

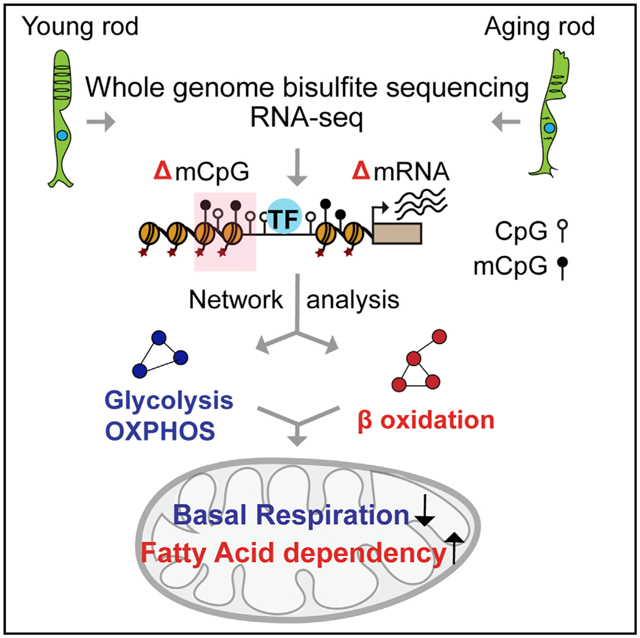
SUMMARY
Aging-associated functional decline is accompanied by alterations in the epigenome. To explore DNA modifications that could influence visual function with age, we perform whole-genome bisulfite sequencing of purified mouse rod photoreceptors at four ages and identify 2,054 differentially methylated regions (DMRs). We detect many DMRs during early stages of aging and in rod regulatory regions, and some of these cluster at chromosomal hotspots, especially on chromosome 10, which includes a longevity interactome. Integration of methylome to age-related transcriptome changes, chromatin signatures, and first-order protein-protein interactions uncover an enrichment of DMRs in altered pathways that are associated with rod function, aging, and energy metabolism. In concordance, we detect reduced basal mitochondrial respiration and increased fatty acid dependency with retinal age in ex vivo assays. Our study reveals age-dependent genomic and chromatin features susceptible to DNA methylation changes in rod photoreceptors and identifies a link between DNA methylation and energy metabolism in aging.
Graphical Abstract
In Brief
The crosstalk between chromatin modifications and aging hallmarks is poorly understood, especially in neurons. Using purified rod photoreceptors, Corso-Díaz et al. identify aging-associated differentially DNA methylated regions that greatly overlap regulatory elements and are linked to lower mitochondrial basal respiration and higher fatty acid dependency.
INTRODUCTION
Advanced age is characterized by progressive decline of physiological functions and increased susceptibility to diseases such as cancer, diabetes, and neurodegeneration. Healthy aging is regulated by a combination of genetic and non-genetic factors (Erikson et al., 2016; Kaeberlein et al., 2015). Multiple interconnected endophenotypes, including mitochondrial dysfunction and epigenomic changes, contribute to the normal aging process (Booth and Brunet, 2016; López-Otín et al., 2013; Sun et al., 2016). Genetic variations, such as those in DNA repair and insulin pathways, have been associated with lifespan differences in humans (Debrabant et al., 2014; Flachsbart et al., 2017; Pan et al., 2016; Ziv and Hu, 2011). Lifestyle and environmental factors, such as caloric-restricted diets and supplementation with naturally occurring molecules, also correlate with increased longevity in various organisms including primates (Kaeberlein et al., 2015). Nonetheless, the response of different tissues to aging likely involves both universal and unique molecular alterations for cellular and functional adaptability.
The epigenome, defined as chromatin regulatory modifications that relate to the memory of past stimuli (Corso-Díaz et al., 2018), is amenable to aging and environmental influences, and could mediate physiological alterations that drive age related decline and disease (Pal and Tyler, 2016; Booth and Brunet, 2016). One of the key epigenetic modifications, methylation of cytosine in the CpG deoxynucleotide context (mCpG), is shown to frequently change with age (Day et al., 2013) and contributes to both epigenetic drift (decreased correlation of epigenomes over time) and epigenetic clock (concordance of specific CpG methylation sites to chronological age) (Jones et al., 2015). DNA methylation changes are predicted to mediate adaptations in cellular homeostasis and are likely associated with functional decline with age. Notably, mCpG alterations have been linked to heterochromatin loss, DNA damage, and chromosome instability in humans (Ciccarone et al., 2018; Rodriguez et al., 2006). However, relevance of specific changes in CpG methylation to aging-associated dysfunctions in different tissues and cell types is still far from clear.
Advanced age is the primary risk factor for multifactorial common neurodegenerative diseases (Wyss-Coray, 2016; Yankner et al., 2008), including those affecting retinal function, such as glaucoma (Chrysostomou et al., 2010) and macular degeneration (Swaroop et al., 2009). Vision decline during normal aging includes impairments in visual acuity, dark adaptation, and contrast sensitivity (Owsley, 2016) that can be caused by alterations in different retinal cell populations (Cavallotti et al., 2004). Rod photoreceptors constitute more than 70% of retinal cells in the human retina and are involved in dim-light vision; their dysfunction is prominent in aging adults and is an early indicator of subsequent vision impairment in both inherited and aging associated retinal degenerative diseases (Jackson et al., 2002). Furthermore, rods are highly vulnerable to metabolic stress and are considered the primary drivers of age-related synaptic remodeling in the retina (Samuel et al., 2014). Thus, elucidation of molecular changes and biological pathways linked with aging of rod photoreceptors would likely stimulate investigations for preventing or delaying vision loss in aging and associated neurodegeneration.
Advent of “omics” technologies has permitted delineation of transcriptional regulatory networks, including the chromatin landscape, that guide rod and cone photoreceptor development (Aldiri et al., 2017; Corso-Díaz et al., 2018; Kim et al., 2016; Mo et al., 2016). We have previously reported aging-associated gene expression changes in rod photoreceptors using microarrays (Parapuram et al., 2010). However, to date, there is no whole-genome study of DNA methylation on a purified neuronal cell type that incorporates multiple stages of aging. To explore the association of DNA methylation with gene regulation during aging, we performed genome-wide base-resolution methylome analysis of purified mouse rod photoreceptors at four different ages and integrated these data with transcriptome (generated by RNA sequencing [RNA-seq]), open chromatin, and histone modification profiles. In addition to unraveling chromatin features and clusters of differentially methylated regions (DMRs) in aging rods, we uncovered established and unique cellular pathways, such as longevity and energy metabolism, which exhibit aging-dependent progressive alterations. We also validated the impact of age on basal mitochondrial oxygen consumption capacity and fatty acid dependency, and propose a link between epigenome and energy metabolism, which can explain, at least in part, the age-related functional decline in rods.
RESULTS
DNA Methylome Analysis Identifies Regulatory Regions in Rod Photoreceptors
We performed whole-genome bisulfite sequencing (WGBS) of rod photoreceptors purified from the retina of young (3-month old), middle-aged (12-month-old), and old (18-and 24-month-old) male pNrl-EGFP (enhanced green fluorescent protein) mice (Figure 1A) (n = 3 for 3, 18, and 24 months; n = 2 for 12 months). This strain expresses an EGFP under the control of the promoter of the rod-specific gene Nrl (Akimoto et al., 2006). We obtained a cumulative 16-to 25-fold CpG coverage genome-wide at all ages (Table S1). In addition, we generated RNA-seq of all time points (n = 4 for 3 months; n = 3 for 12–24 months) and H3K27ac and H3K27me3 profiles on native chromatin of 3-month-old rods (n = 4–6) using male mice to examine the relevance of DNA methylation to gene regulation (Figure 1A; Table S1). All samples showed a high degree of correlation across time points (Pearson’s and Spearman’s rank correlation coefficient > 0.94), although older samples were less correlated in DNA methylation profiles (Figures S1A and S1B).
Figure 1. Epigenomic Characteristics of Young Rod Photoreceptors.
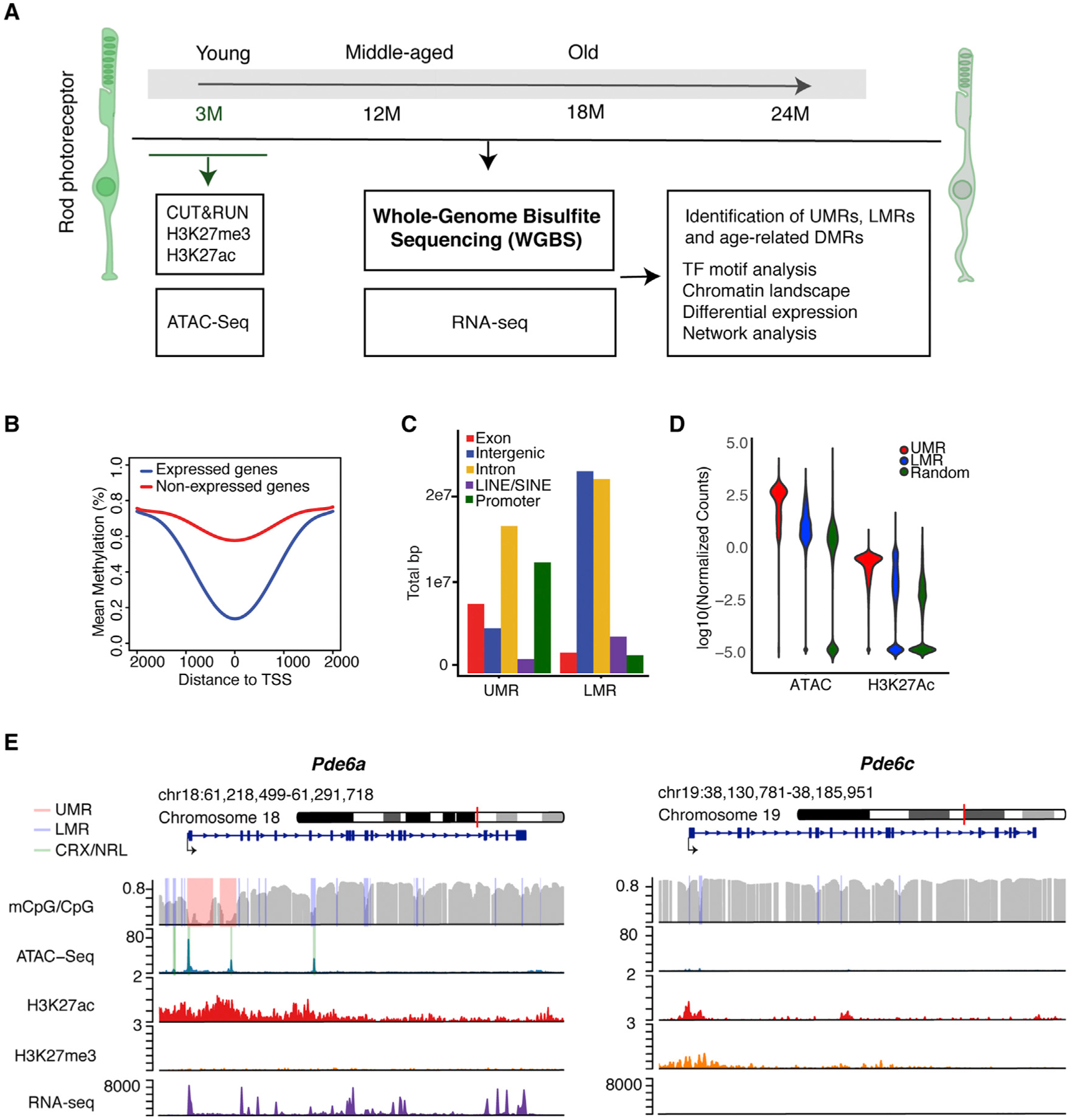
(A) Outline of the experimental paradigm. Integrative analysis of base-resolution DNA methylation and RNA-seq profiles from young (3-month-old), middle-aged (12-month-old), and old (18-and 24-month-old) rods with chromatin accessibility and histone marks from 3-month-old rods.
(B) DNA methylation levels around the TSS of expressed and unexpressed genes in 3-month-old rods.
(C) Distribution of UMRs and LMRs across various genomic regions of 3-month-old rods.
(D) Normalized ATAC-seq and H3K27ac read numbers over total region size of UMRs, LMRs, and random genomic regions of similar size in 3-month-old rods.
(E) Examples of UMRs (pink) and LMRs (purple) in Pde6a and Pde6c. CRX and NRL ChIP-seq peaks are shown in green. ATAC-seq peaks overlap with CRX and NRL binding in both UMRs and LMRs within the rod-specific Pde6a gene. CpG methylation, ATAC-seq, H3K27ac, H3K27me3, and RNA-seq profiles are shown for the 3-month time point over the entire gene and its surroundings.
DMR, differentially methylated region; LMR, low methylated region; TF, transcription factor; TSS, transcription start site; UMR, unmethylated region.
We initially focused on the analysis of 3-month rod methylome that revealed high methylation at most CpGs, as reported for other cell types (Figures S1C and S1D) and similar to levels shown for mature rods and cones (Mo et al., 2016). DNA methylation across genomic regions followed a previously described pattern at promoters of expressed genes, which have lower levels of methylation compared with unexpressed genes (Figure 1B). Even though rods represent about 75% of retinal cells, this phenomenon is more robust in flow-sorted rods than in the whole retina, highlighting the importance of using purified photoreceptors (see Figure S1E for an example of the Pde6a promoter, where lower levels of methylation are detected in rods compared with whole retina). Methylation levels at different genomic contexts were also consistent among cell types (Figure S1F). We then segmented the genome into unmethylated regions (UMRs; <10% average methylation) and low methylated regions (LMRs; 10%–50% average) in the pooled 3-month samples using a Hidden Markov model (Burger et al., 2013) and mapped UMRs and LMRs in different genomic regions (Figure 1C). We detected enrichment of UMRs in promoters and intronic regions, whereas LMRs were over-represented in intergenic and intronic regions, reflecting their potential role in enhancers (Burger et al., 2013). We then integrated methylation levels within these regions to RNA-seq and histone H3K27ac data that we generated from 3-month-old rods, together with published ATAC-seq profiles (Mo et al., 2016). Rod chromatin appears to be highly open and active in UMRs and less open, although higher than in random regions, in LMRs (Figure 1D). Notably, consensus binding sites for rod transcription factors are enriched in UMRs and LMRs, in agreement with their role in rod gene regulation (Figure S1G). As an example, UMRs and LMRs at the rod photo transduction gene Pde6a show chromatin immunoprecipitation sequencing (ChIP-seq) peaks of rod transcription factors NRL and CRX (Corbo et al., 2010; Hao et al., 2012), whereas the cone-specific gene Pde6c that shows very low expression in rods reveals an overlap of LMRs with small ATAC-seq peaks and the inhibitory histone mark H3K27me3 (Figure 1E). Overall, these data demonstrate that DNA methylome profiles can identify regulatory regions critical for rod-specific transcriptional programs.
Age-Related Changes in DNA Methylation Are Distributed Non-randomly in the Genome
Evaluation of global methylation patterns of rods at different stages of aging revealed high correlation among different samples but a somewhat higher increase in variability in older samples (Figure S1A); however, methylation levels at different genomic elements appeared to be similar (Figure S2A). In addition, epigenetic clocks trained on other mouse tissues (Meer et al., 2018; Stubbs et al., 2017) did not appear to work on isolated rod photoreceptors because the correlation between chronological and epigenetic age was low and not statistically significant (Figures S2B and S2C). Therefore, to identify non-stochastic changes in methylation during aging, we profiled three independent samples per age (3 versus 24 months) and used the package BSmooth, which performs a t test on each CpG to identify regions containing differentially methylated neigh boring CpGs (Hansen et al., 2012). By comparing the 3-and 24-month samples and using stringent criteria (methylation difference > 10%, areaStat > 20, t-statistic quantile cutoff of0.001), we identified 2,054 DMRs (Table S2; see Figure 2A for examples of three DMRs identified in Dnajb12); of these, 1,037 DMRs were hypomethylated and 1,017 hypermethylated. A majority of methylation changes were modest (10%–20%) and occurred within 1-kb regions (Figures S2D and S2E).
Figure 2. Age-Dependent Differential Methylation Accumulates in Chromosome 10.
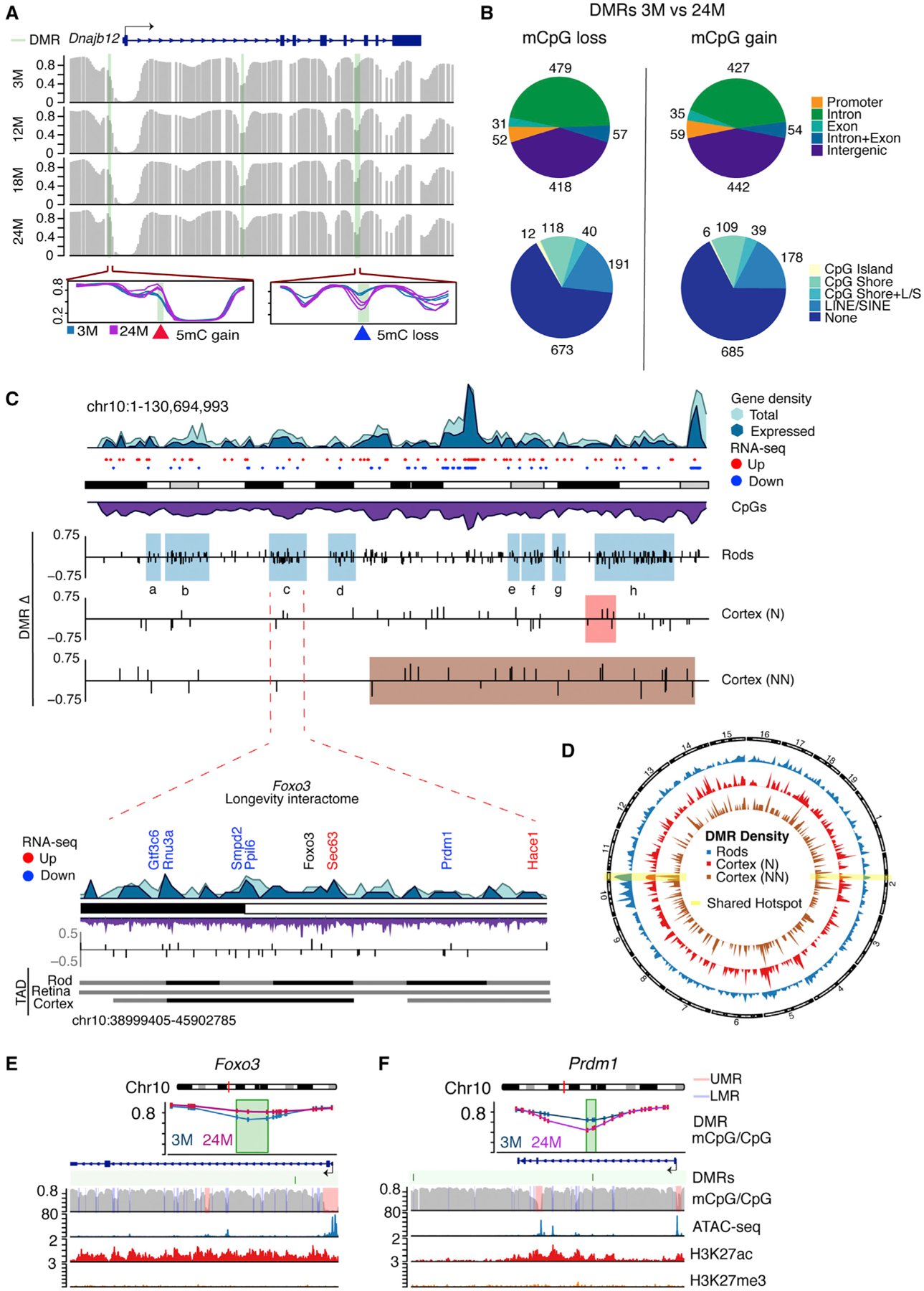
(A) Example of DMRs with loss and gain of DNA methylation (green bars) in Dnajb12 identified by comparing 3 versus 24 months with BSmooth. Individual samples are shown in the zoomed-in regions (n = 3 for 3 and 24 months).
(B) Distribution of DMRs that lose or gain DNA methylation with age in distinct genomic regions. LINE(L), SINE(S).
(C) Idiogram of chromosome 10 showing hotspots of clustered DMRs (colored boxes) in rods (blue shadows) and frontal cortex of neuronal (N, red shadow) and non-neuronal (NN, brown shadow) cell populations. Frontal cortex profiles were obtained from Lister et al. (2013). Gene density is also shown for all genes (light blue) and expressed genes >10 counts per million (CPM) (darker blue) in rods. Upregulated and downregulated genes are shown as red and blue dots, respectively. CpG density is shown in purple. A zoom-in of hotspot c is presented. This region corresponds to a longevity interactome proposed to function as a transcriptional unit centered in Foxo3 (Donlon et al., 2017). Downregulated and upregulated genes are shown in blue and red, respectively. Genes with no change are shown in black. Topological-associated domains (TADs) (Norrie et al., 2019) are shown for rods, whole retina, and frontal cortex.
(D) Circos plot showing the density of DMRs for rods and N and NN cells from the frontal cortex. Highlighted in yellow are regions with shared clusters of DMRs among all cell types.
(E and F) Examples of DMRs (green) in (E) Foxo3 and (F) Prdm1 genes. Methylation levels at the DMR are shown for 3 and 24 months. CpG methylation, ATAC-seq, H3K27ac, and H3K27me3 profiles are shown for the 3-month time point over the gene and its surroundings.
The DMRs associated with rod aging were not evenly distributed across the genome and revealed two distinct characteristics. First, the majority of DMRs localized to coding regions, especially introns (mCpG loss = 479 [45%], gain = 427 [51%]), with less than 6% localizing in promoters (0–1 kb from transcription start site [TSS]; mCpG loss = 52, mCpG gain = 59) (Figure 2B). Nonetheless, DMRs were significantly enriched (false discovery rate [FDR] % 0.05) for promoters, introns, and CpG shore regions, and were depleted in CpG islands, repetitive elements, and intergenic regions (Figures 2B and S2F). Second, DMRs clustered in 13 chromosomal regions, based on the higher density of DMRs compared with the genomic average (Figure 2C; Figures S2G and S3; Table S3). To identify statistically significant enriched regions (hotspots) with high density of DMRs, taking into account the CpG context and gene density, we used an algorithm that implements a generalized linear modeling with local CpG content and gene density as covariates. The probability of observing the actual number of DMRs was determined within the hotspot using an FDR of 0.01 and focused on regions longer than 1 Mb. Notably, a cluster on chromosome 14 contains the Nrl gene, which controls rod cell fate and function (Mears et al., 2001; Figure S3). Another cluster on chromosome 6 includes Hk2, which encodes a key enzyme for photoreceptor function (Petit et al., 2018; Figure S3).
Importantly, 8 of the 13 DMR hotspots were located on chromosome 10 (Figure 2C). One of these hotspots was centered on the Foxo3 gene (Figure 2C), which is proposed to function as part of a longevity interactome, including cell survival genes looping together over a 7-Mb region in humans (Donlon et al., 2017), and belongs to a topological-associated domain (TAD) in the retina (Norrie et al., 2019). Notably, we observed a DMR in the gene body of Foxo3 itself (Figure 2E). Within this hotspot, we identified altered expression of several genes (Figure 2C; blue, downregulated; red, upregulated), including Prdm1, which is crucial for rod development (Brzezinski et al., 2010). A DMR in the gene body of Prdm1 is shown in Figure 2F. Other DMR hotspots on chromosome 10 harbored genes associated with cell survival and rod function; e.g., hotspot h contains the autophagy gene Pawr and several synaptic genes (Gene Ontology [GO]: 0097060; p = 0.04). Differentially expressed genes in this region include Atxn7l3b and Cct2 (Table S2), defects in these can be associated with retinal degeneration (Minegishi et al., 2016; Tan et al., 2014).
To assess whether the identified DMR hotspots are unique to rod aging, we re-analyzed five published aging methylomes of four different cell types (Avrahami et al., 2015; Cole et al., 2017; Lister et al., 2013). We observed chromosomal hotspots of DMRs in other aging cell types with varying degree of overlap with rods (Figures 2C and 2D; Figure S4; Table S3). The hotspot h on chromosome 10 and the hotspot on chromosome 2 containing the mitochondrial ATP synthase Atp5g3 are shared with neuronal and non-neuronal cells from the frontal cortex (Figures 2C and 2D, highlighted in yellow; Figure S4; Table S3). Similarly, hotspots e, f, and g on chromosome 10 are shared with non-neuronal cells from the frontal cortex (Figure 2C). Interestingly, the high density of clusters on chromosome 10 seems to be unique to rod cells and was not influenced by CpG coverage. Overall, our results show that age-related DNA methylation changes are non-random and enriched at specific genomic elements and chromosomal regions.
Age-Related DMRs Occur in Candidate Rod Regulatory Regions
Our analysis revealed that most DMRs were present in and around UMRs and LMRs (UMRs = 179, LMRs = 986), and 733 overlapped with ATAC-seq peaks (±<500 bp from peak) (Figure 3A; Table S2). Gain of DNA methylation was frequently observed within regions spanning LMRs (Figure 3A). In many in stances, DMRs seem to occur at open chromatin boundaries (Figures 3A–3C), which indicate regions involved in the maintenance of open chromatin domains (Chai et al., 2013). Our results therefore suggest that mechanisms that support chromatin stability at regulatory elements may be perturbed with age.
Figure 3. Age-Related DMRs Dominate in Rod Regulatory Regions.
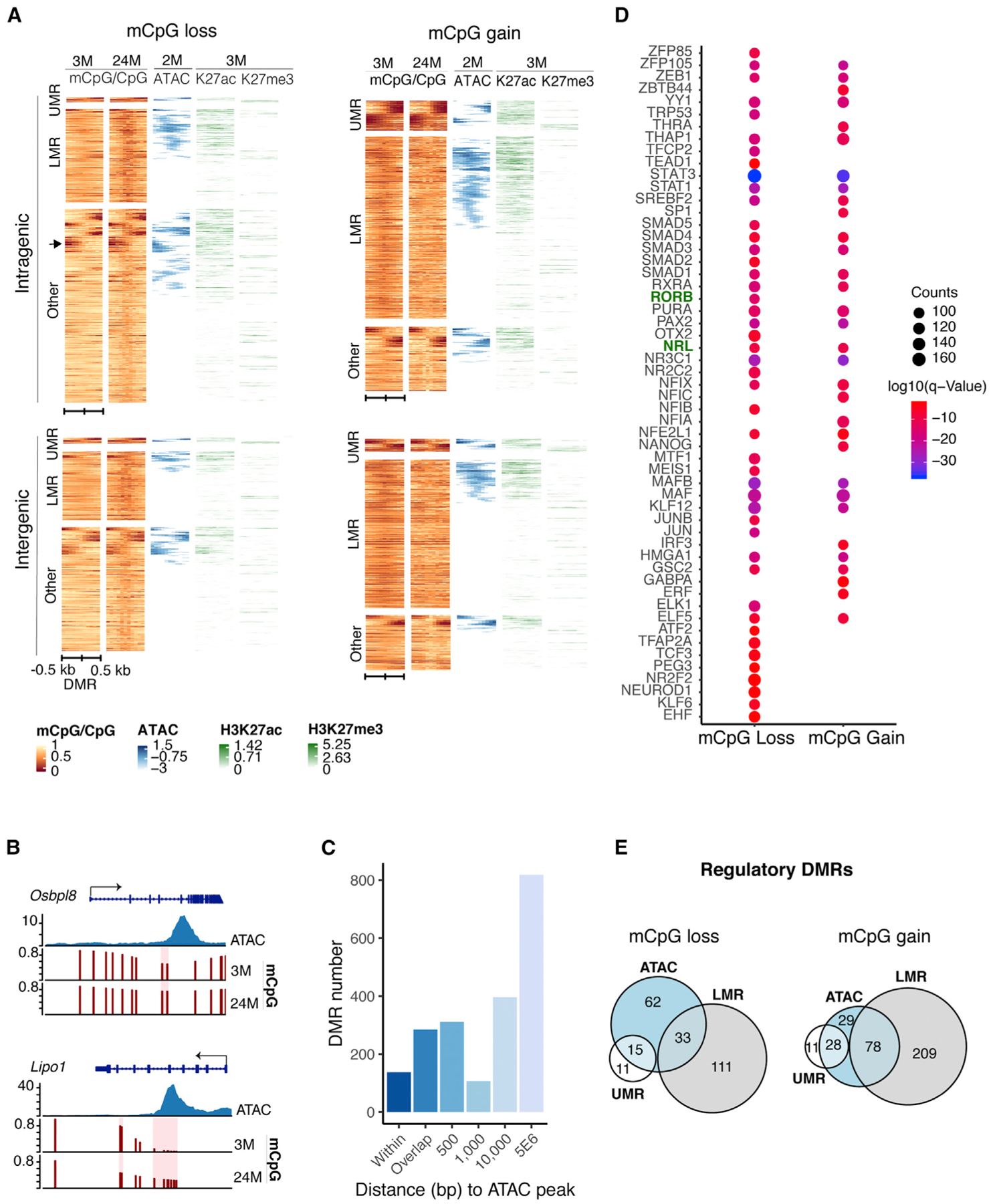
(A) Heatmap showing chromatin features over a 1-kb region centered on intragenic or intergenic DMRs. DMRs often localize to open chromatin boundaries and harbor marks of active (H3K27ac) or repressed/poised (H3K27me3) regulatory elements. Color scale bar represents methylation levels in young (3-month-old) and old (24-month-old) mice, with red being low and yellow being high. Scale for ATAC-seq and histone modifications represents read density, with blue and green being high and white being low in 2-to 3-month-old rods (n = 6 for H3K27me3 and n = 4 for H3K27ac). ATAC-seq data were obtained from Mo et al. (2016). The arrow indicates an example of DMRs located at boundaries of ATAC-seq peaks.
(B) Examples of DMRs at boundary regions of ATAC-seq peaks in Osbpl8 or within ATAC-seq peaks in Lipo1.
(C) Distance of DMRs from ATAC-seq peaks. DMRs shown as “within” represent DMRs completely contained in ATAC-seq peaks.
(D) TF motifs enriched in DMRs. Top 20% enriched TFs are shown. p ≤ 0.01.
(E) Euler diagrams showing the numbers of intergenic DMRs overlapping with UMRs, LMRs, and ATAC-seq peaks (±<500 bp from ATAC-seq peak), herein referred to as regulatory DMRs.
To evaluate whether DMRs were present in active or repressed regulatory regions, we studied the occupancy of H3K27ac, which is associated with active regulatory elements (n = 4), and H3K27me3, which is associated with repressed or poised regions (Young et al., 2011) (n = 6), on native chromatin by Cleavage Under Targets and Release Using Nuclease (CUT&RUN) (Skene and Henikoff, 2017). We noted that most intergenic DMRs overlapping with assay for transposase-accessible chromatin using sequencing (ATAC-seq) peaks contained signatures of H3K27ac, and that only a small number colocalized with H3K27me3 (Figure 3A). In concordance, DMRs were enriched for motifs of key rod transcription factors such as NRL and RORβ (Figure 3D).
Aging Progression in Rods Correlates to Distinct Patterns of DMRs
To identify pathways susceptible to aging-related epigenomic changes, we performed functional annotation of genes associated with DMRs. Intragenic DMRs could be easily assigned to the associated overlapping gene. For intergenic DMRs, we first identified those that intersected UMRs, LMRs, or open chromatin (<500 bp from ATAC-seq peak) as regulatory DMRs (rDMRs; 68% of intergenic DMRs) (Figure 3E). Expressed genes within 100 kb of rDMRs were selected, resulting in 39% functional annotation of all intergenic DMRs. We noted that DMRs are harbored by key genes involved in aging (GeneAge database: https://genomics.senescence.info/genes/, 39 genes, 10%), retinal disease (RetNet database: https://sph.uth.edu/retnet/), 33 genes, 9.5%) (Figure 4A; Table S4), and those associated with environmental factors. These include genes involved in longevity, such as Irs1 and Kl; several genes related to cilia maintenance and retinal dystrophy, such as Bbip1, Bbs2, Bbs7, Bbs10, Ift172, and sdccag8; and a gene highly associated with cigarette smoking, Lrrn3 (Guida et al., 2015; Table S4). Figure 4B shows examples of DMRs in candidate regulatory regions of Irs1 (poised enhancer), Bbs2 (LMR), and Lrrn3 (LMR).
Figure 4. Aging Progression in Rods Correlates with Distinct Patterns of DMRs.
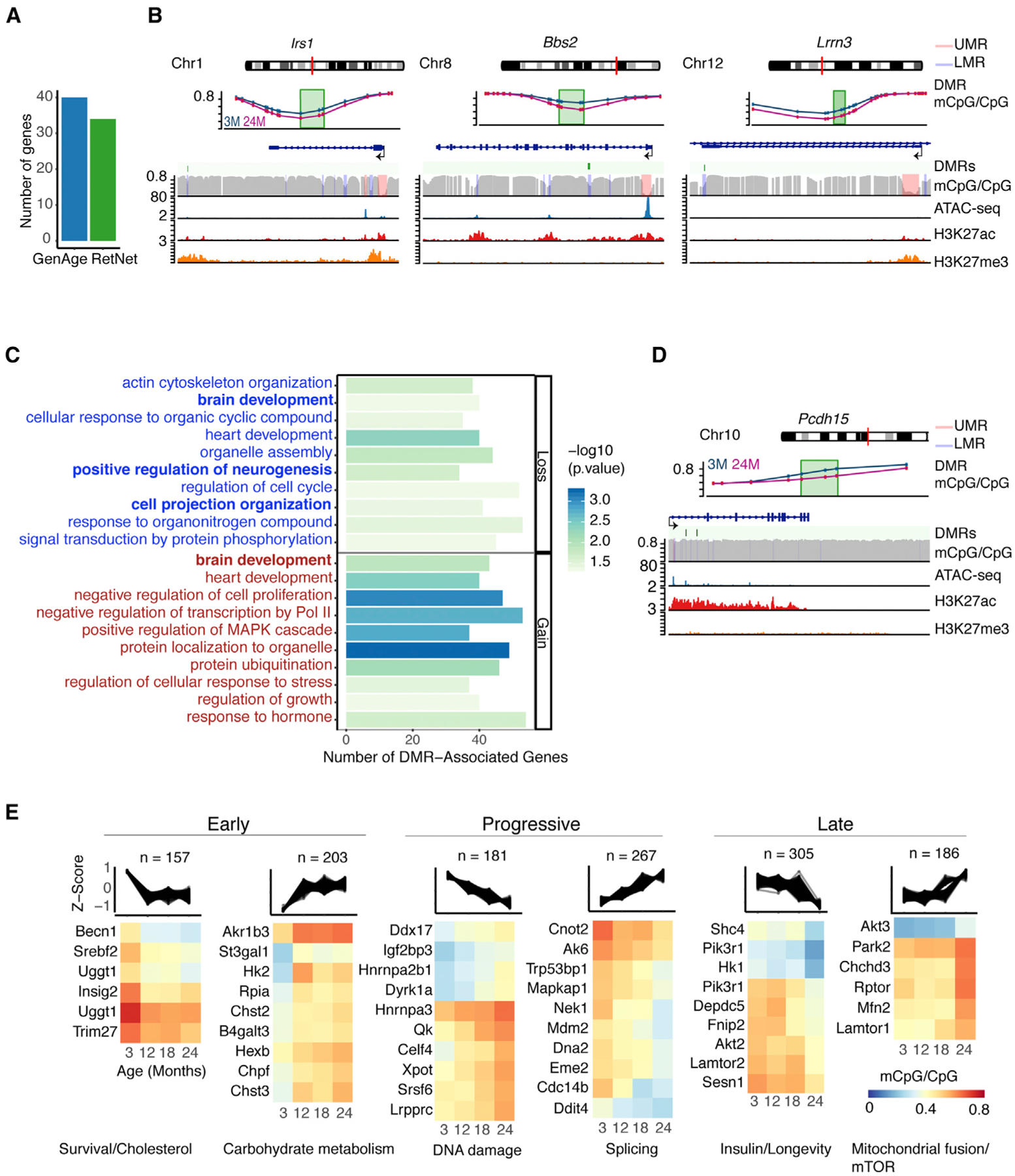
(A) Bar plot showing the number of DMR-associated genes related to aging (as reported in the GeneAge database: https://genomics.senescence.info/genes/) and retinal disease (as reported in the RetNet database: https://sph.uth.edu/retnet/).
(B) Examples of DMRs present in Irs1, Bbs2, and Lrrn3. Methylation levels at DMRs are shown for 3 and 24 months. CpG methylation, ATAC-seq, H3K27ac, and H3K27me3 profiles are shown for the 3-month time point over the respective genes and their surroundings.
(C) GO enrichment of genes associated with age-related DMRs.
(D) DMRs in Pcdh15 similar to those reported in (B).
(E) Age-related rod DMRs showing early, progressive, and late change patterns manually grouped from unsupervised clustering analysis (see Figure S5A). Examples of genes enriched in different pathways after GO analysis using gProfilerR are shown for each pattern of change (see Table S5).
To date, there are no available studies on DNA methylation alterations in the aging retina. However, an analysis of DNA methylation changes in the retinal pigment epithelium (RPE) of individuals with age-associated macular degeneration (AMD) identified hypermethylation at the promoter of glutathione S-transferase 1 (GSTM1) and associated gene expression changes in GSTM1 and GSTM5 involved in detoxification path ways (Hunter et al., 2012). Notably, the promoters of the Gstm2, Gstm5, and Gstm6 genes were all hypermethylated with age in our study (Table S2), suggesting that Gstm genes are highly prone to epigenetic changes in response to homeostatic imbalance in the retina. A global analysis of genes with DNA methylation changes by GO revealed enrichment of genes linked to both intragenic and rDMRs in neuronal function (Figure 4C; Table S5); Figure 4D shows an example of two DMRs harboring H3K27ac in the neuronal gene Pcdh15, which is associated with Usher syndrome (Fuster-García et al., 2018).
To examine whether DNA methylation changes could be correlated to specific stages in aging, we performed unsupervised cluster analysis on all DMRs and identified 30 patterns (Figure S5A) that could be categorized into early, progressive, and late changes (Figure 4E). Surprisingly, most DMRs were detected as early as 12 months regardless of the progression of the change (Figure 4E; Figure S5A). Pathway enrichment analysis using g:Profiler revealed that early and progressive DNA methylation changes were highly associated with neuronal function (Table S5). To examine whether specific differential methylation patterns could be related to rod function, we then evaluated only the DMRs associated with expressed genes. We observed an enrichment of carbohydrate metabolism genes in early DMRs, whereas progressive DMRs included DNA damage and splicing (Figure 4E; Table S5). Insulin/mammalian target of rapamycin (mTOR), neurotrophin signaling, and longevity pathways were enriched in late DMRs (Figure 4E; Table S5). Several of the carbohydrate metabolism genes in the early category, e.g., St3gal1 and Hexb, appear to encode cell-surface proteins, and a few of these are involved in ganglioside biosynthesis (Table S5). Thus, early methylation changes may influence cell adhesion and/or cell-cell communication.
Integration of DMRs with Transcriptome Reveals Dysregulation of Both Established and Unique Aging Pathways in Rods
To assess the potential relationship of DNA methylation changes with gene regulation, we performed RNA-seq analysis of purified rods during aging (Table S1; Figure S1B, 3 months: n = 4, 12–24 months: n = 3) and identified 445 differentially expressed genes (F test, absolute fold change ≥ 1.2, FDR ≤ 0.1) associated with DMRs. Validation of a subset of these genes was performed in sorted rods from a separate group of individuals (Figure S2H). As observed in other genome-wide methylation datasets (Cole et al., 2017; Hahn et al., 2017), the direction of the change in methylation exhibited low correlation with the transcriptional change in aging rods, yet alterations in gene body methylation were more often linked to upregulation of gene expression (Figures S5B and S5C). To capture the biological significance of DNA methylation changes in rods, we then performed an integrated network analysis (Figure S5D) using the proteins encoded by 317 differentially expressed DMR-associated genes that had rod-expressed first-degree interactors from STRING data: https://string-db.org/ (Szklarczyk et al., 2017; see Table S6 for information on the network node and edge list). The resulting network of 3,958 proteins included those encoded by 622 DMR-associated genes and revealed two major modules, both associated with proteostasis: a downregulated hub containing highly connected proteins involved in translation, and an upregulated hub including proteins involved in ubiquitylation and protein degradation (Figure S5D). We further evaluated pathways with a higher proportion of proteins encoded by DMR-associated genes compared with the whole network, which exhibited a DMR/protein ratio of 0.08. Enrichment analysis for a DMR/protein ratio >0.08 and presence of at least two DMRs identified 88 Reactome or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (at FDR ≤ 0.1), corresponding to processes such as proteostasis, energy metabolism, cell communication, and DNA repair (Figure 5A; Table S6). We detected the highest representation of proteins encoded by DMR-associated genes in terms corresponding to carbohydrate metabolism, mitogen-activated protein kinase (MAPK) signaling, and ribosome. In addition to many of these established aging-related pathways, we identified rod-specific aging pathways that include the LKB1-AMPK axis (Samuel et al., 2014), synaptic function, and phototransduction. Thus, altered DNA methylation in rods could be linked to aging hallmarks, as well as specific pathways.
Figure 5. Age-Related Differential Methylation Associates with Expression Changes of Genes Belonging to Aging Pathways.
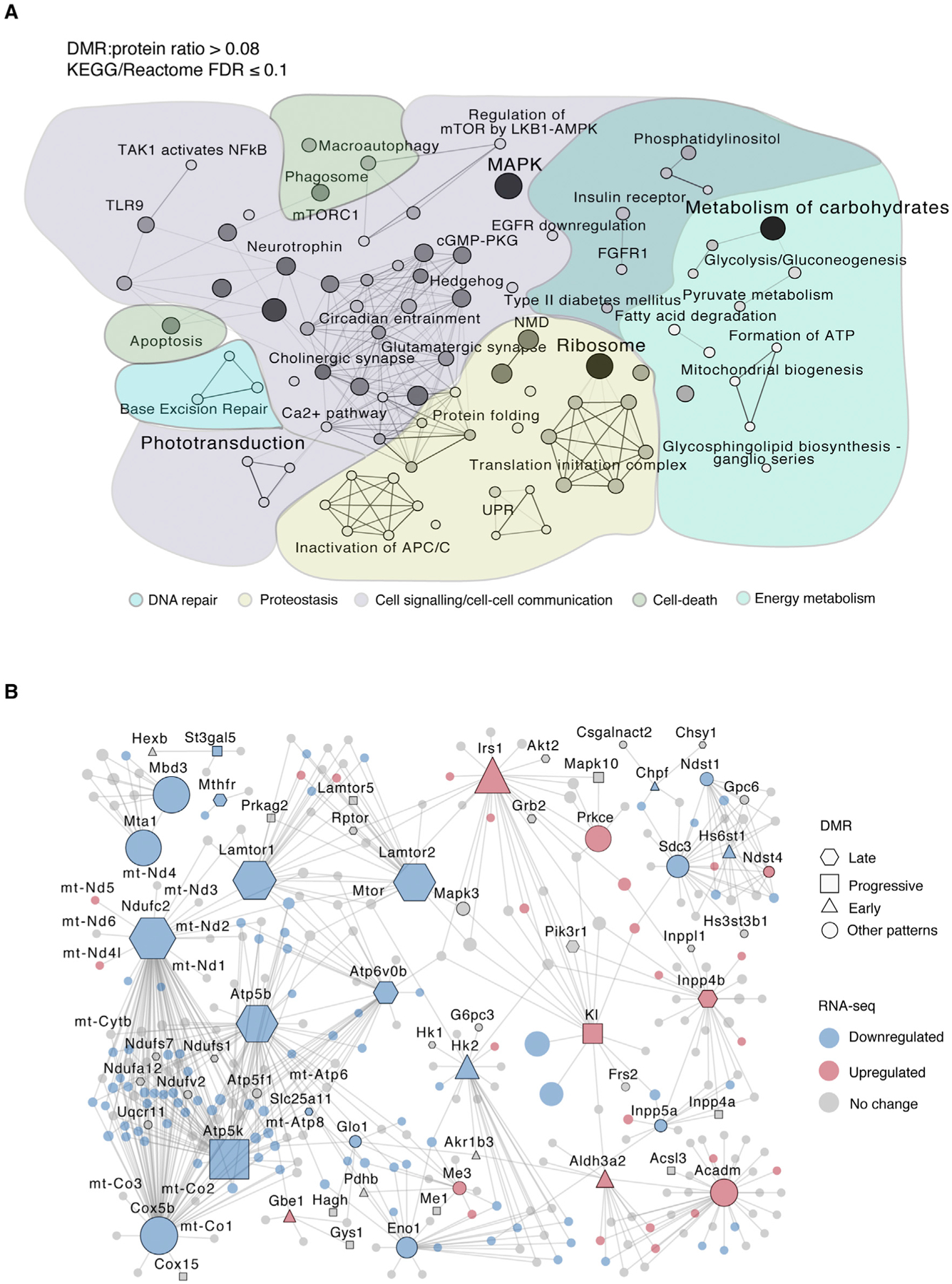
(A) KEGG and Reactome pathways with a higher proportion of DMRs than global average (DMR/protein ratio > 0.08, FDR ≤ 0.1) (see Table S6). Only pathways with at least two DMRs were included. This analysis resulted in 88 pathways summarized using a pathway network with ClueGO for gene similarity calculations based on the entire pathway gene list. Pathways with a kappa score (gene similarity score) of 0.2 or greater are connected. Darker lines represent higher gene similarity scores. Darker node color indicates a higher DMR content.
Aging Rods Show Altered Energy Metabolism
We then specifically examined age-related changes in energy metabolism because of its prominent role in photoreceptor homeostasis and enrichment in our network analysis (see Figure 5A). A subnetwork containing all proteins related to energy metabolism-specific terms, including mitochondrial oxidative phosphorylation (OXPHOS), glycolysis, and insulin signaling, is shown in Figure 5B (see Table S6 for a full list of terms). Importantly, key genes in mitochondrial respiratory chain subunits (complexes I, III, IV, and V) and Acadm, essential for fatty acid beta oxidation, harbored DMRs (Figures 5B and 6A). In addition, we observed a global downregulation of genes related to glucose metabolism, OXPHOS, and tricarboxylic acid (TCA) cycle (Figure S6A), as well as upregulation of genes related to fatty acid metabolism, insulin signaling, and glycogen synthesis (Figure 5B; Figure S6A).
Figure 6. Age-Related Differential Methylation Associates with Alterations in Energy Metabolism.
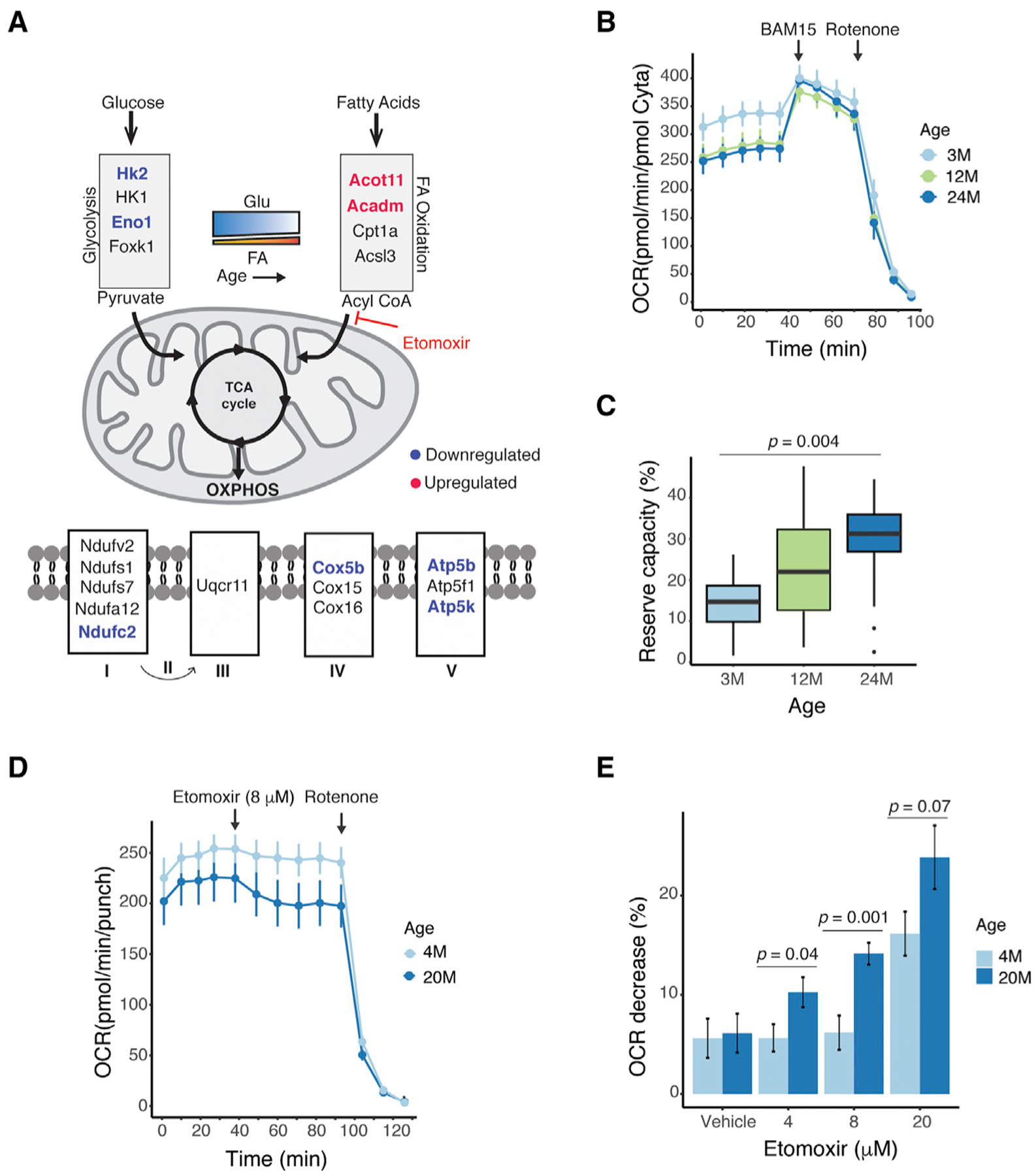
(A) A schematic depicting genes associated with DMRs involved in mitochondrial oxidation. Blue and red represent downregulated and upregulated genes, respectively (absolute fold change [FC] R1.2, FDR % 0.1).
(B) OCR traces from young (3-month-old), middle aged (12-month-old), and old (24-month-old) Nrlp-EGFP mouse retinal punches (n = 11 punches per time point from three individuals). Arrows indicate the injection of a mitochondrial uncoupler (BAM15) or complex I inhibitor (rotenone) in the sample well. Error bars are ± SEM.
(C) A lower basal respiration and a larger mitochondrial reserve capacity can be observed in older mice as defined by 100 × (maximal respiration basal respiration)/maximal respiration. The mitochondrial reserve capacity was compared by one-way ANOVA (post-hoc Tukey 3M vs 24M, p = 0.004). Error bars are ± SEM.
(D) OCR traces from young (4-month-old) and old (20-month-old) Nrlp-EGFP mouse retinal punches after adding etomoxir (8 μM) (n = 7 punches for 4 months; n = 9 punches for 20 months from two individuals each). Error bars are ± SEM.
(E) OCR decrease compared with baseline after adding different concentrations of etomoxir (n = 7–9 punches per condition from two individuals each). OCR decrease was compared by Student’s t test. Error bars are ± SEM. p = 0.8 (vehicle), p = 0.04 (4 μM), p = 0.001 (8 μM), p = 0.07 (20 μM).
BAM15, (2-fluorophenyl) (6-[(2-fluorophenyl) amino] (1,2,5-oxadiazolo[3,4-e] pyrazin-5-yl)) amine; FA, fatty acid; Glu, glucose; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation.
To directly assess whether these alterations have any functional consequence, we measured mitochondrial respiration in mouse retina punches ex vivo using the XF analyzer (Seahorse Bioscience) microplate-based assay. As shown earlier (Kooragayala et al., 2015), the mature 3-month retina exhibited relatively low (~15%) mitochondrial reserve capacity, which primarily indicated a very high demand for ATP from mitochondrial OXPHOS that was close to the maximal possible rate of ATP synthesis; however, basal respiration was reduced and retinal reserve capacity increased to ~30% at 12 and 24 months of age, suggesting a lower energy consumption (and therefore lower ATP requirement from OXPHOS) with age (Figures 6B and 6C). Because maximal mitochondrial respiration is expected to reflect the content of OXPHOS enzymes, normalization to cytochrome a content resulted in the same peak respiration values for all three ages. Unnormalized maximal respiratory rates (data not shown) showed a decrease of ~12% in maximal respiration between 3 and 24 months, suggesting only a slight decrease in the content of OXPHOS complexes with age. Indeed, when analyzing wild-type mouse retinas not expressing GFP (Figures S6B and S6C), a similar pattern was observed in which only basal (but not maximal) respiration decreased, resulting in a higher mitochondrial reserve capacity. Furthermore, higher expression of a subset of genes related to fatty acid detoxification and beta oxidation in rods (Figures 6A and S6A) indicated an increased utilization of fatty acids as a source of energy at an older age. We thus tested the dependency of fatty acid as a fuel for the TCA cycle by blocking the transport of endogenous long-chain fatty acids into the mitochondria using etomoxir (ETO), a specific inhibitor of carnitine palmitoyl-transferase 1A (Cpt1a). Older retinas displayed a dose-dependent reduction in oxygen consumption rate (OCR) upon ETO treatment, and thus a significant dependency on fatty acid as a fuel source, whereas younger retinas did not respond to the drug at lower doses (Figures 6D, 6E, and S6D–S6F). Taken together, these data suggest that older rods display an altered oxidative metabolism linked to DNA methylation changes.
DISCUSSION
Molecular and functional alterations during mammalian aging are progressive, subtle, stochastic, and thereby difficult to dissect. Transcriptomic studies have begun to elucidate global and tissue-specific biological pathways that can in turn be correlated to physiological changes in aging (Benayoun et al., 2019; Parapuram et al., 2010; Park et al., 2009; Stegeman and Weake, 2017). The impact of aging on organismal, tissue, or cellular functions may be largely mediated by changes in the epigenome (Booth and Brunet, 2016); however, most genome-wide epigenetic studies have used tissues with mixed cell populations that could mask cell-type-specific changes. As of yet, no whole-genome DNA methylation study with nucleotide resolution has been performed in purified sensory neurons at multiple stages of aging. Here, we report genome-wide profiles of DNA methylation in purified rod photoreceptors at four stages of aging of the mouse retina and correlate aging DMRs to gene expression changes and biological pathways, which in turn might be relevant for aging-associated retinal dysfunction. Through integration of different datasets, our studies have uncovered a crosstalk between epigenetic alterations and aging hallmarks, highlighting a link between DNA methylation and rod photoreceptor metabolism.
The previously reported multi-tissue epigenetic clocks (Meer et al., 2018; Stubbs et al., 2017) were trained on a limited number of tissues and did not apply to rod photoreceptors, possibly because of CpGs with rod-specific methylation levels. Alternatively, lower coverage of some CpG clock sites in our WGBS data could lead to discordant values. We note that DNA arrays and reduced representation bisulfite sequencing (RRBS) methods employed to develop epigenetic clocks and common methylation screenings do not cover most of the cell-type-specific enhancers, which are usually located in regions of low CpG density (Stadler et al., 2011).
DNA methylation changes are strongly associated with biological age, but we have limited understanding of how these alterations are correlated with cellular function. Some WGBS studies have shown that DNA methylation changes in normal aging cells are modest and can occur at cell-type-specific regulatory regions (Cole et al., 2017; Sun et al., 2014), and unlike in senescent and cancer cells (Cruickshanks et al., 2013), may not affect the integrity of heterochromatic domains. Epigenetic changes at distal regulatory elements have been reported in liver (Cole et al., 2017) and pancreatic beta cells during aging (Avrahami et al., 2015). Our genome-wide profiling demonstrated that age-related DMRs are localized in regions of lower methylation levels and enriched in rod-specific regulatory factors, and thus epigenomic alterations associated with age may represent aberrations in stringently controlled transcriptional programs required for rod homeostasis. Importantly, DNA methylation changes often overlapped with boundaries of open chromatin peaks in our study. We can therefore hypothesize that epigenomic alterations during rod aging can modulate boundaries of accessible chromatin regions and impact finely tuned gene expression patterns, as reported previously in studies on yeast (Chai et al., 2013).
An intriguing observation in our study is that aging-related DNA methylation changes in mouse rods aggregated at specific chromosomal clusters or hotspots, and that many hotspots appeared to be unique to rods. In addition to the hotspot on chromosome 14 harboring the rod differentiation factor Nrl, 1 of the 8 DMR hotspots on chromosome 10 is centered on Foxo3 and localized in a syntenic 7-Mb region involved in longevity (Donlon et al., 2017). Variations at the human Foxo3 locus are suggested to affect chromatin looping and the response of interacting genes to oxidative stress (Donlon et al., 2017). We also identified a DMR in Foxo3 itself, suggesting that epigenomic changes in rods, including those driven by UV or oxidative stress, could influence the stability of this longevity locus. Indeed, one of the genes within this locus, Prdm1, which is associated with stabilization of photoreceptor cell fate (Brzezinski et al., 2010), was differentially expressed with aging in rods. Other chromosomal hotspots are also syntenic with human regions susceptible to disease. For example, hotspot b in Figure 2C is syntenic with human 6q22, which is associated with intellectual disability (Mackenroth et al., 2015) and cancer (Kirchhoff et al., 2009). Hotspot h, syntenic to human 12q14–21, is enriched in synaptic genes and is highly unstable in humans; deletions in this region cause neurodevelopmental disorders and cancer (Andrieux et al., 2002; Rajakulendran et al., 2013; Schneider et al., 2003). In addition, Atxn7l3b and Cct2 genes, which are located in hotspot h, were dysregulated with age and carry mutations associated with retinal degeneration (Minegishi et al., 2016; Tan et al., 2014). Some of the hotspots located in the distal region of chromosome 10, including hotspot h, are shared with other neural cell types, suggesting that DNA methylation can accumulate in regions involved in cell-type-specific functions. We propose that DMR hotspots represent transcriptional hubs that accumulate epigenomic changes with age and modulate responses to stressors, as proposed for the longevity interactome (Donlon et al., 2017).
Integration of DMRs with RNA-seq data and subsequent network analyses revealed dysregulation of both established and rod-specific biological pathways. Notably, 19.8% of rod aging DMRs are associated with subtle but significant changes in gene expression, a higher association than previously observed during aging in whole tissues (Cole et al., 2017; Hahn et al., 2017; Yuan et al., 2015). The most connected pathways identified by protein network analysis represent genes involved in known altered aging pathways, such as metabolism of carbohydrates, MAPK, and proteostasis. Notably, we detected decreased expression of genes related to protein translation and increased expression in those related to protein degradation, a phenome non observed in various aging organisms (López-Otín et al., 2013) and linked to DNA methylation changes in our study.
Given the high energy requirements and metabolic activity in photoreceptors, we were specifically attracted by DMRs and the correspondingly reduced expression of genes belonging to glycolysis and mitochondrial respiration during rod aging. We detected methylation changes in genes related to glucose metabolism and those related to longevity during late stages of rod aging, indicating their contribution to age-related disease susceptibility. Interestingly, DNA methylation changes in Irs1, a part of insulin signaling upstream of glucose metabolism path ways, occurred early in the aging process. Aerobic glycolysis is uniquely critical for maintenance of rod photoreceptors (Chinchore et al., 2017; Hurley et al., 2015). Outer segments of photoreceptors become shorter with age (Cunea and Jeffery, 2007), and thus whether reduced aerobic glycolysis is correlated with this aging-related phenotype requires further exploration. Notably, Hk2, encoding an enzyme crucial for the first step of glycolysis and rod functioning (Petit et al., 2018), was located within a DMR hotspot in addition to harboring a DMR itself, suggesting that Hk2 may be highly susceptible to epigenomic changes that could contribute to its dysregulation.
In our study, alterations in mitochondrial oxidative metabolism were revealed by age-related changes in both DNA methylation and expression of genes associated with mitochondrial respiratory chain and beta oxidation of fatty acids. These changes were consistent with our ex vivo measurements showing an increased mitochondrial reserve capacity and fatty acid dependency. Our results thus suggest that changes in age-related DNA methylation are linked to reduced mitochondrial respiration and increased reliance of beta oxidation with age, and points to alterations in lipid homeostasis. In concordance, we found that fatty acid degradation and glycosphingolipid biosynthesis pathways were enriched in our protein interaction network. Similarly, a recent study showed that DNA methylation changes in a gene encoding the enzyme ELOVL2, involved in elongation of long-chain polyunsaturated fatty acids, plays a role in loss of mouse retinal function during aging (Chen et al., 2020). Furthermore, genes related to peroxisome beta oxidation, including Abcd2, Abcd3, Akt2, and Ppara, also presented DNA methylation changes. Consistent with upregulation of beta oxidation and subsequent generation of reactive oxygen species, we observed DNA methylation changes in genes involved in detoxification pathways, including the aldehyde dehydrogenase Aldh3a2, oxysterol binding proteins Osbp3/8/9/10, and glutathione biosynthesis enzymes Gstm2/5/6. Future studies should thus focus on the relationship between DNA methylation and lipid homeostasis in the aging retina.
One important limitation of our study is the inability to distinguish the mechanistic role of the DNA methylation changes identified. Whether these changes are detrimental, beneficial, or just bystanders requires further studies. However, DMRs occur at regulatory regions and associate with non-random gene expression changes and specific aging pathways, indicating their relevance to alterations in rod function with advancing age. In concordance, defects in enhancer DNA methylation are shown to result in aging phenotypes and impaired energy homeostasis in mice (Schäfer et al., 2018). In this study we used male mice to avoid sex-related confounding factors. Further investigations are needed to evaluate whether the dynamics of DNA methylation patterns with age is different in females.
In brief, our studies demonstrate that age-related epigenetic alterations in rods are linked to aging hallmarks, including neuronal communication, proteostasis, and mitochondrial dysfunction (López-Otín et al., 2013). Specifically, the DNA methylome of aging rods uncovered changes in oxidative metabolism that may contribute to (or reflect) rod functional decline with age. DNA methylation changes in gene bodies, distal regulatory regions, and chromosomal neighborhoods are likely associated with disruptions in stringently controlled patterns of gene expression, rendering cells susceptible to stressors that can precipitate disease at an advanced age.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anand Swaroop (swaroopa@nei.nih.gov). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All procedures involving mice were approved by the Animal Care and Use Committee (NEI-ASP#650). C57BL/6J mice expressing EGFP under the control of the Nrl promoter (Nrlp-EGFP mice) (Akimoto et al., 2006) were used to purify rod photoreceptors and to perform mitochondria oxygen consumption assays in the retina. Wild-type aging mice (C57BL/6J) were obtained from the National Institutes of Aging (MD, USA). Mice were kept in a 12 light/12 dark hour cycle and fed ad libitum at the NEI animal facility. All animals used were male. WGBS: n = 3 for 3M, 18M, 24M; n = 2 for 12M; RNA-seq: n = 4 for 3M and n = 3 for 12M, 18M and 24M; H3K27me3: n = 6 for 3M; H3K27ac: n = 4 for 3M. Mitochondrial oxygen consumption rate: n = 3 per time-point; inhibitor experiments: n = 2 per drug treatment.
METHOD DETAILS
Isolation of rod photoreceptors
Retinas from 3M, 12M, 18M and 24M male mice were dissected in Hank’s Balanced Salt Solution (Life Technologies, NY, USA) and dissociated in Papain solution (Worthington Biochemical, NJ, USA) containing DNase I at 28°C for 8 min with constant agitation, pelleted at 200 g for 5 minutes and resuspended in a solution containing Albumin and DNase I (100 U/ml). Subsequently, cells were pelleted again and resuspended in 1 mL of HBSS. EGFP positive cells were isolated by fluorescence-activated cell sorting (FACS) using FACS Aria II (Becton Dickinson, CA, USA). The purity of isolated EGFP-positive cells was assessed by resorting and only cells that were over 98% pure were used in subsequent experiments. After sorting, cells were flash frozen with or without TRIzol (Invitrogen, CA, USA).
Mitochondria oxygen consumption rate (OCR) assay
Oxygen consumption rate was measured in 1 mm retina punches from 3-month old (M) (n = 11), 12M (n = 11) and 24M (n = 11) Nrlp-EGFP mice using Seahorse XF24 Bioanalyzer (Seahorse Bioscience, North Billerica, MA), as described previously (Kooragayala et al., 2015). Basal oxygen consumption was established after 5 measurements, and then 4 μm of mitochondrial uncoupler (2-fluorophenyl) (6-[(2-fluorophenyl) amino] (1,2,5-oxadiazolo[3,4-e] pyrazin-5-yl)) amine (BAM15) (Timtec, Newark, DE) was added to measure maximum oxygen consumption. Cytochrome a quantification was performed for normalization (Kooragayala et al., 2015) using 6 retinas per time-point. Validation of OCR differences was performed in a wild-type C57BL/6 colony by comparing 4–5M (n = 9) with 23–24M (n = 11). The mitochondrial reserve capacity was defined as 100*(Maximal OCR-Basal OCR)/Maximal OCR. The basal OCR and mitochondrial reserve capacity were compared for significance between different time-points as described (Kooragayala et al., 2015). The inhibitor Etomoxir (ETO) (Agilent Seahorse XF Mito Fuel Flex Test Kit 103260–100) was used to block translocation of long chain fatty acid into mitochondria. Dependency of fatty acid as a fuel in 4M and 20M Nrlp-EGFP mice was calculated by the percentage in OCR decrease after addition of Etomoxir and compared for significance between 4-and 20-month old mice. Vehicle (4M n = 9, 20M n = 6), Etomoxir 4 μM (4M n = 8, 20M n = 8), Etomoxir 8 μM (4M n = 7, 20M n = 9), Etomoxir 20 μM (4M n = 8, 20M n = 7).
WGBS
Genomic DNA was extracted using the PureLink Genomic DNA Mini Kit (Invitrogen, CA, USA) per manufacturer instructions and treated with bisulfite salt using the EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA) on 100 ng of DNA following manufacturer instructions. Libraries were generated from 3M, 12M, 18M and 24M mice (n = 3) using the Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences, MI, USA) according to manufacturer’s instructions. A spike-in of unmethylated lambda DNA was used to determine bisulfite conversion. Paired-end sequencing was performed using the HiSeq 2500 platform (Illumina, CA, USA) with read-length of 125 base pairs. Adapters, reads of low quality (Phred score < 20) and length < 20 bp were trimmed using Trim Galore! version 0.4.5 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore) with the following parameters as suggested by the library kit manufacturer:-clip_R1 10-clip_R2 20-three_prime_clip_R1 10–three_prime_clip_R2 15. Quality control was performed using FastQC version 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). After removal of duplicate reads, Bismark version 0.19.1 (Krueger and Andrews, 2011) was used to align reads to the mouse reference genome Ensembl release 84 (GRCm38.p4) with mapping efficiency between 78.5%–83.7%. CpG methylation was extracted using Bismark and values smoothened by applying a smoothing algorithm with the R package bsseq version 1.14.0 (Hansen et al., 2012). After inspection of M-bias plots, reads were further trimmed 20 base pairs from either end of both reads. One sample from 12M mice was excluded from further analysis as it did not pass the conversion efficiency cutoff of > 99%. External datasets analyzed include wild-type rods, cones and Nr2e3‒/‒ rods (Mo et al., 2016), neural precursor cells (NPC) and embryonic stem cells (ES) (Stadler et al., 2011), cerebellum (Hon et al., 2013), beta cells (Avrahami et al., 2015), liver cells (Cole et al., 2017), and frontal cortex (Lister et al., 2013).
Cleavage under targets and release using nuclease (CUT&RUN)
Flow-sorted rod photoreceptors from 3M, 12M, 18M and 24M male mice (H3K27ac: n = 4, H3K27me3: n = 6) were cryopreserved in a solution of HBSS containing 10% DMSO and slowly frozen using a Mr. Frosty container (Invitrogen, CA, USA). CUT&RUN was performed as previously described (Skene and Henikoff, 2017) using 200–300,000 cells per experiment. Antibodies against H3K27ac (Rabbit, cat.no. ab4729, Abcam, Cambridge, UK), and H3K27me3 (mouse IgG3, cat.no. ab6002, Abcam, Cambridge, UK) were used at a concentration of 1:100 in 100 μl and pA-MNase conjugated to protein A (generous gift of Dr. Steven Henikoff, Howard Hughes Medical Institute, Washington, USA) used at a concentration of 700 ng/ml. Released fragments were purified using QIAquick PCR Purification Kit (QIAGEN, Hilden, Germany). Libraries were generated using SMARTer® ThruPLEX® DNA-Seq Kit (Takara Bio USA, Inc, CA, USA) as per manufacturer instructions with 15 PCR cycles and 60°C extension, and sequenced pair-end using the HiSeq 2500 platform (Illumina, CA, USA) with read-length of 50 base pairs. Reads were quality trimmed 10 bp from the 5ʹend and 5 bp from the 3ʹend using Trim Galore! version 0.4.5. Quality check was performed using FastQC. Reads were aligned against the Mus musculus genome (Ensembl version 84) and the Saccharomyces cerevisiae genome (Ensembl version 93) using bowtie2 version 2.3.4.1 (Langmead and Salzberg, 2012) with additional parameters as detailed in the CUT&RUN protocol (Skene and Henikoff, 2017). Deduplicated and high-quality mapped reads (MAPQ value of at least 30) were extracted using samtools version 1.9 (Li et al., 2009). Cross-correlation analysis implemented in the R package csaw (Lun and Smyth, 2016) was used to determine the enrichment efficiency of each replicate. The deep Tools version 3.1.1 suite was used to generate ‘fingerprint’ plots with an IgG control as a pseudo-input. Visual inspection on IGV (Robinson et al., 2011) of enrichment at positive control regions (i.e., promoters of highly expressed genes for H3K27ac and unexpressed developmental genes for H3K27me3) was also used as a quality control measure. Pearson’s correlation coefficients of high read count bins (identified using csaw) was used to measure similarity between samples. A combination of these quality control measures was used to remove low-quality replicates from further analysis. The samples were TMM normalized against the yeast spike-in as described in the csaw documentation. Read counts were binned into 1 kb windows across the mouse and yeast genomes separately, and a global average read count was determined using the median read count of 5 kb windows across both genomes. Bins were considered enriched if they contained at least two times the global average. TMM normalization was performed on the filtered spike-in bins, and the resulting normalization factors were applied to the endogenous (mouse) data. Normalized read counts were then written to bedgraph files, and biological replicates were pooled for downstream analysis.
RNA-seq
Total RNA from FACS-purified rod photoreceptors was extracted using TRIzol® (Invitrogen, Carlsbad, CA), treated with DNase and cleaned up using the MagMAX mirVana Total RNA Isolation Kit (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions. Libraries were constructed with SMARTer Stranded Total RNA-Seq Kit v2 – Pico Input Mammalian (Takara Bio USA, Mountain View, CA) with 4 ng of RNA and 13 PCR cycles library amplification. Paired-end reads of 125 base pairs were obtained using the HiSeq 2500 platform (Illumina, San Diego, CA). Sequence reads passing chastity filtering were trimmed for Illumina adapters, polyA, and polyT sequences using Trimmomatic v0.36 (Bolger et al., 2014) with the following settings: ILLUMINACLIP:Tru-Seq3-PE-2.fa:2:30:10:1:TRUE HEADCROP:3 TAILCROP:3 MINLEN:42.
Quantitative real-time PCR (qPCR)
RNA from flow-sorted rod photoreceptors (n = 4 for 3M; n = 3 for 24M) was extracted using RNeasy Plus Micro Kit (QIAGEN, Hilden, Germany). Complementary DNA was synthesized using SuperScript II reverse transcriptase (Life Technologies, Carlsbad, CA, USA) according to manufacturer instructions. Quantitative PCR was performed in a QuantStudio 3 instrument (Applied Biosystems, Foster City, CA, USA) using PowerUp SYBR Green master mix (Applied Biosystems, Foster City, CA, USA). Data were analyzed using the delta-delta CT method normalizing against Hnrnpd.
Visualization
All plots and graphs were visualized using any of the following R packages: ggplot2 version 3.2.1 (https://github.com/tidyverse/ggplot2), ComplexHeatmap version 1.17.1 (Gu et al., 2016), pheatmap version 1.0.10 (https://cran.r-project.org/web/packages/pheatmap/index.html), Gviz version 1.22.3 (Hahne and Ivanek, 2016), eulerr version 5.1.0 (https://cran.r-project.org/web/packages/eulerr/index.html), circlize version 0.4.5 (Gu et al., 2014), and karyoploteR version 1.2.2 (https://bioconductor.org/packages/release/bioc/html/karyoploteR.html)
QUANTIFICATION AND STATISTICAL ANALYSIS
Identification of unmethylated regions (UMRs) and low methylated regions (LMRs)
The R package methylSeekR version 1.16.0 (Burger et al., 2013) was used to identify UMRs and LMRs from the pooled unmethylated and methylated read counts of the 3M biological replicates (n = 3). In order to determine if the dataset contained partially methylated domains (PMDs), a hidden Markov model was used to identify such regions, and the quality control measures included in the package were inspected. It was discovered that the rods, as well as all of the other analyzed datasets, showed no signs of harboring large scale PMDs. UMRs and LMRs were subsequently identified without the PMD identification step.
Epigenetic clock analysis
To determine epigenetic age according to Stubbs et al. (2017), the script ‘toRun_Imputation.R’ from https://github.com/EpigenomeClock/MouseEpigeneticClock was used with default parameters and the coverage files output from Bismark. To determine epigenetic age according to Meer et al. (2018), smoothed methylation percentages (regardless of coverage) of the clock CpGs were multiplied by their corresponding weights and summed. Predicted epigenetic age was regressed against chronological age using the base R function lm, and the p-value and correlation coefficient were found from the model summary statistics.
Identification of differentially methylated regions (DMRs)
CpGs with at least 2x coverage in at least 2 biological replicates of each time point were used for analysis. The R package bsseq version 1.14.0 was used to identify differential methylation between 3M and 24M sample (n = 3) using a cutoff of > 10% methylation difference, areaStat > 20, and a quantile cutoff of the t-statistics of 0.001. The same process was used to identify DMRs in aging datasets that had separate biological replicates (liver and beta cells) (Avrahami et al., 2015; Cole et al., 2017). For datasets that performed WGBS on pooled biological replicates (frontal cortex) (Lister et al., 2013), DMRs were identified using the DSS-single statistical method of the R package DSS (Wu et al., 2015) on CpGs with at least 4x coverage. The same statistical cutoffs were applied regardless of the DMR calling method.
Motif enrichment analysis
For UMRs and LMRs, the AME tool from MEME Suite (McLeay and Bailey, 2010) was used to determine enrichment of transcription factor motifs (TRANSFAC release 2017.3) against a shuffled background. For DMRs, the AME tool from MEME Suite was used to determine enrichment of transcription factor motifs (TRANSFAC release 2017.3) against a background of randomly selected 200 base pair regions of the mouse genome with similar CpG content as the DMRs. Enriched motifs were grouped into families according to TRANSFAC’s classification.
DMR Hotspot Modeling
Finding covariates
The genome was binned into 1Mb windows, and the number of DMRs, CpGs, reads, and genes in each of these bins was determined. Using generalized linear modeling implemented in the R package MASS, a negative binomial model of DMR count as a function of any combination of the number of CpGs, reads, and genes was created. The significant covariates of the model with the lowest Akaike information criterion (AIC) were selected for further modeling when defining enriched hotspots. Hotspot defining. A region-growing algorithm was implemented to identify regions of variable length that contained a high density of DMRs. The average distance between DMRs was determined, and DMRs within this distance from the following DMR were considered to be in hotspots. This algorithm yielded many regions of above-average DMR density. Hotspot enrichment. Each potential hotspot was tested in the following method to determine if it contained more DMRs than expected. 1000 random regions of the same length of the hotspot were chosen, and the number of DMRs, CpGs, and genes within them were determined. The DMR count as a function of CpGs and genes within these regions was modeled as a Poisson distribution, a negative binomial distribution, a hurdle negative binomial distribution, and a zero-inflated negative binomial distribution [models were created using the R packages stats version 0.1.0 (https://cran.r-project.org/web/packages/STAT/index.html), MASS version 7.3–50 (https://cran.r-project.org/web/packages/MASS/index.html) and countreg version 0.2–1 (https://r-forge.r-project.org/R/?group_id=522)]. The model with the lowest AIC was chosen to predict the expected number of DMRs within the hotspot based on the number of CpGs and genes within the hotspot. Using the predicted number of DMRs and other parameters of the chosen model, the appropriate probability density function determined the probability of observing the actual number of DMRs within the hotspot. The p-values were adjusted using the Benjamini-Hochberg method, and hotspots with an FDR of at most 0.01 were selected.
Network Analysis
Protein-Protein Interaction Network
Differentially expressed genes associated with DMRs (i.e., genes that overlap DMRs or expressed genes with TSSs within 100 kb of an rDMR) were selected using an absolute fold-change cutoff of 1.2 and an FDR of 0.10. First-order interacting partners of the corresponding gene products were determined using the STRING database (version 11.0) with a combined score cutoff of at least 700. Proteins encoded by genes that were not expressed at any time point were removed from the network. The network was visualized on Cytoscape (Shannon et al., 2003), and the topological analysis was performed using the built-in tool Network Analyzer. Nodes were clustered using the ‘Edge-weighted Spring Embedded Layout’ option. Pathway network generation. KEGG and Reactome pathway enrichment of the entire network was performed using gProfileR version 0.6.7 (Reimand et al., 2007) with 10% FDR. The DMR-to-gene ratio of each pathway was defined as the number of differentially expressed genes associated with DMRs in each pathway divided by the number of overlapping genes in the pathway. The global DMR-to-gene ratio was defined as the total number of differentially expressed genes associated with DMRs to all genes in the network. Any pathway with a DMR-to-gene ratio greater than the global DMR-to-gene ratio was selected for further analysis. The pathways were visualized on Cytoscape version 3.7.1 (https://cytoscape.org/), and the plug-in ClueGO version 2.5.4 (Bindea et al., 2009) was used to calculate the gene similarity kappa score between each pathway.
DMR enrichment
Every tested CpG and every CpG within a DMR was categorized into any of the following regions: promoter, intron, exon, intergenic, CpG island, CpG shore, and LINE/SINE. The odds ratio [implemented in the R package questionr version 0.7.0 (https://cran.r-project.org/web/packages/questionr/index.html)] was calculated on a 2×2 contingency table of CpG counts within each genomic region.
DMR Clustering
The z-score of the methylation levels of each DMR across each time point was calculated using the scale function in base R. Unsupervised clustering of the negative squared distances of z-score values was performed using the R package apcluster version 1.4.7 (Bodenhofer et al., 2011). Clusters were visualized as line plots of z-score values and were manually selected for inclusion in the early, progressive, and late groups.
RNA-seq analysis
Gene-level expression analysis was performed as previously described (Chen et al., 2016) using ENSEMBL v84 annotation (Aken et al., 2017). Genes that were expressed at or above 5 counts-per-million (CPM) in all replicates of at least one time point were used for normalization and differential expression analysis as described (DiStefano et al., 2018), using 3M time point as the reference. Since aging is a subtle phenotype, relatively low absolute cutoffs of fold-change of 1.2 and FDR of 0.1 were used to identify biologically-relevant differentially-expressed genes.
OCR comparisons
OCR measurements between 3M, 12M and 24M were compared using one-way ANOVA using R version 3.4.1 (http://www.R-project.org/). Mitochondrial reserve capacity and treatments with Etomoxir were compared using unpaired two-tailed t test in R.
qPCR analysis
Gene expression levels were analyzed by the delta-delta CT method and differences between 3M and 24M were compared using unpaired two-tailed t test in R.
DATA AND CODE AVAILABILITY
All raw and processed data have been deposited to Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/GEO) with accession GSE134873 and to https://neicommons.nei.nih.gov/#/. In-house codes are deposited at https://github.com/NEI-NNRL/2020-DNA_methylation_signatures_of_aging_in_rods
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Abeam | Cat# ab4729; RRID:AB_2118291 |
| H3K27me3 | Abeam | Cat# ab6002; RRID:AB_305237 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| pA-MNase conjugated to protein A | Steven Henikoff’s Lab | |
| TRIzol® | Invitrogen | Cat# 15596026 |
| MagMAX mirVana Total RNA Isolation Kit | Applied Biosystems | Cat# A27828 |
| Papain | Worthington Biochemical | Cat# LS003118 |
| Superscript II reverse transcriptase | Invitrogen | Cat# 18064 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25741 |
| Etomoxir (Seahorse XF Mito Fuel Flex Test Kit) | Agilent | Cat# 103260 |
| BAM 15 | Timtec | Cat# ST056388 |
| Rotenone | Sigma | Cat# R8875 |
| Critical Commercial Assays | ||
| PureLink Genomic DNA Mini Kit | Invitrogen | Cat# K182001 |
| EZ DNA Methylation-Gold Kit | Zymo | Cat# D5005 |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Cat# 30024 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28104 |
| SMARTer® ThruPLEX® DNA-Seq Kit | Takara Bio | Cat# R400674 |
| SMARTer Stranded Total RNA-Seq Kit v2- Pico Input Mammalian | Takara Bio | Cat# 634411 |
| Oligonucleotides | ||
| Prdm1 Forward ATTAAGCCTATCCCTGCCAAC | This paper | N/A |
| Prdm1 Reverse CTACTGTATTGCTTTGGGTTGC | This paper | N/A |
| Hk2 Forward TCAAAGAGAACAAGGGCGAG | This paper | N/A |
| Hk2 Reverse AGGAAGCGGACATCACAATC | This paper | N/A |
| Irs1 Forward GGATCGTCAATAGCGTAACT | This paper | N/A |
| Irs1 Reverse GAAATAGTTCGAGTCTGGGT | This paper | N/A |
| Hnrnpd Forward CTCCAGACACACCTGAAGAAA | This paper | N/A |
| Hnrnpd Reverse CAGAACCCACGCCTCTTATT | This paper | N/A |
| Plekh2 Forward AGAGACAAGTTATCGATGCAGAG | This paper | N/A |
| Plekh2 Reverse ACTGGTCTGAATATTAGCTGCTT | This paper | N/A |
| Prok1 Forward GCCTGCGGTTGTGTACC | This paper | N/A |
| Prok1 Reverse AGGTATGGTGTTGGCGTTTC | This paper | N/A |
| Immpl2 Forward AAATTGGGCGAGGGTACAATA | This paper | N/A |
| Immpl2 Reverse AGGCCTTGAAGCATCTTCTC | This paper | N/A |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE134873; https://neicommons.nei.nih.gov/#/. |
| Retina topological domains | Norrie et al., 2019 | https://pecan.stjude.cloud/retinalnucleome |
| ATAC-Seq data in mouse rods | Mo et al., 2016 | GEO: GSE72550 |
| Nrl ChIP-Seq in mouse rods | Hao et al., 2012 | https://datashare.nei.nih.gov/nnrlMain.jsp |
| Crx ChIP-Seq in mouse rods | Corbo et al., 2010 | GEO: GSE20012 |
| WGBS in mouse rods, cones, and rd7 model | Mo et al., 2016 | GEO: GSE72550 |
| WGBS in mouse ES cells and NPCs | Stadler et al., 2011 | GEO: GSE30202 |
| WGBS in mouse hepatocytes | Cole et al., 2017 | GEO: GSE89274 |
| WGBS in mouse beta cells | Avrahami et al., 2015 | GEO: GSE68618 |
| WGBS in mouse frontal cortex | Lister et al., 2013 | GEO: GSE47966 |
| WGBS in mouse cerebellum | Hon et al., 2013 | GEO: GSE42836 |
| Mouse genome assembly GRCm38.p4 | Ensembl | http://mar2016.archive.ensembl.org/Mus_musculus/lnfo/lndex |
| Experimental Models: Organisms/Strains | ||
| Nrlp-GFP | Akimoto et al., 2006 | N/A |
| C57BL/6J | National Inst of Aging | https://www.nia.nih.gov/research/dab/aged-rodent-colonies-handbook |
| Software and Algorithms | ||
| R version 3.4.4 | R Core Team | https://www.r-project.org/ |
| samtools version 1.9 | Li et al., 2009 | http://www.htslib.org/ |
| deepTools version 3.1.1 | Max Planck Institute for Immunobiology and Epigenetics | https://deeptools.readthedocs.io/en/develop/ |
| Trim Galore! version 0.4.5 | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic version 0.36 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| FastQC version 0.11.5 | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Bismark version 0.19.1 | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| bowtie2 version 2.3.4.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| bsseq version 1.14.0 | Hansen et al., 2012 | https://www.bioconductor.org/packages/release/bioc/html/bsseq.html |
| ggplot2 version 3.2.1 | Hadley Wickham | https://github.com/tidyverse/ggplot2 |
| csaw version 1.10.0 | Lun and Smyth, 2016 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| DSS version 2.26.0 | Wu et al., 2015 | http://bioconductor.org/packages/release/bioc/html/DSS.html |
| eulerr version 5.1.0 | Larsson J | https://cran.r-project.org/web/packages/eulerr/index.html |
| circlize version 0.4.5 | Gu et al, 2016 | https://cran.r-project.org/web/packages/circlize/index.html |
| apcluster version 1.4.7 | Bodenhofer et al., 2011 | https://cran.r-project.org/web/packages/apcluster/index.html |
| ComplexHeatmap version 1.17.1 | Gu et al., 2016 | https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| pheatmap version 1.0.10 | Raivo Kolde | https://cran.r-project.org/web/packages/pheatmap/index.html |
| Gviz version 1.22.3 | Hahne and Ivanek, 2016 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| karyoploteR version 1.2.2 | Bernat Gel | https://bioconductor.org/packages/release/bioc/html/karyoploteR.html |
| methylSeekR version 1.16.0 | Burger et al., 2013 | https://bioconductor.org/packages/release/bioc/html/MethylSeekR.html |
| Stats version 0.1.0 | Kartikeya Bolar | https://cran.r-project.org/web/packages/STAT/index.html |
| MASS version 7.3–50 | Brian Ripley | https://cran.r-project.org/web/packages/MASS/index.html |
| countreg version 0.2–1 | Achim Zeileis | https://r-forge.r-project.org/R/?group_id=522 |
| gProfileR version 0.6.7 | Reimand et al., 2007 | https://cran.r-project.org/web/packages/gProfileR/index.html |
| questionr version 0.7.0 | Barnier et al., 2018 | https://cran.r-project.org/web/packages/questionr/index.html |
| Cytoscape version 3.7.1 | Shannon et al., 2003 | https://cytoscape.org/ |
| ClueGO version 2.5.4 | Bindea et al., 2009 | http://apps.cytoscape.org/apps/cluego |
| toRun_Imputation.R | Stubbs et al., 2017 | https://github.com/EpigenomeClock/MouseEpigeneticClock |
Highlights.
WGBS identifies 2,054 differentially methylated regions(DMRs) in aging mouse rods
DMRs are present in regulatory regions and cluster at chromosomal hotspots
DMRs are linked to dysregulation of aging pathways and energy metabolism
Old retinas exhibit lower basal respiration and higher fatty acid dependency
ACKNOWLEDGMENTS
We are grateful to Kasper Hansen (Johns Hopkins University, MD, USA) for discussion on differential DNA methylation analysis and Steven Henikoff (Fred Hutchinson Cancer Research Center, WA, USA) for providing CUT&RUN supplies. We thank Vijender Chaitankar for advice on data analysis. This research was supported by the Intramural Research Program of the National Eye Institute (NEI), USA (grants ZIAEY000450 and ZIAEY000456) and utilized the high performance computational capabilities of the Biowulf Linux cluster at the NIH http://biowulf.nih.gov; (https://hpc.nih.gov/systems/).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107525.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, et al. (2017). Ensembl 2017. Nucleic Acids Res 45 (D1), D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, et al. (2006). Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc. Natl. Acad. Sci. USA 103, 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldiri I, Xu B, Wang L, Chen X, Hiler D, Griffiths L, Valentine M, Shirinifard A, Thiagarajan S, Sablauer A, et al. (2017). ). The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron 94, 550–568.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieux J, Demory JL, Morel P, Plantier I, Dupriez B, Caulier MT, Bauters F, and Laï JL (2002). Frequency of structural abnormalities of the long arm of chromosome 12 in myelofibrosis with myeloid metaplasia. Cancer Genet. Cytogenet 137, 68–71. [DOI] [PubMed] [Google Scholar]
- Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schübeler D, Glaser B, and Kaestner KH (2015). Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved b Cell Function. Cell Metab 22, 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnier J, Briatte J, and Larmarange J (2018). questionr: Functions to Make Surveys Processing Easier https://cran.r-project.org/web/packages/questionr/index.html.
- Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, and Brunet A (2019). Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, and Galon J (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhofer U, Kothmeier A, and Hochreiter S (2011). APCluster: an R package for affinity propagation clustering. Bioinformatics 27, 2463–2464. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, and Brunet A (2016). The Aging Epigenome. Mol. Cell 62, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski JA 4th, Lamba DA, and Reh TA (2010). Blimp1 controls photoreceptor versus bipolar cell fate choice during retinal development. Development 137, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger L, Gaidatzis D, Schübeler D, and Stadler MB (2013). Identification of active regulatory regions from DNA methylation data. Nucleic Acids Res 41, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallotti C, Artico M, Pescosolido N, Leali FM, and Feher J (2004). Age-related changes in the human retina. Can. J. Ophthalmol 39, 61–68. [DOI] [PubMed] [Google Scholar]
- Chai X, Nagarajan S, Kim K, Lee K, and Choi JK (2013). Regulation of the boundaries of accessible chromatin. PLoS Genet 9, e1003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Kaya KD, Dong L, and Swaroop A (2016). Three-dimensional retinal organoids from mouse pluripotent stem cells mimic in vivo development with enhanced stratification and rod photoreceptor differentiation. Mol. Vis 22, 1077–1094. [PMC free article] [PubMed] [Google Scholar]
- Chen D, Chao DL, Rocha L, Kolar M, Nguyen Huu VA, Krawczyk M, Dasyani M, Wang T, Jafari M, Jabari M, et al. (2020). The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell 19, e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchore Y, Begaj T, Wu D, Drokhlyansky E, and Cepko CL (2017). Glycolytic reliance promotes anabolism in photoreceptors. eLife 6, e25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysostomou V, Trounce IA, and Crowston JG (2010). Mechanisms of retinal ganglion cell injury in aging and glaucoma. Ophthalmic Res 44, 173–178. [DOI] [PubMed] [Google Scholar]
- Ciccarone F, Tagliatesta S, Caiafa P, and Zampieri M (2018). DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech. Ageing Dev 174, 3–17. [DOI] [PubMed] [Google Scholar]
- Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, Sproul D, Wang T, Brock C, Clark W, Ideker T, et al. (2017). Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol 18, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Lawrence KA, Karlstetter M, Myers CA, Abdelaziz M, Dirkes W, Weigelt K, Seifert M, Benes V, Fritsche LG, et al. (2010). CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res 20, 1512–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso-Díaz X, Jaeger C, Chaitankar V, and Swaroop A (2018). Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res 65, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, et al. (2013). Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol 15, 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunea A, and Jeffery G (2007). The ageing photoreceptor. Vis. Neurosci 24, 151–155. [DOI] [PubMed] [Google Scholar]
- Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, and Absher D (2013). Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrabant B, Soerensen M, Flachsbart F, Dato S, Mengel-From J, Stevnsner T, Bohr VA, Kruse TA, Schreiber S, Nebel A, et al. (2014). Human longevity and variation in DNA damage response and repair: study of the contribution of sub-processes using competitive gene-set analysis. Eur. J. Hum. Genet 22, 1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano T, Chen HY, Panebianco C, Kaya KD, Brooks MJ, Gieser L, Morgan NY, Pohida T, and Swaroop A (2018). Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Reports 10, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlon TA, Morris BJ, Chen R, Masaki KH, Allsopp RC, Willcox DC, Elliott A, and Willcox BJ (2017). FOXO3 longevity interactome on chromosome 6. Aging Cell 16, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA, Topol SE, Wineinger NE, Niederhuber JE, Topol EJ, and Torkamani A (2016). Whole-Genome Sequencing of a Healthy Aging Cohort. Cell 165, 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachsbart F, Dose J, Gentschew L, Geismann C, Caliebe A, Knecht C, Nygaard M, Badarinarayan N, ElSharawy A, May S, et al. (2017). Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat. Commun 8, 2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster-García C, García-García G, Jaijo T, Fornés N, Ayuso C, Fernández-Burriel M, Sánchez-De la Morena A, Aller E, and Millán JM (2018). High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep 8, 17113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Guida F, Sandanger TM, Castagné R, Campanella G, Polidoro S, Palli D, Krogh V, Tumino R, Sacerdote C, Panico S, et al. (2015). Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet 24, 2349–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn O, Grönke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, Andrews S, Zhang Q, Wakelam MJ, Beyer A, et al. (2017). Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol 18, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 13, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H, Kim DS, Klocke B, Johnson KR, Cui K, Gotoh N, Zang C, Gregorski J, Gieser L, Peng W, et al. (2012). Transcriptional regulation of rod photoreceptor homeostasis revealed by in vivo NRL targetome analysis. PLoS Genet 8, e1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, and Ren B (2013). Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet 45, 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter A, Spechler PA, Cwanger A, Song Y, Zhang Z, Ying GS, Hunter AK, Dezoeten E, and Dunaief JL (2012). DNA methylation is associated with altered gene expression in AMD. Invest. Ophthalmol. Vis. Sci 53, 2089–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JB, Lindsay KJ, and Du J (2015). Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res 93, 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Owsley C, and Curcio CA (2002). Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res. Rev 1, 381–396. [DOI] [PubMed] [Google Scholar]
- Jones MJ, Goodman SJ, and Kobor MS (2015). DNA methylation and healthy human aging. Aging Cell 14, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Rabinovitch PS, and Martin GM (2015). Healthy aging: The ultimate preventative medicine. Science 350, 1191–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Yang HJ, Brooks MJ, Zelinger L, Karakülah G, Gotoh N, Boleda A, Gieser L, Giuste F, Whitaker DT, et al. (2016). NRL-Regulated Transcriptome Dynamics of Developing Rod Photoreceptors. Cell Rep 17, 2460–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff T, Chen ZQ, Gold B, Pal P, Gaudet MM, Kosarin K, Levine DA, Gregersen P, Spencer S, Harlan M, et al. (2009). The 6q22.33 locus and breast cancer susceptibility. Cancer Epidemiol. Biomarkers Prev 18, 2468–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooragayala K, Gotoh N, Cogliati T, Nellissery J, Kaden TR, French S, Balaban R, Li W, Covian R, and Swaroop A (2015). Quantification of Oxygen Consumption in Retina Ex Vivo Demonstrates Limited Reserve Capacity of Photoreceptor Mitochondria. Invest. Ophthalmol. Vis. Sci 56, 8428–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub group (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, and Smyth GK (2016). csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res 44, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenroth L, Hackmann K, Beyer A, Schallner J, Novotna B, Klink B, Schröck E, and Di Donato N (2015). 6q22.33 microdeletion in a family with intellectual disability, variable major anomalies, and behavioral abnormalities. Am. J. Med. Genet. A 167A, 2800–2807. [DOI] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, and Swaroop A (2001). Nrl is required for rod photoreceptor development. Nat. Genet 29, 447–452. [DOI] [PubMed] [Google Scholar]
- Meer MV, Podolskiy DI, Tyshkovskiy A, and Gladyshev VN (2018). A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Sheng X, Yoshitake K, Sergeev Y, Iejima D, Shibagaki Y, Monma N, Ikeo K, Furuno M, Zhuang W, et al. (2016). CCT2 Mutations Evoke Leber Congenital Amaurosis due to Chaperone Complex Instability. Sci. Rep 6, 33742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Luo C, Davis FP, Mukamel EA, Henry GL, Nery JR, Urich MA, Picard S, Lister R, Eddy SR, et al. (2016). Epigenomic landscapes of retinal rods and cones. eLife 5, e11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrie JL, Lupo MS, Xu B, Al Diri I, Valentine M, Putnam D, Griffiths L, Zhang J, Johnson D, Easton J, et al. (2019). Nucleome Dynamics during Retinal Development. Neuron 104, 512–528.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owsley C (2016). Vision and Aging. Annu. Rev. Vis. Sci 2, 255–271. [DOI] [PubMed] [Google Scholar]
- Pal S, and Tyler JK (2016). Epigenetics and aging. Sci. Adv 2, e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MR, Li K, Lin SY, and Hung WC (2016). Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci 17, e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parapuram SK, Cojocaru RI, Chang JR, Khanna R, Brooks M, Othman M, Zareparsi S, Khan NW, Gotoh N, Cogliati T, and Swaroop A (2010). Distinct signature of altered homeostasis in aging rod photoreceptors: implications for retinal diseases. PLoS ONE 5, e13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Kim K, Page GP, Allison DB, Weindruch R, and Prolla TA (2009). Gene expression profiling of aging in multiple mouse strains: identification of aging biomarkers and impact of dietary antioxidants. Aging Cell 8, 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit L, Ma S, Cipi J, Cheng SY, Zieger M, Hay N, and Punzo C (2018). Aerobic Glycolysis Is Essential for Normal Rod Function and Controls Secondary Cone Death in Retinitis Pigmentosa. Cell Rep 23, 2629–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran S, Roberts J, Koltzenburg M, Hanna MG, and Stewart H (2013). Deletion of chromosome 12q21 affecting KCNC2 and ATXN7L3B in a family with neurodevelopmental delay and ataxia. J. Neurol. Neurosurg. Psychiatry 84, 1255–1257. [DOI] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, and Vilo J (2007). g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35, W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Bio-technol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capellà G, Ribas M, and Peinado MA (2006). Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res 66, 8462–8468. [DOI] [PubMed] [Google Scholar]
- Samuel MA, Voinescu PE, Lilley BN, de Cabo R, Foretz M, Viollet B, Pawlyk B, Sandberg MA, Vavvas DG, and Sanes JR (2014). LKB1 and AMPK regulate synaptic remodeling in old age. Nat. Neurosci 17, 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Mekker B, Mallick M, Vastolo V, Karaulanov E, Sebastian D, von der Lippen C, Epe B, Downes DJ, Scholz C, and Niehrs C (2018). Impaired DNA demethylation of C/EBP sites causes premature aging. Genes Dev 32, 742–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BG, Rha SY, Chung HC, Bravo JC, Mera R, Torres JC, Plaisance KT Jr., Schlegel R, McBride CM, Reveles XT, and Leach RJ (2003). Regions of allelic imbalance in the distal portion of chromosome 12q in gastric cancer. MP, Mol. Pathol 56, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Stegeman R, and Weake VM (2017). Transcriptional Signatures of Aging. J. Mol. Biol 429, 2427–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs TM, Bonder MJ, Stark AK, Krueger F, BI Ageing Clock Team; von Meyenn F, Stegle O, and Reik W (2017). Multi-tissue DNA methylation age predictor in mouse. Genome Biol 18, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. (2014). Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, and Finkel T (2016). The Mitochondrial Basis of Aging. Mol. Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Chew EY, Rickman CB, and Abecasis GR (2009). Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu. Rev. Genomics Hum. Genet 10, 19–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45 (D1), D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JY, Vance KW, Varela MA, Sirey T, Watson LM, Curtis HJ, Marinello M, Alves S, Steinkraus B, Cooper S, et al. (2014). Cross-talking noncoding RNAs contribute to cell-specific neurodegeneration in SCA7. Nat. Struct. Mol. Biol 21, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Xu T, Feng H, Chen L, Li B, Yao B, Qin Z, Jin P, and Conneely KN (2015). Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res 43, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T (2016). Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T, and Loerch P (2008). The aging brain. Annu. Rev. Pathol 3, 41–66. [DOI] [PubMed] [Google Scholar]
- Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, Oshlack A, and Majewski IJ (2011). ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res 39, 7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, and Teschendorff AE (2015). An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet 11, e1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv E, and Hu D (2011). Genetic variation in insulin/IGF-1 signaling pathways and longevity. Ageing Res. Rev 10, 201–204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed data have been deposited to Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/GEO) with accession GSE134873 and to https://neicommons.nei.nih.gov/#/. In-house codes are deposited at https://github.com/NEI-NNRL/2020-DNA_methylation_signatures_of_aging_in_rods