

Abstract
This article re-evaluates the taxonomy of Hyphoderma macaronesicum based on various strategies, including the cohesion species recognition method through haplotype networks, multilocus genetic analyses using the genealogical concordance phylogenetic concept, as well as species tree reconstruction. The following loci were examined: the internal transcribed spacers of nuclear ribosomal DNA (ITS nrDNA), the intergenic spacers of nuclear ribosomal DNA (IGS nrDNA), two fragments of the protein-coding RNA polymerase II subunit 2 (RPB2), and two fragments of the translation elongation factor 1-α (EF1-α). Our results indicate that the name H. macaronesicum includes at least two separate species, one of which is newly described as Hyphoderma paramacaronesicum. The two species are readily distinguished based on the various loci analysed, namely ITS, IGS, RPB2 and EF1-α.
Keywords: corticioid fungi, Macaronesia, species delimitation, taxonomy
INTRODUCTION
Over the past two decades, the discovery of genetic diversity within morphologically defined species has increased as more and more traditional species become molecularly characterised. Bickford et al. (2006) defined cryptic species as “two or more species erroneously classified (and hidden) under one species name”. They also defined sibling species as “two species that are the closest relative to each other and have not been distinguished from one another taxonomically”. Korshunova et al. (2017) describe different degrees of cryptibility, from a completely indistinguishable pair of taxa (sibling species) to species complexes that have minor differences (e.g. semi-cryptic, pseudocryptic species). The same paper points out, however, that the terms sibling, semi-cryptic, and pseudocryptic can be very confusing, and it recommends the term cryptic species for species where present knowledge cannot define morphological features unambiguously. In a recent paper, Struck et al. (2018) mention that current definitions of cryptic species are inconsistent and propose an approach focused on quantifying the phenotypic disparity of taxa relative to the divergence and exchange of their genes. Thus, these authors consider that cryptic species should derive from diverged genotype clusters, and that when the divergence is recent, cryptic species are sister taxa (the term sibling is not used by these authors). These concepts underpin our study.
According to Bickford et al. (2006), fungi are key target organisms for cryptic species investigations, and as indicated by Taylor et al. (2006), an ideal group to compare species delimitation based on morphological characters (MSR, morphological species recognition), on reproductive isolation (BSR, biological species recognition) and on genetic isolation (PSR, phylogenetic species recognition). Taylor et al. (2006) discussed several fungi thought to be a single species by MSR or BSR, but which molecular analysis has shown are two or more geographically distinct species. For example, James et al. (2001) analysed the rDNA loci in Schizophyllum commune, a model organism of Basidiomycota found throughout the world on woody substrates. The phylogenetic analyses of the internal transcribed spacers (ITS nrDNA; ITS) and the intergenic spacers (IGS nrDNA; IGS) revealed a strong geographic pattern and supported three evolutionarily distinct lineages within the global population. Also, examples are found in corticoid fungi (basidiomycota having effused, smooth basidiocarps), e.g., Serpula hymantioides is considered a species complex of five phylogenetic species (PSR) with divergent substrate affinities (Carlsen et al. 2011). Using Serpula species as a model group, Balasundaram et al. (2015) concluded that at least five markers from independent loci are needed to separate cryptic species in fungi.
While multilocus sequencing will remain the gold standard for an unambiguous definition of new species (Yar et al. 2016), numerous cryptic species have recently been described based on the ITS region. After a phylogenetically wide-ranging test, Schoch et al. (2012) showed that the ITS region discriminates species effectively across more than 70 % of fungi tested, so this region was selected as the first barcode for fungi. Mallo & Posada (2016) clearly state the four different strategies for species assignment using the barcoding approach: a) tree-based strategies, using any classic phylogenetic method to estimate the gene tree, assuming that gene trees and species trees are topologically equivalent; b) sequence-similarity method, assuming that intraspecific similarity is larger than the interspecific, the barcode gap; c) statistical methods, which involve evaluating the statistical evidence, such as posterior probabilities, towards associating a new sequence with one group or another; and, d) diagnostic methods, that depend on finding in the reference sequence a combination of nucleotides to assign potential queries to a given species. Since statistical and diagnostic methods are computationally expensive (Nielsen & Matz 2006, Bertolazzi et al. 2009), the main repositories of fungal sequence data (GenBank and UNITE) facilitate automatic species discrimination with specialised bioinformatics pipelines.
In Telleria et al. (2012) we described a new Hyphoderma species, Hyphoderma macaronesicum from the Canary Islands and Azores Archipelago based on comparison of morphological characters and ITS region sequences, the universal fungal DNA barcode marker (Schoch et al. 2012). Even though the 35 specimens of H. macaronesicum shared similar patterns of morphological variability, molecular analyses yielded two strongly supported clades (clade A with four specimens and clade B with 31 specimens separated into subclade B1 and subclade B2), suggesting that the morphologically defined H. macaronesicum could contain some cryptic species. When we analysed these sequences through the UNITE database/PlutoF (Kõljalg et al. 2013), we obtained two SH (Species Hypothesis). The SH191350.07FU included the sequence from the holotype of H. macaronesicum (reference sequence) and the other sequences from clade B (15 sequences), without discriminating between sequences from subclade B1 and B2; and the SH191352.07FU included the four sequences from clade A (Supplementary Fig. 1).
This work presents a re-evaluation of the taxonomy of H. macaronesicum, based on the clades from our previous study (Telleria et al. 2012), as well as the SHs obtained through the UNITE database. Thus, this paper aims to evaluate a one-species hypothesis (H. macaronesicum) against two other hypotheses, a two-species hypothesis (clades A and B) and a three-species hypothesis (clades A, B1, and B2).
Different strategies for species assignments were applied. A new evaluation of macro- and micro-morphological characters was conducted. A Latin binomial is proposed for the species named in UNITE database as SH191352.07FU, and a description is provided.
MATERIALS AND METHODS
Taxon sampling and morphological studies
Six new specimens initially identified as H. macaronesicum, were analysed morphologically together with 14 specimens from Telleria et al. (2012). Specific information about geographic origin, substrate, and sequences, with the GenBank accession numbers of the specimens, is shown in Table 1. The specimens are deposited in the mycological collections of MA and TFC herbaria (Thiers 2016).
Table 1.
Hyphoderma paramacaronesicum and H. macaronesicum specimens analysed. Clade names based on ITS sequences according to Telleria et al. (2012). The initials for the collections correspond to M. Dueñas (MD) and M.T. Telleria (Tell.). Geographical names are abbreviated as follows: CI, Canary Islands, and AA, Azores Archipelago. (*) New specimens included in this study. In bold, new sequences obtained in this study.
| Species / specimens | Geographical origin | Substrate |
GenBank accession no |
||||
|---|---|---|---|---|---|---|---|
| ITS | IGS | RPB21 | RPB22 | EF1-α | |||
| Clade A | |||||||
| H. paramacaronesicum | |||||||
| TFCMic. 15161 | Fuerteventura (CI) | Nicotiana glauca | HE577025 | KF150075 | - | LT627631 | LT627612 |
| TFCMic. 15831 | Gran Canaria (CI) | Pistacia atlantica | HE577026 | KF150076 | - | - | LT627613 |
| TFCMic. 15981 | Tenerife (CI) | Cistus monspeliensis | HE577027 | KF150077 | - | LT627632 | LT627614 |
| MA-Fungi 87737, 12353MD* | Gran Canaria (CI) | Ocotea foetens | KC984405-KC984407 (Clones A, D, E) | KF150073 | - | LT627633 | LT627615 |
| KC984402-KC984404 (Clones B, C, F) | |||||||
| MA-Fungi 87736, 12262MD* | Fuerteventura (CI) | Launea arborescens | KC984397-KC984401 (Clones A–C, D, F) | KF150074 | - | LT627634 | LT627616 |
| MA-Fungi 87738, 16099Tell., holotype | Faial (AA) | Banksia integrifolia | HE577028 | KF150078 | - | - | - |
| Clade B | |||||||
| H. macaronesicum | |||||||
| Subclade B1 | |||||||
| TFCMic. 8954 | La Palma (CI) | Echium brevirame | HE577003 | KF150040 | - | - | LT627618 |
| TFCMic. 14968 | El Hierro (CI) | Euphorbia lamarckii | HE577006 | KF150045 | KF181105 | LT627637 | LT627619 |
| TFCMic. 14993 | El Hierro (CI) | Euphorbia lamarckii | HE577008 | KF150047 | KF181106 | LT627636 | LT627617 |
| TFCMic. 15019 | El Hierro (CI) | Euphorbia lamarckii | HE577009 | KF150048 | - | LT627638 | LT627620 |
| TFCMic. 15032 | El Hierro (CI) | Schizogyne sericea | HE577010 | KF150049 | KF181107 | LT627639 | LT627621 |
| TFCMic. 15802 | Gran Canaria (CI) | Kleinia neriifolia | HE577015 | KF150061 | KF181108 | LT627640 | LT627622 |
| TFCMic. 15810 | Gran Canaria (CI) | Kleinia neriifolia | HE577016 | KF150062 | KF181109 | LT627641 | LT627623 |
| TFCMic. 15939, holotype | Tenerife (CI) | Plocama pendula | HE577024 | KF150072 | KF181102 | - | LT627624 |
| Subclade B2 | |||||||
| TFCMic. 15115 | Fuerteventura (CI) | Launaea arborescens | HE577011 | KF150050 | KF181118 | - | LT627630 |
| TFCMic. 15917 | La Gomera (CI) | Rumex lunaria | HE577023 | KF150071 | KF181117 | LT627644 | LT627629 |
| MA-Fungi 90387, 12236MD* | Lanzarote (CI) | Euphorbia balsamifera | KC984326 | KF150023 | - | - | LT627625 |
| MA-Fungi 90388, 12241MD* | Lanzarote (CI) | Euphorbia balsamifera | KC984327 | KF150025 | KF181122 | LT627635 | LT627626 |
| MA-Fungi 90389, 12244MD* | Lanzarote (CI) | Euphorbia balsamifera | KC984328 | KF150026 | KF181121 | LT627642 | LT627627 |
| MA-Fungi 90390, 12301MD* | Gran Canaria (CI) | Salvia canariensis | KC984351 | KF150036 | KF181120 | LT627643 | LT627628 |
1 Ribosomal polymerase two, subunit two (RPB2) sequences, obtained through PCR and sequencing with primer pairs fRPB2-7cF/fRPB2-11aR (Liu et al. 1999).
2 RPB2 sequences, obtained through nested PCR with primer pair RPB2- f5F/RPB2–7.1R (Matheny 2005, Binder et al. 2010) to the first amplification, and primer pair RPB2-6R/RPB2-7R.2 (Matheny 2005, Matheny et al. 2007) to the second amplification and sequencing.
Dried specimens were used for light microscopy. Measurements were made from microscopic sections mounted in 3 % KOH solution and examined at up to 1250× with an Olympus BX51 microscope. The length and width of 30 spores, 10 basidia and 10 cystidia were measured from each sample.
DNA extraction, amplification and sequencing
From the 14 specimens of H. macaronesicum representing the three subclades (A, B1 and B2) from Telleria et al. (2012), DNA isolation was not required, since genomic DNA is stored at the Real Jardín Botánico, RJB-CSIC (Madrid, Spain). From the six new collections (Table 1 marked with asterisk), genomic DNA was extracted using a DNeasy™ Plant Mini Kit (Qiagen, Valencia, California, USA), following the instructions of the manufacturers; except that lysis buffer incubation was done overnight at 60 °C.
Amplifications were done using illustra PuReTaq Ready-To-Go PCR beads (GE Healthcare, Buckinghamshire, UK) as described in Winka et al. (1998). Negative controls lacking fungal DNA were run for each experiment to check for contamination of reagents. Results of amplifications were assayed from 5 μL aliquots by gel electrophoresis in 2 % Pronadisa D-1 Agarose (Lab. Conda, Spain).
First, the primer pair ITS1F/ITS4 (White et al. 1990, Gardes & Bruns 1993) was used to amplify the ITS of the six new specimens, as described in Martín & Winka (2000). The second locus analysed was IGS, as in James et al. (2001). The primer pair CNL12/5SA (Anderson & Stasovski 1992) was used to amplify a fragment of the IGS of the 21 isolates included in this study; the amplification cycles were: an initial denaturation at 94 °C for 5 min, 30 cycles of 94 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s, and the final extension of 72 °C for 10 min. Haplotype network analyses were done for both loci.
Second, in addition to ITS and IGS, two more genes were included (RPB2 and EF1-α). To amplify and sequence two regions of the protein-coding RNA polymerase II subunit two (RPB2), two primer pairs were used, fRPB2-7cF/fRPB2-11aR (Liu et al. 1999) and RPB2- f5F/RPB2–7.1R (Matheny 2005, Binder et al. 2010), using the amplification cycles described in Liu et al. (1999) and Matheny (2005), respectively. When necessary, nested-PCR was done following Wilson et al. (2012); thus, one μL of the first PCR (RPB2- f5F/RPB2–7.1R) was used as DNA template for the nested primer pair RPB2-6F/ RPB2-7R.2 (Matheny 2005, Matheny et al. 2007), with the amplification cycles described in Matheny (2005).
To amplify and sequence translation elongation factor 1-alpha (EF1-α), two primer pairs were used EF1–1018F/EF1-1620R and EF-1002F/EF1-1688R (Stielow et al. 2015). The amplification cycles were: an initial denaturation at 94 °C for 5 min, 40 cycles of 94 °C for 1 min s, 48 °C for 1 min s and 72 °C for 2 min, and the final extension of 72 °C for 10 min.
Prior to sequencing, the PCR products were cleaned using a QIAquick Gel PCR Purification kit (Qiagen, Valencia, California) according to the manufacturer’s instructions. Both strands were sequenced separately using primers mentioned above at Macrogen (South Korea). Sequences were edited and assembled using Sequencher™ v. 4.2 (Genes Codes Corporations, Ann Arbor, Michigan, USA).
When electropherograms were of bad quality (no sharp peaks and with background), purified PCR products were cloned using pGEM-T Easy Vector System II cloning kit (Promega, Madison, Wisconsin, USA). From each cloning reaction, up to six clones were selected for sequencing. To confirm that the inserted product was correct, 2 μL of the purified plasmid DNA was digested with EcoRI prior to sequencing following the instructions of the manufacturer. Both strands were sequenced separately using vector specific primers T7 and SP6 at Secugen S.L. (Madrid, Spain) or Macrogen (Seoul, Korea).
All sequences derived in this study were deposited in GenBank and accession numbers are given in Table 1. Sequence data of each locus were aligned separately using Se-Al v. 2.0a11 Carbon (Rambaut 2002) for multiple sequences.
Analyses of ITS and IGS nrDNA, and haplotype network
For the first analyses, ITS and IGS Minimum length Fitch trees were constructed separately using heuristic searches with tree-bisection-reconnection (TBR) branch swapping, collapsing branches if maximum length was zero and with the MulTrees option on in PAUP v. 4.0b10 (Swofford 2003). Gaps were treated as a 5th character state and as a missing character using two different datasets. The robustness of trees was calculated by nonparametric bootstrap (MPbs) support (Felsenstein 1985) for each clade, based on 10 000 replicates using the fast-step option. The consistency index CI (Kluge & Farris 1969), retention index RI (Farris 1989), and rescaled consistency index RC (Farris 1989), were obtained. Phylogenetic trees were viewed with FigTree v. 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/) and edited with Adobe Illustrator CS3 v. 11.0.2 (Adobe Systems). To test for potential conflict among data sets, 75 % bootstrap consensus trees were examined for conflict (Lutzoni et al. 2004); since no conflicts were found, the combined data set was used for a maximum parsimony analysis, as well as to calculate the genetic distances among specimens and in the network analyses.
The second strategy for species assignment using ITS and IGS was to estimate the genetic distances among specimens under the Kimura 2-parameter (K2P) model in PAUP, which is widely used in DNA barcoding analyses to distinguish species (e.g. Neigel et al. 2007). The ITS and IGS intraclade distances were calculated as the mean value of the pairwise distances between the samples of each clade; the interclade distances were calculated as the pairwise distances between the samples of the two clades; if there is no overlap of the two measures, a barcoding gap exists. The genetic distance is sufficient to assign species names to specimens. With genetic distances UPGMA and NJ dendrograms were built from the combined matrix of genetic distances.
To visualise the relationships of the samples, a neighbour-net analysis (Bryant & Moulton 2002) was run using Hamming distances (Hamming 1950), combining ITS and IGS sequences of Hyphoderma specimens from clades A and B (subclade B1 and subclade B2). Also, to display the mutational differences among haplotypes, a second method based on the statistical parsimony method implemented in TCS v. 1.21 (Clement et al. 2000) was carried out using a 90 % or 95 % connection limit; gaps were excluded from the alignments or coded as a 5th character state. In TCS the root haplotype is considered the oldest haplotype in a given network (Crandall & Templeton 1993, Posada & Crandall 2001, Templeton 2001).
Analyses of ITS, IGS, RPB2 and EF1-α, and species tree reconstruction
In order to apply the phylogenetic species concept based on the concordance of gene genealogies (Taylor et al. 2000) – in our case four loci –, three classic phylogenetic methods were used for the tree-based strategy: 1) Minimum length Fitch trees were constructed using the same parameters mentioned above; 2) Maximum likelihood (ML) analyses were done in PAUP, using the GTR+I+G model selected in this program for assessing branch supports, 1000 non-parametric bootstrap replicates (MLbs) were performed with the fast-step option. 3) The Bayesian analysis (Larget & Simon 1999, Huelsenbeck & Ronquist 2001) was done using MrBayes v. 3.2 (Ronquist et al. 2012) with the GTR+I+G model; both the 50 % majority-rule consensus tree and the posterior probability (PP) of the nodes were calculated from the remaining trees with MrBayes. Sequences of Hyphoderma prosopidis (HE577029) were used as outgroup for ITS analyses; Antrodia vaillantii (AM286436) for IGS; Hyphoderma litschaueri (KP134965) for RPB2; and Trametes hirsuta (JN164891) and T. maxima (JN164885) for EF1-α. (Neither IGS nor EF1-α Hyphoderma sequences are located in GenBank). To test for potential conflict among data sets, branches with ≥ 75 % MPbs (Lutzoni et al. 2004), ≥ 90 % MLbs (Wilson et al. 2012) and ≥ 0.95 PP (Wilson et al. 2012) were considered to be strongly supported. Phylogenetic trees were viewed with FigTree v. 1.3.1 and edited with Adobe Illustrator CS5 v. 15.0.2.
Finally, locus alignments were included in a full Bayesian framework for species tree estimation using *BEAST2 v. 2.4.3 (Heled & Drummond 2010, Bouckaert et al. 2014), under a multispecies coalescent model (Dowton et al. 2014); only specimens with the four locus sequences were included. This method co-estimates gene and species trees from sequence data while considering evolutionary processes that could generate species tree/gene tree discordance, as incomplete lineage sorting (Mallo & Posada 2016). The substitution model for each marker was selected according to the model selection obtained from jModelTest2 and BIC indexes. Constant population function (population mean = 1) was used to model the species tree population size. Coalescent constant population prior was used to build the species tree. To visualise the species tree and concordance between the four loci, the Densitree v. 2.01 package (Bouckaert 2010), included in BEAST2 v. 2.4.3, was used.
Three different species delimitation hypotheses were tested. In the first model (1-Species-Model), traditional morphological classification was addressed, including all specimens in a single group, separated from the outgroup. For the second hypothesis (2-Species-Model), specimens were grouped according with the two main clades (A and B) obtained in the barcoding analyses. Finally, for the third hypothesis (3-Species-Model), specimens were grouped according to the clades A, B1 and B2 obtained in the barcoding analyses. Each one of these models was used as input in BEAST v. 2.4.3 to obtain the species tree and were compared using the Bayes factors approach (Grummer et al. 2014). Bayes factors (BF) can be used as a model selection tool, to choose the most probable scenario given your data. To compute the Bayes factor for each model comparison, the marginal likelihood of each hypothesis was calculated through stepping-stone analyses using Path Sampler Analyser (BEAST model-selection 208 package v. 1.0.2; default parameters: alpha = 0,3, steps = 8, chain length = 100 000 and burning = 50 %). The 2-Species-Model was contrasted with the other two models (2-Species-Model vs. 1-Species-Model and 2-Species-Model vs. 3-Species-Model). The BF was calculated by subtracting the marginal L estimates; when the difference is positive, BF is in favour of H1. Following Kass & Raftery (1995), the BF scores are decisive for selecting the most probable scenario; thus, BF from 6 to 10 is strong evidence, and BF > 10 is decisive evidence.
Statistical tests of morphological characters
One-way ANOVA tests were performed to detect significant differences in spores, basidia and cystidia morphology between clades obtained in the genomic analyses. Exploratory plots (i.e. residuals vs fitted values, normal Q-Q plots and residuals vs leverage) were used to assess test assumptions.
RESULTS
A total of 79 sequences were generated for this study and are available under GenBank accession numbers indicated in Table 1: 15 ITS, 20 IGS, 11 RPB2 (fRPB2-7cF/fRPB2-11aR), 14 RPB2 (RPB2-6R and RPB2-7R.2), and 19 EF1-α (primer pair EF1–1018F/EF1-1620R). In two cases, cloning was performed to obtain the ITS sequences; from collection 12262MD (MA-Fungi 87736) all sequences were identical, but from 12353MD (MA-Fungi 87737) two haplotypes were distinguished (Table 1). As indicated in Table 1, using the primer pair fRPB2-7cF/fRPB2-11aR, no RPB2 sequences were obtained from any of the specimens included in clade A, nor from two specimens of subclade B1, and one specimen from subclade B2. For EF1-α using primer pair EF-1002F/EF1-1688R no or weak amplimers were visualised; and no sequences were obtained.
Analyses of ITS and IGS nrDNA, and haplotype network
The first analyses were performed using ITS and IGS sequences from the 20 specimens included in this study.
The topology of the consensus tree resulting from the analysis of the ITS region is nearly identical to the consensus tree topology resulting from the IGS (trees not shown); except the position of specimen TFCMic. 15032 from El Hierro (on Schizogyne sericea), which in the ITS analysis groups in clade B2, but in the IGS analysis was nested in clade B1. With only this phylogenetic conflict between the two loci, MP and UPGMA/NJ analyses on the combined gene alignment were done. The combined dataset had 1 089 characters, 54 were parsimony informative. The MP analysis yielded 100 most parsimonious trees, all with similar topology, consistency index (CI) = 0.8676, and retention index (RI) = 0.9708. Figure 1 shows one of the most parsimonious trees; the parsimony consensus tree (not shown) had the same topology, as well as the UPGMA and NJ dendrograms from the combined matrix of genetic distances (trees not shown). In all analyses, as shown in Fig. 1, samples 16099Tell., TFCMic. 15831 clones BCF, 12353MD, TFCMic. 15161, TFCMic. 15981, 12353MD clones ADE, and 12262MD clones ABCDF form a monophyletic clade with strong bootstrap support (100 %), as in Telleria et al. (2012). The combined alignment and one of the MPtrees derived from MP analyses were deposited in TreeBASE (21649; http://purl.org/phylo/treebase/phylows/study/TB2:S1649).
Fig. 1.
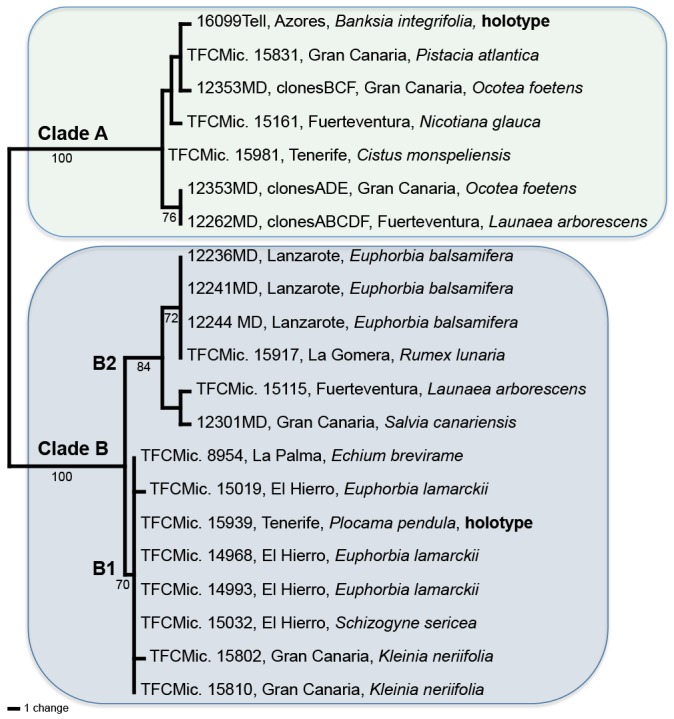
One of the 100 most parsimonious trees inferred from a heuristic search based on concatenated dataset of ITS and IGS nrDNA sequences. The two clades (A and B) and the two subclades (B1 and B2) described in Telleria et al. (2012) are recovered. Bootstrap values (%) are indicated below the branches.
The K2P genetic distance among clade A, and the subclades B1 and B2, in the combined ITS-IGS database revealed values ranging from 0.02809 to 0.03766; however, the maximum values among specimens of clade A, subclade B1 and subclade B2 were 0.000464, 0.00098 and 0.00483 respectively, and between subclade B1 and subclade B2 was 0.00886.
The median-joining network obtained using Hamming distances (Fig. 2) display the mutational differences for all haplotypes. The six haplotypes belonging to clade A (in Fig. 1), are clearly separated from the rest of the haplotypes (clade B in Fig. 1).
Fig. 2.
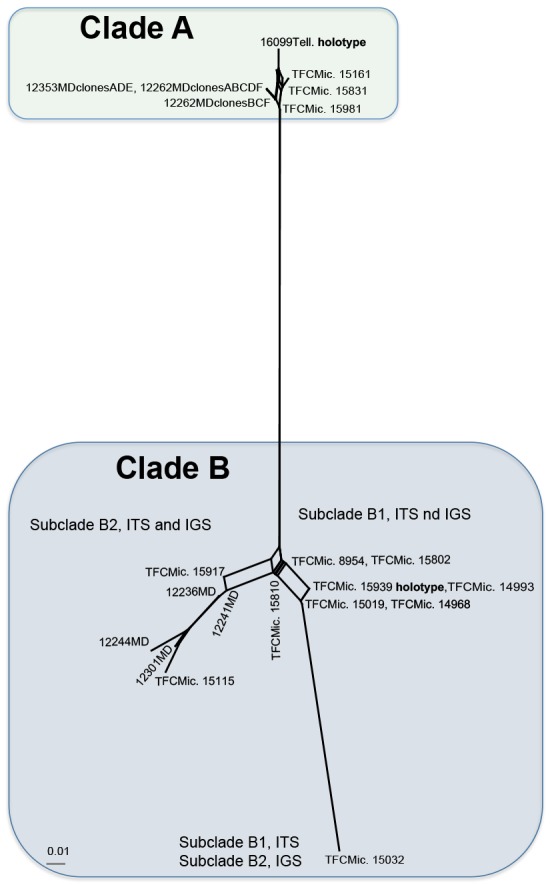
Neighbour-net network based on concatenated dataset of ITS and IGS nrDNA sequences using Hamming distances. For clarity, edges are labelled with the specimen numbers; the two clades (A and B) and the two subclades (B1 and B2) obtained after heuristic search in the parsimony analysis (Fig. 1) are indicated.
Parsimony network analyses (Fig. 3), both using 90 % or 95 % connection limit, and considering gaps as a 5th character state or excluding them, revealed two networks (named A and B in Fig. 1), and 14 haplotypes. The six haplotypes belonging to clade A separated by a maximum of four mutational steps; the highest root probability (Fig. 3 rectangle) was assigned to the haplotype consisting of one isolate from Gran Canaria growing on Pistacia atlantica. The haplotypes of network B (Fig. 3) appeared in two main clusters, corresponding with subclade B1 and B2, separated from each other by at least 15 mutational steps; the highest root probability was assigned to the haplotype consisting of five isolates from Canary Islands (Gran Canaria, El Hierro, La Palma and Tenerife, the type of H. macaronesicum) growing on different substrates. Between these two clusters, appears the haplotype of specimen TFCMic. 15032 at seven mutational steps from subclade B1, and at the same number of mutational steps from clade B2.
Fig. 3.
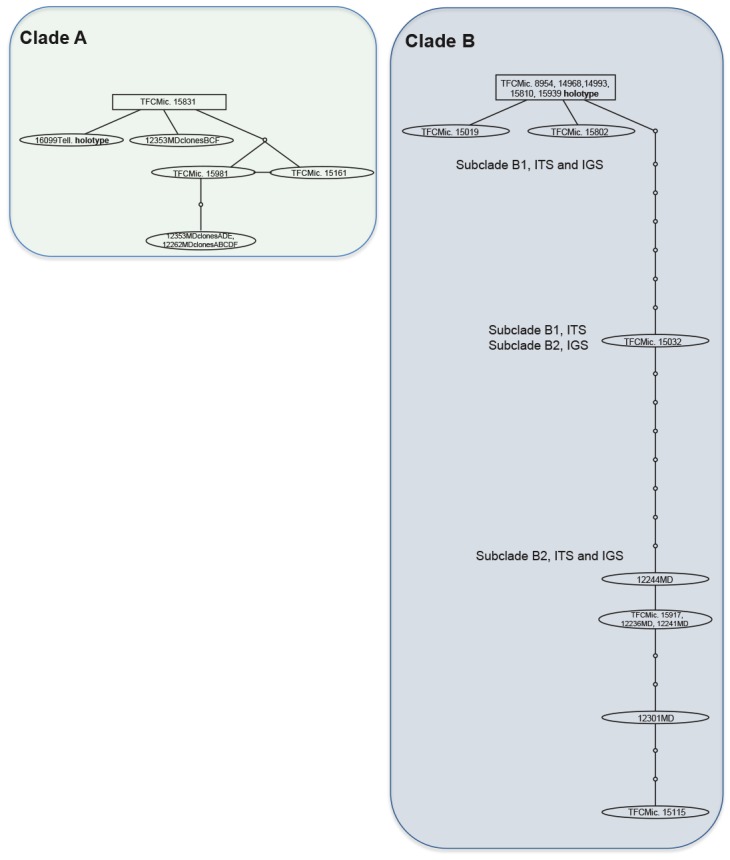
Parsimony network analysis based on concatenated dataset of ITS and IGS nrDNA sequences. Two separate nets were obtained. Specimens from clade A identified as Hyphoderma macaronesicum in Telleria et al. (2012), but here proposed as Hyphoderma paramacaronesicum. Each connecting line represents one substitution and each small circle represents a missing intermediate character. The square connected identify the haplotype considered as ancestral to each net by the analysis.
Analyses of ITS, IGS, RPB2 and EF1-α, and species tree reconstruction
In each locus tree, the clade A is strongly supported as a group apart from specimens of clade B (Supplementary Fig. 2A–D). The protein-code trees do not support the separation of clade B specimens into two subclades B1 and B2.
All alignments and trees deriving from Bayesian analyses were deposited in TreeBASE (21649; http://purl.org/phylo/treebase/phylows/study/TB2:S21649).
Marginal likelihoods obtained for each hypothesis and Bayes factors for model comparisons are shown in Table 2. The species tree (Fig. 4), estimated from the four loci dataset in *BEAST, clearly supports (PP = 1.00) a hypothesis of two evolutionary units (2-Species-Model); also, the BF indicates decisive support for the 2-Species-Model against the 1-Species-Model, and strong support for the 2-Species-Model against the 3-Species-Model. The two evolutionary units are herein considered different species: one includes the six specimens under clade A in barcoding analyses (SH191352.07FU in UNITE database), and its sister clade is composed of all specimens of clade B, without separating two subclades (SH191350.07FU).
Table 2.
Marginal likelihood of each species tree hypotheses, and Bayes factors for model comparison.
| -log (marginal L stimate) | Bayes Factor | ||
|---|---|---|---|
| 1-Species-Model | -5381,00124400668 | 2-Species-Model vs 1-Species-Model | 14,5329173133159 |
| 2-Species-Model | -5366,46832669336 | ||
| 3-Species-Model | -5373,93356887555 | 2-Species-Model vs 3-Species-Model | 7,46524218218565 |
Fig. 4.
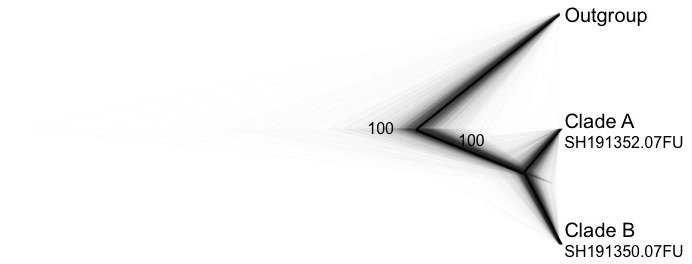
Species tree obtained from *BEAST2 v. 2.4.3 applying multispecies coalescent model for the four loci used in the analysis (ITS, IGS, RPB2 and EF1-α). Species hypothesis obtained from ITS tree is tested and posterior probabilities are show as support of each node. Grey background trees represent bootstrap gene trees topologies obtained from *BEAST2 v. 2.4.3.
Statistical analyses of morphological characters
Morphological studies show no differences between clades A and B in spores and basidia morphology (Table 3, Supplementary Fig. 4). Specimens of clade A share very similar morphological characters as described in Telleria et al. (2012) to H. macaronesicum holotype (Clade B). The only significant differences were found for cystidia width; the cystidia of clade A are thinner than those of clade B (F= 8.16, P-value < 0.05; Table 3, Supplementary Fig. 4).
Table 3.
One-way ANOVA test results showing F statistic and P-values between clade A (Hyphoderma paramacaronesicum) and clade B (H. macaronesicum).
| Characters | F | P-value |
|---|---|---|
| Spores length (L) | F(1.10) = 1.46 | 0.26 |
| Spores width (W) | F(1.10) = 0.18 | 0.68 |
| Spores length/width (Q) | F(1.10) = 1.14 | 0.31 |
| Basidia length | F(1.8) = 2.17 | 0.18 |
| Basidia width | F(1.8) = 0.37 | 0.56 |
| Cystidia length | F(1.10) = 0.17 | 0.69 |
| Cystidia width | F(1.10) = 8.16 | < 0.05 |
TAXONOMY
Here, we formally describe the clade A specimens (SH191352-07FU) as a new species, Hyphoderma paramacaronesicum, following the Melbourne Code (McNeill et al. 2012) and changes adopted by the 19th International Botanical Congress in Shenzhen 2017 (Hawksworth et al. 2017).
Hyphoderma paramacaronesicum Telleria, M. Dueñas, J. Fernández-López & M.P. Martín, sp. nov. MycoBank MB811865. Fig. 5.
Fig. 5.

Hyphoderma paramacaronesicum (16099Tell., MA-Fungi 87738, holotype). A. Section through basidiome. B. Basal hyphae. C. Hymenial layer with basidia and cystidia. D. Spores. Scale bars: A = 50 μm, B–D = 10 μm.
Etymology: Named for its morphological similarity to Hyphoderma macaronesicum.
Holotype: Portugal, Azores Archipelago, Faial, Horta, Ponta do Varadouro, 38°34′10″N 28°46′24″W, 42 msl, on Banksia integrifolia, 23 Feb. 2005, 16099Tell. (MA-Fungi 87738; ITS and IGS sequences GenBank HE577028 and KF150078, respectively).
Description: Basidioma resupinate, adnate, orbicular to confluent, yellowish white to pale orange yellow; hymenophore smooth and margin not clearly differentiated. Hyphal system monomitic with clamps at all septa; cylindrical leptocystidia with several constrictions often moniliform, 70–124 × 8−13 μm; basidia claviform, with oil drops in the protoplasm, basal clamp always present and four sterigmata, 40−48(−56) × 6−9 μm; spores ellipsoid, 12−15(−17) × 5.5−7(−8.5) μm, thin-walled.
Additional material examined: Spain, Canary Islands, Fuerteventura, Pájara, Jandía, barranco de Vinamar, 28°04′52″N 14°20′41″W, 220 msl, on Nicotiana glauca, 8 Feb. 2005, TFCMic. 15161 (paratype); Betancuria, Aula de la Naturaleza, 28°24′16.05″N 14°03′26.12″W, 357 msl, on Launaea arborescens, 4 Dec. 2007, 12262MD, MA-Fungi 87736 (paratype); Canary Islands, Gran Canaria, Las Palmas de Gran Canaria, Barranco de Guiniguada, next to Jardín Botánico ‘‘Viera y Clavijo’’, 28°03′56″N 15°27′49″W, 262 msl, on Pistacia atlantica, 13 Feb. 2006, TFCMic. 15831 (paratype); Barranco de Moya, 28°05′15.33″N 15°35′34.34″W, 560 msl, on Ocotea foetens, 8 Dec. 2007, 12353MD, MA-Fungi 87737 (paratype); Canary Islands, Tenerife, Ladera de Güímar, 28°17′33″N 16°24′31″W, 541 msl, on Cistus monspeliensis, 25 Oct. 2002, TFCMic.15981 (paratype).
Remarks: This new species is closely related to Hyphoderma macaronesicum, but differs in its unique ITS nrDNA, IGS nrDNA, RPB2 and EF1-α sequences (Supplementary Fig. 3). Their morphological characters are, more or less, similar to H. macaronesicum as described in Telleria et al. (2012).
DISCUSSION
Our study indicates that the name H. macaronesicum covers at least two separate species, based on preliminary automatic discrimination under the SH concept adopted in UNITE database and based on ITS sequences. The low genetic variability obtained among specimens of clade A (SH191352.07FU), and the high genetic variability among these specimens and those of clade B (SH191350.07FU where H. macaronesicum holotype is included), as well as the species tree inferred, led us to describe a new species from specimens of clade A, and to provide it a Latin binomial. The two species share the same morphological characters according to H. macaronesicum original description (Telleria et al. 2012), but they can be distinguished based on phylogenetic analyses of ITS, IGS, RPB2 and EF1-α sequences, genetic distances, haplotype networks, multilocus tree and species tree. Salgado-Salazar et al. (2013) calculate the genetic distances of putative species within Thelonectria discophora species-complex, and their values, as in our study, exceeded the standard (0.01–0.03) used to delimit operational taxonomic units (OTU).
Moreover, H. macaronesicum is found in the seven islands of the Canary Archipelago growing mainly on Macaronesian endemic plants, such as Echium brevirame, Euphorbia balsamifera, Euphorbia lamarckii, Kleinia neriifolia, Plocama pendula, Rumex lunaria, Salvia canariensis, and Schizogyne sericea (Telleria et al. 2012); except the specimen from Fuerteventura growing on Launaea arborescens, and the specimen from Gran Canaria in Telleria et al. (2012) growing on Agave americana. One specimen of the new sibling species, H. paramacaronesicum, from Fuerteventura was found growing on Launaea arborescens; but other specimens were collected on introduced plants, such as Nicotiana glauca (Fuerteventura), and Banksia integrifolia in Faial Island (Azores Archipelago), some others were found also on Macaronesian endemisms such as Ocotea foetens, as well as Mediterranean plants e.g. Pistacea atlantica (Gran Canaria), and Cistus monspeliensis (Tenerife).
On the other hand, the genetic divergence among isolates from subclade B1 and B2, the multilocus phylogenetic analyses and the species tree are sufficient to consider that these are not separate taxa; the Bayes Factor indicates strong strength of evidence of two species instead of three. Based on the results of the network analyses between isolates of Phlebia livida ssp. livida and Phlebia livida ssp. tuberculata, that appear in two clusters separated from each other by 12 mutational steps, Ghobad-Nejhad & Hallenberg (2012) raised P. livida ssp. tuberculata to species level. In our study, the number of mutational steps between the two clusters, B1 and B2, is around 17, suggesting that they could belong to two different species; however, we have detected a heteroduplex (TFCMic. 15032). Selosse et al. (1996) were among the first to detect IGS heteroduplex formation. They state that heterozygosity is probably common to many dikaryotic fungi (e.g., Laccaria bicolor, their model organism), and that it can provide helpful information to distinguish between introduced exotic and indigenous populations. Also, the presence of two haplotypes within isolate TFCMic.15032 would be consistent with simple heterozygosity within a panmictic clade B.
ACKNOWLEDGEMENTS
We are grateful to Marian Glenn (Seton Hall University, New Jersey, USA) and John Spouge (NCBI, Bethesda, USA) for checking the English language; to Thorsten Lumbsch (The Field Museum, Chicago, USA) for helping with the neighbour-net analysis; to Kessy Abarenkov (Tartu University, Estonia) for always resolving our doubts with the UNITE database; to Fátima Durán and Eva Ma Rodriguez (RJB–CSIC) for providing technical assistance. Financial support was provided by Plan Nacional I+D+i projects CGL2009–07231, CGL2012–35559 and CGL-2015-67459-P.
Supplementary Material: http://fuse-journal.org/
REFERENCES
- Anderson JB, Stasovski E. (1992). Molecular phylogeny of Northern hemisphere species of Armillaria. Mycologia 84: 505–516. [Google Scholar]
- Balasundaram SV, Engh IB, Skrede I, et al. (2015). How many DNA markers are needed to reveal cryptic fungal species. Fungal Biology 119: 940–945. [DOI] [PubMed] [Google Scholar]
- Bertolazzi P, Felici G, Weitschek E. (2009). Learning to classify species with barcode. BMC Bioinformatics 10 (Suppl. 14): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford D, Lohman DJ, Sodhi NS, et al. (2006). Cryptic species as a window on diversity and conservation. Trends in Ecology and Evolution 22: 148–155. [DOI] [PubMed] [Google Scholar]
- Binder M, Larsson K-H, Matheny PB, et al. (2010). Amylocorticiales ord. nov. and Jaapiales ord. nov.: Early diverging clades of Agaricomycetidae dominated by corticioid forms. Mycologia 102: 865–880. [DOI] [PubMed] [Google Scholar]
- Bouckaert R. (2010). DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26: 1372–1373. [DOI] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kühnert D, et al. (2014). BEAST 2: A software platform for bayesian evolutionary analysis. PLOS Computational Biology 10: e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, Moulton V. (2002). Neighbor-Net: An agglomerative method for the construction of planar phylogenetic networks. In: Algorithms in Bioinformatics Proceedings (Guigo R, Gusfield D, eds), Second International Workshop, WABI, Rome, Italy Lecture Notes in Computer Science 2452: 375–391 [Google Scholar]
- Carlsen T, Engh IB, Decock C, et al. (2011). Multiple cryptic species with divergent substrate affinities in the Serpula himantioides species complex. Fungal Biology 115: 54–61. [DOI] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA. (2000). TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659. [DOI] [PubMed] [Google Scholar]
- Crandall KA, Templeton AR. (1993). Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstructions. Genetics 134: 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M, Meiklejohn K, Cameron Sl, et al. (2014). A preliminary framework for DNA barcoding, incorporating the multispecies coalescent. Systematic Biology 63: 639–644. [DOI] [PubMed] [Google Scholar]
- Farris JS. (1989). The retention index and the rescaled consistency index. Cladistics 5: 417–419. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- Gardes M, Bruns TD. (1993). ITS Primers with enhanced specificity for Basidiomycetes – applications to the identification of mycorrhizae and rusts. Molecular Ecology 1: 113–118. [DOI] [PubMed] [Google Scholar]
- Ghobad-Nejhad M, Hallenberg N. (2012). Multiple evidence for recognition of Phlebia tuberculata, a more widespread segregate of Phlebia livida (Polyporales, Basidiomycota). Mycological Progress 11: 27–35. [Google Scholar]
- Grummer JA, Bryson RW, Reeder TW. (2014). Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae). Systematic Biology 63: 119–133. [DOI] [PubMed] [Google Scholar]
- Hamming RW. (1950). Error detecting and error correcting codes. Bell System Technical Journal 29: 147–160. [Google Scholar]
- Hawksworth DL, May TW, Redhead SA. (2017). Fungal nomenclature evolving: changes adopted by the 19th International Botanical Congress in Shenzhen 2017, and procedures for the Fungal Nomenclature Session at the 11th International Mycological Congress in Puerto Rico 2018. IMA Fungus 8: 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. (2010). Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution 27: 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. (2001). MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- James TY, Moncalvo JM, Li S, Vilgalys R. (2001). Polymorphism at the ribosomal DNA spacers and its relation to breeding structure of the widespread mushroom Schizophyllum commune. Genetics 157: 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. (1995). Bayes Factors. Journal of the American Statistical Association 90: 773–795. [Google Scholar]
- Kõljalg U, Nilsson RH, Abarenkov K, et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Molecular Ecology 22: 5271–5277. [DOI] [PubMed] [Google Scholar]
- Kluge AG, Farris JS. (1969). Quantitative phyletics and the evolution of anurans. Systematic Zoology 18: 1–32. [Google Scholar]
- Korshunova T, Martynov A, Bakken T, et al. (2017). External diversity is restrained by internal conservatism: New nudibranck mollusk contributes to the cryptic species problem. Zoologica Scripta 46: 683–692. [Google Scholar]
- Larget B, Simon DL. (1999). Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Molecular Biology and Evolution 16: 750–759. [Google Scholar]
- Liu YJ, Whelen S, Hall BD. (1999). Phylogenetic relationships among Ascomycetes: Evidence from an RNA polymerase II subunit. Molecular Biology and Evolution 16: 1799–1808. [DOI] [PubMed] [Google Scholar]
- Lutzoni F, Kauff F, Cox CJ, et al. (2004). Assembling the Fungal Tree of Life: progress, classification, and evolution of subcellular traits. American Journal of Botany 91: 1446–1480. [DOI] [PubMed] [Google Scholar]
- Mallo D, Posada D. (2016). Multilocus inference of species trees and DNA barcoding. Philosophical Transactions of the Royal Society B 371: 20150335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín MP, Winka K. (2000). Alternative methods of extracting and amplifying DNA from lichens. The Lichenologist 32: 189–196. [Google Scholar]
- Matheny PB. (2005). Improving phylogenetic inference of mushrooms with RPB1 and RPB2 nucleotide sequences (Inocybe; Agaricales). Molecular Phylogenetics and Evolution 35: 1–20. [DOI] [PubMed] [Google Scholar]
- Matheny PB, Wang Z, Binder M, et al. (2007). Contributions of rpb2 and tef1 to the phylogeny of mushrooms and allies (Basidiomycota, Fungi). Molecular Phylogenetics and Evolution 43: 430–451. [DOI] [PubMed] [Google Scholar]
- McNeill J, Barrie FR, Buck WR, et al. (2012). International Code of Nomenclature for algae, fungi, and plants (Melbourne Code) adopted by the Eighteenth International Botanical Congress Melbourne, Australia, July 2011. [Regnum Vegetabile No. 154.] Königstein: Koeltz Scientific Books. [Google Scholar]
- Neigel J, Domingo A, Satke J. (2007). DNA barcoding as a tool for coral reef conservation. Coral Reefs 26: 487–499. [Google Scholar]
- Nielsen R, Matz M. (2006). Statistical approaches for DNA barcoding. Systematic Biology 55: 162–169. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. (2001). Intraspecific gene genealogies: tress grafting into networks. Trends in Ecology and Evolution 16: 37–45. [DOI] [PubMed] [Google Scholar]
- Rambaut A. (2002). Se-Al: sequences alignment editor v2.0a11. Edinburgh: Institute of Evolutionary Biology, University of Edinburgh, http://tree.bio.ed.ac.uk/software/. [Google Scholar]
- Ronquist F, Teslenko M, Van der Mark P, et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado-Salazar C, Rossman AY, Chaverri P. (2013). Not as ubiquitous as we thought: taxonomic crypsis, hidden diversity and cryptic speciation in the cosmopolitan fungus Thelonectria discophora (Nectriaceae, Hypocreales, Ascomycota). PlosOne 8: e76737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch C, Seifert KA, Huhndorf S, et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal barcode marker for Fungi. Proceedings of the National Academy of Sciences USA 109: 5907–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selosse MA, Costa G, Di Battista C, et al. (1996). Meiotic segregation and recombination of the intergenic spacer of the ribosomal DNA in the ectomycorrhizal basidiomycete Laccaria bicolor. Current Genetics 30: 332–337. [DOI] [PubMed] [Google Scholar]
- Stielow JB, Lévesque CA, Seifert KA, et al. (2015). One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia: Molecular Phylogeny and Evolution of Fungi 35: 242–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck TH, Feder JL, Bendiksby M, et al. (2018). Finding evolutionary processes hidden in cryptic species. Trends in Ecology and Evolution 33: 153–163. [DOI] [PubMed] [Google Scholar]
- Swofford DL. (2003). PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, Massachusetts, Sinauer Associates. [Google Scholar]
- Taylor JW, Jacobson DJ, Kreoken S, et al. (2000). Phylogenetic species recognition and species concepts in fungi. Fungal Genetics and Biology 31: 21–32. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Turner E, Townsend JP, et al. (2006). Eukaryotic microbes, species recognition and the geographic limits of species: examples from the kingdom Fungi. Philosophical Transactions of the Royal Society B: Biological Sciences 361: 1947–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telleria MT, Dueñas M, Beltrán-Tejera E, et al. (2012). A new species of Hyphoderma (Meruliaceae, Polyporales) and its discrimination from closely related taxa. Mycologia 104: 1121–1132. [DOI] [PubMed] [Google Scholar]
- Templeton AR. (2001). Using phylogenetic analyses of gene trees to test species status and processes. Molecular Ecology 10: 779–791. [DOI] [PubMed] [Google Scholar]
- Thiers B. (2016). [continuously updated]. Index Herbariorum: A global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium; http://sweetgum.nybg.org/ih/ [Google Scholar]
- White TJ, Bruns T, Lee S, et al. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols: a guide to methods and applications (Innes MA, Gelfand DH, Sninsky JJ, White TJ, eds.). Academic Press, San Diego, California: 315–322. [Google Scholar]
- Wilson AW, Binder M, Hibbett DS. (2012). Diversity and evolution of ectomycorrhizal host associations in the Sclerodermatineae (Boletales, Basidiomyctota). New Phytologist 194: 1079–1095. [DOI] [PubMed] [Google Scholar]
- Winka K, Ahlberg C, Eriksson OE. (1998). Are there lichenized Ostropales? The Lichenologist 30: 455–462. [Google Scholar]
- Yar R, Schoch CL, Dentinger BTM. (2016). Scaling up discovery of hidden diversity in fungi: impacts of barcoding approaches. Philosophical Transactions Royal Society B 371: 20150336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.