

A strain of bioinorganic hybrid bacteriophage was screened to enhance the antitumor immune response of colorectal cancer.
Abstract
Mounting evidence suggests that the gut microbiota contribute to colorectal cancer (CRC) tumorigenesis, in which the symbiotic Fusobacterium nucleatum (Fn) selectively increases immunosuppressive myeloid-derived suppressor cells (MDSCs) to hamper the host’s anticancer immune response. Here, a specifically Fn-binding M13 phage was screened by phage display technology. Then, silver nanoparticles (AgNP) were assembled electrostatically on its surface capsid protein (M13@Ag) to achieve specific clearance of Fn and remodel the tumor-immune microenvironment. Both in vitro and in vivo studies showed that of M13@Ag treatment could scavenge Fn in gut and lead to reduction in MDSC amplification in the tumor site. In addition, antigen-presenting cells (APCs) were activated by M13 phages to further awaken the host immune system for CRC suppression. M13@Ag combined with immune checkpoint inhibitors (α-PD1) or chemotherapeutics (FOLFIRI) significantly prolonged overall mouse survival in the orthotopic CRC model.
INTRODUCTION
Bacteria are colonized on almost all human body surfaces, and 99% of these are found in the human gastrointestinal tract (1, 2). The gut microbiota plays an important role in human physiological function, especially in modulating the host immune system (3). Accumulating evidence indicates that gut microbiota can profoundly influence the effect of tumor immunotherapy (4, 5). For instance, the antitumor efficacy of cyclophosphamide was dependent on two gut commensal microbiota species, Enterococcus hirae and Barnesiella intestinihominis (6). In addition, Bifidobacterium could specifically enhance the response of immune checkpoint inhibitors and induce intratumoral interferon-γ (IFN-γ)–CD8+ T cell accumulation (7), thus revealing the great potential of gut microbiota in promoting antitumor immune response. However, particular gut bacteria could also exert a detrimental impact by promoting tumor growth (8, 9). Recent studies have shown that colorectal cancer (CRC) development is associated with the gut microbiota, and that Fusobacterium nucleatum (Fn) enrichment in CRC tissue could selectively enlarge immunosuppressive myeloid cells, resulting in an immune-suppressive tumor microenvironment (TME) to further dampen T cell responses (10–15). Thus, given the encouraging immunomodulatory effects of most gut microbiota, an important prerequisite for benefiting the antitumor immunotherapy is how to manipulate the specific gut bacteria, especially those bacteria that could inhibit host antitumor immunity.
Because of the multiple effects of gut microbiota, antibiotics as a typical antibacterial treatment are limited. The reason is that antibiotics would simultaneously remove bacteria that have a positive effect in cancer immunotherapy as well as those with drug resistance and adverse effects (16). Thus far, several inorganic nanomaterials are known to exhibit superior antibacterial activity, such as silver nanoparticles (AgNP), which are widely applied in both the medical field and consumer products (17, 18). Nevertheless, the antibacterial activity of AgNP lacks specificity. Currently, numerous studies are shifting focus toward bacteriophages as promising natural agents. Phages could infect host bacteria with accurate species-specific mechanism, and professionally lytic phages are often used in antibacterial strategies (19, 20). However, these lytic phages integrate their genome into the host cell during the infection process, and that genome could potentially code resistance genes and release toxins (21, 22). Recently, phage display technology has gained attention as a means of developing targeting devices (23–25). Phages with high affinity for a given target of interest can be screened by phage display technology. Besides, temperate phages incapable of replicating themselves compared with lytic phages are currently used for phage display technology, representing a safe therapy option (26). Thus, we hypothesized a rational choice relying on the accurate targeting characteristics of phages and the capacity of killing bacteria assisted by inorganic nanomaterials to optimize the defect of traditional treatments.
Here, we propose a phage-based bioinorganic hybridization system that could rebuild a tumor-immune microenvironment via manipulation of gut microbiota for CRC suppression (Fig. 1). We used a M13 filamentous phage display library (27) to screen a specific Fn-binding phage strain in vitro. Next, AgNP were electrostatically assembled on the surface of antibacterial phages (M13@Ag) (28). After accumulation of M13@Ag in TME, AgNP selectively killed protumoral Fn with the exquisite recognition mechanism of phages and blocked recruitment of immunosuppressive cells. Moreover, phages are highly immunogenic and can directly stimulate the host immune system with their naive coat proteins, which induce dendritic cell (DC) maturation and promote the activation of M1-phenotype tumor-associated macrophages (TAMs) (29). It was demonstrated that such bioinorganic phage-based recombinants could precisely regulate gut microbiota to reverse immune-suppressive TME with the inhibition of myeloid-derived suppressor cell (MDSC) expansion. Both in vitro and in vivo experiments demonstrated that this strategy could markedly enhance the antitumor immune response to achieve satisfactory therapeutic effects in CRC.
Fig. 1. Schematic illustration of phage-based bio/abiotic hybrid system (M13@Ag) to regulate gut microbes for cancer-specific immune therapy.
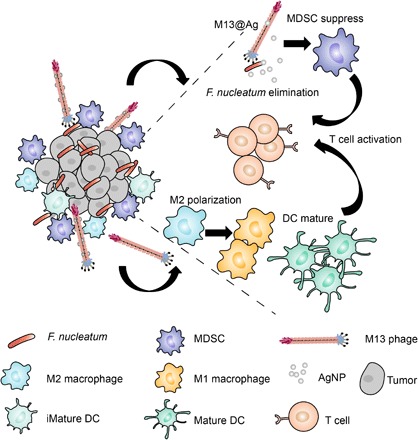
RESULTS
The effect of Fn in the CRC immune microenvironment
To investigate the role of Fn in tumor-immune microenvironment, we first analyzed the expression of cancer immune regulation–related genes in CRC from The Cancer Genome Atlas (TCGA) database. These negative regulation immunity genes (e.g., FOXXP3, TGFβ1, and HMGB1) were significantly up-regulated, while proimmunity genes, like CD3E and CD86, were lowly expressed in CRC (Fig. 2A). Furthermore, from the National Center for Biotechnology Information (NCBI) bio-project (30), the enrichment of Fn was found in fecal samples of patients with CRC (n = 72) and healthy people (n = 52) (Fig. 2B). Meanwhile, a clinical CRC sample of microchip was used to further analyze the correlation between Fn and immunosuppressive TME (Fig. 2C and fig. S3, A and B). Fluorescence in situ hybridization (FISH) was used to label Fn with green fluorescence in tissue sections, and red fluorescent–labeled MDSCs were nearly overlapped with the Fn. This positive correlation was validated in prior reports, which indicated that Fn colonized in CRC to modulate the tumor-immune microenvironment.
Fig. 2. The screen of protumor gut microbiome in CRC.
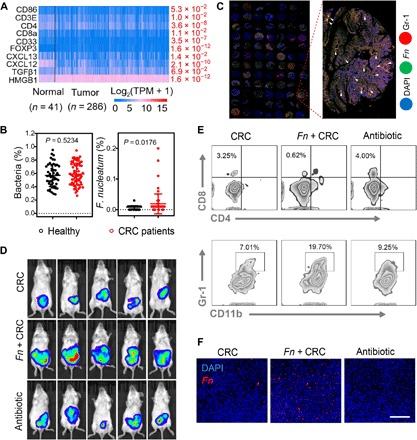
(A) Differential gene expression heat map of identified genes about immunity-associated human CRC gene in the TCGA CRC dataset. (B) Bacteria levels of Fusobacterium colonization in fecal samples of patients with CRC from the NCBI bio-project (PRJEB10878). Significant difference was assessed by using t test. (C) Fluorescence images of microchip from patients with CRC within tumor tissues [Fn visualized with fluorescence in situ hybridization assay is green, tumor cell nucleoid is stained with 4′,6-diamidino-2-phenylindole (DAPI) with blue fluorescence, and Gr-1 antibody–labeled MDSC cell infiltration is red). (D) In vivo bioluminescence imaging of orthotopic CT26-luc tumor-bearing mice with different treatments at 15 days (n = 5). (E) Flow cytometry analysis of the number of CD3+ CD8+ T cells and CD11b+ Gr-1+ MDSC cells at the tumor site after receiving treatment at 10 days (n = 3). Representative dot plots as indicated. (F) Typical images of fluorescence in situ hybridization assay for measuring the Fn level in tumor tissues treated with Fn colonization or antibiotic cocktail. Scale bar, 200 μm. Significant difference was assessed in (B) by using t test. The mean values and SD are presented.
To similarly evaluate the effect of Fn in a clinically relevant in vivo model of CRC, we established an orthotopic CRC model with luciferase transfection murine CT26 cells. After 5 days of continuous oral gavage of Fn, mice were treated with antibiotic cocktail (metronidazole, vancomycin, and ampicillin) every day for a total of five oral administrations. The tumor growth was monitored through bioluminescence on day 15. As expected, the antibiotic cocktail–treated group and the Fn absent group showed the decreased intensity of bioluminescence. In contrast, Fn enrichment in the CRC group had no inhibition effect and even promoted the growth of CRC (Fig. 2D and fig. S1, A to C). To further identify that Fn contributed to tumorigenesis by affecting intratumoral immune cells, the rate of MDSCs in infiltrating cells after different treatments was measured. Compared with the other groups, the Fn highly enriched group showed an increased tendency of the MDSCs, while lower abundance of T cells was detected in tumor tissues (Fig. 2E). We also tested the effect of antibiotic cocktail in cleaning Fn. As shown in Fig. 2F, specific FISH probe with red fluorescence was used to label Fn in tumor, and there was almost no red fluorescence in the antibiotic cocktail–treated group. These consequences revealed that sweeping away the specific bacteria could improve T cell infiltration inside the tumor and restrict MDSC augmentation. The same results were also confirmed by immunofluorescence staining of CD3+ T cells and MDSCs in the orthotopic CRC model (fig. S1D).
Overall, all the above results attested that Fn expanded MDSCs to promote CRC progression. The clearance of Fn could reverse immunosuppressive TME and enhance immunotherapy response for the orthotopic CRC. However, conventional antibiotic treatment was restricted on account of nonspecificity and drug resistance. One strategy that screened a phage strain with natural host-specific character could selectively kill Fn with high efficiency.
Identification of M13@Ag specifically eliminated Fn
Since phage display technology has been widely used for binding to a variety of targets including bacteria, it was speculated that specifically Fn-binding phages also could be found. We used M13 phage library containing abundant random peptide sequences displayed on these temperate filamentous phages to explore a strain of selectively Fn-binding M13 phage. After three rounds of competitive biopanning, phages eluted from Fn were expanded (fig. S2, A to C). Afterward, representative phage clones were separated, and then, the binding ability of M13 phages was determined by enzyme-linked immunosorbent assay (ELISA). As shown in Fig. 3A, M13 phages (named M5) demonstrated the optimal capacity of targeting Fn, whereas control wild-type phages did not bind to Fn. High-throughput sequencing was also used to identify genome information of M13 phage named M5 (fig. S2D).
Fig. 3. Characterization of bioinorganic hybridization phages.
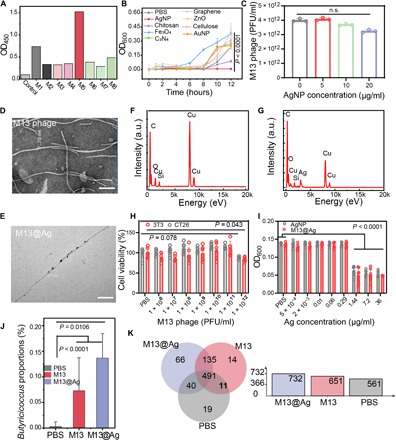
(A) In vitro biopanning of specifically Fn-binding M13 phages by Ph.D.-12 peptide phage display library. Eight kinds of clones were examined by ELISA. (B) Screening of a variety of antibacterial nanoparticles (n = 6). (C) Assessment bioactivity of M13 phages after directly assembling with AgNP (n = 3). TEM images of M13 phages (D) and M13@Ag (E), in which M13 phages were native stained. Scale bars, 100 nm. Analysis of element distribution on the surface of M13 phages (F) and M13@Ag (G) detected with EDX. (H) Cell viability of cancerous CT26 cells and normal 3T3 cells after coculture with M13 phages (n = 6). (I) Antibacterial activity of M13@Ag by incubating with Fn for 8 hours (n = 6). (J) Genus-level analysis in the feces from the Fn-colonization CRC murine model with significant increase in antitumor bacteria of Butyricicoccus after PBS, M13, and M13@Ag treatment (n = 3). (K) Venn diagram of identified fecal bacterial strains in Fn-colonization CRC murine model with different treatments (n = 3). Significant differences were assessed in (C) and (J) using one-way ANOVA and t test and in (B), (H), and (I) using t test. The mean values and SD are presented. n.s., not significant; OD, optical density; a.u., arbitrary units.
In addition, it was necessary to investigate active antibacterial materials that could assist M13 phages in killing Fn effectively. We tested eight kinds of typical nanomaterial impacts on Fn, and it was obviously observed that AgNP showed prominent capability for Fn growth suppression (Fig. 3B). We then performed studies to evaluate the effect of AgNP on M13 phages (31). After being coincubated with various concentrations of AgNP, plaque-forming unit (PFU) changes of M13 phages were determined by double agar layer method. There was no notable difference in the PFU number of M13 phages compared with the control, suggesting that natural liveness of M13 phages was hardly inactivated by AgNP (Fig. 3C). Thereby, bioinorganic hybridization phages (32–34) of M13@Ag were prepared via electrostatic interaction. As outlined in Fig. 3 (D and E), transmission electron microscopy (TEM) characterized the assemblies of AgNP on the surface of M13 coat protein. M13@Ag, with a length of about 800 nm, were almost consistent with M13 phages (35). The assemblies of M13@Ag were also proved by energy-dispersive x-ray spectroscopy (EDX) analysis (Fig. 3, F and G). For biosafety verification, M13 phages showed no evident toxicity toward normal cells (3T3) and tumor cells (CT26) (Fig. 3H). As well as in vivo intravenous injection of phosphate-buffered saline (PBS), M13 phages, AgNP, and M13@Ag, after 24 hours, blood chemistry was analyzed. As presented in fig. S3C, both M13 phages and M13@Ag groups did not cause rapid changes in liver functions and renal functions. The AgNP group exhibited negligible changes as well. The same result of biosafety was tested by hemolytic test in fig. S3D. All the above results manifested that M13@Ag-mediated treatment did not cause serious toxic effect, indicating that M13 phages could be appropriate for further study. We also tested the antibacterial effect and stability of M13@Ag. As depicted in Fig. 3I and fig. S4A, silver ions at 1.44 μg/ml showed desirable bacteria-killing ability, and the concentration of silver ions in M13@Ag was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Meanwhile, M13@Ag showed negative charge and stability in PBS, serum [fetal bovine serum (FBS)], and cell culture media [Dulbecco’s modified Eagle’s medium (DMEM)], which were almost stable in physiological environment (fig. S4, B and C).
Furthermore, we investigated the role of M13@Ag to maintain the balance of gut microbiota. Fecal samples from Fn-colonized CRC murine model were collected after treatment with PBS, M13 phages, and M13@Ag. As illustrated in Fig. 3J and fig. S4 (D and E), an enhanced number of butyrate-producing bacteria, such as of Butyricicoccus (36), were observed by 16S ribosomal DNA (rDNA) sequencing. On account of the antitumor immune effects of butyrate-like short-chain fatty acids produced by fermentative bacteria, protumor bacteria of Fn were eliminated specifically by M13@Ag, while proliferation of antitumor bacteria was facilitated to further regulate the balance of intestinal microbiota. A Venn diagram indicated that OTU (operational taxonomic units) abundance was decreased in the PBS-treated group compared with M13@Ag treatment (Fig. 3K). All the above results pointed the potential competence of M13@Ag for particularly modulating gut microbiota and maintaining gut microbiota diversity.
M13@Ag induced immune activation in vitro
Next, we examined the targeting ability of M13@Ag by various characterization methods. After coculturing with M13 phages or M13@Ag individually, representative TEM images of Fn, M13 phage–binding Fn, and M13@Ag-binding Fn were shown in Fig. 4 (A and B). It was clearly observed that the filamentous structure of M13 phages, as well as M13@Ag, strongly bound to Fn. Meanwhile, the same results were also confirmed in fluorescence colocalization assay by superresolution microscopy. As displayed in Fig. 4C and fig. S4F, red fluorescent (Cy5)–labeled M13 phages and M13@Ag were well adhered to green fluorescent [fluorescein isothiocyanate (FITC)]–labeled Fn. As a comparison, control wild-type phages were nearly off-targeted to Fn, while another control group of Staphylococcus aureus showed only green fluorescence of itself. Flow analysis for quantification of the targeting capacity of M13@Ag was similarly tested (Fig. 4D). Collectively, these results illustrated that M13@Ag, with excellent Fn-binding ability, would manipulate bacteria accurately.
Fig. 4. In vitro activation of anticancer immune responses by M13@Ag.
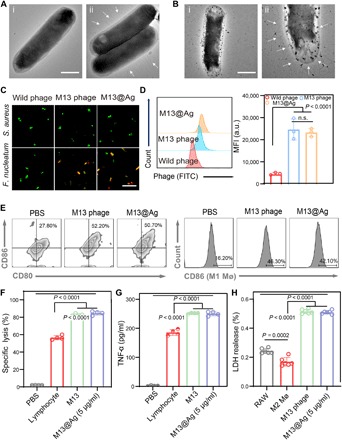
(A and B) TEM images of Fn (A-i), M13 phages targeting Fn (A-ii), and M13@Ag targeting Fn (B-i and B-ii). The filamentous M13 phages were indicated with white arrows. Scale bars, 1 μm. (C) Confocal fluorescence images of phage-binding assays of M13 phages and M13@Ag toward different species of bacteria. Phages were marked with red fluorescent Cy5, and bacteria were labeled with green fluorescent FITC, respectively. Scale bar, 30 μm. (D) Flow cytometry for quantification of the targeting capacity of M13@Ag after coincubating with Fn and M13@Ag for 30 min. Phages were stained with green fluorescent FITC (n = 3). (E) Representative flow cytometry images for evaluating the DC maturation and TAMs toward M1 phenotype. BMDCs and M2 macrophages were incubated with M13 phages or M13@Ag for 24 hours (n = 3). Efficacy of immune responses for CTL-mediated cancer cell–specific lysis (F) and cytokine production levels of TNF-α (G) measured by incubating M13 phage–stimulated splenocytes with CT26 tumor cells for 24 hours. The supernatant was collected to detect the LDH leakage level and TNF-α level, with cytotoxic lymphocytes toward CT26 cells with an effector/target ratio of 20:1 (n = 4). (H) In vitro anticancer effects of M13 phage–polarized M1 macrophages measured by LDH assay after coculture with CT26 cells at the ratio of 20:1 (n = 6). Significant differences were assessed in (F), (G), and (H) using one-way ANOVA and t test and in (D) using t test. The mean values and SD are presented. MFI, mean fluorescence intensity.
It is well known that phage as a type of virus has a natural immunostimulatory effect to trigger specific immune responses (37, 38). For this reason, we evaluated immunogenic properties of M13 phages. The influence of M13@Ag in antigen-presenting cells (APCs) was detected by flow cytometry (Fig. 4E). Upon coincubation with bone marrow–derived dendritic cells (BMDCs), the expression of cell surface markers in mature DCs (CD11c+, CD80+, and CD86+) (39) was analyzed. Flow analysis demonstrated about a twofold enhancement of DC maturation compared with PBS treatment. Next, we assessed the effect of M13 phages on macrophages. RAW 264.7 cells were pretreated with inducers to obtain M2-phenotype macrophages (M2 Mø). Under the same coculture conditions, CD86-positive M1-phenotype macrophages (M1 Mø) (40) were measured. In vitro culture of M2 Mø cells with M13@Ag resulted in switching TAMs from the M2 to the M1 phenotype, and increasing M1-phenotype polarization was about 2.5-fold. These findings revealed the important role of M13@Ag in activating APCs. Subsequently, cytotoxic T lymphocyte (CTL)–mediated cancer cell–specific lysis was assessed by lactate dehydrogenase (LDH) release assay. Splenocytes from BALB/C mice immunized with CT26 cell lysates were preprocessed with concanavalin A (Con A) and M13 phages and then cocultured with CT26 cells. Lymphocytes were preactivated by naive coat proteins of M13 phages and M13@Ag, which showed more virulent toward tumor cells (Fig. 4F). In collected supernatants, tumor necrosis factor–α (TNF-α) secreted by CTL was measured to reconfirm the potent antitumor immune response (Fig. 4G). In addition, cytotoxicity of M1 Mø to CT26 tumor cells was also examined. M13@Ag-polarized M1 macrophages showed much stronger antitumor effect than the other controls expectably (Fig. 4H). These data demonstrated that M13@Ag exerted a potent effect on APC activation and played a pivotal role in boosting the antitumor immune response.
Tumor targeting and tumor treatment effects of M13@Ag in vivo
The tumor-targeting capacity of M13@Ag was studied in the orthotopic CT26 murine model. After 24 hours of Fn colonization in mice gut, M13 phages and M13@Ag were intravenously injected into the mice. Wild-type phages without Fn-binding ability were the control group. As shown in Fig. 5A, the intensity of phage fluorescence (labeled Cy5) at the intestine and tumor site gradually increased with time, prolonging in the M13 phages and M13@Ag groups. Meanwhile, prime overlap between bioluminescence of CT26 tumors and fluorescence of phages was observed, whereas the control group showed uniform distribution of fluorescence and very limited overlap. The major phage accumulation sites were determined to be the liver and the tumor of the intestinal tract at 24 hours after injection by ex vivo fluorescence imaging. In line with the above results, intestinal fluorescence intensity of M13 phages and M13@Ag groups was higher than that of the control group (Fig. 5B and fig. S5A). Together, these results indicated that M13 phages could accurately bind to Fn for realizing specifically eliminating bacteria.
Fig. 5. In vivo M13@Ag mediated tumor targeting and anticancer effect.
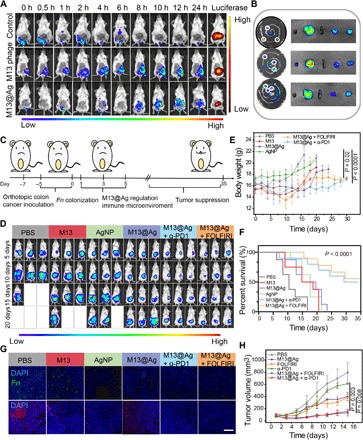
(A) Representative IVIS images for the capacity of phage accumulation in orthotopic CT26-luc tumors after treatment with wild phages (without Fn-binding ability), M13 phages (with Fn affinity), and M13@Ag (n = 3). h, hours. (B) Ex vivo fluorescence imaging of the major organs as well as tumors for measuring the accumulation of M13 phages and M13@Ag after the intravenous injection at 24 hours (n = 3). The tumors were pointed by white circles. (C) Schematic of building orthotopic CRC model and therapeutics-based M13@Ag treatment plan. (D) In vivo bioluminescence imaging of orthotopic CT26-luc tumor-bearing mice after receiving PBS, M13 phages, AgNP, M13@Ag, and M13@Ag combined with α-PD1 or FOLFIRI treatments (n = 5). Mice body weight (E) and survival curves (F) after receiving various treatments (n = 5). (G) Representative fluorescence images of orthotopic tumor-bearing mice showing the number of Fn and Ki67 staining after treatments. (H) Tumor growth curves after different treatments in subcutaneous CT26 tumor model (n = 5). Significant differences were assessed in (F) using one-way ANOVA and in (E) and (H) using t test. The mean values and SD are presented.
To validate the therapeutic effects of M13@Ag, we combined exogenous agents of immune checkpoint inhibitors (α-PD1) and chemotherapy [FOLFIRI, a first-line chemotherapeutics including fluorouracil, leucovorin, and irinotecan (IRT)] for tumor treatment. In orthotopic CT26 model, mice received various treatments, and tumor growths were monitored through the bioluminescence of cancer cells and mice body weight (Fig. 5, C to E, and fig. S5B). It was obviously observed that the M13@Ag-treated group displayed smaller tumor volume compared with the other groups treated with PBS, M13, and AgNP, while the combination treatment of α-PD1 or FOLFIRI resulted in significant tumor growth suppression over other groups with the weakest bioluminescence intensity and lower body weight. From the quantitative analysis of in vivo bioluminescence image in orthotopic CT26 model (fig. S5B), the combination treatment of the α-PD1 or FOLFIRI groups showed about 2.5-fold and 2-fold in enhancing tumor cell suppression compared with M13@Ag-only treatment, which indicates the cooperativeness of exogenous agents like immune checkpoint inhibitors and chemotherapy that could exhibit stronger antitumor response. Moreover, it should be noted that the combination treatment prolonged the survival time (=35 days) in comparison with the M13@Ag treatment alone (=23 days) (Fig. 5F). Accordingly, from FISH and fluorescence staining of Ki67, the M13@Ag-treated group exhibited a significant decrease in Fn load, and M13@Ag combination treatment groups displayed apparent suppression of tumor cells (Fig. 5G and fig. S5C). Hematoxylin and eosin (H&E) staining also yielded the same results (fig. S6, A and B). To further visualize the tumor development in intestinal tracts, mice were euthanized and dissected to count the number of tumors. Similar to the above results, the combination treatments revealed a remarkable anticancer effect (fig. S6, C and D). We also conducted an antitumor study in a subcutaneous tumor model. Mice were treated with PBS, M13@Ag, α-PD1 or FOLFIRI monotherapy, and M13@Ag combination treatments, respectively. Next, the tumor burden and body weight were assessed during 15 days (Fig. 5H and fig. S7, A and B). The tumor volume and H&E staining reconfirmed that M13@Ag combination treatments could effectively elevate tumor therapy (fig. S7C).
M13@Ag mediated immunosuppressive TME reversion in vivo
To further investigate whether M13@Ag could eliminate the immunosuppressive microenvironment by clearing Fn, immune cells were analyzed within the tumor tissues and spleen after various treatments. The aforementioned tests, in which M13 phages influenced APCs, were proved again in vivo. Significant improvement of mature DCs and M1 Mø on the M13 phage–treated group by up-regulation of the cell surface marker is showed in Fig. 6 (A and B). For the M13@Ag treatment groups (M13@Ag, M13@Ag + α-PD1, and M13@Ag + FOLFIRI), the number of activation of APCs was about 1.3-fold and about 3.3-fold higher than the M13 phages group and PBS group. Thereafter, tumor-infiltrating lymphocytes were analyzed at tumor sites. As shown in fig. S8 (A and B), the proportion of tumor-infiltrating CD45+ T cells in M13@Ag combination treatment groups was remarkably enhanced compared with the other groups. The intensity of the antitumor immune response was reflected by the number of CD8+ toxic T cells (Fig. 6C). The percentage of CD8+ T cells in the tumors treated with M13@Ag + α-PD1 was about 7.1-fold compared with the PBS-treated group and about 1.2-fold compared with the group treated with M13@Ag. Next, we quantified the negative regulation immune cells of regulatory T cells (Tregs) and MDSCs around the tumor site and spleen. The reduction in MDSCs and Tregs was shown in mice treated with M13@Ag and M13@Ag + α-PD1, as well as the M13@Ag + FOLFIRI group, markedly lower than the other groups for which the reason was that 5-fluorouracil (5-FU) of the FOLFIRI played an important role in depleting MDSCs (Fig. 6D and fig. S8, C to E). We reconfirmed the conclusion based on the reduction in MDSCs in spleen tissues and immunohistochemistry staining of arginase-1 (a marker of M2 phenotype and MDSC) and immunofluorescence staining of Ly6G and Gr-1 at tumor sites in M13@Ag and M13@Ag combination treatment groups (Fig. 6, E and F, and fig. S9). T cell response through IFN-γ production in the M13@Ag + α-PD1–treated group also increased distinctly by ELISA assay, as tumor-derived factor, interleukin-10 (IL-10) involvement of MDSCs was down-regulated with M13@Ag combination therapy treatment (Fig. 6, G and H). Consequently, these data revealed that rebuilding of the tumor-immune microenvironment by decreasing the frequency of MDSCs and facilitating the APC development could strengthen systemic anticancer immunity.
Fig. 6. M13@Ag reversed immunosuppressive TME for activation of antitumor immune response.
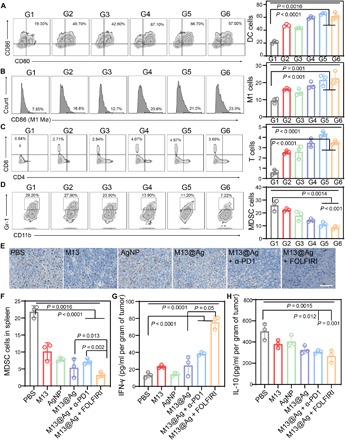
Orthotopic CT26-luc tumors were harvested from mice at 14 days after different treatments. Representative flow cytometric analysis images (left) and relative quantification (right) of infiltrating immune cells within tumor tissues as indicated. G1, PBS; G2, M13; G3, AgNP; G4, M13@Ag; G5, M13@Ag + α-PD1; G6, M13@Ag + FOLFIRI. (A) The mature of double-positive CD80+CD86+ dendritic cell gating on CD11c+ cells (n = 3). (B) TAM of classical activation M1 phenotype highly expressed CD86 (gated on CD11b+ cells) (n = 3). (C) The level of tumor infiltration CD8+ cytotoxic T cells gating on CD3+ cells (n = 3). (D) Percentage of CD11b+Gr-1+ MDSCs in the total of CD45+ lymphocytes (n = 3). (E) Immunohistochemistry staining images for arginase-1 (Arg-1) expression with immunosuppressive cells in different groups. (F) Relative quantification of MDSCs in spleen. (G and H) Secretion levels of IL-10 and IFN-γ in different treated groups (n = 3). Significant differences were assessed in (A) to (D) and (F) to (H) using one-way ANOVA and t test. The mean values and SD are presented.
DISCUSSION
The intestinal microbiome plays a pivotal role in host processes, including the development of cancer. Cancer treatment with antimicrobial agents, such as antibiotics, have displayed therapeutic potential for inhibiting tumor proliferation. However, the effectiveness of antimicrobial agents is limited by the diversity and complexity of microbial communities associated with human physiological function. With the in-depth understanding of the relationship between bacteria and tumor progression, we present a new type of immune treatment strategy against CRC based on the regulation of gut microbiota to reverse immunosuppressive TME by bioinorganic hybridization of M13 phages. In summary, a strain of selectively Fn-binding M13 phage was screened by using a phage display library, and AgNPs were introduced on the surface of phages to realize precise scavenging of pro-tumor bacteria of Fn, thereby blocking MDSC augmentation in the TME. We also showed that M13@Ag not only could boost the host immune response, but also that in vivo studies of M13@Ag combination therapeutics notably delayed orthotopic CRC tumorigenesis as well as prolonged the survival time of mice. Our work highlights the possibility of selectively modulating the gut microbiota using phages for neutralizing the tumor-immunosuppressive microenvironment as a potential treatment for CRC. However, we caution that such treatment could potentially yield only a modest therapeutic effect on CRC if there is no specific colonization of Fn in the tumor. Moreover, further studies focused on the combination of advanced biocomposites would be required to strengthen the therapeutic effect on CRC. Along with this concept, we expect that new efforts targeting specific tumor microbiota for tumor therapy will become an increasingly researched field in both basic and translational research.
MATERIALS AND METHODS
Cell lines and bacterial strains
CT26 cells and 3T3 cells were obtained from the China Center for Type Culture Collection and cultured in Roswell Park Memorial Institute 1640 (RPMI 1640; Gibco, Invitrogen) and DMEM containing 10% FBS (Invitrogen), penicillin (100 U/ml; Invitrogen), and streptomycin (100 U/ml; Invitrogen) in an incubator at 37°C with 5% CO2. All experiments were performed with cells on passages 2 to 10. Fn [American Type Culture Collection (ATCC) 10953] was obtained from the ATCC. S. aureus [CMCC (B) 26003] was obtained from the National Center for Medical Culture Collections (CMCC). Brain heart infusion broth (Qingdao Hope Biotechnology, China) and Luria-Bertani broth were used for the bacteria culture, respectively. All bacteria were purified from colonies isolated from plates for subsequent liquid subcultures.
Animal model establishment
All the animal experiments were performed according to the guidelines for laboratory animals established by the Wuhan University Center for Animal Experiment/A3-Lab. The anticancer effect of M13@Ag combination treatment was assessed in subcutaneous CT26 tumor mode and orthotopic CRC model. In the subcutaneous CT26 tumor model, 5 × 105 CT26 cells per mice were injected into the right flank of the female BALB/c mice of 6 weeks. Tumor size was recorded every 2 days and calculated as V = 0.5 × L × W2, in which L and W are the longest and the shortest lengths of the tumor, respectively. In the orthotopic CRC model, luciferase-transfected CT26 cells (40 μl per mice, 1 × 107 cells/ml) were implanted in the cecum of BALB/c mice. The tumor burden was observed through bioluminescence by the IVIS (PerkinElmer) system.
Materials
FOLFIRI containing 5-FU, IRT, and leucovorin were purchased from Aladdin Reagents. ZnO, graphene oxide, and cellulose were purchased from Xfnano (China). Dextran was purchased from Shanghai Yuanye Biotechnology. RPMI 1640 medium, FBS, penicillin-streptomycin, and trypsin were provided by Invitrogen (USA). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium-bromide (MTT) and LDH release assay kit were supplied by Beyotime Biotechnology Co. Ltd. (China). AgNO3 was purchased from Sinopharm Chemical Reagent Co. Ltd. (China). ELISA kits for TNF-α, IL-10, and IFN-γ were purchased from 4A Biotech Co. Ltd. (Beijing, China). A library of random peptides 12 amino acids long displayed on the minor coat protein, gene III, of the bacteriophage M13 was purchased from New England Biolabs (Ph.D.-12 Phage display library kit).
Antibodies for flow cytometry
The primary fluorescence-labeled antibodies used for immunostaining were CD45 (clone 30-F11), CD11b (clone M1/70), CD80 (clone 16-10A1), Gr-1 (clone RB6-8C5), CD3 (clone 17A2), CD4 (clone GK1.5), CD8 (clone 53-5.8), CD11c (clone N418), CD86 (clone GL-1), CD25 (clone PC61), and CD127 (clone SB/199) following the manufacturer’s instructions.
In vitro phage display
Biopanning of Fn-binding specificity M13 phage experiments were carried out by using Ph.D.-12 Phage display library kit. Briefly, a six-well plate was coated with 1 ml of Fn (100 μg/ml) (OD600 = 0.3) that dispersed in 0.1 M NaHCO3 (pH 8.6) overnight at 4°C. After complete removal of the coating solution, each coated well was filled with blocking buffer [0.1 M NaHCO3 (pH 8.6), bovine serum albumin (BSA; 5 mg/ml), and 0.02% NaN3] and incubated at 4°C for 2 hours. Then, we removed the blocking buffer and washed each well six times with TBST (tris buffered saline tween) [50 mM tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Tween 20]. Phages (10 μl) (approximately 4 × 1010 PFU) from the random display library diluted in TBST were incubated in the Fn-coated well for 1 hour at room temperature. Unbound phages were removed by washing with TBST for 10 times. Phages with high affinity of Fn were eluted with 0.2 M glycine (pH 2.2) containing BSA (1 mg/ml) and neutralized with 1 M tris-HCl (pH 9.1). Subsequently, phage was eluted, titered, and amplified for the next round of screening. After the third round of biopanning, predominant Fn-binding selective phages were isloated and purified for further study.
Preparation of various nanoparticles
AgNP
AgNO3 (0.57 g) was added into 133 ml of dextran T100 [weight % (wt %) = 0.2%] solution and fully mixed for 30 min. The mixture was irradiated for 2 hours by ultraviolet light (12 W) at room temperature to reduce Ag+ and centrifuged (3920g, 30 min) to obtain AgNP.
AuNP
One hundred milliliters of 5 mM HAuCl4 was added to 300 ml of 2 mM ice-cold NaBH4 solution. Then, 50 ml of 1% PVA (polyvinyl alcohol) was added during the reduction and stirred for 3 hours. After completion of the reaction, the mixture was boiled for 1 hour to decompose excess of NaBH4 to obtain the final nanoparticles.
C3N4
Urea (10 g) was added to 5 ml of ultrapure water. Then, the suspension was placed in an alumina crucible and heated to 550°C with a heating rate of 0.5°C min−1 in a muffle furnace. The temperature of the muffle furnace was maintained for 3 hours. Then, the obtained powder was cooled to room temperature and washed with deionized water (DI) water three times. C3N4 was collected by filtration and dried at room temperature.
Fe3O4
FeCl3∙6H2O (13.10 g) and 6.65 g of FeCl2∙4H2O were mixed in 80 ml of DI water. The solution was stirred under nitrogen for 0.5 hour, and 45 ml of NH3∙H2O was added dropwise at 80°C. Citric acid (7.5 g) in 15 ml of water was introduced after the temperature rose to 95°C and stirred for 90 min. The solution was dialyzed against water with a dialysis bag [molecular weight cutoff (MWCO), 14,000 Da] to obtain a stable magneto fluid solution.
Screen out antibacterial nanoparticles
Eight types of nanoparticles (AuNP, Fe3O4, C3N4, cellulose, graphere, ZnO, AgNP, and dextran) were cocultured with Fn [105 colony-forming units (CFU) ml−1] individually. The absorbance at 600 nm was measured for monitoring the bacteria growth.
Assembly of M13 with AgNP
Excess of AgNP was mixed with M13 phage (1010 PFU) via electrostatic interaction for 2 hours. Then, the complexes were precipitated by PEG (polyethylene glycol)/NaCl and centrifuged (15 min, 3920g) to remove residual AgNP. The concentration of M13@Ag was measured by silver content by using ICP-AES spectroscopy.
Bioactivity of M13@Ag assay
To determine bacteriophage inactivation by AgNP, solutions with various concentrations of AgNP (Cmax = 20 μg/ml, Cmin = 5 μg/mL) were incubated with M13 phages (10 μl, 1010 PFU) for 2 hours. After that, phages that contained AgNP were diluted (109-fold) in PBS, and titer was determined by the “double-layer” method.
The antimicrobial effect of M13@Ag
For measuring the ability of killing Fn by M13@Ag, various concentrations of AgNP and M13@Ag were cocultured with Fn (1 × 105 CFU/ml) for 8 hours, individually. After that, the absorbance at 600 nm was measured for monitoring the bacteria growth. The mixture was then serially diluted 100-fold and plated on blood agar plates overnight at 37°C. CFU also determined the inhibition rate of Fn.
Cell viability assay
To evaluate cytotoxicity of M13 phages toward normal cells and tumor cells, CT26 cells and 3T3 cells were seeded in 96-well plates with a density of 104 cells/ml. M13 phage with gradient concentrations (Cmax = 1 × 1012 PFU/ml; Cmin = 1 × 108 PFU/ml) were added into each well and incubated for 24 hours. Subsequently, MTT (5 mg ml−1) was added into each well and coincubated for another 4 hours. The cultured medium in each well was replaced with 150 μl of dimethyl sulfoxide. The relative cell viability was assessed by MTT assay.
In vivo blood biochemistry analysis of M13@Ag
To assess the effects in physiology caused by M13@Ag, blood biochemical indexes were tested in vivo. Healthy female BALB/c mice were intravenously injected with PBS (100 μl), AgNP (100 μg/ml; 100 μl per mice), M13 phages (1 × 1012/PFU; 100 μl per mice), and M13@Ag [1 × 1012/PFU in AgNP (100 μg/ml); 100 μl per mice). After 24 hours of posttreatment, blood samples were collected from the heart, respectively. The blood biochemistry analysis was measured by a biochemical autoanalyzer (MNCHIP, Tianjin, China).
Transmission electron microscopy
For TEM images, 1 × 1010 PFU M13 phages and M13@Ag [1 × 1010 PFU in AgNP (5 μg/ml)] were resuspended in DI water and dropped on a carbon copper holey grid. To demonstrate the M13@Ag-binding Fn, M13@Ag (10 μl; 1 × 1010 PFU) was coincubated with Fn (200 μl; 1 × 105 CFU) for 30 min. Then, the mixture was centrifuged (15 min, 3920g) to remove residual M13@Ag, and the complexes were washed three times. The TEM images were obtained using a JEOL 2000FX instrument.
In vitro M13@Ag selective binding assay
Fn (108 CFU/ml) and S. aureus (108 CFU/ml) were labeled with green fluorescent FITC. Meanwhile, M13 phage and M13@Ag were marked with red fluorescent Cy5. Then, bacteria were coincubated with M13 phage and M13@Ag for 30 min, severally. The mixture was centrifuged (15 min, 3920g) to remove residual phages, and the complexes were washed for three times. The fluorescence colocalization was observed with a superresolution fluorescence microscopy. For flow cytometry analysis, green fluorescent FITC-labeled bacteria were cocultured with M13 phage or M13@Ag (without fluorescence) following the above process.
M13 phage for activating immune cells of DCs and M1 macrophages
To evaluate immunomodulatory activity of M13 phage, BMDCs were seeded in a six-well plate with a density of 5 × 105 per well and incubated with M13 phage (1 × 1010 PFU) or M13@Ag (1 × 1010 PFU) for 24 hours. Then, flow cytometry analysis of mature DCs stained the surface marker with CD11c, CD80, and CD86. For M1-phenotype macrophage polarization, RAW 264.7 cells were seeded in a six-well plate (1 × 104 per well). After 24 hours, IL-4 (20 ng/ml) was added to induce differentiation of M2 macrophages for another 24 hours. M13 phage (1 × 1010 PFU) or M13@Ag (1 × 1010 PFU) was cocultured with M2 macrophages for 24 hours, subsequently. Flow cytometry analysis of M1 phenotype highly expressed CD86.
M13 phage–mediated cytotoxicity of lymphocytes or M1 macrophages assays
CTL-mediated cancer cell–specific lysis was performed as follows. CT26 cells were seeded in a 24-well plate at a density of 1 × 105 per well. Meanwhile, splenocytes were harvested from spleen in female BALB/c mice 6 weeks after being immunized with CT26 cell lysates for generation of CT26-specific T lymphocytes. Cells were then prestimulated with Con A (10 μg/ml) and M13 phages (1 × 1010 PFU) or M13@Ag (1 × 1010 PFU) for 4 hours. Then, activated splenocytes were coincubated with CT26 cells at the effective target cell ratio of 20:1. After 24 hours of incubation, specific-lysed 4T1 cells that released LDH in supernatant was measured by the LDH release assay kit. In the meantime, supernatant was collected for TNF-α quantification by ELISA. Cytotoxicity of M1 macrophage activity was also certified with a coculture system of CT26 cells and M1 macrophages. RAW 264.7 cells were pretreated with IL-4 (20 ng/ml) for 24 hours to prepare M2 macrophages, and then M13 phages (1 × 1010 PFU) or M13@Ag (1 × 1010 PFU) was added for switching TAMs from the M2 to the M1 phenotype. Twenty-four hours later, different types of macrophages (RAW 264.7, M2 macrophages, and M1 macrophages) were cocultured with CT26 cells (effective target cell ratio = 20:1) for another 24 hours to measure LDH release.
In vivo specific targeting capacity of M13@Ag
Luciferase-transfected CT26 tumor cells were transplanted in the cecum of BALB/c mice of orthotopic CRC model. Fn (1 × 108 CFU per mice) was orally administered 24 hours to colonize in the gut before the posttreatment. Cy5-labeled wild-type phage (1 × 1011 PFU per mice), M13 phage (1 × 1011 PFU per mice), and M13@Ag (1 × 1011 PFU per mice) were intravenously injected into mice, respectively. Fluorescence accumulation at the tumor site was imaged with IVIS system at different time points. Orthotopic tumor was imaged by bioluminescence after intraperitoneal injection of d-luciferin into mice (1 mg per mice). At 6 hours of in vivo fluorescence imaging, mice were euthanized, while tumors and organs of these mice were collected for ex vivo imaging.
M13@Ag mediated combination therapy assay in various CRC models
For the orthotopic CRC model, Fn (1 × 108 CFU per mice) was orally administered every day for a total of five times to colonize in the gut before the posttreatment. Mice bearing tumors were divided randomly into six groups (n = 8 per group): PBS (100 μl per mice), M13 phage (1 × 1012/ml PFU; 100 μl per mice), AgNP (100 μg/ml; 100 μl per mice), M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/ml); 100 μl per mice], M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/ml); 100 μl per mice] + α-PD1 (25 μg per mice), and M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/ml); 100 μl per mice] + FOLFIRI [5-FU (30 mg kg−1), leucovorin (90 mg kg−1), and IRT (16 mg kg−1)], respectively. M13 phage, M13@Ag, and α-PD1 were intravenously injected into mice, while FOLFIRI was orally administered. All treatments were proceeded every other day for a total of three injections. The growth of the orthotopic tumors was observed through the bioluminescence of cancer cells by the IVIS (PerkinElmer) system. After various treatments, mice were euthanized and dissected to analyze tumor progression.
For the subcutaneous model, mice with tumors approximately 100 mm3 were blinded to the group allocation. Fn (1 × 108 CFU per mice) was intratumorally injected every day for a total of three times to colonize. Then, PBS (100 μl, intravenously), M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/ml); 100 μl per mice, intravenously], FOLFIRI [5-FU (30 mg kg−1), leucovorin (90 mg kg−1), and IRT (16 mg kg−1), intraperitoneally], α-PD1 (25 μg per mice, intravenously), M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/ml); 100 μl per mice, intravenously] + α-PD1 (25 μg per mice, intravenously) and M13@Ag [1 × 1012/ml PFU in AgNP (100 μg/m); 100 μl per mice, intravenously] + FOLFIRI [5-FU (30 mg kg−1), leucovorin (90 mg kg−1), and IRT (16 mg kg−1), intraperitoneally] were given into mice tumors, respectively. The increase in tumor volumes was monitored every 2 days.
Flow cytometry analysis of immune cells in vivo
In the orthotopic CRC treated model at 14 days, mice were euthanized (n = 3) and tumor tissues and spleen were freshly harvested. Tumor tissues were digested with collagenase type IV, hyaluronidase, and deoxyribonuclease I to prepare single-cell suspensions. Meanwhile, spleen was triturated by using 2% FBS in PBS. Then, cells (1 × 107 cells/ml; 100 μl) were collected and stained with specific fluorescent conjugated antibodies for flow cytometry analysis (BD FACS Aria TM III).
Immunofluorescence staining and FISH assays
In a clinically relevant in vivo model of CRC, mice were euthanized at day 15, and tumors were collected for FISH assay and immunofluorescence staining of CD3 and Gr-1. In orthotopic CRC therapy model, immunofluorescence staining of Ly6G and Gr-1 and immunohistochemistry staining of arginase-1 were performed at posttreatment on day 14, as well as FISH assay and immunofluorescence staining of Ki67 were conducted at the end of treatments. All these antibodies were purchased from Servicebio.
16S ribosomal DNA sequencing assay
Fecal samples of orthotopic CT26-luc tumor-bearing mice were collected after different treatments. Twenty-four hours after the administration of Fn (108 CFU per mice), mice were treated with PBS, M13 phages (1012 PFU/ml; 100 μl per mice, intravenously), and M13@Ag [1 × 1012/PFU in AgNP (100 μg/ml); 100 μl per mice, intravenously]. Then, these fecal samples were collected for 16S rDNA sequencing.
Statistical analysis
The experimental data were presented with average values, expressed as means ± SD. Statistical analysis was conducted using one-way analysis of variance (ANOVA) or Student’s t test of origin 8.5. For in vivo experiments, operations were blinded to treated groups. Animals were randomly divided into different groups.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (51690152, 51873162, and 51833007). Author contributions: X.D. and X.-Z.Z. conceived the project and designed the experiments. X.D. and P.P. synthesized materials. P.P. performed in vitro microbiological experiments. X.D. performed in vitro cell experiments. X.D., P. P., and P.B. performed in vivo experiments. X.D., P.P., and D.-W.Z. collected and analyzed the data. X.D., P.P., D.-W.Z., X.Z., and X.-Z.Z. cowrote the manuscript. All authors discussed the results and reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba1590/DC1
REFERENCES AND NOTES
- 1.Schwabe R. F., Jobin C., The microbiome and cancer. Nat. Rev. Cancer 13, 800–812 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zitvogel L., Galluzzi L., Viaud S., Vétizou M., Daillère R., Merad M., Kroemer G., Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 7, 271ps1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitt J. M., Vétizou M., Daillére R., Roberti M. P., Yamazaki T., Routy B., Lepage P., Boneca I. G., Chamaillard M., Kroemer G., Zitvogel L., Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity 44, 1255–1269 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Vétizou M., Pitt J. M., Daillère R., Lepage P., Waldschmitt N., Flament C., Rusakiewicz S., Routy B., Roberti M. P., Duong C. P., Poirier-Colame V., Roux A., Becharef S., Formenti S., Golden E., Cording S., Eberl G., Schlitzer A., Ginhoux F., Mani S., Yamazaki T., Jacquelot N., Enot D. P., Bérard M., Nigou J., Opolon P., Eggermont A., Woerther P. L., Chachaty E., Chaput N., Robert C., Mateus C., Kroemer G., Raoult D., Boneca I. G., Carbonnel F., Chamaillard M., Zitvogel L., Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gopalakrishnan V., Helmink B. A., Spencer C. N., Reuben A., Wargo J. A., The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33, 570–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daillére R., Vétizou M., Waldschmitt N., Yamazaki T., Isnard C., Poirier-Colame V., Duong C. P. M., Flament C., Lepage P., Roberti M. P., Routy B., Jacquelot N., Apetoh L., Becharef S., Rusakiewicz S., Langella P., Sokol H., Kroemer G., Enot D., Roux A., Eggermont A., Tartour E., Johannes L., Woerther P. L., Chachaty E., Soria J. C., Golden E., Formenti S., Plebanski M., Madondo M., Rosenstiel P., Raoult D., Cattoir V., Boneca I. G., Chamaillard M., Zitvogel L., Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45, 931–943 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Sivan A., Corrales L., Hubert N., Williams J. B., Aquino-Michaels K., Earley Z. M., Benyamin F. W., Lei Y. M., Jabri B., Alegre M.-L., Chang E. B., Gajewski T. F., Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu S., Rhee K. J., Albesiano E., Rabizadeh S., Wu X., Yen H. R., Huso D. L., Brancati F. L., Wick E., McAllister F., Housseau F., Pardoll D. M., Sears C. L., A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geva-Zatorsky N., Sefik E., Kua L., Pasman L., Tan T. G., Ortiz-Lopez A., Yanortsang T. B., Yang L., Jupp R., Mathis D., Benoist C., Kasper D. L., Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tilg H., Adolph T. E., Gerner R. R., Moschen A. R., The intestinal microbiota in colorectal cancer. Cancer Cell 33, 954–964 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Louis P., Hold G. L., Flint H. J., The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12, 661–672 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Kostic A. D., Chun E., Robertson L., Glickman J. N., Gallini C. A., Michaud M., Clancy T. E., Chung D. C., Lochhead P., Hold G. L., El-Omar E. M., Brenner D., Fuchs C. S., Meyerson M., Garrett W. S., Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen T., Li Q., Wu J., Wu Y., Peng W., Li H., Wang J., Tang X., Peng Y., Fu X., Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 67, 1635–1646 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binnewies M., Roberts E. W., Kersten K., Chan V., Fearon D. F., Merad M., Coussens L. M., Gabrilovich D. I., Ostrand-Rosenberg S., Hedrick C. C., Vonderheide R. H., Pittet M. J., Jain R. K., Zou W., Howcroft T. K., Woodhouse E. C., Weinberg R. A., Krummel M. F., Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phuengkham H., Song C., Um S. H., Lim Y. T., Implantable synthetic immune niche for spatiotemporal modulation of tumor-derived immunosuppression and systemic antitumor immunity: Postoperative immunotherapy. Adv. Mater. 30, e1706719 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Peng B., Zhang X., Aarts D. G. A. L., Dullens R. P. A., Superparamagnetic nickel colloidal nanocrystal clusters with antibacterial activity and bacteria binding ability. Nat. Nanotechnol. 13, 478–482 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Sunderland K. S., Yang M., Mao C., Phage-enabled nanomedicine: From probes to therapeutics in precision medicine. Angew. Chem. Int. Ed. Engl. 56, 1964–1992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richter A. P., Brown J. S., Bharti B., Wang A., Gangwal S., Houck K., Cohen Hubal E. A., Paunov V. N., Stoyanov S. D., Velev O. D., An environmentally benign antimicrobial nanoparticle based on a silver-infused lignin core. Nat. Nanotechnol. 10, 817–823 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Ando H., Lemire S., Pires D. P., Lu T. K., Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 1, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dedrick R. M., Guerrero-Bustamante C. A., Garlena R. A., Russell D. A., Ford K., Harris K., Gilmour K. C., Soothill J., Jacobs-Sera D., Schooley R. T., Hatfull G. F., Spencer H., Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilcher S., Loessner M. J., Engineering bacteriophages as versatile biologics. Trends Microbiol. 27, 355–367 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Krom R. J., Bhargava P., Lobritz M. A., Collins J. J., Engineered phagemids for nonlytic, targeted antibacterial therapies. Nano Lett. 15, 4808–4813 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Qin H., Lerman B., Sakamaki I., Wei G., Cha S. C., Rao S. S., Qian J., Hailemichael Y., Nurieva R., Dwyer K. C., Roth J., Yi Q., Overwijk W. W., Kwak L. W., Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 20, 676–681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussain S., Joo J., Kang J., Kim B., Braun G. B., She Z. G., Kim D., Mann A. P., Mölder T., Teesalu T., Carnazza S., Guglielmino S., Sailor M. J., Ruoslahti E., Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat. Biomed. Eng. 2, 95–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin P. P., Sha R., Zhang Y., Liu L., Bian Y., Qian J., Qian J., Lin J., Ishimwe N., Hu Y., Zhang W., Liu Y., Yin S., Ren L., Wen L. P., Blood circulation-prolonging peptides for engineered nanoparticles identified via phage display. Nano Lett. 19, 1467–1478 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Chernousova S., Epple M., Silver as antibacterial agent: Ion, nanoparticle, and metal. Angew. Chem. Int. Ed. Engl. 52, 1636–1653 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Oh J.-W., Chung W.-J., Heo K., Jin H.-E., Lee B. Y., Wang E., Zueger C., Wong W., Meyer J., Kim C., Lee S.-Y., Kim W.-G., Zemla M., Auer M., Hexemer A., Lee S.-W., Biomimetic virus-based colourimetric sensors. Nat. Commun. 5, 3043 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Souza G. R., Molina J. R., Raphael R. M., Ozawa M. G., Stark D. J., Levin C. S., Bronk L. F., Ananta J. S., Mandelin J., Georgescu M. M., Bankson J. A., Gelovani J. G., Killian T. C., Arap W., Pasqualini R., Three-dimensional tissue culture based on magnetic cell levitation. Nat. Nanotechnol. 5, 291–296 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksson F., Tsagozis P., Lundberg K., Parsa R., Mangsbo S. M., Persson M. A., Harris R. A., Pisa P., Tumor-specific bacteriophages induce tumor destruction through activation of tumor-associated macrophages. J. Immunol. 182, 3105–3111 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Yu J., Feng Q., Wong S. H., Zhang D., Liang Q. Y., Qin Y., Tang L., Zhao H., Stenvang J., Li Y., Wang X., Xu X., Chen N., Wu W. K., Al-Aama J., Nielsen H. J., Kiilerich P., Jensen B. A., Yau T. O., Lan Z., Jia H., Li J., Xiao L., Lam T. Y., Ng S. C., Cheng A. S., Wong V. W., Chan F. K., Xu X., Yang H., Madsen L., Datz C., Tilg H., Wang J., Brünner N., Kristiansen K., Arumugam M., Sung J. J., Wang J., Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 66, 70–78 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Scibilia S., Lentini G., Fazio E., Franco D., Neri F., Mezzasalma A. M., Guglielmino S. P. P., Self-assembly of silver nanoparticles and bacteriophage. Sens. Biosensing Res. 7, 146–152 (2016). [Google Scholar]
- 32.Agarwal R., Johnson C. T., Imhoff B. R., Donlan R. M., McCarty N. A., García A. J., Inhaled bacteriophage-loaded polymeric microparticles ameliorate acute lung infections. Nat. Biomed. Eng. 2, 841–849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tawil N., Sacher E., Boulais E., Mandeville R., Meunier M., X-ray photoelectron spectroscopic and transmission electron microscopic characterizations of bacteriophage–Nanoparticle complexes for pathogen detection. J. Phys. Chem. C 117, 20656–20665 (2013). [Google Scholar]
- 34.Yang L., Liang H., Angelini T. E., Butler J., Coridan R., Tang J. X., Wong G. C., Self-assembled virus-membrane complexes. Nat. Mater. 3, 615–619 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Ghosh D., Lee Y., Thomas S., Kohli A. G., Yun D. S., Belcher A. M., Kelly K. A., M13-templated magnetic nanoparticles for targeted in vivo imaging of prostate cancer. Nat. Nanotechnol. 7, 677–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q., Wu Y., Wang J., Wu G., Long W., Xue Z., Wang L., Zhang X., Pang X., Zhao Y., Zhao L., Zhang C., Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci. Rep. 6, 27572 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karimi M., Mirshekari H., Moosavi Basri S. M., Bahrami S., Moghoofei M., Hamblin M. R., Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 106, 45–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao P., Zhu J., Mahalingam M., Batra H., Rao V. B., Bacteriophage T4 nanoparticles for vaccine delivery against infectious diseases. Adv. Drug Deliv. Rev. 145, 57–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng D.-W., Chen Y., Li Z.-H., Xu L., Li C.-X., Li B., Fan J.-X., Cheng S.-X., Zhang X.-Z., Optically-controlled bacterial metabolite for cancer therapy. Nat. Commun. 9, 1680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng R. H., Zou M. Z., Zheng D., Peng S. Y., Liu W., Bai X. F., Chen H. S., Sun Y., Zhou P. H., Zhang X. Z., Nanoparticles from cuttlefish ink inhibit tumor growth by synergizing immunotherapy and photothermal therapy. ACS Nano 13, 8618–8629 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba1590/DC1