developed a protocol for the β-C(sp3)−H arylation of aliphatic alcohols. High yields and high degrees of β-specificity result from the combination of an electron-deficient 2-pyridone ligand and an L,X-type directing group that likely favors the formation of a [6,5]-fused palladacycle intermediate. This protocol, leading notably to the activation of methylene β-C–H bonds, is compatible with a wide range of substrates (including aza-heterocycles), and coupling partners (including heteroaryl iodides). Given the variety of commercially available aliphatic alcohol substrates and suitable coupling partners, we anticipate that this C(sp3)–H functionalization strategy will facilitate the synthesis of novel chemical matter, particularly in the context of drug discovery.
Supplementary Material
Acknowledgements
We gratefully acknowledge Scripps Research and NIH (NIGMS, R01GM084019) for financial support. We thank Dr. Jason Chen, Ms. Brittany Sanchez and Ms. Emily Sturgell (Automated Synthesis Facility, The Scripps Research Institute) for their assistance with HRMS analysis.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].For selected examples, see a) Dai H-X, Stepan AF, Plummer MS, Zhang Y-H, Yu J-Q, J. Am. Chem. Soc 2011, 133, 7222–7228. [DOI] [PubMed] [Google Scholar]; b) Rosen BR, Simke LR, Thuy-Boun PS, Dixon DD, Yu J-Q, Baran PS, Angew. Chem. Int. Ed 2013, 52, 7317–7320. [DOI] [PubMed] [Google Scholar]; c) He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q, Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gong W, Zhang G, Liu T, Giri R, Yu J-Q, J. Am. Chem. Soc 2014, 136, 16940–16946. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Leal RA, Bischof C, Lee YV, Sawano S, McAtee CC, Latimer LN, Russ ZN, Dueber JE, Yu J-Q, Sarpong R, Angew. Chem. Int. Ed 2016, 55, 11824–11828. [DOI] [PubMed] [Google Scholar]; f) Shang M, Feu KS, Vantourout JC, Barton LM, Osswald HL, Kato N, Gagaring K, McNamara CW, Chen G, Hu L, Ni S, Fernandez-Canelas P, Chen M, Merchant RR, Qin T, Schreiber SL, Melillo B, Yu J-Q, Baran PS, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 8721–8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].For recent reviews on C(sp3)−H functionalization, see a) Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ackermann L, Chem. Rev 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]; c) Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) He J, Wasa M, Chan KSL, Shao Q, Yu J-Q, Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For examples on aza-cyclic substrate C(sp3)–H functionalization, see a) Affron DP, Davis OA, Bull JA, Org. Lett 2014, 16, 4956–4959. [DOI] [PubMed] [Google Scholar]; b) Ye S, Yang W, Coon T, Fanning D, Neubert T, Stamos D, Yu J-Q, Chem. Eur. J 2016, 22, 4748–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS, Nature 2016, 531, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Maetani M, Zoller J, Melillo B, Verho O, Kato N, Pu J, Comer E, Schreiber SL, J. Am. Chem. Soc 2017, 139, 11300–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Antermite D, Affron DP, Bull JA, Org. Lett 2018, 20, 3948–3952. [DOI] [PubMed] [Google Scholar]
- [4].For selected examples using heteroaryl iodides as coupling partners, see a) Chen Y-Q, Wang Z, Wu Y, Wisniewski SR, Qiao JX, Ewing WR, Eastgate MD, Yu J-Q, J. Am. Chem. Soc 2018, 140, 17884–17894. [DOI] [PubMed] [Google Scholar]; b) Zhu R-Y, Liu L-Y, Park HS, Hong K, Wu Y, Senanayake CH, Yu J-Q, J. Am. Chem. Soc 2017, 139, 16080–16083. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu R-Y, Li Z-Q, Park HS, Senanayake CH, Yu J-Q, J. Am. Chem. Soc 2018, 140, 3564–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Falbe J, Bahrmann H, Lipps W, Mayer D, Frey GD, Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH, Weinheim, Germany, 2000) [Google Scholar]; b) Mo F, Tabor JR, Dong G, Chem. Lett 2014, 43, 264–271. [Google Scholar]; c) Espino CG, Du Bois J, Angew. Chem. Int. Ed 2001, 40, 598–600. [PubMed] [Google Scholar]; d) Espino CG, Wehn PM, Chow J, Du Bois J, J. Am. Chem. Soc 2001, 123, 6935–6936. [Google Scholar]; e) Liang J-L, Yuan S-X, Huang J-S, Yu W-Y, Che C-M, Angew. Chem. Int. Ed 2002, 41, 3465–3468. [DOI] [PubMed] [Google Scholar]
- [6].a) Lu Y, Wang D-H, Engle KM, Yu J-Q, J. Am. Chem. Soc 2010, 132, 5916–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang X, Lu Y, Dai H-X, Yu J-Q, J. Am. Chem. Soc 2010, 132, 12203–12205. [DOI] [PubMed] [Google Scholar]; c) Lu Y, Leow D, Wang X, Engle KM, Yu J-Q, Chem. Sci 2011, 2, 967–971. [Google Scholar]
- [7].a) Hu A, Guo J-J, Pan H, Tang H, Gao Z, Zuo Z, J. Am. Chem. Soc 2018, 140, 1612–1616. [DOI] [PubMed] [Google Scholar]; b) Li G-X, Hu X, He G, Chen G, Chem. Sci 2019, 10, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Simmons EM, Hartwig JF, Nature 2012, 483, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li B, Driess M, Hartwig JF, J. Am. Chem. Soc 2014, 136, 6586–6589. [DOI] [PubMed] [Google Scholar]; c) Bunescu A, Butcher TW, Hartwig JF, J. Am. Chem. Soc 2018, 140, 1502–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Karmel C, Li B, Hartwig JF, J. Am. Chem. Soc 2014, 140, 1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ren Z, Mo F, Dong G, J. Am. Chem. Soc 2012, 134, 16991–16994. [DOI] [PubMed] [Google Scholar]; b) Xu Y, Yan G, Ren Z, Dong G, Nat. Chem 2015, 7, 829–834. [DOI] [PubMed] [Google Scholar]
- [10].Mao YJ, Lou SJ, Hao HY, Xu DQ, Angew. Chem. Int. Ed 2018, 57, 14085–14089. [DOI] [PubMed] [Google Scholar]
- [11].Jin L, Zeng X, Li S, Hong X, Qiu G, Liu P, Chem. Commun 2017, 53, 3986–3989. [DOI] [PubMed] [Google Scholar]
- [12].Tanaka K, Ewing WR, Yu J-Q, J. Am. Chem. Soc 2019, 141, 15494–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xia G, Weng J, Liu L, Verma P, Li Z, Yu J-Q, Nat. Chem 2019, 11, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang P, Verma P, Xia G, Shi J, Qiao JX, Tao S, Cheng PTW, Poss MA, Farmer ME, Yeung K-S, Yu J-Q, Nature 2017, 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang B-X, Mao Y-J, Hao H-Y, Wu Q-Z, Zhou K, Lou S-J, Xu D-Q, Chem. Commun 2019, 55, 7049–7052. [DOI] [PubMed] [Google Scholar]
- [16].For selected reviews, see a) Taylor AP, Robinson RP, Fobian YM, Blakemore DC, Jonesb LH, Fadeyi O, Org. Biomol. Chem 2016, 14, 6611–6637. [DOI] [PubMed] [Google Scholar]; b) Baumann M, Baxendale IR, Beilstein J. Org. Chem 2013, 9, 2265–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hoshiya N, Kobayashi T, Arisawa M, Shuto S, Org. Lett 2013, 15, 6202–6205. [DOI] [PubMed] [Google Scholar]
- [18].Stevenson RW, McPherson RK, Genereux PE, Danbury BH, Kreutter DK, Metabolism 1991, 40, 1268–1274. [DOI] [PubMed] [Google Scholar]
- [19].Urban FJ, Moore BS, J. Heterocycl. Chem 1992, 29, 431–438. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.