

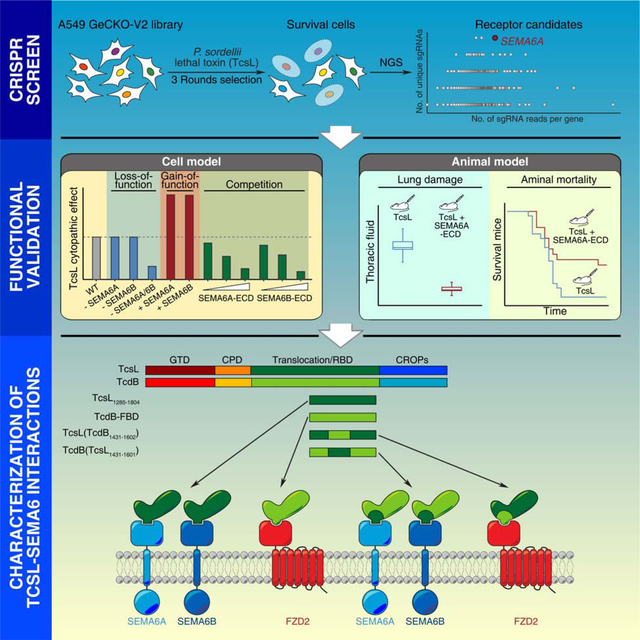
Summary
The exotoxin TcsL is a major virulence factor in Paeniclostridium (Clostridium) sordellii and responsible for the high lethality rate associated with P. sordellii infection. Here we present a genome-wide CRISPR-Cas9-mediated screen using a human lung carcinoma cell line and identify semaphorin (SEMA) 6A and 6B as receptors for TcsL. Disrupting SEMA6A/6B expression in several distinct human cell lines and primary human endothelial cells results in reduced TcsL sensitivity, while SEMA6A/6B over-expression increases their sensitivity. TcsL recognizes the extracellular domain (ECD) of SEMA6A/6B via a region homologous to the receptor-binding site in Clostridioides difficile toxin B (TcdB), which binds the human receptor Frizzled. Exchanging the receptor-binding interfaces between TcsL and TcdB switches their receptor-binding specificity. Finally, administration of SEMA6A-ECD proteins protects human cells from TcsL toxicity and reduces TcsL-induced damage to lung tissues and the lethality rate in mice. These findings establish SEMA6A and 6B as pathophysiologically relevant receptors for TcsL.
Graphical Abstract
eTOC Blurb:
Tian et al. identify semaphorin (SEMA) 6A and 6B as the pathophysiologically relevant receptors for TcsL, a bacterial toxin responsible for the high lethality rate in Paeniclostridium sordellii infection. The recombinant SEMA6A extracellular domain proteins can serve as a receptor decoy and protects human cells and mice from co-administered TcsL.
Introduction
Paeniclostridium sordellii (formerly known as Clostridium sordellii (Sasi Jyothsna et al., 2016)) is a spore-forming anaerobic bacterium and its virulent strains can cause deadly infections in humans and animals (Vidor et al., 2015). P. sordellii infections in humans are often associated with soft tissue trauma and in most cases severe, leading to edema, gangrene, hypotension, and systemic toxic shock with ~70% death rate (Aldape et al., 2006; Vidor et al., 2015). Women have the highest risk to infections, due to gynecologic procedures, childbirth, miscarriage, and abortion that may result in intrauterine infection with a mortality rate close to 100% (Aldape et al., 2006; Fischer et al., 2005; McGregor et al., 1989). P. sordellii infections are more frequent in animals such as sheep and cattle, often resulting in severe enteritis and enterotoxaemia (Vidor et al., 2015).
The major virulence factors of P. sordellii are two exotoxins, the lethal toxin TcsL (~270 kDa) and the hemorrhagic toxin TcsH (~300 kDa) (Arseculeratne et al., 1969; Couchman et al., 2015; Martinez and Wilkins, 1988; Popoff, 1987; Vidor et al., 2015; Vidor et al., 2018). These toxins belong to the large clostridial toxin (LCT) family, which also include the toxin A (TcdA) and toxin B (TcdB) in Clostridioides difficile, Tcnα in Clostridium novyi, and TpeL in Clostridium perfringens (Aktories et al., 2017; Jank and Aktories, 2008; Schirmer and Aktories, 2004; Voth and Ballard, 2005). TcsL and TcsH are particularly related to TcdB and TcdA, with TcsL sharing an overall 76% sequence identity with TcdB and TcsH sharing 77% identity with TcdA, respectively (Bette et al., 1991; Martinez and Wilkins, 1992). Similar to TcdA and TcdB, TcsL and TcsH contain an N-terminal glucosyltransferase domain (GTD) that inactivates small GTPases, followed with a cysteine protease domain (CPD), which releases the GTD through autoproteolytic cleavage (Guttenberg et al., 2011), an intermingled translocation/receptor binding domain, and a C-terminal region known as combined repetitive oligopeptides (CROPs). Small GTPase Rho/Ras family are the targets of TcsL (Genth et al., 1996; Genth et al., 2014; Hofmann et al., 1996; Huelsenbeck et al., 2009; Just et al., 1996; Popoff et al., 1996). GTDs covalently attaches a glucose onto a key threonine residue in these GTPases, inactivating their activity, leading to disruption of actin cytoskeleton and downstream signaling, and resulting in cell rounding and eventual cell death (Geny et al., 2010; Varela Chavez et al., 2016; Voth and Ballard, 2007).
Between TcsL and TcsH, TcsL appears to be the essential and sufficient virulent factor associated with lethal P. sordellii infection (Carter et al., 2011; Hao et al., 2010; Popoff, 1987; Voth et al., 2006). Mouse infection models using isogenic mutant strains of P. sordellii, in which tcsL gene has been inactivated, showed that mice infected with the wild type (WT) strain quickly developed lethal infection with 100% death within 72 h, while mice infected with the mutant strains showed no signs of disease (Carter et al., 2011). Furthermore, in a local intrauterine infection model in mice, severe tissue edema developed with infection of WT P. sordellii, while infection with mutant strains showed no clinical symptoms (Carter et al., 2011). Thus, TcsL is the key virulence factor for both lethality and local edema of P. sordellii infection.
Among all LCTs, TcsL showed the highest degree of lethality in mice. Analysis of mouse tissues after intraperitoneal injection (IP) of TcsL suggests that the major damage occurs on endothelial cells, particularly in lungs, leading to increased vascular permeability and massive edema in lungs (Geny et al., 2007). Death of mice is likely due to edema in lungs and heart, with no obvious inflammatory components (Geny et al., 2007; Popoff, 2018). These data suggest that lung vascular endothelial cells are the primary and pathologically relevant targets in vivo for TcsL.
Receptors dictate the cell type and tissue specificity of TcsL and understanding toxin-receptor interactions may enable development of inhibitors that block toxin binding to target cells. The CROPs in LCTs bear similarity with carbohydrate-binding proteins and are known to mediate attachment to cells via interactions with cell surface carbohydrates (Hartley-Tassell et al., 2019; Krivan et al., 1986; Teneberg et al., 1996; Tucker and Wilkins, 1991; von Eichel-Streiber and Sauerborn, 1990). Whether there is a specific receptor for TcsL besides carbohydrates remains unknown. Here we carried out a genome-wide screen using the CRISPR-Cas9 approach and identified semaphorin (SEMA) 6A and 6B as high-affinity receptors for TcsL. Furthermore, recombinant extracellular domain (ECD) of SEMA6A was able to reduce toxicity of TcsL on both human umbilical vein cells (HUVECs) and lung tissues in vivo in mice, demonstrating the pathological relevance of SEMA6A/6B as toxin receptors in vivo.
Results
CRISPR-Cas9 screen identifies host factors for TcsL
To select suitable cell lines for CRISPR-Cas9 screen, we compared a list of well-established human cell lines for their sensitivity to TcsL using the standard cytopathic cell-rounding assay. TcsL was recombinantly produced using Bacillus megaterium expression system (Figure S1A). Cells were exposed to various concentrations of TcsL for 24 h and the percentages of rounded cells were determined and plotted (Figures S1B and S1C). The toxin concentration that resulted in 50% cells to become round is defined as CR50. A549, a human lung epithelial carcinoma cell line, is among the most sensitive ones to TcsL (Figure S1B). Thus, we established a A549 cell line that stably expresses Cas9 for screen (Figure 1A). A genome-wide sgRNA library targeting all human genes (GeCKO-V2, with each gene targeted by six distinct sgRNAs (Sanjana et al., 2014)) was prepared and transduced into A549-Cas9 cells using lentiviruses (Figure S1D). Cells were subjected to selection with increasing concentrations of TcsL for three rounds (Figure 1A, Round 0 (R0): 0.5 ng/mL, R1: 1 ng/mL, and R3: 2 ng/mL). Integrated sgRNA sequences in cells after each round were obtained by polymerase chain reaction (PCR) and decoded by next generation sequencing (NGS).
Figure 1. Genome-wide CRISPR-Cas9-mediated loss-of-function screen for TcsL.
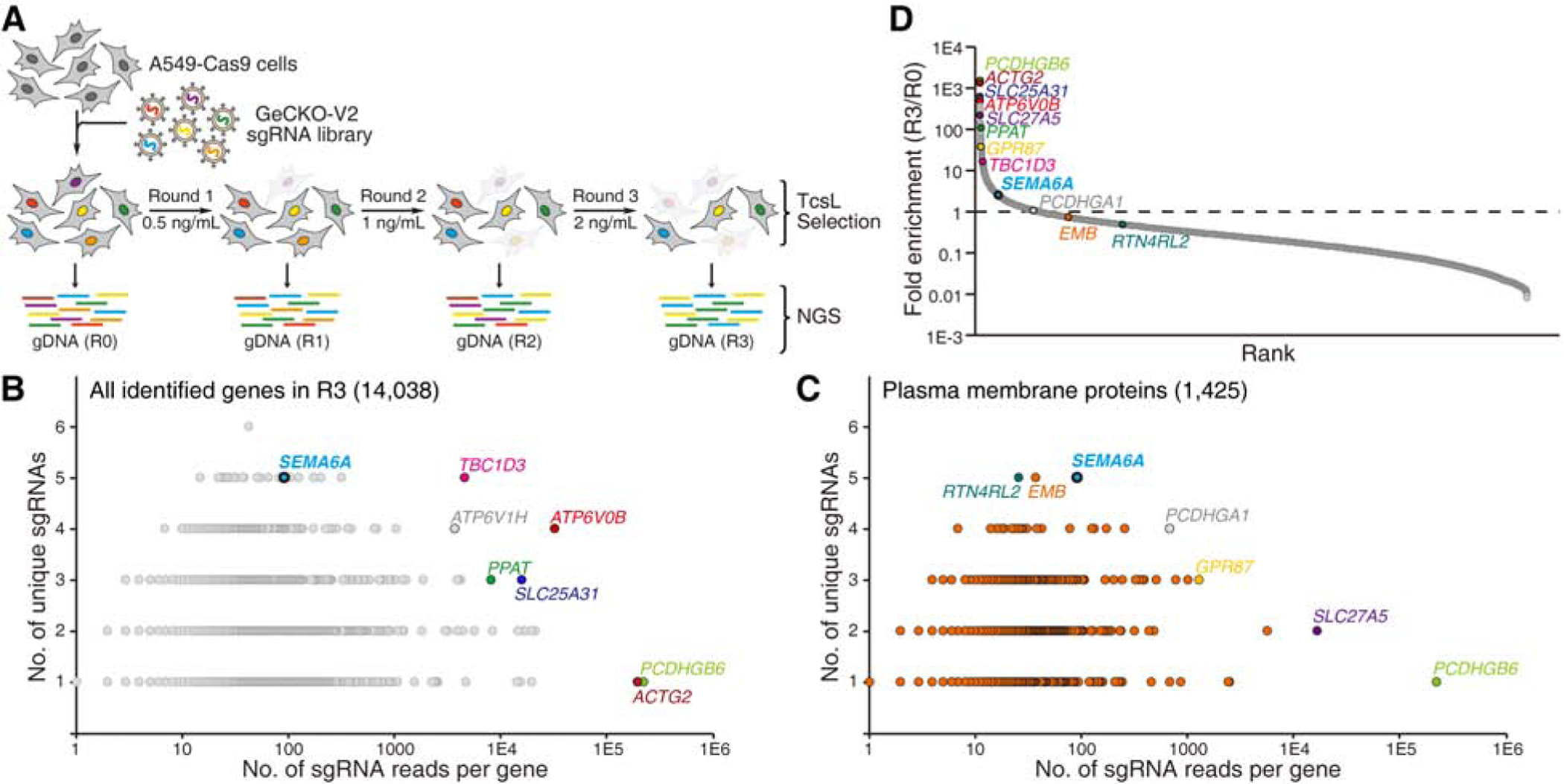
(A) Schematic diagram of the screen process. A549 cells that stably express Cas9 (A549-Cas9) were transduced with lentiviral GeCKO-V2 sgRNA libraries. Cells were then selected with TcsL at 0.5 ng/mL for 24 h (Round 1, R1). Cells were recovered in toxin-free medium and then subjected to two more rounds of selection with TcsL (R2: 1 ng/mL and R3: 2 ng/mL). Cells of each round were harvested and sgRNA sequences were identified by NGS. Cells not treated with toxins served as a control (R0).
(B) Genes identified in R3 are plotted based on the number of unique sgRNAs (y axis) and total sgRNA reads (x axis). Selected top hits are marked and colored.
(C) A total of 1,425 genes identified in R3 are annotated as plasma membrane proteins. These proteins are plotted based on the number of unique sgRNAs (y axis) and total sgRNA reads (x axis). Selected top hits are marked.
(D) Genes identified in R3 are plotted based on their fold-enrichment from R0 to R3 (sgRNA reads of a gene among total sgRNA reads in R3 versus R0). Selected top hits in panels B and C are marked.
We first ranked the identified genes in R3 primarily based on the number of unique sgRNAs (y-axis), followed by the total number of reads (x-axis, Figure 1B, Data S1). One of the top hits is a cytosolic protein TBC1D3 (TBC1 domain member 3), which is potentially involved in Rab GTPase signaling and vesicle trafficking. Two subunits of vesicular proton pump, ATP6V0B (ATPase H+ Transporting V0 Subunit B) and ATP6V1H (ATPase H+ Transporting V1 Subunit H), are also among the top hits, likely because acidification of endosomes is an essential condition to trigger toxin translocation (Qa’Dan et al., 2000, 2001). Among the top-10 ranked proteins, SEMA6A (Semaphorin 6A) stands out as the only transmembrane protein (Figure 1B, Data S1). It is a well-characterized cell surface protein with a single transmembrane domain and well known for its role in repulsive axon guidance during neuronal development (Yazdani and Terman, 2006). SEMA6A is also expressed in endothelial cells and regulates angiogenesis (Segarra et al., 2012; Urbich et al., 2012). Thus, it is a promising receptor candidate.
To further searching for potential receptors, we then ranked the subset of genes annotated as plasma membrane associated proteins in R3 (Figure 1C, Data S1). Besides SEMA6A, other top-ranked plasma membrane proteins include EMB (Embigin, an immunoglobulin superfamily member), RTN4RL2 (Reticulon 4 Receptor Like 2, a GPI-anchored cell surface receptor), PCDHGA1 (Protocadherin gamma-A1), GPR87 (G Protein-Coupled Receptor 87), SLC27A5 (Bile acyl-CoA synthetase), and PCDHGB6 (Protocadherin gamma-B6). We also calculated fold-of-enrichment (the ratio of the sequencing reads of R3 over R0) for each sgRNA (Figures 1D and S1E, S1F, S1G, and S1H), which serves as another parameter to select genes for further validation.
Validation of top hits for TcsL
We next generated mixed population stable knockout (KO) A549 cells using the CRISPR-Cas9 approach to disrupt expression of selected membrane proteins SEMA6A, EMB, RTN4RL2, GPR78, SLC27A5, and PCDHGB6, as well as a few other promising top hits including TBC1D3, ATP6V0B, SLC25A31 (Solute Carrier Family 25 Member 31, ADP/ATP translocase 4), PPAT (Phosphoribosyl Pyrophosphate Amidotransferase), and ACTG2 (actin gamma 2). We also targeted SEMA6B, 6C, and 6D, three homologs of SEMA6A within the class 6 of semaphorin family (Yazdani and Terman, 2006). These mixed KO cells were generated using antibiotic selection after lentiviral transduction; thus, the population contain a mix of mutations within the sgRNA-targeted locus. The KO efficiency was further validated for SEMA6A by immunoblot of cell lysates at protein levels, but not for others due to the lack of suitable antibodies (Figure S2A). To address the concern on potential off-target effects, we utilized two distinct sgRNAs to target SEMA6A, 6B, and 6D, and generated two independent cell populations for each of them.
We first analyzed the sensitivity of these cells to a fixed concentration of TcsL (10 ng/mL) and monitored the percentage of cell-rounding over time. This assay has the advantage to detect small differences on toxin binding and entry that might be masked after the standard 24 h incubation. Disrupting SEMA6A and SEMA6B genes, each with two different sgRNAs, reduced cell-rounding percentages within the first 5 h, although the difference becomes insignificant by 24 h (Figures 2A and S2B). Targeting SEMA6C and 6D did not change the sensitivity of cells to TcsL, neither do targeting all other plasma membrane proteins (Figures 2A, 2B, 2C, and S2B). TBC1D3, ATP6V0B, and SLC25A31 are three other top hits that their disruption reduced cell-rounding percentages within the first 5 h, with TBC1D3 as the only hit that showed reduction in cell-rounding at 24 h (Figures 2B, 2C, and S2B).
Figure 2. Validation of top hits identified in the screen.
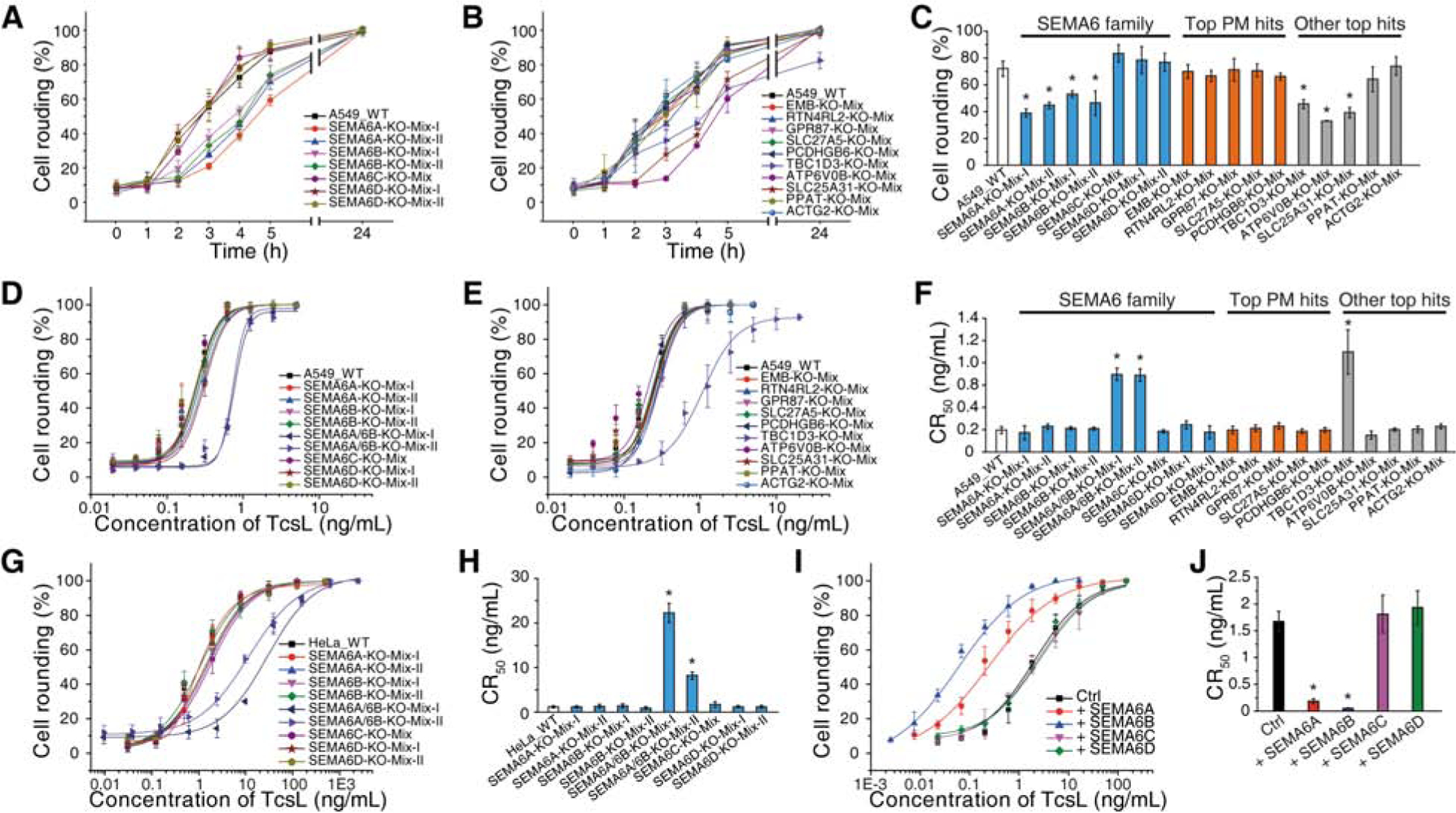
(A-C) Mixed stable A549 KO cells for the indicated genes were generated via the CRISPR-Cas9 approach. Two independent knockout cell lines using two different sgRNAs were generated for SEMA6A, 6B and 6D, one line was generated for 6C and other hits. The sensitivity of these cells to TcsL (10 ng/mL) were examined by quantifying cell rounding over time (A for SEMA6 family, B for other proteins). The percentages of cell rounding at 4 h post TcsL exposure are plotted as bar-chart in panel C. PM, plasma membrane. Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
(D-F) Two independently generated SEMA6A and 6B double KO cell lines (SEMA6A/6B-KO-Mix-I and SEMA6A/6B-KO-Mix-II) and other indicated KO cell lines were exposed to TcsL for 24 h. The percentages of rounded cells were plotted over toxin concentrations. (D for SEMA6 family, E for other proteins). Their CR50 are plotted in a bar-chart (F). Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
(G-H) Mixed stable HeLa KO cells for SEMA6 family genes were generated via the CRISPR-Cas9 approach and exposed to TcsL for 24 h (G). Their CR50 are plotted in a bar-chart (H). Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
(I-J) HeLa cells overexpressing SEMA6 family proteins via lentiviral transduction were exposed to TcsL for 24 h (I). Their CR50 are plotted in a bar-chart (J). Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
As SEMA6A and 6B may function redundantly, we next generated mixed populations of A549 cells using the CRISPR-Cas9 approach to target both SEMA6A and 6B. Two independent cell populations were generated using two distinct sets of sgRNAs (Figure S2A). These cells were subjected to the standard 24 h cytopathic cell-rounding assays with TcsL. Comparing the CR50 revealed that both SEMA6A/6B mixed cell populations showed ~4-fold reduction in sensitivity to TcsL, while cells with disruption in SEMA6C or 6D showed no reduction (Figures 2D, 2E, and 2F). TBC1D3 is the only other top hit that showed reduction in CR50 (~5-fold), while cells with disruptions in other top hits showed no reduction in CR50 (Figures 2E and 2F). None of the cell populations showed any change in sensitivity to TcdB, suggesting that SEMA6A/6B and TBC1D3 are specifically involved in TcsL action (Figures S2C, S2D, and S2E).
To further validate the role of SEMA6A/6B, we also generated mixed populations of HeLa cells using the CRISPR-Cas9 approach to disrupt SEMA6A, 6B, 6C, 6D, as well as both 6A and 6B expression. Among them, two independent populations for targeting SEMA6A, 6B, and 6D were generated using distinct sgRNAs, and the KO efficiency was further confirmed by immunoblot analysis of SEMA6A expression levels (Figure S2A). Cells were analyzed for their sensitivity to TcsL using the standard 24 h cell-rounding assays. Cell populations with both SEMA6A and 6B disrupted showed reduction in sensitivity to TcsL compared with WT cells (Figures 2G and 2H), suggesting that SEMA6A and 6B function redundantly in HeLa cells.
We next analyzed whether ectopic expression of SEMA6A, 6B, 6C, or 6D in cells may increase cell sensitivity to TcsL. Expression of SEMA6A or 6B via lentiviral transduction in HeLa cells increased their sensitivity to TcsL in the cytopathic cell-rounding assays, while expression of SEMA6C and 6D did not change the sensitivity of cells (Figures 2I, 2J, and S2F). Similarly, ectopic expression of SEMA6A or 6B in 5637 cells, a human bladder carcinoma cell line, via lentiviral transduction also increased the sensitivity of these cells to TcsL, while expression of SEMA6C or 6D did not alter the sensitivity (Figures S2G, S2H, and S2I).
Characterization of TcsL-SEMA6 interactions
Semaphorin family is defined by the presence of a common extracellular semaphorin domain (~500 residues) that mediates binding to their cognate receptors (Plexin family). Semaphorin family is divided into eight classes: Classes 3–7 are expressed in vertebrates, classes 1 and 2 in invertebrates, and class V in viruses (Yazdani and Terman, 2006). The class 6 includes SEMA6A, 6B, 6C, and 6D. Their semaphorin domains share 53–64% sequence identity. We next examined whether TcsL directly binds to the recombinantly expressed and purified ECDs of SEMA6A, 6B, 6C, and 6D. Their interactions were evaluated using biolayer interferometry (BLI) assays, with Fc-tagged SEMA6 proteins immobilized on the probe. As shown in Figure 3A, TcsL showed robust binding to both SEMA6A-ECD and SEMA6B-ECD, but not the ECDs of SEMA6C and 6D or the negative control human IgG-Fc.
Figure 3. Characterization of TcsL-SEMA interactions.
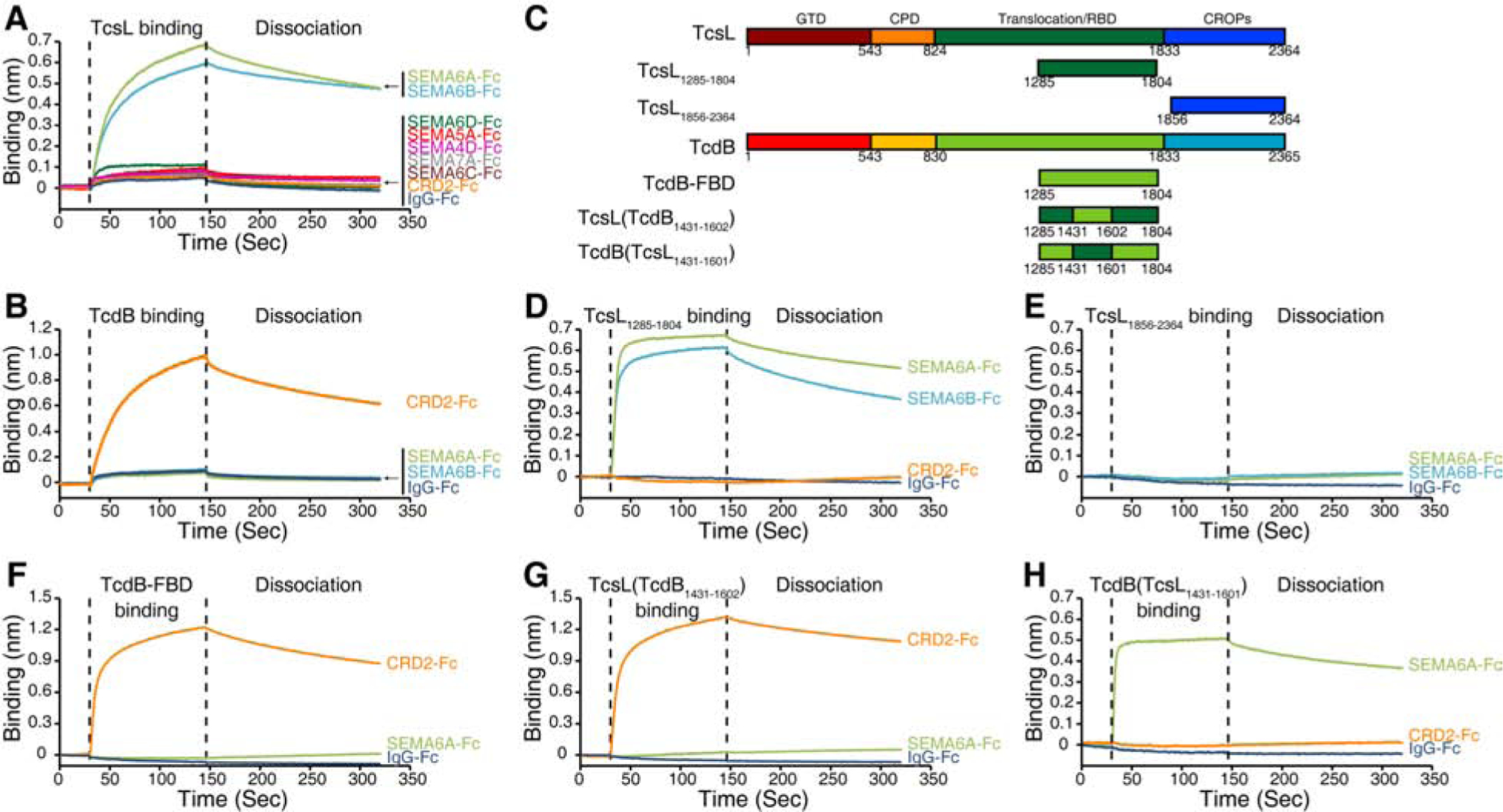
(A) Binding of TcsL (1 μM) to Fc-tagged ECDs of SEMA6A, 6B, 6C, 6D, 4D, 5A, and 7A (immobilized onto capture biosensors) was examined using BLI assays. Fc-tagged CRD2-ECD and IgG-Fc were used as controls. Representative sensorgrams from one of three independent experiments are shown.
(B) Binding of full-length TcdB to CRD2, SEMA6A-ECD, 6B-ECD, and IgG-Fc was examined using BLI assays. Representative sensorgrams from one of three independent experiments are shown.
(C) Schematic diagrams of TcsL, TcsL1285–1804, TcsL1856–2364, TcdB, TcdB-FBD, TcsL(TcdB1431–1602), and TcdB(TcsL1431–1601). The numbers indicate the position of amino acid residues. GTD, glucosyltransferase domain; CPD, cysteine protease domain; RBD, receptor-binding domain; CROPs, combined repetitive oligopeptides.
(D-E) Binding of 1 μM TcsL1285–1804 (D) and TcsL1856–2364 (E) to Fc-tagged ECDs of SEMA6A and SEMA6B was examined using BLI assays. Fc-tagged CRD2-ECD and IgG-Fc were used as controls. Representative sensorgrams from one of three independent experiments are shown.
(F-H) Binding of 1 μM TcdB-FBD (F) and TcsL-TcdB chimeras (G: TcsL(TcdB1431–1602) and H: TcdB(TcsL1431–1601)) to Fc-tagged SEMA6A-ECD and CRD2 was examined using BLI assays. IgG-Fc was used as a control. Representative sensorgrams from one of three independent experiments are shown.
We further screened other representative SEMA class members containing either a transmembrane domain (class 4 and 5: SEMA4D and SEMA5A) or a GPI anchor (class 7: SEMA7A). None of them showed detectable binding to TcsL (Figure 3A). TcsL also showed no binding to the extracellular cysteine-rich domain (CRD) of Frizzled 2 (FZD-CRD2) (Figure 3A), which is a high-affinity receptor for TcdB (Chen et al., 2018; Tao et al., 2016). Consistently, TcdB showed robust binding to CRD2 and no binding to SEMA6A and 6B (Figure 3B). These findings demonstrate the specificity of TcsL binding to SEMA6A and 6B. The binding affinity (KD) was further estimated to be ~ 40 nM for TcsL-SEMA6A-ECD and ~60 nM for TcsL-SEMA6B-ECD interactions (Figures S3A, S3B, and S3E).
To map the binding region for SEMA6A and 6B, we generated two TcsL fragments (Figure 3C). One is the C-terminal CROPs (residues 1856–2364), which may recognize carbohydrate moieties. The other fragment (residues 1285–1804) is within the translocation/receptor-binding domain and modeled based on the homologous region in TcdB that binds to its high-affinity receptors FZD1, 2, and 7 (residues 1285–1804, known as TcdB-FBD) (Chen et al., 2018; Tao et al., 2016). TcsL1285–1804 showed robust binding to the ECDs of SEMA6A and 6B, while TcsL-CROP showed no binding (Figures 3D and 3E). Binding of TcsL1285–1804 to SEMA6A-ECD and 6B-ECD showed slightly higher binding affinity than the full-length TcsL (Figures S3C, S3D, and S3E), suggesting that other regions of TcsL do not contribute to the interactions. SEMA6A is glycosylated on cell surfaces. Treatment of SEMA6A-ECD with endoglycosidase H (Endo H) under non-denature conditions reduced the molecular weight of purified SEMA6A-ECD (Figure S3F), indicating that glycosylation was removed. Binding of this Endo-H-treated SEMA6A-ECD to TcsL is similar to the intact SEMA6A-ECD (Figure S3G), suggesting that glycosylation in SEMA6A is not involved in TcsL-SEMA6A interactions.
As a control, the homologous TcdB-FBD binds to CRD2 but not SEMA6A-ECD (Figure 3F). The crystal structure of TcdB-FBD in complex with CRD2 has defined a binding interface located within the middle portion of TcdB-FBD (Chen et al., 2018). Interestingly, key residues involve in TcdB-CRD2 interactions are different between TcsL and TcdB, despite their high overall sequence identity (Figure S3H). To test whether the similar region in TcsL is involved in binding to SEMA6A-ECD, we generated two chimeric fragments by switching the middle portion of TcdB-FBD (residues 1431–1602) with the homologous region in TcsL1285–1804 (residues 1431–1601) (Figure 3C). The resulting TcsL(TcdB1431–1602) showed robust binding to CRD2, but lost binding to SEMA6A-ECD, while TcdB(TcsL1431–1601) showed strong binding to SEMA6A-ECD and no binding to CRD2 (Figures 3G and 3H). These results demonstrate that TcsL recognizes SEMA6A-ECD via an interface mainly located within the residues 1431–1602, homologous to the CRD-binding interface in TcdB.
SEMA6A and 6B mediate binding and entry of TcsL to cells
We next examined whether TcsL-SEMA6A/6B interactions contribute to toxin binding and entry into cells. Binding of a HA-tagged TcsL1285–1804 to WT Hela cells were not detectable using immunostaining approach, possibly because endogenous expression levels of SEMA6A/6B are low. Transfection of exogenous full-length SEMA6A or 6B resulted in robust binding of TcsL1285–1804 to HeLa cells (Figure 4A). As controls, transfection of SEMA6C, 6D, or FZD2 did not mediate binding of TcsL1285–1804 (Figure 4A).
Figure 4. SEMA6A and SEMA6B mediate binding and entry into cell lines and primary human endothelial cells.
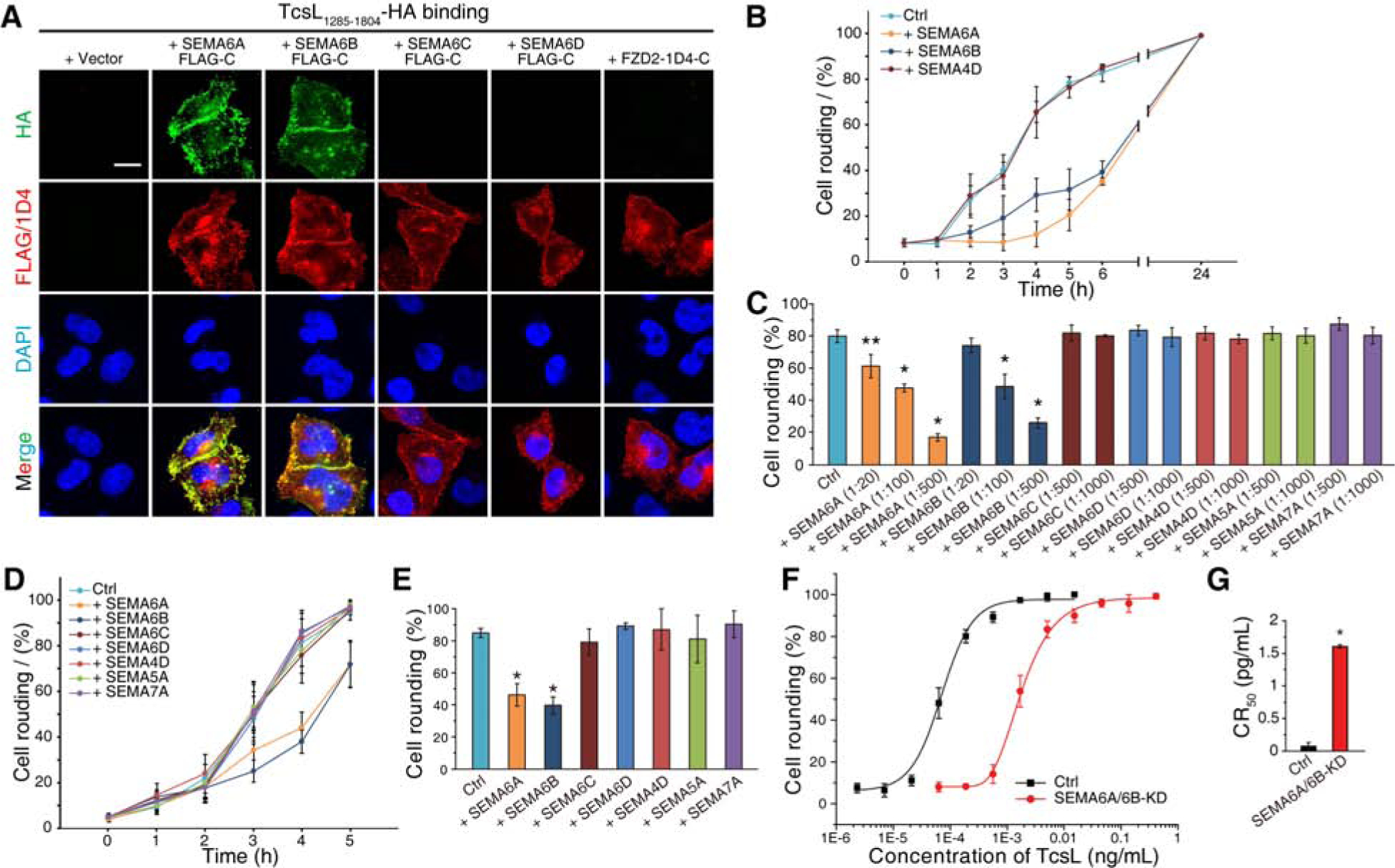
(A) HeLa cells transiently transfected with SEMA6A, 6B, 6C, 6D, or FZD2 were exposed to HA-tagged TcsL1285–1804 (7.5 μg/mL) on ice for 60 min, washed, fixed, permeabilized and subjected to immunostaining analysis. Expression of exogenous SEMA proteins was confirmed by detecting fused FLAG tags. FZD2 was detected by fused 1D4 tag. Nuclei were labeled with DAPI. Scale bar, 5 μm. Representative images were from one of three independent experiments.
(B) A549 cells were exposed to either TcsL (40 pM) alone or TcsL pre-incubated with ECDs of SEMA6A, 6B, or 4D at 1:500 molar ratio on ice for 1 h. The percentages of cell rounding were recorded over time.
(C) Experiments were carried out as described in panel B, except that TcsL was pre-incubated with the indicated proteins at the indicated molar ratios. The degrees of cell-rounding with 5 h incubation were plotted as a bar-chart. Error bars indicate mean ± s.d., N = 3, *, p < 0.01, **, p < 0.05 (one-way ANOVA).
(D-E) HUVECs were exposed to either TcsL (4 pM) alone or TcsL pre-incubated with ECDs of SEMA6A, 6B, 4D, 5A, or 7A at 1:1000 molar ratio on ice for 1 h. The percentages of cell rounding were plotted over time (D). The degrees of cell-rounding with 4 h incubation were plotted in a bar-chart (E). Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
(F-G) The sensitivity of HUVECs transfected with siRNAs targeting SEMA6A and SEMA6B to TcsL was analyzed using the 24 h cell-rounding assay. HUVECs transfected with scrambled siRNAs served as a control. The dose-response curves are plotted in panel F, and their CR50 are plotted in a bar-chart (panel G). Error bars indicate mean ± s.d., N = 3, *, p < 0.01 (one-way ANOVA).
We then examined whether Fc-tagged ECDs of SEMA6A/6B can block binding and entry of TcsL. As shown in Figures 4B and 4C, both SEMA6A-ECD and 6B-ECD showed a concentration-dependent protection of A549 cells from TcsL, as evidenced by reduced cell-rounding, while ECDs of SEMA6C, 6D, 4D, 5A, and 7A showed no protection (Figures 4B, 4C, and S4A). None of SEMA members showed any protection from TcdB on A549 cells (Figure S4B), demonstrating the specificity of SEMA6A-ECD and 6B-ECD toward TcsL. Besides A549 cells, SEMA6A-ECD also offered a degree of protection from TcsL on HeLa cells (Figure S4C).
SEMA6A and 6B mediate TcsL entry into human endothelial cells
As the previous studies suggest that endothelial cells are major pathologically relevant targets for TcsL, we next examined the relevance of SEMA6A/6B for TcsL in HUVECs, which are primary endothelial cells cultured from the endothelium of umbilical cord veins. They can maintain characteristics of endothelial cells within a few passages in vitro and have been widely utilized as an endothelial cell model. It has been previously shown that SEMA6A is expressed in HUVECs (Dhanabal et al., 2005). Fc-tagged ECDs of SEMA6A and 6B offered a partial protection of HUVECs from TcsL, while ECDs of SEMA6C, 6D, 4A, 5A, and 7A showed no effect (Figures 4D, 4E, and S4D).
We further took the siRNA-mediated knockdown approach to acutely reduce expression of endogenous SEMA6A and 6B in HUVECs. SiRNAs targeting human SEMA6A has been previously reported (Hasson et al., 2013). Three sets of siRNAs targeting SEMA6B were designed. We first validated their efficacy in knocking down ectopic SEMA6A and SEMA6B expression in HEK293 cells (Figure S4E). The siRNA targeting SEMA6A and a selected siRNA targeting SEMA6B were co-transfected into HUVECs, which resulted in a ~22-fold increase in resistance to full-length TcsL in the standard cell-rounding assay compared with cells transfected with the control scrambled siRNAs (Figures 4F and 4G).
SEMA6A-ECD reduces the lung toxicity of TcsL in vivo
Previous studies have established that lethality caused by TcsL results from massive edema in the thoracic cavity due to damages on lung endothelial cells and subsequently increased lung vascular permeability (Geny et al., 2007). Recent single-cell RNAseq analysis of mouse lung tissues confirmed that SEMA6A and 6D are strongly enriched in endothelial cells (Figure S5A, S5B, S5C, and S5D) (He et al., 2018; Vanlandewijck et al., 2018). To directly examine whether endothelial cells are major targets of TcsL in lungs in vivo, we injected the HA-tagged TcsL1285–1804 via the IP route and examined the distribution of the protein in lung tissues 30 and 60 min later by immunostaining. The lung endothelial cells were labeled using their specific marker CD31. As shown in Figure 5A, TcsL1285–1804 within lung tissues is largely co-localized with CD31, confirming that TcsL1285–1804 is preferentially delivered to lung endothelial cells following IP injection.
Figure 5. SEMA6A-ECD reduces the toxicity of TcsL on lung tissues in vivo.
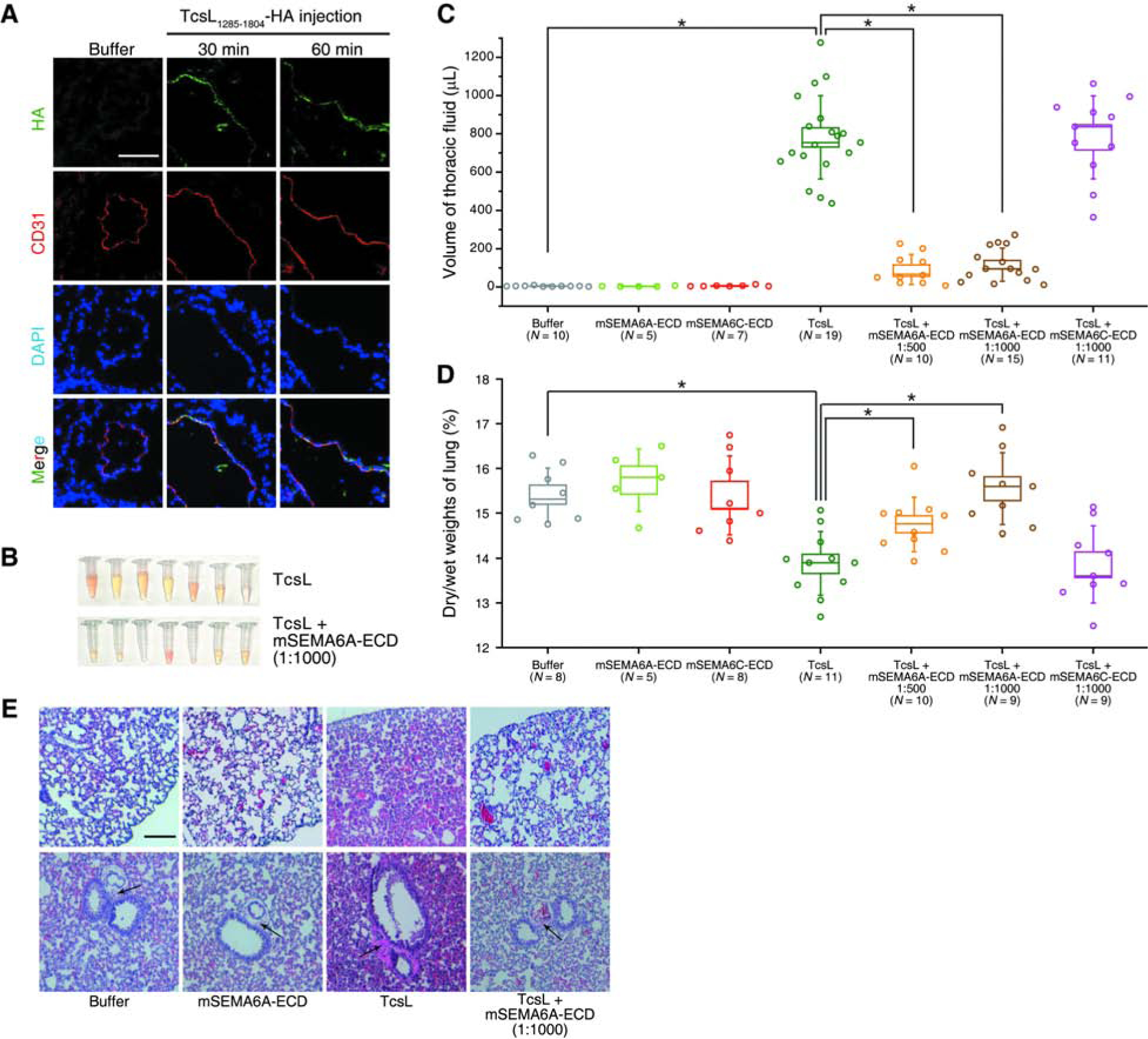
(A) Mice were injected with Buffer (HBSS with 0.1% BSA) or HA-tagged TcsL1285–1804 (200 μg per mice) for 30 min or 60 min. The lung tissue was harvested, washed, fixed, and subjected to immunostaining analysis. The TcsL1285–1804 signal was detected via the HA tag. The endothelial cell marker, CD31, was detected to label endothelial cells. Nuclei were labeled with DAPI. Scale bar, 100 μm. TcsL1285–1804 is largely co-localized with CD31. Representative images were from one of six independent experiments.
(B-C) Massive accumulation of fluid in the thoracic cavity occurred within 4 h after IP injection of TcsL (20 ng per 25 g bodyweight). Co-injection of TcsL with mouse SEMA6A extracellular domain (mSEMA6A-ECD) at 1:1000 and 1:500 (w/w) ratios both reduced accumulation of fluid. Co-injection of TcsL with mouse SEMA6C extracellular domain (mSEMA6C-ECD) at 1:1000 (w/w) ratio did not reduce accumulation of fluid. Injection of Buffer (HBSS, with 0.1% BSA), mSEMA6A-ECD alone, or mSEMA6C-ECD alone were examined in parallel. Boxes indicate ± s.e.m., error bars indicate ± s.d., *, p < 0.01 (one-way ANOVA). The representative picture of the fluid collected from TcsL and TcsL + mSEMA6A-ECD groups are shown in panel B.
(D) Experiments were carried as described in panel C and the edema in lung tissues was evaluated by measuring the dry-to-wet weights. TcsL reduced dry-to-wet weight ratio of lung tissue 4 h after the injection compared with the Buffer group and the mSEMA6A-ECD alone group. Co-injection of TcsL with mSEMA6A-ECD prevented this reduction, while co-injection with mSEMA6C-ECD showed no protection from TcsL. Boxes indicate ± s.e.m., error bars indicate ± s.d., *, p < 0.01 (one-way ANOVA).
(E) Experiments were carried as described in panel C, and the indicated lung tissues were harvested and subjected to histological analysis by H&E staining. TcsL resulted in alveolar hemorrhage (upper panels) and widening of perivascular space (black arrows in lower panels). These pathological changes are reduced in the TcsL + mSEMA6A-ECD groups. Scale bar, 200 μm.
We then evaluated whether SEMA6A-ECD might be able to serve as a receptor decoy and offer a degree of protection for lung tissues in vivo. We produced Fc-tagged mouse SEMA6A-ECD (mSEMA6A-ECD) using mammalian cell expression system. This mSEMA6A-ECD protein was first incubated with TcsL with ratios of 500:1 or 1000:1 (w/w) on ice and then the mixtures were injected into mice via IP route. mSEMA6A-ECD alone did not affect mice, while TcsL alone caused a gradual loss of mobility, labored breath, and eventually reaching the endpoint for euthanization. Previous studies showed massive accumulation of fluid in the thoracic cavity after the injection of the toxin, which is confirmed in our analysis (Figure 5B). Co-injection of mSEMA6A-ECD with TcsL drastically reduced the amount of thoracic fluid compared to TcsL alone analyzed 4 h after injection (Figures 5B and 5C). A Fc-tagged mouse SEMA6C-ECD (mSEMA6C-ECD) was analyzed in parallel as a control, which did not reduce TcsL-induced accumulation of thoracic fluid (Figure 5C). We also produced a His6-tagged mouse SEMA6A-ECD protein without the Fc tag (mSEMA6A-His), which reduced accumulation of thoracic fluid when co-injected with TcsL (Figure S6A).
The edema in lung tissues can also be evaluated by comparing the dry versus wet lung tissue weights. TcsL alone resulted in a greatly decreased dry-to-wet lung tissue weight ratio 4 h after the injection. Co-injection of Fc-tagged mSEMA6A-ECD or mSEMA6A-His with TcsL both prevented this reduction (Figures 5D and S6B), while mSEMA6C-ECD showed no protection (Figure 5D). Histological analysis of the lung tissues at this time point showed that TcsL injection resulted in alveolar hemorrhage and widening of perivascular space, indicating severe alveolar damage and tissue edema around lung vessels, while co-injection of mSEMA6A-ECD reduced these pathological changes (Figure 5E).
To further explore the window of using mSEMA6A-ECD to block TcsL in vivo, we injected TcsL into mice first, followed by injection of mSEMA6A-ECD once 5 min, 20 min, or 60 min later (Figure 6A). Injection of mSEMA6A-ECD 5 min after the initial injection of TcsL still reduced thoracic fluid accumulation and prevented changes in dry-to-wet lung tissue weight ratios (Figures 6B and 6C). Injection of mSEMA6A-ECD 20 min after TcsL injection showed modest degrees of protection of lung tissues, while injection of mSEMA6A-ECD 60 min after TcsL injection showed no protection (Figures 6B and 6C).
Figure 6. Administration of SEMA6A-ECD post-injection of TcsL reduces the toxicity of TcsL on lung tissues in vivo.
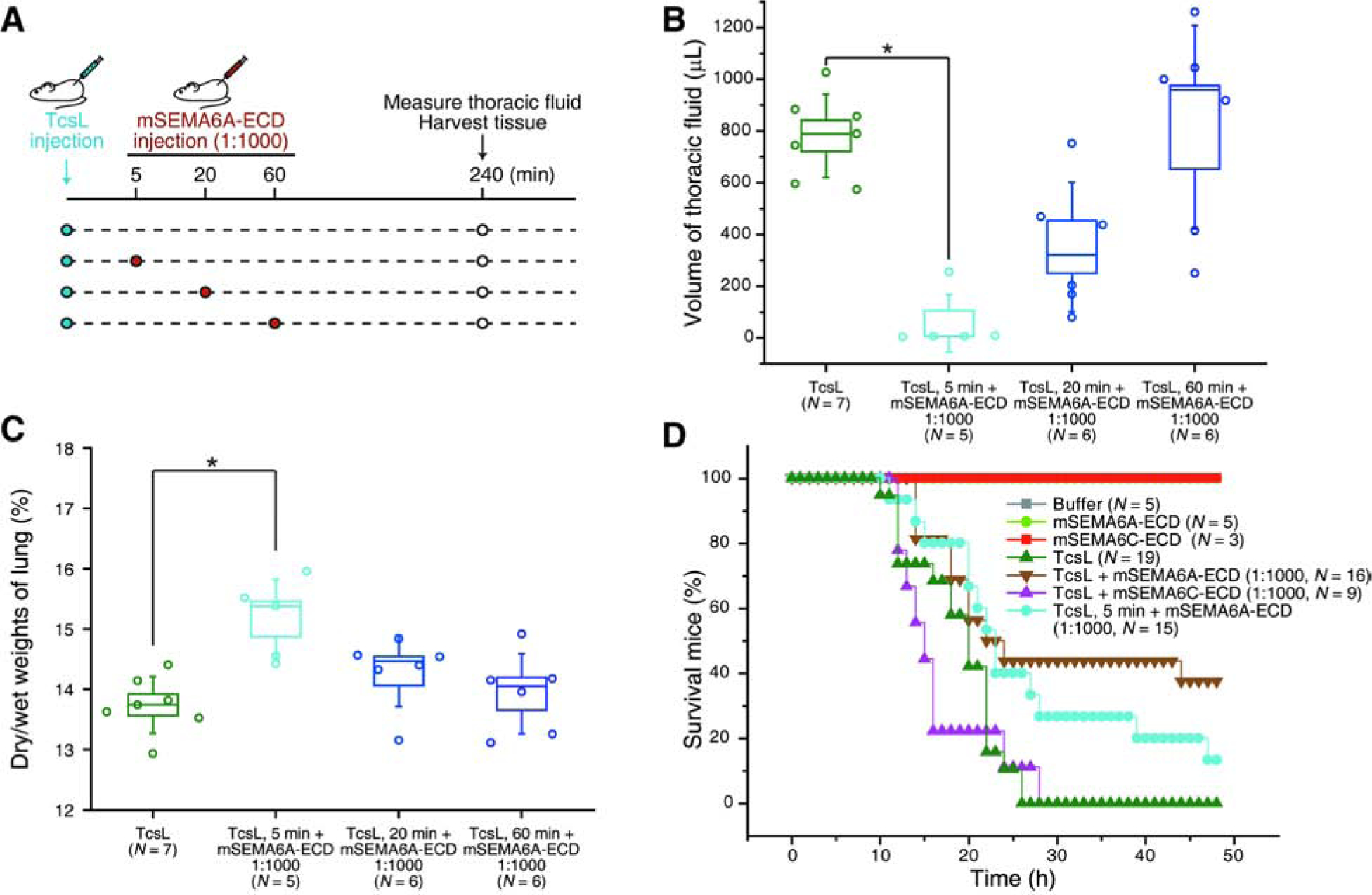
(A-C) Schematic drawing illustrating that TcsL alone was injected first into mice, and mSEMA6A-ECD (1:1000 ratio) was then injected 5 min, 20 min, or 60 min after the injection of TcsL (A). The volume of thoracic fluid was measured 240 min after TcsL injection (B), and the lung tissue was harvested and the dry-to-wet weights of lung tissues were measured (C).
(D) All mice with IP injection of TcsL (20 ng per 25 g bodyweight, N = 19) died within 26 h. Six in a total of sixteen mice with co-injection of TcsL with mSEMA6A-ECD (1:1000 ratio) survived (p < 0.01 comparing with TcsL alone, Kaplan-Meier Log-Rank test). Co-injection of TcsL with mSEMA6C-ECD (1:1000 ratio, N = 9) did not protect mice from death (p > 0.1). Two in a total of fifteen mice injected with mSEMA6A-ECD (1:1000 ratio) 5 min post-injection of TcsL survived (p < 0.01). Injection of Buffer (HBSS with 0.1% BSA), mSEMA6A-ECD, or mSEMA6C-ECD alone showed no toxicity.
We finally assessed whether mSEMA6A-ECD may reduce the lethality of TcsL in vivo. While all mice injected with TcsL alone died within 26 h, ~38% mice co-injected with mSEMA6A-ECD were able to survive and recover, while the control mSEMA6C-ECD showed no protection (Figure 6D). Consistently, co-injection of mSEMA6A-His rescued ~45% of mice from death (Figure S6C), and injection of mSEMA6A-ECD 5 min after the initial injection of TcsL also rescued ~13% mice from death (Figure 6D).
Discussion
Through a genome-wide CRISPR-Cas9 screen, SEMA6A was identified as a candidate receptor for TcsL. Validation assays suggested that both SEMA6A and 6B are redundant receptors. This is confirmed by BLI analysis showing a direct, specific, and high-affinity interaction of TcsL with the ECDs of SEMA6A and 6B. The relevance of these interactions for toxin binding and entry were further validated using loss-of-function and gain-of-function approaches in cells, as well as competition assays using SEMA6A-ECDs on cells and in vivo. Together, these data establish SEMA6A/6B as functionally relevant receptors for TcsL.
Our screen also identified TBC1D3, which is further validated in cell-rounding assays. TBC1D3 is a member of the TBC1 (Tre-2/Bub2/Cdc16) domain family of proteins, which are known to act as GTPase activating protein (GAP). TBC1D3 has been reported to activate the small GTPase Rab5A, which plays critical roles in regulating early endosome trafficking. However, disruption of TBC1D3 did not affect the sensitivity of cells to TcdB, suggesting that its disruption specifically renders cells less sensitivity to TcsL. How TBC1D3 contributes to TcsL action remains to be established.
The binding site in TcsL for SEMA6A/6B is within a region homologous to the FZD-binding region in TcdB. Furthermore, exchanging the middle portion of this binding region containing the FZD-binding interface also switched the binding specificity: the resulting mosaic TcdB(TcsL1431–1601) binds to SEMA6-ECD, while TcsL(TcdB1431–1602) binds to FZD-CRD2. These data demonstrate that the location for the receptor-binding domain is conserved between these two homologous toxins. However, they evolved to recognize distinct receptors, likely due to residue variations within the binding interface. The crystal structure of TcdB-FBD showed that this region forms an overall “L” shape, with the binding interface at the turning point of the “L”, which might be a hotspot for receptor-recognition (Chen et al., 2018). Whether the location of this receptor-binding region is a common theme for the LCT family remains to be further determined. It has been reported that TcdA utilizes sulfated glycosaminoglycans and low-density lipoprotein receptor (LDLR) as CROPs-independent receptors (Tao et al., 2019), while TpeL utilizes a LDLR family member LRP1 as the receptor (Schorch et al., 2014). The receptor-recognition regions in TcdA and TpeL however remain to be defined.
Within SEMA6 class, TcsL showed high-affinity binding to only SEMA6A and 6B. The structural basis for this binding preference within class 6 remains to be established. SEMA6 family act mainly by binding to their cognate receptor class A Plexins, which are large cell surface membrane proteins with a single transmembrane domain and a cytoplasmic domain that induces cytoskeleton changes when Plexin is activated by binding to SEMA6. Binding of PlexinA2 to immobilized SEMA6A-ECD showed a low binding KD ~ 2.3 μM, rendering it difficult to examine whether there is a direct competition between PlexinA2 and TcsL for binding to SEMA6 (Janssen et al., 2010; Nogi et al., 2010). It remains to be determined whether binding of TcsL may inhibit SEMA6-PlexinA2 interactions and disrupt PlexinA2-mediated downstream signaling.
It is likely that TcsL utilize additional receptors and attachment factors besides SEMA6A/6B in cultured cells, which might be the reason for modest changes in toxin sensitivity observed in our loss-of-function approaches and a rather wide range of sensitivity among different cell lines. The CROPs domains in LCT family are well known for recognizing carbohydrates and mediating attachment to cell surfaces. Although the carbohydrate-binding ability of TcsL-CROP remains to be characterized, it is likely that CROPs-carbohydrate interactions contributes to binding and entry of TcsL on cultured cells. The CROPs in TcdB is also essential for binding to a specific receptor CSPG4 (chondroitin sulfate proteoglycan 4), although the exact binding interface for CSPG4 remains to be established (Gupta et al., 2017; Tao et al., 2016; Yuan et al., 2015). Whether TcsL-CROPs is involved in recognizing additional specific protein receptor(s) remains to be determined, and potentially could be investigated with a mutated TcsL that does not bind to SEMA6A/6B. Although it remains possible that there are additional receptors contributing to targeting of TcsL to lung endothelial cells, our findings that SEMA6A-ECDs was able to reduce the edema induced by TcsL on lung tissues and decrease the mortality rate in mice demonstrate that SEMA6A/6B are major pathophysiologically relevant receptors to TcsL toxicity in vivo. Blocking SEMA6A/6B-TcsL recognition thus represents a potential therapeutic approach for neutralizing TcsL in vivo and reducing the high fatality rate associated with P. sordellii infections.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Min Dong (min.dong@childrens.harvard.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
All cell lines were cultured in DMEM media plus 10% fetal bovine serum (FBS) and 100 U penicillin / 0.1 mg/mL streptomycin in a humidified atmosphere of 95 % air and 5 % CO2 at 37 °C. HUVECs were cultured in F-12K media contains 10% FBS, 0.1 mg/mL heparin, and endothelia cell growth supplement (ECGS).
Mice
All the animal studies were conducted according with ethical regulations under protocols approved by the Institute Animal Care and Use Committee (IACUC) at Boston Children’s Hospital (18-10-3794R). Ten to twelve-weeks-old, CD-1 strain mice (both male and female were examined randomly) were purchased from Charles River. Mice were housed with food and water ad libitum and monitored under the care of full-time staff.
METHOD DETAILS
cDNA constructs.
The selected sgRNA sequences (SEMA6A-I: TTGCCATGCGAAATACTGAT; SEMA6A-II: GGCTTGTGGCCCACAAACAC; SEMA6B-I: CGAGTGTCGAAACTTCGTAA; SEMA6B-II: TGGGGGGCTCCAGCTCTACG; SEMA6C: GCTGAATGAGTTCGTTCCAC; SEMA6D-I: AGTGATAGTCGACAGTATTA; SEMA6D-II: GAAAGCTGACTGCCCTCAAC; EMB: AGTCATAACATATCACTGAC; RTN4RL2: ATCGAGACAAGATGCTGCCC; GPR87: GTCTGCGTGTAATGTTTGCC; SLC27A5: GTCGAACTGCACCAGCTCAA; PCDHGB6; TTTCGACCAGACGTCCTACG; TBC1D3: GCTTCCGCTTTGATGTGGCA; ATP6V0B: TTGTTGTTGTAGCTTCGAAA; SLC25A31: CCCTTGAATTGTCGCTCCTC; PPAT: TTCGTTGTTGAAACACTTCA; ACTG2: GTGTGACATTGACATCCGTA) were cloned into LentiGuide-Puro vector (Addgene, #52963). TcsL1285–1804, TcsL1865–2364, TcsL(TcdB1431–1602), and TcdB(TcsL1431–1601) were cloned into pET28a vector (Novagen) with His tag at their C-termini by Gibson Assembly (NEB, E2621). An extra HA-tag was introduced to the C-terminus of TcsL1285–1804 to generate the HA-tagged version. The cDNAs of human SEMA proteins were obtained from the indicated vendors: SEMA6A (Sino Bio, HG11189-M), SEMA6B (GE Dharmacon, 40147342), SEMA6C (Sino Bio, HG23441-UT), and SEMA6D (R&D Systems, RDC2156). Full-length SEMA6A, SEMA6B, SEMA6C, and SEMA6D with triple-FLAG tag at their C-termini (with EFGSGSGS linker) were cloned into either pcDNA3.1 vector (Invitrogen, V800–20) or pLenti-Hygro vector (Addgene, #17484). The mSEMA6A-His construct was generated by subcloning mouse SEMA6A-ECD into pcDNA3.1 vector with a C-terminal His tag. 1D4-tagged full-length mouse FZD2 was obtained from Addgene (pRK5-mFzd2–1D4, #42264).
Recombinant proteins.
Recombinant His-tagged TcsL (from P. sordellii strain 6018) and TcdB was subcloned into pHis1522 vector and expressed in B. megaterium following the supplier’s protocol (MoBiTec GmbH, Germany). TcdB-FBD was constructed and purified as described previously (Chen et al., 2018). TcsL1285–1804, TcsL1865–2364, TcsL(TcdB1431–1602), and TcdB(TcsL1431–1601) were expressed in E. coli (BL21 strain) and purified as His-tagged proteins. The recombinant human Fc-tagged chimera proteins were purchased from R&D Systems: SEMA6A-Fc (1146-S6), SEMA6B-Fc (2264-S6), SEMA6C-Fc (2219-S6), SEMA6D-Fc (2095-S6), SEMA4D-Fc (5235-S4), SEMA5A-Fc (6584-S5), SEMA7A-Fc (1835-S3), CRD2-Fc (1307-FZ), and IgG-Fc (110-HG). The mSEMA6A-ECD and mSEMA6C-ECD constructs were obtained from Addgene (#72163 and #72167). mSEMA6A-ECD, mSEMA6A-His, and mSEMA6C-ECD proteins were expressed using Expi293F cells (Life Technologies). Briefly, 3 × 107 Expi293F cells were transfected with 37.5 μg plasmid using PEIMax (1 mg/mL) (Polysciences Inc.). The culture was harvested 5 days after transfection. The proteins in the culture medium were collected and purified as His-tagged proteins.
Cell-rounding assay.
The cytopathic effect (cell rounding) of TcsL was analyzed using the standard cell-rounding assay. Briefly, cells were exposed to TcsL or TcdB for the indicated time. The phase-contrast images of cells were taken (Olympus IX51, 10~20 × objectives). A zone of 300 × 300 μm was selected randomly, which contains 50~150 cells. Round-shaped and normal-shaped cells were counted manually. The percentage of round-shaped cells was analyzed using the OriginPro (OriginLab, v8.5) and Excel (Microsoft, 2007). Data were represented as mean ± s.d. from three independent biological replicates. Data were considered significant when p-value < 0.01 (one-way ANOVA). Statistical analysis was performed using Excel (Microsoft, 2007).
Genome-wide CRISPR-Cas9 screens.
A549 cells that stably express Cas9 were generated using Lenti-SpCas9-Blast (Addgene, #52962) and selected using 10 μg/mL Blasticidin S (RPI, B12150.01). The GeCKO-V2 sgRNA sub-library A and B were obtained from Addgene (#1000000049) and independently packed into lentiviral libraries. A549-Cas9 Cells were transduced with sgRNA lentiviral library at a MOI (multiplicity of infection) of 0.2. Polybrene (Santa Cruz, sc-134220, 8 μg/mL) was added to the medium to facilitate the viral transduction. Cells were cultured in lentivirus-containing medium for two days. Infected cells were selected with 10 μg/mL Puromycin (Thermo Scientific, A1113830). At least 3.3 × 107 (for sub-library A) or 2.9 × 107 (for sub-library B) cells were plated onto four 15-cm culture dishes to ensure sufficient sgRNA coverage, with each sgRNA being represented around 500 times. These cells were either saved as Round 0 (R0) samples or exposed to TcsL for 24 h. The survival cells were washed and re-seeded within toxin-free medium until ~70% confluence, followed by the next round of selection with TcsL. In total three rounds of selections were performed with 0.5, 1, and 2 ng/mL TcsL, respectively. The genomic DNA of survival cells of each round was extracted using a commercial kit (Qiagen, 13323). The DNA fragments containing the sgRNA sequences were amplified by PCR using primers lentiGP-1_F (AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG) and lentiGP-3_R (ATGAATACTGCCATTTGTCTCAAGATCTAGTTACGC). Next-generation sequencing was performed by a commercial vendor (Genewiz, Illumina MiSeq).
Generating KO cells and lentiviral transduction.
A549-Cas9 and HeLa-Cas9 cells were utilized for generating KO cells via lentiviral transduction of sgRNAs. SEMA6A/6B double KO cells were generated by co-transduction with two viruses (SEMA6A-I + SEMA6B-I, or SEMA6A-II + SEMA6B-II, respectively). Mixed populations of infected cells were selected with puromycin (10 μg/mL for A549, and 5 μg/mL for HeLa, respectively). HeLa or 5637 cells were transduced with lentiviruses expressing SEMA6 family proteins and cells were selected with 200 μg/mL Hygromycin B (EMD Millipore, 400051).
Immunoblot analysis.
Cells were scraped and washed three times with PBS. Cell pellets were lysed with RIPA buffer (50 mM Tris, pH 7.5, 1% NP-40, 150 mM NaCl, 0.5% sodium deoxycholate, 1% SDS, protease inhibitor cocktail) on ice for 30 min. The protein amounts in cell lysate were determined by BCA assay (Thermo Scientific, 23225). The cell lysates were heat for 5 min, analyzed by SDS-PAGE, and transferred onto a nitrocellulose membrane (GE Healthcare, 10600002). The membrane was blocked with TBS-T buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 0.1 % Tween-20) containing 5 % skim milk at room temperature (RT) for 40 min. The membrane was then incubated with the primary antibodies for 1 h, washed, and incubated with secondary antibodies for 1 h. Signals were detected using the enhanced chemiluminescence method (Thermo Fisher Scientific, 34080) with a Fuji LAS3000 imaging system. The images were analyzed and quantified using ImageJ software (Version 1.52o)
Biolayer interferometry (BLI) assay.
The binding affinities (KD) between TcsL and SEMA proteins were measured using the BLI assay with the BLItz system (ForteBio) and were calculated using the BLItz system software. Briefly, 10 μg/mL Fc-tagged proteins were immobilized onto capture biosensors (Dip and Read Anti-Human IgG Fc Capture, ForteBio) and balanced with DPBS (0.5% BSA, w/w). The biosensors were then exposed to 1 μM or the indicated concentrations of TcsL, TcdB, TcsL1285–1804, TcsL1865–2364, TcdB-FBD, or TcsL-TcdB chimera fragments, followed by dissociation in DPBS (0.5% BSA, w/w). The Endo H digestion of SEMA6A-Fc was carried out following the supplier’s protocol (NEB, P0702) under non-denaturing conditions. The experiments were repeated three times.
TcsL binding and immunostaining.
HeLa cells were transfected using PolyJet (SignaGen, SL100688), seeded onto glass coverslips (Hampton, HR3–239) in 24-well plates, and incubated for 48 h until ~ 70% confluence. Cells were washed three times with ice-cold PBS, and were incubated with 7.5 μg/mL TcsL1285–1804-HA in medium on ice for 60 min. Cells were washed, fixed with 4% paraformaldehyde (PFA) for 20 min at RT, permeabilized with 0.3% Triton X-100 for 30 min, blocked with 10% goat serum for 40 min, followed by incubation with primary antibodies for 1 h and fluorescence-labeled secondary antibodies for another 1 h. Slides were sealed within DAPI-containing mounting medium (SouthernBiotech, 0100–20). Fluorescent images were captured using an Olympus DSU-IX81 Spinning Disk Confocal System. Images were pseudo-colored and analyzed using ImageJ.
Competition assays with SEMA proteins.
Toxins (40 pM TcsL for A549, 4 pM TcsL for HUVECs, 4 nM TcsL for HeLa, 16 nM TcsL for 5637, or 0.4 pM TcdB for A549) were pre-mixed with or without recombinant Fc-tagged proteins (SEMA proteins or CRD2) in fresh culture medium and incubated on ice for 1 h. The mixtures were then added into cell culture medium. Cells were further incubated at 37 °C and the percentages of cell rounding were examined.
SEMA6A and 6B knock-down in HUVECs.
The siRNA sequence targeting human SEMA6A was selected from a previous publication (Hasson et al., 2013). Three siRNA sequences targeting human SEMA6B and non-targeting scramble siRNA were designed and ordered from Life Technologies. The knockdown efficiency was validated by immunoblot analysis using HEK293T cells transfected with FLAG-tagged SEMA6A or SEMA6B. The SEMA6B siRNA II was selected to knock down SEMA6B. HUVECs were incubated in 96-well dish plates for 24 h. When the confluency reached 70%, cells were incubated in serum-free medium for 8 h. The SEMA6A siRNA and SEMA6B siRNA II (0.3 μM) were combined and transfected into cells using Lipofectamine RNAiMAX (ThermoFisher). TcsL treatment and cell rounding assays were carried out 48 h later.
Binding of TcsL to lung tissues.
Mice were injected with Buffer (Hank’s balanced salt solution, HBSS, 0.1% BSA, sterilized by filtration) or 0.2 mg TcsL1285–1804-HA via IP route. The lung tissues were harvested 30 min or 60 min later and embedded in O.C.T. compound (Tissue-Tek). Tissues were sectioned, fixed and subjected into immunostaining assays without permeabilization. Binding of toxins was detected using an anti-HA antibody. Endothelial cells were marked using an anti-CD31 antibody. Nuclei were labeled using DAPI. Fluorescent images were captured with an Olympus DSU-IX81 Spinning Disk Confocal System. Images were pseudo colored and quantified using ImageJ.
In vivo competition assays.
Mice (CD-1 strain, 10–12 weeks of age, male and female, randomly separated into experimental groups) were purchased from Charles River and were kept in house for 2 weeks before experiments (bodyweight ~25 g). TcsL (20 ng / 25 g bodyweight) was diluted in 100 μL Buffer (HBSS, 0.1% BSA) and injected into mice via IP route. For the pre-incubation groups, TcsL was premixed with mSEMA6A-ECD, mSEMA6C-His or mSEMA6C-ECD at indicated ratios and incubated on ice for 1 h. The mixtures were then injected into mice via IP route. For the non-preincubation groups, mSEMA6A-ECD was injected into mice 5 min, 20 min, or 60 min after TcsL injection. Buffer, mSEMA6A-ECD alone, mSEMA6A-His alone, and mSEMA6C-ECD alone were used as controls. Mice were observed for 48 h after TcsL injection and the time-of-death was recorded. Subgroups of mice were euthanized 4 h after the injection to collect the fluid in the thoracic cavity, and lung tissues were also harvested and weighted (wet weight). The tissues were then dried in an oven at 100 °C overnight and weighted again (dry weight). Parts of freshly harvested lung tissues were fixed with 10% formalin in phosphate buffer and embedded in paraffin. Tissues were sectioned and histological analysis were carried out with hematoxylin and eosin (H&E) staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were considered statistically significant when p < 0.01 using one-way ANOVA or Kaplan-Meier Log-Rank test as indicated in the Figure legends. Data were represented as mean ± s.d. from at least three independent biological replicates. Statistical analysis was performed using OriginPro and Excel software.
DATA AND SOFTWARE AVAILABILITY
The published article includes all dataset generated or analyzed during this study. The full list of identified genes is included in Data S1.
Supplementary Material
Data S1: Lists of identified genes from R0 to R3 in an Excel file, ranked based on the number of sgRNAs and total reads. Related to Figure 1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Actin | Aves Labs | ACT-1010 |
| Mouse monoclonal anti-HA | BioLegend | 901502 |
| Chicken polyclonal anti-HA | AVES Labs | ET-HA100 |
| Mouse monoclonal anti-FLAG | Sigma | F3165 |
| Mouse monoclonal anti-Rhodopsin (1D4) | ThermoFisher | MA1–722 |
| Rabbit polyclonal anti-SEMA6A | ThermoFisher | PA5–81009 |
| Purified Rat Anti-Mouse CD31 | BD Pharmingen | 553370 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| All the other chemicals | Sigma | N/A |
| PolyJet | SignaGen | SL100688 |
| PEIMax | Polysicences | 24765–1 |
| Lipofectamine RNAiMAX | ThermoFisher | 13778150 |
| Polybrene | Santa Cruz | sc-134220 |
| Dulbecco’s Modified Eagle Medium | Life technologies | Cat#11995–065 |
| Fetal bovine serum | Life technologies | Cat#26140–079 |
| Penicillin/streptomycin | Life technologies | Cat#15140–122 |
| F-12K Medium | ATCC | 30–2004 |
| Heparin | Sigma | H3393 |
| Endothelial cell growth supplement | BD Biosciences | # 354006 |
| Puromycin | ThermoFisher | A1113830 |
| Blasticidin S | RPI | B12150.01 |
| Hygromycin B | EMD Millipore | 400051 |
| Protease Inhibitor Cocktail | Roche | 4693159001 |
| Nitrocellulose membrane | GE Healthcare | 10600002 |
| DAPI-containing mounting medium | SouthernBiotech | 0100–20 |
| Endo H | NEB | P0702 |
| Recombinant human Fc-tagged SEMA6A | R&D Systems | 1146-S6 |
| Recombinant human Fc-tagged SEMA6B | R&D Systems | 2264-S6 |
| Recombinant human Fc-tagged SEMA6C | R&D Systems | 2219-S6 |
| Recombinant human Fc-tagged SEMA6D | R&D Systems | 2095-S6 |
| Recombinant human Fc-tagged SEMA4D | R&D Systems | 5235-S4 |
| Recombinant human Fc-tagged SEMA5A | R&D Systems | 6584-S5 |
| Recombinant human Fc-tagged SEMA7A | R&D Systems | 1835-S3 |
| Recombinant human Fc-tagged CRD2 | R&D Systems | 1307-FZ |
| Recombinant human Fc-tagged IgG | R&D Systems | 110-HG |
| TcsL | This paper | N/A |
| TcsL1285–1804 | This paper | N/A |
| TcsL1285–1804-HA | This paper | N/A |
| TcsL1865–2364 | This paper | N/A |
| TcdB-FBD | Rongsheng Jin | Chen et al., 2018 |
| TcsL(TcdB1431–1602) | This paper | N/A |
| TcdB(TcsL1431–1601) | This paper | N/A |
| mSEMA6A-ECD | This paper | N/A |
| mSEMA6A-His | This paper | N/A |
| mSEMA6C-ECD | This paper | N/A |
| Critical Commercial Assays | ||
| Gibson Assembly | NEB | E2621 |
| Bacillus Expression Systems | MoBiTec GmbH | BMEG04 |
| BCA assay kit | ThermoFisher | 23225 |
| Genomic DNA extraction kit | Qiagen | 13323 |
| Enhanced chemiluminescence kit | ThermoFisher | 34080 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | CCL-2 |
| A549 | ATCC | CRM-CCL-185 |
| 5637 | ATCC | HTB-9 |
| ACHN | ATCC | CRL-1611 |
| Caco-2 | ATCC | HTB-37 |
| U2OS | ATCC | HTB-96 |
| Huh7 | Y. Matsuura | N/A |
| HEK293T | ATCC | CRL-3216 |
| HUVECs | Lonza | 00191027 |
| Expi293F | ThermoFisher | A14527 |
| A549-SEMA6A-KO-Mix-I | This paper | N/A |
| A549-SEMA6A-KO-Mix-II | This paper | N/A |
| A549-SEMA6B-KO-Mix-I | This paper | N/A |
| A549-SEMA6B-KO-Mix-II | This paper | N/A |
| A549-SEMA6C-KO-Mix | This paper | N/A |
| A549-SEMA6D-KO-Mix-I | This paper | N/A |
| A549-SEMA6D-KO-Mix-II | This paper | N/A |
| A549-EMB-KO-Mix | This paper | N/A |
| A549-RTN4RL2-KO-Mix | This paper | N/A |
| A549-GPR87-KO-Mix | This paper | N/A |
| A549-SLC27A5-KO-Mix | This paper | N/A |
| A549-PCDHGB6-KO-Mix | This paper | N/A |
| A549-TBC1D3-KO-Mix | This paper | N/A |
| A549-ATPV0B-KO-Mix | This paper | N/A |
| A549-SLC25A31-KO-Mix | This paper | N/A |
| A549-PPAT-KO-Mix | This paper | N/A |
| A549-ACTG2-KO-Mix | This paper | N/A |
| A549-SEMA6A/6B-KO-Mix-I | This paper | N/A |
| A549-SEMA6A/6B-KO-Mix-II | This paper | N/A |
| HeLa-SEMA6A-KO-Mix-I | This paper | N/A |
| HeLa-SEMA6A-KO-Mix-II | This paper | N/A |
| HeLa-SEMA6B-KO-Mix-I | This paper | N/A |
| HeLa-SEMA6B-KO-Mix-II | This paper | N/A |
| HeLa-SEMA6C-KO-Mix | This paper | N/A |
| HeLa-SEMA6D-KO-Mix-I | This paper | N/A |
| HeLa-SEMA6D-KO-Mix-II | This paper | N/A |
| HeLa-SEMA6A/6B-KO-Mix-I | This paper | N/A |
| HeLa-SEMA6A/6B-KO-Mix-II | This paper | N/A |
| HeLa + SEMA6A-FLAG-C | This paper | N/A |
| HeLa + SEMA6B-FLAG-C | This paper | N/A |
| HeLa + SEMA6C-FLAG-C | This paper | N/A |
| HeLa + SEMA6D-FLAG-C | This paper | N/A |
| 5637 + SEMA6A-FLAG-C | This paper | N/A |
| 5637 + SEMA6B-FLAG-C | This paper | N/A |
| 5637 + SEMA6C-FLAG-C | This paper | N/A |
| 5637 + SEMA6D-FLAG-C | This paper | N/A |
| Experimental Models: Mice | ||
| CD-1 strain | Charles River | #022 |
| Oligonucleotides | ||
| See Table S1 for oligonucleotides used in these studies | This paper | N/A |
| Recombinant DNA | ||
| SEMA6A cDNA | Sino Bio | HG11189-M |
| SEMA6B cDNA | GE Dharmacon | 40147342 |
| SEMA6C cDNA | Sino Bio | HG23441-UT |
| SEMA6D cDNA | R&D Systems | RDC2156 |
| pcDNA3.1 | ThermoFisher | V80020 |
| pET28a | Novagen | 69864 |
| pHIS1522 | MoBiTec GmbH | BMEG12 |
| pMD2.G | Addgene | #12259 |
| pSPAX2 | Addgene | #12260 |
| LentiGuide-puro | Addgene | #52963 |
| Lenti-SpCas9 blast | Addgene | #104997 |
| pLenti-Hygro | Addgene | #17484 |
| Sema6a.a-Fc-His | Addgene | #72163 |
| Sema6c-Fc-His | Addgene | #72167 |
| pRK5-mFzd2–1D4 | Addgene | 42264 |
| GeCKO-V2 sgRNA library | Addgene | #1000000049 |
| pET28a-TcdB-FBD | Liang Tao | Chen et al., 2018 |
| Lentiguide-SEMA6A-I | This paper | N/A |
| Lentiguide-SEMA6A-II | This paper | N/A |
| Lentiguide-SEMA6B-I | This paper | N/A |
| Lentiguide-SEMA6B-II | This paper | N/A |
| Lentiguide-SEMA6C | This paper | N/A |
| Lentiguide-SEMA6D-I | This paper | N/A |
| Lentiguide-SEMA6D-II | This paper | N/A |
| Lentiguide-EMB | This paper | N/A |
| Lentiguide-RTN4RL2 | This paper | N/A |
| Lentiguide-GPR87 | This paper | N/A |
| Lentiguide-SLC27A5 | This paper | N/A |
| Lentiguide-PCDHGB6 | This paper | N/A |
| Lentiguide-TBC1D3 | This paper | N/A |
| Lentiguide-ATP6V0B | This paper | N/A |
| Lentiguide-SLC25A31 | This paper | N/A |
| Lentiguide-PPAT | This paper | N/A |
| Lentiguide-ACTG2 | This paper | N/A |
| pcDNA3.1-SEMA6A-FLAG-C | This paper | N/A |
| pcDNA3.1-SEMA6A-ECD-His | This paper | N/A |
| LentiHygro-SEMA6A-FLAG-C | This paper | N/A |
| pcDNA3.1-SEMA6B-FLAG-C | This paper | N/A |
| LentiHygro-SEMA6B-FLAG-C | This paper | N/A |
| pcDNA3.1-SEMA6C-FLAG-C | This paper | N/A |
| LentiHygro-SEMA6C-FLAG-C | This paper | N/A |
| pcDNA3.1-SEMA6D-FLAG-C | This paper | N/A |
| LentiHygro-SEMA6D-FLAG-C | This paper | N/A |
| pET28a-TcsL-1285-1804 | This paper | N/A |
| pET28a-TcsL-1856-2364 | This paper | N/A |
| pET28a-TcsL-1285-1804-HA | This paper | N/A |
| pET28a-TcsL(TcdB-1431-1602) | This paper | N/A |
| pET28a-TcdB(TcsL-1431-1601) | This paper | N/A |
| Software and Algorithms | ||
| OriginPro v8.5 | OriginLab | https://www.originlab.com/ |
| Excel | Microsoft | https://products.office.com/en-us/home |
| BLItz pro. Software Version 1.1.0.29 | ForteBio | https://www.fortebio.com/products/label-free-bli-detection/personal-assay-blitz-system |
| ImageJ Version 1.52o | Image J | https://imagej.nih.gov/ij/ |
| Others | ||
| Fluorescence microscope | Olympus | IX51 |
| Spinning Disk Confocal Microscope | Olympus | DSU-IX81 |
| Fuji LAS3000 imaging system | Fuji | LAS3000 |
| Personal assay BLItz System | ForteBio | BLItz System |
| Dip and Read Anti-Human IgG Fc Capture | ForteBio | 18–5060 |
Highlights:
Genome-wide CRISPR-Cas9 screen identifies receptors of P. sordellii toxin TcsL
Semaphorin (SEMA) 6A and 6B are pathophysiologically relevant receptors for TcsL
Receptor-binding interface/specificity of TcsL and C. difficile TcdB can be swapped
SEMA6A extracellular domain proteins reduce co-injected TcsL toxicity in mice
Acknowledgments
We thank H. Tatge (Hannover Medical School, Germany) for toxin purification. This study was partially supported by grants from National Institute of Health (NIH) (R01NS080833, R01AI132387, R01AI139087, and R21NS106159 to M.D.; R01HL093242 and R01HL130845 to H.C.), and Intelligence Advanced Research Projects Actvity (IARPA) (grant number W911NF-17-2-0089 to M.D.). R.G. acknowledges support by the Federal State of Lower Saxony, Niedersächsisches Vorab (VWZN3215/VWZN3266, VWZN3380). M.D. acknowledges support by the NIH-funded Harvard Digestive Disease Center (P30DK034854) and Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (P30HD18655). M.D. holds the Investigator in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflicts of interest.
References
- Aktories K, Schwan C, and Jank T (2017). Clostridium difficile Toxin Biology. Annu Rev Microbiol 71, 281–307. [DOI] [PubMed] [Google Scholar]
- Aldape MJ, Bryant AE, and Stevens DL (2006). Clostridium sordellii infection: epidemiology, clinical findings, and current perspectives on diagnosis and treatment. Clin Infect Dis 43, 1436–1446. [DOI] [PubMed] [Google Scholar]
- Arseculeratne SN, Panabokke RG, and Wijesundera S (1969). The toxins responsible for the lesions of Clostridium sordelli gas gangrene. J Med Microbiol 2, 37–53. [DOI] [PubMed] [Google Scholar]
- Bette P, Oksche A, Mauler F, von Eichel-Streiber C, Popoff MR, and Habermann E (1991). A comparative biochemical, pharmacological and immunological study of Clostridium novyi alpha-toxin, C. difficile toxin B and C. sordellii lethal toxin. Toxicon 29, 877–887. [DOI] [PubMed] [Google Scholar]
- Carter GP, Awad MM, Hao Y, Thelen T, Bergin IL, Howarth PM, Seemann T, Rood JI, Aronoff DM, and Lyras D (2011). TcsL is an essential virulence factor in Clostridium sordellii ATCC 9714. Infect Immun 79, 1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Tao L, Wang T, Zhang J, He A, Lam KH, Liu Z, He X, Perry K, Dong M, et al. (2018). Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couchman EC, Browne HP, Dunn M, Lawley TD, Songer JG, Hall V, Petrovska L, Vidor C, Awad M, Lyras D, et al. (2015). Clostridium sordellii genome analysis reveals plasmid localized toxin genes encoded within pathogenicity loci. BMC Genomics 16, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanabal M, Wu F, Alvarez E, McQueeney KD, Jeffers M, MacDougall J, Boldog FL, Hackett C, Shenoy S, Khramtsov N, et al. (2005). Recombinant semaphorin 6A-1 ectodomain inhibits in vivo growth factor and tumor cell line-induced angiogenesis. Cancer Biol Ther 4, 659–668. [DOI] [PubMed] [Google Scholar]
- Fischer M, Bhatnagar J, Guarner J, Reagan S, Hacker JK, Van Meter SH, Poukens V, Whiteman DB, Iton A, Cheung M, et al. (2005). Fatal toxic shock syndrome associated with Clostridium sordellii after medical abortion. N Engl J Med 353, 2352–2360. [DOI] [PubMed] [Google Scholar]
- Genth H, Hofmann F, Selzer J, Rex G, Aktories K, and Just I (1996). Difference in protein substrate specificity between hemorrhagic toxin and lethal toxin from Clostridium sordellii. Biochem Biophys Res Commun 229, 370–374. [DOI] [PubMed] [Google Scholar]
- Genth H, Pauillac S, Schelle I, Bouvet P, Bouchier C, Varela-Chavez C, Just I, and Popoff MR (2014). Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell Microbiol 16, 1706–1721. [DOI] [PubMed] [Google Scholar]
- Geny B, Grassart A, Manich M, Chicanne G, Payrastre B, Sauvonnet N, and Popoff MR (2010). Rac1 inactivation by lethal toxin from Clostridium sordellii modifies focal adhesions upstream of actin depolymerization. Cell Microbiol 12, 217–232. [DOI] [PubMed] [Google Scholar]
- Geny B, Khun H, Fitting C, Zarantonelli L, Mazuet C, Cayet N, Szatanik M, Prevost MC, Cavaillon JM, Huerre M, et al. (2007). Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am J Pathol 170, 1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Zhang Z, Sugiman-Marangos SN, Tam J, Raman S, Julien JP, Kroh HK, Lacy DB, Murgolo N, Bekkari K, et al. (2017). Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor binding determinants. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenberg G, Papatheodorou P, Genisyuerek S, Lu W, Jank T, Einsle O, and Aktories K (2011). Inositol hexakisphosphate-dependent processing of Clostridium sordellii lethal toxin and Clostridium novyi alpha-toxin. J Biol Chem 286, 14779–14786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Senn T, Opp JS, Young VB, Thiele T, Srinivas G, Huang SK, and Aronoff DM (2010). Lethal toxin is a critical determinant of rapid mortality in rodent models of Clostridium sordellii endometritis. Anaerobe 16, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley-Tassell LE, Awad MM, Seib KL, Scarselli M, Savino S, Tiralongo J, Lyras D, Day CJ, and Jennings MP (2019). Lectin Activity of the TcdA and TcdB Toxins of Clostridium difficile. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, et al. (2013). High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Vanlandewijck M, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data 5, 180160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Rex G, Aktories K, and Just I (1996). The ras-related protein Ral is monoglucosylated by Clostridium sordellii lethal toxin. Biochem Biophys Res Commun 227, 77–81. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck SC, Klose I, Reichenbach M, Huelsenbeck J, and Genth H (2009). Distinct kinetics of (H/K/N)Ras glucosylation and Rac1 glucosylation catalysed by Clostridium sordellii lethal toxin. FEBS Lett 583, 3133–3139. [DOI] [PubMed] [Google Scholar]
- Jank T, and Aktories K (2008). Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16, 222–229. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Robinson RA, Perez-Branguli F, Bell CH, Mitchell KJ, Siebold C, and Jones EY (2010). Structural basis of semaphorin-plexin signalling. Nature 467, 1118–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I, Selzer J, Hofmann F, Green GA, and Aktories K (1996). Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem 271, 10149–10153. [DOI] [PubMed] [Google Scholar]
- Krivan HC, Clark GF, Smith DF, and Wilkins TD (1986). Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1–3Gal beta 1–4GlcNAc. Infect Immun 53, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez RD, and Wilkins TD (1988). Purification and characterization of Clostridium sordellii hemorrhagic toxin and cross-reactivity with Clostridium difficile toxin A (enterotoxin). Infect Immun 56, 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez RD, and Wilkins TD (1992). Comparison of Clostridium sordellii toxins HT and LT with toxins A and B of C. difficile. J Med Microbiol 36, 30–36. [DOI] [PubMed] [Google Scholar]
- McGregor JA, Soper DE, Lovell G, and Todd JK (1989). Maternal deaths associated with Clostridium sordellii infection. Am J Obstet Gynecol 161, 987–995. [DOI] [PubMed] [Google Scholar]
- Nogi T, Yasui N, Mihara E, Matsunaga Y, Noda M, Yamashita N, Toyofuku T, Uchiyama S, Goshima Y, Kumanogoh A, et al. (2010). Structural basis for semaphorin signalling through the plexin receptor. Nature 467, 1123–1127. [DOI] [PubMed] [Google Scholar]
- Popoff MR (1987). Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect Immun 55, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff MR (2018). Clostridium difficile and Clostridium sordellii toxins, proinflammatory versus anti-inflammatory response. Toxicon 149, 54–64. [DOI] [PubMed] [Google Scholar]
- Popoff MR, Chaves-Olarte E, Lemichez E, von Eichel-Streiber C, Thelestam M, Chardin P, Cussac D, Antonny B, Chavrier P, Flatau G, et al. (1996). Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J Biol Chem 271, 10217–10224. [DOI] [PubMed] [Google Scholar]
- Qa’Dan M, Spyres LM, and Ballard JD (2000). pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 68, 2470–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qa’Dan M, Spyres LM, and Ballard JD (2001). pH-enhanced cytopathic effects of Clostridium sordellii lethal toxin. Infect Immun 69, 5487–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi Jyothsna TS, Tushar L, Sasikala C, and Ramana CV (2016). Paraclostridium benzoelyticum gen. nov., sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii. Int J Syst Evol Microbiol 66, 1268–1274. [DOI] [PubMed] [Google Scholar]
- Schirmer J, and Aktories K (2004). Large clostridial cytotoxins: cellular biology of Rho/Rasglucosylating toxins. Biochim Biophys Acta 1673, 66–74. [DOI] [PubMed] [Google Scholar]
- Schorch B, Song S, van Diemen FR, Bock HH, May P, Herz J, Brummelkamp TR, Papatheodorou P, and Aktories K (2014). LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc Natl Acad Sci U S A 111, 6431–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segarra M, Ohnuki H, Maric D, Salvucci O, Hou X, Kumar A, Li X, and Tosato G (2012). Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood 120, 4104–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Tian S, Zhang J, Liu Z, Robinson-McCarthy L, Miyashita SI, Breault DT, Gerhard R, Oottamasathien S, Whelan SPJ, et al. (2019). Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat Microbiol 4, 1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X, et al. (2016). Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneberg S, Lonnroth I, Torres Lopez JF, Galili U, Halvarsson MO, Angstrom J, and Karlsson KA (1996). Molecular mimicry in the recognition of glycosphingolipids by Gal alpha 3 Gal beta 4 GlcNAc beta-binding Clostridium difficile toxin A, human natural anti alpha-galactosyl IgG and the monoclonal antibody Gal-13: characterization of a binding-active human glycosphingolipid, non-identical with the animal receptor. Glycobiology 6, 599–609. [DOI] [PubMed] [Google Scholar]
- Tucker KD, and Wilkins TD (1991). Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect Immun 59, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C, Kaluza D, Fromel T, Knau A, Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN, Hergenreider E, et al. (2012). MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood 119, 1607–1616. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Varela Chavez C, Haustant GM, Baron B, England P, Chenal A, Pauillac S, Blondel A, and Popoff MR (2016). The Tip of the Four N-Terminal alpha-Helices of Clostridium sordellii Lethal Toxin Contains the Interaction Site with Membrane Phosphatidylserine Facilitating Small GTPases Glucosylation. Toxins (Basel) 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidor C, Awad M, and Lyras D (2015). Antibiotic resistance, virulence factors and genetics of Clostridium sordellii. Res Microbiol 166, 368–374. [DOI] [PubMed] [Google Scholar]
- Vidor CJ, Watts TD, Adams V, Bulach D, Couchman E, Rood JI, Fairweather NF, Awad M, and Lyras D (2018). Clostridium sordellii Pathogenicity Locus Plasmid pCS1–1 Encodes a Novel Clostridial Conjugation Locus. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eichel-Streiber C, and Sauerborn M (1990). Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 96, 107–113. [DOI] [PubMed] [Google Scholar]
- Voth DE, and Ballard JD (2005). Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, and Ballard JD (2007). Critical intermediate steps in Clostridium sordellii lethal toxin-induced apoptosis. Biochem Biophys Res Commun 363, 959–964. [DOI] [PubMed] [Google Scholar]
- Voth DE, Martinez OV, and Ballard JD (2006). Variations in lethal toxin and cholesterol-dependent cytolysin production correspond to differences in cytotoxicity among strains of Clostridium sordellii. FEMS Microbiol Lett 259, 295–302. [DOI] [PubMed] [Google Scholar]
- Yazdani U, and Terman JR (2006). The semaphorins. Genome Biol 7, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, He R, Li C, Guo S, Li S, et al. (2015). Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Lists of identified genes from R0 to R3 in an Excel file, ranked based on the number of sgRNAs and total reads. Related to Figure 1.
Data Availability Statement
The published article includes all dataset generated or analyzed during this study. The full list of identified genes is included in Data S1.