

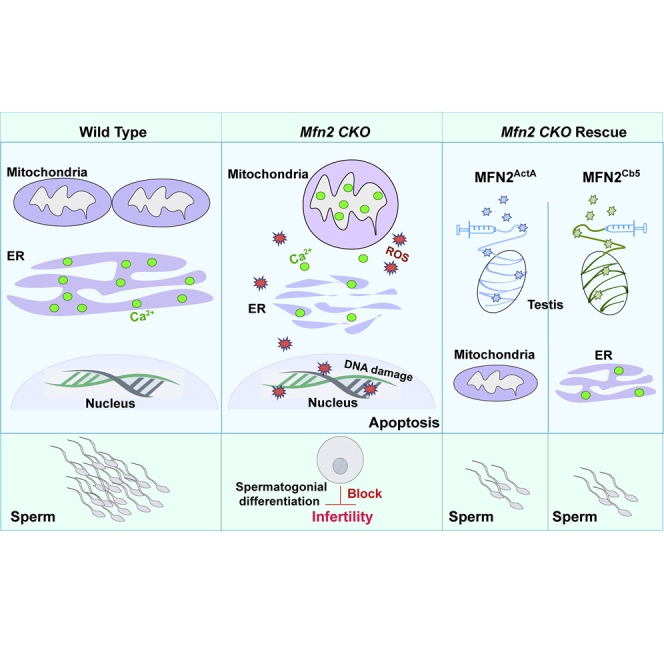
Summary
Although mitochondrial morphology is well-known for its role in cellular homeostasis, there is surprisingly little knowledge on whether mitochondrial remodeling is required for postnatal germ cell development. In this study, we investigated the functions of MFN1 and MFN2, two GTPases in mitochondrial fusion, during early spermatogenesis. We observed increased MFN expressions along with increased mitochondrial and endoplasmic reticulum (ER) activities during spermatogonial differentiation. Deletion of either Mfn led to DNA oxidation and apoptosis specifically in differentiating spermatogonia and spermatocytes, which in turn caused male infertility. We further found MFN2 regulated spermatogenesis by modulating both mitochondrial and ER functions, a mechanism distinct from that of MFN1. Defects of germ cell development in MFN2 mutants were corrected by MFN2 at either mitochondria or ER but not by MFN1. Our study thus reveals an essential requirement of MFN-mediated mitochondrial and ER coordination in spermatogenesis, providing critical insights into mitochondrial determinants of male fertility.
Key Words: mitochondrial fusion, ER stress, mitofusins, spermatogonial differentiation, male fertility
Graphical Abstract
Highlights
-
•
Mitochondrial and ER activities increase during spermatogonial differentiation
-
•
Mfn deletions specifically impair differentiating spermatogonia and spermatocytes
-
•
MFN2 impacts male fertility via regulating mitochondrial fusion and ER homeostasis
-
•
MFN2 functions non-redundantly from MFN1 in spermatogenesis
In this article, Wang and colleagues show that MFN1 and MFN2 are both required for spermatogenesis and proper male fertility. These authors further demonstrate that MFN2 plays a distinct role from MFN1 through regulating both mitochondrial and ER functions during spermatogonial differentiation and meiosis.
Introduction
Mammalian postnatal male germ cell development is known as spermatogenesis, during which haploid spermatozoa are produced through stages of differentiation, meiosis, and maturation (Wylie, 1999). In this process, spermatogonial stem cells maintain a pool of undifferentiated progenitor spermatogonia which will further differentiate, in response to developmental cues, such as retinoic acid, and become KIT+ differentiating spermatogonia (Mecklenburg and Hermann, 2016, Wylie, 1999, Yang and Oatley, 2014). In mice, primary spermatocytes develop from differentiating type B spermatogonia and sequentially become secondary spermatocytes and haploid spermatids via two rounds of meiosis by postnatal day 21 (P21) in the first-wave spermatogenesis (Mecklenburg and Hermann, 2016, Yang and Oatley, 2014). These dramatic changes of germ cells during spermatogenesis, especially in the processes of spermatogonial differentiation and spermatocyte formation, require orchestrated actions of subcellular organelles.
Notably, the architecture and distribution of mitochondria differ in germ cells at different stages of spermatogenesis (Chuma et al., 2009, De Martino et al., 1979, Eddy, 1975). Although mitochondria with less dense matrix and more compact crista compartment exist in spermatogonia, pachytene spermatocytes contain mitochondria that are in a condensed form (Ramalho-Santos and Amaral, 2013). In addition, in spermatogonia and early-stage spermatocytes, perinuclear mitochondria cluster within a germ cell-specific subcellular structure, called intermitochondrial cement (IMC), where primary Piwi-interacting RNA (piRNA) biogenesis occurs (Grivna et al., 2006, Lau et al., 2006). The piRNAs are crucial for maintaining genome integrity of germ cells by inhibiting the expression of transposable elements (Grivna et al., 2006, Lau et al., 2006). However, underlying mechanisms how distinct mitochondrial features are maintained during early spermatogenesis and their developmental importance are yet to be defined.
Mitochondrial features are largely regulated by their fission and fusion, collectively known as mitochondrial dynamics (Chan, 2006). Through mitofusion (i.e., mitochondrial fusion) and fission, mitochondria continuously change between tubular and fragmented morphology, and in turn modify their architecture to respond to various environmental stimuli and cellular requirements (Chan, 2006). More importantly, mitochondrial dynamics ensure exchanging of mitochondrial contents, which allows coordinated responses of individual mitochondria to extracellular cues (Chan, 2006). Mitofusion is mainly regulated by two GTPases, mitofusin 1 (MFN1) and mitofusin 2 (MFN2) (Chen et al., 2003, Santel et al., 2003). Additional factors, such as MitoPLD, also contribute to mitofusion in an MFN1- and MFN2-dependent manner (Huang et al., 2011, Watanabe et al., 2011). Null mutation of MitoPLD in mice leads to mislocalized mitochondria to centrosomes, lack of IMC formation, reduced piRNA biosynthesis, and altered expression of transposable elements, which in turn result in male infertility (Huang et al., 2011, Watanabe et al., 2011). Mfn1 conditional deletion from germ cells also causes male infertility (Zhang et al., 2016). However, IMC remainsintact in Mfn1 knockout mice without alteration in the expression of transposable elements (Zhang et al., 2016), thereby supporting distinct functional mechanisms of different pro-mitofusion molecules in regulating spermatogenesis. It has yet to be fully ascertained how MFN-dependent mitochondrial activities contribute to postnatal germ cell development.
Mitochondrial functions are also regulated by their interaction with other subcellular organelles, including endoplasmic reticulum (ER) (de Brito and Scorrano, 2008). In addition to its roles in protein synthesis, folding, and transport, ER is also the storage site for Ca2+, which is released upon stimulation (Rizzuto et al., 1998). Disturbed ER homeostasis induces ER stress, characterized by Ca2+ release from ER into cytoplasm and a surge of unfolded or misfolded proteins (Bukau et al., 2006, Gaut and Hendershot, 1993). This in turn leads to increased expression of ER chaperon proteins to restore ER homeostasis (Bukau et al., 2006, Gaut and Hendershot, 1993). Increased cytosolic Ca2+ may disturb Ca2+ signaling, upregulate mitochondrial Ca2+ load, and trigger cell death (Bukau et al., 2006, Gaut and Hendershot, 1993, Rizzuto et al., 1998). Moreover, ER membrane connects dynamically to mitochondria and may regulate mitochondrial Ca2+ levels by modulating mitochondrion-ER juxtaposition (de Brito and Scorrano, 2008, Rizzuto et al., 1998, Schneeberger et al., 2013). Currently, it is unclear whether the state of ER contributes to postnatal germ cell development, nor do we know how ER homeostasis is regulated during spermatogenesis.
MFN1 and MFN2 are ubiquitously expressed, but their expression levels and exact functions in regulating mitochondrial and ER activities depend upon specific cell types (Chen et al., 2007, de Brito and Scorrano, 2008, Dietrich et al., 2013, Santel et al., 2003, Schneeberger et al., 2013). We previously demonstrated that GASZ, a germ cell-specific protein, also known as ASZ1 (Ma et al., 2009), interacted with both MFN1 and MFN2 in testes, suggesting an intrinsic contribution of MFN-mediated functions to spermatogenesis (Zhang et al., 2016). To understand the functional importance and underlying mechanisms of mitochondrial and ER functions in postnatal germ cell development, we hereby investigated the roles of MFN1 and MFN2 during spermatogenesis using conditional Mfn knockout mice. Intriguingly, we found that MFN1 and MFN2 affected spermatogenesis in distinct manners. In neonatal pro-spermatogonia, only MFN1 regulated mitochondrial fusion. Later in spermatogenesis, loss of MFN2 notably impaired both mitochondrial functions and increased ER stress. Nevertheless, both MFN1 and MFN2 are required for sustaining spermatogonial differentiation and spermatocyte formation during early spermatogenesis. Our study thus reveals an essential need for MFN-mediated mitofusion and ER homeostasis in sustaining male fertility.
Results
Elevated Mitochondrial and ER Activities during Spermatogonial Differentiation
Cellular reactive oxygen species (ROS) is largely produced from mitochondrial respiration (Dickinson and Chang, 2011), while the ER is the major Ca2+ storage site (Rizzuto et al., 1998) for maintaining a proper cytoplasmic Ca2+ level. To understand changes in mitochondrial and ER activities during early spermatogenesis, we first examined the ROS and Ca2+ levels from CD90+/KIT− undifferentiated spermatogonia and KIT+ differentiating spermatogonia from mice at P12. At this stage, only differentiating spermatogonia and primary spermatocytes before the pachytene stage are formed (Figure S1A). We observed that both cytoplasmic ROS and Ca2+ levels were significantly upregulated in KIT+ cells (Figures 1A and 1B). In addition, the expression levels of ER chaperon proteins were also increased (Figure 1C), indicating elevated activities of both mitochondria and ER.
Figure 1.

Elevated Mitochondrial/ER Activities and MFN Expression during Spermatogonial Development
(A and B) ROS (A) and cellular Ca2+ levels (B) were measured in CD90+/KIT− undifferentiated spermatogonia and KIT+ differentiating spermatogonia collected from wild-type P12 testes by flow cytometry.
(C) Real-time RT-PCR analyses on the expression of ER chaperons in sorted CD9+/KIT− undifferentiated spermatogonia and KIT+ differentiating population from wild-type P12 testes.
(D) Protein levels of MFN1 and MFN2 were measured by western blots in testes collected at various time points during spermatogenesis.
(E) Real-time RT-PCR analyses on Mfn1 and Mfn2 expression in sorted CD9+/KIT− and KIT+ cells from wild-type P12 testes.
(A–C and E) Data are presented as mean ± 1 SEM from three or more biological replicates. Fold changes or expression levels were compared with mean value of CD90+/KIT− or CD9+/KIT− cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; N.S., no statistical significance.
Because MFNs (i.e., MFN1 and MFN2) are key regulators of mitochondrial dynamics, we further examined their expression at various stages during spermatogenesis. Interestingly, we found that protein levels of MFN1 and MFN2 increased between P7 and P14, a period during which spermatogonial differentiation and meiosis I start (Figures 1D and S1A). In addition, we examined Mfn1 and Mfn2 expression in sorted CD9+/KIT− undifferentiated spermatogonia and the KIT+ population by real-time RT-PCR. We found comparable transcript levels between Mfn1 and Mfn2, but for both MFNs the expression levels were higher in KIT+ cells than those in CD9+/KIT− undifferentiated spermatogonia (Figure 1E). These data thus suggest that high levels of MFNs are required for proper spermatogonial differentiation and primary spermatocyte formation.
MFN Deficiency Affects Spermatogonial Differentiation and Spermatocyte Formation
We previously demonstrated that the germline protein GASZ was localized to mitochondrial outer membrane and interacted with both MFN1 and MFN2 to regulate mitochondrial functions (Zhang et al., 2016). In addition, Mfn1 conditional knockout in germ cells resulted in male infertility (Zhang et al., 2016). To understand whether MFN2 also regulates spermatogenesis, we crossed a Mfn2 conditional knockout mice with a Ddx4-Cre line, in which Cre recombinase under the germ cell-specific promoter of Ddx4 (Gallardo et al., 2007) deletes Mfn2 in gonocytes at 15.5 days post coitum (Figures S1A and S1B). Compared with their littermate controls, Mfn2 conditional knockout (Mfn2f/f; Ddx4-Cre, abbreviated as Mfn2 CKO in figures) testes were smaller at P14 (Figure 2A) and were dramatically reduced in size at adulthood (Figure S1D). Histology examination at various time points during spermatogenesis revealed that germ cell formation started to reduce in Mfn2f/f; Ddx4-Cre testes at P10, with much fewer spermatocytes at P14 (Figure 2B). No spermatids were detected in MFN2-deficient testes at week 5 or later (Figure 2B). These data support an essential requirement of MFN2 for sustaining male fertility.
Figure 2.

Loss of Function of Either Mfn Blocks Spermatogonial Differentiation and Spermatocyte Formation
(A) Gross morphology of testes from mice at P14. Right panel: testis weights in P14 Mfn2f/+ and Mfn2f/f; Ddx4-Cre mice were measured. Data represent mean ± 1 SEM. ∗∗p < 0.01; N = 10.
(B) Histological studies on Mfn2f/+ and Mfn2f/f; Ddx4-Cre testes at various ages.
(C and D) IHF assays on testicle sections from Mfn2f/f; Ddx4-Cre mice or Mfn2f/+ littermates at P14 with DDX4 (C) or PLZF (D) antibodies.
(E) Percentage of CD9+/KIT− undifferentiated spermatogonia and KIT+ differentiated spermatogonia in P12 testes were determined by flow cytometry. Data are presented as mean ± 1 SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n ≥ 5.
(F) IHF on Mfn2f/+ and Mfn2f/f; Ddx4-Cre testes from P14 mice with an antibody against SYCP3.
(G) Meiosis chromosome/DNA spreading assay was performed with antibodies against SYCP3 and γ-H2A.X on testes from P14 mice. The percentage of primary spermatocytes at different stages of prophase I was calculated according to the chromatin structure and staining of γ-H2A.X. CKO: conditional deletion of Mfn1 or Mfn2 in germ cells by Ddx4-Cre.
See also Figure S1.
Interestingly, cells positive for DDX4, the germ cell maker, only existed along the base of seminiferous epithelium in Mfn2f/f; Ddx4-Cre testes (Figure 2C). By contrast, in littermate controls, high DDX4 expression was detected in differentiated cells at the lumen of seminiferous tubules (Figure 2C). Undifferentiated spermatogonia which are mainly localized along the base of the seminiferous epithelium appeared to be unaffected by Mfn2 conditional deletion from germ cells at early spermatogenesis (Figures 2B and 2C). To confirm this result, we performed immunohistofluorescence (IHF) assays using an antibody against PLZF that marks undifferentiated spermatogonia. Indeed, PLZF+ cells were not evidently disturbed at P7, and their numbers were increased at P14 and P21 (Figures 2D, S2A, and S2B). Because MFN2 expression levels increased during spermatogonial differentiation (Figure 1E), we next examined whether MFN2 deficiency impairs the formation of differentiating spermatogonia. Flow cytometry analyses demonstrated an increased proportion of CD9+/KIT− undifferentiated spermatogonia in Mfn2 knockout testes accompanied by substantially reduced KIT+ differentiating spermatogonia (Figure 2E). Notably, Mfn1 conditional knockout mice (Mfn1f/f; Ddx4-Cre, abbreviated as Mfn1 CKO in figures) displayed a similar spermatogonial differentiation block with MFN2 mutants (Figure 2E).
We further analyzed spermatocyte formation in Mfn null testes. IHF assays showed a significant reduction in SYCP3+ spermatocytes in Mfn2f/f; Ddx4-Cre mice (Figure 2F), suggesting a meiotic arrest upon MFN2 deficiency, similar to a phenotype we previously observed in Mfn1 conditional knockout mice (Zhang et al., 2016). To identify the exact stages of spermatocyte development that MFNs regulate, meiotic chromosome spreading assays were performed on both MFN1- and MFN2-deficient testes at P14. We found few zygotene spermatocytes in Mfn1f/f; Ddx4-Cre mice, while meiotic arrest in Mfn2f/f; Ddx4-Cre mice occurred at the pachytene stage (Figures 2G and S2C). These data reveal that MFN1 deficiency leads to a relatively earlier blockage of meiosis than that due to Mfn2 conditional knockout in germ cells.
Mfn Conditional Knockout Causes Apoptosis Specifically in Differentiating Spermatogonia and Spermatocytes
To understand how spermatogonial differentiation and spermatocyte formation were blocked upon MFN2 deficiency, we first examined whether the proliferation of germ cells upon Mfn2 deletion was affected by co-staining of PCNA (proliferating cell nuclear antigen) and the germ cell marker DDX4 at P7 and P14. No obvious change in PCNA expression was detected in DDX4+ cells of Mfn2f/f; Ddx4-Cre mice compared with their control littermates (Figure 3A). By contrast, flow cytometry analyses using dual-staining of annexin V and propidium iodide (PI) demonstrated that PI+ apoptotic cells were significantly increased in the KIT+ population (Figure 3B). Annexin V+/PI− early apoptotic cells were slightly increased in CD9+/KIT− cells from Mfn1f/f; Ddx4-Cre (Figure 3B), but cell death (PI+ cells) was negligible in CD9+/KIT− spermatogonia upon deletion of either Mfn1 or Mfn2 (data not shown). TUNEL assays further supported this result that apoptotic cells were mainly differentiated germ cells located in the lumen of testicular tubules (Figure S2D). Taken together, our data suggest that the impaired spermatogenesis upon MFN deficiency is likely due to increased apoptosis in differentiating spermatogonia and spermatocytes.
Figure 3.

MFN Deficiency Leads to Apoptosis in Differentiating Spermatogonia and Spermatocytes
(A) IHF with PCNA and DDX4 co-staining on Mfn2f/+ and Mfn2f/f; Ddx4-Cre testes at P7 and P14.
(B) Percentage of apoptotic cells was analyzed by flow cytometry with FITC-Annexin V and PI co-staining on CD9+/KIT− undifferentiated spermatogonia and KIT+ cells collected from testes of P12 mice. Data represent mean ± 1 SEM from at least four littermate pairs. ∗p < 0.05, ∗∗p < 0.01; N.S., no statistical significance.
See also Figure S2.
MFN Null Mutations Lead to Increased ROS Levels and DNA Oxidation in Differentiating Spermatogonia and Spermatocytes
The diverse mitochondrial functions regulated by mitochondrial dynamics vary by cell types. To determine the extent to which mitochondrial functions are impaired due to MFN deficiency during spermatogenesis, we first examined the mitochondrial membrane potential upon Mfn deletion in non-adherent germ cells collected from dissociated P12 testes using serial adhering procedures (Figure S1F). Indeed, we found the percentage of cells with lower mitochondrial potential was increased (Figures 4A and S3A), suggesting impaired mitochondrial functions upon conditional knockout of either Mfn1 or Mfn2.
Figure 4.

Mfn Deletion Causes Mitochondrial Dysfunction and Elevated DNA Oxidation
(A) Percentage of cells with low mitochondrial membrane potential was measured with flow cytometry in non-adherent germ cells collected from testes of P12 mice.
(B) ATP levels were measured on dissociated cells from P12 testes, calculated in comparison with their littermate controls.
(C) ROS levels were analyzed by flow cytometry in CD9+/KIT− undifferentiated spermatogonia and KIT+ differentiating germ cells from testes of P12 mice.
(D) Immunofluorescence (IF) with an antibody against 8-oxoguanine on sorted KIT+ germ cells from mice at P11. Scale bar, 2 mm.
(A–C) Data are presented as mean ± 1 SEM. N ≥ 3. ∗p < 0.05, ∗∗p < 0.01; N.S., no statistical significance. See also Figure S3.
Because mitochondria serve as a powerhouse for ATP generation, we next assessed ATP generation in MFN mutants. To our surprise, ATP levels were not significantly altered in Mfn-deleted testes at P12 compared with their littermate controls (Figure 4B). Similar results were observed in GC1, an immortalized type B differentiating spermatogonial cell line (Hofmann et al., 1992). No significant alteration in ATP levels was observed upon downregulation of MFN1 or MFN2 expression by RNA interference (Figures S3B and S3C). However, upon the loss of function of either MFN, we observed increased ROS levels specifically in KIT+ cells (Figures 4C and S3D). By contrast, no significant changes in ROS production were detected in CD9+/KIT− undifferentiated spermatogonia (Figures 4C and S3D). Since high ROS levels cause DNA oxidation, we collected KIT+ germ cells from testes at P11 and stained them with an antibody against 8-oxoguanine, which is an indicator for DNA oxidation. Indeed, the 8-oxoguanine+ germ cells were significantly increased upon deletion of either Mfn1 or Mfn2 (Figure 4D). Notably, we did not observe significant changes in autophagy (Figure S4A), piRNA levels (Figure S4B), and expression levels of transposable elements (Figure S4C) upon Mfn2 conditional knockout. Therefore, we conclude that the increased cell death in MFN-deficient germ cells is not directly caused by altered energy production, autophagy, or piRNA biogenesis, but rather due to DNA damage from increased oxidation.
Loss of Function of MFN1 and MFN2 Causes Distinct Defects in Mitochondrial Architecture at Different Stages of Spermatogenesis
As described above, mitochondrial functions were disturbed by either MFN1 or MFN2 deficiency. To directly visualize the underlying mitochondrial defects due to Mfn deletion, we closely examined alterations in mitochondrial morphology and architecture of germ cells by transmission electron microscopy (TEM) at various time points during early spermatogenesis. We found normal mitochondrial morphology at P0 in Mfn2 knockout pro-spermatogonia (Figure 5A). At P5, mitochondrial morphology started to vary, with a small fraction appeared to be rounded up and contained less cristae (Figure 5B, right panel) upon the loss of function of MFN2. Between P8 and P11, the majority of mitochondria in Mfn2f/f; Ddx4-Cre germ cells became swollen and enlarged (Figures 5C and 5D) compared with their littermate controls. In addition, differentiated germ cells that were located toward the lumen of testicular tubules displayed more severe abnormalities than undifferentiated spermatogonia (Figure 5C, right two panels). Intriguingly, mitochondria in Mfn2-deleted germ cells returned to relatively normal size around P13, but few tubular mitochondria were detected (Figure 5D, right two panels).
Figure 5.

Ultrastructural Changes of Mitochondria upon Mfn2 Deletion
Mitochondrial architecture was examined by TEM on testicular sections from Mfn2 conditional knockout testes and littermate controls during spermatogenesis at P0 (A), P5 (B), P8 (C), and P11 and P13 (D). At P5 (B), a small fraction of round mitochondria containing less cristae are shown in the right panel. At P8 (C), the left two panels show mitochondrial morphology in undifferentiated spermatogonia at the basal membrane of the seminiferous tubule. Mitochondria from differentiated cells located toward the lumen of tubules are displayed in the two right panels. N, nucleus. See also Figure S4.
The exact stages that abnormalities of mitochondrial features occurred in Mfn2 knockout appeared to be different from the ones we had previously detected in MFN1 mutants (Zhang et al., 2016). Mfn1 conditional deletion in DDX4+ germ cells caused severely swollen and significantly enlarged mitochondria in neonatal (P0) pro-spermatogonia compared with their littermate controls (Zhang et al., 2016). To further reveal the differential impacts of Mfn1 and Mfn2 knockout on mitochondrial features, we examined mitochondrial morphology and architecture by TEM in Mfn1-deleted germ cells at various time points of early spermatogenesis. Interestingly, mitochondria in Mfn1 knockout germ cells also displayed dynamical alterations during spermatogenesis. They became round mitochondria with normal size at P5 and were then substantially smaller at P8 compared with their littermate controls (Figure S4D). In summary, our results demonstrate that either Mfn1 or Mfn2 deletion causes mitochondrial defects that, however, vary at different time points during early spermatogenesis.
Both Mitochondrial and ER Dysregulation Contribute to Male Infertility Caused by MFN2 Deficiency
Based on our results above, MFN1 appears to play a dominant role in regulating mitochondrial features at early time points of spermatogenesis (i.e., before P5). Compared with Mfn1 knockout, Mfn2 deletion only led to relative mild defects in mitochondrial morphology and architecture at later stages during spermatogenesis (P8–P11). Nevertheless, MFN2 deficiency blocked spermatogonial differentiation and eventually led to male infertility, similar to what we observed in MFN1 null mutants. This indicates the existence of other mechanisms that underlie impaired germ cell development due to MFN2 deficiency. It was reported that, in fibroblasts and certain types of neurons, MFN2 affected mitochondrial functions indirectly by regulating mitochondrion-ER interaction and ER homeostasis (Chen et al., 2003, Chen et al., 2007, de Brito and Scorrano, 2008, Dietrich et al., 2013, Schneeberger et al., 2013). We observed upregulated ER activities and increased expression of MFNs during spermatogonial differentiation (Figure 1). Therefore, we examined whether MFN2 regulates ER functions in germ cell development.
Because ER is the major Ca2+ reservoir, and ER disturbance leads to abnormal Ca2+ homeostasis, we first analyzed cellular Ca2+ levels in germ cells from Mfn2f/+ and Mfn2f/f; Ddx4-Cre mice by flow cytometry with Fluo4-AM, a fluorescent Ca2+ indicator. We also examined Ca2+ levels in Mfn1 conditional knockout mice for comparison purposes. No significant changes were detected in Mfn1f/f; Ddx4-Cre germ cells, while Mfn2 deletion led to a substantial increase in cellular Ca2+ levels compared with their control littermates (Figures 6A and S5A). The increase in Ca2+ levels upon Mfn2 deletion started in CD9+/KIT− undifferentiated spermatogonia and became more dramatic in KIT+ cells (Figure 6B). Similar phenomena were observed in GC1 type B spermatogonial cells (Figure S5B). No significant alteration of cellular Ca2+ levels was observed upon downregulation of MFN1 expression by RNA interference, but substantially increased Ca2+ levels were detected upon MFN2 knockdown (Figure S5B). To assess whether increased cellular Ca2+ levels affects mitochondrial Ca2+ storage, we further examined mitochondrial Ca2+ levels in Mfn1- or Mfn2-deleted germ cells. We found that mitochondrial Ca2+ levels dramatically increased upon Mfn2 knockout, as displayed by increased co-staining of Fluo3-AM, a fluorescent Ca2+ indicator similar to Fluo 4-AM, and a MitoTracker to specifically label mitochondria (Figure 6C). By contrast, no significant alteration in mitochondrial Ca2+ level was found in Mfn1f/f; Ddx4-Cre germ cells compared with their littermate controls (Figure 6C). Notably, we found an increased percentage of fragmented ER in Mfn2-deleted germ cells at P5 by TEM (Figures 6D and S5C). This finding was further supported by multiplexed 3D super-resolution confocal microscopy with Tomm20 (to label mitochondria) and calreticulin (to mark ER) co-staining, which showed swollen mitochondria and fragmented ER in Mfn2-deleted germ cells (Figure S5D). Taken together, these data reveal a disturbed cellular and mitochondrial Ca2+ homeostasis upon Mfn2 but not Mfn1 deletion, probably due to Ca2+ released from ER caused by its fragmentation.
Figure 6.

MFN2 Regulates Both Mitochondrial and ER Functions in Germ Cells
(A and B) Fluo4-AM fluorescent intensity was measured by flow cytometry on non-adherent germ cells (A), as well as sorted CD9+/KIT− undifferentiated and KIT+ differentiating spermatogonia (B) from testes of P11 mice. Data are presented as mean ± 1 SEM from three biological replicates. ∗∗∗p < 0.001; N.S., no statistical significance.
(C) IF on non-adherent germ cells from P11 testes with green Fluo3-AM probe (to indicate Ca2+ level) and a red mitochondrial tracker.
(D) Mitochondrial and ER architectures were examined by TEM on testicular sections from Mfn2f/+ and Mfn2f/f; Ddx4-Cre testes of P5 mice. Red arrows indicate ER. N, nucleus.
(E) Expression levels of chaperon proteins were examined by western blots on testes from P12 mice.
(F) Histological studies of testicle sections from Mfn2f/f; Ddx4-Cre mice at day 30 after viral introduction of MFN2 tagged with mitochondrion (MFN2ActA) or ER (MFN2Cb5) localization signals. Red stars indicate the seminiferous tubules containing haploid spermatids. Scale bar, 50 μm.
(G) IHF analyses with an antibody against acrosin (ACR), counterstained with peanut agglutinin (PNA) and DAPI on Mfn2f/f; Ddx4-Cre testis sections at day 30 after viral introduction with ActA- or Cb5-tagged MFN2 through seminiferous tubules. Ctrl, control viruses produced with the empty vector. Scale bar, 20 μm.
(F and G) Three to four mice per group were used for viral injection. See also Figures S5 and S6.
As disturbed ER homeostasis usually leads to upregulation of chaperone genes to restore ER functions, we next assessed the expression levels of ER stress markers and ER chaperons by real-time RT-PCR and western blotting. Indeed, expression levels of several ER stress markers (calreticulin and CHOP) and ER chaperon GPR78 were significantly increased in Mfn2f/f; Ddx4-Cre testes but not in Mfn1-deleted mutants (Figure 6E). Similar results were observed in Mfn2f/f; Ddx4-Cre non-adherent germ cells (Figure S5E, left panel), sorted spermatogonia (Figure S5E, right panel) from Mfn2f/f; Ddx4-Cre testes, and GC1 cells upon MFN downregulation by RNA interference (Figure S5F), thereby suggesting that ER homeostasis was disturbed upon Mfn2 deletion.
Both mitochondrial and ER functions are impaired by MFN2 deficiency, it is therefore unclear whether the male infertility upon Mfn2 deletion is due to dysfunctional mitofusion, ER stress, or both. Recent data suggest a partial localization of MFN2 at ER and mitochondrion-ER contacts (de Brito and Scorrano, 2008, Schneeberger et al., 2013). To answer this question, lentiviral vectors were constructed to express MFN2 that were specifically tagged either to the surfaces of mitochondria (MFN2ActA) or to ER (MFN2Cb5) by localization signaling peptides (de Brito and Scorrano, 2008, Schneeberger et al., 2013). We then introduced these viruses to Mfn2f/f; Ddx4-Cre mice through testicular seminiferous tubule injection (Gou et al., 2017, Ikawa et al., 2002, Li et al., 2013) and examined the germ cell development around days 30–40 after viral transduction. The MFN2 expression from these viral vectors was confirmed in both immortalized Mfn2−/− mouse embryonic fibroblasts (Chen et al., 2003) with the same viral infection (Figure S6A) and testis after viral injection (Figure S6B). Strikingly, we found fully developed haploid spermatids in seminiferous tubules of Mfn2f/f; Ddx4-Cre testes upon the introduction of MFN2 proteins, either localized at mitochondrial membrane or at ER (Figures 6F and S6C). We further performed IHF with an antibody against acrosin (ACR) or with rhodamine-labeled peanut agglutinin (PNA) to detect acrosome that only exists in haploid spermatids and spermatozoa. Consistent with our histological observations, ACR+ and PNA+ spermatids were clearly observed in Mfn2f/f; Ddx4-Cre testes rescued with either mitochondrion- or ER-localized MFN2 (Figures 6G and S6D). Taken together, our data reveal that MFN2 contributes to male germ cell development by regulating both mitochondrial and ER functions.
Because both MFN1 and MFN2 promote mitochondrial fusion, we further tested whether MFN1 compensates the defects of germ cell development caused by Mfn2 deletion, or vice versa. We introduced the viruses expressing MFN1 into both Mfn1f/f; Ddx4-Cre and Mfn2f/f; Ddx4-Cre testes and examined their germ cell development. ACR+ and PNA+ spermatids were detected in Mfn1f/f; Ddx4-Cre testes but not in Mfn2f/f; Ddx4-Cre mice injected with Mfn1-expressing virus (Figure S6D). Similarly, Mfn2 transgene did not rescue blocked germ cell development from Mfn1f/f; Ddx4-Cre mice (Figure S6D). Therefore, we conclude that MFN1 and MFN2 play non-redundant roles in regulating male germ cell development.
Discussion
Although the machinery for mitochondrial fusion has been extensively investigated in somatic cells, its role in germline stem cell differentiation under developmental settings remains elusive. In this study, along with increased ROS generation during postnatal germ cell development, we detected upregulated expression of both MFN1 and MFN2, two GTPases critical for mitochondrial fusion. We found that loss of function of either MFN blocked spermatogonia differentiation and spermatocyte formation, which in turn led to male infertility. These phenotypes in MFN mutants were accompanied by altered mitochondrial morphology, architecture, and function, supporting a critical requirement of MFN-mediated mitochondrial activities during early spermatogenesis.
The cellular reliance on mitochondrial functions varies by cell type and developmental stage. Between stem cells or progenitors and the cells into which they differentiate, mitochondria have stage-specific functions, as shown by studies on pluripotent stem cells, neural stem cells (NSCs), hematopoietic stem cells, and muscle stem cells (Bracha et al., 2010, Figueroa et al., 2010, Khacho et al., 2016, Mandal et al., 2011, Shyh-Chang et al., 2013, Son et al., 2015, Wanet et al., 2015, Zhou et al., 2012). These changes in mitochondrial functions and activities in turn impact the intrinsic programming of specification of stem cell fate. For example, mitochondrial features of NSCs are remarkably different from their committed progenitors, and disrupting mitochondrial fusion specifically impairs NSC self-renewal via ROS-NRF2-mediated retrograde (mitochondria to nucleus) signaling (Khacho et al., 2016). By contrast, inhibition of mitochondrial fusion during reprogramming upregulates the formation of induced pluripotent stem cells (Son et al., 2015). In our study, we found upregulated mitochondrial functions in KIT+ differentiation spermatogonia, as indicated by increased ROS levels and MFN expression. Although Mfn deletion caused defects in mitochondrial morphology and architecture in spermatogonia, the numbers of CD9+/KIT− or PLZF+ undifferentiated spermatogonia increased upon Mfn knockout compared with their littermate controls. Conversely, the formation of KIT+ differentiating spermatogonia and primary spermatocytes was specifically blocked. Our data, therefore, suggest that spermatogonial differentiation and meiosis are particularly sensitive to mitochondrial defects, likely due to a high basal dependence on mitochondrial functions in differentiated germ cells. The differential sensitivity to disrupted mitochondrial dynamics across hierarchical lineages thus reflects the importance of mitochondrial regulation in germ cell development.
Our data further reveal that the blockage of spermatogonial differentiation and meiosis upon Mfn deletions is likely due to increased apoptosis in differentiated cell populations. Although clustered mitochondria are the essential components of IMC, a unique subcellular structure for piRNA biogenesis, no alteration was observed in the expression of transposable elements and piRNAs from Mfn-deleted germ cells. Furthermore, despite the classic role of mitochondria as a powerhouse, ATP production was not decreased either at the time points when significant apoptosis occurred in Mfn knockout. By contrast, ROS level was significantly upregulated in KIT+ cells, which in turn led to increased DNA damage due to oxidation, as supported by increased 8-oxoguanine formation—an indicator for DNA oxidation in Mfn-deficient germ cells. In addition, Mfn2 deletion also led to increased cellular and mitochondrial Ca2+ levels, which disturbs cellular Ca2+ signaling and sensitizes cells to apoptosis (de Brito and Scorrano, 2008, Filadi et al., 2015). Therefore, we conclude that high DNA oxidation and pathologically increased Ca2+ levels are at least partially responsible for germ cell apoptosis upon MFN2 deficiency. This conclusion does not contradict to our finding that ROS and Ca2+ levels increase during germ cell development. One may speculate that physiologically controlled ROS and Ca2+ elevation from upregulated mitochondrial and ER activities are required for spermatogonial differentiation; however, abnormal ROS or Ca2+ elevation beyond pathological thresholds will cause excessive DNA oxidation/damage and dysfunctional Ca2+ signaling, and in turn lead to cell death. Our findings thus highlight the levels of mitochondrial and ER activities need to be precisely controlled by MFNs to maintain proper germ cell development.
In somatic cells, such as fibroblasts, deleting either Mfn1 or Mfn2 directly leads to mitochondria fragmentation (Chen et al., 2003). Interestingly, in Mfn knockout germ cells we were able to observe a dynamic alteration of mitochondrial morphology and architecture during spermatogenesis. Before fragmentation, mitochondria were first enlarged and swollen with vesiculation of the mitochondrial inner membrane. It is possible that, at early stages of MFN deficiency, impaired mitofusion leads to a feedback downregulation in mitochondrial fission which causes enlarged mitochondria. However, as MFN deficiency continues, reduced mitochondrial fission is no longer able to compensate the severely compromised mitofusion, which eventually leads to small and fragmented mitochondria in Mfn-deleted germ cells.
MFN1 and MFN2 share more than 80% similarity in their protein sequences, and they are expressed in germ cells at comparable levels. However, our data show that deletion of either Mfn leads to male infertility, and they likely play distinctive and non-redundant roles in spermatogenesis. MFN1 re-introduction into testes restored spermatogenesis in Mfn1f/f; Ddx4-Cre mice but not in Mfn2 conditional knockout, and vice versa. Compared with MFN2, MFN1 deficiency affected mitofusion at earlier stages of spermatogenesis. In addition, MFN2 regulated mitochondrial functions directly through mitofusion and possibly indirectly via ER homeostasis, as supported by altered mitochondrial and ER architectures in MFN2-deficient germ cells. Introduction of either mitochondrion- or ER-localized MFN2 rescued the defects in spermatogenesis due to Mfn2 deletion. Previous studies suggest that MFN2 regulates mitochondrion-ER contacts in embryonic fibroblasts and in certain neurons (de Brito and Scorrano, 2008, Schneeberger et al., 2013). However, in germ cells, we did not observe consistent alterations in mitochondrion-ER juxtaposition upon Mfn2 knockout. This may be explained by cell-type-specific regulatory mechanisms of MFN2 on mitochondria and ER activities. For example, MFN2 deficiency in POMC neurons causes diet-induced obesity due to defects in mitochondrion-ER juxtaposition (Schneeberger et al., 2013), whereas in agouti-related peptide neurons or Purkinje cells, MFN2 mutations mainly lead to dysfunctional mitochondrial fusion (Chen et al., 2007, Dietrich et al., 2013). Our data not only delineate crucial and unique contributions of MFN2 to both mitochondrial and ER components during germ cell development, but also indicate an essential coordination between MFN1 and MFN2 during spermatogonial differentiation.
Infertility affects approximately 48.5 million couples worldwide, with half due to male factors (Agarwal et al., 2015). This public health problem is further compounded by an increasing prevalence of metabolic disorders, particularly obesity and diabetes, which are linked to male infertility (Du Plessis et al., 2010). Notably, MFN2 mutations have been identified from patients with metabolic alterations (Bach et al., 2003, Rovira-Llopis et al., 2017). Our data demonstrate that deletion of either Mfn1 or Mfn2 impairs mitochondrial functions and destroys male fertility, providing an important connection between mitochondrial metabolic disorders and impaired spermatogenesis. Further research into this connection may lead to an important gateway to treat male infertility.
Experimental Procedures
Generation of Conditional Mfn2 Knockout Mice
Sperm harboring the floxed Mfn2 allele were obtained from Mutant Mouse Regional Resource Center (MMRRC at UC Davis, USA, resource identifier no. MMRRC_029902-UCD). Mfn2f/+ mice were derived by intracytoplasmic injection of sperm into oocytes and maintained by crossing with C57BL/6 mice. Ddx4-Cre mice (Model Animal Research Center at Nanjing University, China) were used to obtain targeted Mfn2 deletion in germ cells. Mfn2f/f; Ddx4-Cre mice were generated by Mfn2f/f female mice crossing with Mfn2f/+; Ddx4-Cre males. Conditional Mfn1 knockout mice were generated in the same way by intracytoplasmic injection of sperm (MMRRC_029901-UCD) as described previously (Zhang et al., 2016). Animal use protocols for this study were approved by the Institutional Animal Care and Use Committee of East China Normal University (project no. AR201305006) and all experiments were carried out in accordance with approved protocols.
Seminiferous Tubule Injection with Lentivirus
Mfn2 cDNAs tagged with sequences encoding a mitochondrial (ActA) or an ER (Cb5) localization signal (Chen et al., 2003, Schneeberger et al., 2013) (kind gifts from Dr. Luca Scorrano from the Venetian Institute of Molecular Medicine, Italy) were amplified by PCR and cloned into pll3.7-EF1α-Mfn2-IRES-Hygro, a lentiviral vector. All constructs were verified by DNA sequencing. Lentiviral vectors expressing Mfn1 or Mfn2 full-length cDNAs were reported previously (Zhang et al., 2016). Lentiviral injection was performed according to published protocols (Gou et al., 2017, Ikawa et al., 2002, Li et al., 2013). In brief, Mfn1f/f; Ddx4-Cre or Mfn2f/f; Ddx4-Cre mice at 3 weeks were injected with 5 μL of lentivirus through the testicular seminiferous tubule and examined by histology and IHF at days 30–40 after injection.
Histology and IHF Assays
For histology study, mouse testes were fixed in Bouin's fixative at 4°C overnight for staining with hematoxylin and eosin as described previously (Wang et al., 2017). For IHF, testes were fixed with 4% paraformaldehyde in PBS at 4°C overnight and embedded in paraffin. The 4-μm-thick testicular sections were incubated with primary antibodies and washed three times with PBS containing 0.5% Tween 20. Nuclei were stained in 0.5 μg/mL DAPI after blotting with FITC- or TRITC-conjugated second antibodies. Slides were imaged under a fluorescent microscope (Leica, DM400BLED368424) or a Lecia TCS/SP5 confocal microscope and processed with Image-Pro Plus. Antibodies and fluorescent probes used in this study: PLZF (Santa Cruz Biotechnology, SC-28319), DDX4 (Abcam, ab13840), SYCP3 (Abcam, ab15093), PCNA (Abcam, ab29), Acrosin/ACR (Atlas Antibodies, HPA048687), rhodamine-labeled PNA (Vector Laboratories, RL-1072), Alexa Fluor 488- or TRITC-conjugated anti-mouse, or anti-rabbit secondary antibodies (Jackson ImmunoResearch).
TEM
TEM was performed as described previously (Zhang et al., 2016). In brief, mouse testes were fixed with 2.5% TEM-grade glutaraldehyde in 0.1 M PBS (pH 7.4) overnight and incubated in 1% osmium tetroxide for 2 h at 4°C in the dark. After dehydration in ethanol with increasing concentration from 30% to 90%, samples were treated in 45% ethanol and acetone at 4°C for 15 min, and then in 100% acetone three times before further embedding, slide processing, and staining. Sample grids were examined using a Hitachi H7500 transmission electron microscope.
Flow Cytometry
Testes were digested into single cells and cell surface antigens were stained with fluorochrome-conjugated CD90 or CD9 and KIT antibodies for 40 min on ice and washed with PBS before analyses. For ROS detection, cells were stained with 5 μM H2DCFDA (Thermo Fisher Scientific, D399) in PBS containing 1% BSA at 37°C for 40 min in the dark followed by washing with PBS twice. For analyses of mitochondrial membrane potential, cells were stained with JC-1 dye (Beyotime Biotechnology, C2006) at 37°C for 40 min following the manufacturer's instructions. For the apoptosis test, cells were stained with FITC-Annexin V and PI using an FITC-Annexin V Apoptosis Detection Kit (BD Biosciences, 556420). To determine Ca2+ levels, cells were stained with 2 μM Fluo-4 AM (Beyotime Biotechnology, S1060) in PBS containing 1% BSA at 37°C for about 40 min. Flow cytometry analyses and sorting were performed on a Fortessa analyzer and an Aria II cell sorter (BD Biosciences), respectively. Antibodies below were used with 2 μg/mL per 1–2 million cells as a final working concentration: APC-CD9 (17-0091-82, eBioscience, USA); APC-CD90.2 (105311) and FITC-KIT (105806) from BioLegend, USA, and PE-KIT (553355, BD Biosciences, USA).
RT-PCR, PCR, and Real-Time PCR
Total RNA from cultured cells was extracted by using TRIzol (Thermo Fisher Scientific) and reverse-transcribed by using a cDNA Synthesis Kit (Takara Biotechnology). PCR and real-time PCR were performed on a Bio-Rad thermal cycler (Bio-Rad) or on a Stratagene Mx3000P (Stratagene, CA, USA). Three or more technical replicates were conducted for each independent experiment. Sequences of primers used in this study are provided in Table S1.
Meiotic Chromosome Spreading Assays, Immunofluorescence Assays, Measurement of ATP Levels, and Western Blotting
All assays were performed following standard procedures, with details of antibody/probe/dye information in Supplemental Experimental Procedures.
Statistical Analysis
Data are presented as mean ± SEM. All experiments were performed independently three times or more unless otherwise stated. Statistical significance of between-group differences was analyzed with unpaired Student's t test using the Prism Graphic software.
Authors Contributions
W.C., Y.S., Q.S., J.Z., M.J., C.C., X.H., C.W., P.W., and Z.Z. performed the research. W.C., X.C., and Y.W. analyzed the data. Y.W. designed the experiments and wrote the article.
Acknowledgments
We thank Dr. Luca Scorrano from Venetian Institute of Molecular Medicine for providing ActA- and Cb5-tagged MFN2 expression plasmids, and Dr. Mofang Liu from Chinese Academy of Sciences for help in piRNA detection. This work was supported by grants from the Ministry of Science and Technology of the People's Republic of China (2016YFA0100300), the National Natural Science Foundation of China (91854123 and 31771655). C.C., Z.Z., and Y.W. were supported by the NIFA through the AgbioResearch Hatch Fund from Michigan State University.
Published: April 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.03.024.
Supplemental Information
References
- Agarwal A., Mulgund A., Hamada A., Chyatte M.R. A unique view on male infertility around the globe. Reprod. Biol. Endocrinol. 2015;13:37. doi: 10.1186/s12958-015-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach D., Pich S., Soriano F.X., Vega N., Baumgartner B., Oriola J., Daugaard J.R., Lloberas J., Camps M., Zierath J.R. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- Bracha A.L., Ramanathan A., Huang S., Ingber D.E., Schreiber S.L. Carbon metabolism-mediated myogenic differentiation. Nat. Chem. Biol. 2010;6:202–204. doi: 10.1038/nchembio.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B., Weissman J., Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Chan D.C. Dissecting mitochondrial fusion. Dev. Cell. 2006;11:592–594. doi: 10.1016/j.devcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chuma S., Hosokawa M., Tanaka T., Nakatsuji N. Ultrastructural characterization of spermatogenesis and its evolutionary conservation in the germline: germinal granules in mammals. Mol. Cell. Endocrinol. 2009;306:17–23. doi: 10.1016/j.mce.2008.11.009. [DOI] [PubMed] [Google Scholar]
- de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- De Martino C., Floridi A., Marcante M.L., Malorni W., Scorza Barcellona P., Bellocci M., Silvestrini B. Morphological, histochemical and biochemical studies on germ cell mitochondria of normal rats. Cell Tissue Res. 1979;196:1–22. doi: 10.1007/BF00236345. [DOI] [PubMed] [Google Scholar]
- Dickinson B.C., Chang C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich M.O., Liu Z.W., Horvath T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155:188–199. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Plessis S.S., Cabler S., McAlister D.A., Sabanegh E., Agarwal A. The effect of obesity on sperm disorders and male infertility. Nat. Rev. Urol. 2010;7:153–161. doi: 10.1038/nrurol.2010.6. [DOI] [PubMed] [Google Scholar]
- Eddy E.M. Germ plasm and the differentiation of the germ cell line. Int. Rev. Cytol. 1975;43:229–280. doi: 10.1016/s0074-7696(08)60070-4. [DOI] [PubMed] [Google Scholar]
- Figueroa M.E., Abdel-Wahab O., Lu C., Ward P.S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H.F. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R., Greotti E., Turacchio G., Luini A., Pozzan T., Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U S A. 2015;112:E2174–E2181. doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo T., Shirley L., John G.B., Castrillon D.H. Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis. 2007;45:413–417. doi: 10.1002/dvg.20310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut J.R., Hendershot L.M. The modification and assembly of proteins in the endoplasmic reticulum. Curr. Opin. Cell Biol. 1993;5:589–595. doi: 10.1016/0955-0674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- Gou L.T., Kang J.Y., Dai P., Wang X., Li F., Zhao S., Zhang M., Hua M.M., Lu Y., Zhu Y. Ubiquitination-deficient mutations in human Piwi cause male infertility by impairing histone-to-protamine exchange during spermiogenesis. Cell. 2017;169:1090–1104.e13. doi: 10.1016/j.cell.2017.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna S.T., Beyret E., Wang Z., Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M.C., Narisawa S., Hess R.A., Millan J.L. Immortalization of germ cells and somatic testicular cells using the SV40 large T antigen. Exp. Cell Res. 1992;201:417–435. doi: 10.1016/0014-4827(92)90291-f. [DOI] [PubMed] [Google Scholar]
- Huang H., Gao Q., Peng X., Choi S.Y., Sarma K., Ren H., Morris A.J., Frohman M.A. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev. Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa M., Tergaonkar V., Ogura A., Ogonuki N., Inoue K., Verma I.M. Restoration of spermatogenesis by lentiviral gene transfer: offspring from infertile mice. Proc. Natl. Acad. Sci. U S A. 2002;99:7524–7529. doi: 10.1073/pnas.072207299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M., Clark A., Svoboda D.S., Azzi J., MacLaurin J.G., Meghaizel C., Sesaki H., Lagace D.C., Germain M., Harper M.E. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–247. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Lau N.C., Seto A.G., Kim J., Kuramochi-Miyagawa S., Nakano T., Bartel D.P., Kingston R.E. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–367. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- Li X., Mao Z., Wu M., Xia J. Rescuing infertility of Pick1 knockout mice by generating testis-specific transgenic mice via testicular infection. Sci. Rep. 2013;3:2842. doi: 10.1038/srep02842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Buchold G.M., Greenbaum M.P., Roy A., Burns K.H., Zhu H., Han D.Y., Harris R.A., Coarfa C., Gunaratne P.H. GASZ is essential for male meiosis and suppression of retrotransposon expression in the male germline. PLoS Genet. 2009;5:e1000635. doi: 10.1371/journal.pgen.1000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S., Lindgren A.G., Srivastava A.S., Clark A.T., Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. 2011;29:486–495. doi: 10.1002/stem.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenburg J.M., Hermann B.P. Mechanisms regulating spermatogonial differentiation. Results Probl. Cell Differ. 2016;58:253–287. doi: 10.1007/978-3-319-31973-5_10. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos J., Amaral S. Mitochondria and mammalian reproduction. Mol. Cell. Endocrinol. 2013;379:74–84. doi: 10.1016/j.mce.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rovira-Llopis S., Banuls C., Diaz-Morales N., Hernandez-Mijares A., Rocha M., Victor V.M. Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol. 2017;11:637–645. doi: 10.1016/j.redox.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A., Frank S., Gaume B., Herrler M., Youle R.J., Fuller M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003;116:2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- Schneeberger M., Dietrich M.O., Sebastian D., Imbernon M., Castano C., Garcia A., Esteban Y., Gonzalez-Franquesa A., Rodriguez I.C., Bortolozzi A. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N., Daley G.Q., Cantley L.C. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M.J., Kwon Y., Son M.Y., Seol B., Choi H.S., Ryu S.W., Choi C., Cho Y.S. Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ. 2015;22:1957–1969. doi: 10.1038/cdd.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanet A., Arnould T., Najimi M., Renard P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev. 2015;24:1957–1971. doi: 10.1089/scd.2015.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Tang C., Wang Q., Su J., Ni T., Yang W., Wang Y., Chen W., Liu X., Wang S. NRF1 coordinates with DNA methylation to regulate spermatogenesis. FASEB J. 2017;31:4959–4970. doi: 10.1096/fj.201700093R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Chuma S., Yamamoto Y., Kuramochi-Miyagawa S., Totoki Y., Toyoda A., Hoki Y., Fujiyama A., Shibata T., Sado T. MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev. Cell. 2011;20:364–375. doi: 10.1016/j.devcel.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie C. Germ cells. Cell. 1999;96:165–174. doi: 10.1016/s0092-8674(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Yang Q.E., Oatley J.M. Spermatogonial stem cell functions in physiological and pathological conditions. Curr. Top. Dev. Biol. 2014;107:235–267. doi: 10.1016/B978-0-12-416022-4.00009-3. [DOI] [PubMed] [Google Scholar]
- Zhang J., Wang Q., Wang M., Jiang M., Wang Y., Sun Y., Wang J., Xie T., Tang C., Tang N. GASZ and mitofusin-mediated mitochondrial functions are crucial for spermatogenesis. EMBO Rep. 2016;17:220–234. doi: 10.15252/embr.201540846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Choi M., Margineantu D., Margaretha L., Hesson J., Cavanaugh C., Blau C.A., Horwitz M.S., Hockenbery D., Ware C. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.