

Abstract
Direct‐acting antiviral agents (DAAs) represent a class of drugs targeting viral proteins and have been demonstrated to be very successful in combating viral infections in clinic. However, DAAs suffer from several inherent limitations, including narrow‐spectrum antiviral profiles and liability to drug resistance, and hence there are still unmet needs in the treatment of viral infections. In comparison, host targeting antivirals (HTAs) target host factors for antiviral treatment. Since host proteins are probably broadly required for various viral infections, HTAs are not only perceived, but also demonstrated to exhibit broad‐spectrum antiviral activities. In addition, host proteins are not under the genetic control of viral genome, and hence HTAs possess much higher genetic barrier to drug resistance as compared with DAAs. In recent years, much progress has been made to the development of HTAs with the approval of chemokine receptor type 5 antagonist maraviroc for human immunodeficiency virus treatment and more in the pipeline for other viral infections. In this review, we summarize various host proteins as antiviral targets from a medicinal chemistry prospective. Challenges and issues associated with HTAs are also discussed.
Keywords: broad‐spectrum antivirals, direct‐acting antiviral agents, drug resistance, host targeting antiviral agents
1. INTRODUCTIONS
Viral infections still represent a major global public health problem. Although hundreds of viruses are known to be pathogenic, only less than 10 of them can be treated in clinic with available antiviral drugs. For some highly pathogenic virus such as Zika (ZIKV), Ebola (EBOV), severe acute respiratory syndrome (SARS), as well as many others, there is still no effective drugs on market against them. Most of the antiviral drugs approved are designed to target viral proteins to inhibit viral infections, and they are named as direct‐acting antiviral agents (DAAs). 1 DAAs have been demonstrated to be very successful in clinic to combat viral infections, and are generally considered as very safe for human use because most of the targeted viral proteins have no human homologs. However, viral polymerases, one of the hottest targets for antiviral design, do share some structural similarity with their human counterparts, especially at the active sites, which is the major underlying reason for the toxicity observed with nucleoside‐based antivirals. In addition, viral proteins do not normally share structural similarity among different species or even genotypes of virus, and hence one antiviral agent targeting a specific viral protein can hardly impart the same inhibitory effects against the other virus. Consequently, the antiviral drugs available on market can barely be employed to treat newly emerging virus, and the magnitude of this problem is further exacerbated by the lack of broad‐spectrum antiviral drugs on market. The other inherent limitation of DAAs is that DAAs, in most cases, have low genetic barrier to drug resistance because they act directly on viral proteins, and the resulted selective pressure will facilitate the mutations of virus during replication, which will make the virus refractory to the treatment of DAAs. 2 Altogether, there still exist great unmet needs for the treatment of viral infections, and new antivirals with broad‐spectrum antiviral activities as well as new mechanism of action are highly demanded.
It is widely known that viruses are unable to complete their replication without the help of the host. They will hijack various host proteins or pathways throughout their replication cycle to facilitate their replication, and hence the inhibition or knockdown of such host proteins will block viral replication. Therefore, host proteins as such are potential antiviral targets for drug development. The host targeting antivirals (HTAs) are complementary to the DAAs, and are superior to DAAs from several aspects. Chiefly, a specific host protein might play crucial roles in the replication of several types of viruses, and thereby its inhibition will yield broad‐spectrum antiviral activities. HTAs as such are likely to be effective against newly emerging virus, because they may also leverage on the same host protein for replication. For example, heat shock protein 90 (Hsp90), a host chaperone responsible for the folding, assembly, and maturation of endogenous proteins, is also found to be crucial for maturation of many viral proteins, and therefore, Hsp90 inhibitors are endowed with broad‐spectrum antiviral activities. 3 Altogether, targeting host factors is a very promising strategy with possibility to address the critical challenges faced with the DAAs. In this review, we summarize the recent advances made in HTAs from a medicinal chemistry standpoint, and the host targets are generally classified into three different categories based on the development stage of their corresponding inhibitors/modulators, namely the ones which reached Food and Drug Administration (FDA) approval, that have entered clinical trials and those in preclinical studies. The focus is mainly placed on the small‐molecule based HTAs, and the antibody or small interfering RNA (siRNA)‐based strategy will not be included. For several well‐studied host targets with quite a lot of known inhibitors, because each one can stand alone as an independent review, and this review is not intended to be comprehensive, so for such targets, only a selection of representative inhibitors will be presented to discuss related issues.
2. HOST TARGETS WITH MODULATORS APPROVED FOR ANTIVIRAL TREATMENT
2.1. Chemokine receptors type 4 and 5
It should be noted that chemokine receptor type 5 (CCR5) antagonist but not chemokine receptor type 4 (CXCR4) antagonist has reached FDA approval. However, CXCR4 is discussed in this section because it is closely related to CCR5. The chemokine receptors CXCR4 and CCR5 belong to the superfamily of G‐protein‐coupled receptors with seven transmembrane domains, and both are involved in the entrance of human immunodeficiency virus (HIV) virus. 4 The glycoprotein 120 (gp120) on the surface of HIV envelop will bind to the CD4 receptor on the surface of T lymphocytes, leading to the conformation change of gp120. The gp120 will then bind to coreceptor CXCR4, CCR5, or both to facilitate the entrance of HIV virus. 5 HIV‐1 virus can thus be broadly classified into three types based on the coreceptor tropism, namely the X4‐, R5‐, and dual‐tropic viruses. The feasibility of targeting CXCR4 or CCR5 for HIV therapy is supported by the fact that CCR5 mutations confer the host with resistance to HIV infection. 6 Ample studies have demonstrated that CXCR4 antagonists can block HIV‐1 infection through CXCR4. 7 , 8 Generally, CXCR4 antagonist can be divided into two types: peptide and nonpeptide based. The peptide‐based antagonists were designed using the natural chemokines of CXCR4 as template, such as stromal cell‐derived factor 1α (SDF‐1α) and viral macrophage inflammatory protein II (vMIP‐II), both of which can bind to and then activate CXCR4 to initiate downstream signaling pathway. For example, peptide V1 (1; Figure 1) was derived from the first 1 to 21 residues of VMIP‐II, and it can block the interaction between CXCR4 and HIV‐1, and thereby inhibit the entry of both X4‐ and dual‐tropic HIV‐1 virus. Interestingly, replacement of all the residues in 1 with all d‐amino acid lead to much more potent antiviral activities. 9 Due to the inherent limitation with the peptide‐based antagonist, several nonpeptide based antagonists have been developed. AMD3100 (2) was discovered by random screening, and it inhibited HIV‐1 infection by disrupting the interaction between CXCR4 and HIV‐1. 10 Encouragingly, it showed antiviral efficacy in X4 HIV‐1‐infected patients in a phase II clinical trial. However, compound 2 lacks oral bioavailability due to its high positive charge, and cardiotoxicity was also reported in the clinical trial. 11 The structural optimization identified AMD3465 (3) with just one macrocyclic structure retained. This compound retained all the biological profiles of 2, and it showed improved antiviral activity (IC50: 1‐10 nM). It also inhibited the replication of drug‐resistant virus strain (zidovudine). 12 AMD070 (4), a noncyclam derivative, was discovered by AnorMED as a new CXCR4 antagonist. It showed much improved oral bioavailability (rat: 20%; dog: 80%), but hepatotoxicity was observed in preclinical studies. 13 Further structural optimization led to GSK812397 (5) without the basic sidechain. It showed potent anti‐HIV‐1 activity with an IC50 value of 1.5 nM, and it did not show any detectable toxicity (>1000 nM) in in vitro assay. In addition, 5 also presented acceptable pharmacokinetic profiles with the oral bioavailability in the range between 10% and 21% among different species. 14 Further structural modification with constraining conformation (6) and/or opening up the tetrahydroquinoline ring (7) resulted in a series of new derivatives with retained anti‐HIV activities. 15 , 16 However, none of these compounds have been advanced into clinical trials.
Figure 1.
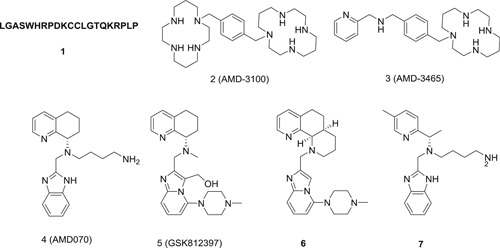
The chemical structures of selected chemokine receptor type 4 antagonists
Although compound 2, also known as plerixafor, was approved by the FDA for autologous transplantation in patients with non‐Hodgkin's lymphoma or multiple myeloma, 17 no CXCR4 antagonist has been approved for the treatment of HIV infections. The underlying reasons are at least twofold: one major concern with the CXCR4 antagonist is the toxicity issue, especially that CXCR4 18 , 19 or SDF‐1α 20 knockout mice die prenatally with multiple neurological, cardiac/vascular, and hematopoietic defects. Such potential adverse outcomes are exacerbated in the case of HIV treatment, wherein a life‐long treatment is required; the other minor issue with these CXCR4 antagonists is the lack of oral bioavailability. IV injection is not practical in the case of HIV treatment, because this will definitely hurt the compliance of the patients.
In comparison, the development of CCR5 antagonists for HIV treatment is much more successful. One CCR5 antagonist maraviroc (8; Figure 2) has been approved by FDA as HIV entry inhibitor in 2007. 21 With a long plasma half‐life (15‐23 hours), 22 8 is administrated orally once daily. The clinical data showed that 8 is well tolerated in patients and it can significantly repress the viral load in R5‐infected HAART‐treatment experienced patients. Moreover, it is active against 200 clinically derived HIV‐1 envelope‐recombinant pseudoviruses, with 100 of them being resistant to existing drug classes, demonstrating the merits of HTAs. 23 In the clinic, it is primarily recommended for the patients infected with R5‐tropic HIV virus, and a tropism test (although not approved by FDA) is highly required to determine the viral tropism population within the patient before the treatment to ensure treatment success. The first generation of the test (known as TROFILE) failed to distinguish R5 from R5/X4 mixed tropism, resulting in significant drug failure in clinic in R5/X4‐infected patients. 24 Therefore, 8 is just designated as a backup regimen, despite that a new reliable and cost‐efficient tropism test is available in clinic now. Interestingly, the combination of 8 with raltegravir/tenofovir/emtricitabine led to faster reduction of 2‐long term repeat (2‐LTR+) newly infected cells and recovery of CD4+ T‐cell counts after 48 weeks of treatment, 25 and 8 in combination with reverse transcriptase inhibitors is being tested in clinic for preexposure prophylaxis. 26 However, the use of 8 is accompanied with severe side effects, and in some cases even life‐threatening conditions such as hepatotoxicity and heart attack were observed. To mitigate these limitations, the second generation of CCR5 antagonists have been developed. Two clinical candidates vicriviroc (9) 27 and aplaviroc (10) 28 have made to phase II trials. However, the studies were halted due to insufficient efficacy and observed hepatotoxicity for 9 and 10, respectively. Much of the subsequent medicinal chemistry effort is devoted to the structural modification toward 8 to 10, aiming to improve either efficacy and/or ADME profiles. For example, Pfizer discovered compound 8's structural analogue PF‐232798 (11), 29 which showed not only potent anti‐HIV activity (IC50: 2.0 nM) and moderate hERG inhibition (IC50: 12 μM), but also superior oral bioavailability as compared to 8. In addition, compound 11 is also active against maraviroc‐resistant HIV‐1 isolate strain CC185. The data from phase‐II trials demonstrated its superior safety in patients with no adverse effects observed at a dosage up to 250 mg. However, no further data were disclosed after that. 30 GSK 163929 (12) with a 4,4‐disubstituted piperidine scaffold exhibited potent anti‐HIV activity and excellent pharmacokinetics (PK) profiles, but it did not progress to clinic trials due to toxicity concerns. 31 The other two representative new scaffolds derived from compound 8 are monocyclic piperidine amides and cyclic and acyclic urea‐piperidines. Some representative candidate compounds from these two series are shown in Figure 2. Among these compounds, only TAK‐220 (13) was progressed to phase‐I clinical trials. 32 However, no further update about the status of compound 13 has been reported. Inspired by the scaffold of 9 and 10, several new CCR5 antagonists have been devised as depicted in Figure 2. 33 The most promising antagonist among this series is the one developed by Incyte based on the structure of 10. INCB9471 (16) showed potent antiviral activity against R5 HIV‐1 strains at IC50 values in sub‐nanomolar range. In addition, it presented excellent PK profiles with oral bioavailability being 100% and 95% in rat and dog, respectively. 34 As such, 16 has been advanced to phase II trials, and the data showed that it was well‐tolerated in humans. However, its clinical studies were halted due to business issues. Besides the chemotypes described above, CCR5 antagonists with miscellaneous scaffolds have also been discovered, yet none of them have made to clinical trials. 35
Figure 2.
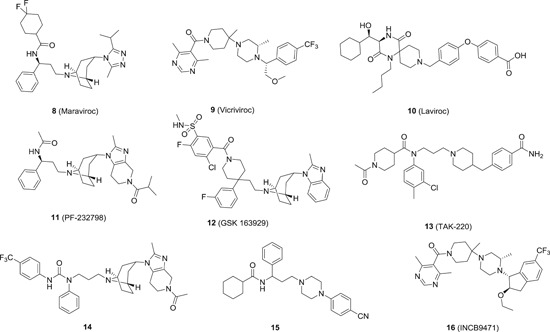
The chemical structures of representative chemokine receptor type 5 antagonists
Targeting CXCR4 or CCR5 alone for HIV treatment possesses several downsides. Chiefly, a tropism test is required before the treatment, and tropism shifts are frequently observed in both treatment‐experienced and treatment‐naive HIV‐infected patients. Therefore, a dual antagonist against both CXCR4 and CCR5 would be perfect to mitigate these limitations. The first dual‐tropic inhibitor AMD3451 (17; Figure 3) with N‐pyridinylmethyl cyclam scaffold was discovered in 2004. It was moderately active against not only X4‐ and R5‐tropic HIV strains but also dual‐tropic HIV strains with IC50 in the range of 1 to 30 μM. 36 Although 17 is not potent enough for further development, it established the proof of concept of targeting CXCR4 and CCR5 simultaneously for HIV‐1 treatment.
Figure 3.
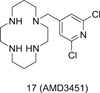
The chemical structure of a dual‐tropic inhibitor AMD3451
2.2. Ribavirin
Ribavirin (18; Figure 4) is an approved antiviral drug used in clinic to treat respiratory syncytial virus (RSV), hepatitis C virus (HCV) infection and viral hemorrhagic fever. Although its specific antiviral mechanism of action remains largely uncertain, it is widely accepted that it elicits its antiviral efficacy via modulating host pathways. For example, 18 showed moderate inhibitory potency against inosine‐5′‐monophosphate dehydrogenase (IMPDH) with a K i value of around 250 nM, which is believed to be highly involved in the replication of various viruses (see below). In addition, several other mechanism of actions have also been proposed and evidenced: (a) direct inhibition of RNA polymerase by converting 18 to its triphosphate form to competitively bind to the nucleotides binding site in RNA polymerase 37 , 38 ; (b) 18 can act as a mutagen by inserting into the viral RNA to push the virus beyond the threshold of error catastrophe 39 , 40 ; (c) 18 shows immunomodulatory effect of shifting a Th2 response in favor of a Th1 phenotype, which helps to clear virus infections. 41 Although the combination of 18 and interferon 2α (IFN‐2α) used to be the SOC for HCV treatment, it is notorious for several severe side effects. One major adverse effect associated with the use of 18 is hemolytic anemia. 42 To alleviate this unwanted effect, taribavirin (19) was developed as the prodrug of 18. Ideally, 19 can be metabolized to 18 mainly in the liver to target HCV‐infected hepatocytes, and hence the distribution of 18 within red blood cells will be significantly decreased, and thereby the development of hemolytic anemia will be subsequently eliminated. Indeed, the clinical data from phase III trials revealed that patients receiving 19 (fixed dosage 600 mg, BID) and IFN showed significantly lower rates of anemia as compared to the ones in 18 (1000‐1200 mg) and IFN group (5.3% vs 24%). 43 However, the sustained virologic response (SVR) rates for 19 and 18 group are 38% and 52%, respectively, failing to demonstrate the noninferiority end point for efficacy. The ViSER‐2 trials also failed to meet the noninferiority end points. 44
Figure 4.
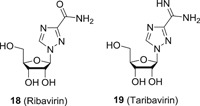
The chemical structures of 18 and 19
With the approval of several DAAs such as sofosbuvir and ledipasvir, the treatment of HCV in clinic has been significantly revolutionized, and several both ribavirin (RBV)‐ and IFN‐free regimens have been approved with better efficacy and safety profiles as compared to the old SOC. Initially, it was expected that the HCV treatment would not benefit much from the combination of RBV with other DAAs due to the safety concern associated with RBV. However, very interestingly, it has been found recently that RBV remained an indispensable component for the optimal treatment for some difficult‐to‐cure subgroups of HCV patients. 45 For example, in a phase 3 C‐EDGE studies, the combination of RBV with elbasvir/grazoprevir achieved much higher SVR (12 weeks, 93% [14/15] vs 78% [7/9]; 16 weeks, 100% [8/8] vs 60% [3/5], respectively) as compared to the group without RBV in patients infected with genotype 4 HCV. 46 In addition, RBV increased the barrier to resistance, especially in patients receiving DAAs with low barriers to resistance. Importantly, the combination of RBV with DAAs showed much improved safety and tolerability as compared with the combination with IFN, and the frequency and severity of anemia is significantly reduced with an adverse effect‐induced discontinuation rate of less than 3%. 45 Nevertheless, the last generations HCV DAAs (including glecaprevir/pibrentasvir, sofosbuvir/velpatasvir, sofosbuvir/velpatasvir/voxilaprevir) are highly effective in most cases without any need to use RBV.
3. HOST TARGETS TESTED IN CLINICAL TRIALS
3.1. Cyclophilins
Cyclophilins are a family of cellular peptidyl‐prolyl cis‐trans isomerases (PPIase) and are involved in many cellular processes. The important roles cyclophilin A (CypA) plays during HCV replication were found by an unexpected clinical finding. Cyclosporin A (CsA; 20; Figure 5), which shows high binding affinity to CypA, is an approved immunosuppressive drug. In a clinical trial for CsA's therapeutic potential against hepatitis‐associated inflammation, it was unexpectedly found that a significantly more potent antiviral response was observed in the combination group of CsA and IFN‐α2b as compared with the IFN‐α2b alone group. 47 The function of CypA in the replication of HCV was subsequently confirmed in vitro. 48 The main cellular function of Cyps is to convert the conformation (trans‐ to cis‐form) of prolines in the protein, which is essential for trafficking of many proteins and forming protein complexes, and these functions are also indispensable for the replication of HCV. Therefore, it is anticipated that the CypA inhibitors will show anti‐HCV activity. Although 20 is an approved drug, its main indication is suppressing immunoreaction after organ transplantation, which is an unwanted “side‐effect” for antivirals. Fortunately, the immnosuppressive effect of 20 is attributed to the inhibition of calcineurin (CN) but not CypA, and hence it is feasible to eliminate the immnosuppressive effect while retaining the anti‐HCV activity by making new CsA analogues. NIM811 (21) is one of such analogues with a methyl‐isoleucine at position 4 of CsA. It showed anti‐HCV activity in vitro with an IC50 value of 0.12 μM, whereas its immunosuppressive effect was completely eliminated. 49 The phase I clinical trial showed that 21 is well‐tolerated with no obvious adverse effects observed. However, in a subsequent double‐blind, placebo‐controlled study, the monotherapy with 21 failed to yield significant viral load reduction in genotype 1 HCV‐infected patients. The underlying reason is attributed to a relatively low trough concentration (0.47 μM) at a dosage of 600 mg (BID), which is lower than the IC50 value (1.5 μM) of 21 against HCV in the presence of serum, and the dosage elevation did not result in proportional exposure. 50 Therefore, the clinical trial with 21 is not continued.
Figure 5.
The chemical structures of cyclosporin A analogues [Color figure can be viewed at wileyonlinelibrary.com]
Alisporivir (22) is a more potent nonimmunosuppressive CsA analogue with an anti‐HCV IC50 values of 0.045 and 0.33 μM in the absence and presence of serum, respectively. In addition, it is much more difficult to develop resistance against 22 both in vitro and in patients, as compared to other DAAs. 51 In phase II trial, 22 was studied in combination with RBV as an IFN‐free regimen in genotype 2 and 3 patients, and about half of the patients receiving 22 (800 mg, QD) plus RBV, and one‐third of those receiving 22 only (1000 mg QD) were cured with a SVR. 52 However, unfortunately, in a pivotal phase III trial designed to study the combination of 22 with RBV and PEG‐IFN‐α2a in treatment‐naive, genotype 1 HCV patients, six cases of severe pancreatitis along with one death were reported, and the clinical trial with 22 was then put on hold. It is worth noting that all the severe side effects were observed in the triple‐therapy aim including RBV and PEG‐IFN‐α2a, both of which are notorious for their adverse effects, while the monotherapy with 22 or the IFN‐free regimen did not result in the same side effects. 53
To mitigate the observed side effects with 21 and 22, the third generation of CsA derivatives have been developed. The representative candidate compound from this series is SCY‐635 (23) with a dimethylaminoethylthio substituent at position 3 and a hydroxyl group at the γ position of the 4‐N‐methyl leucine residue. 23 showed potent anti‐HCV activity in vitro with an EC50 and EC90 value of 0.1 and 0.3 μM, respectively, and exhibited synergistic effect with IFN‐α and additive effect with RBV. It also showed low potential for drug‐drug interaction with no obvious induction on the major cytochrome P450 enzymes 1A2, 2B6, and 3A4. In addition, 23 also showed acceptable PK profiles with an oral bioavailability of around 20% in rat and monkey. 54 As such, 23 has been advanced into clinical trials. In patients with genotype 1 HCV infection, 900 mg/day of 23 achieved a decline in plasma viremia by 2.2log 10 after 15 days. 55 It is interesting to note that 23 showed totally different side effects from those of 21 and 22, indicating that the adverse effects may not be associated with the inhibition of Cyps, but are likely resulted from the off‐target effects of individual inhibitors.
Non‐CsA based Cyps inhibitors have also been discovered. Sanlifehins (SFA) including sanlifehin A (24), B (25), C (26), and D (27) are a class of natural occurring polyketides isolated from the soil bacterium Streptomyces sp. strain A92‐308110. SFAs were identified as Cyps inhibitors with stronger potency as compared to CsA derivatives, particular 25, of which the inhibitory potency against all Cyps was 30‐ to 50‐fold more potent than 20. It also showed much more potent antiviral activity in vitro with an EC50 value of 70 nM against HCV genotype 1b. Interestingly, albeit slightly less potent as compared to against the wild type, 24, 26, and 27 retained inhibitory effect against CsA‐resistant Huh 9 to 13 subgenomic replicon with EC50 values ranging from 3.3 to 6.8 μM. 56 However, the PK studies revealed that SFA suffered from poor water solubility (<25 μM) and poor oral bioavailability (<4%). Moreover, SFA possessed undesirable immunosuppressive activity via an unknown mechanism. 57 Structural modifications have been made to 24, and it was revealed that only the macrocyclic moiety was essential for the Cyps inhibition, and modification on the sidechain had little effect on the binding affinity. 58 Removal of the spirolactam moiety on the sidechain of 24 only led to the loss of immunosuppressive activity but not the Cyps inhibition. Such structure‐activity relationships are very important for further optimization of SFA as anti‐HCV agents (Figure 6).
Figure 6.
The chemical structures of sanlifehin A‐D [Color figure can be viewed at wileyonlinelibrary.com]
Both CsA and SFA derivatives are macrocyclic molecules with large molecular size, and as such both suffered from some limitations, including poor cell membrane permeability, high risk of drug‐drug interactions and off‐target toxicity, and synthetic inaccessibility for structural optimization and manufacturing. In 2016, a new family of nonpeptide based small‐molecule Cyps inhibitors have been designed using fragment‐based strategy. 59 The crystal structure of CypA indicated that its PPIase catalytic site consisted of hydrophobic, aromatic, and polar residues, next to the catalytic site is a deep pocket called gatekeeper site, which might contribute to the substrate binding specificity (Figure 7). A total of 34 409 fragments were docked into these two sites, and several fragments were identified to bind to these two sites separately. Eventually, fragment 28 and 29 from each binding site were selected and connected together by a urea linkage to yield compound 30 (Figure 8), which showed potent inhibitory effect against CypA and B with IC50 values of 13 and 6 μM, respectively. Further structural optimization identified compound 31 with much‐improved potency. Its IC50 values against CypA, B, and D ranged from 0.08 to 0.2 μM, which was around 10‐fold less potent than 20. Compound 31 together with other analogues showed definitive antiviral activity against a panel of viruses including HCV (genotype 1a, 1b, 2a, 3a, 2a/4a, and 5a), HIV, human coronavirus 229E, dengue virus (DENV), ZIKV, and YFV in vitro with IC50 values ranging from 0.4 to 44 μM. 59 Mutations at Ns5A (D320E and R318H) did not increase the IC50 values of compound against HCV, indicating the advantage of high generic barrier to resistance by targeting Cyps. Although compound 31 is a less potent inhibitor against Cyps as compared to 20 and 22, it did not display any immunosuppressive effect and inhibition of IL‐2 production in stimulated immortalized T lymphocytes (EC50 > 20μM). Altogether, these results presented this scaffold of Cyps inhibitors as a very promising starting point for further development.
Figure 7.
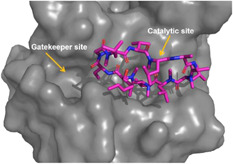
The binding site of 20 in cyclophilin A. PDB: 5HSV. The image was generated with Pymol (https://pymol.org/2/) [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8.
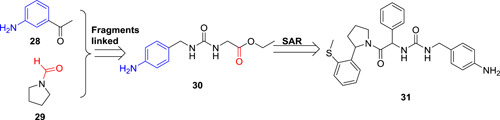
Fragment‐based drug design for Cyps inhibitors. Cyp, cyclophilin [Color figure can be viewed at wileyonlinelibrary.com]
It has been well‐established that Cyps are involved in a broad range of viral infections, including HIV‐1, 60 influenza virus, 61 HBV, 62 SARS coronavirus, 63 human cytomegalovirus (HCMV), 64 papillomavirus, 65 and nidovirus, 66 among others. Therefore, it can be envisioned that Cyps inhibitors should exhibit broad‐spectrum antiviral activity against these viruses. Indeed, Cyps inhibitors were reported to show broad‐spectrum antiviral activities. 67 , 68 Consequently, the development of Cyps inhibitors could benefit the treatment of a variety of viral infections, possibly including the newly emerging unidentified viral infections.
3.2. Eukaryotic initiation factor 2α
It is widely known that virus will hijack a wide range of host factors to facilitate its replication. Meanwhile, the host has also evolved an innate immune system to counteract viral infections. Compounds with the capacity to mediate the host antiviral pathways are expected to confer broad‐spectrum antiviral activity, and nitazoxanide (32; Figure 9) is such a compound that regulates several host antiviral pathways to convey broad‐spectrum antiviral profiles. 32 is originally an FDA approved drug for the treatment of diarrhea caused by Cryptosporidium parvum and Giardia intestinalis in both adults and children, and it has been demonstrated to be well‐tolerated in humans with the gastrointestinal disorders as the most frequently observed side effects. It also showed no effect on cardiac repolarization in a clinical trial for cardiac safety. 69 In recent studies, 32 was revealed as a promising antiviral agent against a wide range of pathogenic viruses including influenza virus, 70 HBV, 71 HCV, 71 EBOV, 72 DENV, 73 JEV, 74 HIV, 75 and ZIKV, 76 among others with IC50 values ranging from 0.06 to 1.0 μg/mL 77 , 78 Its mechanism studies revealed that multiple host antiviral pathways were involved, and as such 32 is widely known as a polypharmacology antiviral agent. 32 activated protein kinase R (PKR), which plays vital roles in innate immune system. The activation of PKR leads to the phosphorylation of eukaryotic initiation factor 2α, an important host restriction factor against viral replications. 79 HBV viral protein HBX was found to interact with host protein damage‐specific DNA‐binding protein 1 (DDB1) to promote transcription of covalently closed circular DNA (cccDNA) and degradation of a host restriction factor chromosomes 5/6 (Smc5/6). 80 32 was reported to disrupt the interaction between HBx and DDB1 to block the transcription from cccDNA. 81 In addition, 32 was also able to activate cellular antiviral response and induce the expression of a subset of IFN‐stimulated genes, especially interferon regulatory factor 1, 82 which is known to block the replication of a wide range of viruses. 83 Other host targeting mechanisms are also involved in the broad‐spectrum antiviral profiles of 32. 72 Since 32 has been safely used in clinic for many years, it has been advanced into clinical trials for the treatment of several viral infections. The most advanced one is the treatment of acute uncomplicated influenza with 32. In a phase 2b/3 trial, significant reductions in TCI50 viral titer and alleviation in symptoms were observed in 32 (600 mg, twice daily) group as compared to the placebo. No resistance was identified in the influenza virus collected from the patients receiving 32, and no adverse effect on humoral immune response was reported. 84 A large global phase 3 trial is being conducted. Several clinical trials of 32 for the treatment of other viral infections are also ongoing. In a pilot clinical trial of 32 for the treatment of chronic HBV, the serum HBV DNA of eight of nine patients became undetectable after 4 to 20 weeks of treatment with 32, and more importantly, three out of nine subjects became HBeAg negative, which is a rare outcome for current standard of care. This proof‐of‐concept study presented 32 as a very promising drug to achieve a HBV cure. 85 32 was also tested in clinical trials against HCV infections, and it showed very pronounced efficacy either as monotherapy or in combination with IFN‐α and RBV. For example, administration of 500 mg 32 twice a day for 48 weeks with 180 μg of IFN‐α once weekly from week 13 to 48 achieved a SVR of 79% in patients infected with chronic hepatitis C (CHC) genotype 4, as compared to 50% for IFN‐α and RBV. However, the development of 32 as an anti‐HCV drug was not further pursued due to the approval of several new DAAs, despite that 32 has much higher genetic barrier to resistance as compared to DAAs.
Figure 9.
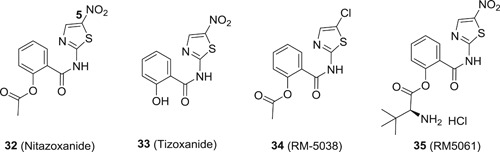
The chemical structures of nitazoxanide analogues
Interestingly, 32 is considered as a prodrug because it is rapidly hydrolyzed to tizoxanide (33) in the plasma with a half‐life of only 6 minutes. Compound 33 showed broad‐spectrum antiviral activity against a panel of viruses both in vitro and in vivo. To obtain new candidate compounds with improved potency and safety profiles, structural modifications were made to 32 primarily on the phenyl ring A and the thiazole moiety. It was revealed that electron‐withdrawing group such as nitro and chloro group at C‐5 position was favorable for the antiviral activity, and replacement of the thiazole ring with phenyl ring retained the activity. The structural optimization led to the identification of a candidate compound RM‐5038 (34) with a chloro group at the C‐5 position, which showed comparable activity to that of 32 with EC50 values in the submicromolar range. 34 was considered to be superior as compared to 32 due to the replacement of the potential cytotoxicity nitro group in 32. 32 also suffers from poor systemic exposure after oral administration, and it is primarily biodistributed in the gastric intestinal tract, and excreted via urine and faeces. For antiviral therapy, it is preferred to increase the systemic exposure of the active drug. For such purpose, amino acid based prodrugs for 33 were devised, 86 and they showed much improved aqueous solubility as compared to the parent drug. As such, prodrug 35 exhibited significantly improved PK profiles with oral bioavailability being around 20% in rats, as compared to 2.8% and 0 for 32 and 33, respectively. In addition, 35 also showed preferable safety profiles in laboratory animals, with a no observed adverse effect level being 25 mg/kg/day for a consecutive 28 days in beagle dogs, and it did not present any obvious toxicity in rats after a single oral dosage of 300 mg/kg, and only minor toxicity on central nervous system and respiratory system was observed at a single dosage of 1000 mg/kg. All these PK results present 35 as a very promising candidate compound for further development, and it is now undergoing phase I clinical trial.
3.3. α‐Glucosidase
α‐Glucosidase is an enzyme removing glucose units from N‐linked glycans attached to a nascent glycoprotein, which is essential for proper folding and functions of many glycoproteins. Most viral envelope glycoproteins contain N‐linked glycans, and α‐glucosidase (especially endoplasmic reticulum [ER] α‐glucosidase) is highly involved in their proper folding and maturation. Therefore, the inhibition of ER α‐glucosidase would yield broad‐spectrum antiviral activity. Indeed, ER α‐glucosidase inhibitors showed pronounced antiviral activity against a series of enveloped viruses both in vitro and in vivo including HIV, 87 HCV, 88 human coronavirus, 89 influenza A virus, 90 and DENV. 91 Although α‐glucosidase is critical in the proper folding of viral envelop glycoproteins, it is less important to host cells, and the host cells can well‐tolerate the complete shutdown of these ER α‐glucosidase. 92 Moreover, several glucosidase inhibitors are being used in clinic for treating type II diabetes and Gaucher disease. Consequently, targeting α‐glucosidase for antiviral therapy would not raise a red flag on toxicity issues.
To date, a wide range of α‐glucosidase inhibitors have been discovered, 93 among which the iminosugars are the most promising inhibitors as antiviral agents. It is widely accepted that the antiviral profiles of iminosugars is attributed to the inhibition of ER α‐glucosidases (I and II). The natural occurring iminosugars 1‐deoxynojirimycin (DNJ; 36; Figure 10) and castanospermine (37; Figure 10) has been used as starting points for further modifications. The modifications to 36 were primarily made at the amino position by introducing an alkyl chain. The n‐butylated DNJ 38 has been advanced to clinical trials for HIV treatment. In a phase II study, although 38 showed some efficacy on viraemia, it failed to maintain a serum concentration needed to inhibit HIV replication in vitro, 87 so the clinical trials of 38 for HIV treatment have been discontinued. The other DNJ analogue Mon‐DNJ (39) also showed broad‐spectrum antiviral activity, and it has recently been tested in human against DENV infection. In the phase I trial, 39 has been demonstrated to be well‐tolerated in human with no severe side effects observed after a single dosage of 1000 mg, and the PK data indicated a low interindividual variability and good linearity over a wide range of dosage (NCT02061358). In a recent study, 39 has been tested in a proof‐of‐concept non‐human primate trial against EBOV infections. However, 39 failed to yield any survival benefit to macaques infected with EBOV‐Makona, despite that 39 showed definitive antiviral activity against EBOV in vitro. 94 Celgosivir (40), a prodrug of 37, has also been tested in human against dengue fever. In a phase Ib, placebo‐controlled study, 40 failed to meet the primary end point in patients infected with uncomplicated dengue fever. However, in a mouse model study, it was confirmed that the dosing regime was crucial for the efficacy, 95 and therefore, a four‐time daily dosing regime is planned for a phase II trial (NCT02569827). 40 was also studied for the treatment of patients infected with genotype I HCV. In a phase II trial, 40 only showed a moderate antiviral effect as a monotherapy, but exhibited synergistic effects with IFN‐based therapies. 88 However, further development of 40 as anti‐HCV agents were discontinued due to inferior efficacy as compared with other approved DAAs.
Figure 10.
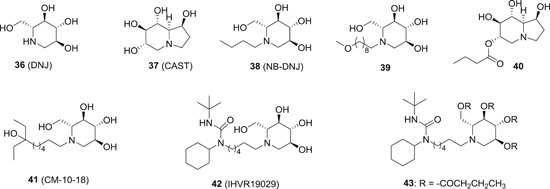
The chemical structures of iminosugars
Although the results from the clinical trials of 39 and 40 demonstrated the safety of iminosugars, they are all limited by poor PK profiles and insufficient efficacy. To address these limitations, further structural modifications were made to 36 by introducing various substituted alkyl sidechain, and several analogues were identified as potent antiviral activity against bovine viral diarrhea virus (BVDV), tacaribe virus, and DENV with EC50 values in the submicromolar range, which is hundreds fold more potent than 38. In addition, these compounds showed much superior PK profiles, especially compound 42, which had an oral bioavailability of 94%. Most importantly, they all demonstrated significant protective effects in both EBOV and BVDV infected mice models, and the in vivo glycan analysis also indicated significant ER α‐glucosidase suppression in the compounds treatment group. 96 It should be noted that the inhibition of other glucosidase such as intestinal glycosidase will lead to unwanted side effects. To avoid such side effects, a prodrug of compound 42 was designed by masking the free hydroxyl group by acylation. The tetrabutyrate prodrug 43 exhibited preferred stability toward simulated gastric and intestinal fluid, and yet was readily converted to the parent drug in the plasma and liver of mice. In a cell‐based assay, compound 43 showed inhibitory activity against EBOV with EC50 value of 15.6 μM, which is slightly less potent as compared with the parent compound (4.1 μM). Compound 43 also showed much improved overall drug exposure after either oral or intravenous administration to mice. 97 Another strategy to alleviate the side effects of iminosugars is to design more specific inhibitors toward ER α‐glucosidase. In 2016, the crystal structure of ER α‐glucosidase II in complex with 36 and 39 has been successfully resolved, which provided significant insights into the interactions between inhibitors and the active site of the enzyme, laying a firm foundation for the structure‐based drug design of more specific inhibitors. 98
3.4. Inosine‐5′‐monophosphate dehydrogenase
IMPDH is an enzyme catalyzing the conversion of inosine monophosphate (IMP) to xanthosine monophosphate, which is a critical step in the de novo biosynthesis of guanine nucleotides. 99 Inhibition of IMPDH will lead to the decrease in the intracellular guanosine‐5'‐triphosphate (GTP) level, which will disrupt the gene synthesis of both DNA and RNA virus, and thereby inhibit viral replication. Therefore, IMPDH inhibitors are expected to show broad‐spectrum antiviral activities. Indeed, IMPDH inhibitors exhibited broad‐spectrum antiviral activities against both DNA and RNA virus in vitro. 100 The approved non‐nucleoside‐based IMPDH inhibitors include mycophenolic acid (44), its prodrug mycophenolate mofetil (45) and mizoribine (46; Figure 11), and all of them were approved for prevention of organ transplant rejection, but not for antiviral therapy, Nevertheless, all these inhibitors were reported to inhibit the replication of a wide range of virus in vitro and even in vivo. For example, 44, an uncompetitive IMPDH inhibitor with respect to IMP and NAD+, is active against flaviviruses, paramyxoviruses, orthopoxviruses, avian reoviruses, and DENVs with EC50 values in low micromolar range, and its antiviral potency is closely correlated to the intracellular GTP levels, indicating the involvement of IMPDH inhibition in its antiviral mechanism. 101 , 102 However, one major limitation with the application of 44 is the presence of a phenolic hydroxyl group, which is prone to glycosylation for excretion, and thereby limits its efficacy. To overcome this limitation, a series of phenyloxazoles were developed by the Vertex group and others. The representative compound from this series is VX‐497 (47), which shows high affinity to IMPDH with a K i value of 10 nM. It shows potent antiviral activity against a wide range of DNA and RNA virus including HBV, HCMV, RSV, and murine encephalomyocarditis virus, HCV, ZIKV, and EBOV, 103 among others with IC50 values ranging from low micromolar to submicromolar levels, and its antiviral activity can be reversed by the addition of guanosine, indicating that its antiviral mechanism is resulted from IMPDH inhibition. Some other VX‐497 derivatives also exhibited similar antiviral profiles. 104 , 105 In light of the successful application of RBV in clinic for HCV treatment, as a more potent IMPDH inhibitor and antiviral agent as compared to RBV, 100 47 has been evaluated in clinic for the treatment of HCV in combination with IFN‐α. In a randomized, double‐blind, placebo‐controlled dose‐escalation trial, IFN‐α (3 MIU/week) plus 47 (100, 300 mg/8 hours) is well‐tolerated among genotype‐1 patients, and patients receiving 47 and IFN‐α demonstrated a greater reduction in mean HCV‐RNA level (−1.78 log vs −0.86 log, P = .037) as compared to the ones treated with IFN‐α only, 106 suggesting that 47 may have additive antiviral activity in combination with other agents. However, in a phase II triple combination study using 47, RBV, and IFN‐α to treat genotype 1 CHC pegylated IFN and RBV nonresponders, the addition of 47 to IFN‐alfa‐2a and RBV failed to increase the proportion of nonresponder patients with genotype 1 CHC achieving an SVR. 107 Due to the unsatisfactory results, the clinical trials of 47 for HCV treatment was discontinued. Although 47 is a very selective IMPDH inhibitor with potent anti‐HCV activity in vitro, it did not achieve similar magnitude of antiviral efficacy in patients. It is speculated that the level and supply of nucleotides varies in vitro and in vivo, and hence the dependence of viral replication on the de novo synthesis of nucleotides may be significantly different between in vitro and in vivo. Therefore, it is expected that IMPDH inhibitors can only show limited efficacy in vivo, albeit with potent antiviral activities in vitro. One may argue that the success of RBV in the treatment of HCV has established IMPDH as a viable antiviral target. It should be noted that RBV is only a weak IMPDH inhibitor with a K i value of around 250 nM, and it is widely accepted that other mechanisms are involved in the antiviral action of RBV. In addition, RBV has to be used in combination with IFN‐alpha or other anti‐HCV agents in clinic, and the monotherapy with RBV failed to yield any antiviral efficacy in patients. 108 Although quite a few other IMPDH inhibitors are under development, they are all intended for other indications. Consequently, special attentions should be paid to this issue in pursuit of IMPDH inhibitors as antiviral agents.
Figure 11.
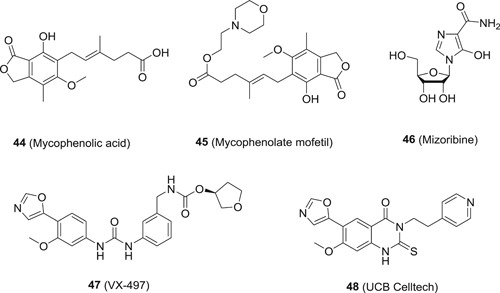
The chemical structures of a subset of inosine‐5′‐monophosphate dehydrogenase inhibitors
3.5. Kinases
Kinase represents a huge family of host proteins, and have been successfully targeted by a myriad of small‐molecule inhibitors for the treatment of cancers and inflammatory diseases in clinic. Mounting evidence have shown that viruses hijack a variety of human kinases throughout the entire viral life cycle to facilitate their replications, and some kinases are broadly required. 109 , 110 , 111 Therefore, it can be anticipated that the inhibition of such kinases would lead to the disruption of a broad spectrum of viral replications. Since tremendous kinase inhibitors have been approved in clinic for treating cancer and inflammatory diseases, and more are under development in the pipelines, increasing efforts have been devoted to repurposing approved kinase inhibitors as broad‐spectrum antiviral agents. In this section, only one type of kinases along with their respective inhibitors are picked for the discussion of related issues, and the selection of these examples is not based on the “importance” of the articles, but rather whether there are appropriate issues to discuss.
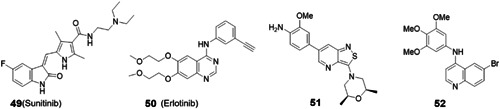
The numb‐associated kinases (NAKs) constitute a diverse family of Ser/Thr kinases with a broad range of cellular functions. NAKs have been found to play key roles in a diverse range of human diseases ranging from Parkinson's and prostate cancer to viral infections. Host kinases adaptor protein 2 (AP2)‐associated protein kinase 1 (AAK1) and cyclin G‐associated kinase (GAK), two kinases from this family, are found to regulate intracellular trafficking of a variety of viruses including DENV and EBOV. Two approved kinase inhibitors sunitinib (49) and erlotinib (50) were found to potently inhibit AAK1 and GAK, respectively, and both are reported to exhibit potent broad‐spectrum antiviral activity in vitro. In addition, the combination of 49 and 50 showed very pronounced protective effects against morbidity and mortality in DENV and EBOV infection mouse models. 112 A cocktail treatment containing 49 and 50 against EBOV infection is being investigated in clinic trial, but no clinical data have been disclosed yet (NCT02380625). GAK and AAK1 were also reported to regulate the binding of AP2M1 to HCV core protein, which is essential to the HCV assembly. Inhibitors of GAK and AAK1 disrupted the interaction between AP2M1 and core, and thus inhibited HCV replication with EC50 values ranging from 0.15 to 1.8 μM. 113 Although 49 and 50 exhibited potent inhibitory activity against GAK and AAK1, both of which are known as pan‐kinase inhibitors, and thus raise off‐target toxicity concerns when used as antiviral agents. In 2015, Herdewijn et al developed a series of highly selective inhibitors against GAK. The crystal structure of GAK in complex with one of these inhibitors has been resolved, showing that inhibitors as such behaved as classic type I adenosine triphosphate (ATP)‐competitive kinase inhibitors. These compounds showed pronounced anti‐HCV activity in vitro with EC50 in the low micromolar range. 114 Further structure optimization lead to the identification of compound 51 with a K d value of 8.9 nM, which is endowed with broad‐spectrum antiviral activity against a panel of viruses including DENV, EBOV, CHIKV with EC50 values in the low micromolar range. The mechanism of action studies confirmed that the inhibition of GAK is an important target underlying the broad‐spectrum antiviral activity of compound 51. 115 , 116 The other chemotype of GAK inhibitor is quin(az)oline, 117 , 118 yet their antiviral activity was not probed.
Many other kinases are also involved and play essential roles in different life cycle of various viruses, and their corresponding kinase inhibitors show broad‐spectrum antiviral activities against a wide range of viruses both in vitro and in vivo. 109 , 110 , 111 Since host kinase is not under the genetic control of virus, kinase inhibitors for antiviral treatment have much higher genetic barrier to resistance. For example, DENV developed resistance against the viral NS4B inhibitor SDM25N after eight passages, yet it remained sensitive to the treatment of 49 and 50 under the same conditions. 119 Despite with advantages of broad‐spectrum antiviral profiles and higher genetic barrier to resistance, there still remain concerns or limitations for repurposing kinase inhibitors as antiviral agents. Chiefly, toxicity is the major concern associated with kinase inhibitor, because most of kinases (if not all) play essential roles to regulate the cellular functions. However, it should be noted that, in most cases, the duration of antiviral therapy can be as short as several weeks or even several days, and hence the incidence of severe side effects can be significantly decreased. In addition, the potential toxicity can be further diminished by operating in a well‐defined therapeutic window. Since most of the kinase inhibitors are designed to target the ATP binding site, which is highly conserved among different kinase families, so nearly all kinase inhibitors possess cross inhibitory activity against other kinases, which could be problematic. First, targeting multiple kinase may result in off‐target side effects; second, it is challenging to understand the mechanism underlying the antiviral action of these inhibitors because multiple kinases are involved. However, one could also argue that pan‐kinase inhibition is favorable for antiviral therapy in that the off‐target kinase(s) may also contribute to the viral replication, and targeting multiple kinases simultaneously may result in not only synergetic antiviral activity but also high genetic barrier to resistance as well. Altogether, targeting kinases represent a promising strategy for the development of antiviral agents, especially the ones with broad‐spectrum antiviral profiles and high genetic barrier to resistance.
3.6. Sodium taurocholate cotransporting polypeptide
Sodium taurocholate cotransporting polypeptide (NTCP) is expressed on the hepatic basolateral membranes specifically and functions as a cotransporter for bile acids and sodium ions. Recently, NTCP has been identified as HBV/HDV infection receptor via interacting with HBV large surface protein, and the silence of NTCP inhibited HBV and HDV infections. 120 Therefore, the inhibition of NTCP would block the entry of HBV/HDV virus, and the subsequent formation of the persistent viral reservoir: cccDNA will also be halted, which cannot be achieved by current therapy. 121 To date, a series of chemotypes have been identified as NTCP inhibitors to inhibit the infection of HBV/HDV. The most advanced inhibitor is Myrcludex B (MyrB), which is a synthetic N‐acylated lipopeptide pre‐S1. It just completed phase 2 clinical trial for the treatment of patients coinfected with HBV and HDV. The results showed that MyrB is well‐tolerated with bile acid increase as the only abnormal observation, and the bile acid level dropped to baseline after follow‐up week 1. The primary endpoint (HDV RNA reduction by 2log or negative) was achieved by 46.4%, 46.8%, 76.6%, and 3.3% of patients in arms A (2 mg), B (5 mg), C (10 mg), and D (tenofovir disoproxil fumarate [TDF]), indicating a dose‐dependent antiviral efficacy of MyrB. The mean liver stiffness values were also significantly declined in all MyrB groups but not the control TDF group, 122 demonstrating superiority by targeting NTCP (Figure 12).
Figure 12.
The chemical structures of a subset of GAK and AAK1 inhibitors. AAAK1, AP2‐associated protein kinase 1; GAK, cyclin G‐associated kinase
The other discovered NTCP inhibitors include FDA approved drugs (ie, CsA, 123 ezetimibe, and ritonavir, 124 etc), fasiglifam, 125 oxysterol, 126 vanitaracin A, 127 NTI007, 128 and among others. 129 The inhibition of NTCP by these inhibitors normally will result in the loss of transporter function of NTCP, leading to the inhibition of bile acid uptake, which might cause unwanted adverse effects. Interestingly, Shimura et al 130 identified several CsA derivatives with anti‐HBV activity in vitro via direct interaction with NTCP to inhibit viral attachment. These compounds inhibited multiple HBV genotypes including one clinically relevant nucleoside analog‐resistant HBV isolate. Importantly, they did not compromise the transporter function of NTCP. Two analogs SCY446 (56) and SCY450 (57; Figure 13) also did not show meaningful inhibition against CN, and therefore, these two compounds did not show any unwanted immunosuppressive effects. Taken together, these results showed that the inhibition of viral attachment via NTCP can be functionally separated from the bile acid uptake, and future efforts should be dedicated to new NTCP inhibitors with transporter function retained to eliminate unwanted adverse effects.
Figure 13.
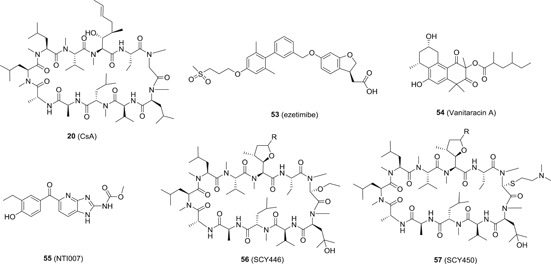
The chemical structures of a subset of sodium taurocholate cotransporting polypeptide inhibitors. The R group in 56 and 57 was not specified in the original paper 130
3.7. Farnesoid X receptor
Farnesoid X receptor (FXR) belongs to the nuclear receptor superfamily and plays regulatory roles in the metabolism of bile acid, lipid, and glucose. Recently, it was identified as a proviral host factor for HBV virus. 131 Silence of FXR by small hairpin RNA resulted in significant decrease in the levels of cccDNA, pregenomic‐ and precore‐RNAs, secreted relaxed circular DNA (rcDNA), and HBsAg by 40%‐70%, and the same effect was observed with the treatment of a FXR agonist GW4064 (58; Figure 14). Importantly, in C3H/HeN adult mice infected with a recombinant AAV2/8‐HBV vector, GW4064 (50 mg/kg/d) significantly suppressed the rcDNA and HBsAg titers (mean rcDNA variation: −0.52 and −0.93log10; mean HBsAg variation −0.89 vs −0.73log10, at day 21 and 28, respectively). 132 Interestingly, 58 together with other FXR agonists such as WAY362450 (59), fexaramine (60), and chenodeoxycholic acid (61) also showed inhibitory effect against HCV replications in Huh7.5 cells with IC50 values ranging from 0.75 to 7.03 μM. The mechanism of action studies showed that compound 58's anti‐HCV activity is FXR‐dependent. It has been reported that FXR agonist downregulated the expression of SRIB, which is a coreceptor for HCV entrance. As expected, the treatment of 92 also dose‐dependently decreased the level of SR‐BI. In addition, 58 also presented synergistic anti‐HCV activity with other approved DAAs, and it remained sensitive against other DAA‐resistant HCV mutations, suggesting the advantages of targeting host factors. 133 Encouragingly, one specific FXR agonist EYP001 (structure not disclosed) has successfully entered clinical trial for HBV treatment, and the data from phase I trial showed that EYP001 is very well‐tolerated in humans with no treatment‐related discontinuations or severe side effects. The PK of EYP001 was linear up to 500 mg with T max and T 1/2 being 2.3‐2.5 and 1.4‐2.4 hours, respectively. 134 The phase II trial of EYP001 is ongoing.
Figure 14.
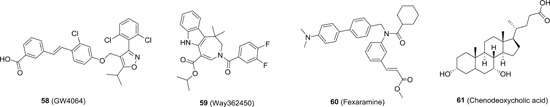
The chemical structures of representative farnesoid X receptor agonists
3.8. Diacylglycerol acyltransferases‐1
Diacylglycerol acyltransferases (DGATs) are the key enzymes catalyzing the biosynthesis of endogenous triglycerides, which are necessary for the biogenesis of lipid droplets in the liver. It has been reported that lipid droplets are the major site for HCV particle assembly and production, and DGATs, especially DGAT‐1, play vital roles during HCV infection. DGAT‐1 forms a complex with nonstructural protein 5A (NS5A) and core protein to enhance the interaction of the latter two, and the trafficking of NS5A to lipid droplets is also highly dependent on DGAT‐1's activity. 135 The silence of DGAT‐1 significantly impaired HCV entry. 136 Therefore, DGAT‐1 could serve as a viable host target for anti‐HCV agents development. One specific DGAT‐1 inhibitor pradigastat (62; Figure 15) developed by Novartis significantly decreased the level of HCV RNA in cell supernatant at a concentration of 10 μM, indicating that compound 62 inhibited the assembly or release of virions. Since compound 62 is in clinical development for the treatment of dyslipidemia, and it has demonstrated convinced efficacy and safety profiles. 137 Therefore, it was directly tested in patients with genotype 1 or 3 HCV infections for safety and efficacy. However, disappointedly, 14 days of treatment with compound 62 failed to afford any significant deduction in serum HCV RNA levels in either GT1 or GT3 patients, and thus the trial was terminated. 138 Since the PK studies showed that the predicted concentration of compound 62 in liver is approximately 10 to 20 μM based on the plasma concentration on day 8 and 14, which is much higher than the concentration needed for DGAT‐1 inhibition (IC50: 66 nM), so the lack of efficacy is not due to PK problem. The other possibility is that HCV virus hijacked other compensate pathway to facilitate the assembly and release while DGAT‐1 activity is inhibited in vivo, or DGAT‐1 plays a nonenzymatic roles during HCV replication, so the inhibition of its enzymatic activity would not yield any inhibitory activity against HCV replication.
Figure 15.
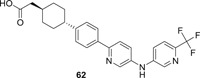
The chemical structure of pradigastat
4. HOST TARGETS IN PRECLINICAL STUDIES
4.1. Heat shock protein 90
HSP90 is a highly conserved chaperone protein that assists the maturation of its clientele proteins. There are four HSP90 isoforms, namely HSP90α, HSP90β, TRAP1, and GRP94, each of which has distinct subcellular distribution and functions. Recently, mounting evidence demonstrate that HSP90 is highly involved in all life cycles of various viral replications. It plays vital roles at different viral replication step by interacting with different viral proteins. At the viral entry and intracellular trafficking stage, HSP90 is reported to be critical for the intracellular translocation of viral proteins as well as other host factors critical for viral replications. For example, HSP90 is required for the nuclear translocation of EBV and HSV‐1 DNA polymerase and 139 , 140 RNA‐dependent RNA polymerase of influenza virus. 141 To facilitate viral gene expression, HSP90 also activate several host signaling pathways. For instance, HSP90 is upregulated to activate Akt and nuclear factor κB for viral gene expression upon HCMV infection. 142 As a chaperone protein, HSP90 is indispensable for the maturation, accurate folding, and maintenance of stability of various proteins including viral proteins. For example, HSP90 facilitates the accurate folding of NS5A of HCV virus in the replication complex to promote viral replication. 143 It can also help to maintain the stability of various viral proteins, including polymerases of VSV, 144 CHIKV, 145 and RSV, 146 and ribonucleoprotein complex 147 to facilitate viral genome replications. In addition, HSP90 is also involved in the formation of viral capsid. Hsp90 can maintain the stability of capsid precursor P1 protein of poliovirus, 147 and it can also increase affinity between core protein dimers to facilitate HBV capsid formation. 148 In summary, HSP90 plays indispensable roles at almost all life cycles of various virus. Therefore, it can be anticipated that HSP90 inhibitors would present broad‐spectrum antiviral activities. Currently, there are up to 17 HSP90 inhibitors in clinical trials for cancer treatment, and most of them are N‐terminal ATP binding domain inhibitors. 149 This type of inhibitors competitively bind to the ATP binding site, and subsequently abolish the hydrolysis of ATP and HSP90's functions. Shown in Figure 9 are some representative HSP90 inhibitors, and the most famous one among this series is geldanamycin (GA; 63; Figure 16). 63 was first isolated from a fermentation broth of Streptomyces hygroscopicus in 1970, and was identified as the first HSP90 inhibitor binding to the N‐terminal ATP binding pocket. It was initially evaluated as an anticancer agent due to the important roles of HSP90 in multiple cancers. Because HSP90 also plays indispensable roles at different life cycle of viral replications, 63 was reported to exhibit antiviral activities against a wide range of viral infections, including HSV‐2, 150 HSV‐1, 151 HCMV, 142 EBOV, 152 HIV‐1, 153 and influenza virus, 154 among others. However, since 63 possesses several inherent limitations, such as poor water solubility and severe hepatotoxicity, structural modifications to 63 have been carried out. The modification was primarily made at the C‐17 position by replacing the methoxyl group with various substituted amines, such as 17‐AAG (64) and 17‐DMAG (65). These two analogues showed improved PK profiles, especially 65, which presented much better water solubility, stability, bioavailability, and safety. Both compounds have entered clinical trials for cancer treatment, but neither of them has made it to market for unknown reasons. 64 was shown to inhibit RSV replication in an in vivo model of well‐differentiated primary human airway epithelial cells at concentration as low as 1.9 nM. Moreover, despite extensive replication in the presence of 64, no resistance against 64 was observed even after 17 passages, which is in direct contrast to previously reported RSV inhibitor. 155 Similarly, 63 was reported to inhibit the replication of poliovirus both in vitro and in vivo, and no resistance against 63 was detected after 10 passages, despite that poliovirus is feature with rapid replication rate and high mutation frequency. 156 These results showcased the advantages of HTAs over DAAs in regard to drug resistance.
Figure 16.
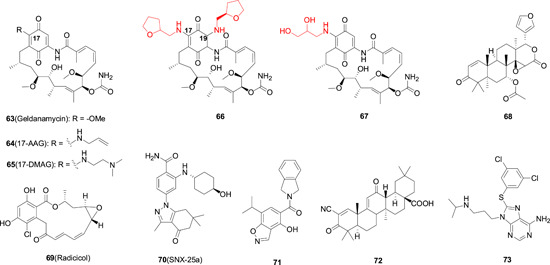
The chemical structures of representative HSP90 inhibitors. HSP, heat shock protein [Color figure can be viewed at wileyonlinelibrary.com]
In 2011, Shan et al 157 synthesized several GA analogues with substituent at C‐17 position and/or C‐19 position, 157 and found that most of the GA analogues with substituent at C‐17 position showed similar magnitude of anti‐HCV activity as compared to 63 with IC50s in the submicromolar range. However, these analogues also showed high toxicity toward the GS4.3 HCV replicon cells with SI values less than 10. One analogue 66 with disubstitution at both C‐17 and C‐19 position exhibited much less cytotoxicity with a CC50 value of 52 μM, but the anti‐HCV activity also decreased significantly with an IC50 value of 58 μM. The same group also synthesized more GA analogues with hydrophilic sidechain at C‐17 position and tested their broad‐spectrum antiviral activities against HBV, HCV, HIV‐1, HSV, Cox, and HCMV in vitro. 158 The results showed that most of the analogues presented potent antiviral activity against all the virus strains tested, albeit with less potency as compared to 63, yet some derivatives were more biocompatible in terms of the cytotoxicity toward the host cells, and the acute toxicity in mice of several selected GA analogues (LD50: 122‐295 mg/kg) was much lower as compared to 63 (LD50: 17 mg/kg). In addition, one representative GA analogue 67 was evaluated in a DHBV infection duckling model, and the results showed that both compound 67 and positive drug 3TC lead to significant decrease in serum HBV DNA level. More importantly, at day 3 after drug withdraw, the serum DHBV DNA level of the 3TC group rebounded to its pretreatment level. However, no rebound was observed with the treatment of compound 67 after drug withdraw, demonstrating the advantage of HTA over DAAs.
Radicicol (69) is another natural occurring HSP90 inhibitor, which was isolated from Monosporium bonorden. 69 was shown to inhibit the replication of paramyxoviruses SV5, HPIV‐2, HPIV‐3, SV41, and La Crosse bunyavirus by destabilizing the newly synthesized L protein (the large subunit of the VSV polymerase) via HSP90 inhibition. Gedunin (68), a noncompetitive HSP90 inhibitor versus ATP was also reported to inhibit DENV in vitro with an EC50 value of 10 μM. SNX‐25a (70) also significantly suppressed the HSV‐1 virus titers, and inhibited nucleocapsid egress from the nucleus. 159 Many other HSP90 inhibitors have also shown definitive antiviral activities against a panel of virus strains. 160 , 161 Although around 17 HSP90 inhibitors are under development in clinical trials, they are all for cancer indication and none is being tested for antiviral purpose albeit with well‐established preclinical results. The major concern associated with the application of HSP90 inhibitors for antiviral treatment is the toxicity. Most of the HSP90 inhibitors in clinical trials are accompanied with some severe side effects including cardiotoxicity, gastrointestinal toxicity, and/or ocular toxicity amongst other side effects. 162 , 163 , 164 It is now widely accepted that these unwanted toxicities are mainly attributed to pan‐inhibition of HSP90 isoforms. For example, the observed cardiotoxicity mainly resulted from the inhibition HSP90α, which is responsible for the maturation of hERG channel. 165 Therefore, HSP90 isoform‐selective inhibitors are expected to side‐step some detrimental toxicity observed with pan‐inhibitors. Indeed, several HSP90 isoform‐selective inhibitors against Grp94 166 , 167 (72‐73) and HSP90β 168 (71) have been successfully developed with much better safety profiles. These inhibitors were devised for cancer treatment, and their antiviral profiles were not investigated. Very recently, two GRP94 selective inhibitors 72 and 73 were found to inhibit the replication of DENV and ZIKV with IC50 values in the low nanomolar range, and it was further confirmed that GRP94 was essential for the replication of DENV and ZIKV. 169 Therefore, it can be deduced that GRP94 might be indispensable for the replications of other viruses as well. It can be envisioned that antiviral therapy with HSP90 inhibitors can benefit from HSP90 isoform inhibition, and isoform‐selective HSp90 inhibitors could be a new strategy for the development of broad‐spectrum antiviral agents.
4.2. Heat shock protein 70
HSP70 is a family of chaperones with diverse cellular functions including protein folding, protein transportation, breakdown of unstable proteins, and removal of protein complexes. There are generally two types of HSP70 protein, namely the inducible (ie, HSP70i) and the constitutive (Hsc70) isoforms, both of which are shown to play an integral role in viral replications. For example, HSP70 is indispensable for the folding of several viral proteins, including HCV NS5A, 170 the L and envelop protein of HBV, 171 G protein of VSV, 172 the envelop protein of HIV, 173 among others. Similarly, Hsc70 is also required for the replication of a wide range of viruses, such as influenza virus, 174 rotavirus, 175 polyomavirus, 176 HCV, 177 among others. Consequently, HSP70 inhibitors are expected to show broad‐spectrum antiviral activities. As compared to HSP90 inhibitors, Hsp70 inhibitors are under developed, and the development of Hsp70 inhibitors is faced with several challenges: HSP70 has a high affinity toward ADP, and the conformation state of HSP70 makes the ATP binding site less accessible. 178 Therefore, it is very hard to design inhibitors targeting the ATP binding site. 179 Due to high sequence similarity between HSP70i and Hsc70, most of the available inhibitors are unable to discriminate between those two isoforms. Shown in Figure 10 are a selected subset of HSP70 inhibitors, and all those inhibitors are intended for cancer indication. MKT‐077 (76; Figure 17) is the most advanced one, which has entered phase I clinical trial for cancer treatment. However, its clinical trial was halted due to severe renal dysfunction observed in patients. 180 , 181 76 inhibits HSP70 by disrupting its interaction with nucleotide exchange factors, which promote the release of Hsp70‐bound substrates. 76 along with its analogue JG‐18 (77) and JG‐40 (80) were reported to inhibit the propagation of DENV, and no toxicity toward the host cells was observed at concentrations inhibiting viral replication. Interestingly, the other two structural analog JG‐19 (78) and JG‐28 (79) did not inhibit viral propagation at the same concentration (1 μM), albeit with highly structural similarity toward 80. 182 In addition, 80 suppressed the inflammation response associated with dengue fever. More importantly, no resistance to 80 was detected even after 10 passages, while significant resistance was observed with a viral NS5 inhibitor under the same conditions, highlighting the advantages of HTAs over DAAs in terms of resistance selection. 80 and its analogues have also exhibited broad‐spectrum antiviral profiles with inhibitory activity against HCV, 183 ZIKV, 184 West Nile, and Japanese encephalitis viruses. 182 The HSP70i is at relatively low level in unstressed cell, while its expression is significantly induced upon viral infections, indicating an integral role of HSP70i in viral infections. To achieve HSP70 isoform inhibition, several inhibitors targeting an allosteric site on HSP70 were elegantly designed with selectivity toward Hsp70i. 185 , 186 HS‐72 (81) is one of such inhibitors, and it was shown to inhibit the entry of DENV mainly by disrupting the association of Hsp70i with the DENV receptor complex. 187 In comparison, Hsc70 selective inhibitors have not been precedented. However, in contrast to direct inhibition, Hsc70 downregulators have been reported with broad‐spectrum antiviral activities. For example, IMB‐DM122 (82), an analogue derived from natural compound oxymatrine, was discovered as a Hsc70 downregulator. The half‐life of Hsc70 messenger RNA (mRNA) was reduced by 78% followed by the treatment of 82 (500 μg/mL) in Huh7.5 cells. As such, 82 is effective to inhibit HCV replication at a concentration of 125 μg/mL. In addition, 82 is well‐tolerated in mice with no obvious toxicity at a single dosage of 1000 mg/kg (intraperitoneal [IP]). 188 Intensive structural modifications were further made to the sidechain and/or substituent at the nitrogen position of 82, yielding several analogues with IC50s against HCV in the low micromolar range (ie, 83). 189 , 190 , 191 The oxymatrine analogues were also shown to inhibit both wild‐type and lamivudine‐resistant HBV infection via downregulating Hsc70 expression with excellent safety profile in mice (LD50 = 750 mg/kg, oral administration). 192 The activity against other virus was also reported with oxymatrine analogues. 193 , 194 The other naturally occurring Hsc70 downregulator is lycorine (84). 121 It was reported to decrease the Hsc70 mRNA level does‐dependently with definitive anti‐HCV activity in vitro. The structural optimization led to several derivatives with IC50 values ranging from low micromolar to submicromolar levels. The SAR study showed that the double bond between C3 and C4 and the basic nitrogen at N‐5 position are crucial to the anti‐HCV activity. 195 Interestingly, its naturally occurring cousin lycoricidine (85) possessed much more potent inhibitory effect against HCV with an EC50 value of 0.55 nM, and the mechanism of action studies revealed that downregulation of Hsc70 expression at least partially account for the observed antiviral activity. 196 Although these compounds are confirmed to downregulate Hsc70 to exert their antiviral efficacy, their respective physical binding protein(s) remain to be clarified, but it can be deduced that their physical binding partner(s) must be host factor(s).
Figure 17.
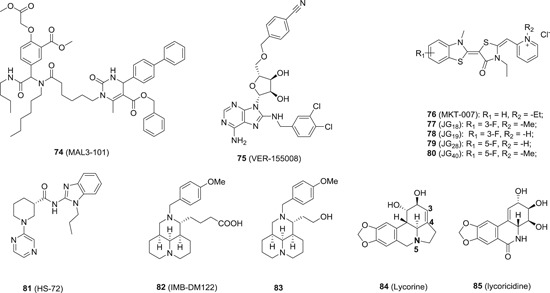
The chemical structures of HSP70 inhibitors and downregulators, HSP, heat shock protein
4.3. Apolipoprotein B mRNA‐editing enzyme catalytic polypeptide‐like 3G
Host cells have developed an innate immune system to act as the first‐line defense against invading virus. In addition, a series of intracellular restriction factors are also expressed endogenously to counteract viral infections, 197 and apolipoprotein B mRNA‐editing enzyme catalytic polypeptide‐like 3G (APOBEC3G, A3G) is one such factor, which possesses the capacity to restrict the replication of a panel of viruses including HIV‐1, HBV, HCV, and EV71 among others via different mechanisms. A3G is known as a cytidine deaminase, catalyzing the irreversible hydrolytic deamination of cytidine and deoxycytidine to uridine and deoxyuridine, respectively. Its anti‐HIV‐1 activity is closely associated with its deaminase function, by which consistent mutation from dG to dA in the positive strand of viral DNA is frequently observed. 198 The high level of G to A mutation is attributed to the C to U transitions occurred on the complementary negative‐strand DNA by the cytidine deaminase activity of A3G. Introduction of deoxyuracils in the proviral DNA can ultimately lead to its cleavage and degradation by specific AP endonucleases. Additionally, high percentages of G to A mutation in the HIV‐1 genome will result in the loss of functions of viral proteins. 199 Interestingly, the antiviral activity of A3G against other virus is independent of its deaminase activity. For example, A3G is reported to inhibit HCV replications by binding to the C‐terminus of HCV NS3 protein to reduce its helicase activity, which is essential for HCV replication. 200 , 201 A3G was also reported to inhibit HBV replication both in vitro and in vivo, but its anti‐HBV mechanism is not related to its deaminase activity, because A3G did not yield any G to A hypermutation within the HBV genome, and the catalytically inactive A3G derivatives were able to result in the same magnitude of HBV inhibition as compared with its wild‐type counterpart. 202 , 203
Although host cells are endowed with antiviral restriction factors such as A3G, viruses are so cunning that they developed their own mechanism to evade host innate immune system. For example, HIV‐1 expresses a viral protein Vif, which binds to A3G and form an ubiquitin ligase complex with cullin 5 (CUL5), elongin B/C (ELOB/C), and CBFβ, leading to the ubiquitination and subsequent proteasomal degradation of A3G. 204 In the cases of HBV and HCV infections, endogenous A3G is also eliminated by some unknown mechanisms. Therefore, agents either disrupting the formation of ubiquitin complex, stabilizing A3G or inducing the expression of A3G are expected to demonstrate antiviral activity. In 2008, Chen et al identified two compounds IMB‐26 (86; Figure 18) and IMB‐35 (87), which directly binds to A3G and disrupted its interaction with Vif, and therefore rescue A3G from Vif‐mediated degradation. Both compounds showed A3G‐dependent anti‐HIV‐1 activity in nonpermissive H9 cells with EC50 values in the low nanomolar range. Moreover, no cytotoxicity was observed at a concentration of around 4 μM, indicating a therapeutic index of greater than 200, and the LD50 value for 87 is as high as greater than 1000 mg/kg (IP). 205 86 also showed strong antiviral activity against HCV in vitro via stabilizing intracellular A3G. 206 Due to the presence of a bromo substituent at the α position of amide, which is highly reactive as a alkylation agent, structural modifications were made to 86, and it turned out that the bromo is not essential for the antiviral activities. Most of the synthesized derivatives showed potent antiviral activity against HCV and EV71 virus with IC50 values ranging from 0.57 to 80 μM. 207 , 208 Since the amide linkage is liable to hydrolysis, a methylene group was inserted between the carbonyl and amino group. However, such modification resulted in complete loss of activity. 207 To further address this issue, a ring formation strategy was employed to generate compound with 2‐aryl‐isoindolin‐1‐ones scaffold (88). These analogues showed much‐enhanced stability toward hydrolysis, and most importantly, the anti‐EV71 activity was retained with IC50 in the low micromolar range. 209 Interestingly, the derivatives of 86 also showed definitive antiviral activity against both wild‐type and Tamiflu resistant influenza virus in vitro with IC50 values in the low micromolar range, 210 despite the fact that A3G does not yield any inhibitory activity against influenza virus replication, 211 indicating that other A3G‐independent antiviral mechanism must be involved.
Figure 18.
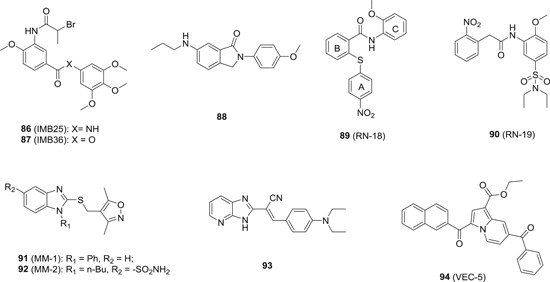
The chemical structures of A3G stabilizer
In 2009, Rana et al identified two Vif inhibitors RN‐18 (89) and RN19 (90) by a fluorescence‐labeled A3G recovery assay. Both compounds inhibited HIV‐1 replication only in A3G positive cells with an IC50 value of 4.5 and 10 μM, respectively, and the IC50 went up to over 100 μM in A3G negative cells. In addition, the treatment of 89 not only increased the A3G level, but also led to the degradation of Vif. 212 The SAR studies based on the scaffold of 89 revealed that the amide of sulfonamide linkage between ring B and C is essential for the anti‐HIV activity, and variations on ring A were well‐tolerated with both electron donating and withdrawing groups. 213 , 214 By a very similar assay, two new Vif inhibitors 91 and 92 were identified to recover A3G expression in the presence of HIV‐1 Vif, and both inhibited viral replication via A3G pathway. The immunoprecipitation experiment confirmed that neither compound disrupted the interaction between Vif and A3G, and thus inhibited the ubiquitination of A3G. In addition, neither compound impaired 20S proteasome activity, suggesting that recovery of A3G is not simply caused by inhibiting the general proteasome activities. The specific mechanism(s) underlying the recovery of A3G need further clarification. 215 Both compounds also showed high cytotoxicity toward 293T cells with IC50 values being 30 and 50 μM, respectively, necessitating further structural optimization. Benzimidazole derivatives were also identified as potent anti‐HIV‐1 agents via stabilizing A3G. 216 The structural modifications led to the identification of compound 93 with an anti‐HIV‐1 IC50 value of 3.4 nM in H9 cells, and no toxicity in mice was observed in 2 weeks after the IP injection of 1000 mg/kg of compound 93. The mechanism of action studies showed that this type of compounds stabilized A3G via disrupting the interaction between Vif and ELOC, which is essential for the Vif‐mediated A3G degradation. 217
In 2012, Zuo et al 218 identified a small‐molecule inhibitor VEC‐5 (94) of Vif‐ELOC interaction by virtual screening. The inhibition of the Vif and ELOC interaction will disrupt the formation of ubiquitin complex, and thus rescue the A3G from Vif‐mediated degradation. Indeed, treatment of 94 resulted in the elevation of A3G level, and reduced viral infectivity. 94 inhibited viral replications only in A3G positive cells with an IC50 value of 24.48 μM, confirming its A3G‐dependent antiviral mechanism. The coimmunoprecipitation and computational analysis suggested that 94 directly bound to ELOC at the interface of ELOC‐Vif interaction to suppress Vif activity. The SAR studies concluded that both the naphthoyl and benzoyl group were essential for the activity, and modifications made to the ester group were well‐tolerated. 219
The abovementioned antiviral strategy is based on the recovery of A3G to inhibit viral infection. Very interestingly, it was reasonably argued that HIV‐1 virus might actually benefit from sub‐lethal levels of A3G‐induced mutation, which might contribute to the high mutation rate of HIV‐1 virus and the capacity of the virus to escape innate immune system and evolve resistance against antiretroviral drugs. Therefore, current antiretroviral treatment regime may benefit from the inhibition of A3G deaminase activity. 220 However, a proof of concept study needs to be established to support such hypothesis.
4.4. Bone marrow stromal cell antigen 2
Bone marrow stromal cell antigen 2 (BST‐2), also known as tetherin or CD317, is a potent IFN‐induced antiviral molecule inhibiting the release of various enveloped virus particles from infected cells, including filoviruses, 221 , 222 arenaviruses, 223 paramyxovirus, 223 γ‐herpesviruses, 224 and among others. 225 The broad‐spectrum antiviral profiles of BST‐2 is attributed to its ability to target a common feature shared by these viruses: host cell‐derived lipid bilayer. Since the target of the BST‐2 is not encoded by viral genome, so the virus cannot mutate the viral protein to evade the antiviral activity of BST‐2. However, viruses have evolved other mechanisms to counteract the action of BST‐2 by expressing different viral proteins to physically bind to BST‐2, and thereby block the interaction between BST‐2 and its target. Such proteins include HIV‐1 Vpu, HIV‐2 Env, SIV Env, SIV Nef, KSHV K5, and the EBOV glycoprotein. For example, HIV‐1 Vpu, a type I integral membrane protein, can either induce the ubiquitination of BST‐2 for degradation or downregulate the cell‐surface BST‐2 level by sequestering the de novo synthesized BST‐2 away from the plasma membrane to counteract its antiviral activity. 226 Therefore, it can be envisioned that disruption of the interaction between BST‐2 and these viral proteins or stabilization of BST‐2 could yield antiviral agents. Zhang et al 227 developed a cell‐based high‐throughput enzyme‐linked immunosorbent assay for the quantification of cell‐surface BST‐2 levels, 227 and a lead compound IMB‐LA (95; Figure 19) was identified to inhibit Vpu‐mediated BST‐2 degradation and recover the expression of BST‐2 at the cell surface. 228 This compound inhibited both the HIV infection and release in a BST‐2 dependent fashion. The mechanism studies showed that compound 95 did not inhibit the interaction between BST‐2 and Vpu, but it blocked the sorting of BST‐2 into lysosomes for degradation. By the same assay, 2‐thio‐6‐azauridine (96) was also identified as a promising agent to inhibit Vpu‐mediated BST‐2 downregulation. Compound 96 showed a BST‐2 dependent anti‐HIV activity with IC50 values being 0.22 and 4.42 μM in BST‐2 positive and negative cells, respectively. 229 The mechanism study revealed that compound 96 did not disrupt the interaction between Vpu and BST‐2, but inhibited Vpu mediated ubiquitination of BST‐2.
Figure 19.
The chemical structures of two known Vpu‐BST‐2 inhibitors. BST‐2, bone marrow stromal cell antigen 2
Since Vpu binds to BST‐2 via interactions between their trans‐membrane domains, Cen et al designed a peptide BST2‐TM‐P1 consisting a seven residues EDGDKRC before TMD and a motif of GGKKKK for solubility improvement. Such a peptide successfully blocked the interaction between BST‐2 and Vpu, and restored the expression of BST‐2 at the plasma membrane. In addition, this peptide also inhibited HIV‐1 replication in Hela cells in a BST‐2 dependent manner with an IC50 value of around 10 μM. 230 Altogether, these examples validated the feasibility of targeting Vpu‐BST‐2 axis as a novel anti‐HIV strategy, and intensive medicinal chemistry efforts are needed to either improve potency or druggability of these lead compounds.
4.5. DEAD‐box polypeptide 3
Human DEAD‐box polypeptide 3, an ATPase/RNA helicase, was founded to be involved in a variety of cellular biogenesis process, including cellular differentiation, cell‐cycle regulation, apoptosis, and cell survival. Recently, it has also been identified as an essential host factors for the replication of both DNA and RNA virus, including HIV, 231 HCV, 232 DENV, 233 and WNV, 234 among others. Although the exact mechanism(s) DDX‐3 employed to facilitate viral replication is yet to be elucidated, DDX‐3 has been proposed as a very promising host target for broad‐spectrum antiviral agent development.
The crystal structure of DDX‐3 in complex with AMP has been resolved, 235 and a pharmacophore has been proposed for virtual screening, 236 which greatly facilitated the design of specific inhibitors targeting the ATP binding site of DDX‐3. To date, a number of DDX‐3 inhibitors targeting the ATP‐binding site have been devised with potency (K i) in the submicromolar range, 237 , 238 , 239 , 240 and some of them have shown definitive anti‐HIV activity in vitro, establishing the proof of concept of targeting DDX‐3 for viral infection treatment. However, inhibitors targeting ATP‐binding site have one major inherent limitation: they are likely to show cross inhibitory activity against other kinases with ATP‐binding site. Although kinases have been proposed as viable targets for broad‐spectrum antiviral agents development, targeting DDX‐3 and multiple kinases simultaneously may lead to some unwanted adverse effects. To side‐step this limitation, other inhibitors were designed to target the RNA binding site of DDX‐3. In 2012, Botta et al discovered the first inhibitor (97 and 98; Figure 20) targeting the RNA binding site of DDX‐3. Further structural modification led to the identification of more potent inhibitors with IC50 values in the low micromolar range. Such inhibitors were able to block both the helicase and ATPase activity of DDX‐3, and thereby inhibited viral replication in HIV‐1 infected peripheral blood mononuclear cell (PBMC) cells with an EC50 value of 10 μM. 241 In a follow‐up study, the structure‐based drug design strategy was employed to devise new inhibitors with more potent activity. The structural modification was mainly made to the two phenyl rings to form more interactions with the other two unexplored binding pockets within the RNA‐binding site, and one inhibitor (99) with submicromolar activity were successfully identified. Compound 99 showed broad‐spectrum antiviral activity against a panel of virus including HIV, HCV, WNV, and DENV with IC50 values ranging from 0.97 to 16.5 μM. In addition, compound 99 retained the same magnitude of inhibitory activity against several clinically relevant mutant HIV‐1 virus, suggesting the advantage of targeting DDX‐3. The preliminary in vivo studies in rats indicated a good safety profile with no obvious side effects and acceptable PK profiles with a half‐life of around 3 hours for compound 99. Altogether, DDX‐3 is a very promising target for broad‐spectrum antiviral drug development, and compound 99 represents a good start point for further in‐depth investigation.
Figure 20.
The chemical structures of DEAD‐box polypeptide 3 inhibitors
4.6. Neural precursor cell expressed developmentally downregulated protein 4‐PPxY
Enveloped RNA virus such as filoviruses and rhabdoviruses are highly pathogenic. These viruses express various matrix proteins to facilitate virus assembly and egress. Although these viruses will express different types of matrix proteins, they all share one common motif referred as Late (l) budding domain consisting of core consensus amino acid motifs such as PPxY, YxxL, P(T/S)AP, or FPIV (where x could be any amino acid). It has been confirmed that the L domain interacts with host protein to facilitate viral egress and spread. For example, the PPxY motif within the matrix proteins of various RNA virus formed a complex with the WW domain of E3 ubiquitin ligase neural precursor cell expressed developmentally downregulated protein 4 (Nedd4) for efficient virus egress. Therefore, the PPxY‐Nedd4 interaction is a feasible target for broad‐spectrum antiviral agents. In 2014, Harty et al identified several small‐molecule inhibitors of PPxY‐Nedd4 interaction by combining virtual screening and biologic assay. The initial hit compound 100 (Figure 21) was able to dose‐dependently block the PPxy‐dependent budding of Marv vp40 virus‐like particles. Further structural modification lead to the identification of lead compound 101 and 102, which showed much more potent antibudding against filoviruses, arenavirus, and rhabdovirus VLPs. For example, both compounds inhibited egress of LFV‐Z VLPs by more than 10‐fold at a concentration of 1 μM, and as expected, showed no inhibitory activity against a PPxY mutant and budding defective virus, confirming the antibudding mechanism via inhibition of PPxY and Nedd4 interaction. In addition, no cytotoxicity toward HEK293T cells was observed for both compounds at concentrations tested (1 μM). 242 In a follow‐up study, an intensive structural modifications were made to the lead compound 102, and several more potent analogues were identified with nanomolar activity in the antibudding assay. For example, compound 103 can achieve more than 90% inhibition of budding of mVP40 VLPs at 10‐fold lower concentration (100 nM) as compared to the lead compound 102, and 30 nM of compound 104 can also lead to 3‐ and 15‐fold of decrease in mVP40 VLP budding. Importantly, compounds 103 and 104 were able to block the budding of a recombinant virus M40‐VSV (EBOV surrogate) from the HEK293T cells. 243 Of note, it still remains unclear which protein (the viral matrix protein or Nedd4) is the physical binding partner of this type of compounds. The antiviral activity of these compounds against live virus has not been tested. Nevertheless, the PPxY‐Nedd4 interaction represents a promising target for the development of broad‐spectrum antiviral agents.
Figure 21.
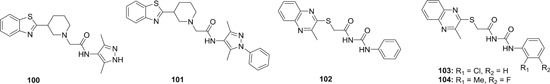
The chemical structures of PPxY‐Nedd4 inhibitors. Nedd4, neural precursor cell expressed developmentally downregulated protein 4
4.7. Chondroitin sulfate N‐acetylgalactosaminyltransferase 1
Li et al identified a natural compound cajanine (105; Figure 22) as a potent HCV inhibitor via a cell‐based phenotype‐screening assay with an IC50 value of 3.12 μM. 244 , 245 The structural‐activity relationships study revealed that both the hydrophobic benzene ring (106) and prenyl group (107) were not essential for the anti‐HCV activity, and several derivatives with more potent anti‐HCV activity (IC50 < 1 μM) were also obtained. The mechanism of action study demonstrated that 105 and its derivatives showed no inhibitory effect against HCV viral proteins, and yet led to the downregulation of a host target chondroitin sulfate N‐acetylgalactosaminyltransferase 1 (CSGalNAcT‐1), which is a key enzyme responsible for the initiation of chondroitin sulfate chain. CSGalNAcT‐1 was shown to play a key role during the replication of HCV virus, and its expression was elevated upon HCV infection. Knockdown of CSGalNAcT‐1 by siRNA led to significant suppression of HCV replications. It is interesting to note that 105 showed no effect on the mRNA level of CSGalNAcT‐1, and the downregulation of CSGalNAcT‐1 protein level was recovered by a general protease inhibitor MG132, indicating that cajanine promoted the degradation of CSGalNAcT‐1 by proteasome pathway. In consistency with such host targeting mechanism, 105 showed the same magnitude of inhibitory effect against both wild‐type and drug resistant HCV virus strains, and it also showed very pronounced synergistic effects with other DAAs to inhibit HCV replications, indicating that 105 and its derivatives are worthy of further studies. Although it is still unclear whether CSGalNAcT‐1 is broadly required for the replication of other viruses, our unpublished data showed that 105 and its derivatives also possessed inhibitory effect against other virus such as influenza virus, HIV‐1, HBV, and coxsackie B virus. Although it is ascertained solidly that 105 inhibits HCV replication via downregulation of CSGalNAcT‐1 protein, it remains unclear how 105 leads to the CSGalNAcT‐1 degradation, and which protein(s) 105 binds to physically. Answering these types of questions would definitely provide new host targets for the development of new antiviral agents.
Figure 22.
The chemical structure of cajanine along with its synthesized analogues
4.8. NS5B‐estrogen receptor α interaction
Throughout a drug repurposing campaign, estrogen receptor α (ERα) inhibitor tamoxifen (108; Figure 23) was identified as an HCV inhibitor. The known targets of 108 include ERα, P‐glycoprotein, calmodulin, and protein kinase C, and so forth. The inhibitors against P‐glycoprotein, calmodulin and protein kinase C failed to inhibit HCV replication, excluding the possibility of these proteins as the anti‐HCV targets for 108. A specific siRNA against ERα significantly suppressed HCV RNA in replicon‐containing cells, and transient transfection with ERα (but not ERβ) expression plasmids also augmented HCV replications. Altogether, these results confirmed the important roles ERα played during HCV replication. The subsequent binding assay showed that domain C of ERα physically bound to NS5B but not NS3, NS4B, and NS5A, and such interaction is essential for the HCV genome replications. The selective estrogen receptor modulators (SERMs) toremifene (109) and clomiphene (110) were also reported to inhibited several other viruses such as HIV‐1, 246 EBOV, 247 and HSV. 248 However, the mechanism studies have excluded ERα as potential antiviral targets, and the observed broad‐spectrum antiviral property for SERMs resulted from the inhibition of other host factors, including protein kinase C (HIV‐1) 249 and chloride channel (HSV‐1). 248 These host proteins together with ERα represent promising targets for the development of novel antiviral agents to combat against drug resistance and newly emerging unknown viruses.
Figure 23.
The chemical structures of the selective estrogen receptor modulators
4.9. Miscellaneous host targets
Quite a few other host proteins or pathways have also been validated as feasible antiviral targets in vitro and even in laboratory animals, such as lipid biosynthesis and metabolism pathways, 250 , 251 PPARα, 252 and HNF4α, 253 among others. 121 , 254 , 255 , 256 Table 1 summarized the host targets included in this review along with the development stages of their respective inhibitors/modulators. It should be noted that the focus of this review is just on small‐molecule based HTAs, and other validated antiviral host targets with only antibody or siRNA‐based modulators were not included. It is reasonable to anticipate that such host proteins can also be targeted by small‐molecule modulators to convey antiviral activity. Notably, the antiviral host targets studied only account for a very small portion of the host factors confirmed to be essential for viral replications. In 2016, Ammari et al 257 released a database for host‐pathogen interactions, with which one can search the known host‐pathogen interactions by inputting either the genes, proteins or the viruses. It can be speculated that most of the recorded host‐virus interactions must play critical roles during viral replications, and thus could serve as new targets for the development of HTAs.
Table 1.
The development stages of various host targets
| Host targets | Representative modulators | Viruses | Development stages |
|---|---|---|---|
| CCR5 | Maraviroc | HIV | Approved |
| Polypharmacology | RBV | HCV/RSV | Approved |
| Cyps | Alisporivir | HCV | Phase 3 (halted) |
| eIF2‐α | Nitazoxanide | Influenza, HBV, HCV, EBOV, DENV, JEV, and HIV | Phase 3 (influenza virus), phase 2 (HCV), and phase 1 (HBV) |
| NTCP | Myrcludex B | HBV/HDV | Phase 2/3 |
| IMPDH | VX‐947 | HCV | Phase 2 (halted) |
| HMGCoA reductase | Statins | HCV | Phase 2 |
| α‐Glucosidase | Iminosugars | DENV, HCV, EBOV, HIV, and influenza virus | Phase 2 (HIV, DENV, and HCV) |
| FXR | EYP001 | HCV/HBV | Phase 2 (HCV), phase 1 (HBV) |
| DGAT‐1 | Pradigastat | HCV | Phase 2 (halted) |
| Kinases (NAKs) | Sunitinib and erlotinib | DENV and EBOV | Phase 1 (EBOV) |
| PPARα | Naringenin | HCV | Phase 1 |
| APOBEC3G | IMB‐35 | HIV, HBV, HCV | Preclinical |
| BST‐2 | IMB‐LZ | HIV | Preclinical |
| HSP90 | GA derivatives | HCV, HIV, EBOV, and DENV, and so forth | Preclinical |
| HSP70 | Oxymatrine derivatives | HCV, DENV, ZIKV, WNV, and JEV | Preclinical |
| CXCR4 | GSK812397 | HIV | Preclinical |
| DDX‐3 | 97‐99 | HIV, HCV, WNV, and DENV | Preclinical |
| Nedd4‐PPxY | 100‐104 | EBOV | Preclinical |
| CSGalNAcT‐1 | Cajanine | HCV, influenza, and HIV | Preclinical |
| NS5B‐ERα | Tamoxifen | HCV | Preclinical |
4.10. Conclusions and perspectives
DAAs have shown great success in combating viral infections in clinic, and they are generally safe to use because they directly target viral proteins, which lack homologs in human. However, DAAs also suffer from several inherent limitations due to its viral protein targeting nature: viral proteins varied among different species and even different genotypes, and thus DAA targeting one specific viral protein is unlikely to exhibit inhibitory effect against viral proteins from other viral species or variants. Therefore, DAAs are normally narrow‐spectrum antiviral agents, and this is also the underlying reason for the lack of effective antiviral drugs against newly emerging viruses; the other major downside for DAAs is they are prone to cause drug resistance, particular among RNA viruses with very high frequency of replication errors. HTA agents perfectly complement to DAAs in regard to narrow‐spectrum activity and drug resistance. Since host factors are probably broadly required for viral replications, HTAs are very likely to show broad‐spectrum antiviral profiles. HTAs also have much higher genetic barrier to resistance as compare to DAAs in that host factors are not under the genetic control of virus. The expected selected mutations are not in the host genome, and should be selected in the viral protein that interacts with the host target. Consequently, HTAs are highly expected to address the problem of drug resistance and provide solution to the treatment of newly emerging viruses.
Although a large number of host factors are known to play vital roles throughout the whole life cycle of virus, only very few of them were explored as antiviral targets. This is primarily attributed to several concerns raised by HTAs. Chiefly, targeting a host protein may potentially lead to unwanted toxicity issues, because the target protein may be indispensable for some cellular functions. However, this may not be intrinsically true for all the host proteins, and many host genes are known to be nonessential for cellular functions. Therefore, the inhibition of such host proteins will be well‐tolerated, and the on‐target based toxicity can be minimized. Even though the target host protein is essential for some cellular functions, the on‐target based toxicity can be mitigated in many ways. First, it is known that the expression level or the activity of many cellular proteins are elevated to facilitate viral replications. Therefore, it is possible to knockdown the target protein to a level, at which the viral replication can be blocked while the normal cellular functions can still be maintained. Second, most of the time, alternative pathways or proteins exist for a cellular function, and hence the inhibition of one of these proteins or pathways may lead to the blockage of viral replication, but not the cellular function. Third, the on‐target based toxicity is closely associated with the duration of treatment, and the treatment of most of viral infections only lasts for several weeks or even days. Therefore, the toxicity concern raised by targeting a host factor for virus treatment can be further alleviated. It is worth noting that quite a few approved drugs targeting host factors for other indications are considered to be generally safe to use in clinic for years. Consequently, toxicity issue is something that one should pay attention to, but not something used to bias against host target antiviral agents.
The other major challenge faced with the development of HTAs is that the antiviral phenotype observed with HTAs is merely in vitro artifact in some cases, and the correlation between in vitro and in vivo or clinical efficacy is very poor as in the case of development of IMPDH inhibitors as anti‐HCV agents. The other example is the repurposement of statins as anti‐HCV drugs. Statins showed very pronounced inhibitory effects against HCV replication in vitro, but yield unsatisfactory outcomes in clinic trials. The lack of reliable predictive in vitro models indeed increased the attrition rates in the development of HTAs. However, despite with these challenges, development of HTAs can be highly rewarding because HTAs can potentially address the unmet needs in the treatment of viral infections.
ACKNOWLEDGMENTS
The work from the Ji lab is financially supported by the National Natural Science Foundation of China (81773608), the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD), the Six Major Talents Peak in Jiangsu Province in China (YY‐050), and CAMS Innovation Fund for Medical Sciences (CIFMS, Grant No: 2017‐I2M‐3‐012).
Biographies
Xingyue Ji is currently a professor and associate Chair of Medicinal Chemistry department at Soochow University. He has been serving as a visiting associate professor at Sichuan University since June 2018. He got both his bachelor's and master's degrees in chemistry from the University of Science and Technology of Beijing in 2006 and 2008, respectively. Thereafter, he moved to Peking Union Medical College Institute of Medicinal Biotechnology, and received his PhD degree in medicinal chemistry under the supervision of Professor Zhuorong Li in 2011. After that, he took an assistant professor position in the same institution, and he worked as a postdoctoral fellow in Dr Binghe Wang's lab from 2013 to September 2018. His current research interests focus on the development of anti‐infection drugs and gasotransmitter prodrugs.
Zhuorong Li is currently a professor and Deputy Director at Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College. She got her bachelor's degree at Peking Medical School in 1984, then she moved to Peking Union Medical College and received her master's degree in Medicinal Chemistry in 1988. Her research interests include the development of anti‐infection, antiosteoporosis, and anticancer drugs. She has published over 150 peer‐reviewed scientific papers and over 30 patents. One candidate compound developed in her lab has successfully entered clinical trials for the treatment of HBV infections.
Ji X, Li Z. Medicinal chemistry strategies toward host targeting antiviral agents. Med Res Rev. 2020;40:1519–1557. 10.1002/med.21664
Contributor Information
Xingyue Ji, Email: jixy@suda.edu.cn.
Zhuorong Li, Email: lizhuorong@imb.pumc.edu.cn.
REFERENCES
- 1. Das D, Pandya M. Recent advancement of direct‐acting antiviral agents (DAAs) in hepatitis C therapy. Mini Rev Med Chem. 2018;18(7):584‐596. [DOI] [PubMed] [Google Scholar]
- 2. Guan Y, Sun H, Li Y, Pan P, Li D, Hou T. The competitive binding between inhibitors and substrates of HCV NS3/4A protease: a general mechanism of drug resistance. Antiviral Res. 2014;103:60‐70. [DOI] [PubMed] [Google Scholar]
- 3. Wang Y, Jin F, Wang R, et al. HSP90: a promising broad‐spectrum antiviral drug target. Arch Virol. 2017;162(11):3269‐3282. [DOI] [PubMed] [Google Scholar]
- 4. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94(5):1925‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8):a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Novembre J, Galvani AP, Slatkin M. The geographic spread of the CCR5 Delta32 HIV‐resistance allele. PLoS Biol. 2005;3(11):e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim MB, Giesler KE, Tahirovic YA, Truax VM, Liotta DC, Wilson LJ. CCR5 receptor antagonists in preclinical to phase II clinical development for treatment of HIV. Expert Opin Investig Drugs. 2016;25(12):1377‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi WT, Yang Y, Xu Y, An J. Targeting chemokine receptor CXCR4 for treatment of HIV‐1 infection, tumor progression, and metastasis. Curr Top Med Chem. 2014;14(13):1574‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou N, Luo Z, Luo J, et al. Exploring the stereochemistry of CXCR4‐peptide recognition and inhibiting HIV‐1 entry with D‐peptides derived from chemokines. J Bio Chem. 2002;277(20):17476‐17485. [DOI] [PubMed] [Google Scholar]
- 10. Donzella GA, Schols D, Lin SW, et al. AMD3100, a small molecule inhibitor of HIV‐1 entry via the CXCR4 co‐receptor. Nat Med. 1998;4(1):72‐77. [DOI] [PubMed] [Google Scholar]
- 11. De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2(7):581‐587. [DOI] [PubMed] [Google Scholar]
- 12. Hatse S, Princen K, Clercq ED, et al. AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIV entry inhibitor. Biochem Pharmacol. 2005;70(5):752‐761. [DOI] [PubMed] [Google Scholar]
- 13. Stone ND, Dunaway SB, Flexner C, et al. Multiple‐dose escalation study of the safety, pharmacokinetics, and biologic activity of oral AMD070, a selective CXCR4 receptor inhibitor, in human subjects. Antimicrob Agents Chemother. 2007;51(7):2351‐2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jenkinson S, Thomson M, McCoy D, et al. Blockade of X4‐tropic HIV‐1 cellular entry by GSK812397, a potent noncompetitive CXCR4 receptor antagonist. Antimicrob Agents Chemother. 2010;54(2):817‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Catalano JG, Gudmundsson KS, Svolto A, et al. Synthesis of a novel tricyclic 1,2,3,4,4a,5,6,10b‐octahydro‐1,10‐phenanthroline ring system and CXCR4 antagonists with potent activity against HIV‐1. Bioorg Med Chem Lett. 2010;20(7):2186‐2190. [DOI] [PubMed] [Google Scholar]
- 16. Skerlj R, Bridger G, McEachern E, et al. Design of novel CXCR4 antagonists that are potent inhibitors of T‐tropic (X4) HIV‐1 replication. Bioorg Med Chem Lett. 2011;21(5):1414‐1418. [DOI] [PubMed] [Google Scholar]
- 17. Wagstaff AJ. Plerixafor: in patients with non‐Hodgkin's lymphoma or multiple myeloma. Drugs. 2009;69(3):319‐326. [DOI] [PubMed] [Google Scholar]
- 18. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595‐599. [DOI] [PubMed] [Google Scholar]
- 19. Tachibana K, Hirota S, Iizasa H, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393(6685):591‐594. [DOI] [PubMed] [Google Scholar]
- 20. Nagasawa T, Hirota S, Tachibana K, et al. Defects of B‐cell lymphopoiesis and bone‐marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF‐1. Nature. 1996;382(6592):635‐638. [DOI] [PubMed] [Google Scholar]
- 21. Rao PKS. CCR5 inhibitors: emerging promising HIV therapeutic strategy. Indian J Sex Transm Dis AIDS. 2009;30(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fätkenheuer G, Pozniak AL, Johnson MA, et al. Efficacy of short‐term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV‐1. Nat Med. 2005;11(11):1170‐1172. [DOI] [PubMed] [Google Scholar]
- 23. Lim JK, Murphy PM. Chemokine control of West Nile virus infection. Exp Cell Res. 2011;317(5):569‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rossetti B, Bianco C, Bellazzi LI, et al. Virological and immunological response to antiretroviral regimens containing maraviroc in HIV type 1‐infected patients in clinical practice: role of different tropism testing results and of concomitant treatments. AIDS Res Hum Retroviruses. 2014;30(1):17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Puertas MC, Massanella M, Llibre JM, et al. Intensification of a raltegravir‐based regimen with maraviroc in early HIV‐1 infection. AIDS. 2014;28(3):325‐334. [DOI] [PubMed] [Google Scholar]
- 26. Herrera C, Armanasco N, Garcia‐Perez J, et al. Maraviroc and reverse transcriptase inhibitors combinations as potential preexposure prophylaxis candidates. AIDS. 2016;30(7):1015‐1025. [DOI] [PubMed] [Google Scholar]
- 27. Tagat JR, McCombie SW, Nazareno D, et al. Piperazine‐based CCR5 antagonists as HIV‐1 inhibitors. IV. Discovery of 1‐[(4,6‐dimethyl‐5‐pyrimidinyl)carbonyl]‐4‐[4‐[2‐methoxy‐1(R)‐4‐(trifluoromethyl)phenyl]ethyl‐3(S)‐methyl‐1‐piperazinyl]‐ 4‐methylpiperidine (Sch‐417690/Sch‐D), a potent, highly selective, and orally bioavailable CCR5 antagonist. J Med Chem. 2004;47(10):2405‐2408. [DOI] [PubMed] [Google Scholar]
- 28. Adkison KK, Shachoy‐Clark A, Fang L, et al. Pharmacokinetics and short‐term safety of 873140, a novel CCR5 antagonist, in healthy adult subjects. Antimicrob Agents Chemother. 2005;49(7):2802‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stupple PA, Batchelor DV, Corless M, et al. An imidazopiperidine series of CCR5 antagonists for the treatment of HIV: the discovery of N‐{(1S)‐1‐(3‐Fluorophenyl)‐3‐[(3‐endo)‐3‐(5‐isobutyryl‐2‐methyl‐4,5,6,7‐tetrahydro‐1H‐imidazo[4,5‐c]pyridin‐1‐yl)‐8‐azabicyclo[3.2.1]oct‐8‐yl]propyl}acetamide (PF‐232798). J Med Chem. 2011;54(1):67‐77. [DOI] [PubMed] [Google Scholar]
- 30. Anti‐HIV agents . Is PF‐232798 a possible successor to maraviroc? TreatmentUpdate. 2008;20(2):8. [PubMed] [Google Scholar]
- 31. Kazmierski WM, Anderson DL, Aquino C, et al. Novel 4,4‐disubstituted piperidine‐based C‐C chemokine receptor‐5 inhibitors with high potency against human immunodeficiency virus‐1 and an improved human ether‐a‐go‐go related gene (hERG) profile. J Med Chem. 2011;54(11):3756‐3767. [DOI] [PubMed] [Google Scholar]
- 32. Imamura S, Ichikawa T, Nishikawa Y, et al. Discovery of a piperidine‐4‐carboxamide CCR5 antagonist (TAK‐220) with highly potent Anti‐HIV‐1 activity. J Med Chem. 2006;49(9):2784‐2793. [DOI] [PubMed] [Google Scholar]
- 33. Hu S, Wang Z, Hou T, et al. Design, synthesis, and biological evaluation of novel 2‐methylpiperazine derivatives as potent CCR5 antagonists. Bioorg Med Chem. 2015;23(5):1157‐1168. [DOI] [PubMed] [Google Scholar]
- 34. Xue CB, Chen L, Cao G, et al. Discovery of INCB9471, a potent, selective, and orally bioavailable CCR5 antagonist with potent anti‐HIV‐1 activity. ACS Med Chem Lett. 2010;1(9):483‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim MB, Giesler KE, Tahirovic YA, Truax VM, Liotta DC, Wilson LJ. CCR5 receptor antagonists in preclinical to phase II clinical development for treatment of HIV. Expert Opin Investig Drugs. 2016;25(12):1377‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Princen K, Hatse S, Vermeire K, et al. Inhibition of human immunodeficiency virus replication by a dual CCR5/CXCR4 antagonist. J Virol. 2004;78(23):12996‐13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eriksson B, Helgstrand E, Johansson NG, et al. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother. 1977;11(6):946‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fernandez‐Larsson R, O'Connell K, Koumans E, Patterson JL. Molecular analysis of the inhibitory effect of phosphorylated ribavirin on the vesicular stomatitis virus in vitro polymerase reaction. Antimicrob Agents Chemother. 1989;33(10):1668‐1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA. 2001;98(12):6895‐6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crotty S, Maag D, Arnold JJ, et al. The broad‐spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6(12):1375‐1379. [DOI] [PubMed] [Google Scholar]
- 41. Hultgren C, Milich DR, Weiland O, Sallberg M. The antiviral compound ribavirin modulates the T helper (Th) 1/Th2 subset balance in hepatitis B and C virus‐specific immune responses. J Gen Virol. 1998;79(Pt 10):2381‐2391. [DOI] [PubMed] [Google Scholar]
- 42. Krishnan SM, Dixit NM. Ribavirin‐induced anemia in hepatitis C virus patients undergoing combination therapy. PLOS Comput Biol. 2011;7(2):e1001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benhamou Y, Afdhal NH, Nelson DR, et al. A phase III study of the safety and efficacy of viramidine versus ribavirin in treatment‐naive patients with chronic hepatitis C: ViSER1 results. Hepatology. 2009;50(3):717‐726. [DOI] [PubMed] [Google Scholar]
- 44. Marcellin P, Gish RG, Gitlin N, et al. Safety and efficacy of viramidine versus ribavirin in ViSER2: randomized, double‐blind study in therapy‐naive hepatitis C patients. J Hepatol. 2010;52(1):32‐38. [DOI] [PubMed] [Google Scholar]
- 45. Feld JJ, Jacobson IM, Sulkowski MS, Poordad F, Tatsch F, Pawlotsky J‐M. Ribavirin revisited in the era of direct‐acting antiviral therapy for hepatitis C virus infection. Liver Int. 2017;37(1):5‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kwo PGE, Peng C, et al. Efficacy and safety of grazoprevir/elbasvir+/− RBV for 12 or 16 weeks in patients with HCV G1, G4 or G6 infection who previously failed peginterferon/RBV: C‐EDGE treatment‐experienced. J Hepatol. 2015;62(Suppl 2):S674‐S675. [Google Scholar]
- 47. Inoue K, Sekiyama K, Yamada M, Watanabe T, Yasuda H, Yoshiba M. Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. J Gastroenterol. 2003;38(6):567‐572. [DOI] [PubMed] [Google Scholar]
- 48. Nakagawa M, Sakamoto N, Enomoto N, et al. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem Biophys Res Commun. 2004;313(1):42‐47. [DOI] [PubMed] [Google Scholar]
- 49. Ma S, Boerner JE, TiongYip C, et al. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50(9):2976‐2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lawitz E, Godofsky E, Rouzier R, et al. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV‐infected patients receiving 14 days of therapy. Antiviral Res. 2011;89(3):238‐245. [DOI] [PubMed] [Google Scholar]
- 51. Li BSJ, Tang Y, Jone JT, et al. Alisporivir—a host‐targeting antiviral, provides low viral breakthrough rate and high barrier to resistance in HCV genotype 1 treatment‐naïve patients in the phase IIb ESSENTIAL study. Hepatology. 2011;54(S1):250A. [Google Scholar]
- 52. P JM, S SK, F GR, et al. Alisporivir plus ribavirin is highly effective as interferon‐free or interferon‐add‐on regimen in previously untreated HCV‐G2 or G3 patients: SVR12 results from VITAL‐1 phase 2b study. J Hepatol. 2012;56(Suppl 2):S553. [Google Scholar]
- 53. Griffel LH, Bao W, Orsenigo R, et al. Interferon (IFN)‐free alisporivir (DEB025) treatment in the VITAL‐1 study has a more beneficial overall safety profile vs IFN‐containing treatment. Hepatology. 2012;56(S1):578A. [Google Scholar]
- 54. Hopkins S, Scorneaux B, Huang Z, et al. SCY‐635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob Agents Chemother. 2010;54(2):660‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hopkins S, DiMassimo B, Rusnak P, et al. The cyclophilin inhibitor SCY‐635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J Hepatol. 2012;57(1):47‐54. [DOI] [PubMed] [Google Scholar]
- 56. Gregory MA, Bobardt M, Obeid S, et al. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob Agents Chemother. 2011;55(5):1975‐1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zenke G, Strittmatter U, Fuchs S, et al. Sanglifehrin A, a novel cyclophilin‐binding compound showing immunosuppressive activity with a new mechanism of action. J Immunol. 2001;166(12):7165‐7171. [DOI] [PubMed] [Google Scholar]
- 58. Sedrani R, Kallen J, Martin Cabrejas LM, et al. Sanglifehrin−cyclophilin interaction: degradation work, synthetic macrocyclic analogues, X‐ray crystal structure, and binding data. J Am Chem Soc. 2003;125(13):3849‐3859. [DOI] [PubMed] [Google Scholar]
- 59. Ahmed‐Belkacem A, Colliandre L, Ahnou N, et al. Fragment‐based discovery of a new family of non‐peptidic small‐molecule cyclophilin inhibitors with potent antiviral activities. Nat Commun. 2016;7:12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. Catalysis of cis/trans isomerization in native HIV‐1 capsid by human cyclophilin A. Proc Natl Acad Sci USA. 2002;99(8):5247‐5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu X, Sun L, Yu M, et al. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol. 2009;11(5):730‐741. [DOI] [PubMed] [Google Scholar]
- 62. Bouchard MJ, Puro RJ, Wang L, Schneider RJ. Activation and inhibition of cellular calcium and tyrosine kinase signaling pathways identify targets of the HBx protein involved in hepatitis B virus replication. J Virol. 2003;77(14):7713‐7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Luo C, Luo H, Zheng S, et al. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem Biophys Res Commun. 2004;321(3):557‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Keyes LR, Bego MG, Soland M St, Jeor S. Cyclophilin A is required for efficient human cytomegalovirus DNA replication and reactivation. J Gen Virol. 2012;93(Pt 4):722‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bienkowska‐Haba M, Patel HD, Sapp M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLOS Pathog. 2009;5(7):e1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. de Wilde AH, Pham U, Posthuma CC, Snijder EJ. Cyclophilins and cyclophilin inhibitors in nidovirus replication. Virology. 2018;522:46‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gallay P, Chatterji U, Bobardt M, et al. P0890: novel cyclophilin inhibitor CPI‐431‐32 shows broad spectrum antiviral activity by blocking replication of HCV, HBV and HIV‐1 viruses. J Hepatol. 2015;62:S677. [Google Scholar]
- 68. Ma C, Li F, Musharrafieh RG, Wang J. Discovery of cyclosporine A and its analogs as broad‐spectrum anti‐influenza drugs with a high in vitro genetic barrier of drug resistance. Antiviral Res. 2016;133:62‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Taubel J, Lorch U, Rossignol JF, Ferber G, Camm AJ. Analyzing the relationship of QT interval and exposure to nitazoxanide, a prospective candidate for influenza antiviral therapy—a formal TQT study. J Clin Pharmacol. 2014;54(9):987‐994. [DOI] [PubMed] [Google Scholar]
- 70. Belardo G, Cenciarelli O, La Frazia S, Rossignol JF, Santoro MG. Synergistic effect of nitazoxanide with neuraminidase inhibitors against influenza A viruses in vitro. Antimicrob Agents Chemother. 2015;59(2):1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Korba BE, Montero AB, Farrar K, et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77(1):56‐63. [DOI] [PubMed] [Google Scholar]
- 72. Jasenosky LD, Cadena C, Mire CE, et al. The FDA‐approved oral drug nitazoxanide amplifies host antiviral responses and inhibits Ebola virus. iScience. 2019;19:1279‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rossignol J‐F. Nitazoxanide: a first‐in‐class broad‐spectrum antiviral agent. Antiviral Res. 2014;110:94‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Matthaei M, Kerr PJ, Read AJ, et al. Comparative quantitative monitoring of rabbit haemorrhagic disease viruses in rabbit kittens. Virol J. 2014;11(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tan X, Hu L, Luquette Iii LJ, et al. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol. 2012;30:1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cao RY, Xu YF, Zhang TH, et al. Pediatric drug nitazoxanide: a potential choice for control of Zika. Open Forum Infect Dis. 2017;4(1):ofx009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Piacentini S, La Frazia S, Riccio A, et al. Nitazoxanide inhibits paramyxovirus replication by targeting the Fusion protein folding: role of glycoprotein‐specific thiol oxidoreductase ERp57. Sci Rep. 2018;8(1):10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rossignol JF. Nitazoxanide, a new drug candidate for the treatment of middle east respiratory syndrome coronavirus. J Infect Public Health. 2016;9(3):227‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Elazar M, Liu M, McKenna SA, et al. The anti‐hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2α via protein kinase activated by double‐stranded RNA activation. Gastroenterology. 2009;137(5):1827‐1835. [DOI] [PubMed] [Google Scholar]
- 80. Decorsiere A, Mueller H, van Breugel PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531(7594):386‐389. [DOI] [PubMed] [Google Scholar]
- 81. Sekiba K, Otsuka M, Ohno M, et al. Inhibition of HBV Transcription From cccDNA With Nitazoxanide by Targeting the HBx‐DDB1 Interaction. Cell Mol Gastroenterol Hepatol. 2019;7(2):297‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dang W, Xu L, Ma B, et al. Nitazoxanide inhibits human norovirus replication and synergizes with ribavirin by activation of cellular antiviral response. Antimicrob Agents Chemother. 2018;62(11):e00707‐e00718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kung H‐C, Evensen Ø, Hong J‐R, et al. Interferon regulatory factor‐1 (IRF‐1) is involved in the induction of phosphatidylserine receptor (PSR) in response to dsRNA virus infection and contributes to apoptotic cell clearance in CHSE‐214 cell. Int J Mol Sci. 2014;15(10):19281‐19306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Haffizulla J, Hartman A, Hoppers M, et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double‐blind, randomised, placebo‐controlled, phase 2b/3 trial. Lancet Infect Dis. 2014;14(7):609‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rossignol J‐F, Bréchot C. A pilot clinical trial of nitazoxanide in the treatment of chronic hepatitis B. Hepatology Commun. 2019;3(6):744‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Stachulski AV, Swift K, Cooper M, et al. Synthesis and pre‐clinical studies of new amino‐acid ester thiazolide prodrugs. Eur J Med Chem. 2017;126:154‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fischl MA, Resnick L, Coombs R, et al. The safety and efficacy of combination N‐butyl‐deoxynojirimycin (SC‐48334) and zidovudine in patients with HIV‐1 infection and 200‐500 CD4 cells/mm3. J Acquir Immune Defic Syndr. 1994;7(2):139‐147. [PubMed] [Google Scholar]
- 88. Durantel D. Celgosivir, an alpha‐glucosidase I inhibitor for the potential treatment of HCV infection. Curr Opin Investig Drugs. 2009;10(8):860‐870. [PubMed] [Google Scholar]
- 89. Zhao X, Guo F, Comunale MA, et al. Inhibition of endoplasmic reticulum‐resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein‐mediated entry by altering the glycan processing of angiotensin I‐converting enzyme 2. Antimicrob Agents Chemother. 2015;59(1):206‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Stavale EJ, Vu H, Sampath A, Ramstedt U, Warfield KL. In vivo therapeutic protection against influenza A (H1N1) oseltamivir‐sensitive and resistant viruses by the iminosugar UV‐4. PLOS One. 2015;10(3):e0121662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Plummer E, Buck MD, Sanchez M, et al. Dengue virus evolution under a host‐targeted antiviral. J Virol. 2015;89(10):5592‐5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81(3):425‐433. [DOI] [PubMed] [Google Scholar]
- 93. Dhameja M, Gupta P. Synthetic heterocyclic candidates as promising α‐glucosidase inhibitors: an overview. Eur J Med Chem. 2019;176:343‐377. [DOI] [PubMed] [Google Scholar]
- 94. Warfield KL, Warren TK, Qiu X, et al. Assessment of the potential for host‐targeted iminosugars UV‐4 and UV‐5 activity against filovirus infections in vitro and in vivo. Antiviral Res. 2017;138:22‐31. [DOI] [PubMed] [Google Scholar]
- 95. Watanabe S, Rathore AP, Sung C, et al. Dose‐ and schedule‐dependent protective efficacy of celgosivir in a lethal mouse model for dengue virus infection informs dosing regimen for a proof of concept clinical trial. Antiviral Res. 2012;96(1):32‐35. [DOI] [PubMed] [Google Scholar]
- 96. Chang J, Warren TK, Zhao X, et al. Small molecule inhibitors of ER α‐glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res. 2013;98(3):432‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ma J, Wu S, Zhang X, et al. Ester prodrugs of IHVR‐19029 with enhanced oral exposure and prevention of gastrointestinal glucosidase interaction. ACS Med Chem Lett. 2017;8(2):157‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Caputo AT, Alonzi DS, Marti L, et al. Structures of mammalian ER α‐glucosidase II capture the binding modes of broad‐spectrum iminosugar antivirals. Proc Natl Acad Sci USA. 2016;113(32):E4630‐E4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Nair V, Ma X, Shu Q, Zhang F, Uchil V, Cherukupalli GR. IMPDH as a biological probe for RNA antiviral drug discovery: synthesis, enzymology, molecular docking, and antiviral activity of new ribonucleosides with surrogate bases. Nucleosides Nucleotides Nucleic Acids. 2007;26(6‐7):651‐654. [DOI] [PubMed] [Google Scholar]
- 100. Markland W, McQuaid TJ, Jain J, Kwong AD. Broad‐spectrum antiviral activity of the IMP dehydrogenase inhibitor VX‐497: a comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob Agents Chemother. 2000;44(4):859‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leyssen P, Balzarini J, De Clercq E, Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol. 2005;79(3):1943‐1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Smee DF, Bray M, Huggins JW. Antiviral activity and mode of action studies of ribavirin and mycophenolic acid against orthopoxviruses in vitro. Antivir Chem Chemother. 2001;12(6):327‐335. [DOI] [PubMed] [Google Scholar]
- 103. Tong X, Smith J, Bukreyeva N, et al. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antiviral Res. 2018;149:34‐40. [DOI] [PubMed] [Google Scholar]
- 104. Zhong Z‐J, Zhang D‐J, Peng Z‐G, et al. Synthesis and antiviral activity of a novel class of (5‐oxazolyl)phenyl amines. Eur J Med Chem. 2013;69:32‐43. [DOI] [PubMed] [Google Scholar]
- 105. Zhang D‐J, Sun W‐F, Zhong Z‐J, et al. Synthesis and broad‐spectrum antiviral activity of some novel benzo‐heterocyclic amine compounds. Molecules. 2014;19(1):925‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. McHutchison JGSM, Cheung RC, Gordon SC, et al. A randomized, double‐blind, placebo‐controlled dose‐escalation trial of merimepodib (VX‐497) and interferon‐alpha in previously untreated patients with chronic hepatitis C. Antivir Ther. 2005;10(5):635‐643. [PubMed] [Google Scholar]
- 107. Rustgi VK, Lee WM, Lawitz E, et al. Merimepodib, pegylated interferon, and ribavirin in genotype 1 chronic hepatitis C pegylated interferon and ribavirin nonresponders. Hepatology. 2009;50(6):1719‐1726. [DOI] [PubMed] [Google Scholar]
- 108. Di Bisceglie AM, Conjeevaram HS, Fried MW, et al. Ribavirin as therapy for chronic hepatitis C. A randomized, double‐blind, placebo‐controlled trial. Ann Intern Med. 1995;123(12):897‐903. [DOI] [PubMed] [Google Scholar]
- 109. Schor S, Einav S. Repurposing of kinase inhibitors as broad‐spectrum antiviral drugs. DNA Cell Biol. 2018;37(2):63‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Perwitasari O, Yan X, O'Donnell J, Johnson S, Tripp RA. Repurposing kinase inhibitors as antiviral agents to control influenza A virus replication. Assay Drug Dev Technol. 2015;13(10):638‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Williams ER. New connections: kinase inhibitors as antivirals. Sci Signal. 2018;11(550):eaau2211. [Google Scholar]
- 112. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad‐spectrum antiviral effects. J Clin Invest. 2017;127(4):1338‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Neveu G, Barouch‐Bentov R, Ziv‐Av A, Gerber D, Jacob Y, Einav S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLOS Pathog. 2012;8(8):e1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kovackova S, Chang L, Bekerman E, et al. Selective inhibitors of cyclin G associated kinase (GAK) as anti‐hepatitis C agents. J Med Chem. 2015;58(8):3393‐3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pu S‐Y, Wouters R, Schor S, et al. Optimization of isothiazolo[4,3‐b]pyridine‐based inhibitors of cyclin G associated kinase (GAK) with broad‐spectrum antiviral activity. J Med Chem. 2018;61(14):6178‐6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Li J, Kovackova S, Pu S, et al. Isothiazolo[4,3‐b]pyridines as inhibitors of cyclin G associated kinase: synthesis, structure‐activity relationship studies and antiviral activity. MedChemComm. 2015;6(9):1666‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Asquith CRM, Laitinen T, Bennett JM, et al. Identification and optimization of 4‐anilinoquinolines as inhibitors of cyclin G associated kinase. ChemMedChem. 2018;13(1):48‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Asquith CRM, Berger B‐T, Wan J, et al. SGC‐GAK‐1: a chemical probe for cyclin G associated kinase (GAK). J Med Chem. 2019;62(5):2830‐2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad‐spectrum antiviral effects. J Clin Invest. 2017;127(4):1338‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dong C, Qu L, Wang H, Wei L, Dong Y, Xiong S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110‐117. [DOI] [PubMed] [Google Scholar]
- 122. Wedemeyer H, Bogomolov P, Blank A, et al. Final results of a multicenter, open‐label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with tenofovir in patients with chronic HBV/HDV co‐infection. J Hepatol. 2018;68:S3. [Google Scholar]
- 123. Nkongolo S, Ni Y, Lempp FA, et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin‐independent interference with the NTCP receptor. J Hepatol. 2014;60(4):723‐731. [DOI] [PubMed] [Google Scholar]
- 124. Blanchet M, Sureau C, Labonté P. Use of FDA approved therapeutics with hNTCP metabolic inhibitory properties to impair the HDV lifecycle. Antiviral Res. 2014;106:111‐115. [DOI] [PubMed] [Google Scholar]
- 125. Nio Y, Akahori Y, Okamura H, Watashi K, Wakita T, Hijikata M. Inhibitory effect of fasiglifam on hepatitis B virus infections through suppression of the sodium taurocholate cotransporting polypeptide. Biochem Biophys Res Commun. 2018;501(3):820‐825. [DOI] [PubMed] [Google Scholar]
- 126. Iwamoto M, Watashi K, Tsukuda S, et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun. 2014;443(3):808‐813. [DOI] [PubMed] [Google Scholar]
- 127. Matsunaga H, Kamisuki S, Kaneko M, et al. Isolation and structure of vanitaracin A, a novel anti‐hepatitis B virus compound from Talaromyces sp. Bioorg Med Chem Lett. 2015;25(19):4325‐4328. [DOI] [PubMed] [Google Scholar]
- 128. Zhang J, Fu L‐l, Tian M, et al. Design and synthesis of a novel candidate compound NTI‐007 targeting sodium taurocholate cotransporting polypeptide [NTCP]–APOA1–HBx–Beclin1‐mediated autophagic pathway in HBV therapy. Bioorg Med Chem. 2015;23(5):976‐984. [DOI] [PubMed] [Google Scholar]
- 129. Fukano K, Tsukuda S, Watashi K, Wakita T. Concept of viral inhibitors via NTCP. Semin Liver Dis. 2019;39(1):78‐85. [DOI] [PubMed] [Google Scholar]
- 130. Shimura S, Watashi K, Fukano K, et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J Hepatol. 2017;66(4):685‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Xu J, Gu W, Li C, et al. Epigallocatechin gallate inhibits hepatitis B virus via farnesoid X receptor alpha. J Nat Med. 2016;70(3):584‐591. [DOI] [PubMed] [Google Scholar]
- 132. Mouzannar K, Fusil F, Lacombe B, et al. Farnesoid X receptor‐alpha is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J. 2019;33(2):2472‐2483. [DOI] [PubMed] [Google Scholar]
- 133. Wu Z‐Y, Li H, Li J‐R, Lv X‐Q, Jiang J‐D, Peng Z‐G. Farnesoid X receptor agonist GW4064 indirectly inhibits HCV entry into cells via down‐regulating scavenger receptor class B type I. Eur J Pharmacol. 2019;853:111‐120. [DOI] [PubMed] [Google Scholar]
- 134. Joly S, Porcherot M, Radreau P, et al. The selective FXR agonist EYP001 is well tolerated in healthy subjects and has additive anti‐HBV effect with nucleoside analogues in HepaRG cells. J Hepatol. 2017;66(1):S690. [Google Scholar]
- 135. Camus G, Herker E, Modi AA, et al. Diacylglycerol acyltransferase‐1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J Bio Chem. 2013;288(14):9915‐9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sung PS, Murayama A, Kang W, et al. Hepatitis C virus entry is impaired by claudin‐1 downregulation in diacylglycerol acyltransferase‐1‐deficient cells. J Virol. 2014;88(16):9233‐9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Meyers CD, Tremblay K, Amer A, Chen J, Jiang L, Gaudet D. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipids Health Dis. 2015;14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Gane E, Stedman C, Dole K, et al. A diacylglycerol transferase 1 inhibitor is a potent hepatitis C antiviral in vitro but not in patients in a randomized clinical trial. ACS Infect Dis. 2017;3(2):144‐151. [DOI] [PubMed] [Google Scholar]
- 139. Burch AD, Weller SK. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol. 2005;79(16):10740‐10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kawashima D, Kanda T, Murata T, et al. Nuclear transport of Epstein‐Barr virus DNA polymerase is dependent on the BMRF1 polymerase processivity factor and molecular chaperone Hsp90. J Virol. 2013;87(11):6482‐6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol. 2007;81(3):1339‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Basha W, Kitagawa R, Uhara M, Imazu H, Uechi K, Tanaka J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir Chem Chemother. 2005;16(2):135‐146. [DOI] [PubMed] [Google Scholar]
- 143. Taguwa S, Okamoto T, Abe T, et al. Human butyrate‐induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J Virol. 2008;82(6):2631‐2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Connor JH, McKenzie MO, Parks GD, Lyles DS. Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology. 2007;362(1):109‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rathore AP, Haystead T, Das PK, Merits A, Ng ML, Vasudevan SG. Chikungunya virus nsP3 & nsP4 interacts with HSP‐90 to promote virus replication: HSP‐90 inhibitors reduce CHIKV infection and inflammation in vivo. Antiviral Res. 2014;103:7‐16. [DOI] [PubMed] [Google Scholar]
- 146. Geller R, Andino R, Frydman J. Hsp90 inhibitors exhibit resistance‐free antiviral activity against respiratory syncytial virus. PLOS One. 2013;8(2):e56762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Vashist S, Urena L, Gonzalez‐Hernandez MB, Choi J, de Rougemont A. Molecular chaperone Hsp90 is a therapeutic target for noroviruses. J Virol. 2015;89(12):6352‐6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shim HY, Quan X, Yi YS, Jung G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology. 2011;410(1):161‐169. [DOI] [PubMed] [Google Scholar]
- 149. Barrott JJ, Haystead TAJ. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013;280(6):1381‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Li Y‐H, Lu Q‐N, Wang H‐Q, Tao P‐Z, Jiang J‐D. Geldanamycin, a ligand of heat shock protein 90, inhibits herpes simplex virus type 2 replication both in vitro and in vivo. J Antibiot. 2012;65:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Li YH, Tao PZ, Liu YZ, Jiang JD. Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother. 2004;48(3):867‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Smith DR, McCarthy S, Chrovian A, et al. Inhibition of heat‐shock protein 90 reduces Ebola virus replication. Antiviral Res. 2010;87(2):187‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Anderson I, Low JS, Weston S, et al. Heat shock protein 90 controls HIV‐1 reactivation from latency. Proc Natl Acad Sci USA. 2014;111(15):E1528‐E1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Chase G, Deng T, Fodor E, et al. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology. 2008;377(2):431‐439. [DOI] [PubMed] [Google Scholar]
- 155. Geller R, Andino R, Frydman J. Hsp90 inhibitors exhibit resistance‐free antiviral activity against respiratory syncytial virus. PLOS One. 2013;8(2):e56762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Geller R, Vignuzzi M, Andino R, Frydman J. Evolutionary constraints on chaperone‐mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007;21(2):195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Shan G‐z, Peng Z‐g, Li Y‐h, et al. A novel class of geldanamycin derivatives as HCV replication inhibitors targeting on Hsp90: synthesis, structure–activity relationships and anti‐HCV activity in GS4.3 replicon cells. J Antibiot. 2010;64:177. [DOI] [PubMed] [Google Scholar]
- 158. Li YP, Shan GZ, Peng ZG, et al. Synthesis and biological evaluation of heat‐shock protein 90 inhibitors: geldanamycin derivatives with broad antiviral activities. Antivir Chem Chemother. 2010;20(6):259‐268. [DOI] [PubMed] [Google Scholar]
- 159. Li F, Jin F, Wang Y, et al. Hsp90 inhibitor AT‐533 blocks HSV‐1 nuclear egress and assembly. J Bio Chem. 2018;164(6):397‐406. [DOI] [PubMed] [Google Scholar]
- 160. Smith DR, McCarthy S, Chrovian A, et al. Inhibition of heat‐shock protein 90 reduces Ebola virus replication. Antiviral Res. 2010;87(2):187‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gao J, Xiao S, Liu X, et al. Inhibition of HSP90 attenuates porcine reproductive and respiratory syndrome virus production in vitro. Virol J. 2014;11:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. Heat shock protein 90: inhibitors in clinical trials. J Med Chem. 2010;53(1):3‐17. [DOI] [PubMed] [Google Scholar]
- 164. Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev. 2013;39(4):375‐387. [DOI] [PubMed] [Google Scholar]
- 165. Peterson LB, Eskew JD, Vielhauer GA, Blagg BS. The hERG channel is dependent upon the Hsp90alpha isoform for maturation and trafficking. Mol Pharm. 2012;9(6):1841‐1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Patel PD, Yan P, Seidler PM, et al. Paralog‐selective Hsp90 inhibitors define tumor‐specific regulation of HER2. Nat Chem Biol. 2013;9(11):677‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Duerfeldt AS, Peterson LB, Maynard JC, et al. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134(23):9796‐9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Khandelwal A, Kent CN, Balch M, et al. Structure‐guided design of an Hsp90beta N‐terminal isoform‐selective inhibitor. Nat Commun. 2018;9(1):425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Rothan HA, Zhong Y, Sanborn MA, et al. Small molecule grp94 inhibitors block dengue and Zika virus replication. Antiviral Res. 2019;171:104590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Choukhi A, Ung S, Wychowski C, Dubuisson J. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J Virol. 1998;72(5):3851‐3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Cho DY, Yang GH, Ryu CJ, Hong HJ. Molecular chaperone GRP78/BiP interacts with the large surface protein of hepatitis B virus in vitro and in vivo. J Virol. 2003;77(4):2784‐2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. de Silva A, Braakman I, Helenius A. Posttranslational folding of vesicular stomatitis virus G protein in the ER: involvement of noncovalent and covalent complexes. J Cell Biol. 1993;120(3):647‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Earl PL, Moss B, Doms RW. Folding, interaction with GRP78‐BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J Virol. 1991;65(4):2047‐2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Li G, Zhang J, Tong X, Liu W, Ye X. Heat shock protein 70 inhibits the activity of Influenza A virus ribonucleoprotein and blocks the replication of virus in vitro and in vivo. PLOS One. 2011;6(2):e16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Guerrero CA, Bouyssounade D, Zarate S, et al. Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol. 2002;76(8):4096‐4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Cripe TP, Delos SE, Estes PA, Garcea RL. In vivo and in vitro association of hsc70 with polyomavirus capsid proteins. J Virol. 1995;69(12):7807‐7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Parent R, Qu X, Petit M‐A, Beretta L. The heat shock cognate protein 70 is associated with hepatitis C virus particles and modulates virus infectivity. Hepatology. 2009;49(6):1798‐1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Qi R, Sarbeng EB, Liu Q, et al. Allosteric opening of the polypeptide‐binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20(7):900‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem. 2010;53(20):7280‐7286. [DOI] [PubMed] [Google Scholar]
- 180. Britten CD, Rowinsky EK, Baker SD, et al. A phase I and pharmacokinetic study of the mitochondrial‐specific rhodacyanine dye analog MKT 077. Clin Cancer Res. 2000;6(1):42‐49. [PubMed] [Google Scholar]
- 181. Propper DJ, Braybrooke JP, Taylor DJ, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo‐resistant solid tumours. Ann Oncol. 1999;10(8):923‐927. [DOI] [PubMed] [Google Scholar]
- 182. Taguwa S, Maringer K, Li X, et al. Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in flavivirus infection. Cell. 2015;163(5):1108‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Khachatoorian R, Riahi R, Ganapathy E, et al. Allosteric heat shock protein 70 inhibitors block hepatitis C virus assembly. Int J Antimicrob Agents. 2016;47(4):289‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Pujhari S, Brustolin M, Macias VM, et al. Heat shock protein 70 (Hsp70) mediates Zika virus entry, replication, and egress from host cells. Emerg Microbes Infect. 2019;8(1):8‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Howe MK, Bodoor K, Carlson DA, et al. Identification of an allosteric small‐molecule inhibitor selective for the inducible form of heat shock protein 70. Chem Biol. 2014;21(12):1648‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Taldone T, Kang Y, Patel HJ, et al. Heat shock protein 70 inhibitors. 2. 2,5'‐thiodipyrimidines, 5‐(phenylthio)pyrimidines, 2‐(pyridin‐3‐ylthio)pyrimidines, and 3‐(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70. J Med Chem. 2014;57(4):1208‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Howe MK, Speer BL, Hughes PF, Loiselle DR, Vasudevan S, Haystead TAJ. An inducible heat shock protein 70 small molecule inhibitor demonstrates anti‐dengue virus activity, validating Hsp70 as a host antiviral target. Antiviral Res. 2016;130:81‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Peng ZG, Fan B, Du NN, et al. Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA. Hepatology. 2010;52(3):845‐853. [DOI] [PubMed] [Google Scholar]
- 189. Li Y‐H, Wu Z‐Y, Tang S, et al. Evolution of matrinic ethanol derivatives as anti‐HCV agents from matrine skeleton. Bioorg Med Chem Lett. 2017;27(9):1962‐1966. [DOI] [PubMed] [Google Scholar]
- 190. Li Y, Peng Z, Gao L, Song D. Synthesis and biological evaluation of sophocarpinic acid derivatives as anti‐HCV agents. Acta Pharm Sin B. 2014;4(4):307‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Du N‐N, Li X, Wang Y‐P, et al. Synthesis, structure−activity relationship and biological evaluation of novel N‐substituted matrinic acid derivatives as host heat‐stress cognate 70 (Hsc70) down‐regulators. Bioorg Med Chem Lett. 2011;21(16):4732‐4735. [DOI] [PubMed] [Google Scholar]
- 192. Gao LM, Han YX, Wang YP, et al. Design and synthesis of oxymatrine analogues overcoming drug resistance in hepatitis B virus through targeting host heat stress cognate 70. J Med Chem. 2011;54(3):869‐876. [DOI] [PubMed] [Google Scholar]
- 193. Yang Y, Xiu J, Zhang X, et al. Antiviral effect of matrine against human enterovirus 71. Molecules. 2012;17(9):10370‐10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Tang S, Li YH, Cheng XY, et al. SAR evolution and discovery of benzenesulfonyl matrinanes as a novel class of potential coxsakievirus inhibitors. Future Med Chem. 2016;8(5):495‐508. [DOI] [PubMed] [Google Scholar]
- 195. Chen D, Cai J, Cheng J, et al. Design, synthesis and structure‐activity relationship optimization of lycorine derivatives for HCV inhibition. Sci Rep. 2015;5:14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Chen D‐Z, Jiang J‐D, Zhang K‐Q, et al. Evaluation of anti‐HCV activity and SAR study of (+)‐lycoricidine through targeting of host heat‐stress cognate 70 (Hsc70). Bioorg Med Chem Lett. 2013;23(9):2679‐2682. [DOI] [PubMed] [Google Scholar]
- 197. Li M, Yan K, Wei L, et al. Zinc finger antiviral protein inhibits coxsackievirus B3 virus replication and protects against viral myocarditis. Antiviral Res. 2015;123:50‐61. [DOI] [PubMed] [Google Scholar]
- 198. Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV‐1 DNA in the absence of the Vif protein. Science. 2003;300(5622):1112. [DOI] [PubMed] [Google Scholar]
- 199. Yu Q, König R, Pillai S, et al. Single‐strand specificity of APOBEC3G accounts for minus‐strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11(5):435‐442. [DOI] [PubMed] [Google Scholar]
- 200. Zhu Y‐P, Peng Z‐G, Wu Z‐Y, et al. Host APOBEC3G protein inhibits HCV replication through direct binding at NS3. PLOS One. 2015;10(3):e0121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Peng ZG, Zhao ZY, Li YP, et al. Host apolipoprotein B messenger RNA‐editing enzyme catalytic polypeptide‐like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology. 2011;53(4):1080‐1089. [DOI] [PubMed] [Google Scholar]
- 202. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303(5665):1829. [DOI] [PubMed] [Google Scholar]
- 203. Lei Y‐C. Inhibition of hepatitis B virus replication by APOBEC3G in vitro and in vivo. World J Gastroenterol. 2006;12(28):4492‐4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Shirakawa K, Takaori‐Kondo A, Kobayashi M, et al. Ubiquitination of APOBEC3 proteins by the Vif–Cullin5–ElonginB–ElonginC complex. Virology. 2006;344(2):263‐266. [DOI] [PubMed] [Google Scholar]
- 205. Cen S, Peng Z‐G, Li X‐Y, et al. Small molecular compounds inhibit HIV‐1 replication through specifically stabilizing APOBEC3G. J Biol Chem. 2010;285(22):16546‐16552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Peng Z‐G, Zhao Z‐Y, Li Y‐P, et al. Host apolipoprotein b messenger RNA‐editing enzyme catalytic polypeptide‐like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology. 2011;53(4):1080‐1089. [DOI] [PubMed] [Google Scholar]
- 207. Jiang Z, Wang H, Li Y, Peng Z, Li Y, Li Z. Synthesis and antiviral activity of a series of novel N‐phenylbenzamide and N‐phenylacetophenone compounds as anti‐HCV and anti‐EV71 agents. Acta Pharm Sin B. 2015;5(3):201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Ji X‐Y, Wang H‐Q, Hao L‐H, et al. Synthesis and antiviral activity of N‐phenylbenzamide derivatives, a novel class of enterovirus 71 inhibitors. Molecules. 2013;18(3):3630‐3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Wang Y, Wang H, Jiang X, et al. Synthesis and broad antiviral activity of novel 2‐aryl‐isoindolin‐1‐ones towards diverse enterovirus A71 clinical isolates. Molecules. 2019;24(5):985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Hao L, Li Y, He WY, et al. Synthesis and antiviral activity of substituted bisaryl amide compounds as novel influenza virus inhibitors. Eur J Med Chem. 2012;55:117‐124. [DOI] [PubMed] [Google Scholar]
- 211. Pauli E‐K, Schmolke M, Hofmann H, et al. High level expression of the anti‐retroviral protein APOBEC3G is induced by influenza A virus but does not confer antiviral activity. Retrovirology. 2009;6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Nathans R, Cao H, Sharova N, et al. Small‐molecule inhibition of HIV‐1 Vif. Nat Biotechnol. 2008;26(10):1187‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ali A, Wang J, Nathans RS, et al. Synthesis and structure‐activity relationship studies of HIV‐1 virion infectivity factor (Vif) inhibitors that block viral replication. ChemMedChem. 2012;7(7):1217‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zhang R‐H, Wang S, Luo R‐H, et al. Design, synthesis, and biological evaluation of 2‐amino‐N‐(2‐methoxyphenyl)‐6‐((4‐nitrophenyl)sulfonyl)benzamide derivatives as potent HIV‐1 Vif inhibitors. Bioorg Med Chem Lett. 2019;29:126638. [DOI] [PubMed] [Google Scholar]
- 215. Matsui M, Shindo K, Izumi T, et al. Small molecules that inhibit Vif‐induced degradation of APOBEC3G. Virol J. 2014;11(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Pan T, He X, Chen B, et al. Development of benzimidazole derivatives to inhibit HIV‐1 replication through protecting APOBEC3G protein. Eur J Med Chem. 2015;95:500‐513. [DOI] [PubMed] [Google Scholar]
- 217. Zhang S, Zhong L, Chen B, et al. Identification of an HIV‐1 replication inhibitor which rescues host restriction factor APOBEC3G in Vif–APOBEC3G complex. Antiviral Res. 2015;122:20‐27. [DOI] [PubMed] [Google Scholar]
- 218. Zuo T, Liu D, Lv W, et al. Small‐molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol. 2012;86(10):5497‐5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Huang W, Zuo T, Luo X, et al. Indolizine derivatives as HIV‐1 VIF–ElonginC interaction inhibitors. Chem Biol Drug Des. 2013;81(6):730‐741. [DOI] [PubMed] [Google Scholar]
- 220. Li M, Shandilya SMD, Carpenter MA, et al. First‐in‐class small molecule inhibitors of the single‐strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012;7(3):506‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Jouvenet N, Neil SJD, Zhadina M, et al. Broad‐spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83(4):1837‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kaletsky RL, Francica JR, Agrawal‐Gamse C, Bates P. Tetherin‐mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci USA. 2009;106(8):2886‐2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Radoshitzky SR, Dong L, Chi X, et al. Infectious Lassa virus, but not filoviruses, is restricted by BST‐2/tetherin. J Virol. 2010;84(20):10569‐10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Mansouri M, Viswanathan K, Douglas JL, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma‐associated herpesvirus. J Virol. 2009;83(19):9672‐9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Sharma A, Lal SK. Is tetherin a true antiviral: the influenza a virus controversy. Rev Med Virol. 2019;29(3):e2036. [DOI] [PubMed] [Google Scholar]
- 226. Goffinet C, Homann S, Ambiel I, et al. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV‐1) particle release and depletion of CD317 are separable activities of HIV‐1 Vpu. J Virol. 2010;84(8):4089‐4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Zhang Q, Liu Z, Mi Z, et al. High‐throughput assay to identify inhibitors of Vpu‐mediated down‐regulation of cell surface BST‐2. Antiviral Res. 2011;91(3):321‐329. [DOI] [PubMed] [Google Scholar]
- 228. Mi Z, Ding J, Zhang Q, et al. A small molecule compound IMB‐LA inhibits HIV‐1 infection by preventing viral Vpu from antagonizing the host restriction factor BST‐2. Sci Rep. 2015;5:18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang Q, Mi Z, Huang Y, et al. 2‐thio‐6‐azauridine inhibits Vpu mediated BST‐2 degradation. Retrovirology. 2016;13(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Mi Z, Wang X, He Y, et al. A novel peptide to disrupt the interaction of BST‐2 and Vpu. Biopolymers. 2014;102(3):280‐287. [DOI] [PubMed] [Google Scholar]
- 231. Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV‐1 Rev‐RRE export function. Cell. 2004;119(3):381‐392. [DOI] [PubMed] [Google Scholar]
- 232. Ariumi Y, Kuroki M, Abe K, et al. DDX3 DEAD‐box RNA helicase is required for hepatitis C virus RNA replication. J Virol. 2007;81(24):13922‐13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Noble CG, Chen YL, Dong H, et al. Strategies for development of dengue virus inhibitors. Antiviral Res. 2010;85(3):450‐462. [DOI] [PubMed] [Google Scholar]
- 234. Chahar HS, Chen S, Manjunath N. P‐body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology. 2013;436(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Högbom M, Collins R, van den Berg S, et al. Crystal structure of conserved domains 1 and 2 of the human DEAD‐box helicase DDX3X in complex with the mononucleotide AMP. J Mol Biol. 2007;372(1):150‐159. [DOI] [PubMed] [Google Scholar]
- 236. Maga G, Falchi F, Garbelli A, et al. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus‐1 replication targeting the human cellular aspartic acid−glutamic acid−alanine−aspartic acid box polypeptide 3. J Med Chem. 2008;51(21):6635‐6638. [DOI] [PubMed] [Google Scholar]
- 237. Maga G, Falchi F, Radi M, et al. Toward the discovery of novel anti‐HIV drugs. Second‐generation inhibitors of the cellular ATPase DDX3 with improved anti‐HIV activity: synthesis, structure‐activity relationship analysis, cytotoxicity studies, and target validation. ChemMedChem. 2011;6(8):1371‐1389. [DOI] [PubMed] [Google Scholar]
- 238. Bol GM, Vesuna F, Xie M, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med. 2015;7(5):648‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Samal SK, Routray S, Veeramachaneni GK, Dash R, Botlagunta M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci Rep. 2015;5:9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Garbelli A, Radi M, Falchi F, et al. Targeting the human DEAD‐box polypeptide 3 (DDX3) RNA helicase as a novel strategy to inhibit viral replication. Curr Med Chem. 2011;18(20):3015‐3027. [DOI] [PubMed] [Google Scholar]
- 241. Radi M, Falchi F, Garbelli A, et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: towards the next generation HIV‐1 inhibitors. Bioorg Med Chem Lett. 2012;22(5):2094‐2098. [DOI] [PubMed] [Google Scholar]
- 242. Han Z, Lu J, Liu Y, et al. Small‐molecule probes targeting the viral PPxY‐host Nedd4 interface block egress of a broad range of RNA viruses. J Virol. 2014;88(13):7294‐7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Loughran HM, Han Z, Wrobel JE, et al. Quinoxaline‐based inhibitors of Ebola and Marburg VP40 egress. Bioorg Med Chem Lett. 2016;26(15):3429‐3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Ji X‐Y, Chen J‐H, Zheng G‐H, et al. Design and synthesis of cajanine analogues against hepatitis C virus through down‐regulating host chondroitin sulfate N‐acetylgalactosaminyltransferase 1. J Med Chem. 2016;59(22):10268‐10284. [DOI] [PubMed] [Google Scholar]
- 245. Ji X, Xue S, Zheng G, et al. Total synthesis of cajanine and its antiproliferative activity against human hepatoma cells. Acta Pharm Sin B. 2011;1(2):93‐99. [Google Scholar]
- 246. Mesange F, Delarue F, Puel J, Bayard F, Faye JC. Ligands of the antiestrogen‐binding site are able to inhibit virion production of human immunodeficiency virus 1‐infected lymphocytes. Mol Pharmacol. 1996;50(1):75‐79. [PubMed] [Google Scholar]
- 247. Johansen LM, Brannan JM, Delos SE, et al. FDA‐approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5(190):190ra179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Zheng K, Chen M, Xiang Y, et al. Inhibition of herpes simplex virus type 1 entry by chloride channel inhibitors tamoxifen and NPPB. Biochem Biophys Res Commun. 2014;446(4):990‐996. [DOI] [PubMed] [Google Scholar]
- 249. Laurence J, Cooke H, Sikder SK. Effect of tamoxifen on regulation of viral replication and human immunodeficiency virus (HIV) long terminal repeat‐directed transcription in cells chronically infected with HIV‐1. Blood. 1990;75(3):696‐703. [PubMed] [Google Scholar]
- 250. Ikeda M, Abe K, Yamada M, Dansako H, Naka K, Kato N. Different anti‐HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44(1):117‐125. [DOI] [PubMed] [Google Scholar]
- 251. Sainz B Jr., Barretto N, Martin DN, et al. Identification of the Niemann‐Pick C1‐like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18(2):281‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Goldwasser J, Cohen PY, Lin W, et al. Naringenin inhibits the assembly and long‐term production of infectious hepatitis C virus particles through a PPAR‐mediated mechanism. J Hepatol. 2011;55(5):963‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Fujita N, Kaito M, Kai M, et al. Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J Viral Hepat. 2006;13(7):441‐448. [DOI] [PubMed] [Google Scholar]
- 254. You H, Yuan H, Fu W, et al. Herpes simplex virus type 1 abrogates the antiviral activity of Ch25h via its virion host shutoff protein. Antiviral Res. 2017;143:69‐73. [DOI] [PubMed] [Google Scholar]
- 255. Zhao P, Guo T, Qian L, et al. Ubiquitin C‐terminal hydrolase‐L3 promotes interferon antiviral activity by stabilizing type I‐interferon receptor. Antiviral Res. 2017;144:120‐129. [DOI] [PubMed] [Google Scholar]
- 256. Li H, Chen X, Zhou SJ. Dauricine combined with clindamycin inhibits severe pneumonia co‐infected by influenza virus H5N1 and Streptococcus pneumoniae in vitro and in vivo through NF‐kappaB signaling pathway. J Pharmacol Sci. 2018;137(1):12‐19. [DOI] [PubMed] [Google Scholar]
- 257. Ammari MG, Gresham CR, McCarthy FM, Nanduri B. HPIDB 2.0: a curated database for host‐pathogen interactions. Database Oxford. 2016;2016:baw103. [DOI] [PMC free article] [PubMed] [Google Scholar]