

Abstract
In the last few years, the unprecedented results of immune checkpoint inhibitors have led to a paradigm shift in clinical practice for the treatment of several cancer types. However, the vast majority of patients with gastrointestinal cancer do not benefit from immunotherapy. To date, microsatellite instability high and DNA mismatch repair deficiency are the only robust predictive biomarkers of response to immune checkpoint inhibitors. Unfortunately, these patients comprise only 5%–10% of all gastrointestinal cancers. Several mechanisms of both innate and adaptive resistance to immunotherapy have been recognized that may be at least in part responsible for the failure of immune checkpoint inhibitors in this population of patients. In the first part of this review article, we provide an overview of the main clinical trials with immune checkpoint inhibitors in patients with gastrointestinal cancer and the role of predictive biomarkers. In the second part, we discuss the actual body of knowledge in terms of mechanisms of resistance to immunotherapy and the most promising approach that are currently under investigation in order to expand the population of patients with gastrointestinal cancer who could benefit from immune checkpoint inhibitors.
Keywords: tumours, immunotherapy, gastrointestinal neoplasms
1. Introduction
In the last few years, immunotherapy has dramatically changed the treatment landscape of several types of solid tumors, such as melanoma and non-small cell lung cancer (NSCLC).1 Immune checkpoint inhibitors (ICI) targeting programmed cell death-1 (PD-1) and programmed cell death ligand-1 (PD-L1) axes are the most widely investigated drugs and the remarkable results obtained with these compounds have led to several US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approvals across different cancer types and numerous indications.2 3 However, identifying tumors which will respond to immunotherapy has remained a key challenge.4
Overall, gastrointestinal (GI) neoplasia and more specifically colorectal cancers (CRC) have been always considered as ‘cold’ tumors, therefore, the vast majority of these patients do not benefit from an immunotherapy approach.5 To date, the only validated predictive biomarkers of response to ICIs used in clinical practice across all solid tumors, including GI cancers, is microsatellite instability high (MSI-H) or DNA mismatch repair deficiency (dMMR). In fact, in 2017 pembrolizumab (Keytruda, Merck)—an anti-PD-1 antibody—was FDA approved for the treatment of metastatic cancers that are characterized by MSI-H status or dMMR, regardless the site of tumor origin. In addition, few months later the FDA granted accelerated approval to nivolumab (Opdivo, Bristol-Myers Squibb) for the treatment of patients with dMMR and/or MSI-H metastatic CRC (mCRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin and irinotecan. Efficacy of PD-1-blockade in MSI-H tumors for patients with CRC, showing an objective response rate (ORR) of 36%, is similar to the efficacy in different cancer type (ORR 46% across 14 other cancer types). However, to date, there is no approved PD-1-blocking antibody for MSI-H cancers in Europe.6 Other predictive biomarkers to better select patients for immunotherapy are being currently studied, such as PD-L1,7–11 tumor mutational burden (TMB)12–14 or different combinations of these,15–17 though showing conflicting results in different tumor types, including GI cancers.
In this review article, we provide an overview of the role of immunotherapy biomarkers in these patients and the main clinical trials with ICI (i.e., anti PD-1/PD-L1) in CRC (table 1), esophageal and gastric (table 2) cancers, and well as other GI cancers. We further discuss the state-of-the-art of the known mechanisms of resistance to ICIs as well as the several methods to overcome resistance which are under investigation, with specific focus on GI cancers. Finally, we provide a perspective on possible future therapeutic approaches that may be implemented in clinical practice.
Table 1.
Main ongoing trials with immune checkpoint inhibitors in colorectal cancer
| Target | Agent | Phase | Setting (treatment line) |
Treatment | Main objectives | Biomarker | NCT identifier |
| PD-1 (±other agents) | Pembrolizumab | II | First line | Pembrolizumab+FOLFOX | ORR=55% | MMR | NCT02375672 |
| III | First line, MSI-H mCRC |
Pembrolizumab vs FOLFOX or FOLFIRI (±cetuximab or bevacizumab) |
Ongoing | MMR |
NCT02563002 (KEYNOTE-177) |
||
| II | Refractory | Pembrolizumab | Ongoing | MRR |
NCT01876511 (KEYNOTE-016) |
||
| II | Refractory | Pembrolizumab | ORR for MSI-H CRC=26.2% | MMR |
NCT02460198 (KEYNOTE-164) |
||
| II | Refractory, advanced solid tumor including MSI-H mCRC | Pembrolizumab | ORR for MSI-H CRC=26.2% | MMR |
NCT02628067 (KEYNOTE-158) |
||
| I/II | Refractory, RAS/BRAF wild-type |
Pembrolizumab+Cetuximab | Ongoing | NA | NCT02318901 | ||
| I/II | Refractory, RAS wild-type |
Pembrolizumab+Cetuximab | Ongoing | NA | NCT02713373 | ||
| II | Refractory pMMR | Pembrolizumab+Radiotherapy or ablation | Interim ORR=9% in the RT cohort | PD-L1 | NCT02437071 | ||
| Ib | Resectable liver mCRC | Pembrolizumab+Stereotactic body Radiotherapy | Ongoing | PD-L1 |
NCT02837263 (KEYNOTE-290) |
||
| II | Refractory | Pembrolizumab+Azacitidine | ORR=3% | MMR | NCT02260440 | ||
| I | Refractory, advanced solid tumors including MSI-H mCRC | Pembrolizumab + Itacitinib (JAK1 inhibitor) |
NA | MMR, TILs and the ratio of CD8 +lymphocytes to FOXP3 +cells infiltrating tumor post-treatment versus pretreatment by IHC |
NCT02646748 | ||
| I/II | After first line, advanced solid tumors including MSI-H mCRC | Pembrolizumab + Epacadostat (IDO-inhibitor) |
Ongoing | PD-L1 |
NCT02178722 (KEYNOTE-037) |
||
| Nivolumab | II | Refractory, first line (cohort C3) |
Nivolumab±other agents Cohort 1: Nivolumab Cohort 2: Nivolumab + Ipilimumab (anti-CTLA-4) (escalation dose) Cohort 3: Nivolumab + Ipilimumab Cohort 4: Nivolumab + Ipilimumab + Cobimetinib Cohort 5: Nivolumab + BMS-986016 (anti-LAG3) Cohort 6: Nivolumab + Daratumumab |
ORR=31% | MMR |
NCT02060188 (CHECKMATE 142) |
|
| I/II | Refractory | Nivolumab+Irinotecan or XELIRI | NCT02423954 | ||||
| II | Refractory, MSS mCRC |
Nivolumab +TAS-102 | ORR=0% | MRR, PD-L1, TILs | NCT02860546 | ||
| II | Refractory | Nivolumab + Ipilimumab + Radiotherapy | ORR=14% | MMR | NCT03104439 | ||
| I/II | Refractory | Nivolumab+Epacadostat (anti-IDO) | Ongoing | PD-L1 | NCT02327078 | ||
| I/II | Refractory | Nivolumab+Varlilumab (anti-CD27) | Ongoing | PD-L1 CD8+ |
NCT02335918 | ||
| II | Stage I–III | Nivolumab+Ipilimumab±Celecoxib | Ongoing | NA | NCT03026140 | ||
| I/II | Neoadjuvant, Rectal cancer |
Chemoradiotherapy followed by Nivolumab before surgery | Ongoing | NA | NCT02948348 | ||
| I/II | Refractory mCRC and GC | Nivolumab+Regorafenib (multi-TKI) | ORR= 29% in MSS CRC |
PD-L1 | NCT03406871 | ||
| PD-L1 (±other agents) | Atezolizumab | III | First line, MSI-H mCRC |
mFOLFOX6/bevacizumab ± Atezolizumab | Ongoing | PD-L1, T cell, cfDNA mutations | NCT02997228 |
| II | Refractory | Atezolizumab+Stereotactic Radiotherapy | NA | NA | NCT02992912 | ||
| II | Refractory | Atezolizumab+Bevacizumab | Ongoing | MRR | NCT02982694 | ||
| III | Stage III, MSI-H CRC |
FOLFOX6± Atezolizumab | Ongoing | MMR | NCT02912559 | ||
| Avelumab | II | After first line, MSI-H mCRC or POLE mut |
Avelumab | Ongoing | POLE mutations, MMR |
NCT03150706 | |
| second line, MSI-H mCRC |
Avelumab versus FOLFOX or FOLFIRI (±targeted treatment) | Ongoing | MSI-H | NCT03186326 | |||
| Durvalumab | II | Refractory, MSI-H mCRC |
MEDI4736 (Durvalumab) | ORR=8% | MSI-H | NCT02227667 | |
| II | Refractory | MEDI4736 +Trametinib (anti-CTLA-4) versus BSC | NA | NA | NCT02870920 | ||
| II | Refractory, MSS mCRC |
MEDI4736 +Trametinib ± Radiotherapy | Ongoing | PD-L1; T-cell infiltration |
NCT02888743 | ||
| II | Refractory, advanced solid tumors including MSS mCRC | MEDI4736 +Azacitidine | Ongoing | MMR | NCT02811497 | ||
| Novel targets | PD-1/PD-L1 inhibitors+novel agents | I/II | Refractory | Pembrolizumab+BBI608 (Napabucasin, STAT3 and cancer cell stemness inhibitor) | ORR=5% in MSI-H | MMR, PD-L1, CD8 |
NCT02851004 |
| I/II | Refractory | Pembrolizumab+Poly ICLC (TLR3 agonist) | Ongoing | NA | NCT02834052 | ||
| I/II | Refractory | Pembrolizumab+AMG820 (anti-CSF1R) | Ongoing | NA | NCT02713529 | ||
| I/II | Refractory, advanced solid tumors | BMS-986015 (KIR inhibitor)+Nivolumab ± Ipilimumab | Ongoing | NA | NCT01714739 | ||
| I | Refractory, advanced solid tumors including mCRC | Nivolumab+Enadenotucirev (oncolytic virus) | NA | NA | NCT02636036 | ||
| I | Refractory, advanced solid tumors including mCRC | TSR-022 (TIM3 inhibitor)±Nivolumab | NA | NA | NCT02817633 | ||
| I | Refractory, advanced solid tumors including mCRC | Atezolizumab+CPI-444 (adenosine-A2A receptor Iinhibitor) | NA | NA | NCT02655822 | ||
| I | Refractory, advanced solid tumors | Atezolizumab+MOXR0916 (OX-40 inhibitor) | NA | NA | NCT02410512 | ||
| I/II | Refractory, advanced solid tumors including MSS mCRC | Durvalumab+Olaparib and/or Cediranib | Ongoing | PD-L1 | NCT02484404 | ||
| I | Refractory | Durvalumab+PEXIDARTINIB (anti-CSF1R) | NA | NA | NCT02777710 | ||
| I | Refractory, advanced solid tumors | MEDI6469 (OX-40 inhibitor)+Durvalumab or Tremelimumab | NA | NA | NCT02705482 |
BSC, best supportive care; CRC, colorectal cancer; CSF1R, colony stimulating factor-1 receptor; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; FOLFOX, 5 fluorouracil, leucovorin, oxaliplatin; GC, gastric cancers; IDO, indoleamine-pyrrole 2,3-dioxygenase; IHC, Immunohistochemestry; LAG3, lymphocyte activation gene 3; mCRC, metastatic CRC; MMR, DNA mismatch repair; MSI-H, microsatellite instablility high; MSS, microsatellite stable; NA, not available/not applicable; NCT, national clinical trial; ORR, objective response rate; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; pMMR, MMR proficient; RT, Radiotherapy; SOC, standard of care; TILs, tumor-infiltrating lymphocytes; TKI, tyrosine kinase inhibitor; TLR, toll-like receptor; XELIRI, capecitabine, irinotecan.
Table 2.
Main ongoing trials with immune checkpoint inhibitors in gastric, GEJ and esophageal cancers
| Target | Agent | Phase | Setting (treatment line) |
Treatment | Main objectives | Biomarker | NCT identifier |
| PD-1 (±other agents) | Pembrolizumab | Ib | Refractory PD-L1 +solid tumors | Pembrolizumab | ORR=30% | PD-L1 ≥1% |
NCT02054806 (KEYNOTE-028) |
| III | Second line | Pembrolizumab versus SOC | mOS=8.2 vs 7.1 months. In PD-L1-positive: mOS=9.3 vs 6.7 months |
PD-L1 (CPS ≥10%) |
NCT03933449 (KEYNOTE-181) |
||
| III | First line | Pembrolizumab+chemotherapy versus placebo +chemotherapy | Ongoing | PD-L1 (CPS ≥10%) |
NCT03189719 (KEYNOTE-590) |
||
| II | Cohort 1: refractory Cohort 2: first line Cohort 3: first line PD-L1+ |
Cohort 1: pembrolizumab monotherapy Cohort 2: pembrolizumab +chemotherapy Cohort: Pembrolizumab monotherapy |
Ongoing | PD-L1 |
NCT02335411 (KEYNOTE-059) |
||
| III | Second line | Pembrolizumab versus paclitaxel | Ongoing | PD-L1 positive |
NCT02370498 (KEYNOTE-061) |
||
| III | First line | Pembrolizumab or pembrolizumab +chemotherapy or placebo plus chemotherapy | Ongoing | PD-L1 positive |
NCT02494583 (KEYNOTE-062) |
||
| III | Neoadjuvant/adjuvant | Pembrolizumab+chemotherapy versus placebo +chemotherapy | Ongoing | NA |
NCT03221426 (KEYNOTE-585) |
||
| III | First line, HER2+ | Pembrolizumab+Trastuzumab + Chemotherapy versus Placebo+Trastuzumab + Chemotherapy | Ongoing | NA |
NCT03615326 (KEYNOTE-811) |
||
| Nivolumab | III | Refractory | Nivolumab versus placebo | mOS=5.26 vs 4.14 months 12 month OS=26.2% vs 10.9% |
NA |
NCT02267343 (ATTRACTION-2) |
|
| III | Refractory | Nivolumab versus chemotherapy | median OS=10.9 vs 8.4 months | PD-L1 |
NCT02569242 (ATTRACTION-3) |
||
| II/III | First line | Nivolumab +chemotherapy (Nivolumab+SOX vs Nivolumab+CapeOX) |
ORR=57.1% with nivolumab plus SOX vs 76.5% with nivolumab plus CapeOX. mPFS=9.7 months vs 10.6 months |
PD-L1 |
NCT02746796 (ATTRACTION-4) |
||
| I/II | Refractory | Nivolumab versus Nivolumab+Ipilimumab | ORR=12% with Nivolumab; vs 24% with Nivo 1+Ipi 3; vs 8% with Nivo 3+Ipi 1 | NA |
NCT01928394 (CHECKMATE 032) |
||
| III | First line | Nivolumab+Ipilimumab or Nivolumab+chemotherapy versus chemotherapy | Ongoing | PD-L1 | NCT02872116 | ||
| III | Adjuvant, resected Esophageal and GEJ cancers | Nivolumab versus placebo | Ongoing | NA |
NCT02743494 (CHECKMATE 577) |
||
| III | First line | O | PD-L1 ≥1% |
NCT03143153 (CHECKMATE 648) |
|||
| PD-L1 (±other agents) | Durvalumab | II | Localized unresectable Esophageal cancer | Durvalumab+chemoradiotherapy followed by durvalumab maintenance | Ongoing | NA | NCT03777813 |
| Avelumab | I | Refractory | Avelumab | NA | NA | NCT01772004 | |
| III | Third line, refractory | Avelumab versus chemotherapy | ORR=2.2% vs 4.3%; mOS=4.6 vs 5 months | PD-L1 |
NCT02625623 (JAVELIN 300) |
||
| III | Maintenance, after first line | Avelumab versus chemotherapy | Ongoing | NA |
NCT02625610 (JAVELIN 100) Gastric and GEJ |
||
| II | Neoadjuvant | Avelumab+chemotherapy | Ongoing | NA | NCT03288350 |
CPS, Combined Positive Score; GEJ, gastroesophageal junction; mOS, median overall survival; mPFS, median progression-free survival; NA, not available; NCT, national clinical trial; ORR, objective response rate; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; SOC, standard of care; SOX, S-1 (tegafur, gimeracil, and oteracil potassium) plus oxaliplatin.
2. Brief overview of immunotherapy biomarkers in GI cancers
PD-L1, also called CD274 or B7 homolog 1, has been identified to be one of the binding and functional partners of PD-1, a cell surface coinhibitory receptor that induces immune inhibition and promotes tumor immune escape from cytotoxic T cells.18 19 Furthermore, PD-L1 has been also shown to collaborate with CD80, a functional ligand for cytotoxic T lymphocyte antigen-4 (CTLA-4), to mediate an immune-inhibitory signal. PD-1 and CTLA-4 mediated modulation of immune checkpoints is critical to regulate the duration and amplitude of immune responses and lead to the development of an exhausted T cell phenotype. PD-1 is expressed on a large proportion of tumor-infiltrating lymphocytes (TILs) from many cancer types, whereas PD-L1 is expressed in 20%–50% of human tumors.20 Hence, immune checkpoint blockade strategies targeting PD-1/PD-L1 have been developed to reduce the effect of tumor immune escape and rescue the cytotoxic cell-induced immune response. The role of PD-L1 as a biomarker of immunotherapy efficacy has been evaluated in several studies across different cancer types, showing conflicting results depending on tumor types21 22 (further discussed in the next paragraphs).
Initial results by Le et al, first published in 2015, reported for the first time the efficacy of PD-1 blockade in MSI-H tumors.23 MMR is a highly conserved DNA repair mechanism which is critical to recognize and correct erroneous nucleotide insertion, deletion and mis-incorporation during DNA replication and recombination, thus ensuring genomic integrity. dMMR determines the accumulation of DNA replication errors which translates into high frequency of frameshift mutations in microsatellite DNA (MSI), leading to a high somatic mutational burden (mutator phenotype).24 25 The role of MSI as a predictive biomarker is now well established, particularly in CRC.26 The prevalence of MSI in CRC varies depending on the tumor stage. The incidence is higher in early stages: about 20% in stage I–II and 12% in stage III, and lower in the metastatic setting: 4%–5%. About one-quarter of dMMR CRCs are due to Lynch syndrome, an autosomal dominant hereditary disease characterized by germline mutations in the MMR genes (ie, MLH1, MSH2, MSH6, PMS2 or EPCAM deletions leading to epigenetic inactivation of MSH2),27 while the remaining cases are defined as ‘sporadic’. The majority (80%–90%) of sporadic cases result from epigenetic silencing of the MLH1 gene promoter by hypermethylation,28–30 a phenomenon associated with a high CpG island methylation phenotype.31 32 The concomitant presence of BRAF V600E mutation can be identified in about 30% of dMMR CRC, limited to sporadic MSI.31 The MSI-H phenotype is characterized by distinct clinical and pathological features compared with those observed in microsatellite stable (MSS) CRC, such as prominent lymphocytic infiltrate, mucinous histology and poor differentiation, and right-sided colon location.33 MSI/dMMR testing is recommended by current international guidelines to assess the eligibility to treatment with ICI in mCRC and other metastatic GI cancers.
An emerging biomarker of response to anti-PD1/PDL1 therapies is the TMB34 35 which quantifies the number of somatic mutations in the tumor. However, tumors containing high mutational burden may exhibit variable responses suggesting that additional factors may contribute to antiPD1/PDL1 response. Lee and Ruppin36 evaluate systematically 36 different variables associated to anti-PD1/PDL1 response of 3 distinct classes: (1) tumor neoantigens, (2) tumor microenvironment and (3) checkpoint target. This analysis of multiomics data from the Cancer Genome Atlas cohort and ORRs to therapy data across 21 cancer types shows that estimated CD8 +T cell abundance is the most predictive biomarker, followed by TMB and the fraction of samples with high PD-1 gene expression. In a recent study within a large cohort of GI cancer, authors aimed to determine TMB, MSI-H and PD-L1 expression interrelationship in GI cancers.17 They found that the TMB-high rate varied widely among GI cancers. Although MSI-H is conceivably the main driver for TMB-high, other factors may be involved and higher PD-L1 expression was more likely to be seen in MSI-H compared with MSS tumors (20.6% vs 7.8%, p<0.0001).
On the other hand, research efforts are underway to identify biomarkers associated with resistance to ICI. The MDM2 proto-oncogene encodes a nuclear localized E3 ubiquitin ligase with the core function of inhibiting the tumor suppressor p53. MDM2 amplification has been reported in multiple tumor types and is a hallmark of tumorigenesis.37 Recently MDM2 amplification also has been implicated as a potential marker for accelerated tumor growth after checkpoint inhibitors treatment, a phenomenon known as hyperprogression, affecting approximately 9% of patients who receive PD-1/PD-L1 inhibitors.38 39 To date, hyperprogression after anti-PD-1/PD-L1 agents has been reported by at least four groups, however, the mechanisms that mediate this phenomenon remain unclear and the only markers that have been shown to correlate with this occurrence are MDM2 family gene amplifications and epidermal growth factor receptor (EGFR) alterations.40
The role of selected biomarkers according to different cancer types will be further addressed in the next paragraphs.
3. Immune checkpoint inhibitors in GI cancers
3.1 Colorectal cancer
The prominent predictive value of MSI assessment in CRC has emerged following the groundbreaking results of immunotherapy with checkpoint inhibitors (ie, anti-CTLA4 and PD-L1/PD-1 inhibitors) in dMMR mCRC.26 First, in the phase II KEYNOTE-016 trial, the anti-PD-1 pembrolizumab demonstrated its activity in 28 MSI-high mCRC patients with chemorefractory disease.23 41 Shortly after, the combination of the anti-CTLA4 ipilimumab and the anti-PD-1 nivolumab, investigated in the phase II Checkmate-142 trial, showed significant results in the same setting.42 43 Complete radiological responses and long-term durable responses were observed in both trials, suggesting an unprecedented rate of long-term survival among heavily pretreated chemorefractory patients. Notably, responses in the Checkmate 142 study were irrespective of immune cell PD-L1 expression, tumor BRAF mutational status and clinical history of Lynch syndrome. Based on these striking results, the FDA granted approval for the use of pembrolizumab44 and nivolumab42 in the treatment of MSI-high or dMMR mCRC. More recently, accelerated approval was granted to ipilimumab in combination with nivolumab for MSI-high or dMMR mCRC that have progressed following treatment with a fluoropyrimidine, oxaliplatin and irinotecan.45
For dMMR CRC, immunotherapy is being explored in front-line, adjuvant and neoadjuvant settings for non-metastatic tumors.46 47 The KEYNOTE-177 is a phase III trial (NCT02563002) evaluating first-line pembrolizumab in stage IV dMMR or MSI-H CRC.48 During the ESMO 2018 Congress, Chalabi et al reported the first neoadjuvant study testing ipilimumab plus nivolumab in 14 early-stage (I–III) dMMR and MMR proficient (pMMR) colon cancers.49 In this study, patients with resectable, early-stage disease received ipilimumab 1 mg/kg on day 1 and nivolumab 3 mg/kg on days 1 and 15, followed by surgery. Treatment was well tolerated, and all patients could undergo radical resection without any delays. More importantly, major pathological responses (defined as <5% viable tumor cells) were observed in 100% of dMMR colon cancers, with 57% complete responses. No major pathological responses were observed in pMMR tumors, which displayed little to no tumor regression. These very promising results warrant further investigation. The Alliance for Clinical Trials in Oncology is currently investigating the benefit of combining the PD-L1 inhibitor atezolizumab with standard FOLFOX compared with FOLFOX alone in the treatment of patients with stage III MSI-H/dMMR colon cancers (NCT02912559).50
Hence, MSI status has become a crucial biomarker to define the therapeutic options in the metastatic setting. On the other hand, studies evaluating the prognostic significance of PD-L1 expression in CRC are limited and remain controversial.51 52
In pMMR or MSS CRC, that comprise the vast majority of patients, immunotherapy have rather limited benefit, if any.53 However, recently, the results of the Cancer Trials Group CO.26 study have shown that the combination of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) prolonged median overall survival (OS) by 2.5 months compared with best supportive care alone in patients with advanced treatment-refractory mCRC.54 More recently, a phase Ib trial (EPOC1603) tested safety and toxicity profile of regorafenib combined with nivolumab in previously treated, advanced gastric cancer (GC) or CRC. Objective tumor response was observed in 19 patients (38%) including 11 MSS GC, 7 MSS CRC and 1 MSI-H CRC for response rates of 44% in GC and 29% in MSS CRC.55 The rather promising results of these studies suggest that the benefit of immune checkpoint blockade therapy, especially in association with targeted therapy, may not be limited to patients with MSI-H or dMMR CRC. Although encouraging, these findings warrant further evaluation in phase II–III confirmatory trials.
Several trials are currently investigating anti PD-1/PD-L1 in different settings for the treatment of mCRC, including earlier lines of treatment, and different combination strategies with or without chemotherapy or other targeted agents. Results from these studies are eagerly awaited.
3.2 Esophageal cancer
Esophageal cancers show a high somatic mutation rate,56 and a high rate of PD-L1/PD-L2 overexpression.57 Several trials with ICIs are ongoing (table 2).
The multicohort phase Ib KEYNOTE-028 trial evaluated the anti PD-1 monoclonal antibody pembrolizumab as single agent in a series of PD-L1-positive esophageal cancer (including both squamous cell carcinomas (SCC), and esophageal adenocarcinomas (EAC)) after failure of standard chemotherapy. Results were promising showing an overall response rate of 30.4% and 52.2% in SCC and EAC, respectively, with a 12-month progression free rate of 21.7%, in a heavily preatreated population.58 More recently, results from the KEYNOTE-181 trial comparing pembrolizumab versus chemotherapy in the second-line treatment of advanced esophageal cancer showed a significantly improved OS in patients whose tumors tested positive for PD-L1 with a Combined Positive Score (CPS) of 10 or greater.59
On the other hand, recently reported results from the randomized phase III ATTRACTION-3 study (NCT02569242) showed a significant improvement in OS and a favorable safety profile from the anti-PD-1 nivolumab compared with chemotherapy in previously treated patients with advanced SCC, irrespective of tumor PD-L1 expression.60 Combined therapy with the anti-CTLA-4 antibody ipilimumab plus nivolumab has been tested in the Checkmate-032 study, enrolling patients with locally advanced or metastatic chemotherapy-refractory gastric, esophageal or gastroesophageal junction (GEJ) cancers. Results from this study showed encouraging antitumor activity, both in terms of durable responses and OS, with a manageable safety profile from the combination treatment. Notably, responses were observed regardless of tumor PD-L1 status.
Phase III studies evaluating anti PD-1 treatments (either nivolumab or pembrolizumab, alone or in combination with chemotherapy) in earlier treatment lines and in the adjuvant setting are underway and will provide additional efficacy and biomarker data (ie, NCT02872116, NCT02743494, NCT03189719, NCT03143153). Another strategy under investigation is the use of the anti PD-L1 durvalumab in combination with definitive radiotherapy delivered with oxaliplatin-based chemotherapy in locally advanced unresectable esophageal cancer (NCT03777813). Ancillary biomarker studies are planned.
3.3 Gastric cancer
The Cancer Genome Atlas network divides GC into four molecular subtypes: Epstein-Barr virus (EBV)-positive tumors (9%), MSI tumors (22%), genomically stable tumors (20%) and tumors with chromosomal instability (50%).61 High PD-L1 expression is characteristic of the EBV and MSI subtypes,62 63 in addition to the high tumor mutational load in MSI-high tumors, which has been shown to correlate with a greater benefit from anti-PD-1/PD-L1 blockade,64 supporting the detection of PD-L1 and the assessment of EBV and MSI status as key biomarkers in immunotherapy for GC. Furthermore, PD-L1 overexpression is associated with large tumors, lymph node metastasis and a poor prognosis in GC.65 66
Based on the positive results of the KEYNOTE-059 trial, pembrolizumab has been approved by the FDA for the treatment of PD-L1-positive (>1%) advanced GC or GEJ adenocarcinoma.67 68 A preplanned analysis of the study, in fact, showed a significantly higher response rate in PD-L1-positive tumors compared with PD-L1 negative ones.
More recently, the Japanese Ministry of Health, Labor and Welfare approved nivolumab for the treatment of unresectable advanced or recurrent GC progressing after at least two lines of chemotherapy. The approval was based on positive results of the phase III ATTRACTION-2 (ONO-4538–12) trial, showing a significant reduction in risk of death and an increased OS rate at 12 months from nivolumab when compared with placebo in 493 Asian patients.69 Notably, no predictive biomarker has been required for this indication. The ATTRACTION-4 phase III trial is currently underway to evaluate nivolumab in combination with chemotherapy in the first-line setting for patients with unresectable advanced or recurrent GC or GEJ cancer irrespective of PD-L1 expression (NCT02746796). Additionally, pembrolizumab is under investigation in several different settings. The phase III KEYNOTE-061 compared pembrolizumab versus paclitaxel as second-line treatment in patients with advanced GC or GEJ cancer, however, no significant OS benefit was observed in this setting for tumors with PD-L1 CPS of 1 or higher (primary endpoint).70 The phase III KEYNOTE-062 tested pembrolizumab as a monotherapy and in combination with chemotherapy in the first-line treatment of PD-L1-positive (CPS >1) advanced GC or GEJ cancer. It has been recently reported that in the monotherapy arm of the study, pembrolizumab showed non-inferiority to chemotherapy in terms of OS in the entire intention-to-treat population. However, in the combination arm pembrolizumab plus chemotherapy was not superior to chemotherapy alone, neither for OS nor for progression-free survival (PFS).71 In an exploratory biomarker analysis, patients with higher levels of PD-L1 (CPS ≥10) showed a clinically meaningful improvement in OS from pembrolizumab versus chemotherapy, and patients with MSI-high tumors experienced a greater clinical benefit from both pembrolizumab alone and in combination with chemotherapy, in terms of response rates, PFS and OS.72 Of interest, the phase III KEYNOTE-585 (NCT03221426) is evaluating the combination of pembrolizumab with chemotherapy in the neoadjuvant and adjuvant settings, while the KEYNOTE-811 trial (NCT03615326) is investigating treatment with pembrolizumab plus trastuzumab and chemotherapy for HER2-positive metastatic GC and GEJ cancer with a stratification based on PD-L1 status.
Another strategy under study for immunotherapy in gastroesophageal cancers is targeting PD-L1. The fully human anti-PD-L1 IgG1 antibody avelumab has been investigated as a first-line maintenance or second-line treatment in patients with advanced GC or GEJ cancer with initial promising results (NCT01772004).73 However, in the phase III JAVELIN Gastric 300 trial (NCT02625623), avelumab as a third-line treatment for advanced GC and GEJ adenocarcinoma, unselected for PD-L1 expression, failed to show improvements in OS or PFS compared with chemotherapy alone.74 Results of the phase III JAVELIN Gastric 100 trial evaluating avelumab as first-line maintenance therapy following induction chemotherapy in gastric or GEJ cancer are awaited.75 In the perioperative setting, encouraging safety and activity results have been recently reported from the interim analysis of a phase II trial testing avelumab plus chemotherapy in locally advanced gastric and esophagogastric adenocarcinoma (NCT03288350).76
3.4 Other GI cancers
Hepatocellular carcinoma (HCC) is the third-leading cause of cancer deaths worldwide and its incidence is increasing. Recently, both pembrolizumab (KEYNOTE-224) and nivolumab (CheckMate 040) showed antitumoral activity in the second-line setting for advanced HCC as monotherapy in phase I/II trials with an ORR of 17% and 20%, respectively, and received FDA approvals.77 78 Accordingly, the association of nivolumab plus ipilimumab showed an ORR of 35%, with complete response of 12% with a manageable toxicity profile.79 Thus, (FDA) approved on 11 March 2020 the combination of nivolumab plus ipilimumab for the treatment of patients with advanced HCC, who were previously treated with sorafenib (Nexavar). Moreover, despite the potential concern for relatively worse toxicity related to ICI due to already poor liver function in HCC patients, overall clinical trials have shown an acceptable safety profile for HCC population.80 81 Recently, immunotherapy showed a significant benefit in HCC patients also in first line: the treatment with atezolizumab combined with bevacizumab (IMbrave150) administered as first line in patients with unresectable HCC significantly prolonged OS (HR=0.58; p = 0.0006), PFS (HR=0.59, p < 0.0001) and improved ORR (27% vs 12%) compared with sorafenib: these results may be practice changing in the first-line setting for HCC treatment.82 Several other approaches are ongoing to demonstrate the activity of ICIs in association with locoregional therapies, including radiofrequency ablation, surgical resection and transcatheter arterial chemoembolization (TACE), which play a pivotal role in the treatment of early stage/locally advanced HCC.83–85
Biliary tract carcinoma is a group of rare GI cancers with poor prognosis with limited treatment options. Biliary tract cancers represent a potentially attractive target for immune-based therapies given the background association with chronic inflammation and conditions such as cholecystitis, sclerosing cholangitis and primary biliary cirrhosis.86 However, to date, immunotherapy has not proven to be effective in these patients and several studies are ongoing with different approaches: combination of immune checkpoints with (1) chemotherapy (NCT04003636, NCT03111732,), (2) Locoregional treatments such as cryoablation or radiofrequency ablation (RFA) (NCT02821754), (3) radiotherapy (NCT03482102), (4) novel target such as DKK1-neutralizing monoclonal antibody DKN-01 (NCT04057365) and CD-40 (NCT03329950); and adoptive transfer of autologous TILs (NCT03801083). Results from these trials are eagerly awaited.
Pancreatic ductal adenocarcinoma (PDAC) is associated with poor prognosis and the ineffectiveness of immunotherapy in pancreatic cancer may be explained by these tumors being non‐immunogenic. Indeed, checkpoint inhibitors such as anti-PD-1 are ineffective as single agents for patients with PDAC. However, recently the combination of APX005M, an anti-CD40 antibody, with nivolumab and standard chemotherapy (gemcitabine with nab-paclitaxel) showed manageable safety profiles and promising antitumor activity in untreated metastatic PDAC patients (ORR 58%).87
Anal SCC (ASCC) is a rare malignancy for which there are limited treatment options for patients with progressive disease after platin-based first-line therapy. Thus, immunotherapeutic approaches have been of great interest in the development of new treatments for this HPV-related driven tumor. After the multicohort phase 1b KEYNOTE-028 trial, which showed the activity of pembrolizumab in patients with heavily pretreated PD-L1-positive advanced or recurrent cancers (including ASCC), the phase 2 KEYNOTE-158 trial was designed to further explore the safety and efficacy of pembrolizumab in pretreated adults with advanced solid tumors. Recently, data were presented at ASCO GI symposium 2020: at a median follow-up of 12 months, the ORR was 11.6%, with 5 and 8 patients achieving a complete response and a partial response, respectively; the median PFS and median OS for all patients with advanced ASCC was 2 months and 12 months, respectively, regardless of PD-L1 status.88 Novel immune-based approaches building on translational work with next-generation immunotherapy trials are warranted, with the goal of broadening the clinical benefit of immunotherapy in this population.89
4. Resistance to immunotherapy: cold and hot tumors
The immune anticancer response is modulated by a complex and dynamic interplay between tumor cells, the host immune system, and the associated microenvironment. ICI have been described to be more active in ‘hot’ tumors, characterized by a prominent lymphoid infiltration of the stroma and high density of TILs coupled with an inflammatory cytokine-rich microenvironment and a T cell-inflamed gene expression profile90 (figure 1). These features suggest the presence of a pre-existing immune response, which the tumor was able to escape employing several different mechanisms including: checkpoints activation leading to T-cell exhaustion; activation of oncogenic pathways; and interaction with the tumor microenvironment to promote an immunosuppressive status.91 An important characteristic of ‘hot’ tumors is the high TMB, which reflects an increased number of nonsynonymous, single nucleotide variants that may lead to an increased neoantigen production promoting the antitumor immune response. Indeed, cancer types harboring high somatic mutational burdens (eg, melanoma, NSCLC and bladder cancer) are among those with the highest response rates to ICI.14 34 92 Genetic instability due to alterations in DNA repair and replication genes can increase immunogenicity through high mutational burden with subsequent neoantigen formation.93 Patients with alterations in BRCA and additional DNA damage response genes, including ATM, POLE, FANCA, ERCC2 and MSH6, have recently shown correlation with high TMB and improved clinical outcomes to immune checkpoints inhibitors.94 Accordingly, dMMR tumors, which display exceptionally high numbers of somatic mutations, show an enhanced response to PD-1 blockade.23 44
Figure 1.
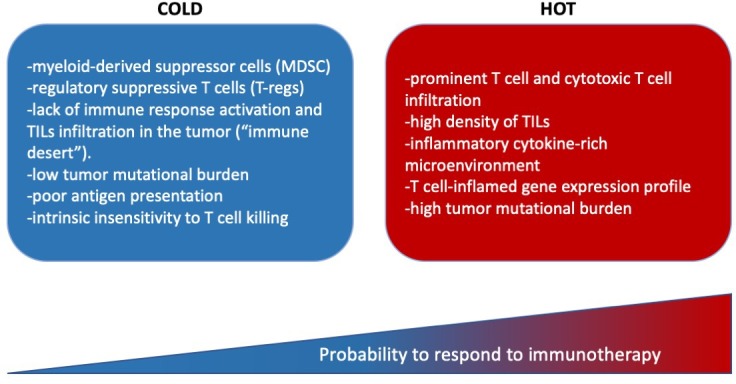
Main characteristic of ‘cold’ and ‘hot’ tumors. TILs, tumor-infiltrating lymphocytes.
On the other hand, cancers with low immune infiltrates are considered ‘cold’ tumors (e.g., pancreas, ovarian and prostate). These cancers are characterized by a tumor microenvironment that is enriched in myeloid-derived suppressor cells (MDSC) and regulatory suppressive T cells (T-regs), leading to a lack of immune response activation and TILs infiltration in the tumor (‘immune desert’).
A deeper understanding of the mechanisms behind ‘hot’ and ‘cold’ immune tumors is critical to overcome primary, adaptive and acquired resistance and to be able to boost a weak antitumor immunity.95 96 In fact, despite the encouraging success of ICI, roughly 60%–70% of advanced tumors are not responsive to single-agent treatment (primary resistance), and often those that initially respond become resistant to treatment over time (adaptive and acquired resistance).97 Extensive research is currently underway to develop novel strategies to overcome resistance to immunotherapies, which include fundamental efforts to transform immunologically ‘cold’ into ‘hot’ tumors.
Many mechanisms of resistance have been characterised to date, and more continue to be uncovered. By elucidating and targeting mechanisms of resistance, treatments can be tailored to improve clinical outcomes.93 Tumor-intrinsic mechanisms of immune evasion comprise genetic and epigenetic alterations (immunoediting) that influence the antitumor immune response, including loss of tumor antigen expression, loss of human leukocyte antigen) complex expression and alterations in the antigen processing machinery. For instance, loss of beta-2-microglobulin expression (due to deletions, point mutations, or loss of heterozygosity) results in impaired cell surface expression of major histocompatibility class I, which in turn impairs antigen presentation to cytotoxic T cells,98 thereby inducing resistance to PD-1 and CTLA-4 blockade.99 In addition, alterations in several key oncogenic signaling pathways, such as MAPK, PI3K, and WNT, can also lead to immune resistance.100 Loss of PTEN, which enhances PI3K signaling, has been found to be associated with resistance to immune checkpoint therapy.101 Constitutive PD-L1 expression and alteration of interferon (IFN) signaling are also considered mechanisms of resistance to immunotherapy.102–104 Finally, there is now emerging evidence that chromatin remodeling is involved in sensitivity and resistance to ICI, through mutations in SWI/SNF (SWItch/Sucrose Non-Fermentable) complexes, such as loss of ARID1A or inactivation of PBAF subunits.105 106
On the other hand, tumor-extrinsic mechanisms also participate in the establishment of immune resistance through processes driven by stromal and immune elements of the tumor microenvironment. Mounting evidence supports the role of the host microbiome in response to cancer treatment, with several recent studies demonstrating the influence of the gut microbiota on the response to immune checkpoint blockade across cancer types.107 108 Other tumor-extrinsic mechanisms include changes in immune checkpoint expression (eg, CTLA-4 and PD-1), T-cell exhaustion and phenotype change, recruitment of immune suppressive cell populations (eg, T-regs, MDSC and type II macrophages), and cytokine or metabolite release in the tumor microenvironment (eg, colony stimulating factor (CSF)-1, tryptophan metabolites, transforming growth factor-β and adenosine).109
5. Overcoming resistance to anti-PD1 and anti-PD-L1 therapy
Several approaches have been proposed and are currently under evaluation in the attempt to overcome the mechanisms of resistance to ICI. Combination strategies using multiple treatment modalities are emerging to surmount the lack of a pre-existing immune response, eventually turning cold tumors into hot tumors. A strong clinical and preclinical rationale support the use of molecularly targeted therapy in combination with immunotherapy. The most extensively studied cancer types treated with this strategy are melanoma and NSCLC, but, theoretically, it could apply to any cancer.110 This approach relies on targeted therapies either (1) inducing a rapid tumor death leading to the release of tumor-associated antigens or (2) modulating key cellular pathways that allow the cancer to maintain an adaptive resistance. For instance, there is accumulating evidence that bevacizumab, an anti-VEGF monoclonal antibody, has immunomodulatory properties, as VEGF exerts immunosuppressive functions through inhibition of dendritic cell maturation, reduction of T-cell tumor infiltration, and promotion of inhibitory cells in the tumor microenvironment. Hence, there is compelling rationale for the combination of ICI with anti-VEGF therapies; several clinical trials are evaluating such combinations across tumor types.111 Similar early favorable results have been reported with anti-BRAF, anti-MEK, anti-EGFR agents, among others. Less commonly targeted pathways, among those that could be involved in immunotherapy resistance, include PI3K-AKT-mTOR, hypoxia-inducible factor, adenosine, JAK/STAT and Wnt/β-catenin. Epigenetic modifications of cancer DNA are known to cause changes in immune-related gene expression, which can impact antigen processing and presentation as well as promote immune evasion. Therefore, demethylating agents may enable re-expression of immune-related genes, with the potential for therapeutic impact, especially in the setting of combination treatment with immunotherapy.112 113
Strategies to promote formation or presentation of suitable neoantigens in tumors with a non-inflamed, non-immune cell infiltrated tumor microenvironment are also being exploited through the use of chemotherapy and radiation (by inducing immunogenic cell death) or by stimulating innate immune responses and dendritic cell function (eg, type I IFN, Toll-like receptor ligands, LIGHT and oncolytic viruses).114 Over the two past decades, a growing body of preclinical and clinical evidence have supported the immunomodulatory effects of radiotherapy.115 116 In fact, the delivery of radiotherapy and the resulting induction of immunogenic cell death pathways can potentially convert the tumor into an in situ (and thus personalized) vaccine. This phenomenon of radiation shrinking the tumor locally while inducing an immune response systemically is known as the ‘abscopal’ effect. Several clinical trials are investigating the efficacy of immunotherapy in combination with radiation therapy across different tumor types, especially in the metastatic setting. Although many questions remain unresolved (eg, sequencing, timing and radiation fractionation) and data analysis is ongoing for this approach that is already being tested in the clinic, immunotherapy combination with radiation therapy may become a novel and effective approach to treat patients with cancer in the near future.
Cancer vaccines have shown promising results as a means of personalizing cancer immunotherapy and potentially enhancing immune memory in a minority of patients, such as NY-ESO-1 cancer vaccines.117 118 True success in this realm may be achieved with the identification of a pan-cancer antigen that can be targeted through vaccination.
6. Future perspectives
Novel combined approaches to enhance immunotherapy exploits the combination of ICI with new negative checkpoint regulators (NCRs), for example, anti-lymphocyte activation gene 3, a coinhibitory receptor on T cells that, among other functions, enhances activity of regulatory T (Treg) cells and regulates T cell proliferation, differentiation and effector function. Other likely targets include additional coinhibitory receptors such as TIM3 (a marker for exhausted T cells); T cell immunoglobulin and ITIM domain (TIGIT, which counterbalances the costimulatory function of CD226, that is rapidly induced following T cell activation); B and T lymphocyte attenuator (also known as CD272), which is expressed by T cells and synergizes with herpesvirus entry mediator (also known as TNFRSF14), expressed on antigen-presenting cells; sialic acid-binding Ig-like lectin 9, which is upregulated in TILs and possibly determines a subclass of tumor-specific CD8 +TILs; and V-domain Ig suppressor of T cell activation (VISTA).119 VISTA is one of the most recently NCRs and can act as both a ligand and a receptor on T cells to inhibit T cell effector function and maintain peripheral tolerance.120 VISTA is produced at high levels in TILs, such as MDSCs and regulatory T cells, and its blockade with an antibody results in delayed tumor growth in mouse models of melanoma121 and SCC.122 A recent study gives a detailed analysis of immune infiltration in primary and metastatic pancreatic tumors compared with melanoma. These data indicate that human pancreatic tumors express CD68 +macrophages and highlight the inhibitory checkpoint molecule VISTA as a potential immunotherapeutic pancreatic cancer.123
Another strategy is the combination of ICI with costimulatory checkpoint molecules, such as anti-ICOS (inducible T cell co-stimulator), OX40 antigen (also known as TNFRSF4 (or CD134)), TNFRSF7 (also known as CD27), CD28, TNFRSF9 (also known as 4-1BB ligand receptor or CD137) and glucocorticoid-induced TNFR-related protein, all of which enhance T cell expansion and effector functions while controlling Treg cell suppressive functions.124 However, despite promising results, the use of costimulatory molecules could be clinically limited by systemic toxicity.
A proposed approach to turn cold tumors into hot tumors is to combine a priming therapy that enhances T cell responses (such as vaccines, adoptive T cell transfer (ACT), oncolytic viruses) with ICI. At present, only one oncolytic virus, talimogene laherparepvec, a genetically modified herpes simplex-1 virus expressing granulocyte-macrophage CSF, has been approved by the FDA for the local treatment of patients with unresectable melanoma. Local administration of therapeutic (oncolytic) viruses, that have a tropism for malignant cells, causes cancer cell lysis and/or death and can induce abscopal effects with reductions in the volume of non-injected lesions.125 Growing preclinical evidence indicates that non-virally induced oncolysis, defined as chemical or physical treatment (electrochemotherapy, locally injected cytotoxins, photodynamic therapy, laser therapy, microwave, radiofrequency or photothermal ablation, high-intensity focused ultrasonography and cryotherapy) administered locally to destroy malignant lesions, can promote a similar effect owing to the release of danger-associated molecular patterns that lead to the recruitment of immune cells, thus inducing a systemic response against tumor antigens that protects against local disease relapse and also mediates distant antineoplastic effects. This abscopal effect is a direct consequence of the immune response triggered by oncolysis and can be supported by localized or systemic immunotherapy.126
Among passive immunization strategies, ACT is becoming an increasingly important component of the antitumor arsenal: examples include TILs, transgenic T cell receptor and chimeric antigen receptor (CAR) T cells—which have been approved by the FDA for the treatment of B cell acute lymphoblastic leukemia and B cell non-Hodgkin's lymphoma.127 128 Several researchers are investigating the use of CAR T-cell therapy for the treatment of solid tumors. These studies require the prior identification of new antigens and the development of preclinical models of solid tumor in which to characterize therapies based on these antigens. Various types of CAR-T cells have been engineered to target HER2, EGFR and CEA, which are overexpressed on the surface of cells in GI tumors and are associated with the development and metastasis of tumors.129
Recent developments in the fields of nanotechnology and bioengineering afford new approaches that can dramatically improve not only the safety but also the efficacy of cancer immunotherapy by modifying their spatiotemporal release profiles.130
There is growing evidence that the gut microbiome plays an important role in cancer treatment. According to experimental studies several bacteria are involved in CRC carcinogenesis, including Fusobacterium nucleatum and certain strains of Escherichia coli and Bacteroides fragilis. The gut microbiota interacts with host cells to regulate many physiological processes, such as metabolism and immune response.131 The gut microbiome was initially found to modulate the response to immune checkpoint blockade in mouse models. Mice with different microbes had significant differences in response to treatment.132 133 Many of these findings have been verified in patients with metastatic melanoma receiving ICI. Several studies demonstrate the significant association between commensal microbial composition and clinical response to anti-PD-1-based immunotherapy.134 Specific bacteria are positively correlated with immunotherapeutic response, including Akkermansia muciniphila,135 Ruminococcus champanellensis, B. fragilis, Bifidobacterium, Eubacterium limosum, Faecalibacterium spp and Alistipes shahii.131 Analysis of the gut microbiota and of the immunological profiling in the tumor microenvironment shows that the expression of cytotoxic T cell markers and antigen processing and presentation are augmented in patients with favorable gut microbiota compared with patients with unfavorable gut microbiota.136 Another study has identified a consortium of 11 bacterial strains that could enhance the action of ICI to inhibit tumor growth in a subcutaneous tumor model derived from a mouse colon adenocarcinoma cell line.137 These findings have led to the possibility of detecting these core bacteria as predictive biomarkers for immunotherapy response.
Furthermore, the gut microbiota can also be associated with the adverse effects of immunotherapy, such as ICI-associated colitis.132 Increased representation of the Bacteroidetes phylum correlates with resistance to ICI-associated colitis, whereas a paucity of genes in polyamine transport and vitamin B biosynthesis pathways are associated with increased susceptibility to colitis.138 A report of two human cases has demonstrated successful treatment of refractory immunotherapy-associated colitis by fecal microbiota transplantation (FMT), with gut microbial changes correlating with complete resolution of colitis up to 53 days after one dose and up to 78 days after two doses of FMT, respectively. This study reports for the first time that modifying gut microbiota by fecal transplantation can ameliorate refractory colitis as an adverse effect of immunotherapy.139
An early phase 1 study (NCT04130763) has been designed to improve the response rate of anti-PD-1 among patients with anti-PD-1 resistant/refractory digestive system cancers (including the esophagus, stomach and intestine) through the intervention on their gut microbiota. Specifically, the study will identify healthy subjects that have the intestine similar to patients with GI cancers who has responded to anti-PD-1 therapy, then extract the gut microbiota of these healthy subjects to product FMT capsule, and perform a rechallenge with anti-PD-1 immunotherapy combined with FMT for GI system patients with cancer whose anti-PD-1 treatment failed.140
7. Conclusion
In the last few years, we witnessed the extraordinary impact that immunotherapy has generated for some patients with selected cancers. Nonetheless, to date, the vast majority of GI cancers do not benefit from this treatment approach, due to either primary or acquired resistance to immunotherapy. Several combination approaches are under investigation to develop novel treatment strategies to enhance immunotherapy efficacy and to expand the available treatment options for patients with GI cancer.
Acknowledgments
Heinz-Josef Lenz has received clinical trial financial support from Merck Serono and Roche, honoraria for advisory board membership and lectures from Bayer, Boehringer Ingelheim, Genentech, Pfizer, Merck Serono and Roche, and travel/accommodations from Bayer, Merck Serono and Roche.
Footnotes
AP and FB contributed equally.
Contributors: Manuscript drafting: AP and FB. Directly provided contributions, read and approved the final manuscript: all authors.
Funding: This manuscript was partly supported by the National Cancer Institute (grant number P30CA014089), the Gloria Borges WunderGlo Foundation-The Wunder Project, the Dhont Family Foundation, the San Pedro Peninsula Cancer Guild, the Daniel Butler Research Fund, the Call to Cure Research Fund, and the Fong Research Project.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:1069–86. 10.1158/2159-8290.CD-18-0367 [DOI] [PubMed] [Google Scholar]
- 2.Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol 2018;62:29–39. 10.1016/j.intimp.2018.06.001 [DOI] [PubMed] [Google Scholar]
- 3.Mayer-Nicolai C, Schmitt E, Hartmann M, et al. Immune checkpoint inhibitors (ICI): An analysis of FDA’s and EMA’s decision patterns and times for initial approvals and extensions of indication. J Clin Oncol 2019;37:e14062. 10.1200/JCO.2019.37.15_suppl.e14062 [DOI] [Google Scholar]
- 4.Riley RS, June CH, Langer R, et al. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov 2019;18:175–96. 10.1038/s41573-018-0006-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer 2017;117:1–7. 10.1038/bjc.2017.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kok M, Chalabi M, Haanen J. How I treat MSI cancers with advanced disease. ESMO Open 2019;4:e000511 10.1136/esmoopen-2019-000511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cottrell TR, Taube JM. Pd-L1 and emerging biomarkers in immune checkpoint blockade therapy. Cancer J 2018;24:41–6. 10.1097/PPO.0000000000000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fusi A, Festino L, Botti G, et al. Pd-L1 expression as a potential predictive biomarker. Lancet Oncol 2015;16:1285–7. 10.1016/S1470-2045(15)00307-1 [DOI] [PubMed] [Google Scholar]
- 9.Sidaway P. Deglycosylated PD-L1 might be a better biomarker. Nat Rev Clin Oncol 2019;16:592. 10.1038/s41571-019-0261-x [DOI] [PubMed] [Google Scholar]
- 10.Ganesan A, Ahmed M, Okoye I, et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci Rep 2019;9:12392 10.1038/s41598-019-48826-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee H-H, Wang Y-N, Xia W, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell 2019;36:168–78. 10.1016/j.ccell.2019.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee M, Samstein RM, Valero C, et al. Tumor mutational burden as a predictive biomarker for checkpoint inhibitor immunotherapy. Hum Vaccin Immunother 2019:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters S, Cho BC, Reinmuth N, et al. Abstract CT074: tumor mutational burden (TMB) as a biomarker of survival in metastatic non-small cell lung cancer (mNSCLC): blood and tissue TMB analysis from MYSTIC, a phase III study of first-line durvalumab ± tremelimumab vs chemotherapy. Cancer Res 2019;79:CT074. [Google Scholar]
- 14.Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morihiro T, Kuroda S, Kanaya N, et al. Pd-L1 expression combined with microsatellite instability/CD8+ tumor infiltrating lymphocytes as a useful prognostic biomarker in gastric cancer. Sci Rep 2019;9:4633 10.1038/s41598-019-41177-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts J, Salaria SN, Cates J, et al. Pd-L1 expression patterns in microsatellite Instability-High intestinal adenocarcinoma subtypes. Am J Clin Pathol 2019;152:384–91. 10.1093/ajcp/aqz052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salem ME, Puccini A, Grothey A, et al. Landscape of tumor mutation load, mismatch repair deficiency, and PD-L1 expression in a large patient cohort of gastrointestinal cancers. Mol Cancer Res 2018;16:805–12. 10.1158/1541-7786.MCR-17-0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2017;19:1189–201. 10.1016/j.celrep.2017.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov 2013;12:130–46. 10.1038/nrd3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou W, Wolchok JD, Chen L. Pd-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv324 10.1126/scitranslmed.aad7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lote H, Cafferkey C, Chau I. Pd-1 and PD-L1 blockade in gastrointestinal malignancies. Cancer Treat Rev 2015;41:893–903. 10.1016/j.ctrv.2015.09.004 [DOI] [PubMed] [Google Scholar]
- 22.Stein A, Moehler M, Trojan J, et al. Immuno-oncology in Gi tumours: clinical evidence and emerging trials of PD-1/PD-L1 antagonists. Crit Rev Oncol Hematol 2018;130:13–26. 10.1016/j.critrevonc.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 23.Le DT, Uram JN, Wang H, et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res 1974;34:2311–21. [PubMed] [Google Scholar]
- 25.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006;7:335–46. 10.1038/nrm1907 [DOI] [PubMed] [Google Scholar]
- 26.Sahin IH, Akce M, Alese O, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–60. 10.1056/NEJMoa043146 [DOI] [PubMed] [Google Scholar]
- 28.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 1998;95:6870–5. 10.1073/pnas.95.12.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003;348:919–32. 10.1056/NEJMra012242 [DOI] [PubMed] [Google Scholar]
- 30.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–87. 10.1053/j.gastro.2009.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch HT, Lynch JF, Lynch PM. Toward a consensus in molecular diagnosis of hereditary nonpolyposis colorectal cancer (Lynch syndrome). J Natl Cancer Inst 2007;99:261–3. 10.1093/jnci/djk077 [DOI] [PubMed] [Google Scholar]
- 32.Kim JH, Bae JM, Cho NY, et al. Distinct features between MLH1-methylated and unmethylated colorectal carcinomas with the CpG island methylator phenotype: implications in the serrated neoplasia pathway. Oncotarget 2016;7:14095–111. 10.18632/oncotarget.7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raut CP, Pawlik TM, Rodriguez-Bigas MA. Clinicopathologic features in colorectal cancer patients with microsatellite instability. Mutat Res 2004;568:275–82. 10.1016/j.mrfmmm.2004.05.025 [DOI] [PubMed] [Google Scholar]
- 34.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrock AB, Ouyang C, Sandhu J, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol 2019;30:1096–103. 10.1093/annonc/mdz134 [DOI] [PubMed] [Google Scholar]
- 36.Lee JS, Ruppin E. Multiomics prediction of response rates to therapies to inhibit programmed cell death 1 and programmed cell death 1 ligand 1. JAMA Oncol 2019;5:1614 10.1001/jamaoncol.2019.2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wade M, Li Y-C, Wahl GM. Mdm2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013;13:83–96. 10.1038/nrc3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Champiat S, Dercle L, Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res 2017;23:1920–8. 10.1158/1078-0432.CCR-16-1741 [DOI] [PubMed] [Google Scholar]
- 39.Kato S, Goodman A, Walavalkar V, et al. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242–50. 10.1158/1078-0432.CCR-16-3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuentes-Antrás J, Provencio M, Díaz-Rubio E. Hyperprogression as a distinct outcome after immunotherapy. Cancer Treat Rev 2018;70:16–21. 10.1016/j.ctrv.2018.07.006 [DOI] [PubMed] [Google Scholar]
- 41.DT L, Uram JN, Wang H, et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J Clin Oncol 2016;34:2016. [Google Scholar]
- 42.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91. 10.1016/S1470-2045(17)30422-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch Repair–Deficient/Microsatellite Instability–High metastatic colorectal cancer. J Clin Oncol 2018;36:773–9. 10.1200/JCO.2017.76.9901 [DOI] [PubMed] [Google Scholar]
- 44.DT L, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Available: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm613227.htm [Accessed 20 Oct 2018].
- 46.Ganesh K, Stadler ZK, Cercek A, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019;16:361–75. 10.1038/s41575-019-0126-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer. Lancet 2019;394:1467–80. 10.1016/S0140-6736(19)32319-0 [DOI] [PubMed] [Google Scholar]
- 48.US national library of medicine. ClinicalTrials.gov, 2019. Available: https://www.clinicaltrials.gov/ct2/show/NCT02997228
- 49.Chalabi M, Fanchi LF, Van den Berg JG, et al. Neoadjuvant ipilimumab plus nivolumab in early stage colon cancer. Ann Oncol 2018;29:viii731 10.1093/annonc/mdy424.047 [DOI] [Google Scholar]
- 50.Sinicrope FA, Ou F-S, Shi Q, et al. Randomized trial of FOLFOX alone or combined with atezolizumab as adjuvant therapy for patients with stage III colon cancer and deficient DNA mismatch repair or microsatellite instability (atomic, alliance A021502). Journal of Clinical Oncology 2017;35:TPS3630 10.1200/JCO.2017.35.15_suppl.TPS3630 [DOI] [Google Scholar]
- 51.Berntsson J, Eberhard J, Nodin B, et al. Expression of programmed cell death protein 1 (PD-1) and its ligand PD-L1 in colorectal cancer: relationship with sidedness and prognosis. Oncoimmunology 2018;59:e1465165 10.1080/2162402X.2018.1465165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, He M, Zhou Y, et al. The prognostic and clinicopathological roles of PD-L1 expression in colorectal cancer: a systematic review and meta-analysis. Front Pharmacol 2019;10 10.3389/fphar.2019.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalyan A, Kircher S, Shah H, et al. Updates on immunotherapy for colorectal cancer. J Gastrointest Oncol 2018;9:160–9. 10.21037/jgo.2018.01.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen EX, Jonker DJ, Kennecke HF, et al. Cctg CO.26 trial: a phase II randomized study of durvalumab (D) plus tremelimumab (T) and best supportive care (BSC) versus BSC alone in patients (PTS) with advanced refractory colorectal carcinoma (rCRC). J Clin Oncol 2019;37:481 10.1200/JCO.2019.37.4_suppl.48130620669 [DOI] [Google Scholar]
- 55.Fukuoka S, Hara H, Takahashi N, et al. Regorafenib plus nivolumab in patients with advanced gastric (GC) or colorectal cancer (CRC): an open-label, dose-finding, and dose-expansion phase 1B trial (REGONIVO, EPOC1603). J Clin Oncol 2019;37:2522. 10.1200/JCO.2019.37.15_suppl.2522 [DOI] [PubMed] [Google Scholar]
- 56.Battaglin F, Naseem M, Puccini A, et al. Molecular biomarkers in gastro-esophageal cancer: recent developments, current trends and future directions. Cancer Cell Int 2018;18:99. 10.1186/s12935-018-0594-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res 2005;11:2947–53. 10.1158/1078-0432.CCR-04-1469 [DOI] [PubMed] [Google Scholar]
- 58.Doi T, Piha-Paul SA, Jalal SI, et al. Safety and antitumor activity of the Anti-Programmed death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. J Clin Oncol 2018;36:61–7. 10.1200/JCO.2017.74.9846 [DOI] [PubMed] [Google Scholar]
- 59.Kojima T, Muro K, Francois E, et al. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: phase III KEYNOTE-181 study. J Clin Oncol 2019;37:2. 10.1200/JCO.2019.37.4_suppl.2 [DOI] [PubMed] [Google Scholar]
- 60.Kato K, Cho BC, Takahashi M, et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2019;20:1506–17. 10.1016/S1470-2045(19)30626-6 [DOI] [PubMed] [Google Scholar]
- 61.Bass AJ, Thorsson V, Shmulevich I, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naseem M, Barzi A, Brezden-Masley C, et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat Rev 2018;66:15–22. 10.1016/j.ctrv.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vranic S. Microsatellite instability status predicts response to anti-PD-1/PD-L1 therapy regardless the histotype: a comment on recent advances. Bosn J Basic Med Sci 2017;17:274–5. 10.17305/bjbms.2017.2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Champiat S, Ferte C, Lebel-Binay S, et al. Exomics and immunogenics: bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014;3:e27817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saito R, Abe H, Kunita A, et al. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod Pathol 2017;30:427–39. 10.1038/modpathol.2016.202 [DOI] [PubMed] [Google Scholar]
- 66.Zhang M, Dong Y, Liu H, et al. The clinicopathological and prognostic significance of PD-L1 expression in gastric cancer: a meta-analysis of 10 studies with 1,901 patients. Sci Rep 2016;6:37933 10.1038/srep37933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuchs CS, Doi T, Jang RW-J, et al. KEYNOTE-059 cohort 1: efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. J Clin Oncol 2017;35:4003 10.1200/JCO.2017.35.15_suppl.400329040031 [DOI] [Google Scholar]
- 68.Wainberg ZA, Jalal S, Muro K, et al. LBA28_PRKEYNOTE-059 update: efficacy and safety of pembrolizumab alone or in combination with chemotherapy in patients with advanced gastric or gastroesophageal (G/GEJ) cancer. Ann Oncol 2017;28:mdx440.020 10.1093/annonc/mdx440.020 [DOI] [Google Scholar]
- 69.Kang Y-K, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461–71. 10.1016/S0140-6736(17)31827-5 [DOI] [PubMed] [Google Scholar]
- 70.Shitara K, Ozguroglu M, Bang Y-J, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018;392:123–33. 10.1016/S0140-6736(18)31257-1 [DOI] [PubMed] [Google Scholar]
- 71.Available: https://www.mrknewsroom.com/news-release/oncology/merck-provides-update-phase-3-keynote-062-trial-evaluating-keytruda-pembrolizu [Accessed 08 May 2019].
- 72.Shitara K, Van Cutsem E, Bang Y-J, et al. Pembrolizumab with or without chemotherapy vs chemotherapy in patients with advanced G/GEJ cancer (GC) including outcomes according to microsatellite Instability-High (MSI-H) status in KEYNOTE-062. Ann Oncol 2019;30:v878–9. 10.1093/annonc/mdz394.035 [DOI] [Google Scholar]
- 73.Chung HC, Arkenau H-T, Wyrwicz L, et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with advanced gastric or gastroesophageal junction cancer from javelin solid tumor phase Ib trial: analysis of safety and clinical activity. J Clin Oncol 2016;34:4009 10.1200/JCO.2016.34.15_suppl.4009 [DOI] [Google Scholar]
- 74.Bang Y-J, Ruiz EY, Van Cutsem E, et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol 2018;29:2052–60. 10.1093/annonc/mdy264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moehler M, Ryu MH, Dvorkin M, et al. Maintenance avelumab versus continuation of first-line chemotherapy in gastric cancer: javelin gastric 100 study design. Future Oncol 2019;15:567–77. 10.2217/fon-2018-0668 [DOI] [PubMed] [Google Scholar]
- 76.Alcindor T, Opu T, Mueller C, et al. Interim analysis of a phase II trial of perioperative chemotherapy plus avelumab in esophagogastric and gastric adenocarcinoma. Ann Oncol 2019;30:v253–324. 10.1093/annonc/mdz247.102 [DOI] [Google Scholar]
- 77.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. 10.1016/S0140-6736(17)31046-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 2018;19:940–52. 10.1016/S1470-2045(18)30351-6 [DOI] [PubMed] [Google Scholar]
- 79.Yau T, Kang Y-K, Kim T-Y, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (PTS) with advanced hepatocellular carcinoma (aHCC): results from CheckMate 040. J Clin Oncol 2019;37:4012. 10.1200/JCO.2019.37.15_suppl.4012 [DOI] [Google Scholar]
- 80.Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J Immunother Cancer 2019;7:267. 10.1186/s40425-019-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iñarrairaegui M, Melero I, Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin Cancer Res 2018;24:1518–24. 10.1158/1078-0432.CCR-17-0289 [DOI] [PubMed] [Google Scholar]
- 82.Cheng A, Qin S, Ikeda M, et al. IMbrave150: efficacy and safety results from a pH III study evaluating atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (SOR) as first treatment (tx) for patients (PTS) with unresectable hepatocellular carcinoma (HCC). Annals of Oncology 2019;30:ix183–202. 10.1093/annonc/mdz446.002 [DOI] [Google Scholar]
- 83.Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545–51. 10.1016/j.jhep.2016.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh P, Toom S, Avula A, et al. The immune modulation effect of locoregional therapies and its potential synergy with immunotherapy in hepatocellular carcinoma. J Hepatocell Carcinoma 2020;7:11–17. 10.2147/JHC.S187121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee HW, Cho KJ, Park JY. Current status and future direction of immunotherapy in hepatocellular carcinoma: what do the data suggest? Immune Netw 2020;20:e11 10.4110/in.2020.20.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2:e000152 10.1136/esmoopen-2016-000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O'Hara MH, O'Reilly EM, Rosemarie M, et al. Abstract CT004: a phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (GEM) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PdaC) patients. Cancer Res 2019;79:CT004. [Google Scholar]
- 88.Marabelle A, Cassier PA, Fakih M, et al. Pembrolizumab for advanced anal squamous cell carcinoma (ASCC): results from the multicohort, phase II KEYNOTE-158 study. J Clin Oncol 2020;38:Abstract 1. [Google Scholar]
- 89.Morris VK, Eng C. Role of immunotherapy in the treatment of squamous cell carcinoma of the anal canal. J Natl Compr Canc Netw 2018;16:903–8. 10.6004/jnccn.2018.7040 [DOI] [PubMed] [Google Scholar]
- 90.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018;362:eaar3593 10.1126/science.aar3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018;18:139–47. 10.1038/nrc.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ott PA, Bang YJ, Piha-Paul SA, et al. T-Cell–Inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 2019;37:318–27. 10.1200/JCO.2018.78.2276 [DOI] [PubMed] [Google Scholar]
- 93.Fares CM, Van Allen EM, Drake CG, et al. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book 2019;39:147–64. 10.1200/EDBK_240837 [DOI] [PubMed] [Google Scholar]
- 94.Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. JCO 2018;36:1685–94. 10.1200/JCO.2017.75.7740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018;118:9–16. 10.1038/bjc.2017.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019. [DOI] [PubMed] [Google Scholar]
- 97.Yan Y, Kumar AB, Finnes H, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol 1739;2018:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–29. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8:1136 10.1038/s41467-017-01062-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grasso CS, Giannakis M, Wells DK, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 2018;8:730–49. 10.1158/2159-8290.CD-17-1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 2016;6:202–16. 10.1158/2159-8290.CD-15-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Albacker LA, Wu J, Smith P, et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One 2017;12:e0176181 10.1371/journal.pone.0176181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu D, Jenkins RW, Sullivan RJ. Mechanisms of resistance to immune checkpoint blockade. Am J Clin Dermatol 2019;20:41–54. 10.1007/s40257-018-0389-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rieth J, Subramanian S. Mechanisms of intrinsic tumor resistance to immunotherapy. Int J Mol Sci 2018;19. 10.3390/ijms19051340. [Epub ahead of print: 02 May 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pan D, Kobayashi A, Jiang P, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359:770–5. 10.1126/science.aao1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen J, Ju Z, Zhao W, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24:556–62. 10.1038/s41591-018-0012-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zitvogel L, Ma Y, Raoult D, et al. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70. 10.1126/science.aar6918 [DOI] [PubMed] [Google Scholar]
- 108.Gopalakrishnan V, Helmink BA, Spencer CN, et al. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80. 10.1016/j.ccell.2018.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. 10.1016/j.cell.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu-Lieskovan S, Mok S, Homet Moreno B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF V600E melanoma. Sci Transl Med 2015;7:279ra241 10.1126/scitranslmed.aaa4691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen DS, Hurwitz H. Combinations of bevacizumab with cancer immunotherapy. Cancer J 2018;24:193–204. 10.1097/PPO.0000000000000327 [DOI] [PubMed] [Google Scholar]
- 112.Loo Yau H, Ettayebi I, De Carvalho DD. The cancer epigenome: exploiting its vulnerabilities for immunotherapy. Trends Cell Biol 2019;29:31–43. 10.1016/j.tcb.2018.07.006 [DOI] [PubMed] [Google Scholar]
- 113.Topper MJ, Vaz M, Marrone KA, et al. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol 2020;17:75-90. 10.1038/s41571-019-0266-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.O'Donnell JS, Long GV, Scolyer RA, et al. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017;52:71–81. 10.1016/j.ctrv.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 115.Marciscano AE, Walker JM, McGee HM, et al. Incorporating radiation oncology into immunotherapy: proceedings from the ASTRO-SITC-NCI immunotherapy workshop. J Immunother Cancer 2018;6:6. 10.1186/s40425-018-0317-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Theelen WSME, Peulen HMU, Lalezari F, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol 2019. 10.1001/jamaoncol.2019.1478. [Epub ahead of print: 11 Jul 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21. 10.1038/nature22991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thomas R, Al-Khadairi G, Roelands J, et al. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol 2018;9:947. 10.3389/fimmu.2018.00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218. 10.1038/s41573-018-0007-y [DOI] [PubMed] [Google Scholar]
- 120.Nowak EC, Lines JL, Varn FS, et al. Immunoregulatory functions of VISTA. Immunol Rev 2017;276:66–79. 10.1111/imr.12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Le Mercier I, Chen W, Lines JL, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res 2014;74:1933–44. 10.1158/0008-5472.CAN-13-1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu W, Hiếu T, Malarkannan S, et al. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol 2018;15:438–46. 10.1038/cmi.2017.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blando J, Sharma A, Higa MG, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A 2019;116:1692–7. 10.1073/pnas.1811067116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018;131:39–48. 10.1182/blood-2017-07-741025 [DOI] [PubMed] [Google Scholar]
- 125.Harrington K, Freeman DJ, Kelly B, et al. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov 2019;18:689–706. 10.1038/s41573-019-0029-0 [DOI] [PubMed] [Google Scholar]
- 126.Kepp O, Marabelle A, Zitvogel L, et al. Oncolysis without viruses - inducing systemic anticancer immune responses with local therapies. Nat Rev Clin Oncol 2020;17:49-64. 10.1038/s41571-019-0272-7 [DOI] [PubMed] [Google Scholar]
- 127.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44. 10.1056/NEJMoa1707447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang Q, Zhang Z, Peng M, et al. CAR-T cell therapy in gastrointestinal tumors and hepatic carcinoma: from bench to bedside. Oncoimmunology 2016;5:e1251539. 10.1080/2162402X.2016.1251539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goldberg MS. Improving cancer immunotherapy through nanotechnology. Nat Rev Cancer 2019;19:587–602. 10.1038/s41568-019-0186-9 [DOI] [PubMed] [Google Scholar]
- 131.Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol 2019;16:690–704. 10.1038/s41575-019-0209-8 [DOI] [PubMed] [Google Scholar]
- 132.Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079–84. 10.1126/science.aad1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084–9. 10.1126/science.aac4255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104–8. 10.1126/science.aao3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91–7. 10.1126/science.aan3706 [DOI] [PubMed] [Google Scholar]
- 136.Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359:97–103. 10.1126/science.aan4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tanoue T, Morita S, Plichta DR, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019;565:600–5. 10.1038/s41586-019-0878-z [DOI] [PubMed] [Google Scholar]
- 138.Dubin K, Callahan MK, Ren B, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 2016;7:10391. 10.1038/ncomms10391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Y, Wiesnoski DH, Helmink BA, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med 2018;24:1804–8. 10.1038/s41591-018-0238-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rao D, Parakrama R, Augustine T, et al. Immunotherapeutic advances in gastrointestinal malignancies. NPJ Precis Oncol 2019;3:4. 10.1038/s41698-018-0076-8 [DOI] [PMC free article] [PubMed] [Google Scholar]