

Abstract
Rare genetic diseases are the result of a continuous forward genetic screen that nature is conducting on humans. Here, we present epistemological and systems biology arguments highlighting the importance of studying these rare genetic diseases. We contend that the expanding catalog of mutations in ∼4,000 genes, which cause ∼6,500 diseases and their annotated phenotypes, offer a wide landscape for discovering fundamental mechanisms required for human development and involved in common diseases. Rare afflictions disproportionately affect the nervous system in children, but paradoxically, the majority of these disease-causing genes are evolutionarily ancient and ubiquitously expressed in human tissues. We propose that the biased prevalence of childhood rare diseases affecting nervous tissue results from the topological complexity of the protein interaction networks formed by ubiquitous and ancient proteins encoded by childhood disease genes. Finally, we illustrate these principles discussing Menkes disease, an example of the discovery power afforded by rare diseases.
Subject Areas: Clinical Genetics, Human Genetics
Graphical Abstract
Clinical Genetics; Human Genetics
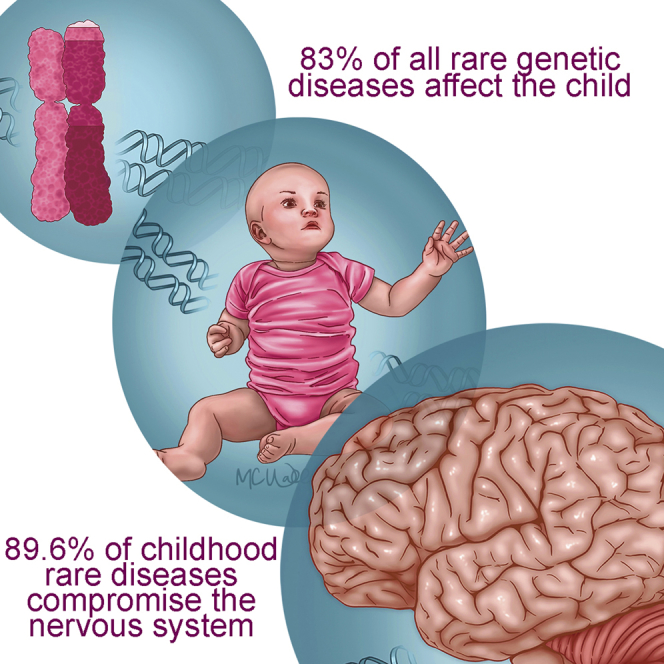
The term “rare disease” describes a group of diseases whose prevalence is so low that they are considered an unviable market for therapeutics in the absence of appropriate incentives and support and too rare to be fully investigated and appropriately managed by professionals (Orphanet, 2020, Waxman, 1983). Among this group, there is a subgroup, the rare genetic diseases target of this article, which frequently affect the nervous system with chronic, progressive, and degenerative pathology that disproportionately affects children (Figure 1). Although the name emphasizes that these diseases are infrequent, the reality is that ∼6,000 rare diseases collectively affect an estimated ∼300 million people worldwide (Nguengang Wakap et al., 2020). In this article, we argue that rare genetic diseases offer a vast landscape for discovery of fundamental and novel biological mechanisms. We will exemplify this concept with three Nobel prizes born out of the study of unique mutations in model genetic organisms. We will discuss examples of how rare monogenic defects have impacted our understanding of important biological problems such as ossification. We will finish this article discussing a rare monogenic disorder of copper metabolism, Menkes disease, because it illustrates how the study of a rare genetic affliction can produce pioneer insight into fundamental biology and prevalent diseases, such as Parkinson disease. The genetic tractability of monogenic defects, their penetrance in early life, and the expanding collection of human mutations and curated phenotypes make rare diseases ideal targets of study for discovering evolutionarily conserved biological mechanisms that go awry in common human diseases. The present and expanding sophistication of genome-wide expression analysis tools, such as genealogical and interactome proteomics (Comstra et al., 2017, Gokhale et al., 2012, Gokhale et al., 2019, Perez-Cornejo et al., 2012, Zlatic et al., 2018), lends itself to comprehensive study of these diseases as a way to deepen our understanding of how a defective protein node participates in the pathogenesis of rare and common diseases.
Figure 1.

Principal Features of Rare Genetic Disorders
What Are Rare Diseases?
Rare diseases are defined by their prevalence rather than by unifying pathological or clinical characteristics. In United States, a disease falls in this category if it affects fewer than 200,000 individuals, corresponding to a prevalence of ∼6.6 in 10,000 subjects. This threshold was adopted by law with the passing of the Orphan Product Act of 1983, a law that successfully achieved its goal of accelerating the therapeutic discoveries for treating rare diseases (Boat, 2015, Haffner, 2006, Waxman, 1983). Other countries have defined rare diseases similarly, with thresholds set at a prevalence of 3.9/10,000 in Japan or 5/10,000 in European countries (Boat, 2015). These definitions capture between 6,000 and 10,000 rare diseases that collectively afflict millions of individuals in the United States and an estimated ∼260–450 million people worldwide (Haendel et al., 2020, Nguengang Wakap et al., 2020).
Because rare diseases are defined by their low prevalence, they encompass a broad spectrum of pathologies and pathogenesis mechanisms (Boat, 2015, Orphanet, 2020). Genetic rare diseases are the focus of our article, accounting for nearly 80% of all rare diseases (Boat, 2015, Wright et al., 2018). However, rare diseases also include non-genetic rare diseases such as autoimmune disorders and rare cancers as well as maladies caused by infectious or toxic agents (Orphanet, 2020). Some are known to the general population, such as rabies, but others are exceptionally rare. This is the case of Lemierre syndrome, a septic thrombophlebitis of the head and neck caused by Fusobacterium necrophorum, an anaerobic oral commensal (Han, 2015). Arsenic and mercury poisoning can also cause rare diseases, and mesothelioma is a neoplasia caused by asbestos exposure. Finally, there is a miscellaneous category that includes some rare nutritional deficiencies, such as beriberi, and complications of trauma or cancer treatments (Orphanet, 2020).
The unmet medical needs and the plight of affected patients and families are powerful arguments for further study of this wide range of rare diseases, as has been discussed previously (Klein and Gahl, 2018, Schieppati et al., 2008, Wastfelt et al., 2006). Here we will focus on biological arguments as justification for the study of rare diseases. Our argument is that rare genetic diseases are a gateway for discovering novel biology with broad impact for common human diseases. We begin to support this contention analyzing three examples where the study of a rare genetic mutation produced biological insight whose fundamental nature was recognized by Nobel awards. We end this article describing the impact of a rare genetic disorder, Menkes disease, in our knowledge of trace metal biology and the intersection of trace metals with common neurodegenerative diseases.
Why Study Rare Genetic Diseases, an Epistemological Perspective
There are contrasting views about the value of a rare event in biology and medicine. On one side for a biologist, a rare mutation and its phenotype are windows into mechanisms governing otherwise inscrutable complex biological processes. This vision motivates biologists to study connections between mutation and phenotype. We often perform these studies in model organisms with identical genomes, or isogenic, such as Saccharomyces cerevisiae, Drosophila melanogaster, or Mus musculus. Forward genetic screens seeking mutants in cell secretion, cell cycle, or circadian rhythms are good examples of how just one or few mutants can open doors to progressively unravel the intricacies of these biological processes. For example, the first cdc genes required for cell-cycle progression were identified in the Baker's yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe (Hartwell, 1974, Hartwell et al., 1970, Nurse, 1975). The sec genes required for protein secretion were identified in the Baker's yeast (Novick et al., 1980, Novick et al., 1981). The first circadian rhythm gene, period, was discovered in the fly (Bargiello et al., 1984, Konopka and Benzer, 1971, Young, 2018). The common thread linking these apparently disparate stories is that a single and rare genetic mutation was sufficient and necessary to begin disentangling these complex biological processes. The knowledge gained in these studies in non-human organisms guided the understanding of disease mechanisms in later discovered rare diseases caused by mutation in human orthologues of these genes (Russo et al., 2013). We document this idea in Table 1, listing the first 23 secretory sec genes and their human orthologues with some of the rare diseases so far identified. These stories of cell cycle, secretion, or circadian rhythms all culminated in Nobel Prizes in 2001, 2013, and 2017, respectively. These stories have spurred considerable research into human diseases ranging from diabetes mellitus to cancer, and these studies span from mechanisms governing insulin secretion by pancreatic beta cells to cell-cycle progression in cancer cells founded on knowledge obtained from these isogenic model genetic organisms (Gerber and Sudhof, 2002, Hartwell and Kastan, 1994). These examples underscore the power of the systematic study of a rare genetic mutation for our understanding of universal biological processes with relevance for prevalent human disease.
Table 1.
List of the 23 Sec Genes, Human Orthologues, and Paralogues plus Rare Diseases
| Yeast Gene | Human Gene | Disease | OMIM # | ORPHANET # | Prevalence |
|---|---|---|---|---|---|
| sec1 | STXBP1 | Epileptic encephalopathy, early infantile, 4 | 612164 | 1934 | 1/100000 |
| STXBP2 | Hemophagocytic lymphohistiocytosis, familial, 5 | 613101 | 540 | 1/100000 | |
| STXBP3 | |||||
| sec2 | RAB3IP | ||||
| RAB3IL1 | |||||
| sec3 | EXOC1 | ||||
| sec4 | RAB8A | ||||
| RAB8B | |||||
| sec5 | EXOC2 | ||||
| sec6 | EXOC3 | ||||
| sec7 | ARFGEF1 | ||||
| ARFGEF2 | Periventricular heterotopia with microcephaly | 608097 | 98892 | ||
| sec8 | EXOC4 | ||||
| sec9 | SNAP25 | Myasthenic syndrome, congenital, 18 | 616330 | 590 | |
| SNAP29 | Cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome | 609528 | 66631 | 7 subjects identified | |
| SNAP23 | |||||
| sec10 | EXOC5 | ||||
| sec11 | SEC11C | ||||
| SEC11A | |||||
| SEC11B | |||||
| sec12 | PREB | ||||
| sec13 | SEC13 | ||||
| sec14 | SEC14L2 | ||||
| SEC14L3 | |||||
| SEC14L1 | |||||
| SEC14L5 | |||||
| SEC14L4 | |||||
| SEC14L6 | |||||
| sec15 | EXOC6B | Spondyloepimetaphyseal dysplasia with joint laxity, type III | 618395 | 93359 | |
| EXOC6 | |||||
| sec16 | SEC16A | ||||
| SEC16B | |||||
| sec17 | NAPA | ||||
| NAPB | |||||
| sec18 | NSF | ||||
| sec20 | BNIP1 | ||||
| sec21 | COPG1 | ||||
| COPG2 | |||||
| sec22 | SEC22B | ||||
| SEC22C | |||||
| SEC22A | |||||
| sec23 | SEC23A | Craniolenticulosutural dysplasia | 607812 | 50814 | 27 cases described |
| SEC23B | Cowden syndrome 7 | 616858 | 201 | 1 in 200,000 to 250,000 | |
| SEC23B | Dyserythropoietic anemia, congenital, type II | 224100 | 98873 |
An alternate view is that the study of natural rare mutations in humans may be a risky choice. Arguments in favor of this view center around three chief arguments. First is the fact that humans are not genetically homogeneous. However, genomic efforts to understand the impact of mouse genetic diversity on phenotypic outcomes will help us to assess the impact of the genetic heterogeneity inherent to the study of human genetic diseases (Srivastava et al., 2017). Second, human mutations can be exceedingly rare, thus preventing a large casuistic for study. Third, some human mutation-associated phenotypes are not systematically annotated or quantitative. Despite these caveats, rare mutations in human models represent an opportunity much like the cdc, sec, or period genes and their mutations. Rare human genetic diseases can be seen as the results of the forward genetic screen that nature has been continuously running on us since the emergence of our species. These mutations and their phenotypes tell us incontrovertibly that these genes matter for a process yet to be discovered. The risk versus reward dilemma in the study of rare human diseases is articulated elegantly by Carl Zimmer in an article describing the rare disease fibrodysplasia ossificans progressiva (Zimmer, 2013). The prevalence of this disease is 1 in 2 million, a fact that could easily become a deterrent for study. Yet the penetrant and severe phenotype and therefore the mechanisms underneath the phenotype were hard to ignore for the pioneers studying this ultra-rare disease. Fibrodysplasia ossificans progressiva results in heterotopic ossification of muscle and connective tissue, a phenotype caused by dominant mutations in the gene ACVR1, encoding the activin A receptor 1 (OMIM 135100). ACVR1 is a widely expressed and evolutionarily recent gene appearing in mammals. The ACVR1 activity is required for controlling growth and development of bones and muscles, including endochondral ossification (Kaplan et al., 2012). The ACVR1 disease-causing mutations increase signal transduction through the bone morphogenetic protein signal transduction pathway whose ligands bind to ACVR1. This biological insight offered by the discovery of ACVR1 mutations in humans precedes information obtained from model genetic organisms (Kaplan et al., 2012, Shore et al., 2006).
Thus, if a mutation causes a strong and penetrant phenotype, the low prevalence of a rare genetic mutation in model genetic organisms or humans should not be a deterrent for their consideration. The frequency of a genetic defect neither foresees the significance of the biological process gone awry nor does it predict the potential for impacting human biology and health. We will discuss this point later using Menkes disease as an example. The pursuit for understanding human mutations with robust and penetrant phenotypes is a demonstrated path for unraveling universal biological principles much like forward genetic screens performed in isogeneic genetic organisms. This idea is not new. It was first formulated by William Harvey in 1657 in a response to John Vlackveld of Harlem, a Dutch physician who was asking Harvey's advice concerning a unique clinical case:
Harvey replied: “It is even so-Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of Nature by careful investigation of cases of rarer forms of disease. For it has been found, in almost all things, that what they contain of useful or applicable is hardly perceived unless we are deprived of them, or they become deranged in some way.”
Harvey's vision was first enunciated in the context of genetic diseases by Archibald Garrod, the father of Medical Genetics in 1928 (Garrod, 1928). Garrod also paraphrased this concept stating:
“The study of nature's experiments is of special value; and many lessons which rare maladies can teach could hardly be learned in other ways.”
In the next sections, we attempt to provide some answers to these questions posed then and now: What lessons can be learned from diseases as individual entities, and what can we learn from their collective study?
Learning from Rare Genetic Diseases through Systems Biology
It is estimated that 6,172 clinically distinct diseases are genetic in nature according to Orphanet, an online database of rare diseases that includes genetic and non-genetic diseases defined according to the European prevalence threshold (Cutillo et al., 2017). As of early 2020, the Online Mendelian Inheritance in Man resource, OMIM, lists 6,594 diseases with known molecular genetic defect, which collectively encompass 4,225 genes (https://www.omim.org/statistics/geneMap). This figure is increasing at a rate of ∼50–60 new genetic diseases per year in the Orphanet and OMIM databases (Boycott et al., 2017). Initiatives such as the NIH Undiagnosed Diseases Program and Network and The International Rare Diseases Research Consortium are accelerating the rate of discovery of novel genetic diseases (Gahl et al., 2016, Kuehn, 2011). Thus, it is conceivable that with time, we may identify sufficient genetic defects in humans so we can come close to the desired goal of all geneticists, a saturation mutagenesis screen to hit all genes and to find phenotypes of every individual gene.
To extract information out of these ∼6,000 rare genetic diseases, we analyzed genetic diseases listed in OMIM using disease descriptors from the Human Phenotype Ontology Database (HPO). The March 2020 release of the HPO offered us ∼156,000 annotations to rare diseases using a palette of over 13,000 ontological descriptors (Kohler et al., 2017). Of the 6,594 OMIM disease entries, 45% correspond to autosomal recessive diseases (HP:0000007), 32% fall into autosomal dominant disease category (HP:0000006), and 10% are diseases linked to the X and Y chromosomes (HP:0010985) (Figure 2A).
Figure 2.

Quantitative Descriptors of Rare Genetic Disorders Curated by OMIM and Annotated by HPO
We used the HPO annotated descriptors for all curated genetic disorders to assess global descriptors of disease.
(A) Describes all diseases and associated genes according to Mode of inheritance HP:0000005.
(B) Presents the distribution of diseases according to the age group in which disease manifestations appear: Onset HP:0003674.
(C) Diseases were classified according to annotated clinical phenotypes.
(D) Graph presents the distribution of disease according to the organ/tissue/system affected. Phenotypic abnormality HP:0000118.
(E–H) Venn diagrams present overlaps between different HPO terms listed in A–D. All bold numbers indicate overlaps of diseases with HPO terms associated to childhood converging on nervous system and behavioral HPO terms. (E and F) present data for recessive disorders. (G and H) depict data for dominant disorders. (A-D) Y axis represent % of curated OMIM diseases and Y1 axis shows the number of genes associated to the HPO terms (red symbols).
Analysis of the age of onset in rare diseases (HP:0003674) reveals the remarkable observation that 55% of all diseases are of pediatric origin (HP:0410280, Figure 2B), an onset category defined as diseases that manifest before the age of 16 years, but excluding neonatal or congenital onset. However, if we pool together pediatric, congenital, and neonatal diseases (referred here as childhood diseases); a staggering 83% of all rare diseases affect the child (Figure 2B). The picture is similar if we consider other ontological terms that capture diverse phenotypic manifestations in all rare diseases. The two top ontological terms encompassing 40% of all rare genetic diseases are by definition ascribed to children. These include global developmental delay and intellectual disability (HP:0001263 and HP:0001249, Figures 2E–2H), which describe delays in achieving motor or mental milestones before puberty. Similarly, analysis of all rare genetic diseases by the organ these diseases affect indicate that, irrespective of age, close to 70% of these diseases produce abnormalities of the nervous system (HP:0000707, Figure 2D). Our findings are in rapport with a recently reported study (Sanders et al., 2019). The compromise of the nervous system is even more pronounced in childhood diseases where 89.6% of childhood diseases compromise the nervous system or behavior irrespective of whether these diseases are dominant or recessive (Figures 2E–2H, bold numbers). These findings show that rare diseases preferentially affect human development and in particular the development of the nervous system.
Unifying Principles of Rare Genetic Diseases, a Systems Biology Perspective
The above findings beg the question of why the most prevalent phenotypes among rare diseases are abnormalities of the nervous system that disproportionally affect the child. We used systems biology analyses of genes affected in diseases of the childhood and compared them with genes associated to diseases of the adult. We chose childhood and adult categories because these disease onset descriptors are annotated in the HPO database (see legend to Figure 3 and HP:0003674). We discriminated among the following hypotheses that could account for this phenotypic bias toward the child nervous system:
-
1)
Genes associated with childhood diseases appeared in evolution together with the emergence of nervous systems in metazoans.
-
2)
Childhood disease genes are preferentially enriched in the developing brain.
-
3)
Childhood disease genes are preferentially expressed in neurons or glia and their subcellular compartments, such as the synapse.
-
4)
Childhood disease genes code for proteins that form nodes of high interconnectivity in protein interaction networks.
Figure 3.

Most Rare Disease Genes are Evolutionarily Ancient
We use the lists of genes associated with childhood diseases (pooled HP:0410280, HP:0030674, HP:0003577, and HP:0003623) and adult genetic diseases (HP:0003581) and analyzed them with the CLIME engine to determine the presence of orthologues and paralogues across species (Li et al., 2014). The presence of paralogues or orthologues is marked by blue cells in the heatmap.
This latter idea is founded on the observation that highly connected nodes tend to enrich gene products in which mutations produce lethality or impair housekeeping functions, suggesting that these genes are physiologically indispensable (Jeong et al., 2001, Lin et al., 2009, Rodrigues and Costa Lda, 2009, Yang et al., 2014, Yang et al., 2016).
Are rare diseases genes associated with childhood or adult genetic diseases a recent or ancient evolutionary occurrence? We used the CLIME engine to analyze the evolution of childhood or adult disease genes (Li et al., 2014)(Figure 3). Sixty percent of all childhood and adult genes possess orthologues in organisms that lack nervous systems including unicellular plants (Chlamydomonas reinhardtii) or unicellular fungi (Saccharomyces cerevisiae). However, just 5% of these childhood and adult genes appeared together with the emergence of metazoans with complex nervous systems, such as Drosophila melanogaster. This enrichment in ancestral genes among rare genetic disorders likely explains the exceedingly low prevalence of these diseases due to early embryonic lethality. In fact, it is estimated that between 50% and 70% of all early miscarriages are associated with aneuploidies and with 66 monogenic defects annotated in the HPO database (HP:0005268) (Hyde and Schust, 2015, Soler et al., 2017, van den Berg et al., 2012). These miscarriage-associated genes are enriched in constituents of the axoneme, an ancient organelle present since the emergence of unicellular plants (GO:0005858, p= 7.603 × 10−12)(Carvalho-Santos et al., 2011). In agreement with these evolutionary findings, we observed that the expression of childhood rare disease genes is not significantly enriched in the brain or any brain regions across development (Figures 4A–4C). Moreover, organelle-based ontologies could not distinguish childhood and adult diseases (Figure 4D). In fact, there is a balanced representation of organelles present in all cell types such as mitochondria, peroxisome, or vacuole. These ubiquitous organelles are similarly represented as compared with neuronal subcompartments (Figure 4D). Childhood diseases genes were overrepresented in all these cellular compartment annotations as compared with genes contributed by adult diseases (Figure 4E). The only exception was the cellular compartment, I-band, a muscle sarcomere structure (GO:0031674, Figure 4D, gray color), where both childhood and adult disease genes were annotated in a 1 to 1 ratio. Genes from childhood and adult diseases were annotated to neuronal compartment terms, such as synapse, in a 4 to 1 ratio (GO:0045202). However, the rare disease gene ratio is even more pronounced with a 1 to 0 and 5.4 to 1 ratio between childhood and adult disease genes for genes that are annotated to the peroxisome (GO:0044439) or mitochondria, respectively (GO:0005739). Genetic defects in these last two ubiquitous cellular organelles have long been recognized as severely affecting the nervous system of the child even though these organelles are present in all eukaryotes and metazoan tissues (Gorman et al., 2016, Koumandou et al., 2013, Waterham et al., 2016). These analyses allow us to draw two general conclusions. First, genes necessary for fundamental cellular processes are likely required throughout the lifespan of the organism, thus providing an explanation to the early life appearance of rare genetic disease phenotypes. Second, our evolutionary, tissue, or subcellular compartment criteria neither explain the preponderance of neurological and behavioral phenotypes in all genetic diseases combined nor in genetic diseases of the childhood.
Figure 4.

Rare Disease Gene Ontologies and Protein-Protein Interaction Topologies
(A–C) We used the childhood and adult gene HPO lists to explore tissue expression in human tissues using the ARCHS4 transcript database (A), the Human Proteome Map Database (B), or de human brain developmental mRNA expression database CSEA (C) (Kim et al., 2014, Lachmann et al., 2018, Wells et al., 2015). A and B present top ranked tissues where gene lists are expressed. Note that only adult disease mRNAs are significantly enriched in categories describing striated muscle. Childhood diseases genes do not enrich nervous tissue or any other tissue ontologies. Figures A–C show Fisher's exact p values, followed by the Benjamini-Hochberg correction.
(D and E) Gene lists for childhood and adult diseases were combined and analyzed using the Cytoscape ClueGo plugin for cellular compartment ontologies (GO:CC) (Bindea et al., 2009). Color in D depicts GO CC terms. Color in E represents the percentage of genes that belong to childhood diseases in the CC term depicted in (D). Note that only one term is equally represented by childhood and adult genes: the I- band belonging to striated muscle. All ontologies are significant with a corrected p value <0.05. Size of circle is proportional to the significance of the term.
(F–H) Protein-protein interaction network data for the childhood (F) and adult disease gene lists (G) were obtained from Genemania (Franz et al., 2018). Networks were built and their topologies analyzed with Cytoscape and the NetworkAnalyzer plugin (Doncheva et al., 2012, Shannon et al., 2003). H presents centrality parameters for the childhood (blue symbols) and adult disease genes (purple symbols).
In seeking answers to the disproportionate effects of rare genetic disease on the nervous system among diseases of the childhood (Sanders et al., 2019), we asked if protein-protein interaction networks constructed with childhood gene products differed in complexity from networks assembled with genes causative of rare diseases of the adult. We used experimentally defined protein-protein interaction networks and integrated them with predicted protein-protein interaction networks generated by yeast two-hybrid analyses. These interaction networks have been curated, maintained, and updated in the Genemania web engine (Franz et al., 2018) (Figures 4F and 4G). The network generated with childhood disease genes has higher network heterogeneity index than the adult network (1.476 versus 0.818), indicating that the childhood disease gene network contains more “hub” protein nodes than the adult network. Furthermore, the childhood disease gene network possesses protein nodes of a higher connectivity to other nodes within the network, as revealed by the average number of neighbors, 4.5 neighbors per node in the childhood disease network versus 3.4 for adult disease network proteins nodes. These global network parameters indicate that the childhood disease protein-protein interaction network is more complex and its nodes are of higher connectivity than those in the adult network.
We further determined the extent of childhood and adult network complexity asking how each protein connected to the rest of protein nodes using centrality parameters (Figure 4H). We described each protein node with betweenness and closeness centrality scores. Betweenness centrality of a protein node quantifies the amount of control that this node exerts over the interactions of other nodes within the network. Closeness centrality is a measure of how close a node is to other nodes, an indication of how quickly information spreads from a given node to other reachable nodes in the network (Doncheva et al., 2012, Dong and Horvath, 2007, Freeman, 1979, Yoon et al., 2006). Childhood disease protein nodes displayed higher centrality indexes as compared with adult disease protein nodes. This demonstrates that childhood disease proteins are biased toward more complex interactions than adult disease proteins. This begs the question, how are the complexity of the childhood disease networks and the preponderance of neurological phenotypes related to each other? One view is that genes expressed in the brain are engaged in networks as nodes of higher connectivity. However, present data argue against this hypothesis, as brain protein-protein interactions are comparable in connectivity to other tissues (Barshir et al., 2014). Rather it seems that widely expressed proteins mutated in genetic diseases are more likely to engage with other proteins involved in tissue-specific protein-protein interactions (Barshir et al., 2014). This observation is in line with recent findings demonstrating that tissue-specific proteins bridge conserved protein complexes present in most tissues (Bossi and Lehner, 2009, Huttlin et al., 2020, Luck et al., 2019). The brain is the most diversified proteome of all organs and the human organ with the second most tissue-enriched/specifically expressed proteins (Fagerberg et al., 2014, Sharma et al., 2015, Sjostedt et al., 2015, Uhlen et al., 2015). Thus, we propose that the prevalence of childhood diseases affecting nervous tissue is the product of the topological complexity of the networks formed by ubiquitous proteins encoded by childhood disease genes plus the abundance of brain-enriched/specific protein. We speculate that brain-enriched/specific proteins would propagate the consequences of genetic defects in a conserved protein complex into other conserved and ubiquitous complexes, thus amplifying the emergence of phenotypes in the brain.
We conclude that investigating rare genetic diseases is in fact a way to study the function of the most evolutionarily conserved genes and cellular mechanisms. Once again, similar to the case with Harvey and Garrod illustrate, this is not a new idea. It has long been recognized that ascertainment of monogenic genetic diseases by rare and early onset cases is a tool to study common yet genetically complex diseases. This is the case of Alzheimer, schizophrenia, or type II diabetes where pedigrees of families with early age onset of disease have been instructive of the mechanisms of common complex diseases that are frequently polygenic and influenced by environmental factors (Fajans et al., 2001, Murrell et al., 1991, Rapoport et al., 2012).
Learning about Common Human Diseases from a Rare Genetic Disease
Menkes disease exemplifies central features of most rare genetic diseases. Menkes is a childhood multisystemic disease that severely affects the nervous system (OMIM 309400). As other examples discussed earlier, the study of Menkes disease has opened the door to fundamental concepts about the molecular biology and physiology of trace metals. Even after 58 years of studying this disease, there are still Menkes-inspired novel biological insights. Recently, we produced evidence of genetic and molecular interactions between the Menkes disease gene, ATP7A, and common diseases such as Parkinson disease (Comstra et al., 2017, Hartwig et al., 2019, Zlatic et al., 2018).
The story of Menkes disease began in New York in 1962 with a family of English-Irish descent. Dr John H. Menkes described the first five patients belonging to this family. His patients were affected by an X-linked recessive disease characterized by failure to thrive, abnormal hair and skin, intellectual disability, as well as cerebral and cerebellar neurodegeneration. Severe neurologic symptoms appeared 1–2 months after birth and progressed rapidly to death (Menkes, 1988, Menkes et al., 1962). The next chapter began in 1972, when David Danks et al. demonstrated low levels of serum copper and ceruloplasmin in seven patients with Australian Menkes disease, leading to Danks' seminal proposition that Menkes disease is a disease of copper absorption (Danks et al., 1972). This hypothesis was definitively tested with an enterocyte-specific knock-out of the murine Atp7a gene that fully recapitulates Menkes disease (Wang et al., 2012). Notably, human-research-founded hypotheses preceded the identification of the metabolic defect in mice that carry Atp7a mutations by decades with the study of the mottled series of mice (Grimes et al., 1997, Hunt, 1974). It took 21 years after the disease's first description for three groups to independently clone the candidate gene of Menkes disease, ATP7A, a finding that spurred our understanding of copper biology (Chelly et al., 1993, Mercer et al., 1993, Vulpe et al., 1993). Even before the identification of the gene mutated in Menkes disease, Danks presciently concluded that the study of this rare disease would lead to “many new lines of research on copper metabolism and trace metal deficiency.” This statement abides by the spirit of Harvey and Garrod dictum about the conceptual gains of studying Nature's defects that fall "apart from the beaten path" (Danks et al., 1972).
Presently, we know that genetic defects affecting the copper-transporter P-ATPase, ATP7A, cause three X-linked recessive rare diseases: occipital horn syndrome (OMIM 304150), spinal muscular atrophy, distal, X-linked 3 (SMAX3, OMIM 300489), and Menkes disease (OMIM 309400) (Kaler, 2011). Approximately 380 different mutations affecting ATP7A gene have been described so far in the Human Gene Mutation Database (Moller et al., 2009, Tumer, 2013). Milder missense mutations in ATP7A generate occipital horn syndrome, which is characterized by ataxia, dysarthria, moderate hypotonia, and intellectual disability in addition to systemic phenotypes (Das et al., 1995, Kaler et al., 1994), and SMAX3, where spinomuscular atrophy occurs independently of cognitive defects (Kennerson et al., 2010, Takata et al., 2004). More severe loss-of-function mutations result in Menkes disease proper, a multisystemic metabolic disease affecting copper homeostasis. Menkes is a rare affliction with an incidence of 1/140,000 to 1/300,000 (Gu et al., 2005, Kaler, 2011). Menkes manifests soon after birth with hypotonia, focal and generalized seizures, impaired cognitive development, and brain atrophy. Systemically, Menkes disease affects hair with multiple defects including pili torti (twisted hairs), monilethrix (beaded hairs), and thickened or weak nodes that cause hair fragility (trichorrhexis nodosa). In addition, hair and skin are hypopigmented. There is laxity of the skin (cutis laxa) and joints, osteoporosis, bladder diverticula, aneurysms, and vascular tortuosity in brain arteries (Kaler, 2011, Lutsenko et al., 2007, Manara et al., 2017, Polishchuk and Lutsenko, 2013).
Menkes disease clinical features have been traditionally attributed to defects in diverse cuproenzymes that traverse the secretory pathway and remain as inactive apoenzymes in the disease state (Kaler, 2011, Lutsenko et al., 2007, Polishchuk and Lutsenko, 2013) (Table 2). This defect in loading copper into apoenzymes results from a defect in copper transport into the Golgi lumen as ATP7A normally is a resident transporter of the Golgi apparatus (Petris et al., 1996). The cutis laxa, bone, bladder, and vascular phenotypes are attributed to the defective activity of enzymes required for the modification of collagen fibers and elastin such as lysyl oxidase (LOX). Hypopigmentation is due to defective tyrosinase activity and hair defects to impaired sulfhydryl oxidase activity (Harris, 2013, Tumer and Moller, 2010, Zlatic et al., 2015). The success of this enzymatic model explaining systemic phenotypes in Menkes disease has been extended to the neurological symptoms in Menkes disease (Zlatic et al., 2015). It has been proposed that defective enzymatic activities of cytochrome c oxidase, dopamine β-monooxygenase, and peptidyl-α−amidating monooxygenase are responsible for the nervous system defects in Menkes disease (Menkes, 1988, Zlatic et al., 2015). These enzymes play major roles in mitochondrial respiration, neurotransmitter, and neuropeptide biosynthesis respectively. We termed this enzymatic model of neurological phenotypes the oligoenzymatic hypothesis, which we consider insufficient to explain Menkes neuropathology (Kaler, 2011, Menkes, 1999, Zlatic et al., 2015). In fact, the interactome of the Menkes ATPase ATP7A and the proteome of ATP7A null cells are enriched in gene products involved in neurodegenerative and neurodevelopmental diseases, suggesting a larger complexity to the pathogenesis mechanisms in Menkes neurological disease (Comstra et al., 2017, Hartwig et al., 2019, Zlatic et al., 2018).
Table 2.
Copper-Dependent Enzymes and Phenotypes Associated in Menkes Disease
| Enzyme | Gene | Compartment | Biological Activity | Symptom |
|---|---|---|---|---|
| Cytochrome c oxidase | Varios Genes | Mitochondria | Cellular respiration | CNS degeneration? |
| See CORUM Complex #6442 | Ataxia? | |||
| Superoxide dismutase | SOD1 | Mitochondria | Free radical scavenging | CNS degeneration? |
| Tyrosinase | TYR | Lysosome-related organelles | Pigment formation | Hypopigmentation |
| Dopamine β-hydroxylase | DBH | Large dense core secretory vesicles | Catecholamine production | Ataxia? |
| Hypothermia | ||||
| Hypotension | ||||
| Peptidylglycine alpha-amidating monooxygenase | PAM | Large dense core secretory vesicles | Activation of peptide hormones | Wide spread effects, vascular and pancreas defects |
| Lysyl oxidase | LOX | Secreted | Collagen and elastin cross-linking | Premature rapture of fetal membranes |
| Cephalohematoma | ||||
| Abnormal facies | ||||
| High-arched palate | ||||
| Emphysema | ||||
| Hernias | ||||
| Bladder diverticula | ||||
| Arterial aneurysms | ||||
| Loose skin and joints | ||||
| Osteoporosis | ||||
| Petechial hemorrhage | ||||
| Poor wound healing | ||||
| CNS degeneration? | ||||
| Sulfhydryl oxidase | Possibly QSOX1 and 2 | Golgi and secreted | Cross-linking of keratin | Abnormal hair |
| Dry skin |
Adapted from (Harris, 2013, Tumer and Moller, 2010, Zlatic et al., 2015).
Despite the fact that Menkes disease is a rare genetic disease, we believe Menkes continues to be a tool to uncover novel and fundamental knowledge regarding mechanisms underlying metal-dependent neurodegeneration in common diseases. Copper imbalances exacerbate the magnitude and progression of neurodegenerative diseases (Davies et al., 2014, Davies et al., 2016, Dusek et al., 2015, Lorincz, 2010), with even minimal exposure to copper at dietary levels sufficient to trigger neuropathology and cognitive decline (Sparks and Schreurs, 2003). Menkes is also a model to identify mechanisms shared with neurodegenerative and neurodevelopmental diseases where environmental exposures, such as redox active metals, are powerful risk factors. For example, copper content alterations are often found in neurodegenerative diseases such as in Parkinson and Parkinsonism (Davies et al., 2014, Davies et al., 2016, Dusek et al., 2015, Genoud et al., 2019, Lorincz, 2010). Thus, as is the case in other rare diseases, the study of Menkes disease has the potential to intersect with and reveal new mechanisms associated with prevalent diseases.
Conclusions
The lessons that rare genetic diseases teach us continue to affirm the dictums of Harvey and Garrod: We can do “discovery of the usual law of Nature by careful investigation of cases of rarer forms of disease”. Rare genetic diseases disproportionately affect the nervous system of children with devastating effects. Paradoxically, the majority of the disease-causing genes affecting the child belong to genes present from the last common eukaryote and ubiquitously expressed in human tissues. Thus, whether we study a mutation in a yeast, fly, or a human, we argue we are in essence studying the same principles across the radiating richness given to us by evolution.
The concept of rare diseases focuses on a seemingly low percentage of people affected by a single affliction. Collectively, however, these rare diseases affect nearly 4% of the people during their lifetime. We predict that the numbers of patients with rare disease will dramatically increase in the new era of the truly rare disease: the disease of one-and-only-one individual. This concept is intrinsic to the idea of personalized medicine (Collins and Varmus, 2015). If personalized medicine delivers on its promises, the future of medicine is bound for the study and treatment of the rarest of all diseases, the disease of the “I” rather than the “us.” The principles that we have articulated for rare diseases here will be equally applicable for personalized medicine. The richest horizon for biological exploration may be just around the corner, where the information of every human genome will be tied to the individual's normal or pathological traits.
Acknowledgments
This work was supported by grants from the National Institutes of Health 1RF1AG060285, the Rett Syndrome Research Trust, and the Loulou Foundation to V.F. We are grateful to the patients affected by Menkes disease, 22q11.2 microdeletion syndrome, Rett syndrome and CDKL5-deficiency syndrome and their families who continually inspire our work. We are indebted to the Faundez lab members for their comments.
Author Contributions
C.E.L., Writing—Review & Editing and Conceptualization.
K.S.S., Writing—Review & Editing.
M.W., Writing—Review & Editing and Visualization.
V.F., Writing—Original Draft, Conceptualization, Investigation, Visualization, Funding Acquisition.
References
- Bargiello T.A., Jackson F.R., Young M.W. Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature. 1984;312:752–754. doi: 10.1038/312752a0. [DOI] [PubMed] [Google Scholar]
- Barshir R., Shwartz O., Smoly I.Y., Yeger-Lotem E. Comparative analysis of human tissue interactomes reveals factors leading to tissue-specific manifestation of hereditary diseases. PLoS Comput. Biol. 2014;10:e1003632. doi: 10.1371/journal.pcbi.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.H., Pages F., Trajanoski Z., Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boat T.F. Natl Academies Press; 2015. RARE DISEASES and ORPHAN PRODUCTS Accelerating Research and Development Summary. [PubMed] [Google Scholar]
- Bossi A., Lehner B. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 2009;5:260. doi: 10.1038/msb.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott K.M., Rath A., Chong J.X., Hartley T., Alkuraya F.S., Baynam G., Brookes A.J., Brudno M., Carracedo A., den Dunnen J.T. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum.Genet. 2017;100:695–705. doi: 10.1016/j.ajhg.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Santos Z., Azimzadeh J., Pereira-Leal J.B., Bettencourt-Dias M. Evolution: tracing the origins of centrioles, cilia, and flagella. J. Cell Biol. 2011;194:165–175. doi: 10.1083/jcb.201011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelly J., Tumer Z., Tonnesen T., Petterson A., Ishikawa-Brush Y., Tommerup N., Horn N., Monaco A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- Collins F.S., Varmus H. A new initiative on precision medicine. N. Engl. J. Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstra H.S., McArthy J., Rudin-Rush S., Hartwig C., Gokhale A., Zlatic S.A., Blackburn J.B., Werner E., Petris M., D’Souza P. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. Elife. 2017;6:e24722. doi: 10.7554/eLife.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutillo C.M., Austin C.P., Groft S.C. A global approach to rare diseases research and orphan products development: the international rare diseases research Consortium (IRDiRC) Adv. Exp. Med. Biol. 2017;1031:349–369. doi: 10.1007/978-3-319-67144-4_20. [DOI] [PubMed] [Google Scholar]
- Danks D.M., Campbell P.E., Stevens B.J., Mayne V., Cartwright E. Menkes's kinky hair syndrome.An inherited defect in copper absorption with widespread effects. Pediatrics. 1972;50:188–201. [PubMed] [Google Scholar]
- Das S., Levinson B., Vulpe C., Whitney S., Gitschier J., Packman S. Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am. J. Hum. Genet. 1995;56:570–576. [PMC free article] [PubMed] [Google Scholar]
- Davies K.M., Bohic S., Carmona A., Ortega R., Cottam V., Hare D.J., Finberg J.P., Reyes S., Halliday G.M., Mercer J.F. Copper pathology in vulnerable brain regions in Parkinson's disease. Neurobiol. Aging. 2014;35:858–866. doi: 10.1016/j.neurobiolaging.2013.09.034. [DOI] [PubMed] [Google Scholar]
- Davies K.M., Mercer J.F., Chen N., Double K.L. Copper dyshomoeostasis in Parkinson's disease: implications for pathogenesis and indications for novel therapeutics. Clin. Sci. (Lond.) 2016;130:565–574. doi: 10.1042/CS20150153. [DOI] [PubMed] [Google Scholar]
- Doncheva N.T., Assenov Y., Domingues F.S., Albrecht M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012;7:670–685. doi: 10.1038/nprot.2012.004. [DOI] [PubMed] [Google Scholar]
- Dong J., Horvath S. Understanding network concepts in modules. BMC Syst. Biol. 2007;1:24. doi: 10.1186/1752-0509-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek P., Roos P.M., Litwin T., Schneider S.A., Flaten T.P., Aaseth J. The neurotoxicity of iron, copper and manganese in Parkinson's and Wilson's diseases. J. Trace Elem. Med. Biol. 2015;31:193–203. doi: 10.1016/j.jtemb.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajans S.S., Bell G.I., Polonsky K.S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 2001;345:971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- Franz M., Rodriguez H., Lopes C., Zuberi K., Montojo J., Bader G.D., Morris Q. GeneMANIA update 2018. Nucleic Acids Res. 2018;46:W60–W64. doi: 10.1093/nar/gky311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman L.C. Centrality in social networks: conceptual clarification. Soc. Netw. 1979;1:215–239. [Google Scholar]
- Gahl W.A., Mulvihill J.J., Toro C., Markello T.C., Wise A.L., Ramoni R.B., Adams D.R., Tifft C.J., Udn The NIH undiagnosed diseases Program and network: applications to modern medicine. Mol. Genet. Metab. 2016;117:393–400. doi: 10.1016/j.ymgme.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod A. The lessons of rare maladies. Lancet. 1928;211:1055–1061. [Google Scholar]
- Genoud S., Senior A.M., Hare D.J., Double K.L. Meta-analysis of copper and iron in Parkinson's disease brain and biofluids. Move.Disord. 2019;35:662–671. doi: 10.1002/mds.27947. [DOI] [PubMed] [Google Scholar]
- Gerber S.H., Sudhof T.C. Molecular determinants of regulated exocytosis. Diabetes. 2002;51:S3–S11. doi: 10.2337/diabetes.51.2007.s3. [DOI] [PubMed] [Google Scholar]
- Gokhale A., Hartwig C., Freeman A.A.H., Bassell J.L., Zlatic S.A., Sapp Savas C., Vadlamudi T., Abudulai F., Pham T.T., Crocker A. Systems analysis of the 22q11.2 microdeletion syndrome converges on a mitochondrial interactome necessary for synapse function and behavior. J. Neurosci. 2019;39:3561–3581. doi: 10.1523/JNEUROSCI.1983-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A., Larimore J., Werner E., So L., Moreno-De-Luca A., Lese-Martin C., Lupashin V.V., Smith Y., Faundez V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J. Neurosci. 2012;32:3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R., Suomalainen A., Thorburn D.R., Zeviani M., Turnbull D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- Grimes A., Hearn C.J., Lockhart P., Newgreen D.F., Mercer J.F. Molecular basis of the brindled mouse mutant (Mo(br)): a murine model of Menkes disease. Hum. Mol. Genet. 1997;6:1037–1042. doi: 10.1093/hmg/6.7.1037. [DOI] [PubMed] [Google Scholar]
- Gu Y.H., Kodama H., Shiga K., Nakata S., Yanagawa Y., Ozawa H. A survey of Japanese patients with Menkes disease from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosis. J. Inherit. Metab. Dis. 2005;28:473–478. doi: 10.1007/s10545-005-0473-3. [DOI] [PubMed] [Google Scholar]
- Haendel M., Vasilevsky N., Unni D., Bologa C., Harris N., Rehm H., Hamosh A., Baynam G., Groza T., McMurry J. How many rare diseases are there? Nat. Rev. Drug Discov. 2020;19:77–78. doi: 10.1038/d41573-019-00180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner M.E. Adopting orphan drugs--two dozen years of treating rare diseases. N. Engl. J. Med. 2006;354:445–447. doi: 10.1056/NEJMp058317. [DOI] [PubMed] [Google Scholar]
- Han Y.W. Fusobacterium nucleatum: a commensal-turned pathogen. Curr. Opin. Microbiol. 2015;23:141–147. doi: 10.1016/j.mib.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E.D. Copper, biological functions. In: Kretsinger R.H., Uversky V.N., Permyakov E.A., editors. Encyclopedia of Metalloproteins. Springer New York; 2013. pp. 718–723. [Google Scholar]
- Hartwell L.H. Saccharomyces cerevisiae cell cycle. Bacteriol. Rev. 1974;38:164–198. doi: 10.1128/br.38.2.164-198.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L.H., Culotti J., Reid B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. U S A. 1970;66:352–359. doi: 10.1073/pnas.66.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L.H., Kastan M.B. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- Hartwig C., Zlatic S.A., Wallin M., Vrailas-Mortimer A., Fahrni C.J., Faundez V. Trafficking mechanisms of P-type ATPase copper transporters. Curr. Opin. Cell Biol. 2019;59:24–33. doi: 10.1016/j.ceb.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D.M. Primary defect in copper transport underlies mottled mutants in the mouse. Nature. 1974;249:852–854. doi: 10.1038/249852a0. [DOI] [PubMed] [Google Scholar]
- Huttlin E.L., Bruckner R.J., Navarrete-Perea J., Cannon J.R., Baltier K., Gebreab F., Gygi M.P., Thornock A., Zarraga G., Tam S. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. bioRxiv. 2020 doi: 10.1101/2020.01.19.905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde K.J., Schust D.J. Genetic considerations in recurrent pregnancy loss. Cold Spring Harb. Perspect..Med. 2015;5:a023119. doi: 10.1101/cshperspect.a023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H., Mason S.P., Barabasi A.L., Oltvai Z.N. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Kaler S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler S.G., Gallo L.K., Proud V.K., Percy A.K., Mark Y., Segal N.A., Goldstein D.S., Holmes C.S., Gahl W.A. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat. Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- Kaplan F.S., Chakkalakal S.A., Shore E.M. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis. Model. Mech. 2012;5:756–762. doi: 10.1242/dmm.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerson M.L., Nicholson G.A., Kaler S.G., Kowalski B., Mercer J.F., Tang J., Llanos R.M., Chu S., Takata R.I., Speck-Martins C.E. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010;86:343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C., Gahl W.A. Patients with rare diseases: from therapeutic orphans to pioneers of personalized treatments. EMBO Mol. Med. 2018;10:1–3. doi: 10.15252/emmm.201708365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S., Vasilevsky N.A., Engelstad M., Foster E., McMurry J., Ayme S., Baynam G., Bello S.M., Boerkoel C.F., Boycott K.M. The human phenotype ontology in 2017. Nucleic Acids Res. 2017;45:D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka R.J., Benzer S. Clock mutants of Drosophila melanogaster. Proc. Natl. Acad. Sci. U S A. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumandou V.L., Wickstead B., Ginger M.L., van der Giezen M., Dacks J.B., Field M.C. Molecular paleontology and complexity in the last eukaryotic common ancestor. Crit. Rev. Biochem. Mol. Biol. 2013;48:373–396. doi: 10.3109/10409238.2013.821444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn B.M. NIH's Undiagnosed Diseases Program reports on successes, challenges. JAMA. 2011;306:2657–2658. doi: 10.1001/jama.2011.1836. [DOI] [PubMed] [Google Scholar]
- Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma'ayan A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018;9:1366. doi: 10.1038/s41467-018-03751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Calvo S.E., Gutman R., Liu J.S., Mootha V.K. Expansion of biological pathways based on evolutionary inference. Cell. 2014;158:213–225. doi: 10.1016/j.cell.2014.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.H., Liu W.C., Hwang M.J. Topological and organizational properties of the products of house-keeping and tissue-specific genes in protein-protein interaction networks. BMC Syst. Biol. 2009;3:32. doi: 10.1186/1752-0509-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz M.T. Neurologic Wilson's disease. Ann. N. Y. Acad. Sci. 2010;1184:173–187. doi: 10.1111/j.1749-6632.2009.05109.x. [DOI] [PubMed] [Google Scholar]
- Luck K., Kim D.-K., Lambourne L., Spirohn K., Begg B.E., Bian W., Brignall R., Cafarelli T., Campos-Laborie F.J., Charloteaux B. A reference map of the human protein interactome. bioRxiv. 2019 doi: 10.1101/605451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsenko S., Barnes N.L., Bartee M.Y., Dmitriev O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- Manara R., D'Agata L., Rocco M.C., Cusmai R., Freri E., Pinelli L., Darra F., Procopio E., Mardari R., Zanus C. Neuroimaging changes in Menkes disease, Part 1. AJNR Am. J. Neuroradiol. 2017;38:1850–1857. doi: 10.3174/ajnr.A5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkes J.H. Kinky hair disease: twenty five years later. Brain Dev. 1988;10:77–79. doi: 10.1016/s0387-7604(88)80074-3. [DOI] [PubMed] [Google Scholar]
- Menkes J.H. Menkes disease and Wilson disease: two sides of the same copper coin. Part I: Menkes disease. Eur. J. Paediatric Neurol. 1999;3:147–158. doi: 10.1016/s1090-3798(99)90048-x. [DOI] [PubMed] [Google Scholar]
- Menkes J.H., Alter M., Steigleder G.K., Weakley D.R., Sung J.H. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- Mercer J.F., Livingston J., Hall B., Paynter J.A., Begy C., Chandrasekharappa S., Lockhart P., Grimes A., Bhave M., Siemieniak D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- Moller L.B., Mogensen M., Horn N. Molecular diagnosis of Menkes disease: genotype-phenotype correlation. Biochimie. 2009;91:1273–1277. doi: 10.1016/j.biochi.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Murrell J., Farlow M., Ghetti B., Benson M.D. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Nguengang Wakap S., Lambert D.M., Olry A., Rodwell C., Gueydan C., Lanneau V., Murphy D., Le Cam Y., Rath A. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 2020;28:165–173. doi: 10.1038/s41431-019-0508-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Ferro S., Schekman R. Order of events in the yeast secretory pathway. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Nurse P. Genetic control of cell size at cell division in yeast. Nature. 1975;256:547–551. doi: 10.1038/256547a0. [DOI] [PubMed] [Google Scholar]
- Orphanet . Orphadata; 2020. Orphadata.http://www.orphadata.org/cgi-bin/index.php [Google Scholar]
- Perez-Cornejo P., Gokhale A., Duran C., Cui Y., Xiao Q., Hartzell H.C., Faundez V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc. Natl. Acad. Sci. U S A. 2012;109:10376–10381. doi: 10.1073/pnas.1200174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petris M.J., Mercer J.F., Culvenor J.G., Lockhart P., Gleeson P.A., Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- Polishchuk R., Lutsenko S. Golgi in copper homeostasis: a view from the membrane trafficking field. Histochem. Cell Biol. 2013;140:285–295. doi: 10.1007/s00418-013-1123-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport J.L., Giedd J.N., Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol. Psychiatry. 2012;17:1228–1238. doi: 10.1038/mp.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F.A., Costa Lda F. Protein lethality investigated in terms of long range dynamical interactions. Mol. Biosyst. 2009;5:385–390. doi: 10.1039/b815702m. [DOI] [PubMed] [Google Scholar]
- Russo R., Esposito M.R., Iolascon A. Inherited hematological disorders due to defects in coat protein (COP)II complex. Am. J. Hematol. 2013;88:135–140. doi: 10.1002/ajh.23292. [DOI] [PubMed] [Google Scholar]
- Sanders S.J., Sahin M., Hostyk J., Thurm A., Jacquemont S., Avillach P., Douard E., Martin C.L., Modi M.E., Moreno-De-Luca A. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat. Med. 2019;25:1477–1487. doi: 10.1038/s41591-019-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieppati A., Henter J.I., Daina E., Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371:2039–2041. doi: 10.1016/S0140-6736(08)60872-7. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K., Schmitt S., Bergner C.G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U.K., Philips M.A. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore E.M., Xu M., Feldman G.J., Fenstermacher D.A., Cho T.J., Choi I.H., Connor J.M., Delai P., Glaser D.L., LeMerrer M. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Sjostedt E., Fagerberg L., Hallstrom B.M., Haggmark A., Mitsios N., Nilsson P., Ponten F., Hokfelt T., Uhlen M., Mulder J. Defining the human brain proteome using transcriptomics and antibody-based profiling with a focus on the cerebral cortex. PLoS One. 2015;10:e0130028. doi: 10.1371/journal.pone.0130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler A., Morales C., Mademont-Soler I., Margarit E., Borrell A., Borobio V., Munoz M., Sanchez A. Overview of chromosome abnormalities in first Trimester miscarriages: aseries of 1,011 Consecutive Chorionic Villi Sample Karyotypes. Cytogenet. Genome Res. 2017;152:81–89. doi: 10.1159/000477707. [DOI] [PubMed] [Google Scholar]
- Sparks D.L., Schreurs B.G. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc. Natl. Acad. Sci.U S A. 2003;100:11065–11069. doi: 10.1073/pnas.1832769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Morgan A.P., Najarian M.L., Sarsani V.K., Sigmon J.S., Shorter J.R., Kashfeen A., McMullan R.C., Williams L.H., Giusti-Rodriguez P. Genomes of the mouse collaborative cross. Genetics. 2017;206:537–556. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata R.I., Speck Martins C.E., Passosbueno M.R., Abe K.T., Nishimura A.L., Da Silva M.D., Monteiro A., Jr., Lima M.I., Kok F., Zatz M. A new locus for recessive distal spinal muscular atrophy at Xq13.1-q21. J. Med. Genet. 2004;41:224–229. doi: 10.1136/jmg.2003.013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mutat. 2013;34:417–429. doi: 10.1002/humu.22266. [DOI] [PubMed] [Google Scholar]
- Tumer Z., Moller L.B. Menkes disease. Eur. J. Hum. Genet. 2010;18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. Proteomics.Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- van den Berg M.M., van Maarle M.C., van Wely M., Goddijn M. Genetics of early miscarriage. Biochim. Biophys. Acta. 2012;1822:1951–1959. doi: 10.1016/j.bbadis.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Vulpe C., Levinson B., Whitney S., Packman S., Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhu S., Hodgkinson V., Prohaska J.R., Weisman G.A., Gitlin J.D., Petris M.J. Maternofetal and neonatal copper requirements revealed by enterocyte-specific deletion of the Menkes disease protein. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G1236–G1244. doi: 10.1152/ajpgi.00339.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wastfelt M., Fadeel B., Henter J.I. A journey of hope: lessons learned from studies on rare diseases and orphan drugs. J. Intern. Med. 2006;260:1–10. doi: 10.1111/j.1365-2796.2006.01666.x. [DOI] [PubMed] [Google Scholar]
- Waterham H.R., Ferdinandusse S., Wanders R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta. 2016;1863:922–933. doi: 10.1016/j.bbamcr.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Waxman H. US Food and Drug Administration; 1983. Orphan Drug Act of 1983. Pub L. No. 97–414, 96 Stat. 2049. [Google Scholar]
- Wells A., Kopp N., Xu X., O'Brien D.R., Yang W., Nehorai A., Adair-Kirk T.L., Kopan R., Dougherty J.D. The anatomical distribution of genetic associations. Nucleic Acids Res. 2015;43:10804–10820. doi: 10.1093/nar/gkv1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.F., FitzPatrick D.R., Firth H.V. Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 2018;19:253–268. doi: 10.1038/nrg.2017.116. [DOI] [PubMed] [Google Scholar]
- Yang L., Wang J., Wang H., Lv Y., Zuo Y., Li X., Jiang W. Analysis and identification of essential genes in humans using topological properties and biological information. Gene. 2014;551:138–151. doi: 10.1016/j.gene.2014.08.046. [DOI] [PubMed] [Google Scholar]
- Yang L., Wang S., Zhou M., Chen X., Zuo Y., Sun D., Lv Y. Comparative analysis of housekeeping and tissue-selective genes in human based on network topologies and biological properties. Mol. Genet. Genomics. 2016;291:1227–1241. doi: 10.1007/s00438-016-1178-z. [DOI] [PubMed] [Google Scholar]
- Yoon J., Blumer A., Lee K. An algorithm for modularity analysis of directed and weighted biological networks based on edge-betweenness centrality. Bioinformatics. 2006;22:3106–3108. doi: 10.1093/bioinformatics/btl533. [DOI] [PubMed] [Google Scholar]
- Young M.W. Time travels: a 40-year journey from drosophila's clock mutants to human circadian disorders (Nobel Lecture) Angew. Chem. Int. Ed. 2018;57:11532–11539. doi: 10.1002/anie.201803337. [DOI] [PubMed] [Google Scholar]
- Zimmer C. The Atlantic; 2013. The Girl Who Turned to Bone. [Google Scholar]
- Zlatic S., Comstra H.S., Gokhale A., Petris M.J., Faundez V. Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease. Neurobiol. Dis. 2015;81:154–161. doi: 10.1016/j.nbd.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic S.A., Vrailas-Mortimer A., Gokhale A., Carey L.J., Scott E., Burch R., McCall M.M., Rudin-Rush S., Davis J.B., Hartwig C. Rare disease mechanisms identified by genealogical proteomics of copper homeostasis mutant pedigrees. Cell Syst. 2018;6:368–380.e6. doi: 10.1016/j.cels.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]