

Abstract
Hypoxia-induced pulmonary vascular constriction and structure remodeling are the main causes of hypoxic pulmonary hypertension. In the present study, an adeno-associated virus vector, containing Tie2 promoter and hypoxia response elements, was designed and named HTSFcAng(1-7). Its targeting, hypoxic inducibility, and vascular relaxation were examined in vitro, and its therapeutic effects on hypobaric hypoxia-induced pulmonary hypertension were examined in rats. Transfection of HTSFcAng(1-7) specifically increased the expression of angiotensin-(1-7) in endothelial cells in normoxia. Hypoxia increased the expression of angiotensin-(1-7) in HTSFcAng(1-7)-transfected endothelial cells. The condition medium from HTSFcAng(1-7)-transfected endothelial cells inhibited the hypoxia-induced proliferation of pulmonary artery smooth muscle cells, relaxed the pulmonary artery rings, totally inhibited hypoxia-induced early contraction, enhanced maximum relaxation, and reversed phase II constriction to sustained relaxation. In hypoxic pulmonary hypertension rats, treatment with HTSFcAng(1-7) by nasal drip adeno-associated virus significantly reversed hypoxia-induced hemodynamic changes and pulmonary artery-wall remodeling, accompanied by the concomitant overexpression of angiotensin-(1-7), mainly in the endothelial cells in the lung. Therefore, hypoxia-inducible overexpression of angiotensin-(1-7) in pulmonary endothelial cells may be a potential strategy for the gene therapy of hypoxic pulmonary hypertension.
Keywords: pulmonary hypertension, hypobaric hypoxia, adeno-associated virus vector, gene therapy, angiotensin-(1-7)
Graphical Abstract
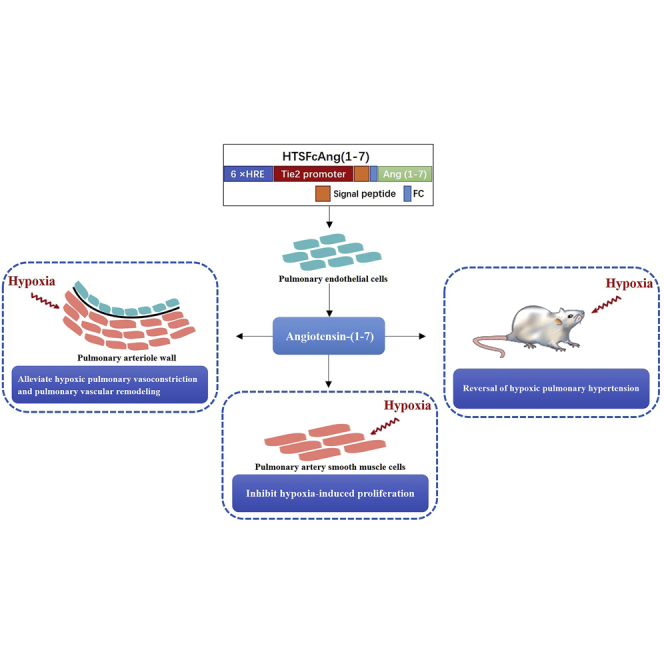
A novel adeno-associated virus vector for the gene therapy of hypoxic pulmonary hypertension is reported that contains the Tie2 promoter, hypoxia response elements, and target gene angiotensin-(1-7). The vector exhibited hypoxia-inducible overexpression of angiotensin-(1-7), mainly in pulmonary endothelial cells, and significantly reversed hypoxic pulmonary hypertension in rats.
Introduction
Hypoxic pulmonary hypertension is highly prevalent in advanced chronic obstructive pulmonary diseases and chronic high-altitude hypoxia.[1],[2] The main pathophysiological characteristics of hypoxic pulmonary hypertension are hypoxia-induced pulmonary vascular constriction and structure remodeling, which are mainly caused by the abnormal activation of local vascular constriction systems in lungs and the aberrant proliferation of pulmonary artery smooth muscle cells (PASMCs), respectively. Although there has been intensive research on hypoxic pulmonary hypertension, the present pharmacological agents can only moderately improve the symptoms and hemodynamic parameters, and there is still a lack of effective agents to improve the long-term prognosis or reduce the mortality. Therefore, it is highly needed to explore novel agents for hypoxic pulmonary hypertension.
The intrapulmonary renin-angiotensin dual-axis system, including vasoconstrictor axis and vasodilatory axis, plays an important role in hypoxic pulmonary hypertension.3, 4, 5 The vasoconstrictor axis is composed of angiotensin-converting enzyme, angiotensin II, and angiotensin II type 1 receptors, whereas the vasodilatory axis is composed of angiotensin-converting enzyme 2 (ACE2), angiotensin-(1-7), and Mas receptors. The inappropriate activation of the vasoconstrictor axis and a lack of compensatory activation of the vasodilatory axis promote the development of hypoxic pulmonary hypertension.6 In the renin-angiotensin system, angiotensin-(1-7) is the main effect molecular to dilate in the renin-angiotensin system. Angiotensin-(1-7) also opposes the trophic, proliferative, and prothrombotic actions of angiotensin II.7 In pulmonary hypertension patients, plasma angiotensin-converting enzyme 2 activity is reduced, and infusion of recombinant human angiotensin-converting enzyme 2 improves pulmonary hemodynamics and reduces the markers of oxidant and inflammatory mediators.8 Therefore, angiotensin-converting enzyme 2/angiotensin-(1-7) may be a potent target for the treatment of hypoxic pulmonary hypertension.3,9, 10, 11, 12
Gene therapy is emerging as an alternative for refractory diseases, such as hypoxic pulmonary hypertension. However, it is troubled by the bottleneck, lacking suitable target genes and effective methods to regulate the expression of transgenes accordingly and specifically.
To determine whether hypoxia-inducible overexpression of angiotensin-(1-7) in the lung can reverse hypoxic pulmonary hypertension, we constructed an adeno-associated virus (AAV) vector, named HTSFcAng(1-7), with the promoter Tie2, hypoxia response elements, and angiotensin-(1-7). Then we explored the endothelial targeting, hypoxic inducibility, and vasodilatory action in vitro and the treatment effect on hypobaric hypoxia-induced pulmonary hypertension in rats. The results showed that HTSFcAng(1-7) exhibited the hypoxia-inducible overexpression of angiotensin-(1-7), mainly in pulmonary endothelial cells, and significantly reversed hypoxic pulmonary hypertension in rats.
Results
HTSFcAng(1-7) Exhibited the Specific Overexpression in Pulmonary Endothelial Cells and Hypoxic Inducibility in Hypoxia
First, we tested the target cell specificity and hypoxic inducibility of the HTSFcAng(1-7) vector in the cultured cells. The HTSFcAng(1-7) or HTSFc was transfected into HEK293 cells, NIH 3T3 cells, A549 cells, NR8383, primary rat pulmonary microendothelial cells (PMVECs), primary pulmonary artery smooth muscle cells (PASMCs), primary bone mesenchymal stem cells (MSCs), peripheral blood neutrophils, and monocytes via Lipofectamine 2000. ELISA was used to measure the content of angiotensin-(1-7) in the cell supernatant, 24 h after transfection. Figure 1B showed that HTSFcAng(1-7) transfection significantly elevated the content of angiotensin-(1-7), only in PMVEC supernatants (p < 0.05, n = 3) but not in the other cell supernatants or after the control vector HTSFc transfection, indicating the targeted expression of HTSFcAng(1-7) in PMVECs.
Figure 1.

HTSFcAng(1-7) Exhibited Specific Overexpression in Pulmonary Endothelial Cells and Hypoxic Inducibility in Hypoxia
(A) Diagram of the constructed HTSFcAng(1-7) and HTSFc. (B) Effects of HTSFcAng(1-7) or HTSFc transfection on the expression of angiotensin-(1-7) in A549 cells, HEK293 cells, NIH 3T3 cells, pulmonary artery smooth muscle cells (PASMCs), pulmonary microendothelial cells (PMVECs), rat peripheral blood neutrophils, monocytes, NR8383 cells, and primary bone mesenchymal stem cells (MSCs). (C and D) Effect of oxygen concentration on the expression of angiotensin-(1-7) (C) and the increment rate of the expression of angiotensin-(1-7) (D) in PMVECs transfected with HTSFcAng(1-7). ∗p < 0.05. Data are represented as mean ± SD from three replicated experiments (B and C, respectively).
The effects of hypoxia on the expression of angiotensin-(1-7) in PMVECs were shown in Figure 1C. In the untransfected PMVECs, hypoxia (5% and 1% O2 concentration) significantly decreased the content of angiotensin-(1-7) in the supernatant of PMVECs (n = 3, p < 0.05), showing that hypoxia inhibited the endogenous expression of angiotensin-(1-7) in PMVECs. HTSFcAng(1-7) transfection significantly increased the expression of angiotensin-(1-7) in PMVECs, both in normoxia and hypoxia (all n = 3, p < 0.05).
The effect of oxygen concentration on the increment rate of the expression of transgene angiotensin-(1-7) was presented in Figure 1D. The increment rate is calculated by normalizing the content of angiotensin-(1-7) in the supernatant of transfected cells to the content of angiotensin-(1-7) in the supernatant of untransfected cells in each oxygen concentration, respectively. The increment rate was shown in an oxygen concentration-dependent manner: the lower oxygen concentration, the higher increment rate of expression of angiotensin-(1-7) (all n = 3, p < 0.05). Therefore, the transgene HTSFcAng(1-7) exhibits the distinct hypoxic inducibility in PMVECs.
Effects of the Condition Medium from PMVECs Transfected with HTSFcAng(1-7) on Hypoxia-Induced Proliferation of Rat Primary PASMCs In Vitro
Then we assessed the effects of the condition medium from PMVECs, which were transfected with HTSFcAng(1-7) and cultured in 10% O2 concentration, on the proliferation of rat primary PASMCs cultured in normoxia (20% O2) and in hypoxia (10%, 5% and 1% O2).
3-(4, 5-Dimethylthiazal-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) experiments showed that hypoxia significantly promoted the proliferation of PASMCs in an oxygen concentration-dependent manner (all p < 0.05, n = 3; Figure 2A). The condition medium from PMVECs, transfected with HTSFcAng(1-7), had no significant effect on the proliferation of PASMCs cultured in normoxia; however, it significantly inhibited the hypoxia-induced proliferation of PASMCs cultured in 10%, 5%, or 1% O2 concentration (p < 0.05, n = 3; Figure 2B), and the inhibition effect was dose dependent (Figures 2C and 2D). Moreover, the condition medium from PMVECs transfected with control vector HTSFc had no such effect. Cell-counting experiments showed the similar results with MTT experiments (Figures S1A–S1D).
Figure 2.

Effects of the Condition Medium from PMVECs Transfected with HTSFcAng(1-7) on Hypoxia-Induced Proliferation of Primary PASMCs
PASMC proliferation was measured by the 3-(4, 5-dimethylthiazal-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. (A) Effects of oxygen concentration on the proliferation of rat primary PASMCs. (B) Inhibition effects of the condition medium from PMVECs transfected with HTSFcAng(1-7) on the proliferation of PASMCs cultured in normoxia (20% O2) and in hypoxia (10%, 5%, and 1% O2). (C and D) Concentration (v/v)-dependent inhibition effects of the condition medium, from the PMVECs transfected with HTSFcAng(1-7), on the proliferation of PASMCs cultured in 10% O2 concentration. CM, condition medium. ∗p < 0.05. Data are represented as mean ± SD from three replicated experiments in each panel.
Effects of the Condition Medium from PMVECs Transfected with HTSFcAng(1-7) on the Constriction and Relaxation of the Isolated Pulmonary Artery Rings in Hypoxia
Then we tested the vascular effects of the condition medium from PMVECs transfected with HTSFcAng(1-7) on the isolated pulmonary artery rings in hypoxia. The experiments were done in the endothelial cell-denuded, phenylephrine-precontracted pulmonary artery rings.
Figure 3A is the representative curve for the effects of acute hypoxia on phenylephrine-precontracted pulmonary artery rings. Acute hypoxia resulted in biphasic vasocontractions: an initial transient contraction (phase I constriction), then a transient relaxation (maximum dilation), and finally, a delayed, sustained contraction (phase II constriction). The effect of acute hypoxia on pulmonary artery ring is consistent with the previous result.13 Figure 3B shows the typical effects of condition medium from PMVECs transfected with HTSFcAng(1-7) pretreatment on the artery ring in hypoxia. The condition medium totally inhibited the hypoxia-induced early contraction, enhanced the following maximum dilation, and reversed the phase II constriction to the sustained relaxation (phase II dilation). The Mas receptor antagonist A-779 at 1 μM eliminated the effects of the condition medium from PMVECs transfected with HTSFcAng(1-7) on pulmonary artery rings in hypoxia (Figure 3C). However, the condition medium from PMVECs transfected with control HTSFc or along with A-779, did not affect the effects of hypoxia on the artery rings (Figures 3D and 3E). The summarized data of maximum dilation, phase II constriction, and phase II dilation were shown in Figures 3F and 3G.
Figure 3.

Effects of the Condition Medium from PMVECs Transfected with HTSFcAng(1-7) on the Constriction and Relaxation of the Isolated Pulmonary Artery Rings in Hypoxia
(A) The representative curve for the effects of acute hypoxia on phenylephrine-precontracted pulmonary artery rings. Acute hypoxia resulted in vascular biphasic contractions: an initial transient contraction, then a transient relaxation (maximum dilation), and finally a delayed, sustained contraction (phase II constriction). (B–E) The representative curve for the effect of the condition medium, which was from PMVECs transfected with HTSFcAng(1-7) (B and C) or HTSFc (D and E), on the artery ring in hypoxia (% O2) with (C and E) or without (B and D Mas receptor antagonist A-779 (1 μM). (F and G) The summarized data of maximum vasodilation (F) and phase II vasoconstriction and phase II vasodilation (G). CM, condition medium. *p < 0.05 versus control and HTSFcAng(1-7). Data are represented as mean ± SD, n = 6.
HTSFcAng(1-7) Reversed Hypobaric Hypoxia-Induced Pulmonary Hypertension in Rats
We then explored the therapeutic effects of HTSFcAng(1-7) administration on hypoxic pulmonary hypertension in rats. Figure 4 showed the dynamic changes of hemodynamic and pulmonary vascular remodeling during 4 weeks of hypoxia exposure in rats. Hypoxia for 4 weeks had no significant effect on the carotid pressures (mean carotid artery pressure [mCAP]; Figure 4A). However, the right ventricle systolic pressure (RVSP; Figure 4B), the ratio of right ventricle weight to left ventricle plus septum weight (RV/(LV + S); Figure 4C), and the pulmonary artery percent wall thickness (WT%) and wall area (WA%; Figures 4D and 4E) progressively and significantly increased during hypoxia exposure (all p < 0.05; n = 6 for RVSP and RV/(LV + S); n = 60 [muscular arteries] for WT% and WA%). The representative hematoxylin and eosin staining of the lung vessels were shown in Figure 4F. Figure 4 showed that hypoxia for 2 weeks induced significant pulmonary hypertension in rats; therefore, to test the therapeutic effect, HTSFcAng(1-7) or HTSFc was administrated 2 weeks after hypoxia, and the rats were kept in hypoxia for another 2 weeks.
Figure 4.

Dynamic Changes of Hemodynamic and Pulmonary Vascular Remodeling during 4 Weeks of Hypoxia Exposure in Rats
(A–E) The dynamic changes of the carotid pressure (A), the right ventricle systolic pressure (RVSP; B), the ratio of right ventricle weight to left ventricle plus septum weight (RV/(LV + S); C), the pulmonary artery percent wall thickness (WT%; D), and percent wall area (WA%; E) during normoxia or hypoxia exposure in rats. (F) Representative hematoxylin and eosin staining of pulmonary vessels. ∗p < 0.05 versus normoxia; n = 6 for RVSP and RV/(LV + S); n = 60 (muscular arteries) for WT% and WA% (for details, see Materials and Methods). Data are expressed as mean ± SD.
Figures 5B and 5C showed that HTSFcAng(1-7) treatment significantly reduced the increased RVSP and RV/(LV + S) (all p < 0.05; n = 6). The representative hematoxylin and eosin staining of lung vessels was shown in Figure 5D. The summarized data of MT% and MA% of pulmonary artery were shown in Figures 5E and 5F. Hypoxia significantly increased MT% and MA%. HTSFcAng(1-7) treatment significantly reduced the increased MT% and MA% (all p < 0.05; n = 60). However, administration of HTSFc had no such effects. In addition, administration of HTSFcAng(1-7) had no significant effects on the mean carotid artery pressure (Figure 5A), and the same dose of HTSFcAng(1-7) or HTSFc did not induce alveolitis in the lungs from rats in normoxia (Figure S2; Table S1). These results demonstrated that HTSFcAng(1-7) significantly reversed hypoxic pulmonary hypertension in rats.
Figure 5.

Therapeutic Effects of HTSFcAng(1-7) Administration on Hypobaric Hypoxia-Induced Pulmonary Hypertension in Rats
(A–C) Administration of adeno-associated virus 9 containing HTSFcAng(1-7) had no significant effects on the mean carotid artery pressure (mCAP; A) but significantly reduced the hypoxia-increased right ventricle systolic pressure (RVSP; B) and the ratio of right ventricle weight to left ventricle plus septum weight (RV/(LV + S); C). (D) Representative hematoxylin and eosin staining of pulmonary vessels. (E and F) The summarized data of pulmonary artery percent wall thickness (WT%; E) and percent wall area (WA%; F). ∗p < 0.05. Data are expressed as mean ± SD, n = 6.
HTSFcAng(1-7) Specifically Increased the Expression of Angiotensin-(1-7) in the Lung Endothelial Cells in Hypoxic Pulmonary Hypertension Rats
Further, we tested the level of angiotensin-(1-7) in lungs, hearts, livers, and kidneys from hypoxic pulmonary hypertension rats administrated with HTSFcAng(1-7) or HTSFc. As shown in Figure 6A, hypoxia progressively and significantly decreased the level of angiotensin-(1-7) in lungs (p < 0.05; n = 6). Hypoxia also significantly decreased the level of angiotensin-(1-7) in hearts, livers, and kidneys (p < 0.05; n = 6) (Figure 6B). However, administration of HTSFcAng(1-7) only significantly increased the level of angiotensin-(1-7) in lungs (p < 0.05; n = 6) and not in livers, hearts, and kidneys. Similarly, administration of the control virus HTSFc did not increase the level of angiotensin-(1-7) in the other four organs.
Figure 6.

HTSFcAng(1-7) Increased the Expression of Angiotensin-(1-7) in the Lungs in Hypobaric Hypoxia-Induced Pulmonary Hypertension Rats
(A) Dynamic changes of the expression of angiotensin-(1-7) in lungs during the development of hypoxic pulmonary hypertension rats. ∗p < 0.05 versus normoxia. (B) Effects of administration of adeno-associated virus 9 containing HTSFcAng(1-7) and HTSFc on the expression of angiotensin-(1-7) in lungs, livers, hearts, and kidneys. ∗p < 0.05 versus normoxia-corresponding organ; #p < 0.05 versus hypoxia lung. Data are expressed as mean ± SD, n = 6.
In the present study, we used the Tie2 promoter to drive the expression of angiotensin-(1-7) in the endothelial cells, but the Tie2 gene also expresses in monocyte, macrophage, and other bone marrow-derived cells. To determine whether HTSFcAng(1-7) specifically increased the expression of angiotensin-(1-7) in the endothelial cells in lungs in hypoxic pulmonary hypertension rats, immunofluorescence costaining was used to detect the colocalization of angiotensin-(1-7) and cluster of differentiation (CD)31 (a PMVEC marker) or CD11b (a marker of monocytes, macrophages, and bone marrow-derived hematopoietic cells). Figure 7A showed that hypoxia decreased the expression of angiotensin-(1-7) and CD31 in the lung endothelial cells (pulmonary alveoli and vessel), which were markedly increased by HTSFcAng(1-7) administration. Angiotensin-(1-7) and CD31 were colocalized mainly in the endothelial cells. Whereas hypoxia had no effect on the expression of CD11b (Figure 7B), HTSFcAng(1-7) only raised the expression of angiotensin-(1-7) but not CD11b, and angiotensin-(1-7) and CD11b had no significant colocalization. Therefore, HTSFcAng(1-7) exhibited targetable overexpression in the pulmonary endothelial cells but not monocytes, macrophages, and bone marrow-derived hematopoietic cells in hypoxic pulmonary hypertension rats.
Figure 7.

Angiotensin-(1-7) Colocalized with CD31 in the Lung Endothelial Cells in Hypobaric Hypoxia-Induced Pulmonary Hypertension Rats
(A and B) Immunofluorescence costaining of angiotensin-(1-7) and CD31 (A) and angiotensin-(1-7) and CD11b (B) was measured in lungs in hypobaric hypoxia-induced pulmonary hypertension rats. Angiotensin-(1-7) is stained green; CD31 and CD11b are stained red. The nucleus was stained blue by DAPI. The images are acquired using a confocal laser microscope with the same exposure time during the same imaging session. Scale bars, 20 μM.
Discussion
In the present study, we developed a novel adeno-associated virus vector composed of endothelial cells promoter Tie2, hypoxia response elements, and target gene angiotensin-(1-7). The constructed vector exhibited specific hypoxia-inducible overexpression of angiotensin-(1-7) in pulmonary endothelial cells in vitro. We first demonstrated that hypoxia-inducible overexpression of angiotensin-(1-7) in pulmonary endothelial cells reversed hypobaric hypoxia-induced pulmonary hypertension in rats.
Gene transferring by viral vector is intensively used in gene therapy research and clinic trials for their high transduction efficiency.14, 15, 16, 17 Lentiviral or adenoviral vector delivery of angiotensin-converting enzyme 2 or bone morphogenetic protein receptor type II prevented and reversed monocrotaline-induced or hypoxia-induced pulmonary hypertension in mice or rats.18,19 Adenovirus-mediated knockdown of pulmonary endothelial Tph1 attenuates hypoxia-induced pulmonary hypertension in rats.14 Vascular sarcoplasmic reticulum Ca(2+)-ATPase 2a (SERCA2a) overexpression by airway-based delivery of AAV vectors ameliorates chronic postcapillary pulmonary hypertension in swine.20
Many of these viral vectors are designed using the universal strong promoter, such as cytomegalovirus (CMV) and thymidine kinase (TK); therefore, the expression of the target gene lacks targetability and inducibility. Some researchers combined the universal strong promoters with hypoxia response elements specifically to enhance the target gene expression in hypoxic cells.21,22 These hypoxia-responsive vectors showed significant hypoxia responsiveness in vitro and in vivo.21, 22, 23
Previously, we constructed a lentiviral vector specifically to drive the expression of the target gene in vascular smooth muscle cells, according to oxygen concentration, by combining hypoxia response elements with the smooth muscle-specific promoter.24 The constructed vector exhibited hypoxic inducibility and relatively targetable expression in pulmonary artery smooth muscle cells. It inhibited the hypoxia-induced proliferation of pulmonary artery smooth muscle cells in vitro and prevented the development of hypoxic pulmonary hypertension in mice.24 In the present study, we adopted the similar strategy to construct an adeno-associated viral vector, named HTSFcAng(1-7), to drive the overexpression of angiotensin-(1-7) in pulmonary endothelial cells in hypoxia by combining hypoxia response elements with promoter Tie2 (Figure 1A). In vitro HTSFcAng(1-7) specifically increased the expression of angiotensin-(1-7) in endothelial cells and showed hypoxic inducibility (Figures 1B–1D). In vivo, treatment with HTSFcAng(1-7) by nasal drip AAV(serotypes 9) significantly reversed hypobaric hypoxia-induced pulmonary hypertension in rats (Figure 5). Therefore, it may be a feasible design to combine the hypoxia response element with the tissue-specific promoter to control the expression of the target gene in the specific tissue or organ in hypoxia.
We determined the content of angiotensin-(1-7) in the tissue homogenate from the heart, liver, kidney, and lung in HTSFcAng(1-7)-treated pulmonary hypertension rats and found that only the angiotensin-(1-7) in the lung was significantly increased (Figure 6). It is interesting to argue that HTSFcAng(1-7) specifically overexpressed in the lung but not in other organs. In the present study, the adeno-associated virus containing HTSFcAng(1-7) was delivered to rats by nasal drip. It may be the main cause for the vector specifically expressed in lungs. In addition, these results may also reflect one of the different characteristics between pulmonary and system circulation when exposed to moderate hypoxia. It is well known that the response of pulmonary vessels to hypoxia is very different to that of circulation vessels. We also found that the proliferation of PASMCs differed from aortic SMCs in moderate hypoxia (5%–10% O2 concentration). Moderate hypoxia significantly promotes the proliferation of PASMCs but not aortic SMCs (unpublished data). It will be explored in our future work.
In the present study, the Tie promoter is used to drive the expression of the target gene in endothelial cells. However, the Tie promoter is not an endothelial cell-specific promoter, because Tie2 is also expressed in the monocyte or macrophage and other bone marrow-derived cells. We did in vitro and in vivo experiments to determine the targeting of HTSFcAng(1-7). In vitro HTSFcAng(1-7) transfection significantly elevated the content of angiotensin-(1-7) only in PMVECs but not in HEK293 cells, NIH 3T3 cells, A549 cells, NR8383, primary PASMCs, primary MSCs, peripheral blood neutrophils, and monocytes (Figure 1); in vivo HTSFcAng(1-7) administration induced the obvious colocalization between angiotensin-(1-7) and CD31, a PMVEC marker, but not between angiotensin-(1-7) and CD11b, a marker of monocytes, macrophages, and other bone marrow-derived hematopoietic cells. Therefore, HTSFcAng(1-7) specifically increased the expression of angiotensin-(1-7) in the lung endothelial cells in hypoxic pulmonary hypertension rats. With the consideration that HTSFcAng(1-7) administration did not induce obvious alveolitis (Figure S2; Table S1) so the bone marrow-derived monocytes were not many in lungs, the beneficial effects of HTSFcAng(1-7) were mediated mainly by the lung endothelial cells but not the hematopoietic cells in the present study.
Previous studies have shown that the plasma ACE2 activity is reduced in human pulmonary arterial hypertension;8 however, in the present study, the level and activity of plasma ACE2 were not measured in an animal model, so it is unknown whether the plasma ACE2 activity is reduced in the present animal model. In addition, the hypoxia-induced overexpression of angiotensin-(1-7) was observed only in cultured primary pulmonary endothelial cells transfected with HTSFcAng(1-7). It has not been proven at a pulmonary endothelial layer in treated model animals, although there was the increased production of angiotensin-(1-7) in lungs. However, in another ongoing study, we designed a similar vector containing the TIE2 promoter and HRE to drive the overexpression of angiotensin-converting enzyme 2, and we found that the overexpression of angiotensin-converting enzyme 2 is mainly distributed in pulmonary endothelial cells in hypoxic pulmonary hypertension rats (unpublished data). Therefore, HTSFcAng(1-7) may work similarly in vivo with in vitro experiments. We hope to explore these questions in future work.
Previous studies showed that endogenous estrogen may attenuate hypoxic pulmonary hypertension,25, 26, 27, 28 and phenobarbital is a potential cardiodepressant.29 In the present study, we chose male animals to avoid the effect of estrogen on the results and used phenobarbital to anesthetize animals for measuring RVSP in all groups. However, it is not known whether animal sex influences the effects of HTSFcAng(1-7) in vivo and whether phenobarbital affects the absolute value of RVSP, so the use of male animals rather than animals of both sexes and phenobarbital as an anesthetic are two limitations of this study. In addition, HTSFcAng(1-7) was constructed with the mouse Tie2 promoter, due to lacking the reference on the rat Tie2 promoter. However, the in vitro experiments showed that the vector worked well in cultured primary rat pulmonary artery endothelial cells (Figure 1B). Therefore, the conclusion from the animal experiment is reliable.
Conclusions
In summary, our study indicates the therapeutic potential of hypoxia-inducible overexpression of angiotensin-(1-7) with the Tie2 promoter and hypoxia response elements in hypoxic pulmonary hypertension and provides evidence supporting angiotensin-(1-7) as a target of gene therapy for hypoxic pulmonary hypertension.
Materials and Methods
Vector Construction and Adeno-Associated Virus Production
The designed sequence is composed of six copies of HREs (derived from the 5′ untranslated region of rat vascular endothelial growth factor), and the sequence is 5′-GACTCCACAGTGCATACGTGGGCTTCCACAGGTCGTCTC-3′), the mouse Tie promoter (Mus musculus receptor tyrosine kinase TIE2 gene, 5′-flanking region, access number AF022456.1, location: AF022456.1:1-223), SPFc (5′-ATGAAACATCTGTGGTTCTTCCTTCTCCTGGTGGCAGCTCCCAGATGGGTCCTGTCC-3′), and the coding region of rat angiotensin-(1-7) (5′-GACCGGGTGTACATACACCCC-3′). The designed sequence is synthesized and cloned into the backbone vector pAAV-MCS. Therefore, the expression of angiotensin-(1-7) is under the control of HREs and the TIE2 promoter (pAAV-HRE-TIE2-SPFc-ANG-(1-7), referred to as HTSFcAng(1-7)). The vector without the coding sequence of angiotensin-(1-7) was used as the control vector (HTSFc) for the vehicle group. The diagram of the vectors is shown in Figure 1A. The AAV (serotype 9) particles, containing HTSFcAng(1-7) or HTSFc, were packaged by biotechnology company Biowit Technologies (Shenzhen, China).
Animal Experiments
All animals were maintained in a 12- to 12-h light-dark cycle in an air-conditioned room (25°C) and had free access to food and water. All of the experiments were approved by the Animal Care and Use Committee of the Fourth Military Medical University and complied with the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Male Sprague-Dawley rats (200–210 g) were randomly divided into four groups (normoxia, hypoxia, hypoxia + HTSFcAng(1-7), and hypoxia + HTSFc; all n = 6) and accordingly maintained in a normoxia (21% O2) or hypobaric hypoxia (depressurized to 380 mmHg corresponding to 10% O2) environment for 28 days. A single injection of the adeno-associated virus (serotype 9) particle (3 × 109 transduction units) containing HTSFcAng(1-7) or HTSFc was given via nasal drop at day 15 accordingly.
Pulmonary Hemodynamics
The rats were anesthetized with phenobarbital (30 mg/kg intraperitoneally [i.p.]). The RVSP was measured according to a right cardiac catheterization procedure and used as an indicator of pulmonary artery pressure. A polyethylene catheter was inserted into the right ventricle via the right external jugular vein. The other end of the catheter was connected to a transducer, and the pressure tracings were simultaneously recorded on a physiologic recorder (PowerLab System; AD Instruments). The data were analyzed using the Chart program supplied by the system. After measurement, rats were euthanized, and the lungs, hearts, kidneys, and livers were harvested for subsequent experiments. The right ventricle and the left ventricle with septum were isolated and individually weighted to calculate RV/(LV + S).
Hematoxylin and Eosin Staining
The left lung was fixed in 4% (w/v) paraform and processed into 5 μm paraffin sections used for morphometric analysis with hematoxylin and eosin staining and immunofluorescence costaining. Then, the wall thickness of pulmonary arterioles with a diameter between 20 and 80 μm was analyzed, according to the previously reported method.18,30, 31, 32 The external diameter and medial wall thickness were measured in 60 muscular arteries from six animal lungs (10 arteries/lung) for each group for analysis of WT% and WA% of the pulmonary arterioles, which were calculated as the following, respectively: WT% = 100 × (medial wall thickness)/(vessel semi-diameter), and WA% = 100 × (cross-sectional medial wall area)/(total cross-sectional vessel area).32
Immunofluorescence Costaining
Immunofluorescence costaining was used to detect the colocalization of angiotensin-(1-7) and CD31 or CD11b. CD31 is regarded as a PMVEC marker and CD11b as the marker of monocytes, macrophage, and many bone marrow-derived hematopoietic cells. The sections were deparaffinized and rehydrated by xylene, ethanol, and water. Antigen retrieval was performed in 10 mM citrate buffer (pH 6.0) at a constant pressure of 20 cm H2O. Subsequently, the sections were blocked with 5% serum in PBS for 20 min at room temperature, followed by incubation with the mixture of anti-angiotensin-(1-7) (Lifespan Bio Sciences, WA, USA; 1:200 dilution) and anti-CD31 (Bioworld Technology, MN, USA; 1:200 dilution) antibodies or anti-angiotensin-(1-7) (1:200 dilution) and anti-CD11b (Proteintech Group, Rosemont, IL, USA; 1:200 dilution) antibodies overnight at 4°C. Anti-angiotensin-(1-7) was detected using a secondary antibody (goat anti-rabbit IgG, Alexa Fluor 488; Abcam, MA, USA; at 1:500 dilution); anti-CD31 and anti-CD11b were detected by the secondary antibody of goat anti-mouse IgG (Alexa Fluor 594; Abcam, MA, USA; at 1:500 dilution). 4′,6-Diamidino-2-phenylindole (DAPI) was used as a nuclear counterstain. PBS, instead of the primary antibody, was used as the secondary antibody-only control. Images were acquired using confocal laser microscope (Leica TCS SP5) with the same exposure time during the same imaging session.
Cell Culture
Rat primary PMVECs and primary PASMCs were cultured by the tissue explant method.33 Rat primary pulmonary microendothelial cells were isolated, according to Sobczak et al.34’s metal method, and cultured in the specific endothelial cell medium (ScienCell Research Laboratories, USA). The PASMCs were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco; Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific). Both cells were cultured in 5% CO2 and 20% O2 at 37°C. At confluence, the cells were trypsinized and split in a 1:3 ratio and recultured in the corresponding medium. The 2nd to 3rd passage PASMCs and the 3rd to 5th passage PMVECs were used for the subsequent experiments. The identity of cultured PASMCs and PMVECs was verified by positive staining for alpha smooth muscle actin and CD31, respectively (>90% of cells stained positive). HEK293, A549, NIH 3T3, and rat alveolar macrophage (NR8383) cell lines were obtained from ATCC (Rockville, MD, USA) and grown in DMEM, supplemented with 10% FBS in 5% CO2 and 20% O2 at 37°C.
The neutrophils and monocytes were isolated from the rat’s peripheral blood samples by using Histopaque 1083 (density = 1.083 g/mL; Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer’s protocols. Then, monocytes were purified by negative selection with magnetic beads (Monocyte Isolation Kit II; Miltenyi Biotec, Bisley, UK). The purity of isolated neutrophils and monocytes was greater than 95%, as assessed by H&E staining and flow cytometry. The neutrophils and monocytes were cultured with DMEM, supplemented with 10% (v/v) FBS in 5% CO2 and 20% O2 at 37°C for the follow-up experiments.
Primary bone MSCs were isolated and cultured, as previously described.35 Briefly, rats were sacrificed, and bone marrow-derived MSCs (BMSCs) were collected from the femur and tibia by flushing the shaft with chilled complete medium, which consisted of DMEM supplemented with 10% FBS. Cells were washed, centrifuged, resuspended, and then cultured with complete medium in 5% CO2 and 20% O2 at 37°C. Nonadherent cells were discarded after 72 h, and the adherent cells were added with fresh DMEM, supplemented with 10% FBS and replaced every 2–3 days. When 80%–90% confluence was reached, BMSCs were trypsinized and passaged. The expressions of cell surface markers, including CD29, CD90, CD45, and CD44, was subsequently analyzed by flow cytometry.
Finally, all of these cells were transfected with HTSFcAng(1-7) or HTSFc via Lipofectamine 2000, respectively. The cell supernatants were collected to measure the content of Ang (1-7) by ELISA, 24 h after transfection. The condition medium preparation from HEK293 cells, NIH 3T3 cells, A549 cells, PMVECs, or PASMCs was seeded in 6-well plates at a density of 105 cells for each well and cultured in 2 mL medium in normoxia (20% O2). At 80% confluence, the cells were transfected with the vector of HTSFcAng(1-7) or HTSFc, respectively. The transfection was performed using Lipofectamine 2000 reagent (Invitrogen, USA). After transfection, the cells were incubated in normoxia (20% O2) or hypoxia (1%, 5%, 10% O2) for 24 h, and then the condition medium was collected to measure the content of angiotensin-(1-7) or immediately used in the PASMC proliferation assay or artery ring experiments.
PASMC Proliferation Assay
PASMC proliferation was measured by the MTT assay and cell counting method, as previously described.36 The PASMCs were seeded in 96-well plates at a density of 5,000 cells each well in DMEM, supplemented with 10% fetal bovine serum in normoxia (20% O2). At 60% confluence, the cells were starved in serum-free medium for 24 h to synchronization. Then, the cells were cultured in DMEM (containing 10% fetal bovine serum) and the freshly collected condition medium of PMVECs, according to the corresponding concentration of condition medium (0%, 25%, 50%, 75%) (v/v) in hypoxia (10% O2) for 24 h. Then, MTT (5 mg/mL, 10 μL/well) was added to the plates. The cells were incubated for another 4 h at 37°C. The supernatant was then carefully removed, and dimethyl sulfoxide (75 μL/well) was added to dissolve the formazan crystals. The absorbance of the solubilized product at 490 nm was measured with a microplate spectrophotometer (Power Wave XS; BioTek, VT, USA).
For cell counting, the cells were seeded in 24-well plates at a density of 5 × 104 cells/ell and then, were treated as the above procedure. At the end of treatment, they were washed with phosphate-buffered solution, harvested by mild trypsinization, and counted with a hematocytometer (QiuJin, Shanghai, China).
Intrapulmonary Artery Ring Isolation and Organ Bath Experiments
Normal rats were anesthetized with sodium pentobarbital (30 mg/kg i.p.). The lungs were rapidly removed and placed in ice-cold Krebs-Henseleit solution, which contained the following (in M): NaCl 118.4, KCl 4.7, CaCl2 2.5, MgCl2 1.2, KH2PO4 1.2, NaHCO3 25.0, and dextrose 11.1. The endothelial cell-denuded intrapulmonary artery (external diameter < 300 μm, 3rd division) rings were prepared. Ring segments (3 mm) were cut and suspended vertically between hooks in the water-jacketed organ chamber (6 mL) containing modified Krebs-Henseleit solution, which was maintained at pH 7.4, heated to 37°C, and bubbled with a mixture gas of 95% O2 and 5% CO2.
Isometric force was recorded with a force-displacement transducer and a PowerLab eight-channel data acquisition system (AD Instruments, Colorado Springs, CO, USA) and then was analyzed by MacLab/400 and Chart software (version 5.5 from ADInstruments). The rings were stretched under a predetermined optimal resting tension of 750 mg. After a 60-min equilibration period with washouts every 15–20 min, phenylephrine (10−6 M) was added to test the viability of rings. The contractile responses of rings to phenylephrine (<300 mg) were considered to be inactive and were discarded. The contractile responses of rings to phenylephrine (>300 mg) were considered to be active and were chosen for subsequent experiments. Next, the rings were treated with KCl (60 mM) again to establish the maximum contractile response. Then, the rings were rinsed with Krebs-Henseleit solution to return the tension to baseline.
To test the protective effects of the condition medium from PMVECs transfected with HTSFcAng(1-7) on hypoxia-induced vasoconstriction, phenylephrine (10−6 M) was added to establish a stable contractile tone again; then, the rings were exposed to hypoxia during the following experiment. Hypoxia was induced by the mixture of gas of 95% N2 and 5% CO2,. Tension changes were measured in the absence or presence of the condition medium from PMVECs transfected with HTSFcAng(1-7) or HTSFc (3.5 mL) or plus A-779 (10−6 M), which was added to the organ baths 20 min before the onset of phenylephrine.
Statistical Analysis
All values were presented as means ± SD. The statistical differences between groups were evaluated by one-way analysis of variance (ANOVA), followed by Dunnett’s test for multiple comparisons. Shapiro-Wilk test was used to test the normality of data. A p value < 0.05 was considered statistically significant.
Author Contributions
M.-L.L., S.-J.X., X.-Q.L., Y.L., and B.Z. conducted the experiments. M.-L.L. and M.-Q.D. analyzed the data. M.-Q.D. and Z.-C.L. designed the experiments. M.-Q.D. wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (nos. 81200036, 81571822, 81570054, 81770056, 31671186, 81571839, and 81800046); Science and Technology Research and Development Program of Shaanxi Province, China (nos. 2019SF-008 and 2019SF-037); and Scientific Research Initiation Funds for the Doctoral Program of Xi’an International University (XAIU2018070106).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.008.
Supplemental Information
References
- 1.Bussotti M., Marchese G. High Altitude Pulmonary Hypertension. Cardiovasc. Hematol. Disord. Drug Targets. 2018;18:187–198. doi: 10.2174/1871529X18666180518085245. [DOI] [PubMed] [Google Scholar]
- 2.Sakao S., Voelkel N.F., Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur. Respir. Rev. 2014;23:350–355. doi: 10.1183/09059180.00007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Passos-Silva D.G., Brandan E., Santos R.A. Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol. Sci. 2015;36:310–320. doi: 10.1016/j.tips.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Tan W.S.D., Liao W., Zhou S., Mei D., Wong W.F. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018;40:9–17. doi: 10.1016/j.coph.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Shenoy V., Ferreira A.J., Qi Y., Fraga-Silva R.A., Díez-Freire C., Dooies A., Jun J.Y., Sriramula S., Mariappan N., Pourang D. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2010;182:1065–1072. doi: 10.1164/rccm.200912-1840OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hampl V., Herget J., Bíbová J., Baňasová A., Husková Z., Vaňourková Z., Jíchová Š., Kujal P., Vernerová Z., Sadowski J., Červenka L. Intrapulmonary activation of the angiotensin-converting enzyme type 2/angiotensin 1-7/G-protein-coupled Mas receptor axis attenuates pulmonary hypertension in Ren-2 transgenic rats exposed to chronic hypoxia. Physiol. Res. 2015;64:25–38. doi: 10.33549/physiolres.932861. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y.X., Li J.F., Yang Y.H., Zhai Z.G., Gu S., Liu Y., Miao R., Zhong P.P., Wang Y., Huang X.X., Wang C. Renin-angiotensin system regulates pulmonary arterial smooth muscle cell migration in chronic thromboembolic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018;314:L276–L286. doi: 10.1152/ajplung.00515.2016. [DOI] [PubMed] [Google Scholar]
- 8.Hemnes A.R., Rathinasabapathy A., Austin E.A., Brittain E.L., Carrier E.J., Chen X., Fessel J.P., Fike C.D., Fong P., Fortune N. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018;51:1702638. doi: 10.1183/13993003.02638-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford C.N., Ely D.R., Raizada M.K. Targeting the vasoprotective axis of the renin-angiotensin system: a novel strategic approach to pulmonary hypertensive therapy. Curr. Hypertens. Rep. 2010;12:212–219. doi: 10.1007/s11906-010-0122-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breitling S., Krauszman A., Parihar R., Walther T., Friedberg M.K., Kuebler W.M. Dose-dependent, therapeutic potential of angiotensin-(1-7) for the treatment of pulmonary arterial hypertension. Pulm. Circ. 2015;5:649–657. doi: 10.1086/683696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai H., Jiang L., Xiao Z., Guang X. ACE2-angiotensin-(1-7)-Mas axis might be a promising therapeutic target for pulmonary arterial hypertension. Nat. Rev. Cardiol. 2015;12:374. doi: 10.1038/nrcardio.2015.6-c1. [DOI] [PubMed] [Google Scholar]
- 12.Li G., Liu Y., Zhu Y., Liu A., Xu Y., Li X., Li Z., Su J., Sun L. ACE2 activation confers endothelial protection and attenuates neointimal lesions in prevention of severe pulmonary arterial hypertension in rats. Lung. 2013;191:327–336. doi: 10.1007/s00408-013-9470-8. [DOI] [PubMed] [Google Scholar]
- 13.Wang J., Dong M.Q., Liu M.L., Xu D.Q., Luo Y., Zhang B., Liu L.L., Xu M., Zhao P.T., Gao Y.Q., Li Z.C. Tanshinone IIA modulates pulmonary vascular response to agonist and hypoxia primarily via inhibiting Ca2+ influx and release in normal and hypoxic pulmonary hypertension rats. Eur. J. Pharmacol. 2010;640:129–138. doi: 10.1016/j.ejphar.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 14.Morecroft I., White K., Caruso P., Nilsen M., Loughlin L., Alba R., Reynolds P.N., Danilov S.M., Baker A.H., Maclean M.R. Gene therapy by targeted adenovirus-mediated knockdown of pulmonary endothelial Tph1 attenuates hypoxia-induced pulmonary hypertension. Mol. Ther. 2012;20:1516–1528. doi: 10.1038/mt.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing Y., Zheng X., Fu Y., Qi J., Li M., Ma M., Wang S., Li S., Zhu D. Long Noncoding RNA-Maternally Expressed Gene 3 Contributes to Hypoxic Pulmonary Hypertension. Mol. Ther. 2019;27:2166–2181. doi: 10.1016/j.ymthe.2019.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claude C., Mougenot N., Bechaux J., Hadri L., Brockschnieder D., Clergue M., Atassi F., Lompré A.M., Hulot J.S. Inhalable delivery of AAV-based MRP4/ABCC4 silencing RNA prevents monocrotaline-induced pulmonary hypertension. Mol. Ther. Methods Clin. Dev. 2015;2:14065. doi: 10.1038/mtm.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacak C.A., Byrne B.J. AAV vectors for cardiac gene transfer: experimental tools and clinical opportunities. Mol. Ther. 2011;19:1582–1590. doi: 10.1038/mt.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazato Y., Ferreira A.J., Hong K.H., Sriramula S., Francis J., Yamazato M., Yuan L., Bradford C.N., Shenoy V., Oh S.P. Prevention of pulmonary hypertension by Angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54:365–371. doi: 10.1161/HYPERTENSIONAHA.108.125468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynolds A.M., Holmes M.D., Danilov S.M., Reynolds P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012;39:329–343. doi: 10.1183/09031936.00187310. [DOI] [PubMed] [Google Scholar]
- 20.Aguero J., Ishikawa K., Hadri L., Santos-Gallego C.G., Fish K.M., Kohlbrenner E., Hammoudi N., Kho C., Lee A., Ibáñez B. Intratracheal Gene Delivery of SERCA2a Ameliorates Chronic Post-Capillary Pulmonary Hypertension: A Large Animal Model. J. Am. Coll. Cardiol. 2016;67:2032–2046. doi: 10.1016/j.jacc.2016.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dachs G.U., Patterson A.V., Firth J.D., Ratcliffe P.J., Townsend K.M., Stratford I.J., Harris A.L. Targeting gene expression to hypoxic tumor cells. Nat. Med. 1997;3:515–520. doi: 10.1038/nm0597-515. [DOI] [PubMed] [Google Scholar]
- 22.Shibata T., Giaccia A.J., Brown J.M. Development of a hypoxia-responsive vector for tumor-specific gene therapy. Gene Ther. 2000;7:493–498. doi: 10.1038/sj.gt.3301124. [DOI] [PubMed] [Google Scholar]
- 23.Kim H.J., Oh J.S., An S.S., Pennant W.A., Gwak S.J., Kim A.N., Han P.K., Yoon D.H., Kim K.N., Ha Y. Hypoxia-specific GM-CSF-overexpressing neural stem cells improve graft survival and functional recovery in spinal cord injury. Gene Ther. 2012;19:513–521. doi: 10.1038/gt.2011.137. [DOI] [PubMed] [Google Scholar]
- 24.Luo Y., Zhang B., Dong H.Y., Liu Y., Li Z.C., Dong M.Q., Gao Y.Q. Prevention of hypoxic pulmonary hypertension by hypoxia-inducible expression of p27 in pulmonary artery smooth muscle cells. Gene Ther. 2014;21:751–758. doi: 10.1038/gt.2014.49. [DOI] [PubMed] [Google Scholar]
- 25.Xu D.Q., Luo Y., Liu Y., Wang J., Zhang B., Xu M., Wang Y.X., Dong H.Y., Dong M.Q., Zhao P.T. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir. Res. 2010;11:182. doi: 10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu D., Niu W., Luo Y., Zhang B., Liu M., Dong H., Liu Y., Li Z. Endogenous estrogen attenuates hypoxia-induced pulmonary hypertension by inhibiting pulmonary arterial vasoconstriction and pulmonary arterial smooth muscle cells proliferation. Int. J. Med. Sci. 2013;10:771–781. doi: 10.7150/ijms.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ou L.C., Sardella G.L., Leiter J.C., Brinck-Johnsen T., Smith R.P. Role of sex hormones in development of chronic mountain sickness in rats. J. Appl. Physiol. (1985) 1994;77:427–433. doi: 10.1152/jappl.1994.77.1.427. [DOI] [PubMed] [Google Scholar]
- 28.Galiè N., Palazzini M., Leci E., Manes A. Current therapeutic approaches to pulmonary arterial hypertension. Rev. Esp. Cardiol. 2010;63:708–724. doi: 10.1016/s1885-5857(10)70145-6. [DOI] [PubMed] [Google Scholar]
- 29.Kawahara Y., Tanonaka K., Daicho T., Nawa M., Oikawa R., Nasa Y., Takeo S. Preferable anesthetic conditions for echocardiographic determination of murine cardiac function. J. Pharmacol. Sci. 2005;99:95–104. doi: 10.1254/jphs.fp0050343. [DOI] [PubMed] [Google Scholar]
- 30.Beppu H., Ichinose F., Kawai N., Jones R.C., Yu P.B., Zapol W.M., Miyazono K., Li E., Bloch K.D. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;287:L1241–L1247. doi: 10.1152/ajplung.00239.2004. [DOI] [PubMed] [Google Scholar]
- 31.Zhang N., Dong M., Luo Y., Zhao F., Li Y. Danshensu prevents hypoxic pulmonary hypertension in rats by inhibiting the proliferation of pulmonary artery smooth muscle cells via TGF-β-smad3-associated pathway. Eur. J. Pharmacol. 2018;820:1–7. doi: 10.1016/j.ejphar.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Luo Y., Dong H.Y., Zhang B., Feng Z., Liu Y., Gao Y.Q., Dong M.Q., Li Z.C. miR-29a-3p attenuates hypoxic pulmonary hypertension by inhibiting pulmonary adventitial fibroblast activation. Hypertension. 2015;65:414–420. doi: 10.1161/HYPERTENSIONAHA.114.04600. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y.F., Liu M.L., Dong M.Q., Yang W.C., Zhang B., Luan L.L., Dong H.Y., Xu M., Wang Y.X., Liu L.L. Effects of sodium tanshinone II A sulphonate on hypoxic pulmonary hypertension in rats in vivo and on Kv2.1 expression in pulmonary artery smooth muscle cells in vitro. J. Ethnopharmacol. 2009;125:436–443. doi: 10.1016/j.jep.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Sobczak M., Dargatz J., Chrzanowska-Wodnicka M. Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 2010:2316. doi: 10.3791/2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Liu Y., Yie S., Lan J., Pi J., Zhang Z., Huang H., Cai Z., Zhang M., Cai K. Characteristics of mesenchymal stem cells isolated from bone marrow of giant panda. Stem Cells Dev. 2013;22:2394–2401. doi: 10.1089/scd.2013.0102. [DOI] [PubMed] [Google Scholar]
- 36.Luo Y., Xu D.Q., Dong H.Y., Zhang B., Liu Y., Niu W., Dong M.Q., Li Z.C. Tanshinone IIA inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via Akt/Skp2/p27-associated pathway. PLoS ONE. 2013;8:e56774. doi: 10.1371/journal.pone.0056774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.