

Abstract
Neurological disorders such as Alzheimer’s disease (AD), Lewy body dementia (LBD), frontotemporal dementia (FTD), and vascular dementia (VCID) have no disease-modifying treatments to date and now constitute a dementia crisis that affects 5 million in the USA and over 50 million worldwide. The most common pathological hallmark of these age-related neurodegenerative diseases is the accumulation of specific proteins, including amyloid beta (Aβ), tau, α-synuclein (α-syn), TAR DNA-binding protein 43 (TDP43), and repeat-associated non-ATG (RAN) peptides, in the intra- and extracellular spaces of selected brain regions. Whereas it remains controversial whether these accumulations are pathogenic or merely a byproduct of disease, the majority of therapeutic research has focused on clearing protein aggregates. Immunotherapies have garnered particular attention for their ability to target specific protein strains and conformations as well as promote clearance. Immunotherapies can also be neuroprotective: by neutralizing extracellular protein aggregates, they reduce spread, synaptic damage, and neuroinflammation. This review will briefly examine the current state of research in immunotherapies against the 3 most commonly targeted proteins for age-related neurodegenerative disease: Aβ, tau, and α-syn. The discussion will then turn to combinatorial strategies that enhance the effects of immunotherapy against aggregating protein, followed by new potential targets of immunotherapy such as aging-related processes.
Electronic supplementary material
The online version of this article (10.1007/s13311-020-00853-2) contains supplementary material, which is available to authorized users.
Key Words: Immunotherapy, vaccination, Aβ, tau, α-synuclein, Alzheimer’s disease
Introduction
Age-related neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), dementia with Lewy bodies (DLB), frontotemporal dementia (FTD), and vascular dementia (VCID) are being increasingly classified as major public health concerns [1]. In a rapidly aging world in which people over the age of 65 are projected to make up a fifth of the population in just 30 years, the prevalence of these dementias is expected to triple within that same time frame [1–3]. However, there are few, if any, disease-modifying treatments to date, making dementia one of the costliest conditions to society [4].
In response to this public health emergency, many countries have established national plans to address the lack of therapies [5, 6]. In 2011, the USA enacted the National Alzheimer’s Project Act (NAPA) (Public Law 111-375). The Act defines “Alzheimer’s” as Alzheimer’s disease and related dementias (ADRDs), including FTD, DLB, and VCID. The law calls for the development of treatments to prevent or slow the rate of AD progression by the year 2025, as well as a national plan and coordination among international bodies to fight AD on a global scale [7]. As a result, funding for AD research via the National Institute on Aging (NIA) has increased substantially within the last 5 years, jumping from 600 million dollars per year to approximately 2.8 billion. The NIA and its sister institutes and centers at the National Institutes of Health (NIH), including the National Institute of Neurological Disorders and Stroke (NINDS), use milestones and recommendations from the AD Summits held at the NIH Bethesda campus to prioritize areas of research.
The most common pathological hallmark of age-related neurodegenerative diseases is the accumulation of proteinaceous deposits in the intra- and extracellular spaces of selected brain regions [8–13]. These proteins have been shown to accumulate several years prior to clinically observable cognitive, behavioral, and motor symptoms [14, 15]. Whereas there remains some debate as to whether protein accumulation is pathogenic or merely a byproduct of disease [16] (Fig. 1), the majority of therapeutic research has been focused on clearing these aggregates [17–20]. AD, DLB, PD, and FTD are thus often defined as proteinopathies of the aging population that display selective degeneration of neuronal circuitries and progressive accumulation of specific proteins such as amyloid beta (Aβ), tau, α-synuclein (α-syn), TAR DNA-binding protein 43 (TDP43), and repeat-associated non-ATG (RAN) [10, 21–25] among many others (Fig. 1). AD plaque and tangle formation are most frequently associated with Aβ and tau, whereas the primary protein component of Lewy bodies in PD and DLB is α-syn [23–29] (Fig. 2). FTD aggregates are generally comprised of tau, TDP-43, or Fused in sarcoma (FUS) [30, 31], but cases with a GGGGCC expansion mutation in intron 1 of the C9ORF72 gene also present with accumulations of TDP43 and repeat-associated non-AUG-dependent (RAN) translation proteins [32]. It must be pointed out, however, that α-syn and TDP43 aggregates are also commonly found in AD, as well as Aβ, tau, and TDP43 in DLB [33–39] (Fig. 2). Moreover, recent studies have shown significant overlap in AD and PD pathology in which a single individual over the age of 80 can present with aggregates of several of the above proteins [36] (Fig. 2). As such, simultaneously targeting multiple aggregating protein species may be more effective at treating these disorders than monotherapy [36, 37].
Fig. 1.
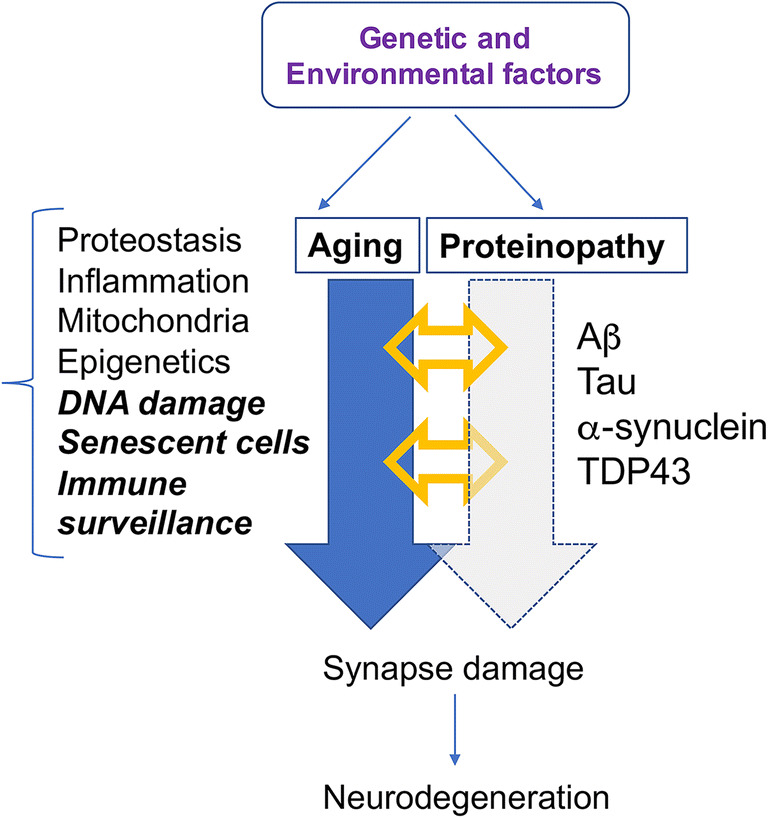
Combined mechanisms of neurodegeneration in AD/ADRD include protein accumulation and aging-related pathways. In the neurodegenerative process of AD/ADRD, synaptic damage and neuroinflammation may lead to neuronal dysfunction which results in dementia and motor deficits. The leading hypothesis is that progressive accumulation of proteins such as amyloid beta (Aβ), tau, α-synuclein (α-syn), TAR DNA-binding protein 43 (TDP43), and repeat-associated non-ATG (RAN) is the primary culprit. However, aging is also likely to play a critical role in pathogenesis, either in synergy with protein accumulation or as an independent pathway. Aging processes relevant to neurodegeneration include inflammation, proteostasis, DNA damage, cell senescence, and mitochondrial alterations
Fig. 2.
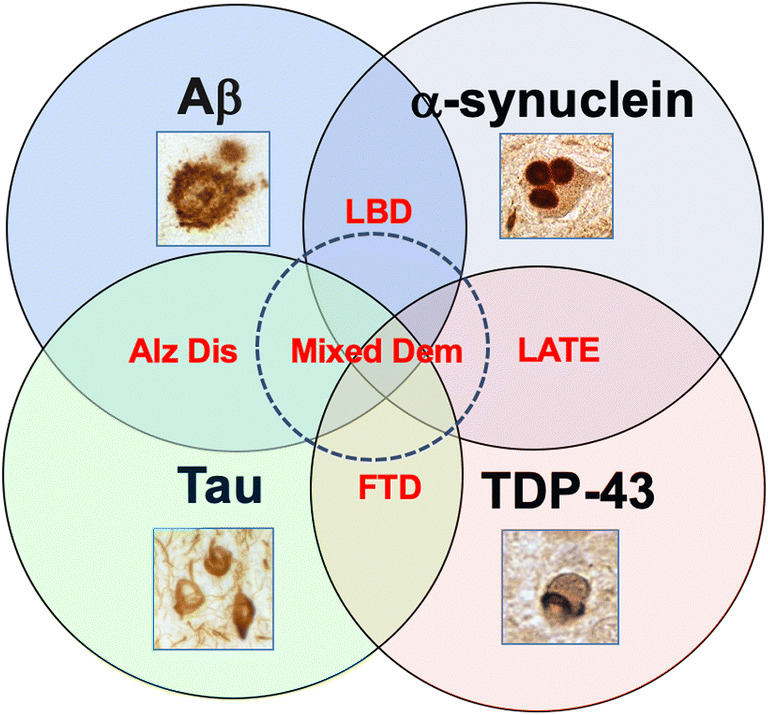
Overlapping protein aggregate pathology in AD/ADRD. Although Aβ and tau are classically associated with AD, α-syn with Lewy body dementia, and TDP43 and tau with FTD and limbic associated TDP43 encephalopathy (LATE), it is becoming increasingly evident that older individuals with dementia (+ 80 years old) display mixed pathology
Abnormal protein accumulation reflects an imbalanced proteostasis network [22, 40, 41]. How these protein aggregates lead to neurodegeneration is unclear but may involve synaptic dysfunction and neuroinflammation triggered by the formation of neurotoxic oligomers and the cell-to-cell propagation of oligomers, protofibrils, and fibrils [21, 22, 40, 42]. Given that age is the greatest risk factor for neurodegenerative disease, age-related alterations in proteostasis, inflammation, stem cell biogenesis, mitochondrial alterations, cell senescence, and DNA damage/repair [43] might also play critical roles in pathogenesis (Fig. 1). Understanding the role of protein homeostasis as it relates to aging could identify new drug targets and delineate reliable markers to accurately determinate a patient’s prognosis and appropriate treatment options [44].
Research on disease-modifying therapies has primarily focused on reducing the accumulation and propagation of protein aggregates by decreasing synthesis and aggregation or enhancing the rate of clearance [22, 45] (Fig. 3), with less emphasis on targeting aging-related processes. These include gene therapy to bolster clearance and degradation (e.g., autophagy, proteolysis, lysosomal degradation) [46], anti-sense technology to block synthesis (e.g., tau and α-syn genes) [47–49], small molecules to decrease aggregation (e.g., inhibitors of Aβ, tau, and α-syn oligomers and higher-order aggregates) [50–53], and immunotherapy to enhance clearance, degradation, and decrease aggregation (Fig. 3). Immunotherapies have garnered particular attention for their specificity [54, 55]. This review will discuss 2 types of immunotherapy: active vaccines, in which inactivated fragments of the pathogenic protein are directly administered to produce a long-lasting immune response, and passive immunization, in which patients are infused with antibodies against the target protein. Both strategies can modulate inflammation, prevent future oligomerization and aggregation activity [56–66], and promote clearance by phagocytic microglia or lysosomal degradation via the endosomal sorting complexes required for transport (ESCRT) pathway [67]. Other immunotherapy modalities, including T-cell modulation or harnessing cellular immunity to target neurodegeneration pathways, are discussed elsewhere [68, 69].
Fig. 3.
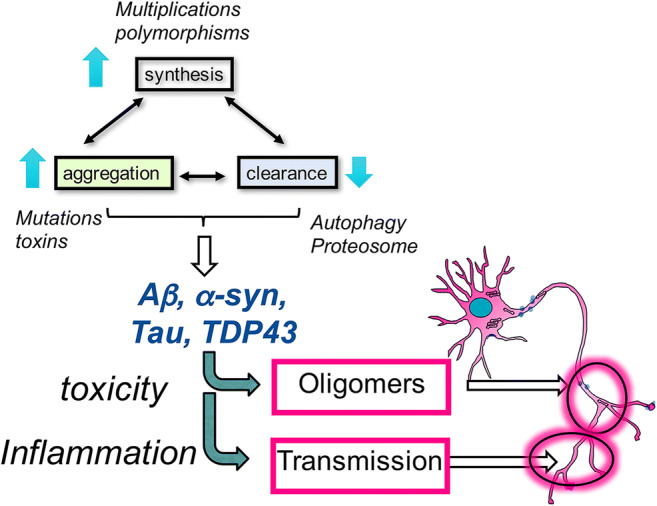
Mechanisms of protein toxicity in AD/ADRD involve oligomerization and propagation of protein aggregates. In neurodegeneration, an imbalance in the synthesis, aggregation, and clearance of proteins results in chronic accumulation, leading to further aggregation and propagation, and eventually inflammation and neurodegeneration
In addition to the various mechanisms above, immunotherapy has many other advantages. Its inherent specificity allows for selective targeting of specific strains and conformations with less off-target effects [61, 70–72] (Fig. 4). Polyvalent single-chain antibodies or combinations of antibodies and vaccines may also allow for the simultaneous targeting of multiple protein aggregate species [60, 73–76] (Fig. 4). Furthermore, immunotherapies can be neuroprotective by neutralizing extracellular protein aggregates and thereby reducing subsequent spread, synaptic damage, and neuroinflammation [77]. The very concept of immunotherapy for AD originated from the observation that the amyloid-β (Aβ) peptide accumulates extracellularly and is therefore accessible to antibodies that can recruit microglia to clear such deposits [78] (Fig. 4). It was accordingly considered unlikely that intracellular protein aggregates such as those of tau, α-syn, TDP43, and RAN would be good targets for immunotherapy. However, the advent of new technology to facilitate intracellular trafficking of antibodies [79–82] and the discovery that these so-called intracellular proteins also exist in cell membranes and extracellular spaces have driven immunotherapy development forward for PD, DLB, and FTD [59, 62, 63, 83, 84].
Fig. 4.

Multiple mechanisms of action of immunotherapy in AD/ADRD. Immunotherapy involves various mechanisms of action to target protein aggregates in the membrane and the intra- and extracellular spaces. Antibodies can block propagation, trigger lysosomal clearance of proteins, reduce inflammation, and promote neuroprotection
In March 2019, 20 years after the original publication by Schenk et al. that propelled the field of immunotherapy for neurodegenerative diseases [78], clinical trials for the Aβ antibody aducanumab were halted early for futility. One of the most promising AD therapies had become yet another failed drug to meet clinical endpoints after reaching phase III. Seven months later, however, the company sponsoring the trial surprised the world by announcing that the results of the futility analysis were premature, and that they would seek US Food and Drug Administration approval by using a larger dataset with longer exposure times to a high dose [85].
The news, while providing renewed hope for AD patients and families, should be received with cautious optimism [86]. Years of clinical trial failures for AD suggest that monotherapy against aggregation-prone proteins may not be enough for clinical efficacy [17]. Identifying an effective treatment for the heterogeneous group of patients affected by neurodegenerative disease may rest on an amalgamation of factors, including developing accurate and early diagnostic techniques, pursuing earlier preventive treatment, simultaneously targeting different proteins, identifying novel targets and age-related pathogenic cascades, and even reducing variability between clinical trial populations such as ApoE4 carrier status [77, 87, 88]. This review will briefly examine the current state of research in immunotherapies against the 3 most commonly targeted proteins for age-related neurodegenerative disease, Aβ, tau, and α-syn. The discussion will then turn to combinatorial strategies and new potential targets for future immunotherapy development.
Immunotherapies Targeting Aβ
Based on the amyloid cascade hypothesis, the transmembrane amyloid precursor protein (APP) can undergo proteolytic cleavage by β-secretase 1 (BACE1) to produce a soluble extracellular fragment and a cell membrane-bound fragment [89–91]. With its catalytic subunit presenilin, γ-secretase further cleaves the cell membrane fragment to release the amyloid-β (Aβ) peptide. Mutations in the genes encoding APP or presenilin carry the greatest incidence of familial AD [92]. Under nonpathological conditions, there is evidence that Aβ is involved in regulating synaptic function and even acting as an antimicrobial peptide to protect against infection and injury [93–95]. The Aβ peptide can be of varying lengths, the most prevalent being Aβ40. Longer forms, such as Aβ42, are less soluble and are prone to accumulate extracellularly to form oligomers, protofibrils, fibrils, and ultimately, plaques [90, 91, 96–98].
In 1999, Schenk et al. published the first immunotherapy for AD: an active vaccine called AN-1792 and comprised of synthetic full-length Aβ42 with QS-21 adjuvant [99]. The vaccine produced long-lasting and nearly complete clearance of Aβ deposits in many patients, but had no impact on the prominent tau pathology and severe dementia [100, 101]. The trial was halted after 4 patients developed meningoencephalitis related to T-cell infiltration. Successful mapping of the B-cell epitope to the N-terminus of Aβ [102–104] and additional work on Th2-biased adjuvants [103–107] were thus essential to the future Aβ vaccine development, allowing for a robust antibody response without the potentially harmful Th1 lymphocyte activation [108]. Vanutide cridificar (ACC-001) was designed as such to include only the B-cell epitope of Aβ plus QS-21 adjuvant [109]. Phase II trials for ACC-001, however, did not reach efficacy endpoints for cognitive evaluations, volumetric brain MRI, and CSF biomarkers, and development was halted in 2013 [110, 111]. Another recent strategy has been to use mimotopes, synthetic peptides that closely resemble the target protein epitope, as active vaccines. AFFITOPE® AD02 by Affiris (Wien, Austria), a 6-amino-acid peptide that mimics the Aβ N-terminus, was regrettably also terminated in phase II for lack of clinical efficacy [112].
There are currently 4 active immunizations being tested in phase II trials. CAD106 from Novartis (Basel, Switzerland) fuses a Qβ virus-like particle to multiple copies of the Aβ N-terminus (a.a. 1-6) [113, 114], and is being tested in homozygous ApoE4 carriers as part of the Alzheimer’s Prevention Initiative (API) program (NCT02565511) [115]. Similarly, ACI-24 by AC Immune (Lausanne, Switzerland) uses Aβ1-15 peptides anchored to the surface of a liposome and is being tested for mild-to-moderate Alzheimer’s disease and Down’s syndrome [116]. Another N-terminus vaccine, UB-311, is comprised of 2 synthetic Aβ1-14-targeting peptides linked to helper T-cell peptide epitopes contained in a Th2-biased formula [117]. After completing phase IIa trials for UB-311, United Neuroscience (Dublin, Ireland) is now assessing long-term safety, tolerability, and immunogenicity in an extension study (NCT03531710). ABVac40 by Araclon Biotech S.L. (Zaragoza, Spain) began phase II trials (NCT03461276) in mild cognitive impairment (MCI) last year and is the only vaccine to use the C-terminal end of Aβ40 [118].
The first passive immunotherapy to reach phase III trials was bapineuzumab, the humanized murine monoclonal antibody 3D6 [119–121] (Table 1). Bapineuzumab targets the Aβ N-terminus to mediate clearance of both soluble and fibrillar forms, but did not meet clinical endpoints in phase III and, moreover, produced amyloid-related imaging abnormalities (ARIA) with edema in patients that received a high dose [122]. Solanezumab, humanized murine antibody m266, is specific for soluble monomeric Aβ and is proposed to operate by the peripheral sink hypothesis, in which removal of Aβ in the periphery also leads to a reduction in the brain by passive diffusion [123]. Multiple phase III trials, including one in early AD patients, either failed to meet clinical efficacy or were terminated early for futility [124, 125] (Table 1). Similarly, phase III trials for crenezumab, human monoclonal IgG4 against oligomeric, fibrillar, and plaque conformations of Aβ, were stopped early in Jan 2019 for futility [126]. However, a phase II prevention trial with crenezumab in Presenilin1 (PSEN1) E280A mutation carriers (NCT01998841) is still underway as part of the API-Autosomal-Dominant Alzheimer’s Disease (ADAD) trial in Medellin, Colombia [127] (Table 1). The study is expected to be completed in February 2022 with the primary outcome being change in API ADAD Composite Cognitive Test Total Score at 260 weeks after baseline. Secondary outcomes include time to MCI or dementia progression because of AD, PET assessment of fibrillar amyloid accumulation, volumetric measurements by MRI, CSF tau biomarker levels, and various measures of memory and neurocognitive function.
Table 1.
Examples of antibodies against Aβ in late clinical development
| Antibody | Phase | Binding | Epitope | Isotype | ARIA-E safety |
|---|---|---|---|---|---|
|
Bapineuzumab (Elan/Pfizer/J&J) |
III |
Monomers ++ Oligomers +++ Fibrils +++ |
N-term aa 1-5 |
IgG1 | 10% |
|
Solanezumab (Eli Lilly) |
III |
Monomers +++ Oligomers 0 Fibrils 0 |
Soluble aa 16-24 |
IgG1 | 0.9% |
|
Crenezumab (Roche/AC Immune) |
III |
Monomers + Oligomers +++ Fibrils ++ |
Conformational aa 12-24 |
IgG4 | 0.3% |
|
Gantenerumab (Roche) |
III |
Monomers 0 Oligomers ++ Fibrils +++ |
aa 3-11 and 19-25 | IgG1 | 10% |
|
Aducanumab (Biogen) |
III |
Monomers 0 Oligomers +++ Fibrils +++ |
Conformational aa 3-6 |
IgG1 | 37-41% |
|
BAN2401 (Eisai/Biogen) |
III |
Monomers 0 Oligomers ++ Fibrils +++ |
Conformational Artic mutation |
IgG1 | < 1% |
Aducanumab was originally slated to take its place among the failed phase III immunotherapy candidates despite being shown to selectively reduce Aβ plaques and slow cognitive decline in earlier trials [128, 129]. Although phase III was halted earlier this year, the company that developed this antibody is now seeking FDA approval with new analysis of a larger data set [85, 130] (Table 1). Similarly, although development of human IgG1 gantenerumab was halted in 2014 for futility, a new phase III trial was initiated at a higher dose (NCT03444870) after the open label extension demonstrated a dramatic decline in Aβ deposition in participants [131]. Both gantenerumab and solanezumab are also being revived as potential preventative therapies by the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) in patients with AD-causing mutations (NCT01760005) [132]. Unfortunately, topline analyses in February 2020 showed that both drugs failed to meet primary endpoints [133]. Preventative trials for sporadic AD are also underway, in which the so-called A4 phase III trial (NCT02008357) tests solanezumab at higher doses and for longer durations in asymptomatic patients with preclinical AD as defined by positive amyloid PET scans [134]. This phase III trial recruited over 1200 participants and will be reporting in the next couple of years. Humanized murine mAb158 BAN2401 (NCT03887455) is the only other passive immunotherapy currently in phase III, but has already produced very promising results in phase II. Specifically, the 18-month study demonstrated a statistically significant slowing in cognitive decline at the highest dose with less than 10% of subjects experiencing ARIA [135]. The antibody is specific to soluble Aβ protofibrils, which have been proposed to be the toxic species rather than the plaques themselves [136] (Table 1).
In conclusion, despite previous failures in phase III immunotherapy trials targeting Aβ, interest has resurged following the recent glimmer of promising results coupled to advances in the early diagnosis and biomarker accessibility of AD. The antibodies currently being tested in clinical trials target a variety of Aβ species, including soluble, oligomeric, and fibrillar Aβ. Mechanisms of action include microglia-facilitated removal of extracellular amyloid oligomers and fibrils, blocking primary and secondary Aβ nucleation, and targeting monomers and soluble Aβ in the periphery that could otherwise trigger Aβ accumulation in the CNS. Rather than for late-stage disease reversal, immunotherapy against Aβ aggregates appears to be more appropriate for prevention initiatives with prolonged treatments past 78 weeks at high doses guided by reliable biomarkers. ApoE4 carriers exposed to high doses, however, are susceptible to complications such as ARIA-E and may need to be closely monitored by MRI [137].
Immunotherapies Targeting α-Synuclein
Diseases characterized by progressive α-syn accumulation in neuronal and non-neuronal cells of cortical and subcortical regions are collectively termed synucleinopathies, and include PD, DLB, multiple system atrophy (MSA), and a subset of AD [22, 26, 138, 139]. DLB/PD is the second most common cause of dementia and parkinsonism in the aging population after AD, affecting about 1 million in the USA [140]. α-syn is a presynaptic protein reported to be involved in endosomal formation and vesicle release at the synapse [141, 142]. The pathological hallmarks of PD and DLB are intraneuronal occlusions called Lewy bodies and Lewy neurites that are composed of fibrillar α-syn along with cytoskeletal and other synaptic proteins [143]. α-syn accumulation can also occur in glia, such as glial cytoplasmic inclusion (GCIs) in MSA patient oligodendrocytes or Lewy body-like structures in the amygdala, hippocampus, and neocortex of AD and Down syndrome patients [143–145]. Although astrocytes and microglia do not constitutively express α-syn, they actively uptake extracellular α-syn which can lead to glial aggregates in conditions of impaired clearance [146–148]. This is supported by increasing evidence that small amounts of α-syn aggregates are released from neurons into the extracellular space and subsequently interact with glial receptors such as Toll-like receptor 2 to trigger a pro-inflammatory response [40, 146, 149–152].
The original approach to use antibodies to target and remove amyloid plaques from the brain made intuitive sense given that Aβ aggregates are found in the extracellular space [99]. It was unclear how this method might be applied to DLB, PD, and other synucleinopathies, as the majority of the proteinaceous aggregates were thought to form within neurons of the striatonigral system, limbic areas, and deep layers of the neocortex. In the early 2000s, however, we showed that immunization of a newly developed α-syn transgenic model (Line D, PDGF-α-syn wt) with recombinant human α-syn mixed with Freund adjuvant resulted in the production of high titer antibodies against C-terminal α-syn that were capable of removing α-syn aggregates in neurons and ameliorating neurodegeneration and functional deficits [153]. Through additional active and passive immunization experiments in transgenic, viral, and preformed fibril injection models [154, 155], we learned that these antibodies had multiple mechanisms of action, such as recognizing α-syn aggregates in the membrane and triggering endocytosis and clearance via autophagy, lysosomal activity, or proteasomal degradation [156–159] (Fig. 4). Moreover, antibodies can be trafficked intracellularly with single-chain antibodies or intrabodies specifically engineered to penetrate the cell membrane via apolipoprotein B (ApoB), TAT fusion proteins, or uptake by endogenous receptors [160–165] (Fig. 4). Single-chain variable fragments (scFvs), for example, are designed to retain the specificity of the original full-length antibody without activating unwanted Fc-mediated responses [160, 161, 166]. Several studies have shown that transgenic mice injected with viral vectors encoding scFvs against Aβ [167–169], α-syn [81, 170], or tau [171, 172] show long-term in vivo scFv expression, improved functional deficits, and reduced pathogenic protein accumulation. Intrabody technology is continuing to be improved, such as in a recent study in which Chatterjee et al. (2018) enhanced the solubility of single-domain immunoglobulin fragments by engineering a polypeptide tether construct, and demonstrated its protective effects on motor function when delivered by gene therapy to a PD rodent model [173].
These intrabodies and cell-penetrating single-chain antibodies can block aggregation and target α-syn for degradation in the lysosomes using the ESCRT system [73, 81, 174, 175]. Antibodies can also block α-syn oligomerization and fibrillation, target specific strains and isoforms, prevent cell-to-cell transmission, and facilitate clearance via microglia [61, 149, 155, 176]. Therefore, antibodies can target both intracellular and extracellular α-syn aggregates as they spread from cell to cell (Fig. 4). This approach has been since applied to other proteinopathies with predominantly intracellular accumulations of tau, TDP43 [177, 178], SOD1 [179–181], RAN peptides [32], and Huntingtin (Htt) [182, 183]. Additional lessons from α-syn immunotherapy studies include the use of antibodies to develop blood, CSF, and tissue biomarkers to monitor the effects of immunotherapy and the ability of C-terminus-specific antibodies to block protease-mediated C-terminus truncation of α-syn and subsequently prevent oligomerization and transmission [59, 153, 156–158] (Fig. 4).
In this context, 2 main strategies for α-syn immunotherapy have been pursued: mimotope vaccines and antibodies against the N- or C-terminal ends of α-syn or specific conformations of oligomeric and fibrillar α-syn (Fig. 4). Whereas most antibodies have been developed with recombinant or synthetic α-syn monomers or aggregates, a recent and novel variant uses antibodies cloned from human healthy volunteers producing high titers of auto-antibodies against α-syn [184–186]. As a result of seminal cell-free in vitro and in vivo studies, several immunotherapies are currently been tested in clinical trial for synucleinopathies. The vaccines AFFITOPE® PD01A and PD03A were well tolerated in phase I and produced a dose-dependent immune response in patients with early MSA, but plans for phase II have yet to be disclosed [187, 188]. For passive immunotherapies, prasinezumab (also known as PRX002 or RO7046015) is a humanized monoclonal antibody against the C-terminus of α-syn undergoing phase II trials in patients with early PD (NCT03100149) [189] (Table 2). A phase I trial for BIIB054, a human monoclonal antibody that preferentially binds to aggregated α-syn, was recently concluded and showed favorable safety, tolerability, and pharmacokinetic profiles [190] (Table 2). MEDI1341 is also in phase I clinical trial testing (NCT03272165) as an antibody that can bind both monomers and aggregates. In preclinical studies, MEDI1341 was shown to readily cross the blood–brain barrier and block transmission of α-syn aggregates in a combined viral vector model [191] (Table 2).
Table 2.
Examples of antibodies against α-synuclein in clinical development
| Antibody | Phase | Binding | Epitope | Isotype | Safety |
|---|---|---|---|---|---|
|
PRX002/RG7935 (Prothena/Roche) |
I-II |
Monomers ++ Oligomers +++ Fibrils + |
C-term aa 118-126 |
IgG1 | Well tolerated |
|
BIIB-054 (Biogen) |
I-II |
Monomers −/+ Oligomers +++ Fibrils +++ |
N-term aa 1-10 |
IgG1 | Well tolerated |
|
MEDI1341 (MedImmune/Astra Zeneca/Takeda) |
Preclinical |
Monomers + Oligomers +++ Fibrils ++ |
C-term aa 103-129 |
IgG1 | In progress |
|
ABBV-0805/BAN0805 (BioArtic/AbbVie) |
I |
Monomers 0 Oligomers ++ Fibrils +++ |
aa ? | IgG? | In progress |
|
Lu AF82422 (Lundbeck) |
I |
Monomers + Oligomers ++ Fibrils ++ |
C-term aa 112-117? |
IgG1 | In progress |
In summary, antibodies against α-syn ameliorate Lewy body pathology by multiple mechanisms such as promoting the clearance of intracellular α-syn and blocking the propagation of extracellular α-syn. Other rising immunotherapy strategies are T-cell modulation, such as that of Copaxone® [192], and combinatorial approaches. We have tested several of such combinations, including simultaneous administration of 2 AFFITOPEs® against Aβ and α-syn [74, 193], nanoparticles containing both recombinant human α-syn and the immunomodulatory drug rapamycin, and the anti-inflammatory drug thalidomide given alongside a single-chain antibody against oligomeric α-syn derived from human DLB/PD brains and conjugated to ApoB [73]. Through these studies in DLB/PD mouse models, we show that combined immunotherapy may be more effective than monotherapy. This topic will be discussed in more detail in the following sections. The main challenge in the field of synucleinopathies is the lack of reliable and accessible biomarkers and the overlapping pathology among neurodegenerative diseases.
Immunotherapies Targeting Tau
Tau is a major member of the Microtubule Associated Proteins (MAP) family and abundantly expressed in neurons [194]. By binding to tubulin dimers, tau can stabilize microtubule formation and modulate cytoskeletal dynamics [195]. The degree to which tau is phosphorylated is an important regulator of microtubule stability [196]. Nonphosphorylated forms preferentially bind to microtubules, whereas hyperphosphorylation is associated with neurofibrillary tangle (NFT) formation from paired helical filaments (PHF) [197, 198]. Intracellular NFTs are a major hallmark lesion of AD and other neurodegenerative tauopathies such as FTD, cortico-basal degeneration, Pick’s disease, and progressive supranuclear palsy (PSP) [199]. The severity of tau pathology has been shown to correlate with the degree of cognitive impairment and neuronal loss [200–203], making the tau protein an attractive target for new immunotherapies.
Axon Neuroscience (Staré Mesto, Slovakia) recently completed a 24-month phase II trial for active tau vaccine AADvac1 (NCT02579252), announcing in September 2019 that the vaccine was safe and well-tolerated, and generated antibodies against pathological tau in over 98% of patients [204]. AADvac1 consists of a B-cell epitope from a cysteinated 12-mer tau peptide conjugated to keyhole limpet hemocyanin, a carrier protein that stimulates a Th2 immune response [205]. Based on the promising trends in cognitive improvement and decelerated accumulation of AD biomarkers in trial participants, Axon Neuroscience plans to move forward with the next phase of clinical trials and is also testing the vaccine for primary progressive nonfluent aphasia (NCT03174886) [206, 207]. In another active vaccine, ACI-35, 16 copies of synthetic tau fragments phosphorylated at Ser396 and Ser404 are embedded to the surface of a liposome [116, 208]. In August 2019, AC Immune announced a phase Ib/IIa trial in collaboration with Janssen Pharmaceuticals (Beerse, Belgium) to assess the safety, tolerability, and immunogenicity of the second generation of this vaccine, AC-35.030 [209].
AC Immune is also conducting phase 2 studies on the anti-tau passive immunotherapy Semorinemab (also called RO7105705, MTAU9937A, and RG6100) for prodromal/mild AD (NCT03289143) and moderate AD (NCT03828747) in partnership with Roche/Genentech (South San Francisco, CA). Other ongoing trials for passive anti-tau immunotherapies include that of Zagotenemab (also called LY3303560) in AD patients (NCT03518073) and ABBV-8E12 (also called Tilavonemab or C2N-8E12) for early AD (NCT02880956). Zagotenemab is the humanized mouse monoclonal antibody MC-1 and targets soluble tau aggregates [210]. Similarly, ABBV-8E12 is the humanized version of mouse monoclonal antibody HJ8.5 that was shown to reduce tau seeding, hyperphosphorylation, and cognitive deficits in P301S transgenic mice [211, 212]. Although phase II trials for ABBV-8E12 in PSP patients were halted in July 2019, favorable safety and tolerability profiles were obtained [213]. Gosuranemab (BIIB092), the humanized antibody against extracellular N-terminal tau from Biogen (Cambridge, MA), was also undergoing phase II trials for both PSP (PASSPORT) and AD (TANGO) [214, 215]. However, Biogen recently announced that topline results from the PASSPORT study failed to meet primary endpoints and that it would no longer pursue development of gosuranemab for PSP and other primary tauopathies [216].
Interestingly, current preclinical studies seem to be stepping away from targeting N-terminal tau in favor of other epitopes. DC8E8 has been recently characterized as a promising antibody that targets tau at 4 homologous epitopes present in each microtubule binding domain repeat [217]. DC8E8 was shown to not only preferentially bind truncated pathological tau over physiological tau but also prevent both the formation of beta sheets and the uptake of tau into neurons via sulfated heparan proteoglycans (HSPGs) [218]. In another study, Courade et al. (2018) identified an antibody against the central region of tau, dubbed “antibody D,” that was effective at blocking tau seeding in vitro [219]. Albert et al. (2019) further observed that this central tau epitope antibody was more efficacious at preventing AD-like pathology and cell-to-cell transfer of tau in mice [84].
In summary, considerable efforts have been made for over 20 years in the development of immunotherapies for neurodegenerative disorders. Antibodies targeting Aβ have led the way with a number of phase III trials and at least one that reported meeting primary outcome measures, followed closely behind by a handful of immunotherapies for α-syn and tau in phase I and II trials. Whereas accessible biomarkers are currently available for Aβ- and tau-related pathologies to guide the immunotherapy trials, there is a pressing lack of such measures for synucleinopathies. The other challenge at hand is that older patients present with protein aggregates that are no longer primarily one pathologic protein species but comprised of Aβ, tau, α-syn, TDP43, and others, in addition to aging-related processes such as inflammation, proteotasis deficits, DNA damage, mitochondrial alterations, and stem cell alterations. Thus, there is a dire need to develop reliable biomarkers and powerful combinatorial therapies to address these polyproteinopathies and age-related pathophysiological alterations, as will be discussed in the following section.
Combinatorial Immunotherapies
Combination therapy is already a standard of treatment for many cancers [220] and chronic diseases including hypertension, CHF, epilepsy, and HIV [221]. Combinations of currently available treatments are already being shown to improve cognition and behavior in AD patients, such as coadministration of cholinesterase inhibitors and memantine [222, 223] and enhancing memantine efficacy with beta-asarone and tenuigenin [224]. Although a few AD mouse studies have tested combinations of lipid mediators [225] or naturally occurring dietary compounds [226], combinatorial therapeutics in neurodegenerative disease remains a largely underexplored field (Fig. 5). Monotherapy alone may be insufficient against the complex and overlapping pathophysiology of age-related neurodegenerative diseases. As such, there is a growing need to simultaneously target multiple aggregating proteins as well as modulate aging-related mechanisms that synergize with protein aggregation to trigger neurodegeneration (Fig. 5).
Fig. 5.

Combination immunotherapies for AD/ADRD targeting protein aggregates and aging-related pathways. Immunotherapy can also be used to target other pathogens and age-related pathogenic processes associated with aging. One such pathogen may be the herpes simplex virus (#1). Aging-related pathways that can be targeted with antibodies include inflammation (e.g., TNF, TLR2, inflammasome) (#2), senescent cells (e.g., DPP4) (#3), and immune surveillance cells (e.g., NK cells) (#4). Immunotherapies in neurodegeneration have been traditionally developed to target protein aggregates (e.g., Aβ, tau, α-syn, TDP43) (#5) alone or in combination (#6). Combination therapies may involve targeting multiple protein aggregates (#6) or protein aggregates (#5) and age-related pathways (#1–4)
Overlapping pathology has been well-documented in age-related neurodegenerative diseases, such as the presence of Lewy body-like pathology in AD [227–232]. One study reported that 30 to 40% of patients with PD copresent with Aβ plaques and NFTs [233, 234], whereas over 70% of DLB patients [235] and more than 50% of AD patients [236] may have overlapping and, as a result, more aggressive pathology [237]. Indeed, a growing amount of evidence suggests that this copathology directly impacts disease progression. For example, dementia in PD is associated with high levels of AD copathology [235, 238–241]. Several groups have also shown that α-syn can contribute to the formation of toxic Aβ and tau species, as well as vice versa [242]. Moreover, as stated earlier, demented individuals over the age of 80 present with multiple pathologies including Aβ, tau, α-syn, TDP43, inflammatory, and vascular alterations [36]. As such, one therapeutic strategy may be to target the clearance of more than one pathologic protein via immunotherapy.
For example, immunotherapies that generate a response against both α-syn and Aβ may be more effective than either alone given that α-syn and Aβ may directly interact in AD patients and APP transgenic models [243–246]. AD patients have been shown to display elevated levels of α-syn in the CSF [247, 248], and further, Aβ may promote α-syn aggregation and toxicity [249, 250]. In mouse models, hippocampal neurons with α-syn accumulations were found to be more susceptible to Aβ-mediated toxicity [251], whereas genetic depletion of α-syn prevented the degeneration of cholinergic neurons and attenuated behavioral deficits [39, 252]. Similarly, α-syn has been shown to directly interact with PSEN1, which is important for the proteolytic processing of the APP to yield Aβ [231]. α-syn infusion in APP Tg mice also blocked Aβ seeding but enhanced synaptic degeneration [253, 254], potentially by blocking SNARE-vesicle fusion in a cooperative manner [255]. We recently covaccinated a double transgenic mouse model of DLB (mThy1-hAPP- and mThy1-α-syn) with AFFITOPE peptides against Aβ (AD02) and α-syn (PD1A) [74]. Remarkably, targeting one protein often concomitantly lowered the accumulation of another, in which AD02 effectively reduced Aβ pathology as well as that of phosphorylated tau and α-syn. This study also noted potential additive effects, particularly in alleviating behavioral deficits, suggesting that a combined immunotherapy approach may be appropriate for the heterogenous pathology of AD and DLB and other age-related neurodegenerative diseases (Fig. 5).
Tau and α-syn were also found to colocalize in the same cellular compartments, particularly in axons [256]. Although both have been shown to form intracellular aggregations, unlike Aβ, α-syn can self-aggregate whereas tau requires an aggregation-inducing agent [257]. Many studies have demonstrated that α-syn interacts with and catalyzes the oligomerization of tau [258–262]. α-syn may also demonstrate 14-3-3 cochaperone activity on 14-3-3 targets, including tau, promoting mutual misfolding [263–265]. α-syn preformed fibrils were shown to induce intracellular tau aggregation in vivo [266], and may be closely involved in the GSK3β-mediated phosphorylation of tau in a reciprocal manner [267–270]. α-syn oligomers may induce a distinctly toxic tau oligomeric strain in a cross-seeding effect [271, 272] or even form coaggregates [273]. A recent paper also reported that an age-dependent reduction in glucocerebrosidase (GCase) activity was associated with accumulations of lipid-stabilized α-syn and phosphorylated tau [274]. As such, a combination of immunotherapies against both tau and α-syn may be especially effective for cases in which both tau and synuclein pathology are present, as it can not only remove existing aggregates and toxic species but also prevent future cross-seeding events.
The ultimate approach would be to develop immunotherapies that simultaneously target Aβ, tau, α-syn, TDP43, and inflammation (Fig. 5). This could be achieved by 1) combining vaccines against Aβ, tau, α-syn, and TDP43 among other proteins; 2) combining antibodies against Aβ, tau, α-syn, and TDP43; 3) combining passive and active immunization; or 4) using multivalent single-chain antibodies that can target 2 or more of these proteins simultaneously. Another approach would be to design antibodies to recognize a conformation that is similar across multiple protein aggregate species, rather than a specific sequence. Interestingly, studies in APP and α-syn transgenic models have demonstrated that some antibodies against oligomeric tau are effective at reducing α-syn, whereas others have suggested that antibodies against Aβ may also target tau [61, 275]. Moreover, a combined vaccination approach targeting Aβ and tau has been shown to decrease disease pathology in Tau22/5xFAD double transgenic mice [276]. As such, a combination of these polyfunctional antibodies is another way of maximizing the effectiveness of immunotherapy (Fig. 5).
Given the aforementioned copathology, immunotherapy may also be effective in combination with gene silencing approaches. Several studies have shown that α-syn and tau deletion in animal transgenic models can delay the onset of disease. For example, α-syn knockout prevented neurotoxin-induced neurodegeneration in MPTP and rotenone PD mouse models [277, 278]. Injecting modified anti-sense oligonucleotides (ASO) targeting SNCA similarly promoted survival of TH-positive neurons and ameliorated motor deficits in mice expressing wild-type or mutant human SNCA [49]. Another recent study found that tau deletion in A53T α-syn tg mice rescued some cognitive and synaptic deficits without affecting α-syn expression or phosphorylation [279]. Gene therapy is also progressing in human neurodegenerative diseases, most notably nusinersen for the neuromuscular disorder spinal muscular atrophy [280, 281]. As such, immunotherapy to clear existing aggregates and anti-sense therapy to prevent further translation of pathologic proteins may be a viable combination, particularly in early stages of the disease.
As pointed out at the beginning of this section, effective combination immunotherapy should target not only the protein aggregates but also age-related pathological processes contributing to neurodegeneration, such as inflammation and cell senescence. In this regard, although this review was focused on antibody-based immunotherapy, targeting cellular immunity is another attractive approach to treat neurodegenerative disorders given its ability to reduce the protein aggregate load by targeting T cells. We developed a mixed cellular and active immunization in which α-syn and rapamycin are simultaneously delivered in an antigen-presenting cell-targeting glucan microparticle (GP) vaccine system [193]. In this case, the α-syn peptide elicits the production of antibodies against α-syn, whereas rapamycin triggered the recruitment of Tregs into the CNS. In turn, the Tregs immunomodulate microglia and induce greater microglial clearance of α-syn aggregates and reduced neurodegeneration and inflammation in α-syn tg mice. This vaccine, collectively termed GP+RAP/α-syn, was capable of triggering neuroprotective Treg responses in synucleinopathy animal models, and the combined vaccine was more effective than the humoral or cellular immunization alone. These results demonstrate the promise of multifunctional vaccine approaches for the treatment of AD and DLB/PD.
Another interesting and novel approach would be to trigger immune surveillance by NK cells to target senescence cells for elimination [282]. Moreover, senescent and immune cells can be targeted with specific antibodies. For example, a recent study used antibodies against the surface molecule DPP4 (dipeptidyl peptidase 4) of senescent cells [283] to facilitate their clearance (Fig. 5). Antibodies against interleukins, tumor necrosis factor (TNF), and Toll-like receptors may also be effective at modulating the immune response in neurodegenerative disease [284–287]. Previous studies have shown that Aβ, tau, and α-syn toxicity are mediated by the inflammasome [288–290]. In the case of synucleinopathies, we have shown that extracellular α-syn aggregates bind to TLR2 to trigger neuroinflammation and neurodegeneration and that selective neutralizing antibodies against TLR2 were effective at blocking these effects and behavior deficits in α-syn tg animals [152, 291]. In fact, TLR2 has been developed as an important novel target for synucleinopathies [57, 292] (Fig. 5).
Pathogens such as viruses or bacteria have been proposed to contribute to progressive aggregate formation and chronic inflammation given the antimicrobial properties of Aβ and age-related impaired clearance mechanisms [293] (Fig. 5). As such, directly targeting these pathogens with immunotherapy may effectively attenuate neuroinflammation, particularly in combination with immunotherapies against aggregating protein. Infection with herpes simplex virus 1 (HSV-1) in AD patients has long received attention for its association with AD pathology and decreased cognitive function [294–297], although increasing work is being performed on other viruses such as herpes zoster, Epstein–Barr virus, and human cytomegalovirus [298–301]. Bacteria of particular interest include Chlamydia pneumoniae, Helicobacter pylori, and Porphyromonas gingivalis [302–307]. For a comprehensive review of the evidence pertaining to pathogenic agents in age-related neurodegenerative disease, please see Panza et al. [293].
In summary, we have described the need and potential directions for combinatorial immunotherapies that include active, passive, and cellular approaches against specific protein aggregates as well as age-related neurodegenerative pathways such as inflammation and cell senescence (Fig. 5). These immunotherapy approaches may certainly also be amenable for combination with gene therapy (e.g., anti-sense), small molecules (e.g., autophagy modulators, antiaggregation, senolytics), and nonpharmacological (e.g., exercise, diet, training) treatments. Combinatorial therapeutics will also open exciting opportunities for personalized medicine, including catering to different stages of disease. For example, immunotherapy with anti-sense as described above may be well-suited to presymptomatic stages of disease. Another early intervention may be to combine immunotherapy with means of upregulating proteostasis components such as chemical chaperones or gene therapy for BiP or XBP1 to prevent further misfolding and aggregation [308–311]. Following the onset of significant pathological changes or clinical symptoms, however, polyvalent immunotherapy may be needed to address the frequent presence of multiple pathogenic proteins. Combining polyvalent antibodies with stress signaling inhibitors or regenerative therapy may prevent further synaptic loss and delay progression of symptoms [312–314].
Furthermore, different combinations may be used to cater to not only the stage of disease but also the patient’s specific symptoms, lifestyle risk factors, and genetic risk factors. For instance, given alone, the therapeutic efficacy of statins or antihypertensives for AD [315–321] and PD [322–328] patients remains largely inconclusive. However, a patient presenting with overlapping pathology from both AD and VCID may benefit from a combination of protein-targeting immunotherapy and such cardiovascular disease treatments. Current preventative immunotherapy trials such as that of the API are also just the beginning of genetics-based treatments. Many key initiatives for genetics research in neurodegenerative disease, such as the International Parkinson Disease Genomics Consortium (IPDGC), are spearheading a movement for accessible analytics tools and diverse and representative data. In the future, we may be able to use this groundwork to identify a patient’s specific risk variants and ultimately design a combinatorial therapy that can address both the protein accumulation and the biological associations for those variants. Let it also not be forgotten, however, that in addition to further development of these therapeutic strategies, there is a dire need for an overhaul of current policy and clinical trial practices in order to truly pursue combinatorial and personalized medicine for diseases as complex as age-related neurodegeneration.
Electronic supplementary material
(PDF 306 kb)
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ageing and health [Fact sheet on the Internet]. Geneva: World Health Organization. Available from: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health. Accessed 10 Nov 2019.
- 2.Alzheimer’s Association 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019;15(3):321–387. [Google Scholar]
- 3.Dementia [Fact sheet on the Internet]. Geneva: World Health Organization. Available from: https://www.who.int/news-room/fact-sheets/detail/dementia. Accessed 10 Nov 2019.
- 4.Hurd MD, Martorell P, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;369(5):489–90. doi: 10.1056/NEJMc1305541. [DOI] [PubMed] [Google Scholar]
- 5.Office of the Assistant Secretary for Planning and Evaluation (ASPE). National Plan to Address Alzheimer’s Disease. Washington, DC: U.S. Department of Health and Human Services. Available from: https://aspe.hhs.gov/report/national-plan-address-alzheimers-disease-2016-update. Accessed 31 Aug 2016.
- 6.National dementia plans [Internet]. London: Alzheimer’s Disease International. Available from: https://www.alz.co.uk/dementia-plans. Accessed 01 Nov 2017
- 7.Office of the Assistant Secretary for Planning and Evaluation (ASPE). National Alzheimer’s Project Act. Washington, DC: U.S. Department of Health and Human Services. Available from: https://aspe.hhs.gov/national-alzheimers-project-act. Accessed 10 Oct 2019.
- 8.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 9.Chung CG, Lee H, Lee SB. Mechanisms of protein toxicity in neurodegenerative diseases. Cell Mol Life Sci. 2018;75(17):3159–80. doi: 10.1007/s00018-018-2854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trojanowski JQ, Lee VM. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann N Y Acad Sci. 2000;924:62–67. doi: 10.1111/j.1749-6632.2000.tb05561.x. [DOI] [PubMed] [Google Scholar]
- 11.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 12.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsh AP. Molecular mechanisms of proteinopathies across neurodegenerative disease: a review. Neurol Res Pract. 2019;1:35. 10.1186/s42466-019-0039-8. [DOI] [PMC free article] [PubMed]
- 14.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 15.Beason-Held LL, Goh JO, An Y, Kraut MA, O'Brien RJ, Ferrucci L, et al. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. 2013;33(46):18008–18014. doi: 10.1523/JNEUROSCI.1402-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92(7):329–337. doi: 10.1212/WNL.0000000000006926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73–88. doi: 10.1038/s41582-018-0116-6. [DOI] [PubMed] [Google Scholar]
- 18.Villemagne VL, Dore V, Burnham SC, Masters CL, Rowe CC. Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol. 2018;14(4):225–236. doi: 10.1038/nrneurol.2018.9. [DOI] [PubMed] [Google Scholar]
- 19.Salardini A. An Overview of Primary Dementias as Clinicopathological Entities. Semin Neurol. 2019;39(2):153–166. doi: 10.1055/s-0039-1683445. [DOI] [PubMed] [Google Scholar]
- 20.Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem Pharmacol. 2014;88(4):508–516. doi: 10.1016/j.bcp.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chornenkyy Y, Fardo DW, Nelson PT. Tau and TDP-43 proteinopathies: kindred pathologic cascades and genetic pleiotropy. Lab Invest. 2019;99(7):993–1007. doi: 10.1038/s41374-019-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pievani M, Filippini N, van den Heuvel MP, Cappa SF, Frisoni GB. Brain connectivity in neurodegenerative diseases--from phenotype to proteinopathy. Nat Rev Neurol. 2014;10(11):620–633. doi: 10.1038/nrneurol.2014.178. [DOI] [PubMed] [Google Scholar]
- 26.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 27.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152(2):367–372. [PMC free article] [PubMed] [Google Scholar]
- 28.Dugger BN, Dickson DW. Pathology of Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035. 10.1101/cshperspect.a028035. [DOI] [PMC free article] [PubMed]
- 29.Hansen LA, Masliah E, Terry RD, Mirra SS. A neuropathological subset of Alzheimer’s disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol. 1989;78(2):194–201. doi: 10.1007/BF00688209. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(2):a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011;8(3):273–294. doi: 10.2174/156720511795563700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110(51):E4968–77. [DOI] [PMC free article] [PubMed]
- 33.Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016;131(4):571–585. doi: 10.1007/s00401-016-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanko V, Apple AC, Alpert KI, Warren KN, Schneider JA, Arfanakis K, et al. In vivo hippocampal subfield shape related to TDP-43, amyloid beta, and tau pathologies. Neurobiol Aging. 2019;74:171–181. doi: 10.1016/j.neurobiolaging.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahoo A, Bejanin A, Murray ME, Tosakulwong N, Weigand SD, Serie AM, et al. TDP-43 and Alzheimer’s Disease Pathologic Subtype in Non-Amnestic Alzheimer’s Disease Dementia. J Alzheimers Dis. 2018;64(4):1227–1233. doi: 10.3233/JAD-180169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141(7):2181–2193. doi: 10.1093/brain/awy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83(1):74–83. doi: 10.1002/ana.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, et al. Hippocampal alpha-Synuclein in Dementia with Lewy Bodies Contributes to Memory Impairment and Is Consistent with Spread of Pathology. J Neurosci. 2017;37(7):1675–1684. doi: 10.1523/JNEUROSCI.3047-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spencer B, Desplats PA, Overk CR, Valera-Martin E, Rissman RA, Wu C, et al. Reducing Endogenous alpha-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer's Disease Transgenic Mouse Model. J Neurosci. 2016;36(30):7971–7984. doi: 10.1523/JNEUROSCI.0775-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6(12):702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK. How is alpha-synuclein cleared from the cell? J Neurochem. 2019;150(5):577–590. doi: 10.1111/jnc.14704. [DOI] [PubMed] [Google Scholar]
- 42.Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16(2):109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrucci L, Gonzalez-Freire M, Fabbri E, Simonsick E, Tanaka T, Moore Z, et al. Measuring biological aging in humans: A quest. Aging Cell. 2019;19(2):e13080. 10.1111/acel.13080. [DOI] [PMC free article] [PubMed]
- 44.Hodgson R, Kennedy BK, Masliah E, Scearce-Levie K, Tate B, Venkateswaran A, et al. Aging: therapeutics for a healthy future. Neurosci Biobehav Rev. 2019;108:453–458. doi: 10.1016/j.neubiorev.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9(5):387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 46.Sudhakar V, Richardson RM. Gene Therapy for Neurodegenerative Diseases. Neurotherapeutics. 2019;16(1):166–175. doi: 10.1007/s13311-018-00694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481. doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed]
- 48.DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33(31):12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uehara T, Choong CJ, Nakamori M, Hayakawa H, Nishiyama K, Kasahara Y, et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting alpha-synuclein as a novel therapy for Parkinson’s disease. Sci Rep. 2019;9(1):7567. doi: 10.1038/s41598-019-43772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price DL, Koike MA, Khan A, Wrasidlo W, Rockenstein E, Masliah E, et al. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson's disease. Sci Rep. 2018;8(1):16165. doi: 10.1038/s41598-018-34490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tatenhorst L, Eckermann K, Dambeck V, Fonseca-Ornelas L, Walle H, Lopes da Fonseca T, et al. Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta Neuropathol Commun. 2016;4:39. doi: 10.1186/s40478-016-0310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wischik CM, Staff RT. Wischik DJ, Bentham P, Murray AD, Storey JM, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 53.Gregori M, Taylor M, Salvati E, Re F, Mancini S, Balducci C, et al. Retro-inverso peptide inhibitor nanoparticles as potent inhibitors of aggregation of the Alzheimer’s Abeta peptide. Nanomedicine. 2017;13(2):723–732. doi: 10.1016/j.nano.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Liu YH, Giunta B, Zhou HD, Tan J, Wang YJ. Immunotherapy for Alzheimer disease: the challenge of adverse effects. Nat Rev Neurol. 2012;8(8):465–469. doi: 10.1038/nrneurol.2012.118. [DOI] [PubMed] [Google Scholar]
- 55.Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272–293. doi: 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin J, Kim HJ, Jeon B. Immunotherapy Targeting Neurodegenerative Proteinopathies: alpha-Synucleinopathies and Tauopathies. J Mov Disord. 2020;13(1):11–9. [DOI] [PMC free article] [PubMed]
- 57.Kwon S, Iba M, Masliah E, Kim C. Targeting Microglial and Neuronal Toll-like Receptor 2 in Synucleinopathies. Exp Neurobiol. 2019;28(5):547–553. doi: 10.5607/en.2019.28.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katsinelos T, Tuck BJ, Mukadam AS, McEwan WA. The Role of Antibodies and Their Receptors in Protection Against Ordered Protein Assembly in Neurodegeneration. Front Immunol. 2019;10:1139. doi: 10.3389/fimmu.2019.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Valera E, Spencer B, Masliah E. Immunotherapeutic Approaches Targeting Amyloid-beta, alpha-Synuclein, and Tau for the Treatment of Neurodegenerative Disorders. Neurotherapeutics. 2016;13(1):179–189. doi: 10.1007/s13311-015-0397-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valera E, Masliah E. Combination therapies: The next logical Step for the treatment of synucleinopathies? Mov Disord. 2016;31(2):225–234. doi: 10.1002/mds.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol Ther. 2013;138(3):311–322. doi: 10.1016/j.pharmthera.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bittar A, Bhatt N, Kayed R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis. 2019;134:104707. doi: 10.1016/j.nbd.2019.104707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colin M, Dujardin S, Schraen-Maschke S, Meno-Tetang G, Duyckaerts C, Courade JP, et al. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2019;139:3–25. [DOI] [PMC free article] [PubMed]
- 64.Novak P, Kontsekova E, Zilka N, Novak M. Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front Neurosci. 2018;12:798. doi: 10.3389/fnins.2018.00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wisniewski T, Goni F. Vaccination strategies. Handb Clin Neurol. 2018;153:419–430. doi: 10.1016/B978-0-444-63945-5.00023-4. [DOI] [PubMed] [Google Scholar]
- 66.Schilling S, Rahfeld JU, Lues I, Lemere CA. Passive Abeta Immunotherapy: Current Achievements and Future Perspectives. Molecules. 2018;23(5):E1068. 10.3390/molecules23051068. [DOI] [PMC free article] [PubMed]
- 67.Spencer B, Kim C, Gonzalez T, Bisquertt A, Patrick C, Rockenstein E, et al. alpha-Synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum Mol Genet. 2016;25(6):1100–1115. doi: 10.1093/hmg/ddv633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liang Z, Zhao Y, Ruan L, Zhu L, Jin K, Zhuge Q, et al. Impact of aging immune system on neurodegeneration and potential immunotherapies. Prog Neurobiol. 2017;157:2–28. doi: 10.1016/j.pneurobio.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Laurie C, Reynolds A, Coskun O, Bowman E, Gendelman HE, Mosley RL. CD4+ T cells from Copolymer-1 immunized mice protect dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neuroimmunol. 2007;183(1–2):60–68. doi: 10.1016/j.jneuroim.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 70.Congdon EE, Lin Y, Rajamohamedsait HB, Shamir DB, Krishnaswamy S, Rajamohamedsait WJ, et al. Affinity of Tau antibodies for solubilized pathological Tau species but not their immunogen or insoluble Tau aggregates predicts in vivo and ex vivo efficacy. Mol Neurodegener. 2016;11(1):62. doi: 10.1186/s13024-016-0126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bittar A, Sengupta U, Kayed R. Prospects for strain-specific immunotherapy in Alzheimer's disease and tauopathies. NPJ Vaccines. 2018;3:9. doi: 10.1038/s41541-018-0046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85(6):1162–1176. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valera E, Spencer B, Fields JA, Trinh I, Adame A, Mante M, et al. Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol Commun. 2017;5(1):2. doi: 10.1186/s40478-016-0409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mandler M, Rockenstein E, Overk C, Mante M, Florio J, Adame A, et al. Effects of single and combined immunotherapy approach targeting amyloid beta protein and alpha-synuclein in a dementia with Lewy bodies-like model. Alzheimers Dement. 2019;15(9):1133–1148. doi: 10.1016/j.jalz.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bednar MM. Combination therapy for Alzheimer’s disease and related dementias. Prog Mol Biol Transl Sci. 2019;168:289–296. doi: 10.1016/bs.pmbts.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Perry D, Sperling R, Katz R, Berry D, Dilts D, Hanna D, et al. Building a roadmap for developing combination therapies for Alzheimer’s disease. Expert Rev Neurother. 2015;15(3):327–333. doi: 10.1586/14737175.2015.996551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kennedy RE, Cutter GR, Wang G, Schneider LS. Post Hoc Analyses of ApoE Genotype-Defined Subgroups in Clinical Trials. J Alzheimers Dis. 2016;50(4):1205–1215. doi: 10.3233/JAD-150847. [DOI] [PubMed] [Google Scholar]
- 78.Overk C, Masliah E. Dale Schenk One Year Anniversary: Fighting to Preserve the Memories. J Alzheimers Dis. 2018;62(1):1–13. doi: 10.3233/JAD-171071. [DOI] [PubMed] [Google Scholar]
- 79.Messer A, Butler DC. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis. 2019;134:104619. doi: 10.1016/j.nbd.2019.104619. [DOI] [PubMed] [Google Scholar]
- 80.Masliah E, Spencer B. Applications of ApoB LDLR-Binding Domain Approach for the Development of CNS-Penetrating Peptides for Alzheimer's Disease. Methods Mol Biol. 2015;1324:331–337. doi: 10.1007/978-1-4939-2806-4_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spencer B, Williams S, Rockenstein E, Valera E, Xin W, Mante M, et al. alpha-synuclein conformational antibodies fused to penetratin are effective in models of Lewy body disease. Ann Clin Transl Neurol. 2016;3(8):588–606. doi: 10.1002/acn3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhatt MA, Messer A, Kordower JH. Can intrabodies serve as neuroprotective therapies for Parkinson’s disease? Beginning thoughts. J Parkinsons Dis. 2013;3(4):581–591. doi: 10.3233/JPD-130252. [DOI] [PubMed] [Google Scholar]
- 83.Wang Z, Gao G, Duan C, Yang H. Progress of immunotherapy of anti-alpha-synuclein in Parkinson's disease. Biomed Pharmacother. 2019;115:108843. doi: 10.1016/j.biopha.2019.108843. [DOI] [PubMed] [Google Scholar]
- 84.Albert M, Mairet-Coello G, Danis C, Lieger S, Caillierez R, Carrier S, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain. 2019;142(6):1736–1750. doi: 10.1093/brain/awz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Third Quarter 2019: Financial Results and Business Update. Cambridge: Biogen. Available from: https://investors.biogen.com/static-files/40565136-b61f-4473-9e58-9be769bbac6c. Accessed 12 Dec 2019.
- 86.Abbott A. Fresh push for ‘failed’ Alzheimer’s drug. Nature. Available from: https://www.nature.com/articles/d41586-019-03261-5. Accessed 12 Dec 2019. [DOI] [PubMed]
- 87.Petersen RC, Thomas RG, Aisen PS, Mohs RC, Carrillo MC, Albert MS, et al. Randomized controlled trials in mild cognitive impairment: Sources of variability. Neurology. 2017;88(18):1751–1758. doi: 10.1212/WNL.0000000000003907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Overk C, Masliah E. Could changing the course of Alzheimer’s disease pathology with immunotherapy prevent dementia? Brain. 2019;142(7):1853–1855. doi: 10.1093/brain/awz165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 90.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 91.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 92.Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017;14(3):e1002270. doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P. Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One. 2012;7(8):e43201. doi: 10.1371/journal.pone.0043201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018;100(6):1527–1532. doi: 10.1016/j.neuron.2018.11.043. [DOI] [PubMed] [Google Scholar]
- 96.Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer's disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991;1(4):241–251. doi: 10.1111/j.1750-3639.1991.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 97.Wiltfang J, Esselmann H, Bibl M, Smirnov A, Otto M, Paul S, et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1–37/38/39 in addition to 1–40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J Neurochem. 2002;81(3):481–496. doi: 10.1046/j.1471-4159.2002.00818.x. [DOI] [PubMed] [Google Scholar]
- 98.Maddalena AS, Papassotiropoulos A, Gonzalez-Agosti C, Signorell A, Hegi T, Pasch T, et al. Cerebrospinal fluid profile of amyloid beta peptides in patients with Alzheimer's disease determined by protein biochip technology. Neurodegener Dis. 2004;1(4–5):231–235. doi: 10.1159/000080991. [DOI] [PubMed] [Google Scholar]
- 99.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 100.Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, et al. Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain. 2019;142(7):2113–2126. doi: 10.1093/brain/awz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6(2):144–51. [DOI] [PMC free article] [PubMed]
- 102.Lemere CA, Maron R, Selkoe DJ, Weiner HL. Nasal vaccination with beta-amyloid peptide for the treatment of Alzheimer’s disease. DNA Cell Biol. 2001;20(11):705–711. doi: 10.1089/10445490152717569. [DOI] [PubMed] [Google Scholar]
- 103.Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Desai R, et al. Nasal A beta treatment induces anti-A beta antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann N Y Acad Sci. 2000;920:328–331. doi: 10.1111/j.1749-6632.2000.tb06943.x. [DOI] [PubMed] [Google Scholar]
- 104.Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, et al. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15(4):505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD. Intranasal immunotherapy for the treatment of Alzheimer's disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002;23(6):991–1000. doi: 10.1016/s0197-4580(02)00127-6. [DOI] [PubMed] [Google Scholar]
- 106.Maier M, Seabrook TJ, Lemere CA. Modulation of the humoral and cellular immune response in Abeta immunotherapy by the adjuvants monophosphoryl lipid A (MPL), cholera toxin B subunit (CTB) and E. coli enterotoxin LT(R192G) Vaccine. 2005;23(44):5149–5159. doi: 10.1016/j.vaccine.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 107.Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, et al. Vaccination of Alzheimer’s model mice with Abeta derivative in alum adjuvant reduces Abeta burden without microhemorrhages. Eur J Neurosci. 2006;24(9):2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu B, Frost JL, Sun J, Fu H, Grimes S, Blackburn P, et al. MER5101, a novel Abeta1-15:DT conjugate vaccine, generates a robust anti-Abeta antibody response and attenuates Abeta pathology and cognitive deficits in APPswe/PS1DeltaE9 transgenic mice. J Neurosci. 2013;33(16):7027–7037. doi: 10.1523/JNEUROSCI.5924-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arai H, Suzuki H, Yoshiyama T. Vanutide cridificar and the QS-21 adjuvant in Japanese subjects with mild to moderate Alzheimer’s disease: results from two phase 2 studies. Curr Alzheimer Res. 2015;12(3):242–254. doi: 10.2174/1567205012666150302154121. [DOI] [PubMed] [Google Scholar]
- 110.Ketter N, Liu E, Di J, Honig LS, Lu M, Novak G, et al. A Randomized, Double-Blind, Phase 2 Study of the Effects of the Vaccine Vanutide Cridificar with QS-21 Adjuvant on Immunogenicity, Safety and Amyloid Imaging in Patients with Mild to Moderate Alzheimer’s Disease. J Prev Alzheimers Dis. 2016;3(4):192–201. doi: 10.14283/jpad.2016.118. [DOI] [PubMed] [Google Scholar]
- 111.Pasquier F, Sadowsky C, Holstein A, Leterme Gle P, Peng Y, Jackson N, et al. Two Phase 2 Multiple Ascending-Dose Studies of Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Mild-to-Moderate Alzheimer’s Disease. J Alzheimers Dis. 2016;51(4):1131–1143. doi: 10.3233/JAD-150376. [DOI] [PubMed] [Google Scholar]
- 112.Schneeberger A, Hendrix S, Mandler M, Ellison N, Burger V, Brunner M, et al. Results from a Phase II Study to Assess the Clinical and Immunological Activity of AFFITOPE(R) AD02 in Patients with Early Alzheimer’s Disease. J Prev Alzheimers Dis. 2015;2(2):103–114. doi: 10.14283/jpad.2015.63. [DOI] [PubMed] [Google Scholar]
- 113.Vandenberghe R, Riviere ME, Caputo A, Sovago J, Maguire RP, Farlow M, et al. Active Abeta immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement (N Y). 2017;3(1):10–22. doi: 10.1016/j.trci.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11(7):597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- 115.Lopez Lopez C, Tariot PN, Caputo A, Langbaum JB, Liu F, Riviere ME, et al. The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimers Dement (N Y) 2019;5:216–227. doi: 10.1016/j.trci.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hickman DT, Lopez-Deber MP, Ndao DM, Silva AB, Nand D, Pihlgren M, et al. Sequence-independent control of peptide conformation in liposomal vaccines for targeting protein misfolding diseases. J Biol Chem. 2011;286(16):13966–13976. doi: 10.1074/jbc.M110.186338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang CY, Wang PN, Chiu MJ, Finstad CL, Lin F, Lynn S, et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y). 2017;3(2):262–72. doi: 10.1016/j.trci.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lacosta AM, Pascual-Lucas M, Pesini P, Casabona D, Perez-Grijalba V, Marcos-Campos I, et al. Safety, tolerability and immunogenicity of an active anti-Abeta40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther. 2018;10(1):12. doi: 10.1186/s13195-018-0340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 120.Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, et al. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25(40):9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zago W, Buttini M, Comery TA, Nishioka C, Gardai SJ, Seubert P, et al. Neutralization of soluble, synaptotoxic amyloid beta species by antibodies is epitope specific. J Neurosci. 2012;32(8):2696–2702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98(15):8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 125.Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med. 2018;378(4):321–330. doi: 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- 126.Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, et al. Structure of Crenezumab Complex with Abeta Shows Loss of beta-Hairpin. Sci Rep. 2016;6:39374. doi: 10.1038/srep39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N Y) 2018;4:150–160. doi: 10.1016/j.trci.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 129.Budd Haeberlein S, O'Gorman J, Chiao P, Bussiere T, von Rosenstiel P, Tian Y, et al. Clinical Development of Aducanumab, an Anti-Abeta Human Monoclonal Antibody Being Investigated for the Treatment of Early Alzheimer’s Disease. J Prev Alzheimers Dis. 2017;4(4):255–263. doi: 10.14283/jpad.2017.39. [DOI] [PubMed] [Google Scholar]
- 130.Selkoe DJ. Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol. 2019;15(7):365–366. doi: 10.1038/s41582-019-0205-1. [DOI] [PubMed] [Google Scholar]
- 131.Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9(1):95. doi: 10.1186/s13195-017-0318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8–19. doi: 10.1016/j.jalz.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lilly Announces Topline Results for Solanezumab from the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) Study [press release]. Indianapolis (IN): PRNewswire. Available from: https://www.investor.lilly.com/node/42631/pdf. Accessed 10 March 2020.
- 134.Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eisai and Biogen Announce Positive Topline Results of the Final Analysis for Ban2401 at 18 Months [press release]. Tokyo: Eisai Co., Ltd. Available from: https://www.eisai.com/news/2018/pdf/enews201858pdf.pdf. Accessed 10 Oct 2019.
- 136.Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther. 2016;8(1):14. doi: 10.1186/s13195-016-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 139.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58(2):186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 140.Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018;4:21. doi: 10.1038/s41531-018-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Villar-Pique A, Lopes da Fonseca T, Outeiro TF. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. 2016;139(Suppl 1):240–255. doi: 10.1111/jnc.13249. [DOI] [PubMed] [Google Scholar]
- 142.Huang M, Wang B, Li X, Fu C, Wang C, Kang X. alpha-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front Neurosci. 2019;13:28. doi: 10.3389/fnins.2019.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251(3):205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 144.Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249(2–3):180–182. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- 145.Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30(18):6236–6246. doi: 10.1523/JNEUROSCI.0567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun. 2008;372(3):423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 148.Kisos H, Pukass K, Ben-Hur T, Richter-Landsberg C, Sharon R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One. 2012;7(10):e46817. doi: 10.1371/journal.pone.0046817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Caplan IF, Maguire-Zeiss KA. Toll-Like Receptor 2 Signaling and Current Approaches for Therapeutic Modulation in Synucleinopathies. Front Pharmacol. 2018;9:417. doi: 10.3389/fphar.2018.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Karpowicz RJ, Jr, Trojanowski JQ, Lee VM. Transmission of alpha-synuclein seeds in neurodegenerative disease: recent developments. Lab Invest. 2019;99(7):971–981. doi: 10.1038/s41374-019-0195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 154.Henderson MX, Covell DJ, Chung CH, Pitkin RM, Sandler RM, Decker SC, et al. Characterization of novel conformation-selective alpha-synuclein antibodies as potential immunotherapeutic agents for Parkinson’s disease. Neurobiol Dis. 2019;136:104712. doi: 10.1016/j.nbd.2019.104712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, et al. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep. 2014;7(6):2054–2065. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34(28):9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, et al. Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun. 2017;5(1):7. doi: 10.1186/s40478-016-0410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, et al. Single-chain antigen-binding proteins. Science. 1988;242(4877):423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 161.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. 1988;85(16):5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Spencer BJ, Verma IM. Targeted delivery of proteins across the blood-brain barrier. Proc Natl Acad Sci U S A. 2007;104(18):7594–7599. doi: 10.1073/pnas.0702170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Spencer B, Marr RA, Gindi R, Potkar R, Michael S, Adame A, et al. Peripheral delivery of a CNS targeted, metalo-protease reduces abeta toxicity in a mouse model of Alzheimer’s disease. PLoS One. 2011;6(1):e16575. doi: 10.1371/journal.pone.0016575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Becker-Hapak M, McAllister SS, Dowdy SF. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24(3):247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 165.Marschall AL, Dubel S. Antibodies inside of a cell can change its outside: Can intrabodies provide a new therapeutic paradigm? Comput Struct Biotechnol J. 2016;14:304–308. doi: 10.1016/j.csbj.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 167.Fukuchi K, Tahara K, Kim HD, Maxwell JA, Lewis TL, Accavitti-Loper MA, et al. Anti-Abeta single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer’s disease. Neurobiol Dis. 2006;23(3):502–511. doi: 10.1016/j.nbd.2006.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ryan DA, Mastrangelo MA, Narrow WC, Sullivan MA, Federoff HJ, Bowers WJ. Abeta-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer’s disease mice. Mol Ther. 2010;18(8):1471–1481. doi: 10.1038/mt.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang J, Pattanayak A, Song M, Kou J, Taguchi H, Paul S, et al. Muscle-directed anti-Abeta single-chain antibody delivery via AAV1 reduces cerebral Abeta load in an Alzheimer’s disease mouse model. J Mol Neurosci. 2013;49(2):277–288. doi: 10.1007/s12031-012-9877-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lynch SM, Zhou C, Messer A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J Mol Biol. 2008;377(1):136–147. doi: 10.1016/j.jmb.2007.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu W, Zhao L, Blackman B, Parmar M, Wong MY, Woo T, et al. Vectored Intracerebral Immunization with the Anti-Tau Monoclonal Antibody PHF1 Markedly Reduces Tau Pathology in Mutant Tau Transgenic Mice. J Neurosci. 2016;36(49):12425–12435. doi: 10.1523/JNEUROSCI.2016-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vitale F, Giliberto L, Ruiz S, Steslow K, Marambaud P, d'Abramo C. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun. 2018;6(1):82. doi: 10.1186/s40478-018-0585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chatterjee D, Bhatt M, Butler D, De Genst E, Dobson CM, Messer A, et al. Proteasome-targeted nanobodies alleviate pathology and functional decline in an alpha-synuclein-based Parkinson’s disease model. NPJ Parkinsons Dis. 2018;4:25. doi: 10.1038/s41531-018-0062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, et al. ESCRT-mediated uptake and degradation of brain-targeted alpha-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther. 2014;22(10):1753–1767. doi: 10.1038/mt.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pul R, Dodel R, Stangel M. Antibody-based therapy in Alzheimer’s disease. Expert Opin Biol Ther. 2011;11(3):343–357. doi: 10.1517/14712598.2011.552884. [DOI] [PubMed] [Google Scholar]
- 176.Shahaduzzaman M, Nash K, Hudson C, Sharif M, Grimmig B, Lin X, et al. Anti-human alpha-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-alpha-synuclein rat model of Parkinson’s disease. PLoS One. 2015;10(2):e0116841. doi: 10.1371/journal.pone.0116841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pozzi S, Thammisetty SS, Codron P, Rahimian R, Plourde KV, Soucy G, et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest. 2019;129(4):1581–1595. doi: 10.1172/JCI123931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tamaki Y, Shodai A, Morimura T, Hikiami R, Minamiyama S, Ayaki T, et al. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci Rep. 2018;8(1):6030. doi: 10.1038/s41598-018-24463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ghadge GD, Pavlovic JD, Koduvayur SP, Kay BK, Roos RP. Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol Dis. 2013;56:74–78. doi: 10.1016/j.nbd.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, et al. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther. 2014;22(3):498–510. doi: 10.1038/mt.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gros-Louis F, Soucy G, Lariviere R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113(5):1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- 182.Snyder-Keller A, McLear JA, Hathorn T, Messer A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J Neuropathol Exp Neurol. 2010;69(10):1078–1085. doi: 10.1097/NEN.0b013e3181f530ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Butler DC, Messer A. Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS One. 2011;6(12):e29199. doi: 10.1371/journal.pone.0029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Schneeberger A, Mandler M, Mattner F, Schmidt W. Vaccination for Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S11–13. doi: 10.1016/S1353-8020(11)70006-2. [DOI] [PubMed] [Google Scholar]
- 185.Schneeberger A, Mandler M, Mattner F, Schmidt W. AFFITOME(R) technology in neurodegenerative diseases: the doubling advantage. Hum Vaccin. 2010;6(11):948–952. doi: 10.4161/hv.6.11.13217. [DOI] [PubMed] [Google Scholar]
- 186.Mandler M, Santic R, Gruber P, Cinar Y, Pichler D, Funke SA, et al. Tailoring the antibody response to aggregated Ass using novel Alzheimer-vaccines. PLoS One. 2015;10(1):e0115237. doi: 10.1371/journal.pone.0115237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10. doi: 10.1186/s13024-015-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol. 2014;127(6):861–879. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018;75(10):1206–1214. doi: 10.1001/jamaneurol.2018.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord. 2019;34(8):1154–1163. doi: 10.1002/mds.27738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schofield DJ, Irving L, Calo L, Bogstedt A, Rees G, Nuccitelli A, et al. Preclinical development of a high affinity alpha-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular alpha-synuclein and attenuate alpha-synuclein spreading in vivo. Neurobiol Dis. 2019;132:104582. doi: 10.1016/j.nbd.2019.104582. [DOI] [PubMed] [Google Scholar]
- 192.Hutter-Saunders JA, Mosley RL, Gendelman HE. Pathways towards an effective immunotherapy for Parkinson's disease. Expert Rev Neurother. 2011;11(12):1703–1715. doi: 10.1586/ern.11.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rockenstein E, Ostroff G, Dikengil F, Rus F, Mante M, Florio J, et al. Combined Active Humoral and Cellular Immunization Approaches for the Treatment of Synucleinopathies. J Neurosci. 2018;38(4):1000–1014. doi: 10.1523/JNEUROSCI.1170-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kolarova M, Garcia-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. 2012;2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116(2):227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 196.Jouanne M, Rault S, Voisin-Chiret AS. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur J Med Chem. 2017;139:153–167. doi: 10.1016/j.ejmech.2017.07.070. [DOI] [PubMed] [Google Scholar]
- 197.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 198.Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17(1):83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rosler TW, Tayaranian Marvian A, Brendel M, Nykanen NP, Hollerhage M, Schwarz SC, et al. Four-repeat tauopathies. Prog Neurobiol. 2019;180:101644. doi: 10.1016/j.pneurobio.2019.101644. [DOI] [PubMed] [Google Scholar]
- 200.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 201.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103(1):26–35. [DOI] [PubMed]
- 202.Haroutunian V, Davies P, Vianna C, Buxbaum JD, Purohit DP. Tau protein abnormalities associated with the progression of alzheimer disease type dementia. Neurobiol Aging. 2007;28(1):1–7. doi: 10.1016/j.neurobiolaging.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 203.Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. 2013;72(7):708–715. doi: 10.1097/NEN.0b013e31829a25b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Axon Announces Positive Results From Phase II ADAMANT Trial for AADvac1 in Alzheimer's Disease [press release]. London: PRNewswire. Available from: https://www.prnewswire.com/news-releases/axon-announces-positive-results-from-phase-ii-adamant-trial-for-aadvac1-in-alzheimers-disease-300914509.html. Accessed 27 Oct 2019.
- 205.Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther. 2014;6(4):44. doi: 10.1186/alzrt278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Novak P, Schmidt R, Kontsekova E, Kovacech B, Smolek T, Katina S, et al. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):108. doi: 10.1186/s13195-018-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Novak P, Zilka N, Zilkova M, Kovacech B, Skrabana R, Ondrus M, et al. AADvac1, an Active Immunotherapy for Alzheimer's Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J Prev Alzheimers Dis. 2019;6(1):63–69. doi: 10.14283/jpad.2018.45. [DOI] [PubMed] [Google Scholar]
- 208.Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One. 2013;8(8):e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.AC Immune Initiates Ph1b/2a Study of Anti-Phospho-Tau Vaccine in Alzheimer’s Disease [press release]. Lausanne: Globe Newswire. Available from: https://ir.acimmune.com/node/8136/pdf. Accessed 27 Oct 2019.
- 210.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48(2):128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 211.Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80(2):402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yanamandra K, Patel TK, Jiang H, Schindler S, Ulrich JD, Boxer AL, et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med. 2017;9(386): eaal2029. 10.1126/scitranslmed.aal2029. [DOI] [PMC free article] [PubMed]
- 213.West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, et al. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J Prev Alzheimers Dis. 2017;4(4):236–241. doi: 10.14283/jpad.2017.36. [DOI] [PubMed] [Google Scholar]
- 214.Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging. 2015;36(2):693–709. doi: 10.1016/j.neurobiolaging.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 215.Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y). 2018;4:746–755. doi: 10.1016/j.trci.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Biogen Reports Top-Line Results from Phase 2 Study in Progressive Supranuclear Palsy [press release]. Cambridge: Globe Newswire. Available from: https://www.globenewswire.com/news-release/2019/12/13/1960373/0/en/Biogen-Reports-Top-Line-Results-from-Phase-2-Study-in-Progressive-Supranuclear-Palsy.htm. Accessed 10 Jan 2020
- 217.Kontsekova E, Zilka N, Kovacech B, Skrabana R, Novak M. Identification of structural determinants on tau protein essential for its pathological function: novel therapeutic target for tau immunotherapy in Alzheimer’s disease. Alzheimers Res Ther. 2014;6(4):45. doi: 10.1186/alzrt277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Weisova P, Cehlar O, Skrabana R, Zilkova M, Filipcik P, Kovacech B, et al. Therapeutic antibody targeting microtubule-binding domain prevents neuronal internalization of extracellular tau via masking neuron surface proteoglycans. Acta Neuropathol Commun. 2019;7(1):129. doi: 10.1186/s40478-019-0770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Courade JP, Angers R, Mairet-Coello G, Pacico N, Tyson K, Lightwood D, et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018;136(5):729–745. doi: 10.1007/s00401-018-1911-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19–34. doi: 10.1016/j.addr.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gauthier S, Alam J, Fillit H, Iwatsubo T, Liu-Seifert H, Sabbagh M, et al. Combination Therapy for Alzheimer’s Disease: Perspectives of the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2019;6(3):164–168. doi: 10.14283/jpad.2019.12. [DOI] [PubMed] [Google Scholar]
- 222.Schmidt R, Hofer E, Bouwman FH, Buerger K, Cordonnier C, Fladby T, et al. EFNS-ENS/EAN Guideline on concomitant use of cholinesterase inhibitors and memantine in moderate to severe Alzheimer’s disease. Eur J Neurol. 2015;22(6):889–898. doi: 10.1111/ene.12707. [DOI] [PubMed] [Google Scholar]
- 223.Matsunaga S, Kishi T, Yasue I, Iwata N. Cholinesterase Inhibitors for Lewy Body Disorders: A Meta-Analysis. Int J Neuropsychopharmacol. 2016;19(2):pyv086. 10.1093/ijnp/pyv086. [DOI] [PMC free article] [PubMed]
- 224.Chang W, Teng J. Combined application of tenuigenin and beta-asarone improved the efficacy of memantine in treating moderate-to-severe Alzheimer’s disease. Drug Des Devel Ther. 2018;12:455–462. doi: 10.2147/DDDT.S155567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kantarci A, Aytan N, Palaska I, Stephens D, Crabtree L, Benincasa C, et al. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Exp Neurol. 2018;300:111–120. doi: 10.1016/j.expneurol.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 226.Mori T, Koyama N, Tan J, Segawa T, Maeda M, Town T. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J Biol Chem. 2019;294(8):2714–2731. doi: 10.1074/jbc.RA118.004280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, et al. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40(1):1–8. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- 228.Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer’s disease. Am J Pathol. 1996;148(1):201–210. [PMC free article] [PubMed] [Google Scholar]
- 229.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3 Suppl):417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 230.Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68(11):812–819. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 231.Winslow AR, Moussaud S, Zhu L, Post KL, Dickson DW, Berezovska O, et al. Convergence of pathology in dementia with Lewy bodies and Alzheimer’s disease: a role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain. 2014;137(Pt 7):1958–1970. doi: 10.1093/brain/awu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, et al. Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015;129(5):729–748. doi: 10.1007/s00401-015-1406-3. [DOI] [PubMed] [Google Scholar]
- 233.Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Irwin DJ, Hurtig HI. The Contribution of Tau, Amyloid-Beta and Alpha-Synuclein Pathology to Dementia in Lewy Body Disorders. J Alzheimers Dis Parkinsonism. 2018;8(4):444. 10.4172/2161-0460.1000444. [DOI] [PMC free article] [PubMed]
- 235.Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65. doi: 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005;64(12):2069–2073. doi: 10.1212/01.WNL.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, et al. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131(3):393–409. doi: 10.1007/s00401-015-1526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, de Smith A, et al. Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol. 2016;42(5):436–450. doi: 10.1111/nan.12294. [DOI] [PubMed] [Google Scholar]
- 240.Howlett DR, Whitfield D, Johnson M, Attems J, O'Brien JT, Aarsland D, et al. Regional Multiple Pathology Scores Are Associated with Cognitive Decline in Lewy Body Dementias. Brain Pathol. 2015;25(4):401–408. doi: 10.1111/bpa.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Compta Y, Parkkinen L, O'Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. 2011;134(Pt 5):1493–1505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jensen PH, Sorensen ES, Petersen TE, Gliemann J, Rasmussen LK. Residues in the synuclein consensus motif of the alpha-synuclein fragment, NAC, participate in transglutaminase-catalysed cross-linking to Alzheimer-disease amyloid beta A4 peptide. Biochem J. 1995;310(Pt 1):91–94. doi: 10.1042/bj3100091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jensen PH, Hojrup P, Hager H, Nielsen MS, Jacobsen L, Olesen OF et al. Binding of Abeta to alpha- and beta-synucleins: identification of segments in alpha-synuclein/NAC precursor that bind Abeta and NAC. Biochem J. 1997;323(Pt 2):539–546. doi: 10.1042/bj3230539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R. Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res. 2006;31(9):1153–1162. doi: 10.1007/s11064-006-9140-9. [DOI] [PubMed] [Google Scholar]
- 246.Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One. 2008;3(9):e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci. 2012;32(30):10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Twohig D, Rodriguez-Vieitez E, Sando SB, Berge G, Lauridsen C, Moller I, et al. The relevance of cerebrospinal fluid alpha-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):130. doi: 10.1186/s40478-018-0624-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98(21):12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kaneko H, Kakita A, Kasuga K, Nozaki H, Ishikawa A, Miyashita A, et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci. 2007;27(48):13092–13097. doi: 10.1523/JNEUROSCI.4244-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Overk CR, Cartier A, Shaked G, Rockenstein E, Ubhi K, Spencer B, et al. Hippocampal neuronal cells that accumulate alpha-synuclein fragments are more vulnerable to Abeta oligomer toxicity via mGluR5--implications for dementia with Lewy bodies. Mol Neurodegener. 2014;9:18. doi: 10.1186/1750-1326-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Khan SS, LaCroix M, Boyle G, Sherman MA, Brown JL, Amar F, et al. Bidirectional modulation of Alzheimer phenotype by alpha-synuclein in mice and primary neurons. Acta Neuropathol. 2018;136(4):589–605. doi: 10.1007/s00401-018-1886-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology. 1998;51(2):351–7. [DOI] [PubMed]
- 254.Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, et al. Inhibition of amyloid-beta plaque formation by alpha-synuclein. Nat Med. 2015;21(7):802–807. doi: 10.1038/nm.3885. [DOI] [PubMed] [Google Scholar]
- 255.Choi BK, Kim JY, Cha MY, Mook-Jung I, Shin YK, Lee NK. beta-Amyloid and alpha-synuclein cooperate to block SNARE-dependent vesicle fusion. Biochemistry. 2015;54(9):1831–1840. doi: 10.1021/acs.biochem.5b00087. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 256.Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. 1999;274(36):25481–25489. doi: 10.1074/jbc.274.36.25481. [DOI] [PubMed] [Google Scholar]
- 257.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383(6600):550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 258.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 259.Esposito A, Dohm CP, Kermer P, Bahr M, Wouters FS. alpha-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis. 2007;26(3):521–531. doi: 10.1016/j.nbd.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 260.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, et al. alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem. 2013;288(30):21742–21754. doi: 10.1074/jbc.M113.451815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, et al. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem. 2004;279(37):39155–39164. doi: 10.1074/jbc.M405456200. [DOI] [PubMed] [Google Scholar]
- 263.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, et al. alpha-Synuclein shares physical and functional homology with 14–3-3 proteins. J Neurosci. 1999;19(14):5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Hashiguchi M, Sobue K, Paudel HK. 14-3-3zeta is an effector of tau protein phosphorylation. J Biol Chem. 2000;275(33):25247–25254. doi: 10.1074/jbc.M003738200. [DOI] [PubMed] [Google Scholar]
- 265.Moussaud S, Jones DR, Moussaud-Lamodiere EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9:43. doi: 10.1186/1750-1326-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gassowska M, Czapski GA, Pajak B, Cieslik M, Lenkiewicz AM, Adamczyk A. Extracellular alpha-synuclein leads to microtubule destabilization via GSK-3beta-dependent Tau phosphorylation in PC12 cells. PLoS One. 2014;9(4):e94259. doi: 10.1371/journal.pone.0094259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23(9):2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kawakami F, Suzuki M, Shimada N, Kagiya G, Ohta E, Tamura K, et al. Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. FEBS J. 2011;278(24):4895–4904. doi: 10.1111/j.1742-4658.2011.08389.x. [DOI] [PubMed] [Google Scholar]
- 270.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9(8):2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154(1):103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Gerson JE, Kayed R. alpha-Synuclein Oligomers Induce a Unique Toxic Tau Strain. Biol Psychiatry. 2018;84(7):499–508. doi: 10.1016/j.biopsych.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nubling G, Bader B, Levin J, Hildebrandt J, Kretzschmar H, Giese A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with alpha-synuclein at the single molecule level. Mol Neurodegener. 2012;7:35. doi: 10.1186/1750-1326-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Brekk OR, Moskites A, Isacson O, Hallett PJ. Lipid-dependent deposition of alpha-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci Rep. 2018;8(1):15207. doi: 10.1038/s41598-018-33474-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Dai CL, Tung YC, Liu F, Gong CX, Iqbal K. Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther. 2017;9(1):1. doi: 10.1186/s13195-016-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Davtyan H, Hovakimyan A, Kiani Shabestari S, Antonyan T, Coburn MA, Zagorski K, et al. Testing a MultiTEP-based combination vaccine to reduce Abeta and tau pathology in Tau22/5xFAD bigenic mice. Alzheimers Res Ther. 2019;11(1):107. doi: 10.1186/s13195-019-0556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Thomas B, Mandir AS, West N, Liu Y, Andrabi SA, Stirling W, et al. Resistance to MPTP-neurotoxicity in alpha-synuclein knockout mice is complemented by human alpha-synuclein and associated with increased beta-synuclein and Akt activation. PLoS One. 2011;6(1):e16706. doi: 10.1371/journal.pone.0016706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, et al. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest. 2015;125(7):2721–2735. doi: 10.1172/JCI64502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Singh B, Covelo A, Martell-Martinez H, Nanclares C, Sherman MA, Okematti E, et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol. 2019;138(4):551–574. doi: 10.1007/s00401-019-02032-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 281.Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. 2018;378(7):625–635. doi: 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 282.Earls RH, Menees KB, Chung J, Gutekunst CA, Lee HJ, Hazim MG, et al. NK cells clear alpha-synuclein and the depletion of NK cells exacerbates synuclein pathology in a mouse model of alpha-synucleinopathy. Proc Natl Acad Sci U S A. 2020; 117(3):1762–71. [DOI] [PMC free article] [PubMed]
- 283.Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31(15):1529–1534. doi: 10.1101/gad.302570.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Liu Z, Qiu AW, Huang Y, Yang Y, Chen JN, Gu TT, et al. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson’s disease. Brain Behav Immun. 2019;81:630–645. doi: 10.1016/j.bbi.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 285.Fiala M, Mizwicki MT, Weitzman R, Magpantay L, Nishimoto N. Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients. Am J Neurodegener Dis. 2013;2(2):129–139. [PMC free article] [PubMed] [Google Scholar]
- 286.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Shi JQ, Shen W, Chen J, Wang BR, Zhong LL, Zhu YW, et al. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011;1368:239–247. doi: 10.1016/j.brainres.2010.10.053. [DOI] [PubMed] [Google Scholar]
- 288.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kim C, Rockenstein E, Spencer B, Kim HK, Adame A, Trejo M, et al. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015;13(4):771–782. doi: 10.1016/j.celrep.2015.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol Neurodegener. 2018;13(1):43. doi: 10.1186/s13024-018-0276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Panza F, Lozupone M, Solfrizzi V, Watling M, Imbimbo BP. Time to test antibacterial therapy in Alzheimer’s disease. Brain. 2019;142(10):2905–2929. doi: 10.1093/brain/awz244. [DOI] [PubMed] [Google Scholar]
- 294.Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol. 2009;217(1):131–138. doi: 10.1002/path.2449. [DOI] [PubMed] [Google Scholar]
- 295.Ball MJ. Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can J Neurol Sci. 1982;9(3):303–306. doi: 10.1017/s0317167100044115. [DOI] [PubMed] [Google Scholar]
- 296.De Chiara G, Piacentini R, Fabiani M, Mastrodonato A, Marcocci ME, Limongi D, et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019;15(3):e1007617. doi: 10.1371/journal.ppat.1007617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Tzeng NS, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics. 2018;15(2):417–429. doi: 10.1007/s13311-018-0611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Strandberg TE, Pitkala KH, Linnavuori KH, Tilvis RS. Impact of viral and bacterial burden on cognitive impairment in elderly persons with cardiovascular diseases. Stroke. 2003;34(9):2126–2131. doi: 10.1161/01.STR.0000086754.32238.DA. [DOI] [PubMed] [Google Scholar]
- 299.Chen VC, Wu SI, Huang KY, Yang YH, Kuo TY, Liang HY, et al. Herpes Zoster and Dementia: A Nationwide Population-Based Cohort Study. J Clin Psychiatry. 2018;79(1):16m11312. 10.4088/JCP.16m11312. [DOI] [PubMed]
- 300.Shim SM, Cheon HS, Jo C, Koh YH, Song J, Jeon JP. Elevated Epstein-Barr Virus Antibody Level is Associated with Cognitive Decline in the Korean Elderly. J Alzheimers Dis. 2017;55(1):293–301. doi: 10.3233/JAD-160563. [DOI] [PubMed] [Google Scholar]
- 301.Nimgaonkar VL, Yolken RH, Wang T, Chang CC, McClain L, McDade E, et al. Temporal Cognitive Decline Associated With Exposure to Infectious Agents in a Population-based. Aging Cohort. Alzheimer Dis Assoc Disord. 2016;30(3):216–222. doi: 10.1097/WAD.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Balin BJ, Gerard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187(1):23–42. doi: 10.1007/s004300050071. [DOI] [PubMed] [Google Scholar]
- 303.Franceschi F, Ojetti V, Candelli M, Covino M, Cardone S, Potenza A, et al. Microbes and Alzheimer disease: lessons from H. pylori and GUT microbiota. Eur Rev Med Pharmacol Sci. 2019;23(1):426–430. doi: 10.26355/eurrev_201901_16791. [DOI] [PubMed] [Google Scholar]
- 304.Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Grigoriadis N, et al. Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease. Int J Neurosci. 2009;119(6):765–777. doi: 10.1080/00207450902782083. [DOI] [PubMed] [Google Scholar]
- 305.Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–677. doi: 10.3233/JAD-121918. [DOI] [PubMed] [Google Scholar]
- 306.Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer’s Disease. Mediators Inflamm. 2015;2015:137357. doi: 10.1155/2015/137357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. doi: 10.1126/sciadv.aau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, et al. Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther. 2012;20(7):1327–1337. doi: 10.1038/mt.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Valenzuela V, Martinez G, Duran-Aniotz C, Hetz C. Gene therapy to target ER stress in brain diseases. Brain Res. 2016;1648(Pt B):561–570. doi: 10.1016/j.brainres.2016.04.064. [DOI] [PubMed] [Google Scholar]
- 310.Valdes P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A. 2014;111(18):6804–6809. doi: 10.1073/pnas.1321845111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, et al. Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- 312.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation. 2009;6:41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Mercado G, Castillo V, Soto P, Sidhu A. ER stress and Parkinson's disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016;1648(Pt B):626–632. doi: 10.1016/j.brainres.2016.04.042. [DOI] [PubMed] [Google Scholar]
- 314.Peretti D, Bastide A, Radford H, Verity N, Molloy C, Martin MG, et al. RBM3 mediates structural plasticity and protective effects of cooling in neurodegeneration. Nature. 2015;518(7538):236–239. doi: 10.1038/nature14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Qiu C, Winblad B, Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005;4(8):487–499. doi: 10.1016/S1474-4422(05)70141-1. [DOI] [PubMed] [Google Scholar]
- 316.Yasar S, Schuchman M, Peters J, Anstey KJ, Carlson MC, Peters R. Relationship Between Antihypertensive Medications and Cognitive Impairment: Part I. Review of Human Studies and Clinical Trials. Curr Hypertens Rep. 2016;18(8):67. doi: 10.1007/s11906-016-0674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Chu CS, Tseng PT, Stubbs B, Chen TY, Tang CH, Li DJ, et al. Use of statins and the risk of dementia and mild cognitive impairment: A systematic review and meta-analysis. Sci Rep. 2018;8(1):5804. doi: 10.1038/s41598-018-24248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yang M, Williamson J. Blood Pressure and Statin Effects on Cognition: a Review. Curr Hypertens Rep. 2019;21(9):70. doi: 10.1007/s11906-019-0973-4. [DOI] [PubMed] [Google Scholar]
- 319.van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell. 2019;24(3):363–375. doi: 10.1016/j.stem.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Mejias-Trueba M, Perez-Moreno MA, Fernandez-Arche MA. Systematic review of the efficacy of statins for the treatment of Alzheimer’s disease. Clin Med (Lond). 2018;18(1):54–61. doi: 10.7861/clinmedicine.18-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Zissimopoulos JM, Barthold D, Brinton RD, Joyce G. Sex and Race Differences in the Association Between Statin Use and the Incidence of Alzheimer Disease. JAMA Neurol. 2017;74(2):225–232. doi: 10.1001/jamaneurol.2016.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Jeong SM, Jang W, Shin DW. Association of statin use with Parkinson’s disease: Dose-response relationship. Mov Disord. 2019;34(7):1014–1021. doi: 10.1002/mds.27681. [DOI] [PubMed] [Google Scholar]
- 323.Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology. 2008;70(16 Pt 2):1418–1422. doi: 10.1212/01.wnl.0000286942.14552.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Friedman B, Lahad A, Dresner Y, Vinker S. Long-term statin use and the risk of Parkinson’s disease. Am J Manag Care. 2013;19(8):626–632. [PubMed] [Google Scholar]
- 326.Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci. 2009;29(43):13543–13556. doi: 10.1523/JNEUROSCI.4144-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Becker C, Jick SS, Meier CR. Use of statins and the risk of Parkinson’s disease: a retrospective case-control study in the UK. Drug Saf. 2008;31(5):399–407. doi: 10.2165/00002018-200831050-00004. [DOI] [PubMed] [Google Scholar]
- 328.Samii A, Carleton BC, Etminan M. Statin use and the risk of Parkinson disease: a nested case control study. J Clin Neurosci. 2008;15(11):1272–1273. doi: 10.1016/j.jocn.2008.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 306 kb)