

An electrochemical oxidation-induced C(sp3)─H etherification strategy by using nonsolvent alcohols has been disclosed.
Abstract
Direct electrochemical construction of C─O bonds through C(sp3)─H functionalization still remains fundamentally challenging. Here, electrochemical oxidation-induced benzylic and allylic C(sp3)─H etherification has been developed. This protocol not only offers a practical strategy for the construction of C─O bonds using nonsolvent amounts of alcohols but also allows direct electrochemical benzylic and allylic C(sp3)─H functionalization in the absence of transition metal catalysis. A series of alcohols and benzylic and allylic C(sp3)─H compounds were compatible with this transformation. Mechanistically, the generation of aryl radical cation intermediates is the key to this C(sp3)─H etherification, as evidenced by radical probe substrate (cyclopropane ring opening) and electron paramagnetic resonance experiments.
INTRODUCTION
Ethers are widely found in pharmaceuticals and medicinally relevant natural products and likewise serve as important intermediates in synthetic chemistry (1–4). Consequently, major efforts have been devoted toward the construction of ethers (Fig. 1). Traditionally, the Williamson reaction (5–7), using alcohols and electrophiles containing leaving groups as coupling partners, has been widely used in primary ether synthesis. Because of the potential competing β-H elimination, low yields of secondary and tertiary ethers were obtained by these methods. Besides, Williamson etherification suffers from undesired waste and multiple steps owing to the need of prefunctionalization of substrates. Subsequently, etherification reactions via cross-coupling of two alcohols have been disclosed (8–10). In this regard, the need for either high temperature or hazardous reagents leads to complicated operation. In addition, acid- or transition metal-catalyzed hydroalkoxylation of alkenes has emerged as a powerful strategy for C─O bond formation, which still maintains considerable room for improvement in functional group tolerance (11–15). Despite the above publications and other published ether synthesis methods (16–19), a simpler and straightforward synthetic strategy for C─O bond formation is fascinating yet elusive (20, 21).
Fig. 1. Previous etherification reactions and research scheme of this work.
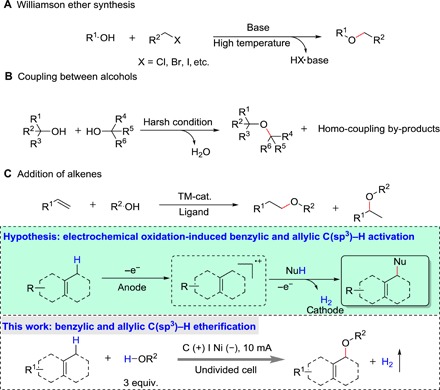
Recently, electrochemical oxidative intermolecular C─H/X─H (X = N, S, O) cross-coupling with release of hydrogen has substantially expanded access to valuable organic molecules (22–37). Electricity, serving as an environmentally friendly alternative to sacrificial oxidants, drives the chemical transformation. Under metal-free electrochemical condition, radicals, radical cations, or cations can be generated through electron transfer on the surface of the anode. Then, the reactions between the generated intermediate and other active substrates enable access to the target molecules along with the release of hydrogen at to cathode, which provides a privileged platform for oxidative C─H/X─H cross-coupling without the use of sacrificial oxidants and toxic catalysts. However, highly reactive intermediates selective for intermolecular cross-couplings under electrochemical conditions are difficult to handle. To address the challenge of selective C─H/X─H cross-coupling, the solvent quantities of one of were substrates were necessary, as exemplified by construction of C(sp2)─O bond and C(sp3)─O bond (30, 33). As a consequence, the art for the electrochemical generation of ethers through intermolecular C(sp3)─H/O─H cross coupling is at the start-up stage.
The disparity of redox potential of reaction substrates offers a possibility of selective activation of substrates. We hypothesized that etherification through C(sp3)─H/O─H cross-coupling under electrochemical conditions can be achieved by using the disparity of redox potential of alcohols and C(sp3)─H compounds, such as benzyl or allyl C(sp3)─H compounds (Fig. 1). In the reaction system, aryl radical cations can be easily formed from C(sp3)─H compounds via single-electron transfer (SET) processes at the anode. Nucleophiles, such as various alcohols, would attack the reactive radical cation to deliver target molecule concomitant with the release of hydrogen gas at cathode through a SET process.
Here, we describe an electrochemical oxidant-free protocol for the benzylic and allylic C(sp3)─H bond etherification using nonsolvent amounts of alcohols as the nucleophile in a simple undivided cell under constant-current conditions. A series of alcohols and benzylic and allylic C(sp3)─H compounds were compatible with this protocol under mild conditions, delivering corresponding products in satisfactory yields. Mechanistic studies demonstrated that this reaction involved a SET process. The formation and resonation of aryl radical cation intermediates are the key to this transformation, which was evidenced by probe substrate (cyclopropane ring opening) and electron paramagnetic resonance (EPR) experiments.
RESULTS AND DISCUSSION
Initially, indan (1a) and phenethyl alcohol (2a) were chosen as standard substrates. The desired product 3aa was obtained in 27% yield by using 10-mA constant current in an undivided cell. Pleasingly, 50% yield was delivered when we used Cs2CO3 as a base (table S1, entry 2). Moreover, after a screening of the electrode materials, a 56% yield of the desired product was obtained with the use of carbon as anode and nickel as cathode (table S1, entries 2 to 4). In addition, the desired product can be acquired in CH3CN (table S1, entry 5). Further experiments have shown that the mixture of 1,2-dichloroethane (DCE) and Et2O increased the yield to 60% (table S1, entry 7), and no obvious by-products were observed. Subsequently, the control experiments were also performed to evaluate the effect of the types of base. Cs2CO3 proved to be an optimal base for this C─H bond etherification (table S1, entries 10 and 11). Thereafter, the electrolytes were investigated. The yield decreased when nBu4NClO4 was replaced with nBu4NBF4 (table S1, entry 12). Furthermore, we could not further improve the reaction yield when we prolonged the reaction time, which might be because the product was decomposed with the extension of time. It is noteworthy that no reaction occurred without electricity (table S1, entry 14). Fifty percent yield of 3aa was obtained in ambient atmosphere, demonstrating that the reaction is not sensitive to the air (table S1, entry 15).
With the optimized conditions, we evaluated the scope of this electrochemical C─O bond formation (Fig. 2). Alcohols with aryl groups were first tested (3aa-3ak). Position of substances on the aromatic ring had no obvious influence on the efficiency of this protocol. 3aa-3ag can be obtained in considerable yield. Halides, such as F, Cl, and Br, which can be transformed into other molecule via coupling reactions, were well tolerated. Moreover, benzyl alcohol, being easily oxidized to benzaldehyde under oxidative conditions (38), was successfully transformed to corresponding products 3ah. β-Methylphenethyl alcohol reacted smoothly with indan (1a) to deliver desired product (3ak) in 62% yield. Subsequently, other alkyl alcohols were evaluated (3al-3az). This transformation was also compatible with a series of cyclic alcohols, affording 3al-3ap in satisfactory yields. Among them, adamantanol offered a lower yield of product because of the steric hindrance (3al). Similarly, other hindered alcohols were also amenable to this C─O bond formation strategy with moderate yields (3ar and 3au). Reductively labile groups, such as alkynes and vinyl, were amenable to this electrochemical strategy. 3ap and 3aw were accessed through C─O bond formation despite the nucleophilic sensitivity of alkyl chloride and cyclopropyl. Alcohols containing multiple F atom reacted smoothly with indan to deliver the desired products (3ax). Alcohols, serving as important structural motifs, have widespread application in medical treatments (39). Pharmaceutical molecules, which contain hydroxyl group such as menthol and epiandrosterone, were suitable coupling partners in this protocol, delivering corresponding products in satisfactory yields (3ay and 3az). With respect to C(sp3)─H compounds, different kinds of substrates were investigated (3ba-3ja). 1,2,3,4-Tetrahydronaphthalene was tolerated (3ba). Furthermore, 3ca and 3da were successfully obtained. Highly selective C─H bond etherification of ethyl groups was achieved rather than of methyl groups when we used 4-ethyltoluene to react with phenethyl alcohol (3ea). Similar result occurred with the use of 2-ethyl-5-methylthiophene as coupling partner (3fa and 3ga), which also means that heterocyclic compounds were well tolerated in this etherification reaction. As is known, activation of allylic C─H bond remains a great challenge. In this work, allylic C─H etherification can be achieved, providing a practical way for the functionalization allylic C─H bond. Cyclohexene reacted smoothly with phenethyl alcohol and hex-3-yn-1-ol, forging 3ha and 3ia in 50 and 46%, respectively. To our delight, cyclododecene was also amenable to this electrochemical etherification reaction (3ja).
Fig. 2. Substrate scope for the electrochemical etherification.
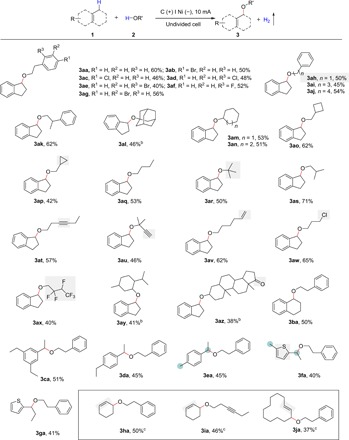
Standard conditions: Carbon rod anode, nickel plate (15 mm by 15 mm by 1 mm) cathode, constant current = 10 mA, 1 (0.5 mmol), 2 (3 equiv.), nBu4NClO4 (0.5 equiv.), Cs2CO3 (2 equiv.), 20°C, in DCE/Et2O (6/0.5 ml) under N2 atmosphere for 3.5 hours, and an undivided cell. bDCE/DCM (4/2 ml). cCarbon rod anode, Pt plate (15 mm by 15 mm by 0.3 mm) cathode, in DCE (6 ml) under N2 atmosphere for 3.5 hours, 12°C.
Subsequently, control experiments were conducted to know more detail about the mechanism of this electrochemical C─H bond reaction, as shown in Fig. 3. Radical clocks have been recognized as efficient tools for radical verification, providing an obvious advantage in experiment design. Cyclopropylbenzene was used as coupling partner, offering a ring-opened product (3la), revealing the existence of an aromatic carbon radical intermediate (Fig. 3A). In addition, 2,4-di-tert-butyl-4-methylphenol could react with 2a, delivering 3ka (Fig. 3B). This result suggests that the mechanism in which the alcohol is oxidized to O-centered radical cannot be excluded in this transformation.
Fig. 3. Control experiments.
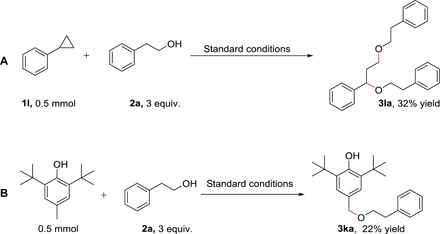
(A) Radical clock experiment. (B) Radical trapping experiment.
To get deeper insight into the mechanism, cyclic voltammetry was carried out to study the redox potential of 1a, 2a, and other alcohols, as shown in Fig. 4A. An oxidation peak of 1a in DCE was detected at 2.22 V. Similarly, oxidation peaks of 2a could also be observed about 2.38 V. On the other hand, other alkyl alcohols have much higher potential than phenethyl alcohol (2a). The results reveal that indan (1a) was more easily oxidized than most of the alkyl alcohols (Fig. 4A). In addition, the potential of 1a and 2a in the presence of Cs2CO3 was also observed, and the result demonstrates that base has no influence on the potential of reaction substrates (for detail, see fig. S2). Thereafter, EPR experiments were performed, as shown in Fig. 4B. Obviously, a distinct signal of a trapped radical by DMPO (5,5-dimethyl-1-pyrroline N-oxide) was detected when the reaction mixture of 1a was carried out under electrochemical conditions. The parameters observed for the spin adduct are g = 2.0068, AH = 21.3 G, and AN = 14.13 G. We proposed that this radical signal belongs to a carbon radical. Moreover, no signal was observed in the reaction mixture of 2a. However, we could not exclude the existence of O-based radical from 2a. This is because the O-based radical from 2a is likely highly reactive and will likely do HAT (hydrogen atom transfer) with the solvent before reacting with DMPO.
Fig. 4. Mechanistic studies.
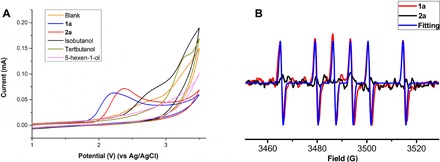
(A) Cyclic voltammograms of 0.01 M related compounds in 0.24 M nBu4NClO4 in DCE, using glass carbon working electrode, Pt wire as counter electrode, and Ag/AgCl as reference electrode at 50-mV/s scan rate. (B) EPR experiments.
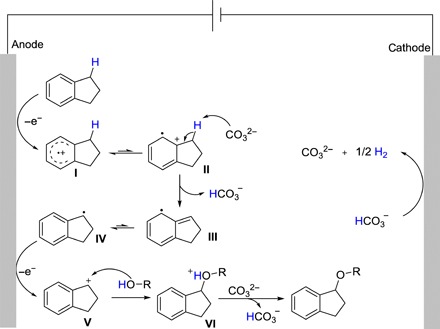
On the basis of the above results, we proposed a mechanism for this electrochemical C(sp3)─H etherification (Fig. 5). Indan is oxidized to aryl radical cation intermediate (I) via SET process. Second, I can resonate to form intermediate II, which generates III through the hydrogen abstraction by base. Subsequently, the resonance of III will form IV. Then, single-electron oxidation of IV can form the cation intermediate (V). Afterward, alcohols react with intermediate V to generate the VI, which will afford the desired product via the proton transfer process. Concomitant cathodic reduction generates hydrogen gas. In addition, an alternative mechanism in which the alcohol is oxidized to O-centered radical followed by HAT with the alkane cannot be excluded in this transformation.
Fig. 5. Proposed mechanism.
CONCLUSION
In conclusion, an electrochemical, oxidant-free strategy for construction of C─O bonds through O─H/C(sp3)─H has been disclosed. A series of ethers were successfully accessed by using nonsolvent amounts of alcohols as coupling partners, averting excessive waste of starting materials. Heteroatom-containing C(sp3)─H compounds and cyclic olefins were also well tolerated. Mechanistically, the generation of aryl radical cation intermediates is the key to this C(sp3)─H etherification, as evidenced by radical probe substrate (cyclopropane ring opening) and EPR experiments. This protocol provides not only a practical methodology for the synthesis of various ethers through C(sp3)─H/O─H cross-coupling but also allows achieving direct electrochemical C(sp3)─H functionalization in the absence of transition metal catalysis. Ongoing research including further mechanistic details and expanding the methodology to other heteroatom substrates is currently underway.
MATERIALS AND METHODS
Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. The instrument for electrolysis is dual display potentiostat (DJS-292B) (made in China). The anodic electrode was graphite rod (ϕ 6 mm), and cathodic electrode was a platinum plate (15 mm by 15 mm by 0.3 mm) or nickel plate electrodes (15 mm by 15 mm by 1 mm). Thin-layer chromatography used glass 0.25-mm silica gel plates. Flash chromatography columns were packed with 200- to 300-mesh silica gel in petroleum (boiling point of 60° to 90°C). Gas chromatographic analyses were performed on SHIMADZU GC-2014 gas chromatography instrument with an flame ionization detector, and biphenyl was added as internal standard. Gas chromatography–mass spectrometry spectra were recorded on Varian GC MS 3900-2100T or SHIMADZU GC MS-2010. 1H and 13C nuclear magnetic resonance data were recorded with Bruker Advance III (400 MHz) spectrometers with tetramethylsilane as an internal standard.
General procedure for electrochemical oxidative etherification of indan
A 6-ml three-necked round-bottomed flask was charged with the Cs2CO3 (1 mmol, 2 equiv.) and nBu4NClO4 (0.25 mmol, 0.5 equiv). The flask was equipped with a carbon rod anode (ϕ 6 mm) and a platinum plate (15 mm by 15 mm by 0.3 mm) or nickel plate electrodes (15 mm by 15 mm by 1 mm) and then flushed with nitrogen. DCE (3 ml), indan (0.5 mmol, 1 equiv), phenethyl alcohol (1.5 mmol, 3 equiv), Et2O (0.5 ml), and DCE (3 ml) were added in order. About 1.2 cm of the graphite rod was under the solution. The constant current (10 mA) electrolysis was carried out at 20°C for 3.5 hours. The reaction mixture was concentrated under reduced pressure. The residue was chromatographed through silica gel eluting with ethyl acetate/hexanes to give the product.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (21390402 and 21520102003) and the Hubei Province Natural Science Foundation of China (2017CFA010). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. Author contributions: K.L., H.W., W.X., S.S., and W.L. performed and analyzed experiments. A.L. and H.W. conceived the project and designed the experiments. A.L. and H.W. wrote the manuscript. All the authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz0590/DC1
REFERENCES AND NOTES
- 1.Enthaler S., Company A., Palladium-catalysed hydroxylation and alkoxylation. Chem. Soc. Rev. 40, 4912–4924 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Evano G., Blanchard N., Toumi M., Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis. Chem. Rev. 108, 3054–3131 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Ma D., Cai Q., Copper/amino acid catalyzed cross-couplings of aryl and vinyl halides with nucleophiles. Acc. Chem. Res. 41, 1450–1460 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Monnier F., Taillefer M., Catalytic C–C, C–N, and C–O Ullmann-type coupling reactions. Angew. Chem. Int. Ed. 48, 6954–6971 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Danishefsky S. J., Hungate R., Schulte G., Total synthesis of octosyl acid A. Intramolecular Williamson reaction via a cyclic stannylene derivative. J. Am. Chem. Soc. 110, 7434–7440 (1988). [Google Scholar]
- 6.Haase R. G., Schobert R., Synthesis of the bioherbicidal fungus metabolite macrocidin A. Org. Lett. 18, 6352–6355 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Williamson A. W., XXII.—On etherification. Q. J. Chem. Soc. 4, 229–239 (1852). [Google Scholar]
- 8.Kim J., Lee D.-H., Kalutharage N., Yi C. S., Selective catalytic synthesis of unsymmetrical ethers from the dehydrative etherification of two different alcohols. ACS Catal. 4, 3881–3885 (2014). [Google Scholar]
- 9.Shintou T., Mukaiyama T., Efficient methods for the preparation of alkyl-aryl and symmetrical or unsymmetrical dialkyl ethers between alcohols and phenols or two alcohols by oxidation-reduction condensation. J. Am. Chem. Soc. 126, 7359–7367 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Xu Q., Xie H., Chen P., Yu L., Chen J., Hu X., Organohalide-catalyzed dehydrative O-alkylation between alcohols: A facile etherification method for aliphatic ether synthesis. Green Chem. 17, 2774–2779 (2015). [Google Scholar]
- 11.Harper M. J., Emmett E. J., Bower J. F., Russell C. A., Oxidative 1,2-difunctionalization of ethylene via gold-catalyzed oxyarylation. J. Am. Chem. Soc. 139, 12386–12389 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Miller Y., Miao L., Hosseini A. S., Chemler S. R., Copper-catalyzed intramolecular alkene carboetherification: Synthesis of fused-ring and bridged-ring tetrahydrofurans. J. Am. Chem. Soc. 134, 12149–12156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sevov C. S., Hartwig J. F., Iridium-catalyzed, intermolecular hydroetherification of unactivated aliphatic alkenes with phenols. J. Am. Chem. Soc. 135, 9303–9306 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Shigehisa H., Aoki T., Yamaguchi S., Shimizu N., Hiroya K., Hydroalkoxylation of unactivated olefins with carbon radicals and carbocation species as key intermediates. J. Am. Chem. Soc. 135, 10306–10309 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Yang C. G., He C., Gold(I)-catalyzed intermolecular addition of phenols and carboxylic acids to olefins. J. Am. Chem. Soc. 127, 6966–6967 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Bunnett J. F., Zahler R. E., Aromatic nucleophilic substitution reactions. Chem. Rev. 49, 273–412 (1951). [Google Scholar]
- 17.Hartwig J. F., Carbon-heteroatom bond formation catalysed by organometallic complexes. Nature 455, 314–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ley S. V., Thomas A. W., Modern synthetic methods for copper-mediated C(aryl)–O, C(aryl)–N, and C(aryl)–S bond formation. Angew. Chem. Int. Ed Engl. 42, 5400–5449 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Liu J., Li M. M., Qu B. L., Lu L. Q., Xiao W. J., A photoinduced Wolff rearrangement/Pd-catalyzed [3+2] cycloaddition sequence: An unexpected route to tetrahydrofurans. Chem. Commun. 55, 2031–2034 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Hu H., Chen S.-J., Krska S., Stahl S., Copper-catalyzed benzylic C–H coupling with alcohols via radical relay. ChemRxiv. Preprint. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang J., Shang M., Kawamata Y., Lundberg H., Reisberg S. H., Chen M., Mykhailiuk P., Beutner G., Collins M. R., Davies A., Del Be M., Gallego G. M., Spangler J. E., Starr J., Yang S., Blackmond D. G., Baran P. S., Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 573, 398–402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng R., Smith J. A., Moeller K. D., Anodic cyclization reactions and the mechanistic strategies that enable optimization. Acc. Chem. Res. 50, 2346–2352 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Jutand A., Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300–2347 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Jiang Y., Xu K., Zeng C., Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 118, 4485–4540 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Sauermann N., Meyer T. H., Qiu Y., Ackermann L., Electrocatalytic C–H activation. ACS Catal. 8, 7086–7103 (2018). [Google Scholar]
- 26.Yoshida J., Kataoka K., Horcajada R., Nagaki A., Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Moeller K. D., Using physical organic chemistry to shape the course of electrochemical reactions. Chem. Rev. 118, 4817–4833 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Zhang S. K., Samanta R. C., Sauermann N., Ackermann L., Nickel-catalyzed electrooxidative C-H amination: Support for nickel(IV). Chemistry 24, 19166–19170 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Sauermann N., Mei R., Ackermann L., Electrochemical C-H amination by cobalt catalysis in a renewable solvent. Angew. Chem. Int. Ed. Engl. 57, 5090–5094 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Sauermann N., Meyer T. H., Tian C., Ackermann L., Electrochemical cobalt-catalyzed C-H oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452–18455 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Yang Q. L., Wang X. Y., Lu J. Y., Zhang L. P., Fang P., Mei T. S., Copper-catalyzed electrochemical C-H amination of arenes with secondary amines. J. Am. Chem. Soc. 140, 11487–11494 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Fu N., Sauer G. S., Saha A., Loo A., Lin S., Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Imada Y., Röckl J. L., Wiebe A., Gieshoff T., Schollmeyer D., Chiba K., Franke R., Waldvogel S. R., Metal- and reagent-free dehydrogenative formal benzyl-aryl cross-coupling by anodic activation in 1,1,1,3,3,3-hexafluoropropan-2-ol. Angew. Chem. Int. Ed. Engl. 57, 12136–12140 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Li J., He L., Liu X., Cheng X., Li G., Electrochemical hydrogenation with gaseous ammonia. Angew. Chem. Int. Ed. Engl. 58, 1759–1763 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Siu J. C., Sauer G. S., Saha A., Macey R. L., Fu N., Chauviré T., Lancaster K. M., Lin S., Electrochemical azidooxygenation of alkenes mediated by a TEMPO-N3 charge-transfer complex. J. Am. Chem. Soc. 140, 12511–12520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong P., Xu H. H., Xu H. C., Metal- and reagent-free intramolecular oxidative amination of tri- and tetrasubstituted alkenes. J. Am. Chem. Soc. 139, 2956–2959 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Siu J. C., Parry J. B., Lin S., Aminoxyl-catalyzed electrochemical diazidation of alkenes mediated by a metastable charge-transfer complex. J. Am. Chem. Soc. 141, 2825–2831 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayeda E. A., Miller L. L., Wolf J. F., Electrooxidation of benzylic ethers, esters, alcohols, and phenyl epoxides. J. Am. Chem. Soc. 94, 6812–6816 (1972). [Google Scholar]
- 39.Ghosh A. K., Cheng X., Enantioselective total synthesis of (−)-zampanolide, a potent microtubule-stabilizing agent. Org. Lett. 13, 4108–4111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz0590/DC1