

The RNA binding protein CPEB2 defines a new posttranscriptional layer in the hormone-driven gene-expression reprogramming.
Abstract
Organogenesis is directed by coordinated cell proliferation and differentiation programs. The hierarchical networks of transcription factors driving mammary gland development and function have been widely studied. However, the contribution of posttranscriptional gene expression reprogramming remains largely unexplored. The 3′ untranslated regions of messenger RNAs (mRNAs) contain combinatorial ensembles of cis-regulatory elements that define transcript-specific regulation of protein synthesis through their cognate RNA binding proteins. We analyze the contribution of the RNA binding cytoplasmic polyadenylation element–binding (CPEB) protein family, which collectively regulate mRNA translation for about 30% of the genome. We find that CPEB2 is required for the integration of hormonal signaling by controlling the protein expression from a subset of ER/PR- regulated transcripts. Furthermore, CPEB2 is critical for the development of ER-positive breast tumors. This work uncovers a previously unknown gene expression regulation level in breast morphogenesis and tumorigenesis, coordinating sequential transcriptional and posttranscriptional layers of gene expression regulation.
INTRODUCTION
The mammary gland develops postnatally and is subjected to marked remodeling in every oestrus cycle and during pregnancy. The mature mammary duct consists of an outer layer of basal myoepithelial cells and a polarized inner layer of luminal epithelial cells, which surround a hollow lumen and include hormone-sensing cells. During lactation, the lobuloalveolar units contain the luminal milk-producing alveolar cells (1, 2). This epithelial ductal tree is embedded within the mammary fat pad, which comprises fibroblasts, adipocytes, blood vessels, nerves, and immune cells (1). The development and remodeling of mammary ducts, through ductal branching and elongation, require epithelial cell proliferation to be coordinated with specification and maintenance of cell differentiation, as well as with tissue and cell polarity. These events are governed by ovarian steroid hormones, which control normal mammary development and lead to the neoplastic conversion of mammary tissue when misregulated. Estrogen is the most potent mitogenic stimulus for mammary ductal elongation during puberty, and it also directs the transcription of progesterone receptor (PR), which, in turn, induces ductal side branching and luminal lineage differentiation (3–5). Hormone-sensing cells, which are positive for estrogen receptor (ER) α and PR, account for only a small fraction (7 to 30%) of the luminal epithelium. These hormone receptor–positive (HR+) cells integrate hormonal cues to signal to adjacent HR-negative (HR−) cells via paracrine communication, which trigger the major proliferative response at the adult stage, mainly through the receptor activator of nuclear factor κB (NFκB) ligand (RANKL) (6–8).
Temporal and spatial control of mRNA translation, coupled to regulation of mRNA stability and localization, link cell proliferation, polarity, and differentiation (9–12). These gene regulation responses and the integration of external signals are coordinated through RNA binding proteins and cognate cis-acting elements to assemble specific ribonucleoprotein complexes. The cytoplasmic polyadenylation element (CPE)–binding (CPEB) family of RNA binding proteins regulates mRNA stability and translation through dynamic changes in their poly(A) tail length (13, 14). The four family members (CPEB1 to CPEB4) competitively recognize the same CPE in the 3′ untranslated region (3′UTR) of target mRNAs (15). CPEs interact with other cis-elements in a “CPE combinatorial code” to define spatiotemporal gene expression patterns (11, 16–19). In turn, individual pairs of CPE/CPEBs assemble into complexes that either repress or activate translation; repressor complexes shorten the poly(A) tail and mediate subcellular localization of repressed mRNAs, while activator complexes elongate the poly(A) tail (13). The switch from repression to activation is regulated by coordinated CPEB-specific posttranslational modifications of all four CPEBs (20). Although most CPEB functions have been studied during early development, CPEB1 in the mammary gland regulates the translation of milk protein transcripts, such as β-casein mRNA (21), and the localization—but not the translational activation—of ZO-1 (Zona Occludens Protein 1) mRNA to the apical surface of epithelial cells for tight junction assembly (22). Changes in poly(A) tail length regulate gene expression, integrating extracellular signals into cellular outcomes, including mitotic cell division and steroid hormone responses (17, 23, 24). Here, we show that the RNA binding protein CPEB2, which regulates the poly(A) tail length of CPE-containing mRNAs, contributes to mammary gland development and luminal breast carcinogenesis by regulating the translation of mRNAs downstream of steroid hormone signaling.
RESULTS
Loss of CPEB2 causes defective mammary gland development
To address how CPEBs could contribute to postnatal mammary gland development, we first determined the relative expression levels of all four CPEB mRNAs in pubertal, adult, pregnant, lactating, and involuted mouse mammary glands (Fig. 1A). Cpeb2 mRNA was the most abundant of the four Cpeb mRNAs in adult virgin mice, and it also peaked at lactation. After cell sorting of mammary epithelial cells (MECs) (fig. S1A), we found that Cpeb2 mRNA was expressed mainly in luminal cells, whereas Cpeb1 was predominant in myoepithelial cells (Fig. 1B). A similar distribution was observed at the protein level (fig. S1B). We next determined the consequences in mammary gland morphogenesis of total loss-of-function mouse models for CPEBs in postpubertal adult nulliparous mice. To this end, we determined the elongation and branching of the epithelial ductal tree in mammary gland whole mounts. We used previously described knockout (KO) mice for CPEB1 and CPEB4 (19, 25) and generated KO mice for CPEB2 and CPEB3 (figs. S2 and S3). CPEB2 and CPEB3 KO mice were viable and fertile and did not show any overt phenotype. While ductal morphogenesis was not affected in CPEB3 KO or CPEB4 KO mice, CPEB1 KO and CPEB2 KO animals displayed reduced branching through the fat pad (Fig. 1C and fig. S4A). Branching was quantified using AngioTool software (fig. S4B). Because of a defect in oogenesis, ovaries from CPEB1 KO females are rudimentary and do not secrete normal levels of reproductive hormones (26). This deficiency, which can be partially rescued by injection of 17β-estradiol (22), limits mammary duct proliferation. Accordingly, we observed reduced ductal expansion through the fat pad only in adult CPEB1-deficient mammary glands (fig. S4). To better define cell-autonomous defects in mammary duct development, we generated CK14-specific KO mice for CPEB1 and CPEB2 (KOCK14), where the CK14 promoter is expressed by all MECs during embryonic development (27). When the KO was restricted to the CK14 lineage, loss of CPEB2 (but not of CPEB1) resulted in reduced number of junctions (Fig. 1D). At earlier developmental times, we also observed a delayed ductal expansion in CPEB2 KO mice, as shown by diminished pubertal invasion of the epithelial tree through the fat pad that was recovered in adulthood (Fig. 1E and fig. S4B). CPEB2 KO mice also showed an increased luminal/myoepithelial cell ratio (Fig. 1F and fig. S4D). Thus, deletion of CPEB2 results in delayed ductal extension and reduced branching, two events sequentially regulated by ER and PR.
Fig. 1. CPEB2 regulates mammary gland postnatal development.
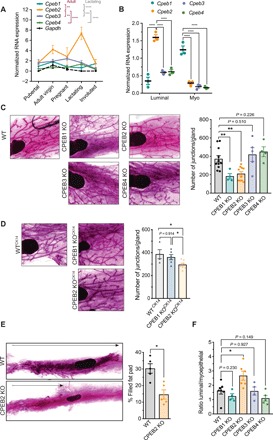
(A) mRNA levels of Cpeb1 to Cpeb4 normalized to Gapdh in whole tissue mammary gland (n = 2; n = 7 for adult nulliparous). Tissue was obtained from mice at puberty (5 weeks old), adult nulliparous (10 weeks old), midpregnancy (day 12 of gestation), lactation (2 weeks of lactation), or involution (6 days after weaning). Gapdh expression is also shown. Statistics were determined using two-way analysis of variance (ANOVA), **P < 0.01, ***P < 0.001, and ****P < 0.0001. (B) mRNA levels of Cpeb1 to Cpeb4 normalized to Gapdh in sorted cells from adult virgin mammary gland (n = 3). Statistics using two-way ANOVA, ****P < 0.0001. Myo, myoepithelial. (C) Representative carmine-stained mammary gland whole mounts and automatic quantification of the number junctions in virgin 10- to 12-week-old WT (n = 11) and constitutive CPEB1 KO (n = 4), CPEB2 KO (n = 10), CPEB3 KO (n = 5), and CPEB4 KO (n = 4) mice. Statistics were determined using the Mann-Whitney test, *P < 0.05 and **P < 0.01. (D) Representative mammary whole mounts and automatic quantification of the number of junctions in virgin 10- to 12-week-old epithelial-specific WTCK14 (n = 4), CPEB1 KOCK14 (n = 6), and CPEB2 KO CK14 (n = 8) mice. Statistics were determined using the Mann-Whitney test, *P < 0.05. (E) Representative mammary whole mounts and quantification of the area of the fat pad filled with epithelial ducts at puberty in WT and CPEB2 KO females (5 weeks old) (n = 5). Statistics were determined using the Mann-Whitney test, *P < 0.05. (F) Ratio between the percentage of luminal and myoepithelial cells gated on lineage-negative (WT, n = 7; CPEB1 KO, n = 4; CPEB2 KO, n = 6; CPEB3 KO, n = 4; and CPEB4 KO, n = 4). Statistics were determined using the Mann-Whitney test, *P < 0.05.
CPEB2 is required for proper differentiation of ductal progenitor cells
To further determine the cell-of-origin of the mammary CPEB2 KO phenotype and given that CPEB2 was mostly expressed in the luminal compartment of the mammary gland (Fig. 1B and fig. S1B), we sorted luminal cell types from adult virgin mammary glands (28). We distinguished the following three cell types [as defined in (28, 29)]: ductal progenitor (DP; Sca1+CD49b+), ductal differentiated (DD; Sca1+CD49b−), and alveolar progenitor (AP; Sca1−CD49b−) (Fig. 2A). We observed a general increase in Sca1 levels in CPEB2 KO mammary glands and increased cell number in the gate for the Sca1+CD49b+ population, concomitant with a reduction in the Sca1+CD49b− window (Fig. 2, A to E). The AP population, on the other hand, did not change significantly upon CPEB2 depletion. To further characterize the effect of CPEB2 loss-of-function in MECs, we studied the transcriptomes of all four wild-type (WT) and CPEB2 KO epithelial populations using DNA microarrays. First, we confirmed our gating strategy through the expression of well-known markers in the expected populations (fig. S5A). Principal components analysis of gene expression profiles further confirmed clustering by populations and showed that the main differences between WT and CPEB2 KO cells affected the Sca1+CD49b+ population, with DPKO placed between DPWT and DDWT (fig. S5B). This was calculated by comparing the distance between centroids of different genotypes on a given population versus the dispersion within the population (see Methods and fig. S5B). Next, on the basis of the genes differentially expressed in the DPWT versus DDWT populations, we generated a WT progenitor signature by selecting candidate genes with the highest and lowest fold change (FC) percentiles and P < 0.01 (1% most up- and down-regulated genes, n = 181 and n = 101, respectively). We found a clear negative enrichment for the WT progenitor signature in DPKO cells, with the genes up-regulated being negatively enriched and vice versa (Fig. 2F nd fig. S5C). Similarly, further filtering using a false discovery rate (FDR) of 0.1 as a threshold (instead of P value) resulted in a more stringent signature with 24 up-regulated and no down-regulated genes (WT DP versus WT DD) that was also negatively enriched in DPKO cells (fig. S5D) (see Methods). These observations suggest that the DPKO cells contained a partially differentiated population. mRNA expression of the luminal progenitor markers Elf5, Kit, Cd14, and Rspo1 (29) was reduced in DPKO cells as compared with the DPWT population (Fig. 2G). Conversely, these luminal progenitor markers were unaffected in APKO cells, with the exception of Rspo1 (see Discussion) (Fig. 2H). Accordingly, DPKO cells showed a reduced capacity to form organoids as compared to DPWT cells (Fig. 2I). Together, these results indicated that CPEB2 might be required for the proper differentiation of DP cells.
Fig. 2. CPEB2 KO females display aberrant Sca1+ ductal luminal progenitors.
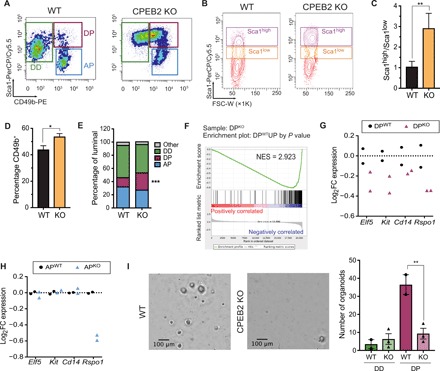
(A) Representative fluorescence-activated cell sorting (FACS) plots gated on luminal cells depicting luminal subpopulations: ductal differentiated (DD; Sca1+CD49b−), ductal progenitors (DPs; Sca1+CD49b+), and alveolar progenitors (APs; Sca1−CD49b+). (B) Representative FACS plots for Sca1 gated on luminal cells. FSC-W, forward scatter width. (C) Ratio of the percentage of Sca1high and Sca1low populations in luminal cells (n = 10). Statistics were determined using the Mann-Whitney test, **P < 0.01. (D) Quantification of the percentage of CD49b+ cells gated on luminal cells (n = 17). Statistics were determined using the Mann-Whitney test, *P < 0.05. (E) Quantification of the luminal subpopulations as in (A). Statistics were determined using two-way ANOVA, ***P < 0.001 (n = 17). (F) Preranked GSEA graphical output for the enrichment in DPKO versus DPWT cells of the up-regulated genes in the “WT progenitor signature” generated by P value (n = 181, see Methods). FDR q ≤ 0.0001. FDR, false discovery rate; NES, normalized enrichment score. (G) Expression of luminal progenitor markers in DPWT and DPKO cells. (H) Expression of luminal progenitor markers in APWT and APKO cells. (I) Representative images of organoids from sorted DPWT and DPKO cells and automatic quantification of the number of organoids from sorted DD or DP cells. Scale bars, 100 μm. Statistics were determined using two-way ANOVA, **P < 0.01.
Proliferation of MECs is decreased in absence of CPEB2
Gene set enrichment analysis (GSEA) showed a clear down-regulation in the gene sets related to cell cycle and proliferation (G2M checkpoint and E2F targets) in all four CPEB2 KO epithelial cell populations (fig. S6A). DD cells are highly proliferative (30). Therefore, we next analyzed MEC proliferation in the CPEB2 KO by Ki67 immunostaining (Fig. 3A) and by 5-ethynyl-2′-deoxyuridine (EdU) incorporation (Fig. 3B). CPEB2 KO mice displayed reduced MEC proliferation. Note that apoptosis was negligible in adult mammary glands, both in WT and CPEB2 KO animals (fig. S6B).
Fig. 3. CPEB2 regulates proliferation in the mammary gland.
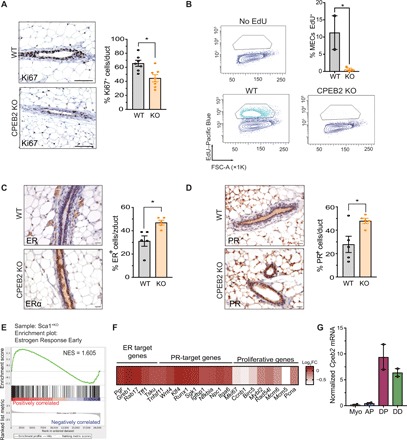
(A) Representative images and automatic quantification of Ki67+ cells by immunohistochemistry in adult virgin mammary gland in WT and CPEB2 KO (n = 7) mice. Statistics were determined using the Mann-Whitney test, *P < 0.05. Scale bars, 50 μm. (B) Representative FACS plots (gated on MECs) and quantification of percentage of EdU incorporation. FSC-A, FSC area. Statistics were determined using two-tailed unpaired Student’s t test, *P < 0.05. MECs, mammary epithelial cells. (C) Representative images and automatic quantification of ER+ cells by immunohistochemistry in adult virgin mammary gland in WT and CPEB2 KO (n = 5). Statistics were determined using the Mann-Whitney test, *P < 0.05. Scale bars, 25 μm. (D) Representative images and automatic quantification of PR+ cells by immunohistochemistry in adult virgin mammary gland in WT and CPEB2 KO (n = 5). Statistics were determined using the Mann-Whitney test, *P < 0.05. Scale bars, 25 μm. (E) Preranked GSEA graphical output for the enrichment in Sca1+KO cells (DPKO + DDKO) of the gene set estrogen response early from the Molecular Signatures Database Hallmarks collection (see Methods). FDR q = 0.0139. (F) Heat map representing the log2FC expression of hormone-driven genes in DPKO compared to DPWT. (G) Cpeb2 expression levels normalized by Gapdh in epithelial subpopulations.
Proliferation in the mammary gland is driven by the action of steroid hormones not only for HR+ but also for HR− cells (including mammary stem cells) through dominant paracrine effects (4, 31). Thus, we first assessed the levels of ER and PR in constitutive and CK14-driven CPEB2 KO mice. Unexpectedly, ER and PR were up-regulated in the absence of CPEB2, both at mRNA and protein levels (Fig. 3, C and D and fig. S6, C to E). Moreover, the hallmark estrogen response early was significantly increased in KO Sca1+ cells (Fig. 3E), suggesting that the ER transcriptional function was not impaired. Direct ER and PR target genes tended to be up-regulated in the absence of CPEB2 at the transcript levels, while downstream proliferative genes were down-regulated (Fig. 3F). These observations suggest that, although hormone-receptor transcriptional activity is normal, or even increased, the downstream effectors of hormone-driven cell proliferation are defective.
We found that, in the absence of CPEB2, there is a delay in ductal elongation at puberty, as well as reduced ductal branching in adulthood, accompanied by decreased epithelial proliferation and impaired differentiation of HR+ cells. All these phenotypes observed in vivo are concordant with blunted HR signaling (4). Given that CPEB2 was expressed mainly in HR+ cells (Fig. 3G and fig. S7A), we hypothesized that CPEB2 may constitute a previously unidentified posttranscriptional layer of regulation in the ER and PR pathways.
CPEB2 posttranscriptionally regulates the expression of hormonal signaling effectors
To identify the CPEB2-target mRNAs that could explain the defective response to hormones in MECs, we performed CPEB2 RNA immunoprecipitation (RIP; Fig. 4A). CPEB2 coimmunoprecipitated 169 mRNAs in MECs, which were significantly enriched in the RIP WT compared with the RIP in CPEB2 KO control cells (see Methods, table S1, and fig. S7B). These CPEB2 targets were enriched in canonical CPEs (UUUUA1–2U), thereby verifying the specificity of the immunoprecipitation (Fig. 4B). Pathway analysis showed that CPEB2-target mRNAs were enriched in breast cancer–related genes (Fzd2, Jag1, Cdk6, Ccnd1, Sp1, Wnt5a, Kit, Kras, and Lrp6) (Fig. 4C). RIP targets were also overrepresented in the phosphoinositide 3-kinase (PI3K)–Akt signaling pathway (Fig. 4C), which has been shown to modulate both genomic and nongenomic activities of the ER and is associated with breast cancer and with endocrine resistance of luminal tumors when mutated (32). The transcription factor 3′,5′-cyclic adenosine monophosphate OR cyclic adenosine monophosphat responsive element binding protein 1 (CREB1), which is activated downstream PI3K-Akt and regulates estrogen signaling (33, 34), was one of the top three enriched transcripts in the RIP WT (table S1 and fig. S7B). Moreover, individual targets included not only Cpeb2 and Cpeb3 mRNAs (suggesting auto- and cross-feedback CPEB loops) but also regulators of cell fate, morphogenesis, and organogenesis in the Wnt and Notch pathways (1, 35), such as the Wnt surface receptors Fzd2 and Lrp6, and the Notch surface ligand Jag1 (table S1 and fig. S7B). Furthermore, although not statistically significant due to low mRNA expression levels, Rankl (Tnfsf11) was enriched in the CPEB2 RIP, and we also found CyclinD1 (Ccnd1) to be a CPEB2 target (fig. S7B). Rankl and Ccnd1 are the key effectors of the autocrine and paracrine proliferative responses to progesterone, respectively. We validated several of these genes as bona fide CPEB2 target mRNAs by RIP–quantitative polymerase chain reaction (qPCR) (Fig. 4D). Given their direct implications on the regulation of hormone-driven proliferation and differentiation in MECs, we further analyzed the regulation of Creb1, Ccnd1, and Rankl. These CPEB2 target mRNAs contained conserved canonical CPEs in their 3′UTRs at optimal distances (17) from the polyadenylation sites (fig. S8A). We found that their protein levels were reduced in the absence of CPEB2, without significant variations in their mRNA levels, thereby suggesting translational changes (Fig. 4, E to H and fig. S8B). This CPEB2-mediated regulation of RANKL appeared to be specific for MECs, given that it was not observed in the immune cells of the mammary lymph node (fig. S8C).
Fig. 4. CPEB2 regulates the synthesis of key effectors in the hormonal signaling pathways.
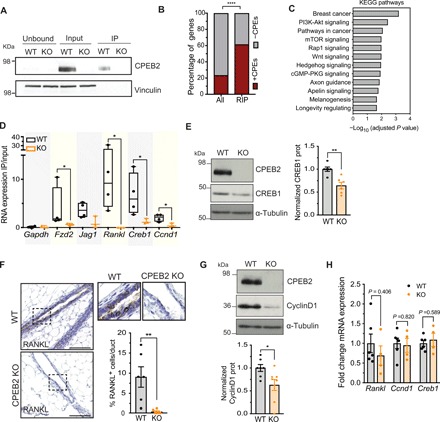
(A) Western blot image for CPEB2 and vinculin (as a control) from unbound, input, and immunoprecipitated fractions with anti-CPEB2 antibody in WT and CPEB2 KO MECs. (B) Percentage of genes with (+CPEs, red) or without (–CPEs, gray) CPEs in the 3′UTR, comparing RIP targets to the mouse transcriptome (all). Statistics were determined using Fisher’s exact test, ****P < 0.0001. (C) Significantly enriched KEGG pathways (adjusted P < 0.05) in the analyzed RIP targets. cGMP-PKG, cyclic guanosine monophosphate (cGMP)–cGMP-dependent Protein Kinase G (PKG). mTOR, mammalian target of rapamycin. (D) RIP-qPCR results showing the RIP values normalized by each input in WT (n = 4) and KO (n = 3) MECs. Gapdh mRNA and RIP in CPEB2 KO MECs are used as negative controls for enrichment in RIP as compared to input. Statistics were determined using the Mann-Whitney test, *P < 0.05. IP, immunoprecipitation. (E) Western blot image for CPEB2, CREB1, and α-tubulin (loading control) and normalized quantification of CREB1 protein levels in WT and KO MECs (n = 6). Statistics were determined using the Mann-Whitney test, **P < 0.01. (F) Representative images and manual quantification of RANKL+ cells by immunohistochemistry in adult virgin mammary gland in WT and CPEB2 KO animals (n = 6). Scale bar, 50 μm. Statistics were determined using the Mann-Whitney test, **P < 0.01. (G) Western blot image for CPEB2, CyclinD1, and α-tubulin (loading control) and normalized quantification of CyclinD1 protein levels in WT and KO MECs (n = 6). Statistics were determined using the Mann-Whitney test, *P < 0.05. (H) mRNA levels of Rankl, Ccnd1, and Creb1 normalized to Gapdh and to WT in MECs (WT, n = 6; KO, n = 4). Statistics were determined using the Mann-Whitney test.
CPEB2 is critical for luminal breast tumorigenesis
As CPEB2 KO mice displayed defective signaling to estrogen and progesterone, both key in breast cancer development (29, 36, 37), and CPEB2-bound mRNAS were components of breast cancer pathways, we next explored whether CPEB2 participates in breast tumorigenesis. Analysis of the expression of CPEB2 mRNA in patient breast tumor samples using the METABRIC cohort determined an association between CPEB2 and ESR1 levels (Fig. 5A). In agreement with the function of CPEB2 in mammary homeostasis, gene expression profiles that classify breast cancer into various subtypes (38) indicate that ER+ primary breast cancer has a characteristic “luminal” transcriptional profile. Using both the METABRIC and The Cancer Genome Atlas RNA sequencing (RNA-seq) dataset, we confirmed that CPEB2 levels were decreased in basal-like and Her2 tumors compared to luminal tumors and to morphologically normal surrounding tissue (Fig. 5B and fig. S9A). This observation was extended to human breast cancer cell lines, with several ER+ (luminal-like) cell lines expressing higher levels of CPEB2 mRNA (Fig 5C).
Fig. 5. Absence of CPEB2 protects against luminal breast cancer.
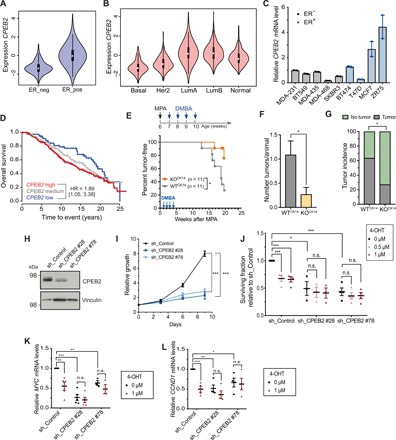
(A) Violin plots for CPEB2 RNA expression depending on ER status; METABRIC cohort (n = 1974). Statistics were determined using the Wald test, P < 10 × 2.22−16. (B) Violin plots for CPEB2 RNA expression in the PAM50 subtypes; METABRIC cohort (n = 1974). Statistics were determined using the Wald test compared to the luminal A subtype: basal-like, P < 10 × 2.22−16; HER2, P < 10 × 2.22−16; and luminal B, P = 0.99003. (C) Quantification of CPEB2 expression levels by RT-qPCR in the indicated breast cancer cell lines. B2M was used as endogenous control. (D) Kaplan-Meier survival curves for patients with luminal A breast cancer [HR (<10 years) = 1.89; P = 0.021; multivariate using tumor size and lymph node as other risk factors n = 550]. (E) Schematic representation of the chemical-induced breast cancer model and kinetics of mammary tumor onset in mice treated with medroxyprogesterone acetate (MPA) and 7,12-dimethylbenz(a)anthracene (DMBA) as indicated. Statistics were determined using the log-rank test, *P < 0.05. (F) Number of macroscopic tumors per animal at time of sacrifice (16 weeks after MPA administration) in WTCK14 (n = 11) and CPEB2 KOCK14 (n = 11) animals. Statistics were determined using the Mann-Whitney test, *P < 0.05. (G) Tumor incidence in WTCK14 (n = 11) and CPEB2 KOCK14 (n = 11) mice. Statistics were determined using chi-square test, *P < 0.05. (H) Western blot image for CPEB2 and vinculin (loading control) in ZR75 cells after KD of CPEB2 using sh_CPEB2 #28 or #78 or in control cells (sh_Control). (I) Relative growth curve of ZR75 cells sh_Control or KD of CPEB2. Cell numbers were quantified relative to day 0 at the indicated time points. Statistics were determined using a two-tailed unpaired Student’s t test, ***P < 0.001. (J) Surviving fraction of CPEB2 KD ZR75 cells (using sh_CPEB2 #28 and #78) or control ZR75 cells treated with vehicle (0 μM), 0.5 μM 4-OHT, or 1 μM 4-OHT. Number of viable cells was quantified 6 days after 4-OHT treatment. Surviving fraction refers to the fraction of cells present after 4-OHT treatment. Statistics were determined using a two-tailed unpaired Student’s t test, *P < 0.05 and ***P < 0.001. n.s., not significant. (K) RT-qPCR quantification of MYC expression levels in CPEB2 KD ZR75 cells (sh_CPEB2 #28 or #78) or control ZR75 cells (sh_Control) treated with vehicle (0 μM) or 1 μM 4-OHT for 48 hours. B2M was used as an endogenous control. Statistics were determined using a two-tailed unpaired Student’s t test, **P < 0.01 and ***P < 0.001. (L) Quantification of CCND1 expression levels by RT-qPCR in CPEB2 KD ZR75 cells (sh_CPEB2 #28 and #78) or control cells (sh_Control) treated with vehicle (0 μM) or 1 μM 4-OHT for 48 hours. B2M was used as an endogenous control. Statistics were determined using a two-tailed unpaired Student’s t test, *P < 0.05, **P < 0.01, and ***P < 0.001.
Next, we explored the association between CPEB2 expression and patient survival at 10 years using the METABRIC public breast cancer primary tumor cohort, for which prognosis annotation was available with sufficient follow-up. We confirmed an interaction between CPEB2 expression and samples classified on the basis of PAM50 molecular subtype (P = 0.0007, continuous model) (39), implying significant differences in prognosis association across biologically diverse tumor subtypes. In luminal A tumors, dependent on ER signaling for growth, high levels of CPEB2 were associated with worse survival compared to samples with the lowest expression [HR (<10 years) = 1.83, P = 0.028, n = 550; Fig. 5D]. No association between CPEB2 expression and tumor size was observed (fig. S9B). Collectively, these findings reveal an association between low CPEB2 expression and survival in patients with luminal ER+ breast cancer.
To experimentally address a potential role of CPEB2 in luminal tumorigenesis, we induced mammary tumor development in WTCK14 and CPEB2 KOCK14 mice, combining the proliferative action of the synthetic progestin medroxyprogesterone acetate (MPA) and the mutagenic agent 7,12-dimethylbenz(a)anthracene (DMBA) (40). Tumor onset was significantly delayed in CPEB2 KOCK14 mice (Fig. 5E), as shown by the higher percentage of tumor-free animals at 20 weeks after MPA treatment, the humane end point determined by the size of WT tumors. Tumor incidence was 63% for WTCK14 animals versus 27% for CPEB2CK14 mice. Moreover, at the end of the experiment, the number of tumors per animal (Fig. 5F) was reduced CPEB2CK14 animals. As previously described (41), these treatments generated hyperplasias, neoplasias, adenomas, adenocarcinomas, and adenosquamous carcinomas. Histopathological analysis of the tumors generated in the CPEB2 KOCK14 and WTCK14 animals revealed no major differences (fig. S9C). Furthermore, we detected lower ER levels in CPEB2 KOCK14 tumors as compared to the WTCK14 ones (fig. S9D), despite the fact that this treatment generates tumors characteristic of the luminal breast cancer subtype with high ER expression (42) (note that determination of significance was limited due to low number of tumors in the CPEB2 KOCK14 mice).
To further explore any functional interactions between ER and CPEB2, we knocked down CPEB2 in ZR75 ER+ luminal human breast cancer cells using two independent short hairpin RNAs (shRNAs; Fig. 5H and fig. S10A). These depletions significantly decreased cell proliferation in vitro but did not increase apoptosis (Fig. 5I and fig. S10B). Next, we treated WT and CPEB2 knockdown (KD) cells with the ER inhibitor 4-hydroxytamoxifen (4-OHT) (Fig. 5J). In contrast to WT ZR75 cells, CPEB2 KD ZR75 cells were insensitive to 4-OHT, thereby indicating that CPEB2 depletion and ER signaling inhibition do not have an additive effect on cell growth and suggesting that CPEB2 and ER act on the same pathway. Consistently, the effects of CPEB2 depletion on MYC and CCND1 expression (genes regulated by ER signaling and mediators of proliferation) were comparable, but not additive, to inhibition of ER signaling by 4-OHT (Fig. 5, K and L). Furthermore, we could also validate the regulation of RANKL by CPEB2 in this breast cancer setting (fig. S10, C and D).
Our results indicate that CPEB2 and ESR1 expression in breast cancer are linked and that high CPEB2 levels are associated with poor prognosis in luminal A tumors. Results of MPA/DMBA tumor generation indicated that high CPEB2 expression promotes luminal tumor development, consistent with the hormone dependence of this breast tumor subtype. On the other hand, ER− tumors (such as basal like) do not seem to require CPEB2; low levels of CPEB2 result in reduced survival (fig. S9E).
DISCUSSION
In this work, we unveil a previously unknown layer of posttranscriptional regulation of gene expression orchestrated by CPEB2 in the mammary epithelia hormone responses. Thus, key HR-driven mediators (both cell autonomous and paracrine) of the differentiation and proliferation pathways (such as RANKL, CyclinD1, or CREB1) are encoded by CPEB2-regulated mRNAs. In the absence of CPEB2, the transcriptional activation of these genes fails to be reflected into increased protein levels. Mammary ductal branching and elongation are coordinated by the ovarian steroid hormones estrogen and progesterone, which activate transcriptional programs resulting in epithelial cell differentiation and proliferation. These hormones are sensed by a minority of HR+ cells, which, in turn, signal to adjacent HR− cells through paracrine signals that coordinate mammary gland development and remodeling. Although CPEB2 can modulate the expression of more than a hundred genes (table S1) rather than switching on a single gene, the depletion of this RNA binding protein shows phenotypic similarities with the depletion of well-characterized HR-activated genes. CyclinD1 and CREB1 determine the proliferative programs of the estrogen signaling in the mammary gland (34, 43). In turn, RANKL is a key paracrine mediator of progesterone-mediated ductal side branching and MEC proliferation (mediated by NFκB and CyclinD1) and differentiation (6, 7, 44, 45). All of these pathways are defective in the absence of CPEB2. In addition to being a CPEB2 target in luminal cells, Ccnd1 is also down-regulated in myoepithelial cells, probably as the result of a paracrine transcriptional effect (fig. S11A). Expression of Rspo1, which was down-regulated in both DPKO and APKO (Fig. 2, G and H), is a RANKL-induced gene (6). Thus, the mammary epithelia defects observed in CPEB2 KO mice could be partly explained by impaired translational activation of Rankl mRNA. However, note that the phenotype of CPEB2 KO mouse model does not phenocopy that of the RANKL KO. RANKL drives mammary alveologenesis (46), which is not defective in CPEB2 KO mice (fig. S11, B and C). Normal alveologenesis in CPEB2 KO mice could be due to a compensatory increase in Cpeb4 mRNA levels, which we observed specifically at the lactating stage but not in adult virgin mammary glands (fig. S11D). Redundancy between CPEB2 and CPEB4 has been reported in other scenarios (47).
In this study, we have focused on the role of CPEB2 in luminal breast cancer as a mediator of ER signaling. Accordingly, CPEB2 is one of the top six genes, together with ESR1, with strongest correlation with ER+ breast cancer prognosis (48). It has been proposed that breast cancer subtypes arise from distinct epithelial differentiation stages and lineages (29). Although the cell-of-origin for luminal tumors has not yet been unambiguously identified, these tumors appear to arise from a population of DPs that not only has clonogenic capacity but also expresses high levels of markers of mature luminal cells, such as ER, PR, GATA3 (GATA binding protein 3), and FOXA1 (Forkhead Box Protein A1) (28, 29, 49, 50). Depletion of CPEB2 generated a differentiation intermediate population with high Sca1/ER levels but low clonogenic capacity and impaired hormonal signaling. Together, our findings reveal a previously unkown posttranscriptional mechanism that regulates mammary gland morphodynamics and influences the outcome of ER+ mammary tumors, which account for 75% of breast cancer cases.
METHODS
Generation of CPEB2 and CPEB3 KO mouse models
To generate a CPEB2 KO mouse model, the vector (EUCOMM, PRPGS00036-W-3-B04) was electroporated in mouse G4 embryonic stem cells (mixed C57BL/6J and 129/Sv). Positive recombinant embryonic stem cells were identified by Southern blotting, transfected in vitro with the FlpO recombinase to remove the geo-cassette, and microinjected into developing blastocysts. Resulting chimeric mice (Cpeb2 lox/lox) were crossed with C57BL6/J mice, and the mouse colony was maintained in a mixed background (70% C57BL/6J and 30% 129/Sv). To generate CPEB3 KO, mouse ES cells carrying a gene-trap lacZ cassette and a promotor-driven neomycin resistance gene in Cpeb3 intron 3 (clones HEPD0670_2_C02 and HEPD0670_2_G03, EUCOMM) were microinjected into developing blastocysts. Resulting chimeric mice were crossed with 129/Sv × C57Bl/6J animals. To obtain a ubiquitous and constitutive depletion, Cpeb2lox/lox mice were crossed with mice expressing DNA recombinase Cre under control of the Sox2 promoter. Excision of exon 4 of Cpeb2 led to a frameshift in the mRNA, generating premature stop codons and resulting in animals that were KO for the CPEB2 protein. For the CPEB3 KO, the Neo cassette and exon 3 were further deleted by crossing Cpeb3loxfrt with transgenic mice expressing Cre under the control of the Sox2 promoter. The mouse colony was maintained in a mixed background (129/Sv × C57Bl/6). Epithelial-specific CPEB1 and CPEB2 KO mice were obtained by crossing Cpeb1lox/lox or Cpeb2lox/lox animals with C57BL/6J transgenic mice expressing Cre under control of the Krt14 promoter. Routine genotyping was performed by PCR; primer sequences are listed in table S2.
Southern blotting
Agarose gels were incubated under soft agitation with depurination solution (0.25 M HCl, 15 min), denaturation solution (1.5 M NaCl and 0.5 M NaOH, 45 min), and neutralization solution (0.5 M tris and 1.5 M NaCl, 30 min). After overnight transfer, DNA was cross-linked (254 nm, 0.12 J) to a nylon membrane (0.45 mm; Pall Corporation). The membrane was prehybridized with Church buffer for 3 hours at 65°C, hybridized with 32P-labeled probes for 12 hours, rinsed with washing buffer (standard saline citrate, 0.1% SDS), and exposed to a phosphorimager screen.
Animal studies
Mice (Mus musculus, C57BL/6J-129/Sv mixed background) were maintained under a standard 12-hour light/12-hour dark cycle at 23°C, with free access to food and water. Female littermates between 10 and 12 weeks of age were used, unless otherwise stated. Mice were staged by histological analysis of ovaries or vaginal cytology and were selected for the follicular phase of the oestrous cycle (51, 52). For tumorigenesis experiments, CK14-Cre–expressing mice were subcutaneously injected with MPA (Depo-Provera) at 7 weeks of age. They were then given DMBA (1 mg) by gavage weekly during the following 4 weeks (53, 54). Tumors were detected and monitored by manual palpation. Mice were sacrificed when a palpable mass exceeded 1 cm in diameter or at 20 weeks after MPA treatment (time for many WT animals to develop tumors reaching this humane end point). End-point tumors were classified on the basis of previously identified pathological nomenclature (55).
MEC isolation and flow cytometry
Thoracic and inguinal mammary glands were dissected, and MECs were prepared as previously described (56). In brief, mammary glands were incubated with a collagenase/hyaluronidase solution (STEMCELL Technologies), red blood cells were lysed, and cells were further dissociated with trypsin (Sigma-Aldrich), dispase II (Sigma-Aldrich), and deoxyribonuclease I (Sigma-Aldrich). In general, fluorescence-activated cell sorting (FACS) analysis and sorting were performed in a FACS Aria Fusion sorter (BD Biosciences), and data were analyzed with the BD FACSDiva software. For four-color FACS analysis, a Gallios flow cytometer (Beckman Coulter) was used, and data were analyzed with the FlowJo software. The following antibodies were used: EpCAM–phycoerythrin (PE) (130-102-265), CD49f–allophycocyamin (APC) (130-100-147), CD45–fluorescein isothiocyanate (FITC) (130-102-778), Ter119-FITC (130-102-257), CD31-FITC (130-102-970), CD49b-PE (130-102-778), EpCAM-APC/Cy7 (BioLegend, 118217), and Ly-6A/E (Sca1) PerCP/Cy5.5 (BioLegend, 108123). Antibodies were purchased from Miltenyi Biotec unless otherwise stated. Gating strategies were adjusted as previously described (28). For EdU incorporation experiments, mice received an intraperitoneal injection of EdU (80 mg kg−1) and were sacrificed 6 hours later, as previously described (57). After isolation of MECs, samples were processed as indicated in the protocol for Click-iT Plus EdU Flow Cytometry Assay (Invitrogen) using Pacific Blue picolyl azide.
Organoid culture
A total of 2000 sorted cells were embedded in one drop of basement membrane extracts (Cultrex) and cultured for 15 days in uncoated 24-well glass plates (no. 242-20, zell-kontakt). The culture protocol was adapted from (58); advanced Dulbecco’s modified Eagle medium (DMEM)/F12 medium was supplemented with penicillin/streptomycin, GlutaMAX, Hepes (Gibco), hydrocortisone (Lonza Bioscience), B27 (Thermo Fisher Scientific), insulin, N-acetylcysteine, epidermal growth factor, fibroblast growth factor 2 (FGF2; Sigma-Aldrich), FGF10 (PeproTech), heparin (STEMCELL Technologies), Y-27632 (ROCK inhibitor, Tocris), Wnt3a, and R-spondin1 (in-house). ROCK inhibitor was added for the first week, and the medium was refreshed every 3 to 5 days. Full drops were scanned with an Olympus IX81 inverted microscope at ×10 magnification (ScanR software). Bright-field Z stacks of each field were projected in a single image, and the full drop was then digitally reconstructed by stitching the different image projections using an ImageJ custom-made macro-developed for this purpose at the Institute of Research in Biomedicine (IRB) Advanced Digital Microscopy Facility.
Immunohistochemistry and whole mounts
For mammary gland whole mounts, inguinal mammary glands were placed on a slide and fixed immediately with Carnoy’s solution overnight. Tissue was then hydrated, stained with carmine alum (Sigma-Aldrich, C1022 and A7167), dehydrated, cleared with xylene, and mounted with Leica CV Mount (14046430011). Images from whole mounts were acquired with an Olympus macroscope (zoom 1.6) and joined with the MosaicJ tool from ImageJ (59). For junction quantification, images were processed using an ImageJ custom-made macro-developed for this purpose and then analyzed using AngioTool (60). For histology and immunohistochemistry, inguinal mammary glands were fixed in 10% neutral-buffered formalin solution and embedded in paraffin. Paraffin-embedded tissue sections (3 μm in thickness) were first air-dried and then dried at 60°C overnight. Immunohistochemistry was performed using Autostainer Plus (Dako, Agilent). Before immunohistochemistry, sections were dewaxed for Ki67 as part of the antigen retrieval process using the low pH EnVision FLEX Target Retrieval Solutions (Dako) for 20 min at 97°C using a PT Link (Dako, Agilent). For caspase 3, samples were dewaxed, and antigen retrieval was performed with citrate buffer (pH 6) for 20 min at 121°C with an autoclave. Endogenous peroxidase was quenched by 10-min incubation with peroxidase blocking solution (Dako REAL, S2023). The rabbit polyclonal primary antibodies anti-Ki67 (Abcam, ab15580) and anti-cleaved caspase 3 (Cell Signaling Technology, 9661S) were diluted 1:1000 and 1:300, respectively, with EnVision FLEX Antibody Diluent (Dako, Agilent, K800621) and incubated for 60 and 120 min, respectively, at room temperature. A biotin-free, ready-to-use BrightVision poly–horseradish peroxidase (HRP)–anti-rabbit immunoglobulin G (Immunologic, DPVR-110HRP) was used as secondary antibody. Immunohistochemistry for ER (clone 1D5; Dako, M7047), PR (Abcam, ab63605), and RANKL (R&D Systems, AF462) was performed as previously described (61, 62). Antigen-antibody complexes were revealed with 3,30-diaminobenzidine tetrahydrochloride (Dako, K3468). Sections were counterstained with hematoxylin (Dako, S202084) and mounted with toluene-free mounting medium (Dako, CS705) using a Dako CoverStainer. Bright-field images were acquired with a NanoZoomer-2.0 HT C9600 scanner (Hamamatsu). All images were visualized with a gamma correction set at 1.8 in the image control panel of the NDP.view software (Hamamatsu, Photonics, France). Image analysis was performed using TMARKER software (63). For immunofluorescence, Alexa secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) were used, and images were obtained on an inverted Leica TCS SP5 confocal microscopy.
Immunoblotting
Beads-homogenized tissue or MECs (EasySep, STEMCELL Technologies) were lysed in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (with phosphatase and protease inhibitors) and sonicated for 5 min at high or low intensity, respectively (Standard Bioruptor Diagenode). Cellular debris was pelleted (15,700g, 15 min, 4°C), and protein concentration was determined by the DC Protein Assay (Bio-Rad). Equal amounts of proteins were separated by SDS–polyacrylamide gel electrophoresis. After transfer onto nitrocellulose membranes (Sigma-Aldrich, GE10600001), membranes were blocked for 1 hour in 5% milk, and specific proteins were labeled with the corresponding primary antibodies against vinculin (Abcam, ab18058), CPEB3 (Abcam, ab10883), CPEB219, CPEB4 (Abcam, ab83009), CPEB1 (Cell Signaling Technology, no. 13583), CyclinD1 (Santa Cruz Biotechnology, sc-717), CREB1 (Cell Signaling Technology, no. 9197), α-tubulin (Sigma-Aldrich, T9026), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Life Technologies, AM-4300). Secondary HRP antibodies were also diluted in 5% milk, and proteins were revealed using enhanced chemiluminescence Western blotting detection reagents (GE Healthcare).
Cell culture and lentiviral infection
Human breast carcinoma cell lines MDA-MB-231, BT549, MDA-MB-435, MDA-MB-468, SKBR3, BT474, T47D, MCF7, and ZR75 were obtained from the American Type Culture Collection–LGC Standards Ltd. Partnership. All cell lines were cultured in DMEM d-glucose medium (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, except BT459 cells, which were cultured in supplemented RPMI medium (Gibco). All cells were cultured at 37°C and in a 5% CO2 humidified atmosphere. For lentiviral infection, human embryonic kidney–293 T cells were transfected with pLKO lentiviral vectors and plasmids encoding lentiviral particles using standard methods. pLKO sh_CPEB2 plasmids were obtained from Sigma-Aldrich MISSION shRNA library (clones TRCN0000149728 and TRCN0000149778). Recipient cells were transduced with the viral medium and selected with puromycin (2 μg ml−1) for 72 hours.
Cell proliferation assay
In vitro cell proliferation was assessed using the CyQUANT Cell Proliferation Kit following the manufacturer’s instructions. For 4-OHT sensitivity experiments, 4-OHT or vehicle (ethanol) was added to the cell culture at the indicated concentrations 24 hours after plating. Cell numbers were quantified after 6 days using BIO-TEK FL600 fluorescence microplate reader at 485 to 530 nm.
Annexin V apoptosis detection and FACS
To detect early apoptosis (APC labeled), cultured cells were trypsinized and processed following the Annexin V Apoptosis Detection Kit (Thermo Fisher Scientific). DAPI solution was also added to the cell suspension to detect the total number of dead cells. A Gallios cytometer (Beckman Coulter) was used for the analysis.
RNA analysis
Total RNA was extracted by TRIzol reagent (Invitrogen). RNA (1 μg) was reverse-transcribed with oligo(dT) and random primers using SuperScript IV (Thermo Fisher Scientific) or RevertAid (Thermo Fisher Scientific), following the manufacturer’s recommendations. Real-time qPCR (RT-qPCR) was performed in a LightCycler 480 (Roche) using PowerUp SYBR Green Master Mix (Roche). Primer sequences are listed in table S2. RNA quantifications were normalized to GAPDH as endogenous control. For human breast carcinoma cell lines, RNA extraction (PureLink RNA Mini Kit, Thermo Fisher Scientific), reverse transcription (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems), and real-time PCR (TaqMan Universal Master Mix, Applied Biosystems) were performed and analyzed as previously described (64). The TaqMan probes (Applied Biosystems) used were Hs0139673_m1 (CPEB2), Hs00153408_m1 (MYC), Hs00765553_m1 (CCND1), and Mm00437762_m1 (B2M). For microarrays, samples in duplicates from sorted cells from WT and CPEB2 KO animals were processed at IRB Barcelona’s Functional Genomics Core Facility following standard procedures. Affymetrix MG-430 PM strip data for DPs, DD, APs, and myoepithelial cell population samples in WT and CPEB2 KO in biological duplicates were processed with Bioconductor (65) using robust multiarray average (RMA) background correction, quantile normalization, and RMA summarization to obtain probeset expression estimates (66). Centroid locations from the principal component for the different combinations between cell populations and genotypes, as well as the resultant Euclidean distances between centroids, were computed. Dispersion within groups (the average Euclidean distance between samples and their corresponding population/genotype centroid) was also measured. Limma 3.22.7 (67) was then used to identify differentially expressed genes between CPEB2 KO and WT in all four cell populations, with P < 0.01 and |FC| > 2. Lists of up- and down-regulated genes between DPWT and DDWT were generated by selecting candidate genes with the highest and lowest FC percentiles and P < 0.01 (1% most up- and down-regulated genes, n = 181 and n = 101, respectively). Alternatively, after selecting with the highest and lowest FC percentiles, we also filtered these using a FDR threshold of 0.1. This resulted in a more stringent list of 24 up-regulated and no down-regulated genes in WT DP versus WT NCL. Enrichment for these gene lists, as well as for Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Broad Institute hallmark gene set categories in whole-genome gene lists ranked by mean log2FC between cell populations and genotypes, was assessed with the GSEA preranked algorithm (68). M. musculus GO and KEGG gene set collections were generated using the org.Mm.eg.db Bioconductor package (October 2014). Homo sapiens Hallmark gene set was downloaded from the Molecular Signatures Database and translated to M. musculus using Ensembl human-mouse homology information (August 2016).
RIP-seq analysis
MECs (EasySep, STEMCELL Technologies) were isolated from WT and CPEB2 KO animals (with two animals pooled per duplicate). Pellets were washed twice with cold Hanks’ balanced salt solution, lysed with RIPA buffer [50 mM tris-HCl (pH 8), 150 mM NaCl, 1 mM MgCl2, 1% NP-40, 1 mM EDTA, 0.1% SDS, protease inhibitor cocktail, and ribonuclease inhibitors] and sonicated for 5 min at low intensity with Standard Bioruptor Diagenode. After centrifugation (10 min, 4°C), supernatants were collected, precleared, and immunoprecipitated (4 hours, 4°C) with 10 μg of anti-CPEB2 antibody (69) bound to 50 μl of Dynabeads Protein G (Invitrogen). Beads were washed and split for either protein or RNA extraction. For RNA isolation, beads were resuspended in 100 μl of proteinase K buffer with 70 μg of proteinase K (Roche) and incubated for 30 min at 42°C and 30 min at 65°C. RNA was extracted following standard phenol/chloroform protocol. Samples were processed at IRB Barcelona’s Functional Genomics Facility following standard procedures: Illumina Hi-Seq 2000 50–base pair single-end RIP-sequencing (RIP-seq) data for WT and CPEB2 KO in biological duplicates, as well as their respective input samples of MECs, were checked for general sequencing quality control and adapter contamination using the FastQC software version 0.11, and no relevant problems were found. Afterward, reads were aligned against the M. musculus University of California, Santa Cruz mm10 ribosomal RNA (rRNA) genome using Bowtie1 0.12.9 (70) with two mismatches and default options to identify and remove reads coming from potential rRNA contamination from downstream analysis. Curated (non-rRNA) reads were then aligned against the M. musculus mm10 reference genome using Bowtie2 2.2.2 (71), allowing for one mismatch and reporting the best alignment site per read. All samples reported >15 million aligned reads. Potential amplification artefacts (duplicated reads) were detected and removed with the sambamba software version 0.5.1 using default options. Binary tiled data file tracks for visual inspection in the Integrative Genomics Viewer (IGV) software were generated using igvtools version 2. Read counts at 3′UTR level (longest 3′UTR per gene, mm10 genome Ensembl, March 2017) were computed using the featureCounts function from the Rsubread package version 1.24.2 with options minMQS = 1. Then, an interaction analysis of WT and CPEB2 KO RIP samples and their respective input controls (RIPWT/InputWT versus RIPKO/InputKO) was performed with DESeq2 (72). Target 3′UTRs were selected using an interaction FC threshold of >1.5 and interaction Benjamini-Hochberg adjusted P < 0.1 (see table S1, high-confidence RIP target genes, n = 169). GO enrichment for selected targets was performed using the online Enrichr (73, 74) tool.
Statistics and reproducibility
For animal experiments, data were expressed as means ± SEM, and statistics were analyzed with the GraphPad Prism software. Experiments were performed following a randomized block design. Littermates kept in the same cage since weaning were used whenever possible. The experiment was blinded before experimental analysis. For human breast carcinoma cell lines, P values were generated using the Student’s t test (unpaired, two tailed); P < 0.05 was considered significant. Error bars were calculated as SE in all the statistical analysis shown. Number of independent experiments is indicated in the figure legends.
Statistical analyses in METABRIC dataset
Transcriptomic and clinical data from the METABRIC breast cancer dataset (75, 76) were downloaded from the cBioPortal for Cancer Genomics database (77). Association of gene expression with molecular features (PAM50 subtype and ER status) was evaluated using a linear model, while a Cox model was fitted to assess association with overall survival. Statistical significance was assessed using the corresponding F tests of log-likelihood ratio tests. A Wald test was used for pairwise comparisons when necessary. In all cases, the cohort of origin of the sample was included as a covariate in the models.
For survival analyses, sample groups of low, medium, and high expression levels were defined using the tertiles of the intensity distribution after correction by cohort effects, as estimated by a linear model in which PAM50 subtypes were included as covariates. Association of gene expression with early relapse was modeled using a step function for a prespecified cutoff of a 10-year follow-up. Hazard ratios and their corresponding 95% confidence intervals were computed as a measure of association. For visualization purposes, Kaplan-Meier curves were estimated for groups of tumors that showed low, medium, or high expression. The threshold for statistical significance was set at 5%. All analyses were conducted with R (78).
Supplementary Material
Acknowledgments
We thank the Advance Digital Microscopy, Biostatistics/Bioinformatics, Histopathology, Mouse Mutant, and Functional Genomics facilities at IRB Barcelona. The Flow Cytometry Facility of the UB/PCB and the CRG Genomic Unit are also acknowledged. We also thank M. Serrano’s laboratory for providing the reagents for the organoid culture media. We thank A. Fernández from CIBERER-ISCII at CNB-CSIC for providing Southern blot images of the Cpeb3 mouse model. Cpeb3 mouse model generation (EMMA deposit: EM 10412) was supported by the EC-funded INFRAFRONTIER-I3 project. We thank S. Aznar-Benitah and members of R. Méndez’s laboratory for discussion. Funding: This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (MINECO, BFU2014-54122-P/BFU2017-83561-P), ERDFs, the Fundación Botín, the Banco Santander through its Santander Universities Global Division, the Scientific Foundation of the Spanish Association Against Cancer (AECC), the Fundación BBVA, the Fundación Bancaria “la Caixa,” and the Worldwide Cancer Research Foundation. R.P. held a la Caixa predoctoral fellowship. IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO (Government of Spain) and is supported by the CERCA Programme (Catalan Government). Author contributions: R.P. performed all studies and contributed to experimental design, data analysis, interpretation, and manuscript and figure preparation. R.M. conceived, directed, and discussed the study and wrote the manuscript. J.M. contributed to vivo mouse experiments. F.S. performed the experiments with the breast cancer cell lines and patient data analysis under the supervision of R.R.G. O.R. performed the bioinformatics analysis of Figs. 2 and 4. A.M.-R. and A.B. performed CPEB overexpression and rRNA coimmunoprecipitation experiments V.C. performed the tissue immunohistochemistry and immunofluorescence. C.S. and G.F-M. generated the CPEB3 KO model. Y.-S.H. provided the anti-CPEB2 antibody. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The CPEB2KO mice can be provided by IRB Barcelona pending scientific review and a completed material transfer agreement. Requests for the CPEB2KO mice should be submitted to innovation@irbbarcelona.org. All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data related to this paper can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115175 or may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaax3868/DC1
REFERENCES AND NOTES
- 1.Brisken C., O’Malley B., Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2, a003178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gjorevski N., Nelson C. M., Integrated morphodynamic signalling of the mammary gland. Nat. Rev. Mol. Cell Biol. 12, 581–593 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Hennighausen L., Robinson G. W., Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 6, 715–725 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Stingl J., Estrogen and progesterone in normal mammary gland development and in cancer. Horm. Cancer 2, 85–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanos T., Rojo L. J., Echeverria P., Brisken C., ER and PR signaling nodes during mammary gland development. Breast Cancer Res. 14, 210 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joshi P. A., Jackson H. W., Beristain A. G., Di Grappa M. A., Mote P. A., Clarke C. L., Stingl J., Waterhouse P. D., Khokha R., Progesterone induces adult mammary stem cell expansion. Nature 465, 803–807 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Asselin-Labat M.-L., Vaillant F., Sheridan J. M., Pal B., Wu D., Simpson E. R., Yasuda H., Smyth G. K., Martin T. J., Lindeman G. J., Visvader J. E., Control of mammary stem cell function by steroid hormone signalling. Nature 465, 798–802 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee A., Soyal S. M., Li J., Ying Y., He B., De Mayo F. J., Lydon J. P., Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J. 24, 4408–4419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robichaud N., Sonenberg N., Translational control and the cancer cell response to stress. Curr. Opin. Cell Biol. 45, 102–109 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Truitt M. L., Ruggero D., New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 17, 332 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Eliscovich C., Peset I., Vernos I., Méndez R., Spindle-localized CPE-mediated translation controls meiotic chromosome segregation. Nat. Cell Biol. 10, 858–865 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Barr J., Yakovlev K. V., Shidlovskii Y., Schedl P., Establishing and maintaining cell polarity with mRNA localization in Drosophila. BioEssays 38, 244–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weill L., Belloc E., Bava F.-A., Méndez R., Translational control by changes in poly(A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 19, 577–585 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Ivshina M., Lasko P., Richter J. D., Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu. Rev. Cell Dev. Biol. 30, 393–415 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Afroz T., Skrisovska L., Belloc E., Guillén-Boixet J., Méndez R., Allain F. H.-T., A fly trap mechanism provides sequence-specific RNA recognition by CPEB proteins. Genes Dev. 28, 1498–1514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belloc E., Méndez R., A deadenylation negative feedback mechanism governs meiotic metaphase arrest. Nature 452, 1017–1021 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Piqué M., López J. M., Foissac S., Guigó R., Méndez R., A combinatorial code for CPE-mediated translational control. Cell 132, 434–448 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Weill L., Belloc E., Castellazzi C. L., Méndez R., Musashi 1 regulates the timing and extent of meiotic mRNA translational activation by promoting the use of specific CPEs. Nat. Struct. Mol. Biol. 24, 672–681 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Maillo C., Martín J., Sebastián D., Hernández-Alvarez M., García-Rocha M., Reina O., Zorzano A., Fernandez M., Méndez R., Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cell Biol. 19, 94–105 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Guillén-Boixet J., Buzon V., Salvatella X., Méndez R., CPEB4 is regulated during cell cycle by ERK2/Cdk1-mediated phosphorylation and its assembly into liquid-like droplets. eLife 5, e19298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi K. M., Barash I., Rhoads R. E., Insulin and prolactin synergistically stimulate β-casein messenger ribonucleic acid translation by cytoplasmic polyadenylation. Mol. Endocrinol. 18, 1670–1686 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Nagaoka K., Udagawa T., Richter J. D., CPEB-mediated ZO-1 mRNA localization is required for epithelial tight-junction assembly and cell polarity. Nat. Commun. 3, 675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novoa I., Gallego J., Ferreira P. G., Mendez R., Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat. Cell Biol. 12, 447–456 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lima S. A., Chipman L. B., Nicholson A. L., Chen Y.-H., Yee B. A., Yeo G. W., Coller J., Pasquinelli A. E., Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 24, 1057–1063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calderone V., Gallego J., Fernandez-Miranda G., Garcia-Pras E., Maillo C., Berzigotti A., Mejias M., Bava F. A., Angulo-Urarte A., Graupera M., Navarro P., Bosch J., Fernandez M., Mendez R., Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology 150, 982–97.e30 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Tay J., Richter J. D., Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev. Cell 1, 201–213 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Van Keymeulen A., Rocha A. S., Ousset M., Beck B., Bouvencourt G., Rock J., Sharma N., Dekoninck S., Blanpain C., Distinct stem cells contribute to mammary gland development and maintenance. Nature 479, 189–193 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Shehata M., Teschendorff A., Sharp G., Novcic N., Russell I. A., Avril S., Prater M., Eirew P., Caldas C., Watson C. J., Stingl J., Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 14, R134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visvader J. E., Stingl J., Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 28, 1143–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giraddi R. R., Shehata M., Gallardo M., Blasco M. A., Simons B. D., Stingl J., Stem and progenitor cell division kinetics during postnatal mouse mammary gland development. Nat. Commun. 6, 8487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beleut M., Rajaram R. D., Caikovski M., Ayyanan A., Germano D., Choi Y., Schneider P., Brisken C., Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. U.S.A. 107, 2989–2994 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Keymeulen A., Lee M. Y., Ousset M., Brohée S., Rorive S., Giraddi R. R., Wuidart A., Bouvencourt G., Dubois C., Salmon I., Sotiriou C., Phillips W. A., Blanpain C., Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525, 119–123 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Lee Y. J., Shin K. J., Park S.-A., Park K. S., Park S., Heo K., Seo Y.-K., Noh D.-Y., Ryu S. H., Suh P.-G., G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget 7, 70898–70911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madak-Erdogan Z., Lupien M., Stossi F., Brown M., Katzenellenbogen B. S., Genomic collaboration of estrogen receptor α and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell. Biol. 31, 226–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pires-daSilva A., Sommer R. J., The evolution of signalling pathways in animal development. Nat. Rev. Genet. 4, 39–49 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Reya T., Morrison S. J., Clarke M. F., Weissman I. L., Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Brisken C., Progesterone signalling in breast cancer: A neglected hormone coming into the limelight. Nat. Rev. Cancer 13, 385–396 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Harris L. N., Ismaila N., McShane L. M., Andre F., Collyar D. E., Gonzalez-Angulo A. M., Hammond E. H., Kuderer N. M., Liu M. C., Mennel R. G., Van Poznak C., Bast R. C., Hayes D. F., Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 34, 1134–1150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker J. S., Mullins M., Cheang M. C. U., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z., Quackenbush J. F., Stijleman I. J., Palazzo J., Marron J. S., Nobel A. B., Mardis E., Nielsen T. O., Ellis M. J., Perou C. M., Bernard P. S., Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aldaz C. M., Liao Q. Y., Paladugu A., Rehm S., Wang H., Allelotypic and cytogenetic characterization of chemically induced mouse mammary tumors: High frequency of chromosome 4 loss of heterozygosity at advanced stages of progression. Mol. Carcinog. 17, 126–133 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Yin Y., Bai R., Russell R. G., Beildeck M. E., Xie Z., Kopelovich L., Glazer R. I., Characterization of medroxyprogesterone and DMBA-induced multilineage mammary tumors by gene expression profiling. Mol. Carcinog. 44, 42–50 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Lanari C., Lamb C. A., Fabris V. T., Helguero L. A., Soldati R., Bottino M. C., Giulianelli S., Cerliani J. P., Wargon V., Molinolo A., The MPA mouse breast cancer model: Evidence for a role of progesterone receptors in breast cancer. Endocr. Relat. Cancer 16, 333–350 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Casimiro M. C., Wang C., Li Z., Sante G. D., Willmart N. E., Addya S., Chen L., Liu Y., Lisanti M. P., Pestell R. G., Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol. Endocrinol. 27, 1415–1428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandez-Valdivia R., Mukherjee A., Ying Y., Li J., Paquet M., De Mayo F. J., Lydon J. P., The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev. Biol. 328, 127–139 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Yoldi G., Pellegrini P., Trinidad E. M., Cordero A., Gomez-Miragaya J., Serra-Musach J., Dougall W. C., Muñoz P., Pujana M.-A., Planelles L., González-Suárez E., RANK signaling blockade reduces breast cancer recurrence by inducing tumor cell differentiation. Cancer Res. 76, 5857–5869 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Fata J. E., Kong Y. Y., Li J., Sasaki T., Irie-Sasaki J., Moorehead R. A., Elliott R., Scully S., Voura E. B., Lacey D. L., Boyle W. J., Khokha R., Penninger J. M., The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 103, 41–50 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Chang Y.-W., Huang Y.-S., Arsenite-activated JNK signaling enhances CPEB4-Vinexin interaction to facilitate stress granule assembly and cell survival. PLOS ONE 9, e107961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao B., Hang J., Lei T., He Y., Kuang Z., Wang L., Chen L., He J., Zhang W., Liao Y., Sun Z., Li L., Identification of key genes relevant to the prognosis of ER-positive and ER-negative breast cancer based on a prognostic prediction system. Mol. Biol. Rep. 46, 2111–2119 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Asselin-Labat M.-L., Sutherland K. D., Barker H., Thomas R., Shackleton M., Forrest N. C., Hartley L., Robb L., Grosveld F. G., van der Wees J., Lindeman G. J., Visvader J. E., Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 9, 201–209 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Kouros-Mehr H., Slorach E. M., Sternlicht M. D., Werb Z., GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041–1055 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bertolin K., Murphy B. D., Reproductive tract changes during the mouse estrous cycle. Guid. to Investig. Mouse Pregnancy , 85–94 (2014). [Google Scholar]
- 52.Byers S. L., Wiles M. V., Dunn S. L., Taft R. A., Mouse estrous cycle identification tool and images. PLOS ONE 7, e35538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aldaz C. M., Liao Q. Y., Bate M. L., Johnston D. A., Medroxyprogesterone acetate accelerates the development and increases the incidence of mouse mammary tumors induced by dimethylbenzanthracene. Carcinogenesis 17, 2069–2072 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Wan L., Lu X., Yuan S., Wei Y., Guo F., Shen M., Yuan M., Chakrabarti R., Hua Y., Smith H. A., Blanco M. A., Chekmareva M., Wu H., Bronson R. T., Haffty B. G., Xing Y., Kang Y., MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell 26, 92–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardiff R. D., Anver M. R., Gusterson B. A., Hennighausen L., Jensen R. A., Merino M. J., Rehm S., Russo J., Tavassoli F. A., Wakefield L. M., Ward J. M., Green J. E., The mammary pathology of genetically engineered mice: The consensus report and recommendations from the Annapolis meeting. Oncogene 19, 968–988 (2000). [DOI] [PubMed] [Google Scholar]
- 56.M. Prater, M. Shehata, C. J. Watson, J. Stingl, Enzymatic dissociation, flow cytometric analysis, and culture of normal mouse mammary tissue, in Methods in Molecular Biology (Springer US, 2012), vol. 531. [DOI] [PubMed] [Google Scholar]
- 57.Giraddi R. R., Shehata M., Gallardo M., Blasco M. A., Simons B. D., Stingl J., Stem and progenitor cell division kinetics during postnatal mouse mammary gland development. Nat. Commun. 6, 8487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jamieson P. R., Dekkers J. F., Rios A. C., Fu N. Y., Lindeman G. J., Visvader J. E., Derivation of a robust mouse mammary organoid system for studying tissue dynamics. Development 144, 1065–1071 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Thévenaz P., Unser M., User-friendly semiautomated assembly of accurate image mosaics in microscopy. Microsc. Res. Tech. 70, 135–146 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Zudaire E., Gambardella L., Kurcz C., Vermeren S., A computational tool for quantitative analysis of vascular networks. PLOS ONE 6, e27385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohammed H., Russell I. A., Stark R., Rueda O. M., Hickey T. E., Tarulli G. A., Serandour A. A., Birrell S. N., Bruna A., Saadi A., Menon S., Hadfield J., Pugh M., Raj G. V., Brown G. D., D’Santos C., Robinson J. L. L., Silva G., Launchbury R., Perou C. M., Stingl J., Caldas C., Tilley W. D., Carroll J. S., Progesterone receptor modulates ERα action in breast cancer. Nature 523, 313–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez-Suarez E., Jacob A. P., Jones J., Miller R., Roudier-Meyer M. P., Erwert R., Pinkas J., Branstetter D., Dougall W. C., RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468, 103–107 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Schüffler P. J., Fuchs T. J., Ong C. S., Wild P. J., Rupp N. J., Buhmann J. M., TMARKER: A free software toolkit for histopathological cell counting and staining estimation. J. Pathol. Inform. 4, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Urosevic J., Garcia-Albéniz X., Planet E., Real S., Céspedes M. V., Guiu M., Fernandez E., Bellmunt A., Gawrzak S., Pavlovic M., Mangues R., Dolado I., Barriga F. M., Nadal C., Kemeny N., Batlle E., Nebreda A. R., Gomis R. R., Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 16, 685–694 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y. H., Zhang J., Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carvalho B. S., Irizarry R. A., A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lai Y.-T., Su C.-K., Jiang S.-T., Chang Y.-J., Lai A. C.-Y., Huang Y.-S., Deficiency of CPEB2-confined choline acetyltransferase expression in the dorsal motor nucleus of vagus causes hyperactivated parasympathetic signaling-associated bronchoconstriction. J. Neurosci. 36, 12661–12676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., Ma’ayan A., Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., Mc Dermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Curtis C., Shah S. P., Chin S.-F., Turashvili G., Rueda O. M., Dunning M. J., Speed D., Lynch A. G., Samarajiwa S., Yuan Y., Gräf S., Ha G., Haffari G., Bashashati A., Russell R., Kinney S. M.; METABRIC Group, Langerød A., Green A., Provenzano E., Wishart G., Pinder S., Watson P., Markowetz F., Murphy L., Ellis I., Purushotham A., Børresen-Dale A.-L., Brenton J. D., Tavaré S., Caldas C., Aparicio S., The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pereira B., Chin S.-F., Rueda O. M., Vollan H.-K. M., Provenzano E., Bardwell H. A., Pugh M., Jones L., Russell R., Sammut S.-J., Tsui D. W. Y., Liu B., Dawson S.-J., Abraham J., Northen H., Peden J. F., Mukherjee A., Turashvili G., Green A. R., Kinney S. M., Oloumi A., Shah S., Rosenfeld N., Murphy L., Bentley D. R., Ellis I. O., Purushotham A., Pinder S. E., Børresen-Dale A.-L., Earl H. M., Pharoah P. D., Ross M. T., Aparicio S., Caldas C., The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N., The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.R Development Core Team, R: A language and environment for statistical computing, R Found. Stat. Comput. Vienna (2008); http://www.R-project.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaax3868/DC1