

Abstract
Novel coronavirus, 2019-nCoV is a danger to the world and is spreading rapidly. Very little structural information about 2019-nCoV make this situation more difficult for drug designing. Benzylidenechromanones, naturally occurring oxygen heterocyclic compounds, having capability to inhibit various protein and receptors, have been designed here to block mutant variety of coronavirus main protease enzyme (SARC-CoV-2 Mpro) isolated from 2019-nCoV with the assistance of molecular docking, bioinformatics and molecular electrostatic potential. (Z)-3-(4′-chlorobenzylidene)-thiochroman-4-one showed highest binding affinity to the protein. Binding of a compound to this protein actually inhibits the replication and transcription of the virus and, ultimately, stop the virus multiplication. Incorporation of any functional groups to the basic benzylidenechromanones enhances their binding ability. Chloro and bromo substitutions amplify the binding affinity. ADME studies of all these compounds indicate they are lipophilic, high gastro intestine absorbable and blood-brain barrier permeable. The outcome reveals that the investigated benzylidenechromanones can be examined in the case of 2019-nCoV as potent inhibitory drug of SARC-CoV-2 Mpro, for their strong inhibition ability, high reactivity and effective pharmacological properties.
Electronic supplementary material
The online version of this article (10.1007/s11224-020-01537-5) contains supplementary material, which is available to authorized users.
Keywords: Novel coronavirus, SARC-CoV-2 Mpro, Benzylidenechromanones, ADME, DFT, Docking
Introduction
In this time, the entire world is facing a threat of a new coronavirus, 2019-nCoV. It first appeared in the Wuhan province of China and spread rapidly in the different parts of the world. According to WHO, there is 1,133,758 confirmed cases of 2019-nCoV infection with 82,061 new case and 62,784 deaths found around the globe, as of 5th April 2020 [1]. Only 9096 patients were recovered and the rate of infection is much higher than that of recovery. At this moment, instigation for effective, safe and easy synthesisable therapeutics is in high demand [2, 3].
The virus 2019-nCoV, a mutant variety of lineage B betacoronavirus, uses angiotensin-converting enzyme2 (ACE2) of human cell as a receptor during infection like other severe acute respiratory syndrome (SARS)-CoV [4]. Coronavirus has largest viral positive-stranded RNA [5] with protein envelop. The maturation process of SARS-CoV is streaming through protein cleavage of polyproteins which influence the replication and transcription of the virus. The CoV main protease (CoV Mpro) is the key enzyme in this event. Inhibition of the protein is an attractive strategy for the coronavirus control. Any kind of mutation of the gene correspond to the CoV Mpro can produce its mutant variety which can partial or complete resistant of these compounds. Development of new anti-SARC-CoV-2 Mpro compounds is very essential for combat such rapid mutation able viruses.
In drug discovery, chromone derivatives have privilege due to their interesting structural features and suitable pharmacokinetics. Many of them are under clinical trial, and hesperetin as one of the members of this family has got clinical approval for cholesterol-lowering medicine [6]. Benzylidene-chromanones, a type of chromone derivative, are very important but a small class of compounds found Hyacinthaceae and Caesalpinioideae plant [7–9]. Several synthetic and naturally occurring 3-benzylidene-4-chromanones are closely resemble with flavones and they are used as anti-mutagenic [10], anti-tumour [11], antioxidant [12] and antiviral [13] activities. It is also found in literature that the benzylidenechromanone has the inhibitory properties to different proteins and receptors like monoamine oxidase, [14] α-glucosidase, [15] efflux pump [16] and 5-lipoxygenase (5-LOX) enzyme [17]. This inspires us to use the benzylidenechromanone in the development of molecules against 2019-nCoV.
In this manuscript, we tested various chromanones designed for the inhibition of the replication and transcription of 2019-nCoV on the basis of different computational studies. The structure of the coronavirus main protease of the mutant variety has been utilised here to designing its inhibitory molecules. The investigation is consist of several phases like modelling of protein structure, pharmacology and pharmacokinetics of 3-benzylidene-4-chromanones, DFT-based study to resolve the structure-activity relations of chromanones with SARC-CoV-2 Mpro and molecular docking study of chromanones with CoV Mpro proteins. From the best of our knowledge, we may say that it is the first approach to develop new drug molecule for treatment of the 2019-nCoV.
Result and discussions
Protein structure
The outbreaks of virus attack at Wuhan region of China is the case of new type of coronavirus from bat origin, 2019-nCoV [4]. For this reason, we have very little information about its genetic material as well as structures of different proteins. Fortunately, in recent time, structure of CoV Mpro protein of 2019-nCoV has been determined using x-ray crystallography [18]. It is interesting to observe that the amino acid sequence of SARC-CoV-2 Mpro protein of 2019-nCoV is highly different from the previously known CoV Mpro. This mutation produces a novel variety of the protein with same activities. The change in the secondary and tertiary structure of the protein is clearly noticeable in the overlying picture of mutant (pink, pdb id: 6lu7) and one of the known protein [19] (cyan; pdb id: 2q6f) is shown below (Fig. 1a). The huge change of the mutant protein in almost every region of it is a key cause for the drug resistance. It creates the scope of development of new compounds against 2019-nCoV.
Fig. 1.

a Overlying picture of mutant (pink) and one of the known proteins (cyan). b Protein sequence alignment of CoV Mpro (pdb ids: 2q6d, 2q6f, 2h2z, 6lu7)
The study has been extended further to identify the extent of mutation of the SARC-CoV-2 Mpro by alignment of multiple protein sequence (Fig. 1b). In this bioinformatics study, more two formerly known CoV Mpro (pdb ids: 2q6d and 2h2z) included to get more clear idea about the new mutation. Taking 2q6d protein as a reference sequence, it was found that the aligning of the length of mutant protein (6lu7) is covered almost 96% which is also found other two proteins. The mutant protein exhibited consensus 70% which is minimum among them. This is the indication of huge mutation (Fig. S1).
Choice of organic compounds
In a critical analysis of the crystal structure of SARC-CoV-2 Mpro, we found that there are two amino acid residues (His41 and Cys145) in the active site, and Cys145 was attached with the peptide-based αβ-unsaturated ketone compound N3 (SARC-CoV-2 Mpro inhibitor, Fig. 2a) through a covalent bond at the β-position by the Michael reaction (Figs. 2a and S2a). Addition to this, it was also observed that the ligand has sufficient hydrogen bonding and stacking interactions with its different hydrophilic and hydrophobic parts (Fig. S2b). Michael reaction of thiol at αβ-unsaturated ketone functional groups is very common in biological systems [20]. This idea insists us to choose such a molecule having the capability of these types of noncovalent interactions and possess αβ-unsaturated ketone functional groups for Michael reaction.
Fig. 2.

a The structural characteristics of peptide-based SARC-CoV-2 Mpro inhibitor N3. b The structural similarity and features flavone and 3-benzylidene-4-chromanones with N3
Chromones are a valid as well as privileged moiety in medicinal chemistry and drug discovery, respectively [21]. Flavones (1), the renowned largest subclass of chromones, belong to a family of natural products with huge structural diversity, having two phenyl rings (A and B) and an oxygen heterocyclic ring (C) with one αβ-unsaturated ketone functional group (Fig. 2b). They are involved in protein binding through hydrogen bonding and different π-staking interactions (as found in flavones-protein complex crystals; pdb id: 4HKI, 2HI4, 4IGE, 6QCD, 4L0S, 4FJ0, 4L31 etc.) which result the protein inhibitory activities of acetylcholinesterase (AChE), microsomal cytochrome P450, NADPH oxidase, Xanthine oxidase, PDE4 and tankyrase 2 etc. (Table S1; entry 1–7). Easy synthesis methods, interesting structural features and protein binding ability make the flavone moiety a privileged drug system in this study. However, Michael reaction at αβ-unsaturated ketone site of flavones 1 is difficult due to the participation of the ring oxygen (major contribution owing to the gaining of aromaticity of the ring C) as well as ring B in extended conjugation with the ketone (Fig. 2b, compound 1 to 1a) which decreases the electrophilicity at β- or C2 position and increase the single bond character of the αβ-double bond (C2 = C3 of 1 to 1a).
3-Benzylidene-4-chromanones (2), a type of natural product and chromone derivative, has a high structural resemblance with flavone (1) (Fig. 2b). They have also protein-binding abilities (Table S1; entry 8–11). In both cases, there is one ketone group (C4), one α,β-unsaturated double bond (C2 = C3 for 1 and C3 = C11 for 2) and one rotatable phenyl ring are present. This is the reason why the class of compounds are often utilised in place of flavones. From the structural point of view, compound 2 has some advantages over 1. In the case of 2, the α,β-unsaturated ketone is more reactive for its exo-cyclic nature and the lack of conjugation with ring oxygen atom (Fig. 2b, structure 2). The presence of exo-cyclic C=C double bond at C3 without the previous type of conjugation enhances its reactivity and allows the compound 2 for facile Michael reaction at the C11 position. Therefore, there is a chance of compound 2 as Michel acceptor during interaction with amino acid residue (Cys145 and His41) at the active site of the protein. These are the reasons for the choice of compound 2 as the best possible lead moiety in this study and compounds 2e-i were also be considered to explore the electronic features of 2 and it is discussed later.
Molecular electrostatic potential (MEP) analysis
It is widely accepted that the molecular recognition between protein and small molecules (ligand) occurs at their surfaces. Hence, the extent of binding forces in the protein-ligand complex depends on two factors, electronic distribution and the shape of the surface of the ligand. These can be predicted from the molecular electrostatic potential surface of the energy-minimised geometry of a molecule by calculating Wang–Ford charges of all atoms [22, 23]. In MEP surface, electronegative atoms show negative values (red, hydrogen bonding acceptor) of charge, and it is positive in value for electron-poor atoms (blue, hydrogen bonding donor). The atoms with zero values (green, π- and other types of staking interactions) are neutral in nature. This molecular detail helps to predict how much they are potential to take part in chemical reaction and to realise their mechanism of interactions. In addition to this, the surfaces provide the lower limit of the proximity of two molecules which describe the steric feature of the molecule [24]. Therefore, the quantum mechanically calculated properties of an energy minimised molecular geometry help to understand the steric, electronic feature and nature of binding forces of molecules.
The MEP of compounds 2a-i show that these molecules are capable to form at least two hydrogen bonds (red patches) during protein binding with the help of ketone and chromanone ring oxygen except 2g (contain S atom in the place of oxygen) and 2h (Fig. 3). The electron density ring A of chromanone is quite higher (green to yellow) due to electron donation ring oxygen through conjugation which can favour the π-staking interaction with aromatic amino acid residues. This effect is enhanced in 2g for the presence of less electronegative atom sulphur in the ring. It is possible to manipulate the electron density at the benzylidene ring and C11 position by adding different functional groups at the benzylidene part. Incorporation of oxygen containing functional groups (OMe) at benzylidene part (2b, 2e and 2h) increases the electron density to the ring (Fig. 3). This enhancement of electron density is also favourable for the π-staking interaction. The case becomes more interesting for halogens because the single atom occupies a large electron density surface with negative values which is also suitable also for multiple π-staking interactions as well as halogen bonding. However, the halogen atoms at this part are unable to influence the electronic behaviour of the ring (green for 2c, 2d and 2g). Therefore, it is expected that these compounds may interact strongly with proteins. In the case of compound 2f, the electron rich oxygen in the furan ring polarises the ring electron cloud towards itself. The compound can bind strongly by hydrogen bonding. The extended conjugated hydrophobic benzylidene system is 2i and is very slightly electron rich with a large surface area (Fig. 3). Therefore, small red, yellow and blue patches on the large green surface of compounds are balancing the hydrophilic and hydrophobic parts which are essential for good binding to the protein.
Fig. 3.

Molecular electrostatic potential surface of compounds 2a–i
Bioavailability
For the assessment of bioavailability, absorption, distribution, metabolism and excretion (ADME) properties and pharmacokinetic studies of small molecules are major concern. To judge the ability of synthetic of isolated molecules as a drug and approval for clinical trials, it should fulfil the ADME requirements [25]. Here, ten molecules have been assessed for their bioavailability.
Lipophilicity
The standard descriptor for lipophilicity is the partition coefficient between n-octanol and water (log Po/w). The partition coefficient is so important for pharmacokinetics drug discovery that SwissADME, a very useful web tool for pharmacokinetic evaluation, provided a section for the properties [26, 27]. Several computational methods for log Po/w estimation have been developed with varying performances on different chemical sets. Normally, multiple predictors have been used to opt for either the most precise method for a given chemical series or to know the consensus evaluation. The models should be diverse enough so that the prediction of the consensus log Po/w will be more accurate [28]. SwissADME provides access to five free predictive models in this connection, i.e. XLOGP3, an atomic method with corrective factors and knowledge-based library [29], WLOGP, a purely atomistic method based upon Wildman and Crippen’s fragmentation method [30], MLOGP is an archetypal topology tool based on a linear relation with 13 molecular descriptors [31, 32], SILICOS-IT, a 27 fragments and seven topological descriptors hybrid system [33] and iLOGP is the physics-based method that uses free solvation energies of n-octanol and water which was calculated using the Generalized-Born and solvent accessible surface area (GB/SA) model.
Each log Po/w values for all the compounds are following same trends. The values are significantly below in the case of 2i which may be due to the enhancement of electron density and polarity in benzylidene part. The negligible change in the log Po/w values for the compound 2a and 2b indicates that the methoxy group cannot increase polarity of these molecules. The presence of halogens (Cl and Br) boosts their lipid solubility (Fig. 4). In case of 2c and 2g molecules, the changes are very high which is manifestation of the decrease in polarity and increase of the lipophilicity of the molecule for sulphur atom replacement. In this study, log Po/w values for designed compounds were found in the range of + 2.29 to + 4.25 (Fig. 4, Table S2). These positive values signify that all the molecules are highly lipophilic and fulfil the essential criteria for drug molecules.
Fig. 4.
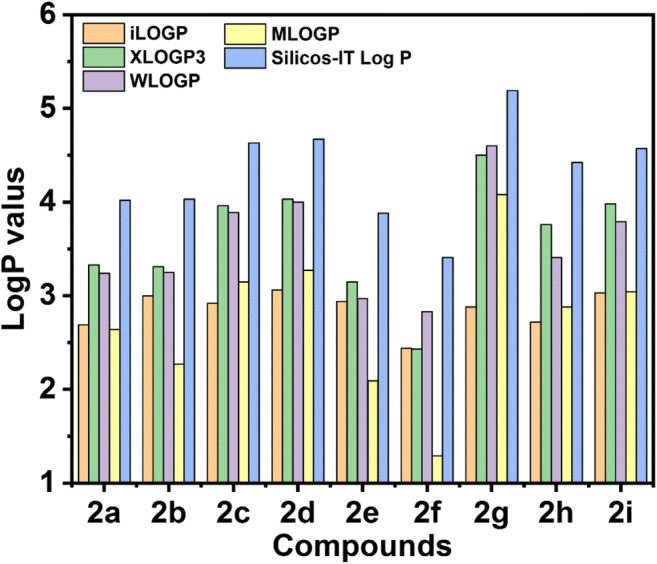
Predicted lipophilicity (log P values) of compounds obtained from different calculation models
Water solubility
Water solubility is an important requirement for any drug molecule intends to administer through the oral or parenteral route as the sufficient quantity of the active pharmaceutical ingredients has to be given in a small volume [34, 35]. SwissADME furnishes three different topological methods for estimating water solubility-Esol model [36], Ali model [37] and SILICOS-IT [33]. The log S values of our compounds were in the range of − 3.19 to − 4.85 using ESOL log S method, − 2.9 to − 4.41 using Ali log S method and − 4.25 to − 6.38 for SILICOS-IT method, which are mentioned in the Table-1 and S3. Considering all the models, we can conclude that the most of the compounds are moderately water-soluble, and some of them show good solubility as the reference value of log S for moderate solubility is between − 4 and − 6 and − 2 to − 4 for good solubility. This suggests that the molecules may be administered in the body through an oral or parenteral route.
Pharmacokinetics
ADME studies of these molecules also give place to these molecules in the approved drug molecule section. All these molecules have high gastrointestinal absorption which allow them for oral dosing and passed the most important and decision-making standard of drug discovery. Blood brain barrier (BBB) permeability test is passed by all of the compounds. Therefore, they are qualified a fundamental index of drug distribution. High negative values of skin permeability also offers their less skin permeation. Binding of CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 metabolic enzymes may cause the degradation of the compounds inside the body or toxicity. The study shows that all the compounds are inhibitor of CYP1A2 and CYP2C19 enzymes (Tables S3 and S4). The compounds 2a, 2f, and 2h are only non-inhibitor of CYP2C9 whereas 2b and 2e can bind with CYP2D6 and CYP3A4 enzymes. Therefore, there is a chance of degradation of some of these molecules inside the body and some may responsible for toxicity. The toxicity of these compounds is further explored in the last section. Bioavailability score of all the compounds is an indication of high potential as a drug molecule. Synthesis of this compounds is very easy as stipulated in the synthetic accessibility of the compounds. All these values of the above-mentioned parameters are in the region found for the successful drug molecule. This makes it very close to drug-like molecules (Table 1).
Table 1.
Predicted data of water solubility, pharmacokinetics, drug likeness and medicinal chemistry of the designed compound 2a-i
| 2a | 2b | 2c | 2d | 2e | 2f | 2g | 2h | 2i | |
|---|---|---|---|---|---|---|---|---|---|
| ESOL log S | − 3.83 | − 3.89 | − 4.41 | − 4.73 | − 3.92 | − 3.19 | − 4.85 | − 4.1 | − 4.29 |
| Ali log S | − 3.56 | − 3.73 | − 4.21 | − 4.29 | − 3.76 | − 2.90 | − 5.11 | − 4.01 | − 4.23 |
| Silicos-IT log Sw | − 5.41 | − 5.53 | − 6.02 | − 6.24 | − 5.15 | − 4.62 | − 6.38 | − 5.8 | − 5.5 |
| GI absorption | High | High | High | High | High | High | High | High | High |
| BBB permanent | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Pgp substrate | No | No | No | No | No | No | No | No | No |
| log Kp (cm/s) | − 5.38 | − 5.57 | − 5.14 | − 5.36 | − 5.77 | − 5.95 | − 4.85 | − 5.16 | − 5.07 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility | 2.74 | 2.87 | 2.73 | 2.76 | 3.00 | 2.77 | 3.17 | 2.55 | 3.05 |
Docking study
A combination of understanding of bioinformatics and molecular docking are also being applied to forecast possible uses and production by industry [21]. Molecular docking has been used here to predict the inhibitory activities of compounds 2a-i against mutant coronavirus main protease enzyme. In this study, we docked all the nine compounds with the SARC-CoV-2 Mpro (pdb id: 6lu7) as well as three previously known coronavirus (pdb ids: 2q6f, 2q6d and 2h2z). The crystal structure of SARC-CoV-2 Mpro contains a peptide fragment in its active site. The peptide-based ligand N3 is also docked and the docking score is considered as the control. All the designed compounds show a much higher binding affinity towards CoV Mpro proteins with respect to the N3 (Table 2). It is so interesting that the compounds bind strongly with the SARC-CoV-2 Mpro in comparison to the previously known proteins (Table 2). The study shows that the compound 2g perform best as anti- SARC-CoV-2 Mpro (Table 2). The more curious fact is that the more lipophilic molecule binds more strongly with the proteins. The presence of halogen atoms increases electron density as well as hydrophobicity of the molecule which goes in favour of their strong binding. The compound exhibited excellent results in all the bioavailability studies as discussed in the previous sections.
Table 2.
Docking score of designed compounds 2a-i and the peptide crystallised with SARC-CoV-2 Mpro
| Compounds | 6lu7 | 2q6f | 2q6d | 2h2z |
|---|---|---|---|---|
| N3 ligand | − 4.47 | – | – | – |
| 2a | − 7.16 | − 6.78 | − 5.16 | − 6.86 |
| 2b | − 7.34 | − 6.56 | − 5.01 | − 6.37 |
| 2c | − 7.58 | − 6.92 | − 5.56 | − 5.23 |
| 2d | − 7.64 | − 7.01 | − 5.87 | − 5.29 |
| 2e | − 7.58 | − 6.91 | − 5.81 | − 6.17 |
| 2f | − 6.53 | − 6.23 | − 5.53 | − 6.09 |
| 2g | − 7.85 | − 7.15 | − 6.19 | − 6.91 |
| 2h | − 7.55 | − 6.89 | − 5.83 | − 6.24 |
| 2i | − 7.23 | − 6.25 | − 5.12 | − 5.87 |
The italic values indicates compound 2g is the best in the docking study
All the compounds are docked inside each of the active site of the CoV Mpro proteins (Fig. 5a) by means of the hydrophobic as well as hydrogen-bonding interactions (Figs. S3-S10). The reddest patch on the electrostatic potential map of 2g (Fig. 2) was on the ketone oxygen, which forms hydrogen bonding with E166 amino acid residue (1.94 Å), observed in the docking study. Amino acid residues H41, Y54 and H163 are involved in π-staking interaction with both phenyl ring separately. Lone pair π-staking between M49 and M165 with Cl containing phenyl ring is also an interesting case. The chlorine is in hydrophobic interactions with C44, M49, P52 and Y54 residues. The π H-N interactions between H163 and thiochromanone benzene ring as well as CH2 hydrogen and H41 are also found (Fig. 5b).
Fig. 5.

a Docking pose. b Binding interactions of 2g inside the binding site of SARC-CoV-2 Mpro
Designing of new compound
The docking study also provides an opportunity to design these compounds in its next level. There is a possibility of rotation of the C–C bond designated by cyan colour in Fig. 5b. This bond rotation can bring the grey-coloured carbon atoms of benzylidene ring closer to the amide carbonyl groups of R188 and Q189 residues (Fig. 5b). Hydroxyl group substitution of the hydrogen atoms at the mentioned positions can involve in hydrogen bonding with the amino acid residue. The grey-coloured carbon atom at thiochromanone ring is closer to P140 and the carboxyl group of E166. Replacement of the attached hydrogen at the marked carbon by NH2 group may render it capable of hydrogen bonding and ionic interactions. If these substitutions occur, the compound 2j can come into existence (Fig. 6a). All the above studies are also performed for the 2j, and it is found that the compound is sufficiently hydrophobic (log P values are in Table S1). The presence of two hydroxyl groups and one amino group reduces the log S values (ESOL log S = − 3.62, Ali log S = − 4.41 and Silicos-IT log Sw = − 4.25) slightly and enhances its water solubility. The compound has high gastrointestinal absorbance without BBB permeability. The compound may behave as Pgp substrate with low skin absorbability (log Kp = − 6.37 cm/s). The compound 2j has the same bioavailability score (0.55) with other and low synthetic accessibility (3.29). Therefore, the designed new compound is well in the pharmacokinetic point of view also.
Fig. 6.

a Molecular electrostatic potential map of 2j and b binding interactions of 2j inside the binding site of SARC-CoV-2 Mpro
The MEP of 2j displays that the electron density increases in both the aromatic rings, in comparison to 2g, due to electron donation of OH and NH2 groups (Fig. 6a) which is favourable to form strong π-staking interactions. The appearance of dark blue patch in the region of the amine group of 2j on the MEP surface is clue for strong hydrogen bonding. The docking of the compound with all the CoV Mpro proteins shows that the compound 2j binds more strongly with the protein than that of 2g in each case. The increment in docking score of 2j (− 7.88, − 7.21, − 6.31 and − 7.13 for the protein with pdb id 6lu7, 2q6f, 2q6d and 2h2z, respectively) is slight which can be because of the compensation of the loss of the huge number of interactions for the elimination of Cl group of 2g by the stabilizing hydrogen bonging of OH and amine groups of 2j with amino acid residues of the protein. Inside the active site, the attached amine and hydroxyl groups of 2j form expected hydrogen bonding interactions as mentioned earlier (Fig. 6b). In addition to this, the hydrogen bonding between the ketone oxygen of 2j and E166 amino acid residue (1.94 Å) is also observed here. Sulphur-π (M94 and 165) and π-π (H41) staking interactions are found with OH containing ring and N-H-π (H163) found at thiochromanone ring. The sulphur–sulphur interactions are also observed between the compound and C145.
Toxicity study
The toxicity of drug molecules is the main cause of the limitation to their wide adequate usefulness. It is very complicatedly related to the patients and show dependency on the type of compounds used, its doses as well as their interaction with the stage of infection. One of the primary tasks in drug discovery is to find a molecule with the least toxicity or non-toxicity from all aspects [38]. Therefore, the prediction of the toxicity of the molecules under investigation is also an essential part of this study. Here, we have judged all these molecules in different toxicity parameters. In this study, we have considered cardiotoxicity, organ toxicity (hepatotoxicity), toxicity endpoints (carcinogenicity, immunotoxicity, mutagenicity and cytotoxicity), nuclear receptor signaling pathways (aryl hydrocarbon receptor (AhR), androgen receptor (AR), androgen receptor ligand-binding domain (AR-LBD), aromatase, estrogen receptor alpha (ER), estrogen receptor ligand-binding domain (ER-LBD) and peroxisome proliferator-activated receptor gamma (PPAR-Gamma)) and stress response pathways (nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE), heat shock factor response element (HSE), mitochondrial membrane potential (MMP), phosphoprotein (tumour suppressor) p53 and ATPase family AAA domain-containing protein 5 (ATAD5)) [39] and details are given in Tables S5 and S6.
Drugs like sertindole, terfenadine and cisapride, can inhibit the human ether-á-go-go related gene (hERG) K+ channels, which have been stopped in use because of heart arrhythmia followed by death. The study shows that all the compounds except 2i and 2j can have an active tendency to bind with hERG K+ channel [40] which results cardiac toxicity (Table S5). Compounds 2a and 2g are almost non-toxic and passed all the above-mentioned toxicity parameters (Table S6). A weak activity against carcinogenicity, immunotoxicity and AhR was predicted for 2a and only AhR activity for 2g. It was also predicted that all other compounds including 2j (cardiac non-toxic) are strongly active only in immunotoxicity. It is also interesting to note that such prediction on the five FDA-approved antiviral drugs (rilpivirine, dolutegravir, glecaprevir, grazoprevir and paritaprevir) also have the same strong immunotoxicity activity (Table S7). Therefore, immunotoxicity may be allowed in some cases. Considering all these factors, it can be concluded that 2j is found to be the best in this case.
Conclusion
In conclusion, the chromone derivatives can be easy synthesisable drug molecule against novel coronavirus treatment. They can inhibit the coronavirus main protease enzyme of the new virus efficiently. In this in silico study, these molecules passed all the essential tests required for a good drug. Based upon DFT analysis, ADMET properties, docking and the toxicity study, all the designed compounds are effective to the inhibition against 2019-nCoV. The binding prospective can guide to the evolution of a powerful drug for humans to combat on going coronavirus threat. Therefore, these non-peptide compounds, mainly 2j, is now subject matter of examine for the in vitro assay to disclose their efficient inhibition power for SARC-CoV-2 Mpro.
Methods
The study focused on the mutant and earlier three CoV Mpro, i.e. 6lu7, 2q6d, 2q6f and 2h2z, respectively, for in silico finding of having the capacity to develop into inhibitors in the near future from chromanone derivatives.
Protein structure modelling
The crystal data of all the protein structures were downloaded from Protein Data Bank (PDB ID: 6lu7, 2q6d, 2q6f and 2h2z). The structures used for further studies were prepared by Discovery studio 2017 R2 client. The modelling of protein structure was done on I-TASSER. All the pictures of the protein were made with MolSoft-ICM browser, Discovery studio 2017 R2 client and Samson core software.
Bioinformatics
The protein sequences of selected four above-mentioned proteins have been obtained from Discovery studio 2017 R2 client. The sequences were aligned using the bioinformatics web server Clustal Omega of European Bioinformatics Institute (EMBL-EBI). The outcome of the alignment protein sequences was analysed in the same web server, m-view and visualised it with the colour coding provided by the server.
Molecular electrostatic potential (MEP) analysis
The compounds 2a–j were optimised with the help of density functional theory using B3LYP functional at 6-311g level of basis set. Molecular electrostatic potential map around the molecular frame work was calculated at the combined B3LYP and 6-31g level of theory with the 0.03 iso values. For all of these DFT calculations, Gaussian 09 W software with D1 revision has been used at windows platform [41].
ADME study
ADME and toxicity of the designed compounds were predicted at the SwissADME web server [25]. Using huge database, the server can speculate physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug likeness and medicinal properties with high precision.
Molecular docking estimation
Energy minimised structure of compounds 2a–j, obtained from DFT optimisation, was used for docking with the mentioned four protein structures. The structure of the compounds and proteins were prepared in appropriate format for the studies with the help of MGL Tools. For docking studies, AutoDock 4.2 was employed by following standard procedure as described [42]. In this case, a 40 × 40 × 40 grid box has been used.
Toxicity
Cardiac toxicity of all the compounds (2a–2j) has been predicted with the help of a web-accessible computational equipment Pred-hERG [40]. Acute toxicity, organ toxicity, toxicological endpoint, nuclear receptor signalling pathways and stress response pathway were also calculated for these compounds and some FDA-approved anti-viral drugs using PROTOX-II [39].
Electronic supplementary material
(PDF 1589 kb)
Acknowledgements
The Department of Chemistry, Jadavpur University is gratefully acknowledged for research facilities. N.S. grateful to RUSA 2.0, New Delhi for fellowship, and A. H. thankful to DST, Government of India for DST Inspire fellowship.
Author contributions
N.S. and U.C.H. designed, evaluated and follow-up the calculations. N.S. and R.M. performed the work associated with bioinformatics to identify mutation in new protein. A.H. and M.M. explain the ADME analysis of compounds. All authors analysed and interpreted the reported data. N.S. and U.C.H. wrote the manuscript with assistance and feedback from all other. All authors checked the theoretical results and approved of the manuscript as the final version.
Funding information
This study is financially supported by University Grant Commission (UGC)-CAS-II and DST-PURSE-II Program of Jadavpur University, Kolkata and King Saud University (RSP-2019/141), Riyadh, Saudi Arabia.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Nayim Sepay, Email: nayimsepay@yahoo.com.
Umesh Chandra Halder, Email: uhalder2002@yahoo.com.
References
- 1.World Health Organization. Coronavirus disease (COVID-2019) situation reports https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed 5th April 2020)
- 2.Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y, Ying T, Liu S, Shi Z, Jiang S, Lu L (2020) Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 10.1038/s41423-020-0374-2 [DOI] [PMC free article] [PubMed]
- 3.Jiang S, Du L, Shi Z. An emerging coronavirus causing pneumonia outbreak in Wuhan, China: calling for developing therapeutic and prophylactic strategies. Emerg Microbes Infect. 2020;9:275–277. doi: 10.1080/22221751.2020.1723441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 10.1038/s41586-020-2012-7
- 5.Ziebuhr J. The coronavirus replicase. Curr Top Microbiol Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drug Bank, http://www.drugbank.ca/drugs
- 7.Lin L-G, Liu Q-Y, Ye Y. Naturally occurring homoisoflavonoids and their pharmacological activities. Planta Med. 2014;80:1053–1066. doi: 10.1055/s-0034-1395069. [DOI] [PubMed] [Google Scholar]
- 8.Yan J, Sun L-R, Zhou Z-Y, Chen Y-C, Zhang W-M, Dai H-F, Tan J-W. Homoisoflavonoids from the medicinal plant, Portulaca oleracea. Phytochemistry. 2012;80:37–41. doi: 10.1016/j.phytochem.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Lin L-G, Xie H, Li H-L, Tong L-J, Tang C-P, Ke C-Q, Liu Q-F, Lin L-P, Geng M-Y, Jiang H, Zhao W-M, Ding J, Ye Y. Naturally occurring homoisoflavonoids function as potent protein tyrosine kinase inhibitors by c-Src-based high-throughput screening. J Med Chem. 2008;51:4419–4429. doi: 10.1021/jm701501x. [DOI] [PubMed] [Google Scholar]
- 10.Perjési P, Das U, De Clercq E, Balzarini J, Kawase M, Sakagami H, Stables JP, Lorand T, Rozmer Z, Dimmock JR. Design, synthesis and antiproliferative activity of some 3-benzylidene-2,3-dihydro-1-benzopyran-4-ones which display selective toxicity for malignant cells. Eur J Med Chem. 2008;43:839–845. doi: 10.1016/j.ejmech.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma L-Y, Zheng Y-C, Wang S-Q, Wang B, Wang Z-R, Pang L-P, Zhang M, Wang J-W, Ding L, Li J, Wang C, Hu B, Liu Y, Zhang X-D, Wang J-J, Wang Z-J, Zhao W, Liu H-M. Design, synthesis, and structure–activity relationship of novel LSD1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents. J Med Chem. 2015;58:1705–1716. doi: 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- 12.Foroumadi A, Samzadeh-Kermani A, Emami S, Dehghan G, Sorkhi M, Arabsorkhi F, Heidari MR, Abdollahi M, Shafiee A. Synthesis and antioxidant properties of substituted 3-benzylidene-7-alkoxychroman-4-ones. Bioorg Med Chem Lett. 2007;17:6764–6769. doi: 10.1016/j.bmcl.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 13.Emamia S, Ghanbarimasir Z. Recent advances of chroman-4-one derivatives: synthetic approaches and bioactivities. Eur J Med Chem. 2015;93:539–563. doi: 10.1016/j.ejmech.2015.02.048. [DOI] [PubMed] [Google Scholar]
- 14.Desideri N, Bolasco A, Fioravanti R, Monaco LP, Orallo F, Yáñez M, Ortuso F, Alcaro S. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-b inhibition properties. J Med Chem. 2011;54:2155–2164. doi: 10.1021/jm1013709. [DOI] [PubMed] [Google Scholar]
- 15.Famuyiwa SO, Ntumy AN, Andrae-Marobela K, Yeboah SO. S Afr J Bot. 2013;88:17–22. doi: 10.1016/j.sajb.2013.04.009. [DOI] [Google Scholar]
- 16.Roy SK, Kumari N, Gupta S, Pahwa S, Nandanwar H, Jachak SM. 7-Hydroxy-(E)-3-phenylmethylene-chroman-4-one analogues as efflux pump inhibitors against Mycobacterium smegmatis mc2 155. Eur J Med Chem. 2013;66:499–507. doi: 10.1016/j.ejmech.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 17.Siddaiah V, Rao CV, Venkateswarlu S, Krishnaraju AV, Subbaraju GV. Bioorg Med Chem. 2006;14:2545–2551. doi: 10.1016/j.bmc.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 18.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H (2020) Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 10.1038/s41586-020-2223-y(2019)
- 19.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravasco J M J M, Faustino H, Trindade A, Gois P M P (2019) Bioconjugation with maleimides: a useful tool for chemical biology 25, 43–59 [DOI] [PubMed]
- 21.Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016;5(e47):1–15. doi: 10.1017/jns.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy K, De AU, Sengupta C. QSAR of antimalarial cyclic peroxy ketals II: exploration of pharmacophoric site using AM1 calculations. Quant Struct-Act Relat. 2001;20:319–326. doi: 10.1002/1521-3838(200111)20:4<319::AID-QSAR319>3.0.CO;2-M. [DOI] [Google Scholar]
- 23.Roy K, Leonard JT. Classical QSAR modeling of anti-HIV 2,3-Diaryl-1,3-thiazolidin-4-ones. QSAR Comb Sci. 2005;24:579–592. doi: 10.1002/qsar.200430901. [DOI] [Google Scholar]
- 24.Sepay N, Mallik S, Guha C, Mallik AK. An efficient synthesis of 1,3-dimethyl-5-(2-phenyl-4H-chromen-4-ylidene)pyrimidine-2,4,6(1H,3H,5H)-triones and investigation of their interactions with β-lactoglobiulin. RSC Adv. 2016;6:96016–96024. doi: 10.1039/C6RA13584F. [DOI] [Google Scholar]
- 25.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pliska V, Testa B, van de Waterbeemd H. Lipophilicity in drug action and toxicology. Verlag GmbH: Wiley-VCH; 1996. pp. 1–6. [Google Scholar]
- 27.Arnott JA, Planey SL. The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discovery. 2012;7:863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 28.Mannhold R, Poda GI, Ostermann C. Calculation of molecular lipophilicity: state-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci. 2009;98:861–893. doi: 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- 29.Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L. Computation of octanol− water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47:2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- 30.Wildman SA, Crippen GM. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Model. 1999;39:868–873. [Google Scholar]
- 31.Moriguchi I, Shuichi H, Liu Q, Nakagome I, Matsushita Y. Simple method of calculating the octanol/water partition coefficient. Chem Pharm Bull. 1992;40:127–130. doi: 10.1248/cpb.40.127. [DOI] [Google Scholar]
- 32.Moriguchi I, Shuichi H, Nakagome I, Hirano H. Comparison of reliability of log P values for drugs calculated by several methods. Chem Pharm Bull. 1994;42:976–978. doi: 10.1248/cpb.42.976. [DOI] [Google Scholar]
- 33.http://silicos-it.be.s3-website-eu-west-1.amazonaws.com/software/filter-it/1.0.2/filter-it.html
- 34.Ottaviani G, Gosling DJ, Patissier C, Rodde S, Zhou L, Faller B. What is modulating solubility in simulated intestinal fluids? Eur J Pharm Sci. 2010;41:452–457. doi: 10.1016/j.ejps.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2010;2012:195727. doi: 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaney JS. ESOL: estimating aqueous solubility directly from molecular structure. J Chem Inf Model. 2004;44:1000–1005. doi: 10.1021/ci034243x. [DOI] [PubMed] [Google Scholar]
- 37.Ali J, Camilleri P, Brown MB, Hutt AJ, Kirton SB. Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J Chem Inf Model. 2012;52:420–428. doi: 10.1021/ci200387c. [DOI] [PubMed] [Google Scholar]
- 38.Cohen J, Opal SM, Powderly WG(2016) Infectious diseases, 4th edn, vol 1 and 2. Elsevier, UK. https://www.elsevier.com/books/infectious-diseases/9780702062858
- 39.http://tox.charite.de/protox_II/index.php?site=home
- 40.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Liȁo LM, Tropsha A, Andrade CH. Pred-hERG: a novel web-accessible computational tool for predicting cardiac toxicity. Mol Inf. 2015;34:698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision D.01. Wallingford CT: Gaussian, Inc.; 2009. [Google Scholar]
- 42.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1589 kb)