

Abstract
The structurally unique “fleximer” nucleosides were originally designed to investigate how flexibility in a nucleobase could potentially affect receptor–ligand recognition and function. Recently they have been shown to have low‐to‐sub‐micromolar levels of activity against a number of viruses, including coronaviruses, filoviruses, and flaviviruses. However, the synthesis of distal fleximers in particular has thus far been quite tedious and low yielding. As a potential solution to this issue, a series of proximal fleximer bases (flex‐bases) has been successfully coupled to both ribose and 2′‐deoxyribose sugars by using the N‐deoxyribosyltransferase II of Lactobacillus leichmannii (LlNDT) and Escherichia coli purine nucleoside phosphorylase (PNP). To explore the range of this facile approach, transglycosylation experiments on a thieno‐expanded tricyclic heterocyclic base, as well as several distal and proximal flex‐bases were performed to determine whether the corresponding fleximer nucleosides could be obtained in this fashion, thus potentially significantly shortening the route to these biologically significant compounds. The results of those studies are reported herein.
Keywords: biocatalysis, deoxyribosyltransferase, fleximers, nucleosides, purine nucleoside phosphorylase
A thieno‐expanded tricyclic base and several distal and proximal flex‐bases have been converted into their corresponding fleximer nucleosides by enzymatic transglycosylation reactions catalyzed by N‐deoxyribosyltransferase from L. leichmannii (LlNDT) or E. coli purine nucleoside phosphorylase (PNP).
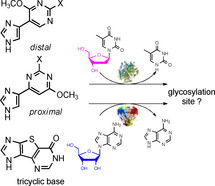
The synthesis of nucleoside analogues has been classically achieved through various chemical methodologies and well‐known coupling reactions.1, 2, 3 However, chemical synthesis typically involves difficult and time‐consuming multistep processes. Moreover, suitable protection of various functional groups is typically required on the nucleos(t)ide sugars and/or on the heterocyclic bases, and subsequent deprotection steps often result in low overall yields. Another significant problem often encountered is the stereospecific control of configuration at the anomeric center (α versus β).2, 3, 4
In contrast, enzymatic syntheses of nucleoside analogues do not typically require protecting groups and are highly stereospecific.5 Nucleoside phosphorylases (NPs) and N‐deoxyribosyl‐transferases (NDTs) are the predominant classes of enzymes used in the synthesis of nucleosides in which they mediate the transglycosylation of a nucleoside sugar to a free heterocyclic base.6, 7, 8, 9, 10
NPs (EC 2.4.2.1) catalyze the reversible phosphorolysis of ribo‐ or 2′‐deoxyribonucleosides to generate a free nucleobase and ribose‐ or 2‐deoxyribose‐1‐phosphate in the presence of inorganic ortho‐phosphate. Addition of a second nucleobase to the reaction mixture can promote the formation of a new nucleoside with the equilibrium in favor of nucleoside formation.11, 12 NPs have been reported to accept modified bases as well as unnatural glycosyl donors.6, 7, 8, 9, 13, 14, 15
NDTs (EC 2.4.2.6) catalyze the transfer of 2‐deoxyribose between purine and pyrimidine bases.16 Two NDT types have been defined based on their substrate specificity: NDT‐I (also named PTD), which is specific for purines,17 and NDT‐II, which accepts either purine or pyrimidine but has a strong preference for 2′‐deoxyribosyl‐pyrimidine as the donor substrate.18 Despite its relatively low acceptance for modified sugar moieties, NDT‐II has served to synthesize some nucleoside analogues of biological interest.19, 20 Interestingly, NDTs tolerate a wide range of modified nucleobases from azole derivatives21, 22 to expanded‐size purines23, 24, 25 with an increased regioselectivity as compared to purine nucleoside phosphorylase (PNP; i.e., N9 versus N7 for purines).6
Fleximers were originally designed to investigate how flexibility in a nucleobase could affect receptor–ligand recognition and function.26, 27, 28, 29, 30, 31 These interesting molecules have several advantages over their analogous natural rigid purine nucleosides. For instance, the distal guanosine fleximer (Figure 1) was found to be an inhibitor of S‐adenosyl‐l‐homocysteine hydrolase (SAHase), an adenosine‐metabolizing enzyme.26
Figure 1.
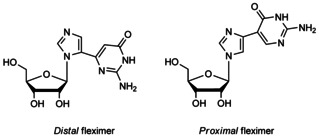
Seley‐Radtke's guanosine fleximers.
By rotation of the hemiaminal bond to place the heterocyclic moieties into a syn‐like conformation rather than the thermodynamically favored anti conformation, the flex‐guanosine's base was able to reposition the amino group or to mimic the adenine nucleobase.26, 32 Furthermore, the flex‐guanosine triphosphate (Flex‐GTP) was shown to be a superior substrate of human GDP‐l‐fucose pyrophosphorylase than the natural substrate GTP,33 likely due to the fleximer's ability to interact with amino acids in the active site not accessible by the natural substrate.34 This also allowed Flex‐GTP to retain all activity when essential catalytic residues needed for GTP binding were mutated.33, 34
Although the biological results for these compounds have been ground‐breaking in some cases, their syntheses are nontrivial. To date, the only approach to the adenosine, guanosine and inosine distal fleximer nucleosides requires the construction of a tricyclic expanded purine nucleoside; the synthesis involves more than seven steps to realize the base itself, with an additional five to seven steps after the coupling reaction to convert the tricyclic nucleoside to the fleximer.28, 29, 35, 36, 37, 38, 39, 40, 41 Attempts to synthesize the tricyclic bases followed by coupling of the base to a ribose resulted only in the undesired N7 isomer, likely due to steric problems caused by the arched shape.29, 41
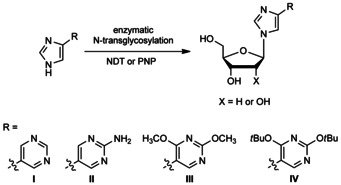
Previously, a series of imidazole nucleosides was successfully synthesized through transglycosylation methodologies.42, 43 Of those, four products resembled the Seley‐Radtke proximal fleximers (I–IV, Scheme 1). The flex‐bases were synthesized by microwave‐assisted Suzuki–Miyaura crosscoupling of 4(5)‐iodoimidazole and the boronic acid pyrimidine partners.44 Subsequent transglycosylation by using Escherichia coli PNP for ribonucleosides and LlNDT for 2′‐deoxyribonucleosides produced the corresponding proximal fleximers as major products.
Scheme 1.
As a few proximal fleximer nucleosides have been realized by enzymatic transglycosylation, the expectation was that the same methodology could produce the desired distal and tricyclic nucleosides from their respective bases. Thus, a series of tricyclic41 and flex‐bases45 (Figure 2) was synthesized as previously reported, and their transglycosylation products were studied.
Figure 2.
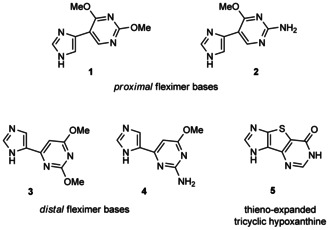
Flex‐ and tricyclic bases used for transglycosylation.
Enzymatic glycosylation mediated by LlNDT
First, we tested the capability of NDT from LlNDT to use these five unnatural nucleobases as substrate. Typically, the flex‐base (1 μmol) and thymidine used as the 2′‐deoxyribose donor (4 μmol) were incubated at 37 °C in the presence of LlNDT (different amounts of enzyme) in a 10 mm citrate buffer (0.1 mL) at the optimum pH of 6.5. Due to the low water solubility of flex‐bases, the transferase reactions were carried out in the presence of 5 % v/v DMSO in the medium.42 The rate of glycosylation (% conversion) was monitored by analytical reversed‐phase HPLC as a function of time and enzyme concentration (Table 1). The incubation temperature (37 in place of 50 °C) was selected in an attempt to optimize the enzyme activity and further isolate all the possible glycosylated products. Indeed, we previously showed that it is possible to manage the regioselectivity of the transferase reaction (N1 vs. N3) of some 4‐substituted imidazole derivatives.43 All the glycosylation products were purified by reversed‐phase HPLC, and their chemical structures, particularly the glycosylation site, were confirmed by NMR analysis.
Table 1.
Transglycosylation reactions of flex‐bases (1–5) catalyzed by LlNDT.[a]
|
|
Acceptor |
LlNDT |
Incubation |
Product [%][b] |
||
|---|---|---|---|---|---|---|
|
|
|
[μL] |
time [h] |
Starting |
N1‐glycosylated |
N3‐glycosylated |
|
|
|
|
|
material |
product |
product |
|
1 |
|
1.25 |
3 |
61 |
17 |
22 |
|
2 |
1.25 |
10 |
53 |
28 |
20 |
|
|
3 |
2.5 |
3 |
40 |
74 |
13 |
|
|
4 |
2.5 |
10 |
24 |
65 |
6 |
|
|
5 |
5.0 |
3 |
15 |
80 |
5 |
|
|
6 |
5.0 |
10 |
10 |
88 |
2 |
|
|
7 |
7.5 |
3 |
12 |
86 |
2 |
|
|
8 |
7.5 |
10 |
10 |
89 |
1 |
|
|
9 |
10.0 |
3 |
11 |
87 |
2 |
|
|
10 |
10.0 |
10 |
10 |
89 |
1 |
|
|
11 |
|
1.25 |
3 |
24 |
30 |
46 |
|
12 |
1.25 |
10 |
17 |
45 |
38 |
|
|
13 |
2.5 |
3 |
18 |
42 |
40 |
|
|
14 |
2.5 |
10 |
10 |
78 |
12 |
|
|
15 |
|
1.25 |
3 |
70 |
30 |
– |
|
16 |
1.25 |
10 |
20 |
80 |
– |
|
|
17 |
2.5 |
3 |
7 |
93 |
– |
|
|
18 |
2.5 |
10 |
4 |
96 |
– |
|
|
19 |
|
1.25 |
0.5 |
0 |
100 |
– |
|
20 |
0.63 |
0.5 |
18 |
82 |
– |
|
|
21 |
0.15 |
3 |
40 |
60 |
– |
|
|
22 |
|
1.25 |
3 |
2 |
62 and 32c |
4 |
|
23 |
1.25 |
10 |
2 |
46 and 44c |
6 |
|
|
24 |
0.63 |
3 |
3 |
71 and 23c |
3 |
|
|
25 |
0.63 |
10 |
2 |
58 and 34c |
6 |
|
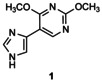
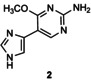
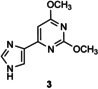
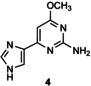
[a] Reaction conditions: 1 μmol acceptor in 5 % v/v DMSO, 4 μmol thymidine in 10 mm citrate buffer (pH 6.5; 0.1 mL) in the presence of LlNDT at 37 °C. [b] Percentage conversion was determined by reversed‐phase HPLC analysis of an aliquot of the incubation mixture monitored at 254 nm. [c] Product from glycosylation at both imidazole and pyrimidine nitrogens of 5.
As reported previously,42 the NDT‐catalyzed transfer reaction between 1 and thymidine led to the simultaneous formation of N1‐ and N3‐glycosylated products (1 a and 1 b, Scheme 2) in a 1:1 ratio after 3 h at 37 °C by using NDT at 1.25 μL/μmol acceptor (Table 1, entry 1). Nucleoside 1 b (i.e., C5 arylimidazole derivative) was progressively converted into the thermodynamically more stable N1 isomer 1 a (i.e., C4 aryl‐imidazole nucleoside; see Figure S1 A in the Supporting Information). By increasing the enzyme concentration (from 1.25 to 10.0 μL/μmol acceptor), the conversion was nearly complete, reaching a plateau at 90 %; nucleoside 1 a was formed as the sole product (Table 1, entries 7 and 8). A catalytic mechanism for LlNDT‐catalyzed glycosylation involving the formation of an oxocarbonium intermediate has recently been proposed.46
Scheme 2.
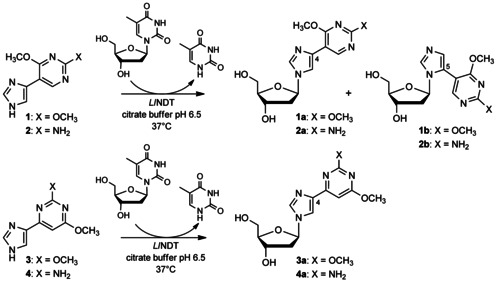
NDT‐catalyzed transglycosylation of flex‐bases 1–4.
A similar trend was observed in the case of flex‐base 2 with the formation of both N1‐ and N3‐glycosylated products (2 a and 2 b, respectively) in a ratio depending on incubation time or enzyme concentration (Figure S1 B). Flex‐base 2 appeared to be a better substrate than flex‐base 1 as a lower enzyme concentration was needed to achieve complete conversion (Table 1, entries 11–14). These results confirmed the easy accessibility of proximal 2′‐deoxy‐fleximers by using trans‐glycosylation reactions.
In the case of flex‐bases 3 and 4, HPLC monitoring showed that the glycosylation reactions were more effective than in the case of 1 (Figure S2 and Table 1, entries 15–21). Only one isomer, the N1‐glycosylated product 3 a or 4 a was formed, even if the enzyme concentration was reduced tenfold (entries 19–21). However, the target distal 2′‐deoxynucleosides (glycosylation at the N3 position of the imidazole ring) were not detected under the conditions tested.
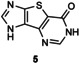
Finally, we tested the tricyclic nucleobase 5 (Scheme 3), which contains, in addition to the imidazole ring N1 and N3, a nitrogen on the pyrimidine ring that is susceptible to being glycosylated, as previously observed in the transglycosylation of 5‐(imidazol‐4‐yl)pyrimidine‐2,4‐(1H,3H)‐dione.42 Heterocycle 5 was rapidly converted into glycosylated products under the conditions used (Table 1, entries 22–25), thus confirming that hindered nucleobases can serve as acceptors for NDT. HPLC monitoring of the reaction showed the formation of two main products 5 a and 5 b, and a third minor one 5 c (Figure 3).
Scheme 3.
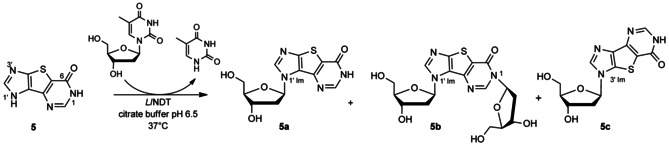
NDT‐catalyzed transglycosylation of flex‐base 5.
Figure 3.
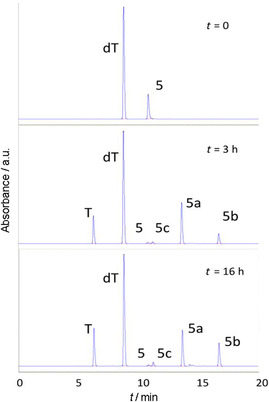
HPLC analysis at t=0, 3 h and 16 h of the transglycosylation reaction of 5 in the presence of thymidine (4 equiv) and NDT (0.63 μL/μmol acceptor) at 37 °C. HPLC conditions: 0–20 % linear gradient of acetonitrile in 10 mm TEAA buffer (pH 6.0) over 20 min at a flow rate of 1 mL min−1. Detection at 254 nm.
Structures of 5 a (glycosylation at imidazole N1, noted N1′‐Im in Scheme 3), 5 b (glycosylation at both imidazole N1 and pyrimidine N1, noted N3′ Im and N1′), and 5 c (glycosylation at imidazole N3, noted N3′ Im) were assigned based on NMR spectra (Figure 4).
Figure 4.
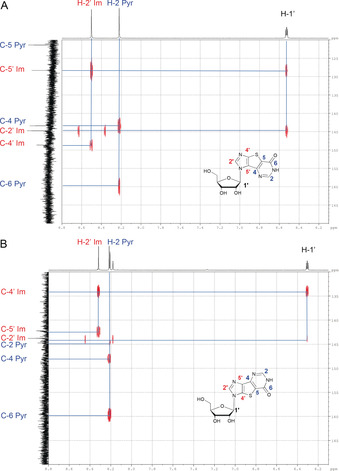
Expansion of the 1H[13C] HMBC NMR spectra of compounds A) 5 a and B) 5 c.
By reducing incubation time and enzyme concentration, it was possible to control further conversion of 5 a into 5 b, and thus isolate 5 a as the main product (Table 1, entry 24). Interestingly, this enzymatic procedure is an attractive route to 5 a over the previously published ones,29, 37 based on the coupling of 4,5‐diiodoimidazole to a protected 2‐deoxysugar chloride, followed by the formation of the tricyclic inosine derivative and sugar deprotection.
Enzymatic glycosylation mediated by PNP
We also tested the ability of PNP to use flex‐bases 1 and 3 and the thieno‐expanded tricyclic base 5 for ribonucleoside synthesis (Scheme 4). Reaction mixtures (0.1 mL) contained the flex‐base (10 mm), adenosine (20 mm) as a donor, and bacterial PNP (0.2 U μmol−1) in 10 mm phosphate buffer (pH 7.4). Incubation was initially performed at 50 °C. Conversion of 3 into the corresponding ribonucleoside 3 c is slow and reached a plateau at 75 % after 16 h at 50 °C, as previously observed for PNP‐catalyzed ribosylation of 1.42
Scheme 4.
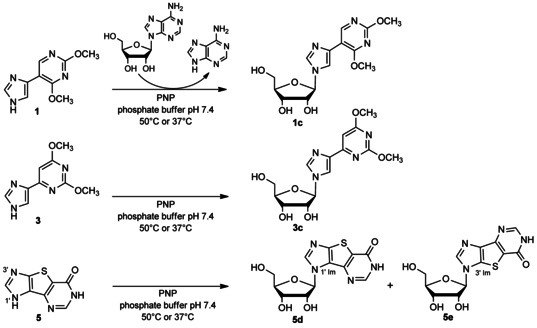
PNP‐mediated transglycosylation of flex bases 1, 3 and 5.
In the case of flex‐base 5, conversion is much more effective (80 % after 2 h at 50 °C), and two monoglycosylated products (5 d and 5 e) were formed, the target compound 5 d being the major one (Scheme 4). When incubation was performed at 37 °C, the formation of 5 e increased as compared to 50 °C, allowing further characterization (Figure S3). The glycosylation sites of 5 d and 5 e at the N1′ or N3′ position of imidazole ring, respectively, were assigned based on NMR analysis (see the Supporting Information). It should be noted that chemical glycosylation of 5 by using classical procedures yielded nucleoside 5 e (glycosylation at N3′ position of imidazole ring) as the sole product.29
In conclusion, the four flex‐bases 1–4 and the tricyclic nucleobase 5 were found to be substrates for the nucleoside 2′‐deoxyribosyltransferase from Lactobacillus leichmannii (LlNDT) and bacterial purine nucleoside phosphorylase (PNP). Experimental conditions were designed to manage the regioselectivity in the NDT‐catalyzed transfer reaction (i.e., at the N1 versus N3 position of the imidazole ring) and also the bis‐glycosylation side reaction (at the pyrimidine nitrogen). Thus, proximal fleximers in both deoxy and ribose series are easily obtained from flex‐bases 1 and 2 by transglycosylation catalyzed by NDT and PNPase, respectively. On the other hand, distal fleximers are not accessible from the corresponding flex‐bases 3 and 4, as glycosylation catalyzed by LlNDT or PNP occurs at the N1 position of the imidazole ring. Importantly, enzymatic glycosylation of the tricyclic base 5 into the deoxy‐ and ribonucleoside 5 a and 5 d should provide a simple and efficient chemoenzymatic access to the target distal deoxy‐ and ribo‐inosine fleximers, thereby overcoming the tedious and low‐yielding approach that has been used to date.
The chemoenzymatic approach compares very well with the classical chemical routes to distal and proximal fleximer nucleosides, as only one anomer and one regioisomer were obtained. This strategy will now be useful for the preparation of other distal fleximers, that is, A and G analogues.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors from the IP would like to thank Frédéric Bonhomme (UMR3523) for assisting with HRMS analysis and Drs. Bruno Vitorge and Inaki Guijarro (BioNMR platform) for providing access to Avance 600 and Avance Neo 800 spectrometers. This work was funded by the Institut Pasteur and the Centre National de la Recherche Scientifique (CNRS; S.V.‐G. and S.P.) and the National Institutes of Health/NIAID R21 AI118470‐01 (K.S.‐R.) and T32 GM066706 (K.S.‐R. and T.K.).
S. Vichier-Guerre, T. C. Ku, S. Pochet, K. L. Seley-Radtke, ChemBioChem 2020, 21, 1412.
Contributor Information
Dr. Sylvie Pochet, Email: sylvie.pochet@pasteur.fr.
Prof. Katherine L. Seley‐Radtke, Email: kseley@umbc.edu.
References
- 1. Townsend L. B., Tipson R. S., Nucleic Acid Chemistry: Improved and New Synthetic Procedures, Methods, and Techniques, Wiley, New York, 1978. [Google Scholar]
- 2. Merino P., Chemical Synthesis of Nucleoside Analogues, Wiley, Hoboken, 2013. [Google Scholar]
- 3. Vorbrüggen H., Ruh-Pohlenz C., Handbook of Nucleoside Synthesis, Wiley, New York, 2001. [Google Scholar]
- 4. Downey A. M., Pohl R., Roithova J., Hocek M., Chem. Eur. J. 2017, 23, 3910–3917. [DOI] [PubMed] [Google Scholar]
- 5. Fernández-Lucas J., Camarasa M.-J., Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives, Wiley-VCH, Weinhein, 2018. [Google Scholar]
- 6. Mikhailopulo I. A., Curr. Org. Chem. 2007, 11, 317–335. [Google Scholar]
- 7. Mikhailopulo I. A., Miroshnikov A. I., Acta Naturae 2010, 2, 36–58. [PMC free article] [PubMed] [Google Scholar]
- 8. Condezo L. A., Fernández-Lucas J., García-Burgos C. A., Alcántara A. R., Sinisterra J. V., Biocatalysis in the Pharmaceutical and Biotechnology Industries; (Ed.: R. N. Patel), CRC Press, Boca Raton, 2006. [Google Scholar]
- 9. Pérez E., Sánchez-Murcia P. A., Jordaan J., Blanco M. D., Mancheño J. M., Gago F., Fernández-Lucas J., ChemCatChem 2018, 10, 4406–4416. [Google Scholar]
- 10. Fresco-Taboada A., de la Mata I., Arroyo M., Fernández-Lucas J., Appl. Microbiol. Biotechnol. 2013, 97, 3773–3785. [DOI] [PubMed] [Google Scholar]
- 11. Krenitsky T. A., Koszalka G. W., Tuttle J. V., Biochemistry 1981, 20, 3615–3621. [DOI] [PubMed] [Google Scholar]
- 12. Hennen W. J., Wong C. H., J. Org. Chem. 1989, 54, 4692–4695. [Google Scholar]
- 13. Slater M. J., Gowrie C., Freeman G. A., Short S. A., Bioorg. Med. Chem. Lett. 1996, 6, 2787–2790. [Google Scholar]
- 14. Fateev I. V., Antonov K. V., Konstantinova I. D., Muravyova T. I., Seela F., Esipov R. S., Miroshnikov A. I., Mikhailopulo I. A., Beilstein J. Org. Chem. 2014, 10, 1657–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fateev I. V., Kharitonova M. I., Antonov K. V., Konstantinova I. D., Stepanenko V. N., Esipov R. S., Seela F., Temburnikar K. W., Seley-Radtke K. L., Stepchenko V. A., Sokolov Y. A., Miroshnikov A. I., Mikhailopulo I. A., Chem. Eur. J. 2015, 21, 13401–13419. [DOI] [PubMed] [Google Scholar]
- 16. Danzin C., Cardinaud R., Eur. J. Biochem. 1976, 62, 365–372. [DOI] [PubMed] [Google Scholar]
- 17. Kaminski P. A., J. Biol. Chem. 2002, 277, 14400–14407. [DOI] [PubMed] [Google Scholar]
- 18. Cardinaud R., Holguin J., Biochim Biophys Acta 1979, 568, 339–347. [DOI] [PubMed] [Google Scholar]
- 19. Van Draanen N. A., Freeman G. A., Short S. A., Harvey R., Jansen R., Szczech G., Koszalka G. W., J. Med. Chem. 1996, 39, 538–542. [DOI] [PubMed] [Google Scholar]
- 20. Carson D. A., Wasson D. B., Biochem. Biophys. Res. Commun. 1988, 155, 829–834. [DOI] [PubMed] [Google Scholar]
- 21. Betbeder D., Hutchinson D. W., Nucleosides Nucleotides 1990, 9, 569–577. [Google Scholar]
- 22. Pochet S., Dugué L., Meier A., Marlière P., Bioorg. Med. Chem. Lett. 1995, 5, 1679–1684. [Google Scholar]
- 23. Ye W. J., Paul D., Gao L. N., Seckute J., Sangaiah R., Jayaraj K., Zhang Z. F., Kaminski P. A., Ealick S. E., Gold A., Ball L. M., PLoS One 2014, 9, e115082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Müller M., Hutchinson L. K., Guengerich F. P., Chem. Res. Toxicol. 1996, 9, 1140–1144. [DOI] [PubMed] [Google Scholar]
- 25. Stachelska-Wierzchowska A., Wierzchowski J., Górka M., Bzowska A., Wielgus-Kutrowska B., Molecules 2019, 24, 1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seley K. L., Quirk S., Salim S., Zhang L., Hagos A., Bioorg. Med. Chem. Lett. 2003, 13, 1985–1988. [DOI] [PubMed] [Google Scholar]
- 27. Seley K. L., Salim S., Zhang L., Org. Lett. 2005, 7, 63–66. [DOI] [PubMed] [Google Scholar]
- 28. Seley K. L., Zhang L., Hagos A., Org. Lett. 2001, 3, 3209–3210. [DOI] [PubMed] [Google Scholar]
- 29. Seley K. L., Zhang L., Hagos A., Quirk S., J. Org. Chem. 2002, 67, 3365–3373. [DOI] [PubMed] [Google Scholar]
- 30. Peters H. L., Ku T. C., Seley-Radtke K. L., Curr. Med. Chem. 2015, 22, 3910–3921. [DOI] [PubMed] [Google Scholar]
- 31. Zucman-Rossi J., Clement B., Buendia M. A., Lerat H., Beers B. V., Bedossa P., Taieb J., Rosenbaum J., Bull. Cancer 2009, 96, 45–50. [DOI] [PubMed] [Google Scholar]
- 32. Polak M., Seley K. L., Plavec J., J. Am. Chem. Soc. 2004, 126, 8159–8166. [DOI] [PubMed] [Google Scholar]
- 33. Quirk S., Seley K. L., Biochemistry 2005, 44, 10854–10863. [DOI] [PubMed] [Google Scholar]
- 34. Quirk S., Seley K. L., Biochemistry 2005, 44, 13172–13178. [DOI] [PubMed] [Google Scholar]
- 35. Ku C. T., Seley-Radtke K. in Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives (Eds.: J. Fernández Lucas, M.-J. Camarasa Rius), Wiley-VCH, Weinheim, 2019, pp. 195–235. [Google Scholar]
- 36. Chen Z., Jochmans D., Ku T., Paeshuyse J., Neyts J., Seley-Radtke K. L., ACS Infect. Dis. 2015, 1, 357 – 366. [DOI] [PubMed] [Google Scholar]
- 37. Wauchope O. R., Velasquez M., Seley-Radtke K., Synthesis 2012, 44, 3496–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wauchope O. R., Tomney M. J., Pepper J. L., Korba B. E., Seley-Radtke K. L., Org. Lett. 2010, 12, 4466–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tomney M., Korba B., Zimmermann S., Seley-Radtke K., Antiviral Res. 2010, 86, A68–A68. [Google Scholar]
- 40. Zhang Z. B., Wauchope O. R., Seley-Radtke K. L., Tetrahedron 2008, 64, 10791–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seley K. L., Januszczyk P., Hagos A., Zhang L., Dransfield D. T., J. Med. Chem. 2000, 43, 4877–4883. [DOI] [PubMed] [Google Scholar]
- 42. Vichier-Guerre S., Dugué L., Bonhomme F., Pochet S., Org. Biomol. Chem. 2016, 14, 3638–3653. [DOI] [PubMed] [Google Scholar]
- 43. Vichier-Guerre S., Dugué L., Bonhomme F., Pochet S., Org. Biomol. Chem. 2017, 15, 8193–8203. [DOI] [PubMed] [Google Scholar]
- 44. Vichier-Guerre S., Dugué L., Pochet S., Tetrahedron Lett. 2014, 55, 6347–6350. [Google Scholar]
- 45. Ku T., Lopresti N., Shirley M., Mori M., Marchant J., Heng X., Botta M., Summers M. F., Seley-Radtke K. L., Bioorg. Med. Chem. 2019, 27, 2883–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. del Arco J., Perona A., González L., Fernández-Lucas J., Gago F., Sánchez-Murcia P. A., Org. Biomol. Chem. 2019, 17, 7891–7899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary