

EccDNAs are hiding in plain sight in conventional ATAC-seq data.
Abstract
Extrachromosomal circular DNAs (eccDNAs) are somatically mosaic and contribute to intercellular heterogeneity in normal and tumor cells. Because short eccDNAs are poorly chromatinized, we hypothesized that they are sequenced by tagmentation in ATAC-seq experiments without any enrichment of circular DNA. Indeed, ATAC-seq identified thousands of eccDNAs in cell lines that were validated by inverse PCR and by metaphase FISH. ATAC-seq in gliomas and glioblastomas identify hundreds of eccDNAs, including one containing the well-known EGFR gene amplicon from chr7. More than 18,000 eccDNAs, many carrying known cancer driver genes, are identified in a pan-cancer analysis of ATAC-seq libraries from 23 tumor types. Somatically mosaic eccDNAs are identified by ATAC-seq even before amplification is recognized by genome-wide copy number variation measurements. Thus, ATAC-seq is a sensitive method to detect eccDNA present in a tumor at the pre-amplification stage and can be used to predict resistance to therapy.
INTRODUCTION
ATAC-seq (assay for transposase-accessible chromatin using sequencing) identifies open chromatin regions all across the genome (1). The method uses the hyperactive transposase Tn5 to cut the accessible chromatin with simultaneous ligation of adapters at cut sites (1). To reduce the contamination of mitochondrial DNA in library preparation, the nuclear pellets are isolated first from cells or tissues before the tagmentation step (2).
We previously reported the presence of tens of thousands of extrachromosomal circular DNA (eccDNA) in the nuclei of human and mouse cell lines as well as normal tissues and cancers using paired-end high-throughput sequencing of circular DNA–enriched preparations (3–5). Several other groups have also used similar approaches to describe the presence of eccDNAs in various eukaryotes ranging from yeasts to humans (6–11). More recently, it has been shown that circular DNA promotes the expression of oncogenes (12). Not only the oncogenes but also the regulatory regions associated with genes are also amplified as eccDNA (13). Since isolated nuclei as a whole are subjected to the transposition reaction in ATAC-seq, we hypothesized that the transposase will also cleave DNA from eccDNAs, so the ATAC-seq libraries will contain fragments of DNA from eccDNA.
To test our hypothesis, we first prepared ATAC-seq libraries using C4-2B (prostate cancer) and OVCAR8 (ovarian cancer) cell lines and identified hundreds of eccDNAs using our newly developed computational pipeline. Inverse polymerase chain reaction (PCR) on exonuclease-resistant eccDNA (highly enriched in circular DNA) and fluorescence in situ hybridization (FISH) on metaphase spreads confirmed the presence of the identified somatically mosaic eccDNA. To provide additional evidence of the success of ATAC-seq in identifying eccDNA, we analyzed an ATAC-seq library generated from patient-derived glioblastoma (GBM) cell lines (14) and identified the eccDNA harboring epidermal growth factor receptor (EGFR) gene, which is amplified through the formation of eccDNA in GBM. Last, we analyzed ATAC-seq data from GBM and low-grade glioma (LGG) generated by The Cancer Genome Atlas (TCGA) consortium to identify hundreds of eccDNAs even before their amplification was apparent as a copy number variation (CNV) by hybridization to single-nucleotide polymorphism arrays. Genes involved in pathways related to nucleosomal events were significantly enriched in these loci.
RESULTS
Principle of circular DNA identification by tagmentation method
eccDNAs are known to have chromosomal origin. A linear DNA fragment is generated either by the chromosome breakage due to adjoining DNA breaks, e.g., in chromothripsis (15), or by DNA synthesis related to DNA replication or repair. The two ends of a linear DNA are ligated to make a circular DNA (Fig. 1A), creating a specific junctional sequence that is not present in the normal reference genome. We have developed a very simple method to identify eccDNAs by collecting all the read pairs where one read of a pair maps uniquely to the genome in a contiguous manner [≤5–base pair (bp) insertions and/or deletions and/or substitutions] and the other read maps as a split read (noncontiguous segments that could be as far apart as a few megabases but usually are much closer) flanking the mapped read (Fig. 1B). The split read maps to the circular DNA ligation junction, and the other (contiguously mapped read) maps to the body of the putative eccDNA. The start of the first split read and the end of the second read are annotated as the start and end of the eccDNA. Tandem duplication of DNA in the genome will also create a similar junctional sequence, but for the purpose of identifying incipient gene amplification, an eccDNA or a tandem duplication of a chromosomal segment is equally important. However, if the aim is to exclusively and comprehensively identify eccDNA, then the ATAC-seq library should be prepared from eccDNA-enriched samples where linear DNA has been removed by exonuclease digestion. The complete pipeline to identify eccDNA coming from one locus (nonchimeric eccDNA) of any length is available through our GitHub page (https://github.com/pk7zuva/Circle_finder and https://github.com/pk7zuva/Circle_finder/blob/master/circle_finder-pipeline-bwa-mem-samblaster.sh). The steps to find a circular DNA from any paired-end high-throughput sequencing library are detailed in Fig. 1 (B and C) and Materials and Methods.
Fig. 1. A schematic to show that a circle could be part of an ATAC-seq library.
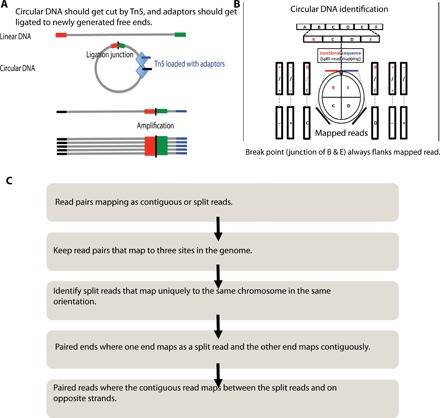
(A) If circular DNA has open chromatin structure near or around the ligation point, then the library preparation method will cut and attach an adaptor into a DNA fragment from eccDNA. (B) One end of paired-end read mapping on the body of a circular DNA with read from the other end mapping on the ligation junction. (C) Detailed steps from mapping to identification of the new Circle_finder pipeline.
Application of ATAC-seq to identify circular DNA in OVCAR8 and C4-2B cell lines
We prepared ATAC-seq libraries from C4-2B prostate cancer and OVCAR8 ovarian cancer cell lines. The sequencing and mapping statistics are given in table S1. Less than 90% of the reads were mapped to human genome, and the computational pipeline identified hundreds of circular DNA. The length distribution of eccDNA is shown in Fig. 2A: Around 68% in C4-2B and 37% in OVCAR8 of eccDNA are <1 kb and, so, are similar to the microDNAs that we identified earlier in normal and cancer cells (4, 5). However, 32% of the eccDNA in C4-2B and 63% in OVCAR8 are >1 kb, including eccDNAs long enough to encode gene segments or even complete genes. The eccDNA are derived from all the chromosomes (Fig. 2B). As a positive control, we identified hundreds of junctional sequences from the circular mitochondrial genome contaminating the nuclear preparations.
Fig. 2. eccDNA in C4-2 and OVCAR8 cell lines.
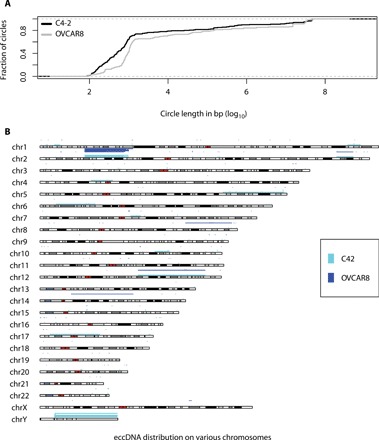
(A) Length distribution of identified eccDNA in C4-2 and OVCAR8 cell lines. (B) Karyotype plot showing chromosomal distribution of C4-2 and OVCAR8 cell lines.
Validation of eccDNA identified in C4-2 and OVCAR8 cells by inverse PCR
To confirm that the identified junctions are genuinely from eccDNA and not from tandem genome duplications, we isolated circular DNA by our previously described method that relies on column chromatography and exonuclease digestion to remove all linear DNA and enrich eccDNA (Fig. 3A; see Materials and Methods for more details) (5). Inverse PCR was performed with primers designed to amplify across the junctions of eccDNAs from C4-2B and OVCAR8 cell lines (Fig. 3B). Eleven eccDNAs from OVCAR8 and six from C4-2B were tested. Nine of the 11 targets from OVCAR8 and 2 of the 6 from C4-2B gave amplicons of expected sizes (Fig. 3, B and C). Sanger sequencing of the amplicons confirmed the junctional sequences identified by ATAC-seq (Fig. 3D). A fraction of the primers (two in OVCAR8 and four in C4-2B) did not give desired amplicons possibly because of their presence in low complexity regions or because they came from tandem linear chromosomal duplications, which did not survive column chromatography and exonuclease digestion.
Fig. 3. Experimental validation of randomly selected eccDNA identified by ATAC-seq in C4-2B and OVCAR8 cells.
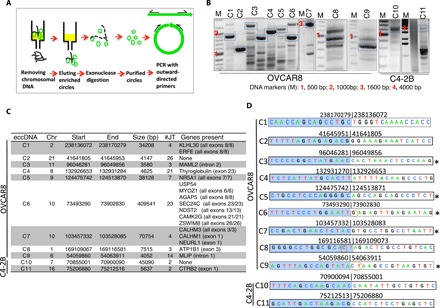
(A) Schematic for isolation and detection of eccDNA. See Materials and Methods for details. (B) PCR detection of eccDNA. DNA bands marked with blue boxes were gel-purified and sequenced. (C) Description of eccDNAs validated in (B) on the basis of analysis of ATAC-seq data from OVCAR8 and C4-2B. (D) Junctional tags obtained after sequencing of PCR products in (B). Shaded (blue) and unshaded sequences depict 15 bases on either side of junctions. Numbers indicate chromosomal location on respective chromosomes. Note the match between numbers for each circle in (C) and (D). Some of the junction sequence identified by Sanger sequencing differ by few bases because of multiple species of eccDNA present in the given cell lines. Oval circles represent insertion, and boxed sequences represent mismatches. *Sequence obtained from the bottom strand.
Validation of eccDNA by metaphase FISH in OVCAR8 cells
An independent method for ascertaining whether a locus identified in this study is in an extrachromosomal DNA is to carry out FISH on metaphase spreads. We performed this analysis with two loci that were predicted to be present as either an eccDNA or a gene duplication in OVCAR8 cells but not in C4-2 cells: chr2:238136071-238170279 and chr10:103457331-103528085. Both were confirmed by inverse PCR in Fig. 3 (C1 and C7). Signal was detected off the main chromosomes in some of the metaphase spreads, but not others (Fig. 4A), consistent with the hypothesis that the junctional sequences identify somatically mosaic eccDNA in this cell line. For negative control C4-2B (Fig. 4B), the spreads do not show an extrachromosomal DNA signal. The 71-kb eccDNA in OVCAR8 (n = 28) and C4-2B (n = 24) (negative control) metaphase spreads were quantified for locus chr10:103457331-103528085 and shown in the graph (Fig. 4C).
Fig. 4. eccDNA in cell lines and LGG or GBM tumors.
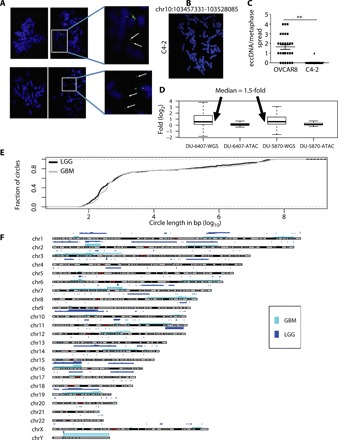
(A) Detection of eccDNA in OVCAR8 cell line by FISH: Metaphase spread of chromosome (blue) from OVCAR8 cells were stained with the probe (green) against the eccDNA locus chr2:238136071-238170279 (top row) or chr10:103457331-103528085 (bottom row). The spreads on the left do not have an extrachromosomal signal, while the spreads on the right have extrachromosomal signals that are better seen in the magnified insets on the extreme right. White arrows mark the eccDNA signals. (B) For the negative control cell lines, C4-2, the spread does not have an extrachromosomal DNA signal. (C) The eccDNA signals in OVCAR8 (n = 28) and C4-2 (n = 24) (negative control) were quantified for locus chr10:103457331-103528085 and shown in the graph. P values were calculated using Student’s t test; **P < 0.01. (D) eccDNA/duplication loci identified in whole-genome sequencing (WGS) libraries show genomic amplification (median, 1.5-fold), suggesting at least one allele is duplicated in all the cells. eccDNA loci identified in ATAC-seq libraries do not show genomic amplification (median close to zero), suggesting that the eccDNA are apparent before a CNV can be detected at the locus. The value of copy number amplification (CNA) in the y axis is in log2. (E) Length distribution of eccDNA identified in LGG and GBM TCGA ATAC-seq data. (F) Karyotype plot showing chromosomal distribution of eccDNA identified in LGG and GBM from TCGA ATAC-seq data.
Identification of eccDNA from ATAC-seq data for GBM cell lines
EGFR was one of the first oncogenes identified in brain cancer and is massively amplified in some patients with GBM (16). This somatic CNV is present in 43% of patients with GBM (17). Recent studies have provided further evidence that this oncogenic amplification occurs on eccDNA (9, 11, 18). To check whether we can detect the eccDNA in ATAC-seq data generated from GBM cell lines, we turned to six ATAC-seq libraries generated from GBM cell lines developed from a single patient with GBM (14). We ran the Circle_finder pipeline combining all the six libraries (GSM3318539, GSM3318540, GSM3318541, GSM3318542, GSM3318543, and GSM3318544) and found 58 eccDNAs varying in size from few hundred bases to few megabases. The length distribution and chromosomal distribution of identified eccDNAs are shown in fig. S1 (A and B). eccDNA harboring the EGFR gene was the most abundant eccDNA. The top five most abundant eccDNAs (or tandem gene duplications) identified were chr4:118591708-119454712 (METTL14, SEC24D, SYNPO2, MYOZ2, USP53, C4orf3, and FABP2), chr7:54590796-55256528 (SEC61G and EGFR), chr7:54771165-54782815(no protein-coding genes), chr7:65038261-65873269 (transcribed unprocessed pseudogenes), and chr7:65038264-65873256 (transcribed unprocessed pseudogenes).
Application to GBM and LGG TCGA ATAC-seq data
Having demonstrated above that ATAC-seq data can be repurposed to identify eccDNA, we turned our attention to ATAC-seq data generated by TCGA consortium (2) with a primary focus on two LGGs for which we have whole-genome sequencing (WGS) data and ATAC-seq data. In the TCGA-DU-5870-02A ATAC-seq library, we found 21 eccDNAs (junctional tag ≥2; 13, >1 kb and 7, >50 kb). In the TCGA-DU-5870-02A WGS library, we found 637 eccDNAs (junctional tag ≥2; 361, >1 kb and 105, > 50 kb). We further compared the eccDNAs identified in ATAC-seq and WGS libraries and found 21 common eccDNAs (junctional tag ≥1; table S2A).
In the ATAC-seq library from TCGA-DU-6407-02B, we found 64 eccDNAs (junctional tag ≥2; 21, >1 kb and 15, >50 kb), and in WGS libraries from the same tumor, we found 455 eccDNAs (junctional tag ≥2; 307, >1 kb and 131, >50 kb). Forty-four common eccDNAs were identified in both libraries (junctional tag ≥1; table 2B). Many of the common eccDNAs had a high number of junctional tags in the WGS library, perhaps a surrogate marker of their abundance.
We see a higher number of eccDNA/duplication events in WGS compared to ATAC-seq, but 21 and 44 eccDNAs were common between ATAC-seq and WGS in TCGA-DU-5870-02A and TCGA-DU-6407-02B libraries, respectively (table S2, A and B). The lack of more overlap between the eccDNAs identified by ATAC-seq and WGS from even the same tumor is most likely due to somatic mosaicism (i) because different sections are used for the two libraries and (ii) because of insufficient depth of sequencing in either library.
As mentioned earlier, the Circle_finder algorithm cannot distinguish between an extrachromosomal circle and chromosomal segmental tandem duplication without experimentally purifying the circles before library preparation, so we will refer to these loci as eccDNA/duplication. The signal for the eccDNA/duplication detected from WGS data was strong in the two tumors and was also evident in a targeted copy number analysis from the WGS data (not a genome-wide analysis). The median sequencing read coverage at the eccDNA/duplication loci was 1.5-fold higher compared to equivalent upstream or downstream regions, suggesting that at least a twofold amplification of one allele occurred in at least 50% of the cells. Unexpectedly, the eccDNA/duplication events detected by ATAC-seq did not show corresponding amplification in WGS (Fig. 4D). This result suggests that as with eccDNAs detected by rolling circle amplification, the eccDNA/duplication events identified by ATAC-seq are somatically mosaic in the GBM cell lines and are detected even before a CNV is apparent from WGS of a large population of tumor cells.
We next analyzed 10 LGG and 8 GBM ATAC-seq libraries and found a total of 2152 and 3147 eccDNA/duplication events in LGG and GBM samples, respectively. The length distribution of eccDNA/duplications is shown in Fig. 4E. Fifty-eight percent of the loci are <1 kb (similar to microDNA), but nearly 41% (2200 eccDNAs in GBM + LGG) are 50 kb to 50 Mb in length, suggesting that they harbor full-length genes. The chromosomal distribution of eccDNA identified (junctional tag ≥2) in LGG and GBM samples is shown in Fig. 4F. The EGFR locus was contained in the eccDNA/duplication identified in patients with GBM, supporting our hypothesis that the use of Circle_finder in ATAC-seq data can identify loci that have been amplified even in a subset of the cells in the tumor.
Cumulative analysis of all small eccDNA (microDNA)
After pooling all the eccDNA identified so far in this paper (OVCAR8 + C4-2 + 8 GBM + 10 LGG), we focused on the ones <1 kb to compare their properties with the microDNA that we have identified earlier by rolling circle amplification (4, 5). We found 4073 eccDNA that were <1kb. The length distribution of these circles reveals characteristic peaks at ~200 and~400 bases (Fig. 5A) that we have noted earlier. The higher GC content relative to the genome average (Fig. 5B) and the enrichment of the microDNA from upstream of genes, 5′ untranslated region (5′UTR), and CpG islands (Fig. 5C) are also similar to our previous reports. Last, around 15% of the small eccDNAs reported here appear to have used flanking sequences of 2- to 15-base microhomology (Fig. 5D) to promote the ligation that gives rise to the circle.
Fig. 5. Properties of microDNA identified in this paper (eccDNA <1 kb) by ATAC-seq.
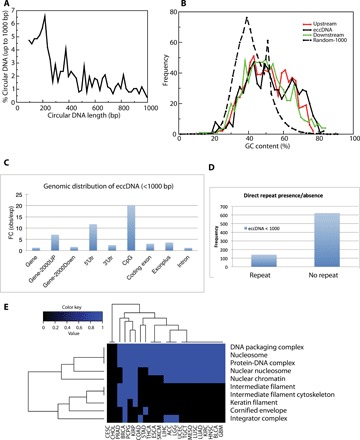
(A) Length distribution of eccDNA shows peaks at 180 and 380 bases. (B) GC content of eccDNA locus and regions immediately upstream and downstream from the eccDNA is higher than genomic average, as calculated from 1000 random stretches of the genome of equivalent length as the eccDNA (Random-1000). (C) The sites in the genome that give rise to small eccDNA are enriched relative to random expectation in genic sites, sequences 2 kb upstream from genes and in CpG islands. FC, Fold Change. (D) Direct repeats of 2 to 15 bp flanking the genomic locus of the eccDNA at ligation point are present for ~20% of the loci. (E) Gene classes enriched in the set of genes found on the circular DNAs in two or more cancers. The color scale indicates enrichment in pathway (blue color indicates pathway was enriched). If the genes found on the eccDNA/duplication loci in a cancer type are significantly enriched in the indicated pathways, then the color in the cell is blue. If the set of genes is not enriched in that cancer, then the cell is black.
Pan-cancer analysis of eccDNA in TCGA ATAC-seq data
Last, we analyzed 360 ATAC-seq libraries from 23 tumor types generated by TCGA consortium (see the Supplementary Materials) (2). We found a total of 18,143 eccDNAs/duplications of which 86% were <1kb. The coordinates of eccDNA identified in each library are available through our laboratory webpage (http://genome.bioch.virginia.edu/TCGA-ATACSEQ-ECCDNA/). The unique eccDNA intervals were used to extract the full-length genes harbored inside the circle. The cancer driver genes (19) amplified as eccDNA/duplication in individual tumor type are shown in Table 1. Gene ontology analysis of all the genes carried on the eccDNA/duplication loci shows that pathways related to nucleosomal events are significantly enriched in these loci (Fig. 5E).
Table 1. Known cancer driver genes amplified in eccDNA/gene duplications (JTGE1) in various tumor types.
| ACC | FGFR2, H3F3A, FOXA1, SMARCA4, NFE2L2, PMS1, SF3B1, SOS1, PCBP1, KIT, EGFR, GNAQ |
| BLCA | ERCC2, GRIN2D, PPP2R1A, SOS1, PCBP1, MSH3 |
| BRCA |
FGFR2, MTOR, WT1, SF1, CCND1, PTPRC, PTPN11, KRAS, H3F3A, ERBB3, KLF5, MACF1, AKT1, FOXA1, MAP2K1, IDH2, ERBB2, SPOP, SETBP1, SMARCA4, CACNA1A, PIK3R2, GNA11, ERCC2, GRIN2D, PPP2R1A, U2AF1, NFE2L2, PMS1, SF3B1, IDH1, MAPK1, SOS1, PCBP1, CTNNB1, RHOA, FBXW7, KIT, MSH3, PIK3CG, UNCX, BRAF, CUL1, EGFR, GTF2I, MYC, SOX17, GNAQ, EIF1AX |
| CESE | MTOR, PTPRC, H3F3A, NFE2L2, PMS1, GNAQ |
| COAD |
FGFR2, MTOR, WT1, SF1, CCND1, PTPRC, KRAS, H3F3A, ERBB3, CDK4, CHD4, ARID1A, KLF5, MACF1, FOXA1, MAP2K1, IDH2, ERBB2, SPOP, SETBP1, SMARCA4, CACNA1A, PIK3R2, GNA11, ERCC2, GRIN2D, PPP2R1A, GNAS, U2AF1, NFE2L2, PMS1, SF3B1, IDH1, MAPK1, PLXNB2, SOS1, PCBP1, PIK3CA, CTNNB1, RHOA, MSH3, EEF1A1, PIK3CG, BRAF, CUL1, EGFR, GTF2I, SOX17, GNAQ, EIF1AX |
| ESCA |
FGFR2, CCND1, PTPN11, KRAS, ERBB3, CDK4, KLF5, FOXA1, MAP2K1, IDH2, ERBB2, SETBP1, NFE2L2, PMS1, SF3B1, IDH1, SOS1, PCBP1, PIK3CA, CTNNB1, RHOA, KIT, MSH3, EEF1A1, EGFR, GTF2I, MYC, SOX17 |
| GBM | H3F3A, CDK4, PIK3R2, ERCC2, GRIN2D, EGFR, MYC |
| HNSC |
FGFR2, MTOR, SF1, CCND1, PTPRC, KRAS, ERBB3, CDK4, KLF5, MAP2K1, ERBB2, SPOP, SETBP1, KEAP1, SMARCA4, CACNA1A, PIK3R2, ERCC2, GRIN2D, NFE2L2, PMS1, SF3B1, IDH1, PCBP1, PIK3CA, EGFR, GTF2I, MYC, GNAQ |
| KIRC |
FGFR2, MTOR, WT1, PTPRC, KRAS, ERBB3, CDK4, FOXA1, MAP2K1, ERBB2, SPOP, SMARCA4, CACNA1A, PIK3R2, ERCC2, GRIN2D, PPP2R1A, NFE2L2, PMS1, SF3B1, IDH1, MAPK1, SOS1, PCBP1, PIK3CG, RAC1, SOX17 |
| KIRP |
FGFR2, PTPRC, FOXA1, IDH2, ERBB2, SPOP, SMARCA4, ERCC2, GRIN2D, PPP2R1A, MAPK1, SMARCB1, PCBP1, PIK3CA, MSH3, PIK3CG, MET, BRAF, CUL1, EGFR, MYC, SOX17, GNAQ |
| LGG | WT1, MACF1, PCBP1, KIT, PIK3CG, MTOR |
| LIHC |
WT1, SF1, CCND1, DHX9, PTPRC, PTPN11, KRAS, CHD4, MACF1, MAP2K1, ERBB2, SPOP, SETBP1, SMARCA4, CACNA1A, PIK3R2, GNA11, ERCC2, GRIN2D, U2AF1, PMS1, SF3B1, IDH1, SOS1, XPO1, PCBP1, PIK3CA, KIT, CDKN1A, EEF1A1, PIK3CG, BRAF, CUL1, GNAQ, EIF1AX |
| LUAD |
FGFR2, MTOR, WT1, SF1, CCND1, PTPRC, KRAS, H3F3A, ERBB3, CDK4, KLF5, MACF1, MAP2K1, ERBB2, SPOP, SETBP1, SMARCA4, CACNA1A, PIK3R2, GNA11, ERCC2, GRIN2D, PPP2R1A, NFE2L2, PMS1, SF3B1, IDH1, SOS1, PCBP1, CTNNB1, RHOA, MSH3, EEF1A1, RAC1, GTF2I, MYC |
| LUSC | NRAS, PTPN11, KRAS, H3F3A, ERBB3, CDK4, ERCC2, GRIN2D, PPP2R1A, U2AF1, SF3B1, IDH1, SOS1, PCBP1, FGFR3, MSH3, GNAQ |
| MESO | FGFR2, PTPRC, PTPN11, ERCC2, GRIN2D, PPP2R1A, IDH1, CTNNB1, RHOA, PIK3CG, RAC1, GTF2I, MYC |
| PCPG | MTOR, NRAS, PTPRC, PMS1, SF3B1, IDH1, SOS1, EPAS1, PIK3CG, EGFR, MYC, SOX17 |
| PRAD |
MTOR, KRAS, ERBB3, CDK4, CHD4, KLF5, FOXA1, ERBB2, SPOP, TP53, SETBP1, SMARCA4, CACNA1A, PIK3R2, GNA11, ERCC2, GRIN2D, PPP2R1A, NFE2L2, PMS1, SF3B1, IDH1, SOS1, PCBP1, KIT, MSH3, PIK3CG, EGFR, GTF2I, MYC, SOX17 |
| SKCM |
CCND1, KRAS, CHD4, FOXA1, SETBP1, SMARCA4, CACNA1A, PIK3R2, ERCC2, GRIN2D, PPP2R1A, MAPK1, CTNNB1, RHOA, EGFR, EIF1AX |
| STAD |
MTOR, NRAS, WT1, SF1, CCND1, KRAS, H3F3A, ERBB3, CDK4, MAP2K1, ERBB2, SPOP, SETBP1, SMAD4, SMARCA4, CACNA1A, PIK3R2, GRIN2D, NFE2L2, PMS1, SF3B1, IDH1, SOS1, PCBP1, PIK3CA, CTNNB1, RHOA, MSH3, CUL1, EGFR, GTF2I, MYC, SOX17, CNBD1, GNAQ, FAM46D |
| TGCT | WT1, KRAS, CHD4, MACF1, MAP2K1, IDH2, SETBP1, ERCC2, GRIN2D, PPP2R1A, SOS1, CTNNB1, SOX17, EIF1AX |
| THCA | NRAS, KLF5, ERBB2, SPOP, ERCC2, GRIN2D, PPP2R1A, PCBP1, CTNNB1, RHOA, PIK3CG, GTF2I |
| UCEC | CCND1, KLF5, ERBB2, SPOP, PIK3R2, U2AF1, PCBP1, PIK3CA, PIK3CG |
DISCUSSION
We demonstrate that the application of Circle_finder to ATAC-seq data can identify eccDNA in cell lines and tissues. Most of the eccDNA thus identified in the cell line could be detected by inverse PCR on DNA enriched for extrachromosomal DNA with disenrichment of linear DNA fragments. The metaphase spreads from OVCAR8 cells showed the presence of these eccDNA loci as a signal of the chromosomes, consistent with the loci being extrachromosomal. Even if ATAC-seq is performed without experimentally disenriching linear chromosomal DNA and/or enriching circular DNA, this approach is useful to identify loci that are either contained in eccDNAs or have suffered a tandem segmental duplication in the chromosome. The identification of eccDNA/duplication in the EGFR locus in GBM cell lines and GBMs suggested that existing ATAC-seq data from other cancers should also be examined closely to find the driver gene amplification on eccDNA/duplication events in each tumor type. We find several cancer driver genes located in such loci (Table 1). These results suggest that deeper sequencing of tumors by ATAC-seq with longer paired-end reads will identify many more clinically important sites involved in eccDNA/duplication in these tumors.
Chromosome ends are protected by telomeres. Once the chromosome suffers a catastrophic fragmentation, as in chromothripsis, some parts of the chromosome may be protected from degradation by eccDNA formation. eccDNA can also be generated from extra linear DNA produced by some kind of copying mechanism as a byproduct of DNA replication or repair. Either way, our results suggest that eccDNAs are very prevalent in cancer cell lines and tumors and that ATAC-seq is an easy method to identify such eccDNAs.
It has been reported that eccDNA longer than a few kilobases may have origins of replication and may get amplified independent of the main chromosome. Thus, if an eccDNA harbors an oncogene, then amplification of such eccDNA in tumor cells will increase the fitness of the tumor cell. In addition, since a centromere is absent in the eccDNA (11), eccDNA may segregate unevenly between daughter cells and result in tumor heterogeneity (9). Both these mechanisms will increase the likelihood that if a particular type of therapy inhibits a gene resident on a preexisting eccDNA, then the tumor is likely to acquire resistance through the selective amplification of that eccDNA.
In this context, it is particularly exciting that circle (or gene duplication) at an important locus in a subset of the tumor cells is identified by ATAC-seq even before the amplification is apparent by a CNV analysis of the whole tumor (Fig. 4D). To estimate whether ATAC-seq can identify loci in early, somatically mosaic states of amplification as eccDNA/segmental duplication, we analyzed the amplicons identified by TCGA from gene array hybridization. It is apparent that an amplicon has to be at least around 1.5 Mb long to be detected as a single-copy amplification by gene microarrays (three copies per cell) (see Materials and Methods and fig. S2). In contrast, we could detect somatically mosaic increase in the copy number of loci far smaller than that length by use of Circle_finder on ATAC-seq or WGS data. For example, Circle_finder on ATAC-seq data identifies sites of incipient amplification of the EGFR gene in a subset of tumor cells even before such amplification is detected by copy number measurements, predicting that even if the tumor responds to anti-EGF therapy, it is likely to recur because of amplification of the EGFR gene.
Many of the abundant eccDNA loci intersect with unprocessed pseudogenes, which are known to have introns and regulatory sequences, but are crippled by stop codons in the open reading frames (20). Since eccDNA evolve and pick up substitution, insertion, and deletion mutations (11, 18), it is tempting to speculate that amplification of unprocessed pseudogenes on eccDNA and their evolution may make these genes translationally competent to give an unknown advantage during tumorigenesis.
Last, we note that a large fraction of eccDNA identified by ATAC-seq have properties similar to the microDNA that we reported earlier: length, <1kb; with peaks at 180 and 380 bases; high GC content; enrichment of their sites of origin in regions upstream of genes and in CpG islands; and the presence of short sequences of homology flanking the chromosomal locus giving rise to the circle. The small size of these circles has thus been confirmed by rolling circle amplification (5), by electron microscopy (5), and, now, by ATAC-seq, ruling out any possibility that the previously reported small size was due to preferential amplification of small circles. Although we observe eccDNA longer than 2 kb in mouse somatic tissue, the majority (>90%) of eccDNA were shorter than 2 kb (3, 5). Turner et al. (11) and deCarvalho et al. (9) have identified long circles of DNA in cancers, called ecDNA. We believe that the long circles identified in tumors by ATAC-seq, e.g., the one containing the EGFR gene, belong to this latter class of circles. The consistent properties of the small circles suggest that common mechanisms are involved in their generation in cell lines and in tumors, although it is unclear whether exactly the same mechanisms are involved in producing the longer circles seen in ecDNAs that give rise to clinically significant gene amplifications.
MATERIALS AND METHODS
ATAC-seq library preparation
ATAC-seq for cell lines was performed as per the Omni-ATAC-seq protocol (21). Briefly, C4-2 and OVCAR8 cells were grown in RPMI 1640 (Corning, no.10-040) supplemented with 10% fetal bovine serum (FBS) to ~80% confluence. Fifty thousand viable cells were lysed in 10 mM tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, and 0.1% Tween 20. Nuclear pellet was then subjected to transposition reaction using Nextera DNA Sample Preparation kit (Illumina, no. FC-121-1030) in the presence of 0.01% digitonin and 0.1% Tween 20 at 37°C for 30 min and cleaned up with DNA Clean and Concentrator-5 Kit (Zymo, no. D4014). For quantitative PCR (qPCR), three to six additional cycles of PCR amplification was performed using NEBNext High-Fidelity 2X PCR Master Mix (NEB, no. M0541L) and Nextera Index Kit (Illumina, no. 15055289). Cleaned up libraries were quantified and pooled for sequencing by Novogene.
Identification of eccDNA from ATAC-seq and WGS libraries
Paired-end reads were mapped to the hg38 genome build using bwa-mem (22) with default setting. The split reads (reads not mapped in contiguous manner) were collected using the tool samblaster (23). If one tag of a paired read is mapped contiguously (one entry in mapped file) and the other tag is mapped in a split manner (two entries in mapped file), then the particular read ID will have three entries in alignment file. We therefore collected all the read pair IDs that mapped to three unique sites in the genome from the alignment file. Next, we collected the split reads that mapped uniquely at two positions on the same chromosome and in the same orientation. Returning to the list of paired-end IDs that mapped uniquely to three sites in the genome, we identified paired-end IDs where the contiguously mapped read is between the two split reads and on the opposite strand. From this list, we annotate a circle if we find at least one junctional sequence. For karyotype and box plot, we considered at least two junctional reads.
Copy number amplification analysis
For each identified eccDNA (JTGE2), an upstream and downstream genomic interval of equivalent length was created. Next, we counted the number of reads that mapped to each of the three intervals (upstream, eccDNA, and downstream). Last, copy number amplification (CNA) was computed by counting the number of mapped read in eccDNA interval divided by the mean of the number of reads in upstream and downstream intervals. A CNA value more than 1 would suggest the amplification of the locus defined by the eccDNA.
eccDNA isolation
eccDNA for Fig. 3 was prepared from the human cancer cell lines. The cells were grown on 150-mm plates until reaching confluence. Approximately 4 × 107 cells were isolated per sample. The cells were trypsinized and then spun down at 300g. The cells were washed with phosphate-buffered saline (PBS), spun down at 300g, and resuspended in 6 ml of resuspension buffer (P1) of the Qiagen HiSpeed Plasmid Midi Kit (catalog no. 12643). Lysis buffer (6 ml) (P2) was added according to the manufacturer’s instructions. The cells were lysed for 5 min before adding the neutralization buffer (P3) and incubated at room temperature for 10 min. The cell lysate was passed through the QIAfilter cartridge. Equilibration buffer (4 ml) (QBT) was added to the HiSpeed Tip and allowed to pass through the resin. The lysate was added to the HiSpeed Tip, and then, the HiSpeed Tip was washed with 20 ml of washing buffer (QC). Then, the DNA was eluted from the HiSpeed Tip with 5 ml of elution buffer (QF), precipitated with 3.5 ml of isopropanol, and incubated at room temperature for 5 min. The DNA was passed through a QIAprecipitator, which was washed with 2 ml of 70% ethanol. The excess ethanol was removed by passing air through the QIAprecipitator five times. The DNA was precipitated from the QIAprecipitator by 1 ml of TE buffer and quantified using a NanoDrop spectrophotometer. The DNA eluted from the QIAprecipitator was then precipitated again by the addition of 2 ml of ethanol and 1 μg of glycogen and centrifuged at 15,000g. The supernatant was removed; the DNA was air-dried for 5 min, resuspended in 20 μl of TE, and warmed to 37°C for 5 min. Then, the DNA was digested with the Lucigen adenosine triphosphate (ATP)–dependent Plasmid-Safe deoxyribonuclease (catalog no. E3101K). The 10× buffer and ATP were added according to the manufacturer’s recommendations. In addition, ribonuclease A was added to the solution to digest RNA concurrently with the linear DNA. The sample was digested overnight and then purified using a Zymo PCR purification kit (catalog no. D4003). Briefly, the DNA binding buffer was added to the DNA solution in a 5:1 ratio. The mixture was then added to a Zymo-Spin column in a collection tube. The sample was centrifuged for 30 s at 10,000g. Then, the column was washed with 200 μl of DNA wash buffer and centrifuged for 30 s at 10,000g. The wash step was repeated. The DNA was eluted by adding 50 μl of DNA elution buffer and centrifuged for 30 s at 10,000g. The DNA was quantified using a NanoDrop spectrophotometer to ensure digestion of the linear DNA, and then, the DNA digestion, purification, and quantification steps were repeated until the DNA concentration no longer decreased after digestion. Together, this process helped ensure that the digestion of linear DNA was complete.
These methods are comparable to the methods previously used where we validated the loss of linear DNA by quantifying the loss of linear DNA with qPCR compared to circular DNA. Electron microscope imaging in those experiments showed that the linear DNA was no longer present in the samples (3, 5).
Outward-directed PCRs (inverse PCR) for detection of eccDNA
Outward-directed primers were designed across the junctional tags identified from ATAC-seq analysis. PCR was done with Phusion High-Fidelity DNA Polymerase (NEB) according to the manufacturer’s instructions. Purified circular DNA (3 ng) was used as template. Unless otherwise stated, all the computation and plots were made of eccDNA present on chr1-22, chrX, and chrY.
Metaphase FISH
OVCAR8 cells were cultured in RPMI medium supplemented with 10% FBS and 1% penicillin-streptomycin in the presence of 5% CO2 in a humidified incubator at 37°C. Cells were treated with 2 mM thymidine for 16 hours and released for 9 hours in a regular medium, followed by another block with 2 mM thymidine to arrest the cells at G1-S boundary. The cells were released from the double-thymidine block for 3 hours in regular medium and 9 hours in colcemid (0.1 μg/ml). Mitotic cells were shaken off, washed twice with 1× PBS, and resuspended in 75 mM KCl for 30 min at 370°C. The cells were centrifuged at 300g for 5 min, fixed with Carnoy’s fixative (3:1 methanol:glacial acetic acid, v/v) on ice for 30 min, and washed twice with fixative, and metaphase spreads were prepared.
The glass slides containing metaphase spreads were immersed in prewarmed denaturation buffer [70% formamide and 2× SSC (pH 7.0)] at 73°C for 5 min, and slides were serially dehydrated with ethanol (70, 85, and 100%) for 2 min each and dried at room temperature until all the ethanol evaporated. The FISH probes (Empire Genomics) were denatured with hybridization buffer at 730°C for 5 min and immediately chilled on ice for 2 min. The probe mixture was added onto the slide, and coverslips were applied onto the slide, sealed with rubber cement, and incubated at 370°C for overnight in a humidified chamber. The coverslips were removed, and slides were washed with prewarmed 0.4× SSC containing 0.3% NP-40 at 73°C for 2 min, followed by washing with 2× SSC buffer containing 0.1% NP-40 at room temperature for 5 min. The slides were dried at room temperature and mounted with VECTASHIELD 4′,6-diamidino-2-phenylindole medium.
List of TCGA IDs that were used for LGG and GBM data analysis
The TCGA IDs used for LGG and GBM data analysis are as follows: LGG: TCGA-P5-A77X-01A, TCGA-DU-5870-02A, TCGA-DB-A75K-01A, TCGA-W9-A837-01A, TCGA-F6-A8O3-01A, TCGA-FG-A4MY-01A, TCGA-E1-A7YI-01A, TCGA-P5-A735-01A, and TCGA-DU-6407-02B and GBM: TCGA-06-A7TK-01A, TCGA-4W-AA9S-01A, TCGA-OX-A56R-01A, TCGA-76-6656-01A, TCGA-RR-A6KB-01A, TCGA-06-A6S1-01A, TCGA-06-A5U0-01A, and TCGA-06-A7TL-01A.
Testing the limit of detection of gene amplification by CNV measurements
We tested whether the detection of eccDNAs from ATAC-seq data can identify somatically mosaic amplifications before they can be detected by CNV analyses from genotyping array data. To determine the sensitivity of detection of an amplicon by genotyping arrays, we downloaded the previously released CNV results generated by the TCGA research network. The algorithm used by the TCGA research network segments the chromosomes into smaller sections where an amplification or deletion is detected. Empirically, the resulting lengths of segments with CNV determined by the algorithm are the result of (i) the true length of the amplified or deleted segment and (ii) the extent to which the segment was amplified or deleted. While we cannot know whether or not a reported CNV segment should have been further segmented, we hypothesized that if we analyzed ten segments with a similar level of amplification, then the smallest length among them approximates the smallest length that can be detected by the algorithm at that level of amplification since the power to detect CNV changes increases as the extent of amplification increases.
The TCGA research network reported amplifications as segment mean > 0, where segment mean is ln(copy number/2). All segments with segment mean > 0.1 were ordered by reported segment mean values. Bins of ten segments were analyzed for the smallest segment in each bin. The median segment mean value of each bin (extent of amplification) is plotted against the log-transformed smallest segment length in that bin (fig. S2).
The correlation between the segment length and segment amplification can be modeled as a linear function with the following formula: ln(minimum segment length) = 15.8304 − 2.7475 × median segment mean. This relatively simple model captured the relationship between the minimum segment length and the extent of amplification as measured by segment mean (adjusted R2 = 0.5442; P < 2.2×10−16).
If one extra copy of an amplicon is present in every single cell of the sample, then the segment mean value is 0.585 [log2(3/2)]. From the linear model in fig. S2, the minimum segment length detectable at this segment mean value is 1.5 Mb. Therefore, most of the somatically mosaic amplifications driven by most of the eccDNAs in our study (median length, ~2 kb) will not be captured using genotyping arrays.
The number of ATAC-seq libraries analyzed in this study for various tumor type is as follows: ACC, 8; BLCA, 10; BRCA, 70; CESC, 3; CHOL, 1; COAD, 38; ESCA, 17; GBM, 8; HNSC, 9; KIRC, 15; KIRP, 29; LGG, 10; LIHC, 15; LUAD, 21; LUSC, 12; MESO, 5; PCPG, 9; PRAD, 21; SKCM, 9; STAD, 19; TGCT, 8; THCA, 12; and UCEC, 10.
Supplementary Material
Acknowledgments
We would like to thank the Dutta laboratory members for thoughtful discussion on this paper. We thank the High Performance Computing team at the University of Virginia for providing all the support with computation. We thank the dbGAP and the TCGA data management teams for data access. We would also like to thank the patients for their participation in TCGA and R. Corces from H. Y. Chang’s group (Howard Hughes Medical Institute, Stanford University, Stanford) for alerting us about raw sequencing data availability through TCGA. We would also like to thank the SOM core facility. Funding: This work was supported by grants from NIH R01 CA60499 and the Owens Foundation to A.D., a fellowship to S.K. from the UVA Cancer Center, and T32 GM007267 grant to A.C. from the NIH. Author contribution: P.K. and A.D. conceived and designed the study and wrote the paper. P.K. collected the data and did most of the analysis. S.K. did the inverse PCR experiment. T.P. isolated circular DNA. S.S. and E.S. performed the FISH experiment. Z.S. prepared the OVCAR8 and C4-2 ATAC-seq library. A.C. performed the cancer driver gene analysis. Y.S. helped P.K. in testing and improving the Circle_finder algorithm. Competing interests: P.K. and A.D. are inventors on a U.S. provisional patent application related to this work filed by University of Virginia (no. 62/832,443, filed 11 April 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The ATAC-seq data generated for OVCAR8 and C4-2B cell lines are deposited to GEO with accession number GSE145409.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba2489/DC1
REFERENCES AND NOTES
- 1.Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J., ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corces M. R., Granja J. M., Shams S., Louie B. H., Seoane J. A., Zhou W., Silva T. C., Groeneveld C., Wong C. K., Cho S. W., Satpathy A. T., Mumbach M. R., Hoadley K. A., Robertson A. G., Sheffield N. C., Felau I., Castro M. A. A., Berman B. P., Staudt L. M., Zenklusen J. C., Laird P. W., Curtis C.; Cancer Genome Atlas Analysis Network, Greenleaf W. J., Chang H. Y., The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dillon L. W., Kumar P., Shibata Y., Wang Y. H., Willcox S., Griffith J. D., Pommier Y., Takeda S., Dutta A., Production of extrachromosomal microDNAs is linked to mismatch repair pathways and transcriptional activity. Cell Rep. 11, 1749–1759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar P., Dillon L. W., Shibata Y., Jazaeri A. A., Jones D. R., Dutta A., Normal and cancerous tissues release extrachromosomal circular DNA (eccDNA) into the circulation. Mol. Cancer Res. 15, 1197–1205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shibata Y., Kumar P., Layer R., Willcox S., Gagan J. R., Griffith J. D., Dutta A., Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 336, 82–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moller H. D., Larsen C. E., Parsons L., Hansen A. J., Regenberg B., Mourier T., Formation of extrachromosomal circular DNA from long terminal repeats of retrotransposons in saccharomyces cerevisiae. G3 6, 453–462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moller H. D., Parsons L., Jorgensen T. S., Botstein D., Regenberg B., Extrachromosomal circular DNA is common in yeast. Proc. Natl. Acad. Sci. U.S.A. 112, E3114–E3122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Møller H. D., Mohiyuddin M., Prada-Luengo I., Sailani M. R., Halling J. F., Plomgaard P., Maretty L., Hansen A. J., Snyder M. P., Pilegaard H., Lam H. Y. K., Regenberg B., Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 9, 1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.deCarvalho A. C., Kim H., Poisson L. M., Winn M. E., Mueller C., Cherba D., Koeman J., Seth S., Protopopov A., Felicella M., Zheng S., Multani A., Jiang Y., Zhang J., Nam D. H., Petricoin E. F., Chin L., Mikkelsen T., Verhaak R. G. W., Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat. Genet. 50, 708–717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoura M. J., Gabdank I., Hansen L., Merker J., Gotlib J., Levene S. D., Fire A. Z., Intricate and cell type-specific populations of endogenous circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 7, 3295–3303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner K. M., Deshpande V., Beyter D., Koga T., Rusert J., Lee C., Li B., Arden K., Ren B., Nathanson D. A., Kornblum H. I., Taylor M. D., Kaushal S., Cavenee W. K., Wechsler-Reya R., Furnari F. B., Vandenberg S. R., Rao P. N., Wahl G. M., Bafna V., Mischel P. S., Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S., Turner K. M., Nguyen N., Raviram R., Erb M., Santini J., Luebeck J., Rajkumar U., Diao Y., Li B., Zhang W., Jameson N., Corces M. R., Granja J. M., Chen X., Coruh C., Abnousi A., Houston J., Ye Z., Hu R., Yu M., Kim H., Law J. A., Verhaak R. G. W., Hu M., Furnari F. B., Chang H. Y., Ren B., Bafna V., Mischel P. S., Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morton A. R., Dogan-Artun N., Faber Z. J., MacLeod G., Bartels C. F., Piazza M. S., Allan K. C., Mack S. C., Wang X., Gimple R. C., Wu Q., Rubin B. P., Shetty S., Angers S., Dirks P. B., Sallari R. C., Lupien M., Rich J. N., Scacheri P. C., Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179, 1330–1341.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Q., Wu T. P., Gimple R. C., Li Z., Prager B. C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D. U., Zhang C., Dowiak A. V., Lin K., Zeng C., Sui Y., Kim L. J. Y., Miller T. E., Jiang L., Lee C. H., Huang Z., Fang X., Zhai K., Mack S. C., Sander M., Bao S., Kerstetter-Fogle A. E., Sloan A. E., Xiao A. Z., Rich J. N., N6-methyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maher C. A., Wilson R. K., Chromothripsis and human disease: Piecing together the shattering process. Cell 148, 29–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libermann T. A., Nusbaum H. R., Razon N., Kris R., Lax I., Soreq H., Whittle N., Waterfield M. D., Ullrich A., Schlessinger J., Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313, 144–147 (1985). [DOI] [PubMed] [Google Scholar]
- 17.Maire C. L., Ligon K. L., Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology 16 (Suppl 8), viii1–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu K., Ding L., Chang T. C., Shao Y., Chiang J., Mulder H., Wang S., Shaw T. I., Wen J., Hover L., McLeod C., Wang Y. D., Easton J., Rusch M., Dalton J., Downing J. R., Ellison D. W., Zhang J., Baker S. J., Wu G., Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol. 137, 123–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey M. H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M. C., Kim J., Reardon B., Kwok-Shing Ng P., Jeong K. J., Cao S., Wang Z., Gao J., Gao Q., Wang F., Liu E. M., Mularoni L., Rubio-Perez C., Nagarajan N., Cortés-Ciriano I., Zhou D. C., Liang W. W., Hess J. M., Yellapantula V. D., Tamborero D., Gonzalez-Perez A., Suphavilai C., Ko J. Y., Khurana E., Park P. J., Van Allen E. M., Liang H.; Group M. C. Working; Network Cancer Genome Atlas Research, Lawrence M. S., Godzik A., Lopez-Bigas N., Stuart J., Wheeler D., Getz G., Chen K., Lazar A. J., Mills G. B., Karchin R., Ding L., Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tutar Y., Pseudogenes. Comp. Funct. Genomics 2012, 424526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corces M. R., Trevino A. E., Hamilton E. G., Greenside P. G., Sinnott-Armstrong N. A., Vesuna S., Satpathy A. T., Rubin A. J., Montine K. S., Wu B., Kathiria A., Cho S. W., Mumbach M. R., Carter A. C., Kasowski M., Orloff L. A., Risca V. I., Kundaje A., Khavari P. A., Montine T. J., Greenleaf W. J., Chang H. Y., An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faust G. G., Hall I. M., SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba2489/DC1