

Abstract
In patients with multiple myeloma, plasmablastic transformation in the bone marrow is rare and associated with poor outcome. The significance of discordant extramedullary plasmablastic transformation in patients with small, mature clonal plasma cells in the bone marrow has not been well studied. Here we report the clinicopathologic, cytogenetic and molecular features of 10 such patients (male/female: 6/4, median age 65 years, range 48–76 years) with an established diagnosis of multiple myeloma in the bone marrow composed of small, mature plasma cells in parallel with a concurrent or subsequent extramedullary plasmablastic transformation. Eight patients with available survival data showed an overall aggressive clinical course with a median survival of 4.5 months after the diagnosis of extramedullary plasmablastic transformation, despite aggressive treatment and even in patients with low-level bone marrow involvement. Pathologically, the extramedullary plasmablastic myeloma were clonally related to the corresponding bone marrow plasma cells, showed high levels of CMYC and/or P53 expression with a high Ki-67 proliferation index by immunohistochemistry and harbored more complex genomic aberrations including frequent mutations in the RAS pathway and MYC rearrangements compared to their bone marrow counterparts. In summary, although genetic and immunohistochemical studies were not uniformly performed on all cases due to the retrospective nature of this study, our data suggest that discordant extramedullary plasmablastic transformation of multiple myeloma has an aggressive clinical course and is characterized by frequent mutations in the RAS pathway and more complex genomic abnormalities.
Keywords: Extramedullary plasmablastic transformation, multiple myeloma, discordance, genetic alteration, RAS, MYC, TP53
Introduction
Multiple myeloma is characterized by clonal plasma cells in the bone marrow (1). Neoplastic plasma cells can show a broad spectrum of cytologic features, including small, mature plasma cells, lymphoplasmacytoid cells and large cells with prominent central nucleoli, high nuclear-to-cytoplasmic ratios and immature cytoplasm lacking a prominent perinuclear hof, designated as plasmablasts. Plasmablastic myeloma, representing the extreme end of the morphologic spectrum of multiple myeloma, is described as a rare and aggressive neoplasm. Formal diagnostic criteria of plasmablastic myeloma are largely lacking in the literature (2). Most studies have used criteria proposed by Bartl, et al or Greipp, et al, in which a designation of plasmablastic myeloma requires plasmablasts to comprise the predominant cell type or at least 2% of total cells by differential count, respectively, based on bone marrow aspirate cytomorphology (3–5).
A subset of patients with multiple myeloma develop extramedullary myeloma, which may demonstrate plasmablastic morphology and is typically associated with poor prognosis (6, 7). Discordant extramedullary plasmablastic transformation has been rarely reported in patients with typical bland/mature plasma cell morphology in the marrow (8–10). In such cases, the clinical significance, pathologic features and molecular pathogenesis are largely unknown. Herein, we report 10 cases of discordant extramedullary plasmablastic transformation of multiple myeloma and characterize their clinicopathologic, cytogenetic and molecular features.
Materials and methods
Case selection
Cases were identified by searching a pathology database at Memorial Sloan Kettering Cancer Center (MSKCC), New York, NY, for samples obtained between 1/2010 and 12/2018 using the search terms “plasmablastic,” “plasma cell neoplasm” and “myeloma.” Cases with plasmablastic morphology (≥2% plasmablasts by aspirate differential) in the bone marrow were excluded. Only cases with typical mature plasma cell morphology in the bone marrow and extramedullary plasmablastic morphology were selected (8 cases). In contrast to the criteria used by Sailer, et al (11) in which the presence of ≥30% plasmablasts defined a plasmablastic subtype of plasma cell myeloma, extramedullary plasmablastic myeloma in the current study was defined as tissue involvement by sheets of plasmablasts (>50% in the biopsy). Additional two cases that met these criteria were submitted from Detroit Receiving Hospital, Detroit, MI. Clinical data was collected from electronic medical records. This study was approved by the Institutional Review Boards of both participating institutions.
Histology
All extramedullary tissue biopsies (hematoxylin and eosin [H&E]-stained slides) and bone marrow samples (H&E-stained core biopsy and/or clot section slides and Wright-Giemsa-stained aspirate smears) were independently reviewed by two hematopathologists.
Immunohistochemistry (IHC)
Immunohistochemical staining was performed on formalin-fixed, paraffin-embedded (FFPE) tissue sections using an automated staining platform (Ventana Medical System, Tucson, AZ) as per the manufacturer’s instructions. The monoclonal antibodies utilized included: CD138, CD56, Cyclin D1, CMYC, TP53, Ki-67 (MIB-1), Kappa and Lambda. EBER in situ hybridization (ISH) was performed using a fluorescein-conjugated EBER oligonucleotide probe and the purified IgG fraction of a mouse monoclonal anti-fluorescein antibody. CMYC was scored as negative (0% positive neoplastic cells), low-level expression (<40% positive neoplastic cells) and high-level expression (≥40% positive neoplastic cells). TP53 was scored as negative (0% positive neoplastic cells), normal (<10% positive neoplastic cells), intermediate-level (10–50% positive neoplastic cells) or high-level (>50% neoplastic cells) expression.
Fluorescence in situ hybridization (FISH) and genomic microarray
FISH analysis was performed on CD138-enriched plasma cell populations from fresh bone marrow specimens as well as on tissue samples following standard protocols. CD138 enrichment was performed using a commercially available magnetic bead-based kit (StemCell Technologies, Vancouver, Canada). The probes used targeted common chromosomal alterations in myeloma, including deletion of 1p, gain of 1q, t(4;14), IGH translocation and deletion or loss of chromosomes 13 and 17 (TP53) (all probes from Abbott Molecular, Des Plaines, IL). In some of the bone marrow specimens, not all FISH probes were applied due to a lack of adequate plasma cells after enrichment. In addition, FISH using a MYC break-apart probe (Abbott Molecular, Des Plaines, IL) was performed on 7 FFPE tissue specimens and on CD138-enriched plasma cells from 2 bone marrow specimens. One hundred cells were evaluated for each probe and results were determined using the corresponding established cut-off values (ranged from 1% to 5%).
Genomic single nucleotide polymorphism (SNP) microarray tests were performed in three patients, including two patients with paired bone marrow samples with myeloma and extramedullary tissue with plasmablastic transformation, following the manufacturers’ instructions. SNP array tests employed a CytoScan HD array with 2.67 million probes, including 750,000 SNP probes, and an OncoScan CNV assay chip with 220,000 SNP probes specifically covering about 900 cancer genes (both from Affymetrix/Thermo Fisher Scientific Inc., Santa Clara, CA). Cytoscan array used DNA from CD138 enriched plasma cells from bone marrow. For cytoscan, the CEL files were converted to CYCHP files while for oncoscan, both CEL files from AT and CG products in combination were converted to a OSCHYP file. Data analysis was performed using ChAS software (version 3.2, Affymetrix) for cytoscan data and Oncoscan Nexus Express (Biodiscovery, Inc., El Segundo, CA) for oncoscan data. All samples were manually reviewed and interpreted with a focus on recurring genomic abnormalities in myeloma and other hematologic malignancies.
Targeted next-generation sequencing
Targeted next-generation sequencing (NGS) analysis was performed on selected bone marrow specimens and tissue samples with available material. NGS utilized a hybridization capture-based assay (MSKCC IMPACT) that targeted 400 genes frequently mutated in hematologic malignancies as previously described (12, 13). For MSKCC IMPACT data analysis, short insert paired-end reads were aligned to the GRCh37 reference human genome with 1000 Genomes decoy contigs using BWA-mem43. After alignment, an average of 500× coverage per sample was obtained. Single base substitutions were called using CaVEMan as described previously (12). Small somatic insertions and deletions (indels) were identified using a modified version of Pindel. The putative variants were further analyzed through a custom annotation pipeline, including VEP, Vagrent, Exac, and Gnomad, to evaluate the likely role of specific mutations. Variants were categorized as pathogenic, likely pathogenic, or unknown. Pathogenic and likely pathogenic variants were reported.
Statistical analysis
Overall survival (OS) was calculated from the date of plasmablastic transformation to the date of last follow-up or death.
Results
Clinical characteristics
The study cohort included 10 patients (6 men and 4 women) with a median age of 65 years (range, 48–76 years) (Table 1). All patients developed either single (4 cases) or multiple (6 cases) extramedullary lesions. Six patients had a well-established prior history of multiple myeloma with the time interval between the initial diagnosis and extramedullary presentation ranging from 1 to 64 months. The other 4 were diagnosed with multiple myeloma concurrently at presentation with extramedullary disease. All 10 patients had end-organ damage (hypercalcemia, renal insufficiency, anemia) with lytic bone lesions (“CRAB”) and an M-spike was detected in 8 cases. None of the patients had a known history of immunodeficiency, HIV infection or body cavity effusions. Initial treatment for multiple myeloma included chemotherapy alone (7 cases) or chemotherapy plus autologous hematopoietic stem cell transplantation (autoHSCT) (3 cases). Chemotherapy regimens for multiple myeloma included a combination of steroids (dexamethasone) and proteasome inhibitors (bortezomib or carfilzomib) in addition to lenalidomide, daratumumab or cyclophosphamide or a combination of dexamethasone, lenalidomide and daratumumab. Treatment regimens for patients with concurrent or subsequent development of extramedullary plasmablastic myeloma included bortezomib and dexamethasone in addition to thalidomide, elotuzumab, PACE (cisplatin, doxorubicin, cyclophosphamide, etoposide) or EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin). At the time of extramedullary plasmablastic transformation, the extent of involvement of the bone marrows by plasma cells was highly variable, ranging from <5% to 95%. All 8 patients with available outcome data had an aggressive clinical course with a median survival of 4.5 months (range 2–31 months) after development of plasmablastic transformation. The poor outcome was independent of the extent of bone marrow involvement.
Table 1.
Clinical features of patients with discordant extramedullary plasmablastic transformation
| Case # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 52 | 67 | 64 | 73 | 59 | 50 | 48 | 76 | 75 | 75 |
| Sex | F | M | M | M | F | F | F | M | M | M |
| Site(s) | Shoulder mass; mediastinal mass | Lymph node | Lymph node; abdominal mass | Liver | Liver | Nasopharynx; abdominal mass | Liver | Lymph node; pleura | Lymph node; shoulder and rib masses | Paraspinal mass; skin |
| BM plasma cell (%)* | 10 | 50 | <5 | <5 | 10 | <5 | 5–10 | 95 | 70 | NA |
| M Spike (g/dl)* | 3.39 | NA | 1.22 | 3.95 | NA | 1.67 | 2.9 | 2.8 | 7.2 | 0.44 |
| CRAB* | + | + | + | + | + | + | + | + | + | + |
| Bone lesions* | + | + | + | + | + | + | + | + | + | + |
| Interval (months)** | 20 | 13 | 64 | 6 | 0 | 0 | 0 | 2 | 1 | 0 |
| Initial therapy | DRd | CyBord | VdT | KRd | Vd, Kd | Vd- PACE | ||||
| Therapy (Post-transformation) | Elo-Vd | NA | Vd-PACE | Vd-PACE | Vd R EPOCH | Vd-PACE R D EPOCH | VdT-PACE | Vd-PACE | Vd- PACE | Vd, Kd D EPOCH |
| HSCT after transformation | + | - | + | + | - | - | - | - | - | - |
| Outcome after transformation (months) | Progressed 4 | NA | Deceased 14 | Deceased 3 | NA | Deceased 12 | Deceased 5 | Deceased 2 | Deceased 2 | Deceased 31 |
At the time of extramedullary plasmablastic transformation.
Between the initial diagnosis of multiple myeloma and extramedullary plasmablastic transformation.
Abrreviation: CRAB: myeloma-defining signs (hypercalcemia, renal insufficiency, anemia, and bone lesions); HSCT: Hematopoietic stem cell transplantation; NA: not available or performed; Vd: bortezomib, dexamethasone; Kd: carfilzomib, dexamethasone; VdT: bortezomib, dexamethasone, thalidomide; Elo: Elotuzumab; PACE: cisplatin, doxorubicin, cyclophosphamide, etoposide; R: Lenalidomide; D: daratumumab; EPOCH: etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; DRd: daratumumab, lenalidomide, and dexamethasone; KRd: carfilzomib, thalidomide, dexamethasone; CyBord: cyclophosphamide, bortezomib and dexamethasone.
Morphologic and immunophenotypic characteristics
Histologically, extramedullary tissue biopsies from all patients showed a diffuse infiltrate of sheets of medium to large sized atypical cells with plasmablastic morphology showing bi/tri-nucleation, large, centrally located nuclei, distinct nucleoli, high nuclear-to-cytoplasmic ratios and scant cytoplasm lacking a perinuclear hof. Abundant mitotic figures and apoptotic bodies were evident in all cases (Fig. 1A&B). Review of the concurrent bone marrow biopsies and aspirate smears showed that all bone marrows were involved by typical small, mature plasma cells without plasmablastic morphology (Fig. 1C&D).
Figure 1.

Morphologic features of extramedullary tissue and bone marrow biopsies from case 1. (A&B). H&E-stained sections (100X and 400X; B, inset, 1000X) of a shoulder mass biopsy showing sheets of atypical cells with plasmablastic morphology exhibiting centrally located, prominent nucleoli, high nuclear-to-cytoplasmic ratios and abundant background mitotic figures and apoptotic bodies. (C&D) H&E -stained sections (100X and 400X) and aspirate smears (D, inset, 1000X) of a bone marrow biopsy showing scattered or clustered mature-appearing plasma cells.
By immunohistochemistry, the extramedullary tissue biopsies were positive for CD138 in 9/10 (90%) cases, CD56 in 8/10 (80%) cases and Cyclin D1 in 5/10 (50%) cases (Fig. 2 & Table 2). Nine of 10 cases showed aberrant expression of either Cyclin D1 or CD56 or both. High-level CMYC expression was seen in 8 of 9 (89%) cases and low-level expression in 1 of 9 (11%) cases. High TP53 expression was identified in 4 of 6 (67%) cases and intermediate expression in 1 of 6 (17%) cases. The Ki-67 proliferation index (MIB-1) was high (median 76%; range 50%−90%). EBER ISH was negative in all cases. HHV8 and ALK were negative in all cases tested (n=6 and n=2, respectively).
Figure 2.
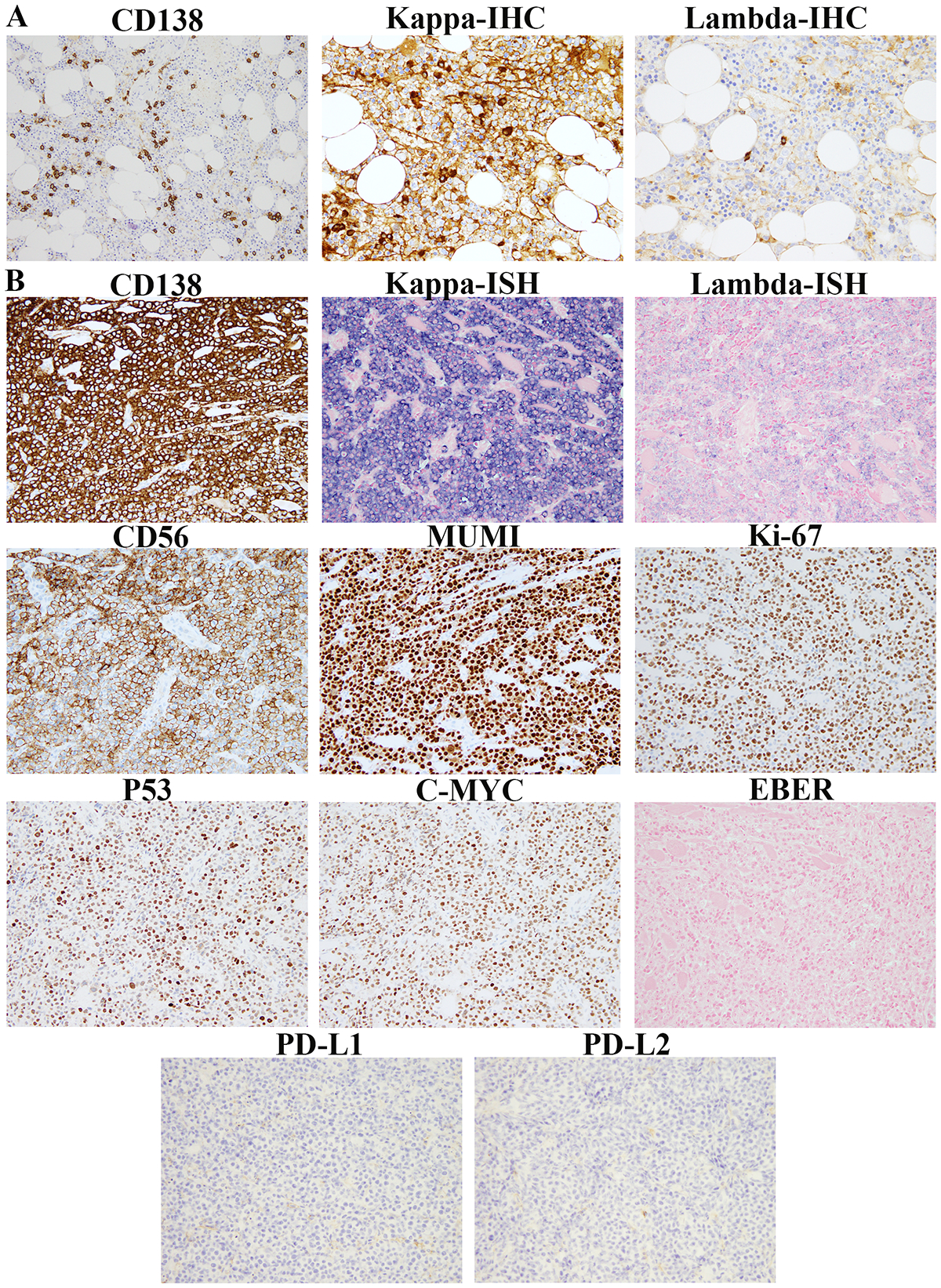
Immunophenotypic features of bone marrow and extramedullary tissue biopsies from case 1. (A). Plasma cells accounting for ~10% of the marrow cellularity (shown by CD138 IHC) with kappa light chain restriction (shown by kappa and lambda IHC) are shown. (B). CD138, kappa, lambda, CD56, MUM1, Ki-67, P53, CMYC, PD-L1 and PD-L2 IHC and in-situ hybridization for Epstein-Barr virus (EBER) performed on the shoulder mass biopsy are shown.
Table 2.
Immunophenotypes of discordant extramedullary plasmablastic transformation
| Case # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| CD138 | + | + | + | + | + | + | + | + | + | - |
| CD56 | + | + | + | + | + | + | - | + | + | - |
| Cyclin D1 | - | + | - | + | - | - | - | + | + | + |
| CMYC | ++ 80% | ++ 50% | + 40% | ++ 90% | ++ 60% | ++ 60% | ++ 50% | NA | + <10% | ++ 60% |
| P53 | +++ 70% | +++ 90% | +++ 60% | NA | +++ 80% | ++ 30% | NA | NA | NA | Normal 5–10% |
| EBER ISH | - | - | - | - | - | - | - | - | - | - |
| HHV8 | - | - | NA | NA | - | NA | - | NA | - | - |
| Light chain | κ | κ | κ | κ | λ | λ | κ | λ | κ | λ |
| Ki67 | 50% | 60% | NA | 90% | 80% | 90% | 80% | NA | 90% | 70% |
EBER ISH: Epstein-Barr virus-encoded RNA in situ hybridization; HHV8: Human herpes virus-8; κ: Kappa light chain; λ: Lambda light chain; NA: not available.
The immunophenotypes of the extramedullary biopsies as determined by immunohistochemistry and/or flow cytometric studies, including kappa and lambda light chain restriction, were consistent with that of their bone marrow counterparts, supporting their clonal relatedness. PD-L1 and PD-L2 immunohistochemical studies were also performed on 4 cases, none of which showed evidence of PD-L1 or PD-L2 overexpression (Fig. 2).
FISH and SNP array analysis
FISH analysis was performed on 5 bone marrows and 1 tissue sample (from 5 patients), which revealed typical chromosomal abnormalities in myeloma, including deletion of 1p (cases 3 and 4), gain of 1q (cases 3, 4, 7 and 8), loss of chromosome 13 (cases 4 and 7), hyperdiploid clones (cases 3 and 8), t(14;16)/IGH/MAF fusion (case 4), and an IGH translocation with an undefined partner gene (cases 3 and 7) (Table 3). No TP53 deletions were observed in the samples analyzed. FISH analysis identified MYC rearrangements in both bone marrow and extramedullary tissue from case 2 and in tissue samples from cases 4 and 5 and a high-level copy gain in tissue sample from case 1 (Fig. 3A). In case 5, a MYC rearrangement, which is a well-defined secondary abnormality acquired during disease progression in myeloma, was detected only in the extramedullary plasmablastic myeloma sample but not in the bone marrow sample.
Table 3.
Summary of SNP-array results, Multiple Myeloma (MM) Fluorescence In Situ Hybridization (FISH), and MYC-FISH findings
| Case | Cytogenetic Analysis | Bone Marrow | Tissue |
|---|---|---|---|
| 1 | SNP-array |
Gain of chromosome 3, 5, 6, 7, 9, 11q12.1-q25, 15, 17p13.3-q12, 17q21.31-q25.3, 19 Loss of chromosome 22 and X CN-LOH of 17q12-q21.31 |
Gain of 1q21.2-qter Gain of chromosome 2, 3, 5, 6p, 7, 8q, 9, 11q12.1-qter, 15, 16, 17pter-q12, 17q21.31-q25.3, 19, 20q Loss of chromosome X and loss of 6q CN-LOH of 17q12-q21.31 |
| MM-FISH | Three copies of IgH(14q32) | NA | |
| MYC-FISH | NA | High copy gain | |
| 2 | SNP-array |
Gain of chromosome 3q24-qter, 5, 6, 9, 11, 15, 17, 19, 20 and 21 CN-LOH of 16q and 18 |
Gain of chromosome 3q22.2-qter, 4q (q25-qter), 5, 6, 7, 8, 8q24-qter, 9, 11, 15q, 17q, 19, 20, 21 and 22 Deletion of 1p and gain of 1q, del(4q), del(8q), del(13q11.2-qter), del(16q), del(17p), del(19p) CN-LOH of 1p, 3, 4, 7, 10q, 16q and 18 |
| MYC-FISH | Positive | Positive | |
| 3 | MM-FISH |
Deletion of 1p32(CDKN2C) Gain of 1q21(CKS1B) |
Deletion of 1p32(CDKN2C) Gain of 1q21(CKS1B), 5 and 15 IgH rearrangement |
| MYC-FISH | NA | Negative | |
| 4 | MM-FISH | Deletion of 1p32(CDKN2C) and gain of 1q21(CKS1B) t(14;16); Loss of 13 Extra copy of MAF(16q23) |
NA |
| MYC-FISH | NA | Positive | |
| 5 | MYC-FISH | Negative | Positive |
| 6 | SNP-array | NA | Gain of chromosomes 3, 7, 9, 11, 18, 19 and gain of 1q, 4q, 5p and 15q CN-LOH of chromosome 1, 2, 4, 5, 6, 8, 10, 12, 13, 14, 15, 16, 17, 20, 21, 22 and X chromosome |
| 7 | MM-FISH | Gain of 1q21.3(CKS1B) Loss of 13 IgH rearrangement |
NA |
| 8 | MM-FISH | Gain of chromosomes 1, 4, 7, 9 and 15 | NA |
| 9 | MYC-FISH | NA | Negative |
| 10 | MYC-FISH | NA | Negative |
Note: Same cytogenetic findings shared between BM and tissue biopsies from the same patients are underlined.
Figure 3.

FISH analysis using MYC break-apart probes (A) and SNP array analysis of bone marrow and extramedullary tissue biopsies (B, C, D). (A). MYC FISH analysis showed multiple copies of MYC in the tissue biopsy of case 1 (Left), a MYC translocation with split signal patterns in the bone marrow (Middle), and a more complex abnormal MYC signal pattern in the tissue biopsy of case 2 (Right). 5’ and 3’ MYC probes are labeled in spectrum green and spectrum red, respectively. Abnormal cells are highlighted by yellow arrows. (B). SNP array analysis of paired bone marrow (Upper) and tissue (Bottom) biopsies from case 1 showed a hyperdiploid genomic profile with gains of chromosomes 3, 5, 6, 7, 9, 15, 17 and 19 in the bone marrow and more complex alterations in the tissue, including gains of 1q, chromosome 2 and 6p, deletions of 6q and 8p, high level gain of 8q including MYC and gain of 11q. (C). SNP array analysis of paired bone marrow (Upper) and tissue (Bottom) biopsies from case 2 showed a hyperdiploid clone with gains of chromosomes 5, 6, 9, 11, 15, 17, 19, 20 and 21, gain of 3q and deletion of 19q in the bone marrow and additional alterations in the tissue, including deletion of 1p, gain of 1q, deletion of 4p, gain of 4q, loss of chromosome 13, gain of 15q, deletion of 17p including TP53 and deletion of 19q. (D). SNP array analysis of case 6 (tissue biopsy only) showed a doubling hypodiploid clone with gains of chromosomes 3, 7, 9, 11, 18 and 19, gains of 1q, 4q, 5p and 15q and copy-neutral loss of heterozygosity (allele differences, bottom part) of all disomic chromosomes/arms of the 1, 2, 4, 5, 6, 8, 10, 12, 13, 14, 15, 16, 17, 20, 21, 22 and X chromosomes. The left Y axis is a log2 ratio (−1.5–1.5), whereas the right Y axis is a smooth signal copy level (0–4).
SNP array studies were performed in 3 patients (cases 1, 2, and 6) with DNA available, including paired bone marrow with myeloma and transformed plasmablastic myeloma samples from 2 patients (cases 1 and 2). In all 3 patients, a hyperdiploid clone with gains of odd numbered chromosomes, such as chromosomes 3 or 3q, 5 or 5p, 7, 9, 11, 15 or 15q and 19, was observed (Fig. 3B–D). In addition, gains of chromosomes 1q, 6 and 17 in case 1, deletion of chromosome 1p and gains of chromosomes 6, 17, 20 and 21 in case 2, and gains of chromosomes 1q, 4q and 18 in case 6 were identified. Interestingly, SNP array revealed copy neutral loss of heterozygosity of most chromosomes in case 6, indicative of a doubling haploid karyotype, which is a rare recurring abnormality in myeloma (Fig. 3D). In cases 1 and 2, clonal relatedness between the bone marrow myelomas and extramedullary plasmablastic myelomas was supported by the identification of gains of chromosomes 3, 5, 7, 9, 11q, 15 and 17 in both marrow and tissue samples from case 1 and gains of chromosomes 3q, 5, 6, 9, 11, 15, 17, 19, 20 and 21 and loss of heterozygosity of chromosomes 16q and 18 in both marrow and tissue samples from case 2 by cytogenetic analysis. In addition, as compared to the bone marrow samples, more complex genomic profiles were identified in the tissue biopsies, indicative of clonal evolution. In case 1, array testing of the tissue revealed high level gains of chromosomes 1q (5 copies) and 8q including MYC (6 copies), loss of chromosome X, gain of chromosome 2, gain of 6p, deletion of 6q, increased gain of 11q, deletion of 15q and gain of 20q (Fig. 3B). In case 2, additional genomic imbalances observed in the tissue included loss of 1p, gain of 1q, deletion of 4p/4q, gains of chromosomes 7 and 8 (with deletion of 8q including MYC), loss of chromosome 10, deletion of 13q, gain of 15q, deletion of 17p including TP53 and gain of 19p (Fig. 3C).
Mutational analysis
NGS analysis was performed on extramedullary plasmablastic myeloma biopsies from 6 cases (Table 4), 4 of which had concurrent bone marrow clot samples sequenced as well (cases 1, 2, 5 and 6). All 6 cases had mutations of genes in RAS pathways (1 KRAS, 3 KRAS, 1 BRAF, 1 ERF and NF1). For case 1, mutaitons in NRAS and ARIDIA (latter is transcriptional regulator) were detected in both tissue and bone marrow biopsies. For case 2, mutations in BRAF, KMT2C and HIST1H1E (latter two genes are epigenetic regulators) were detected in both tissue and bone marrow biopsies. For case 5, variants of PIK3R2 (involved in the PI3K pathway), the epigenetic regulators TET2, DNMT3A, HIST1H2AL, and SAMHD1 were detected in both tissue and bone marrow biopsies. In addition, different variants involved in the RAS pathway (NF1 in bone marrow and ERF in tissue) as well as in epigenetic and transcriptional regulation (ARID4A and NCOR2 in bone marrow and LATS1, YAP1, FAT1 and BCORL1 in tissue) were also detected. Additional TP53 and MGA variants were detected in the tissue only while an additional CYLD (NF-kB pathway) variant was detected in the bone marrow only. For case 6, variants of TP53, KRAS, SGK1 and SH2B3 were detected in the tissue while no variants were detected in the bone marrow, likely due to a low level of disease (<5% involvement). In the remaining 2 cases (cases 3 and 10), NGS studies were available from tissue samples only, which detected variants of KRAS and AURKA of the MAPK pathway, PIK3R2 and MST1R of the PI3K pathway, TRAF5 of the NF-kB pathway and multiple variants in genes involved in epigenetic and transcriptional regulation, the cell cycle, among others (Table 4) in case 3; and KRAS, FLT3 and TET2 variants in case 10. In summary, all 6 cases, including both paired bone marrow and tissue samples, harbored variants involving the RAS pathway. Most tissue biopsies harbored KRAS/NRAS (4/6 cases, 67%) and/or TP53 (2/6 cases, 33%) mutations, which were not mutually exclusive (as seen in case 6).
Table 4.
Summary of detected variants in tested cases
| Case 1 | Case 2 | Case 3 | Case 5 | Case 6 | Case 10 | ||||
|---|---|---|---|---|---|---|---|---|---|
| BM | Tissue | BM | Tissue | Tissue | BM | Tissue | Tissue | Tissue | |
| TP53 | None | None | None | None | None | None | Detected | Detected | None |
| RAS-MAPK | KRAS p.G13R | KRAS p.G13R | BRAF p.V600E | BRAF p.V600E | KRAS p.G12A, AURKA p.F346L | NF1 p.H1826N | ERF p.M76I | KRAS p.Q61L, SGK1 p.N460S | KRAS p.A146P |
| Epigenetic/ transcriptional regulator | ARID1A p.Q288Rfs*75 | ARID1A p.Q288Rfs*75 | KMT2C p.L3046P, HIST1H1E p.L45P | KMT2C p.L3046P, HIST1H1E p.L45P | FAT1 p.E890K, CREBBP p.I1730V, RXRA p.S224N, TFC3 p.A565T, NKX2–1 p.A146T, TET3 p.E653K, KMT2D p.R4282*, KMT2C p.Q3061E, KDM5C p.E23*, HDAC4 p.P522L, HIST1H3A p.E134Q, HIST1H2AM p.E122K | TET2 p.P480S, DNMT3A p.A3S, HIST1H2AL p.A15G, ARID4A p.E872*, NCOR2 p.S1970Y | TET2 p.P480S, DNMT3A p.A3S, HIST1H2AL p.A15G, LATS1 p.E817K, YAP1 p.S163C, FAT1 p.M240I, BCORL1 p.S1970Y | None | TET2 p.L615fs*24, T1592fs*4, FLT3 p.Y572C |
| PI3K | None | None | None | None | PIK3R2 p.S191*, MST1R p.D493Y | PIK3R2 p.V284M | PIK3R2 p.V284M | None | None |
| NF-kB | None | None | None | None | TRAF5 p.E419Rfs*29 | CYLD p.D678Rfs*3 | None | None | None |
| Cell cycle | None | None | None | None | CDKN2C p.A77E, ATM p.E1669K, BARD1 p.E649_E655del, BAP1 p.S596T, RECQL4 p.E1046K | None | None | None | None |
| Others | None | None | None | None | EIF1AX p.E99K, DIS3 p.I779F, IL7R p.L69V, P2RY8 p.P290R, CRBN p.X5_splice | SAMHD1 p.G176E | SAMHD1 p.G176E, MGA p.E1245Q | SH2B3 p.P363S | None |
Discussion
Progression of multiple myeloma generally follows a multi-step process from monoclonal gammopathy of uncertain significance (MGUS) to intramedullary myeloma to extramedullary myeloma (14), despite rare cases can first present with extramedullary plasmacytoma (15). Although primary extramedullary myeloma/plasmacytoma generally has a favorable prognosis due to its exquisite radiosensitivity, extramedullary involvement in conjunction with prior or concurrent multiple myeloma, albeit uncommon, is often associated with poor clinical outcome, particularly in patients with plasmablastic transformation (7, 16, 17). Although our study utilized a limited number of patients, it suggests that discordant extramedullary transformation may have similar clinicopathologic features as marrow-based plasmablastic transformation.
The clonal relatedness between the bone marrow myelomas and extramedullary plasmablastic myelomas in our cohort was supported by shared light chain restriction by IHC/flow cytometry analysis and overlapping cytogenetic and mutational profiles by genomic studies. In addition, our study showed that discordant plasmablastic tumors at extramedullary sites harbored more complex cytogenetic abnormalities than their bone marrow counterparts, a finding consistent with increased genomic instability indicative of progression of multiple myeloma. In both bone marrow and tissue biopsies, in addition to IGH rearrangement, deletion of 1p and/or gain of 1q were present in most cases. These alterations result in deletion of the CDKN2C gene and gain and amplification of the CKS1B gene, respectively, which are considered as “driver” abnormalities in the multi-step progression of myeloma to more aggressive disease with poor prognosis. MYC rearrangements were observed in 43% of the plasmablastic myeloma cases tested, which is higher than the reported frequency (~15%) among typical multiple myeloma cohorts in the literature (18, 19), where MYC rearrangements often occur as secondary abnormalities, as shown in case 5. The overall MYC alteration rate, including rearrangement and gain, was high, present in 57% of tested tissue cases. MYC alterations were not restricted to extramedullary components as a MYC rearrangement was also detected in the bone marrow of 1 case (case 2). Other common cytogenetic abnormalities detected in our cohort included loss of chromosome 13 and gains of chromosomes 5 and 15, all of which appeared to be present at initial diagnosis, as they were seen in the bone marrow of cases 1, 2, 4, 7 and 8, rather than acquired during disease progression. Interestingly, array analysis also revealed a doubling haploid clone in case 6, supported by copy neutral loss of heterozygosity (CN-LOH) of most chromosomes, a rare finding associated with a poor prognosis in myeloma. Only 2 patients had paired bone marrow and tissue samples available for SNP array and 2 patients had paired samples for FISH studies. Nevertheless, our results suggest that accumulated genomic instability may be associated with disease progression in discordant transformation of multiple myeloma.
Mutational profiling of extramedullary plasmablastic myelomas resulting from disease progression has not been extensively performed in the literature, although mutations of genes in the RAS pathway (NRAS, KRAS or BRAF) have been previously shown to correlate with disease progression (<10% in MGUS, 50% in multiple myeloma and 60–70% in plasma cell leukemia) (14, 20–23). In our study, RAS pathway mutations were detected in all bone marrow and tissue biopsies with plasmablastic transformation tested, reaffirming the correlation between RAS mutations and aggressiveness of disease. Relatedly, a recent study showed high frequency of RAS mutations (60–70%) in both intramedullary myeloma and corresponding extramedullary soft tissue plasmacytoma (22, 24). It is unclear if these patients had plasmablastic morphology. In either case, whether RAS mutations predispose or drive the transformation remains uncertain. A higher frequency of TP53 mutations was observed in our extramedullary plasmablastic tumors (~33%) as compared to that reported in typical multiple myeloma cases (~5%) (25–28), suggesting a role for TP53 in disease progression. These findings, suggest KRAS and TP53 mutations as crucial alterations involved in plasmablastic progression. Interestingly, although P53 overexpression by IHC appeared to correspond to TP53 mutations in two cases (cases 5 and 6), complete concordance between TP53 mutational status and P53 overexpression by IHC was not observed in our study as shown by cases 2 and 3, indicating mechanism(s) other than mutation and/or deletion may be responsible for P53 overexpression. In addition, mutations involving epigenetic and transcriptional regulators and the PI3K pathway were detected in both the intra- and extramedullary specimens. Particularly, mutations of LATS1 (large tumor suppressor kinase 1) and YAP1 (Yes-associated protein 1) genes in the Hippo pathway were seen in the extramedullary plasmablastic component but not in the bone marrow component of 1 case (case 5), indicating potential involvement of the Hippo pathway in the pathogenesis of discordant presentation (29, 30). Lastly, NGS studies performed on both components of the discordant cases also identified mutations of ARID1A, KMT2C, TET2, KDM5C, SAMHD1 and DIS3, which have been previously reported as mutational drivers of multiple myeloma (25, 31). It should be noted that the absence of certain mutations in the tissue sample that were identified in the bone marrow of case 5 may be explained by the disappearance of subclones present initially during disease progression.
Discordant extramedullary plasmablastic transformation often poses a diagnostic challenge particularly as a differential diagnosis from plasmablastic lymphoma. Pathologically, both entities show high-grade features such as high proliferation index and high levels of P53 and CMYC expression. Interestingly, 5/10 of our cases showed aberrant expression of Cyclin D1 by IHC, which favored a diagnosis of plasmablastic transformation of myeloma in those cases. Clinical features, such as a history of multiple myeloma, CRAB, diffuse bone marrow involvement and the presence of an M-protein favors a diagnosis of plasmablastic transformation of myeloma. In contrast, the presence of HIV infection, an oral cavity manifestation or EBV positivity of the tumor would favor a plasmablastic lymphoma. All of our cases were negative for EBV by EBER ISH. MYC translocations have been identified in approximately 50% of plasmablastic lymphoma; however, MYC alternations, including translocation, have also been reported in approximately 15% of plasma cells neoplasms, and are present at even higher frequency in extramedullary sites as shown by the current and previous (2, 18) studies. Therefore, high expression of CMYC and detection of MYC translocations in isolation cannot be used to differentiate plasmablastic lymphoma from plasmablastic myeloma. Regarding the molecular characteristics, patterns of genomic aberrations by aCGH in plasmablastic lymphomas were reported to be more closely related to those of diffuse large B-cell lymphoma in one study (32) and more closely related to those of plasma cell neoplasms in another series of 4 patients (33). These conflicting data may be suggestive of disease heterogeneity among plasmablastic lymphomas. More recent studies have shown that plasmablastic lymphoma is characterized by an atypical EBV latency program, an miRNA signature distinct from that of extramedullary plasmacytoma (34) and genetic alterations involving PRDM (35). The high frequency of RAS pathway mutations described in extramedullary plasmablastic myeloma has not be reported in plasmablastic lymphoma and therefore, may be helpful in the differential diagnosis when such data are available. It should be noted that transformation of low grade B-cell lymphomas, such as chronic lymphocytic leukemia and follicular lymphoma, can also present as plasmablastic lymphoma with MYC translocation; therefore, clinical history is also important for accurate diagnosis. (36, 37)
Due to the retrospective nature of this study, genetic and immunohistochemical studies could not be uniformly performed in all cases. Nevertheless, our study showed that discordant extramedullary plasmablastic transformation is a rare phenomenon and has an aggressive clinical behavior. The poor prognosis of extramedullary plasmablastic myeloma prevailed despite the mostly low-level involvement by typical and mature-looking myeloma cells in the bone marrow. Genetically, frequent RAS pathway mutations and accumulation of more complex cytogenetic abnormalities were identified during the transformation. These findings indicate that inhibition of the MAPK pathway might be considered as a therapeutic approach to treat this aggressive condition.
Acknowledgements
This study was supported in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 and MSK SPORE in lymphoma (P50 CA192937).
Footnotes
Part of the data was presented in abstract form at the 106th Annual Meeting of the United States and Canadian Association for Pathologists, Vancouver, Canada in March 2018
Conflict-of-interest disclosure: A.D. has received consultancy fees from Roche, Corvus Pharmaceuticals, Physicians’ Education Resource, Seattle Genetics, Peerview Institute, Oncology Specialty Group, Pharmacyclics, Celgene, Novartis, Takeda, EUSAPharma and research grants from Roche. W.X. has received research support from Stemline Therapeutics. The other authors have nothing to disclose.
References
- 1.Swerdlow SCE, Harris NE, et al. WHO classification of tumours of haematopoietic and lymphoid tissue.. Lyon, IARC press;. 2017. [Google Scholar]
- 2.Lorsbach RB, Hsi ED, Dogan A, et al. Plasma cell myeloma and related neoplasms. American journal of clinical pathology. 2011;136:168–182. [DOI] [PubMed] [Google Scholar]
- 3.Bartl R, Frisch B, Burkhardt R, et al. Bone marrow histology in myeloma: its importance in diagnosis, prognosis, classification and staging. Br J Haematol. 1982;51:361–375. [DOI] [PubMed] [Google Scholar]
- 4.Bartl R, Frisch B, Fateh-Moghadam A, et al. Histologic classification and staging of multiple myeloma. A retrospective and prospective study of 674 cases. American journal of clinical pathology. 1987;87:342–355. [DOI] [PubMed] [Google Scholar]
- 5.Greipp PR, Raymond NM, Kyle RA, et al. Multiple myeloma: significance of plasmablastic subtype in morphological classification. Blood. 1985;65:305–310. [PubMed] [Google Scholar]
- 6.Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol. 2010;21:325–330. [DOI] [PubMed] [Google Scholar]
- 7.Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica. 2012;97:1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Bhutani N, Bhargawa S, et al. Extra-skeletal plasmablastic myeloma presenting as palatal growth - An unusual entity. Int J Surg Case Rep. 2017;41:423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy R, Vohra S, Macurak R. Secondary Extramedullary Plasmablastic Myeloma of the Small Bowel. Clin Gastroenterol Hepatol. 2015;13:A23–24. [DOI] [PubMed] [Google Scholar]
- 10.Srija M, Zachariah PP, Unni VN, et al. Plasmablastic myeloma presenting as rapidly progressive renal failure in a young adult. Indian J Nephrol. 2014;24:41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sailer M, Vykoupil KF, Peest D, et al. Prognostic relevance of a histologic classification system applied in bone marrow biopsies from patients with multiple myeloma: a histopathological evaluation of biopsies from 153 untreated patients. European journal of haematology. 1995;54:137–146. [DOI] [PubMed] [Google Scholar]
- 12.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He J, Abdel-Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 15.Furukawa Y, Kikuchi J. Molecular pathogenesis of multiple myeloma. International journal of clinical oncology. 2015;20:413–422. [DOI] [PubMed] [Google Scholar]
- 16.Touzeau C, Moreau P. How I treat extramedullary myeloma. Blood. 2016;127:971–976. [DOI] [PubMed] [Google Scholar]
- 17.Gagelmann N, Eikema DJ, Iacobelli S, et al. Impact of extramedullary disease in patients with newly diagnosed multiple myeloma undergoing autologous stem cell transplantation: a study from the Chronic Malignancies Working Party of the EBMT. Haematologica. 2018;103:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avet-Loiseau H, Gerson F, Magrangeas F, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98:3082–3086. [DOI] [PubMed] [Google Scholar]
- 19.Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000;97:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen T, Kuehl M, Lodahl M, et al. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood. 2005;105:317–323. [DOI] [PubMed] [Google Scholar]
- 23.Lionetti M, Barbieri M, Todoerti K, et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget. 2015;6:24205–24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Haart SJ, Willems SM, Mutis T, et al. Comparison of intramedullary myeloma and corresponding extramedullary soft tissue plasmacytomas using genetic mutational panel analyses. Blood Cancer Journal. 2016;6:e426–e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker BA, Mavrommatis K, Wardell CP, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Portier M, Moles JP, Mazars GR, et al. p53 and RAS gene mutations in multiple myeloma. Oncogene. 1992;7:2539–2543. [PubMed] [Google Scholar]
- 27.Drach J, Ackermann J, Fritz E, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood. 1998;92:802–809. [PubMed] [Google Scholar]
- 28.Avet-Loiseau H, Li JY, Godon C, et al. P53 deletion is not a frequent event in multiple myeloma. Br J Haematol. 1999;106:717–719. [DOI] [PubMed] [Google Scholar]
- 29.Hao Y, Chun A, Cheung K, et al. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283:5496–5509. [DOI] [PubMed] [Google Scholar]
- 30.Maruyama J, Inami K, Michishita F, et al. Novel YAP1 Activator, Identified by Transcription-Based Functional Screen, Limits Multiple Myeloma Growth. Mol Cancer Res. 2018;16:197–211. [DOI] [PubMed] [Google Scholar]
- 31.Heuck C, Johann D, Walker BA, et al. Characterization of the Mutational Landscape of Multiple Myeloma Using Comprehensive Genomic Profiling. 2014;124:3418–3418. [Google Scholar]
- 32.Chang CC, Zhou X, Taylor JJ, et al. Genomic profiling of plasmablastic lymphoma using array comparative genomic hybridization (aCGH): revealing significant overlapping genomic lesions with diffuse large B-cell lymphoma. Journal of hematology & oncology. 2009;2:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castillo JJ, Winer ES, Stachurski D, et al. HIV-negative plasmablastic lymphoma: not in the mouth. Clinical lymphoma, myeloma & leukemia. 2011;11:185–189. [DOI] [PubMed] [Google Scholar]
- 34.Ambrosio MR, Mundo L, Gazaneo S, et al. MicroRNAs sequencing unveils distinct molecular subgroups of plasmablastic lymphoma. Oncotarget. 2017;8:107356–107373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montes-Moreno S, Martinez-Magunacelaya N, Zecchini-Barrese T, et al. Plasmablastic lymphoma phenotype is determined by genetic alterations in MYC and PRDM1. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2017;30:85–94. [DOI] [PubMed] [Google Scholar]
- 36.Martinez D, Valera A, Perez NS, et al. Plasmablastic transformation of low-grade B-cell lymphomas: report on 6 cases. The American journal of surgical pathology. 2013;37:272–281. [DOI] [PubMed] [Google Scholar]
- 37.Ouansafi I, He B, Fraser C, et al. Transformation of follicular lymphoma to plasmablastic lymphoma with c-myc gene rearrangement. American journal of clinical pathology. 2010;134:972–981. [DOI] [PubMed] [Google Scholar]