

The study produces large amounts of mature human cells in mouse embryos, paving the way for making human organs in animals.
Abstract
It has not been possible to generate naïve human pluripotent stem cells (hPSCs) that substantially contribute to mouse embryos. We found that a brief inhibition of mTOR with Torin1 converted hPSCs from primed to naïve pluripotency. The naïve hPSCs were maintained in the same condition as mouse embryonic stem cells and exhibited high clonogenicity, rapid proliferation, mitochondrial respiration, X chromosome reactivation, DNA hypomethylation, and transcriptomes sharing similarities to those of human blastocysts. When transferred to mouse blastocysts, naïve hPSCs generated 0.1 to 4% human cells, of all three germ layers, including large amounts of enucleated red blood cells, suggesting a marked acceleration of hPSC development in mouse embryos. Torin1 induced nuclear translocation of TFE3; TFE3 with mutated nuclear localization signal blocked the primed-to-naïve conversion. The generation of chimera-competent naïve hPSCs unifies some common features of naïve pluripotency in mammals and may enable applications such as human organ generation in animals.
INTRODUCTION
Mouse embryonic stem cells (mESCs) are in naïve pluripotency (1) that represents the ground state of development (2), from which all cells in the mouse embryo are derived (3). In contrast, human embryonic stem cells (hESCs) (4) are in a primed state of pluripotency with many different properties (5). Despite intense efforts to generate naïve human pluripotent stem cells (hPSCs) (6–12), it has not been possible to derive and maintain naïve hPSCs without relying on chemicals beyond the two inhibitors (2i; PD0325901 and CHIR99021) that confine naïve pluripotency in mouse (2). The resulting hPSCs do not robustly generate mature human cells of identifiable types in the mouse embryo (13, 14). Overexpression of the antiapoptotic factor BMI1 in primed hPSCs supports limited integration of human cells in earlier embryos of mouse, rabbit, and pig (15). Converging evidence suggests that naïve pluripotency has connection to embryonic diapause (16–18), which can be induced in mice by mammalian target of rapamycin (mTOR) inhibition (19). Transcription Factor binding to IGHM Enhancer 3 (TFE3), a transcription factor linking nutrient-sensing, stress, and autophagy (20, 21), plays a critical role in maintaining naïve pluripotency in mice (22). It is located in the cytoplasm of mouse epiblasts (22), primed hESCs (6), and human induced pluripotent stem cells (iPSCs) (23), but resides in the nucleus in naïve mESCs (22) and naïve hPSCs maintained in chemical inhibitors (6) or by transgenes (23).
In this study, we found that a transient inhibition of mTOR by Torin1 converted primed hPSCs to the naïve state, which could be maintained indefinitely in essentially the same condition used to culture mESCs. When injected in mouse blastocysts, naïve hPSCs generated a large amount of mature human cells of all three germ layers, accounting for 0.1 to 4% of cells in mouse embryos at E17.5 (embryonic day 17.5). The conversion was dependent on Torin1-induced nuclear translocation of TFE3, but not on autophagy.
RESULTS
Conversion of hPSCs to the naïve state by transient Torin1 treatment
The cytoplasmic localization of TFE3 in primed hPSCs (6, 23) and its nuclear localization in naïve mESCs (22) and naïve hPSCs maintained in chemical inhibitors (6) or by transgenes (23) led us to search for agents that induce the nuclear translocation of TFE3. We found that inhibition of mTOR by Torin1 (24) (Fig. 1, A to H, and fig. S1, A and B) or rapamycin (fig. S1C) induced rapid translocation of TFE3 from cytoplasm to nucleus in primed H9 hESCs. On the basis of the time course (fig. S1A) and dose response (fig. S1B) of Torin1-induced nuclear translocation of TFE3, we treated primed H9 hESCs with Torin1 (10 μM for 3 hours) in medium for naïve mESCs (2) [50%/50% Dulbecco’s modified Eagle’s medium (DMEM)/F12:Neurobasal with 2i (1 μM PD0325901 and 3 μM CHIR99021), human leukemia inhibitory factor (LIF), N2, and B27 supplements] and then dissociated the cells with TrypLE for replating on mouse embryonic fibroblast (MEF) feeders in the same medium without Torin1. After about 5 days, refractive, dome-shaped colonies containing NANOG+ cells were observed (fig. S1, D to D‴). Replacing N2 and B27 supplements, which contained undisclosed amount of insulin and many other components, with human insulin (18 μg/ml) produced a much higher percentage of NANOG+ cells among all human cells (hNA+) grown on MEF feeders (fig. S1, E to E‴ and M). We named this medium 2iLI. Removing insulin, LIF, 2i, CH, or PD drastically reduced NANOG+ cells and mESC-like colonies (fig. S1, F to J‴ and M). High glucose concentration (21.25 mM) in the naïve mESC medium (2) or 21% O2 markedly decreased the conversion (fig. S1, K to M) compared with 2iLI medium with physiological glucose concentration (5 mM) and O2 tension (5%).
Fig. 1. Converting hPSCs from primed to naïve pluripotency.
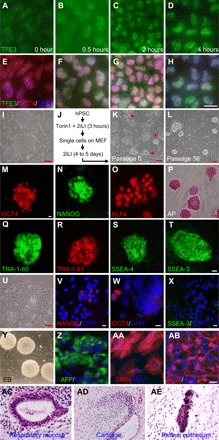
(A to H) Localization of TFE3 in primed H9 hESCs treated with 10 μM Torin1 for the indicated durations. DAPI, 4′,6-diamidino-2-phenylindole. (I to L) Primed H9 (I) were converted with the protocol in (J) to dome-shaped colonies (K; red arrows), which were picked and passaged (L). (M to T) Expression of pluripotency markers in nH9 cultured in 2iLI medium. (U to X) In 2iLI medium, primed H9 differentiated (U) and lost the expression of pluripotency markers (V to X). White scale bars, 10 μm; red scale bars, 100 μm. (Y to AB) Spontaneous differentiation of nH9 in vitro through embryoid bodies (EB) (Y) to cells of all three germ layers (Z to AB). (AC to AE) Naive H9 grafted in kidney capsules of severe combined immunodeficient (SCID) mice produced teratomas.
After primed H9 hESCs (Fig. 1I) were converted with the optimized condition (Fig. 1J), many colonies with mESC morphology were seen (Fig. 1K), picked, and maintained in 2iLI medium (without Torin1) with 5 mM glucose and 5% O2 for at least 56 passages without noticeable differentiation (Fig. 1L). In this condition, naïve H9 (nH9) expressed pluripotency markers (Fig. 1, M to T), while primed H9 differentiated (Fig. 1, U to X). Using the same method, we also converted H1 and RUES2 hESCs as well as C005 and N004 human iPSCs to naïve state (fig. S1, P to S). C005 primed iPSC was originally generated with nonintegrating episomal plasmids (25), while N004 was derived with doxycycline (DOX)–inducible lentiviruses expressing octamer-binding transcription factor 4 (OCT4), SRY-box transcription factor 2 (SOX2), Krüppel-like factor 4 (KLF4), c-MYC, and NANOG (fig. S1N) (26). By optimizing CHIR99021 concentration to 0.8 μM, N004 iPSCs were converted to naïve state with the same method without turning on transgenes (fig. S1, O to S). A transient inhibition of mTOR by rapamycin (10 μM for 3 hours) also converted RUES2 from primed to naïve state (fig. S1T), confirming that the conversion was mediated by mTOR inhibition. Naive hPSCs exhibited normal karyotype at early passages (12 to 15) and late passages (36 to 39) (fig. S1, U to X). They were differentiated to cells of all three germ layers in vitro and in vivo (Fig. 1, Y to AE).
Cellular and transcriptomic properties of naïve hPSCs
Naive hPSCs had much higher clonal efficiency than their parental primed cells (Fig. 2, A to D). Naive state also conferred significantly faster cell proliferation (Fig. 2E). Cell doubling times of nH9 (14.9 ± 3.6 hours) and naïve RUES2 (12.9 ± 4.1 hours) were much shorter than those of primed H9 (35.0 ± 5.3 hours) and primed RUES2 (33.5 ± 6.4 hours) (Fig. 2E, inset). Mitochondrial respiration as revealed by the mitochondrial membrane potential indicator TMRE was nearly absent in primed H9 (Fig. 2F) and became very prominent in nH9 (Fig. 2G). Seahorse analyzer showed that mitochondrial respiration was essentially absent in primed H9 and primed RUES2 but became very prominent in nH9 and naïve RUES2 (Fig. 2, J to M).
Fig. 2. Cellular properties of naïve hPSCs.
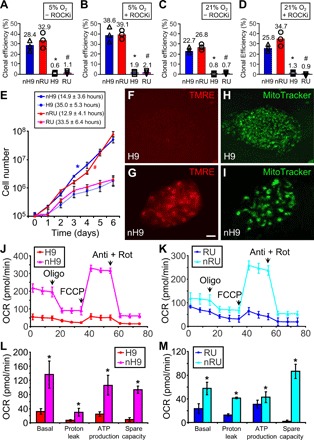
(A to D) Clonal efficiencies of naïve and primed H9 and RUES2 cultured in 5% O2 (A and B) or 21% O2 (C and D) in the absence (A and C) or presence (B and D) of ROCK inhibitor. *P < 0.05 and #P < 0.05, n = 3, unpaired, two-tailed t test versus naïve H9 (nH9) or naïve RUES2 (nRU), respectively. (E) Growth curve and cell doubling time of primed and naïve H9 and RUES2. *P < 0.05 and #P < 0.05, n = 4, repeated-measures analysis of variance (ANOVA) versus primed H9 or primed RUES2 (RU), respectively. (F to I) Primed H9 and nH9 were live stained with TMRE to detect mitochondrial inner membrane potential (F and G) or MitoTracker to locate mitochondria (H and I). (J to M) Mitochondrial respiration in primed versus naïve H9 (J) or RUES2 (K) hESCs was quantified (L and M) in a Seahorse analyzer. *P < 0.05, n = 3, unpaired, two-tailed t test versus primed state. OCR, oxygen consumption rate; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; ATP, adenosine 5′-triphosphate.
Naive H9 used the distal enhancer rather than the proximal enhancer to drive the expression of OCT4, in contrast to the situation in primed H9 (Fig. 3A). Principal components analysis (PCA) of RNA sequencing (RNA-seq) data (Fig. 3B) showed that our naïve hESCs (Hu_N; blue triangles) bore similarities to single cells from human late blastocysts (Ya_LB; black triangles) (27) and the equivalent E5 to E7 preimplantation human embryos (Pe_E5, Pe_E6, and Pe_E7; pink, yellow, and brown pluses, respectively) (28), as well as naïve hESCs established with various chemicals inhibitors (8, 29, 30). They were well separated from the parental primed hESCs (Hu_P; blue circles) and other hESCs (7, 8, 29, 30), which were similar to each other. Clustering analysis of the RNA-seq data (Fig. 3C) showed that our naïve hESCs (Hu_N; red branches) were similar to naïve hPSCs from several other groups [Gr_N (30), Sa_N (29), and Ta_N (8)] but were quite different from our primed hESCs (Hu_P; blue branches) and other primed hPSCs (7, 8, 29, 30). The 1811 coding genes that were differentially expressed between our naïve (underlined green) and primed (underlined red) hPSCs (table S1) showed similar differential expression patterns in naïve and primed hPSCs from other groups (Fig. 3D). We found 310 differentially expressed genes that are changed consistently in these datasets (table S2). Figure S2 showed the expression levels of a subset of these genes encoding for epigenetic modifiers, growth factors, and transcription factors, along with a list of previously identified signature genes differentially expressed in naïve versus primed hPSCs (8, 9, 31, 32). Although there were 310 genes that exhibited consistent changes among naïve hPSCs from this study and others (table S2 and fig. S2, A to C), a number of human naïve pluripotency markers identified in previous studies, such as KHDC1L, KLF4, KLF5, KLF17, DPPA3, TFCP2L1, DPPA5, ARGFX, GDF3, and TBX3, were not significantly up-regulated in our naïve hPSCs (fig. S2D). Primed-to-naïve conversion increases the expression of some transposable elements (TEs) (30, 33). We found that the 889 TEs differentially expressed between our naïve (underlined green) and primed (underlined red) hPSCs (table S3) had similar differential expression patterns in naïve and primed hPSCs from other groups (Fig. 3E). The expression levels of HERVK and LTR5_Hs were significantly increased in naïve hPSCs including ours (Hu_*; blue) (Fig. 3F).
Fig. 3. Transcriptomic profiles of naïve hPSCs.
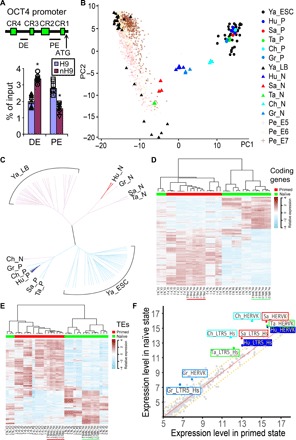
(A) H3K27Ac chromatin immunoprecipitation assays showing the utilization of OCT4 distal enhancer (DE) and proximal enhancer (PE) in primed H9 and nH9. P < 0.05, n = 6 versus H9. (B and C) PCA (B) and clustering analysis (C) of RNA-seq data from naïve (Hu_N; blue triangles) and primed (Hu_P; blue dots) H9 and RUES2 against data on single cells from human late blastocysts (Ya_LB; black triangles) (27); human E5 to E7 embryos (Pe_E5, Pe_E6, and Pe_E7; brown pluses) (28); hESCs (Ya_ESC; black dots) (27); or bulk RNA-seq data from naïve hPSCs maintained in chemical inhibitors [Sa_N (red triangles) (29), Ta_N (green triangles) (8), Ch_N (cyan triangles) (7), and Gr_N (light blue triangles) (30)] and their parental primed hPSCs [Sa_P (red dots) (29), Ta_P (green dots) (8), Ch_P (cyan dots) (7), and Gr_P (light blue dots) (30)]. Primed hPSCs (represented by dots) are named by first two letters of the first author’s family name, followed by _P. Naive hPSCs (represented by triangles) are named accordingly with _N. Matching color of dot and triangle indicates the same genome. (D) Coding genes that were differentially expressed between our naïve (underlined green) and primed (underlined red) H9 and RUES2 as compared with expression patterns in other naïve and primed hPSCs. (E) Transposable elements differentially expressed between our naïve (underlined green) and primed (underlined red) H9 and RUES2 as compared with expression patterns in other naïve and primed hPSCs. (F) Increased expression of TEs such as HERVK and LTR5_Hs in our naïve cells (Hu_*; blue) and other naïve cells as compared with their corresponding primed hPSCs.
Reactivation of X-inactivated genes in female naïve hPSCs
Naive pluripotency in female cells is characterized by two active X chromosomes (XaXa), instead of the inactivation of one X chromosome in primed pluripotency (XaXi) (34). Both histone H3K27me3 staining (6, 35) and XIST RNA fluorescence in situ hybridization (FISH) showed XaXa state in naïve RUES2 and XaXi state in primed RUES2 (Fig. 4, A to H). Because of the substantial variance in XIST expression in human blastocysts (28, 36) and naïve hPSCs (36), which is not correlated with X inactivation status (36), we analyzed the expression levels of genes on the 22 autosomes, X-inactivated (Xi) genes (37), and X chromosome genes that escaped X inactivation (Xe) (37). The ratio of gene expression levels between naïve and primed H9 was significantly increased for the X-inactivated genes but not for the X-escaped genes or autosomal genes (Fig. 4I). Using single-nucleotide polymorphism (SNP)–based allelic expressing analysis, we found that a number of monoallelically expressed X-inactivated genes in primed H9 became biallelically expressed in nH9 (Fig. 4J). In contrast, a sample of X-escaped genes were biallelically expressed in both primed and nH9 (Fig. 4K).
Fig. 4. Reactivation of X-inactivated genes in naïve hPSCs.
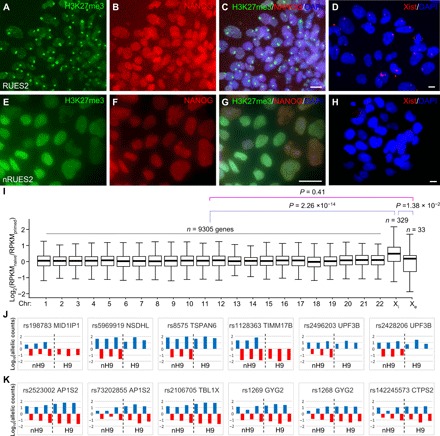
(A to H) X chromosome inactivation in primed RUES2 (A to D) and reactivation in naïve RUES2 (E to H) as revealed by costaining for H3K27me3 (A and E), NANOG (B and F), their merged images with DAPI (C and G), or XIST RNA FISH (D and H). (I) Ratio of the expression level of genes on each chromosome in naïve versus primed hPSCs. Xi, X-inactivated genes (37); Xe, X-escaped genes (37). (J and K) SNP-based allelic expression analysis of X-inactivated genes (J) and X-escaped genes (K) showed that monoallelically expressed X-inactivated genes in primed H9 were expressed biallelically in nH9 (nH9) (J), while genes escaped from X inactivation were always biallelically expressed in H9 and nH9 (K). The two colors represented expression levels of a gene from two different alleles in the three replicates in the RNA-seq data.
DNA hypomethylation in naïve hPSCs
The levels of 5mC and 5hmC were markedly decreased when primed H9 were converted to the naïve state (Fig. 5, A to B‴). Dot blot analysis of genomic DNA isolated from primed H9, nH9, and AB2.2 mESCs showed that 5mC levels were significantly reduced from H9 to nH9 to levels similar to those in mESCs (Fig. 5, C and D). Significant decrease in 5hmC levels was also found from H9 to nH9 (Fig. 5, E and F). PCA of genome-wide methylation data showed a large separation between primed and naïve hESCs along principal component 1 (PC1), explaining 79% of total variance (Fig. 5G). Further analysis identified 128,383 tiling regions (out of a total of 251,092), 24,812 promoters (out of a total of 44,854), and 24,692 gene bodies (out of a total of 34,931) that were differentially methylated between naïve and primed hESCs. More than 93% of the differentially methylated regions (96.1% of tiling regions, 93.5% of promoters, and 96.8% of gene bodies) were demethylated in primed-to-naïve conversion (Fig. 5H). Differential changes in DNA methylation of imprinted regions (38) were observed when H9 or RUES2 were converted to naïve state. In nH9, 51.5% of imprinted regions had unchanged DNA methylation, 28.8% became demethylated, and 19.7% were hypermethylated. The situation in naïve RUES2 was generally similar (28.7% unchanged, 45.5% demethylated, and 25.8% hypermethylated) (Fig. 5I). This contrasts with demethylation of around 70% of imprinted regions in naïve hPSCs maintained in chemical inhibitors (8, 33). All these are different from the situation in vivo, where imprinting is maintained in pluripotent stem cells (39).
Fig. 5. Decreased DNA methylation in naïve hPSCs.
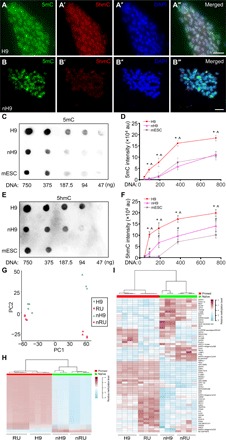
(A to B‴) Costaining for 5mC (A and B), 5hmC (A′ and B′), and DAPI (A″ and B″) in primed H9 (A to A‴) and nH9 (B to B‴). Scale bars, 10 μm. (C to F) Dot blots (C and E) and quantification (D and F) of 5mC (C and D) and 5hmC (E and F) levels in genomic DNA isolated from primed and naïve H9, and AB2.2 mESC. *H9 versus mESC; ^H9 versus nH9; #nH9 versus mESC, all at P < 0.05, n = 3, unpaired, two-tailed t test. au, arbitrary units. (G) PCA of genome-wide DNA methylation in primed and naïve H9 and RUES2 using β values of all probes in Infinium MethylationEPIC BeadChip. (H) Comparison of DNA methylation levels in the 128,383 tiling regions that were differentially methylated between primed and naïve H9 and RUES2. (I) Comparison of DNA methylation levels in imprinted regions (38) between primed and naïve H9 and RUES2. H9, primed H9; nH9, naïve H9; RU, RUES2; nRU, naïve RUES2.
Generation of mature human cells of all three germ layers in mouse embryos by naïve hPSCs
We transferred naïve hPSCs to mouse morulae and blastocysts in 15 rounds of injections (table S4). Mouse morulae were injected with naïve hPSCs and cultured in vitro for 1 day. Some of the morulae developed into blastocysts that contained green fluorescent protein (GFP)–labeled human cells in the inner cell mass (ICM) (fig. S3, A to B″). Mouse blastocysts injected with naïve hPSCs were transferred to gestation carriers. Mouse embryos at different days were retrieved for polymerase chain reaction (PCR) or staining, which showed the presence of GFP-labeled human cells in most embryos (fig. S3, C to Q, with results summarized in table S4). To identify whether injected naïve hPSCs produce human cells of all three germ layers, we retrieved E17.5 mouse embryos, which had normal appearance. In an embryo from mouse blastocysts injected with naïve N004 iPSCs (nN004-2), we found a large amount of GFP+ human cells in the liver (Fig. 6A). At a z-level away from that in Fig. 6A, two neighboring sections of nN004-2 embryo were diaminobenzidine (DAB) stained with anti-GFP (Fig. 6B) or stained with hematoxylin and eosin (H&E) for tissue identification (Fig. 6C, with boxes 1 (heart) and 2 (retina) enlarged in Fig. 6, D and E, respectively). The GFP+ human cells in areas highlighted by arrows and box 1 (Fig. 6B) contained red blood cells (RBCs) (Fig. 6, C and D). The GFP+ human cells in box 2 (Fig. 6B) corresponded to retinal pigmented epithelium (Fig. 6, C and E). At a z-level between Fig. 6A and Fig. 6B, a section of this embryo was DAB stained with an antibody against human RBCs (hRBCs). A large amount of cells, including those corresponding to box 1 (heart) in Fig. 6 (B and C) and to the large block of GFP+ cells in Fig. 6A, were hRBCs (mesoderm) (Fig. 6F). Costaining for GFP, hRBC, and DAPI (4′,6-diamidino-2-phenylindole) confirmed that the GFP+ human cells were enucleated RBCs [Fig. 6, G to G‴, which correspond to white boxes in fig. S4 (A to A‴) for a zoomed-out view]. The finding was substantiated by costaining for GFP, the RBC-specific Band 3 protein, and DAPI (Fig. 6, H to H‴). We found many human CD34 (hCD34)+ hematopoietic stem cells and human stem cell factor (hSCF)+ hematopoietic niche cells in bone marrows and liver (fig. S5), two major hematopoietic organs at this stage of development. In liver, we found that some GFP+ human cells were AFP+ endoderm cells (Fig. 6, I to I‴). Costaining for GFP, recoverin (a protein expressed in photoreceptors), and DAPI identified a large amount of human photoreceptors (ectoderm) (Fig. 6, J to J‴, with the cyan box enlarged in fig. S4B), which corresponded to the GFP+ (Fig. 6B) retinal cells (Fig. 6C) in box 2 (Fig. 6E). Additional costaining found that some of the GFP+ human cells were SMA+ mesoderm cells (fig. S4, C to C″) and vimentin+ cells (fig. S4, D to D″). Thus, the nN004-2 embryo contained human cells of all three germ layers. Similarly, the nC005-1 embryo also contained human cells of all three germ layers (fig. S4, E to J‴). The nRUES2-10 embryo contained large amounts of GFP+ cells (fig. S4, K to O), some of which were SMA+ (fig. S4, P to P″). The specificity of GFP fluorescence and GFP-DAB staining is shown in fig. S4 (Q to S).
Fig. 6. Robust formation of mouse-human chimeric embryos.
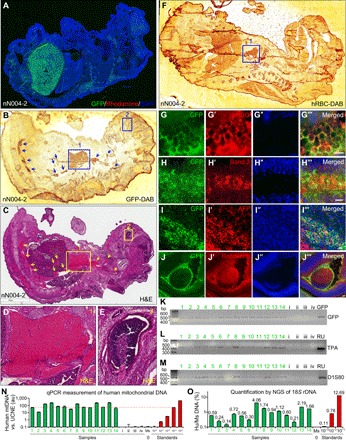
(A) Large amounts of GFP+ human cells were found in an E17.5 mouse embryo (nN004-2) derived from mouse blastocysts injected with GFP-labeled naïve N004 iPSCs (injection #6). (B to E) At a different z-level from (A), two neighboring sections of this embryo were DAB stained with anti-GFP (B) or stained with H&E (C). Boxes 1 (heart) and 2 (retina) in (B) correspond to boxes 1 and 2 in (C), which are enlarged in (D) and (E), respectively. Areas highlighted by arrows and box 1 contained GFP+ (B) RBCs (C and D). Box 2 contained GFP+ (B) retinal pigmented epithelium (C and E). (F to J‴) Different sections of this embryo were DAB stained with antibody against hRBC (F) or immunostained for GFP (G to J), hRBC (G′; mesoderm), the RBC marker Band 3 anion transporter (H′; mesoderm), AFP (I′, endoderm), and the photoreceptor marker recoverin (J′; ectoderm). (G) to (G‴) correspond to boxed areas of fig. S4 (A to A‴). Boxed area in (J‴) is enlarged in fig. S4B. Scale bars, 10 μm. (K to M) PCR detection of GFP (K) or human-specific DNA using DNA fingerprinting primers TPA-25 (L) or D1S80 (M) in genomic DNA isolated from E17.5 mouse embryos derived from mouse blastocysts injected with GFP-labeled naïve RUES2 (embryos 1 to 14; injection #12) or unlabeled naïve RUES2 (embryos i to iv; injection #14). GFP, GFP plasmid as positive control; RU, genomic DNA from RUES2. (N) qPCR measurements of hmtDNA normalized against ultraconserved noncoding element (UCNE) in genomic DNA isolated from the above samples (1 to 14 and i to iv; green bars), C57BL/6 mouse genomic DNA (Ms) and RUES2 human genomic DNA serially diluted in C57BL/6 mouse genomic DNA (red bars). Red dotted line highlights level equivalent to 1:1000 diluted standard. (O) Quantification of human and mouse 18S rDNA by NGS of PCR amplicons of the positive samples in (N) (green bars), C57BL/6 mouse genomic DNA (Ms), and RUES2 human genomic DNA serially diluted in C57BL/6 mouse genomic DNA (red bars). Sequences of the human and mouse amplicons are identical at the primer binding sites on both ends and diverge by 9 bp in the middle of the amplicons. This enables unbiased PCR amplification of human and mouse DNA and their absolute quantification by counting human and mouse reads in NGS.
We detected GFP in genomic DNA isolated from the 14 mouse embryos derived from blastocysts injected with GFP-labeled nRUES2 (green 1 to 14; injection #12 in table S4), but not from the 4 embryos from unlabeled nRUES2 (i to iv; injection #14 in table S4) (Fig. 6K). Individual-specific human genomic DNA was detected in embryos 1 to 14 but not i to iv, using DNA fingerprinting primers for the TPA-25 Alu insert (Fig. 6L) (40) or the D1S80 variable number tandem repeats (VNTRs) (Fig. 6M) (41). Using quantitative PCR (qPCR), we measured the amount of human mitochondrial DNA (hmtDNA) in the above samples, together with serially diluted human genomic DNA as standards (42). Of the 14 positive samples, 11 contained hmtDNA equivalent to human genomic DNA diluted to between 1:1000 and 1:100 in mouse genomic DNA, and 3 (#2, #7, and #14) were between 1:10,000 and 1:1000 (Fig. 6N). To confirm these notable findings, we amplified the V3 region (43) of the gene encoding for 18S ribosomal RNA (18S rDNA), which has high copy numbers (44). The human and mouse amplicons have identical sequences on both ends and only differ in the middle by 9 base pairs (bp). This enables unbiased amplification of human and mouse DNA and absolute quantification of the amplicons by counting the number of human reads and mouse reads by next-generation sequencing (NGS). With this method, we found that the 14 positive samples contained 0.14 to 4.06% human DNA (Fig. 6O and table S5), consistent with the less accurate measurement by qPCR of hmtDNA (Fig. 6N). These quantifications cannot cover the large number of enucleated hRBCs, which do not have mitochondria or nucleus (45). In all 13 rounds of blastocyst injections, 10 rounds generated chimeras and 3 initial rounds failed because of technical unfamiliarity (table S4 and Materials and Methods). The 14 DNA samples from injection #12 were isolated freshly. Other DNA samples were decrosslinked and recovered from fixed embryos, which markedly affected qPCR quantification of hmtDNA and NGS. We did not find a detectable amount of human cells or human DNA in extraembryonic tissues, in contrast to the situation seen with extended pluripotent stem cells, which are cultured in a very different condition (12).
Dependency on nuclear translocation of TFE3 in primed-to-naïve conversion
To explore the mechanism of the conversion, we generated primed H9 stably overexpressing TFE3-GFP fusion proteins (Fig. 7, A to C) or GFP-tagged TFE3 with mutated nuclear localization signal (NLS), which largely resided in the cytoplasm as puncta (Fig. 7, D to F). When both lines of primed hESCs were treated with Torin1 (10 μM for 3 hours) in the conversion protocol (Fig. 1J), TFE3-GFP was enriched exclusively in the nucleus (Fig. 7, G to J), while NLS-GFP remained largely in the cytoplasm (Fig. 7, K to N). NANOG+ naïve hESC colonies were readily obtained from H9 expressing wild-type TFE3 (Fig. 7, G to J, O, and P) but not its NLS mutant (Fig. 7, K to O and Q). The small percentage of NANOG+ cells with the NLS mutant TFE3 did not exhibit mESC morphology and very quickly differentiated. We were unable to establish naïve hESC line from primed H9 overexpressing NLS-GFP with the same condition that readily generated nH9 hESC overexpressing TFE3-GFP. Overexpression of the TFE3 NLS mutant apparently acted in a dominant negative manner to block the action of endogenous TFE3, as the activation of TFE3 requires dimerization (46). Coexpression of the TFE3 NLS mutant with MYC-tagged wild-type TFE3 in human embryonic kidney (HEK) 293 cells significantly reduced nuclear localization of MYC-TFE3, particularly after Torin1 treatment (Fig. 7, R to V). TFE3 was localized in the nucleus in naïve hPSCs, which were maintained in 2iLI media without Torin1 (fig. S6, A to A″). Expression of wild-type TFE3 (fig. S6, B to F) or its NLS mutant (fig. S6, G to K) did not appreciably affect the pluripotency of primed H9 cells, suggesting that TFE3 NLS mutant does not have nonspecific toxicity.
Fig. 7. Nuclear localization of TFE3 is critical for primed to naïve conversion.
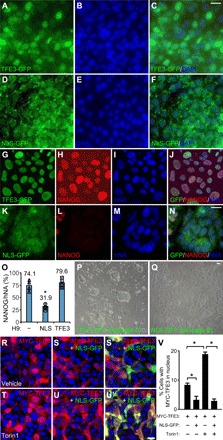
(A to F) Primed H9 hESCs stably expressing TFE3-GFP fusion proteins (A to C) or NLS-mutated TFE3-GFP (NLS-GFP) (D to F) were costained for GFP (A and D) and DAPI (B and E). (G to Q) Primed H9 hESCs stably expressing TFE3-GFP (G to J) or NLS-GFP (K to N) went through the conversion protocol in Fig. 1J and were stained at day 5 for GFP (G and K), NANOG (H and L), and hNA (I and M). Merged images (J and N) were quantified for the percentage of NANOG+ cells in human nuclear antigen (hNA+) cells (O). *P < 0.05, n = 4, one-way ANOVA versus control H9. Phase-contrast images of H9 expressing TFE3-GFP (P) or NLS-GFP (Q) were acquired at day 5 of conversion. Scale bars, 10 μm. (R to V) HEK293 cells transfected with MYC-TFE3 alone (R and T) or together with NLS-GFP (S and U′) were treated with vehicle (R to S′) or Torin1 (10 μM for 3 hours) (T to U′) and stained as indicated. Merged images (S′ and U′) highlighted cells transfected with NLS-GFP. Percentage of cells with MYC-TFE3 in nucleus was quantified for each condition. *P < 0.05, Student’s t test, n = 250 cells per condition. Scale bars, 10 μm.
Torin1 or rapamycin treatment of primed H9 in hESC medium induced autophagy. Autophagy was slightly induced by switching the medium to 2iLI, increased by physiological glucose concentration, and greatly enhanced by the combined treatment with Torin1 (fig. S7, A to G). However, primed-to-naïve conversion was not significantly affected by blocking autophagy with the Ulk1 inhibitor SBI-0206965 or by inducing autophagy with amino acid deprivation (fig. S7, H to O). It suggests that Torin1-induced autophagy is not critical for the conversion. Changes in autophagy in response to treatments in fig. S7 (H to O) were shown in fig. S7 (P to W).
DISCUSSION
In this study, we found a simple and efficient method for the conversion of hPSCs from primed to naïve pluripotency by a 3-hour inhibition of mTOR with Torin1 (Fig. 1) or rapamycin (fig. S1T). Conversion of the cells and their subsequent culture were in medium essentially similar to that used for maintaining mESCs in naïve pluripotency (2). The only differences, which markedly improved the derivation of naïve hPSCs, were the replacement of undefined N2 and B27 supplements with human insulin, as well as the reduction in glucose concentration and O2 tension to physiological levels (fig. S1, D to M). The naïve hPSCs passed many important criteria for naïve pluripotency in human (Figs. 1 to 6 and figs. S1 to S5) (5, 34), although it is unclear why a number of previously identified human naïve pluripotency markers were not up-regulated (fig. S2D) and XIST expression was lost (Fig. 4H), which suggests that our cells may reflect an intermediate state. Nevertheless, the unified culture condition and the robust chimerism distinguish the present study from previous publications that show limited integration of immature human cells in mouse embryos up to E10.5 (6–12, 15). The use of additional inhibitors beyond 2i appears to constrain some of these naïve hPSCs in a state that shares more transcriptomic similarities to cells in human blastocysts (8, 9, 47), but is, however, unable to support robust chimerism in mouse embryos (8, 9). By transiently inhibiting mTOR and capturing the changed cell state in 2iLI media, we confine hPSCs in a state functionally similar to naïve pluripotency in mESCs. It appears that the nuclear translocation of TFE3 induced by Torin1 underlies the conversion (Fig. 7). Consistent with the critical function of TFE3 in naïve pluripotency in mESCs (22), Torin1-induced nuclear translocation of TFE3 activates transcription events (22) that lead to the conversion from primed to naïve pluripotency. Although the exact mechanistic details await further studies, blocking or inducing autophagy did not significantly affect Torin1-induced conversion (fig. S7, H to O).
The transcriptomes of our naïve hPSCs share many similarities and some differences to those of the cells isolated from human blastocysts (Fig. 3, B and C). This mirrors the transcriptomic differences between naïve hPSCs directly derived from the ICM of human blastocysts and the ICM itself (47, 48). Furthermore, transcriptomes of mESCs are different from cells in mouse ICM (49). ESCs have many epigenetic aberrations (50) that may enable indefinite self-renewal in cell culture, while cells in ICM are in a rapid transition state that is poised to execute the developmental program of making the body. Naive hPSCs that are transcriptomically more similar to cells in human blastocysts than ours fail to generate robust chimeras (8, 9), while mESCs, despite their transcriptomic differences to mouse ICM (49), efficiently form chimeras when injected in mouse or rat blastocysts (51). As transcriptomic analysis is restricted to gene expression levels, without considering many other factors, such as activities of the gene products in temporal and spatial domains, it is our own opinion that a functional criteria of naïve pluripotency based on the biology of the cells would be more useful. In this regard, naïve hPSCs generated in the study represent the most similar functional state to mESCs because they were cultured in the same condition and substantially contributed to mouse embryos.
By confining the culture condition of naïve hPSCs to that of mESCs, we unify some common features of naïve pluripotency in mammals. The most notable finding of the study is the robust contribution of naïve hPSCs to human cells of all three germ layers in mouse-human chimeric embryos (Fig. 6 and fig. S4). Absolute quantification by NGS of 18S rDNA showed that the E17.5 mouse embryos contained 0.14 to 4.06% human DNA (Fig. 6O). Both NGS and qPCR (Fig. 6N) quantifications cannot cover the large number of enucleated hRBCs, which do not have mitochondria or nucleus (45). The identification of large amounts of enucleated hRBCs and photoreceptors after 17.5 days of gestation showed that the development of naïve hPSCs was markedly accelerated to match the mouse embryos. Human embryos at this stage do not have these mature cells (www.prenatalorigins.org/virtual-human-embryo/). The detection of a large amount of hRBCs is consistent with the estimate that 70 to 80% of all human cells are RBCs (52, 53). Progenies of naïve hPSCs in a mouse embryo must quickly embark on a developmental program to produce huge amounts of RBCs, which are responsible for O2 delivery and CO2 removal, once the size of the embryo grows beyond gas diffusion limit. The derivation of chimera-competent naïve hPSCs may enable many applications previously impossible in the human system, such as selection-driven heterologous organ generation in chimeric animals (51, 54, 55). The significant utilities of such technologies and their social ramifications call for careful ethical considerations (56, 57).
MATERIALS AND METHODS
Regulatory approvals
The University at Buffalo Institutional Review Board has determined that the use of human cells in the study is not human subject research. The University at Buffalo/Roswell Park Cancer Institute (RPCI) Stem Cell Research Oversight (SCRO) Committee has approved all experiments on hPSCs described in this study. All animal experiments were conducted in the Gene Targeting and Transgenic Resource of RPCI with approval from the Institutional Animal Care and Use Committee (IACUC) of RPCI. The experiments adhere to the 2016 ISSCR Guidelines For Stem Cell Research And Clinical Translation. All mouse embryos were euthanized immediately upon retrieval by fixation in 4% paraformaldehyde (PFA). Animal welfare was not affected in this process. We did not detect contribution of human cells to germline tissues. In nC005-1 embryo, we detected rare Nestin+ or PAX6+ human neural cells. In nN004-2 embryo, we detected substantial amounts of human cells in the retina. These events did not affect the function or welfare of the pregnant mice or the embryos because the embryonic eye was not capable of vision yet and the embryonic brain contained very few human neural cells to affect embryonic mouse brain functions.
Animals
All mice were bred and housed in RPCI following approved IACUC protocols. C57BL/6J female mice at 3 to 4 weeks of age were used to collect morulae and blastocysts. Pseudopregnant CD-1 female mice at 7 to 9 weeks of age were used as gestation carrier of injected blastocysts. Male severe combined immunodeficient (SCID) mice (C.B-Igh-1bIcrTac-Prkdcscid/Ros) at 17 weeks old were used in teratoma formation assay.
Human pluripotent stem cells
hPSCs including hESC lines H1 at passages 40 to 42 (WiCell), H9 at passages 31 to 35 (WiCell), RUES2 at passages 30 to 33 (Rockefeller University), and human iPSC lines C005 at passages 25 to 28 and N004 at passages 21 to 25 were maintained on mitomycin C–treated MEF feeders (2 × 104 to 3 × 104/cm2) in hESC medium [DMEM/F12 containing 20% knockout serum replacement, 2 mM glutamine, 1% nonessential amino acids (NEAA), penicillin (100 U/ml), streptomycin (100 μg/ml), 0.1 mM β-mercaptoethanol, and basic fibroblast growth factor (4 ng/ml)]. Medium was changed daily, and cells were passaged every 6 to 7 days using dispase (1 mg/ml). All hPSCs were cultured in 5% O2 and 5% CO2 unless indicated otherwise. We have eliminated ultraviolet (UV) radiation by using yellow fluorescent bulbs in all cell culture hoods and covering white fluorescent bulbs in all rooms with UV-blocking translucent screens. All cells were tested regularly for mycoplasma contamination by PCR. No mycoplasma was detected.
Naive hPSCs
All established naïve hPSCs were maintained on mitomycin C–treated MEF feeders (5 × 104 to 6 × 104/cm2) in 2iLI medium [50% glucose-free DMEM/F12 and 50% glucose-free Neurobasal, 5 mM glucose, 1 mM glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol, penicillin (100 U/ml), streptomycin (100 μg/ml), bovine serum albumin (BSA; 5 mg/ml) to maintain osmolarity, recombinant human LIF (20 ng/ml), human insulin (18 μg/ml), 1 μM PD0325901, and 3 μM CHIR99021]. Naive hPSCs were passaged every 3 days with TrypLE. All naïve hPSCs were cultured in 5% O2 and 5% CO2 unless indicated otherwise. All cells were tested regularly for mycoplasma contamination by PCR. No mycoplasma was detected.
Mouse ESCs
mESCs (AB2.2) were cultured on gelatin-coated 10-cm dishes in mouse 2iL medium (2) [1:1 mixture of DMEM/F12 with N2 supplements and Neurobasal with B27 supplements, mouse LIF (20 ng/ml), 1 mM glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol, penicillin-streptomycin, BSA (5 mg/ml), 1 μM PD0325901, and 3 μM CHIR99021]. The mESCs were passaged every 2 days with TrypLE.
Converting hPSCs from primed state to naïve state
Primed hPSCs were treated with 10 μM Rho-associated protein kinase (ROCK) inhibitor Y27632 (Abcam) overnight in hESC medium. After being washed twice in phosphate-buffered saline (PBS), primed hPSCs were cultured in 2iLI medium with 10 μM Torin1 (Tocris) for 3 hours. For N004 iPSCs, 2iLI medium with 0.8 μM CHIR99021 was used instead. Then, hPSCs were trypsinized into single cells using TrypLE (Life Technologies) for 5 min at 37°C. The single cells were plated on MEF feeders (5 × 104 to 6 × 104/cm2) in 2iLI medium (without Torin1), which was changed daily. For N004 iPSCs, 2iLI medium with 0.8 μM CHIR99021 was used instead. Small, bright, dome-shaped colonies appeared in 4 to 5 days and were picked manually at days 5 to 7 for dissociation by TrypLE into single cells, which were plated on fresh MEF cells. After several manual passages, mESC-like colonies were uniformly seen and passaged every 3 days with TrypLE and maintained in 2iLI media (without Torin1).
Measurement of clonal efficiency
Naive hESCs (nH9 and nRUES2) or primed hESCs (H9 and RUES2) were trypsinized into single cells and replated on MEF feeders in 2iLI medium or hESC medium without or with the ROCK inhibitor Y27632 (10 μM). They were cultured in incubators with 21 or 5% O2 for 3 days for naïve hESCs or 7 days for primed hESCs. The numbers of naïve or primed hESC colonies were counted by alkaline phosphatase (AP) staining to quantify clonal efficiency, which is the ratio of AP+ colonies over the number of single cells plated, expressed in percentage.
Measurement of cell doubling time
Cell doubling time was measured as previously described (23) by plating 1 × 105 naïve or primed hPSCs on MEF cells in 24-well plates. The numbers of hPSCs in triplicate wells were counted using trypan blue exclusion on a hemocytometer at 1, 2, 3, 4, 5, and 6 days after plating. Cell doubling time was calculated using the calculator at www.doubling-time.com/compute.php.
Reversion of naïve hPSCs to the primed state
Naive hPSC colonies were picked manually, washed once in DMEM/F12 medium, and then plated on MEF feeders in hESC medium. Typical flat hESC colonies appeared in the culture in 7 to 10 days. These reversed primed state hPSCs were passaged every 5 to 7 days using dispase (1 mg/ml).
Spontaneous differentiation of naïve hPSCs in vitro
Naive hPSCs were dissociated into single cells by TrypLE treatment for 5 min at 37°C and cultured in suspension in ultralow attachment 96-well plates (Corning) in differentiation medium [DMEM/F12 with 10% fetal bovine serum (FBS), 1% NEAA, and 1% penicillin-streptomycin) for 4 to 6 days to form embryoid bodies, which were plated on gelatin-coated 24-well plates in differentiation medium for another 2 to 4 weeks of attachment culture.
Teratoma formation assay
Teratoma formation assay was performed by the Mouse Tumor Model Resource at RPCI following approved IACUC protocol. All animal experiments were done by staff members in the core facility in blinded fashion. The persons who performed the animal procedures only knew the codes assigned to the cells, not what kind of cells they were. Briefly, 1 million naïve hPSCs were mixed with collagen at 1:1 ratio to form a 10-μl mixture, which was plated on Parafilm to solidify at room temperature for 1 hour. Three such pellets were grafted under the renal capsule of each kidney in a male SCID mouse (C.B-Igh-1bIcrTac-Prkdcscid/Ros) around 17 weeks of age. Animals were monitored for palpable tumors around the kidney area. Large tumors (~1 cm in size) were generally found 2 to 3 months after grafting. Tumors were harvested and dissected into small pieces and fixed for 24 hours in 10% formalin and processed for paraffin embedding. Tissue sections (5 μm) were stained with H&E for histological identification.
Mitochondrial respiration
Mitochondrial respiration in naïve hPSCs was assessed by measuring oxygen consumption rate in a Seahorse XFe24 analyzer according to the manufacturer’s protocol. Briefly, naïve hPSCs were dissociated with TrypLE and replated at 8 × 105/ml in 2iLI medium in 100-μl volume on laminin-coated XF24 cell culture plates (Seahorse Bioscience) and cultured overnight at 37°C in 5% O2 and 5% CO2. Culture medium was replaced with XF Base Medium (Seahorse Bioscience) supplemented with 2 mM pyruvate and 5 mM glucose at pH 7.4. Cells were incubated at 37°C in the machine for 1 hour to allow the assay medium to preequilibrate. Oligomycin (2 μM), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (0.5 μM), antimycin (1 μM), and rotenone (1 μM) were injected during the assay. The results of the Cell Mito Stress Test were calculated using the manufacturer’s software (Seahorse Bioscience).
PCR detection of reprogramming footprint in the derivation of iPSCs
PCR was performed to detect episomal plasmids in genomic DNA isolated from primed C005 human iPSC, which was generated using episomal plasmids (25) or lentiviral transgene expressing OCT4 in genomic DNA isolated primed N004 human iPSC, which was generated using DOX-inducible lentiviruses expressing OCT4, SOX2, KLF4, c-MYC, and NANOG (26). The primers for detecting episomal plasmids are GCAACATTAGCCCACCGTGCTCTC and GGTTATTAAGATGTGTCCCAGGC (25). The primers for detecting OCT4 lentivirus were CCCCAGGGCCCCATTTTGGTACC and AAAGCAGCGTATCCACATAGCGTA (26).
Dot blot analysis of 5mC and 5hmC
Genomic DNA isolated from different types of cells was denatured at 99°C for 5 min and snap cooled on ice. The sample (5 μl) was spotted on positively charged nylon membrane (Bio-Rad), air dried, and cross-linked by UV. The membrane was washed with 2× SSC buffer, blocked with 5% milk in TBST (1× TBS + 0.1% Tween 20) for 60 min, and incubated with 5mC antibody (1:500) or 5hmC antibody (1:500) in blocking solution at room temperature for 1 hour. The membrane was washed three to four times with TBST at room temperature for 5 to 10 min per wash, incubated with horseradish peroxidase–conjugated anti-rabbit immunoglobulin G (IgG) secondary antibody (1:1000) at room temperature for 60 min, and then washed with TBST at room temperature four times (5 min each). The membrane was treated with enhanced chemiluminescence reagent (Thermo Fisher Scientific). The signals were captured on gel imaging systems (Bio-Rad) and analyzed using the Image Lab software (Bio-Rad).
RNA-seq and bioinformatics analysis
RNA-seq was performed on primed H9 (PH9), naïve H9 (NH9), primed RUES2 (PRUES2), and naïve RUES2 (NRUES2) with three biological replicates for each line. For each sample, colonies of hPSCs were manually picked and homologized in 1 ml of TRIzol reagent (Thermo Fisher Scientific). RNeasy Mini Kit (QIAGEN) was used for RNA extraction. Quality of purified RNA was monitored by agarose gel electrophoresis. Polyadenylated RNA enrichment, cDNA library preparation, sequencing, and Reads Per Kilobase of transcript, per Million mapped reads (RPKM) calculation were performed at the University at Buffalo Genomics and Bioinformatics Core Facility using the published pipeline (58). All bioinformatics analyses were performed using R language. logRPKM was transformed from RPKM by log2(RPKM+0.05). FASTQ data were deposited to Gene Expression Omnibus (GEO) under GSE87452. A list containing 1811 genes named “Coding Genes Differentially Expressed between Naive and Prime States (DENvP_coding)” (table S1) was defined by the following criteria: (i) max(logRPKM) > 1, (ii) |mean(logRPKMnaïve) − mean(logRPKMprime)| > 1, and (iii) Benjamini and Hochberg–adjusted P value [false discovery rate (FDR)] (59) <1% in two-way analysis of variance (ANOVA) on the “Naive versus Prime” factor. RNA-seq data of previously established inhibitor-dependent naïve hPSCs [E-MTAB-2857 (8), E-MTAB-2031 (7), GSE87239 (29), and GSE63570 (30)] and single-cell RNA-seq data from primed hESC and human preimplantation embryo [GSE36552 (27) and E-MTAB-3929 (28)] were downloaded from the GEO or European Bioinformatics Institute databases. RPKM were calculated and log transformed by the same pipeline mentioned above. All datasets were merged by gene names, and each sample was quantile normalized to obtain the same distribution. Rank-based Z score was calculated as well; it gave the similar PCA and clustering results. Expression level of the genes in DENvP_coding were extracted from the full dataset and used in the following analyses. In PCA, single-cell RNA-seq data on preimplantation human blastocysts and primed hESCs were used to define the two-dimensional principle components space. All the other data points of hPSCs and other cells in early embryonic stage (E5 to E7) were projected to this space. Unsupervised clustering was built on Spearman correlation matrix. The result was visualized as phylogenic tree using the ape package (60). Heat map was generated with heatmap.2 function in gplot package. For the gene expression analysis on different chromosomes in Fig. 4I, |mean(logRPKMnaïve)− mean(logRPKMprimed)| was calculated for the genes with min(logRPKM) > 1. Genes in autosomes were grouped by the chromosome in which they locate. Genes in X chromosome were grouped into X-inactivated (Xi) genes and genes escaped from X inactivation (Xe) according to previous report (38). P values in two-tailed t test were adjusted with the Benjamini and Hochberg method. To identify a common signature in gene expression for naïve hPSCs, differentially expressed genes were independently called from datasets of each naïve/primed hPSC pairs with the following criteria: (i) max(logRPKM) > 1, (ii) |mean(logRPKMnaïve) − mean(logRPKMprime)| > 1, and (iii) Benjamini and Hochberg–adjusted P value (FDR) < 5%. Wilcoxon rank-sum test was used when the sample size is bigger than 5, and Student’s t test was used when sample size is only 3. E-MTAB-2031 [for Ch_P (7) and Ch_N (7)] was excluded because of the significant dissimilarity comparing to other naïve cells, as shown in our clustering and heat map analysis (Fig. 3, C and D). GSE87239 [for Sa_P (29) and Sa_N (29)] was excluded since it contains only two replicates for primed cells and, thus, does not have enough power in t test. We found 310 genes whose expression was significantly changed in the same direction across all three datasets [Hu_P (six replicates) versus Hu_N (six replicates) in this study, Ta_P (three replicates) (8) versus Ta_N (three replicates) (8), and Gr_P (three replicates) (30) versus Gr_N (three replicates) (30)] (table S2). Expression levels of the 310 genes in GSE87239 [for Sa_P (29) and Sa_N (29)] are listed in the same Excel file for comparison. Fold changes of the expression levels of those of the 310 genes that encode for transcription factors, epigenetic modifiers, and growth factors, as well as previously identified naïve signature genes, were displayed in fig. S2.
Bioinformatics analysis of TE
Two different mapping and analysis were performed on the basis of previously reported methods (30, 33). First, raw RNA-seq reads were mapped to TE reference map (33) using the following command “tophat -g1 --b2-sensitive --no-novel-juncs --no-novel-indels -o $outputdir --transcriptome-index= $transcriptome $index $reads.” Counts for individual TE were normalized by number of total reads and transformed by log2(Normalized-count+1) to generate relative expression level. A list containing 889 TEs named “Transposable Elements Differentially Expressed between Naive and Prime States (DENvP_TE)” (table S3) was defined with the same criteria used in the analysis of coding genes, except that relative expression level was used instead of RPKM. Relative expression level of these TEs in previously established naïve and primed hPSCs were extracted following the same pipeline. Heat map and clustering were built on Spearman correlation matrix using heatmap.2 function in gplot package. Second, raw RNA-seq reads of our own and other naïve and primed hPSCs were aligned to repbase consensus sequences (downloaded from RepBase) with Bowtie (61) using the command “bowtie -q -p 8 -S -n 2 -e 70 -l 28-maxbts 800 -k 1 -best,” as previously reported (30). Counts for individual TE group were normalized by number of total reads and transformed using log2(Normalized-count+1). Naive and primed hPSCs from the same laboratory were treated as a pair. Each TE group in each naïve and primed hPSCs pairs was plotted on a scatter plot in Fig. 3F. Data points corresponding to HERVK and LTR5_Hs were highlighted.
Determination of allelic expression
Allelic read counts and heterozygotic SNPs were generated with ASEQ (62) using GENOTYPE mode for X chromosome SNVs available in dbSNP (build 146). Heterozygotic SNPs were identified as having a total read coverage above 5 with an alternative base frequency between 0.2 and 0.8. Each RNA-seq dataset was analyzed separately, and the union of all heterozygous SNPs was used to examine allelic expression. Allelic expression for representative genes containing significant coverage (>5) per experimental replicate at known X-inactivated or X-escaped genes is shown in Fig. 4 (J to K, respectively).
Genome-wide DNA methylation study and bioinformatics analysis
Infinium MethylationEPIC Beadchip assay was performed on primed H9 (H9), nH9, primed RUES2 (RUES2), and naïve RUES2 (nRUES2) with four biological replicates for each line at the RPCI Genomics Shared Resource. Genomic DNA was extracted and purified using QIAamp DNA Blood Mini Kit (QIAGEN). DNA methylation was determined using Infinium MethylationEPIC array (Illumina) with the Infinium HD Assay Methylation Protocol. Genomic DNA (500 ng) for each sample is bisulfite converted with EZ DNA Methylation-Gold Kit (Zymo Research); then, 200 ng of each converted sample is amplified, fragmented, loaded into the Methylation EPIC BeadChips and hybridized overnight. Following washing, staining, and addition of a protective coating, the BeadChips are imaged using the Illumina iScan Reader to measure the fluorescence intensity of each probe for both methylation and unmethylated DNA. BeadChip data files are analyzed with Illumina’s GenomeStudio (v2011.1) methylation module (v1.9.0) to report control normalization with background subtraction methylation data. Idat files were input into RnBeads package in R with an additional, customized annotation of imprinting region (38). One sample of nH9 failed in quality control and was discarded in subsequent analysis. Probes were filtered, and the signal of each probe was normalized by default pipeline. PCA was a direct output of RnBeads using beta value of all sites. Averaged beta values for each tiling region (5-kb window by default), promoter, gene body, and imprinting region were calculated using RnBeads. Differentially methylated regions were defined by FDR < 0.01 in two-way ANOVA on naïve versus primed. Differentially methylated tiling regions were rank ordered by the differences between mean of beta values of naïve and primed hPSCs. The scaled beta values of each region were visualized as heat map with the pheatmap function. Heat map for the imprinting regions was built on Pearson correlation matrix. For visualization purpose, averaged beta values were rescaled on each imprinting region in heatmap.2 of gplot.
Incorporation of naïve hPSCs in mouse embryos
All animal experiments on the injection of naïve hPSCs to mouse morulae and mouse blastocysts and the transfer of injected blastocysts to pseudopregnant mice were performed by Gene Targeting and Transgenic Resource of RPCI following IACUC and SCRO approvals. All animal experiments were done by RPCI staff members in the core facility in blinded fashion. The persons who performed the animal procedures only knew the codes assigned to the cells, not what kind of cells they were. The facility had not previously injected any human cells to mouse embryos; we were the only group that requested this service. They did the injection in our absence. Naive hPSCs (nRUES2, nRUES2-GFP, nC005-GFP, and nN004-GFP) were plated at 2 × 105 cells per well on MEF cells in 12-well plates and cultured in 2iLI medium for 2 days. They were dissociated to single cells with TrypLE for 5 to 6 min and placed on 10-cm dishes for 45 min to remove MEF cells through attachment. The supernatant, which contained the hPSCs, was removed from the dish and centrifuged for 10 min at 2000 rpm. Cells in the pellet were resuspended in Mouse Blastocysts Injection Buffer (Hepes-buffered DMEM with 5% FBS). Two to five naïve hPSCs were injected into a mouse morula, which was obtained by superovulation from 3- to 4-week-old C57BL/6J female mice. Injected mouse morulae were incubated for 1 day in vitro to develop into blastocysts. Ten to 12 naïve hPSCs were injected to a C57BL/6J mouse blastocyst, which was obtained by superovulation from 3- to 4-week-old C57BL/6J female mice. Injected blastocysts were transferred bilaterally to the uterus of pseudopregnant CD-1 female mice at 7 to 9 weeks of age, with 14 to 18 blastocysts transferred per mouse. At the indicated days of gestation, mouse embryos were retrieved by the RPCI staff and immediately euthanized by fixation in 4% PFA for us to bring back to our laboratory at State University of New York at Buffalo. The embryos were fixed in 4% PFA at 4°C for 2 days and then transferred to 30% sucrose solution at 4°C for 14 to 24 hours until the embryos sank to the bottom of the tube. The mouse embryos were then embedded in tissue freezing medium (Triangle Biomedical Sciences) and frozen in liquid nitrogen. Frozen embryo blocks were cut on a cryostat into 15-μm-thick sections, which were placed on Ultra Plus Adhesion Slides (Thermo Scientific) for immunostaining. In some experiments, half of each mouse embryo cut sagittally was incubated in proteinase K solution [100 mM tris-HCl (pH8.0), 5 mM EDTA, 0.2% SDS, 200 mM NaCl, and proteinase K (250 μg/ml)] at 55°C for 12 to 18 hours for extraction of genomic DNA. PCR amplification of the GFP gene in genomic DNA isolated from mouse embryos was performed using primers TCACGAACTCCAGCAGGACCATGT and TGACCTACGGCGTGCAGTGCTTCA. Human-specific DNA was detected by DNA fingerprinting using primers for D1S80 VNTR GAAACTGGCCTCCAAACACTGCCCGCCG and GTCTTGTTGGAGATGCACGTGCCCCTTGC (41), or primers for TPA-25 Alu insert GTAAGAGTTCCGTAACAGGACAGCT and CCCCACCCTAGGAGAACTTCTCTTT (40). Details of the 15 rounds of morula or blastocyst injections are listed in table S4. Of the 13 rounds of blastocyst injections, 10 rounds produced chimeras, which accounted for most, if not all, of the embryos generated in each round. The other three rounds failed to produce a chimera (#10, #13, and #14), including one round that did not produce an embryo (#10). The three failed rounds were all done at the beginning, when the core facility was not familiar with injecting naïve hPSCs, which were more sensitive to trypsinization than mESCs. We used naïve hPSCs around passages 15 to 20 for injection to mouse blastocysts and did not systematically study the effect of passage numbers on chimerism.
Quantification of hmtDNA in chimeric mouse embryos
The amounts of hmtDNA and ultraconserved noncoding element (UCNE) were measured by qPCR in triplicates using hmtDNA-specific primers (forward, AATATTAAACACAAACTACCACCTACCT; reverse, TGGTTCTCAGGGTTTGTTATAA) (63) and UCNE primers (forward, AACAATGGGTTCAGCTGCTT; reverse, CCCAGGCGTATTTTTGTTCT) (42) with Fast SYBR Green Master Mix (Thermo Fisher Scientific) on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Human genomic DNA standards were prepared by a 10-fold serial dilution of human genomic DNA in mouse genomic DNA. Genomic DNA (100 ng) was used in each reaction. The relative level of hmtDNA in each sample was calculated by 2Ct(UCNE)−Ct(hmtDNA).
Quantification of human and mouse 18S rDNA by NGS
The high copy numbers of genes encoding the 18S ribosomal RNA (18S rDNA) facilitate quantification of human DNA in mouse DNA. The V3 regions (43) of both human and mouse 18S rDNA contain a stretch with identical sequences at both ends, with the middle sequences diverge by 9 bp. This enables unbiased PCR amplification of the human fragment (134 bp) and the mouse fragment (135 bp), which can be quantified accurately by counting the numbers of human reads and mouse reads on NGS. The sequences of the human/mouse common primers are TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGCTAATACATGCCGACGGG (forward) and GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTCTAGAGTCACCAAAGCCGC (reverse). Underlined sequences are bar code attachment sites for NGS. PCR was performed using Hotstar plus kit (QIAGEN) with an optimized PCR protocol (95°C, 5 min; 94°C, 30s; 58°C, 30s; 72°C, 1 min; 40 cycles; and 72°C, 10 min). PCR products from each sample were gel extracted and purified with QIAquick Gel Extraction Kit (QIAGEN), barcoded and library prepared using NextSeq 500/550 Mid Output Kit v2.5 (Illumina), and then sequenced in duplicates on NextSeq with 150-bp paired sequencing mode. Per-cycle basecall (BCL) was converted to per-read FASTQ files using bcl2fastq version 2.20.0.422 using default parameters. Forward and reverse barcode attachment sites were trimmed using cutadapt (https://doi.org/10.14806/ej.17.1.200) version 1.16. Forward and reverse read pairs were merged using vsearch (64) fastq_mergepairs. Merged reads with greater than one expected error were filtered out. The remaining merged reads were classified as human or mouse using vsearch –usearch_global with the percent identity cutoff set at 99%. The reference sequence used for human 18S rDNA was AGCTAATACATGCCGACGGGCGCTGACCCCCTTCGCGGGGGGGATGCGTGCATTTATCAGATCAAAACCAACCCGGTCAGCCCCTCTCCGGCCCCGGCCGGGGGGCGGGCGCCGGCGGCTTTGGTGACTCTAGA, and the reference sequence used for mouse 18S rDNA was AGCTAATACATGCCGACGGGCGCTGACCCCCCTTCCCGGGGGGGGATGCGTGCATTTATCAGATCAAAACCAACCCGGTGAGCTCCCTCCCGGCTCCGGCCGGGGGTCGGGCGCCGGCGGCTTGGTGACTCTAGA. To remove the influence of copy number variations in different human individuals and mouse strains (44), we used RUES2 human genomic DNA and C57BL/6 mouse genomic DNA to match the source materials (naïve RUES2 and C57BL/6 blastocysts). The ratio of 18S rDNA copy numbers between C57BL/6 and RUES2 was calculated using the mean of [(number of mouse reads/number of human reads) × dilution factor] from the serially diluted standards. This ratio was 83.9 ± 8.0. The percentage of human DNA in mouse DNA was calculated using the following formula: (number of human reads/number of mouse reads) × copy number ratio × 100%. The number of human and mouse reads and the sequences of all human reads are included in table S5. Raw and processed data for NGS of 18S rDNA amplicons were deposited to GEO under GSE125813. Sequences of primers used in the study are listed in table S6.
Immunostaining
Immunostaining using antibodies listed in table S7 was performed using standard protocol to detect various antigens in cultured cells or frozen tissue sections. Briefly, cells or tissue sections were fixed in 4% PFA (Sigma) for 20 min, treated with 0.1% Triton X-100 for 15 min at room temperature for permeabilization, blocked in 3% BSA for 1 hour at room temperature, and then incubated with the indicated primary antibodies overnight at 4°C and secondary antibodies for 1 hour at 37°C. Secondary antibodies were conjugated with Alexa Fluor 488, 594, and 647 (1:1000; Thermo Fisher Scientific). AP staining was performed using the Alkaline Phosphatase Kit (Millipore). DAB staining was also used to detect GFP expression in frozen embryo sections according to the manufacturer’s protocol (Vector Laboratories). Briefly, the frozen sections on slides were thawed at room temperature for 5 min. Then, the slides were treated with 0.3% H2O2 solution in PBS at room temperature for 10 min to block endogenous peroxidase activity and then treated with 3% BSA for 45 min for blocking. The sections were incubated with primary antibody (anti-GFP) in 0.1% BSA for 30 min at room temperature, rinsed briefly in PBS three times (5 min each), and then incubated with biotinylated secondary antibody for 30 min, followed by PBS rinsing three times (5 min each). The sections were incubated with VECTASTAIN ABC reagent (Vector Laboratories) for 30 min and washed with PBS for 5 min. The sections were incubated with 100 μl of peroxidase substrate solution (Sigma, D4293) on the slide under a microscope until desired staining intensity and then washed with PBS to remove the substrate solution. For live imaging of mitochondria, primed or naïve hPSCs were incubated in prewarmed hESC medium (for primed hPSCs) or 2iLI medium (for naïve hPSCs) containing 50 nM MitoTracker Green FM or 100 nM TMRE (Life Technologies) for 15 min at 37°C. Then, the staining medium was replaced with the corresponding prewarmed medium without dyes. Cells were then imaged on a Leica DMI6000B fluorescence microscope. Quantification of LC3II puncta was performed using the National Institutes of Health ImageJ with the AUTOCOUNTER plugin (65), which calculates the percentage of cell area covered by LC3II puncta.
Plasmid constructs and lentiviral labeling of hPSCs
The LV-EF1a-GFP plasmid was provided by S.-c. Zhang at the University of Wisconsin-Madison (66). Lentivirus generated from this construct was used to label naïve-state N004 and C005 iPSCs. In earlier experiments, pLenti6/GFP lentivirus was used to label naïve-state RUES2 and H9. GFP-labeled naïve hPSC lines were derived by picking GFP+ colonies after infected naïve hPSCs were passaged to single cells using TrypLE. pEGFP-N1-TFE3 was purchased from Addgene (plasmid #38120) (67). We mutated the NLS of TFE3 (21), 355ERRRRF to 355EAAAAF. Wild-type (WT) or NLS mutant TFE3-GFP fusion construct was subcloned to pLenti6-V5 (Thermo Fisher Scientific). Lentiviruses generated from these constructs were used to derive stable lines of primed H9 expressing either TFE-GFP or NLS-GFP.
Chromatin immunoprecipitation
Primed H9 or nH9 cells were cross-linked in 1% formaldehyde at room temperature for 10 min. After termination of cross-linking by adding 150 mM glycine, the cells were dissolved in SDS lysis buffer and sonicated on ice. Cleared lysates were used for immunoprecipitation with a ChIP (chromatin immunoprecipitation) assay kit (Millipore, Billerica, MA, USA). Chromatin fragments were immunoprecipitated with 10 μg of anti-H3K27AC antibody (Abcam). After removing proteins from DNA by proteinase K digestion, purified immunoprecipitated DNA was subjected to quantitative real-time PCR. Rabbit IgG was used as control. Primers for OCT4 distal enhancer (−2340/−2142) were as follows: 5′-ACCCCACTGCCTTGTAGACCT-3′ and 5′-CACGCTGACCTCTGTCGACTT-3′ (68). Primers for OCT4 proximal enhancer (−1126/−1040) were as follows: 5′-TCTGTTTCAGCAAAGGTTGGG-3′ and 5′- TTGGTCCCTACTTCCCCTTCA-3′ (69).
XIST RNA FISH
XIST RNA was detected using Stellaris RNA FISH protocol (LGC Biosearch Techanologies, Petaluma, CA). Cells were washed with PBS once, fixed in 3.7% formaldehyde at room temperature for 10 min, washed with PBS twice, and then permeabilized in 0.1% Triton X-100 in PBS for 5 min at room temperature. The cells were washed with PBS once, immersed in Wash Buffer A at room temperature for 5 min, and then incubated in Hybridization Buffer containing 125 nM Stellaris Xist RNA FISH probes (catalog #SMF 2038-1) in the dark at 37°C for 16 hours. After the Hybridization Buffer was aspirated, cells were incubated in Wash Buffer A in the dark at 37°C for 30 min and then incubated in Wash Buffer A containing DAPI (5 ng/ml) in the dark at 37°C for another 30 min. After the DAPI solution was aspirated, cells were incubated in Wash Buffer B at room temperature for 5 min and then immersed in VECTASHIELD antifade mounting medium for imaging.
Quantification and statistical analysis
SPSS 13.0 was used for statistical analysis. All data were expressed as mean ± SE of measurement. Statistical tests used to analyze whether samples are significantly different are indicted in the figure legends. Values of P < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank D. Barnas at the Gene Targeting and Transgenic Shared Resource of RPCI for injections and transfers of mouse embryos; B. Foster, B. Gillard, and E. Karasik at the Mouse Tumor Model Resource at RPCI for teratoma formation assay and tissue processing; the RPCI Genomics Shared Resource for the Infinium MethylationEPIC Beadchip study; J. E. Bard, B. J. Marzullo, and D. Yergeau in the University at Buffalo Genomics and Bioinformatics Core Facility for RNA-seq and NGS of 18S rDNA amplicons; S.-c. Zhang at the University of Wisconsin Madison for the LV-EF1α-GFP plasmid; and R. Puertollano at NIH for TFE3 plasmids. Funding: The work is supported by NYSTEM contracts C028129, C029556, and C30290GG and the Buffalo Blue Sky Initiative (J.F.). Author contributions: Conceptualization: J.F. Methodology: Z.H., H.L., and J.F. Investigation: Z.H. generated all naïve hPSCs. H.L. generated the TFE3 constructs and performed bioinformatics analysis with help from X.Y. and M.J.B. Z.H., H.L., H.J., Y.R., and B.Z. analyzed naïve cells and mouse embryos. J.Q. performed histological identification. A.B.S. performed mouse embryo injections and transfers. Writing: J.F. with input from Z.H. and H.L. Competing interests: J.F. is a cofounder of Vitropy LLC and ASDDR LLC. J.F. is an inventor on a patent application related to this work filed by J.F. (no. 16/346534, filed 30 April 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Raw and processed data for RNA-seq were deposited to GEO under GSE87452. Raw and processed data for Infinium MethylationEPIC Beadchip in DNA methylation study were deposited to GEO under GSE102031. Raw and processed data for NGS of 18S rDNA amplicons were deposited to GEO under GSE125813.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz0298/DC1
REFERENCES AND NOTES
- 1.Nichols J., Smith A., Naive and primed pluripotent states. Cell Stem Cell 4, 487–492 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Ying Q. L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A., The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martello G., Smith A., The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol. 30, 647–675 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., Jones J. M., Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998). [DOI] [PubMed] [Google Scholar]
- 5.De Los Angeles A., Ferrari F., Xi R., Fujiwara Y., Benvenisty N., Deng H., Hochedlinger K., Jaenisch R., Lee S., Leitch H. G., Lensch M. W., Lujan E., Pei D., Rossant J., Wernig M., Park P. J., Daley G. Q., Hallmarks of pluripotency. Nature 525, 469–478 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Gafni O., Weinberger L., Mansour A. A., Manor Y. S., Chomsky E., Ben-Yosef D., Kalma Y., Viukov S., Maza I., Zviran A., Rais Y., Shipony Z., Mukamel Z., Krupalnik V., Zerbib M., Geula S., Caspi I., Schneir D., Shwartz T., Gilad S., Amann-Zalcenstein D., Benjamin S., Amit I., Tanay A., Massarwa R., Novershtern N., Hanna J. H., Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Chan Y. S., Göke J., Ng J. H., Lu X., Gonzales K. A. U., Tan C. P., Tng W. Q., Hong Z. Z., Lim Y. S., Ng H. H., Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 13, 663–675 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W., Reik W., Bertone P., Smith A., Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158, 1254–1269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theunissen T. W., Powell B. E., Wang H., Mitalipova M., Faddah D. A., Reddy J., Fan Z. P., Maetzel D., Ganz K., Shi L., Lungjangwa T., Imsoonthornruksa S., Stelzer Y., Rangarajan S., D’Alessio A., Zhang J., Gao Q., Dawlaty M. M., Young R. A., Gray N. S., Jaenisch R., Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 15, 471–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ware C. B., Nelson A. M., Mecham B., Hesson J., Zhou W., Jonlin E. C., Jimenez-Caliani A. J., Deng X., Cavanaugh C., Cook S., Tesar P. J., Okada J., Margaretha L., Sperber H., Choi M., Blau C. A., Treuting P. M., Hawkins R. D., Cirulli V., Ruohola-Baker H., Derivation of naïve human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, 4484–4489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., Platero-Luengo A., Sakurai M., Sugawara A., Gil M. A., Yamauchi T., Suzuki K., Bogliotti Y. S., Cuello C., Morales Valencia M., Okumura D., Luo J., Vilariño M., Parrilla I., Soto D. A., Martinez C. A., Hishida T., Sánchez-Bautista S., Martinez-Martinez M. L., Wang H., Nohalez A., Aizawa E., Martinez-Redondo P., Ocampo A., Reddy P., Roca J., Maga E. A., Esteban C. R., Berggren W. T., Nuñez Delicado E., Lajara J., Guillen I., Guillen P., Campistol J. M., Martinez E. A., Ross P. J., Izpisua Belmonte J. C., Interspecies chimerism with mammalian pluripotent stem cells. Cell 168, 473–486.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y., Liu B., Xu J., Wang J., Wu J., Shi C., Xu Y., Dong J., Wang C., Lai W., Zhu J., Xiong L., Zhu D., Li X., Yang W., Yamauchi T., Sugawara A., Li Z., Sun F., Li X., Li C., He A., du Y., Wang T., Zhao C., Li H., Chi X., Zhang H., Liu Y., Li C., Duo S., Yin M., Shen H., Belmonte J. C. I., Deng H., Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell 169, 243–257.e25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberger L., Ayyash M., Novershtern N., Hanna J. H., Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Gao S., Human naive embryonic stem cells: How full is the glass? Cell Stem Cell 18, 301–303 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Huang K., Zhu Y., Ma Y., Zhao B., Fan N., Li Y., Song H., Chu S., Ouyang Z., Zhang Q., Xing Q., Lai C., Li N., Zhang T., Gu J., Kang B., Shan Y., Lai K., Huang W., Mai Y., Wang Q., Li J., Lin A., Zhang Y., Zhong X., Liao B., Lai L., Chen J., Pei D., Pan G., BMI1 enables interspecies chimerism with human pluripotent stem cells. Nat. Commun. 9, 4649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angelo G., Van Gilst M. R., Starvation protects germline stem cells and extends reproductive longevity in C. elegans. Science 326, 954–958 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Boroviak T., Nichols J., The birth of embryonic pluripotency. Philos. Trans. R. Soc. Lond B Biol. Sci. 369, 20130541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welling M., Geijsen N., Uncovering the true identity of naïve pluripotent stem cells. Trends Cell Biol. 23, 442–448 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Bulut-Karslioglu A., Biechele S., Jin H., Macrae T. A., Hejna M., Gertsenstein M., Song J. S., Ramalho-Santos M., Inhibition of mTOR induces a paused pluripotent state. Nature 540, 119–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raben N., Puertollano R., TFEB and TFE3: Linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martina J. A., Diab H. I., Lishu L., Jeong-A L., Patange S., Raben N., Puertollano R., The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7, ra9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Betschinger J., Nichols J., Dietmann S., Corrin P. D., Paddison P. J., Smith A., Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153, 335–347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Z., Pu J., Jiang H., Zhong P., Qiu J., Li F., Wang X., Zhang B., Yan Z., Feng J., Generation of naivetropic induced pluripotent stem cells from Parkinson's disease patients for high-efficiency genetic manipulation and disease modeling. Stem Cells Dev. 24, 2591–2604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S., An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K. I., Shibata T., Kunisada T., Takahashi M., Takahashi J., Saji H., Yamanaka S., A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Jiang H., Ren Y., Yuen E. Y., Zhong P., Ghaedi M., Hu Z., Azabdaftari G., Nakaso K., Yan Z., Feng J., Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 3, 668 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J., Huang J., Li M., Wu X., Wen L., Lao K., Li R., Qiao J., Tang F., Single-cell RNA-seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1131–1139 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S. P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F., Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 165, 1012–1026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahakyan A., Kim R., Chronis C., Sabri S., Bonora G., Theunissen T. W., Kuoy E., Langerman J., Clark A. T., Jaenisch R., Plath K., Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell 20, 87–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grow E. J., Flynn R. A., Chavez S. L., Bayless N. L., Wossidlo M., Wesche D. J., Martin L., Ware C. B., Blish C. A., Chang H. Y., Reijo Pera R. A., Wysocka J., Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blakeley P., Fogarty N. M. E., del Valle I., Wamaitha S. E., Hu T. X., Elder K., Snell P., Christie L., Robson P., Niakan K. K., Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development 142, 3151–3165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boroviak T., Loos R., Lombard P., Okahara J., Behr R., Sasaki E., Nichols J., Smith A., Bertone P., Lineage-specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev.Cell 35, 366–382 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Theunissen T. W., Friedli M., He Y., Planet E., O’Neil R. C., Markoulaki S., Pontis J., Wang H., Iouranova A., Imbeault M., Duc J., Cohen M. A., Wert K. J., Castanon R., Zhang Z., Huang Y., Nery J. R., Drotar J., Lungjangwa T., Trono D., Ecker J. R., Jaenisch R., Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell 19, 502–515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boroviak T., Nichols J., Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development 144, 175–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plath K., Fang J., Mlynarczyk-Evans S. K., Cao R., Worringer K. A., Wang H., de la Cruz C. C., Otte A. P., Panning B., Zhang Y., Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Vallot C., Patrat C., Collier A. J., Huret C., Casanova M., Liyakat Ali T. M., Tosolini M., Frydman N., Heard E., Rugg-Gunn P. J., Rougeulle C., XACT noncoding RNA competes with XIST in the control of X chromosome activity during human early development. Cell Stem Cell 20, 102–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrel L., Willard H. F., X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434, 400–404 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Court F., Tayama C., Romanelli V., Martin-Trujillo A., Iglesias-Platas I., Okamura K., Sugahara N., Simon C., Moore H., Harness J. V., Keirstead H., Sanchez-Mut J. V., Kaneki E., Lapunzina P., Soejima H., Wake N., Esteller M., Ogata T., Hata K., Nakabayashi K., Monk D., Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 24, 554–569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plasschaert R. N., Bartolomei M. S., Genomic imprinting in development, growth, behavior and stem cells. Development 141, 1805–1813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valle-Garay E., Montes A. H., Corte J. R., Meana A., Fierer J., Asensi V., tPA Alu (I/D) polymorphism associates with bacterial osteomyelitis. J. Infect. Dis. 208, 218–223 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kasai K., Nakamura Y., White R., Amplification of a variable number of tandem repeats (VNTR) locus (pMCT118) by the polymerase chain reaction (PCR) and its application to forensic science. J. Forensic Sci. 35, 1196–1200 (1990). [PubMed] [Google Scholar]
- 42.Cohen M. A., Wert K. J., Goldmann J., Markoulaki S., Buganim Y., Fu D., Jaenisch R., Human neural crest cells contribute to coat pigmentation in interspecies chimeras after in utero injection into mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 113, 1570–1575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez I. L., Schmickel R. D., The human 18S ribosomal RNA gene: Evolution and stability. Am. J. Hum. Genet. 38, 419–427 (1986). [PMC free article] [PubMed] [Google Scholar]
- 44.Stults D. M., Killen M. W., Pierce H. H., Pierce A. J., Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moras M., Lefevre S. D., Ostuni M. A., From erythroblasts to mature red blood cells: Organelle clearance in mammals. Front. Physiol. 8, 1076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beckmann H., Kadesch T., The leucine zipper of TFE3 dictates helix-loop-helix dimerization specificity. Genes Dev. 5, 1057–1066 (1991). [DOI] [PubMed] [Google Scholar]
- 47.Stirparo G. G., Boroviak T., Guo G., Nichols J., Smith A., Bertone P., Correction: Integrated analysis of single-cell embryo data yields a unified transcriptome signature for the human pre-implantation epiblast (doi: 10.1242/dev.158501). Development 145, dev169672 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo G., von Meyenn F., Santos F., Chen Y., Reik W., Bertone P., Smith A., Nichols J., Naive pluripotent stem cells derived directly from isolated cells of the human inner cell mass. Stem Cell Reports 6, 437–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang F., Barbacioru C., Bao S., Lee C., Nordman E., Wang X., Lao K., Surani M. A., Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 6, 468–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bar S., Benvenisty N., Epigenetic aberrations in human pluripotent stem cells. EMBO J. 38, e101033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu J., Greely H. T., Jaenisch R., Nakauchi H., Rossant J., Belmonte J. C. I., Stem cells and interspecies chimaeras. Nature 540, 51–59 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Bianconi E., Piovesan A., Facchin F., Beraudi A., Casadei R., Frabetti F., Vitale L., Pelleri M. C., Tassani S., Piva F., Perez-Amodio S., Strippoli P., Canaider S., An estimation of the number of cells in the human body. Ann. Hum. Biol. 40, 463–471 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Sender R., Fuchs S., Milo R., Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 14, e1002533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi T., Yamaguchi T., Hamanaka S., Kato-Itoh M., Yamazaki Y., Ibata M., Sato H., Lee Y. S., Usui J. I., Knisely A. S., Hirabayashi M., Nakauchi H., Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 142, 787–799 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Yamaguchi T., Sato H., Kato-Itoh M., Goto T., Hara H., Sanbo M., Mizuno N., Kobayashi T., Yanagida A., Umino A., Ota Y., Hamanaka S., Masaki H., Rashid S. T., Hirabayashi M., Nakauchi H., Interspecies organogenesis generates autologous functional islets. Nature 542, 191–196 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Hyun I., From naïve pluripotency to chimeras: A new ethical challenge? Development 142, 6–8 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Kimmelman J., Hyun I., Benvenisty N., Caulfield T., Heslop H. E., Murry C. E., Sipp D., Studer L., Sugarman J., Daley G. Q., Policy: Global standards for stem-cell research. Nature 533, 311–313 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Sethi I., Romano R.-A., Gluck C., Smalley K., Vojtesek B., Buck M. J., Sinha S., A global analysis of the complex landscape of isoforms and regulatory networks of p63 in human cells and tissues. BMC Genomics 16, 584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 60.Paradis E., Claude J., Strimmer K., APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romanel A., Lago S., Prandi D., Sboner A., Demichelis F., ASEQ: Fast allele-specific studies from next-generation sequencing data. BMC. Med. Genomics 8, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walker J. A., Hedges D. J., Perodeau B. P., Landry K. E., Stoilova N., Laborde M. E., Shewale J., Sinha S. K., Batzer M. A., Multiplex polymerase chain reaction for simultaneous quantitation of human nuclear, mitochondrial, and male Y-chromosome DNA: Application in human identification. Anal.Biochem. 337, 89–97 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Rognes T., Flouri T., Nichols B., Quince C., Mahé F., VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fassina L., Magenes G., Inzaghi A., Palumbo S., Allavena G., Miracco C., Pirtoli L., Biggiogera M., Comincini S., AUTOCOUNTER, an ImageJ javascript to analyze LC3B-GFP expression dynamics in autophagy-induced astrocytoma cells. Eur. J. Histochem. 56, e44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xia X., Zhang Y., Zieth C. R., Zhang S.-C., Transgenes delivered by lentiviral vector are suppressed in human embryonic stem cells in a promoter-dependent manner. Stem Cells Dev. 16, 167–176 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roczniak-Ferguson A., Petit C. S., Froehlich F., Qian S., Ky J., Angarola B., Walther T. C., Ferguson S. M., The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freberg C. T., Dahl J. A., Timoskainen S., Collas P., Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol. Biol. Cell 18, 1543–1553 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.You J. S., Kelly T. K., De Carvalho D. D., Taberlay P. C., Liang G., Jones P. A., OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc. Natl. Acad. Sci. U.S.A. 108, 14497–14502 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz0298/DC1