

Abstract
In 2013, EFSA published a literature review on epidemiological studies linking exposure to pesticides and human health outcome. As a follow up, the EFSA Panel on Plant Protection Products and their residues (PPR Panel) was requested to investigate the plausible involvement of pesticide exposure as a risk factor for Parkinson's disease (PD) and childhood leukaemia (CHL). A systematic literature review on PD and CHL and mode of actions for pesticides was published by EFSA in 2016 and used as background documentation. The Panel used the Adverse Outcome Pathway (AOP) conceptual framework to define the biological plausibility in relation to epidemiological studies by means of identification of specific symptoms of the diseases as AO. The AOP combines multiple information and provides knowledge of biological pathways, highlights species differences and similarities, identifies research needs and supports regulatory decisions. In this context, the AOP approach could help in organising the available experimental knowledge to assess biological plausibility by describing the link between a molecular initiating event (MIE) and the AO through a series of biologically plausible and essential key events (KEs). As the AOP is chemically agnostic, tool chemical compounds were selected to empirically support the response and temporal concordance of the key event relationships (KERs). Three qualitative and one putative AOP were developed by the Panel using the results obtained. The Panel supports the use of the AOP framework to scientifically and transparently explore the biological plausibility of the association between pesticide exposure and human health outcomes, identify data gaps, define a tailored testing strategy and suggests an AOP's informed Integrated Approach for Testing and Assessment (IATA).
Keywords: AOP, Parkinson's disease, childhood leukaemia, infant leukaemia, pesticides, epidemiology
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1190/full
Summary
The European Food Safety Authority (EFSA) asked the Panel on Plant Protection Products and their Residues (PPR Panel) to develop a Scientific Opinion investigating experimental toxicological properties of plant protection products having a potential link to Parkinson's disease (PD) and childhood leukaemia (CHL).
Following a significant association between pesticide exposure, PD and CHL as reported in an external scientific report of EFSA (Ntzani et al., 2013), the PPR Panel analysed the involvement of pesticides exposure as a risk factor in the pathogenesis of these two diseases. This task is required due to the intrinsic weakness of epidemiological studies that do not allow firm conclusions on causal relationships, but still raise a concern and open a question on suitability of regulatory studies to inform on specific and complex human health outcomes.
In addition to epidemiological studies, experimental data, although performed at toxicologically relevant doses, have also provided evidence for neurotoxic effects and biologically plausible mechanisms linking pesticides to PD. Scarce experimental and mechanistic evidence, however, also supports the association between pesticide exposure and paediatric leukaemia.
The definition of biological plausibility in relation to epidemiological studies, if any, was achieved by organising and analysing systematic literature reviews, diseases knowledge and the available experimental data of tool compounds in the Adverse Outcome Pathway (AOP) conceptual framework according to the OECD criteria (OECD 2013, 2014).
An AOP describes the chain of events leading from the first interaction of any chemical with a target (molecular initiating event (MIE)) to an adverse outcome (AO), generally an apical endpoint in accepted regulatory toxicity testing. As such, AOPs are not chemical specific and will not be used to specifically address the issue of linking exposure to a pesticide found to be associated to PD or paediatric leukaemia in epidemiological studies. Rather, the AOP framework will assess the mechanistic plausibility – if any – that pesticides pose a hazard contributing to the pathogenesis of PD or paediatric leukaemia (i.e. CHL and infant leukaemia (IFL)) and therefore represent potential risk factors.
According to the OECD guidelines, MIE and AO are sequentially linked by a series of biologically plausible and essential key events (KEs) and their relationship (key event relationships (KERs)) should be concordant on dose–response, temporality and incidence. The availability and robustness of quantitative experimental data classifies the strength, in a codified assembly of weight of evidence, of the developed AOP. Putative AOPs are based on a hypothesised sequence of KE and KERs supported by biological plausibility and/or statistical inference; qualitative AOPs include assembly and evaluation of the supporting weight of evidence; quantitative AOPs are supported by quantitative relationships and/or computational models that allow quantitative translation of key event measurements into predicted probability or severity of AO.
The Panel adopted the AOP framework to assess biological plausibility of epidemiological studies (i.e. that a plausible mechanism exists linking the cause to the effect). The starting point was the identification of a sequence of events able to (i) link a MIE to an AO, in this case, a relevant complex disease through a series of key events and (ii) describe the KER on the basis of biological plausibility, essentiality and empirical support. It should be noted that biological plausibility in the context of KERs is defined by an established question (i.e. is there a mechanistic relation between KEup and KEdown consistent with established biological knowledge?). The most relevant requisite was to identify a defined symptom for each disease equivalent to an AO for toxicants, reproducible in animal models, and possibly associable to a defined and measurable regulatory apical endpoint also triggered by chemicals in the regulatory or investigative studies. For PD, the application of the above rationale led to the identification of parkinsonian motor symptoms, i.e. the typical motor deficit observed in humans and in experimental conditions, associated with a decrease in number of dopaminergic neurons as a representative AO.
The empirical support for KER was provided by tool compounds (i.e. chemical stressors), which were selected from the literature:
1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), a street drug contaminant, supported by human poisoning data, as well as experimental animal data.
The piscicide/insecticide rotenone, supported by epidemiological and experimental animal data and a well characterised molecular target; the mitochondrial complex I for which human evidence of the involvement in PD exists.
The herbicide paraquat, supported by epidemiological studies and for which experimental data exist.
In line with the selected AO and chemicals and based on the literature retrieved, two qualitative AOPs relevant for PD were built. Two MIEs, binding to mitochondrial complex I and initiation by a chemical of a redox cycling process, were defined. Those MIEs lead to parkinsonian motor deficit converging in a sequence of consequent KEs (summarised as mitochondrial dysfunction, impaired proteostasis and degeneration of dopaminergic neurons of the nigrostriatal pathway). Through a detailed analysis of the KERs, the strength of association was judged by a weight of evidence approach based on modified Bradford–Hill criteria (i.e. based on biological plausibility, essentiality and empirical support of linkage, quantitative understanding of the linkage, evidence supporting taxonomic applicability and evaluation of uncertainties and inconsistencies). The overall weight of evidence indicates a strong link between the identified MIEs and the AO, i.e. parkinsonian motor symptoms.
For CHL, the Panel adopted the same rationale as used for PD, supported both by a systematic literature review commissioned by EFSA (EN‐955, 2016) and expert knowledge. In this case, it became apparent that the term ‘childhood leukaemia’ used in epidemiological studies is general and does not distinguish between infant and childhood leukaemia or other forms of paediatric leukaemia. Although both diseases share in utero exposure to relevant environmental risk factors for the development of the disease, they display distinct pathological pathways. Furthermore, while for CHL, the Panel was not able to identify tool chemicals that were able to induce the disease in the experimental models, for IFL enough evidence supported the applicability of the anticancer drug etoposide as a tool. Symptoms and signs of overt paediatric leukaemia were chosen as AO, although the disease as such is not an apical endpoint in the regulatory toxicity studies. Taking into account the above limitations, it has been considered scientifically acceptable to develop a qualitative AOP relevant for IFL and to design only a putative AOP for CHL. The development of these two different AOPs, also in comparison to AOPs relevant for PD, allowed evaluating the flexibility of such an approach. In line with the selected AO and the prototype chemical etoposide for IFL, a MIE ‘in utero topoisomerase II poisoning’ was defined. It was linked to the selected AO through a single KE summarised as ‘in utero MLL chromosomal rearrangement’. The overall weight of evidence suggests that the link between the MIE and the AO is strong and that the proposed events can be used to explore the IFL‐triggering hazard of chemicals. As stated, the AOP developed for CHL is based on weaker biological plausibility. However, a hypothetical biological plausibility could exist but cannot be convincingly formulated with the currently available circumstantial information. Although epidemiological observations suggest that the association of the disease to in utero exposure to pesticides, complexities in defining a definite MIE and involvement of modulating factors as well as limitations in the standard design of regulatory studies for the exploration of tumour‐related endpoints following in utero exposure prevent building a convincing qualitative AOP. In addition, the Panel recognises that an animal model recapitulating the disease is not available and this is also weakening the assessment.
Based on the results obtained, the Panel supports the use of the AOP framework to scientifically and transparently explore the biological plausibility of the epidemiological association between pesticide exposures and human health outcomes. Moreover, pesticides triggering the MIEs of the proposed AOPs should be considered as potential risk factors with respect to the development of analysed diseases, considering the power of the AOP framework, at its best, to provide quantitative knowledge of biological pathways leading to an AO on a weight of evidence basis.
The Panel also identified a number of uncertainties regarding the three major areas explored during the development of this Scientific Opinion, i.e. epidemiological studies, experimental studies and AOP development.
Although the AOPs developed in the present Scientific Opinion only explain a small fraction of the supposed interactions of pesticides, PD and paediatric leukaemia risk, the Panel considered the outcome of this approach promising. Thus, a multitude of AOPs might be developed to investigate the potential link of various pesticides to the different symptoms of the considered diseases. Beside this very relevant point, the AOP framework also represents a suitable scaffold to help identifying data gaps by analysing the weight of evidence for each KER within the defined AOPs. In addition, by suggesting and providing quantitative and measurable markers for critical biological events leading to the development of an AO, the AOP framework may help in the revision of regulatory studies underlining any limitation in the appropriate identification of effects and mode of actions relevant to complex human diseases, PD and paediatric leukaemia in the specific investigated case.
Summarising, the application of an AOP represents a transparent and weighted approach to define and map the causal linkages between key biological processes (MIE and KEs) to an AO that represents an apical endpoint in accepted regulatory toxicity testing. The design of an AOP, according to the OECD guidelines, identifies data gaps and provides information on the best approach to be adopted to investigate a defined toxicity pathway (representative of a relevant pathway of complex human diseases). This helps in identifying data gaps and in tailoring a tiered testing strategies for hazard identification and characterisation. When quantitative, an AOP would define a threshold able to trigger the sequence of KEs from the MIE to the AO. Because the AOP process as such is ‘chemically agnostic’, it provides indication of the biological plausibility of a hazard.
Based on these considerations, the contribution of the AOP concept has been evaluated by designing a strategy based on the two AOPs relevant for PD due to their strong weight of evidence and the richness of experimental data. In assessment of risk, the AOP framework cannot be used as a stand‐alone procedure but should inform an Integrated Approach for Testing and Assessment (IATA) scheme, integrating the chemical specific toxicokinetic properties. This will enhance confidence that the threshold of activation linking the MIE to the AO indeed triggers the cascade and by this way supports the regulatory process.
1. Introduction
1.1. Background and Terms of Reference as provided by EFSA
According to Regulation (EC) No 1107/2009 on placing of plant protection products on the market, applicants submitting dossiers for approval of active substances shall provide ‘scientific peer‐reviewed open literature […] on the active substance and its relevant metabolites dealing with side‐effects on health […] and published within the last ten years before the date of submission of the dossier’. This should include epidemiological studies, as explicitly listed in Commission Regulation 283/2013 setting out the data requirements for active substances.
In 2013, EFSA published an external scientific report carried out by the University of Ioannina Medical School in Greece on a literature review linking exposure to pesticides and human health effects based on a systematic review of epidemiological studies published between 2006 and 2012 (Ntzani et al., 2013, EFSA 2013:EN‐497). This report summarises the association between pesticide exposure (assessed by different methods) and 23 major categories of human health outcomes. A statistically significant association was observed through fixed and random effect meta‐analyses between pesticide exposure and the following health outcomes: liver cancer, breast cancer, stomach cancer, amyotrophic lateral sclerosis, asthma, type II diabetes, childhood leukaemia (CHL) and Parkinson's disease (PD). The results from the meta‐analysis of the two latter health outcomes were supported by similar findings in previously and subsequent published studies (additional information Sections 1.4.2 and 1.4.4).
Despite the large volume of available research data and the large number (> 6,000) of analyses, firm conclusions could not be drawn for the majority of the outcomes studied. This observation is in line with previous studies on environmental epidemiology and in particular on pesticides which all acknowledge that such epidemiological studies generally suffer from many methodological limitations and large heterogeneities in their conduct. Also, due to the generic terms used for the pesticides assessed in the epidemiological studies, no information could be retrieved on specific pesticides.
In addition, the involvement of pesticide exposure in relation to the aetiology of most of the health outcomes reported by Ntzani et al. (2013) is unknown, and is likely to be influenced by environmental, lifestyle and genetic factors, which may add to the complexity of the interpretation of both epidemiological and experimental data. Consequently, the use of epidemiological studies and their integration in regulatory risk assessment represents a major challenge for scientists, risk assessors and risk managers and the impact of these studies in regulatory risk assessment is still limited.
Nevertheless, the findings observed in the Ntanzi et al., report raise the question on whether the available experimental data and information on mechanisms of toxicity of pesticides can support these observations and if the regulatory risk assessment carried out to authorise the placing of plant protection products on the market covers the hazard assessment of pesticides with regard to these diseases.
The evaluation of the methodological limitations identified in epidemiological studies included in the Ntanzi et al., report (2013) is outside the scope of the mandate and will be addressed in a follow‐up mandate.
1.2. Terms of Reference
The PPR Panel is requested to prepare a Scientific Opinion investigating experimental toxicological properties of plant protection products having a potential link to PD and CHL based on the findings in the Ntanzi et al., report (2013). This opinion will:
-
Review the available data in the open literature and in regulatory toxicological data of pesticide active substances for which a potential link with a Mode of Action (MoA) relevant for the PD and CHL is known to exist to:
-
1
— develop a prototype to assess the risk factor using the principles established for adverse outcome pathways (OECD, 2013);
-
2
— analyse the plausible involvement of pesticide exposure as a risk factor for the development of PD and CHL;
-
3
— evaluate if, how and to what extent the experimental toxicity studies on mechanisms of toxicity cover effects and modes of action that are relevant to Parkinson's disease and childhood leukaemia and are in line with the adverse outcome pathways.
-
1
Make recommendations to address the remaining data gaps for assessing the link between pesticide exposure and PD and CHL, and potential weaknesses in the current regulatory dossiers in supporting the hazard assessment of pesticides with regard to these diseases.
1.3. Interpretation of the Terms of Reference
In the Terms of Reference, EFSA has requested a Scientific Opinion on investigating experimental toxicological properties of plant protection products having a potential link to PD and CHL. The terms of reference further elaborates that the European Food Safety Authority (EFSA) asked the Panel on Plant Protection Products and their Residues (PPR Panel) should develop Adverse Outcome Pathways (AOP)'s for these two diseases on the basis of a systematic literature review and the available toxicological data of pesticide active substances. The PPR Panel is indeed aware that the associations found between pesticide exposure and PD or CHL could be partly due to bias or confounding; also the potential role of pesticides has to be put in context with other risk factors. In the meantime, the Panel wishes to stress that a prominent goal of this opinion is exactly to explore whether the building of AOPs is able to throw light on the biological plausibility underlying the epidemiologically observed associations. The Panel believes that this approach would be beneficial, particularly in pointing out which kind of further studies would be needed to either support or disproof the plausibility of the epidemiological observations.
The AOP framework facilitates functional understanding of complex biological systems and the pathways of toxicity that results in adverse outcomes (AO). The AOP has a broader scope than the WHO International Programme of Chemical Safety (IPCS) MoA concept, which illustrates how to organise and apply mechanistic information on chemical's MoA to understand human relevance of animal data (Meek et al., 2014). In this perspective, the MoA is chemical specific while the AOP is not.
The methodology provides a framework to collect and evaluate relevant chemical, biological and toxicological information in such a way that it is useful for risk assessment (OECD, 2013). The OECD has incorporated the IPCS framework on MoA in its guidance document on developing and assessing AOPs (Handbook series no. 184) in order to evaluate the biological plausibility of the relationships between the identified key events. These key events must be experimentally measurable and causally linked to the AO, which is usually associated with the findings of experiments based on in vivo OECD test guidelines. The AOP identified must not contradict any steps of normal biological processes since they need to be biologically plausible.
The human relevance of the MoA framework has been applied in a number of specific case studies on compounds with a focus on quantitative time– and dose–response relationships. Modified Bradford–Hill's criteria for a causal relationship in epidemiological studies are also applied to the AOP concept as a critical foundation for overall weight of evidence (WoE) evaluation. Therefore, if data are available, the causative link between the identified molecular initiating event (MIE), intermediate key events and final adverse outcome should be described in a quantitative manner, thus increasing the confidence for use in the regulatory context.
For the scope of this scientific opinion, any AOP (e.g. putative, qualitative and/or quantitative) will be useful for hazard identification or priority setting for further testing and development. The Panel understands that the ToR does not encompass full risk assessment (i.e. exposure assessment) of pesticides potentially involved in the diseases. Thus, the opinion will neither address specific exposures to pesticides found to be associated with PD and CHL in epidemiological studies, nor consider exposure scenarios of specific active substance and their uses as specified in dossiers submitted for the European Union (EU) approval and the subsequent evaluation.
The ToR, instead, addresses the potential uses of the AOP concept in the regulatory risk assessment including the definition of biological plausibility in relation to the epidemiological studies i.e. that a plausible mechanism between a cause and an effect exists. The mandate is intended to support the future hazard assessment of pesticides; thus, the AOP conceptual framework will describe the identified MIE and its relationship with intermediate key events leading to a defined AO using a WoE approach based on so called ‘modified Bradford–Hill criteria’. For the empirical support, the panel will use data obtained from experimental studies of tool chemicals to establish response–response, temporality and incidence concordance within the AOP scheme. The mandate will also analyse to what extent the available experimental toxicity studies cover the identified pathways of toxicity that are relevant for the development of the two diseases. Furthermore, the potential gaps of knowledge and uncertainties in the current pesticide data requirements and dossiers will be identified.
By making its evaluation, the Panel realised that the health outcomes from the epidemiological studies did not distinguish between parkinsonian disorders and PD, and between CHL and infant leukaemia (IFL). Conversely, the Panel addressed more specific health outcome in line with the AOP conceptual framework, i.e. parkinsonian motor deficit, CHL and IFL.
In the context of this Scientific Opinion, the Panel made, where possible, a tentative quantitative insight, by estimating the concentration at the target site able to trigger the sequence of events up to the AO.
In conclusion, according to the ToR, the opinion will:
analyse the plausible involvement of pesticide exposure as a risk factor for the development of PD, childhood and infant leukaemia on the basis of AOPs for these diseases;
use the AOP as guidance to evaluate, if the experimental toxicity studies on mechanisms of toxicity cover effects and modes of action relevant to PD and childhood/infant leukaemia;
develop a prototype approach to assess pesticides as risk factors for complex diseases using the principles (OECD, 2013) established for adverse outcome pathways.
1.4. Additional information
This chapter is intended to inform the reader on:
data requirements for pesticide approval in regard to the hazards associated with neurotoxicity, carcinogenicity and haematology as they are expected to include apical endpoints relevant for the diseases considered in this opinion;
a summary of the epidemiological information linking exposure to pesticides and the diseases considered in this opinion;
an introduction to the AOP conceptual framework.
1.4.1. Data requirements for pesticide approval in regard to neurotoxicity
Previous data requirements under Directive 91/414/EEC concerning the placing of plant protection products on the market:
Under Directive 91/414/EEC, in order to apply for the inclusion of an active substance in Annex I, a dossier satisfying the requirements of Annex II has to be submitted.
The toxicological and metabolism requirements, listed in point 5 of the Annex II of the Directive should permit a decision as to whether, or not, the active substance could be included in Annex I, to specify appropriate conditions or restrictions of use, to classify the active substance as to hazard, to establish relevant reference values as regards human health to perform risk assessment for man and to identify relevant first aid measures.
In routine required toxicological studies (acute toxicity studies point 5.2, short‐term toxicity studies point 5.3, long‐term toxicity and carcinogenicity studies point 5.5, and reproductive toxicity studies point 5.6), all potentially adverse effects found should be investigated and reported including neurotoxicity. In cases where specific effects (e.g. neurotoxic effects) are identified, additional studies may be carried out in order to establish a no observed adverse effect level (NOAEL), to assess the significance of these effects and to investigate the probable MoA.
The need for such supplementary studies on the active substance (as indicated in point 5.8.2 of Annex II) must be made on a case‐by‐case basis, taking into account the results of the available toxicological and metabolism studies and the most important exposure routes.
A specific data requirement is dedicated to delayed neurotoxicity (point 5.7). The test submitted should conclude whether the active substance induces delayed neurotoxicity after acute exposure. Such a test is mandatory for substances of similar or related structures to organophosphates.
Current data requirements under REGULATION (EC) No 1107/2009 concerning the placing of plant protection products on the market and repealing Directives 79/117/EEC and 91/414/EEC:
REGULATION (EC) No 1107/2009 came into force on 14 December 2009 and applied from 14 June 2011 replacing Directive 91/414/EEC.
Under REGULATION (EC) No 1107/2009, an active substance is approved at EU level, following assessment against a set of agreed criteria. Those criteria not only cover both the risks arising from the use of plant protection products which contain it as it was already the case under Directive 91/4141/EEC, but also the intrinsic properties of the active substance (i.e. an assessment of its hazard).
Indeed different categories of active substances are defined in REGULATION (EC) No 1107/2009 (active substances candidate for substitution, low risk active substances, basic substances) based on their hazard which impact the conditions of their approval.
Neurotoxicity among other criteria is taken into account to categorise active substances. In this way, an active substance:
shall not be considered of low risk or as basic substance where it has neurotoxic effects (article 22 and 23);
shall be approved as a candidate for substitution, if there are reasons for concern linked to developmental neurotoxic effects (article 24).
For approval of pesticides under REGULATION (EC) No 1107/2009, the data requirements are set out in Regulation (EU) No 283/2013 (replacing Annex II of Directive 91/414/EEC).
As was already the case under Directive 91/414/EEC, potential neurotoxic effects shall be carefully addressed and reported in routine required toxicological studies (acute toxicity studies point 5.2, short‐term toxicity studies point 5.3, long‐term toxicity and carcinogenicity studies point 5.5, and reproductive toxicity studies point 5.6).
Compared to Directive 91/414/EEC, neurotoxicity requirements have been given more importance, the main differences are:
In point 5.6.2 dedicated to developmental toxicity requirements, it is mentioned that information on developmental neurotoxicity may be required when such effects are indicated by observation in other studies or suspected based on the MoA of the active substance.
Point 5.7 is not only restricted to delayed neurotoxicity requirements but also includes both neurotoxicity in rodents (point 7.1) and delayed polyneuropathy studies (point 5.7.2).
Regarding neurotoxicity in rodents, inclusion of neurotoxicity investigations in routine toxicology studies shall also be considered.
1.4.1.1. Triggers for neurotoxicity testing
The circumstances in which neurotoxicity studies should be performed are listed in Regulation (EU) No 283/2013.
-
Specific neurotoxicity studies in rodents (point 7.1) shall be performed in case of one of the following conditions:
-
1
— there is indication of neurotoxicity in routine toxicity studies carried out with the active substance;
-
2
— the active substance is a structurally related to known neurotoxic compound;
-
3
— the active substance has a neurotoxic mode of pesticidal action.
-
1
Delayed neurotoxicity studies shall be performed for active substances with similar or related structures to compounds capable of inducing delayed polyneuropathy such as organophosphates.
Developmental neurotoxicity study may be performed when indication of such effects have been triggered in previous toxicity studies.
As a result, while neurotoxicity screening is part of the requirements in current guidelines of standard toxicity studies (OECD 407, 408 and optionally in OECD 452 see Table 1), specific neurotoxicity studies are not routinely required for all pesticide active substances.
Table 1.
Neurotoxicity test guidelines
| Test procedure | Detailed clinical observations | Functional tests | Pathology | Remarks | |
|---|---|---|---|---|---|
|
Neurotoxicity Study in Rodents OECD 424(1997) |
Animal: Rat young adults 20 (10 M & 10 F)/group 3 doses tested + 1 control group Exposure: Acute or 28 days, 90 days or chronic (1 year or longer) As a stand‐alone study or combined with repeated dose toxicity studies |
In the home cage and open field including: autonomic activity Body position, activity level Gait posture, reactivity to handling, placing or other environmental stimuli, the presence of clonic or tonic movements, convulsions or tremors, stereotypies, behaviour, aggression secretions, excretions Frequency depending on the duration of the study:
|
Sensory reactivity to different stimuli (auditory, visual, proprioceptive stimuli…) Limb grip strength Motor activity measured with an automated device capable of detecting both decreases and increases in activity Frequency depending on the duration of the study:
|
At least 5 M and 5 F/group, perfused in situ and used for detailed neurohistopathology Histopathology of representative sections of:
|
OECD GUIDANCE DOCUMENT FOR NEUROTOXICITY TESTING: In case of stand‐alone study, the remainder of the animals may be used for specific neurobehavioural, neuropathological, neurochemical, electrophysiological procedures If other data available on potential neurotoxicity (e.g. structure–activity, epidemiological data, etc.) Inclusion of more specialised tests of sensory and motor function or learning and memory to be considered |
|
Developmental Neurotoxicity Study OECD 426 (2007) |
Animal: pregnant rats (at least 20 litters/group) At least 3 dose levels + control Exposure: from GD 6 to PND 21 Study termination at PND 70 |
In the home cage and open field (see OECD 424) 20/sex (1/sex per litter) Frequency depending on the duration of the study: Preweaning: weekly Adolescence: at least every 2 weeks Young adults: at least every 2 weeks |
Behavioural ontogeny Frequency: at least 2 measures preweaning) Motor activity Frequency: 1–3 times (preweaning) once (young adults) Motor and sensory function Frequency: once (adolescence) once (young adults) Learning and memory tests Frequency: once (adolescence) once (young adults) |
Brain weights (PND 11–22 & PND 70) Neuropathological examination (at PND 11–22 immersion or perfusion fixation and PND 70 perfusion fixation) Morphometric evaluation Representative sections of Brain: olfactory bulbs, cerebral cortex, hippocampus, basal ganglia, thalamus, hypothalamus, midbrain (tectum, tegmentum and cerebral peduncles), pons, medulla oblongata, cerebellum) In adults, at study termination, eye with optic nerve and retina Spinal cord at the cervical and lumbar swellings, the dorsal and ventral root fibres, the proximal Sciatic nerve, the proximal tibial nerve (at the knee), and the tibial nerve calf muscle branches |
Alternatively OECD 443 Extended One‐Generation Reproductive Toxicity Study could be carried out. In this guideline, cohort is assigned to developmental neurotoxicity testing |
|
Delayed Neurotoxicity of Organophosphorus Substances
|
Animal: hen young adults Acute exposure 1 dose group & vehicle control group & positive control (TOCP) group Exposure: 28 days 3 dose levels + control |
Behavioural abnormalities, ataxia Frequency: immediately after treatment daily |
Forced motor activity, such as ladder climbing Frequency: at least twice a week |
Biochemistry 24 and 48 h after dosing six hens Brain and lumbar spinal cord prepared and assayed for NTE activity Histopathology 21‐day post‐treatment (OECD 418) 14‐day post‐treatment (OECD 419) Six hens Perfusion fixation Sections: include cerebellum (midlongitudinal level), medulla oblongata, spinal cord and peripheral nerves |
Dedicated to organophosphorus compounds TOCP = tri‐o‐cresylphosphate NTE = neuropathy target esterase |
| Repeated dose 28‐day oral toxicity study in rodents OECD 407 (2008) |
Animal: Rat young adults 10 (5 M & 5 F)/group 3 doses tested + 1 control group Exposure: 28 days |
In the home cage and open field (see OECD 424) Frequency:
|
Sensory reactivity Limb grip strength Motor activity Frequency: once May be omitted when the study is conducted as a preliminary study to a subsequent subchronic (90‐day) study |
Brain weight Histopathology of representative sections of: Brain (cerebrum, cerebellum and medulla/pons), Spinal cord Peripheral nerve |
|
|
Repeated dose 90‐day oral toxicity study in rodents OECD 408 (1998) |
Animal: Rat young adults 20 (10 M & 10 F)/group 3 doses tested + 1 control group Exposure: 90 days |
In the home cage and open field (see OECD 424) Frequency:
|
Sensory reactivity Limb grip strength Motor activity Frequency: once not earlier than in week 11 may be omitted when data on functional observations available from other studies and daily observations not revealing functional deficits |
Brain weight Histopathology of representative sections of: Brain (cerebrum, cerebellum and medulla/pons), Spinal cord (at three levels: cervical, mid‐thoracic and lumbar), Peripheral nerve (sciatic or tibial) |
|
|
Repeated dose 90‐day oral toxicity study in non‐rodents OECD 409 (1998) |
Animal: generally Dog 8 (4 M & 4 F)/group 3 doses tested + 1 control group Exposure: 90 days |
In the home cage and open field (see OECD 424) Frequency:
|
No |
Brain weight Histopathology of representative sections of: Brain (cerebrum, cerebellum and medulla/pons), Spinal cord (at three levels: cervical, mid‐thoracic and lumbar), Peripheral nerve (sciatic or tibial) |
|
|
Chronic Toxicity Studies OECD 452 (2009) |
Animal: Rodent young adults 40 (20 M & 20 F)/group Non‐rodent young adults 8 (4 M & 4 F)/group 3 doses tested + 1 control group Exposure: 52 weeks |
In the home cage and open field (see OECD 424) Frequency:
|
Optionally for chemicals where previous repeated dose 28‐day and/or 90‐day toxicity tests indicated the potential to cause neurotoxic effects |
Brain weight Histopathology of representative sections of: Brain (cerebrum, cerebellum and medulla/pons), Spinal cord (at three levels: cervical, midthoracic and lumbar), Peripheral nerve (sciatic or tibial) |
Alternatively OECD 453 Combined Chronic Toxicity/Carcinogenicity Studies Combined Chronic Toxicity/Carcinogenicity Studies could be carried out |
OECD: Organisation for Economic Co‐operation and Development; GD: gestation day; PND: postnatal day.
Triggers to perform those tests are well defined for acetylcholine esterase inhibitors for which delayed neurotoxicity studies are systematically carried out and pesticides with neurotoxic mode of pesticidal action for which at least acute neurotoxicity study in rodent has to be performed.
In other cases, specific neurotoxicity testing becomes obligatory only if neurotoxicity has been observed during organ toxicity testing or in case of structural analogy with a known neurotoxic compound. However, clear and consistent criteria to trigger submission of such data are still lacking and ‘routine’ required in vivo toxicity studies may be not sensitive enough to alert on potential neurotoxicity.
The development of a neurotoxicity testing strategy including robust and reliable in vitro assays along with other alternative methods could be of value, as also raised as one of the main conclusions and recommendations in the EFSA opinion on acetamiprid and imidacloprid (EFSA, 2013). Furthermore, understanding of toxicity mechanisms is given an increasing importance in risk assessment and therefore alternative methods including in vitro assays could also provide useful information on toxicity mechanisms involved.
1.4.1.2. Test guidelines – what do they cover
In the EU, pesticides neurotoxicity testing for regulatory purposes is based on in vivo animal test methods. The Commission Communication provides the list of test methods and guidance documents relevant to the implementation of Regulation (EU) No 283/2013.
In the table below, the test guidelines not only for neurotoxicity testing but also for organ toxicity testing that can highlight neurotoxic effects are summarised (including principle of the assay, the clinical effects, the functional tests and the pathology examinations performed in regard to neurotoxicity).
1.4.2. Epidemiological studies linking pesticide exposure with Parkinson's disease and Parkinsonism
The association between pesticide exposure and PD has been investigated in numerous epidemiological studies. Priyadarshi et al. (2000) conducted the first meta‐analysis on 19 studies published between 1989 and 1999, and found a positive and significant association between pesticide exposure and PD (OR: 1.94; 95% CI: 1.49–2.53), although with significant heterogeneity among studies. Further systematic reviews and meta‐analyses conducted since then have lent support to this observational association (Breckenridge et al., 2016; Hernández et al., 2016a).
The EFSA external scientific report (Ntzani et al., 2013) reviewed 32 studies assessing the association between pesticide exposure and PD published between 2006 and 2012. Most of the studies (80%) involved occupational exposures where general pesticide use was assessed retrospectively by means of questionnaires. Only a minor proportion of studies was prospective in design (10%) or assessed exposure by biomonitoring techniques (particularly for the lipophilic organochlorines DDT and HCB, which represent 10% of the studies). The EFSA external scientific report performed meta‐analyses for general pesticide use, DDT and paraquat exposures (which included 26, 5 and 9 studies, respectively). A significantly increased risk of PD was observed for exposure to pesticides in general, although with high heterogeneity (OR: 1.49; 95% CI: 1.28–1.73, random effect model) and for paraquat exposure (OR: 1.32; 95% CI: 1.09–1.60, fixed effect meta‐analysis), which showed moderate heterogeneity. No significant association was observed for DDT. These results are in accordance with the largest studies carried out on the association between pesticide exposure and PD published from 2000 to 2013. The observed association between pesticides and PD holds true even though the latest meta‐analyses were published considerably later, and contain a large number of additional data, relative to the earlier meta‐analyses. This indicates consistency of results over time. Moreover, different methodologies used to synthesise the available evidence resulted in the same overall result.
Tanner et al. (2011) performed a different kind of analysis in which pesticides were classified by presumed mechanism of toxic action rather than by functional categories or chemical class. Significant associations were found between PD and the use of pesticides grouped as ‘inhibitors of mitochondrial complex I’ or as ‘inducers of oxidative stress’, thus providing support in humans to findings from experimental studies. Use of rotenone, or any of the group of complex I inhibitors, was associated with PD (OR: 2.5 and 1.7, respectively). An interesting subanalysis, intended to provide evidence for temporal concordance, included only studies in which exposure to rotenone was documented up to 15 years before PD diagnosis, and an association of similar magnitude was still observed. Similarly, use of paraquat, or any of the group of oxidative stressors, was associated with PD (OR: 2.5 and 2.0, respectively). Although this study was of considerable size, limitations were reported in terms of variability of exposure, quality of diagnosis, reliability of pesticide exposure information, no separation between prevalent and incident cases, no justification for the subgroup analyses and potential bias could have occurred during selection (Mandel et al., 2012). In addition, the possibility that the results could be attributed to combined exposure or other agents was not considered in the analysis.
A further meta‐analysis on 12 cohort studies published between 1985 and 2011 reported a combined OR of 1.28 (95% CI: 1.03–1.59, random effects model), although with high heterogeneity and inconsistency among studies (van Maele‐Fabry et al., 2012). The 28% increased risk did not vary substantially when omitting studies with extreme weight values, and the highest increased risks were observed for studies with a better design.
The last meta‐analysis conducted so far (Breckenridge et al., 2016) found that most of the studies (88%) of pesticide exposure relied on self‐reported pesticide use obtained either through personal interviews (49%) or by other methods. Despite an extensive effort to correct potential statistical artefacts (correcting for publication bias, stratifying by study characteristics, fixed and random effect models, etc.), the observed association between pesticide use and PD was statistically significant for this meta‐analysis (OR: 1.22; 95% CI: 1.18–1.27 for fixed effects model and OR: 1.56; 95% CI: 1.37–1.77 for the random effects model). Use of herbicides or insecticides was associated with statistically significantly increased PD risk using the fixed effects model (OR: 1.20 and 1.32, respectively). Similar results were obtained with the random effects model. High herbicide and high insecticide use were independently and significantly associated with an increased risk of PD; conversely, use of fungicides failed to be significantly associated with PD. Regarding paraquat use, a statistically significant association was found for PD (OR: 1.69 and 1.47 using the fixed or random effects model, respectively). Moreover, a high paraquat use showed a significantly greater risk of PD as compared to non‐use (OR: 1.75; 95% CI: 1.19–2.57, fixed effects meta‐analysis). ORs for paraquat use, calculated using the fixed effects model, were statistically significant regardless of interview type (in‐person or other), method of paraquat use ascertainment (self‐reported or other) and confounder adjustment (Breckenridge et al., 2016).
An additional factor to take into account is that the use of personal protection measures and hygiene practices are important modifiers of the association between occupational pesticide exposure and PD (Thomas et al., 2009; Hines et al., 2011; Furlong et al., 2015).
The vast majority of studies on PD are case–control in design that rely on prevalent – existing – rather than incident – newly diagnosed – PD cases, potentially leading to recall bias. Besides, the diagnosis of PD is often based on self‐reporting, potentially resulting in outcome misclassification and distortion of the estimated exposure‐disease association. Indeed, the number of prospective cohort studies is much smaller. While almost all studies found a positive association between exposure to pesticides and PD, the association was not always statistically significant. A small number of studies found a negative association; however, none of them reached statistical significance (Breckenridge et al., 2016). The consistency of the size of the effect (OR/RR) between meta‐analyses combining case–control studies and cohort studies (particularly prospective cohorts) strengthens the hypothesis that exposure to pesticides may be an aetiological factor of PD (van Maele‐Fabri et al., 2012). The overall appraisal of meta‐analyses available so far suggests that there is sufficient evidence to conclude an association between pesticide exposure (broad definition) and PD, but not enough to support a causal relationship with specific pesticide classes or compounds. This last concept is also supported by the fact that pesticides are a broad category of functionally diverse substances; rather, toxicity is expected to be related to specific pathways perturbed by a causative agent.
The above observational studies on the relationship of PD and pesticides have intrinsic weaknesses, and their design does not allow conclusions on causal relationships. Limitations include the lack of an accurate exposure estimate (from both a qualitative and quantitative standpoint), the scarcity of information on dose–response relationships (which is difficult to achieve because of the long latency period of PD) and a lack of temporal concordance (most studies are case–control in design). A particular weakness is that exposure is not assessed for defined chemical entities, but rather for broad categories like ‘pesticide’ or functional ‘classes of pesticides’. Even when pesticide subgroups were used, they often provided no useful information and the subgroups herbicides and insecticides cannot be evaluated independently because most of the herbicide‐exposed subjects were also exposed to insecticides. This fact is illustrated by the statistically significant correlations observed between ORs derived from the same studies, e.g. pesticide use and insecticide use (r = 0.82), pesticide use and paraquat use (r = 0.84), herbicide use and insecticide use (r = 0.66), and insecticide use and fungicide use (r = 0.90) (Breckenridge et al., 2016). Another general limitation is that subjects seldom recall the specific class of pesticides used, and when doing so, such statements cannot be validated. The studies found in general that the risk of PD increases with longer exposure durations, but no other indications of a dose–response relation were found. It needs to be noted that environmental, lifestyle and genetic risk factors may exist that have not been corrected for in the epidemiological studies. For instance, allelic variants and single nucleotide polymorphisms (SNP) in certain genes (e.g. ABCB1 transporter (Narayan et al., 2015), nitric oxide synthase (Paul et al., 2016a) can strongly affect the association of pesticide exposure and PD. Thus, effects of environmental chemicals may only get manifest on certain genetic backgrounds (Logroscino, 2005; Hernandez et al., 2016b); in addition, different exposed populations might have unknown differences in the frequency of vulnerable genotypes. This adds a layer of uncertainty for the interpretation of the study data, in addition to the general limitations of study size (power). Concerning the latter, it has been argued that the inconsistency of findings in human populations regarding paraquat exposure and PD might be accounted for by the statistical variation of results in relatively small studies (Tanner et al., 2011).
Major advances in order to reduce uncertainties in epidemiological studies will be obtained by assessment of specific exposure and more accurate diagnostic criteria. Due to the relevance of addressing quality in the epidemiological studies, the PPR Panel is therefore elaborating a Scientific Opinion (Epidemiological Studies in Pesticide Risk Assessment), which will be highly complementary to the present and will specifically address the issues, limitations and uncertainties of epidemiological studies.
More studies are needed to identify individual pesticides that might be associated with PD, in particular with prospective cohort design and with a better characterisation of exposure at the level of individual pesticides. While the available epidemiological studies support an association between pesticides and PD, complementary experimental research is needed to overcome the limitations inherent to those studies. The ultimate goal is that experimental and mechanistic data lend support and biological plausibility to the human epidemiological data. Indeed, the concept of AOP can help in supporting biological plausibility by means of linking a MIE to an AO which is relevant for a given disease. Complex and multihits diseases, like PD, will benefit of this approach by identifying MIE(s) triggering the AO, thus identifying potential risk factors. Moving from identification of a risk factor to a more causal link (i.e. single substance) will need integration of toxicokinetic elements able to support risk assessment.
1.4.3. Data requirements in the pesticide regulations for the exploration of carcinogenicity and haematological endpoints
Under REGULATION (EC) No 1107/2009, an active substance is approved at EU level, following assessment against a set of agreed criteria.
The required toxicological data should permit to identify the hazard of an active substance, to propose a classification according to CLP Regulation, to set relevant reference values as regard human health in order to perform risk assessment and to finally draw a conclusion as to whether, or not, the active substance could be approved with potential appropriate conditions or restrictions of use.
In routine required toxicological studies, all potentially adverse effects observed should be investigated and reported including genotoxicity, carcinogenicity and haematological endpoints.
Furthermore, in REGULATION (EC) No 1107/2009, active substances are categorised according to their intrinsic hazard, which impact the conditions of their approval.
Genotoxicity and carcinogenicity among other criteria are taken into account to categorise active substances. In this way, an active substance:
shall not be approved if it is or has to be classified as mutagen category 1A or 1B, or as carcinogen category 1A or 1B, in accordance with the CLP criteria (Article 4 and Annex II points 3.6.2 and 3.6.3);
shall be approved as a candidate for substitution, if it is or has to be classified as carcinogen category 1A or 1B and has not be excluded (Article 24 and Annex II point 4);
shall not be considered of low risk or as basic substance if it is or has to be classified as mutagen or as carcinogen (Article 22 and Annex II point 5).
For approval of pesticides under REGULATION (EC) No 1107/2009, the data requirements are set out in Regulation (EU) No 283/2013.2
As regard to genotoxicity (point 5.4) and carcinogenicity (point 5.5), specific dedicated studies are routinely performed for all pesticide active substances.
As regard to haematological endpoints, they are investigated in the different repeated dose studies required (i.e. short‐term studies point 5.3, long‐term studies point 5.5).
Commission Communication provides the list of test methods and guidance documents relevant to the implementation of Regulation (EU) No 283/2013.3
1.4.3.1. Genotoxicity testing
The genotoxicity tests should address the three genotoxic endpoints, namely gene mutations, structural and numerical chromosome aberrations. The aims of the tests battery to be performed are to:
predict genotoxic potential of active substances;
identify genotoxic carcinogens at an early stage;
elucidate the mechanism of action of some carcinogens.
In order to address the genotoxicity profile of pesticide substances, a stepwise approach is followed with in vitro testing preceding in vivo testing.
First step: In vitro tests
The basic in vitro tests battery comprises two gene mutation tests (one in bacterial cells and one in mammalian cells) and a test investigating structural and numerical chromosomal alterations.
Studies to investigate gene (point) mutation:
bacterial reverse mutation test (OECD TG 471);
in vitro mammalian cell gene mutation tests using the Hprt or Xprt genes (OECD TG 476);
in vitro mammalian cell gene mutation tests using the thymidine kinase gene (OECD TG 490);
Studies to investigate chromosome aberrations:
in vitro mammalian chromosomal aberration test (OECD TG 473);
in vitro mammalian cell micronucleus test (OECD TG 487).
For active substances harbouring structural alerts not detected by the standard test battery, specific tests investigating properly those alerts may be required.
Second step: In vivo tests
If all the results of the in vitro studies are clearly negative, at least one in vivo study is performed. The appropriate test to be conducted is an in vivo micronucleus assay.
If an equivocal or a positive test result is obtained in any in vitro test, the additional testing needed is considered on a case‐by‐case basis taking into account all relevant information.
In vivo tests performed should cover the genotoxic endpoint(s) identified as positive or equivocal in vitro and investigate appropriate target organs.
Studies to investigate gene mutations:
Transgenic rodent somatic and germ cell gene mutation assays (OECD TG 488)
Studies to investigate chromosome damage:
Mammalian erythrocyte micronucleus test (OECD TG 474)
Mammalian bone marrow chromosome aberration test (OECD TG 475)
Studies to investigate primary DNA damage:
In vivo alkaline mammalian comet assay (OECD TG 489)
Unscheduled DNA synthesis (UDS) test with mammalian liver cells in vivo (OECD TG 486)
In Table 2, the test guidelines for the exploration of genotoxicity under Regulation (EU) No 283/2013 are summarised.
Table 2.
Genotoxicity test guidelines
| Test guideline | Test system | Endpoints | Remarks |
|---|---|---|---|
|
Bacterial reverse mutation test OECD 471 (1997) |
Strains of Salmonella Typhimurium TA1535; TA1537 or TA97a or TA97; TA98, TA100 and Escherichia coli WP2 strains or S. Typhimurium TA102 |
Detection of gene mutations Substitution, addition or deletion, frame‐shift and base‐pair substitutions |
First screening test Easy to use Very large database of results available |
|
In vitro mammalian cell gene mutation tests hprt or xprt genes OECD 476 (2015) |
hprt: CHO, CHL and V79 lines of Chinese hamster cells, L5178Y mouse lymphoma cells and TK6 human lymphoblastoid cells xprt: CHO‐derived AS52 cells |
Detection of gene mutations Including base pair substitutions, frame‐shift, small deletions and insertions |
xprt (contrary to hprt) May allow the detection of large deletions and possibly mitotic recombination due to its autosomal location |
|
In vitro mammalian cell gene mutation tests TK gene OECD 490 (2015) |
L5178Y mouse lymphoma cells and TK6 human lymphoblastoid cells |
Detections of gene mutations Including point mutations, frame‐shift mutations, small deletions |
Preference to the mouse lymphoma assay (MLA) most commonly performed Allows also detection chromosomal events (large deletions, chromosome rearrangements and mitotic recombination) |
|
In vitro mammalian chromosomal aberration test OECD 473 (2014) |
Cell lines including Chinese hamster ovary (CHO), Chinese hamster lung V79, Chinese hamster lung ((CHL)/IU, TK6) or primary cell cultures, including human or other mammalian peripheral blood lymphocytes |
Detection of chromosomes aberrations Chromatid‐ and chromosome‐type aberrations should be recorded separately and classified by subtypes (breaks, exchanges) |
Resource intensive, time consuming and good expertise required Not appropriate to detect aneugens |
|
In vitro mammalian cell micronucleus test OECD 487 (2014) |
Various human or rodent cell lines or primary cell cultures |
Detection of both structural and numerical chromosome aberrations Can be combined with special techniques to add mechanistic information, e.g. fluorescence in situ hybridisation (FISH) |
Rapid and easy to conduct The only in vitro test that can efficiently detect both clastogens and aneugens |
|
Transgenic rodent somatic and germ cell gene mutation assays OECD 488 (2013) |
Transgenic rodents: lacZ mouse (Muta™ Mouse); gpt delta mouse and rat lacI mouse and rat (Big Blue®) |
Detection of gene mutations Base pair substitutions, frameshift mutations, small insertions and deletions |
Allows detection of mutations in both somatic tissues and germ lines |
| Mammalian erythrocyte micronucleus test OECD 474 (2014) | Rodents (usually) |
Detection of both structural and numerical chromosome aberrations Can be combined with special techniques to additional mechanistic information, e.g. FISH |
Detects both clastogens and aneugens Most widely used in vivo test (the only in vivo test performed when in vitro tests all negative) Proof of bone marrow exposure to be provided |
|
Mammalian bone marrow chromosome aberration test OECD TG 475 (2014) |
Rodents (usually) |
Detection of structural chromosomal aberrations Not designed for detection of aneuploidy |
Expertise required |
|
In vivo Alkaline Mammalian Comet assay OECD 489 (2014) |
Rodents (usually) |
Detection of primary DNA damages DNA single‐ and double‐strand breaks |
Allows investigating multiple tissues of animals |
|
Unscheduled DNA synthesis (UDS) test with mammalian liver cells in vivo OECD 486 (1997) |
Rat (commonly used) | Detection of DNA repair | Sensitivity has been questioned |
1.4.3.2. Long‐term toxicity and carcinogenicity testing
The aims of the long‐term toxicity testing are to:
identify adverse effects resulting from long‐term exposure to the active substance;
identify target organs, where relevant;
establish the dose–response relationship;
establish the NOAEL and, if necessary, other appropriate reference points.
As for carcinogenicity testing, it permits one to:
identify carcinogenic effects resulting from long‐term exposure to the active substance;
establish the species, sex and organ specificity of tumours induced;
establish the dose–response relationship;
identify the maximum dose eliciting no carcinogenic effect where possible;
determine the MoA and human relevance of any identified carcinogenic response where possible.
A long‐term oral toxicity study and a long‐term carcinogenicity study (2 years) in rat are to be conducted; where possible these studies shall be combined. A second carcinogenicity study in mouse is to be conducted, unless it can be scientifically justified that this is not necessary. In that case, a scientifically validated alternative carcinogenicity model may be used instead of a second carcinogenicity study.
The relevant regulatory test guidelines are as follows:
Carcinogenicity studies (OECD TG 451);
Chronic toxicity studies (OECD TG 452);
Combined chronic toxicity/carcinogenicity studies (OECD TG 453).
1.4.3.3. Haematological endpoints
No specifically dedicated study is required. However, haematological endpoints among other toxicological endpoints are systematically addressed in routine required repeated dose studies (short‐term toxicity studies, long‐term toxicity and carcinogenicity studies point).
Haematological parameters are also to be investigated in the extended one‐generation reproductive toxicity study while they are not part of the investigated endpoints of the two‐generation reproductive toxicity study. Moreover, when warranted by available information, the extended one‐generation study protocol can include a cohort dedicated to detailed investigation of developmental immunotoxicity.
The haematological parameters monitored in repeated dose studies are:
red blood cells parameters (haematocrit, haemoglobin concentration and erythrocyte count);
total and differential leucocyte count;
platelet count;
blood clotting time/potential.
The relevant regulatory test guidelines are as follows:
-
Short‐term studies:
-
1
— repeated dose 28‐day oral toxicity study in rodents (OECD TG 407);
-
2
— repeated dose 90‐day oral toxicity study in rodents (OECD TG 408);
-
3
— repeated dose 90‐day oral toxicity study in non‐rodents (OECD TG 409).
-
1
-
Long‐term/carcinogenicity studies:
-
1
— carcinogenicity studies (OECD TG 451);
-
2
— chronic toxicity studies (OECD TG 452);
-
3
— combined chronic toxicity/carcinogenicity studies (OECD TG 453).
-
1
-
Reproductive toxicity study:
-
1
— extended one‐generation reproductive toxicity study (OECD TG 443).
-
1
In Table 3, the test guidelines for the exploration of carcinogenicity and haematological endpoints under Regulation (EU) No 283/2013 are summarised (including the test procedure, the haematological parameters investigated and the organs going through to histopathological examination relevant to pick up haematopoietic disorders).
Table 3.
Carcinogenicity test guidelines and haematological endpoints in the regulatory toxicological studies
| Test guideline | Test procedure | Haematology | Histopathology Organs of interest for leukaemia |
|---|---|---|---|
|
Repeated dose 28‐day oral toxicity study in rodents OECD 407 (2008) |
Animals: Rat young adults 5 M & 5 F/group 3 doses tested + 1 control group Exposure: 28 days |
Parameters: RBC parameters Total and differential leucocyte count, Platelet count Blood clotting time/potential Frequency: once at the end of the test period |
Bone Marrow, Thymus, Spleen, LN, liver |
|
Repeated dose 90‐day oral toxicity study in rodents OECD 408 (1998) |
Animals: Rat young adults 10 M & 10 F/group 3 doses tested + 1 control group Exposure: 90 days |
Parameters: RBC parameters Total and differential leucocyte count, Platelet count Blood clotting time/potential Frequency: once at the end of the test period |
Bone Marrow, Thymus, Spleen, LN, liver |
|
Repeated dose 90‐day oral toxicity study in non‐rodents OECD 409 (1998) |
Animals: Dog young adults 4 M & 4 F/group 3 doses tested + 1 control group Exposure: 90 days |
Parameters: RBC parameters Total and differential leucocyte count, Platelet count Blood clotting time/potential Frequency:
|
Bone Marrow, Thymus, Spleen, LN, liver |
|
Chronic Toxicity Studies OECD 452 (2009) |
Animals: Rodent young adults 20 M & 20 F/group Non‐rodent young adults 4 M & 4 F/group 3 doses tested + 1 control group Exposure: 52 weeks |
Parameters: RBC parameters Total and differential leucocyte count, Platelet count Blood clotting time/potential If the chemical has an effect on the haematopoietic system, reticulocyte counts and bone marrow cytology may also be performed although not routinely conducted Frequency: At 3, 6, and 12 months and at the end of test period |
Bone Marrow, Thymus, Spleen, LN, liver |
|
Carcinogenicity Studies OECD 451 (2009) |
Animals: Rodent young adults 50 M & 50 F/group 3 doses tested + 1 control group Exposure: 104 weeks rat 78 weeks mouse |
At the discretion of the study director:
Frequency:
|
Bone Marrow, Thymus, Spleen, LN, liver Non‐neoplastic histopathological findings Neoplastic histopathological findings |
|
Combined Chronic Toxicity/Carcinogenicity Studies OECD 453 (2009) |
Rodent young adults 50 M & 50 F/group (carcinogenicity phase) 10 M & 10 F/group (chronic phase) 3 doses tested + 1 control group Exposure: 52 weeks rat (chronic phase) 104 weeks rat (carcinogenicity phase) |
Parameters: RBC parameters Total and differential leucocyte count, Platelet count Blood clotting time/potential Min 10 M & 10 F/group If the chemical has an effect on the haematopoietic system, reticulocyte counts and bone marrow cytology may also be performed although not routinely conducted Frequency: At 3, 6, and 12 months and at the end of test period |
Bone Marrow, Thymus, Spleen, LN, liver Non‐neoplastic histopathological findings Neoplastic histopathological findings |
|
Extended One‐Generation Reproductive Toxicity Study OECD TG 443 (2012) |
20 M & 20 F (20 litters/group targeted) 3 doses tested + 1 control group Exposure: P: 10 weeks (2 weeks premating, 2 weeks mating 6 weeks post‐mating) F: 6 weeks (in utero +preweaning) + 0–22 weeks according to cohorts F1A: 6 weeks (in utero +preweaning) + 10 week |
Parameters: RBC parameters Total and differential leucocyte count Platelet count Blood clotting time/potential Parents: all Cohort F1A: 10 M & 10 F/group Frequency: Once at the end of the test period |
P and F1A: spleen, liver and thymus all animals Cohort 1A: Bone marrow + lymph nodes of 10 M and 10 F: group Splenic lymphocyte subpopulation analysis (CD4+ and CD8+ T lymphocytes, B lymphocytes, and natural killer cells) Splenic lymphocyte subpopulation analysis (CD4+ and CD8+ T lymphocytes, B lymphocytes, and natural killer cells) → to evaluate if exposure impacts immunological steady state distribution |
LN: Lymph Nodes.
1.4.3.4. Previous data requirements under Directive 91/414/EEC concerning the placing of plant protection products on the market
As detailed above, in the data requirements of the previous regulation 91/414/EEC genotoxicity, carcinogenicity and haematological endpoints are all mandatory to address.
In regard to genotoxicity testing, Salmonella Typhimurium reverse mutation test, in vitro mammalian cytogenetic test and in vitro mammalian cell gene mutation test were the only acceptable tests. Even when all in vitro tests were negative, one in vivo test was to be carried out (the micronucleus test OECD 474). If indicated from the in vitro results, further in vivo testing could be triggered. These are the chromosomal aberration test (OECD 475) or unscheduled DNA synthesis test (OECD 486). Thus, the former data requirements were less comprehensive, in particular in regard to in vivo mutagenicity testing and for most of the in vivo genotoxicity tests, i.e. the in vivo bone marrow micronucleus test, proof of actual bone marrow exposure was often not shown but was only assumed. This is currently being critically assessed in each case during the re‐assessment of active substances.
In relation to carcinogenicity and haematological testing, the former data requirements were as the current except that extended one generation study (OECD 443) was not available and not required.
1.4.4. Epidemiological studies linking pesticide exposure with childhood leukaemia
There is increasing concern about chronic low‐level pesticide exposure during pregnancy or childhood and its influence on childhood cancers. Epidemiological studies have suggested that maternal exposure to certain household pesticides during pregnancy may increase the risk of childhood leukaemia; however, these studies are limited because no specific pesticides were directly associated with the risk of leukaemia, but rather the broad term pesticide exposure was applied (Lu et al., 2015).
The EFSA external scientific report (Ntzani et al., 2013) updated the meta‐analysis conducted by Turner et al. (2010) on residential pesticide exposure during pregnancy and found an increased risk of childhood leukaemia associated with exposure to unspecified pesticides (OR: 1.30; 95% CI: 1.06–1.56. When exposure was restricted to insecticides, a somewhat stronger association was observed (OR: 1.69; 95% CI: 1.35–2.11). In contrast, meta‐analyses on studies examining preconception exposure failed to show statistically significant results. Ntzani et al. (2013) also updated the meta‐analysis of Turner et al. (2010) on pesticide exposure during childhood and found a significant increased risk of childhood leukaemia (OR: 1.36; 95% CI: 1.19–1.55). In spite of these positive associations, the evidence must be carefully interpreted because most studies were of small size, exposure was assessed through non‐validated self‐reported questionnaires (that are prone to misclassification) and concern was raised on publication bias. Also, only few studies included data on leukaemia subtypes.
More recently, meta‐analyses have been carried out on occupational and residential exposure to pesticides and risk of childhood leukaemia. Maternal occupational pesticide exposure during pregnancy and/or paternal occupational pesticide exposure around conception have indicated an increased risk of leukaemia in the offspring. Bailey et al. (2014) pooled data from 13 case–control studies participating in the Childhood Leukaemia International Consortium (CLIC) and found a significant increased risk of acute myeloid leukaemia (AML) in children born from mothers exposed to pesticides during pregnancy (OR: 1.94; 95% CI: 1.19–3.18), which is consistent with previous meta‐analyses; however, no significant risk was found for paternal exposure around conception (OR: 0.91; 95% CI: 0.66–1.24). In relation to acute lymphocytic leukaemia (ALL), Bailey et al. (2014) observed a 20% increased risk with paternal exposure around conception (OR: 1.20; 95% CI: 1.06–1.38), which appeared to be more evident for children with T‐cell ALL; however, no association was found between maternal exposure during pregnancy and risk of ALL (OR: 1.01; 95% CI: 0.78–1.30).
In a separate study investigating residential pesticide exposure, Bailey et al. (2015) pooled data from 12 case–control studies in the CLIC and found an increased risk of ALL associated with exposure to any pesticide shortly before conception, during pregnancy and after birth. The three exposure windows had essentially the same OR: 1.39 (95% CI: 1.25–1.55), 1.43 (95% CI: 1.32–1.54) and 1.36 (95% CI: 1.23–1.51), respectively. Little variation was found by time period, type of pesticide or among other subgroups. Regarding AML, an increased risk was found for exposure to any pesticide in the few months prior to conception (OR: 1.49; 95% CI: 1.02–2.16), and during pregnancy (OR: 1.55, 95% CI: 1.21–1.99); however, exposure after birth did not show a significantly increased risk (OR: 1.08, 95% CI: 0.76–1.53). The relative similarity in ORs between leukaemia types, time periods and pesticide types may suggest similar exposure patterns and effects across the time periods in ALL and AML, exposure to multiple pesticides or recall bias.
The meta‐analysis conducted by Chen et al. (2015) found that children of residents exposed to indoor but not outdoor insecticides had an increased risk of childhood leukaemia (OR: 1.47; 95% CI: 1.26–1.72). A significant association was also found for herbicide exposure during childhood (OR: 1.26; 95% CI: 1.10–1.44).
Almost all the available studies addressing paediatric leukaemia included both infant leukaemia and childhood leukaemia in the same diagnosis. Very few studies examined the risk of pesticide exposure with IFL (< 1 year) as a separate entity. The Brazilian Collaborative Study Group of Infant Acute Leukaemia found an increased risk of IFL in mothers exposed to domestic insecticides during pregnancy (OR: 2.18, 95% CI: 1.53–2.13) with a rather small samples size of 91 cases (Pombo de Oliveira et al., 2006). A further study also conducted in Brazil (Ferreira et al., 2013) found that ever use of pesticides during pregnancy was associated with ALL (OR: 2.10; 95% CI: 1.14–3.86) and AML (OR: 5.01; 95% CI: 1.97–12.7) in children < 1 year of age. In particular, maternal exposure to permethrin was associated with a significantly higher risk of leukaemia in children < 1 year of age (OR: 2.47; 95% CI: 1.17–5.25 for ALL; and OR: 7.28; 95% CI: 2.60–20.38 for AML).
Observational studies on pesticide exposure and paediatric leukaemia have important weaknesses in establishing causal relationships. The consistency of findings across studies may be due to the considerable overlap in the studies included in the different meta‐analyses carried out. Limitations include the lack of an accurate exposure estimate (from both a qualitative and quantitative standpoint), lack of temporal concordance (most studies were case–control in design) and little information on dose–response relationship. In addition, the sound epidemiological evidence available may be challenged by endogenous or exogenous factors, such as genetic polymorphisms, diet, lifestyle and co‐exposure to other environmental agents. Hence, accounting for simultaneous exposure to multiple agents would help to delineate true associations, but this has not been possible for most of the available evidence because of difficulties in properly assessing multiple exposures. The question arises as to whether, and to what extent, experimental and mechanistic data can lend support to the human data.
In evaluating the aetiological role of environmental factors in the pathogenesis of childhood leukaemia, there is a need to know the evidence for an association between exposure to certain environmental factors and the incidence of the disease assessed by epidemiological studies. Furthermore, evidence from experimental research is also required to know the possible mechanisms that would explain an observed or hypothesised association between the exposure to certain environmental factors and the incidence of CHL.
In observational studies, the quality of exposure assessment is crucial, especially in deriving dose–response relations. Moreover, the reduction in bias and the adjustment for confounding factors are important in assessing the evidence for causality of associations. Because of the controversy regarding the role of pesticide exposure in CHL, a WoE analysis based on Bradford–Hill criteria was performed to evaluate the available scientific evidence linking pesticide exposure with CHL (Health Council of the Netherlands. Childhood leukaemia and environmental factors. The Hague: Health Council of the Netherlands, 2012; publication no. 2012/33).
Strength. The observed associations between pesticide exposure through parental occupational exposure or residential exposure and CHL are rather weak (OR/RR < 2–3) and not always statistically significant. However, misclassification of exposure, a real problem in many types of epidemiological studies, leads to underestimation of the real risk when pesticide exposure is categorised in a dichotomous manner, thus decreasing the strength of the association. Conversely, for measures of exposure that are not dichotomous, the bias may result in either under or overestimation of the effect.
Consistency. Despite exposure often not being identical in most situations, almost all meta‐analyses published so far show a trend towards increased risk with minor differences. Overall, pesticide exposure during pregnancy tends to support a positive relationship; however, many individual studies included in the different meta‐analyses are often the same ones.
Specificity. The aetiology of CHL is multifactorial, resulting from the interplay of genetic or environmental factors. It is not possible to associate specific pesticide exposures with CHL because of the low prevalence of this disease and the imprecise exposure assessment. On the other hand, pesticide exposure is associated with many other diseases. While most of the epidemiological studies evaluated are focussed on CHL and other diseases/outcomes are usually not considered, this does not mean that other outcomes do not occur, simply studies were not designed to address them.
Temporality. When risk factors for CHL are investigated in case–control studies, exposure is usually measured retrospectively, so temporality cannot be properly addressed as they can in prospective cohort studies. Besides, responder and recall bias, might influence the accurate timing of exposure. Many epidemiological studies have assessed exposure during pregnancy or even before (prior to conception) such that the risk factor precedes the development of the disease. Nonetheless, the time window at which pesticides might exert its causative action (prior to conception, during early, mid or late pregnancy, or during childhood) is not clear. However, exposures during childhood appear to be less consistently associated with CHL than exposures during pregnancy.
Biological gradients. Exposure–response relationships can only be assessed when exposure is measured adequately and with sufficient precision. However, exposure is often assessed using questionnaires, or at best with biomonitoring techniques on spot‐samples. Accordingly, exposure assessment (and even accumulated exposure to individual chemicals) is difficult to perform and often poorly characterised. In the case of CHL, an additional limitation is that exposure can occur at different stages of the development (early pregnancy, late pregnancy or postnatally) and effects of chemicals at each stage may be different. Additionally, children and their parents are exposed to mixtures of different agents, and chemical interactions are not usually studied as well as the potential combined effect to the same agent(s) between prenatal and postnatal stages.
Many of the epidemiological studies did not assess the risk of CHL in response to the frequency or intensity of pesticide exposures. The only weak support for a positive exposure–response relationship found that the risk of leukaemia increased with the frequency of pesticide use (Van Maele‐Fabry et al., 2011).
Biological plausibility. The growing experimental studies and animal models on the biology of CHL show increased evidence for effects for chemicals, thus strengthening the biological plausibility of an association. However, there are no experimental models on specific pesticides (and hence no dose–response relationship) and the animals used failed to recapitulate all the features of the human disease. Besides, for pesticides and CHL, the qualitative and quantitative evidence on the biological mechanisms underlying the first initiating events at molecular levels is lacking. Pesticides are biologically active molecules that may play some role in cancer aetiology. Consequently, in the European Union, the use of pesticides showing some evidence of carcinogenicity or genotoxicity has been restricted or banned. Nevertheless, potential gaps in the regulatory studies, interspecies variability in target cells, and the use of co‐formulants or potential epigenetic factors cannot be ruled out.
Regarding coherence, the cause and effect interpretation should not seriously conflict with the generally known facts of the natural history and biology of the CHL. However, the natural history of this disease is far from being adequately understood, thus coherence cannot be properly assessed.
Another Bradford–Hill criterion is analogy. This means that if it is known that the effect of one type of exposure can lead to CHL, a similar effect from another type of exposure might also. However, the different variety of types of exposures associated to the disease (ionising radiation, electromagnetic fields, chemicals other than pesticides) are of little help and prevent analogy from being a useful consideration in practice.
In addition to the Bradford–Hill considerations, alternative explanations for epidemiological associations other than causality should be considered: chance, bias (specifically exposure misclassification) and confounding. If these are unlikely, a causal relation is more likely.
Despite the positive associations reported in the meta‐analyses on pesticide exposure and CHL, it is not possible to identify any individual pesticide associated with CHL because of the imprecise exposure assessment and the low prevalence of this complex disease.
1.4.5. The Adverse Outcome Pathway (AOP) framework as a conceptual tool to support the biological plausibility of epidemiology studies
Regulatory studies, traditionally based on animal experimentation, are intended to explore any potential hazard but they are not specifically designed to inform about specific and complex human health outcomes. New data type and methods can be more effective in hazard identification, but there is a need to define which data could be used and/or be more valuable for compound specific risk assessment and which could be informative on data gaps in the standard regulatory assessment or add an insight for their interpretation.
The inclusion of epidemiology findings into risk assessment is an attempt to integrate human data with toxicological data and approaches elucidating mechanisms or pathways of toxicity, rather than relying only on the standard regulatory requirements. Furthermore, human data are compelling and trigger important risk perception opinions that are frequently reported in the media. Many epidemiological studies include pesticides and their integration (why and how) or exclusion in the risk assessment should be legitimate. In this top‐down context, epidemiology findings can be used for validation purposes; however, in the context of risk assessment, they can trigger alternative approaches to investigate the biological plausibility, i.e. that a plausible mechanism between cause and effect exists, overcoming their own limitations or help when human data are not corroborated by the regulatory toxicological studies (Li et al., 2012). Thus, the complex scientific process for the identification of human risk has to involve both epidemiological and experimental data. Furthermore, when epidemiological data are lacking, experimental data are relevant to inform on the biological plausibility as part of the overall WoE.
The AOP is an organisational framework, it combines information from multiple fields of inquiry and provides knowledge of biological pathways, highlight species differences or similarities, identifies research needs and supports regulatory decisions (Villeneuve et al., 2014a,b). In this context, the AOP approach could help in organising all the available experimental knowledge providing a mechanistic based assessment of biological plausibility. The PPR Panel therefore recognises the value of using all the available information when conducting the risk assessment and is considering the AOP framework as a systematic and transparent tool for organising, reviewing and interpreting complex information from different sources. The AOP, being a conceptual framework to mechanistically understand apical endpoints of regulatory relevance, the human health outcome should be included as part of the hazard assessment and the AOP will serve as tool for hazard identification.
The AOP framework is similar to the IPCS Mode of Action/Human Relevance framework (Meek et al., 2003; Seed et al., 2005) with major differences being the dominant applications to which it is applied and the inclusion of the formal incorporation of Bradford–Hill's considerations. Because AOPs are only covering toxicodynamic aspects, they are not intended to be chemical specific in the sense that they are not developed to describe what a single chemical does, but rather to describe how any chemical, sufficiently triggering the MIE might perturb a physiological pathway adversely. The response–response, temporal and incidence concordance will be based on empirical data provided by tool compounds, i.e. the chemical stressor. Consequently, describing an AOP does not require chemical‐specific information but when applying the pathway in a predictive context relevant for risk assessment, such information is needed. Nevertheless, it requires understanding of the chemical tool‐specific properties like potency or absorption, distribution, metabolism and excretion (ADME) properties as these data will be informative for dictating the magnitude and duration of the perturbation at the MIE.
In 2012, the OECD launched the AOP development programme followed by the publication in 2013 of the OECD Guidance Document on Developing and Assessing AOPs, addressing conventions and terminology, information content of an AOP description, WoE evaluation and standardisation and rigour for developing AOPs. Conventionally, an AOP consists of a single sequence of key events connecting the MIE to an AO; the idea is to have a tool that pragmatically simplifies complex biological events (OECD, 2013, 2014).
The MIE is defined as a specialised type of KE that represents the initial point of chemical interaction on the molecular level, within an organism, that results in a perturbation that starts the AOP. The AO is defined as a specialised type of KE that is generally accepted as being of regulatory significance on the basis of correspondence to an established protection goal or equivalence to an apical endpoint in an accepted regulatory guideline toxicity test.
The MIE and the AO are linked by a series of KEs defining a direct relationship among them (KER) where the KE should provide some ability to predict or infer the state of the downstream KEs and their relationships have to be supported by biological plausibility and scientific evidence, with a quantitative understanding in a codified assembly of WoE. The availability and robustness of experimental data will classify the AOP developed into a given category, but the AOP will be considered as a living document that can change in category on the basis of new available data. In moving down from a putative AOP to a quantitative AOP it is expected to see an increase in: strength of evidence, understanding, transparency, defensibility, quantitative precision, cost, data needs and time (Table 4).
Table 4.
AOP categories
| Stages of AOP development | Characteristics |
|---|---|
| Putative AOPs | Hypothesised set of KEs and KERs primarily supported by biological plausibility and/or statistical inference |
| Qualitative AOPs | Include assembly and evaluation of the supporting weight of evidence – developed in AOP knowledgebase in accordance with internationally harmonised OECD guidance |
| Quantitative AOPs | Supported by quantitative relationships and/or computational models that allow quantitative translation of key event measurements into predicted probability or severity of adverse outcome |
AOP: Adverse Outcome Pathway; KE: key event; KER: key event relationship; OECD: Organisation for Economic Co‐operation and Development.
In this context, it is clear that an objective and complete AOP does not exist as methods and/or new experiments can change the existing one. It is also clear that any stage of AOP development has a potential utility as the level of development desired/required depends on its potential application. The PPR Panel has applied the AOP approach to investigate and possibly provide an objective and transparent mechanistic support for the biological plausibility linking a MIE to AO relevant to Parkinson's disease/parkinsonian disorders and childhood/infant leukaemia. Understanding the link between a specific MIE and an AO relevant for a human disease will help in understanding the role of specific pesticides as risk factors and will further develop compound‐specific MoA (with the inclusion of TK/ADME data) informing causality in a relationship.
These human health outcomes were selected because they are consistently observed in different meta‐analyses and represent relevant disease models for the application of the approach. While the link between environmental factors and Parkinson's disease/parkinsonian disorders is relatively data rich, data supporting the link to childhood/infant leukaemia is more scarce. This allows evaluation of the flexibility of the approach and to compare evaluations of data similarity and/or data gaps between the standard regulatory requirements and alternative studies designed to investigate toxicological endpoints specific for the diseases.
2. Introduction to Parkinson's disease, parkinsonian disorders and application of the AOP conceptual framework
Parkinson's disease is a chronic progressive neurodegenerative disorder with a higher prevalence in the aged male population (Cereda et al., 2016). It is a chronic disease as the mean duration is 15 years from the recognition of the disease until death (Shulman et al., 2011) and is progressive as the clinical signs and their severity are linked to the spread and progression of the pathology. Although the clinical symptoms include slowness of movement, resting tremor, rigidity and disturbances in balance, it is now recognised that additional non‐motor symptoms can occur as a result of the progression of the disease. Some or all of the motor symptoms can, however, be observed in different disorders and the resulting syndrome is defined as ‘parkinsonism’. When parkinsonism is the prominent part of the disorder, these are referred to as ‘parkinsonian disorders’ and include PD (Dickinson, 2012). The primary pathology is, however, common to all parkinsonian disorders and is represented by a selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which project mainly to the striatum, in association with the development of cytoplasmic, protein‐rich inclusions, called Lewy bodies (LB) and decreased levels of striatal dopamine. One of the main components of LB is the aberrant oligomeric α‐synuclein (a presynaptic neuronal protein) and a parallelism exist between the presence of motor and non‐motor symptoms and the finding of α‐synuclein pathology beyond the SNpc. This is the basis of the Braak paradigm (Braak et al., 2004, 2009), which proposes a staging system to describe the spread and progression of the pathology resulting from multiple detailed post‐mortem analysis in PD patients. The sequential occurrence of alterations and the involvement of different structures of the nervous system, including the peripheral one, is a key aspect of the disease that is relevant to understand the contribution of environmental factors and their role in the initiation of the disease (Pan‐Montojo et al., 2012).
Complex molecular landscape of PD and parkinsonian disorders
Indeed, although the molecular aetiology of the disease is unknown, it is most likely caused by a complex interplay of genetic and environmental factors with multiple interacting pathways including synaptic and mitochondrial dysfunction, impaired protein degradation, α‐synuclein pathobiology and neuroinflammation (Fujita et al., 2014). Some cases may have a clear genetic cause while others can be caused by effects of toxins (e.g. MPTP) and/or a gene–environment interaction; however, although these degenerative disorders can be inherited or idiopathic they all have as a common denominator the loss of dopaminergic neurons projecting from the substantia nigra to the putamen (Dickinson, 2012). In this context, the role of pesticides as potential environmental risk factors for PD has long been suspected and recurrent through multiple epidemiological meta‐analyses, though the specific causative agents and the mechanisms underlying the disease are not fully understood (Priyadarshi et al., 2000; Franco et al., 2010; Shulman et al., 2011; Ntzani et al., 2013; Baltazar et al., 2014). For this reason, PD and parkinsonian disorders in general is of high interest for the pesticides risk assessment and several experimental models have been proposed (Drechsel and Patel, 2008; Cicchetti et al., 2009; Moretto and Colosio, 2011; Baltazar et al., 2014). However, the linkage of a complex, multihit and unique human disease with experimental toxicological studies is still representing an important challenge for risk assessment. This is because, regulatory toxicology studies, as good as they are for exploring any potential hazards, are not designed to understand relevant mechanisms of toxicity and, particularly, they can be of limited sensitivity when hazards are likely consequent to long‐term, low‐dose exposure to toxicants, or when multiple toxicants are interacting on the same AO through different MIE or when the genetic background is influencing the adverse outcome. Due to the complexity of the disease, multiple MIEs and AOPs can be developed for PD. For this reason, the PPR Panel considered as the initial step in the construction of AOPs of interest for PD the general scientific consensus that mitochondrial and protein dysfunctions, aggregation of toxic oligomers of α‐synuclein, oxidative stress and neuroinflammation are involved in the degeneration of dopaminergic neurons in the SNpc, and that loss of these neurons is leading to parkinsonian motor symptoms. Based on the existing knowledge supporting such a consensus, the PPR Panel built up a number of initial schemes from which two AOPs were selected for further development. Tool chemicals were selected based on data availability and their use as prototype chemicals in experimental models of PD.
Tool chemicals for the AOP building
In this context, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), rotenone and paraquat, are likely to be the most widely used chemical substances to induce loss of dopaminergic neurons. In particular, MPTP is of high interest as it was able to produce Parkinson‐like motor disorders in human a few days following exposure to an illicit substance of abuse containing this compound as a contaminant. MPTP is able to cross the blood–brain barrier and is selectively taken up by dopaminergic neurons after metabolic activation by MAO‐B of astrocytes to MPP+. Rotenone is a highly lipophilic insecticide/piscicide which, unlike MPTP/MPP+, lacks specificity for dopaminergic neurons but this chemical is able to reproduce features of PD when chronically administered to rodents at doses that achieved complex I inhibition, similar to that observed in platelets of PD patients (Betarbet et al., 2000; Cannon et al., 2009). The susceptibility of dopaminergic neurons is likely due to their sensitivity to the toxicity induced by rotenone rather than toxicokinetic (i.e. metabolic) characteristics. For both substances, the neurotoxic effect is considered consequent to inhibition of complex I in the mitochondrial respiratory transport chain leading to mitochondrial dysfunction. However, both MPTP/MPP+ and rotenone can produce neuronal loss by a large number of processes and this was considered an important limitation in the construction of the AOPs (Aguilar et al., 2015). However, at least for rotenone, it was demonstrated that it causes microtubule depolymerisation at a concentration of 10 μM (Ren et al., 2005), while the IC50 for complex I inhibition is approximately 10–20 nM (Degli Espositi, 1998). It is also worth to note that these substances were mainly used as tools to reproduce in vivo and/or in vitro models of PD or to study mechanisms relevant for PD rather than for hazard identification.
Paraquat is an herbicide belonging to the chemical class of bipyridyl quaternary ammonium. Although the general toxicity of paraquat and its target organs is well characterised, its neurotoxic effect has been mainly explored in the last decade (Baltazar et al., 2014). The mechanism underlying paraquat neurotoxicity is not fully elucidated, although several pathways have been proposed the toxicity is essentially linked to its redox potential. Paraquat has complex toxicokinetics and this also includes interaction with microglia (Baltazar et al., 2014). As toxicokinetic and metabolism considerations are not relevant for the construction of the AOPs, in this context, paraquat will be used as a tool chemical to define an AOP dealing with oxidative stress, mitochondrial dysfunction and neuroinflammation (Baltazar et al., 2014).
3. Plausibility of the involvement of pesticide exposure as a risk factor for Parkinson's disease and contribution of the AOP concept to support plausibility
3.1. Biological plausibility in support of pesticide‐associated Parkinson's disease
In addition to the above‐mentioned epidemiological studies, laboratory experiments have provided evidence for neurotoxic effects and biologically plausible mechanisms linking pesticides to PD. Biologically plausible mechanisms for PD causation have been postulated for specific pesticides, including inhibition of mitochondrial complex I by rotenone, induction of oxidative stress by paraquat, and inhibition of aldehyde dehydrogenase by the dithiocarbamate fungicides maneb, ferbam or mancozeb (reviewed in Breckenridge et al., 2016).
Studies with rotenone are consistent with the assumed role of respiratory chain complex I and mitochondrial dysfunction in PD pathogenesis; data on paraquat are in line with the assumed role of oxidative stress in the disease; the toxicity of maneb in experimental animals also involved mitochondrial dysfunction. These experimental toxicants selected from the group of pesticides have triggered many of the features known from PD in animal models. They have in particular been shown to trigger dopaminergic neuronal cell death in the substantia nigra, similar to the pathology observed in PD (Drechsel and Patel, 2008; Hatcher et al., 2008).
Despite the large body of epidemiological and experimental evidence linking pesticide exposure to PD, the exact aetiological factors remain elusive, and pathogenic mechanism(s) triggering neuronal loss and PD progression are not completely known. Advances concerning the plausibility of the association have been made in the following areas:
a) Repeated and multiple chemical exposures
Pesticides currently used do not strongly bioaccumulate in the human body, whereas in the past this was not the case. Prolonged effects may therefore arise from long exposure periods or previous exposure to more bioaccumulating compounds. Alternatively, single exposure may cause minute, clinically undetectable neurotoxic effects that, if accumulated over the course of decades, might lead to triggering of disease or to the enhancement of ongoing endogenous disease progression. In this context, it is important that PD symptoms become clinically apparent only after considerable dopaminergic cell death has been ongoing. Most likely, it takes years of only a few individual neurons dying per day or month, until the threshold for clinical symptoms is reached.
The majority of work identifying potential dopaminergic toxicants associated with PD comes from studies examining mechanisms and risks arising from a single chemical. However, human environmental exposures are much more dynamic and they likely involve numerous risk modifiers including multiple chemicals or chemical mixtures. Pesticides consist of a wide range of chemical structures with diverse mechanisms of toxicity and not necessarily all of them contribute to the development of PD. The effect of pesticide mixtures has to be considered for risk assessment. The multihit hypothesis supporting neurodegeneration and PD, suggests that the brain may be capable of withstanding the effects of an individual chemical targeting dopaminergic neurons. However, when multiple chemicals target numerous sites within the dopaminergic system, defence mechanisms may be compromised resulting in cumulative damage and neuronal death (Hatcher et al., 2008). Furthermore, exposure to different pesticides may initiate a number of neurotoxic mechanisms that may converge later in a chain of linked events eventually leading to nigrostriatal dopaminergic cells death and impaired motor function. This might explain why pesticides dissimilar in their chemical structure and unlikely affecting the same cellular structure, trigger similar downstream events (e.g. mitochondrial dysfunction and oxidative stress).
Paraquat is a herbicide that has long been considered a potential risk factor for PD because of its structural similarity to MPP+, the active metabolite of MPTP. While much of the focus has been put on paraquat, other classes of pesticides are also known to impair dopaminergic neurons. Exposure to maneb, a dithiocarbamate fungicide, has been linked to neurological impairments in agricultural workers, and there are epidemiological data showing that neurodegeneration occurs more frequently in environments where workers are co‐exposed to paraquat and maneb (Thrash et al., 2007). Paraquat and maneb administered individually to mice caused no neurological damage, but when administered as a mixture, produced traits characteristic of PD (Thiruchelvam et al., 2000). A further study on mice demonstrated enhanced sensitivity of the ageing nigrostriatal dopaminergic pathway to the combination of paraquat and maneb, resulting in irreversible and progressive neurotoxicity (Thiruchelvam et al., 2003). These results were partially supported by a case–control study on 362 incident PD cases recruited between 2001 and 2007, where ambient exposures to the pesticides paraquat, maneb and ziram were estimated. The combined exposure to these pesticides at workplaces increased threefold the risk of PD, whereas the combined exposure to only ziram and paraquat, excluding maneb exposure, was still associated with a 80% increase in risk (Wang et al., 2011).
A further type of chemical interaction may occur, if one chemical, given at an early time in life, sensitises to another chemical, given at a later time point. This was observed in an experiment where (perinatal exposure during gestation and lactation exposure (i.e. developmental) to low dieldrin levels altered dopaminergic neurochemistry in offspring and exacerbated MPTP toxicity later in life (Richardson et al., 2006)).
Potentially interaction of chemicals may also occur through the links of epigenetic changes (Balmer and Leist, 2014) or of neuroinflammation, even in cases in which exposure periods are far apart. For instance, the short exposure to MPTP/MPP+ in humans resulted in an injury that initiated self‐perpetuating pathological processes, and neuroinflammation persisted for many years (Langston et al., 1999), making the respective brain regions vulnerable to potential second hits.
b) Genetic factors and gene–environment interactions
There is growing evidence suggesting that genetics may affect susceptibility to PD among the subgroup of people exposed to pesticides. Exposure to pesticides (or to specific pesticides) over the course of decades could initiate or accelerate the underlying neurodegenerative process; however, without concurrent genetic or metabolic risk factors pesticides may not necessarily lead to the disease. While a minority of PD cases may be primarily due to a specific genetic or environmental risk factor, most cases are likely due to gene–environment interactions (Fujita et al., 2014). These kinds of interactions may explain why despite the large number of people regularly exposed to pesticides not everyone develops the disease; it may only affect those carrying a genetic vulnerability. Highly penetrant mutations in some genes (SNCA, Parkin, DJ‐1, PINK 1, LRRK2 and VPS35) produce rare, monogenic forms of PD, while unique variants within LRRK2 and GBA show incomplete penetrance and are strong risk factors for the disease (Hernandez et al., 2016b). On the other hand, polymorphisms of genes encoding enzymes involved in the metabolism of pesticides or in cell damage mechanisms, in particular PON1, PON2, NQO1, NAT2, NOS and ALDH‐2 may point towards an inherent population‐specific genetic predisposition (Fong et al., 2005; Manthripragada et al., 2010; Punia et al., 2011; Wang et al., 2011; Furlong et al., 2016; Paul et al., 2016a). However, most studies addressing gene–environment interactions are limited by the small sample size and recall bias inherent to case–control studies.
c) Oxidative stress
The role of oxidative stress in the aetiopathology of PD is well established (Zhou et al., 2008; Surmeier et al., 2011; Schildknecht et al., 2013). The metabolism of DA can lead to the generation of hydrogen peroxide (H2O2) and other reactive oxygen species (ROS). This exposes dopaminergic neurons of the SNpc to a higher level of oxidative stress than other brain regions. There is evidence that some pesticides would enhance these oxidative stress events. Structurally diverse pesticides can do this, based on several different mechanisms that eventually converge on a shift of the redox balance of the dopaminergic cell. For instance, paraquat toxicity is related to its ability to redox cycle, accepting an electron from an appropriate donor with subsequent reduction in oxygen to produce superoxide while also regenerating the parent compound. Moreover, paraquat may enhance oxidative stress by activating the NADPH oxidase of microglia cells (Drechsel and Patel, 2008). A third way to increase oxidative stress would be to activate glial cells (neuroinflammation), which may directly mediated neurotoxicity or exacerbate the toxic outcomes already initiated within neurons following exposure to toxic chemicals (Ramsey and Tansey, 2014). Moreover, part of the toxic mechanism of dithiocarbamate fungicides (e.g. maneb) has been associated with the dopamine oxidation and chelation of metals, leading to alterations in cellular redox status. Permanent parkinsonism has been reported following chronic occupational exposure to maneb (Meco et al., 1994), which supports the potential role of this fungicide in the aetiology of PD. Although dopaminergic areas of the brain (striatum, substantia nigra and nucleus accumbens) have the highest levels of the antioxidant enzyme paraoxonase (PON2), levels in males are two‐ to threefold lower than in females. These lower PON2 levels may provide weaker defences against oxidative stress in male dopaminergic neurons and may support the higher incidence of PD in males (Furlong et al., 2016).
d) Aldehyde dehydrogenase (ALDH) inhibition
ALDH enzymes are responsible for detoxification of exogenous and endogenous aldehydes by oxidising aldehydes to carboxylic acids. Aldehyde metabolites have been suggested to be involved in the pathogenesis of PD; for instance, 4‐hydroxy‐nonenal (4‐HNE), a common aldehyde product of lipid peroxidation, promotes the formation of α‐synuclein oligomers (Zhang et al., 2007). ALDH also continuously detoxifies 3,4‐dihydroxyphenylacetaldehyde (DOPAL). This degradation product of dopamine is generated in neurons by monoamine oxidase (MAO), and has been involved in the loss of dopaminergic neurons in PD as a result of generating hydroxyl radicals. Under specific experimental conditions, ALDH activity can be inhibited by pesticides such as the metal‐complexed dithiocarbamates (e.g. maneb, ziram), imidazoles (benomyl, triflumizole), phthalimides (captan, folpet) and organochlorines (dieldrin) (Fitzmaurice et al., 2013, 2014).
e) Mitochondrial dysfunction
Inhibition of complex I of the mitochondrial electron transport chain is a biologically plausible mechanism for the development of PD that has gained growing relevance. Damaged mitochondrial DNA, as evidence of mitochondrial oxidative stress is, e.g. found in PD brains (Sanders et al., 2014). Both intoxication with MPTP/MPP+ and that with rotenone directly result in inhibition of complex I and in mitochondrial dysfunction (reviewed in Breckenridge et al., 2016). Dopaminergic neurons of the SNpc have been shown to be uniquely sensitive because of their higher production of mitochondrial H2O2 in response to complex I inhibition as compared to cortical neurons (Sanders et al., 2014). Inhibition of complex I activity can lead to the generation of ROS, which then target and inhibit the respiratory chain leading to subsequent ROS production and further mitochondrial damage. The consequent failure in energy production may disrupt the vesicular storage of dopamine, leading to increased free cytosolic concentrations of this auto‐oxidisable neurotransmitter (Drechsel and Patel, 2008). Two other pesticides, maneb and dieldrin, have been suggested to also inhibit the respiratory chain. For instance, exposure to maneb has been found to result in inhibition of mitochondrial complex III. This contributes to ROS production and mitochondrial dysfunction (Zhang et al., 2003; Drechsel and Patel, 2008). Organochloride pesticides related to dieldrin have been suggested to impair sequestration of dopamine into neurotransmitter vesicles, and the resultant increase in cytosolic dopamine may increase the risk of oxidative stress (Miller et al., 1999; Vergo et al., 2007).
f) Congruence of clinical features
Parkinsonism is a complex syndrome with a heterogeneous set of clinical features. For instance, parkinsonism observed in humans due to high‐dose exposure to manganese or carbon monoxide, has clinical features that differ from those that are normally related to idiopathic PD. For instance, there is a poor response to dopaminergic therapy. This situation is different for cases of PD associated to pesticides (Tanner et al., 2011). Similar clinical features were found in PD cases that did or did not have exposure to rotenone, paraquat, or groups of pesticides with similar mechanisms. This observation suggest that PD associated with these agents is clinically typical, and this provides further plausibility for a role of pesticide exposure in the aetiology of typical PD.
3.2. To what extent do experimental toxicity studies on mechanisms of toxicity cover mechanisms relevant for PD, and what is the contribution of the AOP in supporting biological plausibility
3.2.1. Rationale of the working approach
At present, different and separate sets of information exist concerning the following five domains: pesticide exposure, toxicant MoA, experimental studies, disease pathogenesis and the occurrence of PD. The combination of information from these domains may shed light on the questions (i) whether the statistical correlation of pesticide exposure and occurrence of PD is mechanistically plausible, (ii) whether there are causal links and (iii) if such links can be confirmed or refuted by experimental testing.
As a starting point, data are available from regulatory toxicity studies that link pesticides to traditional endpoints (e.g. histopathology). In addition, for some toxicants a mechanism of action is known. The first question relevant to the working group's mission was to investigate, whether a mechanism of pathogenesis could be assigned to PD in form of an AOP. The second open issue addressed was to investigate whether experimental studies would yield information concerning the mechanism of action of toxicants. The third step was then to investigate whether mechanisms of action of toxicants overlapped with mechanisms of disease pathogenesis (AOPs) relevant for PD. Finally, the answer to these questions was used to establish plausible links between the exposure to pesticides and the risk of developing PD.
3.2.2. Capturing of a complex disease (PD) by AOP
The AOP concept has been developed by toxicologists to describe the hazard of toxicants. The concept has not been envisaged to cover complex human disease. This has several reasons: defined MIE may not exist for diseases; diseases may follow a multihit principle instead of linear chains of events; diseases have multiple symptoms instead of one final unhealthy outcome; pathogenesis of chronic degenerative diseases is likely to be based on cyclic events; KEs of chronic disease are difficult to capture or to be modified experimentally; data on diseases and disease pathogenesis are different in type and in the way they can be obtained than data of poisonings with toxicants; experimental data on disease are either difficult to obtain and to reproduce or they cannot be obtained at all.
Considering the above arguments, the development of ‘AOP for diseases’ will only be possible in some favourable situations. ‘AOP relevant for a certain disease’ is a more exact definition than the more superficial but easy to remember term ‘disease AOP’. The process of AOP development is greatly facilitated, if the disease has a variant that is known to be induced by a defined toxicant; if defined molecular interventions are known to block the pathogenesis of the disease; if complete sets of data are known on defined stages of the disease; and if biomarkers or measures obtainable by non‐invasive methods describe the progression of the disease.
The most important restriction is that the AOP should not be defined for the disease as such, but for a sharply defined symptom of the disease (as equivalent to an adverse outcome for toxicants). A second important condition is that this endpoint can be reproduced in animal models, and that chemicals exist that trigger the same endpoint in the animal models; this implies that example (tool) chemicals are available that are likely to trigger the envisaged ‘disease AOP’. As such conditions were fulfilled here, it was scientifically acceptable to work on model AOPs relevant to PD.
3.2.3. Selection of the AO
Parkinson's disease is a human‐specific clinical syndrome, usually not observed in animals. The key clinical signs are bradykinesia, rigidity, resting tremor and postural instability. In addition, the disease may be associated with vegetative symptoms (intestinal disturbances, disturbed sleep pattern), cognitive decline and affective symptoms (most frequently depression). Many different AOPs may thus be associated to the disease. This is because first, the disease has several adverse outcomes, and second, several AOP may converge onto each of these AO, according to the OECD definition of AOP. For proof of concept, ‘parkinsonian motor symptoms’, i.e. what is described in patients mainly as bradykinesia and rigour, were chosen as AO. Parkinsonian motor symptoms were defined here as the typical motor deficit observed in human disease and in experimental conditions, as a result of the loss of dopaminergic neurons of the nigrostriatal pathway. Other AO could have been chosen. For instance, cognitive function (Paul et al., 2016b) and tremor would have been candidate endpoints. The choice of the Panel was driven by the relative specificity of the endpoint for PD, by the possibility to associate the AO to known and defined pathologic changes, and by the transferability to animal models. Parkinsonian motor symptoms were considered to be relatively specific, to be found universally in all cases of PD, have a well‐defined underlying pathology, and to be measurable and modifiable (by drugs) in experimental animals. Notably, parkinsonian motor symptoms are not 100% specific for PD, but this is not a necessary condition for an AO.
3.2.4. Choice of example AOP relevant both for parkinsonian motor symptoms and for pesticides as risk factors
Having chosen the AO, the next question was which types of assumed pathological sequences were to be reflected by a proof‐of‐concept AOP. The decision was taken to consider only pathological processes occurring during adult life. It has been hypothesised that PD may also have developmental origins (Landrigan et al., 2005), and pesticides may have effects on early brain development, but this potential aetiology was deliberately not considered here. For practical reasons, pathological processes were preferred for which there was sound and ample evidence that they were triggered by chemicals in experimental animals, and preferably also in humans. Having decided on these criteria, and on the AO, the literature was screened for chemicals that triggered parkinsonian motor symptoms. For this, on the one hand, a systematic literature review commissioned by EFSA (EN‐955, 2016) was consulted, and on the other hand, expert knowledge on the state of experimental parkinsonism research was used. On this basis, the Panel decided to develop two relevant AOP up to a quality level sufficient for submission to the OECD. These two AOPs (described in detail in Appendix A) are mainly based on data for three chemicals (MPTP/MPP+, rotenone, paraquat) that had particularly abundant documentation and that could be used to define the corresponding AOP.
This decision process has some important implications for the interpretation of this opinion. The most important one is that the AOP developed here may only explain a small fraction of the supposed interaction of pesticides and PD risk. As the initial molecular structures and biochemical pathways disturbed by a toxicant are highly compound‐specific, there is no such thing as a ‘pesticide AOP’ or a MoA that makes the connection of pesticide exposure and PD risk plausible. This also applies to smaller subclasses, such as herbicides, fungicides or insecticides (Breckenridge et al., 2016). The aim of the Panel was to test whether the hazard posed by individual pesticides could be linked to the pathogenesis of PD via AOP. If the outcome of this approach is considered promising, then a multitude of AOPs would need to be developed to allow linking of many different pesticides to various symptoms of PD. Some pesticides may fail to fit any of these AOPs, which could be an interesting finding as such. On the other hand, several of these AOP may share common key events, such as oxidative stress, and this would considerably reduce the development work to the definition of partial AOP and their connection to common KE.
3.2.5. Use of tool chemicals to determine whether their mechanism of action overlaps with AOP for PD
The three most data rich chemicals were selected from the literature to build AOP that would describe their hazard. MPTP/MPP+ was chosen, as there are well‐documented human poisoning data, large sets of primate data and very extensive sets of rodent data, documented by several hundred publications per year (Daneshian et al., 2015). Rotenone was chosen because of the numerous data from rodent models, and because its molecular target, the mitochondrial complex I is particularly well‐characterised. Notably, MPTP/MPP+ is assumed to have the same target, and also for human disease pathology there is good evidence that this target plays a role (Schildknecht et al., 2015). Paraquat was chosen, first as there is good evidence for its toxicity in animal models, and second as this has been an individual compound (as opposed to the group ‘pesticides’) that was associated to PD in epidemiological studies. In line with the example chemicals chosen, two MIE were defined: binding to mitochondrial complex I and initiation of a redox cycling process. These were linked to the AO via two AOP.
This process was fundamentally different from biomedical and systems biology initiatives to define disease pathogenesis. For instance, a universal PD map has been developed (Fujita et al., 2014) that incorporates the biomedical knowledge on disease processes relevant to PD. This map takes into account multiple genetic susceptibility factors and modulating events, and its organisation is non‐linear. Nevertheless, the two AOP chosen by the panel can be identified also on this complex map as relevant pathways (among others) and thus are consistent with current medical knowledge on the disease process of PD. The prototype AOPs are fully reported in the Appendix.
3.2.6. Evaluation of the AOP concerning consistency and strength of evidence
A large part of the effort to develop AOPs was used for their evaluation, and the documentation of this process. The strength of association was judged by a WoE approach based on modified Bradford–Hill criteria. This is fully described in Appendix A.
Based on the overall WoE, the Panel concluded that the link between the MIEs and the AOs as proposed in the developed AOPs is strong and that the proposed KEs (including the MIEs and the AO) can be used as a tool for exploring the hazard of a chemical to trigger parkinsonian motor deficits.
One key conclusion from this is that, if a chemical triggers the MIE or an intermediate KE of such an AOP to a sufficiently large extent, it is likely that it will also trigger the downstream KE, including the AO. This would be a large conceptual advance in predicting chemical hazard in terms of increasing the risk for chronic human disease. Another important feature also resulted from the evaluation: it is highly important to obtain as much quantitative data as possible on the KE relationships in order to practically apply hazard predictions based on AOP.
3.2.7. Support of mechanistic plausibility by AOP
Based on above considerations, the Panel is supporting the use of the AOP framework to explore a plausible mechanism linking pesticide exposure and Parkinson's disease. The recommendation is that pesticides affecting the AOPs developed here should be considered as potential risk factors (with respect to the development of PD). The same would apply to other AOPs linked to PD, and that would need to be developed in the future.
To avoid misunderstandings, it needs to be stressed that the Panel pursued the development of AOP and the recommendations of their use specifically for the identification of risk factors, and not for risk assessment. This is fully in line with the standard backbone of risk assessment, i.e. to evaluate whether there is any hazard at all, and if so, to proceed with more complex evaluation of the risk.
This exclusive focus on hazard is logical and necessary, as the AOP framework does not consider (external or internal) exposure data or any toxicokinetic and metabolic processes. To fully rationalise this, it needs to be recalled that an AOP is a ‘pathway’, i.e. a series of biochemical interactions and pathological events. From this, it becomes evident that the pathway as such cannot have pharmacokinetic parameters. These latter ones are associated with individual compounds that trigger the pathway, and they are evidently unique for each chemical, i.e. cannot be associated to the AOP as such. For practical risk assessment, this means that potential triggering of an AOP by a chemical corresponds to the step of hazard evaluation. The next step within the MoA framework of risk assessment would then be the consideration of exposure and specific ADME properties of a given compound to come to an overall conclusion on the likelihood of a pesticide to trigger PD.
3.2.8. Conclusions from AOPs on suitability of current testing methods
The Panel is interpreting the AOP as a practical, transparent and pragmatic tool to integrate knowledge on mechanisms of toxicity with the measurement of apical endpoints of toxicity. In the case of ‘AOPs relevant for human disease’, as developed here, the integration of different levels of information goes one step further. The AOP integrates mechanistic knowledge on disease pathogenesis, apical endpoints, as measured in experimental toxicity studies and clinical symptoms of the disease. This situation allows solutions to the question, in how far the apical endpoints measured in animal studies adequately reflect endpoints of disease. Already during the process of development of the small number of AOPs of this Panel assignment, it became obvious that there are limitations of the standard regulatory studies when dealing with hazards linked to human complex multihit diseases like PD and parkinsonian disorders in general. The AO (parkinsonian motor deficits) and the KE linked to degeneration of DA neurons of the nigrostriatal pathway (which are common to both AOPs developed by the Panel) are typical apical endpoints that would in theory be identifiable in the regulatory toxicity studies. However, a review of the standard technology and approach used for such studies, showed that changes in these endpoints would most likely be missed, even if large adverse effects were present (e.g. loss of 30% of all nigral dopaminergic neurons). The identification of neuropathology would require specific sectioning of the respective area (which is not done in standard OECD 90‐day guideline studies), and it would require immunohistochemical approaches instead of standard H/E staining. The motor deficit would also not be identifiable if neuronal loss in the nigrostriatal pathway was below the threshold activating motor deficits (i.e. below 50–70% loss).
The lessons learned from the AOP suggest that even if histological sectioning of the substantia nigra and staining for dopaminergic markers were included in a guideline study, severe adverse effects of test chemicals may still be missed. Both AOPs indicate that the perturbation of the key events shows not only a response–response concordance, but also that triggering of some downstream KE requires disturbance of the upstream KE for a prolonged period of time. This has major implications for the study design. For instance, dosing should be tailored in a way to continuously trigger the MIE for a long time. This may not be the case, if toxicants are dosed only once or twice a week, and only three to four times altogether. With an inappropriate dosing schedule, changes in the downstream KE or AO (i.e. the apical endpoints of regulatory studies) may be very low, or even absent. In view of these considerations, AOPs, and the mechanistic information derived from them should be used. Optimising the design of hazard identification studies according to the expected mechanisms of toxicity can then be achieved. Moreover, AOP can be used to indicate data gaps in cases of inconsistent experimental studies, and to provide guidance for improved study design to address data gaps, inconsistencies and uncertainties. This also comprises suggestions on additional endpoints to be assessed, either as direct indicators of hazard or as mechanistic support to improve data interpretation and species extrapolation.
3.3. AOPs as informative sources for appropriate identification of data gaps and testing strategy
3.3.1. AOP as a scaffold to help identifying data gaps
Due to the nature of the AOP that is building KERs and thus supporting causality of events with a WoE approach, the AOP concept is very well suited for identifying data gaps. Based on the epidemiological data linking pesticide exposure to PD and the definition of the AO being ‘parkinsonian motor deficits’, several modes of action were identified linking an initiating event to the KE essential for the AO. This essential KE is the death of dopaminergic neurons of the nigrostriatal pathway with decrease in DA, which is essential for motor control. Thus, for the AOPs developed by the Panel, the causality of substance binding to and subsequent inhibition of complex I or mitochondrial ROS formation by redox cycling both leading to mitochondrial dysfunction, impaired proteostasis, death of DA neurons of the nigrostriatal pathway and parkinsonian motor deficits is biologically plausible and essential.
Assessment of data gaps within an AOP is feasible by analysing the WoE for each KER within an AOP. In the case of KER ‘Binding of inhibitor to NADH‐ubiquinone oxidoreductase (complex I) leads to its inhibition, the WoE is strong. Despite this high level certainty, there are several open questions within this KER: (1) low doses of complex I inhibitors with only partial inhibitory function do not compromise cellular ATP levels suggesting an alternative mechanism contributing to long‐term, low‐dose nigrostriatal toxicity; (2) few data on complex I inhibitor concentration–response using human brain cells/mitochondria thus lacking sufficient quantitative human data. Also, the KER ‘A Redox Cycling compound leads to mitochondrial ROS formation and dysfunction’ has a high WoE. This is especially true for substances with an electron reduction potential more negative than O2, which effectively produce superoxide. Generation of superoxide and subsequent mitochondrial dysfunction has been well described in different taxa. The second level of KER ‘Inhibition of Complex I leads to mitochondrial dysfunction’ also has a strong WoE as complex I inhibition causes loss in mitochondrial membrane potential with decrease in ATP production, elevated levels of ROS, followed by reduced activities of enzymes of the mitochondrial respiratory chain causing ultimate mitochondrial dysfunction: a process, which is also very well studied.
Although there is the notion that other mechanisms than complex I inhibition might be responsible for dopaminergic cell death by complex I inhibiting substances, the overall data supporting this KER is outweighing. The KER ‘Mitochondrial dysfunction results in an impaired proteostasis’ has a strong WoE because there is a high biological plausibility that proteasome activity is dependent on mitochondrial function and that increased ROS formation interferes with proteasomal function. However, there is data gap on the sequence of events triggering proteasomal dysfunction. This is the case as there is a vicious cycle concerning α‐synuclein aggregation and proteasomal dysfunction and it is not clear which one is occurring in a first instance. Some studies suggest that induced oxidative stress leads to α‐synuclein aggregation that triggers proteasomal dysfunction. Other studies report that initial proteasomal dysfunction induced by ROS causes α‐synuclein aggregation. Moreover, the role of alterations in the cytoskeleton contributing to proteasomal dysfunction is not clear. For example, tubulin colocalises with α‐synuclein in Lewy bodies and tubulin function is ATP dependent.
WoE for the KER ‘Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway’ is strong. Yet, the essentiality for impaired proteostasis for nigrostriatal cell death is moderate, e.g. acute MPTP exposure leads to specific DA cell death without the formation of Lewy bodies. This might be due to the acute exposure scheme followed in the assay. Effects of long‐term and low‐dose exposure on proteostasis would be of interest and is representing a data gap in that it is not know how long this KE needs to be perturbated to trigger DA neuronal death. In addition, different features of imbalanced proteostasis can trigger one another (e.g. disturbed protein degradation, pathological protein aggregation, microtubule dysfunction); and each of them can lead to cell death. Therefore, the ‘single’ event triggering axonal degeneration or neuronal death is not known. For instance, for α‐synuclein aggregation, it is not clear whether this causes death because some vital function of neurons is lost, or whether some protein increases, e.g. because of inhibited chaperone‐mediate autophagy.
The involvement of the KER ‘neuroinflammation leading to nigrostriatal cell death and vice‐versa’ by interaction of a chemical with microglia/astrocyte cells as a MIE is discussed controversially. Some compounds like paraquat might directly activate microglia/astrocyte cells by ROS production through redox cycling by interaction with inflammatory cells NADPH oxidase. Moreover, neuroinflammation is debated as a modulatory KE possibly enhancing nigrostriatal toxicity of chemicals. In the two AOPs related to PD, neuroinflammation was placed as a late event, paralleling degeneration of dopaminergic cells of the nigrostriatal pathway. More exactly, the placement of neuroinflammation in the AOP assumes that degeneration is an important trigger of neuroinflammation, and that neuroinflammation contributes to degeneration. This cyclic nature of events is common to many chronic disease processes. In the case of neuroinflammation, even further cycles may be involved that have not been considered here: (i) possibly some features of neuroinflammation are already triggered by earlier KE and (ii) neuroinflammation may further enhance early KE of the AOPs. This complex relationship of neuroinflammation to other KE makes it difficult to define thresholds for its activation.
Furthermore, there is a data gap in the precise understanding on how activation of neuroinflammatory cells might contribute to DA toxicity and how to quantify it. There is strong WoE for the KER ‘Degeneration of DA neurons of nigrostriatal pathway leads to parkinsonian motor symptoms’. Impaired motor symptoms are expected to be clinically visible when striatal dopamine levels drop by approximately 80%, corresponding to a DA neuronal loss of approximately 60%. However, in vivo experimental studies gave inconsistent results upon compound treatment. Yet, the precise reasons for inconsistencies in results in well‐performed in vivo studies are not known, indicating a data gap.
3.3.2. Present data gaps in regulatory studies
In humans, the main neurological symptoms of PD are tremor, rigidity, bradykinesia and postural instability, which can be accompanied by non‐motor symptoms such as olfactory deficits/anosmia, sleep impairments, depression, cognitive impairment, constipation, incontinence and autonomic dysfunctions.
Pathologically, PD is characterised by the loss of dopaminergic neurons in the SNpc and the presence of cytoplasmic protein aggregates, Lewy bodies (LB), in remaining dopaminergic cells and a loss of dopamine (DA) in the striatum. Although PD animal models developed for better understanding of the disease and development of new therapeutics do not exactly reproduce the human disease, they exhibit some of the hallmarks of PD (both motor dysfunction and pathological outcomes). With regard to neurotoxicity requirements for pesticides regulatory assessment, the question is if the guidelines followed may identify these specific motor dysfunction and pathological outcomes.
Motor dysfunction
Detailed clinical observations including: autonomic activity, body position, activity level, gait, posture, reactivity to handling, placing or other environmental stimuli, the presence of clonic or tonic movements have to be performed in all OECD toxicity guidelines.
Motor activity should be measured once in short‐term repeated dose toxicity studies (OECD 407, 408 and 422) and several times in specific neurotoxicity studies (OECD 424, OECD 426 and cohort 2 of OECD 443). The same measurements (of horizontal and/or vertical movements in a test chamber) are implemented in both routine studies and neurotoxicity studies.
However, this is not a requirement in chronic toxicity studies unless neurotoxic effects have been reported in the shorter studies.
In PD animal models, co‐ordination and balance are evaluated by rotation, rotarod or pole tests, and gait abnormalities by forepaw stride length test (Li et al., 2014). Those tests are not required by any repeated dose toxicity OECD guidelines and they can be optionally incorporated in the design of neurotoxicity studies OECD 424 and OECD 426.
Pathology outcomes
Brains should be weighed and histopathological examination performed on the brain, spinal cord and peripheral nerves in all OECD guidelines.
Perfusion fixation of brains for neuropathology evaluation is only required in both OECD 424 and 426, while morphometric evaluation should be performed in OECD 426 but is only optional in OECD 424.
In order to detect damage on the substantia nigra, appropriate samples of the brain should be obtained (i.e. rostral midbrain section through the anterior colliculus).
The standard three brain sections performed in repeated dose toxicity studies do not contain the substantia nigra while in OECD 424 and 426 adequate samples from all major brain regions should be taken (e.g. olfactory bulbs, cerebral cortex, hippocampus, basal ganglia, thalamus, hypothalamus, midbrain (tectum, tegmentum, and cerebral peduncles), pons, medulla oblongata, cerebellum) to ensure a thorough examination.
Both the Society of Toxicologic Pathology (Bolon et al., 2012) and the National Toxicology Program (Rao et al., 2011) recommend routine sampling of six to seven coronal sections of the brain in standard general toxicity studies including a section through the midbrain, with the substantia nigra listed as one of the specific areas of interest. However, those recommendations are not included in current OECD 407, 408 or 452 guidelines. Furthermore, studies complying to former guidelines constitute the dossiers of many pesticides for the renewal process, thus, the recommendations are most likely not applied in the studies.
Furthermore, in order to capture the hallmarks of PD, specific procedures could be necessary (see also Appendix A) as:
detailed neuropathology assessment of the SNpc and striatum for cell death and neuroinflammation (Breckenridge et al., 2013; Smeyne et al., 2016);
detection of the total number of TH (the enzyme responsible for catalysing the conversion of the amino acid l‐tyrosine to l‐3,4‐dihydroxyphenylalanine (l‐DOPA)) positive neurons in SNpc and TH positivity in the striatum by immunocytochemistry. Stereological protocols for cell counting should be applied (Tieu et al., 2003; Breckenridge et al., 2013; Smeyne et al., 2016);
it is important that an unbiased assessment of neuronal cell loss must include carefully conducted stereology (not simply morphometry) and pathology studies where the pathologist is ‘blinded’ to the treatment regimen of each experimental animal assessed (Breckenridge et al., 2013; Smeyne et al., 2016);
neurochemistry toxicity endpoints: Dopamine (DA) and dopamine metabolites (DOPA and homovanillic acid);
immunostaining to detect α‐synuclein (AS) aggregates.
All such procedures are not routinely carried out in a standard toxicological data package submitted for pesticide approval.
With regard to the regulatory requirements, identifying hallmarks of PD may be challenging for active substance for which no previous data indicating potential neurotoxic effect is available. Indeed, only motor activity measurements performed in short‐term organ toxicity studies could give rise to a presumption. If there are no signals of neurotoxic effect in those studies, then specific neurotoxicity studies will not be required and motor activity will not been assessed in chronic toxicity study (although longer exposure may lead to different results). In the same way, histopathological measures carried out in routine studies may be not specific enough to stress PD outcomes.
In case of suspected neurotoxicity (like pesticidal mode of action or structural similarity to known neurotoxicants, neurotoxicity study (OECD 424) is required and inclusion of more specialised tests of sensory, motor function or learning and memory, specific pathological procedures should be considered in order to examine these possible effects (in this case PD) in greater detail.
Neuroinflammation
The identification of the several different features of neuroinflammation during the AOP construction process showed an important shortcoming of regulatory experimental test procedure: the lack of specific methods to assess neuroinflammation. The standard neurotoxicity testing does not require measurements of any marker of neuroinflammation, except for fuel additives, where testing for a potential increase in glial fibrillary acidic protein (GFAP), as marker of astrocyte reactivity, is mandatory according to the US EPA (40 CFR 79 67). This is a deficiency for two reasons: (i) neuroinflammation is not easily identified by standard histopathological methods (e.g. neutrophil infiltration as in many peripheral tissues is rarely observed in the brain); (ii) neuroinflammation is obviously a good indicator of a multitude of different damage processes., i.e. it indicates a toxic action of a compound even if other damage parameters are only slightly affected (and thus remain undetected by standard methods). The latter point is related to the relatively low specificity of neuroinflammation. Indeed, this process is not only exclusively observed in PB, but also observed in most neurodegenerative diseases (Whitton, 2007; Tansey and Goldberg, 2009; Niranjan, 2014; Verkhratsky et al., 2014; Sampson et al., 2016). Neuroinflammation can also be triggered by several classes of toxicants (Monnet‐Tschudi et al., 2007). This relative non‐specificity (i.e. the capture of many different AOPs with one apical endpoint) makes the testing of neuroinflammation an interesting additional endpoint in regulatory toxicology to provide an alert of ongoing damage that may otherwise have been missed. Nevertheless, neuroinflammation testing is still a challenging issue since it requires multiple endpoints and careful consideration of the test data. This is because neuroinflammation is a complex event (not a single biochemical reaction), involving different cell types (mainly microglial cell and astrocytes), responding to diverse (sometimes yet unknown) inflammogens or signals from injured neurons (Graeber and Streit, 1990; Monnet‐Tschudi et al., 2007; Kraft and Harry, 2011; Claycomb et al., 2013). Activated glial cells release a large panel of mediators, which can (i) have positive or negative consequences on the adjacent neurons; (ii) change composition during the long duration of the neuroinflammatory process; or (iii) lead to a self‐sustained vicious circle (Carson et al., 2006; Glass et al., 2010; Aguzzi et al., 2013). Thus, neuroinflammation depends strongly on the pathogenic context. The problem is that the negative/neurodegenerative consequences of neuroinflammation do not only depend on the intensity of the glial reaction (quantity), but also rather on the type of the neuroinflammatory process (quality). For instance, activated microglia can be in the M1 (prodegenerative) or the M2 (protective) state (Ponomarev et al., 2007; Maresz et al., 2008; Kigerl et al., 2009; Perego et al., 2011). Both phenotypes can be observed concomitantly (von Tobel et al., 2014) and the features of neuroinflammation can change over time, e.g. with a neurodegenerative phenotype appearing late, after cessation of exposure, as observed after repeated treatments with the herbicide paraquat (Sandstrom von Tobel et al., 2014). Therefore, it is not possible to define a threshold that should be reached to trigger the next key event, but the phenotype, the production and the composition of the inflammatory mediators, such as pro‐inflammatory cytokines, reactive oxygen (ROS) or nitrogen species (RNS) (Dong and Benveniste, 2001; Brown and Bal‐Price, 2003) should rather be considered in order to predict the consequences of the neuroinflammatory process. In addition, as inhibition of one or two features of neuroinflammation leads only to partial protection of dopaminergic neurons and terminals following rotenone, MPTP/MPP+, or paraquat exposure (for references, see table of quantitative relationships in KER neuroinflammation to neurodegeneration of nigrostriatal pathway), it is a combination of several factors and not a single one that trigger the neurodegenerative process. Therefore, neuroinflammation cannot be sufficiently characterised by measurement of a single parameter. All these considerations make it, for the time being, a challenge to include neuroinflammation in the standard regulatory studies. However, the future mechanistically driven hazard identification approaches implies also the development of in vitro testing and several test systems for neuroinflammation have been developed, based on cocultures of neurons and glial cells in 2D and 3D, using human or rodent cells as a starting point (Monnet‐Tschudi et al., 2007; Alépée et al., 2014; Sandstrom von Tobel et al., 2014; Efremova et al., 2015, 2016).
4. Introduction to Childhood Leukaemia
Paediatric leukaemia is a common childhood cancer (representing 30% of all cancers in children under the age of 15) with an incidence peak between 3 and 5 years of age. The disease is phenotypically and genetically heterogeneous, targeting B‐cell, T cell or myeloid progenitors and can be additionally stratified according to the differentiation stage at which the haematopoietic stem and progenitor cells (HSPC) are blocked. The HSPC being the target cell, fetal haematopoiesis and in utero exposure are key elements that have to be considered for the assessment of the relationship between pesticide exposure and the disease. Fetal haematopoiesis starts in the aorta gonad‐mesonephros region and yolk‐sac (ref) and colonises the fetal liver and eventually, just before birth, the bone marrow (Wang et al., 2011). The fetal liver haematopoiesis is therefore representing the sensitive target as it is entailing a massive active proliferation of progenitor cells, rendering the HSPC susceptible to oncogenic transformation following DNA damage during pregnancy (Emerenciano et al., 2007). Although the aetiology of the acute leukaemia is not defined, in utero exposure to environmental factors represents a relevant aetiological suspect; nevertheless, the paucity of mechanistic data is still representing a major obstacle to understand which toxicological pathways are involved. This is also corroborated by the likely multifactorial origin of the disease with the risk derived from environmental exposure and influenced by genetic susceptibility (Hernández and Menéndez, 2016). In addition, recent mechanistic data has supported previous epidemiological data on the role of late infections in clonal evolution of ALL. Whether environmental (i.e. pesticides) cues are affecting infection development during childhood remains unknown (Greaves, 2006).
Of note, almost all the available epidemiological evidences are not making a distinction between infant and childhood leukaemia which are two distinct aetiological and pathological entities and this is complicating the interpretation of the epidemiological outcome where the terms paediatric or childhood leukaemia is frequently generalised. Although chromosomal translocation is likely representing the common initiating oncogenic event for both disease, IFL shows a unique biological feature which is the common association with the rearrangements of the MLL gene, a master gene that regulates the normal progression of the human haematopoietic development and differentiation (Ernst et al., 2002). It has to be recognised, however, that there are ALLs with normal karyotypes, i.e. no translocation detected. It is obvious that the MIE in these cases is more obscure. Although the MLL (and analogous gene) rearrangement is representing (one of) the key events for the initiation of the disease in the HSPC (or an earlier mesenchymal cell), it is likely that it is sufficient only for the development of the overt IFL and additional factors would contribute to the aberrant proliferation of the initiated cells in CHL. These might depend on alternative (epi)genetic cooperating lesions at a critical developmental window. In addition, and relevant for this Scientific Opinion, epidemiological and genetic studies suggest that MLL rearrangement may result from in utero exposure to DNA topoisomerase‐II poisons, including but not limited to the chemotherapeutic agent etoposide (Emerenciano et al., 2007; Sanjuan‐Pla et al., 2015). A chain of pathogenetic events linking the in utero exposure to Topo‐II poisons to IFL is fully reported in Appendix B and is representing the attempt made by the Panel to build up a qualitative AOP to mechanistically support the biological plausibility that exposure to pesticides could be linked to the development of IFL.
In utero exposure to environmental risk factors is also relevant for the development of CHL. However, for the CHL, although the initiation event still involves a structural or numerical chromosomal alteration, the development of the leukaemia requires the activation of cell proliferation. The longer latency period for the CHL (when compared to the IFL) clearly indicates that the initiating event is not enough for the conversion of a preleukaemic clone into cancer, strongly suggesting that a second, very likely postnatal, hit is necessary. Dysfunction of the immune system and delayed infections have been frequently linked to CHL by means of mechanistic considerations like a dysregulated immune response consequent to a low repertoire of infections during the early development of the immune system and an aberrant congenital response to infections (Greaves, 2006). In its attempt to build an AOP specific for the CHL, the Panel found insufficient evidence to identify a mechanistically plausible MIE and no chemicals were identified to empirically support the toxicity pathway. Nevertheless, considering the relevance of the debate linking pesticide exposure and potential development of CHL, the Panel developed a hypothetical AOP which is fully reported in Appendix.
5. Plausibility of the involvement of pesticide exposure as a risk factor for infant and childhood leukaemia and contribution of the AOP concept to support plausibility
5.1. Biological plausibility in support of pesticide‐associated IFL and CHL
In contrast to the epidemiological studies mentioned above, there is scarce experimental and mechanistic evidence supporting an association between exposure (at any stage of development) to pesticides (or any other chemical excluding benzene and related compounds) and paediatric leukaemia. While for CHL, there is no tool chemical capable of inducing the disease under experimental conditions, for IFL there is enough evidence for the anticancer drug etoposide. Despite the distinct natural history and pathogenesis of infant and childhood leukaemia, both entities share a chromosomal translocation as the major initiating oncogenic event.
Most of the studies available in the open literature pertaining to pesticides do not directly link pesticide exposure to development of CHL. Nevertheless, they do provide some evidence of the genotoxic or cancer‐promoting capacities of some pesticides based on cellular studies, suggesting the potential of these compounds to trigger leukaemogenesis. An in vitro study showed that a human leukaemic (K562) cell line exposed to 1 μg/mL isofenphos for 72 h exhibited an enhanced proliferation and poor cellular differentiation (Boros and Williams, 2001). In addition, human peripheral lymphocytes exposed to 0.1–10 μg/mL isofenphos for 1 h exhibited dose‐dependent damage to chromosomal DNA (using the comet assay) as well as disruption of the cholinergic nuclear signalling pathway, which collectively could lead to genomic instability and leukaemogenesis (Williams et al., 2004). On the other hand, human K562 cells exposed to 0.1 μM of diazinon resulted in hypermethylation of several genes involved in cell cycle arrest such as cyclin‐dependent kinase inhibitors (CDKN1A and CDKN1C) as well as tumour suppressor genes such as p53 and PTEN (Zhang et al., 2012). Furthermore, human mammary carcinoma MCF‐7 cells exposed to low concentrations of diazinon http://www.who.int/foodsafety/areas_work/chemical-risks/jmpr/en/) or fenitrothion (0.001–10 nM) for 24 h exhibited a higher degree of micronucleus formation (Ukpebor et al., 2011).
Lu et al. (2015) found that human fetal liver HSPCs exposed to chlorpyrifos for 24 h resulted in MLL re‐arrangements and double‐strand DNA breaks in a dose‐ and time‐dependent manner. This study suggested that chlorpyrifos might act as a topoisomerase II (TOP2) poison similar to benzoquinone (a benzene metabolite), etoposide and bioflavonoids, thus belonging to chemicals capable of interacting with TOP2, the MIE target in the AOP IFL. However, given some limitations of the study, firm conclusions can for the time being not be drawn based on this one study.
Pesticides other than OPs have been demonstrated in in vitro studies to show genotoxic properties, which could be linked to the MIE of the AOP CHL and IFL. For instance, human neuroblastoma SH‐SY5Y cells and human T‐cell leukaemia Jurkat cells exposed to methyl‐pyrazole insecticides (tebufenpyrad, bixafen, fenpyroximate or tolfenpyrad) for 1 h showed increased induction of γ‐H2AX (a marker of double strand DNA breaks) attributed to the generation of oxidative stress as a result of impairment of the mitochondrial electron transport chain (Graillot et al., 2012). Furthermore, exposure of the CEM x 174 cell line, a hybrid of human T and B cells, to 50 μM heptachlor, chlordane or toxaphene for 24–36 h showed decreased protein levels of the tumour suppressors p53 and Rb (Rought et al., 1998, 1999). Low concentrations of heptachlor (5–10 μM) suppressed doxorubicin‐induced caspase‐3 activity and subsequent activation of apoptosis in this cell line (Rought et al., 2000). Human peripheral lymphocytes exposed to 20 μg/mL of a commercial formulation of the fungicide dinocap for 24 h exhibited increased chromosomal aberrations, formation of sister chromatid exchanges and decreased mitotic index (Celik et al., 2005). In vitro studies with the chloroalkylthiocarboximide fungicides captan and captafol at a concentration of 1 μM have shown to decrease the activity of murine TOP2 by 50% and 20%, respectively (Rahden‐Staroń, 2002). Similarly, thiram (a dithiocarbamate fungicide) inhibits TOP at 10 μM (Rahden‐Staroń et al., 1993). However, the in vivo genotoxic potential of these fungicides (i.e. genetic deletions and/or mutations) occurred only at very high doses in Drosophila (10–100 mM) (Rahden‐Staroń, 2002).
In assessing the above studies coming from the open literature, findings from regulatory studies should also be taken into account. Tebufenpyrad, bixafen, fenpyroximate, captan, chlorpyrifos and thiram are approved in the EU, and none of them are classified for genotoxicity, thus the mandatory regulatory studies did not show genotoxic potential. Captan is classified for carcinogenicity (duodenal tumours in mice). For the rest of the pesticides not approved in the EU, none of them are currently classified as being genotoxic, while two are classified being carcinogenic; namely chlordane and captafol. Thus, although a thorough assessment of the genotoxic potential of the mentioned pesticides have not been undertaken, the Panel finds that the few in vitro studies available from the open literature so far to support the epidemiological evidence for the association between childhood leukaemia and exposure (in utero and/or after birth) to some classes of pesticides is limited. Also, there is limited evidence from in vivo studies. However, it remains uncertain whether this association arises from a causal or non‐causal relationship and biological studies to provide evidence for a potential mechanism have been inconclusive. Almost all in vitro studies used immortalised cell lines or primary human lymphocytes from adults and 3‐week‐old mice, which are not appropriate cell models for studying CHL. The only one study using fetal liver HSPCs can be considered as the best cell model for this purpose.
This clearly indicate how complex is to define and weight biological plausibility when both regulatory studies and experimental studies from the open literature are contradicting.
The mechanisms underlying the association between pesticides and CHL are currently poorly understood and more studies are needed to better understand this association. There is agreement in the scientific community that a well defined key event involved in paediatric leukaemogenesis is the induction of chromosomal rearrangements. The hypothetical mechanistic linkage between some pesticides and this genetic damage may be accounted for by TOP2 poisoning (for IFL) or generation of oxidative stress leading directly or indirectly to DNA damage.
a) TOP2 poisoning (inhibition)
TOP2 has critical functions in both DNA replication and transcription processes. Under physiological circumstances, the active site tyrosine in TOP2 serves as a nucleophile to initiate the first transesterification reaction to form a covalent adduct with the backbone phosphate in DNA, thus generating a transient break. The second transesterification reseals the DNA break and regenerates the free tyrosine (Chen et al., 2013). In contrast, exposure to TOP2 poisons can lead to the stabilisation of the transient DNA/Top2 cleavage complex resulting in an increased frequency of DNA double‐strand breaks and error‐prone non‐homologous end‐joining (NHEJ) repair. For this reason, these chemicals are called TOP2 poisons to distinguish them from catalytic inhibitors of the enzyme. Cells harbouring accumulated breaks in DNA are not able to enter into the mitotic phase of the cell cycle, thus undergoing cell death.
Some anticancer drugs (i.e. etoposide, doxorubicin), environmental chemicals (i.e. benzene, some pesticides) and natural substances (i.e. bioflavonoids) are TOP2 poisons with DNA cleavage activity (Pendleton et al., 2014). Among the TOP2‐poison chemicals, only etoposide has strong evidence for causing acute leukaemia in human via the general process of the AOP described herein. For the other Top2 poisons, including bioflavonoids, the evidence is weaker.
Etoposide is a semisynthetic derivative of podophyllotoxin that exhibits cytotoxicity by inhibiting DNA synthesis as described above. However, if cells manage to bypass cell death, the accumulation of DNA double‐strand breaks (DSB)s can lead to chromosomal translocations and further generation of fusion gene products (particularly MLL rearrangement). Evidence supporting the causal relationship between etoposide‐induced TOP2 inhibition and the MLL rearrangement is strong regarding treatment‐related acute leukaemia (Cowell and Austin, 2012; Pendleton et al., 2014). Between 2% and 12% of patients that receive epipodophyllotoxin develop secondary AML, with the mean latency period from drug administration to the onset of secondary leukaemia being about 2 years. The risk of secondary AML appears to be dependent on both treatment schedule and dose. Typically, epipodophyllotoxin‐induced AML occurs after multiple doses administered in brief intravenous infusions with cumulative doses ranging from 5,200 to 19,200 mg/m2 (Ezoe, 2012). Dose–response relationships between etoposide and treatment‐related leukaemia are difficult to unravel, but risk of leukaemia seems to increase with larger total exposure to etoposide. There is no doubt that the fusion genes are caused by etoposide treatment because MLL rearrangements have not been detected in bone marrow samples banked before the start of the treatment of the first malignancy (Pendleton et al., 2014).
Chemical‐induced DNA breakpoints are associated with predicted Top2 cleavage sites (i.e. MLL), supporting an essential role for TOP2‐mediated breakage. The high frequency of Top2 recognition sites in specific DNA regions and the high expression of this enzyme in human CD34+ HSPCs represent favourable conditions for breakage following exposure to Top2 poisons. Because CD34+ HSPCs appear to be more sensitive to DNA damage than committed progenitor cells, exposure to low levels of different chemicals may induce DNA breakage at certain sites in HSPCs, increasing the risk of chromosomal rearrangements (Bueno et al., 2009; Montecucco et al., 2015; Thys et al., 2015; Hernández and Menéndez, 2016).
Studies on identical twins and neonatal blood samples strongly implicate an in utero occurrence of the key events (Sanjuan‐Pla et al., 2015). Furthermore, a study in pregnant mice demonstrated that in utero exposure of the fetus to etoposide causes the MLL chromosomal translocation analogously as in humans but with different gene fusion partners (Nanya et al., 2015). Indirect evidence from human prehaematopoietic/mesenchymal stem cells and fetal liver HSPCs strengthens the biological plausibility. Experimental evidence in these cell lines has demonstrated that etoposide causes DSBs in MLL and partner genes, which leads to the formation of fusion genes and their products (Sanjuan‐Pla et al., 2015).
Nanya et al. (2015) have shown that in utero exposure to etoposide induces MLL translocations in ATM‐knockout mice, which are defective in the DNA damage response, but not in wild‐type mice. Moreover, fetal liver HSPCs were more susceptible to etoposide than maternal bone marrow mononuclear cells, pointing out the life stage‐related susceptibility in regard to Top2 inhibition also in the mouse. However, in utero exposure to etoposide failed to induce leukaemogenesis (Nanya et al., 2015). However etoposide can induce a large number of MLL rearrangements, most of them occur within non‐coding regions, without eliciting direct oncogenic consequences. The appropriate oncogenic event needs to occur in a target cell within a relatively small and spatially restricted cell population during the appropriate and epigenetically plastic developmental window. Thus, it is a very rare event and difficult to support empirically.
Li et al. (2014) developed a cell model based on the hypothesis that cells are capable of clearing low‐level DNA damage with existing repair capacity. When the number of DSBs exceeds a certain value, ATM and p53 become fully activated through reversible mechanism, leading to elevated repair capacity. The dose–response relationships for activation of p53 and the formation of micronuclei in the target cell model indicate that critical concentrations of etoposide are in the range of 0.01–0.1 μM (Li et al., 2014). This range is in agreement with the increased levels of DSBs observed in human fetal liver CD34+ cells at a concentration of 0.14 μM of etoposide; however, MLL translocations were detectable at higher concentrations (Moneypenny et al., 2006; Bueno et al., 2009).
Despite the limited number of studies investigating the role of chemicals in the pathogenesis of paediatric leukaemia, the consistent observation of the inhibition of Top2 activity suggests that this might be a key mechanism induced by chemicals with leukaemogenic potential. Apart from this, there are also other common mechanisms observed among the studies involving chemicals, such as oxidative stress.
b) Oxidative stress
Oxidative stress has been implicated in haematotoxicity induced by benzene and pesticides (Choi et al., 2016).
Under some circumstances, oxidative lesions can lead to DNA DSB formation in HSPCs. Environmental exposures to numerous chemicals, including many pesticides, have been shown in vivo and in vitro to generate ROS that can ultimately induce DNA oxidative damage, leading to single‐strand breaks (SSBs) and DSB formation in the DNA (Sedelnikova et al., 2010). For example, OP insecticides (chlorpyrifos, methyl‐parathion, malathion), methyl‐carbamates (methomyl) and the herbicide paraquat all cause oxidative DNA damage followed by DNA SSBs and DSBs (Muniz et al., 2008; Guanggang et al., 2013; Ojha and Srivastava, 2014; Esperanza et al., 2015). There is also evidence of pesticide‐induced oxidative stress and DNA damage in agricultural workers (Muniz et al., 2008). Additionally, oxidative species may interact with biological molecules to disrupt normal DNA synthesis and repair, and so inhibition/inactivation of antioxidant proteins or DNA repair enzymes may also be an underlying molecular mechanism (Kryston et al., 2011).
ROS are not known to directly cause DSBs; however, DSBs could be generated if two SSBs oppose each other on complementary strands or could occur as secondary lesions at the replication fork or during an intermediate step in a repair process (Li et al., 2013). DNA DSBs are the most harmful initial event in molecular and cell carcinogenesis. Unrepaired DSBs may result in structural chromosomal abnormalities, whole or partial chromosome loss and genetic recombination, but can also lead to the breakdown of DNA replication, causing apoptosis to prevent a possible mutation being passed or during replication (Ap Rhys and Bohr, 1996).
Specific oncogene activation in different tumour models has been linked to DNA DSBs and the activation of DNA‐damage checkpoints. Efficient DSB repair is crucial for the maintenance of genomic integrity. In response to DNA damage, phosphatidylinositol‐3 kinase‐related kinases ATM (ataxia telangiectasia mutated) and ATR (ATM‐ and Rad3‐related) are initially activated and subsequently phosphorylate a variety of proteins that regulate the DNA‐damage response. DNA DSBs observed in some studies with chlorpyrifos and atrazine may be due to the ability of these compounds to generate highly reactive molecules/radicals (Huang et al., 2015; Lu et al., 2015). Aldicarb has caused a dose‐dependent DNA damage, with single‐strand breaks being produced by low concentrations during short time, which could be repaired, whereas high concentration led to DSBs which were difficult to repair (Li et al., 2003). Tetrachlorohydroquinone, the major toxic metabolite of pentachlorophenol, can induce DNA lesions as this metabolite contributes to the release of free radicals which have been linked to tumour promotion (Chen et al., 2015).
When the ROS are elevated beyond physiological levels, oxidative stress can cause HSPC dysfunction. These cells are extremely sensitive to oxidative stressors, such as anticancer agents, radiation, and the extensive accumulation of ROS. NADPH oxidase has been proposed for ionising radiation‐induced persistent and prolonged intracellular ROS generation in human CD34+ HSPCs, such that the resulting oxidative stress is associated with inhibition of the clonogenic potential of these cells (Yamaguchi and Kashiwakura, 2013).
ROS‐induced DSBs in human fetal liver CD34+ HSPCs following maternal exposure to chemicals trigger recombination/repair pathways by NHEJ. The majority of damaged HSPCs may either successfully repair the DNA DSBs or fail to do so and undergo apoptotic cell death. If the DNA DSBs within particular breakpoint cluster regions (bcr) are not correctly repaired, chromosomal translocations or deletions may occur, although this possibility is very unlikely and would only happen in a small fraction of cells. For fusion genes to be leukaemogenic, DSBs must occur simultaneously in two chromosomes and must also involve the coding region of the genes to generate an exon‐exon in‐frame functional chimeric gene product. Importantly, this has to occur in a HSPC that has managed to bypass cell death and displays a sustainable lifespan and clonal potential to propagate the chimeric gene product (Hernández and Menéndez, 2016).
In the case of infant leukaemia, and based on the very short latency of the disease, it is not completely clear whether the fusion gene generated from chromosomal translocations requires additional cooperating oncogenic hits for leukaemogenesis. Recurrent activating mutations of genes associated with cellular proliferation, such as components of the RAS signalling pathway, are important for tumour maintenance rather than initiation in human HSPCs (Prieto et al., 2016). The transformation mediated by the aberrant proteins encoded by fusion genes might depend on alternative (epi)genetic cooperating lesions at a critical developmentally earlier window of stem cell vulnerability to develop overt leukaemia (Sanjuan‐Pla et al., 2015).
For CHL, chromosomal translocations resulting in aberrant chimeric proteins alter the normal transcriptional program of HSPCs and block normal B‐cell and/or myeloid differentiation. In contrast to MLL‐associated infant leukaemia, chromosomal translocations linked to childhood leukaemia (i.e. TEL‐AML1) are not sufficient to cause the disease by themselves. As TEL‐AML1 fusion gene is observed in cord blood from about 1% of normal newborns, a significant proportion of the population carries self‐limiting preleukaemic clones, the majority of which do not result in disease. The longer latency and that only a fraction of children carrying the translocation develop the disease unequivocally indicates that the initiating chromosomal translocation per se is unlikely to convert a preleukaemic clone into an overt disease, consequently secondary cooperating (epi)genetic events are needed. In this regard, developmental dysfunction of the immune system followed by an aberrant immune response upon delayed infections has been linked to the development of CHL (Greaves, 2002; Pui et al., 2008; Teitell and Pandolfi, 2009; Wiemels, 2012).
5.2. To what extent do experimental toxicity studies on mechanisms of toxicity cover mechanisms relevant for IFL and CHL, and what is the contribution of the AOP in supporting biological plausibility
5.2.1. Rationale of the working approach
At present, the scientific evidence available sheds light on the following topics: pesticide exposure, toxicant MoA (e.g. etoposide), experimental studies, disease biology and the occurrence of paediatric leukaemia (including both infant and childhood leukaemia). The combined information allows understanding better whether the statistical association of pesticide exposure with the development of paediatric leukaemia is mechanistically plausible; whether causal links are indicated; and if such links can be confirmed or refuted by experimental testing.
As a starting point, there is sound epidemiological evidence in support of the association between pesticide exposure (either occupational or environmental/residential, either pre‐ or postnatal) and paediatric leukaemia. However, positive data from regulatory toxicity studies linking pesticides to traditional endpoints (e.g. genotoxicity) are lacking. In addition, for some chemicals, a mechanism of action is known (e.g. etoposide), but this is not the case for pesticides for which the evidence is weak. The first question to be addressed was to evaluate whether a pathogenic mechanism could be assigned to paediatric leukaemia in form of an AOP. This could only be achieved for IFL but not for CHL because of the lack of clearly delineated molecular initiating events. The second issue raised was to ascertain whether experimental studies provide information concerning the mechanism of toxic action of chemicals. The next step was to investigate whether toxicity pathways of chemicals overlapped with the mechanisms supporting the disease pathogenesis (AOPs) relevant for infant and childhood leukaemia. Finally, the answer to these questions was used (see Section 8.4) to establish plausible links between exposure to pesticides and the risk of developing paediatric leukaemia.
5.2.2. Capturing complex diseases (IFL and CHL) by AOP
A major obstacle in developing AOP of events leading to AO in paediatric leukaemia stems from the complex nature of this disease. The MIE and the adverse outcome highly depend on the subtypes of the disease (e.g. infant or childhood leukaemia, lymphoblastic or myeloid leukaemia).
Furthermore, the most important restriction is that the AOP should not be defined for the disease as such, but for a sharply defined feature of the disease (as equivalent to an endpoint or adverse outcome for toxicants). A second important condition is that this endpoint can be reproduced in animal models, and that there exist chemicals that trigger the endpoint in the animal models. This implies that example chemicals are available that are likely to trigger the envisaged ‘disease AOP’. As such conditions were partially fulfilled herein (only for IFL), it was scientifically acceptable to work on model AOPs relevant to IFL but only an hypothetical AOP could have been developed for CHL. Although the apical outcome is the same for those clinical conditions, the natural history of the disease is different for IFL and CHL and the MIE has been identified only for IFL. This can be considered as a developmental disease with all the relevant pathogenic steps occurring in utero and with an exceptionally short latency. In contrast, childhood leukaemia fits better to the two‐hit model confirmed in the natural history of several cancers and hypothesised for leukaemogenesis. The initiating event would occur in utero and the promoting event would occur postnatally. Besides, whereas a single event is enough for IFL (as exemplified by MLL rearrangement), a similar event is not enough to trigger childhood leukaemia.
5.2.3. Selection of the AO
Leukaemia is a group of cancers that presents with the proliferation of immature, clonal, myeloid or lymphoid precursors leading to progressive marrow failure and ultimately death. Clinical symptoms and signs of paediatric leukaemia usually reflect bone marrow infiltration by leukaemic blasts and/or extramedullary disease. The major clinical signs consist of neutropaenia, thrombocytopenia and anaemia, with these signs being responsible for symptoms such as increased susceptibility to infections (with fever), bruising, bleeding, pallor and fatigue. Accordingly, many different AOPs might be associated with the disease because of the several adverse outcomes of the disease, as well as pathway may converge into the same AO or share the same KE. Despite this, leukaemia has been identified in several animals and animals’ models have been developed to study the disease, the disease as such is not an apical endpoint in the regulatory toxicity studies and a mechanistic understanding of apical hazard suggestive of the AO is needed.
For proof of concept, symptoms and signs of overt paediatric leukaemia were chosen as AO. However, human leukaemia features have not been fully recapitulated in experimental animal models and the major candidate endpoints consist of chromosomal translocations that show similarities with those found in humans (sometimes paediatric patient‐derived leukaemia xenografts with MLL or TEL/AML1 translocations). The wide range of genetic and epigenetic changes needed for the expansion of preleukaemic clones prevents from delineating a sharp definition of paediatric leukaemia, either IFL or CHL.
5.2.4. Choice of example AOP relevant both for IFL/CHL and for pesticides as risk factors (see Appendix B)
Once the AO had been defined, the next question was to describe the sequence of pathogenic events that could be incorporated into a proof‐of‐concept AOP. For practical reasons, etoposide (a non‐pesticide chemical; a chemotherapeutic drag currently used for the treatment of cancer at various sites) was chosen as a tool example for IFL leukaemia because of the sound and consistent evidence in humans and experimental animals pointing out the pathophysiological processes triggered by this chemical. However, no chemical has been identified so far with the capability of triggering the toxicological pathway leading to childhood leukaemia. Notwithstanding this limitation for building an AOP, the systematic literature review commissioned by EFSA (Ntzani et al., 2013) concluded that there was sound epidemiological evidence linking pesticide exposure at diverse developmental stages and paediatric leukaemia. Moreover, expert knowledge on the state of experimental paediatric leukaemia research was used. On this basis, the Panel decided to develop two relevant AOPs one of them based on data for etoposide and the second AOP was a putative one because of the lack of empirical data at clinical, cellular, mechanistic or regulatory level that support any particular chemical with the onset of childhood leukaemia. However, there is abundant data that could be used to define the rest of the corresponding AOP.
This decision process has some important implications for the interpretation of this scientific opinion. The most important one is that the AOPs developed herein fail to support strongly how the different pesticides can lead to any of the different types of paediatric leukaemia. Besides, as the initial molecular targets and biochemical pathways disturbed by a toxicant are highly chemical‐specific, a ‘pesticide AOP’ cannot be defined. Likewise, there is no a plausible MoA that relates exposure to any individual pesticide (or pesticide classes) with paediatric leukaemia. Therefore, the PPR Panel decided to test whether the hazard posed by pesticides could be linked to the pathogenesis of paediatric leukaemia via AOPs. However, a single AOP may not capture all events that contribute to the relevant adverse outcome, instead sets of AOPs sharing at least one common event may capture more realistically potential toxic effects. If this approach is considered useful, then a multitude of AOPs could be developed for the many different pesticides currently used for improving our knowledge on their mechanism of toxic action. It can be anticipated that not all pesticides will fit any of these AOPs and also that several of these AOPs may share common KEs, or may converge into common intermediate key events, which would allow the definition of partial AOPs and their connection to a common KE.
5.2.5. Use of tool chemicals to check whether their mechanism of action overlaps with AOP for IFL and CHL
Etoposide was selected from the biomedical literature as the most promising tool chemical to build an AOP that would describe its hazard. The rationale is that well‐documented human and experimental data, both in vivo and in vitro, support the involvement of etoposide in the development of leukaemia. Its molecular target, TOP2, is particularly well‐characterised. Also, for infant leukaemia biology, there is good evidence that this target plays a key role (Pendleton et al., 2014). In contrast, no tool chemical has been identified in the open literature for CHL. Since data are lacking to delineate the MIE for this disease, efforts were made to fill a conceptual gap by presenting a hypothetical framework that provides sufficient biological plausibility based on an analogy approach derived from toxic mechanism following exposure to ionising radiation. Accordingly, the example chemical chosen for IFL allowed to define a MIE (TOP2 inhibition/poisoning), whereas the lack of MIE for CHL restricted the exercise to develop a putative AOP linked to the AO. The approach followed takes into account multiple genetic susceptibility factors and modulating events such that it falls within ‘system toxicology’. By analogy to systems biology, this approach is intended to decode the toxicological blueprint of an active substance that interact with biological targets that function as a network in cells, tissues or organisms (Sturla et al., 2014).
The two AOPs chosen by the PPR Panel can be fitted on this complex scenario as relevant pathways and are consistent with current medical knowledge on the disease biology of paediatric leukaemia. The prototype AOPs are fully reported in Appendix B.
5.2.6. Evaluation of the AOP concerning consistency and strength of evidence
Data generated from experimental models collectively contribute to the WoE supporting the proposed AOP. A large part of the effort to develop AOPs was used for their evaluation, and the documentation of this process. The strength of association was judged by a WoE approach based on modified Bradford–Hill criteria. This is fully described in Appendix B.
Based on the overall WoE, the PPR Panel concluded that the link between the MIE and the AO as proposed in the developed AOPs for IFL is strong and that the proposed key events (together with the MIE and the AO) can be used as a tool for exploring the IFL‐triggering hazard of a chemical. Once an AOP has been established, the MIE can be used to develop screening assays for compounds that might affect the AOP and networks of interacting AOPs (Knudsen et al., 2015). However, this framework is not valid for CHL because of the lack of a clearly defined MIE as aforementioned. This limitation is partially overcome by a set of hypothetical mechanisms that can provide a plausible biological basis for the epidemiological evidence gathered on the association between pesticides exposure and CHL.
One key conclusion that can be drawn from this approach is that any chemical triggering the MIE, or an intermediate key event of the proposed AOP, to a sufficiently quantitative extent, may also be expected to trigger the downstream key events and eventually the AO under certain qualified and specified conditions. This represents an important conceptual advance in predicting chemical hazards in terms of increasing the risk for human disease. Another important conclusion is the need of obtaining and using quantitative data as much as possible on the key event relationships for a practical application of hazard predictions based on AOPs.
5.2.7. Support of hazard plausibility by AOP
Based on the above considerations, the Panel is supporting the use of the AOP framework to explore the biological plausibility of the epidemiological association between pesticide exposure and paediatric leukaemia. The recommendation is that pesticides affecting the AOPs proposed in this opinion should be considered as potentially hazardous with respect to the development of paediatric leukaemia. The same would apply to other AOPs linked to this disease that could be developed in the future.
It is stressed again that the PPR Panel recommended the use of AOP specifically for hazard identification, and not for the assessment of risk. According to the risk assessment process, once any hazard has been identified, there is a need to proceed with a more complex evaluation of the risk. The AOP framework is exclusively focused on hazard and considers neither exposure data nor toxicokinetic (including metabolism) processes. The series of biochemical reactions and pathological events of an AOP by definition cannot have pharmacokinetic parameters. Doses of chemicals are taken into account for defining a threshold above which individual compounds may trigger the pathway, and they are evidently unique for each chemical but are not associated with the AOP as such.
Nevertheless, certain features of paediatric leukaemia should be considered for the hazard plausibility. The scientific evidence undoubtedly indicates that exposure to chemicals takes place in utero (prenatal) and even prior to conception. This assumption implies the need to account for toxicokinetic factors as chemicals will need to go across the placenta to reach fetal targets. In addition, many compounds are not only active by themselves but also need to be bioactivated by maternal (and to a lower extent fetal) biotransformation processes as occurs with etoposide for IFL or with benzene for adult myeloid leukaemia. The active chemical, either the parent compound or a metabolite, needs to reach the proper target in the embryo/fetus at a proper time window of development and at a concentration high enough to trigger the initiating events defined in the AOP. However, these considerations are not directly related to the chain of pathogenic events involved in the AOP as these are meaningful only for hazard plausibility. Another feature of paediatric leukaemia is related to toxicodynamic factors since differences in susceptibility regarding ontogeny processes may be relevant. For instance, IFL is considered a ‘developmental disease’ showing different features and pathogenesis than CHL, as more immature haematopoietic precursors are involved. The physiological role and susceptibility of these precursors to chemicals may vary depending on the embryonic/fetal stage of development.
5.3. Data gaps and suggestion for testing strategy. Also include the AOP as informative source for appropriate identification of data gaps and testing strategy
5.3.1. AOP as a scaffold to help identifying data gaps
Since the AOP concept shows causality of events under a weight of evidence approach, it is very well suited for identifying data gaps. Based on the epidemiological data linking pesticide exposure to paediatric leukaemia, and the AO being defined as ‘overt leukaemia’, several modes of action were identified linking an initiating event to the key event essential for the AO. This essential KE is the chromosomal translocations within HSPCs, which are cells essential for haematopoiesis. Thus, for the AOPs developed, the causality of substance binding to and subsequent inhibition of Top2, non‐repaired DNA DSBs or leading to chromosomal translocations, differentiation block of HSPCs, clonal expansion of preleukaemic clones and overt paediatric leukaemia is biologically plausible and essential. However, the main challenge of developing AOPs for leukaemia is the complex nature of the disease. For example, a tumour suppressor gene could be mutated or transcriptionally inactivated while in another instance an oncogene could be activated to trigger leukaemogenesis. Different genetic aberrations are associated with different subtypes of leukaemia. In addition, although leukaemia is a cancer with a low mutation rate, paediatric (childhood and infant) leukaemia are the second cancer with the least somatic mutations of all cancers sequenced so far (Bardini et al., 2010, 2011; Dobbins et al., 2013; Andersson et al., 2015). This stable genome makes difficult to unravel the aetiology and pathogenesis of paediatric cancer and there are no many genetic tags to be traced back for associating exposure to specific compounds and then validate the pathway.
Assessment of data gaps within an AOP is feasible by analysing the weight of evidence for each KER within an AOP. For IFL, the KER ‘In utero exposure to DNA Top2 poisons leading to MLL chromosomal translocation’ has a strong WoE, although there are still some open questions. For instance, the appropriate target cell model that recapitulates the production of DSB as a result of Top2 ‘poisoning’ has not been identified so far. Approximately 80% of IFL cases have the MLL rearrangement, but the remaining 20% carry other chromosomal aberrations leading to different fusion genes that eventually result in the same leukaemia phenotype. In utero etoposide‐treatment in a murine model, failed to induce leukaemogenesis because the appropriate chromosomal rearrangement is a rare event that needs to occur in a target cell within a relatively small and spatially restricted cell population during the appropriate, epigenetically plastic, developmental window. Moreover, although the risk of IFL seems to increase with larger total exposure to etoposide, dose–response relationships between etoposide and treatment‐related leukaemia are difficult to unravel. In contrast, for CHL, there is no evidence at the molecular level as to how some chemicals interact with biological targets to elicit DNA damage. This is not a straightforward question as genetic damage in HSPCs may be properly repaired in most cases, but if not cells undergo apoptosis. The exact nature of how and when this damage is not repaired has not yet been clarified as many factors are involved, thus contributing to a stochastic process with the final occurrence of the disease being very unlikely. The genetic damage (i.e. chromosomal translocations) has to occur in a particularly vulnerable genetic locus, within the proper cell, and in a specific time window; however, details of this entire process and how it happens are not clear.
The second level of KER for IFL ‘In utero MLL chromosomal translocation leading to infant leukaemia’ also has a high weight of evidence as the potential of both differentiation blockage and clonal expansion are inherent properties of the MLL‐rearranged fusion product. Thus, WoE indicates that IFL originates from one ‘big‐hit’ occurring during a critical developmental window of stem cell vulnerability. However, although the MLL rearrangement is essential to develop leukaemia, it alone may not be sufficient and further (epi)genetic factors would contribute to convey a proliferative advantage to preleukaemic clones to develop overt leukaemia. On the other hand, the MLL‐AF4 knock‐in mice developed leukaemia only after a prolonged latency, thus not recapitulating an important feature of IFL.
For CHL, the KE ‘In utero chromosomal translocations leading to differentiation arrest of HSPCs’ has a high WoE as this process has been very well studied, although the identity of leukaemia‐initiating mutations that result in preleukaemic clones is still an important open question. The block of differentiation of HSPCs confers self‐renewal properties to these cells and provides proliferative advantage to lymphoid progenitors. However, chromosomal translocations are insufficient by themselves to cause overt disease. Additional postnatal events are needed for the development of full‐blown disease, but they are not yet sufficiently well understood. Experimental models should be developed in cell lines and in mice to accurately recapitulate human leukaemogenesis. Additionally, oncogenes and chromosomal translocations should be studied in the appropriate cellular context, which consist of primary human haematopoietic cells. If the initiating oncogenic alteration is not occurring in the right cell, mouse models would be unlikely to recapitulate the human disease and would constitute an inaccurate model of human leukaemia.
The KE ‘Differentiation arrest of HSPCs leading to clonal expansion of leukaemogenic cells’ has a sound WoE as murine models with human precursor cells harbouring the TEL‐AML1 fusion gene need the acquisition of additional genetic abnormalities to result in overt leukaemia. However, the reproducibility and accuracy of these models have yet to be validated for humans, providing a data gap. Functional studies are needed to unveil the key mechanisms driving the evolution of these progenitors/stem cells into the appropriate type of leukaemia. Besides, individual patients harbour multiple genetic subclones of leukaemia‐initiating cells, with a complex clonal architecture which limits to build a consistent AOP. Owing to the technical challenge of distinguishing and isolating distinct cancer subclones, many aspects of clonal evolution are poorly understood. For instance, it remains to be demonstrated to what extent epigenetic diversity contributes to subclonal heterogeneity in acute leukaemia.
There is scarce scientific evidence for the KER ‘clonal expansion of leukaemogenic cells leading to overt childhood leukaemia’ since there are data gaps in the precise understanding on how leukaemic clones grow and expand. However, the biological plausibility of this KER is large as the pathobiology of the disease together with its evolutionary genetic landscape clearly indicates a causal linkage between the expansion of leukaemic clones within either the myeloid or lymphoid lineage and the onset of clinical phenotype of the disease.
5.3.2. Conclusions from AOP on suitability of current testing methods and present data gaps in regulatory studies
According to Regulation 1107/2009 on placing of plant protection products on the market, carcinogenicity and haematological endpoints must be evaluated for hazard identification and characterisation of active substances in order to decide on their approval. In addition, Regulation 283/2013 setting out the data requirements for active substances indicates that genotoxicity and carcinogenicity studies are always required and that haematological endpoints will be addressed and reported in general toxicity studies (repeated doses – short‐term and long‐term – and reproductive toxicity studies). Haematological endpoints addressed in these studies include red blood cell parameters, total and differential leucocyte count, platelet count and blood clotting time among others. In addition, haematopoietic organs are investigated in repeated dose and carcinogenicity studies, so that substances inducing leukaemia in rodents are expected to be identified in the basic data set.
The adequate evaluation of the genotoxic potential of a chemical is consistently addressed in regulatory dossiers by the assessment of different endpoints, i.e. induction of gene mutations, structural and numerical chromosomal alterations. These endpoints can only be covered by the use of diverse test systems as no individual test can simultaneously provide information on all of them. The bacterial reverse mutation assay covers gene mutations and the in vitro micronucleus test allows for the identification of both structural and numerical chromosome aberrations. If all these endpoints are clearly negative, the substance can be reasonably regarded as devoid of any genotoxic potential. Conversely, in the case of inconclusive, contradictory or equivocal results from this basic battery of tests, further in vitro testing must resolve the situation. In the case of positive results, further tests in vitro are appropriate either to optimise any subsequent in vivo testing or to provide additional useful mechanistic information. In vivo tests (mammalian erythrocyte micronucleus or transgenic rodent gene mutation assays) should relate to the genotoxic endpoint(s) identified as positive in vitro and to appropriate target organs or tissues (EFSA Scientific Committee, 2011). According to this testing strategy, a substance inducing leukaemia by a genotoxic mode of action is supposed to be captured by the genotoxicity tests battery.
However, the cell lines or primary cell cultures routinely used for regulatory in vitro genotoxicity testing may not be representative of HSPCs. The cell systems used in the different tests consist of adult cells and it is a widely recognised assumption that exposure of these cell systems at high dosages (until cytotoxicity occurs) would stress the cells to the extent that genotoxic properties would be found if the chemical was actually genotoxic. However, very early HSPCs have been considered particularly sensitive to genotoxicity because inter alia of the unfolded nature of their DNA, immature repair systems, high division rate, high TOP2 expression and activity levels.
Concerning assessment of carcinogenicity and haematological end‐points, the design of regulatory studies does not include a prenatal exposure. While a specific window of exposure is explored in the two‐generation reproduction toxicity studies, haematological measurements and histopathology of haematopoietic organs are not performed.
The only regulatory test guideline where haematology and histopathology of haematopoietic organs are investigated in animals exposed in utero to the testing chemicals is the extended one‐generation reproduction toxicity study. However, only 10 male and 10 female rats per group are examined for those parameters in order to assess the potential impact of the substance on the immune system. Nonetheless, this regulatory study is not intended to explore any carcinogenic event, which would require 50 animals/sex per group. Since this guideline has been adopted recently (2011), this kind of study has been submitted in very few cases compared to the two‐generation reproduction toxicity study.
For adult leukaemogenesis induced by external factors, the following four main patterns were outlined by the US‐EPA (Eastmond, 1997):
the primary type of lymphohaematopoietic cancer induced by chemicals and ionising radiation in humans is myeloid leukaemia;
potent human leukaemia‐inducing agents produce significant myelotoxicity and structural chromosomal aberrations in humans and animal models;
administration of human leukaemia‐inducing agents to mice results in more lymphohaematopoietic tumours that, unlike to happen in humans, are primarily lymphoid in origin;
the rat is considerably less responsive than the mouse to the induction of lymphohaematopoietic neoplasia following administration of human leukaemogenic agents.
Chemical‐ and radiation‐induced lymphohaematopoietic tumours are complex processes involving multiple genes, chromosomal alterations and altered differentiation. In addition, other factors such as metabolic capabilities, DNA repair and genetic susceptibilities may influence cancer incidence. Given the complexity and multiplicity of steps, animal models are unlikely to reproduce precisely all the critical stages involved in development of chemical‐induced leukaemias or lymphomas in humans (Eastmond, 1997).
Other limitations of rodent models for assessing the risk of leukaemias are represented by the different classification schemes of haematopoietic neoplasms used for rodents and humans. In mouse, histopathological distinction between malignant and non‐malignant myeloproliferations is also hard to establish. Likewise, distinction between lymphoma and leukaemias is often difficult, particularly in mouse, which can lead to misclassifications. Rats are relatively resistant to chemically induced leukaemogenesis; however, Fischer 344 rats, which are commonly used in carcinogenesis studies, exhibit a high incidence of spontaneous large granular lymphocyte leukaemia (LGLL). In contrast, spontaneous leukaemias are rare in other rat strains. The background incidence of LGLL in F344 rats has increased over time reaching more than 50% in males (Irons, 1991). Therefore, the usefulness of this strain for haematopoietic neoplasms exploration is questionable. Despite significant interspecies differences, rodents are valuable models for immunotoxic and myelotoxic effects, including leukaemogenesis. Furthermore, chronic animal bioassays using mouse models have been shown to be effective in identifying human (adult) leukaemia‐inducing agents (Eastmond, 1997).
Paediatric leukaemias represent a diverse group of diseases with distinct biological features compared with adult leukaemias. B‐cell acute lymphoblastic leukaemia, the most frequent leukaemia found in children, is characterised by an uncontrolled expansion of immature B cell (pre‐B phenotype). However, in the particular case of infant B‐cell acute lymphoblastic leukaemia, a very early haematopoietic precursor (pro‐B phenotype) is involved. These progenitor cells are initiated in utero, usually as a result of structural or numerical chromosomal aberrations and/or gene mutations. A wide range of acquired chromosomal translocations have been associated with early stages of acute leukaemias pathogenesis, with MLL gene being frequently involved. Special techniques not routinely performed, such as fluorescence in situ hybridisation (FISH), can be combined to classic genotoxicity protocols in order to identify specific translocations with dedicated probes. This would gain additional mechanistic information.
In summary, assuming that the critical events of paediatric leukaemia consist of in utero induction of chromosome aberrations followed in cases of CHL by an aberrant postnatal immune response to common infections, it is evident that the current animal tests/models do not cover these critical events.
5.3.3. Consideration on testing strategy
The above considerations point out that the current testing paradigm is not designed to detect those leukaemogenic agents that act only during early life stages (prenatal and early postnatal). There appear to be different sensitivities between cells for in vitro genotoxicity testing (notably HSPCs are considered more sensitive to genotoxic damage than other cells) and some in vivo tests (i.e. the chromosomal aberration test and the micronucleus test) have shown a poor sensitivity, likely because of the low exposure of haematopoietic cells in vivo. Besides, the carcinogenicity study design does not cover the relevant window of exposure and the model does not include a second hit that has been captured in experimental models (i.e. aberrant immune response to delayed infections). The only test guideline that covers this developmental period of susceptibility is the extended one generation test, in which haematology parameters and histopathology of haematopoietic organs are assessed on animals exposed in utero and during the juvenile period. However, this guideline has a low power to detect the leukaemogenic potential of a chemical because of the low number of animals examined. Besides, since this is a recent testing protocol there is scarce data on chemical substances already on the market.
The EFSA Scientific Committee recommended a stepwise approach for the generation and evaluation of data on genotoxic potential. This approach consist of: (a) a basic battery of in vitro tests; (b) consideration of whether specific features of the test substance might require substitution of some of the recommended in vitro tests by other in vitro or in vivo tests in the basic battery; (c) in the event of positive results from the basic battery, review of all the available relevant data on the test substance; and (d) where necessary, conduct of an appropriate in vivo study (or studies) to assess whether the genotoxic potential observed in vitro is expressed in vivo (EFSA Scientific Committee, 2011).
For a practical testing approach to leukaemogenesis potential of chemicals, in vitro genotoxicity should be tested in the relevant cells, particularly HSPCs, and technologies should be applied to detect structural or numerical chromosomal abnormalities (i.e. FISH). Besides this, evidence of target cell exposure is necessary for in vivo tests. In relation to whether or not the carcinogenicity studies (or the combined chronic toxicity/carcinogenicity studies) are appropriate to capture the carcinogenic potential of chemical exposures during developmental phases, consensus among the scientific (and regulatory) community is needed to reach sound and feasible recommendations as to how to proceed. It seems reasonable that improved models for in vitro testing should be used as a screening tool and that optimised carcinogenicity studies should be triggered when sufficient positive evidence has been obtained from lower tier tests. AOP can be used to design a testing strategy.
6. Application of the AOP concept to support the regulatory process; using parkinsonian motor deficits as an example
AOPs are not chemical specific and do not include ADME data of the chemical. Therefore, the Panel decided to evaluate the effective application of the AOP concept to the regulatory process by designing an exemplary strategy based on the two AOPs relevant for PD. The choice of the Panel was mainly motivated by the completeness of these two AOPs. In this process, the Panel is proposing to use the AOPs to build up an IATA strategy to evaluate whether the exposure to a pesticide (e.g. triggering mitochondrial dysfunction) causes dopaminergic neurodegeneration and ultimately parkinsonian motor deficits.
The AOP‐based KE testing cannot be used as a stand‐alone approach but needs integration of ADME data and embedding into an IATA framework. This will give the confidence that the threshold of KEs activation will indeed trigger the full cascade of events and that a dose, temporal and response concordance is maintained.
In designing a test battery for parkinsonian motor deficits in the context of IATA, the following considerations should be taken into account:
duration of exposure, e.g. how long proteostasis needs to be impaired for inducing DA neuronal death;
in vitro concentrations relevant to hazard assessment;
metabolic capacity of the test system;
assays permitting evaluation of KERs and modulatory factors or measuring recognised biomarkers reflecting KERs activation;
predictive capacity of the assays, e.g. taking into account a role of glial cells, species differences, etc.
6.1. Which types of data are needed to predict risk of parkinsonian motor deficits applying an AOP‐informed IATA
Elaboration on the IATA proposal is not formally part of this mandate; however, for understanding the value of an AOP‐based testing strategy and placing the AOP in a larger context for any potential application in risk assessment, the Panel considered an introduction to the AOP‐based IATA as necessary. For the proposed AOP‐informed IATA ‘Assessment of nigrostriatal toxicity’, the Panel adapted the general IATA concept from the AOP‐informed IATAs for skin irritation and corrosion (NV/JM/MONO(2014)19). Ten modules were identified for assessing if a compound poses a hazard for inducing Parkinsonian motor deficits in humans (Table 5).
Table 5.
Parts and Modules of the proposed IATA for assessment of nigrostriatal toxicity (adapted from NV/JM/MONO(2014)19)
| Part | Module | Data |
|---|---|---|
|
Part 1 (Existing information, physicochemical properties and non‐testing methods) |
1 2 3 |
|
|
4 5 |
|
|
| 6 |
|
|
| Part 2 (WoE analysis) | 7 |
|
|
Part 3 (Additional testing, if required) |
8 9 10 |
|
OECD: Organisation for Economic Co‐operation and Development; ADME: absorption, distribution, metabolism and excretion; (Q) SAR: quantitative structure–activity relationship; WoE: weight of evidence; AOP: Adverse Outcome Pathways; KE: key event.
The first step consists in collecting all available data (including regulatory toxicology studies) and human epidemiological information (Modules 1 & 2) followed by integration of toxicokinetic information (information on ADME, Module 3; Table 1, Figure 1). Moreover, information on physicochemical properties, e.g. compound's redox potential, are gathered (Module 5). Here, the focus lies on the compounds’ abilities to (a) be taken up by dopamine transporter or other transporters, (b) generate ROS, or (c) interfere with complexes of the mitochondrial respiratory chain. Apply (Q)SAR and read across where possible (module 6). All these data should be evaluated by a WoE approach (module 7). The WoE approach should be structured and possibly quantitative, and should inform on data gaps for decision making. At this stage, decision should be taken if new data are needed. However, it is beyond the scope of this opinion to detail on WoE analysis in general, but guidance on transparent WoE analysis is being developed by EFSA.
Figure 1.
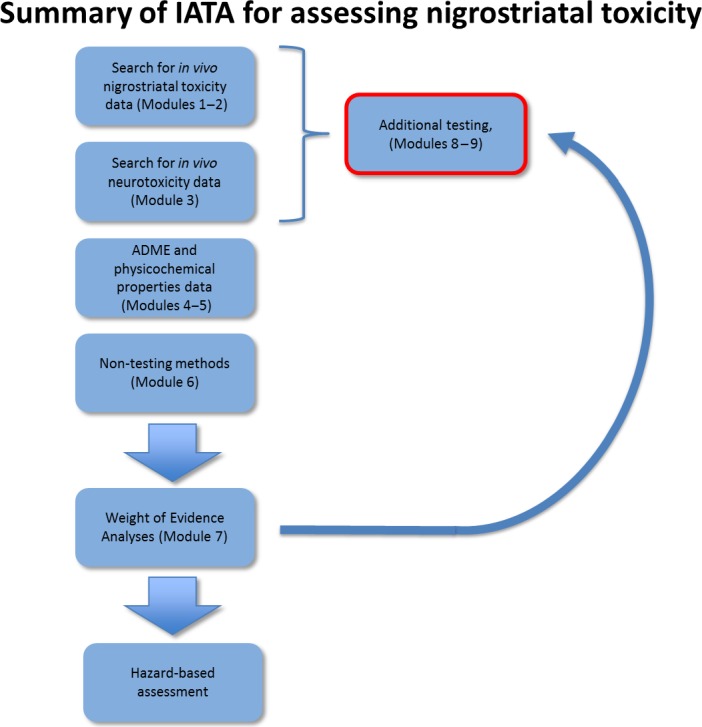
Schematic representation of proposal for IATA process
Part 3; if new data are required, an AOP‐informed IATA will be used to set‐up the most appropriate testing approach (modules 8–10 and Figure 1). After data generation, the WoE has to be re‐evaluated and decision taken on hazard assessment.
The Panel recommends that the following points are considered for an AOP informed IATA for ‘Parkinsonian motor deficits’.
should be based on human cells when necessary;
testing for neurotoxicity performed only on neuronal monoculture might not be sufficient because glial cells could be modifiers of the toxicity; either by promoting toxicity through, e.g. pro‐inflammatory stimuli or by buffering toxicity through e.g. activation of the cellular antioxidative defence (Henn et al., 2009; Efremova et al., 2015, 2016);
dopaminergic neurons should be the preferred neuronal subtype for testing;
in cases where cell–cell contacts affect cellular responses a 3D format may be considered (Alépée et al., ALTERNATIVES TO ANIMAL EXPERIMENTATION (ALTERNATIVES TO ANIMAL EXPERIMENTATION (ALTEX) 2014; Yamada & Cuckierman Cell, 2007);
cells used for testing should be without ethical concern and of high availability. The human induced pluripotent stem cells (hiPSC) are envisioned to solve the ethical issues and limitation of material when working with cells of human origin. They can be differentiated into (dopaminergic) neurons and astroglia functioning also in vivo when transplanted into rodent brains (Wernig et al., 2008; Palm et al., 2015);
the result of the tests must be in line with the temporal and concentration concordance as described in the AOP;
apply quality control and good cell culture practice (GCCP) principles to ensure reproducibility of the results.
Initially, the testing strategy should consider all KEs followed by selection of the most predictive assays.
According to the proposed AOPs, the following endpoints could be considered for testing: (1) oxidative stress, (2) mitochondrial dysfunction, (3) proteasomal dysfunction, (4) α‐synuclein accumulation and (5) dopaminergic cell death (Figure 2). Moreover, the testing strategy should include considerations on the temporality of events as described in the AOPs, by selecting the appropriate timing for endpoint measurements which has to reflect the time concordance. For example, oxidative stress as KE1 should occur before disturbance of proteasomal function as KE3.
Figure 2.
Proposed KERs‐based testing strategy established on the AOPs for ‘Parkinsonian Motor Deficits’. Assay 1: measurement of oxidative stress; Assay 2: assessment of mitochondrial function; Assay 3: determination of proteasomal function as a measure of proteostasis; Assay 4: identification of α‐synuclein accumulation; and Assay 5: assessment of dopaminergic cell death
The Panel also recommends that in the context of these AOPs, length of treatment/exposure should allow for triggering the KEdown (Betarbet et al., 2000; Fornai et al., 2003). The Panel does not expect this battery consisting of, for example, five assays (Figure 2) measuring the endpoints mentioned above, to be the final testing strategy, but recommends a statistical analysis of data with a test set of compounds for selecting a minimum amount of tests that would predict the adverse outcome with the highest predictivity value. The Panel is also recommending that a tiered approach should be considered.
The structure provided by the 10 modules described above (Table 1) allow for composing an IATA. Ideally, this IATA should make the maximum use of existing data, being resource efficient and minimising or eliminating the requirement for animal experiments.
7. Uncertainties
A number of uncertainties were identified by the Panel for each AOP developed; such uncertainties are reported in detail in the Appendices. The Panel has, however identified general uncertainties concerning human epidemiological studies, experimental evidences and AOP development methodologies.
Epidemiological studies
Human diseases from epidemiological studies and meta‐analyses have been used in this Scientific Opinion to define the adverse outcome (AO) and this is considered an uncertainty per se due to the known intrinsic limitations of the epidemiological studies.
Definition of the human health outcome, when this is a complex, multifactorial human disease was considered by the Panel as an uncertainty. This issue is exemplified by epidemiological studies showing an association with ‘childhood leukaemia’ (i.e. the diagnosis of leukaemia in infants and children), whereas the AOP development led to the identification of two distinct diseases, i.e. IFL and CHL. The current epidemiological data do elucidate whether the association with pesticide exposure concerns infant and/or childhood leukaemia. This problem leads to significant data heterogeneity in the epidemiological studies.
Exposure estimates was considered by the Panel as a major limitation and uncertainty of the epidemiological studies. Indeed, this uncertainty includes two components: (a) the generic definition of the substances of concern which in most cases refers to large usage groups of diverse pesticides (e.g. ‘insecticides’) and only in some cases refers to pesticide structural groups (‘organophosphates’, ‘chloro‐S‐triazines’). This generic definition of exposure cannot identify the individual substances contributing to the risk; (b) lack of detailed quantitative information concerning internal exposure. For chronic diseases, such as PD, it is difficult to integrate biomonitoring (biomarkers of exposure) and epidemiology (health outcomes) information, leading the investigators to use methodologies with low accuracy, e.g. pesticide usage. Furthermore, in a realistic field scenario, humans are exposed to several substances and coformulants contained in actual pesticide products, representing an additional source of uncertainties. It should be also noted that epidemiological studies are referring to formulated products while experimental studies are conducted with the active substance and this should be considered as an uncertainty.
The design of epidemiological studies and the heterogeneity of the target populations may introduce a considerable heterogeneity among studies that apparently investigate the same outcomes weakening the reliability of meta‐analyses and this was considered by the Panel as an uncertainty.
An uncertainty directly relevant to AOP development is the limitation of knowledge about the aetiology of multifactorial human diseases, involving the presence of different phenotypes, genetic factors and environmental factors, other than pesticides. This limitation affects the characterisation of KEs as well as the estimation of the effects of modulating factors on the KERs.
A second opinion will further elaborate on general uncertainties of epidemiological studies (Q……).
Animal studies
Regulatory toxicity studies have shown intrinsic limitations leading to uncertainties (Paparella et al., 2013). These are also relevant in relation to this scientific opinion as regulatory toxicity studies may not include endpoints that model or predict the relevant adverse outcomes deriving from the mechanistic understanding of human health outcomes. The Panel also recognises lack of (human) disease‐specific animal models as an uncertainty. This lack is due to the problem that the adverse outcome is complex and multifactorial and poorly understood. In particular, the Panel notes the deficiency in predictive in vivo models for both IFL and CHL.
AOP development
Some uncertainties in the process of AOP development were identified by the Panel.
Most of the empirical support for the on MIEs and KEs is currently derived from in vitro assays using ad‐hoc non‐standardised models, whereas MIE and KEs can be accurately identified, their full description and characterisation may be affected by such factors as the available models in the literature (e.g. life stage of primary cell cultures) or the use of tool chemicals at very high concentrations.
In general, there are uncertainties in correlating in vitro to in vivo concentration. However, the AOP conceptual framework, should be regarded as a tool for building evidence of response–response and temporal concordance.
Correlation between the duration of the exposure in vitro and in vivo is also considered an uncertainty. Similarly, methods of administering the chemical stressors in experimental conditions are an uncertainty when considering the presumed routes and duration of exposure in humans. Nevertheless, at a minimum, the AOP model can provide proof of concept that environmental chemicals can cause the AOs.
An AOP illustrated by a single chemical stressor or on a limited number of chemical stressors (for empirical supports) is recognised as an uncertainty.
The extent of which the study(ies) supporting the empirical evidences has/have been reproduced is recognised as an uncertainty.
The Panel recognise that the AOPs proposed for the parkinsonism motor symptoms are very specific and do not cover all the complexity of PD. In particular, the pathology evaluation reflected in these AOPs is only detailed for the SNpc and different brain areas can be involved in PD‐related pathology.
Lack of criteria for data selection (assembling evidence) might account for differences in the strength of an AOP. In addition, there is uncertainty about the consistent way to include the role of modulating factors into AOP development, particularly when dealing with AOs derived from complex human diseases.
Finally, there is uncertainty whether the developed AOPs are the only ones linking PD, IFL and CHL to pesticide toxicological MoAs.
8. Discussion and Conclusions
Human health risk assessment for pesticides is mostly based on experimental toxicity studies performed in laboratory animals by oral dosing. These studies are conducted at relatively high doses with the highest dose expected to be the maximum tolerated dose for the experimental animal species and strain to be tested and for the predefined study design. The outcome of these pivotal studies is extrapolated to humans, to other exposure routes and to relatively low environmental doses. With some exceptions, human data are only available through the epidemiological studies which are required to be incorporated into the risk assessment according to Regulation No 1107/2009 when available, and as indicated in Regulation 283/2013 setting out data requirements for active substances. It is then essential for the evaluation of epidemiological data to weigh, integrate and make use of all the available information coming from multiple experimental studies. This complex task becomes more difficult when epidemiological data deals with multifactorial, multihit, chronic diseases, for which toxicological models or disease‐specific animal models are limited. This Scientific Opinion exploring method and principles to guide and investigate the use of experimental data and available knowledge; it aims to develop a mechanistically driven approach that aims to evaluate the evidence of cause–effect relationship i.e. biological plausibility and coherence.
In the context of the current opinion, the outcome from an epidemiological observation is biological plausible if a mechanistic link between cause and effect exists and can be demonstrated using a coherent framework linking a MIE (cause) to an AO (effect) through a sequence of KEs at tissue, cellular and population level. This is done in the AOP conceptual framework by assembling and weighing the evidence using modified Bradford–Hill criteria. These criteria focus on the biological plausibility of the KERs (i.e. biology of the pathway), essentiality of the KEs within the AOP (i.e. necessity of the KEs) and empirical support of the KERs (i.e. quantitative associations among KEs tested through application of stressors/tool compounds) in a ranking order of importance. The empirical support should provide evidence for the response–response, incidence and temporal concordance of the KE relationships. Consequently, in the context of the AOP framework, any chemical triggering the MIE with a sufficient intensity and time has the potential to lead to the AO. Therefore, an AOP supporting biological plausibility (i.e. the plausibility that a chemical triggering a MIE can lead to a specific AO) will represent relevant information for the inclusion of human data from epidemiological studies in the process of hazard identification. As described in the mandate, the Panel selected PD and CHL as human health outcomes based on the associations observed for these diseases and exposure to pesticides which are consistently reported in multiple meta‐analyses (Ntzani et al., 2013).
In this attempt to integrate epidemiological studies on pesticide exposure and human diseases by developing AOPs to assess the biological plausibility of the associations, the Panel recognised a number of limitations. The premise of the mandate, i.e. that pesticide exposure is associated with PD and CHL, in itself may constitute uncertainty to the epidemiological conclusions. Other uncertainties arise especially from the lack of detailed quantitative information regarding exposure and data heterogeneity.
In regard to the link between pesticide exposure and PD, since the Ntzani report (Ntanzi et al., 2012) the association has been further consolidated in a systematic review by Hernández et al. (2016a), Hernandez et al. (2016b). In this work, the authors could confirm that PD is significantly associated with pesticide exposure; however, they could not conclusively identify a single pesticide, or a specific pesticide group, associated with a significant risk of PD other than paraquat. Regarding the epidemiological evidence of pesticide exposure and the risk of CHL since the Ntanzi report (Ntzani et al., 2012), the association has been investigated by additional meta‐analyses. Again the association between prenatal exposure (via occupational exposure of the parents was observed both for ALL and AML, but recognising the considerable uncertainty in regard to the assessment of pesticide exposure. Again, besides an association with ‘generic insecticides’ the data did not allow to conclude about a single pesticide, or a specific pesticide group, associated with a significant risk of CHL.
To summarise, assuming that the observed outcomes are not due to other confounders, to date they cannot be linked to a specific pesticide active substance, but only to the exposure to pesticides. However, exposure is rarely to a pesticide active substance alone, but to pesticide formulations and/or multiple active substances. In reality, a plethora of coformulants that are largely uncharacterised in regard to toxicological effects (other than acute toxicity by different routes, irritation and sensitisation) should be considered. The recent assessment and discussion of the epidemiological findings between exposure to glyphosate and different cancer outcomes (RAR glyphosate 2015, Addendum 2015) also highlighted the major uncertainties in associating exposure to a certain pesticide active substance with an adverse outcome. Uncertainty from the lack of quantitative exposure estimates was compounded by the simultaneous co‐exposure to other coformulants. Another critical issue, that was also relevant for the conclusion of the EU assessment in regard to the carcinogenic potential of glyphosate, is the strength of the biological plausibility of the epidemiological observations.
Why an AOP?
The terms of reference of this mandate included exploration of the use of the AOP framework for supporting both a mechanistic‐derived hazard identification and biological plausibility of epidemiological associations, in order to incorporate the human health adverse outcome as part of the hazard identification process. The AOP framework was thus selected as a flexible and transparent tool for the review, organisation and interpretation of complex information coming from different sources. In this perspective, the AOP was intended to overcome one of the major limitations of many epidemiological studies, i.e. lack of understanding of biological plausibility and this aspect has been investigated in this opinion.
The Panel, in proposing the AOP framework as an integral part of this mandate also considered several relevant aspects namely: exploring how to improve toxicity testing in regard to effects involved in complex and multifactorial diseases such as PD and CHL; integration of in vitro predictive tools into testing in order to shift towards a more ‘toxicity pathways’ ‐based framework; highlight species differences or similarities; identify data gaps, research needs and requirements for development of toxicological assays and IATA. All these aspects have to be dealt with in order to support regulatory decision in a scientifically robust framework.
To support his mandate, the Panel committed a systematic review specifically tailored to serve as a basis for defining and mapping the causal linkages between an MIE and a final AO and possibly identify all relevant publications related to the mechanisms and chemicals involved in the pathogenesis of PD and CHL. In case of PD, this turned out to be a considerable number, making it a challenge to select the relevant publications but at the same time avoid ‘cherry‐picking’. The Panel noted that the use of the systematic review framework also showed some limitations. Using the outcome of the systematic literature review in building an AOP, it was evident that relevant information was sometimes not captured despite the very large number of publications retrieved by the search. The most likely reason was explored and identified as the use of a structured search protocol with strict inclusion and exclusion criteria. Although it was recognised that an extensive literature search would have been more appropriate, the relevant scientific expertise present in the working group in the field of neurodegenerative diseases and childhood leukaemia with knowledge of the relevant available literature, overcame this potential limitation. As part of the process the working group of the Panel also met with authors of relevant publications to gain methodological details which were considered important in the context of the AOP framework.
Indeed, an AOP need not necessarily be based on a systematic literature review. The quality assessment of the literature is filtered and guaranteed by the strength of the structured weight of evidence analysis proposed in the AOP framework; this includes the evaluation of the empirical support to assess the reproducibility, time and response concordance for the selected chemical tools.
It was noted that the regulatory acceptance of an AOP where the majority of the components, e.g. the empirical data supporting a KER, is not supported by reproduced data would be very limited. Such an AOP could, however, serve the purpose of identifying where more data should be generated.
An additional consideration for the regulatory use of AOPs is variability. Variability (due to intrinsic factors such as genetic polymorphisms, species, age, gender, as well as ‘environmental’ factors such as diet, lifestyles, etc.) is likely to be a considerable contributing factor for some KEs. To some extent this is already considered and discussed in building an AOP, but it is recognised that modulating factors should be given more space in the AOP development. However, the role of modulating factors is a general issue in toxicology (e.g. the use of uncertainty factors), to understand how modulating factors can impact on a threshold is a specific challenge for AOP development. Indeed, by properly accounting for modulating factors, AOP might provide a scientific background to build up specific uncertainty factors.
The Panel also observed that the studies included in the empirical support can be quite heterogeneous in terms of design and route of exposure, and this can be interpreted as a source of inconsistencies. However, some elements of the study design are important for hazard characterisation and ultimately for risk assessment. For example the intraperitoneal route of administration is usually not considered a relevant exposure route for pesticide risk assessment since this route would be most unlikely for humans. The current toxicological studies conducted in laboratory animals are designed for hazard identification and then for hazard characterisation, in order to determine a suitable point of departure (NOAEL or benchmark dose). Accordingly, in the context and scope of this Scientific Opinion, the Panel intended the development of AOPs for the purpose of solely hazard identification to support biological plausibility based on mechanistic knowledge. If hazard identification was based on a route of administration not relevant for risk assessment, the Panel still considered this to be acceptable and in line with the principle for developing AOPs. However, for a quantitative AOP with the purpose of being used for hazard characterisation, this would need different considerations, again depending on problem formulation. If, for example, the scope is to define a threshold able to trigger the sequence of KEs from the MIE to the AO, the route of administration of the tool compound used will still not be relevant; instead, the concentration(s) at the target(s) able to activate the MIE and KE(s) will be relevant, independently of the route of administration. Toxicokinetic and metabolism information will be indeed very important when dealing with compound specific hazard characterisation by applying the MOA and/or IATA framework; this information will tell the risk assessor if the concentration of the specific compound at the target MIE will be relevant or not for its activation. In any case, the concentrations used in the empirical support with the tool chemicals should always be assessed to define the strength of the response–response concordance. In addition, as well as for the animal studies, effects detected at excessive doses – close to the maximal tolerated dose/cytotoxicity – would always require a careful assessment of the biological relevance of the observed finding.
As detailed in this opinion, the core studies of the regulatory dossiers do not necessarily capture the potential hazard of pesticides in regard to PD and CHL. This is not unexpected when considering the complexity of these diseases and the fact that regulatory toxicology studies are intended and designed to explore multiple hazards and should be considered as stand‐alone experiment, i.e. one species, one strain, one NOAEL for the endpoints explored in the context of the study design. For the purpose of analysing the plausible mechanisms linking pesticides to human health outcomes, AOPs can serve as an important tool, particularly when the regulatory animal toxicological studies are negative but the evaluation of the apical endpoint (or relevant biomarkers) was considered inadequate based on the AOP.
The scientific and regulatory relevance of AOP at the different levels of maturity (whether putative, qualitative or quantitative) would depend on it being fit‐for‐purpose in a given context; the problem formulation will therefore drive the building of an AOP, with the expectation that the AOP reflects the current knowledge and the WoE evaluation is transparent and complete.
A putative AOP is intended as a set of hypothesised KEs and KERs primarily supported by biological plausibility. The Panel considered that for the problem formulation, as expressed in the terms of reference, a putative AOP can be useful in order to give indications about the strength of the relationship between the AO (intended as a human health outcome) and pesticides affecting the pathway. In addition, by detecting and/or identifying data gaps and/or research needs, putative AOPs could serve to inform IATA or give guidance for further works.
The Panel considered that qualitative AOPs (intended as an AOP including the assembly and evaluation of the supporting weight of evidence following the OECD guidance for AOP development) should be the starting and standard approach in the process of integration of the epidemiological studies into risk assessment by supporting (or identifying the lack of support for) that a plausible mechanistic link exists between pesticides affecting the pathway and the AO, intended as the human health outcome. This should be based on the agreement of the current understanding of the AO and the strength of the weight of evidence will define the boundaries of its scientific validity. In developing qualitative AOPs, the Panel realised that these can be also used as screening tools.
Quantitative AOPs (intended as supported by quantitative relationship that allow quantitative translation of KE measurement into predicted probability or severity of AO) can cover any need, including a complete hazard assessment of a chemical, by identifying regulatory relevant point of departures for reference values; the quantitative AOP can also support the inclusion of chemical specific factors like internal exposure and metabolism. Fully quantitative AOP's are very data demanding; thus, it is envisaged that they would be a second step in a regulatory prioritisation process. However, the Panel recognised that moving from qualitative to quantitative AOPs, would potentially represent an important step forward to a more effective use of pathway‐based data to support risk assessment and build up a predictive network.
The Panel considered that the use of properly developed AOPs is important to guide on future tailored and tiered testing strategies for hazard identification and characterisation, and consequently, a proposal for an AOP‐based IATA framework for identifying the risk of PD was made. The most sensitive, robust, reliable test for a KE can be proposed to be further validated and, if needed, ultimately might become a part of the OECD testing programme for chemicals. This is considered a very important element of integrating non‐animal data for regulatory use (NRC, [Link]). The Panel further concluded that the AOP framework is a powerful tool to support the most appropriate design for in vitro and in vivo studies, and increase the sensitivity of methods and experimental designs for capturing and possibly characterising a given hazard. As an AOP is expected to reflect the current knowledge, this would imply that a large number of studies and methods will be included in the description and empirical support for KE and KER. In this perspective, the quality of the studies and scientific validation of the methods are relevant for regulatory purposes; in the meantime, the Panel considered that decisions on how to make the best use of the available information should depend on the problem formulation. For the scope of this Scientific Opinion, i.e. to explore the AOP framework as a tool for supporting biological plausibility in relation to epidemiological studies, it is essential that the description of the WoE would be complete and transparent; merging in the WoE of the biological plausibility, essentiality and strength of empirical support in a ranking order of relevance for each KE and KER will foster the decision made. If AOPs are intended to inform IATA or define the optimal study design for hazard identification/characterisation, elements of scientific validation and study quality should be taken into consideration.
The Panel identifies AOPs as a critical element to facilitate the move towards a mechanism‐based risk assessment instead of the current testing paradigm relying heavily on apical effects observed in animal studies (as recommended by EFSA, as well as by ECHA and OECD). Shifting the risk assessment paradigm and mechanistic understanding would reduce limitations of the animal data in predicting human health effects for the single pesticide, and also support the current efforts on carrying out cumulative risk assessment of pesticide exposure. Regarding the grouping of pesticides for cumulative risk assessment and in particular the refining of groups, the Panel concluded in 2013 that the current read‐out from animal studies (apical endpoints) were not tailored for this purpose and recommended that a better mechanistic understanding of toxicity should be achieved, and in particular recommended development of AOPs (EFSA, 2013 – SO on dissimilar MoA). In this perspective, AOP networks represent the functional unit of prediction of the AO as AOPs are not triggered in isolation but they rather interact. Indeed, KEs and KERs are shared by multiple AOPs. Rather than looking at hazards or MIEs in isolation, developing AOPs in a modular approach gradually describes the complexity of potential interactions at cell, tissue, organ, system and organism levels, thus meeting the concept of systems biology. The Panel recommends that the use of AOP's should be explored for the purpose of refining cumulative assessment groups; considering this specific mandate, this opinion underlines how biological plausibility can support the link between pesticide and complex human health outcomes (e.g. parkinsonian disorders) by evidencing how multiple MIEs can lead to the same AO. The Panel also appreciated that interactions between AOPs can be easily made visible if AOPs are downloaded in the AOP‐Wiki.
In regard to the future process of AOP development, the collaborative AOP‐Wiki of the AOP knowledge base (https://aopkb.org) serves as a useful platform and the upload of AOPs into the AOP‐Wiki is encouraged by the Panel. The use of the AOP‐Wiki would provide multiple benefits in terms of easy approach to AOPs network, transparency and accurate peer‐review will be ensured by the formal review process of the proposed AOPs. This effort is still under development and with the experience of this mandate future work and improvements may be suggested. Common KEs should be shared only if approved following peer‐review processes. Overall, to increase the future regulatory acceptance of AOPs and the key events based testing, more emphasis should be put on the transparent data selection for building AOPs; accordingly, weight of evidence analysis should be conducted and built into the AOP‐Wiki. Collier et al. (2016) have recently suggested a framework which also includes quantitative considerations, especially on the strength of linkages between key events: such considerations would include criteria for soundness, applicability and utility, clarity and completeness, uncertainty and variability and last evaluation and review.
The Panel concluded that:
The AOP framework is a useful tool for risk assessment to explore if an AO is biologically plausible or not; by means of mechanistically describing apical endpoints, the AOP contributes to the hazard identification and characterisation steps in risk assessment. As the AOP framework is chemically agnostic, it will consolidate the chemical specific risk assessment with the aid of, and within, the MOA and/or IATA framework.
The prototype AOPs developed by the Panel support the view that pesticides affecting the proposed MIEs and the pathways are risk factors for the development of the diseases, i.e. PD and IFL. This conclusion is based on a weight of evidence assessment appropriately characterised by defining questions on biological plausibility and empirical support for the KER and on the essentiality of the KEs. In addition, inconsistencies and uncertainties were identified and reported for each of the developed AOP.
The Panel recognised that the systematic literature review and meta‐analysis indicated that the plausible involvement of pesticide exposure as a risk factor for the development of PD and IFL could by linked to further AOPs development in addition to the ones developed by the Panel. This limitation in the current Scientific Opinion does not weaken the overall conclusion about the plausible involvement of pesticides in the pathogenesis of PD and IFL.
The Panel concluded that the AOP developed for CHL does not yield definitive evidence of biological plausibility. However, circumstantial evidence indicates that a hypothetical biological plausibility could exist but cannot be formulated with the current available information. These circumstantial evidences are mainly derived by the epidemiological observation that the disease is prevalent following in utero exposure to pesticides and that exploration of tumour related endpoints following in utero exposure has limitations in the standard design of regulatory studies. In addition, the Panel recognise that an animal model recapitulating the disease is not available and this is also weakening the assessment.
The AOP framework was considered by the Panel as an appropriate tool to understand if chemical hazards related to the relevant human diseases (PD, infant and childhood leukaemia) can be explored and detected in the standard regulatory studies. Although apical endpoints in the regulatory studies, i.e. histological evaluation of the nigrostriatal pathway and neurological examination (for Parkinson diseases), blood analysis, genotoxicity testing, immunological parameters in reproductive studies and carcinogenicity assays (for infant and childhood leukaemia's), can potentially inform KEs or AOs, the mechanistic understanding of the apical endpoints indicate that the regulatory toxicological studies have limitations because of the study design or because of the sensitivity of the test system. Tailored studies or more sensitive tools should be considered to prove that a chemical has negligible hazard if it is affecting the pathway.
9. Recommendations
Overall, the Panel recommends the AOP conceptual framework to assess the biological plausibility, or lack of biological plausibility, of the association between pesticide exposure and human health effects reported in epidemiological studies by means of including the observed effect in the AO and consequently in the hazard identification process.
The AOPs should stimulate regulators to ask for the application of additional testing based on the mechanistic understanding of the MIEs and KEs. Therefore, the Panel recommends that the AOP should be used as a mechanistic tool to support biological plausibility and mechanistic understanding of apical hazards when toxicity studies are considered insufficient, inconclusive or inadequate, but the substance is known to affect the pathway.
The AOPs should stimulate additional research work in order to provide a more robust quantitative evaluation of the threshold effects for the different KEs, using the same tool compounds used for their development. Quantitative evaluation should foster the regulatory use of the AOP and should include, where possible, a concentration response analysis by means of identification of a non‐effect threshold and a minimum threshold effect able to trigger the pathway. A biologically relevant battery of assays preferably based on human cells, able to recapitulate the key events of the AOP and predictive of the concentration of the chemical leading to the AO should be developed.
The systematic literature review indicated that the plausible involvement of pesticide exposure as a risk factor for the development of PD and infant/childhood leukaemia could by linked to additional AOPs other than the ones developed by the Panel. The Panel recognises this limitation of the current Scientific Opinion and recommends that additional AOPs should be developed with the intent of using the AOP to support the biological plausibility of additional MIEs and pathways but also to develop an AOP network to be used as a functional unit for the prediction of the diseases.
The Panel recommends that the AOP network should be considered as a tool for the refinement of the Cumulative Assessment Groups to be used in cumulative risk assessment of pesticides.
The Panel recommends that for compounds affecting the AOPs developed for parkinsonian motor symptoms, the evaluation of the nigrostriatal pathway should be performed by means of application of proper stereology protocols and detailed neuropathology assessment with inclusion of special stain procedure in addition to H/E to examine whether there is evidence for neuronal cell loss, cell damage and any neuroinflammatory response. It is important that an unbiased assessment of neuronal cell loss must include carefully conducted stereology and pathology studies where the pathologist is ‘blinded’ to the treatment regimen of each experimental animal assessed. Furthermore, relevant neurochemistry toxicity endpoints should also be examined. The Panel also recommends that biomarkers, e.g. α‐synuclein could be considered to help in the study design, i.e. dose selection and length of the treatment, when compounds are known to affect the pathway but the regulatory endpoints are negative.
In addition, the Panel recommends investigation and evaluation of the use of mixed neurons/glial cocultures in the development of in vitro batteries for neurotoxicity screening.
The Panel proposes neuroinflammation as a key event for the two AOPs developed for the AO ‘parkinsonian motor symptoms’. The Panel does not, however, recommend the use of neuroinflammatory biomarkers for the time being. Although the Panel is supporting their utility and validity from the scientific point of view, they are still too challenging and complex for regulatory uses. However, this does not include the use of immunomarkers for cell phenotyping.
The Panel recognises the limitations of the standard regulatory studies as evidenced by the AOP developed in this Scientific Opinion and recommends that an AOP‐informed IATA should be developed to support the testing strategy. The Panel also recommends that an AOP‐informed IATA framework should be developed for the IFL and the CHL and that the testing strategy should be based on non‐animal testing as a first approach for new data generation.
The Panel recommends that the standard OECD guidance on histological evaluation of the brain in the 90‐day toxicity study (OECD TG 408) and in general in the toxicity studies performed in vivo, should be revised in order to include a more in depth evaluation of brain structures involved in Parkinson disease i.e. the nigrostriatal pathway.
The Panel recommends that genotoxicity assays should consider the sensitive detection of TOP2 poisons. In particular, the Panel recommends to the investigation of the differential sensitivity (also in regard to single vs repeat exposure) of embryonic cell systems such as liver HSPCs compared to established systems (e.g. CHO, V79, peripheral blood lymphocytes), in regard to clastogenicity (including TOP2 poisons); this could be achieved effectively by means of a multicentre collaborative study.
The Panel recognises that the use of a non‐validated cell system needs more scientific work in order to provide robust data on specificity and sensitivity for an appropriate use in risk assessment.
The Panel recommends that the epidemiological studies and meta‐analysis should make a distinction between infant and childhood leukaemia which are aetiologically and pathologically different entities.
The Panel recommends that tailored and tiered testing strategies should be developed and the assays should be anchored to the KEs identified in the AOPs. Accordingly, the test system should be selected to model the human biology of KEs.
In order to facilitate the regulatory relevance, the AOPs developed for this mandate should be submitted to the AOP‐Wiki and undergo the rigorous peer‐review by the OECD.
Based on the experience gained in developing AOPs, the Panel recommends that the transparency and WoE in building AOP's should be strengthened in the future. An agreed approach during the process of AOP preparation (assembling evidence) for the evaluation of data quality of individual studies and for aggregating lines of evidence, possibly in a more quantitative and structured way, is recommended. The framework suggested by Collier et al. (2016) could serve this purpose. Also, the Panel notes that for the future development of AOPs and AOP‐network of the AOP‐Wiki, that careful updates of KER of common KE must be implemented.
The Panel recommends applying in vitro methods as a first approach for gaining mechanistic information to support the biological plausibility linking exposure to pesticide to human health outcomes.
Abbreviations
- ADME
absorption, distribution, metabolism and excretion
- ALDH
aldehyde dehydrogenase
- ALL
acute lymphocytic leukaemia
- AML
acute myeloid leukaemia
- AO
adverse outcome
- AOP
Adverse Outcome Pathway
- AS
α‐synuclein
- ATM
ataxia telangiectasia mutated
- CHL
childhood leukaemia
- CHO
Chinese hamster ovary
- CI
confidence interval
- CLIC
Childhood Leukaemia International Consortium
- CLP
Classification, Labelling and Packaging
- DA
dopamine
- DDT
dichlorodiphenyltrichloroethane
- DOPA
3,4‐dihydroxyphenylalanine
- DOPAL
3,4‐dihydroxyphenylacetaldehyde
- DSB
double‐strand breaks
- FISH
fluorescence in situ hybridisation
- GCCP
good cell culture practice
- GD
gestation day
- GFAP
glial fibrillary acidic protein
- HCB
hexachlorobenzene
- hiPSC
human induced pluripotent stem cells
- HNE
hydroxy‐nonenal
- HSPC
haematopoietic stem and progenitor cells
- IATA
Integrated Approach for Testing and Assessment
- IFL
infant leukaemia
- IPCS
International Programme of Chemical Safety
- KE
key event
- KER
key event relationship
- LB
Lewy bodies
- LC50
half‐maximal inhibitory concentration
- LGLL
large granular lymphocyte leukaemia
- LN
lymph nodes
- MAO
monoamine oxidase
- MIE
molecular initiating event
- MLA
mouse lymphoma assay
- MoA
Mode of Action
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- NHEJ
non‐homologous end‐joining
- NOAEL
no observed adverse effect level
- NTE
neuropathy target esterase
- OECD
Organisation for Economic Co‐operation and Development
- OP
organophosphate
- PD
Parkinson's disease
- PND
postnatal day
- PON
paraoxonase
- PPR
Panel Panel on Plant Protection Products and their Residues
- RBC
red blood cell
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphisms
- SNpc
substantia nigra pars compacta
- SSB
single‐strand break
- TOCP
tri‐o‐cresylphosphate
- TOP2
topoisomerase II
- ToR
Term of Reference
- UDS
unscheduled DNA synthesis
- WHO
World Health Organization
- WoE
weight of evidence
Appendix A – AOP developed for parkinsonian motor deficit
AOP1: Inhibition of the mitochondrial complex I of nigrostriatal neurons leads to parkinsonian motor deficits
Abstract
This AOP describes the linkage between inhibition of complex I (CI) of the mitochondrial respiratory chain and motor deficit as in parkinsonian disorders. Binding of an inhibitor to complex I has been defined as the molecular initiating event (MIE) that triggers mitochondrial dysfunction, impaired proteostasis, which then cause degeneration of dopaminergic (DA) neurons of the nigrostriatal pathway. Neuroinflammation is triggered early in the neurodegenerative process and exacerbates it significantly. These causatively linked cellular key events result in motor deficit symptoms, typical for parkinsonian disorders, including Parkinson's disease (PD), described in this AOP as an Adverse Outcome (AO). Since the release of dopamine in the striatum by DA neurons of the substantia nigra pars compacta (SNpc) is essential for motor control, the key events refer to these two brain structures. The weight‐of‐evidence supporting the relationship between the described key events is based mainly on effects observed after an exposure to the chemicals rotenone and 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), i.e. two well‐known inhibitors of complex I.
Data from experiments with these two chemicals reveal a significant response–response concordance between the MIE and AO and within KEs. Also, essentiality of the described KEs for this AOP is strong since there is evidence from knock out animal models, engineered cells or replacement therapies that blocking, preventing or attenuating an upstream KE is mitigating the AO. Similarly, there is proved experimental support for the KERs as multiple studies performed with modulating factors that attenuate (particularly with antioxidants) or augment (e.g. overexpression of viral‐mutated α‐synuclein) a KEup show that such interference leads to an increase or attenuation/prevention of KEdown or the AO.
Information from in vitro and in vivo experiments is complemented by human studies in brain tissues from individuals with sporadic Parkinson's disease (Keeney et al., 2006) to support the pathways of toxicity proposed in this AOP.
This AOP is reported in line with the OECD Guidance Document for developing and assessing AOPs [ENV/JM/MONO(2013)6] and with the supplemented user's handbook.
A.1. Molecular Initiating Event (MIE): Binding of an inhibitor to NADH ubiquinone oxidoreductase (complex I)
A.1.1. How this Key Event works
Electron transport through the mitochondrial respiratory chain (oxidative phosphorylation) is mediated by five multimeric complexes (I–V) that are embedded in the mitochondrial inner membrane (Figure A.1). NADH‐ubiquinone oxidoreductase is the Complex I (CI) of the electron transport chain (ETC). It is a large assembly of proteins that spans the inner mitochondrial membrane. In mammals, it is composed of about 45‐47 protein subunits (45 in humans) of which 7 are encoded by the mitochondrial ge nome (i.e. ND1, ND2, ND3, ND4, ND4L, ND5 and ND6) and the remaining ones by the nuclear ge nome (Greenamyre, 2001). CI oxidises NADH elevating the NAD+/NADH ratio by transferring electrons via a flavin mononucleotide (FMN) cofactor and several iron‐sulfur centres to ubiquinone (Friedrich et al., 1994) (Figure A.2).
Figure A.1.
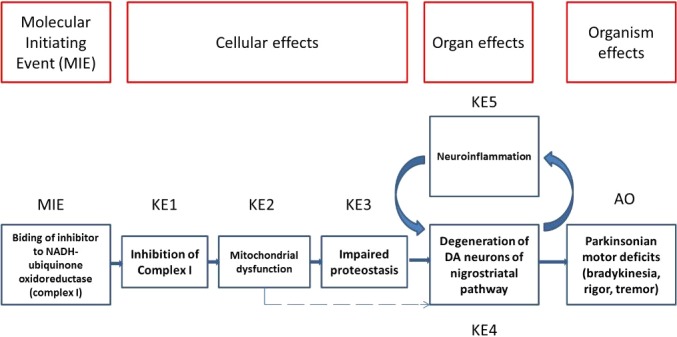
AOP scheme
Figure A.2.
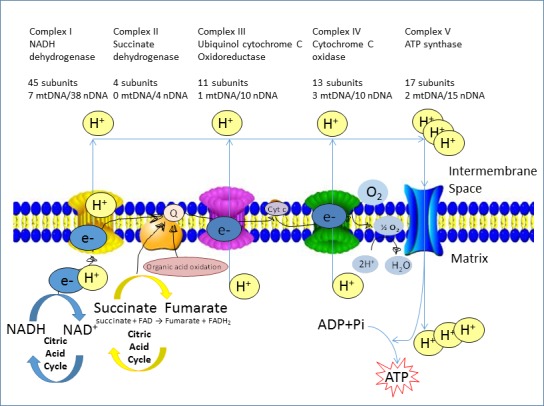
The electron transport chain in the mitochondrion. CI (NADH‐coenzyme Q reductase or NADH dehydrogenase) accepts electrons from NADH and serves as the link between glycolysis, the citric acid cycle, fatty acid oxidation and the electron transport chain. Complex II also known as succinate‐coenzyme Q reductase or succinate dehydrogenase, includes succinate dehydrogenase and serves as a direct link between the citric acid cycle and the electron transport chain. The coenzyme Q reductase or Complex III transfers the electrons from CoQH2 to reduce cytochrome c, which is the substrate for Complex IV (cytochrome c reductase). Complex IV transfers the electrons from cytochrome c to reduce molecular oxygen into water. Finally, this gradient is used by the ATP synthase complex (Complex V) to make ATP via oxidative phosphorylation. mtDNA: mitochondrial DNA; nDNA: nuclear DNA
Binding of an inhibitor to CI inhibits the NADH‐ubiquinone oxidoreductase activity, i.e. blocks the electron transfer. Recent studies suggest that a wide variety of CI inhibitors share a common binding domain at or close to the ubiquinone reduction site (Ino et al., 2003). Furthermore, the structural factors required for inhibitory actions have been characterised on the basis of structure–activity relationships (Miyoshi, 1998, Hideto, 1998).
Based on molecular docking simulations, in silico models mimicking the binding of chemicals to the pocket of NADH ubiquinone oxidoreductase have been created according to the crystal structure of mitochondrial CI. To investigate the ability of chemicals to bind to the active pocket, around 100 individual docking simulations have been performed. These confirmed the possible site of interaction between the chemical and the pocket of CI. In particular, Miao YJ and co‐workers recently investigated the IC50 values of 24 chemicals (annonaceous acetogenins) for inhibition of mitochondrial CI (Miao et al., 2014).
Based on their binding sites, CI inhibitors are classified as follows (Degli Esposti, 1998) (Figure A.3):
type A inhibitors are antagonists of fully oxidised ubiquinone binding;
type B inhibitors displace the partially reduced ubisemiquinone intermediate;
type C inhibitors are antagonists of the fully reduced ubiquinol product.
Figure A.3.
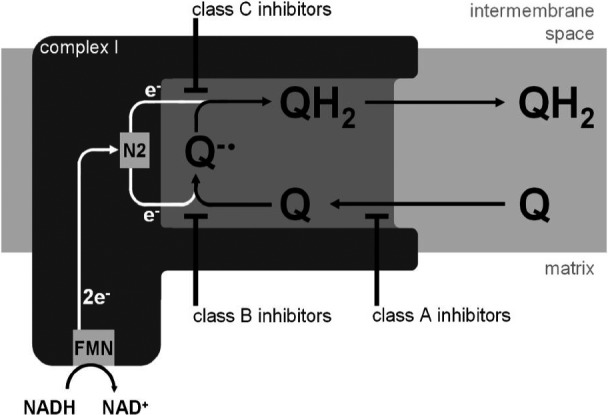
Schematic representation of CI and proposed inhibition binding sites by inhibitors of class A, B and C. Nicotinamide adenine dinucleotide (NADH, reduced and NAD, oxidised), flavin mononucleotide (FMN) and ubiquinone (Q) (Haefeli et al., 2012, fig. 46, CC‐BY‐ND)
The affinity of the different types of CI inhibitors to their diverse CI binding sites is described in the paragraph Evidence for Chemical Initiation of this Molecular Initiating Event (see below) in the context of a specific type of inhibitor.
A.1.2. How it is measured or detected
Two different types of approaches have been used. The first is to measure binding as such, and the corresponding assays are described below; the second is to infer binding indirectly from assays that quantify e.g. CI activity and to assume that the activity can only be altered upon binding.
The second type of approach is dealt with in the chapter entitled KE1: Inhibition of NADH ubiquinone oxidoreductase (complex I). However, it has to be noted here that indirect assays can lead to wrong conclusions. For instance, some compounds may trigger oxidative stress without actually binding to CI. Such compounds, by triggering the generation of reactive oxygen species (ROS), may damage CI protein components, thus causing a reduction of CI activity.
A.1.3. Measurement of binding by quantitative autoradiography
To assess binding of an inhibitor at the rotenone binding site of CI in tissues (e.g. in the substantia nigra or in the striatum), the standard approach is to quantify the displacement of a radioactively labelled ligand of this binding site by the toxicant under evaluation. Most commonly, binding of [3H]‐labelled dihydrorotenone (DHR) is measured and compared in control tissue and treated tissue. Binding of this rotenone‐derivative is detected by autoradiography. Unselective binding is determined by measurement of [3H]‐DHR binding in the presence of an excess of unlabelled rotenone. Since a rotenone‐derivative is used for the assay, only CI inhibitors that bind to the rotenone‐binding site in CI are detected. This was observed, for e.g. meperdine, amobarbital or MPP+. This method allows a spatial resolution of CI expression and the mapping of the binding of a competitive inhibitor on CI.
The method can be used for (a) in vitro measurements and for (b) ex vivo measurements:
In vitro measurements. Tissues are embedded in a matrix for cutting by a cryostat. The tissue slices are then mounted onto slides. For the binding experiment, they are incubated with the test compound in the presence of labelled [3H]‐DHR. Then the tissue slices are washed and prepared for autoradiographic detection (Greenamyre et al., 1992; Higgins and Greenamyre, 1996).
Ex vivo measurements. As rotenone can pass the blood brain barrier, the in vitro method was further extended for in vivo labelling of CI in the brains of living animals, and detection of binding after preparation of the tissue from such animals. Animals are exposed to test compounds and [3H]‐DHR is applied intraventricularly for 2–6 h before the brain is dissected and arranged for the preparation of tissue slices (Talpade et al., 2000). In untreated animals, this method allows a precise spatial resolution of the expression pattern of CI. In animals with impaired CI activity, either as a result of CI deficiencies, or upon treatment with CI inhibitors, the assay allows an assessment of the degree of CI inhibition.
A.1.4. Complex I Enzyme Activity (Colorimetric)
The analysis of mitochondrial OXPHOS CI enzyme activity can be performed using human, rat, mouse and bovine cell and tissue extracts (abcam: http://www.abcam.com/complex-i-enzyme-activity-microplate-assay-kit-colorimetric-ab109721). Capture antibodies specific for CI subunits are precoated in the microplate wells. Samples are added to the microplate wells which have been precoated with a specific capture antibody. After the target has been immobilised in the well, CI activity is determined by following the oxidation of NADH to NAD+ and the simultaneous reduction of a dye which leads to increased absorbance at OD=450 nm. By analysing the enzyme's activity in an isolated context, outside of the cell and free from any other variables, an accurate measurement of the enzyme's functional state can be evaluated.
A.1.5. Evidence supporting taxonomic applicability (tissue type, taxa, life stage, sex)
CI has a highly conserved subunit composition across species, from lower organisms to mammals (Cardol, 2011). Fourteen subunits are considered to be the minimal structural requirement for physiological functionality of the enzyme. These units are well conserved among bacterial (Escherichia coli), human (Homo sapiens) and bovine (Bos taurus) (Vogel et al., 2007b; Ferguson, 1994). However, the complete structure of CI is reported to contain between 40 to 46 subunits and the number of subunits differs, depending on the species (Gabaldon, 2005; Choi et al., 2008). In vertebrates CI consists of at least 46 subunits (Hassinen, 2007), particularly, in humans 45 subunits have been described (Vogel et al., 2007b). Moreover, enzymatic and immunochemical evidence indicate a high degree of similarity between mammalian and fungal counterparts (Lummen, 1998). Mammalian CI structure and activity have been characterised in detail (Vogel et al., 2007a,b), referring to different human organs including the brain. There is also a substantial amount of studies describing CI in human muscles, brain, liver, as well as bovine heart (Janssen et al., 2006; Mimaki et al., 2012) (Okun et al., 1999).
A.1.6. Evidence for Chemical Initiation of this Molecular Initiating Event (MIE)
The most studied examples of chemicals that inhibit CI are: rotenone and 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) (Sava et al., 2007; Desplats et al., 2012; Lin et al., 2012). Both, rotenone (pesticide) and MPP+ (the active metabolite of MPTP) are well known to reproduce the anatomical, neurochemical, behavioural and neuropathological features of PD‐like syndrome (Betarbet et al., 2000; Greenamyre et al., 2001). Indeed, overwhelming evidence has accumulated in the existing literature suggesting such a link, and therefore these two inhibitors of CI will be discussed in the context of all the KEs identified in this AOP.
A.1.6.1. Rotenone affinity to complex I binding sites
Rotenone, a colourless, odourless, crystalline ketonic chemical compound (a flavonoid) naturally occuring in the seeds and stems of several plants, such as the jicama vine plant, and the roots of several members of Fabaceae, is a classical, high affinity and irreversible inhibitor of CI and is typically used to define the specific activity of this enzymatic complex. Rotenone is extremely lipophilic, it crosses biological membrane easily and can get into the brain very rapidly. Dose‐dependent relative affinities of rotenone to the inhibitory site of CI is shown in Figure A.4B (for more details, see Grivennikova et al., 1997).
Figure A.4.
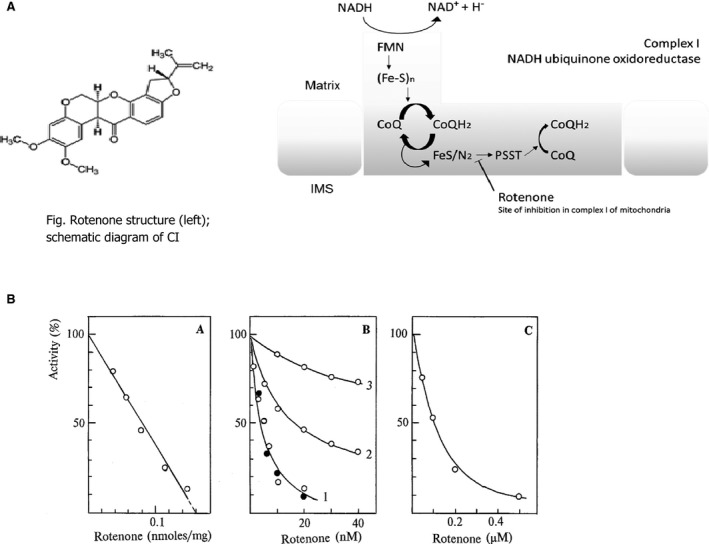
(A) NADH ubiquinone oxidoreductase, illustrating molecular mode of action and binding site of Rotenone (and Rotenone‐like compounds) IMS: inter‐membrane space (based on Lummen, 1998). (B) Dose‐dependent relative affinities of rotenone to the inhibitory site of CI (for more details, see Grivennikova et al., 1997. Reprinted from Grivennikova et al., 1997, Fig. 1, copyright (1997) with permission from Elsevier)
Most of the studies suggest that hydrophobic inhibitors like rotenone or Piericidin A most likely disrupt the electron transfer between the terminal Fe‐S cluster N2 and ubiquinone (Figure A.4A).
A.1.6.2. MPTP affinity to complex I binding sites
MPTP is not directly binding to CI and it is therefore non‐toxic to DA neurons. MPTP exerts its toxicity after it is metabolised by mono‐amino‐oxidase, type B (MAO B) in astrocytes to 1‐methyl‐4‐phenylpyridinium (MPP+). This metabolite binds to CI, and is toxic. MPP+ is a good substrate for dopamine transporters (DAT), expressed selectively by DA neurons (Greenamyre et al., 2001). Due to both a positive charge and an amphoteric character, MPP+ specifically accumulates in mitochondria, where despite a lower affinity to the binding site of CI than rotenone, it reaches high enough intra‐mitochondrial concentrations to inhibit CI activity (Ramsay et al., 1991). The binding affinity of MPP+ is low (mM range), and it can be totally reversed by washing out. However, prolonged treatment results in a severe, progressive and irreversible inhibition of CI, most likely by indirect mechanisms involving oxidative damage (Cleeter et al., 1992). Competitive binding experiments with rotenone and MPP+ suggest that the two compounds bind to the same site of the CI (Ramasay et al., 1991).
A.1.6.3. General characteristics of other complex I inhibitors
Besides rotenone, there is a variety of CI inhibitors, both naturally occurring, such as Piericidin A (from Streptomyces mobaraensis), acetogenins (from various Annonaceae species), as well as their derivatives, and synthetically manufactured compounds, like pyridaben and various piperazin derivatives (Ichimaru et al., 2008). They have been used to probe the catalytic activity of CI especially in order to clarify its ubiquinone binding site and indeed, most of these compounds inhibit the electron transfer step from the Fe‐S clusters to ubiquinone (Friedrich et al., 1994).
Therefore, classification of CI inhibitors is based on their types of action. Type A inhibitors, like piericidin A, 2‐decyl‐4‐quinazolinyl amine (DQA), annonin VI and rolliniastatin‐1 and ‐2, are considered to be antagonists of the ubiquinone substrate. For piericidin A, it has been shown that it inhibits NADH:Q2 activity in a partially competitive manner. Contrary to type A, type B inhibitors, like the commonly used rotenone, have hydrogen‐bonding acceptors only in the cyclic head of the molecule and are non‐competitive towards ubiquinone, but are believed to displace the semiquinone intermediate during the catalysis (Figure 2). Finally, inhibitors classified as type C, like stigmatellin and capsaicin, form a third group of hydrophobic CI inhibitors that are believed to act as antagonists of reduced ubiquinone (Friedrich et al., 1994; Degli Esposti, 1998; Haefeli 2012) (Figure 2).
Competition studies with representatives of all three different types of inhibitors revealed that type A and B, and type B and C, but not type A and C, compete with each other for binding. This led to a suggestion that all CI inhibitors acting at the ubiquinone binding pocket share a common binding domain with partially overlapping sites (Okun et al., 1999).
Some inhibitors bind to the outside of the ubiquinone reduction site and do not fit the preceding classification. Examples of such compounds are ADP‐ribose, which competes for substrate binding at the NADH site (Zharova and Vinogradov, 1997), and diphenyleneiodonium (DPI) that covalently binds to reduced flavin mononucleotide (FMN) in the hydrophilic part of the enzyme, blocking the electron transfer to the Fe‐S clusters (Majander et al., 1994).
There are also new, commercially available insecticides/acaricides with potential to inhibit mitochondrial respiration, such as benzimidazole, bullatacin, 6‐chlorobenzothiadiazole, cyhalothrin, fenazaquin, fenpyroximate, Hoe 110779, pyridaben, pyrimidifen, Sandoz 547A, tebufenpyrad and thiangazole (Greenamyre et al., 2001). It is clear that they are capable of inhibiting the mammalian CI by binding to and blocking ubiquinone‐dependent NADH oxidation with high efficacy (Lummen, 1998).
References
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Cardol P, 2011. Mitochondrial NADH: ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases. Biochimica et Biophysica Acta, 1807, 1390–1397.
Choi WS, Kruse SE, Palmiter R, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP, or paraquat. Proceedings of the National Academy of Sciences, 105, 39, 15136–15141.
Cleeter MW, Cooper JM, Schapira AH, 1992. Irreversible inhibition of mitochondrial complex I by 1‐methyl‐4‐phenylpyridinium: evidence for free radical involvement. Journal of Neurochemistry, 58, 786–789.
Degli Esposti, 1998. Inhibitors of NADH‐ubiquinone reductase: an overview. Biochimica et Biophysica Acta, 1364, 222–235.
Desplats P, Patel P, Kosberg K, Mante M, Patrick C, Rockenstein E, Fujita M, Hashimoto M, Masliah E, 2012. Combined exposure to Maneb and Paraquat alters transcriptional regulation of neurogenesis‐related genes in mice models of Parkinson's disease. Molecular Neurodegeneration, 7, 49.
Ferguson SJ, 1994. Similarities between mitochondrial and bacterial electron transport with particular reference to the action of inhibitors. Biochemical Society Transactions, 22, 181–183.
Friedrich T, van Heek P, Leif H, Ohnishi T, Forche E, Kunze B, Jansen R, TrowitzschKienast W, Hofle G, Reichenbach H, 1994. Two binding sites of inhibitors in NADH: ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone‐binding site of bacterial glucose: ubiquinone oxidoreductase. European Journal of Biochemistry, 219, 691–698.
Gabaldon T, Rainey D, Huynen MA, 2005. Tracing the evolution of a large protein complex in the eukaryotes, NADH: ubiquinone oxidoreductase (Complex I). Journal of Molecular Biology, 348, 857–870.
Greenamyre JT, Sherer TB, Betarbet R, Panov AV, 2001. Critical review Complex I and Parkinson's Disease Life, 52, 135–141.
Greenamyre JT, Higgins DS, Eller RV, 1992. Quantitative autoradiography of dihydrorotenone binding to complex I of the electron transport chain. Journal of Neurochemistry, 59(2), 746–749.
Grivennikova VG, Maklashina EO, Gavrikova EV, Vinogradov AD, 1997. Interaction of the mitochondrial NADH‐ubiquinone reductase with rotenone as related to the enzyme active/inactive transition. Biochimica et Biophysica Acta, 1319, 223–232.
Haefeli RH, 2012. Molecular Effects of Idebenone. Doctoral thesis http://edoc.unibas.ch/19016/1/Molecular_Effects_of_Idebenone_Roman_Haefeli.pdf fig46, p. 89. CC‐BY‐ND
Hassinen I, 2007. Regulation of mitochondrial respiration in heart muscle. In: Schaffer and Suleiman (eds.). Mitochondria – The Dynamic Organelle. Springer ISBN‐13: 978‐0‐387‐69944‐8.
Hideto M, 1998. Structure–activity relationships of some complex I inhibitors. Biochimica et Biophysica Acta. 1364, 236–244.
Higgins DS Jr1, Greenamyre JT, 1996. [3H]dihydrorotenone binding to NADH: ubiquinone reductase (complex I) of the electron transport chain: an autoradiographic study. Journal of Neuroscience, 16, 3807–3816.
Ichimaru N, Murai M, Kakutani N, Kako J, Ishihara A, Nakagawa Y, Nishioka T, Yagi T, Miyoshi H, 2008. Synthesis and Characterization of New Piperazine‐Type Inhibitors for Mitochondrial NADH‐Ubiquinone Oxidoreductase (Complex I). Biochemistry, 47(40), 10816–10826.
Ino T, Takaaki N, Hideto M, 2003. Characterization of inhibitor binding sites of mitochondrial complex I using fluorescent inhibitor. Biochimica et Biophysica Acta, 1605, 15–20.
Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA, 2006. Mitochondrial complex I: structure, function and pathology. Journal of Inherited Metabolic Disease, 29, 499–515.
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr, 2006. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. Journal of Neuroscience, 10, 26, 5256–5264.
Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, Chen TH, Shih CM, 2012. Inhibition of Mitochondria‐ and Endoplasmic Reticulum Stress‐Mediated Autophagy Augments Temozolomide‐Induced Apoptosis in Glioma Cells. Public Library of Science (PLoS ONE), 7, e38706.
Lümmen P, 1998. Complex I inhibitors as insecticides and acaricides 1. Biochimica et Biophysica Acta (BBA) – Bioenergetics, 1364, 2, 287–296.
Majander A, Finel M, Wikstrom M, 1994. Diphenyleneiodonium inhibits reduction of iron–sulfur clusters in the mitochondrial NADH–ubiquinone oxidoreductase (complex I). Journal of Biological Chemistry, 269, 21037–21042.
Miao YJ, Xu XF, Xu F, Chen Y, Chen JW, Li X, 2014. The structure–activity relationships of mono‐THF ACGs on mitochondrial complex I with a molecular modelling study. Natural Product Research, 28, 1929–1935.
Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT, 2012. Understanding mitochondrial complex I assembly in health and disease. Biochimica et Biophysica Acta, 1817, 851–862. doi: 10.1016/j.bbabio.2011.08.010
Miyoshi H, 1998. Structure–activity relationships of some complex I inhibitors. Biochimica et Biophysica Acta, 6, 236–244.
Okun JG, Lümmen P, Brandt U, 1999. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:Ubiquinone Oxidoreductase). Journal of Biological Chemistry, 274, 2625–2630. doi: 10.1074/jbc.274.5.2625
Ramsay R, Krueger MJ, Youngster SK, Gluck MR, Casida JE, Singer TP, 1991. Interaction of 1‐methyl‐4‐phenylpyridinium ion (MPP+) and its analogs with the rotenone/piericidin binding site of NADH dehydrogenase. Journal of Neurochemistry, 56, 1184–1190.
Sava V, Velasquez A, Song S, Sanchez‐Ramos J, 2007. Dieldrin elicits a widespread DNA repair and antioxidative response in mouse brain. Journal of Biochemical and Molecular Toxicology, 21, 125–135.
Talpade DJ, Greene JG, Higgins DS Jr, Greenamyre JT, 2000. In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. Journal of Neurochemistry, 75, 2611–21.
Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG, 2007a. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Molecular Genetics and Metabolism, 91, 176–182.
Vogel RO, Smeitink JA, Nijtmans LG, 2007b. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochimica et Biophysica Acta, 1767, 1215–1227.
Zharova TV, Vinogradov A, 1997. A competitive inhibition of the mitochondrial NADH‐ubiquinone oxidoreductase (Complex I) by ADP‐ribose. Biochimica et Biophysica Acta, 1320, 256–264.
A.2. KE1: Inhibition of NADH ubiquinone oxidoreductase (complex I)
A.2.1. How this Key Event works
Under physiological conditions complex I (CI) couples the oxidation of NADH to NAD+ by reducing flavin mononucleotide (FMN) to FMNH2. FMNH2 is then oxidised through a semiquinone intermediate. Each electron moves from the FMNH2 to Fe‐S clusters, and from the Fe‐S clusters to ubiquinone (Q). Transfer of the first electron results in the formation of the free‐radical (semiquinone) form of Q, and transfer of the second electron reduces the semiquinone form to the ubiquinol form (CoQH2). Altogether, four protons are translocated from the mitochondrial matrix to the inter‐membrane space for each molecule of NADH oxidised at CI. This leads to the establishment of the electrochemical potential difference (proton‐motive force) that may be used to produce ATP (Garrett and Grisham, 2010).
Binding of an inhibitor attenuates or completely blocks the activity of CI, i.e. the oxidation of NADH is impaired and protons are not moved. This causes two major consequences: first, electrons are channelled towards oxygen instead Q. This impairs normal oxygen reduction into water at complex IV and leads to the formation of the ROS superoxide at other sites of the respiratory chain. Superoxide may cause damage of proteins, lipid and DNA of the cell, or damage components of the mitochondria after transformation into e.g. hydrogen peroxide. These processes result in mitochondrial dysfunction (Voet and Voet, 2008). The second consequence is the increase of the NADH/NAD+ ratio in mitochondria. This affects the function of key dehydrogenase enzymes in the citric acid cycle and can lead to its block, resulting in an inhibition of mitochondrial ATP production and mitochondrial respiration.
The functional consequences of CI inhibition have been titrated in a time‐ and dose‐dependent manner (Barrientos and Moraes, 1999), with mitochondrial dysfunction measured by a range of different assays (Barrientos and Moraes, 1999; Greenamyre et al., 2001). These included quantification of ROS derived from mitochondria, and of cellular respiration (see KE2: Mitochondrial dysfunction).
A.2.2. How it is measured or detected
As CI has an enzymatic function as such, but also contributes to the overall function of oxidative phosphorylation, there are two fundamental approaches to assess CI inhibition. The first approach measures the enzymatic activity of the complex itself; the second one assesses the overall activity of oxidative phosphorylation of entire mitochondria, and indirectly infers from this a potential dysfunction of CI.
A.2.3. Direct detection of complex I activity
This type of assay is always performed in homogenates of cells or tissues, and requires at least a partial purification of mitochondria or respiratory chain components. In order to focus on CI activity, the activities of complex III (e.g. antimycin A) and complex IV (e.g. cyanide) need to be blocked by pharmacological inhibitors in these setups.
A.2.3.1. Forward Electron Transfer
Submitochondrial particles or intact isolated mitochondria are incubated with NADH as electron donor and with an electron acceptor to measure the flow of electrons from NADH, through CI to the acceptor. As readout, either the consumption of NADH, or the reduction of the electron acceptor is followed photometrically or fluorometrically (Lenaz et al., 2004; Kirby et al., 2007; Long et al., 2009; Spinazzi et al., 2012). The physiological electron acceptor of CI is Coenzyme Q10 (CoQ10). Due to its hydrophobicity, it is not suitable for use in an experimental in vitro setup. Short‐chain analogs of CoQ10, such as CoQ1 or decylubiquinone (DB) with a 10 carbon‐atom linear saturated side chain are hence applied as alternatives. With these non‐physiological electron acceptors, it is important to consider that the activity of CI can easily be underestimated. As water‐soluble electron acceptors, either ferricyanide or 2,6‐dichlorophenolidophenol (DCIP) are used. However the reduction of such compounds is not strictly coupled to the transduction of energy. To identify the portion of rotenone‐inhibitable CI activity, all samples investigated are assayed in parallel following treatment with rotenone. In contrast to the autoradiography assays, direct CI activity detection allows the identification also of CI inhibitors that bind to sites of CI different from the rotenone binding site.
A.2.3.2. Reverse Electron Transfer
An alternative setup for the direct measurement of CI activity with minimal interference by the activities of complex III and complex IV make use of the observation of a general reversibility of oxidative phosphorylation and electron flow across the mitochondrial respiratory chain (Ernster et al., 1967). With this method, electrons enter the respiratory chain via complex II. Based on the reverse flux, this method allows the complete circumvention of complexes III and IV. As electron donor, succinate is applied, together with NAD+ as electron acceptor. Formation of NADH from NAD+ can be determined photometrically. The succinate‐linked NAD+ reduction can be performed either with intact isolated mitochondria or with submitochondrial particles. For the direct assessment of CI activity, submitochondrial particles are used. For assays with intact mitochondria, the succinate‐linked reduction of NAD+ is performed in the presence of ATP as energy source. Potassium cyanide (KCN) is added for inhibition of forward electron transport towards complex IV.
A.2.3.3. Complex I activity dipstick assay
To assess CI activity and its inhibition in cell or tissue homogenates without interference by other components of the respiratory chain, CI‐selective antibodies attached to a matrix (e.g. multiwell plates) are used (Willis et al., 2009). Homogenised tissue can directly be added for capturing of CI, the unbound supernatant is washed away and leaves a complex of the antibody and mitochondrial CI. For activity determination, NADH as electron donor and nitroblue tetrazolium (NBT) as acceptor are added. Reduced NBT forms a coloured precipitate, its signal intensity is proportional to the amount of CI bound to the antibody. CI inhibitors can directly be added for an assessment of their inhibitory potential. This method, when applied in e.g. 96‐well or 384‐well plates, allows screening of large sets of potential CI inhibitors without any interference by other elements of the mitochondrial respiratory chain.
A.2.4. Indirect measurements of complex I activity
Such assays mostly require/allow the use of live cells.
A.2.4.1. Oxygen consumption
Electrons, fed into the mitochondrial respiratory chain either by CI or complex II, ultimately reduce molecular oxygen to water at complex IV. In a closed system, this consumption of oxygen leads to a drop of the overall O2 concentration, and this can serve as parameter for mitochondrial respiratory activity. Measurements are traditionally done with a Clark electrode, or with more sophisticated optical methods. At the cathode of a Clark electrode, oxygen is electrolytically reduced, which initiates a current in the electrode, causing a potential difference that is ultimately recorded. Clark electrodes however have the disadvantage that oxygen is consumed. Furthermore, interferences with nitrogen oxides, ozone, or chlorine are observed (Stetter et al., 2008). To circumvent these limitations, optical sensors have been developed that have the advantage that no oxygen is consumed, combined with a high accuracy and reversibility. Optical oxygen sensors work according to the principle of dynamic fluorescence quenching. The response of the respective fluorescence dye is proportional to the amount of oxygen in the sample investigated (Wang and Wolfbeis, 2014). In a model of isolated mitochondria in the absence of complex II substrates, oxygen consumption can serve as surrogate readout for the assessment of the degree of CI inhibition. It is however essential to realise that also complex III and complex IV activities are involved and their inhibition also results in a decline in O2 consumption. In addition to that, CI inhibitors can lead to a one‐electron reduction of molecular oxygen at the site of CI to yield superoxide. The amount of superoxide formed hence contributes to the consumption of oxygen, but this must not be interpreted as oxygen consumption as a result of controlled and coupled electron flux through the complexes of the mitochondrial respiratory chain. A modern convenient method to measure oxygen consumption is provided by the Seahorse technology of extracellular flux (XF) analysis, in which cells are kept in a very small volume, so that changes of oxygen levels can be detected very sensitively by an oxygen sensor. To allow manipulation of the mitochondria in cells, the cell membrane can be permeabilised with saponin (SAP), digitonin (DIG) or recombinant perfringolysin O (rPFO) (XF‐plasma membrane permeabiliser (PMP) reagent), to allow addition of specific substrates to measure activity of different respiratory chain complexes, including CI (Salabei et al., 2014).
A.2.4.2. Intracellular ATP levels
Intracellular ATP levels originate both from mitochondria and from glycolysis. If glycolytic ATP production is impaired or inhibited, the cellular production of ATP is a measure of mitochondrial function. If it is assumed that the ATP consumption remains constant, then the steady state ATP levels can serve as indirect readout for mitochondrial activity, and the latter depends on the functioning of CI. Inhibitors of CI reduce cellular ATP levels, but it has to be remembered that intracellular ATP levels are also affected by inhibitors of other parts of the respiratory chain, of the citric acid cycle or of the transport of energy substrates. For a proper interpretation of assay results, it has to be ascertained in each particular test system, that ATP production from other sources is excluded and that the cellular ATP consumption remains constant. ATP levels can be easily measured from lysates of in vitro cell cultures or from tissues by a luminometric luciferase/luciferin assay. The amount of light emitted is proportional to the amount of ATP in the sample (Nguyen et al., 1988; Leist, 1997).
A.2.4.3. Other approaches
As mitochondrial activity is coupled to many cellular functions, there is a multitude of other indirect assays that are sensitive to inhibitors of CI. Some of these tests may indeed be very sensitive, while they have a low specificity. Thus, their application requires usually a good control of the experimental system and care with the interpretation of the data. One exemplary approach is the measurement of NADH/NAD+ ratios in mitochondria by imaging methods. This provides resolution on the level of individual mitochondria within a living cell (van Vliet et al., 2014)
A.2.5. Evidence Supporting Taxonomic Applicability
The CI is well‐conserved across species from lower organisms to mammals. The central subunits of CI harbouring the bioenergetic core functions are conserved from bacteria to humans. CI from bacteria and from mitochondria of Yarrowia lipolytica, a yeast genetic model for the study of eukaryotic CI (Kerscher et al., 2002) was analysed by x‐ray crystallography (Hofhaus et al., 1991; Baradaran et al., 2013; Zickermann et al., 2015). The CI of the mitochondria of eukaryotes and in the plasma membranes of purple photosynthetic bacteria are closely related to respiratory bacteria and the close homology of sequences, function, and prosthetic groups shows a common ancestry (Friedrich et al., 1995).
References
Baradaran R, Berrisford JM, Minhas GS and Sazanov LA, 2013. Crystal structure of the entire respiratory complex I. Nature, 494, 443–448.
Barrientos A, Moraes CT, 1999. Titrating the effects of mitochondrial complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197.
Degli Esposti, 1998. Inhibitors of NADH‐ubiquinone reductase: an overview. Biochimica et Biophysica Acta, 1364, 222–235.
Ernster L, Lee C, 1967. Energy‐linked reduction of NAD+ by succinate. Methods in Enzymology, 10, 729–738.
Friedrich T, Steinmüller K, Weiss H, 1995. The proton‐pumping respiratory complex I of bacteria and mitochondria and is homologue of chloroplasts. Federation of the European Biochemical Societies's Letters (FEBS)ers(Minireview), 367, 107–111.
Garrett and Grisham, 2010. Biochemistry, Brooks/Cole, pp 598–611.
Greenamyre JT, Sherer TB, Betarbet, R, Panov AV, 2001. Critical review complex I and Parkinson's disease life, 52, 135–141.
Hofhaus G, Weiss H and Leonard K, 1991. Electron microscopic analysis of the peripheral and the membrane parts of mitochondrial NADH dehydrogenase (Complex I). Journal of Molecular Biology, 221, 1027–1043.
Kerscher S, Dröse S, Zwicker K, Zickermann V, Brandt U, 2002. Yarrowia lipolytica, a yeast genetic system to study mitochondrial complex I. Biochimica et Biophysica Acta, 1555, 83–91.
Kirby DM, Thorburn DR, Turnbull DM, Taylor RW, 2007. Biochemical assays of respiratory chain complex activity. Methods in Cell Biology, 80, 93–119.
Leist M, Single B, Castoldi AF, Kühnle S, Nicotera P, 1997. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. Journal of Experimental Medicine, 185, 1481–6.
Leist M, 2014. Current approaches and future role of high content imaging in safety sciences and drug discovery. Alternatives to Animal Experimentation (ALTEX), 31, 479–493.
Lenaz G, Fato R, Baracca A, Genova ML, 2004. Mitochondrial quinone reductases: complex I. Methods in Enzymology, 382, 3–20.
Long J, Ma J, Luo C, Mo X, Sun L, Zang W, Liu J, 2009. Comparison of two methods for assaying complex I activity in mitochondria isolated from rat liver, brain and heart. Life Science Journal, 85, 276–280.
Nguyen VT, Morange M, Bensaude O, 1988. Firefly luciferase luminescence assays using scintillation counters for quantitation in transfected mammalian cells. Analytical Biochemistry, 171, 404–408.
van Vliet E, Daneshian M, Beilmann M, Davies A, Fava E, Fleck R, Julé Y, Kansy M, Kustermann S, Macko P, Mundy WR, Roth A, Shah I, Uteng M, van de Water B, Hartung T, Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C, 2012. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nature Protocols, 7, 1235–1246.
Salabei JK, Gibb AA, Hill BG, 2014. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nature Protocols, 9, 421–438.
Stetter JR, Li J, 2008. Amperometric gas sensors–a review. Chemical Reviews, 108, 352–366.
Wang XD, Wolfbeis OS, 2014. Optical methods for sensing and imaging oxygen: materials, spectroscopies and applications. Chemical Society Reviews, 43, 3666–3761.
Voet DJ, Voet JG, Pratt CW, 2008. Chapter 18, Mitochondrial ATP synthesis. Principles of Biochemistry. 3rd Edition, Wiley. p. 608. ISBN 978‐0‐470‐23396‐2.
Willis JH, Capaldi RA, Huigsloot M, Rodenburg RJ, Smeitink J, Marusich MF, 2009. Isolated deficiencies of OXPHOS complexes I and IV are identified accurately and quickly by simple enzyme activity immunocapture assays. Biochimica et Biophysica Acta, 1787, 533–538.
Zickermann V, Christophe Wirth, Hamid Nasiri, Karin Siegmund, Harald Schwalbe, Carola Hunte, Ulrich Brandt, 2015. Mechanistic insight from the crystal structure of mitochondrial complex I. Science, 347, 44–49.
A.3. KE2: Mitochondrial dysfunction (ENV/JM/WRPR(2016)34; 2016)
A.3.1. How this Key Event works
Mitochondria play a pivotal role in cell survival and cell death because they are regulators of both energy metabolism and apoptotic/necrotic pathways (Fiskum, 2000; Wieloch, 2001; Friberg and Wieloch, 2002). The production of ATP via oxidative phosphorylation is a vital mitochondrial function (Kann and Kovács, 2007; Nunnari and Suomalainen, 2012). The ATP is continuously required for signalling processes (e.g. Ca2+ signalling), maintenance of ionic gradients across membranes, and biosynthetic processes (e.g. protein synthesis, haem synthesis or lipid and phospholipid metabolism) (Green, 1998; Hajnóczky et al., 2006; McBride et al., 2006; Kang and Pervaiz, 2012). Inhibition of mitochondrial respiration contributes to various cellular stress responses, such as deregulation of cellular Ca2+ homoeostasis (Graier et al., 2007) and ROS production (Nunnari and Suomalainen, 2012; reviewed in Mei et al., 2013).
It is well established in the existing literature that mitochondrial dysfunction may result in: (a) an increased ROS production and a decreased ATP level, (b) the loss of mitochondrial protein import and protein biosynthesis, (c) the reduced activities of enzymes of the mitochondrial respiratory chain and the Krebs cycle, (d) the loss of the mitochondrial membrane potential, (e) the loss of mitochondrial motility, causing a failure to re‐localise to the sites with increased energy demands, (f) the destruction of the mitochondrial network, (g) increased mitochondrial Ca2+ uptake, causing Ca2+ overload (reviewed in Lin and Beal, 2006; Graier et al., 2007), (h) the rupture of the mitochondrial inner and outer membranes, leading to the release of mitochondrial pro‐death factors, including cytochrome c (Cyt. c), apoptosis‐inducing factor, or endonuclease G (Martin, 2011; Braun, 2012; Correia et al., 2012; Cozzolino et al., 2013), which eventually leads to apoptotic, necrotic or autophagic cell death (Wang and Qin, 2010). Due to their structural and functional complexity, mitochondria present multiple targets for various compounds.
A.3.2. How it is measured or detected
Mitochondrial dysfunction can be detected using isolated mitochondria, intact cells or cells in culture as well as in vivo studies. Such assessments can be performed with a large range of methods (revised by Brand and Nicholls, 2011) for which some important examples are given. All approaches to assess mitochondrial dysfunction fall into two main categories: the first approach assesses the consequences of a loss‐of‐function, i.e. impaired functioning of the respiratory chain and processes linked to it. Some assays to assess this have been described for KE1, with the limitation that they are not specific for CI. In the context of overall mitochondrial dysfunction, the same assays provide useful information, when performed under slightly different assay conditions (e.g. without addition of complex III and IV inhibitors). The second approach assesses a ‘non‐desirable gain‐of‐function’, i.e. processes that are usually only present to a very small degree in healthy cells, and that are triggered in a cell upon mitochondria failure.
A.3.2.1. Mitochondrial dysfunction assays assessing a loss‐of function
A.3.2.1.1. Cellular oxygen consumption
See KE1 for details regarding oxygen consumption assays. The oxygen consumption parameter can be combined with other endpoints to derive more specific information on the efficacy of mitochondrial function. One approach measures the ADP‐to‐O ratio (the number of ADP molecules phosphorylated per oxygen atom reduced (Hinkle, 1995 and Hafner et al., 1990). The related Phosphate/Oxygen (P/O) ratio is calculated from the amount of ADP added, divided by the amount of O consumed while phosphorylating the added ADP (Ciapaite et al., 2005).
A.3.2.1.2. Mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential (Δψm) is the electric potential difference across the inner mitochondrial membrane. It requires a functioning respiratory chain in the absence of mechanisms that dissipate the proton gradient without coupling it to ATP production. The classical, and still most quantitative method uses a tetraphenylphosphonium ion (TPP+)‐sensitive electrode on suspensions of isolated mitochondria.
The Δψm can also be measured in live cells by fluorimetric methods. These are based on dyes which accumulate in mitochondria because of Δψm. Frequently used are tetramethylrhodamine ethyl ester (TMRE), tetramethylrhodamine methyl ester (TMRM) (Petronilli et al., 1999) or 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazole carbocyanide iodide (JC‐1). In particular, mitochondria with intact membrane potential concentrate JC‐1, so that it forms red fluorescent aggregates, whereas de‐energised mitochondria cannot concentrate JC‐1 and the dilute dye fluoresces green (Barrientos et al., 1999). Assays using TMRE or TMRM measure only at one wavelength (red fluorescence), and depending on the assay setup, de‐energised mitochondria become either less fluorescent (loss of the dye) or more fluorescent (attenuated dye quenching).
A.3.2.1.3. Enzymatic activity of the electron transport system (ETS)
Determination of ETS activity can be determined following Owens and King's assay (1975). The technique is based on a cell‐free homogenate that is incubated with NADH to saturate the mitochondrial ETS and an artificial electron acceptor [l‐(4‐iodophenyl)‐3‐(4‐nitrophenyl)‐5‐phenyltetrazolium chloride (INT)] to register the electron transmission rate. The oxygen consumption rate is calculated from the molar production rate of INT‐formazan which is determined spectrophotometrically (Cammen et al., 1990).
A.3.2.1.4. ATP content
For the evaluation of ATP levels, various commercially available ATP assay kits are offered (e.g. Sigma, http://www.abcam.com/atp-assay-kit-colorimetricfluorometric-ab83355.html), based on luciferin and luciferase activity. For isolated mitochondria various methods are available to continuously measure ATP with electrodes (Llaudet et al., 2005), with luminometric methods, or for obtaining more information on different nucleotide phosphate pools (e.g. Ciapaite et al., 2005).
A.3.2.2. Mitochondrial dysfunction assays assessing a gain‐of function
A.3.2.2.1. Mitochondrial permeability transition pore (PTP) opening
The opening of the PTP leads to the permeabilisation of mitochondrial membranes (Lemasters et al., 2009; Fiskum, 2000), so that different compounds and cellular constituents can change intracellular localisation. This can be measured by assessment of the translocation of cytochrome c, adenylate kinase or the apoptosis‐inducing factor (AIF) from mitochondria to the cytosol or nucleus. The translocation can be assessed biochemically in cell fractions, by imaging approaches in fixed cells or tissues, or by life‐cell imaging of GFP fusion proteins (Single et al., 1998; Modjtahedi et al., 2006). An alternative approach is to measure the accessibility of cobalt to the mitochondrial matrix in a calcein fluorescence quenching assay in live permeabilised cells (Petronilli et al., 1999).
A.3.2.2.2. mtDNA damage as a biomarker of mitochondrial dysfunction
Various quantitative polymerase chain reaction (QPCR)‐based assays have been developed to detect changes of DNA structure and sequence in the mitochondrial genome (mtDNA). mtDNA damage can be detected in blood after low‐level rotenone exposure, and the damage persists even after CI activity has returned to normal. With a more sustained rotenone exposure, mtDNA damage can be also detected in skeletal muscle. These data support the idea that mtDNA damage in peripheral tissues in the rotenone model may provide a biomarker of past or ongoing mitochondrial toxin exposure (Sanders et al., 2014).
A.3.2.3. Generation of ROS and resultant oxidative stress
A.3.2.3.1. General approach
Electrons from the mitochondrial ETS may be transferred ‘erroneously’ to molecular oxygen to form superoxide anions. This type of side reaction can be strongly enhanced upon mitochondrial damage. As superoxide may form hydrogen peroxide, hydroxyl radicals or other ROS, a large number of direct ROS assays and assays assessing the effects of ROS (i.e. indirect ROS assays) are available. Direct assays are based on the chemical modification of fluorescent or luminescent reporters by ROS species. Indirect assays assess cellular metabolites, the concentration of which is changed in the presence of ROS (e.g. glutathione, malonaldehyde, isoprostanes, etc.). In living animals, the effects of oxidative stress can be detected by analysis of specific biomarkers in the blood or urine.
A.3.2.3.2. Measurement of the cellular glutathione (GSH) status
GSH is regenerated from its oxidised form (GSSH) by the action of a NADPH‐dependent reductase (GSSH + NADPH + H+ → 2 GSH + NADP+). The ratio of GSH/GSSG is therefore a good indicator for the cellular NADP+/NADPH ratio (i.e. the redox potential). GSH and GSSH levels can be determined by HPLC, capillary electrophoresis, biochemically with DTNB (Ellman's reagent, 5,5’‐dithio‐bis‐[2‐nitrobenzoic acid]) or by mean of luminescence‐based assays (for example, GSH‐Glo™ Glutathione Assay, https://www.promega.co.uk/resources/protocols/technical-bulletins/101/gsh-glo-glutathione-assay-protocol/). As excess GSSG is rapidly exported from most cells to maintain a constant GSH/GSSG ratio, a reduction of total glutathione levels is often considered a good surrogate measure for oxidative stress.
A.3.2.3.3. Quantification of lipid peroxidation
Measurement of lipid peroxidation has historically relied on the detection of thiobarbituric acid (TBA)‐reactive compounds, such as malondialdehyde generated from the decomposition of cellular membrane lipid under oxidative stress (Pryor et al., 1976). This method is quite sensitive, but not highly specific. A number of commercial assay kits are available for this assay using absorbance or fluorescence detection technologies. The formation of F2‐like prostanoid derivatives of arachidonic acid, termed F2‐isoprostanes (IsoPs) has been shown to be more specific for lipid peroxidation. A number of commercial ELISA kits have been developed for IsoPs, but interfering agents in samples requires partial purification before analysis. Alternatively, gas chromatography–mass spectrometry (GC‐MS) may be used as a robust, specific and sensitive method.
A.3.2.3.4. Detection of superoxide (O2 −) production
Generation of superoxide by inhibition of CI and the methods for its detection are described by Grivennikova and Vinogradov (2006). A range of different methods is also described by BioTek (http://www.biotek.com/resources/articles/reactive-oxygen-species.html). The reduction of ferricytochrome c to ferrocytochrome c may be used to assess the rate of superoxide formation (McCord and Fidovich, 1968). Like in other superoxide assays, specificity can only be obtained by measurements in the absence and presence of superoxide dismutase. Chemiluminescent reactions have been used for their increased sensitivity. The most widely used chemiluminescent substrate is lucigenin. Coelenterazine has also been used as a chemiluminescent substrate. Hydrocyanine dyes are fluorogenic sensors for superoxide and hydroxyl radical, and they become membrane impermeable after oxidation (trapping at the site of formation). The best characterised of these probes are Hydro‐Cy3 and Hydro‐Cy5. Generation of superoxide in mitochondria can be visualised using fluorescence microscopy with MitoSOX™ Red reagent (Life Technologies). MitoSOX™ Red reagent is a cationic derivative of dihydroethidium that permeates live cells and accumulates in mitochondria.
A.3.2.3.5. Detection of hydrogen peroxide (H2O2) production
There are a number of fluorogenic substrates, which serve as hydrogen donors that have been used in conjunction with horseradish peroxidase (HRP) enzyme to produce intensely fluorescent products in the presence of hydrogen peroxide (Ruch et al., 1983; Zhou et al., 1997). The more commonly used substrates include diacetyldichloro‐fluorescein, homovanillic acid, and Amplex® Red (https://www.thermofisher.com/order/catalog/product/A22188). In these assays, increasing amounts of H2O2 leads to increasing amounts of fluorescent product (Tarpley et al., 2004).
A.3.3. Evidence Supporting Taxonomic Applicability
Mitochondrial dysfunction is a universal event occurring in cells of any species (Farooqui and Farooqui, 2012). Many invertebrate species (e.g. Drosophila melanogaster and Caenorhabditis elegans) are considered as potential models to study mitochondrial functionality. New data on marine invertebrates, such as molluscs and crustaceans and non‐Drosophila species, are emerging (Martinez‐Cruz et al., 2012). Mitochondrial dysfunction can be measured in animal models used for toxicity testing (Waerzeggers et al., 2010; Winklhofer and Haass, 2010) as well as in humans (Winklhofer and Haass, 2010).
References
Adam‐Vizi V, 2015. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non‐electron transport chain sources. Antioxid Redox Signal, 7, 1140–1149.
Bal‐Price A, Brown GC, 2000. Nitric‐oxide‐induced necrosis and apoptosis in PC12 cells mediated by mitochondria. Journal of Neurochemistry, 75, 1455–1464.
Bal‐Price A, Matthias A, Brown GC, 2002. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. Journal of Neurochemistry, 80, 73–80.
Brand MD, Nicholls DG, 2011. Assessing mitochondrial dysfunction in cells. Biochemical Journal, 435, 297–312.
Braun RJ, 2012. Mitochondrion‐mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Frontiers in Oncology, 2,182.
Barrientos A, Moraes CT, 1999. Titrating the effects of mitochondrial complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197.
Cammen M Corwin, Susannah Christensen, John P, 1990. Electron transport system (ETS) activity as a measure of benthic macrofaunal metabolism. Marine Ecology Progress Series, 65, 171–182.
Ciapaite, Lolita Van Eikenhorst, Gerco Bakker, Stephan JL Diamant, Michaela, Heine, Robert J Wagner, Marijke JV Westerhoff, Hans and Klaas Krab, 2005. Modular kinetic analysis of the adenine nucleotide translocator‐mediated effects of palmitoyl‐CoA on the oxidative phosphorylation in isolated rat liver mitochondria diabetes, 54, 944–951.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA, 2012. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Advances in Experimental Medicine and Biology, 724, 205–221.
Cozzolino M, Ferri A, Valle C, Carri MT, 2013. Mitochondria and ALS: implications from novel genes and pathways. Molecular and Cellular Journal of Neuroscience, 55, 44–49.
Diepart C, Verrax J, Calderon PB, Feron O, Jordan BF, Gallez B, 2010. Comparison of methods for measuring oxygen consumption in tumor cells in vitro. Analytical Biochemistry, 396, 250–256.
ENV/JM/WRPR(2016)34. 2016. Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, Pesticides and Biotechnology. Adverse Outcome pathway on ionotropic glutamatergic receptors and cognition.
Farooqui T, Farooqui AA, 2012. Oxidative Stress in Vertebrates and Invertebrate: Molecular Aspects of Cell Signalling. Wiley‐Blackwell, Chapter 27, pp: 377–385.
Fan LM, Li JM, 2014. Evaluation of methods of detecting cell reactive oxygen species production for drug screening and cell cycle studies. Journal of Pharmacological and Toxicological Methods, 70, 40–47.
Fiskum G, 2000. Mitochondrial participation in ischemic and traumatic neural cell death. Journal of Neurotrauma, 17, 843–855.
Friberg H, Wieloch T, 2002. Mitochondrial permeability transition in acute neurodegeneration. Biochimie, 84, 241–250.
Fujikawa DG, 2015. The role of excitotoxic programmed necrosis in acute brain injury. Computational and Structural Biotechnology Journal, 13, 212–221.
Graier WF, Frieden M, Malli R, 2007. Mitochondria and Ca2+ signaling: old guests, new functions. Pflügers Archiv, 455, 375–396.
Green DR, 1998. Apoptotic pathways: the roads to ruin. Cell, 94, 695–698.
Grivennikova VG, Vinogradov AD, 2006. Generation of superoxide by the mitochondrial Complex I. Biochimica et Biophysica Acta, 1757, 553–561.
Hafner RP, Brown GC, Brand MD, 1990. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and proton motive force in isolated mitochondria using the ‘top‐down’ approach of metabolic control theory. European Journal of Biochemistry, 188, 313–319.
Hinkle PC, 1995. Measurement of ADP/O ratios. In: Brown GC, Cooper CE (eds.). Bioenergetics: A Practical Approach, Energy Dispersive Spectroscopy, Oxford, U.K., IRL Press, p. 5–6.
Hynes J, Marroquin LD, Ogurtsov VI, Christiansen KN, Stevens GJ, Papkovsky DB, Will Y, 2006. Investigation of drug‐induced mitochondrial toxicity using fluorescence‐based oxygen‐sensitive probes. Journal of Toxicological Sciences, 92, 186–200.
James PE, Jackson S, Grinberg OY, Swartz HM, 1995. The effects of endotoxin on oxygen consumption of various cell types in vitro: an EPR oximetry study. Free Radical Biology and Medicine, 18, 641–647.
Kang J, Pervaiz S, 2012. Mitochondria: redox metabolism and dysfunction. Biochemistry Research International, 896751.
Kann O, Kovács R, 2007. Mitochondria and neuronal activity. American Journal Of Physiology‐Cell Physiology, 292, C641–C657.
Knott Andrew B, Guy Perkins, Robert Schwarzenbacher, Ella Bossy‐Wetzel, 2008. Mitochondrial fragmentation in neurodegeneration. Nature Reviews Journal of Neuroscience, 229, 505–518.
Llaudet E, Hatz S, Droniou M, Dale N, 2005. Microelectrode biosensor for real‐time measurement of ATP in biological tissue. Analytical Chemistry, 77, 3267–73.
Lee HC, Wei YH, 2012. Mitochondria and aging. Advances in Experimental Medicine and Biology, 942, 311–327.
Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, 2003. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. Journal of Biological Chemistry, 278, 8516–8525.
Lin MT, Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787–795.
Martin LJ, 2011. Mitochondrial pathobiology in ALS. Journal of Bioenergetics and Biomembranes, 43, 569–579.
Martinez‐Cruz, Oliviert Sanchez‐Paz, Arturo Garcia‐Carreño, Fernando Jimenez‐Gutierrez, Laura Ma de los Angeles Navarrete del Toro, Adriana Muhlia‐Almazan, 2012. Invertebrates mitochondrial function and energetic challenges (http://www.intechopen.com). In: Dr Kevin Clark (ed.). Bioenergetics, ISBN 978‐953‐51‐0090‐4, Publisher InTech, 181–218.
McBride HM, Neuspiel M, Wasiak S, 2006. Mitochondria: more than just a powerhouse. Current Biology, 16, R551–R560.
McCord JM, Fidovich I, 1968. The reduction of cytochrome C by milk xanthine oxidase. Journal of Biological Chemistry, 243, 5733–5760.
Mei Y, Thompson MD, Cohen RA, Tong X, 2013. Endoplasmic reticulum stress and related pathological processes. Journal of Pharmacology and Biomedical Analysis, 1, 100–107.
Modjtahedi N, Giordanetto F, Madeo F, Kroemer G, 2006. Apoptosis‐inducing factor: vital and lethal. Trends in Cell Biology, 16, 264–272.
Nunnari J, Suomalainen A, 2012. Mitochondria: in sickness and in health. Cell 148:1145–1159. Hajnóczky G, Csordás G, Das S, Garcia‐Perez C, Saotome M, Sinha Roy S, Yi M, 2006. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2 + uptake in apoptosis. Cell Calcium, 40, 553–560.
Oliviert Martinez‐Cruz, Arturo Sanchez‐Paz, Fernando Garcia‐Carreño, Laura Jimenez‐Gutierrez, Ma de los Angeles Navarrete del Toro and Adriana Muhlia‐Almazan, 2012. Invertebrates mitochondrial function and energetic challenges (http://www.intechopen.com). In: Dr Kevin Clark (ed.). Bioenergetics, ISBN 978‐953‐51‐0090‐4, Publisher InTech, 181–218.
Owens RG, King FD, 1975. The measurement of respiratory electron‐transport system activity in marine zooplankton. Marine Biology, 30, 27–36.
Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F, 1999. Transient and long‐lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophysical Journal 76, 725–734.
Promega GSH‐Glo Glutathione Assay Technical Bulletin, TB369, Promega Corporation, Madison, WI.
Pryor WA, Stanley JP and Blair E, 1976. Autoxidation of polyunsaturated fatty acids: II. a suggested mechanism for the formation of TBA‐reactive materials from prostaglandin‐like endoperoxides. Lipids, 11, 370–379.
Radkowsky AE, Kosower EM, 1986. Bimanes 17 (Haloalkyl)‐1,5‐diazabicyclo[3.3.O]octadienediones (halo‐9,10‐dioxabimanes): reactivity toward the tripeptide thiol, glutathione. Journal of the American Chemical Society, 108, 4527–4531.
Ruch W, Cooper PH, Baggiollini M, 1983. Assay of H2O2 production by macrophages and neutrophils with Homovanillic acid and horseradish peroxidase. Journal of Immunological Methods, 63, 347–357.
Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT, 2014. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiology of Disease, 70, 214–223.
Sanders LH, Howlett EH2, McCoy J, Greenamyre JT, 2014b. Mitochondrial DNA damage as a peripheral biomarker for mitochondrial toxin exposure in rats. Toxicological Sciences, 142, 395–402.
Single B, Leist M, Nicotera P, 1998. Simultaneous release of adenylate kinase and cytochrome c in cell death. Cell Death & Differentiation, 5, 1001–1003.
Tarpley MM, Wink DA, Grisham MB, 2004. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. American Journal of Physiology. Regulatory integrative and comparative Physiology, 286, R431–R444.
von Heimburg D, Hemmrich K, Zachariah S, Staiger H, Pallua N, 2005. Oxygen consumption in undifferentiated versus differentiated adipogenic mesenchymal precursor cells. Respiratory Physiology & Neurobiology, 146, 107–116.
Waerzeggers, Yannic Monfared, Parisa Viel, Thomas Winkeler, Alexandra Jacobs, Andreas H, 2010. Mouse models in neurological disorders: applications of non‐invasive imaging. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, 1802, 819–839.
Walker JE, Skehel JM, Buchanan SK, 1995. Structural analysis of NADH: ubiquinone oxidoreductase from bovine heart mitochondria. Methods in Enzymology, 260, 14–34.
Wang A, Costello S, Cockburn M, Zhang X, Bronstein J, Ritz B, 2011. Parkinson's disease risk from ambient exposure to pesticides. European Journal of Epidemiology, 26, 547–555.
Wang Y and Qin ZH, 2010. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis, 15, 1382–1402.
Wieloch T, 2001. Mitochondrial involvement in acute neurodegeneration, 52, 247–254.
Winklhofer K, Haass C, 2010. Mitochondrial dysfunction in Parkinson's disease. Biochimica et Biophysica Acta (BBA) –Molecular Basis of Disease, 1802, 29–44.
Zhou M, Diwu Z, Panchuk‐Voloshina N, Haughland RP, 1997. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: application in detecting the activity of phagocyte NADPH oxidase and other oxidases. Analytical Biochemistry, 253, 162–168.
A.4. KE3: Impaired proteostasis
A.4.1. How this key Event works
The concept of proteostasis refers to the homoeostasis of proteins in space and time, i.e. the correct balance between protein synthesis, modification, transport and degradation. Disturbance of proteostasis results in pathological changes either by loss of function events (lack of a pivotal protein/protein function) or by a gain of undesired functions (aggregation of a protein leading to the formation of inclusions and new structures in cells and disturbing turnover of many unrelated proteins).
Proteostasis regulation is the main defence mechanism against toxic proteins, whose accumulation could greatly compromise normal cellular function and viability. Therefore, the chaperone and degradation systems assuring the removal of misfolded and aggregated proteins, as well as damaged, dysfunctional cellular organelles (e.g. defective mitochondria) play a key role in cellular homoeostasis (Lee et al., 2012).
The two major degradation systems are the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathway (ALP) (Korolchuk et al., 2010; Kroemer et al., 2010; Ravikumar et al., 2010). The UPS works through the attachment of multiple ubiquitin molecules to a protein substrate, followed by the subsequent degradation of the tagged polyubiquitinated protein by the proteasome (Ciechanover, 1998; Ciechanover and Brundin, 2003). A compromised function of the UPS leads to the accumulation of ubiquitylated proteins, such as α‐synuclein (Ii et al., 1997; Spillantini et al., 1997; Sulzer and Zecca 2000). The accumulation of polyubiquitinated proteins, as a consequence of a dysfunctional proteasome activity, is observed in some pathologies, and experimental inhibition of the proteasome has been shown to trigger parkinsonian neurodegeneration (Hardy et al., 2001; McNaught and Jenner 2001).
ALP involves the engulfment of cytoplasmic materials into autophagosomes, which are degraded by lysosomal enzymes after fusion of autophagosomes with lysosomes (Kuma et al., 2004) or direct import of proteins into lysosomes (Cuervo, 2004; Mizushima et al., 2008). Autophagy also plays an essential role for the removal of damaged organelles, such as mitochondria. Both, excessive autophagy or reduced autophagic flux can compromise cell survival (Rothermel and Hill, 2007), and several genetic forms of PD are linked to the autophagy‐related genes Pink1, Parkin or Uchl1.
Autophagy enables cell survival during mitochondrial stress by clearing the damaged organelles (Lee et al., 2012).
One of the main aggregated proteins found to accumulate in nigrostriatal cells during Parkinson's disease is α‐synuclein. Aggregation of α‐synuclein can obstruct normal cellular transport, leading to impaired intracellular trafficking and/or trapping of cellular organelles in inappropriate locations, this resulting in synaptic and cell dysfunctions (Cookson, 2005; Bartels et al., 2011; Bellucci et al., 2012; Games et al., 2013; Hunn et al., 2015).
Importantly, accumulation of α‐synuclein affects mitochondrial trafficking. The polarity and correct function of different types of cells depend on an efficient transport of mitochondria to areas of high energy consumption (Sheng, 2014). Therefore, the correct distribution of mitochondria to various parts of a cell is essential to preserve cell function (Zhu et al., 2012; Schwarz, 2013).
A.4.2. How it is measured or detected
A.4.2.1. Evaluation of UPS function
A.4.2.1.1. General turnover assays
Quantitative evaluation can be based on the detection of increased ubiquitin or ubiquinated proteins, as well as proteasomal subunits, either by immunocyto/histochemistry or by western blotting (Rideout et al., 2001; Ortega and Lucas, 2014). UPS activity can be continuously monitored by quantitating (by mean of flow cytometry or microscopy) the level of e.g. EGFP‐degron fusion proteins that are selectively degraded by the proteasome (Bence et al., 2001).
A.4.2.1.2. Proteasome activity assay
Various fluorogenic substrates (e.g. Suc‐Leu‐Leu‐Val‐Tyr‐AMC for the chymotrypsin‐like activity) can be used for the determination of proteasomal activity in in vivo or in vitro applications. These substrates may be applied to tissue or cell homogenates, but specific measurements require partial purification of the proteasome (Kisselev and Goldberg, 2005).
A.4.2.1.3. Detection of α‐synuclein (AS) aggregates
The most common methods to detect AS aggregates use immunostaining for AS (in cells or in tissues). In cell culture, AS may also be epitope‐tagged or coupled to GFP to allow an indirect detection. The detection of small, not microscopically visible AS aggregates is indicative of protease‐resistance. Tissue slices may be exposed to proteases before immunostaining for AS. Alternatively, small or large aggregates may be biochemically enriched by differential centrifugation and proteolytic treatment, and then analysed, e.g. by western blot, mass spectrometry or ELISA‐like immunoquantification.
A.4.2.2. Evaluation of ALP function
A.4.2.2.1. Quantification of lysosomes or autophagosomes
Disturbances of ALP often result in counter‐regulations that can be visualised by staining of lysosomes or parts of the autophagy system. Several weakly basic dyes can be used to stain acidic organelles (lysosomes) in live cells. For example, the dye LysoTracker Red stains lysosomes and can be used to monitor autophagy (Klionsky et al., 2007, 2008). The autofluorescent drug monodansylcadaverine (MDC) has also been used as autophago‐lysosome marker (Munafó and Colombo, 2002). A convenient way to stain lysosomes in tissue or fixed cells is the use of antibodies against the Lysosomal‐Associated Membrane Protein 1 (LAMP‐1) (Rajapakshe et al., 2015) or against cathepsins (Foghsgaard et al., 2001).
For qualitative or semiquantitative estimates of lysosomes and related organelles, transmission electron microscopy has been frequently used (Barth et al., 2010).
A.4.2.2.2. Monitoring of autophagy‐related molecules
The amount and the localisation of autophagy‐related proteins can change during disturbance of the ALP. Especially in cell culture, but also in transgenic mice, various techniques have been used to monitor autophagy by mean of fluorescence‐tags or other substrates, e.g. ATG, autophagy‐related protein or autophagy substrates, to monitor their fate in cells and thus provide information on disturbed ALP, or the overexpression of GFP–LC3, in which GFP (green fluorescent protein) is expressed as a fusion protein at the amino terminus of LC3 (microtubule‐associated protein 1A/1B‐light chain 3), which is the a mammalian homologue of Saccharomyces cerevisiae ATG8 (Kadowaki and Karim, 2009).
A.4.2.2.3. Monitoring autophagic flux
The lysosomal degradation of the autophagic cargo constitutes the autophagic flux, which can be measured by assessing the rate of turnover of long‐lived proteins that are normally turned over by autophagy (Bauvy et al., 2009) This is performed by labelling intracellular proteins with either [14C]‐leucine or [14C]‐valine, followed by a long culture period in standard medium. The release of radioactive leucine or valine into the culture medium corresponds to the protein degradation rate in cells, and it may be measured by liquid scintillation counting.
A.4.2.2.4. Monitoring the conversion of LC3‐I to LC3‐II
The progression of autophagy (autophagic flux) can be studied by the conversion of LC3‐I into LC3‐II (i.e. a post‐translational modification specific for autophagy) by mean of Western blot analysis. The amount of LC3‐II correlates with the number of autophagosomes. Conversion of LC3 can be used to examine autophagic activity in the presence or absence of lysosomal activity (Klionsky et al., 2007, 2008). The technology can also be used in vivo, e.g. by the use of transgenic mice that overexpress GFP–LC3 (Kuma et al., 2004).
A.4.2.2.5. Evaluation of intracellular transport of mitochondria and other organelles
A range of technologies has been used to visualise mitochondrial dynamics in live cells (Jakobs, 2006; Grafstein and Forman, 1980). They usually employ a combination of mitochondrial labelling with fluorescent dyes (e.g. DiOC6 (3,3′‐dihexyloxacarbocyanine iodide), JC‐1 (5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimida‐zolylcarbo‐cyanine iodide), MitoTracker, MitoFluor probes, etc.), followed by video‐ or confocal microscopy for live cell imaging (Pool et al., 2006; Schwarz, 2013). Most frequently, mitochondrial mobility is observed along neurites, and measurable endpoints may be mitochondrial speed and direction with regard to the cell soma (Schildknecht et al., 2013). Additionally, also mitochondrial fusion and fission have been monitored by such methods (Exner et al., 2012). The transport of other organelles along neurites may be monitored using similar methods, and the microtubule structures that serve as transport scaffold may be co‐stained.
A.4.3. Evidence supporting taxonomic applicability
The ubiquitin proteasome system is highly conserved in eukaryotes, from yeast to human. Ubiquitin is a small (8.5 kDa) regulatory protein that has been found in almost all tissues of eukaryotic organisms. For instance, drosophila has been used as PD model to study the role of ubiquitin in α‐synuclein induced‐toxicity (Lee et al., 2009). Human and yeast ubiquitin share 96% sequence identity. Neither ubiquitin nor the ubiquitination machinery is known to exist in prokaryotes.
Autophagy is ubiquitous in eukaryotic cells and is the major mechanism involved in the clearance of oxidatively or otherwise damaged/worn‐out macromolecules and organelles (Esteves et al., 2011). Due to the high degree of conservation, most of the knowledge on autophagy proteins in vertebrates is derived from studies in yeast (Klionsky et al., 2007).
Autophagy is seen in all eukaryotic systems, including fungi, plants, slime mould, nematodes, fruit flies and insects, rodents (i.e. laboratory mice and rats), and humans. It is a fundamental and phylogenetically conserved self‐degradation process that is characterised by the formation of double‐layered vesicles (autophagosomes) around intracellular cargo for delivery to lysosomes and proteolytic degradation.
References
Barth S, Danielle Glick and Kay F Macleod, 2010. Autophagy: assays and artifacts. Journal of Pathology, 221, 117–124.
Bartels T, Choi JG, Selkoe DJ, 2011. “α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation”. Nature, 477, 107–110.
Bauvy C, Meijer AJ, Codogno P, 2009. Assaying of autophagic protein degradation. Methods in Enzymology, 452, 47–61.
Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P, 2012. “From α‐synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson's disease.” Brain Research, 1476, 183–202.
Bence NF, Sampat RM, Kopito RR, 2001. Impairment of the ubiquitin–proteasome system by protein aggregation. Science, 292, 1552–1555.
Ciechanover A, 1998. The ubiquitin‐proteasome pathway: on protein death and cell life. European Molecular Biology Organization (EMBO), 17, 7151–7160.
Ciechanover A, Brundin P, 2003. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron, 427–446.
Cookson MR, 2005. “The biochemistry of Parkinson's disease.” Annual Review of Biochemistry, 74, 29–52.
Cuervo AM, 2004. “Autophagy: many paths to the same end.” Molecular and Cellular Biochemistry, 263, pp. 55–72.
Exner N, Lutz AK, Haass C, Winklhofer KF, 2012. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. European Molecular Biology Organization (EMBO), 31, 3038–3062.
Esteves AR, Arduíno DM, Silva DF, Oliveira CR, Cardoso SM, 2011. Mitochondrial dysfunction: the road to alpha‐synuclein oligomerization in PD. Journal of Parkinson's Disease, 693761.
Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M, Jäättelä M, 2001. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. Journal of Cell Biology, 153, 999–1010.
Games D, Seubert P, Rockenstein E. “Axonopathy in an α‐synuclein transgenic model of Lewy body disease is associated with extensive accumulation of c‐terminal‐truncated α‐synuclein.” American Journal of Pathology, 182, pp. 940–953.
Grafstein B, Forman DS, 1980. Intracellular transport in neurons. Physiological Reviews Published, 60.
Hardy J Rideout, Kristin E Larsen, David Sulzer and Leonidas Stefanis, 2001. Proteasomal inhibition leads to formation of ubiquitin/a‐synuclein‐immunoreactive inclusions in PC12 cells. Journal of Neurochemistry, 78, 899–908.
Hunn BH, Cragg, SJ, Bolam JP, Spillantini MG, Wade‐Martins R, 2015. “Impaired intracellular trafficking defines early Parkinson's disease.” Trends in Journal of Neurosciences, 38, 178–188.
Ii K, Ito H, Tanaka K, Hirano A, 1997. Immunocytochemical co‐localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. Journal of Neuropathology & Experimental Neurology, 56, 125–131.
Jakobs S, 2006. High resolution imaging of live mitochondria. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1763, 561–575.
Kadowaki M, Karim MR, 2009. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods in Enzymology, 452, 199–213. [PubMed]
Kisselev AF, Goldberg AL, 2005. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods in Enzymology, 398, 364–378.
Klionsky DJ, Ana Maria C, Per OS, 2007. Methods for monitoring autophagy from yeast to human. Autophagy, 3, 181–206.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy, 4, 151–175.
Korolchuk VI, Menzies FM, Rubinsztein DC, 2010. Mechanisms of cross‐talk between the ubiquitin–proteasome and autophagy–lysosome systems. Federation of the European Biochemical Societies's Letters (FEBS), 584, 1393–1398.
Kroemer G, Mariño G, Levine B, 2010. Autophagy and the integrated stress response. Journal of Molecular and Cellular Cardiology, 40, 280–293.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, 2004. The role of autophagy during the early neonatal starvation period. Nature, 432, 1032–1036.
Lee J, Giordano S, Zhang J, 2012. “Autophagy, mitochondria and oxidative stress: cross‐talk and redox signalling”. Biochemical Journal, 441, 523–540.
Lee FK, Wong AK, Lee YW, Wan OW, Chan HY, Chung KK, 2009. The role of ubiquitin linkages on alpha‐synuclein induced‐toxicity in a Drosophila model of Parkinson's disease. Journal of Neurochemistry, 110, 208–219.
McNaught KS and Jenner P, (2001) Proteasomal function is impaired in substantia nigra in Parkinson's disease. Journal of Neuroscience Letters 297, 191–194.
Mizushima N, 2008. Autophagy fights disease through cellular self‐digestion. Nature, 451, 1069–75. Review.
Munafó DB, Colombo MI, 2002. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP‐Rab24. Traffic, 3, 472–482.
Ortega Z, Lucas JJ, 2014. Ubiquitin–proteasome system involvement in Huntington's disease. Frontiers in Molecular Journal of Neuroscience, 7, 77.
Pool M, Rippstein P, Mcbride H, Kothary R, 2006. Trafficking of macromolecules and organelles in cultured dystonia musculorum sensory neurons is normal. Journal of Comparative Neurology, 494, 549–558.
Rajapakshe AR, Podyma‐Inoue KA, Terasawa K, Hasegawa K, Namba T, Kumei Y, Yanagishita M, Hara‐Yokoyama M, 2015. Lysosome‐associated membrane proteins (LAMPs) regulate intracellular positioning of mitochondria in MC3T3‐E1 cells. Experimental Cell Research, 331, 211–222. doi: 10.1016/j.yexcr.2014.09.014
Ravikumar B, Sarkar S, Davies JE, 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological Reviews, 90, 1383–1435. doi: 10.1152/physrev.00030.2009
Rothermel BA, Hill JA, 2007. Myocyte autophagy in heart disease: friend or foe? Autophagy, 3, 632–634.
Rideout HJ, Larsen KE, Sulzer D, Stefanis L, 2001. Proteasomal inhibition leads to formation of ubiquitin/a‐synuclein‐immunoreactive inclusions in PC12 cells. Journal of Neurochemistry, 78, 899–908.
Schildknecht S, Karreman C, Pöltl D, Efrémova L, Kullmann C, Gutbier S, Krug A, Scholz D, Gerding HR, Leist M, 2013. Generation of genetically‐modified human differentiated cells for toxicological tests and the study of neurodegenerative diseases. Alternatives to Animal Experimentation (ALTEX), 30, 427–444.
Schwarz TL, 2013. Mitochondrial trafficking in neurons. Cold Spring Harbor Perspectives in Biology, 5, a011304.
Sheng ZH, 2014. Mitochondrial trafficking and anchoring in neurons: new insight and implications. Journal of Cell Biology, 204, 1087–1098.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R and Goedert M, 1997. Alpha‐synuclein in Lewy bodies. Nature, 388, 839–840.
Sulzer D, Zecca L, 2000. Intraneuronal dopamine‐quinone synthesis: a review. Neurotoxicity Research, 1, 181–195.
Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W, 2012. Quantitative imaging of energy expenditure in human brain. Neuroimage, 60, 2107–17.
A.5. KE4: Degeneration of dopaminergic neurons of the nigrostriatal pathway
A.5.1. How this Key Event works
Degeneration of dopaminergic neurons (DA neurons) within the Substantia Nigra pars compacta (SNpc), i.e. the nigrostriatal pathway, paralleled by the formation of cytoplasmic fibrillar inclusions called Lewy bodies (LB), is regarded as a key event in Parkinson's disease (PD) and is in a quantitative manner directly linked to the occurrence of clinical signs indicative of PD, i.e. impaired motor behaviour (Dauer et al., 2003; Jellinger et al., 2009; Shulman et al., 2011; Dickinson, 2012). The severity of the clinical signs correlates with the degree of nigral cell loss, and the reduced level of dopamine in the striatum. It is estimated that at the onset of clinical signs, 60% of SNpc neurons are lost, corresponding to an 80% depletion of striatal dopamine (Jellinger et al., 2009).
PD is clinically and pathologically defined as a progressive disorder: There is a temporally progress, according to a specific pattern, from the brain stem to the nigrostriatal areas and to cortical locations (Braak et al., 2004, 2009) and there is a temporal increase in the occurrence of Lewy bodies, of dopamine depletion in the striatum and of loss of DA neurons in the SNpc (Shulman et al., 2012). Indeed, in patients dying with PD there is a more evident loss of dopamine in striatum compared to SNpc, indicating that striatal dopaminergic nerve terminals are the primary target of the degenerative process in the nigrostriatal pathway and that neuronal loss in SNpc would result as a final outcome (Bernhaimer et al., 1973; Hornykiewicz et al., 1966; Dauer et al., 2003; Pavese et al., 2009). Post‐mortem studies in PD patients and experimental models are also suggesting that progression from striatal terminal to loss of DA neurons occurs through a ‘dying back’ axonopathy pathology and that axonal dysfunction may be an important hallmark in PD (Raff et al., 2002, Orimo et al., 2005, O'Malley 2010, Kim‐Han et al., 2011).
In human brain, the classical Lewy body (LB) is characterised at light microscopy by eosinophilic, spherical, intracytoplasmic inclusion and it stains for α‐synuclein and ubiqutin proteins which form the ultrastructural fibrillar core of LB visible at transmission electron microscopy. On autopsy, from individuals affected by PD, accumulation of aggregates positive for α‐synuclein protein are also observed within neuronal processes, called Lewy neurites, as well as by neurons showing a more diffuse or granular perinuclear pattern (Dickson, 2012). Because dopaminergic cells are rich in melanin, their loss is detectable by depigmentation of the midbrain at gross pathology examination (Shulman et al., 2010; Dickson 2012). However, it should be noted that, although LB are recognised as characteristic of PD, they are not found in a minority of clinically defined PD cases (Dauer, 2003) and they can also be observed in other diseases (Dickson, 2012).
The biological function of the nigrostriatal pathway depends on the intactness of its anatomical structure. Preservation of the striatum terminals and of neuronal cell bodies of DA neurons in the SNpc is a prerequisite for the maintenance of the physiological function (Fujita et al., 2014). The nigrostriatal system is anatomically located in the basal ganglia circuit which comprises the motor system structures caudate nucleus, putamen, globus pallidum and substantia nigra. The caudate nucleus and the putamen are collectively called striatum (David Robinson in: Neurobiology, Springer edition, 1997). The system plays a unique integrative role in the control of movement as part of a system called the ‘basal ganglia motor loop’. This anatomical loop includes structures in the thalamus, motor and somatosensory cortex and wide regions of surrounding cortex. Neurons of the SN produce dopamine (DA) and project to the striatum. They give dopaminergic excitatory (D1 receptors) and inhibitory (D2 receptors) inputs to striatal interneurons (GABAergic). These control thalamic output to the motor cortex. Degeneration within the SNpc leads to a decreased thalamic activation of the motor cortex (Shulman et al., 2011).
The dopaminergic cells localised in the SNpc synthesise the transmitter substance dopamine (DA) and make extensive contacts within the caudate and putamen (the striatum). These DA neurons have a complex morphology and high energy demand. They are provided with very long and dense arborisations projecting into the striatum where DA is released. This unique morphological characteristics demand a high level of energy to maintain the activity at the synaptic level, to compensate for the risk of depolarisation of the unmyelinated fibres and to support a long distance axonal transport. This puts a tremendous burden on mitochondrial functions (Pissadaki et al., 2013). SNpc neurons are provided with specific calcium channels, the L‐type Cav 1.3 which are intended to regulate the autonomous firing as ‘pacemaker’. The high demand of calcium buffering arising from this is handled by the endoplasmic reticulum (ER) and by the mitochondria. This is a function specific for SNpc DA neurons, as the dopaminergic neurons belonging to the ventral tegmental area (VTA) are using Na+ channels as a pacemaker. Additional peculiarities of the neurons of the nigrostriatal pathway are the high number of synapses and the higher probability of these neurons to accumulate misfolded proteins, including α‐synuclein. Furthermore, the nigrostriatal metabolic pathway of DA is known to induce oxidative and nitrative stress (Asanuma et al., 2003, Cantuti‐Castelvetri et al., 2003, Pissadaki et al., 2013; Fujita et al., 2014) making DA neurons particularly sensitive to oxidative stress (Lotharius and Brundin, 2002). DA neurons in SNpc also have a relatively low mitochondria mass which may contribute to the vulnerability of these neurons (Liang et al., 2007). In addition, increased levels of iron have been observed in SN of PD patients (Gotz et al., 2004) and the high content of iron in dopamine neurons has been reported to trigger oxidative/nitrosative stress and subsequent neurodegeneration (Benshachar et al., 1991; Ayton and Lei 2014). As a consequence, these neurons are particularly sensitive to various stressors that can contribute to their preferential loss (Fujita et al., 2014).
A.5.2. How it is measured or detected
The presence of DA cells in the SNpc and DA terminals in the striatum can be visualised using different phenotypic histological markers. Changes can be captured by measurement of markers specific for dopaminergic neurons such as tyrosine hydroxylase (TH), dopamine transporter (DAT) and vesicular monoamine transporter type 2 (VMAT2). Degenerating and/or degenerated neurons can be detected by the silver stains and the Fluoro‐Jade stains.
The silver degeneration stain is considered as the best method to trace degeneration of axons. By this matter, products from disintegrated cells are visualised (Betarbet et al., 2000; Switzer, 2000). Evaluation of neuronal degenerative changes in the SNpc and in the striatum is complex and should be performed by a detailed neuropathological evaluation complemented by unbias stereological count of neurons and by the evaluation of neurochemistry endpoints of toxicity, i.e. DA, DOPAC and HVA. Neuropathology and stereological counts should be done using a ‘blind’ protocol for all the samples from treated animals (Beckenridge et al., 2013; Smeyne et al., 2016)
Fluoro Jade stain is a fluorochrome derived from fluorescein used in neuroscience disciplines to label degenerating neurons. It is an alternative technique to traditional methods for labelling degenerating neurons such as silver degeneration staining. Fluoro‐Jade may be preferred to other degeneration stains due to the simplicity of staining procedures, which are a common drawback of conventional stains. However, the mechanism by which Fluoro‐Jade labels degenerating neurons is unknown (Schmued et al., 1997; Betarbet et al., 2000).
Detection of TH, the enzyme responsible for catalysing the conversion of the amino acid L‐tyrosine to L‐3,4‐dihydroxyphenylalanine (L‐DOPA), a precursor for dopamine. Detection of TH can be done either by immunocytochemistry (at the protein level) followed by cell counting (quantitative evaluation) or by western blot followed by densitometry analysis (Lee 1987; Fetissov 1999; Betarbet et al., 2000).
Counting of cells, immunostained for TH, or counting of nuclei by e.g. with Nissel's, DAPI (Kapuscinski, 1995) or Hoechst stain (Latt et al., 1976) should be done following standard morphometric protocols. However, inclusion of stereological cell counts to assess neurodegeneration represents the most sensitive method to confirm quantitatively this specific morphological change (Brooks, 1992; Thiruchelvam, 2000a,b; Dauer, 2008; Baquet, 2009; Tapias et al., 2013; Smyne, 2016).
Quantification of dopaminergic neurons in SNpc: the average number of DA neurons in adult mouse SN is approximately 8.000 to 14.000, depending on strain (Zaborszky and Vadasz, 2001). Their distribution is not homogeneous with difference in density between the caudal and rostral part of the SN. The gold standard for counting neurons is then to use an unbiased stereological protocol for cell counting with an optical dissector system (Tieu et al., 2003). This requires a computerised stereology software. The count should include TH+ neurons as well the total count of neurons using a non‐specific cell stain (e.g. Nisell's, Fox3).
Quantification of dopaminergic terminals in the striatum: the density of dopaminergic terminals is not homogeneous in the striatum, increasing from the rostral to the caudal part and representative regions of the striatum should be assessed. This can be done by digitalisation of the fibres and quantification by optical density or quantification of the fibre density identified by TH+ immunoreactivity (Tieu et al., 2003; Fernagut et al., 2007). Alternatively, striatal tissue can be isolated for immunoblotting of TH or DAT.
DA transporters (DAT) and vesicular monoamine transporter type 2 (VMAT2) can be visualised and quantified using immunocytochemistry (single cell levels) or western blot followed by densitometry analysis, to quantify the changes in their expression (Ciliax et al., 1995; Fornai et al., 2003; Hirata et al., 2007; Tong et al., 2011).
DA, DOPAC (DA metabolite) and HVA (homovanillic acid), formed from dopamine that escapes conversion to noradrenaline in noradrenergic neurons throughout the body as well as from dopamine synthesised in dopaminergic neurons that are mainly in brain(Kopin et al., 1988)) content in the striatum can be quantified through several methodologies such as capillary electrophoresis, spectrofluorimetry and high performance liquid chromatography (HPLC). The commonly used detectors for chromatography include MS, UV, optical fibre detector, electrochemical detector and fluorescence detector (Magnusson et al., 1980; Fornai et al., 2005; Zhao et al., 2011).
Identification of LB in standard histological sections stained with haematoxylin and eosin, they are characterised by the presence of pale eosinophilic vacuoles (Pappolla, 1988; Dale, 1992; Betarbet, 2000,2006).
Immunostaining for α‐synuclein and ubiquitin to identify and quantify Lewy bodies presence. In vivo, α‐synuclein and ubiqutin can be evaluated in the fixed tissue and quantified for fluorescence intensity (Kuzuhara, 1988; Tiller‐Borcich, 1988; Galloway, 1992; Forno, 1996; Betarbet, 2000,2006; Kuusisto, 2003).
Imaging techniques: 18‐fluorodopa positron emission tomography (PET) quantification of various dopamine presynaptic markers (e.g. dopamine transporter DAT, vesicular monoamine transporter type 2 VMAT2) identified by single photon emission tomography (SPECT). They permit to visualise the loss of nigrostriatal DA neurons in patients (Shapira, 2013).
A.5.3. Evidence Supporting Taxonomic Applicability
Parkinson's disease (PD) is a progressive age‐related human neurodegenerative disease with a multi‐factorial pathogenesis implicating various genetic and environmental factors and is more prevalent in males (Fujita et al., 2014). However, the anatomy and function of the nigrostriatal pathway is conserved across mammalian species (Barron et al., 2010). Pathological changes, similar to the one described in PD, have been reproduced with chemicals such as rotenone and MPTP. These chemicals have been tested successfully in primates and mice. The mouse C57BL/6 strain is the most frequently used strain in the reported experiments. A difference in vulnerability was observed, particularly for rats, depending on the strain and route of administration. The Lewis strain gives more consistency in terms of sensitivity when compared to the Sprague–Dawley. A genetic‐based susceptibility has been also described for mice following paraquat treatment, underlying the relevance of the genetic component in Parkinsonism syndromes with the C57BL/6J strain resulting the more susceptible (Yin et al., 2011; Jiao et al., 2014). In addition to rodents, the pesticide rotenone has been also studied in C. elegans, Drosophila, zebrafish and Lymnaea stagnalis (Johnson et al., 2015).
References
Ayton S, Lei P, 2014. Nigral iron elevation is an invariable feature of Parkinson's disease and is a sufficient cause of neurodegeneration. BioMed Research International, 1–9.
Asanuma M, Miyazaki I, Ogawa N, 2003. Dopamine or L‐DOPA‐induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson's disease. Neurotoxicity Research, 165–176.
Baquet ZC, Williams D, Brody J, Smeyne RJ, 2009. A comparison of model‐based (2D) and design‐based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Journal of Neuroscience, 161, 1082–1090.
Barron AB, Søvik E, Cornish JL, 2010. The roles of dopamine and related compounds in reward‐seeking behavior across animal phyla”. Frontiers in Behavioral Journal of Neuroscience, 4, 163.
Ben‐Shachar D, Youdim MBH, 1991. Intranigral iron injection induces behavioral and biochemical “Parkinsonism” in rat. Journal of Neurochemistry, 57, 2133–2135.
Bernhaimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F, 1973. Brain dopamine and the syndrome of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. Journal of the Neurological Sciences, 415–455.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Betarbet R, Canet‐Aviles RM, Sherer TB, Mastroberardino PG, Mc Lendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinfelter G, Cookson MR, Greenmyre JT, 2006. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ‐1, α‐synuclein, and the ubiquitin‐proteasome system. Neurobiology Disease, 404–420.
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K, 2004. Stages in the development of Parkinson's disease‐related pathology. Cell and Tissue Research, 121–124.
Braak H, Del Tredici K, 2009. Neuroanatomy and pathology of sporadic Parkinson's disease. Advances in Anatonomy Embryology Cell Biology, 1–119.
Brooks AI, Chadwick CA, Gelbard HA, Cory‐Slechta DA, Federoff HJ, 1999. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Research, 1–10.
Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, Cook AR, Botham PA, Smith LL, 2013. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology, 37, 1–14. doi: 10.1016/j.neuro.2013.03.005
Canuti‐Silvestri I, Shukitt‐Hale B, Joseph JA, 2003. Dopamine neurotoxicity: age dependent behavioural and histological effects. Neurobiology of Aging, 697–6.
Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN, 1992. Relationships between Lewy bodies and pale bodies in Parkinson's disease. Acta Neuropathologica, 83, 525–529.
Dauer W, Przerdborski S, 2003. Parkinson's disease: mechanisms and models. Neuron 39, 889–909.
Dickinson D, 2012. Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harbor Perspectives in Medicine, 2, a009258.
Efremova L, Scildknecht S, Adam M, Pape R, Gutbier S, Hanf B, Burkle A, Leist M, 2015. Prevention of the degeneration of dopaminergic neurons in an astrocyte co‐culture system allowing endogenous drug metabolism. British Journal of Pharmacology, 172, 4119–4132.
Fetissov SO, Marsais F, 1998. Combination of immunohistochemical and in situ hybridization methods to reveal tyrosine hydroxylase and oxytocin and vasopressin mRNA in magnocellular neurons of obese Zucker rats. Brain Research Protocols, 4, 36–43.
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti C, Ruffoli R, Soldani P, Ruggieri S, Alessandri’ MG, Paparelli A, 2003. Fine structure and mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. The Journal of Neuroscience, 23, 8955–8956.
Forno LS, DeLanney LE, Irwin I, Langston JW, 1992. Electron microscopy of lewy bodies in the amigdala‐parahippocampal region. Comparison with inclusion bodies in the MPTP‐treated squirrel monkey. Advances in Neurology, 217–218.
Forno LS, 1969. Concentric hyaline intraneuronal inclusion of Lewy type in brains of elderly person (50 incident cases): relationship to parkinsonism. Journal of the American Geriatrics Society, 557–575.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Galloway PG, Mulvihill P, Perry G, 1992. Filaments of Lewy bodies contain insoluble cytoskeletal elements. American Journal of Pathology, 809–822.
Gotz ME, Double K, Gerlach M, Youdim MBH, Riederer P, 2004. The relevance of iron in the pathogenesis of Parkinson's disease. Annals of the New York Academy of Sciences, 193–198.
Hirata Y, Suzuno S, Tsuruta T, OH‐hashi K, Kiuchi K, 2008. The role of dopamine transporter in selective toxicity of manganese and rotenone. Toxicology, 249–256.
Hornykiewicz O, Kish SJ, 1987. Biochemical pathophysiology of Parkinson's disease. In Parkinson's Disease. Yahr M and Bergmann KJ, Energy Dispersive Spectroscopy. Raven Press, New York, pp. 19–34.
Kapuscinski J, 1995. Biotechnic and histochemistry, 70, 220–3.
Kim‐Han JS, Dorsey JA, O'Malley KL, 2011. The parkinsonian mimetic MPP+, specifically impairs mitochondrial transport in dopamine axons. The Journal of Neuroscience, 31, 7212–7221.
Kopin IJ, Bankiewicz KS, Harvey‐White J, 1988. Assessment of brain dopamine metabolism from plasma HVA and MHPG during debrisoquin treatment: validation in monkeys treated with MPTP. Neuropsychopharmacology, 1, 119–125.
Kuusisto E, Parkkinen L, Alafuzoff I, 2003. Morphogenesis of Lewy bodies: dissimilar incorporation of α‐synuclein, ubiquitin and 62. Journal of Neuropathology & Experimental Neurology, 62, 1241–1253.
Kuzuhara S, mori H, Izumiyama N, Yoshimura M, Ihara Y, 1988. Lewy bodies are ubiquinated. A light and electron microscopic immunocytochemical study. Acta Neuropathologica, 75, 345–353.
Jellinger KA, 2009. A critical evaluation of current staging of α‐synuclein pathology in Lewy body disorders. Biochemica and Biophysica Acta, 730–740.
Johnson ME, Bobrovskaya L, 2015. An update on the rotenone models of Parkinson's disease: Their ability to reproduce features of clinical disease and model gene‐environment interactions, 946, 101–116.
Latt SA, Stetten G, Juergens LA, Willard HF, Scher CD, 1975. Recent developments in the detection of deoxyribonucleic acid synthesis by 33258 Hoechst fluorescence. The Journal of Histochemistry and Cytochemistry, 23, 493–505.
Liang CL, Wang TT, Luby‐Phelps K, German DC, 2007. Mitochondria mass is low in mouse substantia nigra dopamine neurons: implication for Parkinson's disease. Experimental Neurology, 203, 370–380.
Lotharius J, Brundin P, 2002. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha‐synuclein. Nature Reviews Journal of Neuroscience, 3, 932–942.
Magnusson O, Nilsson LB, Westerlund D, 1980. Simultaneous determination of dopamine, DOPAC, and homovanilic acid. Direct injection of supernatants from the brain tissue homogenates in a liquid chromatography‐electrochemical detection system. Journal of Chromatography, 221, 237–247.
Minnema DJ, Travis KZ, Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck MJ, Mathews JM, Tisdel MO, Cook AR, Botham PA, Smith LL, 2014. Dietary administration of paraquat for 13 weeks does not result in a loss of dopaminergic neurons in the substantia nigra of C57BL/6J mice. Regulatory Toxicology and Pharmacology, 68, 250–258.
O'Malley KL, 2010. The role of axonopathy in Parkinson's disease. Experimental Neurobiology, 19, 115–119.
Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H, 2005. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathologica, 109, 583–588.
Ossowska K, Wardas J, Smialowska M, Kuter K, Lenda T, Wieronska JM, Zieba B, Nowak P, Dabrowska J, Bortel A, Kwiecinski A, Wolfarth S, 2005. A slowly developing dysfunction of dopaminergiv nigrostriatal neurons induced by long‐term paraquat administration in rats: an animal model of preclinical stages of Parkinson's disease? European Journal of Neurosciences, 22, 1294–1304.
Pappolla MA, Shank DL, Alzofon J, Dudley AW, 1988. Colloid (hyaline) inclusion bodies in the central nervous system: their presence in the substantia nigra is diagnostic of Parkinson's disease. Human Pathology, 19, 27–31.
Pavese N, Brooks DJ, 2009. Imaging neurodegeneration in Parkinson's disease. Biochimica and Biophysica Acta, 1792, 722–729.
Pissadaki EK, Bolam JP, 2013. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Frontiers in Computational Neuroscience, 7, 1–17.
Raff MC, Whitemore AV, Finn JT, 2002. Axonal self‐destruction and neurodegeneration. Science, 296, 868–871.
Robinson D, 1997. Neurobiology. Springer‐Verlag. pp. 245–247.
Schapira AHV, 2013. Recent developments in biomarkers in Parkinson disease. Neurology, 26, 395–400.
Shmued LC, Albertson C, Sikker W, 1997. Fluoro‐jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Research, 751, 37–46.
Smeyne RJ, Breckenridge CB, Beck M, Jiao Y, Butt MT, Wolf J, Zadory D, Minnema D, Sturgess NC, Travis KZ, Cook AR, Smith LL, Botham PA, 2016. Assessment of the effects of MPTP and Paraquat on dopaminergic neurons and microglia in the substantia nigra pars compacta of C57BL/6 mice. Public Library of Science (PLOS ONE). doi:10.1371/journal.pone0164094
Shulman JM, DeJager PL, Feany MB, 2011. Parkinson's disease: genetics and pathogenesis. Annual Review of Pathology Mechanisms of Disease, 6, 193–222.
Switzer RC, 2000. Application of silver degeneration stains for neurotoxicity testing. Toxicologic Pathology, 28, 70–83.
Tapias V, Greenamyre JT, Watkins SC, 2013. Automated imaging system for fast quantification of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiology of Disease, 54, 158–168.
Thiruchelvam M, Brockel BJ, Richfield EK, Bags RB, Cory‐Slechta DA, 2000a. Potential and preferential effects of combined paraquat and maneb on nigrostriatal dopamine system: environmental risk factor for Parkinson's disease? Brian Research, 873, 225–234.
Thiruchelvam M, Richfield EK, Bags RB, Tank AW, Cory‐Slechta DA, 2000b. The nigrostriatal dopaminergic system as a potential target of repeated exposures to combined paraquat and maneb: implication for Parkinson's disease. Journal of Neuroscience, 20, 9207–9214.
Tong J, Boileau I, Furukawa Y, Chang LJ, Wilson AA, Houle S, Kish S, 2011. Distribution of vesicular monoamine transporter 2 protein in human brain: implications for brain imaging studies. Journal of Cerebral Blood Flow & Metabolism, 31, 2065–2075.
Tiller‐Borcich JK, Forno LS, 1988. Parkinson's disease and dementia with neuronal inclusions in the cerebral cortex: Lewy bodies or Pick bodies. Journal of Neuropathology & Experimental Neurology, 47, 526–535.
Zhao HX, Mu H, Bai YH, Hu Y, Hu YM, 2011. A rapid method for the determination in porcine muscle by pre‐column derivatization and HPLC with fluorescence detection. Journal of Pharmaceutical Analysis, 1, 208–212.
A.6. KE5: Neuroinflammation (ENV/JM/WRPR(2016)34; 2016)
A.6.1. How this KE works
Neuroinflammation or brain inflammation differs from peripheral inflammation in that the vascular response and the role of peripheral bone marrow‐derived cells are less conspicuous. The most‐easily detectable feature of neuroinflammation is the activation of microglial cells and astrocytes. It is evidenced by changes in shape, increased expression of certain antigens, and accumulation and proliferation of these glial cells in affected regions (Graeber and Streit, 1990; Aschner, 1998; Streit et al., 1999; Monnet‐Tschudi et al., 2007; Kraft and Harry, 2011; Claycomb et al., 2013). Upon stimulation by cytokines or inflammogens (e.g. from pathogens or from damaged neurons), both glial cell types activate inflammatory signalling pathways, which result in increased expression and/or release of inflammatory mediators such as cytokines, eicosanoids and metalloproteinases (Dong and Benveniste, 2001), as well as in the production of reactive oxygen (ROS) and nitrogen species (RNS) (Brown and Bal‐Price, 2003). Different types of activation states are possible for microglia and astrocytes, resulting in different responses concerning pro‐inflammatory/anti‐inflammatory signalling and other cellular functions (such as phagocytosis) (Streit et al., 1999; Nakajima and Kohsaka, 2004).
Therefore, neuroinflammation can have both neuroprotective/neuroreparative and neurodegenerative consequences (Carson et al., 2006; Monnet‐Tschudi et al., 2007; Glass et al., 2010; Aguzzi et al., 2013). Under normal physiological conditions, microglial cells scan the nervous system for neural integrity (Nimmerjahn et al., 2005) and for invading pathogens (Kreutzberg, 1995, 1996; Aloisi, 2001; Rivest, 2009). They are the first type of cell activated (first line of defence), and can subsequently lead to astrocyte activation (Falsig, 2008). Two distinct states of microglial activation have been described (Gordon, 2003; Ponomarev et al., 2005; Maresz et al., 2008; Mosser and Edwards, 2008; Kigerl et al., 2009; Perego et al., 2011): The M1 state is classically triggered by interferon‐gamma and/or other pro‐inflammatory cytokines, and this state is characterised by increased expression of integrin alpha M (Itgam) and CD86, as well as the release of pro‐inflammatory cytokines (TNF‐alpha, IL‐1beta, IL‐6), and it is mostly associated with neurodegeneration. The M2 state is triggered by IL‐4 and IL‐13 (Ponomarev et al., 2007; Maresz et al., 2008; Perego et al., 2011) and induces the expression of mannose receptor 1 (MRC1), arginase 1 (Arg 1) and Ym1/2; it is involved in repair processes. The activation of astrocytes by microglia‐derived cytokines or TLR agonists resembles the microglial M1 state (Falsig, 2006).
A.6.2. How it is measured or detected
Neuroinflammation, i.e. the activation of glial cells can be measured by quantification of cellular markers (most commonly), or of released mediators (less common). As multiple activation states exist for the two main cell types involved, it is necessary to measure several markers of neuroinflammation:
Microglial activation can be detected based on the increased numbers of labelled microglia per volume element of brain tissue (due to increase of binding sites, proliferation, and immigration of cells). A specific microglial marker, used across different species, is CD11b. Alternatively, various specific carbohydrate structures can be stained by lectins (e.g. IB4). Beyond that, various well‐established antibodies are available to detect microglia in mouse tissue (F4/80), phagocytic microglia in rat tissue (ED1) or more generally microglia across species (Iba1). Transgenic mice are available with fluorescent proteins under the control of the CD11b promoter to easily quantify microglia without need for specific stains.
The most frequently used astrocyte marker is GFAP (99% of all studies) (Eng et al., 2000). This protein is highly specific for astrocytes in the brain, and good clinically validated antibodies are available for immunocytochemical detection. In neuroinflammatory brain regions, the stain becomes more prominent, due to an upregulation of the protein, a shape change/proliferation of the cells, or better accessibility of the antibody. Various histological quantification approaches can be used. Occasionally, alternative astrocytic markers, such as vimentin of the S100beta protein have been used for staining of astrocytes (Struzynska et al., 2007).
All immunocytochemical methods can also be applied to cell culture models.
In patients, microglial accumulation can be monitored by PET imaging, using [11C]‐PK 11195 as microglial marker (Banati et al., 2002).
Activation of glial cells can be assessed in tissue or cell culture models also by quantification of sets of activation markers. This can for instance be done by PCR quantification of inflammatory factors, of by measurement of the respective mediators, e.g. by ELISA‐related immunoquantification. Such markers include:
Pro‐ and anti‐inflammatory cytokine expression (IL‐1β; TNF‐α, Il‐6, IL‐4); or expression of immunostimmulatory proteins (e.g. MHC‐II)
Itgam, CD86 expression as markers of M1 microglial phenotype
Arg1, MRC1, as markers of M2 microglial phenotype
(for description of techniques, see Falsig, 2004; Lund, 2006; Kuegler, 2010; Monnet‐Tschudi et al., 2011; Sandstrom von Tobel et al., 2014; von Tobel et al., 2014).
A.6.3. Evidence supporting taxonomic applicability
Neuroinflammation is observed in human, monkey, rat, mouse, and zebrafish, in association with neurodegeneration or following toxicant exposure. Some references (non‐exhaustive list) are given below for illustration:
In human: Vennetti et al., 2006 in monkey (Macaca fascicularis): Charleston et al., 1994, 1996 in rat: Little et al., 2012; Zurich et al., 2002; Eskes et al., 2002 in mouse: Liu et al., 2012 in zebrafish: Xu et al., 2014.
6.4. Regulatory examples using the KE
Measurement of glial fibrillary acidic protein (GFAP), whose increase is a marker of astrocyte reactivity, is required by the US EPA for fuel additives (40 CFR 79.67), but is optional for other toxicant evaluation.
References
Aschner M, 1998. Immune and inflammatory responses in the CNS: modulation by astrocytes. Toxicology Letters, 103, 283–287.
Banati, RB, 2002. “Visualising microglial activation in vivo.” Glia, 40, 206–217.
Brown GC, Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355.
Charleston JS, Body RL, Bolender RP, Mottet NK, Vahter ME, Burbacher TM. 1996. Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long‐term subclinical methylmercury exposure. NeuroToxicology, 17, 127–138.
Charleston JS, Bolender RP, Mottet NK, Body RL, Vahter ME, Burbacher TM, 1994. Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long‐term methyl mercury exposure. Toxicology and Applied Pharmacology, 129, 196–206.
Dong Y, Benveniste EN, 2001. Immune function of astrocytes. Glia, 36, 180–190.
ENV/JM/WRPR(2016)34, 2016. Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, Pesticides and Biotechnology. Adverse outcome pathway on ionotropic glutamatergic receptors and cognition.
Eng LF, Ghirnikar RS, Lee YL, 2000. Glial fibrillary acidic protein: GFAP‐thirty‐one years (1969–2000). Neurochemical Research, 25, 1439–1451.
Eskes C, Honegger P, Juillerat‐Jeanneret L, Monnet‐Tschudi F, 2002. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL‐6 release. Glia, 37, 43–52.
Falsig J, Latta M, Leist M, 2004. Defined inflammatory states in astrocyte cultures correlation with susceptibility towards CD95‐driven apoptosis. Journal of Neurochemistry, 88, 181–193.
Falsig J, Pörzgen P, Lund S, Schrattenholz A, Leist M, 2006. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. Journal of Neurochemistry, 96, 893–907.
Falsig J, van Beek J, Hermann C, Leist M, 2008. Molecular basis for detection of invading pathogens in the brain. Journal of Neuroscience Research, 86, 1434–1447.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH, 2010. Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934.
Gordon S, 2003. Alternative activation of macrophages. Nature Reviews Immunology, 3, 23–35.
Graeber MB, Streit WJ, 1990. Microglia: immune network in the CNS. Brain Pathology, 1, 2–5.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG, 2009. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. Journal of Neuroscience, 29, 13435–13444.
Kuegler PB, Zimmer B, Waldmann T, Baudis B, Ilmjärv S, Hescheler J, Gaughwin P, Brundin P, Mundy W, Bal‐Price AK, Schrattenholz A, Krause KH, van Thriel C, Rao MS, Kadereit S, Leist M, 2010. Markers of murine embryonic and neural stem cells, neurons and astrocytes: reference points for developmental neurotoxicity testing. Alternatives to Animal Experimentation (ALTEX). 27, 17–42.
Kreutzberg GW, 1995. Microglia, the first line of defence in brain pathologies. Arzneimttelforsch, 45, 357–360.
Kreutzberg GW, 1996. Microglia: a sensor for pathological events in the CNS. Trends in Journal of Neurosciences, 19, 312–318.
Little AR, Miller DB, Li S, Kashon ML, O'Callaghan JP, 2012. Trimethyltin‐induced neurotoxicity: gene expression pathway analysis, q‐RT‐PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicology and Teratology, 34, 72–82.
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, et al., 2012. alpha7 nicotinic acetylcholine receptor‐mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. Journal of Neuroinflammation, 9, 98.
Lund S, Christensen KV, Hedtjärn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Pörzgen P, Leist M, 2006. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. Journal of Neuroimmunology, 180, 71–87.
Maresz K, Ponomarev ED, Barteneva N, Tan Y, Mann MK, Dittel BN, 2008. IL‐13 induces the expression of the alternative activation marker Ym1 in a subset of testicular macrophages. Journal of Reproductive Immunology, 78, 140–148.
Monnet‐Tschudi F, Zurich MG, Honegger P, 2007. Neurotoxicant‐induced inflammatory response in three‐dimensional brain cell cultures. Human and Experimental Toxicology, 26, 339–346.
Monnet‐Tschudi F, Defaux A, et al., 2011. Methods to assess neuroinflammation. Current Protocols in Toxicology, Chapter 12: Unit 12 19.
Mosser DM, Edwards JP, 2008. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology, 8, 958–969.
Nakajima K, Kohsaka S, 2004. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Current Drug Targets – Cardiovascular & Hematological Disorders, 4, 65–84.
Perego C, Fumagalli S, De Simoni MG, 2011. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. Journal of Neuroinflammation, 8, 174.
Ponomarev ED, Maresz K, Tan Y, Dittel BN, 2007. CNS‐derived interleukin‐4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. Journal of Neuroscience, 27, 10714–10721.
Ponomarev ED, Shriver LP, Maresz K, Dittel BN, 2005. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of Neuroscience Research, 81, 374–389.
Sandstrom von Tobel J, Zoia D, et al., 2014. Immediate and delayed effects of subchronic Paraquat exposure during an early differentiation stage in 3D‐rat brain cell cultures. Toxicology Letters doi: 10.1016/j.toxlet.2014.02.001
Struzynska L, Dabrowska‐Bouta B, Koza K, Sulkowski G, 2007. Inflammation‐like glial response in lead‐exposed immature rat brain. Journal of Toxicological Sciences, 95, 156–162.
von Tobel JS, Antinori P, et al., 2014. Repeated exposure to Ochratoxin A generates a neuroinflammatory response, characterized by neurodegenerative M1 microglial phenotype. Neurotoxicology, 44C, 61–70.
Venneti S, Lopresti BJ, Wiley CA, 2006. The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: from pathology to imaging. Progress in Neurobiology, 80, 308–322.
Xu DP, Zhang K, Zhang ZJ, Sun YW, Guo BJ, Wang YQ, et al., 2014. A novel tetramethylpyrazine bis‐nitrone (TN‐2) protects against 6‐hydroxyldopamine‐induced neurotoxicity via modulation of the NF‐kappaB and the PKCalpha/PI3‐K/Akt pathways. Neurochemistry International, 78, 76–85.
Zurich M‐G, Eskes C, Honegger P, Bérode M, Monnet‐Tschudi F, 2002. Maturation‐dependent neurotoxicity of lead acetate in vitro: implication of glial reactions. Journal of Neuroscience Research, 70, 108–116.
A.7. Adverse Outcome: parkinsonian motor deficits
A.7.1. How this key events works
A large number of neurological disorders are characterised by a clinical syndrome with motor symptoms of bradykinesia, tremor, rigidity and postural instability. As these clinical features are common to multiple disorders, the clinical syndrome is referred as ‘parkinsonism’ and when parkinsonism is representing the prevalent part of the syndrome, these are referred as ‘parkinsonian disorders’. Parkinson's disease (PD) is one of parkinsonian disorders and can have an idiopathic, genetic or toxic (i.e. MPTP‐induced parkinsonism) cause (Dickson, 2012). The pyramidal motor system comprises bundles of neurons originating in the motor centres of the cerebral cortex to terminate in the brainstem or in the spinal cord where they are responsible for voluntary control of motor functions (Brooks, 1971). The extrapyramidal system, which is in the centre of AO, is the part of the motor system primarily involved in the control and regulation of involuntary motor control, and in fine tuning (Barnes, 1983). Especially the initiation and maintenance of complex movement patterns or of neuronal regulatory pathways involved in postural control of the body are regulated by the nigrostriatal system that is affected in parkinsonian states. The CNS input is modulated by extrapyramidal circuits before the execution of complex motor movements. The modulated information from the basal ganglia is looped back through the thalamus to the cortex, from where final motor signals are sent via the pyramidal system; i.e. the basal ganglia system is not involved in the control of motor neurons and striatal muscles, but it modulates the signals from the cortex to these systems. Thus, an impaired input of dopamine into the striatum leads to an impairment of this modulation loop, and a disturbance of basal ganglia feedback to the thalamus and cortex. This ultimately manifests in key parkinsonian symptoms such as tremor, rigidity, or bradykinesia (Bernheimer, 1973). These conditions can be generated experimentally by dopamine depletion with reserpine (Carlsson), by inhibition of dopamine receptors, by mechanical or chemical ablation of nigrostriatal dopamine neurons (cut of the median forebrain bundle or injection of the toxicant 6‐OH‐dopamine) or the application of toxicants that leading to a relatively selective death of dopaminergic neurons in the substantia nigra (e.g. MPTP) and therefore a reduction of dopamine in the striatum (Kolata, 1983).
The basal ganglia loop include the ventral striatum, the neostriatum composed of the putamen and the caudate nucleus, the globus pallidus pars externa (GPe), the globus pallidus pars interna (GPi), the subthalamic nucleus (STN), the substantia nigra pars reticulata (SNpr) and the substantia nigra pars compacta (SNpc) (Obeso, 2008). The main input sites into basal ganglia are the striatum and the STN where cortical (glutamatergic) innervations terminate in a topographically organised manner that largely reflects the organisation in the cortex (Fallon, 1978; Takada, 1998). Both the GPi and the SNpr represent the main output nuclei projecting into the thalamus (Alexander, 1990; Parent, 1999). The connection between input and output nuclei is functionally organised into a ‘direct’ and an ‘indirect’ pathway (Silverdale et al., 2003). These two pathways in parallel regulate the activity of the basal ganglia output neurons of the GPi and STN and are modulated by dopamine in the striatum. The dopaminergic terminals in the striatum originate from dopaminergic projections from the SNpc. Striatal dopamine modulates the activity of inhibitory GABAergic medium spiny neurons that make up 90% of all neurons in the striatum (Smith, 1994). Medium spiny neurons that preferentially express the D1 dopamine receptor are involved in the direct pathway and directly project into the two main output nuclei (GPi and SNpr). Activation of the D1 medium spiny neuronal direct pathway results in a reduction of the inhibitory basal ganglia output (GPi and SNpr) leading to a disinhibition of thalamic target neurons (Bolam, 2000). These events ultimately lead to an elevated activity in the respective cortical neurons, i.e. D1 signalling in the striatum leads to an increase in motor activity.
Medium spiny neurons predominantly expressing the D2 dopamine receptor mostly project to the GPe (Gerfen, 1990). Activation of D2 expressing neurons leads to an inhibition of their activity. D2 neurons of the indirect pathway connect the striatum with GPi/SNpr via synaptic connections in the GPe and the STN. Activating neurons originating in the STN project into the GPi/SNpr are glutamatergic. From the STN, activating glutamatergic neuronal projections into the GPi/SNpr lead to a basal, low activation. Activation of the indirect pathway by striatal dopamine from the substantia nigra hence leads to a low basal inhibitory GABAergic output into thalamic structures, and thus allows a strong motor cortex activation of the thalamus.
Figure A.5.
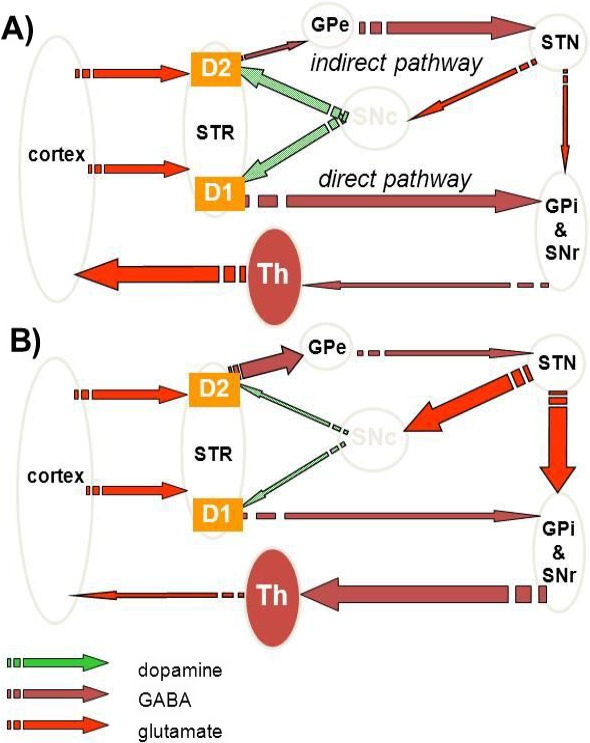
Functional anatomy of basal ganglia. (A) Normal conditions. Striatal (STR) dopamine mainly originates from projections originating in the substantia nigra pars compacta (SNc). The STR is mainly composed of inhibitory GABAergic medium spiny neurons (MSN). MSN involved in the direct pathway directly project to the globus pallidus pars interna (GPi) and the sunstantia nigra pars reticulata (SNpr) leading to a basal inhibition of these output nuclei. MSN involved in the indirect pathway send inhibitory projections to the globus pallidus pars externa (GPe). Their activity is dampened by dopamine binding to D2 receptor expressing MSN in the striatum. (B) Lack of striatal dopamine. Under conditions of a lack of striatal dopamine, inhibitory GABAergic neurons, originating in the striatum, receive less activation, resulting in a declined inhibition of GPi and SNpr inhibitory output. In the indirect pathway, the lack of dopamine causes a lack of its inhibitory influence on inhibitory GABAergic projections into the GPe. This accelerated inhibition of the GPe results in a decline in its inhibitory output into the STN. According to Silverdale et al., 2003, the decline in STN inhibition allows its overactivation, resulting in an excessive activation of stimulatory glutamatergic projections into the GPi and SNpr
Parkinson's disease is characterised by a decline in striatal dopamine input from the substantia nigra pars compacta (Smith, 1994). Under normal conditions, ganglial output via GPi/SNpr nuclei causes a moderate inhibitory influence on cortical and brainstem motor neurons. A reduction in striatal dopamine leads to an underactivation of D1 receptor‐expressing medium spiny neurons of the direct pathway. This insufficient activation of the inhibitory GABAergic medium spiny neurons results in a reduction of its normal inhibitory influence on GPi and SNpr output nuclei. As a consequence, dopamine depletion leads to the overactivation of the inhibitory GABAergic GPi/SNpr output via the direct pathway (Mitchell, 1989).
In the indirect pathway, the reduced activation of D2 receptors expressing neurons leads to an overactivation of inhibitory output nuclei projecting into the GPe. The resulting inhibitory output of the GPe is hence reduced, thus leading to a declined inhibition of the STN. Overactivation of the stimulatory glutamatergic projections originating in the STN leads to the hyperactivation of the output GPi/SNpr nuclei. As a consequence of striatal dopamine depletion, the direct pathway becomes underactivated and the indirect pathway becomes overactivated. This leads to an overactivation of the basal ganglia output nuclei. Due to their inhibitory influence on thalamocortical motor centres, the resulting reduced cortical activation leads to the prominent impairment of motor functions in parkinsonian states (Silverdale et al., 2003).
The model of direct and indirect pathways linking striatal dopamine content with the basal ganglia output nuclei has been criticised in recent years as it ignores the influence of extrastriatal dopamine (Smith 2000), or the fact that some medium spiny neurons express dopamine receptors of both the D1 and of the D2 type (Surmeier, 1996). Principal validity of the model and the central role of striatal dopamine was e.g. demonstrated by l‐DOPA‐mediated supplementation of striatal dopamine content in unprimed PD patients that causes a partial reduction in the overactivation of GPi/SNpr output (Heimer, 2006; Yuan, 2010). As an alternative way for symptomatic treatment of parkinsonian conditions, deep brain stimulation of either the STN or the GPi was shown to relieve from parkinsonian motor features (Mazzone, 2003; Odekerken, 2013).
A.7.2. How is it measured or detected
For the analysis of striatal dopamine content and its correlation with motor control, both biochemical analysis methods on the cellular and tissue level as well as behavioural tests are required. Available test models are mice and rats on the one hand and non‐human primates and humans on the other. Motor impairment features associated with parkinsonian states in man serve as reference standard. Monkey models have the advantage to largely reflect complex motor impairment patterns observed in humans which are rather difficult to assess in rodents. Rodent models in contrast are cost‐efficient and allow both biochemical analysis that require major invasive methods as well as basic behavioural tests. Due to the limitations in the assessment of moderate motor impairment in rodents and the well‐established correlation between striatal dopamine content and impaired motor output, analysis of striatal dopamine is often applied as surrogate readout for the assessment of motor deficits.
A.7.2.1. Detection of striatal dopamine (total or extracellular)
The standard method used in the majority of experimental work is the determination of total contents of dopamine and its two degradation metabolites HVA and DOPAC. For this purpose, the striatum is quickly removed from experimental animals, homogenised in a suitable acidic buffer, and the dopamine (metabolites) determined by HPLC with electrochemical detector or by HPLC‐MS.
For live in vivo detection of extracellular dopamine levels, a microdialysis probe is inserted into the striatum. Microdialysis can be performed in anesthetised animals or freely moving animals; basal dopamine levels or stimulated levels (amphetamine, KCl) can be recorded. Dopamine and its metabolites are detected in the dialysate either by HPLC or by HPLC–mass spectrometry analysis (Saraswat, 1981; Cui, 2009; Gonzalez, 2011).
A.7.2.2. Detection of dopamine neuron terminals in the striatum
As alternative to the detection of striatal dopamine that is to a large extent limited to live detection setups due to its instability in tissues, the number of remaining dopamine neurons in the substantia nigra pars compacta was suggested as alternative readout (Burns, 1983). It allows the analysis of ex vivo samples without the limitations associated with the instability and reactivity of extravesicular dopamine. Although the number of surviving dopamine neurons in the SNpc in PD or in complex‐I inhibitor challenged test animals is a valuable parameter on its own, it was discovered that the number of DA neurons in the SNpc not necessarily correlates with the amount of dopamine released in the striatum. Tyrosine hydroxylase (TH) was regularly stained as marker for DA neurons, however it was observed that TH expression was very variable following MPTP intoxication in the absence of cell death and therefore has only limited suitability for the assessment of DA neuronal numbers (Aznavour, 2012). Second, many DA neurites and terminals displayed damage or degradation in the absence of death of the corresponding neuronal cell (Ling, 2015). Hence, even in the presence of viable DA neurons in the SNpc, their corresponding terminals could no longer be able to release dopamine into the striatum. Staining of DA neuronal terminals in the striatum is therefore used as a more reliable indirect marker for striatal dopamine content. For the analysis of nigrostriatal terminals, the dopamine transporter (DAT) is visualised either by antibody‐mediated staining in tissue slices or by the application of radioactively labelled DAT ligands that allow their application both in vivo and in ex vivo samples (Morris, 1996).
A.7.2.3. Behavioural tests: Rodent models
Rotation: the rotation model of Ungerstedt et al. (1970) is based on the unilateral lesion of the nigrostriatal dopamine neuron system either in rodents or in non‐human primates. The lesion can be produced either surgically, or by stereotaxic infusion of e.g. 6‐OHDA into the nigrostriatal system of one hemisphere, or by infusion of MPTP through one carotid (single sided). After the lesion, animals are left to recover, then the dopamine system is stimulated by injection of amphetamine. The asymmetry of remaining dopamine neurons (only on one side) triggers spontaneous asymmetric motor behaviour, i.e. rotations of the animals. Each full turn of an animal is recorded, the respective numbers of left‐ and right turns are plotted versus time, respectively. In the standard rotation model, monkeys become hypokinetic in the limbs on the contralateral side of the brain hemisphere treated. Rats preferentially rotate towards the side of the lesion upon treatment with drugs that trigger activation of the remaining dopamine neurons.
Rotarod: assessment of motor coordination. The animals are placed on a rotating rod that is subjected to linear acceleration. The latency to fall from the rod is detected (Jones 1968).
Hang test: Detection of neuromuscular strength. Mice are placed on a horizontal grid. When the animals grabbed the grid with their fore‐ and hindpaws, the grid is inverted with the animal hanging upside down. In a typical setup, mice are required to remain on the grid for at least 30 s (Tillerson 2002).
Forepaw Stride length during walking: Ink is applied to the forepaws and the mice walk across a blank sheet of paper. Training of the animals to walk across the white paper in a straight line without stopping is performed before the respective treatment. The distance between single steps on each side are measured (Klapdor, 1997).
Grid test: Mice hang upside down for 30 s on the grid that is also used for the Hang test and are recorded on video for closer analysis. With this method, the average forepaw distance is measured by assessing the distance covered, divided by the number of successful forepaw steps. In the course of the analysis, the number of unsuccessful forepaw steps are detected and displayed as percentage of the total number of steps performed (Crawley, 1999).
Akinesia: the animal is placed on a flat surface and the latency until it has moved all of its four limbs is assessed.
Open field test: Infrared beams detect the animals activity for the determination of parameters such as the time spent locomoting, the distance travelled, or the number of rearings.
Pole test: the animal is placed on a gauze‐taped pole with the head upwards below the top of the pole. Two parameters are detected: 1) time until animals turn by 180°; 2) time until the animals reach the floor.
A.7.2.4. Non‐invasive imaging of DA neuron terminals
Positron emission tomography (PET): Based on its appropriate half life time of ca. 2 h for clinical investigations, fluorine‐18 labelled L‐[18F]‐fluorodopa is routinely used in trace amounts for intravenous administration. Striatal uptake of L‐[18F]‐fluorodopa is followed by applying positron emission tomography (PET) (Leenders, 1986).
Single photon emission computed tomography (SPECT): monitoring of dopamine transporter (DAT). Iodine‐123‐β‐CIT is used as a sensitive ligand for dopamine and serotonin transporters and was applied in monkeys and humans (Winogrodzka, 2003).
A.7.2.5. Human neurological tests
A recent systematic review and evaluation of currently used rating scales for the assessment of motor impairment and disability in PD patients identified the (1) Columbia University rating scale, (2) the Northwestern University Disability Scale and (3) the Unified Parkinson's Disease rating scale as the most evaluated and reliable scales available (Ramaker, 2002). All scales evaluate several parameters, some of which are not motor related. Thus, only subscales are useful for readout of motor symptoms (e.g. 13 of the 42 UPDRS parameters). Of these, not all are equally dependent on nigrostriatal dopamine. Examination needs to be done by a trained neurologist.
A.7.2.6. Regulatory examples using this Adverse Outcome
Neurotoxic effects shall be carefully addressed and reported in routine required regulatory toxicological studies (acute toxicity studies, short‐term toxicity studies, long term toxicity and carcinogenicity studies and reproductive toxicity studies). Regarding neurotoxicity in rodents, inclusion of neurotoxicity investigations in routine toxicology studies shall also be considered. For pesticide active substances the circumstances in which neurotoxicity studies should be performed are listed in Regulation (EU) No 283/2013:
Specific neurotoxicity studies in rodents shall be performed in case the following conditions:
there is indication of neurotoxicity in routine toxicity studies carried out with the active substance;
the active substance is a structurally related to known neurotoxic compound;
the active substance has a neurotoxic mode of pesticidal action.
As a result, specific neurotoxicity studies are not routinely required for all pesticide active substances. Specific neurotoxicity testing becomes obligatory only if neurotoxicity has been observed during histopathological evaluation or in case of structural analogy with a known neurotoxic compound. Motor activity should be measured once in short‐term repeated dose toxicity studies (OECD 407, 408 and 422) and several times in specific neurotoxicity studies (OECD 424, OECD 426 and cohort 2 of OECD 443). However, this is not a requirement in chronic toxicity studies unless neurotoxic effects have been reported in the shorter studies. The same test (measures horizontal and/or vertical movements in a test chamber) is implemented in both routine studies and neurotoxicity studies. Coordination and balance are evaluated by rotation or rotarod or pole tests, and gait abnormalities by forepaw stride length test. Those tests are not required by any repeated dose toxicity OECD guidelines and they can be optionally incorporated in the design of neurotoxicity studies OECD 424 and OECD 426.
Although motor deficits is the AO in this AOP, degeneration of DA neurons, is also considered an adverse effect in the regulatory framework, even in the absence of clear clinical symptoms or motor deficits. Morphological assessment of brain structures is a standard requirement in the regulatory toxicological studies supporting the risk assessment of chemical substances and it is a regulatory expectation that the anatomical structures belonging to the nigrostriatal pathway would be included and evaluated as part of the standard evaluation of the brain. Treatment related neuronal degeneration, when occurring as a consequence of the treatment, is generally dose‐dependent in incidence and severity. However, if not accompanied by clinical signs or behavioural changes indicative of central nervous system pathology, minimal loss of DA neurons would likely remain undetected in the standard histological evaluation, due to the presence of non DA neurons or as a consequence of the subjectivity of non‐quantifiable analysis, unless specific markers are used. As multiple forms of perturbation can affect the neurons, some changes are potentially still reversible (e.g. loss of TH or DA) and irreversibility should be confirmed as part of the assessment. It is then important to apply a sensitive and appropriate method (Switzer, 2000) and evaluation of the phenotypic markers in the striatum and in the SNpc should be always performed as a minimum standard (Minnema et al., 2014) when investigating perturbation of the nigrostriatal pathway. It should additionally considered that rat is likely to be a poor model to capture this kind of hazard, as demonstrated by the poor sensitivity of rat to MPTP or related compounds and this should be taken into account for the design and interpretation of the studies.
Dissimilarities of chemical‐induced animal models to human disease are also important and should be carefully weighted when considering the duration and schedule of the study/treatment. Differently from the human disease, with the MPTP animal model, the damage occurs rapidly, is hardly progressive, is little age‐dependent and formation of Lewy bodies is sometime not occurring (Efremova et al., 2015, 2016). Therefore, for different animals models, the standard 90 days toxicity study could not match with the chronic and progressive characteristics of the human disease and compensatory changes influencing DA metabolism and turnover and protein catabolism can occur during the treatment period with an impact on the time of onset of the lesion (Ossowska et al., 2005).
References
Aschner M, 1998. Immune and inflammatory responses in the CNS: modulation by astrocytes. Toxicology Letters, 103, 283–287.
Banati RB, 2002. Visualising microglial activation in vivo. Glia, 40, 206–217.
Brown GC, Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355.
Charleston JS, Body RL, Bolender RP, Mottet NK, Vahter ME, Burbacher TM. 1996. Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long‐term subclinical methylmercury exposure. NeuroToxicology, 17, 127–138.
Charleston JS, Bolender RP, Mottet NK, Body RL, Vahter ME, Burbacher TM, 1994. Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long‐term methyl mercury exposure. Toxicology and Applied Pharmacology, 129, 196–206.
Dong Y, Benveniste EN, 2001. Immune Function of Astrocytes. Glia, 36, 180–190.
Eng LF, Ghirnikar RS, Lee YL, 2000. Glial fibrillary acidic protein: GFAP‐thirty‐one years (1969–2000). Neurochemical Research, 25, 1439–1451.
Eskes C, Honegger P, Juillerat‐Jeanneret L, Monnet‐Tschudi F, 2002. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL‐6 release. Glia, 37, 43–52.
Falsig J, Latta M, Leist M, 2004. Defined inflammatory states in astrocyte cultures correlation with susceptibility towards CD95‐driven apoptosis. Journal of Neurochemistry, 88, 181–93.
Falsig J, Pörzgen P, Lund S, Schrattenholz A, Leist M, 2006. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. Journal of Neurochemistry, 96, 893–907.
Falsig J, van Beek J, Hermann C, Leist M, 2008. Molecular basis for detection of invading pathogens in the brain. Journal of Neuroscience Research, 86, 1434–1447.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH, 2010. Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934.
Gordon S, 2003. Alternative activation of macrophages. Nature Reviews Immunology, 3, 23–35.
Graeber MB, Streit WJ, 1990. Microglia: immune network in the CNS. Brain Pathology, 1, 2–5.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG, 2009. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. Journal of Neuroscience, 29, 13435–13444.
Kuegler PB, Zimmer B, Waldmann T, Baudis B, Ilmjärv S, Hescheler J, Gaughwin P, Brundin P, Mundy W, Bal‐Price AK, Schrattenholz A, Krause KH, van Thriel C, Rao MS, Kadereit S, Leist M, 2010. Markers of murine embryonic and neural stem cells, neurons and astrocytes: reference points for developmental neurotoxicity testing. Alternatives to Animal Experimentation (ALTEX), 27, 17–42.
Kreutzberg GW, 1995. Microglia, the first line of defence in brain pathologies. Arzneimttelforsch 45, 357–360.
Kreutzberg GW, 1996. Microglia: a sensor for pathological events in the CNS. Trends in Journal of Neurosciences, 19, 312–318.
Little AR, Miller DB, Li S, Kashon ML, O'Callaghan JP, 2012. Trimethyltin‐induced neurotoxicity: gene expression pathway analysis, q‐RT‐PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicology and Teratology, 34, 72–82.
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, et al., 2012. alpha7 nicotinic acetylcholine receptor‐mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. Journal of Neuroinflammation, 9, 98.
Lund S, Christensen KV, Hedtjärn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Pörzgen P, Leist M, 2006. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. Journal of Neuroimmunology, 180, 71–87.
Maresz K, Ponomarev ED, Barteneva N, Tan Y, Mann MK, Dittel BN, 2008. IL‐13 induces the expression of the alternative activation marker Ym1 in a subset of testicular macrophages. Journal of Reproductive Immunology, 78, 140–148.
Monnet‐Tschudi F, Zurich MG, Honegger P, 2007. Neurotoxicant‐induced inflammatory response in three‐dimensional brain cell cultures. Human and Experimental Toxicology, 26, 339–346.
Monnet‐Tschudi F, Defaux A, et al., 2011. Methods to assess neuroinflammation. Current Protocols in Toxicology Chapter 12: Unit12 19.
Mosser DM, Edwards JP, 2008. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology, 8, 958–969.
Nakajima K, Kohsaka S, 2004. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Current Drug Targets – Cardiovascular & Hematological Disorders, 4, 65–84.
Perego C, Fumagalli S, De Simoni MG, 2011. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. Journal of Neuroinflammation, 8, 174.
Ponomarev ED, Maresz K, Tan Y, Dittel BN, 2007. CNS‐derived interleukin‐4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. Journal of Neuroscience, 27, 10714–10721.
Ponomarev ED, Shriver LP, Maresz K, Dittel BN, 2005. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of Neuroscience Research, 81, 374–389.
Sandstrom von Tobel J, Zoia D, et al., 2014. Immediate and delayed effects of subchronic Paraquat exposure during an early differentiation stage in 3D‐rat brain cell cultures. Toxicology Letters. doi: 10.1016/j.toxlet.2014.02.001
Silverdale MA, Fox SH, Crossman AR, Brotchie JM, 2003. Potential dopaminergic drugs for Parkinson's disease. Advances in Neurology, 91:273–291.
Struzynska L, Dabrowska‐Bouta B, Koza K, Sulkowski G, 2007. Inflammation‐like glial response in lead‐exposed immature rat brain. Journal of Toxicological Sciences, 95, 156–162.
von Tobel JS, Antinori P, et al., 2014. Repeated exposure to Ochratoxin A generates a neuroinflammatory response, characterized by neurodegenerative M1 microglial phenotype. Neurotoxicology, 44C, 61–70.
Venneti S, Lopresti BJ, Wiley CA, 2006. The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: from pathology to imaging. Progress in Neurobiology, 80, 308–322.
Xu DP, Zhang K, Zhang ZJ, Sun YW, Guo BJ, Wang YQ, et al., 2014. A novel tetramethylpyrazine bis‐nitrone (TN‐2) protects against 6‐hydroxyldopamine‐induced neurotoxicity via modulation of the NF‐kappaB and the PKCalpha/PI3‐K/Akt pathways. Neurochemistry International, 78, 76–85.
Zurich M‐G, Eskes C, Honegger P, Bérode M, Monnet‐Tschudi F, 2002. Maturation‐dependent neurotoxicity of lead acetate in vitro: implication of glial reactions. Journal of Neuroscience Research 70, 108–116.
KEY EVENTS RELATIONSHIPS (KERs)
1st KER: Binding of inhibitor to NADH‐ubiquinone oxidoreductase (complex I) leads to its inhibition
A.1.1. How does this Key Event Relationship work
It is well documented that binding of an inhibitor to CI inhibits its activity (see MIE). Naturally occurring and synthetic CI inhibitors have been shown to inhibit the catalytic activity of CI, leading to partial or total inhibition of its activity in a dose response manner (Degli Esposti and Ghelli, 1994; Barrientos and Moraes, 1999; Betarbet et al., 2000; Ichimaru et al., 2008). Indeed, binding of inhibitors stops the electron flow from CI to ubiquinone. Therefore, the Fe‐S clusters of CI become highly reduced and no further electrons can be transferred from NADH to CI. This leads to the inhibition of the NADH oxidoreductase function, i.e. CI inhibition.
A.1.2. Weight of Evidence for the KER
The weight of evidence supporting the relationship between binding of an inhibitor to NADH‐ubiquinone oxidoreducatse and its inhibition is strong.
A.1.2..1.
A.1.2.1. Biological Plausibility
There is an extensive understanding of the functional relationship between binding of an inhibitor to NADH‐ubiquinone oxidoreductase (CI) and its inhibition. As the first entry complex of mitochondrial respiratory chain, CI oxidises NADH and transfers electrons via a flavin mononucleotide cofactor and several Fe‐S complexes to ubiquinone. The electron flow is coupled to the translocation of protons from the matrix to the intermembrane space. This helps to establish the electrochemical gradient that is used to fuel ATP synthesis (Greenamyre et al., 2001). If an inhibitor binds to CI, the electron transfer is blocked. This compromises ATP synthesis and maintenance of Δψm, leading to mitochondrial dysfunction. As CI exerts a higher control over oxidative phosphorylation in synaptic mitochondria than in non‐synaptic mitochondria in the brain (Davey and Clark, 1996), specific functional defects observed in PD may be explained.
It is well documented that CI inhibition is one of the main sites at which electron leakage to oxygen occurs. This results in a production of ROS, such as superoxide (Efremov and Sazanow, 2011) and hydrogen peroxide, which are main contributors to oxidative stress (Greenamyre et al., 2001).
A.1.2.2. Empirical support for linkage
A variety of studies show a significant correlation between binding of an inhibitor to CI and its inhibition, usually measured by the decreased mitochondrial respiration. Different classes of CI inhibitors, such as rotenone, MPP+, piericidin A, acetogenins, pyridaben, and various piperazin derivatives (Ichimaru et al., 2008) have been shown to bind to the ubiquitin site of CI, leading to a partial or total inhibition of oxidoreductase activity in a dose response manner (Grivennikova et al., 1997; Barrientos and Moraes, 1999; Betarbet et al., 2000).
The reduction of CI activity is well documented in a variety of studies using isolated mitochondria or cells, as well as in in vivo experiments and in human post mortem PD brains. Usually it is measured by assays described in 2nd Key Event Relationship (KER): Inhibition of complex I leads to mitochondrial dysfunction.
It has been shown that binding of rotenone to CI (e.g. Betarbet et al., 2000, Greenamyre et al., 2001) or MPP+ (e.g. Langston, 1996; Krug et al., 2014) can reproduce the anatomical, neurochemical, behavioural and neuropathological features of PD. Therefore, the empirical support for this KER will be mainly based on the experiments performed after exposure to rotenone or MPP+.
The binding of rotenone to CI resulted in time‐ and dose‐dependent inhibition of CI activity measured in submitochondrial particles. The kinetics of the active CI inhibition was determined after exposure to rotenone at 20, 30 and 40 nM at different times of exposure (30 s, 1 min or 2 min) (Grivennikova et al., 1997). This study suggests that two rotenone binding sites exist in CI: one affecting NADH oxidation by ubiquinone and the other one operating in ubiquinol‐NAD+ reductase action.
Partial inhibition of CI produces a mild, late‐onset mitochondrial damage. The threshold effect seen in brain mitochondria (25–50% decrease in activity) may not directly impact ATP levels or Δψm but could have long‐term deleterious effects triggered by oxidative stress, as it has been shown that an electron leak upstream of the rotenone binding site in CI leads to ROS production (Greenamyre et al., 2001).
Exposure of rats to rotenone (2 days, 2 mg/kg) produced free brain rotenone concentration of 20–30 nM and resulted in 73% inhibition of specific binding to CI of [3H] dihydrorotenone (Betarbet et al., 2000). However, oximetry analysis indicated that in brain mitochondria (but not liver mitochondria) this rotenone concentration (30 nM maximum) was insufficient to inhibit glutamate (CI substrate)‐supported respiration (Betarbet et al., 2000) suggesting that this rotenone concentration did not alter mitochondrial oxygen consumption in isolated brain mitochondria.
Rotenone has been reported to be a specific and potent mitochondrial CI inhibitor with IC50 values from 0.1 to 100 nM depending on the system and methods used (Chinopoulos and Vizi, 2001; Lambert and Brand, 2004; Beretta et al., 2006; Ichimaru et al., 2008).
Mesencephalic cultures prepared from C57BL/6 mice and treated with 5, or 10 nM rotenone for 24 h inhibited CI activity by 11% or 33%, respectively (Choi et al., 2008).
The inhibition of CI was studied in the human osteosarcoma‐derived cell line (143B) after the exposure to rotenone or using a genetic model (40% loss of CI activity in human xenomitochondrial cybrids (HXC) lines). Different degrees of CI inhibition were quantitatively correlated with levels of decreased cellular respiration (Barrientos and Moraes, 1999). Only when CI was inhibited by 35–40% (< 5 nM rotenone), cell respiration decreased linearly until 30% of the normal rate. Increasing concentrations of rotenone produced further but slower decrease in CI activity and cell respiration (Figure 1). Cells with the complete rotenone‐induced CI inhibition still maintain a cell respiration rate of approximately 20% because of an electron flow through complex II. At high concentrations (five‐ to sixfold higher than the concentration necessary for 100% CI inhibition), rotenone showed a secondary, toxic effect at the level of microtubule assembly (Barrientos and Moraes, 1999).
Bovine submitochondrial particles were used to test rotenone affinity binding at 20 nM. This concentration of rotenone reduced the NADH oxidation rate by approximately 50% (Okun et al., 1999)
MPP+ (an active metabolite of MPTP) is an inhibitor of CI (Sayre et al., 1986; Nicklas et al., 1987; Mizuno et al., 1989). Inhibition of the mitochondrial CI by MPP+ suppresses aerobic glycolysis and ATP production (Book chapter in Cheville, 1994).
MPP+ binds loosely to CI and causes reversible inhibition of its activity: approximately 40% inhibition was observed at 10 mM concentration within 15 min of incubation. However, prolonged incubation (> 15 min) produces up to 78% of irreversible inhibition of CI (Cleeter et al., 1992).
Human studies
There are many studies that show impaired catalytic activity of CI in multiple PD post‐mortem brain tissues. For example (Parker and Swerdlow, 1998), five PD brains were used to measure activities of complexes I, III, IV, and of complexes I/III together (NADH: cytochrome c reductase). These measurements were performed in purified frontal cortex mitochondria and revealed a significant loss of CI activity in these PD samples as compared to controls.
Human data indicate that impairment of CI activity may contribute to the pathogenic processes of PD (e.g. Schapira et al., 1989; Greenamyre et al., 2001; Shults, 2004).
A.1.3. Uncertainties or inconsistencies
It is not clear the number of subunits constituting CI in mammals, as according to the existing literature different numbers are cited (between 41 and 46) (Hassinen, 2007; Vogel et al., 2007a). The majority of data claims that mammalian CI is composed of 46 (Greenamyre et al., 2001; Hassinen, 2007) or 45 subunits (Vogel et al., 2007). It is not sure whether there may exist tissue‐specific subunits of CI isoforms (Fearnley et al., 2001). It is unclear, which subunit(s) bind rotenone or other inhibitors of CI.
Additionally, it is not clear whether CI has other uncharacterised functions, taking into consideration its size and complexity (43–46 subunits vs. 11 subunits of complex III or 13 subunits of complex IV) (Greenamyre et al., 2001).
There is no strict linear relationship between inhibitor binding and reduced mitochondrial function. Low doses of rotenone that inhibit CI activity partially do not alter mitochondrial oxygen consumption. Therefore, bioenergetic defects cannot account alone for rotenone‐induced neurodegeneration. Instead, under such conditions, rotenone neurotoxicity may result from oxidative stress (Betarbet et al., 2000). Few studies used human brain cells/human brain mitochondria. Therefore, full quantitative data for humans are not available.
A.1.4. Quantitative understanding
The kinetics of binding and CI inhibition by rotenone has been quantitatively evaluated in a dose‐dependent manner using the submitochondrial particles (Grivennikova et al., 1997) The consequences of CI inhibition were quantitatively measured by a variety of assays that are used to study mitochondrial dysfunction (see Key Event Relationship (KER): Inhibition of Complex I leads to mitochondrial dysfunction). There are also many in vitro and in vivo studies combining the quantification of CI inhibition and DA cell death (e.g. Betarbet et al., 2000; Choi et al., 2008; see KER Mitochondrial dysfunction induces degeneration of nigrostriatal pathway).
The binding of different classes of inhibitors (e.g. pesticides, drugs and other toxins) to CI has been determined quantitatively and I 50, and K I values are available. Potency relative to that of rotenone has been determined under the same conditions in beef mitochondria or submitochondrial particles using the ratio of the K I values, when they were available (Degli Esposti, 1998; Okun et al., 1999). Rotenone I50 value is defined as 20 nM (Okun et al., 1999).
Example of a quantitative evaluation of concentration‐dependent CI inhibition by rotenone (from Barrientos and Moraes, 1999, Figure A.6).
Figure A.6.
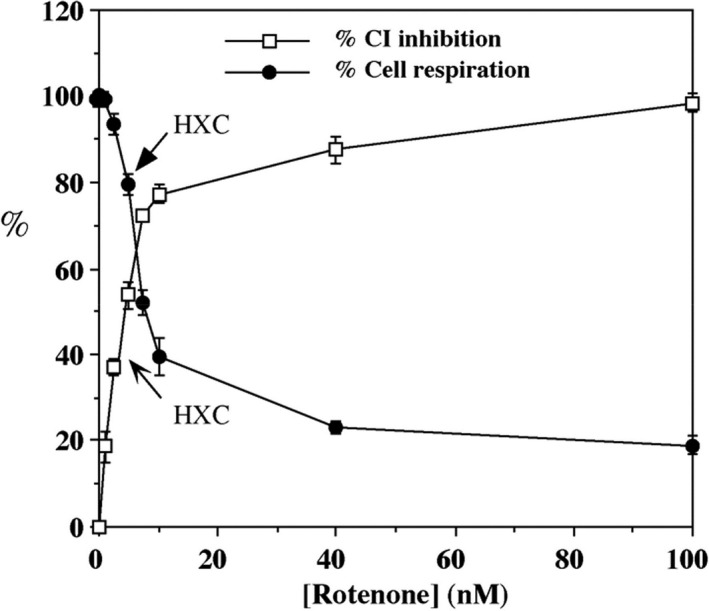
Effect of CI (NADH decylubiquinone reductase) inhibition on endogenous cell respiration. Cells were treated with different concentrations of rotenone for 4 h before measuring cell respiration in whole cells and CI activity in isolated mitochondria. Complete CI inhibition was achieved with 100 nM rotenone. The cell respiration was inhibited also in a dose‐dependent manner but showed different inhibition kinetics and a saturation threshold. For comparison, the genetically altered cell line HXC had an approximately 40% CI reduced activity and an approximately 80% residual cell respiration. HXC, human xenomitochondrial cybrids (This research was originally published in the Journal of Biological Chemistry, Barrientos and Moraes, 1999, copyright the American Society for Biochemistry and Molecular Biology)
Time‐ and concentration‐relationship of NADH oxidase inhibition by rotenone (Figure A.7 from Grivennikova et al., 1997).
Figure A.7.
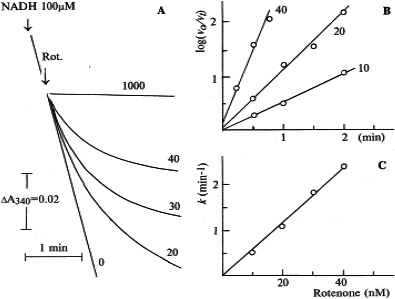
Panel A and B: Time‐ and concentration‐relationship of NADH oxidase inhibition by rotenone. The numbers on the curves indicate the final concentrations of rotenone (0, 20, 30, 40, 1,000 nM). In Panel B: v o, zero‐order rate of NADH oxidation in the absence of rotenone; v t, the `instant’ values of the rates approximated within 10 s time intervals. Panel C: The dependence of first‐order inhibition rate constant on the concentration of rotenone (for further description see Grivennikova et al., 1997. Reprinted from Grivennikova et al., 1997, copyright (1997) with permission from Elsevier)
Table A.1.
Quantitative evaluation of the 1st KER: Binding of inhibitor to NADH‐ubiquinone oxidoreductase (MIE; KE upstream) leads to its inhibition (KE downstream)
| MIE (KE upstream) Binding of inhibitor to NADH‐ubiquinone oxidoreductase (nM) | KE (downstream) Inhibition of CI (%, approximately) | Comments (in vivo, in vitro or human studies) | References |
|---|---|---|---|
| Administration of rotenone at 2 mg/kg per day for 2 days resulted in free rotenone concentration of 20–30 nM in the brain | 75% | DA neuronal cell death determined after rotenone administration at 1–12 mg/kg per day, Sprague–Dawley and Lewis rats infused continuously by jugular vein, 7 days up to 5 weeks | Betarbet et al. (2000) |
|
20 nM rotenone Direct binding studies using bovine and Musca domestica submitochondrial particles |
50% | Binding studies that defined the I50 and K d values for three classes of CI inhibitors (12 chemicals) including rotenone | Okun et al. (1999) |
| Human skin fibroblasts exposed to 100 nM Rotenone for 72 h | 20% | In the same experiment mitochondria morphology, motility was also evaluated | Koopman et al. (2007) |
|
0–2.5 nM Rotenone 5/10 nM Rotenone Mesencephalic neurons were cultured from E14 C57BL/6 mouse embryos for 6 days and then treated with rotenone for 24 h |
No effect 11% and 33%, respectively |
Treatments with 5 or 10 nM rotenone killed 50% or 75% DA neurons respectively | Choi et al. (2008) |
|
1–2.5–5–7.5–10–20 nM 1–10–20–80 nM |
10–20–35–50–65–80% 5–75% |
In this study time course of the active and deactivated enzymes inhibition by rotenone and piericidin A is study in a dose‐dependent manner Binding studies in submitochondrial particles prepared from bovine heart after 20 min of exposure to rotenone |
Grivennikova et al. (1997) |
|
5–10 nM 20 nM 40 nM 100 nM 143B Cells (human osteosarcoma), exposed for 4 h to rotenone |
55–78% 80% 87% 100% |
In the same study similar experiments were performed using HXC cell line (see Figure A.1 above) | Barrientos and Moraes (1999) |
A.1.5. Evidence Supporting Taxonomic Applicability
The CI is well‐conserved across species from lower organism to mammals. The central subunits of CI harbouring the bioenergetic core functions are conserved from bacteria to humans. CI from bacteria and from mitochondria of Yarrowia lipolytica, a yeast genetic model for the study of eukaryotic CI (Kerscher et al., 2002) was analysed by x‐ray crystallography (Zickermann et al., 2015).
However, the affinity of various chemicals to cause partial or total inhibition of CI activity across species is not well studied (except for rotenone).
References
Barrientos A, Moraes CT, 1999. Titrating the effects of mitochondrial complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197. http://www.jbc.org/content/274/23/16188.full.pdf Fig 1 p.16190. © the American Society for Biochemistry and Molecular Biology
Beretta S, et al., 2006. Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Relevance to Leber Hereditary Optic Neuropathy (LHON). Neurobiology of Disease, 24, 308–317.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Cheville NF, 1994. Ultrastructural Pathology: The Comparative Cellular Basis of Disease. Wiley John Wiley & Sons, 09 dic 2009 ‐ 1000 pagine
Chinopoulos C, Adam‐Vizi V, 2001. Mitochondria deficient in complex I activity are depolarized by hydrogen peroxide in nerve terminals: relevance to Parkinson's disease. Journal of Neurochemistry, 76, 302–306.
Choi WS, Kruse SE, Palmiter R, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP, or paraquat. Proceedings of the National Academy of Sciences, 105, 15136–15141.
Cleeter MW, Cooper JM, Schapira AH, 1992. Irreversible inhibition of mitochondrial complex I by 1‐methyl‐4‐phenylpyridinium: evidence for free radical involvement. Journal of Neurochemistry, 58, 786–789.
Davey GP, Clark JB, 1996. Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. Journal of Neurochemistry, 66, 1617–1624.
Degli Esposti M, Ghelli A, 1994. The mechanism of proton and electron transport in mitochondrial complex I. Biochimica et Biophysica Acta, 1187, 116–120.
Degli Esposti, 1998. Inhibitors of NADH‐ubiquinone reductase: an overview. Biochimica et Biophysica Acta, 1364, 222–235.
Efremov RG, Sazanov LA, 2011. Structure of the membrane domain of respiratory complex I. Nature, 476, 414–420.
Fearnley IM, Carroll J, Shannon RJ, Runswick MJ, Walker JE, Hirst J, 2001. GRIM‐19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH:ubiquinone oxidoreductas (complex I). Journal of Biological Chemistry, 276, 38345–38348.
Greenamyre TJ, Sherer TB, Betarbet R, Panov AV, 2001. Complex I and Parkinson's Disease. Critical Review. International Union of Biochemistry and Molecular Biology (IUBMB Life), 52, 135–141.
Grivennikova VG, Maklashina EO, Gavrikova EV, Vinogradov AD, 1997. Interaction of the mitochondrial NADH‐ubiquinone reductase with rotenone as related to the enzyme active/inactive transition. Biochimica et Biophysica Acta, 1319, 223–232.
Hassinen I, 2007. Regulation of Mitochondrial Respiration in Heart Muscle. In Mitochondria – The Dynamic Organelle Edited by Schaffer & Suleiman. Springer ISBN‐13: 978‐0‐387‐69944‐8.
Ichimaru N, Murai M, Kakutani N, Kako J, Ishihara A, Nakagawa Y, Miyoshi H, 2008. Synthesis and characterization of New Piperazine‐type inhibitors for mitochondrial NADH‐ubiquinone oxidoreductase (Complex I). Biochemistry, 47, 10816–10826.
Koopman W, Hink M, Verkaart S, Visch H, Smeitink J, Willems P, 2007. Partial complex I inhibition decreases mitochondrial motility and increases matrix protein diffusion as revealed by fluorescence correlation spectroscopy. Biochimica et Biophysica Acta, 1767, 940–947.
Krug AK, Gutbier S, Zhao L, Pöltl D, Kullmann C, Ivanova V, Förster S, Jagtap S, Meiser J, Leparc G, Schildknecht S, Adam M, Hiller K, Farhan H, Brunner T, Hartung T, Sachinidis A, Leist M, 2014. Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP(+). Cell Death and Disease, 8, e1222. doi: 10.1038/cddis.2014.166
Lambert AJ, Brand MD, 2014. Inhibitors of the quinone‐binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). Journal of Biological Chemistry, 279, 39414–39420.
Langston JW, 1996. The etiology of Parkinson's disease with emphasis on the MPTP story. Neurology, 47, S153–S160.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y, 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications, 163, 1450–1455.
Nicklas WJ, Yougster SK, Kindt MV, Heikkila RE, 1987. MPTP, MPP+ and mitochondrial function. Life Science Journal, 40, 721–729.
Okun JG, Lummen PL, Brandt U, 1999. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:Ubiquinone Oxidoreductase), 274, 2625–2630.
Parker Jr WD, Swerdlow RH, 1998. Mitochondrial dysfunction in idiopathic Parkinson disease. American Journal of Human Genetics, 62, 758–762.
Sayre LM, Arora PK, Feke SC, Urbach FL, 1986. Mechanism of induction of Parkinson's disease by I‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Chemical and electrochemical characterization of a geminal‐dimethyl‐blocked analogue of a postulated toxic metabolite. Journal of the American Chemical Society, 108, 2464–2466.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD, 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet, 1, 1269.
Shults CW, 2004. Mitochondrial dysfunction and possible treatments in Parkinson's disease–a review. Mitochondrion, 4, 641–648.
Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG, 2007. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Molecular Genetics and Metabolism, 91, 176–182.
2nd KER: Inhibition of Complex I lead to mitochondrial dysfunction
A.2.1. How does this Key Event Relationship work
Inhibited CI is unable to pass off its electron to ubiquinone and it cannot translocate protons across the mitochondrial inner membrane. This creates a back‐up of NADH within the mitochondrial matrix (Brown and Borutaite, 2004). This leads to an arrest of the citric acid cycle and a failure to build a proton gradient (mitochondrial membrane potential, Δψm) across the inner membrane. This results in impaired ATP production. In addition, the direct transfer of electrons from CI to oxygen is increased. This leads to oxidative stress as ROS (e.g. superoxide, hydrogen peroxide) are produced, which can damage DNA, proteins, lipids and other cell components and function (Sanders et al., 2014).
A.2.2. Weight of Evidence
The weight of evidence supporting the relationship between inhibition of CI and mitochondrial dysfunction is strong. The mechanisms behind this KER are partially understood and well documented based on in vitro as well as in vivo experiments (e.g. Sanders et al., 2014), complemented by data from human post‐mortem PD brain evaluations (Parker et al., 1989; Schapira et al., 1989; Sherer et al., 2003).
A.2.2.1. Biological Plausibility
The biological plausibility that inhibition of CI activity triggers mitochondrial dysfunction is strong. It is well understood, how the inhibition of CI can lead to mitochondrial dysfunction as measured by: a) decreased oxygen consumption, b) decrease or loss of ATP production, c) decrease of Δψm, d) the loss of mitochondrial protein import and protein biosynthesis, e) reduced activities of enzymes of the mitochondrial respiratory chain and the Krebs cycle, f) elevated levels of ROS, g) the loss of mitochondrial motility, causing a failure of mitochondria to re‐localise to sites of increased energy demands (such as synapses), h) destruction of the mitochondrial network, i) increased mitochondrial uptake of Ca2+ causing mitochondrial Ca2+ overload (Graier et al., 2007) and opening of mitochondrial PTP, (j) rupture of the mitochondrial inner and outer membranes, leading to release of mitochondrial pro‐death factors, including cytochrome c, AIF and endonuclease G (Braun, 2012; Martin, 2011; Correia et al., 2012; Cozzolino et al., 2013). These pathological mechanisms are extremely well studied.
A.2.2.2. Empirical support for linkage
Many studies show that the pathophysiological consequences of a partial or total CI inhibition are linked to mitochondrial dysfunction. In many of these experiments the cellular damage caused by mitochondrial dysfunction is reduced (or entirely prevented) by treatment with antioxidants.
Different degrees of CI inhibition by rotenone have been studied in the human osteosarcoma‐derived cell line (143B). A quantitative correlation between increasing inhibition of CI and mitochondrial dysfunction (as shown by inhibition of mitochondrial respiration, reduced ATP production, increased ROS release and lipid peroxidation, as well as decreased Δψm) was established (Figure 1 and Table 1 based on Barrientos and Moraes, 1999).
Based on the existing literature it is suggested that rotenone exerts toxicity via oxidative stress, rather than via decrease of ATP synthesis (bioenergetics effects).
A few examples illustrating mitochondrial damage and oxidative stress in animal model of PD and human cells induced by:
Rotenone
Rotenone administered subcutaneously for 5 weeks (2.5 mg/kg/d) caused a selective increase (by ~ 2 fold) in oxidative damage in the striatum, as compared to the hippocampus and cortex, accompanied by massive degeneration of DA neurons (~ 80% decrease) in the substantia nigra. Rotenone reduced intracellular ATP levels in the striatum (by > 40%), increases malondialdehyde (MDA, indicative of lipid peroxidation, by ~ 60%), reduced GSH levels (by ~ 20%), thioredoxin (by ~ 70%), and manganese superoxide dismutase (SOD, by ~ 15%) (all parameters significantly changed in the striatum). Antioxidant polydatin (Piceid) treatment significantly prevented the rotenone‐induced changes by restoring the above parameters to control levels, confirming that rotenone‐induced mitochondrial dysfunction resulted in oxidative stress (Chen et al., 2015).
Rotenone was administered 2.5 mg/kg body weight to male Wistar rats for 4 weeks in the presence or absence of ferulic acid (FA, at the dose of 50 mg/kg) that has antioxidant and anti‐inflammatory properties. Rotenone administration caused DA neuronal cell death (~ 50%), significant reduction in endogenous antioxidants, such as superoxide dismutase (~ 75%), catalase (~ 40%), and glutathione (~ 50%), and induced lipid peroxidation evidenced by increased MDA formation (~ 2 folds). Treatment with FA rescued DA neurons in substantia nigra pars compacta area and nerve terminals in the striatum, as well as restored antioxidant enzymes, prevented depletion of glutathione, and inhibited lipid peroxidation induced by rotenone (Ojha et al., 2015).
Many studies have shown that mitochondrial aldehyde dehydrogenase 2 (ALDH2) functions as a cellular protector against oxidative stress by detoxification of cytotoxic aldehydes. Dopamine is metabolised by monoamine oxidase to yield 3,4‐dihydroxyphenylacetaldehyde (DOPAL) then converts to a less toxic acid product by ALDH. The highly toxic and reactive DOPAL has been hypothesised to contribute to the selective neurodegeneration of DA neurons. In this study, rotenone (100 nM, 24 h) in both SH‐SY5Y cells and primary cultured substantia nigra (SN) DA neurons, was shown to reduce DA cell viability (~ 40%), reduce Δψm (~ 40%, as shown by TMRM), induce mitochondrial ROS production (~ 30%, as shown by increase of MitoSox Red), and increased cytosolic protein levels of proteins related to the mitochondrial apoptotic pathway (i.e. Bax, cytochrome c, active caspase‐9 and active caspase‐3) (~ 2 folds for all proteins).
The neuroprotective mechanism of ALDH2 was observed as overexpression of wild‐type ALDH2 gene (but not the enzymatically deficient mutant ALDH2*2 (E504K)) reduced rotenone‐induced DA neuronal cell death, prevented rotenone‐induced reduction in TMRM signal (95.7 ± 1.6% vs 67 ± 3.5%), and prevented rotenone‐induced increase in MitoSox Red intensity (103.1 ± 1% vs 133.4 ± 0.8%). Additionally, pretreatment of cells with Alda‐1 (activator of ALDH2) (1–10 μM, for 24 h) prevented rotenone‐induced loss of Δψm and ROS production in a dose‐dependent manner. These results were confirmed by in vivo studies. Rotenone (50 mg/kg per day, oral administration for 14 days) or MPTP (40 mg/kg per day, i.p. for 14 days) both administered to C57BL/6 mice caused significant SN TH+ DA neuronal cell apoptosis (~ 50%). Alda‐1 attenuated rotenone‐induced apoptosis by decreasing ROS accumulation, reversing Δψm depolarisation, and inhibiting the activation of proteins related to mitochondrial apoptotic pathway. The present study demonstrates that rotenone or MPP+ induces DA neurotoxicity through oxidative stress. Moreover, Alda‐1 is effective in ameliorating mitochondrial dysfunction by inhibiting rotenone‐ or MPP+‐induced mitochondria‐mediated oxidative stress that leads to apoptosis (Chiu et al., 2015).
Rotenone‐induced mitochondrial dysfunction was observed in human neuroblastoma cells exposed to 5 nM rotenone for 1–4 weeks. After 3–4 weeks of treatment, rotenone‐treated cells showed evidence of oxidative stress, including loss of GSH (by 5%) and increased oxidative DNA (qualitative, measured by using antibodies to 8‐oxo‐dG) and protein damage (223 ± 29% of control, as shown by the large increase in protein carbonyls in the insoluble fraction) (Sherer et al., 2002). This chronic rotenone treatment markedly sensitised cells to further oxidative challenge since in response to H2O2 cytochrome c release from mitochondria and caspase‐3 activation occurred earlier and to a greater extent in rotenone‐treated cells vs Ctr (1.44 ± 0.02% vs 0.38 ± 0.07% apoptosis/h). This study indicates that chronic, low‐level CI inhibition by rotenone induces progressive oxidative damage, and caspase‐dependent neuronal cell death (Sherer et al., 2002).
By using antioxidant, kaempferol (6 μM, 1 h prior addition of rotenone) and rotenone (50 nM, max up to 24 h) on SH‐SY5Y cells, kaempferol was found to counteract rotenone‐induced ROS production (especially superoxide: with kaempferol, ethidium fluorescence decreased below the control (Ctr) levels), rotenone‐induced mitochondrial oxidative dysfunction (protein carbonyls values: 2.5 in Ctr, 6.2 with rotenone, 2.7 with kaempferol + rotenone), rotenone‐induced oxygen respiration (values of nmol of atomic oxygen/minute per mg protein: 5.89 Ctr, 0.45 with rotenone, 2.47 with kaempferol + rotenone), rotenone‐induced Δѱm decrease (~ 70% cells of with rotenone only vs ~ 30% with kaempferol + rotenone) (Filomeni et al., 2012).
To model the systemic mitochondrial impairment, rats were exposed to rotenone. A single rotenone dose (10 nM, for 24 h) induced mtDNA damage in midbrain neurons (> 0.4 lesions/10 kb vs 0 lesions/10 kb in vehicle), but not in cortical neurons; similar results were obtained in vitro in cultured neurons. Importantly, these results indicate that mtDNA damage is detectable prior to any signs of neuronal degeneration and is produced selectively in midbrain neurons. The selective vulnerability of midbrain neurons to mtDNA damage was not due to differential effects of rotenone on CI since rotenone suppressed respiration equally (~ 60%) in midbrain and cortical neurons compared to vehicle. However, in response to CI inhibition, midbrain neurons produced more mitochondrial H2O2 (5 min of rotenone increased MitoPY1 fluorescence of ~ 10% in midbrain mitochondria vs vehicle, and progressively for the duration of measurement), than cortical neurons. The selective mtDNA damage in midbrain could serve as a molecular marker of vulnerable nigral neurons in PD. Oxidative damage to cell macromolecules in human PD and the rotenone model have been recently reviewed (Sanders et al., 2014).
Adult male Sprague–Dawley rats were intranigrally infused with rotenone (6 μg in 1 μl) alone or in the presence of L‐deprenyl (0.1, 1, 5 and 10 mg/kg; i.p.) at 12 h intervals for 4 days. Rotenone alone (100 μM, 30 min) increased the levels of hydroxyl radials in the mitochondrial P2 fraction 2,3‐DHBA (122.90 ± 5.4 pmol/mg protein) and 2,5‐DHBA (146.21 ± 6.3 pmol/mg protein). L‐deprenyl (100 nM–1 mM) dose‐dependently attenuated rotenone‐induced ·OH generation in the mitochondrial P2 fraction. L‐deprenyl‐induced attenuation in the rotenone‐mediated 2,3‐DHBA generation was from 17 ± 1.1% to 67 ± 4.3%, respectively, for 100 nM–1 mM of the MAO‐B inhibitor. Also, rotenone caused about 51 ± 3.3% reduction in GSH levels in the cell body region, SN and 34 ± 1.1% decrease in the nerve terminal region, NCP (nucleus caudatus putamen). L‐deprenyl alone did not cause any significant difference in the GSH content in either region. L‐deprenyl treatment dose‐dependently attenuated the rotenone‐induced GSH depletion in SN from 51 ± 3.1% to 44 ± 2.1%, 32 ± 1.7% and 9 ± 1.0%, respectively, for doses of 1, 5 and 10 mg/kg. Additionally, SOD activity was assayed in rotenone‐lesioned animals, which were treated with l‐deprenyl at different doses (1–10 mg/kg). SN exhibited two‐ and threefold activity of Cu/Zn‐SOD (i.e. cytosolic SOD fraction) and Mn‐SOD (i.e. particulate SOD fraction), respectively, compared to the nerve terminal region, NCP. L‐deprenyl (5 and 10 mg/kg) in rotenone‐lesioned animals caused a significant increase in the cytosolic Cu/Zn SOD activity in SN of both the sides. Intranigral infusion of rotenone alone caused a significant increase in the enzyme activity in SN of the side of infusion as compared to the non‐infused side (~ 20%). L‐deprenyl (5 and 10 mg/kg) further increased catalase activity in both ipsilateral SN and striatum, as compared to the contralateral side of infusion. Finally, rotenone caused a 74% reduction in the striatal TH staining intensity, which was partially recovered by L‐deprenyl. These results showed that oxidative stress is one of the major causative factors underlying DA neurodegeneration induced by rotenone and they support the view that L‐deprenyl is a potent free radical scavenger and an antioxidant (Saravanan et al., 2006). Similar results were obtained after exposure to MPP+ (Wu et al., 1994).
Antioxidant (piperaceae; PLL) with some anti‐inflammatory activities demonstrated in preclinical studies protective effects in PD animal models. Rats treated with rotenone and PLL‐derived alkaloids showed decreased ROS, stabilised Δψm, and the opening of the mitochondrial PTP – which is triggered by ROS production – was inhibited. In addition, rotenone‐induced apoptosis was abrogated in the presence of these alkaloids (Wang et al., 2015b).
In SK‐N‐MC human neuroblastoma cells, rotenone (10 nM – 1 μM, 48 h) caused dose‐dependent ATP depletion (~ 35% reduction by 100 nM rotenone vs Ctr), oxidative damage (100% increase of carbonyls levels upon 100 nM rotenone), and death (100 nM rotenone after 48 h caused 1.1 AU (arbitrary units) increase of cell death vs untreated – 0.00 AU ‐). α‐Tocopherol pretreatment (62.5 or 125 μM 24 h before rotenone (10 nm)) attenuated rotenone toxicity (Sherer et al., 2003).
MPTP (1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine) or MPP+ (1‐methyl‐4‐phenyl‐pyridinium ion)
MPTP converted into MPP+ inhibits mitochondrial CI activity, resulting in excessive intracellular ROS production followed by further mitochondrial dysfunction leading to mitochondrial‐dependent apoptosis. Lutein, a carotenoid of xanthophyll family (antioxidant) reversed MPTP‐induced mitochondrial dysfunction, oxidative stress, apoptotic cell death and motor abnormalities. These results revealed that antioxidant protected DA neurons and diminished mitochondrial dysfunction and apoptotic death (Nataraj et al., 2015).
Antioxidant (salidroside; Sal) pretreatment protected DA neurons against MPTP/MPP+‐induced toxicity in a dose‐dependent manner by: (1) reducing the production of ROS, (2) regulating the ratio of Bcl‐2/Bax, (3) decreasing cytochrome‐c and Smac release, and inhibiting caspase‐3, caspase‐6, and caspase‐9 activation, which are known to trigger apoptosis following mitochondrial dysfunction. Sal acted as an effective neuroprotective agent through modulation of the ROS‐induced mitochondrial dysfunction in vitro and in vivo (Wang et al., 2015a).
In an in vitro study, MPP+ (1 mM, 24 h) was found to elicit production of ROS (by 2 fold vs Ctr) and reduce by 50% SOD (by about 50%) and catalase (by about 65%) activity in SH‐SY5Y human neuroblastoma cells. Pretreatment with the antioxidant astaxanthin (AST; 50 μM, 24 h) inhibited MPP+‐induced production of ROS and attenuated both SOD and catalase activity decrease. Furthermore, MPP+ (1 mM, 48 h) increased caspase‐3 activity to 243% of the Ctr and also increased cleaved caspase‐3 in the cells (qualitative). Addition of 50 μM AST attenuated MPP+‐induced caspase‐3 activation (57% suppression). MPP induced also a 70% reduction of Δψm and cytochrome c release (qualitative), while AST prevented both these effects. The protective effects of AST on MPP+‐induced mitochondrial dysfunction was due to its antioxidative properties and antiapoptotic activity via induction of expression of SOD and catalase (as shown above) and regulating the expression of Bcl‐2 and Bax (Bax/Bcl‐2 ratio increased to 1.6‐fold vs Ctr upon treatment with MPP+, while AST prevented the MPP+‐induced increase of the Bax/Bcl‐2 ratio). These results were confirmed by in vivo studies (Lee et al., 2011).
DA neurons in primary mesencephalic cultures treated with MPP+ (100 μM, for 48 h) underwent reduction of cell viability (~ 55% MTT reduction), LDH release (~ 90%), about 60% reduction of TH+ cells, disruption of Δψm (~ 45% decline) and ROS production (~ 60% increase), upregulation of Nox2 (~ 45%) and Nox4 (~ 60%), while promoting a decrease of both SOD (~ 45%) and GSH activity (~ 85%). Additionally, MPP‐induced apoptosis via mitochondrial dysfunction, as shown by induction of cytochrome c (~ 55%), cleaved‐caspase‐3 (~ 75%), upregulation of Bax expression (~ 55%), and downregulation of Bcl2 (~ 60%). Liuwei dihuang (LWDH), a widely used traditional Chinese medicine (TCM), has antioxidant characteristics. LWDH‐WH, derivative of LWDH (0.01–10 μg/ml, added 1 h prior to MPP+ addition) reduced oxidative damage via increasing antioxidant defence (SOD, GSH), decreasing ROS production, and downregulating NADPH oxidases (Nox2 and Nox4). LWDH‐WH also inhibited neuronal apoptosis by increasing antiapoptotic protein Bcl‐2 expression, and downregulating apoptotic signalling (Bax, cytochrome c, cleaved‐caspase‐3) in MPP+‐treated neurons. All these protective effects were induced in a dose‐dependent manner (Tseng et al., 2014).
PC12 cells treated with MPP+ (500 μM, for 24 h) underwent reduction of viability (~ 55% MTT reduction), oxidative stress (~ 160% increase in ROS production) and downregulation of haem oxygenase‐1 expression (~ 2‐fold). Pretreatment with edaravone, a novel free radical scavenger, (25, 50, 75, 100 μM, for 1 h prior MPP+ treatment) protected PC12 cells against MPP+‐cytotoxicity via inhibiting oxidative stress and upregulating haem oxygenase‐1 expression in a dose‐dependent manner (Cheng et al., 2014).
The protective effects of antioxidant, apigenin (AP), naturally occurring plant flavonoids were observed on the MPP+–induced cytotoxicity in cultured rat adrenal phaeochromocytoma cells (PC12 cells). The PC12 cells were pretreated with various concentrations of the test compound for 4 h, followed by the challenge with 1,000 μM MPP+ for 48 h. Pretreatment with AP (3–6–12 μM) before MPP+ significantly reduced the level of intracellular ROS and elevated Δψm in the MPP+–treated PC12 cells. In addition, AP markedly suppressed the increased rate of apoptosis and the reduced Bcl–2/Bax ratio induced by MPP+ in the PC12 cells. The findings demonstrated that AP exerts neuroprotective effects against MPP+–induced neurotoxicity in PC12 cells, at least in part, through the inhibition of oxidative damage and the suppression of apoptosis through the mitochondrial pathway (Liu et al., 2015).
Brain mitochondria isolated from ventral midbrain of mitochondrial matrix protein cyclophilin D (CYPD) knockout mice were significantly less sensitive to acute MPP+ (20 μM)‐induced effects. CYPD ablation attenuated in vitro Ca2+ ‐induced mitochondrial dysfunction and ROS generation upon Ca2+ loading, both in the absence and in the presence of MPP+, compared to wild‐type mice. CYPD ablation conferred a protection to mitochondrial functions upon in vivo treatment with MPTP.
Ventral midbrain mitochondria (that constitutes < 5% of SNpc DA neurons) isolated from brains of wild type (wt) mice acutely treated with MPTP (single MPTP 20 mg/kg injection, analysis done after 4 h), as compared with saline‐treated mice, showed a reduction of CI (by 53%), a reduced rate of phosphorylating respiration (by 38%), a reduced respiratory control index (by 37%), and a decreased ADP/O ratio (by 18%).
Ventral midbrain mitochondria isolated from brains of CYPD knockout mice acutely treated with MPTP, as compared with MPTP‐treated wt mice, exhibited higher activity of CI (~ 80%, vs 53% wt), higher rate of phosphorylating respiration (~ 82%, vs 62% wt), a better respiratory control index (~ 79%, vs 63% wt), and a higher ADP/O ratio (~ 90% vs 82% wt) (Thomas et al., 2012).
CYP plays as a regulatory component of a calcium‐dependent permeability transition pores (PTP), and the data suggest that PTP is involved in MPP+‐induced mitochondrial damage. Under oxidative stress, the prolonged opening of the PTP results in calcium overload and with time mitochondrial dysfunction as they get de‐energised, depolarised, triggering apoptotic or necrotic cell death (Bernardi, 1999).
There are many other studies showing that MPP+ induces NADH‐dependent SOD formation and enhances NADH‐dependent lipid peroxidation in submitochondrial particles, confirming that oxidative stress is induced by MPP+ (e.g. Ramsay and Singer, 1992; Takeshige, 1994).
Based on the human post mortem studies of PD brains it is well established that oxidative stress and mitochondrial dysfunction accompany the pathophysiology of PD (e.g. Hartman et al., 2004; Zhu and Chu, 2010; Dias et al., 2013; Fujita et al., 2014).
Examples of human data confirming the presence oxidative stress and mitochondrial dysfunction in PD post mortem brains:
A significant decrease in CI activity has been identified in a large study of post‐mortem PD brains, specifically in substantia nigra compared with age matched controls. In idiopathic PD all 10 patients studied had significant reductions of CI activity (Parker et al., 1989). It is hypothesise that the CI dysfunction may have an aetiological role in the pathogenesis of PD (Schapira et al., 1989; Greenamyre et al., 2001; Sherer et al., 2003).
The structure and function of mitochondrial respiratory‐chain enzyme proteins were studied post‐mortem in the substantia nigra of nine patients with PD and nine matched controls. Total protein and mitochondrial mass were similar in the two groups. CI and NADH cytochrome c reductase activities were significantly reduced, whereas succinate cytochrome c reductase activity was normal. These results indicated a specific defect of CI activity in the substantia nigra of patients with PD (Schapira et al., 1990).
Post mortem human studies show that CI deficiency in PD is anatomically specific for the substantia nigra, and they are not present in another neurodegenerative disorder involving the substantia nigra. These results suggest that CI deficiency may be the underlying cause of DA cell death in PD (Schapira et al., 1990; Schapira, 1994).
The mitochondrial respiratory chain function was studied in various brain regions as well as in skeletal muscle and in blood platelets from patients with idiopathic PD and from matched controls. The evidence suggests that the CI deficiency in PD is limited to the brain and that this defect is specific for the substantia nigra (Mann et al., 1992).
Immunoblotting studies on mitochondria prepared from the striata of patients who died of PD were performed using specific antisera against Complexes I, III and IV. In four out of five patients with PD, the 30, 25 and 24 kDa subunits of CI were moderately to markedly decreased. No clear difference was noted in immunoblotting studies on subunits of Complexes III and IV between the control and PD. The authors claim that deficiencies in CI subunits seem to be one of the most important clues to elucidate pathogenesis of PD (Mizuno et al., 1989).
Redox markers have been found unchanged in PD patient‐derived vs Ctr‐derived fibroblasts at baseline. Basal mitochondrial respiration and glycolytic capacity resulted similar at baseline between PD and Ctr fibroblasts, while rotenone‐sensitive respiration (analysed by using 0.5 μM rotenone) resulted lower in PD fibroblasts vs Ctr (174.74 ± 48.71 vs 264.68 ± 114.84) (Ambrosi et al., 2014).
Augmented oxidative metabolism has been detected in PD brains by magnetic resonance studies, in conjunction with energy unbalance. Decreased glucose consumption (22% mean reduction), likely reflecting a decrease in neuronal activity, has been reported in the nigrostriatal system of PD patients (Piert et al., 1996). These symptoms were hypothesised to be indicative of mitochondrial dysfunction as early markers, present in the brain of patients with PD even in the absence of overt clinical manifestations (Rango et al., 2006). In particular, by using high temporal and spatial resolution 31P magnetic resonance spectroscopy (31P MRS) technique authors studied mitochondrial function by observing high‐energy phosphates (HEPs) and intracellular pH in the visual cortex of 20 PD patients and 20 normal subjects at rest, during, and after visual activation. In normal subjects, HEPs remained unchanged during activation, but rose significantly (by 16%) during recovery, and pH increased during visual activation with a slow return to rest values. In PD patients, HEPs were within the normal range at rest and did not change during activation, but fell significantly (by 36%) in the recovery period; pH did not reveal a homogeneous pattern with a wide spread of values. Energy unbalance under increased oxidative metabolism requirements, that is, the post‐activation phase, discloses a mitochondrial dysfunction that is present in the brain of patients with PD even in the absence of overt clinical manifestations (Rango et al., 2006).
There are many other studies providing evidence that oxidative stress and mitochondrial dysfunction play an important role in PD pathophysiology (see indirect KER Mitochondrial dysfunction induced DA neuronal cell death of nigrostriatal pathway).
A.2.3. Uncertainties or inconsistencies
Some studies suggest that rotenone may have effects other than CI inhibition, and it has been claimed that rotenone induces microtubule disruption, rather than ETC CI inhibition (Ren et al., 2005; Feng, 2006).
Some studies suggested that there was no evidence for significant change in mitochondrial CI function in PD patients’ brains (Jenner et al., 1992).
It is still unclear whether the site of superoxide production in CI inhibited mitochondria is CI itself or not (Singer and Ramsay, 1994).
A.2.4. Quantitative Understanding of the Linkage
Based on the available data, the threshold effect seen in brain mitochondria indicates that modest CI inhibition (~ 25–50% decrease in activity) may not directly impact ATP levels or Δψm. Indeed, low levels of CI inhibition produces an oxidative stress without any significant changes in mitochondrial respiration (Betarbet et al., 2000; Greenamyre et al., 2001) or causes not significant changes in ATP levels (Sherer et al., 2003).
In particular, in rotenone‐infused animals (2.0 mg/kg per day for 2 days), [3H] dihydrorotenone binding to CI in brain was reduced by about 73%. Based on this degree of binding inhibition, the rotenone concentration in brain was estimated to be between 20 and 30 nM. Complexes II and IV were unchanged by rotenone infusion (Betarbet et al., 2000).
However, such defects have long‐term deleterious effects. It is well documented that that there is a site of electron leak upstream of the rotenone binding site in CI (i.e. on the ‘NADH side’ of the complex) (Hensley et al., 1998) leading to the superoxide (O2 −) and followed up by H2O2 production by CI (Greenamyre et al., 2001). The relative role of each ETC complex in forming superoxide differs by tissue; however CI is a major source of O2 − in the brain (Halliwell, 2007).
Thus, a low inhibition of CI activity that is insufficient to affect cell respiration may lead to mitochondrial damage and chronic upregulation of ROS production. Therefore, it is suggested that rotenone that binds to CI with an affinity of 10–20 nM induces toxicity not by bioenergetics effects but rather via accumulative oxidative stress. Sustained oxidative stress leads to decrease levels of reduced glutathione; activation of superoxide dismutase (SOD) (scavenger of O2 −), catalase and indeed, treatments with antioxidants reduce the oxidative stress‐induced damage. Such data are abounded in the existing literature based both on in vivo and in vitro studies and a few examples are described.
A.2.5. Empirical support for linkage
The selective CI defects (other complexes were unaffected) (Schapira et al., 1990a) and induced mitochondrial damage followed by oxidative stress is also described in PD patients brains as documented by: (a) reduced glutathione levels (Jenner et al., 1992); (b) increased content of 8‐oxo‐deoxyguanine, a marker of oxidatively damaged nucleic acids (Mecocci et al., 1993; Alam et al., 1997); (c) increased level of malondialdehyde (marker of lipid peroxidation) (Navarro et al., 2009); (d) increased cholesterol lipid hydroperoxide (Dexter et al., 1994); (e) increased protein oxidation measured, e.g. by elevated levels of methionine sulfoxide formation or protein carbonyl content (Alam et al., 1997). These studies in human brain present a semiquantitataive evaluation of the oxidative stress, as there is no data showing KER between the various degrees of CI inhibition and mitochondrial damage (ROS production) and the parameters described above. However, these studies clearly confirmed that oxidative stress in PD patient brain is increased as shown by the measured biomarkers (Sanders and Greenamyre, 2013).
In in vitro and in in vivo animal studies there are some data showing the quantitative relationship between the oxidative stress produced by inhibition of CI and mitochondrial damage measured by the same assays, as described in human studies, and a few examples of such experiments are discussed below.
The quantitative evaluation of the causative relationship between the CI inhibition (KEup) induced by rotenone (4 h exposure) and mitochondrial dysfunction (KEdown) measured in human‐chimpanzee isolated mitochondria (xenomitochondrial cybrids; HXC) by a decreased cell respiration and Δψm, increased ROS production and lipid peroxidation showed linear, time‐ and concentration‐dependent effects (below Figure A.8 from Barrientos and Moraes, 1999).
Figure A.8.
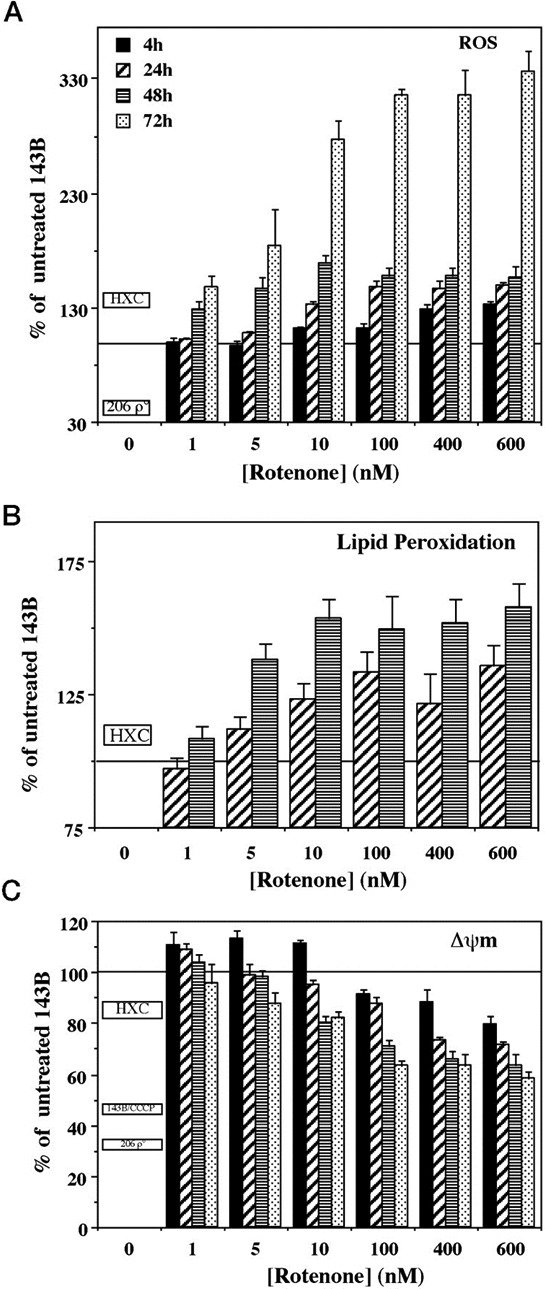
A dose‐ and time‐dependent effect of CI inhibition by rotenone on (A) reactive oxygen species production (ROS), (B) Lipid peroxidation and (C) mitochondrial membrane potential (Δψm) studied in the human osteosarcoma‐derived cell line (143B) or using a genetic model (40% CI inhibited in HXC lines) (This research was originally published in the Journal of Biological Chemistry, Barrientos and Moraes, 1999, fig. 5, copyright the American Society for Biochemistry and Molecular Biology)
The endogenous respiration was inhibited in a dose‐dependent manner but showed different inhibition kinetics. Only when CI was inhibited by 35–40% (< 5 nM rotenone), cell respiration started decreasing (a threshold for inhibition for cell respiration triggered by rotenone). Between 40 and 60% of CI inhibition (5–10 nM), cell respiration decreased linearly until 30% of the normal rate. Increasing concentrations of rotenone produced further but slower decrease in CI activity and cell respiration. 100% CI inhibition was achieved with 100 nM rotenone but the cells still maintained a cell respiration rate (through complex II), approximately 20% and the rate of ROS production increased by a maximum of 20–25% (4 h treatment). ROS production was saturated at 100 nM rotenone but an initial effect was observed already at 1–5 nM (Barrientos and Moraes, 1999). Inhibition of CI activity triggered decrease of cell respiration by different concentrations of rotenone and resulted in mitochondrial damage measured not only by ROS production, but also by lipid peroxidation and decreased Δψm. Inhibition of CI by 25, 50, 75 and 100% decreased cell respiration by 5, 20, 53, 81%, increased ROS production by 48, 81, 157, 216%, increased lipid peroxidation by 8, 27, 45, 55% and decreased Δψm by 6, 13, 20, and 37% respectively (approximately).
Similar studies were also performed using different types of neuronal cells.
Hoglinger and colleagues, by using DA neurones derived from the rat (embryonic day 15.5) ventral mesencephalon, showed that CI inhibition by rotenone at 30 nM, (or MPP+ 3 μM) for 24 h decreased ATP levels (by > 80%) within the first 6 h, and neuronal cell death within 24 h. When residual ATP levels remained above 20%, there was no or little neuronal loss, suggesting that 20% of normal ATP level was the minimum compatible with neuronal survival. Rotenone (and MPP+) increased ROS (≥ 40% over control levels) already at low concentrations that were subtoxic or only moderately toxic (i.e. 10–30 nM for rotenone, 10–30 μM for MPP+) (Figure A.9) (Hoglinger et al., 2003).
Figure A.9.
ATP levels, ROS production and neuronal surviving cells in mesencephalic cultures treated with CI inhibitors (rotenone and MPP+) (from Hoglinger et al., 2003, fig. 4a,b, copyright John Wiley and Sons)
Shamoto‐Nagai and colleagues showed that 25 or 50 nM rotenone decreased ATP levels over time. In particular, the intracellular ATP level was reduced to 18.0% and 19.6% of control after 44 h of treatment with 25 and 50 nM of rotenone, respectively, and thereafter the decreased level was sustained (Figure A.10, left) (Shamoto‐Nagai et al., 2003). Also, The production of ROS‐RNS increased 6 h after the rotenone treatment, and the increase was about 1.5‐fold of the basal value. With treatment with the higher (50 nM) concentration of rotenone, DCF production level was restored to the basal level after 48 h, whereas, at the lower concentration (25 nM), DCF production increased again at 48 h and then declined to the basal value after 90 h (Figure A.10, right) (Shamoto‐Nagai et al., 2003, copyright John Wiley and Sons).
Figure A.10.
Effect of rotenone on ATP level (left) and on ROS and RNS production (right) in SH‐SY5Y cells. SH‐SY5Y cells were treated with 25 nM (circles) or 50 nM (triangles) of rotenone. *Indicates significant difference from control (p < 0.05) (from Shamoto‐Nagai et al., 2003, figs. 2, 3)
Human neuroblastoma cell line (SK‐N‐MC) exposed to 5 nM rotenone chronically, for 4 weeks caused reduction in GSH by 44%, GSSG by 40%. These effects were not observed after 2 weeks of exposure. Total cellular GSH levels were reduced after 4 weeks of exposure by 50% (Sherer et al., 2002). Similarly, in the same study, 1–2 weeks of treatment did not alter protein carbonyl levels (oxidative protein damage) but exposure for 3–4 weeks caused a large increase in carbonyls in the insoluble fraction by approximately 223% of control. Systemic in vivo rotenone infusion (up to 5 weeks, 3.0 mg/kg per day) modestly elevated soluble protein carbonyls in the rat cortex by approximately 19%, in the striatum by 27% and the largest elevation occurred in the DA neurons of midbrain, around 41% (no effect in cerebellum or hippocampus) (Sherer et al., 2003).
The prolonged treatment with rotenone (3–4 weeks, not 1–2 weeks) caused also a marked increase in 8‐oxo‐dG immune‐reactivity (i.e. oxidative DNA damage) and redistribution of cytochrome c (Sherer et al., 2002).
The same group showed that exposure of SK‐N‐MC cells for 6–8 h to low concentrations of rotenone (100 pM, 1 nM, 10 nM and 100 nM) produced a concentration‐dependent decrease in ATP levels by 0, 2.5, 10, and 32.2% respectively (Sherer et al., 2003).
The oxidative stress (mitochondrial damage) induced by rotenone exposure was confirmed in ex‐vivo studies using brain sections at the level of the substantia nigra that were treated with 50 nM rotenone over 1 week. A significant increase of protein carbonyls (indicative of oxidative damage to proteins; biomarkers of oxidative stress) was observed (~ 25%) when compared to the untreated slices. Exposure to 100 M tocopherol, antioxidant (vitamin E) significantly protected the neurons from the oxidative damage induced by 50 nM rotenone over 1 week (~ 25%), as shown by lower protein carbonyl levels (~ 3%), with very similar effects observed with 20 nM rotenone over 2 weeks (Testa et al., 2005).
The same assays for mitochondrial dysfunction evaluation after exposure to rotenone, MPTP or other chemicals were used through a range of different studies (Betarbet et al., 2000; Sherer et al., 2003) and the role of CI inhibition in PD is discussed in many published reviews (Schapira et al., 1990a,b; Greenamyre et al., 2001; Sanders and Greenamyre, 2013).
Conclusions: It is well documented in human PD brain studies as well as in vivo and in vitro existing data that CI inhibition induces mitochondrial dysfunction as shown by measuring the decreased cellular respiration and induced oxidative damage to protein, lipids and nucleic acids, as well as compromised function of antioxidant defence mechanisms (e.g. decreased levels of reduced glutathione). As discussed above, oxidative damage is largely reversed by antioxidants treatments. These data are largely semi quantitative only, as the full dose‐ and time response curves are available. They indicate that low levels of CI inhibition for long periods of time (4–5 weeks) mostly increase ROS production, having negative effects on DA neurons in SNpc, which seem to be affected more than other neuronal cell types in other brain structures (reviews e.g. by Schapira et al., 1990a,b; Greenamyre et al., 2001; Sanders and Greenamyre, 2013).
A.2.6. Evidence Supporting Taxonomic Applicability
Mitochondrial CI in eukaryotes has highly conserved subunit composition based on protein databases (Cardol, 2011).
The characterisation of induced mitochondrial dysfunction phenotypes in zebrafish was studied in the presence of CI and CII inhibitors (Pinho et al., 2013).
Exposure of Caenorhabditis elegans (C. elegans) to rotenone, reduced bioluminescence (an assay for mitochondrial dysfunction) after both relatively short (2 h) and longer exposures (24 h) to a range of concentrations. A sharp decline in bioluminescence (maximal inhibition) relative to controls occurred at the lowest rotenone concentration of 2.5 μM. This decline in bioluminescence was consistent with reduced cellular ATP (Lagido et al., 2015).
The results obtained from C. elegans exposed to rotenone suggested that chronic exposure to low concentration (2 or 4 μM) caused mitochondrial damage through persistent suppression of mitochondrial biogenesis and mitochondrial gene expression leading to mitochondrial dysfunction that contributed to DA neuron degeneration (Zhou et al., 2013).
Drosophila melanogaster has been proven suitable to study signalling pathways implicated in the regulation of mitochondrial function and integrity, such as the PINK1/parkin pathway (controlling mitochondrial integrity and maintenance), DJ‐1 and Omi/HtrA2 genes (associated with the regulation of mitochondrial functionality). Notably, PINK1, PARKIN, and DJ‐1 genes are associated with recessive forms of PD (Guo, 2012). Drosophila flies lacking DJ‐1 result to be viable, but show an increased sensitivity to oxidative stress induced upon rotenone or Paraquat (an herbicide inducer of CI‐dependent ROS) feeding (Menzies et al., 2005; Meulener et al., 2005, 2006). Moreover, it has been reported in Drosophila that inhibition of CI by mean of sublethal chronic exposure to rotenone (< 750 μM) via the feeding medium caused a selective loss of DA neurons in all of the brain regions and locomotor impairments, while L‐dopa (3,4‐dihydroxy‐l‐phenylalanine) rescued the behavioural deficits (but not neuronal death) (Coulom and Birman, 2004).
MPTP causes Parkinsonism in primates including humans. However, rodents (rats) are much less susceptible to MPTP+ but are fully susceptible to MPP+ (due to the differences in toxicokinetics). In all species, CI inhibition leads to mitochondrial dysfunction. Mitochondrial dysfunction is an universal event occurring in cells of any species (Farooqui and Farooqui, 2012).
References
Alam ZI, et al., 1997. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8‐hydroxyguanine levels in substantia nigra. Journal of Neurochemistry, 69, 1196–203.
Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan‐Gomez C, Bombardieri CR, Armentero MT, Zangaglia R, Pacchetti C, Mastroberardino PG, Blandini F, 2014. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson's disease. Biochimica et Biophysica Acta, 1842, 1385–1394.
Barrientos A, Moraes CT, 1999. Titrating the effects of mitochondrial Complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197. http://www.jbc.org/content/274/23/16188.full.pdf Fig 5 p.16193 © the American Society for Biochemistry and Molecular Biology
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Bernardi P, 1999. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiological Reviews, 79, 1127–55.
Braun RJ. 2012. Mitochondrion‐mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Frontiers in Oncology, 2, 182.
Brown GC, Borutaite V, 2004. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S‐nitrosothiols, Biochimica et Biophysica Acta (BBA) – Bioenergetics, 1658, 1–2.
Cardol P, 2011. Mitochondrial NADH:ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases”. Biochimica et Biophysica Acta, 1807, 1390–1397.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP, 2015. Anti‐oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Molecular Neurodegeneration, 10, 4.
Cheng B, Guo Y, Li C, Ji B, Pan Y, Chen J, Bai B, 2014. Edaravone protected PC12 cells against MPP(+)‐cytoxicity via inhibiting oxidative stress and up‐regulating heme oxygenase‐1 expression. Journal of the Neurological Sciences, 343, 115–119.
Chiu CC, Yeh TH, Lai SC, Wu‐Chou YH, Chen CH, Mochly‐Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS, 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone‐induced cellular and animal models of parkinsonism. Experimental Neurology, 263, 244–253.
Coulom H, Birman S, 2004. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. Journal of Neuroscience, 24, 10993–10998.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA, 2012. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Advances in Experimental Medicine and Biology, 724, 205221.
Cozzolino M, Ferri A, Valle C, Carri MT, 2013. Mitochondria and ALS: implications from novel genes and pathways. Molecular and Cellular Journal of Neuroscienceence, 55, 44–49.
Dexter DT, et al., 1994. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Movement Disorders, 9, 92–97.
Dias V, Junn E and Mouradian MM, 2013. “The role of oxidative stress in parkinson's disease”. Journal of Parkinson's Disease, 3, 461–491.
Farooqui T, Farooqui AA, 2012. Oxidative stress in vertebrates and invertebrate: molecular aspects of cell signalling. Wiley‐Blackwell, Chapter 27, 377–385.
Feng JM, 2006. A common target for parkin and Parkinson's disease toxins. Neuroscientist, 12, 469–476.
Filomeni G, Graziani I, de Zio D, Dini L, Centonze D, Rotilio G, Ciriolo MR, 2012. Neuroprotection of kaempferol by autophagy in models of rotenone‐mediated acute toxicity: Possible implications for Parkinson's disease. Neurobiology of Aging, 33, 767–785.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Graier WF, Frieden M, Malli R, 2007. Mitochondria and Ca2+ signaling: old guests, new functions. Pflügers Archiv, 455, 375–396.
Greenamyre JT, Sherer TB, Betarbet R, Panov AV, 2001. Critical review Complex I and Parkinson's Disease. Life, 52, 135–141.
Guo M, 2012. Drosophila as a model to study mitochondrial dysfunction in Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, 2, pii: a009944.
Halliwell B, 2007. Free Radicals in Biology and Medicine. Journal of Medicinal Chemistry. 4. Oxford University Press.
Hartman P, Ponder R, Lo HH, Ishii N, 2004. Mitochondrial oxidative stress can lead to nuclear hypermutability. Mechanisms of Ageing and Development, 125, 417–420.
Hensley, K, Pye, QN, Maidt ML, Stewart, CA, Robinson KA, Jaffrey F, Floyd, RA, 1998. Interaction of alpha‐phenyl ‐N‐tert‐butyl nitrone and alternative electron acceptor s with complex I indicates a substrate reduction site upstream from the rotenone binding site. Journal of Neurochemistry. 71, 2549–2557.
Höglinger GU, Carrard G, Michel PP, Medja F, Lombès A, Ruberg M, Friguet B, Hirsch EC, 2003. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson's disease. Journal of Neurochemistry, 86, 1297–1307.
Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD, 1992. Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Annals Of Neurology, 32 Suppl, S82‐S87.
Lagido C, McLaggan D, Glover LA, 2015. A screenable in vivo assay for mitochondrial modulators using transgenic bioluminescent caenorhabditis elegans. Journal of Visualized Experiments, 104, 53083.
Lee DH, Kim CS, Lee YJ, 2011. Astaxanthin protects against MPTP/MPP+‐induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food and Chemical Toxicology, 49, 271–280.
Liu W, Kong S, Xie Q, Su J, Li W, Guo H, Li S, Feng X, Su Z, Xu Y, Lai X, 2015. Protective effects of apigenin against 1‐methyl‐4‐phenylpyridinium ion induced neurotoxicity in PC12 cells. International Journal of Molecular Medicine, 35, 739–746.
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD, 1992. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain, 115(Pt 2), 333–342.
Martin LJ, 2011. Mitochondrial pathobiology in ALS. Journal of Bioenergetics and Biomembranes, 43, 569–579.
Mecocci P, et al., 1993. Oxidative damage to mitochondrial DNA shows marked age‐dependent increases in human brain. Annals of Neurology, 34, 609–616.
Menzies FM, Yenisetti SC, Min KT, 2005. Roles of Drosophila DJ‐1 in survival of dopaminergic neurons and oxidative stress. Current Biology, 15, 1578–1582.
Meulener M, Whitworth AJ, Armstrong‐Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM, 2005. Drosophila DJ‐1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Current Biology, 15, 1572–1577.
Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM, 2006. Mutational analysis of DJ‐1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proceedings of the National Academy of Sciences, 103, 12517–12522.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y, 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications, 163, 1450–1455
Nataraj J, Manivasagam T, Justin Thenmozhi A, Essa MM, 2015. Lutein protects dopaminergic neurons against MPTP‐induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutritional Journal of Neuroscience. [Epub ahead of print].
Navarro A, et al., 2009. Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radical Biology and Medicine, 46, 1574–1580.
Ojha S, Javed H, Azimullah S, Abul Khair SB, Haque ME, 2015. Neuroprotective potential of ferulic acid in the rotenone model of Parkinson's disease. Drug Design Development and Therapy, 9, 5499–5510.
Parker WD Jr, Boyson SJ, Parks JK, 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Annals of Neurology, 26, 719–723.
Piert M, Koeppe RA, Giordani B, Minoshima S, Kuhl DE, 1996. Determination of regional rate constants from dynamic FDG‐PET studies in Parkinson's disease. Journal of Nuclear Medicine, 37, 1115‐1122.
Pinho BR, Santos MM, Fonseca‐Silva A, Valentão P, Andrade PB, Oliveira JM, 2013. How mitochondrial dysfunction affects zebrafish development and cardiovascular function: an in vivo model for testing mitochondria‐targeted drugs. British Journal of Pharmacology, 169, 1072–1090.
Rango M, Bonifati C, Bresolin N, 2006. Parkinson's disease and brain mitochondrial dysfunction: a functional phosphorus magnetic resonance spectroscopy study. Journal of Cerebral Blood Flow & Metabolism, 26, 283–290.
Ren Y. et al., 2005. Selective vulnerability of dopaminergic neurons to microtubule depolymerisation. Journal of Biological Chemistry, 280, 434105–434112.
Ramsay RR, Singer TP, 1992. Relation of superoxide generation and lipid peroxidation to the inhibition of NADH‐Q oxidoreductase by rotenone, piericidin A, and MPP+. Biochemical and Biophysical Research Communications, 189, 47–52.
Sanders LH, Greenamyre JT, 2013. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radical Biology and Medicine, 62, 111‐120.
Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT, 2014. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiology of Disease, 70, 214–223.
Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP, 2006. L‐deprenyl protects against rotenone‐induced, oxidative stress‐mediated dopaminergic neurodegeneration in rats. Neurochemistry International, 49, 28–40.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD, 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet, 1, 1269.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD, 1990a. Mitochondrial complex I deficiency in Parkinson's disease. Journal of Neurochemistry, 54, 823–827.
Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD, 1990b. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. Journal of Neurochemistry, 55, 2142–2145.
Schapira AH, 1994. Evidence for mitochondrial dysfunction in Parkinson's disease–a critical appraisal. Movement Disorders, 9, 125–138.
Shamoto‐Nagai M, Maruyama W, Kato Y, Isobe K, Tanaka M, Naoi M, Osawa T, 2003. An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH‐SY5Y cells. Journal of Neuroscience Research, 74, 589–597.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT, 2002. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha‐synuclein metabolism and oxidative damage. Journal of Neuroscience, 22, 7006–7015.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al., 2003. Mechanism of toxicity in rotenone models of Parkinson's disease. Journal of Neuroscience, 23, 10756–10764.
Singer TP, Ramsay RR, 1994. The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochimica et Biophysica Acta, 1187, 198–202.
Takeshige K, 1994. Superoxide formation and lipid peroxidation by the mitochondrial electron‐transfer chain. Rinsho Shinkeigaku, 34, 1269–1271.
Testa CM, Sherer TB, Greenamyre JT, 2005. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Researchearch. Molecular Brain Research, 134, 109–118.
Thomas B, Banerjee R, Starkova NN, Zhang SF, Calingasan NY, Yang L, Wille E, Lorenzo BJ, Ho DJ, Beal MF, Starkov A, 2012. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxid Redox Signal, 16, 855–868.
Tseng YT, Chang FR, Lo YC2, 2014. The Chinese herbal formula Liuwei dihuang protects dopaminergic neurons against Parkinson's toxin through enhancing antioxidative defense and preventing apoptotic death. Phytomedicine, 21, 724–733.
Wang S, He H, Chen L, Zhang W, Zhang X, Chen J, 2015a. Protective effects of salidroside in the MPTP/MPP(+)‐induced model of Parkinson's disease through ROS‐NO‐related mitochondrion pathway. Molecular Neurobiology, 51, 718–728.
Wang H, Liu J, Gao G, Wu X, Wang X, Yang H, 2015b. Protection effect of piperine and piperlonguminine from Piper longum L. alkaloids against rotenone‐induced neuronal injury. Brain Research. pii: S0006‐8993(15)00558‐2. doi: 10.1016/j.brainres.2015.07.029. [Epub ahead of print].
Wu RM, Mohanakumar KP, Murphy DL, Chiueh CC, 1994. Antioxidant mechanism and protection of nigral neurons against MPP+ toxicity by deprenyl (selegiline). Annals of the New York Academy of Sciences, 738, 214–221.
Zhou S, Wang Z, Klaunig JE, 2013. Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. International Journal of Biochemistry and Molecular Biology, 4, 191–200. eCollection 2013.
Zhu J, and Chu CTT, 2010. Mitochondrial dysfunction in Parkinson's disease. Journal of Alzheimer's Disease, 20, S325–S334.
3rd KER: Mitochondrial dysfunction results in an impaired proteostasis
A.3.1. How this Key Event Relationship work
In any cell type, including neurons, the protein homoeostasis (proteostasis) plays a key role in cellular functions. There are two major systems involved in the removal of damaged cellular structures (e.g. defective mitochondria) and misfolded or damaged proteins, the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathway (ALP). These processes are highly energy demanding and highly susceptible to oxidative stress. Upon mitochondrial dysfunction UPS and ALP functions are compromised resulting in increased protein aggregation and impaired intracellular protein/organelles transport (e.g. Sherer et al., 2002; Esteves et al., 2011; Fujita et al., 2014; Song and Cortopassi, 2015; Zaltieri et al., 2015).
A.3.2. Weight of Evidence
The weight of evidence supporting the relationship between mitochondrial dysfunction and impaired proteostasis, including the impaired function of UPS and ALP that results in decreased protein degradation and increase protein aggregation is strong.
A.3.2.1. Biological Plausibility
The biological relationship between Mitochondrial dysfunction and Impaired proteostasis (unbalanced protein homoeostasis) that involves dysregulation of proteins degradation (misfolded or damaged) as well as removal of cell organelles is partly understood. Under physiological conditions, mechanisms by which proteostasis is ensured include regulated protein translation, chaperone assisted protein folding and functional protein degradation pathways. Under oxidative stress, the proteostasis function becomes burdened with proteins modified by ROS (Powers et al., 2009; Zaltieri et al., 2015). These changed proteins can lead to further misfolding and aggregation of proteins (especially in non‐dividing cells, like neurons). Particularly in DA cells, oxidative stress from dopamine metabolism and dopamine auto‐oxidation may selectively increase their vulnerability to CI inhibitors (such as rotenone) and cause additional deregulation of protein degradation (Lotharius and Brundin, 2002; Esteves et al., 2011). As most oxidised proteins get degraded by UPS and ALP (McNaught and Jenner, 2001), mitochondrial dysfunction and subsequent deregulation of proteostasis play a pivotal role in the pathogenesis of PD (Sherer et al., 2002; Fornai et al., 2005; Pan et al., 2008; Dagda et al., 2013).
It is also well documented that increased oxidative stress changes the protein degradation machinery and leads to a reduction of proteasome activity (Lin and Beal, 2006; Schapira, 2006).
A.3.2.2. Empirical support for linkage
Based on the existing in vitro and in vivo data it is suggested that mitochondrial dysfunction impairs protein homoeostasis through oxidative and nitrosative stress resulting in protein aggregation, disruption of microtubule assembly and damaged intracellular transport of proteins and cell organelles.
Mitochondrial dysfunction by rotenone or MPP+ reduces UPS activity
Mitochondrial dysfunction induced by systemic and chronic CI inhibition by rotenone, results in a selective inhibition of proteasomal function in the midbrain (not in cortical or striatal homogenates) of rats that had lost the TH‐positive terminals in the striatum. Initially, proteasomal activity showed an acute increase prior to a decrease by 16–31%, during chronic rotenone exposure (3.0 mg/kg per day, through osmotic pump during 5 weeks). In the same animals a significant and selective increase in ubiquitinated proteins (~ 25%) was observed in the ventral midbrain of lesioned rats, indicating an increase in the proteins levels that have been marked for degradation by UPS. These results were confirmed immunocytochemically, pointing out that ubiquitin levels were elevated selectively in DA neurons present in SNpc (Betarbet et al., 2006).
Nigral neurons in chronically rotenone‐treated rats (up to 5 weeks, infusion of rotenone at 2.5 mg/kg per day) accumulate fibrillar cytoplasmic inclusions that contain ubiquitin and α‐synuclein (the main protein of Lewy bodies observed in PD) (qualitative data, obtained by immunoelectron microscopy) (Betarbet et al., 2000).
Inhibition of proteasomal function was also observed in in vitro systems using SK‐N‐MC human neuroblastoma. Exposure to 5 nM rotenone, for up to 4 weeks caused 60% increase in the levels of ubiquinated proteins, suggesting that chronic exposure to rotenone increased the level of misfolded or oxidised proteins targeted for degradation by UPS (Betarbet et al., 2006).
To determine whether rotenone‐induced proteasomal inhibition was due to CI inhibition or direct effects of rotenone on the UPS, proteasomal activity was determined in SKN‐MC cells expressing the rotenone‐insensitive single‐subunit NADH dehydrogenase of Saccharomyces cerevisiae (NDI1), which acts as a ‘replacement’ for the entire CI in mammalian cells (Seo et al., 2000, 2002; Bai et al., 2001). The obtained results confirmed that rotenone‐induced proteasomal dysfunction is due to CI inhibition and not to direct effects of rotenone on proteasomal function (Betarbet et al., 2006). In the same study the decreased proteasomal activity and an accumulation of ubiquitinated proteins was completely prevented by continuous treatment with α‐tocopherol (62.5 μM added 1 week prior to and continuously thereafter along with 5 nM rotenone) (qualitative data), confirming that oxidative damage played a major role in rotenone‐induced proteasomal dysfunction rather than bioenergetic defects. Indeed, chronic, low levels of rotenone exposure did not changed significantly ATP levels (111.5 ± 1.5% of control), but produced ROS (data not shown in this study). Similar results were published by Shamoto‐Nagai's group (Shamoto‐Nagai et al., 2003).
Rotenone significantly lowered UPS activity in a concentration dependent manner in HEK (human embryonic kidney cells) and SK‐N‐MC human neuroblatoma cells even after 24 h exposure to doses as low as 10 nM. It caused a reduction in the 20S proteasome activity (by 5–25%) and of the 20S proteasome subunit (by 20–60%) (as shown by increase of GFP‐U fluorescence) (Chou et al., 2010). Similar results were obtained using other pesticides that inhibit CI, including pyridaben and fenazaquin (Wang et al., 2006). This effect was mediated by oxidative stress as antioxidants, such as butylated‐hydroxy toluene (BHT), and catalase attenuated rotenone‐induced UPS inhibition. Additionally, nitric oxide (NO) and peroxynitrite contributed to this effect as well, since neuronal nitric oxide synthase (nNOS) inhibitor (LNMMA) attenuated rotenone‐induced proteasome inhibition by 20% (Chou et al., 2010) indicating that both oxidative and nitrative stress can directly inhibit the proteasome activity through increased degradation of proteasome subunits. The same mechanisms of proteasome inhibition were suggested by many other studies (e.g. Szweda et al., 2002; Shamoto‐Nagai et al., 2003; Osna et al., 2004).
CI inhibition‐induced proteasomal dysfunction has been reported in ventral mesencephalic cultures following acute rotenone or MPP+ exposure (Hoglinger et al., 2003). In DA neurones derived from rat (embryonic day 15.5) ventral mesencephalon, it has been shown that proteasome inhibition (by 100 nm epoxomicin) exacerbated the neurotoxicity of CI inhibitors (by mean of rotenone 30 nM, or MPP+ 3 μM, for 24 h). All three proteasomal peptidase activities (i.e. chymotrypsin (CT)‐like, trypsin (T)‐like, and peptidylglutamyl‐peptide hydrolase (PGPH) activity) significantly decreased in cultures upon 6 h treatment with 30 nM rotenone (by 50 + ‐60%) or 30 μM MPP+ (by 25–30%) (Hoglinger et al., 2003).
CI inhibition‐induced proteasomal dysfunction has been reported in human SH‐SY5Y neuroblastoma cells following acute rotenone exposure (Shamoto‐Nagai et al., 2003). After 96 h of incubation with 25 or 50 nM rotenone, the activity was reduced respectively to 28.7% and 21.9% of control, and adding ATP did not increase the activity. After 120 h, the activity was virtually undetectable (with or without added ATP). On the contrary, the levels of the proteins composing proteasome did not change with rotenone treatment (Shamoto‐Nagai et al., 2003).
The ability of rotenone to cause proteasome inhibition via disruption of microtubules (MT) assembly has been also documented. In human embryonic kidney (HEK) and neuroblastoma SK‐N‐MC cells rotenone (10–100–100 nM, 24 h) was found to inhibit 26S UPS activity (by 25%, at 10 nM) (Chou et al., 2010). Rotenone was found to interfere with MT assembly at concentrations as low as 10 nM, providing evidence that there could be additional mechanisms implicated in the rotenone induced UPS inhibition, possibly mediated by nitric oxide (NO). In the same study, nocodazole, a MT disrupter (positive control), strongly inhibited the UPS activity (e.g. 10 μM nocodazole caused ~ 80% decrease of 26S UPS activity) (Chou et al., 2010).
Oxidative stress triggered by the MPP+ inhibited CI (1 mM, for 2–6–24 h) led to a decrease in proteolytic activity, as shown in NT2 human teratocarcinoma cells containing mitochondrial DNA (ρ+) and NT2 cells depleted of mtDNA (ρ0) (Domingues et al., 2008). In particular, MPP+ (1 mM, 2 h) elevated ubiquitinylated protein content (by ~ 3 fold compared to untreated Ctr), and after 24 h induced a significant decrease of chymotrypsin‐like activity (by ~ 30%) and peptidyl‐glutamyl peptide hydrolytic‐like activity (by ~ 75%) compared to untreated cells (Domingues et al., 2008).
Mice following continuous MPTP infusion (1–5–30 mg/kg daily) exhibited inhibition of the UPS (respectively by 40–50–60%) and increased inclusions of ubiquitin and α‐synuclein in the neurons in the substantia nigra (Fornai et al., 2005).
A mouse model of mitochondrial CI deficiency (Ndufs4−/− mice) showed an impaired 20S proteasomal activity (by ~ 50%), leading to increased ubiquitin protein levels (by ~ 40%) in the substantia nigra (not in cortex and hippocampus), increased of ubiquitin+/TH+ neurons (by ~ 2 fold, compared to WT mice), and increased ubiquitinated neurofilaments in the midbrain (values of 1.2–2.8 vs 1.0 in WT) (Song and Cortopassi, 2015).
Human studies
PD patients appear to have an impaired UPS. The presence of aggregated, poly‐ubiquitinated proteins in Lewy Bodies indicates that proteolytic dysfunction and proteotoxicity are critical steps in the pathogenic cascade of PD (Betarbet et al., 2005). In this regard, impairment of proteasomal activity and reduced expression of proteasomal subunits have been reported selectively in substantia nigra of sporadic PD post‐mortem brains (McNaught and Jenner, 2001; McNaught et al., 2003). In particular, in PD, there was a 40.2% reduction in the amount of α‐subunits in the SNc. On the opposite α‐subunits levels were increased by 9.2% in the cerebral cortex and by 29.1% in the striatum in PD compared to Ctr (McNaught et al., 2003). Chymotrypsin‐like, trypsin‐like, and peptidyl glutamyl‐peptide hydrolytic (PGPH) 20/26S proteasomal activities were significantly decreased in the substantia nigra (by 43.9%, 45.9%, and 44.6% respectively) (not in the cortex or striatum) in PD patients. At the same time, in PD there was a marked increase in the levels of PA700 subunits (the 19S regulatory complex of the 26S proteasome) in the frontal cortex and/or the striatum compared to controls, while in the SNpc PA700 subunits resulted decreased up 33%, whereas levels of nigral PA28 were almost undetectable in both normal and PD subjects (McNaught et al., 2003).
Steady‐state levels of soluble AF‐6 (modulates parkin ubiquitin‐ligase activity) have been found significantly lower in the caudate/putamen (~ 66% lower) as well as in the SN of PD patients (~ 66% lower). AF‐6 was also detected in ~ 25% of mature Lewy bodies and in occasional Lewy neurites in the substantia nigra of the four PD brains analysed, and may contribute to the disruption of mitochondrial homoeostasis (Haskin et al., 2013).
HDAC6 has recently been identified by immunocytochemistry as a constituent in Lewy bodies of PD and dementia with LBs (DLB), as well as in glial cytoplasmic inclusions in multiple system atrophy (MSA) (Kawaguchi et al., 2003; Miki et al., 2011; Chiba et al., 2012). HDAC6 is considered a sensor of proteasomal inhibition and a cellular stress surveillance factor. Upon proteasomal inhibition, HDAC6 is relocated and recruited to polyubiquitin‐positive aggresomes. HDAC6 inhibition elicits tubulin acetylation and restores microtubule (MT)‐dependent transport mechanisms in neurons (Richter‐Landsberg and Leyk, 2013).
Basal activity of 20S proteasome was significantly reduced (by ~ 33%) in PD as compared to control fibroblasts. Higher accumulation of ubiquitinated proteins (by ~ 2 fold), representative of impaired 26S proteasome function, were found in PD as compared to Ctr cells at baseline. In the presence of rotenone (20 and 500 μM, 6 h) PD‐derived fibroblasts showed a higher induction of 20S proteasome activity (~ 15% higher) as compared to Ctr fibroblasts, with no significant changes in autophagy (except from increased LC3‐II accumulation in both groups after exposure to 500 μM rotenone) (Ambrosi et al., 2014).
Mitochondrial dysfunction by rotenone or MPP+ deregulates ALP activity
Exposure to rotenone (10 μM, 24 h) induced neurotoxicity in human neuronal SH‐SY5Y cells (number of dead cells was 8 folds higher than Ctr group) and pretreatment with rapamycin (3 μM, 48 h) (strong inducers of autophagy) robustly protected against rotenone‐mediated toxicity (number of dead cells was 3 folds higher than Ctr group) and this was due to the induction of autophagy. Indeed, suppression of autophagy (by silencing of Atg5) blocked the neuroprotection of rapamycin (Pan et al., 2009).
Similar results were produced using kaempferol (6 μM, 1 h prior addition of rotenone) and rotenone (50 nM, max up to 24 h) on SH‐SY5Y cells. Kaempferol was found to counteract rotenone‐induced effects (see KER2) and these protective effects were related to induction of autophagy (6 h kaempferol induced LC3‐II formation, as shown by Western blot) (Filomeni et al., 2012).
Treatment of SH‐SY5Y cells with high doses of rotenone (500 nM, 48 h) induced Atg5–Atg12 dependent autophagy, which leads to lysosomal dysfunction, increased p62 levels, and an aberrant accumulation of α‐synuclein (Pan et al., 2009; Dadakhujaev et al., 2010). In particular, in α‐synuclein expressing SH‐SY5Y cells Atg5–Atg12 were increased by addition of rotenone and rapamycin (100 nM, 48 h). Co‐treatment with rotenone and autophagy inhibitors (e.g. 3‐MA, bafilomycin or wortmannin) similarly diminished the level of Atg5–Atg12 in α‐synuclein expressing cells (western blot analyses) (Dadakhujaev et al., 2010).
A few studies have suggested that rotenone can act as an inducer of autophagic flux. For instance, treating human embryonic kidney cells (HEK 293) and U87 glioma cells with rotenone (50 μM, for 0–72 h) caused cell death (in HEK 293 cells, rotenone induced 30% cell death, after 72 h; in U87 cells, 40%) by upregulating autophagy and mitophagy (as shown by increase of cells with AVOs (indicative of autophagosomes and autolysosomes, analysed by flow cytometry): by ~ 14% in HEK 293 cells, and by ~ 20% in U87 cells, as compared to untreated cells, 0%), a process that is supposed to be triggered by mitochondrial superoxide (Chen et al., 2007).
Increased autophagic flux has been observed in SH‐SY5Y cells and primary cortical neurons treated respectively with 1 μM and 250 nM of rotenone. Rotenone elicited increases in autophagy (~ 2 folds vs Ctr) and mitophagy (i.e. as shown by the percentage of GFP‐LC3 puncta colocalising with mitochondria (~ 4 folds vs Ctr), indicating a preferential increase in ‘mitophagosomes’ relative to total autophagosomes. Additionally, rotenone induced a decrease in p62 (SQSMT1), levels (~ 40% decrease with 250 nM), consistent with increased autophagic flux. This effect was reversed by co‐treating cells with bafilomycin A2, a specific inhibitor of vacuolar‐type H(+)‐ATPase, or by RNAi (knockdown of ATG7 and ATG8/LC3). The mechanism by which LC3 recognises damaged mitochondria in rotenone‐treated neurons involves, among others, the externalisation of cardiolipin and recruitment of LC3 at the mitochondria initiating rotenone induced‐mitophagy and lysosomal‐mediated degradation of mitochondria (Chu et al., 2013).
In the study by Wu et al. (2015) chronically rotenone‐treated rats (male Lewis rats received rotenone 1 mg/kg subcutaneously twice a day for 8 weeks) had a robust loss of TH+ neurons in striatum (~ 50%) and in SNpc (~ 30%). However, in the remaining DA neurons of SNpc, cytoplasmic inclusions containing α‐synuclein were observed (~ 7% of α‐synuclein+/TH+ cells vs ~ 2% in Ctr), probably due to rotenone‐induced decreased degradation of the autophagosomes (upregulation of LC3‐II by ~ 30%, Beclin 1 by ~ 10%, and p62 by ~ 150%, after 24 h rotenone) indicating decreased ALP function. Compared with the control group, the nigral DA neurons of the rotenone‐treated group exhibited an increased diffuse distribution of LAMP2 (~ 15% vs ~ 25% Ctr) and cathepsin D (~ 22% vs ~ 60% Ctr) instead of punctuate pattern, indicating impaired lysosome integrity and a redistribution of cathepsin D from lysosomes to the cytosol. In parallel in vitro studies by the same group showed that PC12 cells exposed to rotenone (500 nM for 24 h) underwent increased protein levels (but not mRNA levels) of α‐synuclein (~ 4.5 folds vs Ctr), indicating an impairment of protein degradation. In TEM pictures, the majority of neurons displayed mitochondrial swelling, crista fragmentation, and accumulation of double membrane structures containing damaged mitochondria, which were stalled autophagosomes (Wu et al., 2015).
Similar results, showing impaired autophagic flux resulting in α‐synuclein accumulation and the rupture of lysosomes in neuronal cell lines exposed to rotenone have been described in many other studies (e.g. Mader et al., 2012; Sarkar et al., 2014).
Rotenone produced bidirectional effects on macroautophagy (decrease or increase). This may be attributed to differences in the dosage, the duration, and cell type which can produce variable levels of ROS and mitochondrial damage (Chen et al., 2007; Pan et al., 2009; Dadakhujaev et al., 2010; Filomeni et al., 2012; Mader et al., 2012).
MPP+ (2.5 mM, 24–48 h) increased autophagy (~ 14 folds increase vs Ctr, of LC3‐II) and mitochondrial loss in SH‐SY5Y cells (a DA neuronal cell line widely used as a cell culture model of PD) by increased MAP kinase signalling (MEK inhibition by UO126 reversed by both autophagy and mitochondrial loss elicited by MPP+) (Zhu et al., 2007).
Another study from the same group showed that longer MPP+ treatment (250 μM, 2 weeks) induced formation of enlarged, coarse GFP‐LC3 puncta, in a time‐ and dose‐dependent manner (~ 1.8% of cells presenting coarse GFP‐LC3 puncta, vs ~ 0.2% in Ctr, at 14 days with 250 μM rotenone) (Zhu et al., 2012).
An in vitro study on MN9D cells (a fusion of embryonic ventral mesencephalic and neuroblastoma cells, used as a model of DA neurons) showed that MPP+ (50 μM, for 24 h) blocked autophagic flux, as evidenced by increased steady‐state levels of p62 (qualitative data, Western blot), increased of authophagic vacuoles numbers (~ 3 folds vs Ctr) along with lysosomal depletion and dysfunction presumably due to leakage of lysosomes, impaired lysosomal biogenesis, and increased proteasomal‐mediated degradation of proteins (as shown by time‐dependent increase of ubiquitinated proteins, by IC) (Lim et al., 2011).
In another study human neuroblastoma BE‐M17 cells were treated with MPP+ (0.25–2.5 mM, 24 h); Lamp1 protein levels were decreased in a dose‐dependent manner in MPP+‐treated cells (by ~ 40% at 2.5 mM), without concomitant decreases in mRNA expression levels. Also, LC3‐II increased in a dose‐dependent manner with MPP+ treatment (~ 3000% increase at 2.5 mM vs Ctr), indicating lysosome depletion and autophagosome accumulation upon MPP+ treatment. These data were confirmed in vivo: lysosomal depletion and accumulation of autophagosomes (as shown by ~ 600% increase of LC3‐II, and ~ 40% decrease of Lamp1, after 1 day of MPTP injection compared to saline) occurred also in MPTP‐intoxicated mice (30 mg/kg per day, for five consecutive days) (Dehay et al., 2010).
Other in vivo data support a negative role of MPTP on autophagic flux. Mice were i.p. injected with 2 mg/ml MPTP (30 mg/kg) for 7 days. Suppression of autophagic flux induced by MPTP (~ 20% reduction vs Ctr) was detrimental to neuronal survival (as shown by ~ 60% decrease of TH+ neurons). Treating mice with the autophagy inducer rapamycin after 7 days of MPTP treatment (daily i.p. injections of 2 mg/mL MPTP (30 mg/kg) for 7 days, followed by 0.1 ml of 20 μg/ml rapamycin by i.v. for an additional 7 days), significantly increased the number of surviving dopamine neurons (~ 60% TH+ neurons vs ~ 30% with MPTP alone, as compared to Ctr 100%) and the levels of TH protein (~ 75% vs ~ 60% with MPTP alone, as compared to Ctr 100%) and decreased the levels of α‐synuclein aggregates (~ 210% of α‐synuclein protein level, vs ~ 300% with MPTP alone, as compared to Ctr 100%) (Liu et al., 2013).
Treating mice with the autophagy inducer rapamycin after 7 days of MPTP treatment (daily i.p. injections of 2 mg/ml MPTP (30 mg/kg) for 7 days, followed by 0.1 mL of 20 μg/mL rapamycin by i.v. for an additional 7 days), significantly increased the number of surviving dopamine neurons (~ 75% of TH protein level vs ~ 60% with MPTP alone) and decreases the levels of α‐synuclein aggregates (~ 210% of α‐synuclein protein level, vs ~ 300% with MPTP alone) (Liu et al., 2013).
MPP+ induced dysregulation of macroautophagy in neurons is discussed in recently published reviews (e.g. Cherra et al., 2010; Jiang et al., 2010).
The potential other mechanisms by which rotenone or MPTP induce mitochondrial dysfunction are furEsteves et al., 2011; ther discussed in recent publications (e.g. Esteves et al., 2011; Dagda et al., 2013).
Human studies
In PD patient postmortem cortical tissues, levels of oligomeric α‐synuclein in SNpc (~ 1000% vs Ctr samples) and expression of LC3‐II levels (~ 130% vs Ctr samples) were upregulated (Yu et al., 2009) (for further info, see the review from Vekrellis et al., 2011).
The pathological observations in PD autopsy brains showed that LC3‐II levels were elevated in the SNpc and amygdala of PD brain samples, suggesting an increase in macroautophagy (but they did not reach statistical significance). LC3 colocalised with α‐synuclein in most LBs and Lewy neurites in PD SNpc as well as in small punctate α‐synuclein immunoreactive inclusions (IC images) (Alvarez‐Erviti et al., 2010).
Analogously, another study reported that brain homogenates derived from the temporal cortex of dementia with LB (DLB) patients vs non‐demented controls were characterised by higher levels of both mTor (~ 130% vs Ctr) and p‐mTor (~ 10 folds higher than Ctr), and levels of Atg7 (molecular initiator of autophagy) were moderately reduced in DLB cases compared to Ctr (~ 40% lower than Ctr). Consistent with the studies in human brains, levels of both mTor and p‐mTor were increased in the membrane fractions from brains of α‐synuclein tg mice compared to non tg controls (respectively, by ~ 250% and ~ 200% vs Ctr), and levels of Atg7 were reduced in α‐synuclein tg brains compared to non tg controls (~ 75% less than Ctr) (Crews et al., 2010).
Another study showed that post‐mortem brain samples derived from PD patients, compared to age‐matched controls, presented significant reductions of LAMP1, CatD, HSP73, and 20S proteasome (calculated by optic density (OD) measures) (Chu et al., 2009).
| Group | LAMP1 OD | CatD OD | HSP73 OD | 20S proteasome OD |
|---|---|---|---|---|
| Age‐matched control | 2069.10 ± 329.52 | 1809.35 ± 533.47 | 2604.92 ± 494.56 | 1660.84 ± 229.87 |
| PD | 1261.54 ± 107.77 | 1094.64 ± 378.10 | 1799.27 ± 376.19 | 1172.65 ± 273.28 |
These data globally indicate that the functions of both the UPS and ALP systems seem compromised in PD patients.
HDAC6, which plays a central role in autophagy by controlling the fusion process of autophagosomes with lysosomes, has recently been identified as a constituent in Lewy bodies of PD and glial cytoplasmic inclusions of multiple system atrophy (Richter‐Landsberg and Leyk, 2013).
Impaired UPS and ALP function leads to α‐synuclein aggregation
α‐synuclein is one of the most abundant neuronal proteins (Vekrellis et al., 2011). Several PD‐related mutations and environmental toxicants cause autophagy dysfunction and lead to the accumulation of misfolded proteins in DA neurons, including α‐synuclein. Both monomeric and aggregated forms of α‐synuclein can be degraded by macroautophagy, whereas only wild‐type α‐synuclein (not Ala30Pro, Ala53Thr and Glu46Lys mutant forms) is degraded by the process of chaperone‐mediated autophagy (CMA) (Vekrellis et al., 2011).
Rotenone‐induced α‐synuclein aggregation has the ability to inhibit proteasome activity due to its propensity to assemble into filaments (as reviewed in Zaltieri et al., 2015). In particular, expression of α‐synuclein was found to inhibit proteasome activity in SH‐SY5Y cells. Increased levels of GFP‐CL1 band were observed in cells coexpressing GFP‐CL1 and α‐synuclein (~ 9000 arbitrary units (au) vs ~ 500 au in DMSO‐Ctr), indicating that proteasome activity is inhibited effectively by expression of α‐synuclein (Nonaka and Hasegawa, 2009).
By using stable PC12 cell lines expressing wild‐type (WT) or A53T mutant human α‐synuclein it has been shown that cells expressing mutant α‐synuclein showed: (1) disruption of the ubiquitin‐dependent proteolytic system, manifested by small cytoplasmic ubiquitinated aggregates and by an increase in polyubiquitinated proteins (qualitative data); (2) marked accumulation of autophagic‐vesicular structures (qualitative data); (3) reduction of lysosomal hydrolysis and chymotrypsin‐like proteasomal function (by ~ 30%, compared to WT) (Stefanis et al., 2001).
Rotenone‐ (or MPP+)‐induced inhibition of CI results in calcium (Ca2+) release from mitochondria. Calcium rise and oxidative stress cooperatively can promote α‐synuclein aggregation (Nath et al., 2011; Follett et al., 2013; Goodwin et al., 2013).
For instance, to investigate the influence of raised Ca2+ in response to plasma membrane depolarisation on the aggregation of a‐synuclein, HEK293T and SH‐SY5Y neuroblastoma cells have been used and depolarised by addition of KCl to the cell culture medium. After KCl treatment (50 mM) increase of cellular Ca2+ was observed (~ 90% increase 20 min after KCl treatment), leading to the formation of frequent perinuclear a‐synuclein focal aggregates at 26–74 h post‐treatment (qualitative IC images). By adding TMO (a selective T‐type Ca2+ channel blocker) no a‐synuclein aggregates were detected (Follett et al., 2013).
Similarly, increased intracellular free Ca2+ (obtained by treating cells with either calcium ionophore or thapsigargin) induced the formation of α‐synuclein aggregates in α‐synuclein‐GFP‐transfected 1321N1 glioma cells (~ 65% increase compared to Ctr‐untreated cells) (Nath et al., 2011).
On the other hand, α‐synuclein can control mitochondrial calcium homoeostasis by enhancing endoplasmic reticulum‐mitochondria interactions. Silencing of endogenous α‐synuclein (siRNA‐α‐syn) in HeLa cells was found to impair mitochondrial Ca2+ transients (~ 35% decrease compared to Ctr‐scrambled siRNA) and morphology (Calì et al., 2012). Also, α‐synuclein oligomerisation exacerbates calcium dysregulation by increasing mitochondria permeability transition (Danzer et al., 2007). Therefore, it is possible that mitochondrial dysfunction‐induced calcium rise precede the onset of α‐synuclein accumulation leading to UPS inhibition (Chou et al., 2010).
It has been demonstrated that rotenone increased the intracellular calcium levels, triggering aggregation and phosphorylation of α‐synuclein in a calcium‐dependent manner. The aggregation of α‐synuclein in PC12 cells following rotenone exposure was observed in a dose and time‐dependent manner (1, 10 and 100 nM for 48 h, 3 days, 1 and 3 weeks) (~ 4 fold increase of α‐syn with 100 nM rotenone for 48 h, vs Ctr; and also, ~ 2.5 fold increase of α‐syn with 1 nM rotenone for 1 week, vs Ctr) as evaluated via a variety of methods, including western blotting, immunofluorescence and electron microscopy. The observed attenuation of autophagy and α‐synuclein aggregation was reversed by scavenging calcium (by using the calcium chelator BAPTA at 10 μM). Aggregated α‐synuclein is typically degraded by autophagy, but rotenone impaired this process (Yuan et al., 2015).
Under physiological conditions, α‐synuclein is degraded by both the proteasome and autophagy. Mutant α‐synuclein inhibits ALP functioning by tightly binding to the receptor on the lysosomal membrane for autophagy pathway control (e.g. Betarbet et al., 2000; Pan et al., 2009).
The strongest evidence supporting that mitochondrial dysfunction precedes the onset of α‐synuclein pathology derives from studies on rotenone and MPTP in which repetitive exposure of rodents and monkeys to these chemicals via oral, intraperitoneal, intragastric, or nasal administration resulted in the pathological accumulation of α‐synuclein in central as well as peripheral neurons (Mandel et al., 2004; Cannon et al., 2009; Drolet et al., 2009; Pan‐Montojo et al., 2012, 2010; Tristão et al., 2014). For example, male Lewis rats were injected with rotenone (2.0 mg/kg, i.p.) and sacrificed at 0, 4, 8, 16, or 32 h after injection and showed α‐synuclein and poly‐ubiquitin accumulation and aggregation (as shown by IHC data) (Cannon et al., 2009).
Drolet and colleagues injected rats with rotenone (2.0 mg/kg, 1.0 ml/kg, i.p. 5 injections/week for 6 weeks) and found formic acid‐resistant α‐synuclein aggregates in the small intestine myenteric plexus, particularly 6‐months after the last rotenone injection (3.5 median, vs 2.0 in Ctr) (Drolet et al., 2009).
Mandel et al., injected male C57BL mice with MPTP (24 mg/kg per day, i.p. for 5 days) and found α‐synuclein aggregates (IHC data), which were decreased by using the radical scavengers apomorphine (injected s.c. at 10 mg/kg per day) or epigallocatechin‐3‐gallate (EGCG, given alone orally, 2 mg/kg per day) for 10 days) or a combination of both (Mandel et al., 2004).
Inhibition of the mitochondria respiratory chain induces oxidative stress that in turn leads to lipid peroxidation of cellular and vesicular membranes at synaptic sites, resulting in dysfunction of neurotransmitter release. These effects facilitate α‐synuclein conformational changes, such as accumulation, and aggregation. It has been demonstrated that synaptic dysfunction (caused by mitochondrial dysfunction) triggered the accumulation of α‐synuclein (Nakata et al., 2012).
Also, alterations of mitochondrial fission or dynamics can reduce synaptic mitochondrial load and impair neuronal function by hindering the proper energy demand to ensure synaptic function. Mitochondrial behaviours, especially those regulated by neuronal activity and synapse location, determine their distribution in the axon (Obashi and Okabe, 2013). These observations support the idea that mitochondrial dysfunction can affect synaptic environment and consequently result in α‐synuclein accumulation at synapses (Zaltieri et al., 2015).
It was found that continuous administration of MPTP produced formation of nigral inclusions immunoreactive for ubiquitin and α‐synuclein (Fornai et al., 2005). Mice were implanted with osmotic pump to deliver MPTP‐HCl. Delayed and prolonged inhibition of striatal proteasome activity (i.e. 40–50‐60% inhibition of UPS) occurred after continuous MPTP administration (respectively, 1–5–30 mg/kg MPTP daily) for the indicated time periods (Figure A.8) (Fornai et al., 2005). Continuous MPTP infusions caused also a long‐lasting activation of glucose uptake. Additionally, in mice lacking α‐synuclein, the MPTP‐induced inhibition of the UPS system and the production of inclusion bodies were reduced (e.g. Ctr mice showed ~ 40% inhibition of post‐glutamyl peptidase (PGPH) activity, vs ~ 13% inhibition observed in α‐synuclein KO mice) (Figure A.9), suggesting that α‐synuclein could play an important role in UPS inhibition induced by MPP+ (Fornai et al., 2005). These data suggest that continuous, low‐level exposure of mice to MPTP causes a Parkinson‐like syndrome in a α‐synuclein‐dependent manner (Fornai et al., 2005).
These results are supported by other studies showing that α‐synuclein−/− mice are resistant to MPTP toxicity (Dauer et al., 2002; Drolet et al., 2004). MPTP exposure (0.5, 5, 50 μM, 48 h) increases in a dose‐dependent manner the α‐synuclein protein level in mesencephalic neurons in culture (e.g. ~ 70% increase at 5 μM vs Ctr) (Duka et al., 2006).
Increased expression of α‐synuclein predisposes DA neuronal cells to proteasomal dysfunction (~ 50% decrease compared to Ctr‐vector cells) (Sun et al., 2005).
Accumulation/overexpression of α‐synuclein, both wild type and mutant, potentiates inhibition of proteasomal activity. Cells expressing mutant α‐synuclein showed a reduction of lysosomal hydrolysis and chymotrypsin‐like UPS function (by ~ 30%, compared to WT) (Stefanis et al., 2001).
Proteasomal inhibition (by mean of lactacystin, a proteasome inhibitor, used at different concentrations for 24 h) contributes to the accumulation of α‐synuclein as it has been described by immunostaining in PC12 cells (Rideout et al., 2001) and in primary mesencephalic neurons (McNaught et al., 2002).
α‐Synuclein levels were selectively increased in the ventral midbrain (VMB) region of rotenone‐infused rats with or without lesion (~ 110% increase vs Ctr) (Figure A.3) (Betarbet et al., 2006). Rotenone was administered up to 5 weeks, at 2.5 mg/kg per day. Additionally, 4 weeks of in vitro rotenone exposure (5 nM, on SK‐N‐MC human neuroblastoma cells) increased α‐Synuclein levels by 24%, while lactacystin (9 μM, overnight) did not induce any detectable changes in α‐synuclein levels. α‐Tocopherol attenuated the rotenone‐induced increase in α‐synuclein (comparable to Ctr) (Figure A.10). Furthermore, levels of ubiquitinated proteins detected in solubilised protein fractions from SK‐N‐MC cells resulted increased (by 60%) with rotenone treatment (5 nM), and even more (by 484%) with rotenone combined with lactacystin (Figure A.11) (Betarbet et al., 2006).
CI inhibition‐induced proteasomal dysfunction has been reported in human SH‐SY5Y neuroblastoma cells following acute rotenone exposure (Shamoto‐Nagai et al., 2003). The proteasome activity decreased in the cells treated with rotenone (25 or 50 nM) in a time‐ and dose‐dependent way. ATP addition restored the reduction of proteasome activity in the cells treated with 25 nM rotenone for 72 h. However, after 96 h of incubation with 25 or 50 nM rotenone, the activity was reduced respectively to 28.7% and 21.9% of control, and adding ATP did not increase the activity. After 120 h, the activity was virtually undetectable (with or without added ATP) (Figure A.6). On the contrary, the levels of the proteins composing proteasome did not change with rotenone treatment (Shamoto‐Nagai et al., 2003).
Figure A.11.
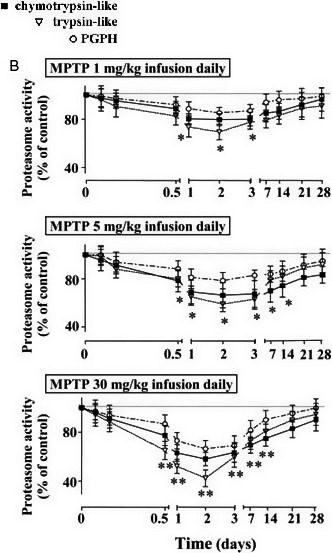
Dose‐ and time‐dependent striatal proteasome activity after MPTP continuously infused up to 28 days measured by relative chymotrypsin‐like, trypsin‐like, and peptidyl‐glutamyl‐peptide hydrolysing (PGPH) proteasome activities in mice. Delayed and prolonged inhibition of proteasome activity after continuous MPTP administration (1, 5 or 30 mg/kg MPTP daily) for the indicated time periods. Asterisks indicate statistically significant differences (p < 0.05) from baseline proteasome activity (single asterisk) or from both baseline proteasome activity and activity after lower MPTP doses (1 and 5 mg/kg, daily, double asterisk; n = 5 mice) (Fornai et al., 2005, fig. 2B, copyright The National Academy of Sciences)
Cytoskeletal damage further enhances disturbed proteostasis
α‐Synuclein can trigger hyperphosphorylation of Tau. Treatment of primary mesencephalic neurons acutely (48 h) or subchronic treatment of wild‐type (WT) mice with MPP+/MPTP results in selective dose‐dependent hyperphosphorylation of Tau at Ser396/404 (p‐Tau). The presence of α‐synuclein was absolutely mandatory to observe MPP+/MPTP‐induced increases in p‐Tau levels, since no alterations in p‐Tau were seen in transfected cells not expressing α‐synuclein or in α‐synuclein−/− mice. MPP+/MPTP also induced a significant accumulation of α‐synuclein in both mesencephalic neurons and in WT mice striatum. Subchronic MPTP exposure increased phosphorylated‐Tau in striatum of WT (but not α‐syn−/− mice) causing microtubule (MT) cytoskeleton instability that affects cellular microtubule transport (including axonal transport) (Duka et al., 2006; Qureshi and Paudel, 2009). For instance, MPTP was found to elicit an increase of phosphorylated Tau at Ser262 by 2.8‐, 4.5‐, 4.6‐, and 4.0‐fold higher in 1, 5, 25, and 50 μM MPTP‐treated cells than the basal level observed in Ctr/vehicle‐treated cells, respectively. Additionally, MPTP caused a dose‐dependent increase in the intracellular α‐synuclein level in M17 human neuroblastoma cells (~ 3.5 fold increase in cells treated with 25 μM MPTP vs Ctr) (Qureshi and Paudel, 2009). These results were confirmed by other studies (e.g. Dauer et al., 2002; Drolet et al., 2004).
α‐Synuclein accumulation followed by MT depolymerisation induces disruption in axonal transport, which leads to an accumulation of damaged organelles, aggregated/misfolded proteins and impaired vesicular release. Dopamine is leaking from the vesicles to the cytosol promoting an increase in oxidative stress, potentiated by dopamine oxidation (Feng, 2006; Kim et al., 2007). When microtubule network is disrupted, the amount of free tubulin increases, triggering α‐synuclein fibrillisation (Payton et al., 2001).
Axonal transport might be impaired by misfolded α‐synuclein through perturbation of microtubule assembly (Lee et al., 2002; Chen et al., 2007; Esposito et al., 2007), especially together with MAPT protein (Giasson et al., 2003; Qureshi and Paudel, 2011). It induces not only microtubule disruption but also impairs microtubule‐dependent trafficking (Lee et al., 2006). MT‐dependent transport is important for maintaining the Golgi structure, and thus, depolymerisation of the MT leads to a specific pattern of Golgi fragmentation (Cole et al., 1996). When the MT network was disrupted by nocodazole treatment (5 μg/mL) or α‐synuclein was overexpressed, this normally compact organelle was fragmented and dispersed (IC images) as shown in COS‐7 cells (Lee et al., 2006). Similarly, overexpression of α‐synuclein in differentiated SH‐SY5Y cells caused Golgi fragmentation (e.g. ~ 190% increased fragmented Golgi at 12 m.o.i. (multiplicity of infection) of α‐synuclein vs Ctr) (Lee et al., 2006).
It was found that α‐synuclein mutants associated with PD exhibit reduced transport in neurons, as shown in rat primary neuronal cortical cultures transfected with wild‐type (WT), A53T or A30P α‐synuclein. For instance, the rate of transport (expressed in μm/h) was reduced of ~ 55% and ~ 60% after 3–4 h for A30P and A53T respectively (vs Ctr‐WT) (Saha et al., 2004).
Damaged cytoskeletal proteins disrupt also mitochondrial trafficking. Mitochondria use cytoskeletal proteins as tracks for their directional movement (Nogales, 2000). The cytoskeletal system regulates not only mitochondrial movement but also their morphology and function. Therefore, damage to microtubules perturbs transport of mitochondria through axons, increasing their retrograde movement. These changes in mitochondria dynamics lead to a decrease of mitochondria numbers in axons and mitochondria accumulation in cell bodies (Miller and Sheetz, 2004; De vos et al., 2007). Depletion of mitochondria quantity and function in axons occurs in neurodegenerative disorders (Brownlees et al., 2002; Stamer et al., 2002).
Since mitochondria are ATP suppliers and microtubules need ATP to accomplish their function, mitochondrial dysfunction has a profound effect on axonal transport and function (De Vos et al., 2008).
Mitochondrial dysfunction may damage mitochondrial trafficking through calcium dysregulation. Cytosolic Ca2+ is one of the best‐studied regulators of mitochondrial movement. Elevation of cytosolic Ca2+ stops both the anterograde and retrograde trafficking of mitochondria in neurons and in many cell lines (Chang et al., 2006; Szabadkai et al., 2006). In H9c2 cells simultaneous measurements of free Ca2+ levels and mitochondrial dynamics showed that 50% reductions in mitochondrial movement occurred at concentrations of approximately 400 nM Ca2+, and a complete arrest in the low micromolar range (Yi et al., 2004; Saotome et al., 2008). These are indirect proofs suggesting that inhibition of CI, followed by mitochondrial dysfunction, could damage mitochondrial trafficking. Also, chronic exposure to rotenone (50 nM at different times of exposure) was reported to reduce mitochondrial movement in differentiated SH‐SY5Y cells (e.g. ~ 30% reduction of mitochondrial movement (μm/s) after 8 days of rotenone treatment vs Ctr) (Borland et al., 2008).
A.3.3. Uncertainties or inconsistencies
The exact molecular link from mitochondrial dysfunction to disturbed proteostasis is not known. It is not clear which is the oxidative modification that drives the process.
The sequence of events taking place after inhibition of CI is not entirely clear (Zaltieri et al., 2015). Some studies suggest that induced oxidative stress leads to α‐synuclein aggregation that triggers proteosomal dysfunction (Betarbet et al., 2006). Such order of events is suggested to take place in vivo (McNaught and Jenner, 2001). However, in other studies opposite sequence of events is proposed suggesting that first proteosomal dysfunction take place that leads to α‐synuclein aggregation.
A vicious circle is observed here as α‐synuclein aggregation potentiates proteosomal dysfunction and v/v. In this vicious cycle it is difficult to establish exact quantitative relationship of these two events.
Whether α‐synuclein is a substrate for proteasome remains controversial since both positive and negative data have been reported (Paxinou et al., 2001). Furthermore, polyubiquitination of α‐synuclein, a prerequisite for 26S proteasomal degradation has yet to be reported (Stefanis et al., 2001). It is also not clear whether polyubiquitination of α‐synuclein is necessary for its degradation. However, α‐synuclein gets targeted by the UPS in the SHSY5Y neuroblastoma cell line. Phosphorylated α‐synuclein gets targeted to mono‐ or di‐ubiquitination in synucleinopathy brains (Hasegawa et al., 2002), but it is not clear if this modification can play any role in proteasomal degradation since monoubiquitination of proteins serves mainly as a signal for endocytosis or membrane trafficking.
On the contrary to the increased α‐synuclein levels observed in the midbrain, decreased α‐synuclein levels were found in the cerebellums of PD patients when compared to controls, suggesting an imbalance of α‐synuclein levels in different parts of the brain (Westerlund et al., 2008).
Although mitochondrial alterations have been reported in PD patients (Ikawa et al., 2011) and disease models, it is not clear whether they represent a primary pathogenic mechanism. In particular, the critical interplay between mitochondrial dysfunction and oxidative stress, which has been widely reported in PD (Dias et al., 2013) and could constitute either a cause or a consequence of mitochondrial damage, hampers an effective comprehension of the above‐mentioned studies. Oxidative stress can constitute a bridge connecting mitochondrial dysfunction to the induction of α‐synuclein misfolding, aggregation, and accumulation, but otherwise it may be also triggered by these latter events that in turn could induce mitochondrial alterations (Zhu and Chu, 2010; Dias et al., 2013).
It is still unclear whether the involvement of α‐synuclein in chronic MPTP toxicity reflects a physiological function for α‐synuclein that has been activated in the wrong context, or whether α‐synuclein produces an accidental pathogenicity that contributes to MPTP toxicity but is unrelated to the normal function of α‐synuclein (Fornai et al., 2005).
The inconsistent effects of MPP+ on autophagy (up or down regulation) are reported. It may be attributed to differences observed between immortalised cell lines and primary neurons, different timing or dose. While dysregulation of autophagy is always described, the direction is not clear. Further studies are required to clarify this issue.
MPTP administration does not induce Lewy body formation (in contrast to rotenone) characteristic of PD, even after repeated injections (Dauer et al., 2002; Drolet et al., 2004).
There is also controversy over whether the increase in autophagic markers is protective or, on the contrary, causative of neuronal death.
MPP+ may have effects apart from CI inhibition, e.g. on microtubules but it is still unclear whether this is a primary effect. Indeed, MPP+ binds to microtubules in PC12 cells and inhibits their polymerisation and stability (Cappelletti et al., 1999, 2001).
It is not clear whether microtubules disruption may be associated with α‐synuclein aggregation since tubulin was shown to co‐localise with α‐synuclein in Lewy bodies. Furthermore, tubulin folding is dependent on ATP and GTP hydrolysis, and mitochondrial dysfunction with subsequent energy failure could trigger microtubules disruption. Cytoskeletal microtubule (MT) injury is likely to be responsible for altered rearrangement and movement of cell organelles, being a common feature of several neurodegenerative diseases including PD (Mattson et al., 1999; Wade, 2009).
It is not clear whether rotenone could cause microtubules depolymerisation in vivo and in vitro (Brinkley et al., 1974) by binding to the colchicine site on tubulin heterodimers (Marshall et al., 1978). Ren and Feng (2007) found that microtubule depolymerisation induced by rotenone caused vesicle accumulation in the soma and kills neurons.
A.3.4. Quantitative evaluation of KERs
As described in the studies above (Empirical support for linkage) a quantitative or semiquantitative relationship has been established between rotenone‐induced mitochondrial dysfunction and the impairment of UPS/ALP function. Below some representative studies are reported as examples for how such quantitative evaluations can be performed.
Human neuroblastoma SK‐N‐MC or human embryonic kidney (HEK) cells were exposed to rotenone at 100 nM for 24 or 48 h (for further details, see Chou et al., 2010).
Examples of quantitative evaluation of this KER
Figure A.12.
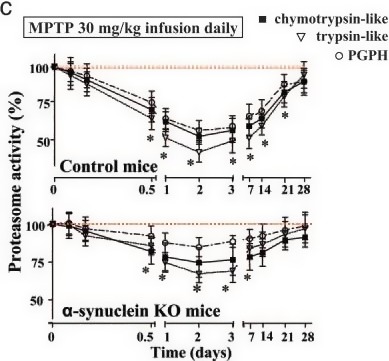
Effect of a α‐synuclein deletion on MPTP toxicity. Proteasome activity in control and α‐synuclein KO mice continuously infused for 28 days with MPTP (30 mg/kg of body weight daily, striatum concentration approximately 13 μM). Proteasome activities in the substantia nigra are depicted as per cent of control (means ± SEMs) as a function of time after beginning of the infusions (five mice per group). Asterisks indicate statistically significantly different values (p < 0.05) from controls (Fornai et al., 2005, fig. 5c, copyright The National Academy of Sciences)
Figure A.13.
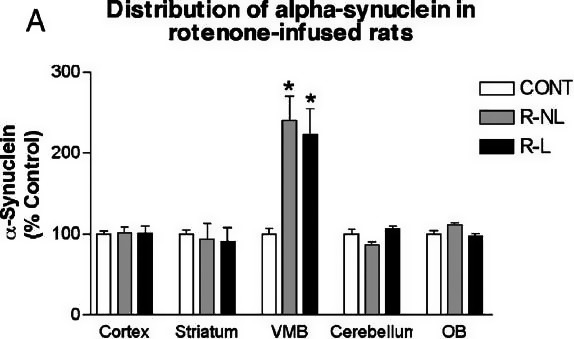
α‐Synuclein levels were selectively increased in the ventral midbrain (VMB) region of rotenone‐infused rats with or without lesion. α‐Synuclein levels, as determined from Western blot analysis, from rotenone‐treated rats were expressed as a percentage of values from control vehicle‐infused rats. Results are mean ± SEM (n = 3 control, 6 rotenone with lesion, 3 rotenone with no lesion) *p < 0.05 vs. vehicle‐infused rats (Reprinted from Betarbet et al., 2006, fig. 3A, copyright (2006) with permission from Elsevier)
Figure A.14.
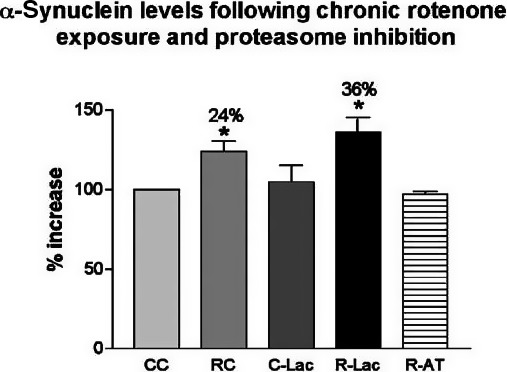
Bar graph showing the effects of rotenone and lactacystin on α‐synuclein levels after 4 weeks of rotenone exposure (5 nM) in vitro, on SK‐N‐MC human neuroblastoma cells. Rotenone alone increased α‐synuclein levels, but lactacystin alone did not. α‐Tocopherol attenuated the rotenone‐induced increase in α‐synuclein. Results are mean ± SEM (n = 4). *p < 0.05 vs. solvent‐treated cells. CC, control cells; RC, rotenone‐treated cells; C‐Lac or CL, lactacystin‐treated cells; R‐lac or RL, rotenone and lactacystin‐treated cells; R‐AT, rotenone and α‐tocopherol‐treated cells (Reprinted from Betarbet et al., 2006, fig. 5B, copyright (2006) with permission from Elsevier)
Figure A.15.
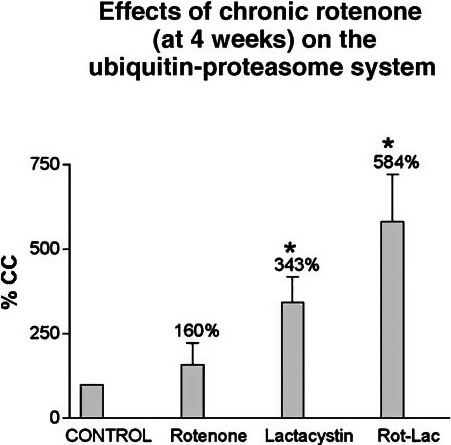
Levels of ubiquitinated proteins were estimated in solubilised protein fractions from SK‐N‐MC cells collected at the end of each week of rotenone treatment (5 nM), using gel electrophoresis and immunoblotting. Quantitative analysis demonstrated significant increases in ubiquitinated protein levels 4 weeks after rotenone treatment and after proteasomal inhibition with lactacystin. Band intensities were expressed as % of control. Results represent mean ± SEM. *p < 0.05 compared to control (Reprinted from Betarbet et al., 2006, fig. 8C, copyright (2006) with permission from Elsevier)
Figure A.16.
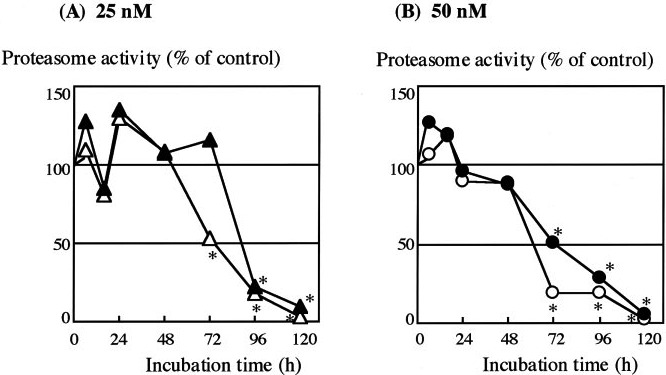
Effects of rotenone on the activity of proteasome. Proteasome activity in the cytoplasmic fraction of cells treated with 25 nM (A) or 50 nM (B) rotenone was measured fluorometrically in the absence (open triangles and circles) or presence (solid triangles and circles) of exogenously added ATP (2 mM) (Reprinted from Shamoto‐Nagai et al., 2003, fig. 6, copyright (2003) John Wiley and Sons)
Table A.2.
Quantitative evaluation of the KER
| KE (upstream) Mitochondrial dysfunction (rotenone, nM) | KE3 (downstream) Impaired proteostasis UPS inhibition (% approx.) measured by: | Comments | References | |
|---|---|---|---|---|
| 26S UPS activity | + catalase (antioxidant) | HEK cells exposed for 24 h | Chou et al. (2010) | |
| 10 | 24 | Not done | ||
| 100 | 48 | Increased UPS activity by 40% | ||
| 1000 | 60 | Not done | ||
| 20S proteasome activity | SK‐N‐MC human neuronal cell line (exposed for 24 h) | Chou et al. (2010) | ||
| 1 | 8 | |||
| 50 | 4 | |||
| 100 | 18 | |||
| 500 | 22 | |||
| 1000 | 24 | |||
| 20S proteasome immune‐reactivity decrease | ||||
| 10 | 22 | |||
| 100 | 48 | |||
| 100 | 70 | |||
| MPTP administration | Chymotrypsin‐like UPS activities (at day 2) | |||
| 1 mg/kg daily | 20 | Mice continuously infused with MPTP for 28 days | Fornai et al. (2005) | |
| 5 mg/kg daily | 30 | |||
| 30 mg/kg daily | 40 | |||
| Trypsin‐like UPS activities (at day 2) | ||||
| 1 mg/kg daily | 30 | |||
| 5 mg/kg daily | 40 | |||
| 30 mg/kg daily | 60 | |||
| Peptidyl‐glutamyl‐peptide hydrolysing (PGPH) UPS activities (at day 2) | ||||
| 1 mg/kg daily | 20 | |||
| 5 mg/kg daily | 20 | |||
| 30 mg/kg daily | 30 | |||
The studies presented in the above Table showed that rotenone caused a reduction in UPS activity (measured by 26S and 20S proteasome activity) in a dose‐dependent manner. Further studies showed that rotenone increases proteasome subunit degradation, but does not alter synthesis (Western blot and RT‐PCR studies, reviewed in Chou et al., 2010). Dose‐ and time‐dependent striatal proteasome activity is also shown after MPTP continuously infused up to 28 days measured by relative chymotrypsin‐like, trypsin‐like, and peptidyl‐glutamyl‐peptide hydrolysing (PGPH) proteasome activities in mice (Fornai et al., 2005).
PD patient‐derived fibroblasts (vs Ctr fibroblasts) showed reduction of UPS function (by ~ 33%) and higher accumulation of ubiquitinated proteins (by ~ 2 fold) in PD as compared to control fibroblasts at baseline. Treatment with rotenone (20, 500 μM, 6 h) caused a higher induction of 20S proteasome activity in PD fibroblasts vs Ctr. An increase of LC3‐II accumulation (indicative of autophagic vesicle accumulation) in both groups (PD and Ctr) after exposure to 500 μM rotenone was observed (Ambrosi et al., 2014).
Human neuroblastoma cells (SK‐N‐MC) after short treatment with rotenone (1 week) elevated soluble α‐synuclein protein (41 ± 16% increase) levels without changing mRNA levels, suggesting impairment of α‐synuclein degradation via UPS. Chronic rotenone exposure (4 weeks) increased levels of insoluble α‐synuclein (29 ± 9% increase) and ubiquitin (87 ± 14% increase) (Sherer et al., 2012).
SHSY‐5Y cells treated with rotenone (500 nM, 24 h) showed a ~ 2 fold increase in DCF fluorescence compared to untreated cells (indicative of intracellular ROS). Additionally, rotenone elevated cytosolic calcium (about 35–40% increase vs Ctr), ER‐stress (about 45% increase vs Ctr), impaired UPS function (~ 3‐fold increase of insoluble protein aggregate vs Ctr). Inhibition of Rac1 (Rho‐like GTPase) mitigated the oxidative/nitrosative stress, prevented calcium‐dependent ER‐stress, and partially rescued UPS function (Pal et al., 2014).
Human neuronal SH‐SY5Y cells treated with rotenone (10 μM, for 24 h showed accumulation of high molecular weight ubiquitinated bands (by immunoblotting qualitative assay), and increase of both mitochondrial‐ (~ 5 fold increase vs Ctr) and cytosolic‐ cytochrome c fractions (~ 1.2‐fold increase vs Ctr). Rapamycin pretreatment (3 μM, for 48 h prior addition of rotenone) diminished rotenone‐induced effects, as shown by enhanced degradation of ubiquitinated proteins, and reduced levels of cytosolic cytochrome c. Also, rapamycin promoted mitophagy (as shown by lysosome and mitochondria co‐localisation within the cells) (Pan et al., 2009).
References
Alvarez‐Erviti L, Rodriguez‐Oroz MC, Cooper JM, et al., 2010. Chaperone‐mediated autophagy markers in Parkinson disease brains. Archives of Neurology, 67, 1464–1472.
Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan‐Gomez C, Bombardieri CR, Armentero MT, Zangaglia R, Pacchetti C, Mastroberardino PG, Blandini F, 2014. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson's disease. Biochimica et Biophysica Acta, 1842, 1385–1394.
Bai Y, Hajek P, Chomyn A, Chan E, Seo BB, Matsuno‐Yagi A,Yagi T, Attardi G, 2001. Lack of complex I activity in human cells carrying a mutation in MtDNA‐encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH‐quinone oxidoreductase (NDI1) gene. Journal of Biological Chemistry, 276, 38808–38813.
Betarbet R, Canet‐Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, et al., 2006. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ‐1, α‐synuclein, and the ubiquitin–proteasome system. Neurobiology of Disease, 22, 404–20.
Betarbet R, Sherer TB, Greenamyre JT, 2005. Ubiquitin‐proteasome system and Parkinson's diseases. Experimental Neurology, 191(Suppl.), S17–S27.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Borland MK, Trimmer PA, Rubinstein JD, Keeney PM, Mohanakumar KP, Liu L and Bennett JP, 2008. Chronic, low‐dose rotenone reproduces Lewy neurites found in early stages of Parkinson's disease, reduces mitochondrial movement and slowly kills differentiated SH‐SY5Y neural cells. Molecular Neurodegeneration, 3, 21.
Bove J, Martinez‐Vicente M, Vila M, 2011. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nature Reviews Journal of Neuroscience, 12, 437–452.
Brinkley BR, Barham SS, Barranco SC, Fuller GM, 1974. Rotenone inhibition of spindle microtubule assembly in mammalian cells,” Experimental Cell Research, 85, 41–46.
Brownlees J, Ackerley S, Grierson AJ, Jacobsen NJ, Shea K, Anderton BH, Leigh PN, Shaw CE, Miller CC, 2002. Charcot‐ Marie‐Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Human Molecular Genetics, 11, 2837–2844.
Calì T, Ottolini D, Negro A, Brini M, 2012. α‐Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum‐mitochondria interactions. Journal of Biological Chemistry, 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT, 2009. “A highly reproducible rotenone model of Parkinson's disease”. Neurobiology of Disease, 34, 279–290.
Cappelletti G, Maggioni MG, Maci R, 1999. Influence of MPP+ on the state of tubulin polymerisation in NGF‐differentiated PC12 cells. Journal of Neuroscience Research, 56, 28–35.
Cappelletti G, Pedrotti B, Maggioni MG, Maci R, 2001. Microtubule assembly is directly affected by MPP(+)in vitro. Cell Biology International, 25, 981–984.
Chang DT, Honick AS, Reynolds IJ, 2006. Mitochondrial trafficking to synapses in cultured primary cortical neurons. Journal of Neuroscience, 26, 7035–7045.
Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, Lockhart PJ, Zhang J, 2007. Oligomeric α‐synuclein inhibits tubulin polymerization. Biochemical and Biophysical Research Communications, 356, 548–553.
Chen Y, McMillan‐Ward E, Kong J, Israels SJ, Gibson SB, 2007. Mitochondrial electron‐transport‐chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. Journal of Cell Science, 120, 4155–4166.
Cherra SJ, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT, 2010. Regulation of the autophagy protein LC3 by phosphorylation. Journal of Cell Biology, 190, 533–539.
Chiba Y, Takei S, Kawamura N, Kawaguchi Y, Sasaki K, Hasegawa‐Ishii S, Furukawa A, Hosokawa M, Shimada A, 2012. Immunohistochemical localization of aggresomal proteins in glial cytoplasmic inclusions in multiple system atrophy. Neuropathology and Applied Neurobiology, 38, 559–571.
Chou AP, Li S, Fitzmaurice AG, Bronstein JM, 2010. Mechanisms of rotenone‐induced proteasome inhibition. NeuroToxicology, 31, 367–372.
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein‐Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayır H, Kagan VE, 2013. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology, 15, 1197–1205.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH, 2009. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to α‐synuclein inclusions. Neurobiology of Disease, 35, 385–398.
Cole NB, Sciaky N, Marotta, A, Song J, Lippincott‐Schwartz J, 1996. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Molecular Biology of the Cell, 7, 631–650.
Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E, 2010. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α‐synucleinopathy. Public Library of Science (PLoS ONE), 5, e9313.
Dadakhujaev S, Noh HS, Jung EJ, Cha JY, Baek SM, Ha JH, Kim DR, 2010. Autophagy protects the rotenone‐induced cell death in α‐synuclein overexpressing SH‐SY5Y cells. Journal of Neuroscience Letters, 472, 47–52.
Dagda RK, Banerjee TD, Janda E, 2013. How parkinsonian toxins dysregulate the autophagy machinery. International Journal of Molecular Sciences, 14, 22163–22189.
Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M, 2007. Different species of α‐synuclein oligomers induce calcium influx and seeding. The Journal of Neuroscience, 27, 9220–9232.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson‐Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R, 2002. Resistance of alpha ‐synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences of the United States of America, 99, 14524–14529.
Domingues AF, Arduíno DM, Esteves AR, Swerdlow RH, Oliveira CR, Cardoso SM, 2008. Mitochondria and ubiquitin‐proteasomal system interplay: relevance to Parkinson's disease. Free Radical Biology and Medicine, 45, 820–825.
De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ, 2008. Role of axonal transport in neurodegenerative diseases. Annual Review of Neuroscience, 31, 151–173.
De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ, 2007. Familial amyotrophic lateral sclerosis‐linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content”. Human Molecular Genetics, 16, 2720–2728.
Dehay B, Bove J, Rodriguez‐Muela N, Perier C, Recasens A, Boya P, Vila M, 2010. Pathogenic lysosomal depletion in Parkinson's disease. Journal of Neuroscience Methods, 30, 12535–12544.
Dias V, Junn E, and Mouradian MM, 2013. The role of oxidative stress in parkinson's disease. Journal of Parkinson's Disease, 3, 461–491.
Drolet RE, Cannon JR, Montero L, Greenamyre JT, 2009. Chronic rotenone exposure reproduces Parkinson's disease gastrointestinal neuropathology. Neurobiology of Disease, 36, 96–102.
Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL, 2004. Mice lacking α‐synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology, 25, 761–769.
Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, Sidhu A, 2006. α‐synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. Federation of American Societies for Experimental Biology (FASEB), 20, 2302–2312.
Esposito A, Dohm CP, Kermer P, Bähr M, Wouters FS, 2007. α‐synuclein and its disease‐related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiology of Disease, 26, 521–531.
Esteves AR, Arduíno DM, Silva DF, Oliveira CR, Cardoso SM, 2011. Mitochondrial dysfunction: the road to alpha‐synuclein oligomerization in PD. Journal of Parkinson's Disease, 2011, 693761.
Feng J, 2006. Microtubule: a common target for Parkin and Parkinson's disease toxins. Neuroscientist, 12, 469–476.
Filomeni G, Graziani I, de Zio D, Dini L, Centonze D., Rotilio G, Ciriolo MR, 2012. Neuroprotection of kaempferol by autophagy in models of rotenone‐mediated acute toxicity: Possible implications for Parkinson's disease. Neurobiology of Aging, 33, 767–785.
Follett J, Darlow B, Wong MB, Goodwin J, and Pountney DL, 2013. Potassium depolarization and raised calcium induces α‐synuclein aggregates. Neurotoxicity Research, 23, 378–392.
Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC, 2005. Parkinson‐like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitinproteasome system and _α‐synuclein. Proceedings of the National Academy of Sciences, 102, 3413–3418 http://www.pnas.org/content/102/9/3413.full Fig 2b; http://www.pnas.org/content/102/9/3413.full Fig 5c © National Academy of Sciences
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's Disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM, 2003. Initiation and synergistic fibrillization of tau and α‐synuclein. Science, 300, 636–640.
Goodwin J, Nath S, Engelborghs Y, Pountney DL, 2013. Raised calcium and oxidative stress cooperatively promote α‐synuclein aggregate formation. Neurochemistry International, 62, 703–711.
Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H. Lee VMY, Trojanowski JQ, Mann D, and Iwatsubo T, 2002. Phosphorylated R‐synuclein is ubiquitinated in R‐synucleinopathy lesions. Journal of Biological Chemistry, 277, 49071–49076.
Haskin J, Szargel R, Shani V, Mekies LN, Rott R, Lim GG, Lim KL, Bandopadhyay R, Wolosker H, Engelender S, 2013. AF‐6 is a positive modulator of the PINK1/parkin pathway and is deficient in Parkinson's disease. Hum Molecular Genetics and Metabolism, 22, 2083–2096.
Höglinger GU, Carrard G, Michel PP, Medja F, Lombès A, Ruberg M, Friguet B, Hirsch EC, 2003. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson's disease. Journal of Neurochemistry, 86, 1297–1307.
Ikawa M, Okazawa H, Kudo T, Kuriyama M, Fujibayashi Y, Yoneda M, 2011. “Evaluation of striatal oxidative stress in patients with Parkinson's disease using [62Cu]ATSM PET” Nuclear Medicine and Biology, 38, 945–951.
Jiang H, Cheng D, Liu W, Peng J, Feng J, 2010. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochemistry and Biophysics Research Communication, 395, 471–476.
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP, 2003. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell, 115, 727–738.
Kim I, Rodriguez‐Enriquez S, and Lemasters JJ, 2007. Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics, 462, 245–253.
Lee HJ, Khoshaghideh F, Lee F, and Lee SJ, 2006. Impairment of microtubule‐dependent trafficking by overexpression of α‐synuclein. European Journal of Neuroscience, 24, 3153–3162.
Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ, 2002. Formation and removal of α‐synuclein aggregates in cells exposed to mitochondrial inhibitors. Journal of Biological Chemistry, 277, 5411–5417.
Lim J, Kim HW, Youdim MB, Rhyu IJ, Choe KM, Oh YJ, 2011. Binding preference of p62 towards LC3‐ll during dopaminergic neurotoxin‐induced impairment of autophagic flux. Autophagy, 7, 51–60.
Lin MT and Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787–795.
Liu K, Shi N, Sun Y, Zhang T, Sun X, 2013. Therapeutic effects of rapamycin on MPTP‐induced Parkinsonism in mice. Neurochemical Research, 38, 201–207.
Lotharius J, and Brundin P, 2002. Pathogenesis of Parkinson's disease: dopamine, vesicles and α‐synuclein. Nat. Rev., Journal of Neuroscience, 3, 932–942.
Mader BJ, Pivtoraiko VN, Flippo HM, Klocke BJ, Roth KA, Mangieri LR, Shacka JJ, 2012. Rotenone inhibits autophagic flux prior to inducing cell death. American Chemical Society Chemical Neuroscience, 3, 1063–1072.
Mandel S, Maor G, and Youdim MBH, 2004. Iron and α‐synuclein in the substantia nigra ofMPTP‐treated mice: effect of neuroprotective drugs R‐apomorphine and green tea polyphenol (−)‐epigallocatechin‐3‐gallate. Journal of Molecular Neuroscience, 24, 401–416.
Marshall LE, Himes RH, 1978. “Rotenone inhibition of tubulin self‐assembly,” Biochimica et Biophysica Acta, 543, 590–594.
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S, 1999. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Annals of the New York Academy of Sciences, 893, 154–175.
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW, 2003. Altered proteasomal function in sporadic Parkinson's disease. Experimental Neurology, 179, 38–46.
McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O, 2002. Selective loss of 20S proteasome alpha‐subunits in the substantia nigra pars compacta in Parkinson's disease. Journal of Neuroscience Letters, 326, 155–158.
McNaught KS, Jenner P, 2001. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Journal of Neuroscience Letters, 297, 191–194.
Miki Y, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K, 2011. Accumulation of histone deacetylase 6, an aggresome‐related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology, 31, 561–568.
Miller KE, Sheetz MP, 2004. “Axonal mitochondrial transport and potential are correlated, Journal of Cell Science, 117, 2791–2804.
Müftüoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogüs H, Dalkara T, Ozer N, 2004. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Movement Disorders, 19, 544–548.
Nakata Y, Yasuda T, Fukaya M, Yamamori S, Itakura M, Nihira T, Hayakawa H, Kawanami A, Kataoka M, Nagai M, Sakagami H, Takahashi M, Mizuno Y, Mochizuki H, 2012. Accumulation of α‐synuclein triggered by presynaptic dysfunction. The Journal of Neuroscience, 32, 17186–17196.
Nath S, Goodwin J, Engelborghs Y, and Pountney DL, 2011. Raised calcium promotes α‐synuclein aggregate formation. Molecular and Cellular Neuroscience, 46, 516–526.
Nogales E, 2000. Structural insights into microtubule function. Annual Review of Biochemistry, 69, 277–302.
Nonaka T, and Hasegawa M, 2009. A cellular model to monitor proteasome dysfunction by α‐synuclein. Biochemistry, 48, 8014–8022.
Obashi K, Okabe S, 2013. Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. European Journal of Neuroscience, 38, 2350–2363.
Osna NA, Haorah J, Krutik VM, Donohue TM Jr, 2004. Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology, 40, 574–82.
Pal R, Monroe TO, Palmieri M, Sardiello M, Rodney GG, 2014. Rotenone induces neurotoxicity through Rac1‐dependent activation of NADPH oxidase in SHSY‐5Y cells. Federation of the European Biochemical Societies's Letters (FEBS)ers, 588, 472–481.
Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W, 2009. Rapamycin protects against rotenone‐induced apoptosis through autophagy induction. Journal of Neuroscience, 164, 541–551.
Pan T, Kondo S, Le W, Jankovic J, 2008. The role of autophagy‐lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain, 131, 1969–1978.
Pan‐Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo‐Angulo M, Gille G, Funk RH, Reichmann H, 2012. Environmental toxins trigger PD‐like progression via increased alphasynuclein release from enteric neurons in mice. Scientific Reports, 2, 898.
Pan‐Montojo FJ, Funk RHW, 2010. Oral administration of rotenone using a gavage and image analysis of α‐synuclein inclusions in the enteric nervous system. Journal of Visualized Experiments, no. 44, article 2123.
Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, 2001. Ischiropoulos H. Induction of alpha‐synuclein aggregation by intracellular nitrative insult. Journal of Neuroscience, 21, 8053–8061.
Payton JE, Perrin RJ, Clayton DF, George JM, 2001. Protein‐protein interactions of α‐synuclein in brain homogenates and transfected cells. Molecular Brain Research, 95, 138–145.
Powers ET1, Morimoto RI, Dillin A, Kelly JW, Balch WE, 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annual Review of Biochemistry, 78, 959–91.
Qureshi HY, Paudel HK, 2011. Parkinsonian neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) and alphasynuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. Journal of Biological Chemistry, 286, 5055–5068.
Ren Y, Feng J, 2007. Rotenone selectively kills serotonergic neurons through a microtubule‐dependent mechanism. Journal of Neurochemistry, 103, 303–311.
Richter‐Landsberg C, Leyk J, 2013. Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathologica, 126, 793–807.
Rideout HJ, Larsen KE, Sulzer D, Stefanis L, 2001. Proteasomal inhibition leads to formation of ubiquitin/a‐synuclein‐immunoreactive inclusions in PC12 cells. Journal of Neurochemistry, 78, 899–908.
Saha AR, Hill J, Utton MA, Asuni AA, Ackerley S, Grierson AJ, Miller CC, Davies AM, Buchman VL, Anderton BH, Hanger DP, 2004. Parkinson's disease α‐synuclein mutations exhibit defective axonal transport in cultured neurons. Journal of Cell Science, 117, 1017–1024.
Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G, 2008. Bidirectional Ca2 + ‐dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences, 105, 20728–20733.
Sarkar S, Chigurupati S, Raymick J, Mann D, Bowyer JF, Schmitt T, Beger RD, Hanig JP, Schmued LC, Paule MG, 2014. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP‐induced Parkinson's disease mouse model. Neurotoxicology, 44C, 250–262.
Scarffe LA, Stevens DA, Dawson VL, Dawson TM, 2014. Parkin and PINK1: much more than mitophagy. Trends in Journal of Neurosciences, 37, 315–324.
Schapira AH, 2006. Etiology of Parkinson's disease. Neurology, 66, S10–S23.
Seo BB, Nakamaru‐Ogiso E, Flotte TR, Yagi T, Matsuno‐Yagi A, 2002. A single‐subunit NADH‐quinone oxidoreductase renders resistance to mammalian nerve cells against complex I inhibition. Molecular Therapy, 6, 336–341.
Seo BB, Wang J, Flotte TR, Yagi T, Matsuno‐Yagi A, 2000. Use of the NADH‐quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae as a possible cure for complex I defects in human cells. Journal of Biological Chemistry, 275, 37774–37778.
Shamoto‐Nagai M, Maruyama W, Kato Y, Isobe K, Tanaka M, Naoi M, Osawa T, 2003. An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH‐SY5Y cells. Journal of Neuroscience Research, 74, 589–97.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT, 2002. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha‐synuclein metabolism and oxidative damage. Journal of Neuroscience, 22, 7006–7015.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno‐Yagi A, Greenamyre JT, 2003. Mechanism of toxicity in rotenone models of Parkinson's disease. Journal of Neuroscience, 23, 10756–10764.
Song L, Cortopassi G, 2015. Mitochondrial complex I defects increase ubiquitin in substantia nigra. Brain Research, 1594, 82–91.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM, 2002 Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. “Journal of Cell Biology, 156, 1051–1063.
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA, 2001. Expression of A53T mutant but not wild‐type α‐synuclein in PC12 cells induces alterations of the ubiquitindependent degradation system, loss of dopamine release, and autophagic cell death. Journal of Neuroscience, 21, 9549–9560.
Sun F, Anantharam V, Latchoumycandane C, Kanthasamy A, Kanthasamy AG, 2005. Dieldrin induces ubiquitin‐proteasome dysfunction in α‐synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. Journal of Pharmacology and Experimental Therapeutics, 315, 69–79.
Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R, 2006. Mitochondrial dynamics and Ca2 + signaling. Biochimica et Biophysica Acta, 1763, 442–449.
Szweda PA, Friguet B, Szweda LI, 2002. Proteolysis, free radicals, and aging. Free Radical Biology and Medicine, 33, 29–36.
Thomas B, Banerjee R, Starkova NN, Zhang SF, Calingasan NY, Yang L, Wille E, Lorenzo BJ, Ho DJ, Beal MF, Starkov A, 2012. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxid Redox Signal, 16, 855–868.
Tristão FS, Amar M, Latrous I, Del‐Bel EA, Prediger RD, Raisman‐Vozari R, 2014. Evaluation of nigrostriatal neurodegeneration and neuroinflammation following repeated intranasal 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) administration in mice, an experimental model of Parkinson's disease. Neurotoxicity Research, 25, 24–32.
Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L, 2011. Pathological roles of α‐synuclein in neurological disorders. Lancet Neurology, 10, 1015–1025.
Wade RH, 2009. On and around microtubules: an overview. Molecular Biotechnology, 43, 177–191.
Wang XF, Li S, Chou AP, Bronstein JM, 2006. Inhibitory effects of pesticides on proteasome activity: implication in Parkinson's disease. Neurobiology of Disease, 23, 198–205.
Westerlund M, Belin AC, Anvret A, Håkansson A, Nissbrandt H, Lind C, Sydow O, Olson L, Galter D, 2008. Cerebellar alpha‐synuclein levels are decreased in Parkinson's disease and do not correlate with SNCA polymorphisms associated with disease in a Swedish material. FEDERATION OF AMERICAN SOCIETIES FOR EXPERIMENTAL BIOLOGY (FASEB), 22, 3509–3514.
Wu F., Xu HD, Guan JJ, Hou YS, Gu JH, Zhen XC and Qin ZH, 2015. Rotenone impairs autophagic flux and lysosomal functions in Parkinsons's disease. Journal of Neuroscience, 284, 900–911.
Yi M, Weaver D, Hajnoczky G, 2004. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. Journal of Cell Biology, 167, 661–672.
Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Clark LN, Duff KE, 2009. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α‐synuclein. American Journal Of Pathology, 175, 736–747.
Yuan YH, Yan WF, Sun JD, Huang JY, Mu Z, Chen NH, 2015. The molecular mechanism of rotenone‐induced α‐synuclein aggregation: emphasizing the role of the calcium/GSK3β pathway. Toxicology Letters, 233, 163–171.
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A, 2015. Mitochondrial dysfunction and α‐synuclein synaptic pathology in Parkinson's Disease: who's on first? Journal of Parkinson's Disease, 2015, 108029.
Zhang H, Duan C, Yang H, 2015. Defective autophagy in Parkinson's disease: lessons from genetics. Molecular Neurobiology, 51, 89–104. doi: 10.1007/s12035‐014‐8787‐5
Zhu J and Chu CT, 2010. Mitochondrial dysfunction in Parkinson's disease. Journal of Alzheimer's Disease, 20, S325–S334.
Zhu JH, Gusdon AM, Cimen H, van Houten B, Koc E, Chu CT, 2012. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death and Disease, 3, e312.
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT, 2007. Regulation of autophagy by extracellular signal‐regulated protein kinases during 1‐methyl‐4‐phenylpyridinium‐induced cell death. American Journal of Pathology, 170, 75–86.
4th KER: Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway
A.4.1. How this key event relationship works
One of the critical functions in the long‐lived cells such as neurons is the clearing system for the removal of the unfolded proteins. This function is provided by two major systems, the ubiquitin proteosome system (UPS) and the autophagy‐lysosome pathway (ALP) (Tai, 2008; Korolchuck VI et al., 2010; Ravikumar et al., 2010). Impaired proteostasis with formation of misfolded α‐synuclein aggregates deregulates microtubule assembly and stability with reduction in axonal transport and impairment of mitochondrial trafficking and energy supply (Chen et al., 2007; Esposito et al., 2007, Borland et al., 2008; Weihofen et al., 2009; O'Malley, 2010; Fujita et al., 2014). Pathological consequences of these deregulated process include interference with the function of synapses, formation of toxic aggregates of proteins, impaired energy metabolism and turnover of mitochondria and chronic endoplasmic reticulum stress; all eventually leading to degeneration of DA neurons in the nigrostriatal pathway (Dauer et al., 2003; Raff et al., 2005; Orimo et al., 2008; Fujita et al., 2010; Shulman et al., 2011; Schwarz, 2015).
A.4.2. Weight of evidence
The weight of evidence for the relationship between impaired proteostasis and degeneration of dopaminergic neurons of the nigrostriatal pathway is strong. The biological plausibility is based on the knowledge of the physiological cellular process governing the cleaning processes of degraded proteins and organelles and on the observations done in genetic and idiopathic forms of Parkinson's disease. Dose and time concordance support a strong response–response relationships which is also supported by the very well known chronic and progressive behaviour of the Parkinson's disease. Although essentiality has been demonstrated in multiple models and lines of evidence, including knockout animals, a single molecular chain of events cannot be established; therefore essentiality for this KEs relationship was considered moderate.
A.4.2.1. Biological plausibility
The fact that impaired proteostasis can induce degeneration of DA neurons of the nigrostriatal pathway is well known and based on the understanding of the physiological cellular processes involved in removing degraded/misfolded proteins as they are critical for normal mitochondria and axonal transport. Accumulation of misfolded and/or aggregated α‐synuclein and the presence of abnormal mitochondria is a consequence of deregulation of this clearing process, and the Lewy bodies, a pathological hallmark of sporadic PD, stain specifically for proteins associated with UPS (Gai et al., 2000; McNaught et al., 2002; Fornai et al., 2003). Impaired proteostasis has been described in humans affected by sporadic PD (McNaught et al., 2001, 2003), and changes induced by excess cellular levels of degraded proteins in nigral dopaminergic neurons cause a progressive decline in lysosome function, i.e. ALP system, contributing to neurodegeneration (Decressac, 2013). In this context, the ALP system is likely working in a complementary way, with the UPS being the major cleaning system in the soma and the ALP playing a role at presynaptic sites (Friedman et al., 2012). Pathological observations from patients affected by PD and from animal models show an increased number of autophagic vacuoles or autophagic markers (Alvarez‐Erviti et al., 2010; Crews et al., 2010). Additional observations support the role of impaired proteostasis in nigrostriatal toxicity such as: several genetic variants of sporadic PD are due to susceptible genes able to participate in or modify proteostasis (Leroy et al., 1998; Shimura et al., 2000; Fornai et al., 2003; Shulman et al., 2011) and striatal microinfusion of proteasome inhibitors induce selective nigrostriatal toxicity with loss of DA and DA metabolites (DA, DOPAC and HVA) in the striatum, retrograde loss of nigral DA cell and intracytoplasmic inclusions positive for protein of the UPS (Fornai et al., 2003).
Transgenic overexpression of mutant or wild‐type forms of α‐synuclein in mice causes neuropathological changes including dystrophic neurites and α‐synuclein positive LB‐inclusion (Masiliah et al., 2000; Dauer et al., 2003). However, they fail to reproduce specific cell death in the nigrostriatal pathway. In contrast, injection of human α‐synuclein expressing viral vectors into the SN of adult rats causes a selective death of dopaminergic neurons and formation of LB inclusions (Kirik et al., 2002; Lo Bianco et al., 2002; Dauer et al., 2003). These effects were observed with adeno‐associated virus‐mediated expression of A30P α‐synuclein and with lentiviral‐mediated expression of α‐synuclein in rats, mice and non‐human primates (Klein, 2002; Lo Bianco, 2002, 2004; Kirk, 2003; Lauwers, 2003; Shulman, 2010).
Impaired proteostasis and formation of proteins aggregates also affect the axonal transport and mitochondrial trafficking. α‐synuclein mutants accumulate in the neuronal soma when overexpressed, reducing the axonal transport (Saha, et al., 2004; Kim‐Han, 2011); in addition, overexpressed vesicle‐associated α‐synuclein binds to the microtubules with a detrimental role on axonal transport (Yang et al., 2010; Kim‐Han 2011). Postmortem studies on PD patients are indicative of axonal damage. It appears that axonal changes precede neuronal loss, supporting the idea that axonal impairments are early events in neurodegenerative disorders (Orimo, 2005, 2008; Raff, 2002; Braak et al., 2004). These changes, and observation from animals models using the chemical stressor MPTP (Serra et al., 2002; Meissner et al., 2003; Hasbani et al., 2006) are supporting the notion that DA neurons of the nigrostriatal pathway degenerate through a ‘dying back’ axonopathy (Raff et al., 2002). It was demonstrated that axonal degeneration follows an active process distinct from cell body loss in a Wallerian degeneration slow (WldS) mutant mouse transgenic model. In this model, axonal degeneration in a variety of disorders is inhibited. In WldS mice, acute treatment with MPTP (20 mg/kg i.p. for 7 days) resulted in attenuated nigrostriatal axon degeneration and attenuated DA loss, but cell bodies were not rescued (Hasbani et al., 2006). Indeed, multiple evidences from genetic and experimental models (particularly using MPTP as a stressor) support an early and critical role of axonal impairment with early occurrence of Lewy neurites preceding Lewy bodies formation and cell death (O'Malley, 2010).
In addition, a strong link between mitochondrial dysfunction and PD came from the discovery that mutations in PINK1, α‐synuclein, LRRK2, parkin and DJ‐1, all linked with genetic causes of PD, can affect mitochondrial function (O'Malley, 2010; Rappold et al., 2014). Deregulation of mitochondrial dynamics (fission, fusion and movement of mitochondria) can affect neuronal activity and viability and imbalance of mitochondrial dynamics have been reported in experimental models of PD with mutated α‐synuclein (Tieu, 2014) or chronic model of primary neuronal cells treated with low concentrations (0.1–1 nM) of rotenone (Arnold et al., 2011). Progression of neuronal changes with formation of Lewy neurites and reduction of mitochondrial movement leading to cell death has been also observed in‐vitro in a chronic cell‐based model (SH‐SY5Y neuroblastoma cell line) treated with low concentration of Rotenone (50 nM for 21 days). In this assay, reduction in mitochondrial movement was associated with a progressive damage, first including formation of Lewy neurites, followed by cell death (Borland et al., 2008).
A.4.2.2. Empirical support for linkage
Degeneration of DA neurons of the nigrostriatal pathway, similar to the one observed in PD, have been reproduced in human and experimental animal models following exposure to MPTP (Langston, 1983; Forno, 1993; Irwin, 1993; Rose, 1993; Ovadia, 1995; Kitamura, 2000; Serra, 2002; Dauer, 2003; Meissner, 2003 and Porras et al., 2012) and in animals following administration of rotenone through multiple routes of exposure (Betabret, 2000, 2006; Schmidt, 2002; Sherer, 2003; Fleming, 2004, Saravanan, 2005; Inden, 2007; Pan‐Montojo, 2010; Johnson, 2015). This indicates that both chemicals can be used as a tool compound for experimental investigations on PD and to explore the key event relationship between impaired proteostasis and degeneration of DA neurons of nigrostriatal pathway. Also, similar to PD, susceptibility to MPTP increases with age in both non‐human primates and mice (Irwin et al., 1993; Rose et al., 1993, Ovadia et al., 1995).
Neurotoxic external doses of both rotenone and MPTP are well characterised and reported; however, the corresponding brain concentration is much less frequently quoted. In order to understand the brain concentration for both compounds, data were retrieved from Betarbet et al. (2000 and 2006) for rotenone and from Fornai et al. (2005) and Thomas et al. (2012) for MPTP. In all cases, the compounds were administered by infusion and, at least for MPTP, the brain concentrations were taken after chronic infusion and are expected to be at the steady state. For MPTP only, brain concentration was expressed as ng/mg protein (Fornai et al., 2005) or as ng/mg weight tissue (Thomas et al., 2012). To do the final estimate we assumed a density for protein as 1.4 (Quillin and Matthews, 2000) and a protein content in the brain of about 10% (Schwartz et al., 2012). Density for brain tissue was assumed to be 1. The final concentration was 12 μM (Fornai et al., 2005) and 47 μM (Thomas et al., 2012).
It should be noted that the upstream key event includes multiple pathological events, eventually leading to the downstream key event. As it is difficult to assess real time changes for a series of complex and dynamic events in a single experiment, most of the empirical supporting evidences are performed by exploring single factors (e.g. impairment of ALP or UPS or axonal transports) and their role in the degeneration of DA neurons.
MPTP/MPP+
Inhibition of the UPS was observed following continuous infusion of MPTP at 1, 5 and 30 mg/kg per day for 28 days in mice. A dose related decrease in the enzyme activity of the UPS was observed and this effect was associated with a dose‐related decrease of TH positive terminals (densitometry analysis) in the dorsal and ventral striatum. This effect was accompanied by a dose‐related cell loss in the SN (counting of TH positive cells) at 5 and 30 mg/kg per day. At 30 mg/kg per day the authors reported cytoplasmic inclusions positively staining for ubiquitin and α‐synuclein in neurons of the SN (and locus coeruleus). In the same experiment, acute administration of MPTP (single injection of 30 mg/kg/ or 4 separate injections of 20 mg/kg) induced a transient inhibition of the UPS activity, neuronal loss but no intracytoplasmatic inclusions, indicating that a continuous infusion is necessary to induce permanent inhibition and pathological changes similar to the one observed in PD (Fornai et al., 2005).
In mice lacking α‐synuclein, continuous infusion of up to 30 mg/kg per day for 28 days of MPTP neuronal cell death and behavioural symptoms were almost alleviated (Dauer et al., 2002, Fornai et al., 2005).
Administration of MPTP to mice (30 mg/kg per day i.p. for 5 days) produced autophagosome (AP) accumulation (increase in LC3II) and dopaminergic cell death which was preceded by a decrease in the amount of lysosomes in DA neurons. MPTP also induced mitochondrial‐derived ROS and permeabilisation of the lysosomal membrane. This resulted in a decrease in Lamp 1 lysosome structural protein and accumulation of undegraded AP and release of lysosomal enzymes into the cytosol. The effect observed in‐vivo was quantitatively confirmed in‐vitro (human neuroblastoma cell line BEM17(M17EV)). MPP+ was tested in‐vitro at the concentrations of 0.25–2.5 μM and induced a concentration‐related decrease in Lamp1, increase in LC3II, increase in cell death and decrease in lysotracker. In the same in‐vitro system, MPP+ also induced lysosome membrane permeabilisation. In the same experiment, induction of lysosome biogenesis by the autophagy‐enhancer compound rapamycin attenuated the dopaminergic neurodegeneration, both in vitro and in vivo, by restoring lysosomal levels (Dehay et al., 2010).
In an in‐vitro microchamber that allowed specific exposure of neuritis of murine mesencephalic neurons, treatment with 1–5 μM of MPP+ induced impairment of mitochondrial transport, neurite degeneration (degeneration of proximal dendrites) and autophagy, before cell death (Kim‐Han et al., 2011). The number of TH positive cell bodies and neurites was reduced at 1 μM, and axonal fragmentation and LC3 dots increased while tubulin density decreased (Kim‐Han et al., 2011).
Mice treated with MPTP at 20 mg/kg per day i.p. for 5 days showed loss of DA neurons in SN which was attenuated by the pharmacological block of mitochondrial fission protein Drp1. Drp1 blockade also promoted mitochondrial fusion and enhanced the release of DA from the striatal terminals in a PINK1 knockout model showing a defective DA release (Rappold et al., 2014; Tieu et al., 2014).
In differentiated (d6) LUHMENS cell system stably expressing eGFP/mito‐tRFP, treatment with MPP+ (5 μM) for 24 h revealed a reduction in the total number of mitochondria in neuritis and a significant reduction in velocity. Partial protection from MPP+ dependent mitochondrial immobilisation in neuritis as well as from drop in mitochondria numbers in neuritis was detects following co‐treatment with the antioxidant vitamin C (Schildknecht et al., 2013)
Proteasome inhibitors
Intracerebral microinfusion of proteasome inhibitors (lactacystein or epoxomycin at, 100 and 1,000 μM) induced loss of TH and DAT immunostaining and decrease in DA and DOPAC in DA terminals in the striatum and loss of nigral cells in SN (counting of TH positive cells). Formation of cell inclusions (positively immunostained for α‐synuclein and ubiquitin) and apoptosis were observed after treatment with proteasome inhibitors (0.1–50 μM) in an in‐vitro system (PC 12 cells). The concentration response curve for apoptosis was shifted to the right compared to the concentration response curve for cellular inclusions indicating that inclusions occurred earlier and independently of cell death. A maximum effect was reached between 1 and 10 μM (Fornai et al., 2003).
Rotenone
Administration of rotenone, via osmotic mini pumps implanted to rats (3 mg/kg per day for 7 days) induced decrease of TH in substantia nigra and striatum and decrease in α‐synuclein, in its native form, in substantia nigra and striatum, while monoubiquitinated alpha‐synuclein increased in the same regions. Valproic acid (VPA) treatment (effective inhibitor of histone deacetylases) significantly counteracted the death of nigral neurons and the 50% drop of striatal dopamine levels caused by rotenone administration. VPA treatment also counteracted both type of α‐synuclein alterations. Furthermore, monoubiquitinated alpha‐synuclein increased its localisation in nuclei isolated from substantia nigra of rotenone‐treated rats, an effect also prevented by VPA treatment. Nuclear localisation of alpha‐synuclein has been recently described in some models of PD and its neurodegenerative effect has been ascribed to histone acetylation inhibition (Monti et al., 2010).
Chronic oral administration of rotenone at 30 mg/kg per day in mice produced neuronal loss and degeneration of TH positive terminals in the striatum accompanied by an increase in α‐synuclein, ubiquinated proteins and decrease in proteasomal activity. Concomitant treatment with 4‐PBA (a chemical chaperone able to reverse the mislocalisation and/or aggregation of proteins) inhibited rotenone‐induced neuronal death and decreased protein level of α‐synuclein (Inden et al., 2007).
Treatment of Lewis rat with 2 mg/kg per day of rotenone, administered sc for 8 weeks impaired autophagic flux, induced lysosomal dysfunction and degeneration of DA neurons (decrease in number of TH positive cells and decrease in density of TH positive fibres) in SNpc. The effect of rotenone was paralleled by an increase in LC3 immunopositive dots and upregulation of the LC3II in DA neurons. A concomitant effect was observed and characterised by a decrease in LAMP2 and catepsin immunodots with a diffuse morphological pattern, possibly indicative of decreased lysosomal membrane integrity and leaking to cytosol. In‐vitro (PC12 cells) at 500 nM, rotenone also induced increases in α‐synuclein, microtubule associated protein 1, light chain 3‐II, Beclin 1, p62, increased lysosome permeability and induced cell death. In PC12 cell, the concomitant treatment with trehalose (autophagic inducer) attenuated the rotenone‐induced cell death while in‐vivo trehalose treatment decreased the rotenone‐induced dopaminergic neurons loss (Wu et al., 2015).
Rotenone LD50 of 10 nM in differentiated SH‐SY5Y cells decreased autophagic flux at both 2 and 24 h. Upregulation of autophagy by rapamycin protected against cell death while inhibition of autophagy by 3‐methyladenine exacerbated cell death (Giordano et al., 2014)
Treatment of embryonic midbrain neuronal cells with 0.1–10 μM rotenone for 30 min induces a decrease in polymerised tubulin and increased the number of apoptotic TH+ cells. Similar effects were observed with colchicine treatment, a well‐known microtubule‐depolarising agent and prevented by taxol, a well‐known microtubule‐stabilising agent. The effect was considered specific to DA neurons as the effect on apoptosis and cell death was much less evident in GABAergic and glutamatergic neurons (Ren et al., 2005).
Human evidences
Inclusion bodies in DA neurons (i.e. Lewy bodies), a pathological hallmark for sporadic PD, stains specifically for proteins associated with the UPS (Gai et al., 2000; Mcnaught et al., 2002; Fornai et al., 2003), including α‐synuclein, parkin and ubiquitin; possibly indicating that failure of the UP system represents a common step in the pathogenesis of PD and impairment of the proteasome system was found in humans affected by sporadic PD (McNaught et al., 2001, 2003).
Lysosomal breakdown and autophagosome (AP) accumulation with co‐localisation of lysosomal markers in Lewy Bodies is reported to occur in PD brain samples where Lewy bodies were strongly immunoreactive for the autophagosome markers (LC3II) (Dehay et al., 2010).
Postmortem studies on PD patients show axonal pathology that is likely to precede the loss of neuronal bodies In this investigation, TH immunoreactive fibres had almost entirely disappeared with preservation of neuronal bodies (Orimo et al., 2005, 2008).
A.4.3. Uncertainties or inconsistencies
MPTP can induce damage to nigrostriatal neurons without formation of Lewy bodies (hall mark of PD). Acutely intoxicated humans and primates with MPTP lack LB‐like formation (Forno, 1986, 1993; Dauer, 2003). Similarly, discontinuous administration of rotenone, even at high doses, damages the basal ganglia but produce no inclusions (Heikkila et al., 1985; Ferrante et al., 1997; Lapontine, 2004). To reproduce the formation of neuronal inclusions, continuous infusion of MPTP or rotenone is necessary.
Acute intoxication with rotenone seems to spare dopaminergic neurons (Ferrante, 1997, Dauer et al., 2003). In addition, in rats chronically infused with rotenone showed a reduction in striatal DARPP‐32‐positive, cholinergic and NADPH diaphorase‐positive neurons (Hoglinger, 2003) or in other brain regions. These results would suggest that Rotenone can induce a more widespread neurotoxicity (Aguilar, 2015).
The vulnerability of the dopaminergic pathway still remains circumstantial. The selectivity of MPP+ for dopaminergic neurons is due to its selective uptake via dopamine transporter (DAT), which terminates the synaptic actions of dopamine (Javitch, 1985; Pifl, 1993; Gainetdinov, 1997; Hirata, 2008). Selectivity of Rotenone for dopaminergic neurons is not fully understood (Hirata, 2008).
Transgenic overexpression of α‐synuclein induces neurotoxicity (i.e. neuronal atrophy, distrophic neuritis, astrocytosis and LB‐like formation). However they fail to cause death of dopaminergic neurons. Nevertheless, injection of the human protein or mutated form expressing viral vectors into the SN, are able to induce all the pathological changes characteristic of PD. This discrepancy could be due to the higher expression of α‐synuclein in the viral vector model or because in these models, α‐synuclein overexpression would occur suddenly in adult animals (Dauer et al., 2003). In addition, transgenic expression of C‐terminal truncated α‐synuclein also leads to motor symptoms but neuronal degeneration is not reported (Halls et al., 2015).
There is conflicting literature on whether increased autophagy would be protective or enhances damage. Similarly, a conflicting literature exists on extent of inhibition or activation of different protein degradation system in PD and a clear threshold of onset is unknown (Fornai et al., 2005).
Several mechanisms may affect the axonal transport in neurons showing swelling of neurites positive for α‐synuclein. These include e.g. ROS production, lysosome and mitochondria membranes depolarisation, increased permeability and microtubule depolymerisation (Borland, 2008; Choi, 2008; Kim‐Ham 2011). As both MPTP and Rotenone could directly trigger these effects, a clear mechanistic understanding leading to cell death is difficult to identify (Aguilar et al., 2015).
Different features of imbalanced proteostasis can trigger one another (e.g. disturbed protein degradation, pathological protein aggregation, microtubule dysfunction); and each of them can lead to cell death. Therefore, the ‘single’ triggering event triggering axonal degeneration or neuronal death is not known. For instance, for α‐synuclein aggregation, it is not clear whether this causes death because some vital function of neurons is lost, or whether some protein increases e.g. because of inhibited chaperone‐mediate autophagy (Kaushik et al., 2008; Cuervo et al., 2014).
Real‐time changes in DA axons are difficult to assess, accounting for the limitation of testing models of structural or trafficking impairment in vivo.
A.4.4. Quantitative evaluation of KER
As described in the empirical support, a quantitative relationship has been established between chemical stressors inducing impaired proteostasis and loss of DA neurons of nigrostriatal pathway. The response–response relationship was evident in most of the studies and, where possible a relationship in dose–response could be also observed. A chronic dose regimen for the chemical stressor was necessary in most of the studies and this is confirming that a long lasting perturbation of the key event up is necessary to affect neuronal loss consistent with the presence of intracytoplasmatic inclusions. However, some inconsistency in the measurement of the endpoints relevant for impaired proteostasis were observed, probably because they also act as compensatory factors (Betarbet et al., 2006). The acute administration of MPTP (single injection of 30 mg/kg/ or 4 separate injections of 20 mg/kg) induced a transient inhibition of the UPS activity and neuronal loss but no intracytoplasmatic inclusions i.e. Lewy body were observed, supporting the temporal relationship among the two events (Fornai et al., 2005).
Table A.3.
Quantitative evaluation of the KER
| Measured endpoint relevant for the KEup (KE3) | Measured endpoint relevant for the KEdown (KE4) | Model | Reference |
|---|---|---|---|
| Approx. 40% inhibition of UPS | Approx. 38% decrease in TH density in dorsal striatum | MPTP 1 mg/kg per day IV infusion for 28 days in mice | Fornai et al. (2005) |
| Approx. 50% inhibition of UPS | Approx. 40% decrease in number of TH positive cells/mm2 in SN and approx. 25% decrease in TH in dorsal striatum | MPTP 5 mg/kg per day IV infusion for 28 days in mice | |
| Approx. 60% inhibition of UPS | Approx. 86% decrease in number of TH positive cells/mm2 in SN and approx. 50% decrease in TH in dorsal striatum and approx. 50% in ventral striatum | MPTP 30 mg/kg per day IV infusion for 28 days in mice | |
| Approx. 40% proteasome inhibition | Approx. 70% decrease in DA and 50% decrease in DOPAC in striatum and 30% cell loss in SN | i.c. infusion of lactacystin (proteasome inhibitors) in rats 100 μM | Fornai et al. (2003) |
| Approx. 50% increase in mRNA expression for α‐synuclein | Decrease in TH immunoreactivity (approx. 50%), in TH‐positive nerve terminals in the striatum | Transgenic model overexpressing α‐synuclein | Kirk et al. (2002) |
| Approx. 16–13% reduction in proteosomal activity | Degeneration of nigrostriatal dopaminergic neurons in 50% of animals | Chronic i.v. treatment (up to 5 weeks) of Lewis rat with rotenone at 2–3 mg/kg day (free brain Rotenone 20–30 nM) | Betabret et al. (2000, 2006) |
| Approx. 50% increase in α‐synuclein |
Approx. 57% reduction in TH immunoreactivity in SNpc neurons at 30 mg/kg per day Decrease in TH and DAT in the striatum (approx. 30% and 70% respectively) and ventral midbrain area (approx. 60%) at 30 mg/kg per day |
Oral chronic administration (28 days) of rotenone (0.25, 1, 2.5, 5, 10 or 30 mg/kg per day) to mice | Inden et al. (2007) |
| Increase in LC3 positive dots in nigral DA neurons (approx. 380%), upregulation of LC3II (approx. 40%), Beclin 1 (approx. 33%) and P62 (approx. 50%) autophagic substrate | Approx. 40% decrease in the number of TH neurons (SNpc) and density of TH positive fibres (approx. 50%) (striatum) | 2 mg/kg per day for 8 weeks sc of Rotenone in Levis rats | Wu et al. (2015) |
| Approx. 8 fold increase in the number of TH+ neurons with granular LC3 | Approx. 40% decrease in the number of TH immunoreactive neurons | Primary dopaminergic neurons following treatment with MPP+ (LD50 of 5 μM/L) | Zhu et al. (2007) |
| Decrease in mitochondrial speed (approx. 100% decrease in anterograde speed and approx. 28% increase in retrograde speed) | Approx. 70% decrease in positive TH neuronal bodies at 48 h | Treatment with up to 5 μM (1–5 μM) of MPP+ in TH positive murine mesencephalic neurons in an in‐vitro microchamber segregating system | Kim‐Ham et al. (2011) |
| Reduction in mitochondrial movement was statistically significant from day 8 and was greatest on day 16 at 50 nM (approx. day 3 19%, day 6 7%, day 8 62%, day 14 37%, day 16 200%) | Approx 60% of cell loss by day 21 | In vitro SH‐SY5Y neural cells treated with 50 nM rotenone for 21 days | Borland et al. (2008) |
| 30% increase over control in static mitochondria and 50 decrease over control in number of mitochondria | Significant decline of intracellular ATP at 24 h | Differentiated (d6) LUHMENS stably expressing eGFP/mito‐tRFP, treated with MPP+ (5 μM) for 24 h | Schildknecht et al. (2013) |
Figure A.17.
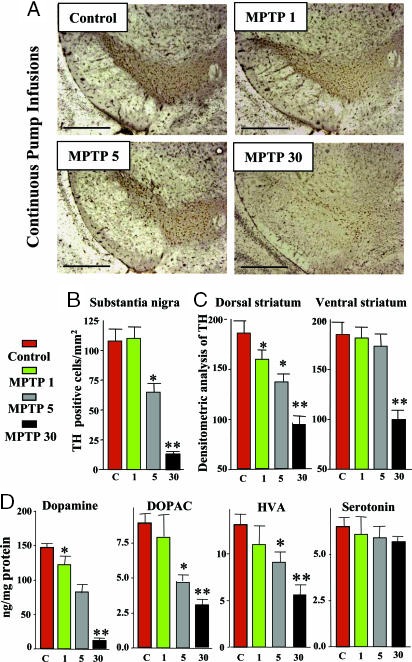
Neurotoxicity induced by continuous MPTP administration. (A) Representative tyrosine hydroxylase (TH)‐stained sections of the substantia nigra from mice that were continuously treated for 28 days with control pump infusions or with infusions of 1, 5, or 30 mg MPTP/kg daily. (Scale bar, 600 μm) (B and C) TH‐positive cell counts in the substantia nigra (B) and semiquantitative densitometric measurements of the TH signal in striatum (C) (n = 10 mice per group). (D) Striatal monoamine levels in MPTP‐treated mice (n = 10 mice per group). Asterisks indicate statistically significant differences (p < 0.05) of a sample compared to control (single asterisks) or to both the control and the lower MPTP dose (double asterisks) (Fornai et al., 2005. fig. 3, copyright The National Academy of Sciences)
Figure A.18.
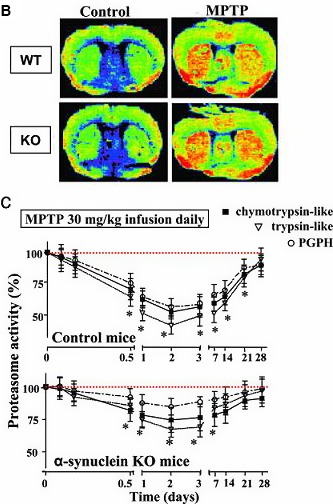
Effect of an α‐synuclein deletion on MPTP toxicity. (B) Uptake of [14C]2‐DG in littermate wild‐type and α‐synuclein KO mice that were continuously infused for 7 days with control or MPTP (30 mg/kg daily) solution. Pictures display false‐colour autoradiograms. (C) Proteasome activity in control and α‐synuclein KO mice continuously infused with MPTP (30 mg per kg of body weight daily). Proteasome activities in the substantia nigra are depicted as per cent of control (means ± SEMs) as a function of time after beginning of the infusions (five mice per group). In A and C, asterisks indicate statistically significantly different values (p < 0.05) from controls (Fornai et al., 2005. fig. 5, copyright The National Academy of Sciences)
Evidence Supporting Taxonomic Applicability
Multiple animal models have been used to mimic PD (Johnson et al., 2015). There are no sex restriction; however, susceptibility to MPTP increases with age in both non‐human primates and mice (Rose et al., 1993, Irwin et al., 1993, Ovadia et al., 1995).
References
Agiular JS, Kostrzewa RM. Neurotoxin mechanisms and processes relevant to parkinson's disease: un update. Neurotoxicity Research. doi 10.1007/s12640‐015‐9519‐y
Alvarez‐Erviti L, Rodriguez‐Oroz MC, Cooper JM, Caballero JD, Ferrer I, Obeso JI, Schapira AHV, 2010. Chaperone‐Mediated Autophagy Markers in Parkinson Disease Brains. Archives of Neurology, 67, 1464–1472.
Arnold B, et al., 2011. “Integrating multiple aspects of mitochondrial dynamics in neurons: age‐related differences and dynamic changes in a chronic rotenone model.” Neurobiology of Disease, 41, 189–200.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Neuroscience, 3, 1301–1306.
Betarbet R, Canet‐Aviles RM, Sherer TB, Mastroberardino PG, Mc Lendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinfelter G, Cookson MR, Greenmyre JT, 2006. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ‐1, α‐synuclein, and the ubiquitin‐proteasome system. Neurobiology Disease, 22, 404–420.
Borland MK, Trimmer PA, Rubinstein JD, et al., 2008. Chronic, low dose rotenone reproduces Lewy neuritis found in early stages of Parkinson's disease, reduces mitochondrial movement and slowly kill differentiated SH‐SY5Y neural cells. Molecular Neurodegeneration, 3–21.
Chen L, Jin J, Davis J, 2007. Oligomeric α‐synuclein inhibits tubulin polymerization. Biochemical and Biophysical Research Communications, 356, 548–553.
Choi WS, Kruse SE, Palmiter RD, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, 105, 15136–15141.
Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Malsiah E, 2010. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α‐synucleinopathy. 5, 1–16.
Cuervo AM, Wong E, 2014. Chaperone‐mediated autophagy: roles in disease and aging. Cell Research, 24, 92–104.
Decressac M, Björklund A, 2013. Pathogenic role and therapeutic target in Parkinson disease. Autophagy, 9, 1–3.
Dehay B, Bove J, Rodriguez‐manuela N, perier C, Recasens A, Boya P, Vila M, 2010. Pathogenic lysosomal depletion in Parkinson's disease. The Journal of Neuroscience, 30, 12535–12544.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Lewis VJ, Hersch S, Sulzer D, Przedborski S, burke R, Hen R, 2002. Resistance of α‐synuclein null mice to the parkinsonian neurotoxicity MPTP. Proceedings of the National Academy of Sciences, 99, 14524–14529.
Dauer W, Przerdborski S, 2003. Parkinson's disease: Mechanisms and Models. Neuron, 39, 889–899.
Esposito A, Dohm CP, Kermer P, 2007. α‐synuclein and its disease‐related mutants interact differentially with the microtubule protein tau and associate with actin cytoskeleton. Neurobiology of Disease, 26, 521–531.
Fleming SM, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman R, Chesselet MF, 2004. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusion of varying doses of rotenone. Experimental Neurology, 187, 418–429.
Ferrante RJ, Schulz JB, Kowall NW, Beal MF, 1997. Systematic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Research, 753, 157–162.
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti C, Ruffoli R, Soldani P, Ruggieri S, Alessandri’ MG, Paparelli A, 2003. Fine structure and mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. The Journal of Neuroscience, 23, 8955–8966.
Fornai F, Schluter OM, lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggeri S, Paparelli A, Sudhof TC. 2005. Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin‐proteasome system and α‐synuclein. Proceedings of the National Academy of Sciences, 102, 3413–3418. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC552938/ Fig. 3; Fig. 5; © National Academy of Sciences
Forno LS, DeLanney LE, Irwin I, Langston JW, 1993. Similarities and differences between MPTP‐induced parkinsonsim and Parkinson's disease. Neuropathologic considerations. Advances in neurology, 600–608.
Forno LS, Langston JW, DeLanney LE, Irwin I, Ricaurte GA, 1986. Locus coeruleus lesions and eosinophilic inclusions in MPTP‐treated monkeys, 20, 449–455.
Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z, 2012. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α‐synuclein and LRRK2 in the brain. The Journal of Neuroscience, 32, 7585–7593.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH, 2000. In situ and in vitro study of colocalization and segregation of alpha‐synuclein, ubiquitin, and lipids in Lewy bodies. Experimental Neurology, 166, 324–333.
Gainetdinov RR, Fumagalli F, Jones SR, Caron MG, 1997. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. Journal of Neurochemistry, 69, 1322–1325.
Giordano S, et al., 2014. “Bioenergetic Adaptation in Response to Autophagy Regulators During Rotenone Exposure.” Journal of Neurochemistry, 131, 625–633.
Hall K, Yang S, Sauchanka O, Spillantini MG, Anichtchik O, 2015. Behavioural deficits in transgenic mice expressing human truncated (1‐120 amino acid) alpha‐synuclein. Experimental Neurology, 264, 8–13.
Hasbani DM, O'malley KL, 2006. Wlds mice are protected against Parkinsonisn mimetic MPTP. Experimental Neurology, 202, 93–99.
Heikkila RE, Nicklas WJ, Vyas I, Duvoisin RC, 1985. Dopaminergic toxicity of rotenone and the 1‐methyl‐4‐phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine toxicity. Journal of Neuroscience Letters, 62, 389–394.
Hirata Y, Suzuno H, Tsuruta T, Oh‐hashi K, Kiuchi K, 2008. The role of dopaminergic transporter in selective toxicity of manganese and rotenone. Toxicology, 244, 249–256.
Hoglinger GU, Feger J, Annick P, Michel PP, Karine P, Champy P, Ruberg M, Wolfgang WO, Hirsch E, 2003. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. Journal of Neurochemistry, 84, 1–12.
Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH, 1985. Parkinson inducing neurotoxin, MPTP,: uptake of the metabolite MPP+ by dopamine neurons explains selective toxicity. Proceedings of the National Academy of Sciences, 82, 2173–2177
Johnson ME, Bobrovskaya L, 2015. An update on the rotenone models of Parkinson's disease: Their ability to reproduce features of clinical disease and model gene‐environment interactions. 946, 101–116.
Kaushik S, Cuervo AM, 2008. Chaperone Mediated Autophagy. Methods in Molecular Biology, 445, 227–244.
Kim‐Han JS, Dorsey JA, O'Malley KL, 2011. The parkinsonian mimetic MPP+, specifically impairs mitochondrial transport in dopamine axons. The Journal of Neuroscience, 31, 7212–7221.
Kirk D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel R, Bijorklund A, 2002. Parkinson‐like neurodegeneration induced by targeted overexpression of α‐synuclein in the nigrostriatal system, 22, 2780–2791.
Kirk D, Annett L, Burger C, Muzyczka N, Mandel R, Bijorklund A, 2003. Nigrostriatal α‐synucleinopathy induced by viral vector‐mediated overexpression of human α‐synuclein: A new primate model of Parkinson's disease. Proceedings of the National Academy of Sciences, 100, 2884–2889.
Kitamura Y, Shimohama S, Akaike A, Taniguchi T, 2000. The Parkinsonian Models: invertebrates to mammals. The Japanese Journal of Pharmacology, 84, 237–243.
Klein RL, King MA, Hamby ME, Meyer EM, 2002. Dopaminergic cell loss induced by human A30P α‐synuclein gene transfer to the rat substantia nigra. Human Gene Therapy, 13, 605–612.
Korolochuk VI, Menzies FM, Rubinsztein DC, 2010. Mechanism of cross‐talk between the ubiquitin‐proteasome and autophagy‐lysosome systems. Federation of the European Biochemical Societies Letters (FEBS), 584, 1393–1398.
Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, Taira T, Ariga H, Shimohama S, 2007. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4‐phenylbutyrate, a chemical chaperone. Journal of Neurochemistry, 101, 1491–1494.
Irwin JK, 1993. Parkinson's disease: Past, Present and Future. Neuropsycopharmacology, 9, 1–1.
Langston JW, Ballard P, Irwin I, 1983. Chronic parkinsonism in human due to a product of meperidine‐analog synthesis. Science, 219, 979–980.
Lapointe N, St‐Hilaire M, martinoli MG, Blanchet J, gould P, Rouillard C, Cicchetti F, 2004. Rotenone induces non‐specific central nervous system and systemic toxicity. The Federation of American Societies for Experimental Biology (FASEB). Journal express article, doi: 10.1096/fj.03‐0677fje
Lauwers E, Debyser Z, Van Drope J, DeStrooper B, Nuttin B, 2013. Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector‐mediated overexpression of α‐synuclein. Brain Pathology, 13, 364–372.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, gasser T, Steinbach PI, Wilkinson KD, Polymeopoulos MH, 1998. The ubiquitin pathway in Parkinson's disease. Nature, 395, 451–462.
Lo Bianco C, Ridet JL, Deglon N, Aebischer P, 2002. Alpha‐synucleopathy and selective dopaminergic neuron loss in a rat lentiviral‐based model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 10813–10818.
Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, 2004. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in a α‐synuclein rat model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 17510–17515.
Masiliah E, Rockenstein E, Veibergs I, Malloty M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L, 2000. Dopaminergic loss and inclusion body formation in α‐synuclein mice: implications for neurodegenerative disorders. Science, 287, 1265–1269.
Meissner W, Prunier C, Guilloteau D, Chalon S, Gross CE, Bezard E, 2003. Time‐course of nigrostriatal degeneration in a progressive MPTP‐lesioned macaque model of Parkinson's disease. Molecular Neurobiology, 3, 209–218.
McNaught KSC, Olanow W, Halliwell B, 2001. Failure of the ubiquitin‐proteasome system in Parkinson's disease. Nature Reviews Neuroscience, 2, 589–594.
McNaught KSP, Belizaire R, Isacson O, Jenner P, Olanow CW, 2002. Altered proteasomal function in sporadic Parkinson's disease. Experimental Neurology, 179, 38–46.
McNaught KSP, Olanow CW, 2003. Proteolytic stress: a unifying concept for the etiopathogenesis of Parkinson's Disease. Annals of Neurology, 53, 73–76.
Monti, B. Gatta V, Piretti F, Raffaelli S, Virgili M, Contestabile A, 2010. “Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha‐synuclein.” Neurotoxicity Research, 17, 130–141.
O'Malley KL, 2010. The role of axonopathy in Parkinson's disease. Experimental Neurobiology, 19, 115–119.
Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H, 2005. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathologica, 109, 583–598.
Ovadia A, Zhang Z, Gash DM, 1995. Increased susceptibility to MPTP in middle‐aged Rhesus Monkeys. Neurobiology of aging, 16, 931–947.
Pan‐Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RHW, 2010. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. Public Library of Science (PLoS ONE), 5, 1–10.
Pifl C, Giros B, Caron MG, 2004. Dopamine transporter expression confers cytotoxicity to low doses of the parkinsonism‐inducing neurotoxin MPTP. Journal of Neuroscience, 13, 4246–4253.
Porras G, Bezard E, 2012. Modelling Parkinson's disease in primates: The MPTP model. Cold Spring Harbor Perspectives in Medicine, 2, 1–10.
Raff MC, Whitemore AV, Finn JT, 2002. Axonal self‐destruction and neurodegeneration. Science, 296, 868–871.
Rappold PM et al., 2014. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nature Communications, 5, 5244 doi: 10.1038/ncomms6244
Ravikumar BB, Sarkar S, Davies JE, et al., 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological Reviews. 90, 1383–1435.
Ren Y, Liu W, Jiang H, Jiang Q, Feng J, 2005. Selective vulnerability of dopaminergic neurons to microtubule depolymerisation, 280, 34105–34112.
Rose S N M, Jackson EA, Gibb WR, Jaehnig P, Jenner P, Marsden CD, 1993. Age‐related effects of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine treatment of common marmosets. European Journal of Pharmacology, 230, 177–185.
Saha AR, Utton MA, Asuni AA, Ackerley S, Grierson AJ, Miller CC, Davies AM, Bucham VI, Anderton BH, Hanger DP, 2004. Parkinson's disease alpha‐synuclein mutation exhibit defective axonal transport in cultured neurons Journal of Cell Science, 117, 1017–1024.
Saravan KS, Sindhu K, Mohanakumar P, 2005. Acute intranigral infusion of rotenone in rats causes progressive biochemical lesions in the striatum similar to Parkinson's disease. Brian research, 1049, 147–155.
Serra PA, Sciola L, Delogu MR, Spano A, Monaco G, Miele E, Rocchitta G, Miele M, Migheli R, Desole MS, 2002. The neurotoxin MPTP induces apoptosis in mouse nigrostriatal glia. The Journal of Biological Chemistry, 277, 34451–34461
Sherer TB, Kim JH, Betarbet R, Greenmayre JT, 2002. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and α synuclein aggregation. Experimental Neurology, 179, 9–16.
Schildknecht S, Karreman C, Pöltl D, Efrémova L, Kullmann C, Gutbier S, KrugA, Scholz D, Gerding HR, Leist M, 2013. Generation of genetically‐modified human differentiated cells for toxicological tests and the study of neurodegenerative diseases. Alternatives to Animal Experimentation (ALTEX), 30, 427–444.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Chiba IK, Tanaka K, Suzuki T, 2000. Familial Parkinson's disease gene product, parkin, is an ubiquitin‐protein ligase. Nature Genetics, 25, 302–305.
Schmidt MA, 2002. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behavioural Brain Research, 136, 317–324.
Schwarz TL, 2015. Mitochondrial trafficking in neurons. Cold Spring Harbor Perspectives in Biology, 2013–2015.
Shulman JM, DeJager PL, Feany MB, 2011. Parkinson's disease: genetics and pathogenesis. Annual Review of Pathology Mechanisms of Disease, 6, 193–202.
Tai HC, Schuman EM, 2008. Ubiquitin, the proteasome and the protein degradation in neuronal function and dysfunction. Nat. Rev. Journal of Neuroscience, 9, 826–838.
Tieu Kim, Imm Jennifer, 2014. Mitochondrial dynamics as potential therapeutic target for Parkinson's disease? Advances in Clinical Neuroscience and Rehabilitation, 14, 6–8.
Yang MI, Hasasdri L, Woods WS, George JM, 2010. Dynamic transport and localization of alpha‐synuclein in primary hippocampal neurons. Molneurodegener, 5–9.
Weihofen A, Thomas KJ, Ostaszewski BL, 2009. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry, 48, 2045–2052.
Wu F, Xu HD, Guan JJ, Hou YS, Gu JH, Zhen XC, Qin ZH, 2015. Rotenone impairs autophagic flux and lysosomal functions in Parkinson's disease. 284, 900–911.
5th KER: Neuroinflammation leads to degeneration of the dopaminergic neurons of nigrostriatal pathway
A.5.1. How this KER works
Cells of the innate (microglia and astrocytes) and adaptive (infiltrating monocytes and lymphocytes) immune system of the brain have, like other immune cells (in peripheral tissues), various ways to kill neighbouring cells. This is in part due to evolutionary‐conserved mechanisms evolved to kill virus‐infected cells or tumour cells; in part it is a bystander phenomenon due to the release of mediators that should activate other cells and contribute to the killing of invading microorganisms. An exaggerated or unbalanced activation of immune cells can thus lead to parenchymal (neuronal) cell death (Gehrmann et al., 1995). Mediators known to have such effects, and that are also known to be produced during inflammation in the brain comprise components of the complement system and cytokines/death receptor ligands triggering programmed cell death (Dong and Benveniste, 2001). Besides these specific signals, various secreted proteases (e.g. matrix metalloproteases), lipid mediators (e.g. ceramide or gangliosides) or reactive oxygen species can contribute to bystander death of neurons (Chao et al., 1995; Nakajima et al., 2002; Brown and Bal‐Price, 2003; Kraft and Harry, 2011; Taetzsch and Block, 2013). Especially the equimolar production of superoxide and NO from glial cells can lead to high steady state levels of peroxynitrite, which is a very potent cytotoxicant (Yuste et al., 2015). Already damaged neurons, with an impaired antioxidant defence system, are more sensitive to such mediators.
An important role of microglia in the brain is the removal of cell debris (Xu et al., 2015). Healthy cells continuously display anti‐‘eat me’ signals, while damaged and stressed neurons/neurites display ‘eat‐me’ signals that may be recognised by microglia as signal to start phagocytosis (Neher et al., 2012), thus accelerating the loss of DA neurites in the striatum.
Activated microglia surrounding DAergic neurons in PD express the M1 neurodegenerative phenotype (Hunot et al., 1999), which promote proliferation and function of CD4+ T cells (for review Appel et al., 2010), which in turn induce DA neuron toxicity, as assessed by experiments with immunodeficient mice (Brochard et al., 2009). Possible infiltration of other myeloid cells, such as monocytes or macrophages through a compromised blood‐brain barrier, may also be involved in phagocytosis and neurodegeneration (Depboylu et al., 2012; Pey et al., 2014).
A.5.2. Weight of evidence
A.5.2.1. Biological plausibility
Histopathological studies have shown that glial activation is a hallmark of every neurodegenerative disease, including Parkinson's disease (Whitton, 2007; Tansey and Goldberg, 2009; Niranjan, 2014; Verkhratiky et al., 2014). PET studies in PD patients have revealed that microglial activation in the substantia nigra is an early event in the disease process (Iannaccone et al., 2012), and that it is extremely persistent. The role of astrocytes is less clear than the one of microglia, but reactive astrocytes are able to release neurotoxic molecules (Mena and Garcia de Ybenes, 2008; Niranjan, 2014). However, astrocytes may also be protective due to their capacity to quench free radicals and secrete neurotrophic factors. The activation of astrocytes reduces neurotrophic support to neurons, and the proportion of astrocytes surrounding dopaminergic neurons in the substantia nigra is the lowest for any brain area suggesting that dopaminergic neurons are more vulnerable in terms of glial support (for review, Mena and Garcia de Ybenes, 2008).
In vitro co‐culture experiments have demonstrated that reactive glial cells (microglia and astrocytes) can kill neurons (Chao et al., 1995; Brown and Bal‐Price, 2003; Kraft and Harry, 2011; Taetzsch and Block, 2013), and that interventions with e.g. i‐NOS inhibition can rescue the neurons (Yadav et al., 2012; Brzozowski et al., 2015). Direct activation of glial cells with the inflammogen LPS has also resulted in vivo in the death of DA neurons (Li et al., 2009; Zhou et al., 2012; Sharma and Nehru, 2015).
Circulating monocytes and lymphocytes
Neuroinflammation can disrupt blood‐brain barrier integrity (Zhao et al., 2007), facilitating infiltration of circulating monocytes and lymphocytes (Qian et al., 2010; Machado et al., 2011). T cell infiltration has been found in CNS tissue of PD patients (Miklossy et al., 2006; Qian et al., 2010), and in animal models, in which depletion or inactivation of lymphocytes has been found to protect striatal DA terminals (for review, Appel et al., 2010).
A.5.2.2. Empirical support for linkage
LPS injections
Lipopolysaccharide (LPS, a known activator of microglia) injected into the substantia nigra successfully replicated the pathogenic features of Parkinson's disease in rats. An increase in the mRNA expression of pro‐inflammatory cytokines (TNF‐alpha, IL‐1 beta) was observed 7 days post‐injection; alterations in oxidative stress markers (ROS, lipid peroxidation, NO formation, NADPH oxidase activity, GSH system, SOD and catalase) became significant 14 days post‐injection, and this was followed by a significant decline in tyrosine hydroxylase (TH), as marker of dopaminergic neurons (Sharma and Nehru, 2015). LPS‐induced downregulation of TH expression seemed to depend on the pro‐inflammatory cytokine IL‐1 beta, since it was not observed in LPS‐injected IL‐1 knockout mice (Tanaka et al., 2013).
Progressive hypokinesia, selective loss of dopaminergic neurons in substantia nigra and reduction of striatal dopamine content, as well as alpha‐synuclein aggregation in substantia nigra was also achieved by unilateral intranasal instillation of LPS every other day for 5 months, mimicking a progressive inflammation‐mediated chronic pathogenesis of Parkinson's disease (He et al., 2013). It is important to note that LPS administrated either directly in the brain, intraperitoneally or in utero results in a delayed and progressive loss of nigral DA neurons that persists well after the initial inflammatory stimulus (for review, Taetzsch and Block, 2013).
Rotenone
Chronic systemic rotenone exposure reproduces features of Parkinson's’ disease with loss of DA neurons and putative Lewis bodies in substantia nigra, accompanied by neuroinflammation and oxidative stress, and reduction of TH immunoreactivity in striatum together with an increase in reactive astrocytes (Betarbet et al., 2000; Ferris et al., 2013). In this chronic rotenone model (2–3 mg/kg per day up to 4 weeks), microglia activation precedes neuronal death (Sherer et al., 2003). Several interventions aiming at blocking several features of microglial activation (NADPH oxidase, myeloperoxidase, phagocytosis, opening of KATP channels, etc.) protected DA neurons from death (Gao et al., 2003; Zhou et al., 2007; Chang et al., 2013; Emmrich et al., 2013; Salama et al., 2013; Wang et al., 2014). An enhanced sensitivity of dopaminergic neurons to rotenone‐induced toxicity was observed with aging, in parallel with the increase of glial cell activation in older rats (Phiney et al., 2006).
In vitro, little neurotoxicity was detected in primary DA neuron cultures (low glia‐content) exposed to rotenone, whereas significant and selective dopaminergic neurodegeneration was observed in neuron/glia cultures (Gao et al., 2002).
MPTP/MPP+
Following MPTP treatment, microglial cells are activated by a mechanism secondary to dopaminergic neuron injury (Zhou et al., 2005). However, elevation of interferon‐gamma and TNFalpha in substantia nigra was detected before the death of DAergic neurons (Barcia et al., 2011); and serum levels of INF‐gamma and TNFalpha remain elevated for years in monkeys exposed to MPTP (Barcia et al., 2011). The role of microglia in the progression of DA neurodegeneration is suggested by in vivo and in vitro experiments in which feature of microglial reactivity (TNF‐alpha, i‐NOS, NADPH‐oxidase, ROS generation) were blocked (Dehmer et al., 2000; Feng et al., 2002; Sriram et al., 2002; Ferger et al., 2004; Wang et al., 2006; Chung et al., 2011; Bodea et al., 2014; Borrajo et al., 2014; Wang et al., 2014; Brzozowski et al., 2015; Liu et al., 2015). Some evidence from above studies also extends to astrocytes (Sathe et al., 2012; Khan et al., 2014). For instance, systemic administration of nicotine (stimulating the anti‐inflammatory role of alpha 7 nicotinic acetylcholine receptors on astrocytes and microglia) reduced MPTP‐induced motor symptoms, and protected against neurodegeneration in the substantia nigra by (Liu et al., 2012, 2015).
Entrance into the brain of bone marrow‐derived cells expressing i‐NOS may also play a deleterious role in neurodegeneration (Kokovay and Cunningham, 2005). Indeed, pharmacological inhibition or deletion of CD95 in peripheral myeloid cells hampered brain infiltration and was protective for MPTP‐induced DA loss in striatum (Chung et al., 2015; Gao et al., 2015). Similarly, therapies aiming at suppressing immune reactivity, such as administration of Treg cells (CD4+ CD25+ regulatory T cells) lead in MPTP treated mice, to a robust nigrostriatal protection associated to an inhibition of microglial reactivity (Reynolds et al., 2010).
Paraquat
Paraquat alone (10 mg/kg, 2x/week, for 4 weeks) or in combination with maneb (30 mg/kg) induces a loss of DAergic neurons in the substantia nigra paralleled by an increase in microglial reactivity (Cicchetti et al., 2005; Mitra et al., 2011). In a paraquat rat model, microglial reactivity was observed 4 weeks post‐injection, whereas degeneration of DAergic neurons was only observed 2 weeks later (Sant‐Pierre et al., 2006).
Direct treatment of primary microglial cells with paraquat (5–15 μM) showed no morphological change and no upregulation of IL‐10, IL‐1beta, IL‐2, IL‐4, TNF‐alpha, GM‐CSF or INF‐gamma, suggesting that paraquat cannot activate directly microglial cells (Klintworth et al., 2009), despite contrasting observations in the microglial cell lines BV2 (Miller et al., 2007) or N9 (Bonneh‐Barkay et al., 2005). But « priming » of microglial cells by a first exposure to paraquat (10 mg/kg) (Purisai et al., 2007), by LPS (2–4 mg/kg) (Purisai et al., 2007), or by a viral mimic (Bobyn et al., 2012) increased the vulnerability of DA neurons to further paraquat treatments. Interestingly, if minocycline (45 mg/kg), an antibiotic known to decrease microglial reactivity, was applied together and after the first priming paraquat treatment, subsequent exposure to paraquat failed to cause DA neurodegeneration (Purisai et al., 2007). If paraquat treatments were made in mice lacking functional NADPH oxidase, no DA neurodegeneration was detected (Purisai et al., 2007), identifying again NADPH‐oxidase as a key factor (Wu et al., 2005).
In particular, the NADPH oxidase isoform NOX2 located on microglia plasma membranes transfers electrons to paraquat inducing the formation of the paraquat radical cation (Rappold et al., 2011). Radical paraquat may then (i) react with oxygen efficiently producing superoxide and regenerating paraquat, and/or (ii) enter DA neurons being a substrate for the dopamine transporter (DAT) (Rappold et al., 2011). This second possibility is supported by the observation that cells expressing DAT efficiently uptake paraquat only in the presence of microglia, but not when NOX2 activity is specifically abolished (Rappold et al., 2011). Neurodegeneration may be then triggered (i) by the amplification of the extracellular redox signalling (Bonneh‐Barkay et al., 2005; Purisai et al., 2007) and/or (ii) establishing a new round of redox cycling once paraquat is taken up into DA neurons. Accordingly, expression of DAT sensitise HEK293 cells to paraquat (50 microM) induced intracellular ROS production and cell death as well as mutant mice with hypomorphic DAT are resistant to paraquat neurotoxicity (Rappold et al., 2011).
Besides NADPH‐oxidase, other inflammatory factors are involved in DA neurodegeneration: for example, iNOS, NF‐kappaB or p38 MAPK, since their blockade reverted the 50% decrease of TH immunoreactivity, as well as IL1‐beta and NO increased expression in striatum observed following paraquat or paraquat and maneb treatments (Yadav et al., 2012). Similarly, IFN‐gamma silencing prevented the paraquat‐induced morphological signs of microglial activation, the NADPH‐oxidase expression, as well as the time‐dependent changes in the pro‐inflammatory enzymes i‐NOS and COX‐2, of cytokines (IL‐1beta, TNF alpha), and of signalling molecules (JNK and p38 MAPK), and protected against paraquat‐induced DA neurodegeneration (Mangano et al., 2012).
Protection against paraquat‐induced DA neurodegeneration can also be achieved by providing trophic support (intranigral or peripheral injection of GDNF or GM‐CSF, respectively), which is reduced upon paraquat treatment (Mangano et al., 2011).
A.5.3. Uncertainties or inconsistencies
Mice deficient in microglia (depletion by a ganciclovir‐thymidine kinase system under the CD11b promoter) were still susceptible to MPTP toxicity, while mixed cell cultures prepared from these deficient mice showed partial protection (Kinugawa et al., 2013).
Although some publications show strong protection by COX‐2 inhibition/deletion, others showed that mice deficient for COX‐2 were partly protected against MPTP‐induced decrease of DAergic neurons in substantia nigra, but not against DA terminal loss in striatum (Feng et al., 2000).
Mice deficient in IL6 (IL6−/−) showed an increased vulnerability of the nigrostriatal pathway following MPTP treatment associated to a normal astrogliosis but a transient microgliosis, suggesting that transient microgliosis and IL6 may have also protective effects (Cardenas and Bolin, 2003).
MMTV integration site 1 (Wnt 1) is a key transcript involved in DAergic neurodevelopment, and is dynamically regulated during MPTP‐induced DA degeneration and glial activation. MPTP‐activated astrocytes of the ventral midbrain were identified as candidate source of Wnt 1 by in situ hybridisation and RT‐PCR in vitro, suggesting that reactive astrocytes may be rather involved in neuroprotective/neurorescue pathways, as further demonstrated by deletion of Wnt 1 or pharmacological activation of Wnt/catenin signalling pathway (L'Episcopo et al., 2011).
The role of microglia, NADPH‐oxidase and oxidative stress in paraquat‐induced neurodegeneration is well established. Nevertheless, the mechanism connecting these three elements remain poorly understood since direct evidence for extracellular and/or intracellular formation of radical paraquat and superoxide is controversial.
Rotenone (1–3 nM) applied directly on BV2 microglial cells increased their phagocytosis and the release of pro‐inflammatory cytokines (TNF‐alpha, IL‐1 beta), suggesting that microglial cell can also be a primary target of rotenone (Zhang et al., 2014). However, these results in a transformed microglial cell line contrast with the experiments performed on isolated primary microglial cells, where rotenone (10–50 nM) was not able to trigger a direct activation (Klintworth et al., 2009).
The regulation of inducible nitric oxide synthase (for production of peroxynitrite) differs strongly between rodents and human, and thus, the role of NO in human remains unclear (Ganster et al., 2001).
While in human long‐term use of anti‐inflammatory drugs (NSAIDs, aspirin, iboprufen) for preventing PD onset or for slowing the progression is still controversial, a new strategy is emerging aiming at targeting microglial cells by modulating their activity, rather than simply trying to counteract their inflammatory neurotoxicity. The advantage of this therapeutic approach could be to reduce neuroinflammation and neurotoxicity, while at the same time strengthening intrinsic neuroprotective properties (Pena‐Altamira et al., 2015)
A.5.4. Quantitative relationship
As it is rather the features and the duration of the inflammatory response that determine the extent of the nigrostriatal pathway neurodegeneration, the best way to propose a quantitative or semiquantitative evaluation of the links between KEup and KEdown is to use studies where any feature of neuroinflammation is inhibited and to quantify the protection of the Daergic neurons and terminals. Thus it will give an evaluation of how much neurodegeneration depends on the neuroinflammatory process. Below are some examples for illustration.
Table A.4.
Quantitative evaluation of the KER
| KE upstream Neuroinflammation | KE downstream Neurodegeneration of dopaminergic nigrostriatal pathway | Reference | Type of study | Comment |
|---|---|---|---|---|
| Inhibition of any feature of neuroinflammation (microglia/astrocyte) | How much nigrostriatal pathway degeneration depends on KE up as assessed by protection when any KE up feature is inhibited | |||
| K ATP channel opener (iptakalim) induced decrease of TNF‐alpha and COX2 mRNA expression and TNF‐alpha content, as well as microglial reactivity (OX42, ED1) | TH immunoreactivity: Total recovery | Zhou et al. (2007) |
In vivo Rotenone 2.5 mg/kg per day + in vitro |
|
|
NADPH oxidase Neuron enriched cultures Neuron‐Glia co‐cultures +apocynin |
DA uptake TH immunoreactivity About 50% more neuronal death in the presence of glia (80% of protection with apocynin) |
Gao et al. (2002) |
In vitro Rotenone 5–20 nM |
|
|
NADPH oxidase Mice knockout for NADPH ox gp91−/− Co‐culture neuron‐glia |
DA uptake: 40% protection TH immuno: 20% protection |
Gao et al. (2003) |
In vitro Rotenone 5–10 nM |
|
| Phagocytic signalling between neuron and microglia i.e. block of vitronectin and P2Y6 on microglia or annexin or phophatidylserine on neuron (eat‐me signal) | About 20% neuronal protection | Emmrich et al. (2013) |
In vitro Co‐cultures of cerebellum Rotenone 2.5 nM |
|
| Decrease in the number of activated microglia by L‐thyroxin in substantia nigra, not in striatum | Protection of DA terminals in striatum, but no effect in substantia nigra | Salama et al. (2012) |
In vivo Rotenone 3 mg/kg per day |
|
|
Myeloperoxidase (HOCl from H2O2) Resveratrol decreased NO, ROS, phagocytosis in microglia and astrocytes |
Protection of neuron: 40% cell viability 50–60% TH immuno + number of dendrites |
Chang et al. (2013) |
In vitro Rotenone 30 nM MPP+ 0.1 μM |
|
|
NADPH oxidase: NOX2 Diphenyleneiodonium: long acting NOX2 inhibitor |
DA uptake and TH immuno: 30–40% of protection | Wang et al. (2014) |
In vitro LPS 20 ng/mL MPP+ 0.15 μM |
|
|
Control of microglial and astrocyte reactivity by Alpha 7 nicotinic Ach receptor present on microglia and astrocyte Its activation decreased microglial and astrocyte reactivity |
MPP+ caused 40% decrease of TH+ neurons Nicotine induced a 30% recovery |
Liu et al. (2012, 2015) |
In vivo MPTP 20 mg/kg Nicotine 5 mg/kg In vitro on isolated microglia and astrocytes |
|
|
TNF‐alpha of microglial origin By blocking angiotensin‐1 receptors, NADPH‐oxidase, Rho‐kinase and NF.kB |
20% of recovery of TH immunoreactivity | Borrajo et al. (2013) |
In vitro + in vivo MPP+ 0.25 μM |
|
| Infusion of the anti‐inflammatory cytokine TGF beta protects from MPP+‐induced cell loss by decreasing CD11b, i‐NOS, TNFalpha, IL+ beta, and increasing IGF‐1. Silencing of TGFbR1 gene abolished the protective effect | MPP+ caused 60% decrease of TH immuno, and TGFbeta induced a dose‐dependent recovery (5–20 ng/mL) | Liu et al. (2015) |
In vitro Co‐cultures MPP+ 5 μM |
Indirect |
|
i‐NOS inhibition caused a decrease of astrocyte and microglial reactivity as assessed by GFAP and OX6, respectively (n‐NOS inhibition had no effect) |
TH immunoreactivity Dose‐dependent recovery with 1,400 W (0.1–100 μM) |
Brzozowski et al. (2015) |
In vitro MPP+ 43 μM |
|
|
Inhibition of laminin receptor on microglia i.e. regulating cell‐ECM interactions induced a decrease of microglia phagocytosis and of O2 − production |
Dose‐dependent partial recovery (about 35% of TH immunoreactivity | Wang et al. (2006) |
In vitro MPP+ 0.1–0.5 μM |
|
| Inhibition of glial activation‐mediated oxidative stress by Fluoxetine, antidepressant) | 30% of recovery of TH immunoreactivity in Substantia nigra and total recovery of DA terminals in striatum | Chung et al. (2011) |
In vivo MPTP 20 mg/kg i.p. |
|
|
Mice lacking both TNFR Induced a decrease of GFAP in striatum Double KO, if only KO for TNFR1 or TNFR2, no protection |
TH staining in striatum, DA content and GFAP staining, all returned to control level | Sriram et al. (2014) |
In vivo MPTP 12.5 mg/kg sc |
|
|
Mice‐deficient for COX2 Microglial cells are the major cells expressing COX2 |
MPTP caused in substantia nigra 40% loss in wild type 45% loss in COX1−/− 20% loss in COX2−/− in striatum 70% loss of DA in all 3 types of mice |
Feng et al. (2002) |
In vivo MPTP 20 mg/kg sc |
|
| S100B −/− in astrocytes caused decreased microgliosis, TNF‐alpha and RAGE |
12% of protection for TH+ neuron 30% of protection for Nissl‐labelled neurons |
Sathe et al. (2012) |
In vivo MPTP 30 mg/kg i.p. |
|
|
Glia Maturation Factor (GMF) overexpression or GMF−/− showed decreased TNF‐alpha, IL‐1beta, ROS and NFkappaB downregulation |
Overexpression of GMF exacerbate DA neuron degeneration GMF−/− induced a protection of 40% of TH+ neurons |
Khan et al. (2014) |
In vitro Mesencephalic neuron/glia cultures MPP+ 5, 10, 20 μM |
|
|
Pharmacological inhibition or deletion of CD95 in peripheral myeloid cells (monocytes, macrophages, microglia, leucocytes) hampered infiltration in the brain of peripheral myeloid cells |
Total preservation of DA level in striatum Total protection of TH+ neurons in S. nigra (25% affected in wild type mice) |
Gao et al. (2015) |
In vivo MPTP 30 mg/kg i.p. |
|
|
Glucocorticoid receptor (GR) deletion in microglia increased their reactivity and induced a persistent activation |
2X aggravation of TH+ neuronal loss in S. nigra | Ros‐Bernal et al. (2011) |
In vivo MPTP 20 mg/kg i.p. |
|
| TNF−/− mice |
No protection in substantia nigra TH density in striatum: return to control level |
Ferger et al. (2004) |
In vivo MPTP 20 mg/kg i.p. |
|
|
Intra‐venous transplantation of mesenchymal stem cells Cell migration in substantia nigra and release of TGFbeta (anti‐inflammatory) Reparation of BBB, decreased infiltration and microglial activation |
About 15% protection of TH+ neurons in S. nigra | Chao et al. (2009) |
In vivo MPTP 20 mg/kg i.p. |
|
|
Nrf2−/− Increase in microgliosis and astrogliosis Microglial M1 phenotype Nrf2 involved in tuning microglial activation, switch M1/M2 phenotypes |
40% more DA neurons loss in substantia nigra (TM immunostaining) | Rojo et al. (2010) |
In vivo MPTP 20 mg/kg i.p. |
Indirect |
| Beta2 adrenergic receptor activation decreased microglial activation | 20% protection of TH+ neurons in Substantia nigra | Qian et al. (2011) |
In vivo MPTP 15 mg/kg sc |
|
|
Deficiency in i‐NOS blocks MPTP‐induced increase of i‐NOS, but not morphological microglial activation (IB4) |
Rescue of TH+ neurons in substantia nigra to control level, but no protection for striatal DA content | Dehmer et al. (2000) |
In vivo MPTP 30 mg/kg per day i.p., 5 days |
|
|
C3‐deficient mice Inhibition of complement‐phagossome pathway Induced a decrease in several markers of microglial activation |
Loss of DA neurons induced by repeated systemic LPS application is rescued to control level | Bodea et al. (2014) |
In vivo 4 days injection of LPS 1 μg/gbw LPS |
|
|
Minocyline or silencing of NADPH oxidase Microglial priming by a single injection of paraquat (PQ) (10 mg/kg) or by LPS (2–4 mg/kg) increased the vulnerability of DA neurons |
Blockade of priming by minocycline or by silencing NADPH oxidase prevent DA neurodegeneration by subsequent exposure to PQ | Purisai et al. (2007) |
In vivo Paraquat 10 mg/kg |
|
|
Interferon‐gamma knockout prevented PQ‐induced microglial activation as evidenced by morphological changes, i‐NOS, COX2, IL1beta, TNFalpha, overexpression |
In the knockout mice, DAergic neurons were protected from PQ‐induced neurodegeneration | Mangano et al. (2012) |
In vivo Paraquat 10 mg/kg |
|
|
Absence of microglia or NADPH silencing No effect of PQ on DA uptake and TH immunoreactivity in cultures depleted of microglia. No effect of PQ in neuron‐glia co‐cultures prepared from NADPH oxidase‐deficient mice |
Microglial NADPH oxidase as essential factor for mediating DA neurodegeneration | Wu et al. (2005) |
In vitro Paraquat 0.5–1 μM |
|
|
Blockade of i‐NOS, NF‐kB or p38 MAPK Cause a significant decrease of microglial reactivity, NO and IL‐1beta |
TH immunoreactivity, recovery of 20% | Yadav et al. (2012) |
In vivo Paraquat 10 mg/kg i.p. ± maneb 30 mg/kg |
Evidence Supporting Taxonomic Applicability
Rodent models have been mainly used to study the impact of neuroinflammation on DAergic nigrostriatal pathway degeneration, without any sex restriction. Neuroinflammation preceding neuronal death was detected in monkeys exposed to MPTP (Barcia et al., 2011); and in human, neuroinflammation is considered as an early event in the disease process (Innaccone et al., 2012).
References
Appel SH, Beers DR, Henkel JS, 2010. T cell‐microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends in Immunology, 31, 7–17.
Barcia C, Ros CM, Annese V, Gomez A, Ros‐Bernal F, Aguado‐Yera D, et al., 2011. IFN‐gamma signaling, with the synergistic contribution of TNF‐alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson's disease. Cell Death & Disease, 2, e142.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Bobyn J, Mangano EN, Gandhi A, Nelson E, Moloney K, Clarke M, et al., 2012. Viral‐toxin interactions and Parkinson's disease: poly I:C priming enhanced the neurodegenerative effects of paraquat. Journal of Neuroinflammation, 9, 86.
Bodea LG, Wang Y, Linnartz‐Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al., 2014. Neurodegeneration by activation of the microglial complement‐phagosome pathway. Journal of Journal of Neuroscience, 34, 8546–8556.
Bonneh‐Barkay D, Reaney SH, Langston WJ, Di Monte DA, 2005. Redox cycling of the herbicide paraquat in microglial cultures. Brain Research. Molecular Brain Research, 134, 52–56.
Borrajo A, Rodriguez‐Perez AI, Villar‐Cheda B, Guerra MJ, Labandeira‐Garcia JL, 2014. Inhibition of the microglial response is essential for the neuroprotective effects of Rho‐kinase inhibitors on MPTP‐induced dopaminergic cell death. Neuropharmacology, 85, 1–8.
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray‐Berthat V, et al., 2009. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. Journal of Clinical Investigation, 119, 182–192.
Brown GC, Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355.
Brzozowski MJ, Jenner P, Rose S, 2015. Inhibition of i‐NOS but not n‐NOS protects rat primary cell cultures against MPP(+)‐induced neuronal toxicity. Journal of Neural Transmission, 122, 779–788.
Cardenas H, Bolin LM, 2003. Compromised reactive microgliosis in MPTP‐lesioned IL‐6 KO mice. Brain Research, 985, 89–97.
Chang CY, Choi DK, Lee DK, Hong YJ, Park EJ, 2013. Resveratrol confers protection against rotenone‐induced neurotoxicity by modulating myeloperoxidase levels in glial cells. Public Library of Science (PLoS ONE), 8, e60654.
Chao CC, Hu S, Peterson PK, 1995. Glia, cytokines, and neurotoxicity. Critical Reviews TM in Neurobiology, 9, 189–205.
Chao YX, He BP, Tay SS, 2009. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson's disease. Journal of Neuroimmunology, 216, 39–50.
Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, et al., 2011. Fluoxetine prevents MPTP‐induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology, 60, 963–974.
Chung ES, Lee G, Lee C, Ye M, Chung HS, Kim H, et al., 2015. Bee venom phospholipase A2, a novel Foxp3 + regulatory T cell inducer, protects dopaminergic neurons by modulating neuroinflammatory responses in a mouse model of Parkinson's disease. Journal of Immunology.
Cicchetti F, Lapointe N, Roberge‐Tremblay A, Saint‐Pierre M, Jimenez L, Ficke BW, et al., 2005. Systemic exposure to paraquat and maneb models early Parkinson's disease in young adult rats. Neurobiology of Disease, 20, 360–371.
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB, 2000. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. Journal of Neurochemistry, 74, 2213–2216.
Depboylu C, Stricker S, Ghobril JP, Oertel WH, Priller J, Hoglinger GU, 2012. Brain‐resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T‐lymphocytes in the MPTP mouse model of Parkinson disease. Experimental Neurology, 238, 183–191.
Dexter DT, Jenner P, 2013. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biology and Medicine, 62, 132–144.
Dong Y, Benveniste EN, 2001. Immune Function of Astrocytes. Glia, 36, 180–190.
Emmrich JV, Hornik TC, Neher JJ, Brown GC, 2013. Rotenone induces neuronal death by microglial phagocytosis of neurons. The FEBS Journal, 280, 5030–5038.
Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B, et al., 2002. Cyclooxygenase‐2‐deficient mice are resistant to 1‐methyl‐4‐phenyl1, 2, 3, 6‐tetrahydropyridine‐induced damage of dopaminergic neurons in the substantia nigra. Journal of Neuroscience Letters, 329, 354–358.
Ferger B, Leng A, Mura A, Hengerer B, Feldon J, 2004. Genetic ablation of tumor necrosis factor‐alpha (TNF‐alpha) and pharmacological inhibition of TNF‐synthesis attenuates MPTP toxicity in mouse striatum. Journal of Neurochemistry, 89, 822–833.
Ferris CF, Marella M, Smerkers B, Barchet TM, Gershman B, Matsuno‐Yagi A, et al., 2013. A phenotypic model recapitulating the neuropathology of Parkinson's disease. Brain and behavior, 3, 351–366.
Gao HM, Hong JS, Zhang W, Liu B, 2002. Distinct role for microglia in rotenone‐induced degeneration of dopaminergic neurons. Journal of Journal of Neuroscience, 22, 782–790.
Gao HM, Liu B, Hong JS, 2003. Critical role for microglial NADPH oxidase in rotenone‐induced degeneration of dopaminergic neurons. Journal of Journal of Neuroscience, 23, 6181–6187.
Gao L, Brenner D, Llorens‐Bobadilla E, Saiz‐Castro G, Frank T, Wieghofer P, et al., 2015. Infiltration of circulating myeloid cells through CD95L contributes to neurodegeneration in mice. Journal Of Experimental Medicine, 212, 469–480.
Gehrmann J, Banati RB, Wiessnert C, Hossmann KA, Kreutzberg GW, 1995. Reactive microglia in cerebral ischaemia: An early mediator of tissue damage? Neuropathology and Applied Neurobiology, 21, 277–289.
He Q, Yu W, Wu J, Chen C, Lou Z, Zhang Q, et al., 2013. Intranasal LPS‐mediated Parkinson's model challenges the pathogenesis of nasal cavity and environmental toxins. Public Library of Science (PLoS ONE), 8, e78418.
Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, et al., 1999. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor‐alpha in glial cells. Journal of Neuroscience, 19, 3440–3447.
Iannaccone S, Cerami C, Alessio M, Garibotto V, Panzacchi A, Olivieri S, et al., 2013. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson's disease. Parkinsonism & Related Disorders, 19, 47–52.
Khan MM, Zaheer S, Nehman J, Zaheer A, 2014. Suppression of glia maturation factor expression prevents 1‐methyl‐4‐phenylpyridinium (MPP(+))‐induced loss of mesencephalic dopaminergic neurons. Journal of Neuroscience, 277, 196–205.
Kinugawa K, Monnet Y, Bechade C, Alvarez‐Fischer D, Hirsch EC, Bessis A, et al., 2013. DAP12 and CD11b contribute to the microglial‐induced death of dopaminergic neurons in vitro but not in vivo in the MPTP mouse model of Parkinson's disease. Journal of Neuroinflammation, 10, 82.
Klintworth H, Garden G, Xia Z, 2009. Rotenone and paraquat do not directly activate microglia or induce inflammatory cytokine release. Journal of Neuroscience Letters, 462, 1–5.
Kokovay E, Cunningham LA, 2005. Bone marrow‐derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson's disease. Neurobiology of Disease, 19, 471–478.
Kraft AD, Harry GJ, 2011. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International Journal of Environmental Research and Public Health, 8, 2980–3018.
L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Cossetti C, et al., 2011. Reactive astrocytes and Wnt/beta‐catenin signaling link nigrostriatal injury to repair in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine model of Parkinson's disease. Neurobiology of Disease, 41, 508–527.
Li XZ, Bai LM, Yang YP, Luo WF, Hu WD, Chen JP, et al., 2009. Effects of IL‐6 secreted from astrocytes on the survival of dopaminergic neurons in lipopolysaccharide‐induced inflammation. Journal of Neuroscience Research, 65, 252–258.
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, et al., 2012. alpha7 nicotinic acetylcholine receptor‐mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. Journal of Neuroinflammation, 9, 98.
Liu Z, Chen HQ, Huang Y, Qiu YH, Peng YP, 2015. Transforming growth factor‐beta1 acts via TbetaR‐I on microglia to protect against MPP‐induced dopaminergic neuronal loss. Brain, Behavior, and Immunity.
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, et al., 2015. Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress‐induced apoptosis: implications for Parkinson's disease. Neuropharmacology, 91, 87–96.
Machado A, Herrera AJ, Venero JL, Santiago M, De Pablos RM, Villaran RF, et al., 2011. Peripheral inflammation increases the damage in animal models of nigrostriatal dopaminergic neurodegeneration: possible implication in Parkinson's disease incidence. Parkinson's disease, 2011, 393769.
Mangano EN, Peters S, Litteljohn D, So R, Bethune C, Bobyn J, et al., 2011. Granulocyte macrophage‐colony stimulating factor protects against substantia nigra dopaminergic cell loss in an environmental toxin model of Parkinson's disease. Neurobiology of Disease, 43, 99–112.
Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, et al., 2012. Interferon‐gamma plays a role in paraquat‐induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiology of Aging, 33, 1411–1426.
Mena MA, Garcia de Yebenes J, 2008. Glial cells as players in parkinsonism: the “good,” the “bad,” and the “mysterious” glia. Neuroscientist, 14, 544–560.
Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL, 2006. Role of ICAM‐1 in persisting inflammation in Parkinson disease and MPTP monkeys. Experimental Neurology, 197, 275–283.
Miller RL, Sun GY, Sun AY, 2007. Cytotoxicity of paraquat in microglial cells: involvement of PKCdelta‐ and ERK1/2‐dependent NADPH oxidase. Brain Research, 1167, 129–139.
Mitra S, Chakrabarti N, Bhattacharyya A, 2011. Differential regional expression patterns of alpha‐synuclein, TNF‐alpha, and IL‐1beta; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Nakajima K, Tohyama Y, Kohsaka S, Kurihara T, 2002. Ceramide activates microglia to enhance the production/secretion of brain‐derived neurotrophic factor (BDNF) without induction of deleterious factors in vitro. Journal of Neurochemistry, 80, 697–705.
Neher JJ, Neniskyte U, Brown GC, 2012. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Frontiers in pharmacology, 3, 27.
Niranjan R. 2014. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's disease: focus on astrocytes. Molecular Neurobiology, 49, 28–38.
Pena‐Altamira E, Prati F, Massenzio F, Virgili M, Contestabile A, Bolognesi ML, et al., 2015. Changing paradigm to target microglia in neurodegenerative diseases: from anti‐inflammatory strategy to active immunomodulation. Expert Opinion on Therapeutic Targets: 1–14.
Pey P, Pearce RK, Kalaitzakis ME, Griffin WS, Gentleman SM, 2014. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer's and Parkinson's disease. Acta Neuropathologica communications, 2, 21.
Phinney AL, Andringa G, Bol JG, Wolters E, van Muiswinkel FL, van Dam AM, et al., 2006. Enhanced sensitivity of dopaminergic neurons to rotenone‐induced toxicity with aging. Parkinsonism & Related Disorders, 12, 228–238.
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat‐induced dopaminergic cell degeneration. Neurobiology of Disease 25, 392–400.
Qian L, Flood PM, Hong JS, 2010. Neuroinflammation is a key player in Parkinson's disease and a prime target for therapy. Journal of Neural Transmission, 117, 971–979.
Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, et al., 2011. beta2‐adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. Journal of Immunology, 186, 4443–4454.
Rappold PM, Cui M, Chesser AS, Tibbett J, Grima JC, Duan L, et al., 2011. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter‐3. Proceedings of the National Academy of Sciences USA, 108, 20766–20771.
Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE, 2010. Regulatory T cells attenuate Th17 cell‐mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. Journal of Immunology, 184, 2261–2271.
Rojo AI, Innamorato NG, Martin‐Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A, 2010. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia, 58, 588–598.
Ros‐Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, et al., 2011. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proceedings of the National Academy of Sciences of the United States of America, 108, 6632–6637.
Saint‐Pierre M, Tremblay ME, Sik A, Gross RE, Cicchetti F, 2006. Temporal effects of paraquat/maneb on microglial activation and dopamine neuronal loss in older rats. Journal of Neurochemistry, 98, 760–772.
Salama M, Helmy B, El‐Gamal M, Reda A, Ellaithy A, Tantawy D, et al., 2013. Role of L‐thyroxin in counteracting rotenone induced neurotoxicity in rats. Environmental Toxicology and Pharmacology, 35, 270–277.
Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al., 2012. S100B is increased in Parkinson's disease and ablation protects against MPTP‐induced toxicity through the RAGE and TNF‐alpha pathway. Brain, 135(Pt 11), 3336–3347.
Sharma N, Nehru B, 2015. Characterization of the lipopolysaccharide induced model of Parkinson's disease: Role of oxidative stress and neuroinflammation. Neurochemistry International, 87, 92–105.
Sherer TB, Betarbet R, Kim JH, Greenamyre JT, 2003. Selective microglial activation in the rat rotenone model of Parkinson's disease. Journal of Neuroscience Letters, 341, 87–90.
Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP, 2002. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. Federation of American Societies for Experimental Biology (FASEB), 16, 1474–1476.
Taetzsch T, Block ML, 2013. Pesticides, microglial NOX2, and Parkinson's disease. Journal of Biochemical and Molecular Toxicology, 27, 137–149.
Tanaka S, Ishii A, Ohtaki H, Shioda S, Yoshida T, Numazawa S, 2013. Activation of microglia induces symptoms of Parkinson's disease in wild‐type, but not in IL‐1 knockout mice. Journal of Neuroinflammation, 10, 143.
Tansey MG, Goldberg MS, 2009. Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease
Verkhratsky A, Parpura V, Pekna M, Pekny M, Sofroniew M, 2014. Glia in the pathogenesis of neurodegenerative diseases. Biochemical Society Transactions, 42, 1291–1301.
Wang T, Zhang W, Pei Z, Block M, Wilson B, Reece JM, et al., 2006. Reactive microgliosis participates in MPP+‐induced dopaminergic neurodegeneration: role of 67 kDa laminin receptor. Federation of American Societies for Experimental Biology (FASEB), 20, 906–915.
Wang Q, Chu CH, Qian L, Chen SH, Wilson B, Oyarzabal E, et al., 2014. Substance P exacerbates dopaminergic neurodegeneration through neurokinin‐1 receptor‐independent activation of microglial NADPH oxidase. Journal of Neuroscience, 34, 12490–12503.
Wang Q, Chu CH, Oyarzabal E, Jiang L, Chen SH, Wilson B, et al., 2014. Subpicomolar diphenyleneiodonium inhibits microglial NADPH oxidase with high specificity and shows great potential as a therapeutic agent for neurodegenerative diseases. Glia, 62, 2034–2043.
Whitton PS. 2007. Inflammation as a causative factor in the aetiology of Parkinson's disease. British Journal of Pharmacology, 150, 963–976.
Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, et al., 2005. The role of microglia in paraquat‐induced dopaminergic neurotoxicity. Antioxidants & Redox Signaling, 7, 654–661.
Xu L, He D, Bai Y. 2015. Microglia‐Mediated Inflammation and Neurodegenerative Disease. Molecular Neurobiology.
Yadav S, Gupta SP, Srivastava G, Srivastava PK, Singh MP, 2012. Role of secondary mediators in caffeine‐mediated neuroprotection in maneb‐ and paraquat‐induced Parkinson's disease phenotype in the mouse. Neurochemical Research, 37, 875–884.
Yuste JE, Tarragon E, Campuzano CM, Ros‐Bernal F, 2015. Implications of glial nitric oxide in neurodegenerative diseases. Frontiers in cellular Journal of Neuroscienceence, 9, 322.
Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM, 2007. TNF‐alpha knockout and minocycline treatment attenuates blood‐brain barrier leakage in MPTP‐treated mice. Neurobiology of Disease, 26, 36–46.
Zhang XY, Chen L, Yang Y, Xu DM, Zhang SR, Li CT, et al., 2014. Regulation of rotenone‐induced microglial activation by 5‐lipoxygenase and cysteinyl leukotriene receptor 1. Brain Research, 1572, 59–71.
Zhou Y, Wang Y, Kovacs M, Jin J, Zhang J, 2005. Microglial activation induced by neurodegeneration: a proteomic analysis. Molecular & Cellular Proteomics: MCP, 4, 1471–1479.
Zhou F, Wu JY, Sun XL, Yao HH, Ding JH, Hu G, 2007. Iptakalim alleviates rotenone‐induced degeneration of dopaminergic neurons through inhibiting microglia‐mediated neuroinflammation. Neuropsychopharmacology, 32, 2570–2580.
Zhou Y, Zhang Y, Li J, Lv F, Zhao Y, Duan D, et al., 2012. A comprehensive study on long‐term injury to nigral dopaminergic neurons following intracerebroventricular injection of lipopolysaccharide in rats. Journal of Neurochemistry, 123, 771–778.
6th KER: Degeneration of dopaminergic neurons of the nigrostriatal pathway directly leads to neuroinflammation
A.6.1. How does this KER work?
Several chemokines and chemokines receptors (fraktalkine, CD200) control the neuron‐microglia interactions and a loss of this control on the side of neurons can trigger microglial reactivity without any further positive signal required (Chapman et al., 2000; Streit et al., 2001; Blank and Prinz, 2013). Upon neuronal injury, signals termed ‘Damage‐Associated Molecular Patterns (DAMPs)’ are released by damaged neurons to promote microglial reactivity (Marin‐Teva et al., 2011; Katsumoto et al., 2014). These are for instance detected by Toll‐like receptors (TLRs) (for review, see Hayward and Lee, 2014). TLR‐2 functions as a master sensing receptor to detect neuronal death and tissue damage in many different neurological conditions including nerve transection injury, traumatic brain injury and hippocampal excitotoxicity (Hayward and Lee, 2014). Astrocytes, the other cellular actor of neuroinflammation besides microglia (Ranshoff and Brown, 2012) are also able to sense tissue injury via e.g. TLR‐3 (Farina et al., 2007; Rossi, 2015), and neuronal injury can result in astrocytic activation (Efremova et al., 2015).
A.6.2. Weight of evidence
A.6.2.1. Biological Plausability
Kreutzberg and co‐workers (1995, 1996) showed that neuronal injury generally leads to activation of microglia and astrocytes. This is a general phenomenon: for instance it is always observed in ischaemic damage (stroke; often in the form of glial activation following neuronal injury (Villa 2007)) as well as in stab or freeze injuries (Allahyari and Garcia, 2015). It is also observed regularly when neurons are killed by highly specific neurotoxicants that do not affect glia directly, such as injection of quinolinic acid or of 6‐hydroxydopamine into the striatum (Hernandez‐Baltazar et al., 2013; Arlicot et al., 2014). The vicious circle of neuronal injury triggering glial activation and glial activation triggering/enhancing neurodegeneration is often assumed to be a key element in the pathogenesis of neurodegenerative diseases, not just PD, but also (Alzheimer's disease, prion disease and many others) (Griffin et al., 1998; McGeer and McGeer, 1998; Blasko et al., 2004; Cacquevel et al., 2004; Hirsch and Hunot, 2009; Tansey and Goldberg, 2009; Barbeito et al., 2010; Rubio‐Perez and Morillas‐Ruiz, 2012; Thundyil and Lim, 2014).
A.6.2.2. Empirical support for linkage
MPP+
The chemokine fractalkine (regulating neuron‐glia interactions) was found to be released by neurons after unilateral injection of MPP+ in substantia nigra. It induced microglial activation by binding on the microglial receptor CXCR1 (Shan et al., 2011). Similarly, in chronically MPTP‐injected macaques, metalloproteinases‐9 (MMP‐9) released by injured neurons favour glial activation (Annese et al., 2015). Advanced glycation endproducts (AGEs), which are endproducts of reactions involving ROS, colocalised with DAergic neurons 2 days post last MPTP injection, suggesting neuronal injury (Teismann et al., 2012). In contrast, the receptors for AGEs (RAGEs) were found on microglial cells and astrocytes (Teismann et al., 2012). RAGE can activate NF‐kappaB, the transcription factor involved in the inflammatory response (Abdelsalam and Safar, 2015). Ablation of RAGE proved to be protective against MPTP‐induced decreases of TH+ neurons, by decreasing NF‐kappaB p65 nuclear translocation and by mitigating microglia and astrocyte reactivities (Teismann et al., 2012).
Rotenone
Rotenone‐induced neurotoxicity was less pronounced in neuron‐enriched cultures, than in neuron‐glia co‐cultures (Gao et al., 2002), suggesting that neuron‐glia interactions are critical for rotenone‐induced neurodegeneration. Indeed, CD200‐CD200R signalling regulates neuron‐glia interactions and holds microglia in a quiescent state (Biber et al., 2007). Therefore, inhibition of CD200R by blocking antibodies increased rotenone‐induced DA neurotoxicity in neuron‐glia mesencephalic cocultures (Wang et al., 2011). Ageing is associated with a decrease of CD200 expression (Wang et al., 2011) and deficits in neuronal CD200 production is also observed in several animal models of Parkinson's disease (Wang et al., 2011; Zhang et al., 2011; Sung et al., 2012). Inhibition of RAGE, which is upregulated in the striatum following rotenone exposure and in response to neuroinflammation, decreases rotenone‐induced apoptosis by decreasing mitochondrial cytochrome c release and caspase‐3 activation and suppresses NF‐kappaB activation, as well as the downstream inflammatory markers TNF‐alpha, i‐NOS and myeloperoxidase (Abdelsalam and Safar, 2015), showing again intermingled links between neuronal injury/death and neuroinflammation.
Paraquat
Non‐lethal neuronal damage is sufficient to trigger neuroinflammation: in 3D rat brain cell cultures, repeated treatment with concentrations of paraquat that did not kill the neurons, microglia and astrocytes were activated (Sandstrom von Tobel et al., 2014). Paraquat alone (10 mg/kg, 2x/week, for 4 weeks) or in combination with maneb (30 mg/kg) induces a loss of DAergic neurons in the substantia nigra paralleled by microglial activation (Cicchetti et al., 2005; Mitra et al., 2011). Neuronal injury is facilitated by uptake of paraquat via DA transporters (Rappold et al., 2011). In this model, paraquat‐induced neuronal perturbations are sufficient to induce neuroinflammation, but then neuroinflammation exacerbates the neurodegenerative process (Purisai et al., 2007).
A.6.3. Inconsistencies and uncertainties
Triggering of glia by injured neurons may not necessarily be due to the damage of neurons, but it may also be due to released synuclein (Sanchez‐Guajardo, 2010). In a AAV alpha‐synucleinopathy model, it was shown that cytoskeletal perturbation and accumulation of alpha‐synuclein were sufficient to induce microglial reactivity, suggesting that neuroinflammation appears early in the disease process and is not a result triggered by cell death (Chung et al., 2009). Direct effects of toxicants on glia cannot be completely excluded. They have been reported for most toxicants in one or the other publication (rotenone, paraquat, MPP+) (Brooks et al., 1989; Rappold et al., 2011; Zhang et al., 2014). The overwhelming evidence speaks against such effects for rotenone and MPP+ (Klintworth et al., 2009), but for paraquat there is evidence of direct interaction with microglial membrane NADPH oxidase (Rappold et al., 2011).As paraquat has several MIE (Rappold et al., 2011; Czerniczyniec et al., 2015), these may involve both neurons and microglia.
A.6.4. Quantitative relationship
Some examples of quantitative relationships between KEup and KEdown are given below. For KEdown Neuroinflammation, only the features measured are cited, as neuroinflammation is a complex KE involving several cell types and measured by changes in the expression /release of several markers
Table A.5.
Quantitative evaluation of the KER
| KE upstream Degeneration of DAergic nigrostriatal pathway | KE downstream Neuroinflammation | Compound | Reference | Comment |
|---|---|---|---|---|
| About 25% decrease of TH+ neurons 24–72 h post‐injection | Microglial and astroglial reactivities in substantia nigra and striatum |
MPTP 20 mg/kg i.p. 4 injections at 2 h intervals |
Annese et al. (2013) | MMP‐9 released by neurons as trigger of neuroinflammation |
| About 60% decrease of TH+ neurons in substantia nigra and of DA terminals in striatum 7 days post‐injection | Increase in ED1+ cells (macrophagic microglia or invading monocytes) |
MPTP 20 mg/kg i.p. 4 injections at 2 h intervals |
Chung et al. (2013) | MMP‐3‐induced disruption of BBB |
| About 50% decrease of TH+ neurons | Microglial and astroglial reactivity in substantia nigra and striatum |
MPTP 30 mg/kg i.p. each day during 5 days |
Teisman et al. (2012) | RAGE as trigger of neuroinfl. |
| About 50% decrease of DA content in striatum | Increase of TNF‐alpha (about 5X) and of i‐NOS (about 8X) in striatum |
Rotenone 1.5 mg/kg s.c. for 21 days |
Abdesalam and Safar (2015) | |
|
About 40% decrease of TH+ neurons about 50% decrease of TH+ neurons |
About 20% increase in microglial diameter as sign of activation microglial reactivity in substantia nigra |
Paraquat 0.5–2 μM (neuron‐glia cocultures) 10 mg/kg i.p. twice a week for 4 weeks |
Cicchetti et al. (2005) | |
|
Decrease of TH immuno‐reactivity of about 50% in subst. nigra 60% in frontal cortex 60% in hippo‐campus |
IL‐1beta immunoreactivity increased in frontal cortex and hippocampus TNFalpha immunoreactivity increased in all 3 regions Iba+ immunoreactivity increased in substantia nigra and decreased in frontal cortex and hippocampus |
Paraquat 10 mg/kg i.p., twice a week for 4 weeks |
Mitra et al. (2011) |
Evidence Supporting Taxonomic Applicability
Beside the rodent models, the concept of vicious circle with neuronal injury leading to neuroinflammation and neuroinflammation triggering or enhancing neurodegeneration is described in several neurodegenerative diseases in human, without any sex restriction (Griffin et al., 1998; McGeer and McGeer, 1998; Blasko et al., 2004; Cacquevel et al., 2004; Hirsch and Hunot, 2009; Tansey and Goldberg, 2009; Barbeito et al., 2010; Rubio‐Perez and Morillas‐Ruiz, 2012; Thundyil and Lim, 2014). Aging is an aggravating factor and increases the risk for developing a neurodegenerative disease (Kawas et al., 2000; Blasko et al., 2004).
References
Abdelsalam RM, Safar MM, 2015. Neuroprotective effects of vildagliptin in rat rotenone Parkinson's disease model: role of RAGE‐NFkappaB and Nrf2‐antioxidant signaling pathways. Journal of Neurochemistry, 133, 700–707.
Allahyari RV, Garcia AD, 2015. Triggering Reactive Gliosis In Vivo by a Forebrain Stab Injury. Journal of Visualized Experiments: JoVE, 100, e52825.
Annese V, Herrero MT, Di Pentima M, Gomez A, Lombardi L, Ros CM, et al., 2015. Metalloproteinase‐9 contributes to inflammatory glia activation and nigro‐striatal pathway degeneration in both mouse and monkey models of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced Parkinsonism. Brain Structure & Function, 220, 703–727.
Arlicot N, Tronel C, Bodard S, Garreau L, de la Crompe B, Vandevelde I, et al., 2014. Translocator protein (18 kDa) mapping with [125I]‐CLINDE in the quinolinic acid rat model of excitotoxicity: a longitudinal comparison with microglial activation, astrogliosis, and neuronal death. Molecular Imaging, 13, 4–11.
Barbeito AG, Mesci P, Boillee S, 2010. Motor Neuron‐immune interactions: the vicious circle of ALS. Journal of Neural Transmission, 117, 981–1000.
Biber K, Neumann H, Inoue K, Boddeke HW, 2007. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in Journal of Neurosciences, 30, 596–602.
Blank T, Prinz M. 2013. Microglia as modulators of cognition and neuropsychiatric disorders. Glia, 61, 62–70.
Blasko I, Stampfer‐Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck‐Loebenstein B, 2004. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell, 3, 169–176.
Brooks WJ, Jarvis MF, Wagner GC, 1989. Astrocytes as a primary locus for the conversion MPTP into MPP+. Journal of Neural Transmission, 76, 1–12.
Cacquevel M, Lebeurrier N, Cheenne S, Vivien D, 2004. Cytokines in neuroinflammation and Alzheimer's disease. Current Drug Targets, 5, 529–534.
Chung CY, Koprich JB, Siddiqi H, Isacson O, 2009. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha‐synucleinopathy. Journal of Neuroscience, 29, 3365–3373.
Cicchetti F, Lapointe N, Roberge‐Tremblay A, Saint‐Pierre M, Jimenez L, Ficke BW, et al., 2005. Systemic exposure to paraquat and maneb models early Parkinson's disease in young adult rats. Neurobiology of Disease, 20, 360–371.
Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJLM, 2000. Fractalkine Cleavage from Neuronal Membrans Represents an Acute Event in Inflammatory Response to Excitotoxic Brain Damage. Journal of Neuroscience, 20 RC87, 1–5.
Czerniczyniec A, Lanza EM, Karadayian AG, Bustamante J, Lores‐Arnaiz S, 2015. Impairment of striatal mitochondrial function by acute paraquat poisoning. Journal of Bioenergetics and Biomembranes, 47, 395–408.
Efremova L, Schildknecht S, Adam M, Pape R, Gutbier S, Hanf B, et al., 2015. Prevention of the degeneration of human dopaminergic neurons in an astrocyte co‐culture system allowing endogenous drug metabolism. British Journal of Pharmacology, 172, 4119–4132.
Farina C, Aloisi F, Meinl E, 2007. Astrocytes are active players in cerebral innate immunity. Trends in Immunology, 28, 138–145.
Gao HM, Hong JS, Zhang W, Liu B, 2002. Distinct role for microglia in rotenone‐induced degeneration of dopaminergic neurons. Journal of Neuroscience, 22, 782–790.
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al., 1998. Glial‐neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathology, 8, 65–72.
Hayward JH, Lee SJ, 2014. A decade of research on TLR2 discovering its pivotal role in glial activation and neuroinflammation in neurodegenerative diseases. Experimental Neurobiology, 23, 138–147.
Hernandez‐Baltazar D, Mendoza‐Garrido ME, Martinez‐Fong D, 2013. Activation of GSK‐3beta and caspase‐3 occurs in Nigral dopamine neurons during the development of apoptosis activated by a striatal injection of 6‐hydroxydopamine. Public Library of Science (PLoS ONE), 8, e70951.
Hirsch EC, Hunot S, 2009. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurology, 8, 382–397.
Katsumoto A, Lu H, Miranda AS, Ransohoff RM, 2014. Ontogeny and functions of central nervous system macrophages. Journal of Immunology, 193, 2615–2621.
Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A, 2000. Age‐specific incidence rates of Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology, 54, 2072–2077.
Klintworth H, Garden G, Xia Z, 2009. Rotenone and paraquat do not directly activate microglia or induce inflammatory cytokine release. Journal of Neuroscience Letters, 462, 1–5.
Kreutzberg GW, 1995. Microglia, the first line of defence in brain pathologies. Arzneimttelforsch, 45, 357–360.
Kreutzberg GW, 1996. Microglia: a sensor for pathological events in the CNS. Trends in Journal of Neurosciences, 19, 312–318.
Marin‐Teva JL, Cuadros MA, Martin‐Oliva D, Navascues J, 2011. Microglia and neuronal cell death. Neuron Glia Biology, 7, 25–40.
McGeer PL, McGeer EG, 1998. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Disease and Associated Disorders, 12(Suppl. 2), S1–S6.
Mitra S, Chakrabarti N, Bhattacharyya A, 2011. Differential regional expression patterns of alpha‐synuclein, TNF‐alpha, and IL‐1beta; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat‐induced dopaminergic cell degeneration. Neurobiology of Disease, 25, 392–400.
Ransohoff RM, Brown MA, 2012. Innate immunity in the central nervous system. Journal of Clinical Investigation, 122, 1164–1171.
Rappold PM, Cui M, Chesser AS, Tibbett J, Grima JC, Duan L, et al., 2011. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter‐3. Proceedings of the National Academy of Sciences U S A, 108, 20766–20771.
Rossi D, 2015. Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Progress in Neurobiology, 130, 86–120.
Rubio‐Perez JM, Morillas‐Ruiz JM, 2012. A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal, 2012, 756357.
Sanchez‐Guajardo V, Febbraro F, Kirik D, Romero‐Ramos M, 2010. Microglia acquire distinct activation profiles depending on the degree of alpha‐synuclein neuropathology in a rAAV based model of Parkinson's disease. Public Library of Science (PLoS ONE), 5, e8784.
Sandstrom von Tobel J, Zoia D, Althaus J, Antinori P, Mermoud J, Pak HS, et al., 2014. Immediate and delayed effects of subchronic Paraquat exposure during an early differentiation stage in 3D‐rat brain cell cultures. Toxicology Letters 10.1016/j.toxlet.2014.02.001
Shan S, Hong‐Min T, Yi F, Jun‐Peng G, Yue F, Yan‐Hong T, et al., 2011. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson's disease. Neurobiology of Aging, 32, 443–458.
Streit WJ, Conde J, Harrison JK, 2001. Chemokines and Alzheimer's disease. Neurobiology of Aging, 22, 909–913.
Sung YH, Kim SC, Hong HP, Park CY, Shin MS, Kim CJ, et al., 2012. Treadmill exercise ameliorates dopaminergic neuronal loss through suppressing microglial activation in Parkinson's disease mice. Life Science Journal, 91, 1309–1316.
Tansey MG, Goldberg MS, 2009. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease
Teismann P, Sathe K, Bierhaus A, Leng L, Martin HL, Bucala R, et al., 2012. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiology of Aging, 33, 2478–2490.
Thundyil J, Lim KL, 2014. DAMPs and Neurodegeneration. Ageing Research Reviews.
Villa P, van Beek J, Larsen AK, Gerwien J, Christensen S, Cerami A, Brines M, Leist M, Ghezzi P, Torup L, 2007. Reduced functional deficits, neuroinflammation, and secondary tissue damage after treatment of stroke by nonerythropoietic erythropoietin derivatives. Journal of Cerebral Blood Flow & Metabolism, 27, 552–563.
Wang XJ, Zhang S, Yan ZQ, Zhao YX, Zhou HY, Wang Y, et al., 2011. Impaired CD200‐CD200R‐mediated microglia silencing enhances midbrain dopaminergic neurodegeneration: roles of aging, superoxide, NADPH oxidase, and p38 MAPK. Free Radical Biology and Medicine, 50, 1094–1106.
Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, et al., 2011. CD200‐CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson's disease. Journal of Neuroinflammation, 8, 154.
Zhang XY, Chen L, Yang Y, Xu DM, Zhang SR, Li CT, et al., 2014. Regulation of rotenone‐induced microglial activation by 5‐lipoxygenase and cysteinyl leukotriene receptor 1. Brain Research, 1572, 59–71.
7th KER: Mitochondrial dysfunction leads to the degeneration of dopaminergic neurons of the nigrostriatal pathway
A.7.1. How this key event relationship works
Neurons are characterised by the presence of neurites, the formation of action potentials, and the release and re‐uptake of neurotransmitters into the synaptic cleft. The presence of long extensions implies a significant enlargement of total cell surface. In combination with the transmission of action potentials that require a continuous maintenance of active transport processes across the membrane, the steady state energy demand of these neurons is significantly higher compared with non‐neuronal cells. Dopaminergic (DA) neurons located in the substantia nigra pars compacta (SNpc) that project into the striatum are unique with respect of the total length of their neurites and the number of synapses that are significantly higher compared with other neuronal cell types (Bolam et al., 2012). Besides this complex morphology DA neurons have a distinctive physiological phenotype that could contribute to their vulnerability (Surmeier et al., 2010). Other features such as high energy demand, high calcium flux, dopamine autoxidation process as well as high content of iron and high content of microglia makes these DA neurons at vulnerable population of cells to oxidative stress produced by mitochondrial dysfunction. These architectural features of SNpc DA neurons render this cell type as particularly vulnerable to impairments in energy supply. Mitochondrial dysfunction, either evoked by environmental toxins such as the complex I inhibitor rotenone or MPTP, by oxidative modifications of components of the mitochondrial respiratory chain, or by genetic impairments of mitochondrial ATP generation hence have direct influence on the function and integrity of SNpc DA neurons.
A.7.2. Weight of evidence for the KER
A.7.2.1. Biological plausibility
Mitochondria are organelles essentials for multiple cellular processes, including production of ATP, maintenance of calcium homeostasis, management of ROS production and apoptosis. Mitochondrial dynamics are also critical for the maintenance of cellular homeostasis, which involve multiple factors controlling mitophagy (Youle et al., 2012). Deregulation of mitochondrial functions may impact any neuronal population; however, SNpc DA neurons are indeed the most vulnerable population in PD. Multiple factors are related to their vulnerability: These include autonomous activity, broad action potentials, low intrinsic calcium buffering capacity, poorly myelinated long highly branched axons and terminal fields, and use of a catecholamine neurotransmitter, often with the catecholamine‐derived neuromelanin pigment (Surmeier et al., 2010; Sulzer et al., 2013).
The above mentioned factors imply a significantly higher total cell surface and a high energy requirement in order to maintain the re‐distribution of ions across the membrane following an action potential. In addition, SNpc DA neurons are characterised by significantly higher numbers of synapses compared with other neuronal types or with DA neurons of different anatomical localisations (Anden et al., 1966; Kawaguchi et al., 1990; Kita et al., 1994; Bevan et al., 1998; Wu et al., 2000; Tepper et al., 2004). In humans, ca. 10 times higher numbers of synapses compared with rats are expected, making human DA neurons particularly vulnerable (Matsuda et al., 2009; Bolam et al., 2012). These extreme bioenergetics demands pose SNpc DA neurons energetically ‘on the edge’. Any stressor that might perturb energy production would hence lead to conditions under which the energy demand would exceed energy supply, resulting in cell damage and ultimately to cell death
The mechanistic link between mitochondrial dysfunction and loss of SNpc DA neurons also comes from evidence of mutated proteins related to mitochondrial function in familial PD, resulting in reduced calcium capacity, increased ROS production, increase in mitochondrial membrane permeabilisation and increase in cell vulnerability (Gandhi et al., 2009; Koopman et al., 2012). In addition, excessive ROS production can damage mitochondrial DNA and activate the intrinsic pathway of apoptosis (Tait et al., 2010). Additional sources of oxidative stress come from the autoxidation of dopamine and the active generation of ROS by activated glia cells; however, the mitochondrial respiratory chain itself represents a source of constant superoxide formation, even under normal conditions (Moosmann et al., 2002).
Furthermore, imbalance of mitochondrial dynamics have been reported in a wide range of experimental models of PD and inhibition of the mitochondrial fission proteins (i.e. Drp1) promote mitochondrial fusion and fission and enhanced the release of dopamine from the nigrostriatal terminals (Tieu et al., 2014).
Additional link between mitochondrial dysfunction and the degeneration of DA neurons of the nigrostriatal pathway comes from studies indicating a reduced activity of mitochondrial complex I in human idiopathic PD cases in the substantia nigra (Parker et al., 1989, 2008; Swerdlow et al., 1996; Keeney et al., 2006). The impairment in complex I activity was directly correlated with an elevated sensitivity of SNpc DA neurons and their demise. Transfer of mitochondria from human platelets collected from idiopathic PD subjects into fibroblasts or neuronal cells resulted in elevated levels of basal oxidative stress, a declined supply with ATP, and an elevated vulnerability towards exogenous stressors such as the complex I inhibitors rotenone or the redox cycler paraquat (Swerdlow et al., 1996; Gu et al., 1998). Systemic application of complex I inhibitors such as rotenone or MPTP lead to a preferential loss of nigrostriatal DA neurons, while other brain areas or peripheral cells are not affected to the same degree (Langston et al., 1983).
A.7.2.2. Empirical support for linkage
The experimental support linking mitochondrial dysfunction with the degeneration of DA neurons of the nigrostriatal system is based on the analysis of mitochondria from PD patients, from genetic mouse models, from in vitro knockdown and overexpression systems, and from in vitro and in vivo toxin models.
In vitro/rotenone: Prevention of ROS formation protects from cell death. The concept of mitochondrial dysfunction as a consequence of defects in complex I has been fuelled by observations of impaired complex I activity in the SNpc, muscle, and in platelets of PD patients. Human neuroblastoma SK‐N‐MC cells, exposed to rotenone, displayed a time‐ and concentration‐dependent decline in viability. Transfection of rotenone‐insensitive single subunit NADH dehydrogenase (NDI 1) allowed a replacement of endogenous complex I activity. NDI 1 transfected cells showed no oxidative damage, no declined mitochondrial activity, or cell death. A significant amount of endogenously formed ROS at complex I was identified in SK‐N‐MC cells and in a chronic midbrain slice culture exposed to rotenone. Antioxidants such as α‐tocopherol prevented cell death evoked by rotenone, but not the rotenone‐induced drop in ATP (Sherer et al., 2003).
In vitro/rotenone/MPP+: Antioxidants protect from rotenone/MPP+ cell death. Analysis of post mortem nigrostriatal material from PD patients regularly revealed the presence of elevated levels of oxidative modified proteins, lipids, and DNA. These observations indicate an elevated formation of ROS in the cells affected by the disease and triggered the concept of antioxidants as a potential intervention strategy to slow down the progression of PD. In MES23.5 cells, a reduction in viability, DA content, NADH levels, as well as an increase in ROS formation and elevated nuclear condensation was observed upon treatment with MPP+. Rosmarinic acid is well known for its radical scavenging activities and displayed a complete protection from MPP+‐mediated cell death and rescued NADH levels. In addition, it lead to a partial protection from the loss of DA and resulted in a rate of nuclear condensation that was about half of that observed with MPP+ alone (Du et al., 2010).
The flavonoid rutin has been demonstrated to protect from oxidative stress in 6‐OHDA induced motor deficits in rats as well as to inhibit the formation of nitric oxide and proinflammatory cytokines (Khan et al., 2012). In a model of SH‐SY5Y cells, exposure to rotenone lead to a reduction in viability by ca. 50% that was almost completely protected in the presence of rutin. Rotenone‐dependent increase of ROS formation and an elevation of intracellular Ca2+ was significantly dampened by the presence of rutin, similar to its rescue from rotenone‐dependent decrease in mitochondrial membrane potential (Park et al., 2014).
Comparable observations were made with the quinone triterpene celastrol that protected SH‐SY5Y cells exposed to rotenone almost completely from cell death, from a rotenone‐dependent elevation in ROS levels, and from a rotenone‐dependent loss of the mitochondrial membrane potential (Choi et al., 2014).
In vitro/different complex I inhibitors: Inhibition of complex I triggers oxidant formation and cell death. The majority of experimental PD studies were either conducted using rotenone or MPP+. In order to demonstrate that the concept of complex I inhibition and its ROS‐mediated triggering of mitochondrial dysfunction and cell demise can be regarded as a general principle, alternative complex I inhibitors were applied to substantiate previous observations made with rotenone. In human SK‐N‐MC neuroblastoma cells, rotenone as well as the pesticides fenazaquin, fenpyroximate, pyridaben, tebufenpyrad, pyridaben were tested. In all cases, a time‐ and concentration‐dependent decline in intracellular ATP and cell viability was observed. Expression of the rotenone‐insensitive NADH dehydrogenase from Saccharomyces cerevisiae (NDI 1) prevented from the toxicity of the different complex I inhibitors completely. Rotenone‐ and pyridaben‐dependent cell death was prevented by ca. 75% by the presence of the antioxidant α‐tocopherol (Sherer et al., 2007).
In vitro/rotenone: Mitochondrial dysfunction‐dependent cell death is prevented by antioxidants. In a human neuroblastoma SH‐SY5Y model, exposed either to the complex I inhibitors MPP+ or rotenone, the imine antioxidants iminostilbene, phenothiazine, phenoxazine in the low nanomolar concentration range partially protected from MPP+ or rotenone toxicity. A reduction in the membrane potential evoked by MPP+ and rotenone was completely prevented by these antioxidants (Hajteva et al., 2009)
In vitro/rotenone: Circumvention of dysfunctional mitochondria protects from cell death. Assuming a direct causal relationship between complex I inhibition, mitochondrial dysfunction, and the demise of DA neurons, the circumvention of endogenous complex I by expression of the NADH dehydrogenase of Saccharomyces cerevisiae (NDI 1) provided initial evidence for the essential role of complex I inhibition in this sequence of events. As an alternative electron carrier, capable of transferring electrons from NADH to cytochrome c, methylene blue was identified. In hippocampal HT‐22 cells, a rotenone‐mediated reduction in the oxygen consumption rate was completely reversed by the addition of methylene blue. A rotenone‐mediated decline in cell viability by 70% was almost completely prevented by 0.1 μg/ml methylene blue. In rats, rotenone‐mediated decline in striatal DA was entirely prevented by methylene blue, the observed elevation of ROS formation evoked by rotenone was reduced to control levels, and rotarod performance impairments evoked by rotenone were completely avoided by administration of methylene blue. These observations illustrate a causal relationship between dysfunctional mitochondria, the degeneration of nigrostriatal DA neurons, and impaired motor performance (Wen et al., 2011).
In vivo/rotenone: Circumvention of dysfunctional mitochondria prevents from nigrostriatal cell degeneration. Circumvention of a dysfunctional complex I by the rotenone‐insensitive NADH dehydrogenase NDI 1 in vivo and its influence on nigrostriatal DA neuron integrity was demonstrated in a rat model with an unilateral injection of a recombinant adeno‐associated virus, carrying the NDI 1 gene into close special vicinity to the SNpc. The animals were treated with rotenone after the unilateral expression of NDI 1. NDI 1 almost completely prevented from the rotenone‐mediated loss of TH staining in the SNpc and the striatum. Striatal DA levels that were reduced by ca. 50% by rotenone, in the presence of NDI 1, DA levels were also almost identical to the values of untreated controls. These observations highlight a causal relationship between the inhibition of complex I and the degeneration of nigrostriatal DA neurons (Marella et al., 2008).
In vitro/DA: Exogenously added oxidants lead to mitochondrial dysfunction and cell death. Next to an elevated formation of reactive oxygen species evoked by endogenous defects in complex I or in response to pharmacological inhibitors of complex I, nigrostriatal DA neurons are characterised by the neurotransmitter dopamine and its tendency to undergo autooxidation when exposed to physiological pH and oxygen tension conditions. To assess the role of DA‐mediated oxidative stress as a cause of mitochondrial dysfunction and its influence on cell viability, PC12 cells were exposed to DA. The observed increase in intracellular ROS was completely reversed by the presence of the antioxidant N‐acetyl‐cysteine (NAC). The amount of oxidative modified protein increased by DA treatment, its rise was completely prevented by the presence of NAC, and partially prevented by the presence of exogenously added GSH. DA‐dependent PC12 cell death, decline in the transmembrane potential and in intracellular ATP, and decline in complex II/III activities were observed and were all completely prevented by the presence of NAC. (Jana et al., 2011).
In vitro/ GSH depletion: Oxidative stress causes mitochondrial dysfunction and neurodegeneration. Several reports indicated a declined activity of complex I in the brain, but also in muscle and platelets of PD patients. In order to investigate the mutual interaction between pro‐oxidative conditions and complex I activity, a PC12 subclone was generated, allowing the inducible downregulation of γ‐glutamyl‐cysteine synthase involved in the synthesis of glutathione (GSH). This system allows a controlled decrease of intracellular GSH by ca. 50% and a decrease in mitochondrial GSH by ca. 40%. Under these conditions, intracellular and intramitochondrial ROS increased by ca. one third, mitochondrial complex I activity and ATP levels were reduced by ca. two thirds. The observed inhibition of complex I was completely reversed by DTT. These observations indicate that an impairment of complex I activity as a key event in the initiation of mitochondrial dysfunction and ultimately cell death, can be evoked by elevated levels of oxidants, respectively by a declined cellular antioxidant capacity (Jha et al., 2000).
In vitro/ GSH depletion: Oxidative stress causes mitochondrial dysfunction and neurodegeneration. PD is characterised by the depletion of glutathione (GSH) in the SNpc. Declined cellular levels of GSH were reported to be associated with morphological changes of mitochondria (Perry et al., 1982; Jain et al., 1991). To investigate the influence of declined GSH levels, N27 cells were exposed to buthionine‐S‐sulfoximine (BSO), an inhibitor of glutamate cysteine ligase and hence of de novo GSH synthesis. The BSO concentration chosen allowed a reduction in intracellular GSH levels by 50% in the absence of cell death. Chronic GSH depletion resulted in the S‐nitrosation of complex I and its inhibition. Both effects were completely reversed by the addition of DTT (Chinta et al., 2006).
Isolated mitochondria: Exogenous oxidants cause mitochondrial dysfunction. In order to further address the aspect on how DA autoxidation contributes to mitochondrial dysfunction and DA neurodegeneration, isolated rat brain mitochondria were exposed to DA, resulting in an inhibition of complex I by ca. 30% and in an inhibition of complex IV by ca. 50%. Both activities of complex I and complex IV were completely protected from DA‐dependent inactivation by the presence of GSH. These observations point to a direct inhibitory action of endogenous DA and its autoxidation derivatives on the activity of the mitochondrial respiratory chain. (Khan et al., 2005)
In vitro/cybrid cells: Sensitisation of neuronal cells for degeneration by transfer of dysfunctional mitochondria. In a subclone of human neuroblastoma cells (SH‐SY5Y), devoid of mitochondrial DNA, mitochondria from platelets of PD patients were transplanted. Analysis after 5–6 weeks in culture after transplantation of mitochondria indicated a 20% reduction in complex I activity, a twofold increase in the basal formation of reactive oxygen species, and a ca. twofold higher sensitivity towards the mitochondrial PD toxin MPP+ (Swerdlow et al., 1996)
In vitro/cybrid cells: Sensitisation of neuronal cells for degeneration by transfer of dysfunctional mitochondria. In a subclone of the human A549 cell line, devoid of mitochondrial DNA, mitochondria of platelets from PD patients were transplanted. Complex I activity in platelets of PD patients displayed a reduction of 25% compared with age‐matched controls. After transplantation into the A549 cells, complex I activity was reduced by 25% in its activity (Gu et al., 1998)
In vivo: Induction of mitochondrial dysfunction by Drp1 deletion leads to neuronal cell loss. Maintenance of functional mitochondria in a cell is regulated by fission/fusion processes that allow the elimination of damaged mitochondria and the spreading of intact mitochondria. Deletion of the central fission protein dynamin related protein 1 (Drp1) leads to an elimination in DA neuron terminals in the caudate putamen and to a loss of DA neuron cell bodies in the midbrain. In Drp1 deficient mice, mitochondrial mass decreases, particularly in axons (Berthet et al., 2014)
In vivo: Induction of mitochondrial dysfunction by Tfam knockdown leads to neuronal cell loss. Mitochondrial transcription factor A (Tfam) is a key regulator of mitochondrial biogenesis. Conditional knockout mice with a selective disruption of the gene for mitochondrial Tfam in DA neurons indicated a reduction in mtDNA levels and deficiencies in the respiratory chain in midbrain DA neurons that progressed to DA cell death. The demise of DA neurons in the SNpc was associated with the onset of PD symptoms such as a reduction in locomotor activity of these mice by ca. 30%. The decrease in locomotor activity was reversed by L‐DOPA treatment (Ekstrand et al., 2007)
In vivo: MPTP dependent mitochondrial dysfunction and cell death is protected by PGC‐1α overexpression. Peroxisome proliferator‐activated receptor gamma coactivator 1α (PGC‐1α) is a key regulator of mitochondrial biogenesis and metabolism. Transgenic mice overexpressing PGC‐1α show protection against MPTP intoxication (50%). The SNpc in these mice is characterised by elevated levels of SOD2, Trx2. Resveratrol is a known activator of SIRT1, leading to enhanced PGC‐1α gene transcription. In MPTP mice, resveratrol protected TH‐positve neurons by 80% from cell loss (Mudo et al., 2012)
In vivo: Prevention of mitochondrial dysfunction protects from nigrostriatal cell loss. In order to demonstrate the causative connection between complex I‐dependent mitochondrial dysfunction and the degeneration of DA neurons, a series of in vivo experiments were conducted that indicated partial restoration by antioxidants or by compounds supporting a dysfunctional mitochondrial ATP generation. In MPTP challenged mice that additionally received Q10 treatment, a 37% higher striatal DA level compared with the MPTP group was detected. TH positive staining in the striatum dropped by ca. 65% after MPTP. In the MPTP + Q10 group, the loss in striatal TH staining was reduced to ca. 40% compared with the untreated controls. (Beal et al., 1998).
In MPTP challenged marmosets, TH positive cell body numbers were reduced by ca. 60%, co‐administration with ebselen resulted in a reduction of TH staining of only ca. 25% (Moussaoui et al., 2000).
In MPTP challenged mice, a reduction of striatal DA by ca. 70% was detected. Cotreatment with creatine resulted in a reduction of DA levels of only 42%. In the same setup, TH positive neuron number in the SNpc was reduced by 70% in response to MPTP, in the presence of creatine, a drop of only 4% was observed (Matthews et al., 1999).
In vivo/rotenone: Antioxidants prevent from rotenone‐dependent nigrostriatal cell death. Rotenone administered subcutaneously for 5 weeks (2.5 mg/kg per day) caused a selective increase in oxidative damage in the striatum as compared to the hippocampus and cortex, accompanied by massive degeneration of dopaminergic neurons in the substantia nigra. Antioxidant polydatin (Piceid) treatment significantly prevented the rotenone‐induced changes in the levels of glutathione, thioredoxin, ATP, malondialdehyde and the manganese superoxide dismutase (SOD) in the striatum, confirming that rotenone‐induced mitochondrial dysfunction resulted in oxidative stress (Chen et al., 2015).
In vivo/rotenone: Degeneration of DA neurons depends on oxidative stress evoked by mitochondrial dysfunction. Many studies have shown that mitochondrial aldehyde dehydrogenase 2 (ALDH2) functions as a cellular protector against oxidative stress by detoxification of cytotoxic aldehydes. Dopamine is metabolised by monoamine oxidase to yield 3,4‐dihydroxyphenylacetaldehyde (DOPAL) then converts to a less toxic acid product by ALDH. The highly toxic and reactive DOPAL has been hypothesised to contribute to the selective neurodegeneration of dopamine (DA) neurons. In this study, the neuroprotective mechanism of ALDH2 was observed as overexpression of wild‐type ALDH2 gene, but not the enzymatically deficient mutant ALDH2*2 (E504K), reduced rotenone‐induced DA neuronal cell death. Application of a potent activator of ALDH2, Alda‐1, was effective in protecting against rotenone‐induced (100 nM, 24 h exposure) apoptotic cell death in both SH‐SY5Y cells and primary cultured substantia nigra (SN) DA neurons. These results were confirmed by in vivo studies. Intraperitoneal administration of Alda‐1 to C57BL/6 mice treated with rotenone (50 mg/kg per day, oral administration for 14 days) or MPTP (40 mg/kg per day, i.p. for 14 days) significantly reduced death of SN tyrosine hydroxylase‐positive dopaminergic neurons. The attenuation of rotenone‐induced apoptosis by Alda‐1 resulted from decreasing ROS accumulation, reversal of mitochondrial membrane potential depolarisation, and inhibition of activation of proteins related to mitochondrial apoptotic pathway. The present study demonstrates that rotenone or MPP+ induces DA neurotoxicity through oxidative stress. Moreover, Alda‐1 is effective in ameliorating mitochondrial dysfunction by inhibiting rotenone‐ or MPP+‐induced mitochondria‐mediated oxidative stress that leads t0 apoptosis (Chiu et al., 2015).
A.7.3. Uncertainties or inconsistencies
Several in vitro studies applying rotenone to evoke mitochondrial dysfunction came to the conclusion that rotenone‐dependent ROS formation, and not the rotenone‐evoked drop in ATP is the primary cause for cell degeneration. These observations are largely based on experimental systems employing the rotenone insensitive NADH dehydrogenase NDI 1. Expression of NDI 1 protected rotenone exposed cells from degeneration. The presence of NDI 1 however results in a substitution of ATP. Endogenously expressed complex I is still present in these models and it can be assumed that rotenone exposure would still lead to a complex I‐dependent formation of ROS that precludes the modelling of a precise cause‐consequence relationship between either ATP depletion or elevated ROS levels with the demise of DA neurons.
Several studies indicate a dominant role of ROS in the degeneration of DA neurons, based on models in which rotenone/MPP+ mediated mitochondrial dysfunction and cell degeneration was protected by the presence of exogenously added antioxidants. Maintenance of the endogenous redox potential however is a highly ATP‐dependent process. Clear‐cut separations between the respective contribution of ROS or the role of an inhibited mitochondrial ATP synthesis on the degeneration of DA neurons is hence difficult to postulate.
Studies with chronic partial GSH depletions indicated that an experimental reduction of GSH/GSSG by ca. 50% has no influence on cell viability. Reports involving rotenone and MPP+ however regularly observe degeneration of DA neurons under conditions of GSH depletion around 50%. These observations indicate a more prominent role of the intracellular drop of ATP evoked by the complex I inhibitors in the process of cell degeneration.
Studies in which oxidative stress is generated e.g. by the application of DA or 6‐OHDA not only observed a challenge of the cellular redox potential, but also reversible and irreversible inhibitory mechanisms of mitochondrial respiratory chain complexes (nitration, S‐nitrosation) that are accompanied by an inhibition of the respiratory chain in the absence of pharmacological complex I inhibitors. These observations illustrate the close mutual interaction between oxidative stress and the inhibition of mitochondrial respiration and point to a profound role of direct mitochondrial inhibition also under oxidative stress conditions.
Mitochondrial dysfunction is generally associated with conditions of oxidative stress. Dysfunctional mitochondria can act as potent source of superoxide. Oxidative stress associated with PD however not only originates from mitochondrial ROS, but also from DA autoxidation and the Fenton reaction, as well as from inflammatory activated adjacent glia. Interpretations on the role of oxidative stress in DA neurons and its role in DA neurodegeneration is hence hampered by the fact that the respective origin of the reactive oxygen species formed (mitochondria, DA autoxidation, inflammation of glia cells) is rather difficult to identify and often shows overlappings (Starkov et al., 2008; Murphy et al., 2009; Cebrian et al., 2015).
In PD patients, a reduction in complex I activity in the SNpc, but also in peripheral tissue and cells such as platelets, was reported. Studies with isolated mitochondria indicated that for efficient inhibition of mitochondrial ATP formation, an inhibition of complex I by ca. 70% is necessary (Davey et al., 1996). Reports on the reduction of complex I activity in PD patients however repeatedly indicated an inhibition of only 25–30% (Schapira et al., 1989; Schapira et al., 1990; Janetzky et al., 1994).
Data available on the respective inhibition of the components of the respiratory chain are highly dependent on the experimental setup used. Analysis of mitochondrial respiratory chain complex activities in mitochondrial homogenates provide results different from data obtained with intact, isolated mitochondria. These aspects need to be considered in the interpretation of such data (Mizuno et al., 1989; Schapira et al., 1990; Mann et al., 1992; Cardellach et al., 1993; Parker et al., 2008)
A.7.4. Quantitative understanding
Quantitative understanding for this KE relationship mainly comes from in‐vitro and engineered systems, using rotenone and MPTP as main chemical stressors. A clear response–response effect is evident as well as temporality was mainly supported by evidence that modulation of the KEup was attenuating or preventing the KEdown. Evidence of dose relationship was limited, as most of the time a single, generally high, concentration was used.
Table A.6.
Quantitative evaluation of the KER
| KE 2 upstream | KE 4 downstream | Comments | Reference |
|---|---|---|---|
| Rotenone experiments | |||
|
Mitochondrial membrane potential reduced by 50% upon rotenone treatment. Back to 80% compared to controls in the presence of the flavonoid rutin Intracellular Ca2+ elevated by a factor of 3 by rotenone, reduction to an increase of 1.5 in the presence of rutin ROS increased by a factor of 6.5; increase of ROS by a factor of 2 in the presence of rutin |
Rotenone (10 μM) resulted in a reduction of cell viability by 50% In the presence of rutin, cell viability was only reduced by 10% upon rotenone treatment |
SH‐SY5Y cells exposed to rotenone (10 μM) for 24 h When applied alone, rutin displayed no toxic effects, up to 100 μM Rutin was added to the cells 30 min prior rotenone at concentrations from 0–10 μM |
Park et al. (2014) |
|
Mitochondrial membrane potential reduced by ca. 66% upon rotenone treatment; in the presence of celastrol, reduction by ca. 55% ROS formation increased by a factor of 2 in the presence of rotenone; ROS increase by a factor of 1.5 in the presence of celastrol |
Cell viability was reduced by 50% by rotenone; In the presence of the triterpene celastrol, cell viability was only reduced by ca. 10% |
SH‐SY5Y + rotenone (10 μM). Celastrol (2.5 nM) was applied 90 min prior to rotenone Cells were incubated with the two compounds for a period of 24 h |
Choi et al. (2014) |
|
TH staining in the SNpc in arbitrary units: Control (25) Rotenone (14) Rotenone + NDI 1(22) TH staining in the striatum Control (70) Rotenone (40) Rotenone + NDI 1 (65) DA levels in the striatum: Control (2.5) Rotenone (1.3) Rotenone + NDI 1 (2.2) |
5 month old male Sprague–Dawley rats (ca. 500 g) received intracerebral injection of recombinant adeno‐associated virus with the NADH dehydrogenase NDI 1 gene 45 days after virus injection, rats were treated with rotenone‐loaded microspheres (poly(DL‐lactide‐co‐glycolide) 100 mg rotenone /kg body weight s.c With this method, HPLC analysis of plasma rotenone revealed levels of 2 μM 14 days after microsphere treatment, and 1 μM 60 days after microsphere treatment Behavioural experiments and brain sample collection was conducted 30 days after rotenone treatment |
Marella et al. (2008) | |
| MPP + experiments | |||
|
Decline in mitochondrial transmembrane potential by MPP+; 50% prevention from this decline by rosmarinic acid NADH levels were reduced by ca. 50% in the presence of MPP+; loss of NADH was completely prevented by the presence of rosmarinic acid ROS levels increased by 50% in the presence of MPP+. Rosmarinic acid lead to a reduced increase of ROS by only 20% compared with the untreated control |
Cell viability reduced by MPP+ by 30%, complete protection by the presence of the antioxidant rosmarinic acid Striatal DA content reduced by 40% by MPP+ treatment, partially protected by rosmarinic acid back to a value of 25% reduction compared with the untreated control |
MES23.5 cells exposed to MPP+ (200 μM) for 24 h Rosmarinic acid (1 nM) was applied 30 min prior to MPP+ treatment |
Du et al. (2010) |
| Reduction in mitochondrial membrane potential by 60% (MPP+), by 50% (rotenone), complete recovery by the co‐incubation with ISB, PHT, PHO |
SH‐SY5Y + MPP+: Cell viability reduced by 66%; ISB, PHT, PHO partially protected from cell death with a reduction in cell viability by ca. 20% SH‐SY5Y + rotenone: reduction in cell viability by 60% Partial protection by ISB, PHT, PHO to a reduction in cell viability by 25–50% SH‐SY5Y + BSO: Reduction in cell viability by 80% ISB, PHT, PHO partially protected with a residual decline in cell viability by ca. 20% |
SH‐SY5Y + MPP+ (200 μM) or rotenone (150 nM) or BSO (150 μM) for 60 h and 72 h Antioxidants tested: Iminostilbene (ISB) Phenothiazine (PHT) Phenoxazine (PHO) The antioxidants were applied 2 h prior to rotenone, MPP+, or BSO treatment |
Hajieva et al. (2009) |
| Circumvention of endogenous complex I | |||
|
Wt cells exposed to rotenone: increase in carbonyl content as marker of oxidative stress by 100%; completely prevented in NDI 1 expressing cells In midbrain slice cultures exposed to rotenone: increase in carbonyl content by 20% Rats exposed to rotenone: increase in carbonyl content: 27% in the striatum, increase by 41% in the midbrain |
SK‐N‐MC cells: rotenone evoked cell death protected by ca. 90% in NDI 1 expressing cells Rotenone induced cell death prevented by 80% by α‐tocopherol (62.5 μM and 125 μM) |
SK‐N‐MC human neuroblastoma cells transfected with the rotenone insensitive NADH dehydrogenase NDI 1; Cells were treated with rotenone (100 nM) for 48 h or with BSO (10 μM) for 24 h When both compound were used in a combined experiment, cells were first treated with BSO (10 μM) for 24 h, then rotenone (10 nM) was added for additional 36 h |
Sherer et al. (2003) |
|
Application of the complex I inhibitors: Rotenone Fenazaquin Fenpyroximate Pyridaben Tebufenpyrad Pyridaben |
Time and concentration‐dependent cell death with rotenone and a series of other complex I inhibitors NDI 1 expressing cells were resistant towards the different complex I inhibitors |
SK‐N‐MC human neuroblastoma cells expressing the rotenone‐insensitive NADH dehydrogenase NDI 1 from Saccharomyces cerevisiae All complex I inhibitors applied were added at the concentrations: 10 nM, 100 nM, 1 μM Pyridaben was applied at 1 μM, 10 μM, 100 μM Viability was assessed after 48 h, ATP was detected after 6 h. Carbonyl content was detected after 24 h |
Sherer et al. (2007) |
|
Oxygen consumption rate doubled by MB in the absence of complex I inhibitor Oxygen consumption reduced by 50% by rotenone; completely reversed to control levels by the presence of MB Complex I‐III activity reduced by 95% by rotenone. Reversed to control levels by the presence of MB |
HT22 cell viability reduced by 70% by rotenone In the presence of MB, reduction by only 10% of cell viability was observed In rats treated with rotenone, rotarod retention time was reduced by 50% by rotenone. Completely reversed to control levels by the co‐administration of MB In rats, rotenone evoked a reduction of striatal DA by 50%; completely reversed to control levels by MB Complex I‐III activity in the striatum of rats was reduced by 50%, residual inhibition of 10% observed in rats that were additionally treated with MB |
The study included:
Test of methylene blue (MB) (10 and 100 ng/mL in isolated mitochondria; 1 and 10 µg/ml in HT 22 cells) to circumvent the complex I/III blockade |
Wen et al. (2011) |
| Cybrid cells with PD mtDNA display a reduction in complex I activity by 20% |
Cybrid cells: increase in basal formation of reactive oxygen species by 80% 2‐times higher sensitivity towards MPP+ as stressor |
SH‐SY5Y cells devoid of mtDNA; fused with platelets from PD patients for mitochondria transfer: cybrid cells Treatment with MPP+ (40 or 80 μM) for 24 h or 48 h |
Swedlow et al. (1996) |
| Oxidative stress causes mitochondrial dysfunction | |||
|
Isolated mitochondria: Exposure to DA: loss of ca. 50% membrane potential. Completely protected by GSH or N‐acetyl‐cystein (NAC) Decline of mitochondrial respiration capacity by 90% In the presence of NAC or GSH, only a reduction by 25–30% was observed PC12 cells exposed to DA, then isolation and analysis of mitochondria: inhibition of complex I activity by ca. 50%, prevented by co‐incubation with NAC Inhibition of complex II and III; prevented by NAC Intact PC12 exposed to DA: Mitochondrial transmembrane potential reduced by ca. 50%; prevented by NAC Intracellular ATP reduced by ca. 50%; Cell death increased by DA by ca. 30%, caspase 3 activity increased by a factor of 3; all increases prevented by the presence of NAC |
PC12 cells exposed to DA: Increase in intracellular ROS by a factor of 2; completely reversed by NAC Quinoprotein formation increased by a factor of 3; completely prevented by the presence of NAC or GSH Cell death increased from 3% (control) to 37% (DA). Reduced to 10% in the presence of NAC |
PC12 cells and isolated rat brain mitochondria exposed to dopamine (100–400 μM) N‐acetyl cysteine or GSH for protection were added at a concentration of 2.5 mM In experiments including isolated mitochondria, NAC and GSH were added 2 h prior to DA. In experiments including PC12 cells, NAC and GSH were added 1 h prior DA Isolated mitochondria were exposed to DA for 2 h; PC12 cells were expose to DA for 24 h |
Jana et al. (2011) |
|
Reduction of intracellular GSH by 50% and of intramitochondrial GSH by 60% leads to: Mitochondrial ROS increased by 30% ATP levels reduced by 66% Mitochondrial activity reduced by 66% State 3 respiration reduced by 60% Complex I activity inhibited by 60% |
Whole cell ROS increased by 30% |
PC12 cells with inducible knockdown of glutamyl cysteine synthetase (inhibition of GSH synthesis) by addition of 25 μg/mL doxycycline Treatment for 24 h with doxycycline resulted in a GSH decline by ca. 50% |
Jha et al. (2000) |
|
Reduction of GSH levels by ca. 50% result in: Complex I inhibition by 40%; completely reversed by DTT |
No cell toxicity under the applied conditions |
N27 cells exposed to BSO (2.5 μM) for 7 days: Total glutathione was declined by ca. 50% by this chronic treatment; absence of cell toxicity under these conditions. DTT for restoration of complex I activity was added at 1 mM |
Chinta et al. (2006) |
Evidence Supporting Taxonomic Applicability
There are no sex or age restictions for the applicability of this KE and mitochondria are essential for most of eukaryotic cells. Rotenone and MPTp have been tested successfully in primates and mice. The mouse C57BL/6 strain is the most frequently used strain in the reported experiments. A difference in vulnerability was observed, particularly for rats, depending on the strain and route of administration. The Lewis strain gives more consistency in terms of sensitivity when compared to the Sprague–Dawley. In addition to rodents, the pesticide rotenone has been also studied in Caenorhabditis elegans (C. elegans), Drosophila, zebrafish and Lymnaea stagnalis (L. stagnalis) (Johnson et al., 2015), indicating that the system is preserved across species.
References
Andén NE, Hfuxe K, Hamberger B, Hökfelt T, 1966. A quantitative study on the nigro‐neostriatal dopamine neuron system in the rat. Acta Physiologica Scandinavica, 67, 306–312.
Antunes F, Han D, Rettori D, Cadenas E, 2002. Mitochondrial damage by nitric oxide is potentiated by dopamine in PC12 cells. Biochimica et Biophysica Acta, 1556, 233–238.
Beal MF, Matthews RT, Tieleman A, Shults CW, 1998. Coenzyme Q10 attenuates the 1‐methyl‐4‐phenyl‐1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Research, 783, 109–114.
Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K, 2014. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. Journal of Neuroscience, 34, 14304–14317.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Bevan MD, Booth PA, Eaton SA, Bolam JP, 1998. Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. Journal of Neuroscience, 18, 9438–9452.
Bolam JP, Pissadaki EK, 2012. Living on the edge with too many mouths to feed: why dopamine neurons die. Movement Disorders, 27, 1478‐83.
Cardellach F, Martí MJ, Fernández‐Solá J, Marín C, Hoek JB, Tolosa E, Urbano‐Márquez A, 1993. Mitochondrial respiratory chain activity in skeletal muscle from patients with Parkinson's disease. Neurology, 43, 2258–2262.
Cebrián C, Loike JD, Sulzer D, 2015. Neuroinflammation in Parkinson's disease animal models: a cell stress response or a step in neurodegeneration? Curr Top Behav Journal of Neuroscience, 22, 237–270.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP, 2015. Anti‐oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Molecular Neurodegeneration, 10, 4. doi: 10.1186/1750‐1326‐10‐4
Chinta SJ, Andersen JK, 2006. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radical Biology and Medicine, 41, 1442–1448.
Chiu CC, Yeh TH, Lai SC, Wu‐Chou YH, Chen CH, Mochly‐Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS, 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone‐induced cellular and animal models of parkinsonism. Experimental Neurology, 263, 244–53.
Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ, 2014. Celastrol from ‘Thunder God Vine’ protects SH‐SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochemical Research, 39, 84–96.
Davey GP, Clark JB, 1996. Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. Journal of Neurochemistry, 66, 1617–1624.
Du T, Li L, Song N, Xie J, Jiang H, 2010. Rosmarinic acid antagonized 1‐methyl‐4‐phenylpyridinium (MPP+)‐induced neurotoxicity in MES23.5 dopaminergic cells. International Journal of Toxicology, 29, 625–633.
Ekstrand M, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG, 2007. Progressive parkinsonism in mice with respiratory‐chain‐deficient dopamine neurons. Proceedings of the National Academy of Sciences USA, 104, 1325–1330.
Gandhi S, Wood‐Kaczmar A, Yao Z, et al., 2009. PINK1‐associated Parkinson's disease is caused by neuronal vulnerability to calcium‐induced cell death. Molecular Cell, 33, 627–638.
Gu M, Cooper JM, Taanman JW, Schapira AH, 1998. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson's disease. Annals of Neurology, 44, 177–186.
Hajieva P, Mocko JB, Moosmann B, Behl C, 2009. Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. Journal of Neurochemistry, 110, 118–132.
Höglinger GU, Féger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC, 2003. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. Journal of Neurochemistry, 84, 491–502.
Jain A, Mårtensson J, Stole E, Auld PA, Meister A, 1991. Glutathione deficiency leads to mitochondrial damage in brain. Proceedings of the National Academy of Sciences U S A, 88, 1913–1917.
Jana S, Sinha M, Chanda D, Roy T, Banerjee K, Munshi S, Patro BS, Chakrabarti S, 2011. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson's disease. Biochimica et Biophysica Acta, 1812, 663–673.
Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, Zöchling R, Boissl KW, Reichmann H, 1994. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Journal of Neuroscience Letters, 169, 126–128.
Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK, 2000. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. Journal of Biological Chemistry, 275, 26096‐26101.
Kawaguchi Y, Wilson CJ, Emson PC, 1990. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. Journal of Neuroscience, 10, 3421–3438.
Johnson ME, Bobrovskaya L, 2015. An update on the rotenone models of parkinson's disease: Their ability to reproduce features of clinical disease and model gene‐environment interactions. 946, 101–116.
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr, 2006. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. Journal of Neuroscience, 26, 5256–5264.
Khan FH, Sen T, Maiti AK, Jana S, Chatterjee U, Chakrabarti S, 2005. Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: implications for Parkinson's disease. Biochimica et Biophysica Acta, 1741, 65–74.
Khan MM, Raza SS, Javed H, Ahmad A, Khan A, Islam F, Safhi MM, Islam F, 2012. Rutin protects dopaminergic neurons from oxidative stress in an animal model of Parkinson's disease. Neurotoxicity Research, 22, 1–15.
Kita H, Kitai ST, 1994. The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Research, 636, 308–319.
Koopman W, Willems P, 2012. Monogenic mitochondrial disorders. New England Journal of Medicine, 366, 1132–1141. doi: 10.1056/NEJMra1012478
Langston JW, Ballard PA Jr, 1983. Parkinson's disease in a chemist working with 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine. New England Journal of Medicine, 309, 310.
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD, 1992. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease, 115(Pt 2), 333–342.
Marella M, Seo BB, Nakamaru‐Ogiso E, Greenamyre JT, Matsuno‐Yagi A, Yagi T, 2008. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. Public Library of Science (PLoS ONE), 3, e1433.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T, 2009. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. Journal of Neuroscience, 29, 444–453.
Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah‐Daouk R, Beal MF, 1999. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Experimental Neurology, 157, 142–149.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y, 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications, 163, 1450–1455.
Moosmann B, Behl C, 2002. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs, 11, 1407–1435.
Moussaoui S, Obinu MC, Daniel N, Reibaud M, Blanchard V, Imperato A (2000) The antioxidant ebselen prevents neurotoxicity and clinical symptoms in a primate model of Parkinson's disease. Experimental Neurology, 166, 235–245.
Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D, 2012. Transgenic expression and activation of PGC‐1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Science Journal, 69, 1153–1165.
Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochemical Journal, 417, 1–13.
Orimo S, Uchihara T, Kanazawa T, Itoh Y, Wakabayashi K, Kakita A, Takahashi H, 2011. Unmyelinated axons are more vulnerable to degeneration than myelinated axons of the cardiac nerve in Parkinson's disease. Neuropathology and Applied Neurobiology, 37, 791–802.
Park SE, Sapkota K, Choi JH, Kim MK, Kim YH, Kim KM, Kim KJ, Oh HN, Kim SJ, Kim S, 2014. Rutin from Dendropanax morbifera Leveille protects human dopaminergic cells against rotenone induced cell injury through inhibiting JNK and p38 MAPK signaling. Neurochemical Research, 39, 707–718.
Parker WD Jr, Boyson SJ, Parks JK, 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Annals of Neurology, 26, 719–723.
Parker WD Jr, Parks JK, Swerdlow RH, 2008. Complex I deficiency in Parkinson's disease frontal cortex. Brain Research, 1189, 215–218.
Perry TL, Godin DV, Hansen S, 1982. Parkinson's disease: a disorder due to nigral glutathione deficiency? Journal of Neuroscience Letters, 33, 305–310.
Rangaraju V, Calloway N, and Ryan TA, 2014. Activity‐driven local ATP synthesis is required for synaptic function. Cell, 156, 825–835.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD, 1990. Mitochondrial complex I deficiency in Parkinson's disease. Journal of Neurochemistry, 54, 823–827.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD, 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet, 1, 1269.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno‐Yagi A, Greenamyre JT, 2003. Mechanism of toxicity in rotenone models of Parkinson's disease. Journal of Journal of Neuroscience, 23, 10756–10764.
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno‐Yagi A, Miller GW, Greenamyre JT, 2007. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. Journal of Neurochemistry, 100, 1469–1479.
Starkov AA, 2008. The role of mitochondria in reactive oxygen species metabolism and signaling. Annals of the New York Academy of Sciences, 1147, 37–52.
Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP Jr, Davis RE, Parker WD Jr, 1996. Origin and functional consequences of the complex I defect in Parkinson's disease. Annals of Neurology, 40, 663–671.
Surmeier DJ1, Guzman JN, Sanchez‐Padilla J, Goldberg JA, 2010. What causes the death of dopaminergic neurons in Parkinson's disease? Prog Brain Research, 183, 59–77. doi: 10.1016/S0079‐6123(10)83004‐3
Sulzer D, Surmeier DJ, 2013. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Movement Disorders, 28, 715–724.
Tait SWG, Green DR, 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature Reviews Molecular Cell Biology, 11, 621–632.
Tepper JM, Bolam JP, 2004. Functional diversity and specificity of neostriatal interneurons. Current Opinion in Neurobiology, 14, 685–692.
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH, 2011. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. Journal of Biological Chemistry, 286, 16504–16515.
Wu Y, Richard S, Parent A, 2000. The organization of the striatal output system: a single‐cell juxtacellular labeling study in the rat. Journal of Neuroscience Research, 38, 49–62.
Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion and stress. Science, 337, 1062–1065.
Zhu C, Vourc'h P, Fernagut PO, Fleming SM, Lacan S, Dicarlo CD, Seaman RL, Chesselet MF, 2004. Variable effects of chronic subcutaneous administration of rotenone on striatal histology. Journal of Comparative Neurology, 478, 418–426.
8th KER: Degeneration of DA neurons of nigrostriatal pathway leads to parkinosnian motor deficits (bradykinesia, rigor, and tremor)
A.8.1. How Does This Key Event Relationship Work
Degeneration of dopaminergic (DA) neuron terminals in the striatum and the degeneration of DA neurons in the substantia nigra pars compacts (SNpc) are the defining histopathological events observed in idiopathic, familial, and toxicant‐evoked cases of Parkinson's Disease (PD) (Tolwani et al., 1999; Bove et al., 2012). The loss of nigrostriatal DA neurons leads to a decline in the levels of DA in the striatum (Koller et al., 1992). Striatal DA is involved in the modulation of extrapyramidal motor control circuits. A decline in striatal DA leads to an overactivation of the two principal basal ganglia output nuclei (GPi/STN). Therefore, the inhibitory GABAergic neurons that project to thalamocortical structures are overactivated and inhibit cortical pyramidal motor output performance. This inhibited output activity is responsible for key clinical symptoms of PD such as bradykinesia and rigor.
A.8.2. Weight of Evidence
A.8.2.1. Biological Plausibility
The mechanistic understanding of striatal DA and its regulatory role in the extrapyramidal motor control system is well established (Alexander et al., 1986; Penney et al., 1986; Albin et al., 1989; DeLong et al., 1990; Blandini et al., 2000; Obeso et al., 2008). The selective degeneration of DA neurons in the SNpc (and the subsequent decline in striatal DA levels) have been known to be linked to PD symptoms for more than 50 years (Ehringer et al., 1960). The reduction of DA in the striatum is characteristic for all aetiologies of PD (idiopathic, familial, chronic manganese exposure) and related parkinsonian disorders (Bernheimer et al., 1973), and it is not observed in other neurodegenerative diseases, such as Alzheimer's or Huntington's Diseases (Reynolds et al., 1986). In more progressive stages of PD, not only a loss of DA neuronal terminals in the striatum, but also a degeneration of the entire DA neuron cell bodies in the substantia nigra pars compacta (SNpc) was detected (Bernheimer et al., 1973; Leenders et al., 1986). The different forms of PD exhibit variations in the degradation pattern of the SNpc DA neurons. In idiopathic PD, for example, the putamen is more severely affected than the caudate nucleus (Moratalla et al., 1992; Snow et al., 2000). All different PD forms however are characterised by the loss in striatal DA that is paralleled by impaired motor output (Bernheimer et al., 1973). Characteristic clinical symptoms of motor deficit (bradykinesia, tremor, or rigidity) of PD are observed when more than 80% of striatal DA is depleted (Koller et al., 1992). These findings on the correlation of a decline in striatal DA levels as a consequence of SNpc DA neuronal degeneration with the onset of clinical PD symptoms in man provide the rationale for the current standard therapies that aim to supplement striatal DA, either by the application of L‐DOPA, or by a pharmacological inhibition of the endogenous DA degradation‐enzyme monoamine oxidase B (MAO‐B). These treatments result in an elevation of striatal DA that is correlated with an improvement of motor performance (Calne et al., 1970). The success of these therapies in man as well as in experimental animal models clearly confirms the causal role of dopamine depletion for PD motor symptoms.
A.8.2.3. Empirical Support for Linkage
The experimental support linking the degeneration of DA neurons of nigrostriatal pathways with the manifestation of motor symptoms characteristics of parkinsonian disorders comes from human clinical observations as well as from primates, mice and rat in vivo models using DA neuron ablation by toxicants. The levels of striatal DA corrected with the onset of PD symptoms, and dopaminergic degeneration precede the onset of motor symptoms. The exemplary animal studies selected here are based on the use of MPTP or rotenone. The efficacy of MPTP or rotenone treatment depends on the regimen applied (acute, subacute, chronic administration), the age of the animals, and the strains used. For the interpretation of the studies, it is important that in some animal models the initial depletion of DA is only partially explained by neurite degeneration. The other contributing factors are downregulation of TH, and depletion of DA from synaptic terminals. These effects recover after 1–2 weeks. This makes the time point of measurement important for the correlation of effects. Moreover, the mouse brain has a very high plasticity after damage, so that motor deficits can recover after several weeks although there is pronounced dopaminergic neurodegeneration.
Rat in vivo models
Rat/rotenone: Correlation between striatal DA, SNpc DA neurons, and motor deficits. Lewis rats exposed to systemic rotenone (3 mg/kg per day i.p.) exhibited a loss of TH positive neurons in the SNpc by 45%. Motor deficits were assessed by the postural instability test and by the rearing test. While 3 month old animals developed motor symptoms after 12 days of rotenone exposure, 7 month and 12 month old animals developed motor symptoms already after 6 days of exposure. Rotenone treatment elicited a progressive development of motor deficits that was reversible when treated with a DA agonist. Similar to that, the loss of rearing performance evoked by rotenone was reversed by the DA agonist apomorphine. Rotenone elicited terminal loss in the dorsolateral structures. While in the dorsolateral striatum, a significant loss of TH‐positive neurites was detected, striatal cell bodies were spared from degeneration. Initial striatal DA levels (75 ng/mg protein) dropped to 45% following rotenone treatment (Cannon et al., 2009).
Rat/6‐OHDA: Destruction of nigrostriatal DA neurons. Unilateral injection of 6‐OHDA into the dopaminergic nigrostriatal pathway leads to a preferential loss of DA neurons that is correlated with the onset of rotational motor deficits (Luthman et al., 1989; Perese et al., 1989; Przedborski et al., 1995).
Rats/rotenone: Correlation between striatal dopamine and motor symptoms; partial reversibility by L‐DOPA. Rats were exposed to 2.5 mg/kg rotenone, daily, for 48 days. Dopamine detected in the anterior striatum and posterior striatum was reduced by ca. 50% after rotenone treatment. Rotenone treatment resulted in a significantly prolonged descent latency compared to control in the bar test and grid test. In the catalepsy test, descent latency dropped from 35 s of the controls to 5 s. In the grid test, a reduction from 30 s (control) down to 4 s (rotenone) was observed. The average distance travelled within 10 min by the animals was reduced from 37 m to 17 m in the rotenone group. Average number of rearings declined from 65 to 30; the time of inactive sitting of 270 s in controls was increased to 400 s in the rotenone group (Alam et al., 2004).
Rat/rotenone: Correlation between striatal dopamine and motor symptoms. Rats were treated with rotenone either at doses of 1.5 mg/kg or 2.5 mg/kg over 2 months with daily i.p. injections. In the 2.5 mg/kg group, striatal DA levels dropped from 6,400 pg/mg in the controls to 3500 pg/mg in the rotenone group. Rotenone treated animals showed an extended descent latency (5 to 50). In a vertical grid test, latency time increased from 9–72 s (Alam et al., 2002).
Rats/rotenone: Correlation between nigrostriatal TH intensity and motor symptoms. Rats were treated with different doses of rotenone for 21 days with daily i.v. or s.c. injections. In the 2.5 mg/kg group, TH intensity in the striatum dropped from 0.2 to 0.12. The average time to initiate a step increased from 5 s in the controls to 11 s in the rotenone group. Spontaneous rearing scores dropped from 80% of the vehicle‐treated controls to 20% in the rotenone group (Fleming et al., 2004).
Rat/rotenone: In middle‐aged rats exposed to rotenone (3 mg/kg per day for 6 days), a reduction of striatal DA levels and TH positive neurons by ca. 50% correlated with impairments rearing performance and postural instability tests (Cannon et al., 2009).
Rat/rotenone: In rats, exposed to rotenone (2.5 mg/kg per day), spontaneous locomotor activity was reduced by ca. 50% after 1 week of rotenone treatment. This impaired motor performance was correlated with a loss of striatal DA fibres by 54% and a loss of nigral DA neurons by 28.5% (Höglinger et al., 2003).
Mouse in vivo models
Mouse/MPTP: In mice exposed to MPTP in combination with probenecid, both a chronic treatment scheme (MPTP 25 mg/kg, in 3.5 day intervals for 5 weeks) as well as a subacute treatment scheme (25 mg/kg, 1x per day for 5 days) resulted in a deletion of striatal DA that was directly correlated with impairments in motor symptoms (Petroske et al., 2001).
Mouse/MPTP: In a mouse model exposed to MPTP at 15 day intervals (36 mg/kg), lower rotarod performance was observed after the fourth injection. The decline in motor performance was correlated with the decline in TH‐immunoreactivity in the striatum (r2 = 0.87) (Rozas et al., 1998).
Monkey in vivo models
Monkey/MPTP: Correlation between striatal DA, SNpc DA neuron number and PD symptoms. Macaca exposed to MPTP (i.v) (0.2 mg/kg, daily) display signs of PD at day 15, including motor abnormalities. The transition between the presymptomatic and symptomatic period occurred between day 12 and day 15 of MPTP exposure. At day 15, TH neurons in the SNpc were reduced by 50%, DAT binding autoradiography studies revealed a decline in binding also by 50% at day 15. Compared with control values of 150 pg/μg protein, the DA content of the caudate nucleus dropped to values < 10 pg/μg protein at day 15. In the putamen, DA levels dropped from 175 pg/μg protein to 20 pg/μg protein at day 15 (Bezard et al., 2001).
Monkey/MPTP: Correlation between striatal DA, SNpc DA neurons, and PD symptoms. Monkeys display a motor symptom pattern similar to that observed in humans. In order to optimise a MPTP intoxication protocol that allows a gradual development of nigral lesion, different states of PD symptom severity were defined and correlated with the amount of striatal DA and the number of TH‐positive neurons in the SNpc. Asymptomatic monkeys displayed a reduction in striatal DA by 30%, a neuronal loss in the SNpc by 40%, and a decline in striatal expression of TH, DAT and VMAT2 by 50–60%. Monkeys that recovered from early PD symptoms displayed a reduction of striatal DA of 50%, a loss of TH neurons in the SNpc and a loss of DAT and VMAT2 expression up to 60%. In animals with moderate PD symptoms, striatal DA levels as well as TH positive neurons and DAT and VMAT 2 expression were reduced by 70–80%. Animals with severe PD symptoms displayed remaining levels of striatal DA and SNpc expression of TH, DAT and VMAT2 of around 20% compared to untreated controls (Blesa et al., 2012).
Monkey/MPTP: The established model of basal ganglia wiring received ample experimental support in recent years. For instance, an increase in the inhibitory output by GPi/STN has been observed in MPTP treated monkeys, similar to the situation in idiopathic PD patients. These findings were corroborated by observations indicating an elevated mitochondrial activity and an elevated firing rate of the inhibitory output nuclei detected on the level of individual neurons (Mitchell et al., 1989; Filion et al., 1991). Lesions in the output ganglia of monkeys lead to a reduction in the output and to an improvement in motor control (Bergman et al., 1990; Aziz et al., 1991). In analogy to these lesion experiments, deep brain stimulation of these regions results in a profound improvement of motor performance in PD patients (Ceballos‐Baumann et al., 1994; Limousin et al., 1999).
Human data
Human PD: Association of PD phenotype with impaired striatal DA. In the brains of human PD patients, a significant decrease of striatal DA was observed (Lloyd et al., 1975). In the caudate nucleus, levels of DA dropped from control values of 4 μg/g tissue to levels of 0.2 μg/g. In the putamen, control values were in the range of 5 μg/g and 0.14 μg/g in the PD patient group. The levels of DA in the striata of DA patients that received L‐DOPA treatment was 9–15 times higher compared with non‐treated PD cases.
Human PD: Correlation between striatal DA loss and degeneration of DA neurons in the SNpc. Examinations of the brains of PD patients revealed morphological damage in the SNpc, accompanied by the degeneration of DA neurons (Earle et al., 1968).
Human: Association of striatal DA levels and motor performance. In order to substitute degenerated DA neurons in the SNpc, human fetal tissue from the ventral mesencephalon was transplanted to the caudate and putamen in idiopathic cases PD as well as in patients that developed PD‐related motor deficits as a consequence to MPTP intoxication. Transplanted cells led to a reinnervation of the striatum with DA projections (Widner et al., 1992; Kordower et al., 1995, 1998). In these case studies, patients demonstrated a sustained improvement in motor function (decline in rigidity score by more than 80%).
Human PD: correlation between nigrostriatal DA neuron content and motor symptoms. Imaging of DAT was performed by the use of 123I‐FP‐CIT SPECT (single photon emission computed tomography). Clinical PD severity was determined by using the Unified Parkinson's Disease Rating Score (UPDRS). In PD patients, DAT binding in the striatum, caudate, and putamen correlated with disease severity and duration of disease (Benamer et al., 2000).
Human PD: correlation between 18F‐dopa uptake measured by PET and the onset of motor symptoms detected according the UPDRS. 18F‐dopa influx rate constants (Ki/min) were reduced in the midbrain from 0.008 to 0.006, in the right putamen from 0.017 to 0.0036, and in the left putamen from 0.017 to 0.005 (Rakshi et al., 1999).
Human PD: correlation between putamen influx rate (Ki/min). Ki (control): 0.0123; asymptomatic PD (no observable motor deficits): 0.0099; symptomatic PD (clinically evident motor deficits): 0.007. Mean UPDRS value was 15.1 ± 7.5. A correlation coefficient of ‐0.41 was detected between motor UPDRS and putamen influx (Ki) (Morrish et al., 1995).
Human PD: Correlation of the degree of monoaminergic degeneration in early PD with motor symptoms assed by the UPDRS and the Hoehn and Yahr Stage scale. For PET imaging, 18F‐9‐fluoropropyl‐dihydrotetrabenzazine that targets VMAT2 was used. Uptake of the tracer was reduced by 20–36% in the caudate, by 45–80% in the putamen, and by 31% in the substantia nigra. This correlated with a total UPDRS value of 12.1 ± 7.1 in the PD group, respectively with a HY value of 1.0 ± 0.1 in the PD group compared to controls (Lin et al., 2014).
Human PD: Correlation between the decline in 18F‐dopa rate constant (Ki) and the onset of motor deficits. The 18F‐dopa rate constant Ki was reduced in the caudate nucleus (0.011 down to 0.0043) and inversely correlated with an increase in the UPDRS from 11.9 ± 5.2 to 50 ± 11.6 (Broussolle et al., 1999).
Human PD: Correlation between striatal DAT binding measured by the use of 123I‐CIT SPECT and motor deficits. A correlation coefficient between 123I‐CIT binding and UPDRS motor scale of ‐0.56 was detected. A correlation coefficient of ‐0.64 between 123I‐CIT binding and Hoehn and Yahr stage scale was detected. Motor symptoms in the clinically less affected body side show a closer correlation with striatal DAT binding (Pirker et al., 2003).
Human PD: Correlation between the reduction in the putamen uptake of 18F‐CFT and the severity of PD motor symptoms. 18F‐CFT uptale was reduced to 18% in the putamen, to 28% in the anterior putamen, and to 51% in the caudate nucleus (Rinne et al., 1999).
Human PD: Reduction in 123I‐CIT binding in the putamen by 65% correlated with a mean UPDRS score of 27.1 (Tissingh et al., 1998).
Association between striatal DA and motor performance. Application of L‐DOPA leads to a substitution of DA in the striatum and improves motor performance (Hutchonson et al., 1997; Boraud et al., 1998; Papa et al., 1999; Levy et al., 2001; Heimer et al., 2002; Gilmour et al., 2011).
A.8.3. Uncertainties or Inconsistencies
Motor abnormalities observed in PD display large interindividual variations.
The model of striatal DA loss and its influence on motor output ganglia does not allow to explain specific motor abnormalities observed in PD (e.g. resting tremor vs bradykinesia) (Obeso et al., 2000). Other neurotransmitters (Ach) may play additional roles
There are some reports indicating that in subacute rotenone or MPTP models (non‐human primates), a significant, sometimes complete, recovery of motor deficits can be observed after termination of toxicant treatment. While the transient loss of striatal DA can be explained by an excessive release of DA under acute toxicant treatment, the reported losses of TH‐positive neurons in the SNpc and their corresponding nerve terminals in the striatum are currently not explained (Petroske et al., 2001).
In MPTP treated baboons, the ventral region of the pars compacta was observed to be more severely degenerated that the dorsal region. This pattern is similar to the degeneration pattern in idiopathic PD in humans. These observations indicate that two subpopulations of nigrostriatal DA neurons with different vulnerabilities might exist (Varastet et al., 1994).
According to the classical model of basal ganglia organisation, DA is assumed to have a dichotomous effect on neurons belonging either to the direct or indirect pathway. More recent evidence however rather indicates that D1 and D2 receptors are expressed on most striatal neurons in parallel (Aizman et al., 2000).
A.8.4. Quantitative Understanding of the Linkage
An example of quantitative analysis is reported in Table A.7. The analysis of the empirical data produced with the chemical toxicants supports a strong response–response relationship between the KEup and the KEdown which also indicative of the temporal progression and relationship between the degeneration of striatal terminals of DA neurons, loss of DA neurons in the SNpc and the occurrence and severity of the motor deficits. This is also quantitatively supported by studies conducted in human PD patients.
Table A.7.
Quantitative evaluation of the KER
| Upstream key event (KE 4) | Downstream key event (AO) | References | Comments |
|---|---|---|---|
| Rat models | |||
|
45% loss of TH‐positive SNpc neurons in 7 month old rats, ca. 40% loss in 12 month old rats Striatal DA reduced from 90 ng/mg (control) down to 45 ng/mg TH pos. neuron number 18,000 (control) 10,000 (rotenone) |
Bradykinesia, postural instability, rigidity observed in 50% of cases: 3 month old rats: after 12 days of rotenone 7 + 12 month old rats. After 6 days of rotenone Postural instability test: Distance required for the animal to regain postural stability: 3.5 cm (control) 5 cm (rotenone) Rearing test (rears/5 min): 10 (control 3 (rotenone) Loss of rearing performance evoked by rotenone was reversed by the DA agonist Apomorphine in 3 month old rats |
Cannon et al. (2009) | Lewis rats + rotenone (3 mg/kg per day, i.p. daily) |
| Dopamine in the anterior and posterior striatum reduced by ca. 50% |
Catalepsy test: decline from 35 s to 5 s Grid test: decline from 30 s to 4 s Distance travelled in 10 min: reduction from 37 m to 17 m Number of rearings: decline from 65 to 30 Inactivity time increased from 270 to 400 s Partial reversibility by L‐DOPA treatment: L‐DOPA: number of rearings increased from 16 to 30 L‐DOPA: inactivity time reduced from 450 to 360 s L‐DOPA: increase in the distance travelled from 12 to 16 m |
Alam et al. (2004) | Rats + rotenone (2.5 mg/kg) daily over the course of 48 days |
| TH staining intensity reduced from 0.2 to 0.12 |
Rearing scores reduced from 80% (vehicle controls) to 20% (rotenone group) Increase in the average time to initiate a step from 5 to 11 s. |
Fleming et al. (2004) | Rats + rotenone 2.5 mg/kg for 21 days i.v. or s.c. |
|
Loss of striatal DA fibres by 54% Loss of DA neurons by 28.5% |
Spontaneous locomotor activity after 1 week 100% (control) 55% (rotenone) |
Höglinger et al. (2003) | Rats + rotenone (2.5 mg/kg per day for 28 days |
| Mouse models | |||
|
Subacute model: Striatal DA dropped from 11 (control) to 2.5 ng/mg (MPTP) after 3 days 3H‐DA striatal uptake reduced from 2.9 (control) to 1.3 pmol/mg after 3 days of MPTP Total nigrostriatal TH cell count was not affected Chronic model: Striatal DA content reduced from 13 down to 0.5 ng/mL at 1 week after MPTP treatment 3H‐DA uptake in the striatum reduced from 3 to 1 pmol/mg 1 week after start of MPTP treatment TH staining in the nigrostriatal system reduced by ca. 50% 1 week after initiation of MPTP treatment |
Subacute model: Rotarod performance reduced from 1,800 AUC (control) down to 1,500 AUC (MPTP) Chronic model: Rotarod performance reduced from 1,800 AUC (control) to 1,250 AUC (1 week after initiation of MPTP treatment) |
Petroske et al. (2001) |
Mouse + MPTP Subacute model: 25 mg/kg MPTP 1x days for 5 days Chronic model: MPTP (25 mg/kg + 250 mg/kg probenizid) in 3.5 day intervals for maximal 5 weeks |
|
Reduction in TH staining intensity of at least 50% required for detectable influence on motor performance TH density in the nigrostriatal system correlated with the decline of rotarod performance (r2 = 0.87) |
Rotarod performance reduced from 1,250 AUC to 200 AUC Time on rod at a speed of 20 rpm: 125 s in controls, 25 s in MPTP animals |
Rozas et al. (1998) | Mouse + MPTP |
| Monkey models | |||
| Approx. 50% loss of TH positive neurons in the SNpc. DA content in the caudate nucleu reduced to < 10%; DA content of the putamen ca. 10% compared with control | Mean duration in the bradykinesia test increased from 3 s (day 0) to 19 s at day 15 | Bezard et al. (2001) | Macaca + MPTP i.v. 0.2 mg/kg daily for 15 days |
| Human | |||
|
18F‐dopa influx rate constants (Ki) Midbrain: Control: 0.008 Early PD: 0.008 Adv. PD: 0.006 Right putamen: Control: 0.017 Early PD: 0.006 Adv. PD: 0.0036 Left putamen: Control: 0.017 Early PD: 0.0096 Adv. PD: 0.005 |
Early PD: UPDRS: 9 ± 3 Adv. PD: UPDRS: 41 ± 15 |
Rakshi et al. (1999) | Human PD patients |
|
Putamen influx (Ki/min) detected by 18F‐dopa control: 0.0123 asympt. PD: 0.0099 symptom. PD: 0.007 |
Symptom. PD patients: mean UPDRS: 15.1 ± 7.5 Correlation between total UPDRS and putamen Ki: r = −0.41 |
Morrish et al. (1995) | Human PD |
|
Uptake of 18F‐DTBZ (VMAT2 tracer) reduced by: 20–36% (caudate) 45–80% (putamen) 31% (SN) |
UPDRS total: 12.1 ± 7.1 Hoehn and Yahr: 1.0 ± 0.1 |
Lin et al. (2014) | Human PD |
|
Caudate nucleus Ki/min Control: 0.011 PD group 3: 0.0067 Putamen Ki/min Control: 0.011 PD group 3: 0.0043 |
UPDRS: 50 ± 11.6 in PD group 3 | Broussolle et al. (1999) | Human PD |
| Reduction in 18F‐CFT uptake in the posterior putamen (by 18%); in the anterior putamen (by 28%); in the caudate nucleus (by 51%) |
Correlation between total motor score of the UPDRS and 18F‐CFT uptake: Posterior putamen: r = −0.62 Anterior putamen: r = −0.64 Caudate nucleus: r = −0.62 |
Rinne et al. (1999) | Human PD |
|
123I‐CIT SPECT values in controls and PD cases with a Hoehn and Yahr rating of 2–2.5: Putamen (ipsilateral): Control: 6.13 PD: 1.84 Caudate (ipsilateral): Control: 6.93 PD: 3.66 Striatum (ipsilateral): Control: 6.28 PD: 2.33 |
Correlation coefficient between striatal 123I‐CIT binding and: Str. (ipsilateral) and Bradykinesia: r = −0.61 Str. (ipsilateral) and Rigidity: r = −0.46 Str. (ipsilateral) and UPDRS: r = −0.79 |
Tissingh et al. (1998) | Human PD |
|
Binding ration striatum/cerebellum detected by 123I‐CIT/SPECT Control: 8.71 ± 1.54 PD: 4.49 ± 1.86 |
Correlation between 123I‐CIT binding to DAT and PD motor symptoms rated according to the Hoehn and Yahr scale: r = −0.75 Correlation according to the UPDRS: r = −0.49 |
Asenbaum et al. (1997) | Human PD |
| Uptake of 123I‐CIT in the putamen reduced to 54%; uptake into the caudate nucleus reduced to 65% |
Correlation between CIT uptake in the putamen and Hoehn and Yahr stage: r = −0. 79 |
Rinne et al. (1995) | Human PD |
| Decline in nigrostriatal DAT assed by 123I‐CIT SPECT in PD patients |
Correlation coefficients for 123I‐CIT uptake in the striatum and: UPDRS: r = −0.54 Bradykinesia: r = −0.5 Rigidity: r = −0.27 Tremor: r = −0.3 Correlation coefficients for 123I‐CIT uptake in the caudate and: UPDRS: r = −0.5 Bradykinesia: r = −0.43 Rigidity: r = −0.27 Tremor: r = −0.26 Correlation coefficients for 123I‐CIT uptake in the putamen and: UPDRS: r = −0.57 Bradykinesia: r = −0.53 Rigidity: r = −0.29 Tremor: r = −0.37 |
Benamer et al. (2000) | Human PD |
A.8.5. Evidence Supporting Taxonomic Applicability
Parkinsonian disorders are generally recognised as progressive age‐related human neurodegenerative diseases more prevalent in males. However, the anatomy and function of the nigrostriatal pathway is conserved across mammalian species (Barron et al., 2010) and no sex and species restrictions were evidenced using the chemical stressors rotenone and MPTP. It should be noted that animal behaviour models can only be considered as surrogates of human motor disorders as occuring in Parkinson's disease.
References
Aizman O, Brismar H, Uhlén P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A, 2000. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nature Journal of Neuroscience, 3, 226–230.
Alam M, Schmidt WJ, 2002. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behavioural Brain Research, 136, 317–324.
Alam M, Schmidt WJ, 2004. L‐DOPA reverses the hypokinetic behaviour and rigidity in rotenone‐treated rats. Behavioural Brain Research, 153, 439–446.
Albin RL, Young AB, Penney JB, 1989. The functional anatomy of basal ganglia disorders. Trends in Journal of Neurosciences, 12, 366–375.
Alexander GE, DeLong MR, Strick PL, 1986. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annual Review Journal of Neuroscience, 9, 357–381.
Asenbaum S, Brücke T, Pirker W, Podreka I, Angelberger P, Wenger S, Wöber C, Müller C, Deecke L, 1997. Imaging of dopamine transporters with iodine‐123‐beta‐CIT and SPECT in Parkinson's disease. Journal of Nuclear Medicine, 38, 1–6.
Aziz TZ, Peggs D, Sambrook MA, Crossman AR, 1991. Lesion of the subthalamic nucleus for the alleviation of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced parkinsonism in the primate. Movement Disorders, 6, 288–292.
Benamer HT, Patterson J, Wyper DJ, Hadley DM, Macphee GJ, Grosset DG, 2000. Correlation of Parkinson's disease severity and duration with 123I‐FP‐CIT SPECT striatal uptake. Movement Disorders, 15, 692–698.
Bergman H, Wichmann T, DeLong MR, 1990. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science, 249, 1436–1438.
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F, 1973. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. Journal of the Neurological Sciences, 20, 415–455.
Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE, 2001. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐lesioned macaque model of Parkinson's disease. Journal of Neuroscience, 21, 6853–6861.
Blandini F, Nappi G, Tassorelli C, Martignoni E, 2000. Functional changes of the basal ganglia circuitry in Parkinson's disease. Progress in Neurobiology, 62, 63–88.
Blesa J, Pifl C, Sánchez‐González MA, Juri C, García‐Cabezas MA, Adánez R, Iglesias E, Collantes M, Peñuelas I, Sánchez‐Hernández JJ, Rodríguez‐Oroz MC, Avendaño C, Hornykiewicz O, Cavada C, Obeso JA, 2012. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiology of Disease, 48, 79–91.
Boraud T, Bezard E, Guehl D, Bioulac B, Gross C, 1998. Effects of L‐DOPA on neuronal activity of the globus pallidus externalis (GPe) and globus pallidus internalis (GPi) in the MPTP‐treated monkey. Brain Research, 787, 157–160.
Bové J, Perier C, 2012. Neurotoxin‐based models of Parkinson's disease. Journal of Neuroscience, 211, 51–76.
Broussolle E, Dentresangle C, Landais P, Garcia‐Larrea L, Pollak P, Croisile B, Hibert O, Bonnefoi F, Galy G, Froment JC, Comar D, 1999. The relation of putamen and caudate nucleus 18F‐Dopa uptake to motor and cognitive performances in Parkinson's disease. Journal of the Neurological Sciences, 166, 141–151.
Calne DB, Sandler M, 1970. L‐Dopa and Parkinsonism. Nature, 226, 21–44.
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT, 2009. A highly reproducible rotenone model of Parkinson's disease. Neurobiology of Disease, 34, 279–290.
Ceballos‐Baumann AO, Obeso JA, Vitek JL, Delong MR, Bakay R, Linazasoro G, Brooks DJ, 1994. Restoration of thalamocortical activity after posteroventral pallidotomy in Parkinson's disease. Lancet, 344, 814.
DeLong MR, 1990. Primate models of movement disorders of basal ganglia origin. Trends in Journal of Neurosciences, 13, 281–285.
Earle KM, 1968. Studies on Parkinson's disease including x‐ray fluorescent spectroscopy of formalin fixed brain tissue. Journal of Neuropathology & Experimental Neurology, 27, 1–14.
Ehringer H, Hornykiewicz O, 1960. Distribution of noradrenaline and dopamine (3‐hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klinische Wochenschrift, 38, 1236–1239.
Filion M, Tremblay L, 1991. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP‐induced parkinsonism. Brain Research, 547, 142–151.
Fleming SM, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman RL, Chesselet MF, 2004. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Experimental Neurology, 187, 418–429.
Gilmour TP, Lieu CA, Nolt MJ, Piallat B, Deogaonkar M, Subramanian T, 2011. The effects of chronic levodopa treatments on the neuronal firing properties of the subthalamic nucleus and substantia nigra reticulata in hemiparkinsonian rhesus monkeys. Experimental Neurology, 228, 53–58.
Heimer G, Bar‐Gad I, Goldberg JA, Bergman H, 2002. Dopamine replacement therapy reverses abnormal synchronization of pallidal neurons in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine primate model of parkinsonism. Journal of Neuroscience, 22, 7850–7855.
Höglinger GU, Féger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC, 2003. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. Journal of Neurochemistry, 84, 491–502.
Hutchinson WD, Levy R, Dostrovsky JO, Lozano AM, Lang AE, 1997. Effects of apomorphine on globus pallidus neurons in parkinsonian patients. Annals of Neurology, 42, 767–775.
Koller WC, 1992. When does Parkinson's disease begin? Neurology, 42(4 Suppl 4), 27–31.
Kordower JH, Freeman TB, Chen EY, Mufson EJ, Sanberg PR, Hauser RA, Snow B, Olanow CW, 1998. Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson's disease. Movement Disorders, 13, 383–393.
Kordower JH, Freeman TB, Snow BJ, Vingerhoets FJ, Mufson EJ, Sanberg PR, Hauser RA, Smith DA, Nauert GM, Perl DP, 1995. Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson's disease. New England Journal of Medicine, 332, 1118–1124.
Leenders KL, Palmer AJ, Quinn N, Clark JC, Firnau G, Garnett ES, Nahmias C, Jones T, Marsden CD, 1986. Brain dopamine metabolism in patients with Parkinson's disease measured with positron emission tomography. Journal of Neurology, Neurosurgery, & Psychiatry, 49, 853–860.
Levy R, Dostrovsky JO, Lang AE, Sime E, Hutchison WD, Lozano AM, 2001. Effects of apomorphine on subthalamic nucleus and globus pallidus internus neurons in patients with Parkinson's disease. Journal of Neurophysiology, 86, 249–260.
Limousin P, Brown RG, Jahanshahi M, Asselman P, Quinn NP, Thomas D, Obeso JA, Rothwell JC, 1999. The effects of posteroventral pallidotomy on the preparation and execution of voluntary hand and arm movements in Parkinson's disease. Brain, 122(Pt 2), 315–327.
Lin SC, Lin KJ, Hsiao IT, Hsieh CJ, Lin WY, Lu CS, Wey SP, Yen TC, Kung MP, Weng YH, 2014. In vivo detection of monoaminergic degeneration in early Parkinson disease by (18)F‐9‐fluoropropyl‐(+)‐dihydrotetrabenzazine PET. Journal of Nuclear Medicine, 55, 73–79.
Lloyd KG, Davidson L, Hornykiewicz O, 1975. The neurochemistry of Parkinson's disease: effect of L‐dopa therapy. Journal of Pharmacology and Experimental Therapeutics, 195, 453–464.
Luthman J, Fredriksson A, Sundström E, Jonsson G, Archer T, 1989. Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: motor behavior and monoamine alterations at adult stage. Behavioral Brain Research, 33, 267–277.
Mitchell IJ, Clarke CE, Boyce S, Robertson RG, Peggs D, Sambrook MA, Crossman AR, 1989. Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2‐deoxyglucose in monkeys exposed to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Journal of Neuroscience, 32, 213–226.
Moratalla R, Quinn B, DeLanney LE, Irwin I, Langston JW, Graybiel AM, 1992. Differential vulnerability of primate caudate‐putamen and striosome‐matrix dopamine systems to the neurotoxic effects of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Proceedings of the National Academy of Sciences of the United States of America, 89, 3859–3863
Morrish PK, Sawle GV, Brooks DJ, 1995. Clinical and [18F] dopa PET findings in early Parkinson's disease. Journal of Neurology, Neurosurgery, & Psychiatry, 59, 597–600.
Obeso JA, Rodríguez‐Oroz MC, Benitez‐Temino B, Blesa FJ, Guridi J, Marin C, Rodriguez M, 2008. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Movement Disorders, 23(Suppl 3), S548–S559.
Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, Lanciego JL, Artieda J, Gonzalo N, Olanow CW, 2000. Pathophysiology of the basal ganglia in Parkinson's disease. Trends in Journal of Neurosciences, 23(10 Suppl), S8–S19.
Papa SM, Desimone R, Fiorani M, Oldfield EH, 1999. Internal globus pallidus discharge is nearly suppressed during levodopa‐induced dyskinesias. Annals of Neurology, 46, 732–738.
Penney JB Jr, Young AB, 1986. Striatal inhomogeneities and basal ganglia function. Movement Disorders, 1, 3–15.
Perese DA, Ulman J, Viola J, Ewing SE, Bankiewicz KS, 1989. A 6‐hydroxydopamine‐induced selective parkinsonian rat model. Brain Research, 494, 285–293.
Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS, 2001. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Journal of Neuroscience, 106, 589–601.
Pirker W, 2003. Correlation of dopamine transporter imaging with parkinsonian motor handicap: how close is it? Movement Disorders, 18(Suppl 7), S43–S51.
Przedborski S, Levivier M, Jiang H, Ferreira M, Jackson‐Lewis V, Donaldson D, Togasaki DM, 1995. Dose‐dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6‐hydroxydopamine. Journal of Neuroscience, 67, 631–647.
Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ, 1999. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson's disease A 3D [(18)F]dopa‐PET study. Brain, 122(Pt 9), 1637–1650.
Reynolds GP, Garrett NJ, 1986. Striatal dopamine and homovanillic acid in Huntington's disease. Journal of Neural Transmission, 65, 151–155.
Rinne JO, Kuikka JT, Bergström KA, Rinne UK, 1995. Striatal dopamine transporter in different disability stages of Parkinson's disease studied with [(123)I]beta‐CIT SPECT. Parkinsonism & Related Disorders, 1, 47–51.
Rinne JO, Ruottinen H, Bergman J, Haaparanta M, Sonninen P, Solin O, 1999. Usefulness of a dopamine transporter PET ligand [(18)F]beta‐CFT in assessing disability in Parkinson's disease. Journal of Neurology, Neurosurgery, & Psychiatry, 67, 737–741.
Rozas G, López‐Martín E, Guerra MJ, Labandeira‐García JL, 1998. The overall rod performance test in the MPTP‐treated‐mouse model of Parkinsonism. Journal of Neuroscience Methods, 83, 165–175.
Snow BJ, Vingerhoets FJ, Langston JW, Tetrud JW, Sossi V, Calne DB, 2000. Pattern of dopaminergic loss in the striatum of humans with MPTP induced parkinsonism. Journal of Neurology, Neurosurgery, & Psychiatry, 68, 313–316.
Tissingh G, Bergmans P, Booij J, Winogrodzka A, van Royen EA, Stoof JC, Wolters EC, 1998. Drug‐naive patients with Parkinson's disease in Hoehn and Yahr stages I and II show a bilateral decrease in striatal dopamine transporters as revealed by [123I]beta‐CIT SPECT. Journal of Neurology, 245, 14–20.
Tolwani RJ, Jakowec MW, Petzinger GM, Green S, Waggie K, 1999. Experimental models of Parkinson's disease: insights from many models. Laboratory Animal Science, 49, 363–371.
Varastet M, Riche D, Maziere M, Hantraye P, 1994. Chronic MPTP treatment reproduces in baboons the differential vulnerability of mesencephalic dopaminergic neurons observed in Parkinson's disease. Journal of Neuroscience, 63, 47–56.
Widner H, Tetrud J, Rehncrona S, Snow B, Brundin P, Gustavii B, Björklund A, Lindvall O, Langston JW, 1992. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). New England Journal of Medicine, 327, 1556–1563.
Overall assessment of the AOP
A.1. Concordance of dose–response relationship
Data from experiments with the stressor compounds rotenone and MPTP (known inhibitors of the mitochondrial Complex I (CI)) reveal a good concordance of the dose–response relationships between the MIE and AO and within KEs. Although the different KEs have been measured using different methodologies, comparison of data from multiple in‐vitro/in‐vivo studies shows a general agreement in dose relationship (see Tables 1 and 2). There is a good consistency when comparing data on KE4 and the AO after exposure to rotenone and MPTP. However, in vivo rodent studies proved that only exposure to low concentrations of rotenone (rat brain concentration between 20–30 nM of rotenone; Betrabet et al., 2000) or MPTP (mice striatum concentration of approximately 12–47 μM MPP+; Fornai et al., 2005; Thomas et al., 2012) after chronic exposure (approximately 5 weeks) reproduced the anatomical, neurochemical behavioural and neuropathological features similar to the ones observed in Parkinson's disease (PD). Because of the variability of experimental protocols used, a clear no‐effect threshold could not be established; nevertheless, these brain concentrations of rotenone (20–30 nM) and MPP+ (approximately 12–47 μM) could serve as probabilistic thresholds for chronic exposure that could reproduce features of PD as both concentrations trigger approximately a 50% inhibition of Complex I (see Table 3). Generally, a strong response–response relationship is observed within studies. Some exceptions for this rule are observed between KE3/KE5 and KE4, likely because of the all biological complexity associated with these KEs. In this AOP, neuroinflammation was considered to have a direct effect on degeneration of DA neurons. However, it was not clear at which conditions it would become a modulatory factor and for practical reasons was not included in Tables 1, 2 and 3 but considered in the weight of evidence analysis.
A.2. Temporal concordance among the MIE, KEs and AO
There is a strong agreement that loss of DA neurons of the SNpc that project into the putamen is preceded by reduction in DA and degeneration of DA neuronal terminals in the striatum (Bernheimer et al., 1973). The clinical symptoms of a motor deficit are observed when 80% of striatal DA is depleted (Koller et al., 1992) and the sequence of pathological events leading to the adverse outcome has been well‐documented (O'Malley, 2010; Dexter et al., 2013; Fujita et al., 2014). Temporal concordance (see Tables 1 and 2) among the KEs can be observed in the experimental models of PD using the chemical stressors rotenone and MPTP (Betarbet, 2000, 2006; Sherer et al., 2003; Fornai et al., 2005). The acute administration of the chemical stressors can trigger a dose‐related change from the MIE to impaired proteostasis; however, to trigger KE4 (i.e. degeneration of DA neurons in SNpc with presence of intracytoplasmatic Lewy‐like bodies) and motor deficits (AO), proteostasis needs to be disturbed for a minimum period of time (Fornai et al., 2005).
Table A.8.
Response–Response and Temporality concordance table for the tool compound rotenone
| (a): Concentration at the target site | (b): KE1c (c): Inhibition of C I | (d): KE2c (e): Mitochondrial dysfunction | (f): KE3c (g): Impaired proteostasis | (h): KE4 (i): Degeneration of DA neurons of nigrostriatal pathway | (j): AO (k): Parkinsonian motor symptoms |
|---|---|---|---|---|---|
|
5–10 nM in‐vitro [1] |
+ 4–72 h [1] |
+ 4–72 h [4] |
+ 24 h [3] |
– | – |
|
20–30 nM ex‐vivo, rat brain concentration [4–5–2–6] |
++ 4–72 h (4–5) |
++ 4–72 h [4–5] |
++ 24 h [3–2–6] |
++a 5 weeks [2–6] |
+++b 5 weeks [2–6] |
|
100 nM in‐vitro [4] |
+++ 4–72 h [4] |
+++ 4–72 h [4] |
+++ 24 h [3] |
Above the maximum tolerated dose [2–6] | Above the maximum tolerated dose [2–6] |
References: Choi et al., 2008 [1]; Betarbet et al., 2006 [2]; Chou et al., 2010 [3]; Barrientos and Moraes 1999 [4]; Okun et al., 1999 [5]; Betarbet et al., 2000 [6].
–: No data available.
+: Low severity score, ++ intermediate severity score, +++ high severity score.
50% of treated animals showed loss of DA neurons in SNpc.
All animals affected in KE4 showed impaired motor symptoms.
KE 1, 2 and 3 showed a dose‐related severity in the effect and the score ++ was normalised vs. the KE4.
Table A.9.
Response–Response and Temporality concordance table for the tool compound MPTP/MPP+
| (l): Dose | (m): Brain concentration | (n): KE1e (o): Inhibition of C I | (p): KE2e (q): Mitochondrial dysfunction | (r): KE3d (s): Impaired proteostasis | (t): KE4 (u): Degeneration of DA neurons of nigrostriatal pathway | (v): AO (w): Parkinsonian motor symptoms |
|---|---|---|---|---|---|---|
| 1 mg/kg infusion [1] | – | – | – |
+ 4 weeks [1] |
+c 4 weeks [1] |
No effect |
| 5 mg/kg infusion [1] | – | – | – |
++ 4 weeks [1] |
++b 4 weeks [1] |
+++ 4 weeks [1] |
|
20–30 mg/kg infusion [2, 1] |
47 μM [2]f 12 μM [1] |
+++ 4 h [2] |
+++ 4 h [2] |
+++ 4 weeks [1] |
+++a 1–4 weeks [2, 1] |
+++ 4 weeks [1] |
References: Fornai et al., 2005 [1]; Thomas et al., 2012 [2].
–: No data available.
Approx 50% loss of DA neurons in SNpc.
Approx 30% loss of DA neurons SN pc.
No loss of DA neurons in SN pc. Reduced level of striata DA.
For KE3, a dose response effect was observed.
For KE 1 and 2 the severity of the effect was normalised vs. the KE4.
After single dose MPTP administration, brain concentration was approx. 5.15 μM.
A.3. Strength, consistency, and specificity of association of AO and MIE
Strength and consistency of the association of the AO with the MIE is strong. There is a large body of evidence from in‐vitro and in‐vivo studies with chemical stressors, showing association between the MIE that triggers an inhibition of CI and the AO (Betarbet et al., 2000, 2006; Sherer et al., 2003; Fornai et al., 2005; Thomas et al., 2012). Human data also suggest a link between inhibition of CI and AO (Schapira et al., 1989; Greenamyre et al., 2001; Shults, 2004). Using the two different chemical stressors, rotenone and MPTP, data are consistent and the pattern of activation of the MIE leading of the AO is similar. For rotenone and MPTP, specificity is high; however, there are many inhibitors of the mitochondrial CI without evidence of triggering the AO. When considering these chemicals specificity is low; therefore, kinetic and metabolic considerations should be taken into account to fully demonstrate specificity for these compounds.
A.4. Weight of Evidence (WoE)
A.4.1. Biological plausibility, coherence, and consistency of the experimental evidence
The biological plausibility of this AOP is considered strong overall. When using multiple stressors in different studies and assays, the coherence and consistency of the experimental data is well established. Furthermore, in‐vivo and in‐vitro studies are also in line with the human evidence from PD patients. In addition, although the mechanistic understanding of parkinsonian disorders (and PD in particular) are not fully clear, the KEs and KERs described in this AOP are considered critical for the development of the disease (Dauer et al., 2003; Shulman et al., 2011; Dexter et al., 2013; Fujita et al., 2015).
Table A.10.
Biological Plausibility of KERs; WoE analysis
| Support for biological plausibility of KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Is there a mechanistic (i.e. structural or functional) relationship between KEup and KEdown consistent with established biological knowledge? | Extensive understanding of the KER based on extensive previous documentation and broad acceptance | The KER is plausible based on analogy to accepted biological relationships, but scientific understanding is not completely established | There is empirical support for a statistical association between KEs but the structural or functional relationship between them is not understood | |
|
MIE => KE1 Binding of inhibitor to NADH‐ubiquinone oxidoreductase leads of complex I |
Strong | Rationale: As describe in this KER there is an extensive understanding of the functional relationship between binding of an inhibitor to NADH‐ubiquinone oxidoreductase (CI) and its inhibition. Different complex I ligands, both naturally occurring, like rotenone (from Derris scandens), piericidin A (from Streptomyces mobaraensis), acetogenins (from various Annonaceae species) and their derivatives, and synthetically manufactured like pyridaben and various piperazin derivatives inhibit the catalytic activity of complex I (Degli Esposti, 1994; Barrientos and Moraes, 1999; Betarbet et al., 2000; Ichimaru et al., 2008) | ||
|
KE1 => KE2 Inhibition of complex I leads to mitochondrial dysfunction |
Strong | Rationale: There is extensive understanding of the mechanisms explaining how the inhibition of complex I lead to mitochondrial dysfunction (i.e. failure to produce ATP, increase in production of ROS etc). It is well documented that CI inhibition is one of the main sites at which electron leakage to oxygen occurs resulting in oxidative stress (Greenamyre et al., 2001; lauren et al., 2010; Efremov and Sazanow, 2011). These pathological mechanisms are well studied as they are used as readouts for evaluation of mitochondrial dysfunction (Graier et al., 2007; Martin, 2011; Braun, 2012; Correia et al., 2012; Cozzolino et al., 2013) | ||
|
KE2 => KE3 Mitochondrial dysfunction results in impaired proteostasis |
Moderate | Rationale: The weight of evidence supporting the biological plausibility behind the relationship between mitochondrial dysfunction and impaired proteostasis, including the impaired function of UPS and ALP that results in decreased protein degradation and increase protein aggregation is well documented but not fully understood. It is well established that the two main mechanisms that normally remove abnormal proteins (UPS and ALP) rely on physiological mitochondrial function. The role of oxidative stress, due to mitochondrial dysfunction, burdens the proteostasis with oxidised proteins and impairs the chaperone and the degradation systems. This leads to a vicious circle of oxidative stress inducing further mitochondrial impairment (McNaught and Jenner, 2001; Powers et al., 2009; Zaltieri et al., 2015). Therefore, the interaction of mitochondrial dysfunction and UPS/ALP deregulation plays a pivotal role in the pathogenesis of PD (Sherer et al., 2002; Fornai et al., 2005; Pan et al., 2008; Dagda et al., 2013) | ||
|
KE2 => KE4 Mitochondrial dysfunction leads to the degeneration of dopaminergic neurons of the nigrostriatal pathway |
Strong | Rationale: Mitochondrial are essential for ATP production, ROS management, calcium homeostasis and control of apoptosis. Mitochondrial homeostasis by mitophagy is also an essential process for cellular maintenance (Fujita et al., 2014). Because of their anatomical and physiological characteristics, SNpc DA neurons are considered more vulnerable than other neuronal populations (Surmeier et al., 2010; Sulzer et al., 2013). Mechanistic evidence of mutated proteins relate the mitochondrial dysfunction in familial PD with reduced calcium capacity, increased ROS production, increase in mitochondrial membrane permeabilisation and increase in cell vulnerability (Gandhi et al., 2009; Koopman et al., 2012). Human studies indicate mitochondrial dysfunction in human idiopathic PD cases in the substantia nigra (Parker et al., 1989, 2008; Swerdlow et al., 1996; Keeney et al., 2006). In addition, systemic application of mitochondrial neurotoxicants such as rotenone or MPTP leads to a preferential loss of nigrostriatal DA neurons (Langston et al., 1983) | ||
|
KE3 => KE4 Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
Moderate | Rationale: It is well known that impaired proteostasis refers to misfolded and aggregated proteins including alpha‐synuclein, deregulated axonal transport of mitochondria and impaired trafficking of cellular organelles. Evidences are linked to PD and experimental PD models as well as from genetic studies (McNaught et al., 2001, 2003; Arnold, 2011; Rappold et al., 2014; Tieu et al., 2014). Strong evidence for degeneration of the nigrostriatal pathway comes from the experimental manipulations that directly induce the same disturbances of proteostasis as observed in PD patients (e.g. viral mutated alpha‐synuclein expression) or in chronic rotenone/MPTP models trigger degeneration of the nigrostriatal pathway (Betarbet et al., 2000, 2006; Kirk et al., 2003; Fornai et al., 2005). However, a clear mechanistic proof for the understanding of the exact event triggering cell death is lacking. There is only moderate evidence showing that interventions that correct disturbances of proteostasis after exposure to rotenone would prevent neuronal degeneration and that the disturbances of proteostasis correlate quantitatively under many conditions with the extent of nigrostriatal neuronal degeneration | ||
|
KE4 ⇔ KE5 Neuroinflammation |
Moderate | Rationale: The fact that reactive glial cells (microglia and astrocytes) may kill neurons is well accepted. The mechanisms underlying this effect may include the release of cytotoxic signals (e.g. cytokines) or production of ROS and RNS (Chao et al., 1995; Brown and Bal‐Price, 2003; Kraft and Harry, 2011; Taetzsch and Block, 2013). However, the responsible mediators differ from model to model. The fact that neuronal injury/death can trigger neuroinflammation is supported by evidence in human and experimental models. The evidence that neuroinflammation triggered by neuronal damage can cause neuronal death (vicious circle), is mostly indirect or by analogy (Griffin et al., 1998; McGeer and McGeer, 1998; Blasko et al., 2004; Cacquevel et al., 2004; Hirsch and Hunot, 2009; Tansey and Goldberg, 2009; Barbeito et al., 2010; Rubio‐Perez and Morillas‐Ruiz, 2012; Thundyil and Lim, 2014) | ||
|
KE4 => AO Degeneration of DA neurons of the nigrostriatal pathway leads to parkinsonian motor symptoms |
Strong | Rationale: The mechanistic understanding of the regulatory role of striatal DA in the extrapyramidal motor control system is well established. The loss of DA in the striatum is characteristic of all aetiologies of PD and is not observed in other neurodegenerative diseases (Bernheimer et al., 1973; Reynolds et al., 1986). Characteristic motor symptoms such as bradykinesia, tremor, or rigidity are manifested when more than 80% of striatal DA is depleted as a consequence of SNpc DA neuronal degeneration (Koller et al., 1992) | ||
A.4.2. Essentiality
Essentiality of KEs for this AOP is strong. There is ample evidence from knock out animal models, engineered cells or replacement therapies that blocking, preventing or attenuating an upstream KE is mitigating the AO. In addition, there is experimental support for the KERs as multiple studies performed with modulating factors that attenuate (particularly with antioxidants) or augment (e.g. overexpression of viral‐mutated α‐synuclein) a KE show that such interference leads to an increase of KEdown or the AO.
Table A.11.
Essentiality of KEs; WoE analysis
| Support for essentiality of KEs | Defining question: Are downstream KEs and/or the AO prevented if an upstream KE is blocked? | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs (e.g. stop/reversibility studies, antagonism, knock out models, etc.) | Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE leading to increase in KEdown or AO | No or contradictory experimental evidence of the essentiality of any of the KEs | ||
|
KE1 Inhibition of complex I |
Strong | Rationale: Inactivation of the Ndufs 4 gene (knockout mice) that produces CI deficiency causes encephalomyopathy, including ataxia and loss of motor skills (Kruse et al., 2008). NDI1‐transducted SK‐N‐MC cells expressing the rotenone‐insensitive single subunit NADH dehydrogenase of yeast (NDI1) that acts as a replacement for the entire CI in mammalian cells were completely resistant to 100 nM rotenone‐mediated cell death (at 48 h of exposure) indicating that rotenone – induced toxicity requires rotenone biding of CI (Sherer et al., 2003). In all rotenone models, mitochondria CI is inhibited at the dose that cause neurodegeneration (Betarbet et al., 2000, 2006) | ||
|
KE2 Mitochondrial dysfunction |
Strong | Rationale: Many studies showing that antioxidants protect the cells against rotenone or MPTP induced oxidative stress are published (Wu et al., 1994; Sherer et al., 2003; Saravanan et al., 2006; Li et al., 2010; Kim‐Han et al., 2011; Tseng et al., 2014; Chen et al., 2015; Chiu et al., 2015; Lu et al., 2015; Nataraj et al., 2015). This provides (indirect) evidence for essentiality of KE2, if production of ROS is assumed as direct consequence/sign of mitochondrial dysfunction. Additional evidence comes from experiments with overexpression or activation of antioxidative enzymes (e.g. SOD or ALDH2), which also prevent rotenone and MPTP induced neurotoxicity (Mudo et al., 2012; Ciu et al., 2015). Furthermore, promotion of mitochondrial fusion or blocking of mitochondrial fission prevents or attenuates rotenone and MPTP induced neurotoxicity (Tieu et al., 2014) | ||
|
KE3 Impaired proteostasis |
Moderate |
Rationale: Indirect evidence for the role of disturbed alpha‐synuclein proteostasis: Lacking of alpha‐synuclein expression in mice prevented induction of behavioural symptoms, neuronal degeneration in the nigrostriatal pathway and loss of DA neurons after chronic treatment with MPTP (Dauer et al., 2002; Fornai et al., 2004). Injection of adeno/lenti‐associated virus that expresses wild‐type or mutant α‐syn into rat, mice or non‐human primate SN produced loss of dopaminergic neurons, but the effect is not easily reproduced in transgenic mice overexpressing alpha‐synuclein (Kirk, 2002, 2003; Klein, 2002; Lo Bianco, 2002; Lauwers, 2003) Rationale for the role of autophagy: Early dendritic and axonal dystrophy, reduction of striatal dopamine content, and the formation of somatic and dendritic ubiquitinated inclusions in DA neurons were prevented by ablation of Atg7 (an essential autophagy gene (Friedman et al., 2012)) Rationale for the role of UPS/ALP: Protection from DA neuronal death was also observed in multiple experiments through the pharmacological modulation of the UPS, ALP system; however, there are also contradicting data in the literature (Fornai et al., 2003, 2005; Inden et al., 2007; Zhu et al., 2007; Dehay et al., 2010) However, although many lines of evidence exist to support essentiality of impaired proteostasis, a single molecular chain of events cannot be established |
||
|
KE4 Degeneration of DA neurons of nigrostriatal pathway |
Strong |
Rationale: Clinical and experimental evidences show that the pharmacological replacement of the DA neurofunction by allografting fetal ventral mesencephalic tissues is successfully replacing degenerated DA neurons resulting in the total reversibility of motor deficit in animal model and partial effect is observed in human patient for PD (Freed et al., 1990; Henderson et al., 1991; Lopez‐Lozano et al., 1991; Spencer et al., 1992; Widner et al., 1992; Peschanski et al., 1994) Also, administration of L‐DOPA or DA agonists results in an improvement of motor deficits (Calne et al., 1970; Fornai et al., 2005). The success of these therapies in man as well as in experimental animal models clearly confirms the causal role of dopamine depletion for PD motor symptoms (Cotzias et al., 1969; Matsumoto et al., 1976; Narabayashi et al., 1984; Kelly et al., 1987; Freed et al., 1990; Henderson et al., 1991; Lopez‐Lozano et al., 1991; Spencer et al., 1992; Widner et al., 1992; Peschanski et al., 1994; Uitti et al., 1996, 1997; Silva et al., 1997; Lang et al. 1998; Scott et al., 1998; De Bie et al., 1999; Walter et al., 2004; Deuschl et al., 2006; Ferrari‐Tonielli et al., 2008; Fasano et al., 2010; Castrito et al., 2011; Connolly et al., 2014; Liu et al., 2014; Moldovan et al., 2015) Furthermore, experimental evidence from animal models of PD and from in‐vitro systems indicate that prevention of apoptosis through ablation of BCL‐2 family genes prevents or attenuates neurodegeneration of DA neurons (Offen et al., 1998; Dietz et al., 2002). Rotenone treated rats developed bradykinesia, postural instability and/or rigidity consequently to a 45% loss of TH+ neurons in SN and a commensurate loss of striatal dopamine and the clinical signs were reversed by apomorphine, consistent with a lesion of the nigrostriatal dopamine system (Cannon et al., 2009) |
||
|
KE5 Neuroinflammation |
Moderate | Rationale: Following treatment with Rotenone or MPP+, protection of DA neurons and terminals was observed in vivo and in vitro by inhibiting different feature of neuroinflammation (microglia/astrocyte); however, inhibition was different in different models and considered as an indirect evidence of essentiality (Dehmer et al., 2000; Gao et al., 2002, 2003, 2015; Feng et al., 2002; Ferger et al., 2004; Wu et al., 2005; Wang et al., 2006; Purisai et al., 2007; Zhou et al., 2007; Chao et al., 2009; Rojo et al., 2010; Chung et al., 2011; Ros‐Bernal et al., 2011; Qian et al., 2011; Liu et al., 2012, 2015; Mangano et al., 2012; Sathe et al., 2012; Yadav et al., 2012; Salama et al., 2012; Borrajo et al., 2013; Chang et al., 2013; Emmrich et al., 2013; Bodea et al., 2014; Khan et al., 2014; Sriram et al., 2014; Wang et al., 2014; Brzozowski et al., 2015). It should be noted that this KE, depending on the situation, can be bypassed | ||
A.4.3. Empirical support
Empirical support is strong. Many studies show evidence for the KERs by showing temporal concordance and dose concordance when using different stressors.
Table A.12.
Empirical support for the KERs; WoE analysis
| Empirical support for KERs | Defining question: Does the empirical evidence support that a change in the KEup leads to an appropriate change in the KEdown? Does KEup occur at lower doses and earlier time points than KEdown and is the incidence of KEup higher than that for KEdown? Are inconsistencies in empirical support cross taxa, species and stressors that don't align with expected pattern of hypothesised AOP? | High (Strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Multiple studies showing dependent change in both exposure to a wide range of specific stressors (extensive evidence for temporal, dose–response and incidence concordance) and no or few critical data gaps or conflicting data | Demonstrated dependent change in both events following exposure to a small number of specific stressors and some evidence inconsistent with expected pattern that can be explained by factors such as experimental design, technical considerations, differences among laboratories, etc | Limited or no studies reporting dependent change in both events following exposure to a specific stressor (i.e. endpoints never measured in the same study or not at all); and/or significant inconsistencies in empirical support across taxa and species that don't align with expected pattern for hypothesised AOP | ||
|
MIE => KE1 Binding of inhibitor to NADH‐ubiquinone oxidoreductase leads to partial or total inhibition of complex I |
Strong | Rationale: The inhibition of complex I is well documented in a variety of studies using isolated mitochondria or cells as well as in in vivo experiments and in human post mortem PD brains. In many experiments using different inhibitors i.e. rotenone and MPTP, the observed relationship between the two events was temporal, response and dose concordant (Grivennikova et al., 1997; Barrientos and Moraes, 1999; Okun et al., 1999; Betarbet et al., 2000, 2006; Koopman et al., 2007; Choi et al., 2008) | ||
|
KE1 => KE2 Inhibition of complex I leads to mitochondrial dysfunction |
Strong | Rationale: There is a large amount of studies showing that the inhibition of CI inhibition results in mitochondrial dysfunctions in a response and dose dependent manner (Barriento and Moraes, 1999) | ||
|
KE2 => KE3 Mitochondrial dysfunction results in impaired proteostasis |
Strong | Rationale: Based on the existing in vitro and in vivo data it is suggested that mitochondrial dysfunction impairs protein homeostasis (impairment of the UPS and ALP system) through oxidative and nitrosative stress resulting in accumulation of misfolded proteins (including α‐synuclein), disruption of microtubule assembly and damaged intracellular transport of proteins and cell organelles. A number of studies performed with chemical stressors showed evidence of temporal, response and dose concordance (Chou et al., 2010; Betarbet et al., 2000, 2006; Fornai et al., 2005) | ||
|
KE2 => KE4 Mitochondrial dysfunction directly leads to degeneration of DA neurons of nigrostriatal pathway |
Strong | Rationale: Multiple in vitro studies indicate dose and response–response concordance. As most of the studies were conducted in vitro, the temporal concordance is difficult to establish; however, can be expected based on the well know temporal sequence of the two KEs. (Swedlow et al., 1996; Jha et al., 2000; Sherer et al., 2003, 2007; Chinta et al., 2006; Marella et al., 2008; Hajieva et al., 2009; Du et al., 2010; Jana et al., 2011; Wen et al., 2011; Choi et al., 2014; Park et al., 2014) | ||
|
KE3 => KE4 Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
Strong | Rationale: The empirical support linking impaired proteostasis with degeneration of DA neurons of the nigrostriatal pathway is strong and comes from in‐vivo and in‐vitro studies performed with different stressor (i.e. Rotenone, MPTP or proteasome inhibitors) and post‐mortem human evidences in PD patients supporting a causative link between the two key events. Temporal, effect and dose concordance was established in a number of experiments (Betabret et al., 2000, 2006; Fornai et al., 2003, 2005) | ||
|
KE4 <=> KE5 Neuroinflammation directly leads to degeneration of DA neurons of the nigrostriatal pathway |
Moderate | Rationale: multiple in vivo and in vitro experiments support the link between neuroinflammation and degeneration of DA neurons in the nigrostriatal pathway as well as vice versa. The observation of concomitant presence of glial and astrocytic cells and degenerated/degenerating DA neurons is also reported in many studies with a good temporal and response concordance | ||
|
KE4 => AO Degeneration of DA neurons of nigrostriatal pathway leads to parkinsonian motor symptoms |
Strong | Rationale: The experimental support linking the degeneration of DA neurons of nigrostriatal pathways with the manifestation of motor symptoms of PD comes from human in vivo observations as well as from monkey, mice and rat in vivo models exposed to an experimental toxin i.e. rotenone and MPTP. Observations in human allow defining correlation between the levels of striatal DA with the onset of motor dysfunction (Bernheimer et al., 1973; Lloyd et al., 1975; Hornykiewicz et al., 1986). Temporal, effect and dose concordance comes from studies performed in multiple animal species following administration of rotenone and MPTP (Lloyd et al., 1975; Bezard et al., 2001; Petroske et al., 2001; Alvarez‐Fischer et al., 2008; Cannon et al., 2009; Blesa et al., 2012) | ||
A.5. Uncertainties and Inconsistencies
There is no strict linear relationship between inhibitor binding and reduced mitochondrial function. Low doses of rotenone that inhibit CI activity partially do not alter mitochondrial oxygen consumption. Therefore, bioenergetics defect cannot account alone for rotenone‐induced neurodegeneration. Instead, under such conditions, rotenone neurotoxicity may result from oxidative stress (Betarbet et al., 2000). Few studies used human brain cells/human brain mitochondria. Therefore, full quantitative data for humans are not available.
It is molecularly unclear how rotenone binding alter CI function, switching it to ROS production. It is still unclear whether the site of superoxide production in CI inhibited mitochondria is complex I itself or not (Singer and Ramsay, 1994).
Some studies suggest that rotenone and MPTP may have effects other than CI inhibition, e.g. MPTP and rotenone can induce microtubule disruption (Brinkley et al., 1974; Cappelletti et al., 1999, 2001; Ren et al., 2005; Feng, 2006; Aguilar et al., 2015)
There are additional feedback possible between KEs, e.g. ROS production from KE2 may damage CI, this leads to enhancement of KE1.
Some KEs e.g. KE 2, 3, 5 pool molecular processes that may need to be evaluated individually at a later stage.
The exact molecular link from mitochondrial dysfunction to disturbed proteostasis is still unclear (Malkus et al., 2009; Zaltieri et al., 2015).
The role of ATP depletion vs. other features of mitochondrial dysfunction is not clear.
The role of a α‐synuclein in neuronal degeneration is still unclear as well as the mechanisms leading to its aggregation.
It is not clear under which conditions KE3 and KE5 become modulatory factors, and when they are essential. MPTP can induce damage to nigrostriatal neurons without formation of Lewy bodies (Forno 1986, 1993; Dauer 2003). Similarly, discontinuous administration of rotenone, even at high doses, damages the basal ganglia but produce no inclusions (Heikkila et al., 1985; Ferrante et al., 1997; Lapontine 2004). To reproduce the formation of neuronal inclusions, continuous infusion of MPTP or rotenone is necessary. Acute intoxication with rotenone seems to spare dopaminergic neurons (Ferrante 1997; Dauer et al., 2003). In addition, in rats chronically infused with rotenone showed a reduction in striatal DARPP‐32‐positive, cholinergic and NADPH diaphorase‐positive neurons (Hoglinger, 2003) or in other brain regions. These results would suggest that Rotenone can induce a more widespread neurotoxicity (Aguilar, 2015) or the model is not reproducible in all laboratories.
Inconsistent effects of MPP+ on autophagy (up or down regulation) are reported (Dauer et al., 2002, Drolet et al., 2004). There is conflicting literature on whether increased autophagy would be protective or enhances damage. Similarly, a conflicting literature exists on extent of inhibition or activation of different protein degradation system in PD and a clear threshold of onset is unknown (Fornai et al., 2005; Malkus et al., 2009).
The selective vulnerability of the SN pc dopaminergic pathway does not have a molecular explanation.
Priority of the pattern leading to cell death could depend on concentration, time of exposure and species sensitivity; these factors have to be taken into consideration for the interpretation of the study's result and extrapolation of potential low‐dose chronic effect as this AOP refers to long‐time exposure.
The model of striatal DA loss and its influence on motor output ganglia does not allow to explain specific motor abnormalities observed in PD (e.g. resting tremor vs bradykinesia) (Obeso et al., 2000). Other neurotransmitters (Ach) may play additional roles. Transfer to animal models o PD symptoms is also representing an uncertainty.
There are some reports indicating that in subacute rotenone or MPTP models (non‐human primates), a significant, sometimes complete, recovery of motor deficits can be observed after termination of toxicant treatment. The role of neuronal plasticity in intoxication recovery and resilience is unclear.
This AOP is a linear sequence of KEs. However, mitochondrial dysfunction (and oxidative stress) and impaired proteostasis are influencing each other and this is considered an uncertainties (Malkus et al., 2009).
A.6. Quantitative Considerations
The quantitative understanding of this AOP includes a clear response–response relationship and the identification of a threshold effect. The WoE analysis clearly supports the qualitative AOP as a means to identify and characterise the potential of a chemical to induce DA neuronal loss and the AO. Importantly, both the AO and the KE4 are considered relevant regulatory endpoints for this AOP. The empirical evidence supports existence of a response–response relationship. This response–response is likely triggered by a the brain concentrations of approximately 20–30 nM and 17–47 μM of rotenone and MPP+ respectively and both concentrations trigger approx. a 50% inhibition of mitochondrial complex I and this could be considered as a ‘threshold’. However, a more detailed dose–response analysis for each KE is lacking as well as it is not clear which temporal relationship exists for lower CI inhibitory effects. It is clear from the analysis of the AOP that for the identification of these AOs, the design of the in‐vivo studies should be tailored as to a MIE which leads to a long‐lasting perturbation of the KEs. This provides the most specific and definite context to trigger neuronal death. To observe KEs relevant for this AOP, new endpoints need to be introduced. Although a dose, response and temporal relationship is evident for most KEs, the quantitative relationship between impaired proteostasis and degeneration of DA neurons has yet to be elucidated. Moving from a qualitative AOP to quantitative AOP would need a clear understanding of effect thresholds and this is still representing a major hurdle for several KEs of this AOP.
Table A.13.
Concordance table for the tool compounds rotenone and MPTP/MPP+
| Concentration | KE1 Inhibition of C I | KE2 Mitochondrial dysfunction | KE3 Impaired proteostasis | KE4 Degeneration of DA neurons of nigrostriatal pathway | AO Parkinsonian motor symptoms |
|---|---|---|---|---|---|
|
Rotenone 20–30 nM rat brain concentration [1–2] |
Approx. 53% [4–5] |
Approx. 20–53% (decrease in respiration rate) [1–2] |
Approx. 20–60% (decrease in UPS (26S) activity) [3] |
Neuronal loss (50% of animal affected) [2] |
Motor impairment (100% of animals with neuronal loss) [2] |
|
MPP+ 12–47 μM rat brain concentration [4–5] |
Approx. 50–75% [5] |
Approx. 38% (reduction in phosphorylating respiration) [5] |
Approx. 60% (decrease in UPS activity) [4] |
Approx. 50% of neuronal loss [4–5] |
Motor impairment [4] |
References: [1] Okun et al. (1999); [2] Barrientos and Moraes (1999); [3] Borland et al. (2008); [4] Thomas et al. (2012); [5] Betarbet et al., (2000, 2006).
A.7. Applicability of the AOP
This proposed AOP is neither sex‐dependent nor associated with certain life stage; however, aged animals may be more sensitive. The relevance of this AOP during the developmental period has not been investigated.
In vivo testing has no species restriction. The mouse was the species most commonly used in the experimental models conducted with the chemical stressors; though experimental studies using alternative species have been also performed. (Johnson et al., 2015). However, animal models (rodents in particular) would have limitations as they are poorly representative of the long human life‐time as well as of the human long‐time exposure to the potential toxicants. Human cell‐based models would likely have better predictivity for humans than animal cell models. In this case, toxicokinetics information from in‐vivo studies would be essential to test the respective concentrations in‐vitro on human cells.
A.8. Schematic summary of the AOP
Chronic, low level of exposure to environmental chemicals that inhibit complex I could result in mitochondrial dysfunction and oxidative stress, triggering proteasomal dysfunction strongly implicated in parkinsonian disorders, including aggregation/modifications in α‐synuclein protein and organelles trafficking. These cellular key events cause DA terminals degeneration in striatum and progressive cell death of DA neurons in SNpc. Important to notice that at each step, the effects become regionally restricted such that systemic complex I inhibition eventually results in highly selective degeneration of the nigrostriatal pathway.
Figure A.19.
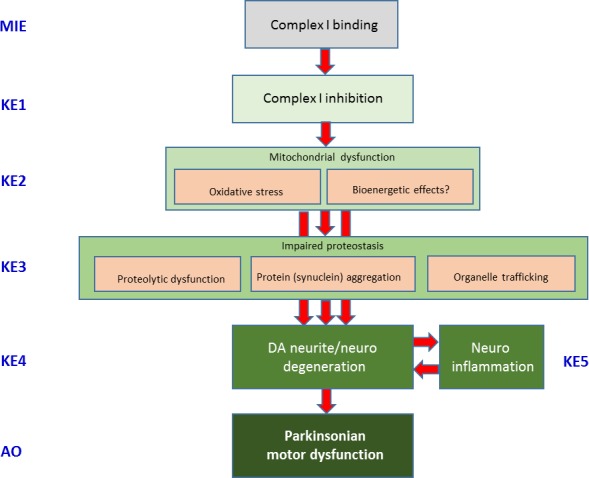
Schematic summary of the AOP
A.9. Potential application of the AOP
This AOP has been developed in order to evaluate the biological plausibility that the adverse outcome i.e. parkinsonian motor deficits, is linked to a MIE that can be triggered by chemical substances i.e. pesticides and chemicals in general. The relevance of the AOP has been documented by tools compounds known to trigger the described AOP. By means of using a human health outcome that has been shown in epidemiological studies to be association with pesticide exposure, the authors intend to draw attention on this AO in the process of hazard identification. This AOP can be used to support the biological plausibility of this association during the process of evaluation and integration of the epidemiological studies into the risk assessment. It is biologically plausible that a substance triggering the pathway, can be associated with the AO and ultimately with the human health outcome, pending the MoA analysis. In addition, this AOP can be used to support identification of data gaps that should be explored when a chemical substance is affecting the pathway. Moreover, the AOP provides a scaffold for recommendations on the most adequate study design to investigate the apical endpoints. It is important to note that, although the AO is defined in this AOP as parkinsonian motor deficits, degeneration of DA neurons is already per se an adverse outcome even in situations where it is not leading to parkinsonian motor deficits, and this should be taken into consideration for the regulatory applications of this AOP.
The MIE and KEs identified in this AOP could serve as a basis for assays development that could contribute to an AOP informed‐IATA construction which can be applied for different purposes such as: screening and prioritisation of chemicals for further testing, hazard characterisation or even risk assessment when combined with exposure and ADME information.
References
Aguilar JS, Kostrzewa RM, 2015. Neurotoxin mechanisms and processes relevant to parkinson's disease: an update. Neurotoxicity Research. doi: 10.1007/s12640‐015‐9519‐y
Alvarez‐Fischer D, Guerreiro S, Hunot S, Saurini F, Marien M, Sokoloff P, Hirsch EC, Hartmann A, Michel PP, 2008. Modelling Parkinson‐like neurodegeneration via osmotic minipump delivery of MPTP and probenecid. Journal of Neurochemistry, 107, 701–711. doi: 10.1111/j.1471‐4159.2008.05651.x
Arnold, B., et al. 2011. “Integrating multiple aspects of mitochondrial dynamics in neurons: age‐related differences and dynamic changes in a chronic rotenone model.” Neurobiology of Disease, 41, 189–200.
Barbeito AG, Mesci P, Boillee S, 2010. Motor neuron‐immune interactions: the vicious circle of ALS. Journal of Neural Transmission, 117, 981–1000.
Barrientos A, and Moraes CT, 1999. Titrating the effects of mitochondrial Complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197.
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F, 1973. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. Journal of the Neurological Sciences, 20, 415–455
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Betarbet R, Canet‐Aviles RM, Sherer TB, Mastroberardino PG,Mc Lendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinfelter G, Cookson MR, Greenmyre JT, 2006. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ‐1, α‐synuclein, and the ubiquitin‐proteasome system. Neurobiology Disease, 22, 404–420.
Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE, 2001. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐lesioned macaque model of Parkinson's disease. Journal of Neuroscience, 21, 6853–6861.
Blasko I, Stampfer‐Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck‐Loebenstein B, 2004. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell, 3, 169–176.
Blesa J, Pifl C, Sánchez‐González MA, Juri C, García‐Cabezas MA, Adánez R, Iglesias E, Collantes M, Peñuelas I, Sánchez‐Hernández JJ, Rodríguez‐Oroz MC, Avendaño C, Hornykiewicz O, Cavada C, Obeso JA, 2012. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiology of Disease, 48, 79–91.
Bodea LG, Wang Y, Linnartz‐Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al. 2014. Neurodegeneration by activation of the microglial complement‐phagosome pathway. Journal of Neuroscience, 34, 8546–8556.
Borrajo A, Rodriguez‐Perez AI, Villar‐Cheda B, Guerra MJ, Labandeira‐Garcia JL, 2014. Inhibition of the microglial response is essential for the neuroprotective effects of Rho‐kinase inhibitors on MPTP‐induced dopaminergic cell death. Neuropharmacology, 85, 1–8.
Brinkley BR, Barham SS, Barranco SC, and Fuller GM, 1974. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Experimental Cell Research, 85, 41–46.
Brown GC, Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355.
Braun RJ, 2012. Mitochondrion‐mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Frontiers in Oncology, 2, 182.
Brzozowski MJ, Jenner P, Rose S, 2015. Inhibition of i‐NOS but not n‐NOS protects rat primary cell cultures against MPP(+)‐induced neuronal toxicity. Journal of Neural Transmission, 122, 779–788.
Cacquevel M, Lebeurrier N, Cheenne S, Vivien D, 2004. Cytokines in neuroinflammation and Alzheimer's disease. Current Drug Targets, 5, 529–534.
Calne DB, Sandler M, 1970. L‐Dopa and Parkinsonism. Nature, 226, 21–24.
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT, 2009. A highly reproducible rotenone model of Parkinson's disease. Neurobiology of Disease, 34, 279–290.
Cappelletti G, Maggioni MG, Maci R, 1999. Influence of MPP+ on the state of tubulin polymerisation in NGF‐differentiated PC12 cells. Journal of Neuroscience, 56, 28–35.
Cappelletti G, Pedrotti B, Maggioni MG, Maci R, 2001. Microtubule assembly is directly affected by MPP(+)in vitro. Cell Biology International, 25, 981–984.
Castrioto A, Lozano AM, Poon YY, Lang AE, Fallis M, Moro E, 2011. Ten‐year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Archives of Neurology 68, 1550–1556.
Chang CY, Choi DK, Lee DK, Hong YJ, Park EJ, 2013. Resveratrol confers protection against rotenone‐induced neurotoxicity by modulating myeloperoxidase levels in glial cells. Public Library of Science (PLoS ONE), 8, e60654.
Chao YX, He BP, Tay SS, 2009. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson's disease. Journal of Neuroimmunology, 216, 39–50.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP, 2015. Anti‐oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Molecular Neurodegeneration, 10, 4.
Chinta SJ, Andersen JK, 2006. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radical Biology and Medicine, 41, 1442–1448.
Choi WS, Kruse SE, Palmiter R, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP, or paraquat. Proceedings of the National Academy of Sciences, 105, 15136–15141.
Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ, 2014. Celastrol from ‘Thunder God Vine’ protects SH‐SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochemical Research, 39, 84–96.
Chiu CC, Yeh TH, Lai SC, Wu‐Chou YH, Chen CH, Mochly‐Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS, 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone‐induced cellular and animal models of parkinsonism. Experimental Neurology, 263, 244–253.
Chou AP, Li S, Fitzmaurice AG, Bronstein JM, 2010. Mechanisms of rotenone‐induced proteasome inhibition. NeuroToxicology, 31, 367–372.
Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, et al. 2011. Fluoxetine prevents MPTP‐induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology, 60, 963–974.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA, 2012. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Advances in Experimental Medicine and Biology, 724, 205–221.
Cotzias GC, Papavasiliou PS, Gellene R, 1969. L‐dopa in parkinson's syndrome. New England Journal of Medicine, 281, 272.
Cozzolino M, Ferri A, Valle C, Carri MT, 2013. Mitochondria and ALS: implications from novel genes and pathways. Molecular and Cellular Journal of Neuroscience, 55, 44–49.
Dagda RK, Banerjee TD, Janda E, 2013. How Parkinsonian toxins dysregulate the autophagy machinery. International Journal of Molecular Sciences, 14, 22163–22189.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson‐Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R, 2002. Resistance of alpha ‐synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences U S A, 99, 14524–14529.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson‐Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R, 2002. Resistance of alpha ‐synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences U S A, 99, 14524–14529.
Dauer W, Przerdborski S, 2003. Parkinson's disease: Mechanisms and models. Neuron, 39, 889–909.
De Bie RM, de Haan RJ, Nijssen PC, Rutgers AW, Beute GN, Bosch DA, Haaxma R, Schmand B, Schuurman PR, Staal MJ, Speelman JD, 1999. Unilateral pallidotomy in Parkinson's disease: a randomised, single‐blind, multicentre trial. Lancet, 354, 1665–1669.
Degli Esposti M, Ghelli A, 1994. The mechanism of proton and electron transport in mitochondrial complex I. Biochimica et Biophysica Acta, 1187, 116–120.
Dehay B, Bove J, Rodriguez‐Muela N, Perier C, Recasens A, Boya P, Vila M, 2010. Pathogenic lysosomal depletion in Parkinson's disease. Journal of Neuroscience Methods, 30, 12535–12544.
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB, 2000. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. Journal of Neurochemistry, 74, 2213–2216.
Deuschl G, Schade‐Brittinger C, Krack P, Volkmann J, Schäfer H, Bötzel K, Daniels C, Deutschländer A, Dillmann U, Eisner W, Gruber D, Hamel W, Herzog J, Hilker R, Klebe S, Kloss M, Koy J, Krause M, Kupsch A, Lorenz D, Lorenzl S, Mehdorn HM, Moringlane JR, Oertel W, Pinsker MO, Reichmann H, Reuss A, Schneider GH, Schnitzler A, Steude U, Sturm V, Timmermann L, Tronnier V, Trottenberg T, Wojtecki L, Wolf E, Poewe W, Voges J, German Parkinson Study Group, Neurostimulation Section, 2006. A randomized trial of deep‐brain stimulation for Parkinson's disease. New England Journal of Medicine, 355, 896–908.
Dexter DT, Jenner P, 2013. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biology and Medicine 62, 132–144.
Dietz GPH, Stockhausen KV, Dietz B et al. 2008. Membrane‐permeable Bcl‐xL prevents MPTP‐induced dopaminergic neuronal loss in the substantia nigra. Journal of Neurochemistry, 104, 757–765. doi: 10.1111/j.1471‐4159.2007.05028.x
Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL, 2004. Mice lacking α‐synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology, 25, 761–769.
Du T, Li L, Song N, Xie J, Jiang H, 2010. Rosmarinic acid antagonized 1‐methyl‐4‐phenylpyridinium (MPP+)‐induced neurotoxicity in MES23.5 dopaminergic cells. International Journal of Toxicology, 29, 625–633.
Efremov RG, Sazanov LA, 2011. Respiratory complex I: ‘steam engine’ of the cell? Current Opinion in Structural Biology, 21, 532–540. doi: 10.1016/j.sbi.2011.07.002
Efremov RG, Sazanov LA, 2011. Structure of the membrane domain of respiratory complex I. Nature, 476, 414–420. doi: 10.1038/nature10330
Emmrich JV, Hornik TC, Neher JJ, Brown GC, 2013. Rotenone induces neuronal death by microglial phagocytosis of neurons. The FEBS Journal, 280, 5030–5038.
Fasano A, Romito LM, Daniele A, Piano C, Zinno M, Bentivoglio AR, Albanese A, 2010. Motor and cognitive outcome in patients with Parkinson's disease 8 years after subthalamic implants. Brain, 133, 2664–2676.
Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B, et al., 2002. Cyclooxygenase‐2‐deficient mice are resistant to 1‐methyl‐4‐phenyl1, 2, 3, 6‐tetrahydropyridine‐induced damage of dopaminergic neurons in the substantia nigra. Journal of Neuroscience Letters, 329, 354–358.
Feng J, 2006. Mictrotubule. A common target for parkin and Parkinson's disease toxins. Neuroscientist, 12, 469–476.
Ferger B, Leng A, Mura A, Hengerer B, Feldon J, 2004. Genetic ablation of tumor necrosis factor‐alpha (TNF‐alpha) and pharmacological inhibition of TNF‐synthesis attenuates MPTP toxicity in mouse striatum. Journal of Neurochemistry, 89, 822–833.
Ferrante RJ, Schulz JB, Kowall NW, Beal MF, 1997. Systematic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Research, 753, 157–162.
Ferrari‐Toninelli G, Bonini SA, Cenini G, Maccarinelli G, Grilli M, Uberti D, Memo M, 2008. Dopamine receptor agonists for protection and repair in Parkinson's disease. Current Topics in Medicinal Chemistry, 8, 1089–1099.
Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z, 2012. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α‐synuclein and LRRK2 in the brain. The Journal of Neuroscience, 32, 7585–7593.
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti C, Ruffoli R, Soldani P, Ruggieri S, Alessandri’ MG, Paparelli A, 2003. Fine structure and mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. The Journal of Neuroscience, 23, 8955–8966.
Fornai F, Lenzi p, Gesi M, et al., 2004. “Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells.” Journal of Neurochemistry, 88, 114–123.
Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC, 2005. Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitinproteasome system and _α‐synuclein. Proceedings of the National Academy of Sciences, 102, 3413–3418.
Freed CR, Breeze RE, Rosenberg NL, Schneck SA, Wells TH, Barrett JN, Grafton ST, Huang SC, Eidelberg D, Rottenberg DA, 1990. Transplantation of human fetal dopamine cells for Parkinson's disease. Results at 1 year. Archives of Neurology, 47, 505–512.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Gandhi S, Wood‐Kaczmar A, Yao Z, et al. 2009. PINK1‐associated Parkinson's disease is caused by neuronal vulnerability to calcium‐induced cell death. Molecular Cell, 33, 627–638.
Gao HM, Hong JS, Zhang W, Liu, 2002. Distinct role for microglia in rotenone‐induced degeneration of dopaminergic neurons. Journal of Neuroscience, 22, 782–790.
Gao HM, Liu B, Hong JS, 2003. Critical role for microglial NADPH oxidase in rotenone‐induced degeneration of dopaminergic neurons. Journal of Neuroscience, 23, 6181–6187.
Gao L, Brenner D, Llorens‐Bobadilla E, Saiz‐Castro G, Frank T, Wieghofer P, et al., 2015. Infiltration of circulating myeloid cells through CD95L contributes to neurodegeneration in mice. Journal Of Experimental Medicine, 212, 469–480.
Graier WF, Frieden M, Malli R, 2007. Mitochondria and Ca2+ signaling: old guests, new functions. Pflügers Archiv, 455, 375–396.
Greenamyre JT, Sherer TB, Betarbet R, Panov AV, 2001. Critical Review Complex I and Parkinson's Disease Life, 52, 135–141.
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al., 1998. Glial‐neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathology, 8, 65–72.
Grivennikova VG, Maklashina EO, Gavrikova EV, Vinogradov AD, 1997. Interaction of the mitochondrial NADH‐ubiquinone reductase with rotenone as related to the enzyme active/inactive transition. Biochimica et Biophysica Acta, 1319, 223–232.
Hajieva P, Mocko JB, Moosmann B, Behl C, 2009. Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. Journal of Neurochemistry, 110, 118–132.
Hornykiewicz O, Kish SJ, 1987. Biochemical pathophysiology of parkinson's disease. In: Yahr M and Bergmann KJ (eds.). Parkinson's Disease. Energy Dispersive Spectroscopy. Raven Press, New York, pp. 19–34.
Jana S, Sinha M, Chanda D, Roy T, Banerjee K, Munshi S, Patro BS, Chakrabarti S, 2011. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson's disease. Biochimica et Biophysica Acta, 1812, 663–673.
Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK, 2000. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. Journal of Biological Chemistry, 275, 26096–26101.
Johnson ME, Bobrovskaya L, 2015. An update on the rotenone models of parkinson's disease: Their ability to reproduce features of clinical disease and model gene‐environment interactions, 946, 101–116.
Heikkila RE, Nicklas WJ, Vyas I, Duvoisin RC, 1985. Dopaminergic toxicity of rotenone and the 1‐methyl‐4‐phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine toxicity. Journal of Neuroscience Letters, 62, 389–394.
Henderson BT, Clough CG, Hughes RC, Hitchcock ER, Kenny BG, 1991. Implantation of human fetal ventral mesencephalon to the right caudate nucleus in advanced Parkinson's disease. Archives of Neurology, 48, 822–827.
Hirsch EC, Hunot S, 2009. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurology, 8, 382–397.
Hoglinger GU, Feger J, Annick P, Michel PP, Karine P, Champy P, Ruberg M, Wolfgang WO, Hirsch E, 2003. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. Journal of Neurochemistry, 84, 1–12.
Ichimaru N, Murai M, Kakutani N, Kako J, Ishihara A, Nakagawa Y, Miyoshi H, 2008. Synthesis and Characterization of New Piperazine‐Type Inhibitors for Mitochondrial NADH‐Ubiquinone Oxidoreductase (Complex I). Biochemistry, 47, 10816–10826.
Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, Taira T, Ariga H, Shimohama S, 2007. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4‐phenylbutyrate, a chemical chaperone. Journal of Neurochemistry, 101, 1491–1494.
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr, 2006. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. Journal of Neuroscience, 10, 26, 5256–5264.
Kelly PJ, Ahlskog JE, Goerss SJ, Daube JR, Duffy JR, Kall BA, 1987. Computer‐assisted stereotactic ventralis lateralis thalamotomy with microelectrode recording control in patients with Parkinson's disease. Mayo Clinic Proceedings, 62, 655–664.
Khan MM, Kempuraj D, Zaheer S, Zaheer A, 2014. Glia maturation factor deficiency suppresses 1‐methyl‐4‐phenylpyridinium‐induced oxidative stress in astrocytes. Journal of Molecular Neuroscience, 53, 590–599.
Kim‐Han JS, Dorsey JA, O'Malley KL, 2011. The parkinsonian mimetic MPP+, specifically impairs mitochondrial transport in dopamine axons. The Journal of Neuroscience, 31, 7212–7221.
Kirk D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel R, Bijorklund A, 2002. Parkinson‐like neurodegeneration induced by targeted overexpression of α‐synuclein in the nigrostriatal system. 22, 2780–2791.
Kirk D, Annett L, Burger C, Muzyczka N, Mandel R, Bijorklund A, 2003. Nigrostriatal α‐synucleinopathy induced by viral vector‐mediated overexpression of human α‐synuclein: a new primate model of parkinson's disease. Proceedings of the National Academy of Sciences, 100, 2884–2889.
Klein RL, King MA, Hamby ME, Meyer EM, 2002. Dopaminergic cell loss induced by human A30P α‐synuclein gene transfer to the rat substantia nigra. Human Gene Therapy, 13, 605–612.
Koller WC, 1992. When does Parkinson's disease begin? Neurology, 42(4 Suppl 4), 27–31.
Koopman W, Hink M, Verkaart S, Visch H, Smeitink J, Willems P, 2007. Partial complex I inhibition decreases mitochondrial motility and increases matrix protein diffusion as revealed by fluorescence correlation spectroscopy. Biochimica et Biophysica Acta, 1767, 940–947.
Koopman W, Willems P, 2012. Monogenic mitochondrial disorders. New England Journal of Medicine, 366, 1132–1141. doi: 10.1056/NEJMra1012478
Kraft AD, Harry GJ, 2011. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International Journal of Environmental Research And Public Health, 8, 2980–3018.
Lang AE, Lozano AM, 1998. Parkinson's disease. Second of two parts. New England Journal of Medicine, 339, 1130–1143.
Langston JW, ballard P, Irwin I, 1983. Chronic parkinsonism in human due to a product of meperidine‐analog synthesis. Science, 219, 979–980.
Lapointe N, StHilaire M, martinoli MG, Blanchet J, gould P, Rouillard C, Cicchetti F, 2004. Rotenone induces non‐specific central nervous system and systemic toxicity. The Federation of American Societies for Experimental Biology (FASEB)Journal express article 10.1096/fj.03‐0677fje
Lauwers E, Debyser Z, Van Drope J, DeStrooper B, Nuttin B, 2003. Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector‐mediated overexpression of α‐synuclein. Brain Pathology, 13, 364–372.
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, et al. 2012. alpha7 nicotinic acetylcholine receptor‐mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. Journal of Neuroinflammation, 9, 98.
Liu Y, Li W, Tan C, Liu X, Wang X, Gui Y, Qin L, Deng F, Hu C, Chen L, 2014. Meta‐analysis comparing deep brain stimulation of the globus pallidus and subthalamic nucleus to treat advanced Parkinson disease. Journal on Neurosurgery, 121, 709–718.
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, et al. 2015. Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress‐induced apoptosis: implications for Parkinson's disease. Neuropharmacology, 91, 87–96.
Liu W, Kong S, Xie Q, Su J, Li W, Guo H, Li S, Feng X, Su Z, Xu Y, Lai X, 2015. Protective effects of apigenin against 1‐methyl‐4‐phenylpyridinium ion induced neurotoxicity in PC12 cells. International Journal of Molecular Medicine, 35, 739–746.
Lloyd KG, Davidson L, Hornykiewicz O, 1975. The neurochemistry of Parkinson's disease: effect of L‐dopa therapy. Journal of Pharmacology and Experimental Therapeutics, 195, 453–464.
Lo Bianco C, Ridet JL, Deglon N, Aebischer P, 2002. Alpha‐synucleopathy and selective dopaminergic neuron loss in a rat lentiviral‐based model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 10813–10818.
López‐Lozano JJ, Bravo G, Abascal J, 1991. Grafting of perfused adrenal medullary tissue into the caudate nucleus of patients with Parkinson's disease. Clinica Puerta de Hierro Neural Transplantation Group. Journal on Neurosurgery, 75, 234–243.
Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, et al. 2012. Interferon‐gamma plays a role in paraquat‐induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiology of Aging, 33, 1411–1426.
Marella M, Seo BB, Nakamaru‐Ogiso E, Greenamyre JT, Matsuno‐Yagi A, Yagi T, 2008. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. Public Library of Science (PLoS ONE), 3, e1433.
Martin LJ, 2011. Mitochondrial pathobiology in ALS. Journal of Bioenergetics and Biomembranes, 43, 569–579.
Matsumoto K, Asano T, Baba T, Miyamoto T, Ohmoto T, 1976. Long‐term follow‐up results of bilateral thalamotomy for parkinsonism. Applied neurophysiology, 39, 257–260.
McGeer PL, McGeer EG, 1998. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Disease and Associated Disorders, 12(Suppl. 2), S1–S6.
McNaught KS, Jenner P, 2001. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Journal of Neuroscience Letters, 297, 191–194.
McNaught KSC, Olanow W, Halliwell B, 2001. Failure of the ubiquitine‐proteasome system in parkinson's disease. Nature Rev. Journal of Neuroscience, 2, 589–594.
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW, 2003. Altered proteasomal function in sporadic Parkinson's disease. Experimental Neurology, 179, 38–46.
Moldovan AS, Groiss SJ, Elben S, Südmeyer M, Schnitzler A, Wojtecki L, 2015. The treatment of Parkinson's disease with deep brain stimulation: current issues. Neural Regeneration Research, 10, 1018–1022.
Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D, 2012. Transgenic expression and activation of PGC‐1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Science Journal, 69, 1153–1165.
Narabayashi H, Yokochi F, Nakajima Y, 1984. Levodopa‐induced dyskinesia and thalamotomy. Journal of Neurology, Neurosurgery, & Psychiatry, 47, 831–839.
Nataraj J, Manivasagam T, Justin Thenmozhi A, Essa MM, 2015. Lutein protects dopaminergic neurons against MPTP‐induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutritional Journal of Neuroscience 2015 Mar 2. [Epub ahead of print].
Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, Lanciego JL, Artieda J, Gonzalo N, Olanow CW, 2000. Pathophysiology of the basal ganglia in Parkinson's disease. Trends in Journal of Neurosciences. 23(10 Suppl), S8–S19.
Offen D, Beart PM, Cheung NS et al. 1998. Transegnic mice expressing human Bcl‐2 in their neurons are resistant to 6‐hydroxydopamine and 1‐methyl‐4‐phenyl‐1,2,3,6‐ tetrahydropyridine neurotoxicity. Proceedings of the National Academy of Sciences, 95, 5789–5794.
O'Malley KL, 2010. The role of axonopathy in Parkinson's disease. 2010. Experimental Neurobiology, 19, 115–119.
Okun JG, Lümmen P and Brandt U, 1999. Three classes of inhibitors share a common binding domain in mitochondrial Complex I (NADH:Ubiquinone Oxidoreductase). Journal of Biological Chemistry, 274, 2625–2630. doi: 10.1074/jbc.274.5.2625
Pan T, Kondo S, Le W, Jankovic J, 2008. The role of autophagy‐lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain, 131, 1969–1978.
Park SE, Sapkota K, Choi JH, Kim MK, Kim YH, Kim KM, Kim KJ, Oh HN, Kim SJ, Kim S, 2014. Rutin from Dendropanax morbifera Leveille protects human dopaminergic cells against rotenone induced cell injury through inhibiting JNK and p38 MAPK signaling. Neurochemical Research, 39, 707–18.
Parker WD Jr, Boyson SJ, Parks JK, 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Annals of Neurology, 26, 719–723.
Peschanski M, Defer G, N'Guyen JP, Ricolfi F, Monfort JC, Remy P, Geny C, Samson Y, Hantraye P, Jeny R, 1994. Bilateral motor improvement and alteration of L‐dopa effect in two patients with Parkinson's disease following intrastriatal transplantation of foetal ventral mesencephalon. Brain, 117 (Pt 3), 487–499.
Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS, 2001. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Journal of Neuroscience, 106, 589–601.
Powers ET1, Morimoto RI, Dillin A, Kelly JW, Balch WE, 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annual Review of Biochemistry, 78, 959–991.
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat‐induced dopaminergic cell degeneration. Neurobiology of Disease, 25, 392–400.
Parker WD Jr, Parks JK, Swerdlow RH, 2008. Complex I deficiency in Parkinson's disease frontal cortex. Brain Research, 1189, 215–218.
Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, et al. 2011. beta2‐adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. Journal of Immunology, 186, 4443–4454.
Rappold PM, et al., 2014. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nature Communications, 5, 5244. doi: 10.1038/ncomms6244
Ren Y, et al., 2005. Selective vulnerability of dopaminergic neurons to microtubule depolymerisation. Journal of Biological Chemistry, 280, 434105–434112.
Reynolds GP, Garrett NJ, 1986. Striatal dopamine and homovanillic acid in Huntington's disease. Journal of Neural Transmission, 65, 151–155.
Rojo AI, Innamorato NG, Martin‐Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A, 2010. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia, 58, 588–598.
Ros‐Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, et al., 2011. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proceedings of the National Academy of Sciences USA, 108, 6632–6637.
Rubio‐Perez JM, Morillas‐Ruiz JM, 2012. A review: inflammatory process in Alzheimer's disease, role of cytokines. Scientific World Journal, 2012, 756357.
Salama M, Helmy B, El‐Gamal M, Reda A, Ellaithy A, Tantawy D, et al., 2013. Role of L‐thyroxin in counteracting rotenone induced neurotoxicity in rats. Environmental Toxicology and Pharmacology, 35, 270–277.
Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP, 2006. L‐deprenyl protects against rotenone‐induced, oxidative stress‐mediated dopaminergic neurodegeneration in rats. Neurochemistry International, 49, 28–40.
Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al., 2012. S100B is increased in Parkinson's disease and ablation protects against MPTP‐induced toxicity through the RAGE and TNF‐alpha pathway. Brain, 135(Pt 11), 3336–3347.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, and Marsden CD, 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet, 1, 1269.
Scott R, Gregory R, Hines N, Carroll C, Hyman N, Papanasstasiou V, Leather C, Rowe J, Silburn P, Aziz T, 1998. Neuropsychological, neurological and functional outcome following pallidotomy for Parkinson's disease. A consecutive series of eight simultaneous bilateral and twelve unilateral procedures. Brain, 121(Pt 4), 659–675.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT, 2002. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha‐synuclein metabolism and oxidative damage. Journal of Neuroscience, 22, 7006–7015.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al., 2003. Mechanism of toxicity in rotenone models of Parkinson's disease. Journal of Neuroscience, 23, 10756–10764.
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno‐Yagi A, Miller GW, Greenamyre JT, 2007. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. Journal of Neurochemistry, 100, 1469–1479.
Shulman JM, DeJager PL, Feany MB, 2011. Parkinson's disease: genetics and pathogenesis. Annual Review of Pathology Mechanisms of Disease, 6, 193–222
Shults CW, 2004. Mitochondrial dysfunction and possible treatments in Parkinson's disease–a review. Mitochondrion, 4, 641–648.
Singer TP, Ramsay RR, 1994.The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochimica et Biophysica Acta, 1187, 198–202.
Spencer DD, Robbins RJ, Naftolin F, Marek KL, Vollmer T, Leranth C, Roth RH, Price LH, Gjedde A, Bunney BS, 1992. Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson's disease. New England Journal of Medicine, 327, 1541–1548
Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP, 2002. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. Federation of American Societies for Experimental Biology (FASEB), 16, 1474–1476.
Sulzer D, Surmeier DJ, 2013. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Movement Disorders, 28, 715–724.
Surmeier DJ, Guzman JN, Sanchez‐Padilla J, Goldberg JA, 2010. What causes the death of dopaminergic neurons in Parkinson's disease? Prog Brain Research, 183, 59–77. doi: 10.1016/S0079‐6123(10)83004
Silva MA, Mattern C, Häcker R, Tomaz C, Huston JP, Schwarting RK, 1997. Increased neostriatal dopamine activity after intraperitoneal or intranasal administration of L‐DOPA: on the role of benserazide pretreatment. Synapse, 27, 294–302.
Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP Jr, Davis RE, Parker WD Jr, 1996. Origin and functional consequences of the complex I defect in Parkinson's disease. Annals of Neurology, 40, 663–671.
Taetzsch T, Block ML, 2013. Pesticides, microglial NOX2, and Parkinson's disease. Journal of Biochemical and Molecular Toxicology, 27, 137–149.
Tansey MG, Goldberg MS, 2009. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease.
Thundyil J, Lim KL, 2014. DAMPs and Neurodegeneration. Ageing research reviews.
Thomas B, Banerjee R, Starkova NN, Zhang S, Calingasan NY, Yang L, Wille E, Lorenzo B, Ho D, Beal M, Starkov A, 2012. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxidants & Redox Signaling, 16, 855–868.
Tieu Kim, Imm Jennifer, 2014. Mitochondrial dynamics as potential therapeutic target for Parkinson's disease? Advances in Clinical Neuroscience and Rehabilitation, 14, 6–8.
Tseng YT, Chang FR, Lo YC, 2014. The Chinese herbal formula Liuwei dihuang protects dopaminergic neurons against Parkinson's toxin through enhancing antioxidative defense and preventing apoptotic death. Phytomedicine, 21, 724–733.
Uitti RJ, Ahlskog JE, 1996. Comparative review of dopamine receptor agonists in Parkinson's Disease. CNS Drugs, 5, 369–388.
Uitti RJ, Wharen RE Jr, Turk MF, Lucas JA, Finton MJ, Graff‐Radford NR, Boylan KB, Goerss SJ, Kall BA, Adler CH, Caviness JN, Atkinson EJ, 1997. Unilateral pallidotomy for Parkinson's disease: comparison of outcome in younger versus elderly patients. Neurology, 49, 1072–1077.
Walter BL, Vitek JL, 2004. Surgical treatment for Parkinson's disease. Lancet Neurology, 3, 719–728.
Widner H, Tetrud J, Rehncrona S, Snow B, Brundin P, Gustavii B, Björklund A, Lindvall O, Langston JW, 1992. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). New England Journal of Medicine, 327, 1556–1563.
Wu RM, Mohanakumar KP, Murphy DL, Chiueh CC, 1994. Antioxidant mechanism and protection of nigral neurons against MPP+ toxicity by deprenyl (selegiline). Annals of the New York Academy of Sciences, 738, 214–221.
Wang Q, Chu CH, Oyarzabal E, Jiang L, Chen SH, Wilson B, et al. 2014. Subpicomolar diphenyleneiodonium inhibits microglial NADPH oxidase with high specificity and shows great potential as a therapeutic agent for neurodegenerative diseases. Glia, 62, 2034–2043.
Wang T, Zhang W, Pei Z, Block M, Wilson B, Reece JM, et al. 2006. Reactive microgliosis participates in MPP+‐induced dopaminergic neurodegeneration: role of 67 kDa laminin receptor. Federation of American Societies for Experimental Biology (FASEB), 20, 906–915.
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH, 2011. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. Journal of Biological Chemistry, 286, 16504–16515.
Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, et al., 2005. The role of microglia in paraquat‐induced dopaminergic neurotoxicity. Antioxidants & Redox Signaling, 7, 654–661.
Yadav S, Gupta SP, Srivastava G, Srivastava PK, Singh MP, 2012. Role of secondary mediators in caffeine‐mediated neuroprotection in maneb‐ and paraquat‐induced Parkinson's disease phenotype in the mouse. Neurochemical Research, 37, 875–884.
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A, 2015. Mitochondrial dysfunction and α‐synuclein synaptic pathology in parkinson's disease: who's on first? Journal of Parkinson's Disease, 2015, 108029.
Zhou F, Wu JY, Sun XL, Yao HH, Ding JH, Hu G, 2007. Iptakalim alleviates rotenone‐induced degeneration of dopaminergic neurons through inhibiting microglia‐mediated neuroinflammation. Neuropsychopharmacology, 32, 2570–2580.
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT, 2007. Regulation of autophagy by extracellular signal‐regulated protein kinases during 1‐methyl‐4‐phenylpyridinium‐induced cell death. American Journal of Pathology, 170, 75–86.
AOP 2: Redox‐cycling of a chemical initiated by electrons released by the mitochondrial respiratory chain leading to parkinsonian motor deficits
Abstract
This Adverse Outcome Pathway (AOP) describes the linkage between excessive ROS production at the level of the mitochondrial respiratory chain and parkinsonian motor deficits, including Parkinson's disease (PD). Interaction of a compound with complex I and/or III of the mitochondrial respiratory chain has been defined as the molecular initiating event (MIE) that triggers mitochondrial dysfunction, impaired proteostasis, which then cause degeneration of dopaminergic (DA) neurons of the nigra‐striatal pathway. These causatively linked cellular key events result in motor deficit symptoms, typical of parkinsonian disorders including PD, described in this AOP as an Adverse Outcome (AO). This AOP also includes neuroinflammation as a KE and is intending the KER with degeneration of dopaminergic neurons as a causative link but the priority of the temporal sequence is not defined as neurodegeneration can be the cause as well the consequence of the KE neuroinflammation.
Since the role DA neurons of the Substantia Nigra pars compacta (SNpc) projecting into the striatum is essential for motor control, the key events refer to these two brain structures, i.e. SNpc and striatum. The weight‐of‐evidence supporting the relationship between the described key events is mainly based on effects observed after an exposure to the well‐known pesticide paraquat which will be used as a tool chemical to support this AOP.
Schematic representation of the proposed AOP:
Figure A.20.
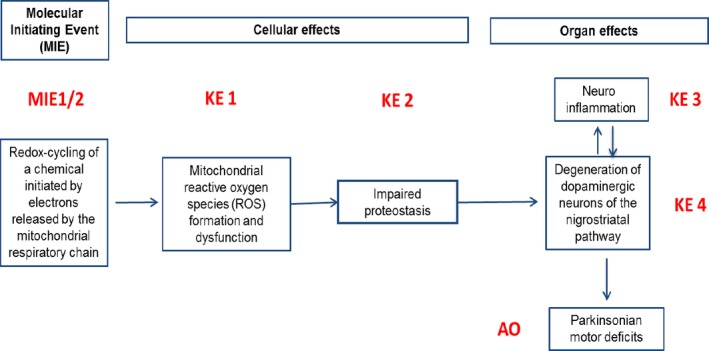
AOP scheme
MIE: Redox cycling of a chemical initiated by electrons released by the mitochondrial respiratory chain
How this Key Event works:
Redox cycling is a process of alternate reduction and reoxidation steps. It is triggered in the presence of chemicals able to accept an electron from a reductant to form free radicals (Figure A.21). These radicals due to their high reactivity may undergo electron transfer to molecular oxygen generating superoxide anion radical (O2 °−) (Kappus, 1986). As a result of electron transfer, the parent compound is regenerated and able to catalyse further O2 .− production. Extent and direction of this reaction depend on both the concentration of the reactants and their reduction potentials relative to the O2/ O2 .− (E0= −160 mV at pH7 for a standard state of 1M O2; Sawer and Valentine, 1981). Compounds with more negative electron reduction potential will react faster being thus effective redox cycler. In addition, very negative E0 limit the pool of possible reductants, which have a sufficiently low reduction potential to donate an electron.
Figure A.21.
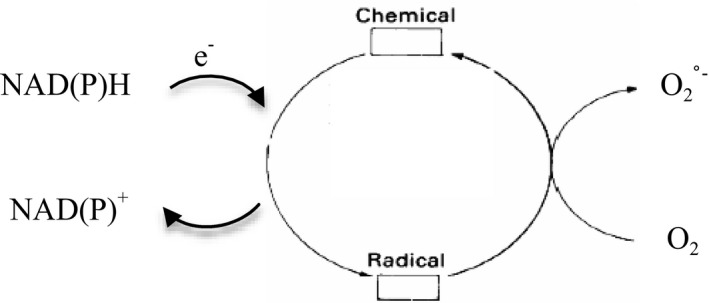
Schematic representation of the mechanism of chemicals redox cycling. (Adapted by permission from Macmilllan Publishers Ltd, Cohen and Doherty, 1987, copyright (1987))
Chemicals radicalisation appears to be the consequence of one electron reduction often catalysed by a flavoprotein (Cohen and Doherty, 1987). A number of different enzymes are involved, including mitochondrial reductases.
Electron transport through the mitochondrial respiratory chain (oxidative phosphorylation) is mediated by five multimeric complexes (I–V) that are embedded in the mitochondrial inner membrane (Figure A.22).
Figure A.22.
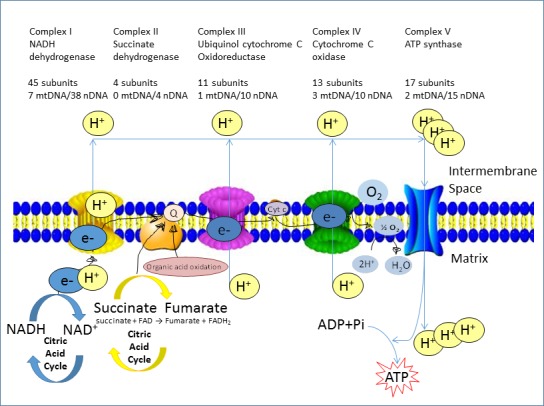
The electron transport chain in the mitochondrion. Complex I (NADH‐coenzyme Q reductase or NADH dehydrogenase) accepts electrons from NADH and serves as the link between glycolysis, the citric acid cycle, fatty acid oxidation and the electron transport chain. Complex II also known as succinate‐coenzyme Q reductase or succinate dehydrogenase, includes succinate dehydrogenase and serves as a direct link between the citric acid cycle and the electron transport chain. The coenzyme Q reductase or Complex III transfers the electrons from CoQH2 to reduce cytochrome c which is the substrate for Complex IV (cytochrome c reductase). Complex IV transfers the electrons from cytochrome c to reduce molecular oxygen into water. Finally, this gradient is used by the ATP synthase complex (Complex V) to make ATP via oxidative phosphorylation
Under physiological conditions 1–5% of the oxygen is converted to O2 .− by mitochondria due to electron leakage from the respiratory chain (Wei et al., 2001). Although different respiratory complexes and individual mitochondrial enzymes are sources of O2 .− (Figure A.23), leaking electron are primarily produced at two discrete points in the electron‐transport chain namely at CI (NADH) and CIII (ubiquinone‐cytochrome c reductase) (Selivanov et al., 2011).
Figure A.23.
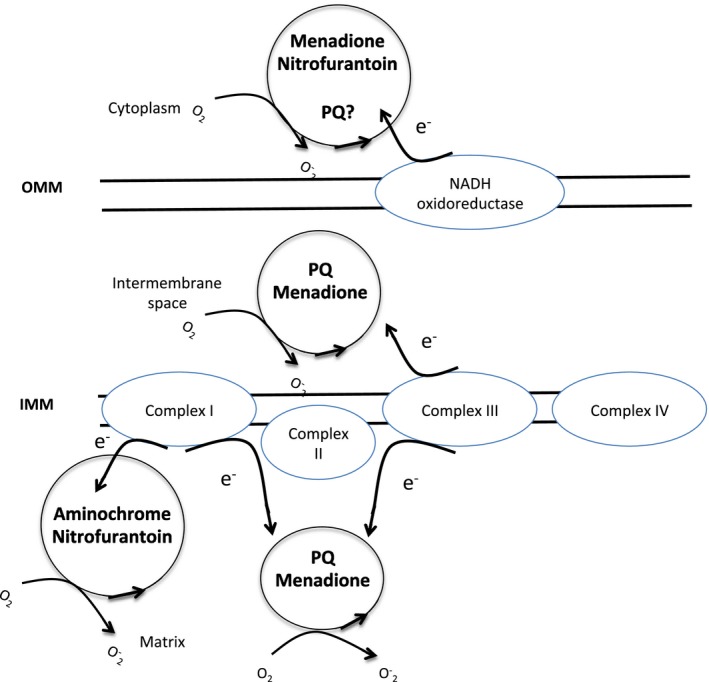
Chemical redox cycling in mitochondria. Complex I and Complex III start PQ redox cycle in bovine heart and brain mitochondria respectively, while the involvement of outer mitochondrial membrane NADH‐oxidoreductase is controversial. OMM: outer mitochondrial membrane, IMM: inner mitochondrial membrane
NADH‐ubiquinone oxidoreductase is the Complex I (CI) of electron transport chain (ETC). It is a large assembly of proteins that spans the inner mitochondrial membrane. In mammals, it is composed of about 45–47 protein subunits (human 45) of which 7 are encoded by the mitochondrial genome (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6) and the remainder by the nuclear genome (Greenamyre, 2001). Complex I oxidises NADH elevating the NAD+/NADH ratio by transferring electrons via a flavin mononucleotide (FMN) cofactor and several iron‐sulfur centres to ubiquinone (Friedrich et al., 1994).
Complex III (CIII) of the ETC is the ubiquinol cytochrome C oxidoreductase, or coenzyme Q reductase. Like CI, CIII is also an assembly of multiple proteins spanning the inner mitochondrial membrane. One of the 11 CIII subunits is encoded by mtDNA, while nuclear DNA codes the remaining 10 proteins. CIII transfers electrons from CoQH2 to reduce cytochrome C, which is the substrate for Complex IV (Figure A.1).
In the presence of a redox cycling chemical, the leaking electrons from these complexes are readily accepted and transferred to molecular oxygen starting the redox cycling and boosting O2 .− production.
How it is measured or detected
Redox cycling of a chemical can be measured directly or indirectly by different methods.
A.1. Direct detection of redox cycling by electron paramagnetic resonance (EPR):
A radical with an unpaired electron, like the PQ•+ radical, has a distinctive EPR spectrum because of the delocalisation of the unpaired electron across the conjugated ring system. Thus, it can be measured by EPR, which is a sensitive, specific method for studying radicals formed in chemical reactions and the reactions themselves (Schweiger and Jeschke, 2001 Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press). An example of an EPR spectrum of the PQ•+ radical is shown in Figure A.24.
Figure A.24.
Detection and quantification of the PQ þ radical by EPR spectroscopy. (A) Typical EPR spectrum of the PQ þ radical (100 mM; trace (a)) generated in vitro by reduction of PQ 2þ with a twofold excess of sodium dithionite. EPR signal of the SO 2 radical present in the dithionite solution (10 mM; trace (b)). Modified after Cochemé and Murphy, 2009 Methods in Enzymology, Copyright (2009)) Cochemé and Murphy, 2009
A.2. Direct detection of chemical radical formation by spectrophotometry:
Each chemical radical with a distinct absorbance spectrum than the parent compound can be measured spectrophotometrically in isolated mitochondria. However, due to the fast reaction of the chemical radical with oxygen, these measures have to be performed under anaerobic conditions (Cochemé and Murphy, 2009).
A.3. Direct detection of chemical radical formation (aromatic cations) by selective electrodes:
Selective electrodes were constructed and used for measuring the concentration of lipophilic cations in real time in mitochondrial incubations (Brand, 1995; Murphy and Smith, 2007; Cochemé and Murphy, 2009).
A.4. Direct detection of superoxide anione formation
The methods for superoxide detection are described by Grivennikova and Vinogradov (2013). A range of different methods is also described by BioTek (http://www.biotek.com/resources/articles/reactive-oxygen-species.html). The reduction of ferricytochrome c to ferrocytochrome c may be used to assess the rate of superoxide formation (McCord, 1968). Like in other superoxide assays, specificity can only be obtained by measurements in the absence and presence of superoxide dismutase. Oxidation of hydroethidine (HE) to 2‐OH‐E+, together with non specific oxidation to ethidium and dimeric ethidium products to exclude the formation of oxidants other than superoxide, is also used as an indicator of superoxide anion formation (Dranka et al., 2012). Chemiluminescent reactions have been used for their increased sensitivity with lucigenin or coelenterazine as substrates. Hydrocyanine dyes are fluorogenic sensors for superoxide and hydroxyl radical, and they become membrane impermeable after oxidation (trapping at sit of formation). The best characterised of these probes are Hydro‐Cy3 and Hydro‐Cy5. Generation of superoxide in mitochondria can be visualised using fluorescence microscopy with MitoSOX™ Red reagent (Life Technologies). MitoSOX™ Red reagent is a cationic derivative of dihydroethidium that permeates live cells and accumulates in mitochondria.
A.5. Indirect detection of superoxide anione formation
The enzyme aconitase contains an iron‐sulfur cluster at its active site, which is highly sensitive to inactivation by O2 .− (Gardner, 2002). Levels of O2 .− production can, therefore, be inferred from the rate of aconitase inactivation during mitochondrial incubations. Aconitase activity is measured spectrophotometrically by a coupled enzyme assay, linking isocitrate production by aconitase to NADPH formation by isocitrate dehydrogenase (Gardner, 2002; Cochemé and Murphy, 2009).
Evidence supporting taxonomic applicability (tissue type, taxa, life stage, sex)
Isolated mitochondria, cultured cells and whole organisms like yeast, worms, flies, rodents and plants generate O2 °− in the presence of redox chemicals like Paraquat mostly increasing mitochondrial oxidative damage (Mason, 1990; Vanfleteren, 1993, Sturz and Culotta, 2002; Van Remmen et al., 2004; Bonilla et al., 2006).
Mitochondria as a major site of mitochondrial superoxide production by PQ are supported in rodents, flies and yeast. Thus, mice heterozygous for MnSOD (the isoform of superoxide dismutase locate in the mitochondrial matrix) (Van Remmen et al., 2004) and flies silenced for MnSOD (Kirby et al., 2002) show greater sensitivity to PQ than the control; flies overexpressing catalase in mitochondria are resistant to PQ, whereas enhancement of cytosolic catalase was not protective (Mockett et al., 2003); human peroxiredoxin 5 in mitochondria protects yeast more efficiently against PQ than expression in the cytosol (Tien Nguten‐nhu et al., 2003).
Complex I has a highly conserved subunit composition in eukaryotes (Cardol, 2011). Fourteen subunits are considered to be the minimal structural requirement for physiological functionality of the enzyme. These units are well conserved between bacterial (E. coli), human (H. sapiens), and bovine (B. Taurus) (Ferguson, 1994; Vogel et al., 2007). However, the complete structure of Complex I is reported to contain between 40 to 46 subunits and the number of subunits differs, depending on the species (Gabaldon 2005; Choi et al., 2008).
Complex I is well‐conserved across species, from lower organism to mammals. In vertebrates it consists of at least 46 subunits (Hassinen, 2007), including human in which 45 subunits were found (Vogel et al., 2007). Moreover, enzymatic and immunochemical evidence indicate a high degree of similarity between mammalian and fungal counterparts (Lummen, 1998). Mammalian complex I structure (Vogel et al., 2007) and activity is characterised in detail, referring to different human organs including brain. There is also substantial amount of studies performed on human muscles, brain, liver as well as bovine heart (Okun et al., 1999).
Yeasts lack Complex I but reduce PQ in dependence on NADPH by intramitochondrial NADPH dehydrogenases (Cochemé and Murphy, 2008).
Cytochrome bc1 complexes (Complex III) are found in the plasma membranes of photosynthetic and respiring bacteria and in the inner mitochondrial membrane of all eukaryotic cells (Trumpower, 1990). In all of these species the bc1 complex contain three electron transfer proteins and transfer electrons from a low‐potential quinol to a higher‐potential c‐type cytochrome (Trumpower, 1990). The number of subunits in the bc1 varies between 3 catalytic subunits in some bacteria and 11 subunits in the mitochondrial bc1 (Trumpower, 1990).
Evidence for Chemical Initiation of this Molecular Initiating Event (MIE)
The most studied examples of chemicals that accept an electron from the mitochondrial respiratory chain and undergo redox cycling in dopaminergic neurons are the three bipyridyl herbicides paraquat, diquat and benzyl viologen. Substantial evidence has accumulated in the existing literature suggesting a role for these chemical, and paraquat in particular, and this AOP. Therefore, the redox cycler parquat will be discussed in the context of all KEs identified in this AOP.
A.1. Paraquat as a mitochondrial electron acceptor
The cellular toxicity of PQ is essentially due to its redox cycling abilities. Mitochondria are a major source of PQ‐induced ROS production in brain (Castello et al., 2007; Figure A.25).
Figure A.25.
PQ 2 ‐induced H2O2 production in cellular fractions from the brain. Fluorometric (A) and polarographic (B) assays were used to measure H2O2 in the presence of malate and glutmate following the addition of 250 mM PQ 2 to equal amounts of protein from each rat brain fraction: mitochondria (solid line), cytosol (dotted line), and homogenate (dashed line) (from: Castello et al. 2007, fig. 2)
The early involvement of mitochondria in PQ‐induced oxidative stress has been also demonstrated in whole cells overexpressing reduction‐oxidation sensitive fluorescent proteins targeted to mitochondria or the cytosol (Rodriguez‐Rocha, 2013; Filograna et al., 2016). PQ (0.1–1 mM) dose‐dependently increases oxidative stress in SK‐N‐SH cells mitochondrial matrix at 24 h with no changes in the cytosol (Figure A.26) (Rodriguez‐Rocha et al., 2013). Accordingly, PQ 0.5 mM increases mitochondrial ROS production in SH‐SY5Y after 6 and 12 h with no evidence in the cytosol (Filograna et al., 2016). Significant cytoplasmic oxidative stress is evident only after 48 h starting from PQ 0.5 mM, but not for lower concentrations (Figure A.26) (Rodriguez‐Rocha et al.,, 2013). A selective involvement of mitochondria is thus dose‐ and time‐dependent.
Figure A.26.
Alterations of mitochondrial and cytosol redox state following exposure to PQ of cells expressing fluorescent redox probe targeted to mitochondria (Mito‐roGFP) or cytosol (roGFP). Cells were co‐stained with PI and only viable cells were analysed. Alteration in the redox state were determined by ratiometric analyses of changes in (Mito‐)roGFP fluorescence at 407/488 ex and 530 em normalised with respect to control values. Data represents means + SE of at least five independent experiments. *p < 0.05 vs control values (Reprinted from Rodriguez‐Rocha et al., 2013, copyright (2013) with permission from Elsevier)
In addition, higher protection against PQ toxicity is reached with mitochondrial, rather than cytosolic, expression of antioxidant enzymes (Mockett et al., 2003; Tien Nguyen‐nhu and Knoops, 2003; Rodriguez‐Rocha et al., 2013; Filograna et al., 2016). Accordingly, the deficiency of the isoform of mitochondrial superoxide dismutase (MnSOD) or mitochondrial thioredoxin reductase (necessary to maintain the H2O2 detoxifying thioreduxin/peroxiredoxin system) increases sensitivity to PQ (Kirby et al., 2002; Van remmen et al., 2004; Lopert et al., 2012).
Both Complex I (Cochemé and Murphy, 2008) and Complex III (Castello et al., 2007; Drechsel and Patel, 2009) have been involved in PQ radicalisation. In Castello et al. (2007), the redox cycle‐initiating electrons are accepted from complex III and to a minor part by complex I as inhibition of complex I by rotenone only partially inhibited PQ‐induced ROS formation in isolated brain mitochondria or rat midbrain cultures, while PQ‐induced ROS formation in these systems was completely blocked after inhibition of complex III by using antimycin A (Castello et al., 2007; Drechsel and Patel, 2009). That complex I is not the major source of electrons triggering PQ toxicity is supported by Choi et al., (2008) who demonstrated that silencing a major component of complex I abolishing its activity does not protect against PQ‐dependent dopaminergic cell death. On the other hand, Cochemé and Murphy (2008) demonstrated that PQ accumulates into yeast and bovine heart mitochondrial matrix in dependence on mitochondrial membrane potential. In heart mitochondria, PQ is then reduced mainly by Complex I forming the radical which rapidly react with O2 to give O2 .−. The Authors explain this discrepancy with differences existing between brain and heart mitochondria (Cochemé and Murphy, 2008; Drechsel and Patel, 2009). The involvement of mitochondrial enzymes other than Complex I and III (VDAC and Cytb5, located at the external mitochondrial membrane) remains controvertial (Shimada et al., 2009; Nikiforova et al., 2014) and potentially excluded by the recent observation that the main site of PQ reduction is inside mitochondria (Nikiforova et al., 2014).
A.2. General characteristics of other mitochondrial redox cyclers
Other mitochondrial redox cyclers include two other bipyridyl herbicides, diquat and benzyl viologen (Figure A.27A and B). These share common structural features with paraquat (Figure A.27C): all compounds are composed of two aromatic rings containing a positively charged nitrogen and are thus good electron acceptors and redox cyclers (Drechsel and Patel, 2009; Sandy et al., 1986).
Figure A.27.

Molecular structures of: (A) diquat, (B) benzyl viologen, (C) paraquat (Copyright Drechsel and Patel, 2009, Fig. 1. Published by Oxford University Press on behalf of the Society of Toxicology)
Quinones (i.e. menadione, Adriamycin) and nitroaromatic compounds (i.e. nitrofurantoin) also radicalise following one electron reduction by mitochondrial reductases (complex I and III and external mitochondria NADH‐oxidoreductase) establishing a redox cycle (Frei et al., 1986; Nikiforova et al., 2014). Intriguingly, free cytosolic dopamine spontaneously oxidises to produce different quinones like dopamine‐o‐quinone and aminochrome. Aminochrome can undergo a one‐electron reduction by NAD(P)H flavoproteins generating a leukoaminochrome‐o‐semiquinone radical and giving rise to redox cycle with production of superoxide anion (Figure A.28) (Zoccarato et al., 2005; Muñoz et al., 2012).
Figure A.28.
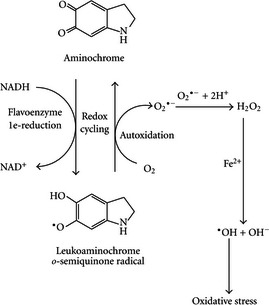
One electron reduction of aminochrome (adapted from Muñoz et al., 2012, fig. 6, CC‐BY)
Aminochrome has been recently suggested to play a role in the death of dopaminergic neurons containing neuromelanin triggering oxidative stress/mitochondrial dysfunction, the formation of α‐synuclein and impaired protein degradation (Sandy et al., 1986; Drechsel and Patel, 2009).
References
Bonila E, Medina‐Leendertz S, Villalobos V, Molero L, Bohorquez A, 2006. Paraquat‐induced oxidative stress in Drosophila melanogaster: effects of melatonin, glutathione, serotonin, minocycline, lipoic acid and ascorbic acid. Neurochemical Research, 31, 1425–1432.
Brand MD, 1995. Measurement of mitochondrial protonmotive force. In: Brown GC and Cooper CE (eds.). ‘‘Bioenergetics—A Practical Approach.’’ Energy Dispersive Spectroscopy. IRL Press, Oxford, pp. 39–62.
Cardol P, 2011. Mitochondrial NADH: ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases. Biochimica et Biophysica Acta, 1807, 1390–1397.
Castello PR, Drechsel DA, Patel M, 2007. Mitochondria are a major source of paraquat‐induced reactive oxygen species production in the brain. Journal of Biological Chemistry, 282, 14186–14193. http://www.jbc.org/content/282/19/14186.full.pdf Fig 2, p.14188.
Cochemé HM, Murphy MP, 2008. Complex I is the major site of mitochondrial superoxide production by paraquat. Journal of Biological Chemistry, 283, 1786–1798.
Cochemé HM, Murphy MP, 2009. Chapter 22 The uptake and interactions of the redox cycler paraquat with mitochondria. Methods in Enzymology, 456, 395–417. doi: 10.1016/S0076‐6879(08)04422‐4
Cohen GM, d'Arcy Doherty M, 1987. Free radical mediated cell toxicity by redox cycling chemicals. British Journal of Cancer. Supplement, 8, 46–52.
Choi WS, Kruse SE, Palmiter R, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP, or paraquat. Proceedings of the National Academy of Sciences, 105, 15136–15141.
Dranka BP, Zielinka J, Kanthasamy AG, Kalyanaraman B, 2012. Alterations in bioenergetics function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. Journal of Neurochemistry, 122, 941–951.
Drechsel DA, Patel M, 2009. Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicological Sciences, 112(2), 427–434. http://doi.org/10.1093/toxsci/kfp223
Ferguson SJ, 1994. Similarities between mitochondrial and bacterial electron transport with particular reference to the action of inhibitors. Biochemical Society Transactions, 22, 181–193. http://www.ncbi.nlm.nih.gov/pubmed/8206221
Filograna R, Godena VK, Sanchez‐Martinez A, Ferrari E, Casella L, Beltramini M, Bubacco L, Whitworth AJ, Bisaglia M, 2016. SOD‐mimetic M40403 is protective in cell and fly models of paraquat toxicity: implications for Parkinson disease. Journal of Biological Chemistry.
Friedrich T, van Heek P, Leif H, Ohnishi T, Forche E, Kunze B, Jansen R, TrowitzschKienast W, Hofle G, Reichenbach H, 1994. Two binding sites of inhibitors in NADH: ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone‐binding site of bacterial glucose: ubiquinone oxidoreductase. European Journal of Biochemistry, 219, 691–698.
Frei B, Winterhalter KH, Richter C, 1986. Menadione‐ (2‐methyl‐1,4‐naphthoquinone‐) dependent enzymatic redox cycling and calcium release by mitochondria. Biochemistry, 25, 4438–4443.
Gabaldon T, Rainey D, Huynen MA, 2005. Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (Complex I). Journal of Molecular Biology, 348, 857–870.
Gardner PR, 2002. Aconitase: sensitive target and measure of superoxide. Methods in Enzymology, 349, 9–23.
Greenamyre JT, Sherer TB, Betarbet R, and Panov AV, 2001. Critical review Complex I and Parkinson's Disease. Life, 52, 135–141.
Grivennikova VG, Vinogradov AD, 2013. Mitochondrial production of reactive oxygen species. Biochemistry (Mosc), 78, 1490–1511.
Kappus H, 1986. Overview of enzyme systems involved in bio‐reduction of drugs and in redox cycling. Biochemical Pharmacology, 35, 1–6.
Hassinen I, 2007. Regulation of mitochondrial respiration in heart muscle. In: Schaffer S. (ed.). Mitochondria – The Dynamic Organelle. Springer ISBN‐13: 978‐0‐387‐69944‐8.
Lim LO, Bortell R, Neims AH, 1986. Nitrofurantoin inhibition of mouse liver mitochondrial respiration involving NAD‐linked substrates. Toxicology and Applied Pharmacology, 84, 493–499.
Lopert P, Day BJ, Patel M, 2012. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. Public Library of Science (PLoS ONE), 7, e50683.
Lümmen P, 1998. Complex I inhibitors as insecticides and acaricides. Biochimica et Biophysica Acta, 1364, 287–296.
Kirby K, Hu J, Hilliker AJ, Phillips JP, 2002. RNA interference‐mediated silencing of Sod2 in Drosophila leads to early adult‐onset mortality and elevated endogenous oxidative stress. Proceedings of the National Academy of Sciences of the United States of America, 99, 16162–16167.
Mason RP, 1990. Redox cycling of radical anion metabolites of toxic chemicals and drugs and the Marcus theory of electron transfer. Environmental Health Perspectives, 87, 237–243.
McCord JM and Fidovich I, 1968. The reduction of cytochrome c by milk xanthine oxidase. Journal of Biological Chemistry, 243, 5733–5760.
Mockett RJ, Bayne AC, Kwong LK, Orr WC, Sohal RS, 2003. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radical Biology and Medicine, 34, 207–217.
Muñoz P, Huenchuguala S, Paris I, Segura‐Aguilar J, 2012. Dopamine oxidation and autophagy. Journal of Parkinson's Disease, 2012, 920953. https://www.researchgate.net/publication/230830981_Dopamine_Oxidation_and_Autophagy P.6, Fig. 6
Murphy MP, Smith RA, 2007. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annual Review of Pharmacology and Toxicology, 47, 629–656.
Nikiforova AB, Saris NE, Kruglov AG, 2014. External mitochondrial NADH‐dependent reductase of redox cyclers: VDAC1 or Cyb5R3? Free Radical Biology and Medicine, 74, 74–84.
Okun JG, Lümmen P, Brandt U, 1999. Three classes of inhibitors share a common binding domain in mitochondrial Complex I (NADH:Ubiquinone Oxidoreductase). Journal of Biological Chemistry, 274, 2625–2630. doi:10.1074/jbc.274.5.2625
Rodriguez‐Rocha H, Garcia‐Garcia A, Pickett C, Li S, Jones J, Chen H, Webb B, Choi J, Zhou Y, Zimmerman MC, Franco R, 2013. Compartmentalized oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: distinct roles of superoxide anion and superoxide dismutases. Free Radical Biology and Medicine, 0, 370‐383.
Sandy MS, Moldeus P, Ross D, Smith MT, 1986. Role of redox cycling and lipid peroxidation in bipyridyl herbicide cytotoxicity. Studies with a compromised isolated hepatocyte model system. Biochemical Pharmacology, 35, 3095–3101.
Sawyer DT, Valentine JS, 1981. How super is superoxide? Accounts of Chemical Research, 14, 393–400.
Selivanov VA1, Votyakova TV, Pivtoraiko VN, Zeak J, Sukhomlin T, Trucco M, Roca J, Cascante M, 2011. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLOS Computational Biology, 7, e1001115. doi: 10.1371/journal.pcbi.1001115
Shimada H, Hirai K, Simamura E, Hatta T, Iwakiri H, Mizuki K et al., 2009. Paraquat toxicity induced by voltage‐dependent anion channel 1 acts as an NADH‐dependent oxidoreductase. Journal of Biological Chemistry, 284, 28642–28649.
Schweiger, Jeschke, 2001. Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press.
Sturtz LA, Culotta VC, 2002. Superoxide dismutase null mutants of baker's yeast, Saccharomyces cerevisiae. Methods in Enzymology, 349, 167–172.
Tien Nguyen‐nhu N, Knoops B, 2003. Mitochondrial and cytosolic expression of human peroxiredoxin 5 in Saccharomyces cerevisiae protect yeast cells from oxidative stress induced by paraquat. Federation of the European Biochemical Societies's Letters (FEBS), 544, 148–152.
Trumpower BL, 1990. Cytochrome bc1 complexes of microorganisms. Microbiol Reviews, 54, 101–129.
Van Remmen H, Qi W, Sabia M, Freeman G, Estlack L, Yang H, Mao Guo Z, Huang TT, Strong R, Lee S, Epstein CJ, Richardson A, 2004. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radical Biology and Medicine, 36, 1625–1634.
Vanfleteren JR, 1993. Oxidative stress and ageing in Caenorhabditis elegans. Biochemical Journal, 292, 605–608.
Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG, 2007. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Molecular Genetics and Metabolism, 91, 176–182.
Zoccarato F, Toscano P, Alexandre A, 2005. Dopamine‐derived dopaminochrome promotes H(2)O(2) release at mitochondrial complex I: stimulation by rotenone, control by Ca(2+), and relevance to Parkinson disease. Journal of Biological Chemistry, 280, 15587–15594.
KE1: Mitochondrial reactive oxygen species (ROS) formation and dysfunction
How this Key Event works:
O2, generated by redox cycling drives a cascade of active oxygen species (ROS). O2 may:
spontaneously or in a reaction catalysed by mitochondrial superoxide dismutase (MnSOD) and CuZnSOD (primarily cytoplasmic but also present in the peroxisome, lysosome, nucleus and mitochondrial intermembrane space) lead to the production of hydrogen peroxide (H2O2), which in turn will favour the formation of hydroxyl anion and hydroxyl radical through the Fenton reaction (Figure 1; Turrens, 2003).
react with nitric oxide (NO), which can be simultaneously produced in mitochondria by a unique form of nitric oxide synthase located at the mitochondrial matrix (Turrens 2003), to form peroxynitrite which may further convert to peroxynitrous acid and may yield nitrogen dioxide and hydroxyl radical (Pryor et al., 1995).
Other possible reactions triggered by free radicals originating from redox‐cycle are: hydrogen atom abstraction and covalent binding to tissue macromolecules by radical addition to carbon‐carbon double bonds or by radical combination.
Figure A.29.
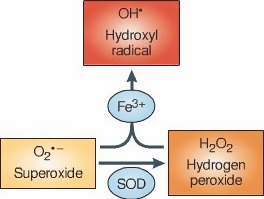
Reactive oxygen species. Two molecules of superoxide can react to generate hydrogen peroxide (H2O2) in a reaction known as dismutation, which is accelerated by the enzyme superoxide dismutase (SOD). In the presence of iron, superoxide and H2O2 react to generate hydroxyl radicals. In addition to superoxide, H2O2 and hydroxyl radicals, other reactive oxygen species (ROS) occur in biological systems., which can be generated from singlet oxygen by antibody molecules 65,66. The colour coding indicates the reactivity of individual molecules (yellow, limited reactivity; orange, moderate reactivity; red, high reactivity and non‐specificity) (Adapted by permission from Macmillan Publishers Ltd, Lambeth, 2004, copyright (2004))
ROS amount can be counter‐balanced by natural antioxidants under physiological conditions. As such, mitochondria are equipped with several ontioxidants systems (vitamin E, phospholipid hydroperoxide glutathione peroxide, MnSOD, cytochrome C, catalase, glutathione, glutathion‐S‐transferase, glutathione reductase, glutathione peroxidase, peroxiredoxins) (Andreyev et al., 2004). Nevertheless, antioxidant response might be overwhelmed by aberrant augmented levels of ROS that react with mitochondrial macromolecules such as lipids, proteins, nucleic acids and carbohydrates (Murphy, 2009) leading to mitochondrial dysfunction, cell death and subsequently to organ pathogenesis. Indeed, oxidative stress is considered as a contributor to the pathogenesis of chronic health problems among which neurodegenerative conditions (Halliwell and Gutteridge, 2007).
It is well established in the existing literature that mitochondrial dysfunction may result in: (a) an increased ROS production and a decreased ATP level, (b) the loss of mitochondrial protein import and protein biosynthesis, (c) the reduced activities of enzymes of the mitochondrial respiratory chain and the Krebs cycle, (d) the loss of the mitochondrial membrane potential, (e) the loss of mitochondrial motility, causing a failure to re‐localise to the sites with increased energy demands, (f) the destruction of the mitochondrial network, (g) increased mitochondrial Ca2+ uptake, causing Ca2+ overload (reviewed in Lin and Beal, 2006; Graier et al., 2007), (h) the rupture of the mitochondrial inner and outer membranes, leading to the release of mitochondrial pro‐death factors, including cytochrome c (Cyt. c), apoptosis‐inducing factor, or endonuclease G (Martin, 2011; Braun, 2012; Correia et al., 2012; Cozzolino et al., 2013), which eventually leads to apoptotic, necrotic or autophagic cell death (Wang and Qin, 2010). Due to their structural and functional complexity, mitochondria present multiple targets for various compounds.
How it is measured or detected
A.I. Reactive oxygen species production
Different ROS including hydrogen peroxide and the hydroxyl radical or the consumption of the ROS detoxifying substance glutathione as well as ROS‐dependent cellular damage like lipid peroxidation or oxidation of protein or DNA can be measured by a variety of assays. Direct assays are based on the chemical modification of fluorescent or luminescent reporters by ROS species. Indirect assays assess cellular metabolites, the concentration of which is changed in the presence of ROS (e.g. glutathione, malondialdehyde, 4‐hydroxynonenal, isoprostanes, etc.). The assays described below are not comprehensive.
A.1. Detection of hydrogen peroxide (H2O2) production
There are a number of fluorogenic substrates, which serve as hydrogen donors that have been used in conjunction with horseradish peroxidase (HRP) enzyme to produce intensely fluorescent products in the presence of hydrogen peroxide (Ruch et al., 1983; Zhou et al., 1997). The more commonly used substrates include diacetyldichloro‐fluorescein, homovanillic acid, and Amplex® Red (https://www.thermofisher.com/order/catalog/product/A22188). In these assays, increasing amounts of H2O2 leads to increasing amounts of fluorescent product (Tarpley et al., 2004).
A.2. Measurement of the cellular glutathione (GSH) status
GSH is regenerated from its oxidised form (GSSH) by the action of a NADPH‐dependent reductase (GSSH + NADPH + H+ → 2 GSH + NADP+). The ratio of GSH/GSSG is therefore a good indicator for the cellular NADP+/NADPH ratio (i.e. the redox potential). GSH and GSSH levels can be determined by HPLC, capillary electrophoresis, biochemically with DTNB (Ellman's reagent, 5,5’‐dithio‐bis‐[2‐nitrobenzoic acid]) or by mean of luminescence‐based assays (for example, GSH‐Glo™ Glutathione Assay, https://www.promega.co.uk/resources/protocols/technical-bulletins/101/gsh-glo-glutathione-assay-protocol/). As excess GSSG is rapidly exported from most cells to maintain a constant GSH/GSSG ratio, a reduction of total glutathione levels is often considered a good surrogate measure for oxidative stress.
A.3. Measurement of ROS‐scavenging enzymes activity
Increased activity of scavenging enzymes like catalase, superoxide dismutase (SOD) and glutathione‐S‐transferase activity (GST) is indicative of ROS‐production. The enzymes are recovered both in cells and tissues homogenates, thus providing a tool to measure the occurrence of ROS ex‐vivo (Mitra et al., 2011) as an alternative to the measurement of lipid peroxidation. Measurements are based on the detection of chromogen, sensitive to the ROS specifically produced by the investigated enzyme, by spectrofluorimetric methods as described by Shina et al. (1972; for catalase); Pabst et al. (1974, for GST) and Kakkar et al. (1984, for SOD).
A.4. Quantification of lipid peroxidation
Measurement of lipid peroxidation has historically relied on the detection of thiobarbituric acid (TBA)‐reactive compounds, such as malondialdehyde generated from the decomposition of cellular membrane lipid under oxidative stress (Pryor et al., 1976). This method is quite sensitive, but not highly specific. A number of commercial assay kits are available for this assay using absorbance or fluorescence detection technologies. The formation of F2‐like prostanoid derivatives of arachidonic acid, termed F2‐isoprostanes (IsoPs) has been shown to be more specific for lipid peroxidation. A number of commercial ELISA kits have been developed for IsoPs, but interfering agents in samples requires partial purification before analysis. Alternatively, gas chromatography–mass spectrometry (GC‐MS) may be used as a robust, specific and sensitive method.
A.5. Detection of peroxynitrite
There are three major approaches for peroxynitrite detection, including electrochemical sensors, detection of nitrotyrosine formation, and fluorescent probes (Chen X, Biomed J. 2014).
A.II. Mitochondrial dysfunction assays assessing a loss‐of function
A.1. Cellular oxygen consumption
Electrons, fed into the mitochondrial respiratory chain either by complex I or complex II, ultimately reduce molecular oxygen to water at complex IV. In a closed system, this consumption of oxygen leads to a drop of the overall O2 concentration, and this can serve as parameter for mitochondrial respiratory activity. Measurements are traditionally done with a Clark electrode, or with more sophisticated optical methods. At the cathode of a Clark electrode, oxygen is electrolytically reduced, which initiates a current in the electrode, causing a potential difference that is ultimately recorded. Clark electrodes however have the disadvantage that oxygen is consumed. Furthermore, interferences with nitrogen oxides, ozone, or chlorine is observed (Stetter et al., 2008). To circumvent these limitations, optical sensors have been developed that have the advantage that no oxygen is consumed, combined with a high accuracy and reversibility. Optical oxygen sensors work according to the principle of dynamic fluorescence quenching. The response of the respective fluorescence dye is proportional to the amount of oxygen in the sample investigated (Wang et al., 2014). In a model of isolated mitochondria in the absence of complex II substrates, oxygen consumption can serve as surrogate readout for the assessment of the degree of complex I inhibition. It is however essential to realise that also complex III and complex IV activities are involved and their inhibition also results in a decline in O2 consumption. In addition to that, CI inhibitors can lead to a one‐electron reduction of molecular oxygen at the site of CI to yield superoxide. The amount of superoxide formed hence contributes to the consumption of oxygen, but must not be interpreted as oxygen consumption as a result of controlled and coupled electron flux through the complexes of the mitochondrial respiratory chain. A modern convenient method to measure oxygen consumption is provided by the Seahorse technology of extracellular flux (XF) analysis, in which cells are kept in a very small volume, so that changes of oxygen levels can be detected very sensitively by an oxygen sensor. To allow manipulation of the mitochondria in cells, the cell membrane can be permeabilised with saponin (SAP), digitonin (DIG) or recombinant perfringolysin O (rPFO) (XF‐plasma membrane permeabiliser (PMP) reagent), to allow addition of specific substrates to measure activity of different respiratory chain complexes, including complex I. (Salabei et al., 2014).
The oxygen consumption parameter can be combined with other endpoints to derive more specific information on the efficacy of mitochondrial function. One approach measures the ADP‐to‐O ratio (the number of ADP molecules phosphorylated per oxygen atom reduced (Hinkle, 1995 and Hafner et al., 1990). The related Phosphate/Oxygen (P/O) ratio is calculated from the amount of ADP added, divided by the amount of O consumed while phosphorylating the added ADP (Ciapaite et al., 2005).
A.2. Mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential (Δψm) is the electric potential difference across the inner mitochondrial membrane. It requires a functioning respiratory chain in the absence of mechanisms that dissipate the proton gradient without coupling it to ATP production. The classical, and still most quantitative method uses a tetraphenylphosphonium ion (TPP+)‐sensitive electrode on suspensions of isolated mitochondria.
The Δψm can also be measured in live cells by fluorimetric methods. These are based on dyes which accumulate in mitochondria because of Δψm. Frequently used are tetramethylrhodamineethylester (TMRE), tetramethylrhodamine, methyl ester (TMRM) (Petronilli et al., 1999) or 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazole carbocyanide iodide (JC‐1). In particular, mitochondria with intact membrane potential concentrate JC‐1, so that it forms red fluorescent aggregates, whereas de‐energised mitochondria cannot concentrate JC‐1 and the dilute dye fluoresces green (Barrientos et al., 1999). Assays using TMRE or TMRM measure only at one wavelength (red fluorescence), and depending on the assay setup, de‐energised mitochondria become either less fluorescent (loss of the dye) or more fluorescent (attenuated dye quenching).
A.3. Enzymatic activity of the electron transport system (ETS)
Determination of ETS activity can be determined following Owens and King's assay (1975). The technique is based on a cell‐free homogenate that is incubated with NADH to saturate the mitochondrial ETS and an artificial electron acceptor [l‐(4‐iodophenyl)‐3‐(4‐nitrophenyl)‐5‐phenyltetrazolium chloride (INT)] to register the electron transmission rate. The oxygen consumption rate is calculated from the molar production rate of INT‐formazan which is determined spectrophotometrically (Cammen et al., 1990).
A.4. ATP content
For the evaluation of ATP levels, various commercially available ATP assay kits are offered (e.g. Sigma, http://www.abcam.com/atp-assay-kit-colorimetricfluorometric-ab83355.html), based on luciferin and luciferase activity. For isolated mitochondria various methods are available to continuously measure ATP with electrodes (Llaudet et al., 2005), with luminometric methods, or for obtaining more information on different nucleotide phosphate pools (e.g. Ciapaite et al., 2005).
A.III. Mitochondrial dysfunction assays assessing a gain‐of function
A.1. Mitochondrial permeability transition pore (PTP) opening
The opening of the PTP leads to the permeabilisation of mitochondrial membranes (Fiskum, 2000; Lemasters et al., 2009), so that different compounds and cellular constituents can change intracellular localisation. This can be measured by assessment of the translocation of cytochrome c, adenylate kinase or the apoptosis‐inducing factor (AIF) from mitochondria to the cytosol or nucleus. The translocation can be assessed biochemically in cell fractions, by imaging approaches in fixed cells or tissues, or by life‐cell imaging of GFP fusion proteins (Single et al., 1998; Modjtahedi et al., 2006). An alternative approach is to measure the accessibility of cobalt to the mitochondrial matrix in a calcein fluorescence quenching assay in live permeabilised cells (Petronilli et al., 1999).
A.2. mtDNA damage as a biomarker of mitochondrial dysfunction
Various quantitative polymerase chain reaction (QPCR)‐based assays have been developed to detect changes of DNA structure and sequence in the mitochondrial genome (mtDNA). mtDNA damage can be detected in blood after low‐level rotenone exposure, and the damage persists even after CI activity has returned to normal. With a more sustained rotenone exposure, mtDNA damage can be also detected in skeletal muscle. These data support the idea that mtDNA damage in peripheral tissues in the rotenone model may provide a biomarker of past or ongoing mitochondrial toxin exposure (Sanders et al., 2014).
Evidence Supporting Taxonomic Applicability
Redox cycling is a universal event occurring in any cells of any species as well as in bacteria and yeast (Cochemé and Murphy, 2008).
Mitochondrial dysfunction is a universal event occurring in cells of any species (Farooqui and Farooqui, 2012). Many invertebrate species (e.g. D. melanogaster and C. elegans) are considered as potential models to study mitochondrial functionality. New data on marine invertebrates, such as molluscs and crustaceans and non‐Drosophila species, are emerging (Martinez‐Cruz et al., 2012). Mitochondrial dysfunction can be measured in animal models used for toxicity testing (Waerzeggers et al., 2010; Winklhofer and Haass, 2010) as well as in humans (Winklhofer and Haass, 2010).
However, there seem to be different susceptibilities towards mitochondrial toxins between mitochondria of different organs. For example, rotenone (complex I inhibitor) severely damage brain mitochondria, whereas liver mitochondria remained virtually unaffected (Panov et al., 2005). Moreover, liver mitochondria have much lower Ca2 + capacities, they evidently undergo mitochondrial permeability transition (mPT), followed by apoptosis much easier than brain mitochondria, which can withstand much higher Ca2 + concentrations than liver (Panov et al., 2007). Not only do mitochondria differ between organs, but also between brain regions. Work from Dubinsky's group found that striatal mitochondria isolated from rats were more sensitive than cortical mitochondria in their response to calcium, perhaps due to increased amounts of cyclophilin D, a mitochondrial permeability transition pore component (Brustovetsky et al., 2003; LaFrance et al., 2005). Independent of the mitochondrial transition pore, brain region‐specific mitochondrial membrane potential and susceptibility towards dysfunction of mitochondrial oxidative phosphorylation (OXPHOS) was also observed by Pickrell et al. (2011). Here the striatum was found to be especially sensitive towards disturbance of OXPHOS due to the high striatal mitochondrial OXPHOS and membrane potential, which is prone to collapse when OXPHOS activity is reduced. This instance becomes important when studies on compound effects on isolated mitochondria are not of the correct origin, which would – for studying Parkinsonism – be the brain, and here the nigrostriatal area. In addition to mitochondrial differences between organs and intra‐organ regions, species‐specific mitochondrial activity was also measured. For example, inhibition of complex III with Antimycin A causes significantly higher ROS formation in mouse than rat brain mitochondria suggesting a species‐specific susceptibility to compounds interfering with complex III across species (Panov et al., 2007). If human brain mitochondria are more similar to mouse or rat mitochondria remains so far enigmatic.
References
Andreyev AY, Kushnareva YE, Starkov AA, 2005. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc), 70, 200–214.
Barrientos A, Moraes CT, 1999. Titrating the effects of mitochondrial Complex I impairment in the cell physiology. The Journal of Biological Chemistry, 274, 16188–16197.
Braun RJ, 2012. Mitochondrion‐mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Frontiers in Oncology, 2, 182.
Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM, 2003. Increased susceptibility of striatal mitochondria to calcium‐induced permeability transition. Journal of Neuroscience, 23, 4858–4867.
Cammen M, Corwin S, Christensen JP, 1990. Electron transport system (ETS) activity as a measure of benthic macrofaunal metabolism. Marine Ecology Progress Series, 65, 171–182.
Chen X, Chen H, Deng R, Shen J, 2014. Pros and cons of current approaches for detecting peroxynitrite and their applications. Biomedical Journal, 37, 120–126.
Ciapaite J, Lolita Van Eikenhorst G, Bakker SJ, Diamant M, Heine RJ, Wagner MJ, Westerhoff HV, Krab K, 2005. Modular kinetic analysis of the adenine nucleotide translocator–mediated effects of palmitoyl‐coa on the oxidative phosphorylation in isolated rat liver mitochondria. Diabetes, 54, 944–995.
Cochemé HM, Murphy MP, 2008. Complex I is the major site of mitochondrial superoxide production by paraquat. Journal of Biological Chemistry, 25, 283, 1786–1798.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA, 2012. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Advances in Experimental Medicine and Biology, 724, 205–221.
Cozzolino M, Ferri A, Valle C, Carri MT, 2013. Mitochondria and ALS: implications from novel genes and pathways. Molecular and Cellular Journal of Neuroscienceence, 55, 44–49.
Farooqui T, Farooqui AA, 2012. Oxidative stress in Vertebrates and Invertebrate: Molecular Aspects of Cell Signalling. Wiley‐Blackwell, Chapter 27, pp. 377–385.
Graier WF, Frieden M, Malli R, 2007. Mitochondria and Ca2 + signaling: old guests, new functions. Pflügers Archiv, 455, 375–396.
Hafner RP, Brown GC, Brand MD, 1990. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ‘top‐down’ approach of metabolic control theory. European Journal of Biochemistry, 188, 313–319.
Halliwell B & Gutteridge JMC, 2007. Free Radicals in Biology and Medicine. Oxford University Press, Oxford. pp. 1–677.
Hinkle PC, 1995. Measurement of ADP/O ratios. In: Brown GC, Cooper CE (eds.). Bioenergetics: A Practical Approach. Energy Dispersive Spectroscopy Oxford, IRL Press, UK, pp. 5–6.
Kakkar P, Das B, Vishwanathan PN, 1984. A modified spectrophotometric assay of superoxide dismutase. Indian Journal of Biochemistry and Biophysics, 21, 130–132.
LaFrance R, Brustovetsky N, Sherburne C, Delong D, Dubinsky JM, 2005. Age‐related changes in regional brain mitochondria from Fischer 344 rats. Aging Cell, 4, 139–145.
Lambeth JD, 2004. NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology, 4, 181–189.
Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL, 2009. Mitochondrial calcium and the permeability transition in cell death. Biochimica et Biophysica Acta, 1787, 1395–1401.
Llaudet E, Hatz S, Droniou M, Dale N, 2005. Microelectrode biosensor for real‐time measurement of ATP in biological tissue. Analytical Chemistry, 77, 3267–3273.
Lin MT, Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787–795.
Martin LJ, 2011. Mitochondrial pathobiology in ALS. Journal of Bioenergetics and Biomembranes, 43, 569–579.
Martinez‐Cruz O, Sanchez‐Paz A, Garcia‐Carreño F, Jimenez‐Gutierrez L, Navarrete del Toro Mde L and Muhlia‐Almazan A, 2012. Invertebrates Mitochondrial Function and Energetic Challenges (http://www.intechopen.com). In: Clark K (ed.). Bioenergetics, ISBN 978‐953‐51‐0090‐4, InTech, pp. 181–218.
Mitra S, Chakrabarti N, Bhattacharyy A, 2011. Differential regional expression patterns of α‐synuclein, TNF‐α, and IL‐1β; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Modjtahedi N, Giordanetto F, Madeo F, Kroemer G, 2006. Apoptosis‐inducing factor: vital and lethal. Trends in Cell Biology, 16, 264–272.
Pabst JM, Habig WH, Jakoby WB, 1974. Glutathione‐S‐transferase. A Journal of Biological Chemistry, 249, 7140–7150.
Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT, 2005. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. Journal of Biological Chemistry, 280, 42026–42035.
Panov A, Dikalov S, Shalbuyeva N, Hemendinger R, Greenamyre JT, Rosenfeld J, 2007. Species‐ and tissue‐specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. American Journal of Physiology‐Cell Physiology, 292, C708–C718.
Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F, 1999. Transient and long‐lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophysical Journal, 76, 725–734.
Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT, 2011. The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. Journal of Neuroscience, 31, 9895–9904.
Pryor WA, Stanley JP, Blair E, 1976. Autoxidation of polyunsaturated fatty acids: II. A Suggested mechanism for the Formation of TBA‐reactive materials from prostaglandin‐like Endoperoxides. Lipids, 11, 370–379.
Pryor WA, Squadrito GL, 1995. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. American Journal of Physiology, 268, L699–L722.
Ruch W, Cooper PH, Baggiollini M, 1983. Assay of H2O2 production by macrophages and neutrophils with Homovanillic acid and horseradish peroxidase. Journal of Immunological Methods 63, 347–357.
Sanders LH, Howlett EH, McCoy J, Greenamyre JT, 2014. Mitochondrial DNA damage as a peripheral biomarker for mitochondrial toxin exposure in rats. Toxicological Sciences, 142, 395–402.
Single B, Leist M, Nicotera P, 1998. Simultaneous release of adenylate kinase and cytochrome c in cell death. Cell Death & Differentiation, 5, 1001–1003
Sinha AK, 1972. Colorimetric assay of catalase. Analytical Chemistry, 47, 389–394.
Tarpley MM, Wink DA, Grisham MB, 2004. Methods for detection of reactive Metabolites of Oxygen and Nitrogen: in vitro and in vivo considerations. American Journal of Physiology. Regulatory integrative and comparative Physiology, 286, R431–R444.
Turrens JF, 2003. Mitochondrial formation of reactive oxygen species. Journal of Physiology, 552, 335–344.
Wang Y and Qin ZH, 2010. Molecular and cellular mechanisms of excitotoxic neuronal death, Apoptosis, 15, 1382–1402.
Waerzeggers, Yannic Monfared, Parisa Viel, Thomas Winkeler, Alexandra Jacobs, Andreas H, 2010. Mouse models in neurological disorders: applications of non‐invasive imaging. Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease, 1802, 819–839.
Winklhofer K, Haass C, 2010. Mitochondrial dysfunction in Parkinson's disease. Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease, 1802, 29–44.
Zhou M, Diwu Z, Panchuk‐Voloshina N, Haughland RP, 1997. A Stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: application in detecting the activity of phagocyte NADPH oxidase and other oxidases. Analytical Biochemistry 253, 162–168.
KE2: Impaired proteostasis
See AOP 1 (p. 87) for) for the description of this KE
KE3: Neuroinflammation
See AOP 1 (p. 95) for the description of this KE
KE4: Degeneration of dopaminergic neurons of nigrostriatal pathway
See AOP 1 (p. 90) for the description of this KE
Adverse Outcome: Parkinsonian motor deficits
See AOP 1 (p. 98) for the description of the AO
KEY EVENTS RELATIONSHIPS (KERs)
1st KER: Chemical redox cycling in mitochondria leads to mitochondrial reactive oxygen species (ROS) production and dysfunction
How this Key Event Relationship works
Chemical redox cycling is triggered in the presence of chemicals able to accept an electron from a reductant to form a mono‐cation free radical. Compounds with a lower electron reduction potential than O2 will react fastest and the newly formed free radical, in the presence of oxygen, will re‐oxidise generating the superoxide radical O2°− (Kappus, 1986). The radical species may then be reformed from the parent compound reacting with oxygen again and establish a futile redox cycle boosting O2°− production (Cohen and Doherty, 1987). Mitochondria may represent the major site of chemical redox cycling, although several membrane and cytosolic enzymes may trigger this reaction. This has been demonstrated for PQ where alterations of mitochondrial redox state occurs earlier in mitochondria than in the cytosol (Castello et al., 2007; Rodriguez‐Roche et al., 2013; Filograna et al., 2016) and higher protection from its toxicity is reached with mitochondrial, rather than cytosolic, expression of antioxidant enzymes (Mockett et al., 2003; Tien Nguyen‐nhu and Knoops, 2003; Rodriguez‐Roche et al., 2013; Filograna et al., 2016). Excessive generation of superoxide within mitochondria, as it occur in the presence of a chemical redox cycler like PQ, will start a cascade of active oxygen species that will overwhelm antioxidant response and damage DNA, proteins, lipids and other mitochondrial components and function (Turrens, 2003; Murphy, 2009; Andreyev et al., 2014) (Figure A.30).
Figure A.30.
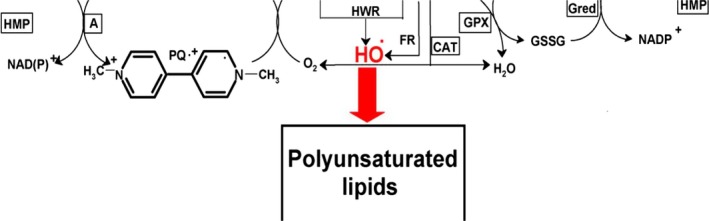
Schematic representation of the mechanism of paraquat toxicity. A, cellular diaphorases; SOD, superoxide dismutase; CAT, catalase; GPX, glutathione peroxidase; Gred, glutathione reductase; PQ2+ , paraquat; PQ+, paraquat cation radical; HMP, hexose monophosphate pathway; FR, Fenton reaction; HWR, Haber–Weiss reaction (Reprinted from Dinis‐Oliveira et al., 2006, copyright (2006), with permission from Elsevier)
Weight of Evidence
That PQ‐dependent superoxide anion formation causes H2O2 in mitochondria is promoted by concentration‐response relationships, e.g. in Cochemé and Murphy, 2009 (Figure A.4). In addition, the observation that higher protection against PQ toxicity is reached with mitochondrial, rather than cytosolic, expression of antioxidant enzymes (Mockett et al., 2003; Tien Nguyen‐nhu and Knoops, 2003; Rodriguez‐Rocha et al., 2013; Filograna et al., 2016), supports the evidence as these enzymes are involved in mitochondrial ROS detoxification, which occurs e.g. after paraquat exposure.
Figure A.31.
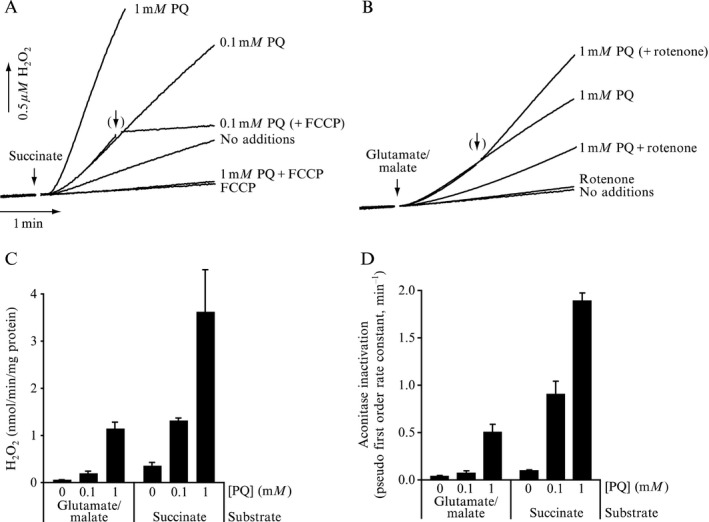
Production of H2O2 and O2 by PQ in mammalian mitochondria – effect of respiratory substrate, uncoupler, and respiratory inhibitors. (A–B) Example traces from the Amplex Red assay. Rat heart mitochondria (0.2 mgprotein/mL) were incubated at 37 in KCl buffer supplemented with 0.01% [w/v] BSA. Substrate (5 mM succinate or 5 mM glutamate/malate), PQ (0.1 or 1 mM), the uncoupler FCCP (1 mM), and the complex I inhibitor rotenone (4 mg/mL) were added as indicated. (C) Rates of H2O2 efflux determined from the above traces. Data are the means SD of three‐four determinations. (D) O2 production in rat heart mitochondria determined by the aconitase inactivation assay. Heart mitochondria (2 mg protein/mL) were incubated for 10 min at 37 in KCl buffer supplemented with 0.1% [w/v] BSA. Substrate (5 mM succinate or 5 mM glutamate/malate) and PQ (0.1 or 1 mM) were present as indicated. Data are the means SD of three determinations (Cochemé and Murphy, 2009. Copyright (2009) Cochemé and Murphy, 2009)
Biological Plausibility
The biological plausibility evolves from the measured that (i) PQ is reaching the brain (Prasad, 2007; Yin, 2011; Breckenridge, 2013, 2014; Liang, 2013), (ii) PQ is taken up into nigrostriatal neurons (Rappold, 2011) and mitochondria (Castello et al., 2007; Cochemé and Murphy, 2009) (iii) PQ is a redox cycler inducing O2°− production and a cascade of ROS in isolated rat brain mitochondria (Castello et al., 2007; Cochemé and Murphy, 2008) brain homogenates (Castello et al., 2007), yeast (Cochemé and Murphy, 2008) and brain cell cultures mitochondria (Castello et al., 2007; Cantu et al., 2011; Dranka et al., 2012; Huang et al., 2012; Rodriguez‐Rocha et al., 2013) (iv) Uncontrolled O2°− production and oxidative stress, due to chemical redox‐cycling or endogenous O2°− overproduction, results in mitochondrial dysfunction, namely decreased activity of enzymes of the respiratory chain (Hinerfeld et al., 2014; De Oliveira et al., 2016), diminished ATP production (De Oliveira et al., 2016), decrease in mitochondrial membrane potential (Huang et al., 2012; De Oliveira et al., 2016), mitochondrial DNA damage (Murphy, 2009). (v) This ROS formation can be blocked by inhibition of complex III activity (and to a lesser extent by complex I inhibition; Castello et al., 2007; Drechsel & Patel 2009), (v) the deficiency of the isoform of mitochondrial superoxide dismutase (MnSOD) or mitochondrial thioredoxin reductase (necessary to maintain the H2O2 detoxifying thioreduxin/peroxiredoxin system) increases sensitivity to PQ (Kirby et al., 2002; Van Remmen et al., 2004; Lopert et al., 2012). Mitochondrial oxidative stress and mitochondrial dysfunction is a contributing factor in the aetiology of PD.
Empirical support for linkage
Existing in vitro and in vivo data shows that compound‐induced mitochondrial redox cycling causes mitochondrial ROS formation and dysfunction. PQ dose‐dependently increases mitochondrial O2°− production both in isolated mitochondria and brain cells. The effect occurs within minutes in isolated mitochondria exposed to PQ and hours in cell cultures. In any biological context the effect is dose dependent and cumulative in time. In vivo evidence supporting PQ‐induced oxidative stress exists and is mainly based on the occurrence of lipoperoxidation. It has been demonstrated that PQ induces an early increase in oxidative stress in the mitochondrial matrix due to O2°− formation that is followed by subsequent oxidative stress in the cytosol (Rodiriguez‐Rocha et al., 2013). In both in vivo and in vitro studies mitochondrial dysfunction and cell death is reduced/prevented by overexpression.
Paraquat redox cycling with superoxide anion formation causes ROS formation and mitochondrial dysfunction.
In Vitro
Incubation of rat primary mesencephalic cells or a dopaminergic cell line, N27, with PQ 0.250–1 mM for 3 or 4 h resulted in a dose‐dependent reduction of aconitase activity significant for all the tested doses (Tab. 1) (Cantu et al., 2009, 2011). Aconitase is uniquely sensitive to O2°− mediated oxidative inactivation thus being an indirect marker of O2°− production. O2°− formation was coupled to a dose dependent H2O2 production after 2–6 h exposure of both cell type to PQ. The effect was significant only for PQ 1mM at 2 h (Tab.1, 17%), 0.5 and 1 mM at 4 h (Tab. 1) and 0.25–1 mM at 6 h (Tab. 1) (Cantu et al., 2009, 2011). Cell death occurred only 18 h after PQ exposure (i.e. after 4–6 h) (Cantu et al., 2009, 2011). Mitochondrial aconitase has also been shown to be a source of °OH, probably Via Fenton chemistry initiated by the co‐released Fe2+ and H2O2 (Vasquez‐Vivar et al., 2000). 60–70% reduction of mitochondrial aconitase expression in N27 cells resulted in a decreased H2O2 production, attenuation of respiratory capacity deficiency and death after PQ exposure (Cantu et al., 2011). On the contrary, overexpression of m‐aconitase resulted in exacerbation of H2O2 production and increased primary mesencepahlic neuron death (Cantu et al., 2009). Aconitase inhibition by PQ (0.1 and 1 mM) has been reported also in yeast and bovine heart mitochondrial within minutes from the exposure (Cochemé and Murphy, 2008). This effect is coupled as well to a dose dependent (PQ 0.1, 0.5 and 1 mM) mitochondrial H2O2 formation and is a consequence of a mitochondrial membrane potential‐dependent uptake of PQ dication (Cochemé and Murphy, 2008).
In another study performed on primary mesencephalic neurons (Cantu et al., 2009) exposure to PQ 0.25 and 0.5 mM reduced aconitase activity of 43% and 58% respectively. A dose– and time–response increase in H2O2
Exposure of human neuroblastoma SK‐N‐SH cells to PQ dose (0.2–1 mM) and time (6–72 h) dependently increases the production of O2°−, as measured by mitosox and electron paramagnetic resonance. PQ (0.5 mM)‐induced O2°− production up to 48 h was due to mitochondria, being prevented by MnSOD (located in the mitochondrial matrix) but not by CuZnSOD (primarily localised in the cytosol). In addition PQ dose‐dependently increases oxidative stress in the mitochondrial matrix at 24 h and both in mitochondrial matrix and cytosol at 48 h. A mitochondrial restricted ROS production after SH‐SY5Y cell exposure to PQ 0.5 mM for 6 and 12 h was also observed in another study (Filograna et al., 2016). MnSOD pretreatment significantly reduced mitochondrial oxidative stress and neuronal cell death induced by PQ 0.5 mM at 48 h, while CuZnSOD had no effect (Rodriguez‐Rocha et al., 2013). Similar results were obtained by Filograna et al. (2016) in SH‐SY5Y after 24 h exposure to PQ. All together these data shows that PQ induces an early increase in oxidative stress in the mitochondrial matrix associated with O2°− production, which is followed by subsequent oxidative stress in the cytosol and is a trigger to neural cell death (Rodriguez‐Rocha et al., 2013).
Paraquat (250 μM) induced H2O2 in the mitochondrial, but not in the cytosolic fraction of rat brain homogenates (Castello et al., 2007). These data indicate again that the mitochondrion is the primary place of PQ‐induced ROS production in the cell.
Redox cycling of Paraquat (250 μM) involves complex III of the MRC as PQ‐dependent H2O2 production of isolated rat brain mitochondria (2–3 min) or primary mid brain cell cultures (6 h) is antagonised by co‐treatment with the complex III inhibitor Antimycin A and to a lesser extent by rotenone (inhibitor of complex I; Castello et al., 2007). These data are supported by Drechsel and Patel (2009), who confirmed that complex III of the MRC is the major player in PQ‐induced ROS production in Malate and Glutamate‐stimulated rat brain mitochondria (100 and 300 μM; measurements over 15 min) and primary midbrain cultures (300 μM, 8 h) by co‐treatment with Antimycin A, while this group measured involvement of MRC complex I in PQ‐induced ROS formation in isolated rat brain mitochondria only after exposure to 1 or 3 mM PQ (15 min measurement).
A neurotoxic concentration of PQ 0.1 mM induces production of O2°−,H2O2 and NO after 24 h in SH‐SY5Y. Oxidative stress is coupled to impairment of complex I and complex V activity, to a decrease mitochondrial potential and ATP production. All these effects are prevented by a 12 h pretreatment with carnosic acid, a diterpene with antioxidant properties (de Oliveira, 2016). ROS production coupled to reduced ATP production and lipid peroxidation were also observed in SH‐SY5Y differentiated cells exposed to PQ 10 μM for 48 h (McCarthy et al., 2004), indicating that PQ ability to trigger an oxidative damage is function of dose and time of exposure.
In vitro, PQ toxicity both in terms of ROS production, mitochondrial dysfunction and neuronal death is rescued by several antioxidants namely EUK 134 and 189 (synthetic SOD/catalase mimetics) (Peng et al., 2005; Hinerfeld et al., 2014), Coenzyme Q10 (McCarthy et al., 2004), rasagiline and cabergoline through their ability to increase the expression of gluthatione (Chau et al., 2009, 2010), carnosic acid through the increased expression of both mitochondrial and total glutathione and several other antioxidant enzymes (de Oliveira, 2016). Similar results are obtained by decreasing the expression of mitochondrial enzymes involved in ROS production (i.e. mitochondrial aconitase) prior to PQ exposure (Cantu et al., 2011) or by overexpressing enzymes involved in O2°− dismutation (i.e. mitochondrial superoxide dismutase) (Choi et al., 2006; Rodriguez‐Rocha et al., 2013). Accordingly, decreased expression or inhibition of detoxifying enzymes like thioredoxin reductase (involved in the conversion of H2O2 in H2O) potentiates synergistically increase H2O2 levels and decreased maximal and reserve respiratory capacity following incubation with PQ oxidative stress and mitochondrial dysfunction in dopaminergic cells (Lopert et al., 2012).
Ex Vivo
Mitochondria isolated from the striatum of Sprague Dawley rats 24 h after exposure to PQ 25 mg/kg produce a significant higher amount of H2O2 compared to controls (+150%) and display decreased complex I and IV activity (−37 and −21%), increased mitochondrial membrane potential, increased lipid peroxidation (+42%) and increased cardiolipin oxidation/depletion (+12%). No changes were observed in cortical mitochondria from PQ treated animals. (Czerniczyniec et al., 2015). Increased O2°− production (50% and 20% for cortical and striatal mitochondria respectively), decreased aconitase activity (30% Cx, 50% Str), increased lipid peroxidation (20% Cx, 30% Str) and release of cytochrome c and AIF were also observed in mitochondria isolated from the cortex and the striatum of Sprague Dawley exposed to PQ (10 mg/kg) over 4 weeks (one injection weekly) (Czerniczyniec et al., 2013). These results show that both acute and prolonged in vivo exposure to PQ promotes mitochondrial O2°−and ROS production coupled to mitochondrial dysfunction with the striatum more sensitive than the cortex.
In Vivo
Paraquat (10 mg/kg i.p.) once a week for 3 weeks causes loss of dopaminergic neurons (TH+) after 2 weeks in mice in vivo. In parallel, 4‐hydroxynonenal (4‐HNE, time course) and nitrotyrosine proteins (single time point) (as markers of PQ‐induced oxidative stress) were measured in TH+ cells of these animals. Lipid peroxidation at TH+ neurons is already significant after the 1st PQ injection (+200%) and increases up to 600% on the 2nd PQ injection. No nigral dopaminergic cell loss occurs after the 1st PQ injection, while a significant reduction of neurons is triggered by the 2nd injection (30%), suggesting a relationship with lipid peroxidation. That ROS was involved in the dopaminergic cell death was not only shown by these markers of peroxidation, but also shown by transgenic, human ferritin overexpressing mice (characterised by a decreased susceptibility to oxidative stress), which were protected against PQ‐induced dopaminergic cell death and 4‐HNE generation (McCormack et al., 2005).
Mice exposed to PQ (5, 10, 20, 40, 80 mg/kg, twice a week for 4 weeks, i.p.) displayed a dose‐dependent increase in superoxide, catalase and glutathione s‐transferase activity as measured in homogenate obtained from substantia nigra, (SN) frontal cortex and the hippocampus. ROS‐scavenging activity dose dependently increased in all the three areas both at sublethal (PQ 5–10 mg/kg) and lethal doses (PQ 20–80 mg/kg) (Tab.1, data referred only to SN and non lethal doses). At PQ 5 and 10 mg/kg, ROS scavenging enzyme activity was specific for the brain since no increase was observed in peripheral organs. Part of the mice exposed to PQ 10/mg kg were also supplemented with α‐tocopherol (20 mg/kg, after the last dose of PQ for five consecutive days, i.p.), which decreased SOD, catalase and GST activity in all three brain areas. All the animals displayed significant DA neuronal death, microglia activation and motor dysfunction at PQ 10 mg/kg (Mitra et al., 2011).
In vivo administration of synthetic superoxide dismutase/catalase mimetics like EUK‐134, 189, (mice, PQ 7 mg/kg, i.p. every 2 days for 10 times; Peng et al., 2005), M40401 (rats, PQ 50 □g infused in the SN); PEP‐SOD (mice, PQ 10 mg/kg, i.p.,once; Choi 2006) fusion protein protects against PQ neurotoxicity. On the other hand, depletion of antioxidants systems exacerbates PQ toxicity.
Subcutaneous administration of PQ (10 mg/kg, twice/week, 3 weeks) to null mice for glutathione (major antioxidant to maintain redox equilibrium in cells) significantly decreased aconitase activity (20%) and complex I activity (20%) in the striatum but not in the cortex. PQ has no effect in wild type mice (Liang et al., 2013).
Quantitative Understanding of the Linkage
PQ ability to trigger mitochondrial ROS production (O2°− and correlated species) by redox cycling has been demonstrated in vitro, both in isolated mitochondria, mitochondrial brain homogenates and cells and ex‐vivo from brain mitochondria isolated from PQ‐treated rats. In vivo evidence of oxidative stress, as a consequence of PQ exposure, is mainly supported by the occurrence of lipoperoxidation, accumulation of oxidised protein or by mean of sodium salicylate molecular trap.
PQ (0.1–1 mM) induces ROS production within minutes in isolated mitochondria and mitochondrial brain fraction (Castello et al., 2007; Cochemé and Murphy, 2008), while in cells this process is detectable after 2–6 h from the exposure in dependence on the dose (Cantu et al., 2011, Dranka et al., 2012; Huang et al., 2012; Rodriguez‐Rocha et al., 2013). Based on the work of Cantu et al. (2009), which compare O2 and H2O2 production by PQ (0.25–0.5 mM) along different time points, O2 formation slightly precedes H2O2 production at the lowest PQ concentrations (Tab.1). In addition, at these time points no death is usually detected in cells exposed to PQ up to 1 mM, pointing at ROS production as an early event preceding cell death.
Table A.14.
Quantitative evaluation of the KER
| Treatment | PQ redox cycling with superoxide formation | ROS formation (KE1) | Reference |
|---|---|---|---|
| Rat primary mesencephalic cell culture PQ at 0.25–1 mM |
Inhibition of aconitase after 3: 43% at 0.25 mM 58% at 0.5 mM |
Increase in H2O2 At 2 h 17% at 1 mM At 4 h 28% at 0.5 mM and 64% at 1 mM At 6 h 31% at 0.25 mM, 59% at 0.5 mM and 119% at 1 mM |
Cantu (2009) |
| N27 cell culture, PQ at 0.3–1 mM | Inhibition of aconitase, 80% at 0.5 mM, 98% at 1 mM at 4 h |
Increase in H2O2 at 4–6 h 25% at 0.3 mM and 33% at 1 mM |
Cantu (2011) Dose‐dependent neuronal cell death occurring at 18 h but not at 4–6 h |
| SK‐N‐SH human neuroblastoma cells treated with PQ 0.2 mM up to 1 mM. 6–62 h sampling | Dose and time related increase of O2 by electro paramagnetic resonance spectroscopy. 50% at 0.2 mM, 80% at 0.5 mM and 150% at 1 mM at 24 h |
Increase in DHE ROS production 800% at 0.5 mM at 48 h |
Rodriguez Rocha (2013) |
| SD rat treated at 25 mg/kg and observed 24 h later |
H2O2 increase of 150% in isolated mitochondria from SN neurons corresponding to 42% mitochondrial lipid peroxidation Decrease in Complex I 33% and Complex IV 21% Increase mitochondrial membrane potential |
Czerniczyniec (2015) | |
| SD rat treated at 10 mg/kg weekly for 4 weeks |
Increase in O2 production in isolated mitochondrial of 20% Decrease in aconitase activity in mitochondrial of 50% in striatum |
Increase in lipid peroxidation in isolated mitochondria of 30% | Czerniczyniec (2015) |
| C57BL/6 mice treated with 10 mg/kg PQ i.p. once a week for 3 weeks |
Increased neuronal lipid peroxidation measured 1 day after weekly injection each: 10 mg kg i.p. 200% increase in lipid peroxidation at 2 and 4 days post‐inj 500–600% in lipid peroxidation after 2nd injection 2/4 days after After third injection limited response due to significant neuronal cell loss |
McCormack et al. (2005) Neuronal cell loss up to 30% in mid brain sections |
|
| Swiss albino mice i.p. at 5 and 10 mg kg twice a week for 4 weeks |
SOD activity ex vivo At 5 mg/kg increase of 42% At 10 mg/kg increase of 75% |
Glutatathione s transferase activity ex vivo At 5 mg/kg increase of 25% At 10 mg/kg increase of 75% Catalase activity ex vivo At 5 increase of 17% At 10 increase of 50% |
Mitra (2011) Neuronal cell loss of 40% TH positive and Fox 3 positive and motor dysfunction symptoms at 10 mg/kg and 10% at 5 mg/kg. Motor symptoms only at 5 mg/kg |
Uncertainties or inconsistencies
Besides mitochondria, NADPH‐oxidase 1 (NOX1) (Cristovao et al., 2012) and plasma membrane microglia NOX (Rappold et al., 2011) also contribute to PQ‐induced ROS production. Furthermore, in vitro data suggest that for time points of exposure longer than 48 h oxidative stress occurs both at mitochondria and cytosol in dependence to the dose. Thus it is difficult to discriminate the source of PQ‐induced ROS and the early involvement of mitochondria in vivo due to the extensive treatments and to the indirect detection of oxidative stress mainly by mean of lipoperoxidation, protein oxidation. Mitochondrial involvement is suggested by ex‐vivo studies (Czerniczyniec et al., 2013, 2015).
Mitochondrial loss of function (i.e. decrease in mitochondrial membrane) might sometimes be the consequence of cell death rather than directly resulting from oxidative stress. This is due to the estimation of this parameter at time points already characterised by a significant cell death without a double staining, which allow discriminating between alive and dead cells. The observation that loss of mitochondrial membrane potential on PQ exposure is only detected in the population of dead cells when cells are double stained for mitochondrial membrane potential and plasma membrane integrity (Rodriguez‐Rocha, 2013) support this uncertainty.
References
Cantu D, Schaack J, Patel M, 2009. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2‐mediated neurotoxicity in rat primary mesencephalic cultures. Public Library of Science (PLoS ONE), 4, e7095.
Cantu D, Fulton RE, Drechsel DA, Patel M, 2011. Mitochondrial aconitase knockdown attenuates paraquat‐induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2. Journal of Neurochemistry, 118, 79–92.
Castello PR, Drechsel DA, Patel M, 2007. Mitochondria are a major source of paraquat‐induced reactive oxygen species production in the brain. Journal of Biological Chemistry, 282, 14186–14193
Cohen GM, d'Arcy Doherty M, 1987. Free radical mediated cell toxicity by redox cycling chemicals. British Journal of Cancer. Supplement 8, 46–52.
Cochemé HM, Murphy MP, 2008. Complex I is the major site of mitochondrial superoxide production by paraquat. Journal of Biological Chemistry, 283, 1786–1798.
Chau KY, Korlipara LV, Cooper JM, Schapira AH, 2009. Protection against paraquat and A53T alpha‐synuclein toxicity by cabergoline is partially mediated by dopamine receptors. Journal of the Neurological Sciences, 278, 44–53.
Chau KY, Cooper JM, Schapira AH, 2010. Rasagiline protects against alpha‐synuclein induced sensitivity to oxidative stress in dopaminergic cells. Neurochemistry International, 57, 525–529.
Choi HS, An JJ, Kim SY, Lee SH, Kim DW, Yoo KY, Won MH, Kang TC, Kwon HJ, Kang JH, Cho SW, Kwon OS, Park J, Eum WS, Choi SY, 2006. PEP‐1‐SOD fusion protein efficiently protects against paraquat‐induced dopaminergic neuron damage in a Parkinson disease mouse model. Free Radical Biology and Medicine, 41, 1058–1068.
Cristóvão AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS, 2012. NADPH oxidase 1 mediates α‐synucleinopathy in Parkinson's disease. Journal of Neuroscience, 32, 14465–14477
Czerniczyniec A, Lores‐Arnaiz S, Bustamante J, 2013. Mitochondrial susceptibility in a model of paraquat neurotoxicity. Free Radical Research, 47, 614–623.
Czerniczyniec A, Lanza EM, Karadayian AG, Bustamante J, Lores‐Arnaiz S, 2015. Impairment of striatal mitochondrial function by acute paraquat poisoning. Journal of Bioenergetics and Biomembranes, 47, 395–408.
de Oliveira MR, Ferreira GC, Schuck PF, 2016. Protective effect of carnosic acid against paraquat‐induced redox impairment and mitochondrial dysfunction in SH‐SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicology in Vitro.
Dinis‐Oliveira RJ, Remião F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F, 2006. Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology, 27, 1110–1122.
Dranka BP, Zielonka J, Kanthasamy AG, Kalyanaraman B, 2012. Alterations in bioenergetic function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. Journal of Neurochemistry, 122, 941–951.
Drechsel DA, Patel M, 2009. Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicological Sciences, 112, 427–434.
Filograna R, Godena VK, Sanchez‐Martinez A, Ferrari E, Casella L, Beltramini M, Bubacco L, Whitworth AJ, Bisaglia M, 2016. SOD‐mimetic M40403 is protective in cell and fly models of paraquat toxicity: implications for Parkinson disease. Journal of Biological Chemistry, pii: jbc.M115.708057.
Liang LP, Kavanagh TJ, Patel M, 2013. Glutathione deficiency in Gclm null mice results in complex I inhibition and dopamine depletion following paraquat administration Toxicological Sciences, 134, 366–373.
McCormack AL1, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA, 2005. Role of oxidative stress in paraquat‐induced dopaminergic cell degeneration. Journal of Neurochemistry, 93, 1030–1037.
Mitra S, Chakrabarti N, and Bhattacharyy A, 2011. Differential regional expression patterns of α‐synuclein, TNF‐α, and IL‐1β; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S, 2004. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. Journal of Neurochemistry, 88, 657–667.
Huang CL, Lee YC, Yang YC, Kuo TY, Huang NK, 2012. Minocycline prevents paraquat‐induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicology Letters, 209, 203–210.
Kappus H, 1986. Overview of enzyme systems involved in bio‐reduction of drugs and in redox cycling. Biochemical Pharmacology, 35, 1–6.
Lopert P, Day BJ, Patel M, 2012. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. Public Library of Science. PLoS ONE, 7, e50683.
McCarthy S1, Somayajulu M, Sikorska M, Borowy‐Borowski H, Pandey S, 2004. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water‐soluble Coenzyme Q10. Toxicology and Applied Pharmacology, 201, 21–31.
Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochemical Journal, 417, 1–13.
Peng J, Stevenson FF, Doctrow SR, Andersen JK, 2005. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat‐mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. Journal of Biological Chemistry, 280, 29194–29198.
Rappold PM, Cui M, Chesser AS, Tibbett J, Grima JC, Duan L, Sen N, Javitch JA, Tieu K, 2011. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter‐3. Proceedings of the National Academy of Sciences USA, 108, 20766–20771.
Rodriguez‐Rocha H, Garcia‐Garcia A, Pickett C, Li S, Jones J, Chen H, Webb B, Choi J, Zhou Y, Zimmerman MC, Franco R, 2013. Compartmentalised oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: distinct roles of superoxide anion and superoxide dismutases. Free Radical Biology and Medicine.
Vasquez‐Vivar J, Kalyanaraman B, Kennedy MC, 2000. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. Journal of Biological Chemistry, 275, 14064–14069.
2nd KER: Mitochondrial dysfunction results in an impaired proteostasis
How this Key Event Relationship work
See AOP 1 (p. 124)
Weight of Evidence
See AOP 1 (p. 124)
Biological Plausibility
See AOP 1 (p. 124)
Empirical support for linkage
Based on the existing in vitro and in vivo data it is suggested that mitochondrial dysfunction impairs protein homoeostasis through oxidative and nitrosative stress resulting in protein aggregation, damaged intracellular transport of proteins and cell organelles.
Paraquat 0.5 mM decreases mitochondrial complex V activity, ATP production and proteasome activity in SH‐SY5Y cells. All these effects increase in time (from 6 to 48 h) and are significant at 24 and 48 h of treatment. In addition, PQ significantly decreases proteasome 19S subunit – but not 20S – only at 48 h. However, since this 19S subunit drops later than proteasome activity decrease, it could not have caused proteasome dysfunction. Significant increased levels of a‐syn and ubiquitinated proteins are also evident at 24 and 48 h following PQ exposure. SH‐SY5Y death occurred only at 48 h. Cell death is dose dependent (PQ 0.05 – 1 mM) and is significant at 0.5 and 1 mM (57 and 75% respectively). PQ induces mitochondrial dysfunction and proteasome impairments leading to neuronal death (Yang and Tiffany‐Castiglioni, 2007).
Reduced mitochondrial membrane potential and proteasome inhibition has been also observed for 0.2 mM PQ as early as 3 h after exposure in SH‐SY5Y cells. A slight but significant effect also occurs at 0.02 mM PQ at longer time (6 h). 0.2 mM PQ‐induced effects precede neuronal death (12 h; no death observed at 0.02 mM). Transfection of the heat shock protein HDJ‐1 (that attenuate protein aggregation without altering ROS production, as measured by DCF) in SH‐SY5Y cells attenuates 0.2 mM PQ‐induced mitochondrial membrane potential decrease at 6 h (from 50% to 80%). This suggests that protein aggregation also contribute to the loss of mitochondrial membrane potential (Ding and Keller, 2001).
Paraquat (10 mg/kg, once a week for 3 weeks) in combination with DJ‐1 deficiency decreases ATP levels, proteasome activities, proteasome subunits levels and increases ubiquitinated proteins in the ventral midbrain including SNpc. None of these effects is observed at the striatum (Yang et al., 2007). DJ‐1 has been suggested to contribute to mitochondrial integrity due to its localisation in the mitochondrial matrix and inter‐membrane space (Zhang et al., 2005) and its antioxidant action (Taira et al., 2004). Likewise, exposure to PQ and deficiency of DJ‐1 might cooperatively induce mitochondrial dysfunction resulting in ATP depletion and contribute to proteasome dysfunction in the brain.
Paraquat (10 mg/kg i.p.) induced significant increase in lipid peroxides (LPO) in ventral midbrain (VM), striatum (STR) and frontal cortex (FCtx), maximum in VM after 5 doses (2.4 times the control). An elevated LPO level was still present in VM after 28 days. Moreover, the activity of 20S proteasome in STR was altered (increased 40–50%) after a single dose and slightly reduced after 5 doses (Prasad et al., 2007). The temporal activation of proteasomal activity at 1 and 24 h after single dose was explained by the fact that carbonylated proteins moderately undergo degradation by UPS (Poppek and Grune, 2006). Sublethal proteasome inhibition induces neurons to increase proteasome activity and promotes resistance to oxidative injury (Lee et al., 2004).
Quantitative evaluation of KERs
Table A.15.
Quantitative evaluation of the KER
| Treatment | Mitochondrial dysfunction (KE2) | Impaired protein degradation (KE3a) | Reference |
|---|---|---|---|
| SHSY5Y cells, PQ 0.5 mM, 12, 24 and 48 h |
Decreased activity of complex V (% of control; significant): 12 h ne 24 h 70% 48 h 50% decreased ATP levels (% of control): 12 h ne 24 h 76% 48 h 39% |
Decreased proteasome activity (% of control): 12 h ne 24 h 40% 48 h 23% Decreased protein level of 19S subunit (% of control): 12 h ne 24 h ne 48 h 32% ne on 20S a and b at any time Increased level of ubiquitinated proteins (% of control): 12 h ne 24 h 154.5% 48 h 167% Increased protein level of a‐syn: 12 h ns 24 h 236% 48 h 305% |
Yang and Tiffany‐Castiglioni (2007) Comments: PQ induced significant SHSY5Y cells death only at 48 h thus mitochondrial dysfunction and impaired protein degradation occurs before neurons die. Furthermore, the lack of effect on 20S subunits suggests that the observed paraquat effects were not nonspecific cytotoxic events Levels of 19S dropped at 48 but not 24 h after paraquat treatment, and therefore could not have caused the proteasome dysfunction observed |
|
SHSY5Y cells, PQ 20 and 200 μM, different time points SHSY5Y transfected with HDJ‐1 (member of the Hsp40 family, attenuate protein aggregation), PQ 200 μM for 6 h |
Reduced mitochondrial membrane potential (% of control): 20 μM‐ 6 h approx. 80%? Reduced of 20% vs control 200 μM‐ 3 h approx. 60%? Reduced 40% vs control 6 h approx 40% reduced 60% vs control Partial significant (20% vs PQ treated only) recovery of mitochondrial membrane potential |
Reduced proteasome activity (% of control) 20 μM‐ 6 h 85% significant reduced of 15% vs control 200 μM‐ 1 h approx. 80% reduced of 20% vs control 3 h approx 60% reduced of 40% vs control 6 h approx. 55% reduced of 65% vs control Partial significant (25% vs PQ treated only) recovery of proteasome activity |
Ding and Keller (2001) Comments: Death at 6 h not measured, significant death at 24 h for 20 μM and 12 h for 200 μM Co‐treatment with 20 μM PQ + epoxomycin 1 μM (proteasome inhibitor) exacerbate PQ‐induced mitochondrial membrane potential decrease (to 75% vs control or 60% vs 20 nM PQ treated only) and cell death. The ability of increased levels of HDJ‐1 to attenuate proteasome inhibition did not appear to be due to a decrease in ROS levels, or altered levels of proteasome subunits |
| Mice WT and DJ‐deficient, 10 mg/kg PQ, once a week for 3 weeks | ATP levels in VMB decreased of 30% in DJ deficient (vs control) |
|
Yang et al. (2007) Effects evident only in VMB (include SNpC) and not in striatum and only in DJ‐deficient mice. DJ‐deficient as WT for all the parameters. Additional measurements:
Thus concordance motor symptoms and decreased dopamine, but not effect on neurons: authors suggested that behavioural and neurochemical consequences manifest before dopamine neuron degeneration |
| PQ 10 mg/kg i.p. (administered 3 times/week for a total of 1, 3 or 5 doses) in C57BL/6J mice | Increased tissue level of lipid peroxides (LPO) after a single (and persistent up to 28 days) and repeated doses, maximum in VM after 5 doses (2.4 times the control, lower in STR (80%) and least (66%) in FrCtx |
Increased activity 20S proteasome in STR (not quant in other tissues) at 1 (40%) and 24 h (50%) after single i.p. dose 20S activity was reduced in STR after 5 doses (15%) |
Prasad et al. (2007) |
ne: negative.
Uncertainties or inconsistencies
The exact molecular link from mitochondrial dysfunction to disturbed proteostasis is not known. It is not clear which is the oxidative modification that drives the process.
Proteostasis incidence is higher than mitochondrial dysfunction at PQ 0.5 mM (Yang and Tiffany‐Castiglioni, 2007) but not at PQ 0.2 mM (Ding and Keller, 2001) at the same time point in SH‐SY5Y cells. These results suggest that, in vitro, at doses higher than 0.2 mM PQ might involve mechanisms other than mitochondrial dysfunction.
The sequence of events that link mitochondrial dysfunction to proteases inhibition is not entirely clear, proteosomal dysfunction might contribute to mitochondrial dysfunction (Ding and Keller, 2001). On the other side, sublethal proteasome inhibition induces neurons to increase pretoasome activity and promotes resistance to oxidative injury (Lee et al., 2004), whereas oxidative stress can increase proteasome activity early in the sequence leading to cell death in vitro (Holtz et al., 2006).
A vicious circle is observed that make it difficult to establish an exact quantitative relationship between mitochondrial and proteosomal dysfunction. This task needs a better dose‐ and time‐related definition of PQ effect on those two events that is actually lacking.
Lack of evidences of the link between mitochondrial dysfunction and disturbed proteostasis in WT animals exposed to PQ.
Distinct unfolded protein response (UPR) signalling branches could have specific and even opposite consequences on neuronal survival depending on the disease input (Hetz and Mollereau, 2014). Proteostasis impairment at the level of the endoplasmic reticulum (ER) is emerging as a driving factor of dopaminergic neuron loss in PD. ER stress engages the activation of the UPR adaptive reaction to recover proteostasis or trigger apoptosis of damaged cells. PQ may induce ER stress (Huang et al., 2012).
A genetic screening in yeast revealed that one of the major physical targets of α‐Synuclein is Rab1, an essential component of the ER‐to‐Golgi trafficking machinery (Cooper et al., 2006; Gitler et al., 2008). Overexpression of Rab1 in animal models of PD reduced stress levels and protected dopaminergic neurons against degeneration (Coune et al., 2011).
References
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, LaBaer J, Rochet JC, Bonini NM, Lindquist S, 2006. α‐Synuclein blocks ER‐Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science, 313, 324–328.
Coune PG, Bensadoun JC, Aebischer P, Schneider B, 2011. Rab1A over‐expression prevents Golgi apparatus fragmentation and partially corrects motor deficits in an alpha‐synuclein based rat model of Parkinson's disease. Journal of Parkinson's Disease, 1, 373–387. doi: 10.3233/JPD‐2011‐11058
Ding Q, Keller JN, 2001. Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. 2001. Journal of Neurochemistry, 77, 1010–1017.
Hetz C, Mollereau B, 2014. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nature Reviews Journal of Neuroscience, 15, 233–249.
Holtz WA, Turetzky JM, O'malley KL, 2008. Oxidative stress triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. Journal of Neurochemistry, 99, 54–69.
Huang CL, Lee YC, Yang YC, Kuo TY, Huang NK, 2012. Minocycline prevents paraquat‐induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicology Letters, 209, 203–210. doi: 10.1016/j.toxlet.2011.12.021
Lee CS, Tee LY, Warmke T, Vinjamoori A, Cai A, Fagan AM, Snider BJ, 2004. A proteasomal stress response: pre‐treatment with proteasome inhibitors increases proteasome activity and reduces neuronal vulnerability to oxidative injury. Journal of Neurochemistry, 91, 996–1006.
Poppek D, Grume T, 2006. Proteosomal defense of oxidative protein modifications. Antioxidants & Redox Signaling, 8, 173–184.
Prasad K, Winnik B, Thiruchelvam MJ, Buckley B, Mirochnitchenko O, 2007. Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain, 115, 1448–1453.
Taira T, Saito Y, Niki T, Iguchi‐Ariga SM, Takahashi K, Ariga H, 2004. DJ‐1 has a role in antioxidative stress to prevent cell death. European Molecular Biology Organization Reports (EMBO), 5, 213–218.
Yang W, Tiffany‐Castiglioni E, 2007. The bipyridyl herbicide paraquat induces proteasome dysfunction in human neuroblastoma SH‐SY5Y cells. Journal of Toxicology and Environmental Health. Part A, 70, 1849–1857.
Yang W, Chen L, Ding Y, Zhuang X, Kang UJ, 2007. Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ‐1 deficient mice. Human Molecular Genetics, 16, 2900–2910.
Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL, 2005. Mitochondrial localization of the Parkinson's disease related protein DJ‐1: implications for pathogenesis. Human Molecular Genetics, 14, 2063–2073.
3rd KER: Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway
How this key event relationship works
See AOP 1 (p.143)
Weight of evidence for the KER
1. Biological plausibility
See AOP 1 (p.143)
2. Empirical support for linkage
Large part of the empirical evidence supporting this KEs relationship comes from observational studies conducted in human affected by PD, from in in‐vitro and in‐vivo studies conducted with the chemical stressors MPTP and rotenone or from experiments conducted with proteasome inhibitors. With the chemical stressor paraquat, used for the empirical support of this AOP, most of the studies where providing evidence that in the same experiments impaired proteostasis and neuronal degeneration where co‐existing. Although different concentrations of paraquat wher used in the in‐vitro assays, in‐vivo studies where generally conducted at fixed dose though different doses scheduling could have been applied.
Paraquat is an herbicide for which a unique sensitivity of dopaminergic neuronal cells was also observed (Brooks et al., 1999; McCormack et al., 2002; Uversky, 2004). Similarly to MPTP and Rotenone, also in paraquat treated mice, an up regulation and aggregation of α synuclein and inhibition of the proteosomal pathway was demonstrated in DA neurons in SN (Manning‐Bog et al., 2002; Wills et al., 2012). Additionally, paraquat is able to reduce proteosomal function in DJ‐I deficient mice with an impaired clearance of altered proteins (Yang et al., 2007). Paraquat is clearly more toxic in aged animal or when co‐administered with the fungicide maneb (McCormack et al., 2002; Thiruchelvam et al., 2003). Duration of treatment could also impact neuronal loss (Ossowska et al., 2005).
Human evidences
Human data from PD patients are indicative of an overall inhibition of axonal autophagy with an increased level of mTor (a major protein involved in autophagy) which was accompanied with an impairment to form autophagosome. The observed increase in m TOR levels was of 63% (Willis et al., 2012).
Inclusion bodies in DA neurons (i.e. Lewy bodies), a pathological hallmark for sporadic PD, stains specifically for proteins associated with the UPS (McNaught et al., 2002; Fornai et al., 2005), including α‐synuclein, parkin and ubiquitin; possibly indicating that failure of the UP system represents a common step in the pathogenesis of PD and impairment of the proteasome system was found in humans affected by sporadic PD (McNaught et al., 2001, 2003).
Lysosomal breakdown and autophagosome (AP) accumulation with co‐localisation of lysosomal markers in Lewy Bodies is reported to occur in PD brain samples where Lewy bodies were strongly immunoreactive for the autophagosome markers (LC3II) (Dehay et al., 2010).
Postmortem studies on PD patients show axonal pathology that is likely to precede the loss of neuronal bodies In this investigation, TH immunoreactive fibres had almost entirely disappeared with preservation of neuronal bodies (Orimo et al., 2005, 2008).
Paraquat
Paraquat (10 mg/kg i.p. once a week for 3 consecutive weeks) exposure in male mice (control mice and transgenic mice expressing either wild type human α‐synuclein or mutant form of the human protein) induced in control mice accumulation of intracellular α‐synuclein‐immunoreactive deposits in 30% of dopaminergic neurons and decreases by 25–35% the number of TH positive and Nissl‐stained neurons in SNpc following stereological evaluation. A protective effect (presence of intracellular protein positive deposits –36% with lack of neurodegeneration) was observed in animal overexpressing the wild as well as a mutated form of α‐synuclein. In these animals a concomitant increase of HSP70 chaperone protein was observed (Manning‐Bog et al., 2003). Heat shock proteins has been reported to play a protective role against PQ toxicity (Ding and Keller, 2001; Minois et al., 2001) and its increase may represent an adaptive change to high intraneuronal α‐synuclein concentrations.
Weekly i.p. injection of 10 mg/kg of paraquat for 3 weeks in male mice overexpressing α‐synuclein induced loss of dopaminergic neurons in SNpc and decrease in TH optical density (slight) in the striatum which was accompanied by an increase of intracytoplasmic insoluble α‐synuclein (Fernagut et al., 2007). (Similar decrease in dopaminergic neurons, without α‐synuclein accumulation, was observed also in PQ‐treated WT animals.)
Administration of 10 mg/kg i.p. twice a week for 4 weeks to adult Swiss albino mice induced dopaminergic neuronal loss (ca. 40% reduction) in SN (also in FC and hippocampus) which was associated with a decrease in α‐synuclein expression (ca. 50% reduction) (increased in hippocampus) and reduction of TH levels (ca. 50% reduction) in SN (and hippocampus) (Mitra et al., 2011). The reduced α‐synuclein expression in SN, increased expression in hippocampus, and aggregated forms in FC might correlate with α‐synuclein gene polymorphism associated with PQ‐mediated neurotoxicity and the differential time frames necessary to initiate neurodegeneration in the different regions.
In male Wistar rat receiving four i.p. injections, separated by 1 day, of paraquat at 10 mg/kg per day, showed a 50% increase of α‐synuclein immunoreactivity and protein level (by Western blot) in SN. The stereological count of TH‐positive neurons showed that Nox 1 knockdown animals (stereotaxically injected with a viral constructed expressing Nox 1 or ablated for it) treated with Paraquat, significantly reduced PQ‐elicited dopaminergic neuronal loss from 37% in the group treated with vector and PQ to 13% in the Nox 1 KO treated with PQ. Nox 1 knockdown reduced by 37% the PQ‐mediated α‐synuclein levels, compared to vector plus PQ, as well as α‐synuclein aggregation and it was accompanied by a reduction in α‐synuclein immunoreactivity and protein level as well as a decrease in α‐synuclein aggregation (Cristovao et al., 2012).
Proteasome activity was investigated in dopaminergic SH‐SY5Y cells treated with paraquat. Results showed that at a concentration of paraquat that reduced viability by about 60% at 48 h (0.5 mM) loss of proteasome activity occurred. Furthermore, paraquat‐treated cells showed decreased protein levels of proteasome 19S subunits, but not 20S alpha or beta subunits, suggesting that the effects observed were not the result of general cytotoxicity. Paraquat also increased levels of alpha‐synuclein and ubiquitinated proteins, suggesting that paraquat‐induced proteasome dysfunction leads to aberrant protein accumulation (Yang et al., 2007).
Low concentration of paraquat (10 μM) induced autophagy in human neuroblastoma cells line (SH‐SY5Y). Paraquat induced autophagic vacuoles (AV) and recruitment of LC3‐GFP fusion protein to AV. Finally, cell death with hallmarks of apoptosis was observed. Paraquat also increased long‐lived protein degradation which was blocked by the autophagy inhibitor 3‐methyladenine (3‐MA). While caspase inhibition retarded cell death, autophagy inhibition accelerated the apoptotic cell death induced by paraquat (Gonzalez‐Polo et al., 2007).
SH‐SY5Y cell transfected with DJ‐1‐specific siRNA and exposed to paraquat showed additive effect on apoptotic cell death, inhibition of the cytoplasmic accumulation of autophagic vacuoles as well as recruitment of LC3 fusion protein to the vacuoles. The effect was time and dose related (25–500 μM) (Gonzalez‐Polo et al., 2009). Apoptotic cell death was accelerated by treatment with the autophagy inhibitor 3‐methyladenine (3‐MA). Findings suggest an active role for DJ‐1 in the autophagic response produced by Paraquat, providing evidence for the role of PD‐related proteins in the autophagic degradation pathway.
Paraquat (500 μM) triggers endoplasmic reticulum stress and cell death (70% reduction in cell survival) and inhibits proteosomal activity (60–70% reduction) in a rat N27 mesencephalic dopaminergic cells system (Shankar et al., 2008).
Males C57BL/6NCrlVr mice received i.p. injections of 10 mg/kg of paraquat twice a week for 4 weeks showed a decrease in TH+ neurons of approximately 43%. This was accompanied by: increased of 133% of α‐synuclein, increased by 13% (not statistically significant) in 19S proteasome function and decrease of 5% 20S proteasome function (not stat significant), increase by 43% in mTOR (autophagy inhibitor), increase by 81% of beclin‐1 (autophagy inducer) and increase in Atg12 of 36% (Su et al., 2015).
Quantifiable understanding
A quantitative relationship has been established between the chemical stressor paraquat inducing impaired proteostasis and loss of DA neurons of nigrostriatal pathway. A response concordance was observed for the quoted studies; however dose and time relationship could be only established in a limited number of in‐vitro studies as the in‐vivo studies were conducted at single dose and single evaluation time‐point.
| Impaired proteostasis | DA neurons degeneration | Treatment | References |
|---|---|---|---|
| Intracellular deposit of in α‐synuclein observed in 30% of DA neurons | Approx. 30 (25–35%) % of cell loss (TH positive cells) in SNpc | C57BL/6 mice treated with Paraquat once a week for 3 weeks at 10 mg/kg i.p. |
Manning –Bog et al. (2003) Fernagut (2007) |
| Increase of approx. 91% of α‐synuclein inclusion (proteinase‐K‐resistant α‐syn aggregates) only observed in α‐synuclein overexpressing animals | Approx. 25% loss of DA neurons (stereological analysis TH‐positive neurons) in both WT as well as α‐synuclein overexpressing animals | Weekly i.p. administration of 10 mg/kg paraquat for 3 weeks in mice WT and overexpressing α‐synuclein | |
| Approx. 50% reduction in α‐synuclein expression in SN | Approx. 40% loss of DA neurons (TH+ and FOX3 + neurons) | Paraquat 10 mg/kg i.p. twice a week for 4 weeks to adult Swiss albino mice | Mitra et al. (2011) |
| Approx. 50% increase of α‐synuclein expression (immunoreactivity and protein) | Paraquat significantly reduced PQ‐elicited dopaminergic neuronal loss from 37% | Wistar rat receiving four i.p. injections, separated by 1 day, of paraquat at 10 mg/kg per day | Cristovao et al. (2012) |
|
Proteasome inhibition (approx. 60% at 24 h and 80% at 48 h) Increased protein levels of α‐synuclein (2.3‐fold at 24 h and 3‐fold at 48 h) Increased ubiquinated protein levels (1.5‐fold at 24 h and 1.7‐fold at 48 h) |
Reduction of 60% in cell viability at 48 h | DA SH‐SY5Y cells treated with paraquat 0.5 mM | Yang et al. (2007) |
|
Accumulation of AV (%vacuolated cell volume) at 6, 15 and 24 h was 20, 40 and 45% respectively. Inhibition of PQ‐induced autophagic vacuolisation and protein degradation after treatment with 3‐MA |
25% of nuclear apoptosis at 24 h (caspase‐3 maximum level) Apoptosis cell death was accelerated and caspase‐3 activation increased after 3‐MA treatment |
DA SH‐SY5Y cells treated with paraquat 10 μM DA SH‐SY5Y cells treated with paraquat 10 μM were then treated with prototypic autophagy inhibitor 3‐MA 10 mM |
Gonzalez‐Polo et al. (2007) |
|
SiRNA knockdown of DJ‐1 has no effect alone on the formation of autophagic vacuoles. In the presence of PQ (250 μM), DJ‐1 knockdown significantly inhibited cytoplasmic accumulation of autophagic vacuoles, with an additive increase in apoptotic chromatin condensation |
SiRNA knockdown of DJ‐1 induces apoptotic death (25–30%) The combination of DJ‐1 si RNA and Paraquat induces additive apoptotic death (more significant in the range 250–500 μM PQ) and caspase‐3 activation. Apoptosis cell death was accelerated after 3‐MA treatment |
DA SH‐SY5Y cells transfected with DJ‐1 si RNAs and exposed to paraquat 250–500 μM DA SH‐SY5Y cells transfected with DJ‐1 si RNAs exposed to paraquat 250–500 μM treated with prototypic autophagy inhibitor 3‐MA 10 mM |
Gonzalez‐Polo et al. (2009) |
| Increased expression of ER stress proteins and inhibition proteosomal activity (60–70% reduction at 500 μM) | Time and concentration‐dependent cell death (70% with 500 μM of PQ for 48 h) reduction in cell survival | Rat N27 mesencephalic dopaminergic cells treated with Paraquat (100–500 μM) for 12–48 h | Chinta et al. (2010) |
|
Increased 133% of α‐synuclein Increased by 13% (not statistically significant) in 19S proteasome function and decrease of 5% 20S proteasome function (not stat signif) Increase by 43% in mTOR (autophagy inhib) Increase 81% beclin‐1 (autophagy inducer) Increased in Atg 12 of 36% Decrease of appr 24% in LC3 II to LC3 I ratio |
C57BL/6 mice treated with Paraquat twice a week for 6 weeks at 10 mg/kg i.p. (12 doses) | Wills et al. (2012) | |
|
Increase 115% α‐synuclein in striatum 10% decrease in 19S proteasome function and 5% in 20S proteasome function (both not statist significant) Increase 47% in mTOR and stat sig in beclin‐1(81–95%) Increase in Atg12 (40%) LC3 II to LC3 I ratio decreased up to 25% |
TH neuronal loss 43% | C57BL/6NC mice treated with Paraquat twice a week for 4 weeks at 10 mg/kg i.p. | Su et al. (2015) |
Uncertainties or inconsistencies
The ability of paraquat to induce loss of DA neurons in SN in vivo is sometime equivocal. Loss of 60% of DA neurons in SN and 90% of their striatal terminals are reported (Brooks et al., 1999) following repeated treatment with paraquat but less significant evidence, or no evidence, has been reported in later studies (Thiruchelvam et al., 2000; McCormack et al., 2002). No effect of paraquat on dopaminergic neurons has been reported by some authors (Widdowson et al., 1996; Breckenridge et al., 2013; Minnema et al., 2014). However, the applied dose, the treatment scheduling, the route of administration as well as the animal age, species and strain (McCormack et al., 2002; Thiruchelvam et al., 2003; Yin et al., 2011; Jiao et al., 2012; Tieu, 2016) are all important factor to be considered in the evaluation of the study's outcome.
Dopaminergic neurons in SN and VTA seem to have a different susceptibility to the damage induced by paraquat (McCormack et al., 2006). However, whether impaired proteostasis and protein aggregation would cause the selective death of DA neurons in the SN still remain an uncertainties.
Selectivity of paraquat‐induced DA neuronal cell death still remains uncertain. Similar effect on other brain region i.e. frontal cortex and hippocampus) are also affected (Mitra et al., 2011).
The vulnerability of the dopaminergic pathway still remains circumstantial. Paraquat has been proposed to pass the blood‐brain‐barrier by mediation of neutral amino acid transportation (Shimizu et al., 2001; McCormack et al., 2003). Accumulation of paraquat in the brain is reported to be age dependent, possibly indicating a role for the blood‐brain‐barrier permeability (Corasaniti et al., 1991); however, paraquat is not a substrate for dopamine transporter (Richardson et al., 2005), and hence how the toxicant enters into dopaminergic neurons still remain uncertain.
References
Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, Cook AR, Botham PA, Smith LL, 2013. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology, 37, 1–14. doi: 10.1016/j.neuro.2013.03.005
Brooks AI, Chadwick CA, Gelbard HA, Cory‐Slechta DA, Federoff HJ, 1999. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Research, 823, 1–10.
Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, Rao RV, 2009. Endoplasmic reticulum stress–induced cell death in dopaminergic cells: effect of resveratrol. Journal of Molecular Neuroscience, 39, 157–168.
Corasaniti MT, Defilippo R, Rodino P, Nappi G, Nistico G, 1991. Evidence that paraquat is able to cross the bloodbrainbarrier to a different extent in rats of various age. Functional Neurology, 6, 385–391.
Cristóvão AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS, 2012. NADPH oxidase 1 mediates α‐synucleinopathy in Parkinson's disease. Journal of Neuroscience, 32, 14465–14477. doi: 10.1523/JNEUROSCI.2246‐12.2012
Dehay B, Bové J, Rodríguez‐Muela N, Perier C, Recasens A, Boya P, Vila M, 2010. Pathogenic lysosomal depletion in Parkinson's disease. Journal of Neuroscience, 30, 12535–12544. doi: 10.1523/JNEUROSCI.1920‐10.2010
Ding Q, Keller JN, 2001. Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. 2001. Journal of Neurochemistry, 77, 1010–1017.
Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF, 2007. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha‐synuclein over‐expression. Synapse, 61, 991–1001.
Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, et al., 2005. Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin‐proteasome system and alpha‐synuclein. Proceedings of the National Academy of Sciences, 102, 3413–3418.
González‐Polo RA, Niso‐Santano M, Ortíz‐Ortíz MA, Gómez‐Martín A, Morán JM, García‐Rubio L, Francisco‐Morcillo J, Zaragoza C, Soler G, Fuentes JM, 2007. Inhibition of paraquat‐induced autophagy accelerates the apoptotic cell death in neuroblastoma SH‐SY5Y cells. Toxicological Sciences, 97, 448–458.
González‐Polo R, Niso‐Santano M, Morán JM, Ortiz‐Ortiz MA, Bravo‐San Pedro JM, Soler G, Fuentes JM, 2009. Silencing DJ‐1 reveals its contribution in paraquat‐induced autophagy. Journal of Neurochemistry, 109, 889–898.
Jiao Y, Lu L, Williams RW, Smeyne R, 2012. Genetic dissection of strain dependent paraquat‐induced neurodegeneration in the substantia nigra pars compacta. PUBLIC LIBRARY OF SCIENCE (PLOS ONE), 7, e29447.
Manning‐Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, DiMonte D, 2002. The herbicide paraquat causes up‐regulation and aggregation of α‐synuclein in mice. The Journal of Biological Chemistry, 277, 1641–1644.
McCormack AL, Thiruchelvam M, Manning‐Bog AB, Thiffault C, Langston JW, Cory‐Slechta DA, Di Monte DA, 2002. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiology of Disease, 10, 119–127.
McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P, 2001. Failure of the ubiquitin‐proteasome system in Parkinson's disease. Nature Reviews Neuroscience, 2, 589–594.
McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O, 2002. Selective loss of 20S proteasome alpha‐subunits in the substantia nigra pars compacta in Parkinson's disease. Journal of Neuroscience Letters, 326, 155–158.
McNaught KS, Belizaireb R, Isacsonb O, Jennerc P, Olanowa CW, 2003. Altered proteasomal function in sporadic Parkinson's disease. Experimental Neurology, 179, 1, 38–46.
Minnema DJ, Travis KZ, Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck MJ, Mathews JM, Tisdel MO, Cook AR, Botham PA, Smith LL, 2014. Dietary administration of paraquat for 13 weeks does not result in a loss of dopaminergic neurons in the substantia nigra of C57BL/6J mice. Regulatory Toxicology and Pharmacology, 68, 250–258. doi: 10.1016/j.yrtph.2013.12.010
Minois N, 2001. Resistance to stress as a function of age in transgenicDrosophila melanogaster overexpressing Hsp70. Journal of Insect Physiology, 47, 1007–1012.
Mitra S, Chakrabarti N, Bhattacharyya A, 2011. Differential regional expression patterns ofa‐synuclein, TNF‐a, and IL‐1b; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H, 2005. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathologica, 109, 583–588.
Ossowska K, Wardas J, Smialowska M, Kuter K, Lenda T, Wieronska JM, Zieba B, Nowak P, Dabrowska J, Bortel A, et al., 2005. A slowly developing dysfunction of dopaminergic nigrostriatal neurons induced by long‐term paraquat administration in rats: An animal model of preclinical stages of Parkinson's disease? European Journal of Neuroscience, 22, 1294–1304.
Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, Suno M, Ogawa K, Hayase N, Kimura K, et al., 2001. Carrier‐mediated processes in blood–brain barrier penetration and neural uptake of paraquat. Brain Research, 906, 135–142.
Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory‐Slechta DA, 2003. Age‐related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. European Journal of Neuroscience, 18, 589–600.
Tieu K, 2011. A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, 1, a009316.
Uversky VN, 2004. Neurotoxicant‐induced animal models of Parkinson's disease: understanding the role of otenone, maneb and paraquat in neurodegeneration. Cell and Tissue Research, 318, 225–241.
Widdowson PS, Farnworth MJ, Upton R, Simpson MG, 1996. No changes in behaviour, nigro‐striatal system neurochemistry or neuronal cell death following toxic multiple oral paraquat administration to rats. Human and Experimental Toxicology, 15, 583–591.
Wills J, Credle J, Oaks AW, Duka V, Lee JH, Jones J, Sidhu A, 2012. Paraquat, but not Maneb, induces synucleinopathy and taupathy in striata of mice through inhibition of proteosomal and autophagy pathways. Public Library of Science (PLoS ONE), 7, 1, e30745.
Yang W, Tiffany‐Castiglioni E, 2007. The bipyridyl herbicide paraquat induces proteasome dysfunction in human neuroblastoma SH‐SY5Y cells. Journal of Toxicology and Environmental Health. Part A, 70, 1849–1857.
Yin L, Lu L, Prasad K, Richfield EK, Unger EL, Xu J, Jones BC, 2011. Genetic‐based, differential susceptibility to paraquat neurotoxicity in mice. Neurotoxicology and Teratology, 33, 415–421. doi: 10.1016/j.ntt.2011.02.012
4th KER: Neuroinflammation leads to degeneration of the dopaminergic neurons of nigrostriatal pathway
See AOP1 (p. 154)
5th KER: Degeneration of dopaminergic neurons of the nigrostriatal pathway directly leads to neuroinflammation
See AOP1 (p. 165)
6th KER: Degeneration of DA neurons of nigrostriatal pathway leads to motor symptoms of PD
How this key event relationship works
See AOP1 (p. 169)
Weight of evidence for the KER
Biological plausibility
See AOP 1 (p. 170)
Empirical support for linkage
Paraquat treatment (10 mg/kg twice a week for 4 weeks) of young adult Sprague‐Dawley rats (2 months old) induced a significant loss of nigral dopaminergic neurons (by Nissl staining and TH immunostaining) in SNpc of 15% and a mixed pattern of motor impairments (postural deficit, decrease in speed and mobility), which may have been related to early effects of nigral dopaminergic neuronal loss (Cicchetti et al., 2005).
Adult C57 BL/6 mice treated i.p. with paraquat (5 and 10 mg/kg) showed a dose‐dependent decrease in substantia nigra dopaminergic neurons (36% and 61%, respectively, assessed by Fluoro‐gold prelabelling method), a decline in striatal dopamine nerve terminal density (87% and 94%, respectively, assessed by TH immunoreactivity) and neurobehavioural syndrome characterised by reduced ambulatory (locomotor) activity (Brooks et al., 1999).
Paraquat treatment (i.p. 10 mg/kg twice a week for 4 weeks) of male Swiss Albino mice, 22–14 weeks old, induced progressive motor dysfunction with severe postural instability and gait impairment. A concomitant decrease in the expression levels of TH in SN (approximately 60%), FC (frontal cortex) and hippocampus and a decrease (approx. 40%) in TH+ and FOX3+ neurons in SN were observed (stereological evaluation). As part of the toxicological evaluation of the most suitable sublethal dose, mice were also treated at 5 mg/kg by i.p. twice a week for 4 weeks. In addition, a decrease in DOPA‐decarboxylase was observed in the SN and FC. The only endpoint measured (in addition to the general toxicity endpoints) was the neuronal count in the SN. A statistical significant decrease (approximately 15%) in TH+ and FOX3+ neurons was observed (Mitra et al., 2011).
Male C57BL/6 mice, 6 weeks, 5 months and 18 months old, were i.p. treated with paraquat at 10 mg/kg twice a week for 3 weeks (6 injections in total). Age‐dependent reduction in locomotor activity and motor coordination was observed. The 18‐month old mice were the most severely affected and failed to recover 24 h post treatment. Progressive reduction in dopamine metabolites and turnover were greatest in the 18‐month old group of animals. Increased in striatal TH activity was observed in the 6‐week‐old and 5‐month‐old animals but not in 18‐month‐old mice. The number of nigrostriatal dopaminergic neurons was reduced in all age group animals but these losses, along with the decreases in striatal TH protein levels, were progressive in 18‐month‐old paraquat groups between 2 weeks and 3 months post‐exposure. (Thiruchelvam et al., 2003).
Intracerebral injection of 1–5 μg paraquat in male Wistar rats (3 months old) for 16 weeks caused dose‐dependent depletion DOPA in the ipsilateral striatum starting 2 weeks after treatment (long‐lasting and irreversible) up to 91.5% at 3 μg paraquat. Paraquat induced marked loss of Nissl substances and severe loss of neurons at 3 μg. PQ caused dose‐dependent rotational behaviour in rats, contralateral to the lesion side, in response to apomorphine administration (inducing circling behaviour) (Liou et al., 1996).
Male Wistar rats were injected with 10 mg/kg paraquat i.p. for 4–24 weeks. Paraquat induced reduction in TH+ neurons of the SN (17% at 4‐week mainly in the rostral region, up to 37% at 24 weeks expanding to the whole length of SN; evaluated by stereology). DOPA levels increased in the caudate‐putamen (4–8 weeks) then returned to control values and dropped (25–30%) after 24 weeks. This seems to result from degeneration of DA neurons. TH level (Western blot) decreased in the caudate‐putament after 24 weeks (55%) but this effect was not reflected by the loss in TH‐ir neurons (being already dropped in the rostral part of SN after 4 weeks) (Ossowska et al., 2005). Clinical signs were not recorded in this study; however the study design was considered of relevance for the evaluation of the progression of the finding associated with neuronal loss.
Paraquat treatment (i.p. injection 10 mg/kg bw every 5 days over 20 days) of Long Evans Hooded rats induced progressive (TH positive neurons stereology counted) loss in dopaminergic neurons up to 47% (end of week 8 post PQ exposure) and deficiency in behavioural motor function (horizontal beam walking test) (after 4 and 8 weeks). Ubisol‐Q10 (6 mg/bw) administration after completion of paraquat injections (when the degenerative process had already began (20% TH positive neurons lost)) was effective in blocking the progression of neurodegeneration and improved motor skills. To maintain this neuroprotection, continuous Ubisol‐Q10 supplementation was required. Discontinuation of treatment resulted in neuronal death, suggesting that the presence of the antioxidant was essential for blocking the pathway (Muthukumaran et al., 2014).
In Fernagut (2007) experiment, male mice overexpressing human α‐syn under the Thy 1 promoter (Thy 1‐aSYN) and WT were i.p. injected PQ 10 mg/kg once a week for 3 weeks. Despite degeneration of dopaminergic neurons (densitometric measurement and stereological analysis for counting TH+ neurons) in both Thy 1‐aSYN mice and WT PQ‐treated mice, behavioural impaired sensimotor performance was observed in non‐treated Thy 1‐aSYN mice only, remaining unchanged after PQ administration. The sensimotor abnormalities in Thy 1‐aSYN were observed in a previous work (Fleming et al., 2004) and the lack of behavioural deficits after PQ administration was commented by the author as not surprising in the view of small magnitude neuronal loss TH‐positive terminals in striatum (25%).
Quantifiable understanding
Table A.16.
Quantitative understanding of the KER
| DA neurons degeneration | Parkinsonian motor symptoms | Treatment | References |
|---|---|---|---|
| 15% DA neuronal loss (Nissl staining and TH immunostaining) in SNpc | Mixed pattern of motor impairment observed for testing posture and speed but not for mobility (approx. 3 times the control, as average for total score‐from Figure A.5) | Young adult Sprague‐Dawley rats (2 months old) i.p. injected with PQ 10 mg/kg, twice a week for 4 weeks | Cicchetti et al. (2005) |
|
Decrease in SN dopaminergic neurons of 36% and 61%, respectively (assessed by Fluoro‐gold prelabelling method) Decline in striatal dopamine nerve terminal density of 87% and 94%, respectively (assessed by TH immunoreactivity) |
Neurobehavioural syndrome characterised by reduced ambulatory (locomotor) activity 48 h after final treatment (during the course of 60 min experimental session) observed at both doses (reduction approx. 45% after 60 min. Figure A.5A) | Adult C57 BL/6J mice i.p. injected with PQ 5 and 10 mg/kg, 3 doses separated by 1 week each | Brooks et al. (1999) |
|
Differential immunolocalisation and decreased expression levels of TH in SN (60%), FC (50%) and hippocampus (30%) (only measured at 10 mg/kg) Decrease in TH+ and FOX3+ neurons in SN (stereological count) of approximately 40% at 10 mg/kg and of approximately 10–15% at 5 mg/kg |
Motor dysfunction (only observed at 10 mg/kg) after 2 weeks of treatment (progressive over the next days) with severe postural instability and gait impairment consistent with a unilateral lesion:
|
Adult male Swiss Albino mice i.p. treated with 5 and 10 mg/kg PQ twice a week for 4 weeks | Mitra et al. (2011) |
|
Dose‐dependent DA depletion in ipsilateral striatum 2 weeks after treatment. 26. 7, 60.3 and 91.5% at 1, 2 and 3 μg PQ respectively. The effect lasted up to 16 weeks Marked loss of Nissl substances and severe loss of neurons at 3 μg PQ (2 weeks after injection). The effect was considered moderate at 2 μg PQ (2 weeks after injection) |
Circling behaviour (direction of the lesioned side) due to the imbalance of dopaminergic activity in striata (unilateral lesion) at 3 μg PQ Dose‐dependent rotational behaviour in rats contralateral to the lesion side in response to apomorphine s.c. administration 0.5 mg/kg (inducing circling behaviour) at 3 μg PQ (2 weeks after injection) |
Intracerebral (unilateral intranigral) injection of 1, 2 and 3 μg PQ in male Wistar rats for 16 weeks | Liou et al. (1996) |
| Progressive TH positive neurons (stereology count) loss up to 47% at the end of week 8 post PQ exposure | Deficiency in behavioural motor function (horizontal beam walking test) after 4 and 8 weeks | Long Evans Hooded rats i.p. injected PQ 10 mg/kg bw, every 5 days over 20 days | Muthukumaran et al. (2014) |
| Nigrostriatal dopaminergic neurons reduced in all age groups but progressive in 18‐month‐old PQ groups between 2 weeks and 3 months post‐exposure | Reduction in locomotor activity and motor coordination, age dependent with 18‐month old mice most affected and failing to recover 24 h post treatment | Male C57BL/6 mice (6 weeks, 5 months and 18 months old) i.p. treated with PQ 10 mg/kg twice a week for 3 weeks (6 injections in total) | Thiruchelvam et al. (2003) |
Uncertainties or inconsistencies
Exposure to paraquat may decrease the number of nigral neurons without triggering motor impairment (Fernagut, 2007). This can be consequent to the low level of DA reduction or limited neuronal loss observed following the treatment.
The impact of paraquat upon the striatum appears to be somewhat less pronounced than the effects of the pesticide upon SNc DA neuronal soma (Mangano et al., 2012). As well, some authors have failed to find changes in striatal DA levels or behavioural impairment, even in the presence of loss of DA soma (Thiruchelvam et al., 2003). It is conceivable that compensatory/buffer downstream processes provoked by soma loss, variations in experimental design (e.g. route of administration, dosing regimen, sacrifice interval, striatal subregions tested, age of mice) can possibly contribute to some of the inconsistency observed across studies (Rojo et al., 2007; Prasad et al., 2009; Kang et al., 2010; Rappold et al., 2010).
The effects on nigral dopaminergic neurons appear to be specific (Tieu et al., 2011). However, damage in dopaminergic cell bodies and terminal has not been consistently observed (Thiruchelvam et al., 2000b; Cicchetti et al., 2005). In addition, even in studies in which a loss of nigral dopaminergic neurons is detected, PQ does not have an effect on striatal dopamine level (Thiruchelvam et al., 2000b; McCormack et al., 2002). This lack of dopamine reduction might be related to the compensatory upregulation of tyrosine hydroxylase activity in the striatum after PQ injection (Thiruchelvam et al., 2000b; McCormack et al., 2002; Ossowska et al., 2005; Tieu 2011).
The repeat dose administration of 10 mg/kg i.p. is likely representing the maximum tolerated dose of the chemical stressor. The observed movement disorders can, at least in part, come from systemic illness and the contribution of systemic pathological changes to the observed movement disorders cannot ruled out (Cicchetti et al., 2005).
In‐vivo, experimental reproducibility of the estimated mean number of TH positive neurons in the SNpc or TH positive axons and terminals in the striatum is weak and representing an uncertainty. In particular, Breckenridge et al. (2013) and Smyne et al. (2016) conducted in vivo experiments where the administration of the chemical stressor Paraquat at an expected neurotoxic dose and treatment schedule showed no effect. The experiments were conducted following a very thorough and comprehensive protocol. In addition, Smyne et al. (2016), conducted a systematic review of the published literature that has evaluated the effects of paraquat on the SNpc and striatum in male mice. In order to evaluate potential sources of variability, multiple information was extracted and evaluated. A number of experimental limitations were identified which can reduce the strength of positive outcomes observed with paraquat. Nevertheless, some positive studies could not be dismissed and the differences in host susceptibility (e.g. dose, timing of treatment, species or strain, age of animal, iron accumulation, strain specific gene ontology) can explain contradictory effects observed following treatment with the chemical stressor paraquat in mice.
References
Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, Cook AR, Botham PA, Smith LL, 2013. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology, 37, 1–14. doi: 10.1016/j.neuro.2013.03.005
Brooks AI, Chadwick CA, Gelbard HA, Cory‐Slechta DA, Federoff HJ, 1999. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Research, 823, 1–10.
Cicchetti F, Lapointe N, Roberge‐Tremblay A, Saint‐Pierre M, Jimenez L, Ficke BW, Gross RE, 2005. Systemic exposure to paraquat and maneb models early Parkinson's disease in young adult rats. Neurobiology of Disease, 20, 360–371.
Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF, 2007. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha‐synuclein over‐expression. Synapse, 61, 991–1001.
Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF, 2004. Early and progressive sensorimotor anomalies in mice overexpressing wild‐type human alpha‐synuclein. Journal of Neuroscience, 24, 9434–9440.
Liou HH, Chen RC, Tsai YF, Chen WP, Chang YC, Tsai MC, 1996. Effects of paraquat on the substantia nigra of the Wistar rats: neurochemical, histological, and behavioral studies. Toxicology and Applied Pharmacology, 137, 34–41.
Minnema DJ, Travis KZ, Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck MJ, Mathews JM, Tisdel MO, Cook AR, Botham PA, Smith LL, 2014. Dietary administration of paraquat for 13 weeks does not result in a loss of dopaminergic neurons in the substantia nigra of C57BL/6J mice. Regulatory Toxicology and Pharmacology, 68, 250–258.
Mitra S, Chakrabarti N, and Bhattacharyy A, 2011. Differential regional expression patterns of α‐synuclein, TNF‐α, and IL‐1β; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Muthukumaran K, Leahy S, Harrison K, Sikorska M, Sandhu JK, Cohen J, Keshan C, Lopatin D, Miller H, Borowy‐Borowski H, Lanthier P, Weinstock S, Pande S, 2014. Orally delivered water soluble Coenzyme Q10 (Ubisol‐Q10) blocks on‐going neurodegeneration in rats exposed to paraquat: potential for therapeutic application in Parkinson's disease. Journal of Neuroscience, 15, 21.
Smeyne RJ, Breckenridge CB, beck M, Jiao Y, Butt MT, Wolf J, Zadory D, Minnema D, Sturgess NC, Travis KZ, Cook AR, Smith LL, Botham PA, 2016. Assessment of the effects of MPTP and Paraquat on dopaminergic neurons and microglia in the substantia nigra pars compacta of C57BL/6 mice. Public Library of Science (PLOS ONE). doi: 10.1371/journal.pone0164094
Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory‐Slechta DA, 2003. Age‐related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. European Journal of Neuroscience, 18, 589–600.
Overall assessment of the AOP
A.1. Response–response and incidence concordance (Table 1)
Toxicity mediated by redox cycling is based on acceptance of an electron by a chemical from a reductant, formation of a radical, and transfer of an electron to molecular oxygen. The process is leading to the generation of superoxide and mitochondria are one of the presumed sites where the chemical is initially reduced within the cell to form superoxide. This is the chemical based mechanism of action of the herbicide paraquat (PQ), which is therefore considered a suitable chemical tool/stressor for exploring the link between the MIE and the AO. In animal models, PQ susceptibility is known to act synergistically with microglia leading to its activation (Purisai et al., 2007; Mitra et al., 2011). Microglia through plasma‐membrane NADPH‐oxidase may also activate the extracellular redox cycling of PQ favouring its transport within dopaminergic neurons (Rappold et al., 2011). The kinetic and metabolism of PQ is complex and the amount of PQ entering and accumulating into the brain is dependent on dose, route of administration, expression of transporters, animal age and strain.
Multiple genetic factors are also involved in host susceptibility which is likely to represent an important source of variability (Corasaniti et al., 1991; Youdim, 2003; Li et al., 2005; Tieu, 2011; Yin et al., 2011; Jiao et al., 2012). These elements are the possible/likely reason of lack of reproducibility of apical endpoints as observed in some studies conducted with this stressor (Breckenridge et al., 2013; Smyne et al., 2016).
In‐vivo, the commonly used dose of 10 mg/kg administered i.p. leads to a brain concentration of around 3 μM after 6 doses (Prasad et al., 2009; Smeyne et al., 2016) and around 6–10 μM in striatum after 24 doses (Prasad et al., 2009). A single s.c. administration of 10 mg/kg leads to 3.88 ± 0.79 μM serum concentration after 3 h reaching 0.36 ± 0.09 μM in the extracellular space of the striatum (Shimizu et al., 2001). At this dose level (10 mg/kg i.p.), ambiguous results in terms of neuronal loss and occurrence of parkinsonian motor symptoms are reported in C57BL/6J mice (Thiruchelvam et al., 2000b; McCormack et al., 2002; Barlow et al., 2004; Li et al., 2005, 2012; Khwaja et al., 2007; Prasad et al., 2007, 2009; Fei et al., 2008; Cristovao et al., 2009; Fernagut et al., 2009; Mangano et al., 2009, 2011, 2012; Breckenridge et al., 2013; Watson, 2013; Minnema, 2014; Smeyne et al., 2016).
In order to integrate data on paraquat toxicity from widely different experimental models, ranging from cell cultures to repeat dose animal studies, the concentrations close to the target site were compared by classical approaches of in vitro in vivo extrapolation (IVIVE) based on the available publications. The lowest observed effect concentrations in vitro were in the range of 20 μM. Concentrations up to 200 μM have been used for some studies. A variability of observed concentrations is expected from the fact that PQ needs a transporter to cross cell membranes, and that different oxidation states of PQ have different affinities for transporters (Rappold et al., 2011). Thus, the levels of transporter expression, the presence of microglia and the oxidation state of PQ determine its intracellular concentration. This may explain variations of effective in vitro concentrations by at least 10‐fold.
For the in vivo situation, different sets of data are available to estimate the PQ concentration in DA neurons. A study on average brain concentrations after 6 doses found an average brain concentration of 0.54 μg/g brain tissue, corresponding to an average brain tissue concentration of 3 μM (Smeyne et al., 2016). It needs to be assumed that some cells accumulate PQ, while others do not. Thus, intracellular concentrations in DA neurons may be considerably higher than 3 μM, considering the uptake of PQ. In another study, plasma concentrations and extracellular brain concentrations were measured 3 h after a single dose. Extracellular concentrations of 0.4 μM were reached in the brain (Shimizu et al., 2001). It needs to be assumed that this would be higher after multiple dosing. Indeed a study by Prasad et al., 2009 showed that the extracellular concentration in the striatum was around 3.3 μM after 6 doses and around 10 μM after 24 doses of 10 mg/kg i.p.
These data suggest that the brain concentration at the target site (inside DA neurons) is in the 1–2 digit μM range. This range overlaps well with the effective in vitro concentrations of 20–50 μM. Within the limits of accuracy possible (the intracellular concentrations have not been measured within DA neurons) this IVIVE shows that in vitro and in vivo active concentrations are within the same order of magnitude and thus aligned.
In a conservative approach, the range of brain concentration expected to represent the steady state (3–10 μM) of PQ can be considered only indicative and will be used in this AOP to define a possible probabilistic threshold of activation of the MIE leading to the AO. With the limited number of doses known from in‐vivo studies, an intra (in the same) and inter (between) KE response–response relationship can be observed. A response–response relationship in the increase in activity of ROS scavenging enzymes can be observed (KE 1) in‐vivo; however, the KE degeneration of dopaminergic neurons and the AO can only be seen at the threshold concentration in the brain of about 3–10 μM reached after repeated exposure to PQ 10 mg/kg bw, and not for higher doses due to the marked general toxicity (Mitra et al., 2011). The frequency of the KE degeneration of dopaminergic neurons and the AO were less than for the other key events. In this AOP, neuroinflammation was considered to have a direct effect on paraquat activation and on loss of DA neurons (Purisai et al., 2007; Rappold et al., 2011). However, in addition to neurodegenerative consequences, neuroinflammation can have also protective effects. Therefore, due to this complexity this key events was not included in Tables 1 and 2.
In vitro, an intra KEs response–response is evident, with some evidences of inter KEs response–response concordance. However, when multiple time of sampling are applied to the experimental design, the inter KEs response–response concordance is stronger. In‐vitro, a strong response–response concordance between KE1 and cell death (KE 3) is evident (Figure A.32). Overall, the response–response and incidence concordance was considered moderate.
Figure A.32.

PQ‐induced ROS and Cell death (% over controls) at 24 h in neuronal cells. Points in the figure derive from the listed papers, targeted by the number associated to each symbol (1‐de Oliveira 2016, 2‐González‐Polo 2007, 3‐Lopert 2012, 4‐Rodriguez‐Rocha 2013, 5‐ Huang 2012, 6‐Ding 2001, 7‐Yang and Tiffany Castiglioni 2007) Data refers to different neuronal cell lines and primary cultures, and to different methods of detection. As such, single results have been calculated over their control to allow comparison between different studies
A.2. Temporal concordance among the MIE, KEs and AO
There is a strong agreement on the sequence of pathological events linking the MIE to the adverse outcome (Fujita et al., 2014). The temporal concordance is strong when considering the chronicity and progressive nature of the pathology of parkinsonian disorders. Temporal concordance among the KE 1, 2, 3 and AO can be observed in the experimental models of PD using the chemical stressors rotenone and MPTP (Betarbet, 2000, 2006; Sherer et al., 2003, Fornai et al., 2005) which are sharing the same KEs with this AOP but are caused by a different MIE. With the chemical stressor MPTP, to trigger the KE3 (i.e. degeneration of DA neurons in SNpc with presence of intracytoplasmic Lewy‐like bodies) and motor deficits (AO), proteostasis needs to be disturbed for a minimum period of time (Fornai et al., 2005) and this is similarly expected with chemicals inducing redox cycling like PQ (Ossowska et al., 2005). In vivo, with the chemical stressor PQ, evidence of temporal concordance is limited by the study design using single time‐point descriptive assessment. In vitro, evidence of temporal concordance is limited by the fact that 24 and 48 h were the most investigated time points. Nevertheless, those papers taking into account shorter time points show that a good temporal concordance exist between MIE (4 h), KE2 (6 h) and KE3 (12–24 h) (Ding, 2001; González‐Polo, 2007; Cantu, 2011; de Oliveira, 2016). Based on the established knowledge on chronicity and progression of parkinsonian disorders, the temporal concordance is considered strong for this AOP up to the KE 3 (degeneration of DA neurons of nigrostriatal pathway). The occurrence of the AO outcome is strongly linked to the amount of DA in the striatum and to the loss of DA neurons in the SNpc. Following treatment with the chemical stressor/s the key events are observed in the proposed order in this AOP.
Table A.17.
Response–Response and temporality concordance table
| Concentration at the target site, in‐vitro | MIE |
KE1 Mitochondrial ROS production and dysfunction |
KE2 Impaired proteostasis |
KE3 Degeneration of DA neurons of nigrostriatal pathway |
AO Parkinsonian motor symptoms |
| 10 μM [1, 10, 12] | No data |
± (at 24 h) |
+ (ALP 6 h, time‐dependent at 24 h) |
± (at 24 h) |
|
| 20 10 to 50 μM [1, 2, 10] | No data |
+ (at 6 and 24 h; ne 3 h) |
+ (UPS at 6 h) |
+ (at 24 h) |
|
| 50–200 μM [1, 2, 8, 10, 13] |
++ (at 24 and 48 h) |
++ (at 6 and 24 h) |
++ (UPS at 6 h) |
++ (at 24 and 48 h) |
|
| 200 μM to 1 mM [3, 4, 7, 8, 9, 10, 12] |
+++ (at 4, 6, 24 and 48 h) |
++ (at 4 and 6 h) +++ (at 24 h) |
+++ (UPS at 48 h) ++ (ALP at 24 h) |
++/+++ (at 18, 24 and 48 h) |
|
| Concentration at the target site, in vivo | MIE |
KE1 Mitochondrial dysfunction (ROS production) |
KE2 Impaired proteostasis |
KE3 Degeneration of DA neurons of nigrostriatal pathway |
AO Parkinsonian motor symptoms |
| Below 3 μM [15, 16] |
+ (4 weeks) |
Increase activity in ROS‐scavenging enzymes + (4 week) |
No data |
Decrease in number of TH+ in SN ± (at 4 week) |
No locomotor deficit |
| About 3–10 μM [6,5, 13,14, 15] |
++ weeks) |
Increased lipid peroxidation − (1 week) ++/+++ (2 week) +++ (6–9 week) Increase activity in ROS‐scavenging enzymes (4 week) ++ |
Impaired proteostasis and autophagy ++ |
Decrease in number of TH+ in SN − (1 week) + (at 2–4 week) |
Locomotor deficit ± (at 2–4 week) |
González‐Polo 2007 [1]; Ding and Keller, 2001 [2]; Yang and Tiffany‐Castiglioni, 2007 [3]; Cantu 2011 [4]; Breckenridge 2013 [5]; Prasad 2007 and 2009 [6]; Huang 2012 [7]; Lopert 2012 [8] *LDH as % of control and not of maximal release; Chau 2009 [9]; de Oliveira 2016 [10]; Garcia‐Garcia 2013 [11]; Rodriguez‐Rocha 2013 [12]; Patel 2006 [13]; McCormack 2005 [14]; Mitra et al. 2011 [15]; Brooks et al. 1999 [16].
+, ++, +++ are intended only to demonstrate intra and inter KEs relationship.
A.3. Strength, reproducibility of the experimental evidence, and specificity of association of AO and MIE
There is a strong agreement that ROS production and mitochondrial dysfunction can lead to neurodegeneration and motor symptoms of parkinsonian disorders and familial PD genes are also implicated in ROS production by mitochondria (Gandhi et al., 2009; Yao et al., 2011; Sanders et al., 2013; Fujita et al., 2014). The chemical stressor used for the empirical support, PQ, is a well‐known substance with a toxicity primarily mediated by redox cycling (Day et al., 1999; Tieu, 2011). With PQ, ROS production and oxidative stress, impaired proteostasis, and loss of nigral dopaminergic neurons are reported (Brooks et al., 1999; McCormack et al., 2002; Mitra et al., 2011; Su et al., 2015). Some uncertainties on the initial mitochondrial involvement in triggering PQ redox‐cycle in vivo exists due to the prolonged (consistent with a generalised oxidative stress) and repeated exposure and the use of general indicators of oxidative stress like lipid and protein oxidation. Degeneration of dopaminergic neurons (KE3) was tested in‐vivo in many studies conducted in rat and mice with an higher prevalence of testing in mice. The estimated mean number of TH+ neurons in the SNpc or TH+ axons and terminals in the striatum was the common endpoint measured, with detailed neuropathology, neurochemistry and behavioural evaluation conducted more seldomly. Reproducibility of the effect of PQ on these parameters in mice was questioned by some authors (Thiruchelvam et al., 2000b; Cicchetti et al., 2005; Breckenridge et al., 2013; Minnema et al., 2014; Smeyne et al., 2016). For studies conducted in mice, a systematic review of the published literature that has evaluated the effects of PQ on the SNpc and striatum was included in Smeyne et al. (2016). The potential impact of animal, dose variables, stereological and methodological factors was evaluated. Despite the thorough evaluation conducted and the identification of some methodological biases potentially affecting a number of studies, a significant controversy remains regarding the reproducibility of the finding using PQ as a chemical stressor and a clear methodological bias affecting it cannot be fully established.
In addition in a number of studies performed with the chemical stressor PQ, loss of dopaminergic neurons (KE3)was not associated with an effect on striatal dopamine levels (Thiruchelvam et al., 2000b; McCormack et al., 2002). Although this can be due to the activation of compensatory effects or compensatory upregulation of TH activity in the striatum following PQ treatment (Thiruchelvam et al., 2000b; McCormack et al., 2002; Ossowska et al., 2005), this effect is known to be quantitatively linked to the loss of DA neurons and a threshold is likely to exists. The role and influence of the animal species, strain, age, route of administration, dose scheduling and susceptibility of neuronal population to the noxa on the outcome of the studies cannot be completely ruled out.
The occurrence of parkinsonian motor symptoms was not consistently reported for the chemical stressor PQ. Evidence on the occurrence of the AO can however be observed with PQ following unilateral intranigral administration where loss of neuronal cells was marked (ca. 90%). For most of the studies conducted with the tool chemical PQ administered by i.p. the amount of DA neuronal loss was relatively limited (e.g. 20–30%) and the AO i.e. parkinsonian motor symptoms, was therefore not consistently reported. These observations are in line with the human evidence that parkinsonian motor symptoms are only evident in PD when striatal DA drops approximately 80% (corresponding to a 60% DA neuronal cells loss (Jellinger et al., 2009). Indeed, the variability observed in the latest KE and in the AO, not clearly associated with methodological biases in some cases, could be well reflecting the variability in host susceptibility. This is particularly true when considering the complexity of the factors influencing the local disposition i.e. concentration at the target site of the chemical stressor.
Considering the relevance of ROS production and oxidative damage in Parkinson's models, it is expected that the specificity of this AOP would be high. However, with the use of PQ as a unique chemical stressor supporting the empirical evidence, judging specificity was not possible. Overall, the strength linking the MIE to the AO was considered high for this AOP; however, using PQ as a chemical tool, the reproducibility of some experimental evidence and the specificity of the association was considered moderate.
A.4. Weight of Evidence (WoE)
A.4.1. Biological plausibility, analogy between chemical stressors, and species consistency of the experimental evidence
ROS generation and deregulation of ROS management by dysfunctional mitochondria is known to be a crucial event in neurodegeneration in general and for dopaminergic neurons in SNpc in particular when considering the unique susceptibility of these neurons (Fujita et al., 2014; Sanders et al., 2013). Familial forms of PD include genes (i.e. PINK1 and DJI) that are implicated in ROS management by mitochondria resulting in mitochondrial DNA damage and inflammation as a downstream effect (Gandhi et al., 2009; Horowitz et al., 2010; Yao et al., 2011; Fujita et al., 2014). The biological plausibility for the KEs relationship linking the MIE to the AO is strong based on the existing knowledge of PD pathogenesis and parkinsonian syndrome. As PQ is the only tool compound so far analysed and comprehensively studied, analogy is considered moderate as the KE relationship is only plausible based on the supporting analogy with PD, but a scientific understanding on the relationship between a chemically induced redox cycler and parkinsonian motor deficits is not completely established. ROS generation is mechanistically recognised as a cause of PD and parkinsonian syndrome. Mouse and rat are the most frequently used animal models to support this AOP using the tool compound PQ. Strain differences in susceptibility are reported for the experiments conducted with the chemical stressor PQ (Prasad et al., 2009; Smyne et al., 2016). The same pattern of effects has been observed in a different test species i.e. drosophila. Overall the consistency of this AOP was considered moderate to high.
Table A.18.
Biological plausibility of the KERs; WoE analysis
| 1 Support for biological plausibility of KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Is there a mechanistic (i.e. structural or functional) relationship between KEup and KEdown consistent with established biological knowledge? | Extensive understanding of the KER based on extensive previous documentation and broad acceptance | The KER is plausible based on analogy to accepted biological relationships, but scientific understanding is not completely established | There is empirical support for a statistical association between KEs but the structural or functional relationship between them is not understood | |
|
MIE to KE1 Chemical redox cycling to mitochondrial reactive oxygen species (ROS) production and dysfunction |
Strong | Chemical redox cycler with electron reduction potential more negative than O2 are effective superoxide producer (Cohen and Doherty, 1987; Mason 1990). Based on the properties of the chemical redox cycler, mitochondria may represent the primary site for chemical redox cycling due to the electron release mainly from complex I and complex III (Selivanov et al., 2011). This has been clearly demonstrated for the chemical tool paraquat, a known redox cycler inducer, in isolated mitochondria, cells, rodents, flies and yeast (Mockett et al., 2003; Tien Nguyen‐nhu and Knoops, 2003; Castello et al., 2007; Rodriguez‐Rocha et al., 2013). It is well established that superoxide formation will give rise to the production of different reactive oxygen species at the mitochondria, which in turn will lead to mitochondrial dysfunction (Turrens, 2003; Andreyev et al., 2005; Murphy, 2009). Mitochondria isolated from the striatum of PQ‐exposed rats overproduce ROS and are dysfunctional (Czerniczyniec et al., 2013, 2015). Similarly, knocking down mitochondrial SOD induces an excessive endogenous production of superoxide (mimicking the effect or redox cycler compounds) and alters the activity of tricarboxylic acid cycle enzymes and respiratory complexes (Hinerfeld et al., 2004). The activity of most of these enzymes is rescued via antioxidant treatment linking endogenous mitochondrial oxidative stress to mitochondrial dysfunction (Hinerfeld et al., 2004). Diquat, a potent redox cycling compound can induce ROS production following intrastriatal administration (Djukic et al. 2012), enhance reactive oxygen species production and elicits an antioxidant response in SH‐SY5Y neuroblastoma cell line (Slaughter et al., 2001; Nisar et al., 2015) | ||
|
KE1 to KE2 Mitochondrial dysfunction (ROS production) to impaired proteostasis |
Moderate | The weight of evidence supporting the biological plausibility behind the relationship between mitochondrial dysfunction and impaired proteostasis, including the impaired function of UPS and ALP that results in decreased protein degradation and increase protein aggregation is well documented but not fully understood. It is well established that the two main mechanisms that normally remove abnormal proteins (UPS and ALP) rely on physiological mitochondrial function. The role of oxidative stress, due to mitochondrial dysfunction, burdens the proteostasis with oxidised proteins and impairs the chaperone and the degradation systems. This leads to a vicious circle of oxidative stress inducing further mitochondrial impairment (McNaught and Jenner, 2001; Moore et al., 2005; Powers et al., 2009; Zaltieri et al., 2015). Furthermore, the interaction of mitochondrial dysfunction and UPS /ALP deregulation plays a pivotal role in the pathogenesis of PD (Sherer et al., 2002; Fornai et al., 2005; Pan et al., 2008; Dagda et al., 2013) | ||
|
KE2 to KE3 Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
Moderate | It is well known that impaired proteostasis refers to misfolded and aggregated proteins including alpha‐synuclein, autophagy, deregulated axonal transport of mitochondria and impaired trafficking of cellular organelles. Evidences are linked to PD and experimental PD models as well as from genetic studies (McNaught et al., 2001, 2003, 2004; Matsuda and Tanaka, 2010; Rappold et al., 2014; Tieu et al., 2014). Strong evidence for degeneration of the nigrostriatal pathway comes from the experimental manipulations that directly induce the same disturbances of proteostasis as observed in PD patients (e.g. viral mutated alpha‐synuclein expression). However, a clear mechanistic proof for the understanding of the exact event triggering cell death is lacking | ||
|
KE3 ⇔ KE4 Neuroinflammation |
Moderate | The fact that reactive glial cells (microglia and astrocytes) may kill neurons is well accepted. The mechanisms underlying this effect may include the release of cytotoxic signals (e.g. cytokines) or production of ROS and RNS (Chao et al., 1995; Brown and Bal‐Price, 2003; Kraft and Harry, 2011; Taetzsch and Block, 2013). However, the studied mediators differ from model to model. The fact that neuronal injury/death can trigger neuroinflammation is supported by evidence in human and experimental models. The evidence that neuroinflammation triggered by neuronal damage can cause neuronal death (vicious circle), is mostly indirect or by analogy (Griffin et al., 1998; McGeer and McGeer, 1998; Blasko et al., 2004; Cacquevel et al., 2004; Hirsch and Hunot, 2009; Tansey and Goldberg, 2009; Barbeito et al., 2010; Rubio‐Perez and Morillas‐Ruiz, 2012; Thundyil and Lim, 2014) | ||
|
KE3 to AO Degeneration of DA neurons of the nigrostriatal pathway leads to parkinsonian motor symptoms |
Strong | The mechanistic understanding of the regulatory role of striatal DA in the extrapyramidal motor control system is well established. The loss of DA in the striatum is characteristic of all aetiologies of PD and is not observed in other neurodegenerative diseases (Bernheimer et al., 1973; Reynolds et al., 1986). Characteristic motor symptoms such as bradykinesia, tremor, or rigidity are manifested when more than 80% of striatal DA is depleted as a consequence of SNpc DA neuronal degeneration (Koller et al., 1992), possibly corresponding to approximately 60% of neuronal cell loss (Jellinger et al., 2009). However, when considering these quantitative thresholds, experimental evidences with the tool chemical paraquat are inconsistent | ||
A.4.2. Essentiality
Direct essentiality evidence is coming from experiments conducted with antioxidant agents or following manipulation of the biological systems protecting from or regulating ROS production and oxidative stress. Manifestation of motor symptoms differs in rodents and human and for this reason their value should depend upon its relationship to striatal dopaminergic function. Study designed to demonstrate recovery of clinical signs following DA replacement are limited with the chemical tool paraquat. Evidence of essentiality is however provided following unilateral intranigral injection of paraquat or in drosophila models. The overall WoE for the essentiality is strong.
Table A.19.
Essentiality of the KE; WoE analysis
| 2 Support for essentiality of KEs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Are downstream KEs and/or the AO prevented if an upstream KE is blocked? | Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs (e.g. stop/reversibility studies, antagonism, knock out models, etc.) | Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE leading to increase in KEdown or AO | No or contradictory experimental evidence of the essentiality of any of the KEs | |
|
MIE Redox‐cycling (of a chemical) initiated by electrons of the mitochondrial respiratory chain |
Strong | Overexpressing enzymes involved in O2 dismutation specifically located at the mitochondria prevents neuronal cell death in vitro (Rodriguez‐Rocha et al., 2013; Filograna et al., 2016). Accordingly, depletion of mitochondrial SOD2 exacerbate PQ‐toxicity in Drosophila (Kirby et al., 2002), while mitochondrial enzymes activity is restored and neuronal death in cortex reduced in SOD2 knock out animals treated with antioxidants (Hinerfeld et al., 2004). Mitochondrial aconitase knock down attenuated PQ induced H2O2 production and respiratory capacity deficiency in neuronal cells (Cantu, 2011) | ||
|
KE1 Mitochondrial reactive oxygen species (ROS) formation and dysfunction |
Strong | In vitro, PQ toxicity both in terms of ROS production, mitochondrial dysfunction and neuronal death is rescued by several antioxidants (Hinerfeld et al., 2004; McCarthy et al., 2004; Peng et al. 2005; Chau et al., 2009, 2010; de Oliveira, 2016). Most of these drugs, like synthetic superoxide dismutase/catalase mimetics or SOD‐fusion proteins also protect against PQ‐induced oxidative damage and/or DA neurons degeneration in vivo (Hinerfeld et al., 2004; Peng et al., 2005; Choi, 2006) improving motor skills (Somayajulu‐Nitu et al., 2009; Muthukumaran et al., 2014). Overexpression of antioxidant enzymes specifically at the mitochondria protects Drosophila and yeast from PQ‐toxicity at low doses (Mockett et al., 2003; Tien Nguyen‐nhu and Knoops, 2003). On the other hand, depletion of antioxidant systems exacerbates PQ toxicity both in vitro and in vivo (Van Remmen et al., 2004; Lopert et al., 2012; Liang et al., 2013). USP30, a deubiquitinase localised to mitochondria, antagonises mitophagy. Overexpression of USP30 removes ubiquitin in damaged mitochondria and blocks mitophagy. Reducing USP30 activity enhances mitochondrial degradation in neurons. Knockdown of USP30 in dopaminergic neurons protects flies against paraquat toxicity in vivo, ameliorating defects in dopamine levels, motor function and organismal survival in Drosophila (Bingol et al., 2014). More in general, overexpression of Sods in DA neurons counteracts PQ‐induced oxidative damage and reduces motor dysfunction in Drosophila (Filograna et al., 2016). Similarly, the use of antioxidants also restores PQ‐induced motor activity in Drosophila (Jimenez‐Del‐Rio et al., 2010) | ||
|
KE2 Impaired proteostasis |
Moderate |
Most of the experimental evidence supporting the essentiality is coming from experiments conducted in transgenic animals or studies conducted with the chemical stressor Rotenone and MPTP, known chemical toxins used to mimic PD (Dauer et al., 2002; Kirk, 2002, 2003; Klein, 2002; Lo Bianco, 2002; Lauwers, 2003; Fornai et al., 2005) Exposure to the chemical stressor paraquat of mice with inducible overexpression of familial PD‐linked mutant α‐syn in dopaminergic neurons of the olfactory bulb exacerbate the increase of soluble and insoluble α‐syn expression, accumulation of α‐syn at the dendritic terminals, reduction of autolysosomial clearance, mitochondrial condensation and damage. None of these effects occurs in PQ‐treated mice with suppressed α‐syn expression. Loss of DA neurons in the olfactory bulb is evident in PQ‐treated mutant mice but not in both PQ‐treated mice with suppressed α‐syn expression (after doxycycline administration) and untreated mutant mice (Nuber et al., 2014) In vitro system overexpressing the neuroprotective molecular chaperone human DJ‐1, showed more resistance to the proteasome impairment induced by paraquat. Similarly, preservation was observed in the same system following treatment with a known proteasome inhibitors (epoxomycin) (Ding and Keller, 2001) However, although evidence exists to support some essentiality of impaired proteostasis, a single molecular chain of events cannot be established |
||
|
KE3 Degeneration of DA neurons of nigrostriatal pathway |
Strong | Clinical and experimental evidences show that the pharmacological replacement of the DA neurofunction by allografting fetal ventral mesencephalic tissues is successfully replacing degenerated DA neurons resulting in the total reversibility of motor deficits in an animal model and a partial effect is observed in human PD patients (Freed et al., 1990; Henderson et al., 1991; López‐Lozano et al., 1991; Spencer et al., 1992; Widner et al., 1992; Peschanski et al., 1994; Han et al., 2015). Concomitant administration of selective type B monoamine oxidase inhibitor slowed the progression parkinsonian motor symptoms induced by unilateral intranigral injection of paraquat which is expected to induce approximately 90% of neuronal loss. It provides a protective effect on the moderate injury elicited by PQ toxicity. A post‐treatment administration of apomrphine, a DA agonist, induced controlateral circling behaviour which correlated well with the decrease of striatal DA (Liou, 1996, 2001). However, for most of the experiments conducted with paraquat, the amount of DA neuronal cell loss and drop in striatal DA was not consistent or below the threshold for triggering motor symptoms In addition, studies showing an altered behaviour resulting from striatal drop in DA, lack a DA replacement strategy | ||
|
KE4 Neuroinflammation |
Moderate | Protection from neuronal cell loss following treatment with 10 mg/kg bw of paraquat was observed in interferon‐gamma KO animals or blockade of i‐NOS, NF‐kB or p38 MAPK. In both cases a decrease of microglial reactivity or prevention of microglia activation was observed (Mangano et al., 2012; Yadav et al., 2012). Minocyline or silencing of NADPH oxidase prevented DA neurodegeneration subsequent to the administration of 10 mg/kg bw of paraquat (Purisai et al., 2007). Essentiality of microglial NADPH oxidase for mediating DA neurodegeneration was observed in vitro in neuron‐glia co‐cultures prepared from NADPH oxidase‐deficient mice (Wu et al., 2005). However, inhibition was different in different models and considered as an indirect evidence of essentiality | ||
A.4.3. Empirical support
The empirical support provides evidence that the KEup is linked to KEdown. With PQ, as the only available chemical tool, the strength of this relationship is limited by the fact that the large majority of studies are conducted at fixed doses and single time‐point descriptive assessment. This affected the dose response and incidence concordance analysis and the overall concordance for empirical support was considered moderate. The empirical support in‐vivo is mainly provided by studies conducted with PQ in rodents species and drosophila aimed to model PD. In vitro the concentration‐response concordance was more evident.
Table A.20.
Empirical support for the KERs; WoE analysis
| 3 Empirical support for KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Does the empirical evidence support that a change in the KEup leads to an appropriate change in the KEdown? Does KEup occur at lower doses and earlier time points than KEdown and is the incidence of KEup higher than that for KEdown? Are inconsistencies in empirical support cross taxa, species and stressors that don't align with expected pattern of hypothesised AOP? | Multiple studies showing dependent change in both exposure to a wide range of specific stressors (extensive evidence for temporal, dose–response and incidence concordance) and no or few critical data gaps or conflicting data | Demonstrated dependent change in both events following exposure to a small number of specific stressors and some evidence inconsistent with expected pattern that can be explained by factors such as experimental design, technical considerations, differences among laboratories, etc | Limited or no studies reporting dependent change in both events following exposure to a specific stressor (i.e. endpoints never measured in the same study or not at all); and/or significant inconsistencies in empirical support across taxa and species that don't align with expected pattern for hypothesised AOP | |
| MIE to KE1 | Moderate | With the chemical tool paraquat, studies are mainly conducted at fixed doses and dose relationships studies are very limited for the O2 production, which is relevant for the intra MIE dose relationship (Cantu et al., 2011; Mitra et al. 2011; Dranka et al., 2012; Huang et al., 2012; Rodriguez‐Rocha et al., 2013; De Oliveira et al., 2016). However, intra KE1 dose relationship is observable for ROS production/lipid peroxidation using the same stressor compound (McCormack et al., 2005; Mitra et al., 2011; Lopert et al., 2012; de Oliveira et al., 2016). In‐vitro, high concentrations of PQ showing activation of the MIE are showing the most pronounced ROS production indicating that a concordance in dose and response relationship exists between the MIE and KE1 and cell death (Chau et al., 2009; Lopert et al., 2012; Rodriguez‐Rocha et al. 2013; de Oliveira et al., 2016). Temporal relationship between MIE and KE1 is indistinguishable due to the fast conversion of O2 to H2O2 and other ROS species (Cohen and Doherty, 1987). However, when considering cell death as the observational end point, a dose response and time concordance exists. PQ (0.1–1 mM) induces O2°− and H2O2 production within minutes in isolated mitochondria and mitochondrial brain fraction (Castello et al., 2007; Cochemé and Murphy, 2008), while in cells this process is detectable after 4–6 h from the exposure (Cantu et al., 2011; Dranka et al. 2012; Huang et al., 2012; Rodriguez‐Rocha et‐al., 2013). At these time points no death is generally detected. In‐vivo, there is limited evidence of intra MIE dose relationship with paraquat and temporal concordance cannot be defined as the experiments are conducted at single time point descriptive assessment (Mitra et al., 2011). However, circumstantial evidences are supported by the knowledge on the chronic and progressive nature of parkinsonian syndromes | ||
|
KE1 to KE2 Mitochondrial dysfunction (ROS production) results in impaired proteostasis |
Low |
Evidence is provided that exposure to PQ and deficiency of DJ‐1 might cooperatively induce mitochondrial dysfunction resulting in ATP depletion and contribute to proteasome dysfunction in mouse brain (Yang et al., 2007). Moreover, exacerbation of Paraquat effect on the autophagic degradation pathway is observed in an in vitro system with silenced DJ‐1 (González‐Polo et al., 2009). In C57BL/6J mice 10 mg/kg i.p. for 1–5 doses, increased level of lipid peroxides in ventral midbrain was associated impaired proteostasis (Prasad et al., 2007) Temporal and dose concordance cannot be elaborated from in vivo studies as they are conducted at the same dose and observational time‐point. However, in vitro studies are indicative of a temporal and concentration concordance, evidencing concentration‐and/or time‐dependent effects on mitochondrial and proteasome functions (Ding and Keller, 2001; Yang and Tiffany‐Castiglioni, 2007) |
||
|
KE2 to KE3 Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
Moderate |
The empirical support linking impaired proteostasis with degeneration of DA neurons of the nigrostriatal pathway comes from post‐mortem human evidences in PD patients supporting a causative link between the two key events.. With paraquat, a response concordance was observed in multiple in vivo studies (Manning‐Bog, 2003; Fernagut, 2007; Mitra, 2011).Temporal and dose concordance cannot be elaborated from these studies as they are conducted at the same dose and observational time‐point. Some inconsistencies were observed, i.e. partial effect on proteasomal inhibition which is likely due to compensatory effects and/or lower toxicity of PQ when compared to other chemical stressor (e.g. rotenone, MPTP) In vivo studies with Paraquat are showing a more relevant effect on the ALP and α‐synuclein overexpression with a less evident effect on proteasome inhibition. A dose and temporal concordance was more consistently observed in in vitro studies (Gonzalez‐Polo, 2009; Chinta, 2010) |
||
|
KE3 <=> KE4 Neuroinflammation |
Moderate | Multiple in vivo and in vitro experiments support the link between neuroinflammation and degeneration of DA neurons in the nigrostriatal pathway as well as vice versa. The observation of concomitant presence of glial and astrocytic cells and degenerated/degenerating DA neurons is also reported in many studies (Cicchetti et al., 2005; Wu et al., 2005; Purisai et al., 2007; Mitra et al., 2011; Mangano et al., 2012). A similar relationship was observed with compounds like rotenone and MPTP | ||
|
KE3 to AO Degeneration of DA neurons of nigrostriatal pathway leads to parkinsonian motor symptoms |
Moderate to low | PQ is reported to induce motor deficits and loss of nigral dopaminergic neurons in a dose‐(Brooks et al., 1999) and age (Thiruchelvam et al., 2003) dependent manner. The concomitant observation of dopaminergic neuronal loss and parkinsonian motor symptoms has been confirmed by other authors (Cicchetti et al., 2005; Prasad et al., 2009; Mitra et al., 2011). However, at similar doses and experimental design a number of inconsistencies or lack of reproducibility were noted and described in the uncertainties (Smyne et al., 2016). In human (and animal models using rotenone and MPTP), motor symptoms are expected to be clinically visible when striatal dopamine levels drop of approximately 80%, corresponding to a DA neuronal loss of approximately 60% (Bernheimer et al., 1973; Lloyd et al., 1975; Hornykiewicz et al., 1986; Jellinger et al., 2009). This threshold of pathological changes was only achieved when paraquat was administered directly in the SN and the link between neuronal loss and clinical symptoms was empirically consolidated by the following treatment with apomorphine or the concomitant treatment with the MAOB inhibitors (Liou, 1996, 2001). When different routes of administration were applied, neuronal loss was below this pathological threshold, not consistently related to drop in striatal DA and motor symptoms were only occasionally observed (Smyne et al., 2016). Some methodological biases can explain the inconsistency in the lack of reproducibility observed for some experiments conducted in‐vivo with the chemical stressor PQ (Smyne et al., 2016); however, when considering the multiple factors associated with host susceptibility and the complexity of the local disposition of PQ i.e. concentration and activation of PQ in the SNpc and striatum, the biological variability can account for the different outcomes observed in the studies | ||
A.5. Uncertainties and Inconsistencies
No direct evidence exists in the literature of PQ‐induced O2°− production in vivo. The involvement of O2°− production is deduced by the efficacy of superoxide dismutase analogues to prevent/reduce PQ neurotoxicity.
Besides mitochondria, cellular NADPH‐oxidases (Rappold et al., 2011; Cristóvão et al., 2012) also contribute to PQ‐induced ROS production. Furthermore, in vitro data suggest that for time points of exposure longer than 48 h oxidative stress occurs both at mitochondria and cytosol. This makes it difficult to discriminate the source of PQ‐induced ROS and the early involvement of mitochondria in vivo due to the extensive treatments and to the indirect detection of oxidative stress mainly by mean of lipid peroxidation and/or protein oxidation. Mitochondrial involvement is suggested by ex‐vivo studies (Czerniczyniec et al., 2013, 2015).
The exact molecular link from mitochondrial dysfunction to disturbed proteostasis is still unclear (Malkus et al., 2009; Zaltieri et al., 2015). Furthermore, whether impaired proteostasis and protein aggregation would cause the selective death of DA neurons in the SN still remains an uncertainty.
The role of α‐synuclein in neuronal degeneration is still unclear as well as the mechanisms leading to its aggregation.
Priority of the pattern leading to cell death could depend on concentration, time of exposure and species/strain sensitivity; these factors have to be taken into consideration for the interpretation of the study's result and extrapolation of potential low‐dose chronic effect as this AOP refers to chronic exposure.
Animal studies, conducted with the chemical stressor PQ, show little consistency in dose, timing of treatment, species, strain and age of animals. All these factors are included in the host susceptibility, possibly contributing to the variability observed. In particular, age and iron accumulation in SNpc are additional putative risk factors for reproducing degeneration of dopaminergic neurons in SNpc (Youdim et al., 2003; Li et al., 2005; Yin et al., 2011; Jiao et al., 2012).
The ability of PQ to induce loss of DA neurons in SNpc in vivo is sometime equivocal and reproducibility of the finding is representing an uncertainty (Smyne et al., 2016). Some methodological biases could account for the lack of reproducibility observed in some studies (Smeyne et al., 2016); however, the same cannot be applied to all studies and the a significant controversy still remains. The role of host susceptibility factors as a matter of variability should be therefore considered.
The model of striatal DA loss and its influence on motor output ganglia does not explain specific motor abnormalities observed in PD (e.g. resting tremor vs bradykinesia) (Obeso et al., 2000). Other neurotransmitters (ACh) may play additional roles in other brain areas like the olfactory bulb. Transfer to animal models of PD symptoms also represents an uncertainty.
The role of neuronal plasticity in intoxication recovery and resilience is unclear.
This AOP is a linear sequence of KEs. However, ROS production, mitochondrial dysfunction and impaired proteostasis are influencing each other and these are considered as uncertainties (Malkus et al., 2009).
When measurement of loss of mitochondrial membrane potential is performed together with cell viability, the former is detected only in dead cells in PQ treated cells (on the contrary for MPTP and Rotenone is detected in alive cells suggesting this event precedes cell death). It is suggested that decrease PQ‐induced neuronal death is independent on mitochondrial membrane potential (mmp) decrease. As such overexpression of Mn SOD (in mitochondria) prevents PQ‐induced cell death but not mmp decrease (Rodriguez‐Rocha, 2013).
Paraquat‐induced neurotoxicity could affect a subpopulation of DA neurons. This might explain why, once the maximum effect is reached, no further neuronal death occurs after supplementary exposures (McCormack et al., 2005). Another possibility is the development of defensive mechanisms, which preserve neurons from further toxicity. This hypothesis is consistent with the in vitro observation of an increased transcriptional activation of redox‐sensitive antioxidant response elements and NF‐κB, specifically induced from paraquat but not from rotenone and MPTP (Rodriguez‐Rocha et al., 2013).
The impact of paraquat upon the striatum appears to be somewhat less pronounced than the effects upon SN dopaminergic neuronal soma (Mangano et al., 2012). In addition, some authors have failed to find changes in striatal DA levels or behavioural impairment, even in the presence of loss of dopaminergic soma (Thiruchelvam et al., 2003). It is conceivable that compensatory/buffer downstream processes provoked by soma loss or variations in experimental design can possibly contribute to some of the inconsistencies observed across studies (Rojo et al., 2007; Prasad et al., 2009; Kang et al., 2010; Rappold et al., 2011).
Dopaminergic neurons in SN and VTA seem to have a different susceptibility to the damage induced by paraquat (McCormack et al., 2006).
Few hypothesis have been put forward to explain the selective vulnerability of the SN pc dopaminergic pathway, although a defined molecular mechanism remains elusive. Elevated iron content in this region, that increase sensitivity to redox‐damage catalysing the generation of ROS (Liddell et al., 2013) and regional distribution of transporters able to uptake PQ (i.e. DAT and Oct3) in combination with a high microglia population in the nigra (Rappold et al., 2011) have been evoked.
The vulnerability of the dopaminergic pathway still remains circumstantial. Paraquat has been proposed to pass the blood‐brain‐barrier by mediation of neutral amino acid transportation (Shimizu et al., 2001; McCormack and DiMonte 2003). Accumulation of paraquat in the brain is reported to be age dependent, possibly indicating a role for the blood‐brain‐barrier permeability (Corasaniti et al., 1991); Dication paraquat has been reported not to be a substrate for dopamine transporter (Richardson et al., 2005). Nevertheless, Rappold et al. (2011) demonstrated that radical paraquat is transported by DAT and hence how the toxicant enters into dopaminergic neurons is still unclear. One possibility is extracellular paraquat reduction by membrane‐bound NADPH oxidase with the formed paraquat monocation radical entering DA neurons by neuronal DAT (Rappold et al., 2011).
Exposure to paraquat may decrease the number of nigral neurons without triggering motor impairment (Fernagut, 2007). This can be consequent to the low level of DA reduction or limited neuronal loss observed following the treatment.
The repeat dose administration of 10 mg/kg i.p. is likely representing the maximum tolerated dose of the chemical stressor. The observed movement disorders can, at least in part, come from systemic illness and the contribution of systemic pathological changes to the observed movement disorders cannot ruled out (Cicchetti et al., 2005).
There is uncertainty concerning the real brain concentration that is triggering this AOP. In addition, because of the complexity of the kinetic e metabolism, including local disposition of PQ in the SNpc, extrapolation of the in vitro concentration to in vivo scenario is an uncertainty.
A.6. Quantitative Considerations
The quantitative understanding of this AOP includes evidence of response–response relationship and the identification of a putative threshold effect. This is because the triggering effect at MIE level was explored in only few studies and the reproducibility of the KE 3 is likely depending by multiple host susceptibility factors. More evidence exists that an increase from 200% to 600% of lipid peroxidation (endpoint of KE1) in DA neuronal cells can be used as a probabilistic threshold triggering the degeneration of DA neurons of the nigrostriatal pathway. In line with others chemical tools that can induce DA neuronal loss through different MIE (i.e. rotenone and MPTP), for the identification of the AO the design of the in‐vivo studies should be tailored as to a MIE which leads to a long‐lasting perturbation of the KEs. This provides the most specific and definite context to trigger neuronal death in vivo. A major hurdle for this AOP is represented by the AO. With PQ, the low level of reported DA neuronal loss (ca. 20–30%) is not expected to induce parkinsonian motor symptoms and essentiality data (i.e. recovery of motor symptoms following treatment with DA) is limited. Moving from a qualitative AOP to quantitative AOP would need a clear understanding of effect thresholds for the different KEs.
Table A.21.
Concordance table for the tool compound paraquat
| Dose/concentration at the target site | MIE | KE1 Mitochondrial dysfunction (ROS production) | KE2 Impaired proteostasis | KE3 Degeneration of DA neurons of nigrostriatal pathway | AO Parkinsonian motor symptoms |
|---|---|---|---|---|---|
| 0.78 μM brain concentration [1 and 2] | No data | 200% increase in lipid peroxidation [4] | No data | No effect [4] | No data |
| Below 3 μM (intended as a cumulative concentration; 8 doses) [8] | 42% increase in SOD activity [8] | No data | 10% decrease in TH+ neurons [8] | No data | |
| 3–10 μM (cumulative concentration after multiple doses) [1 and 2] | 75% increase in SOD activity [8] | 500–600% (cumulative effect) lipid peroxidation [2, 4] |
50% increase in 20S proteasome fraction at 24 h [2] Intracellular deposits of α‐synuclein in 30% of DA neurons [6] 50% increase in α‐synuclein expression [8] |
30–50% (cumulative effect) decrease in TH+ neurons [2,4, 6, 7, 8] | Motor impairment[2, 8, 10] |
References: Breckenridge 2013 [1]; Prasad 2007 and 2009 [2]; Castello 2007 [3]; Mc Cormack 2005 [4]; Cantu 2011 [5]; Manning‐Bog 2003 [6]; Fernagut 2007 [7]; Mitra 2011 [8]; Yang 2007 [9]; Brooks 1999 [10].
A.7. Applicability of the AOP
This proposed AOP is neither sex‐dependent nor associated with certain life stage; however, male aged animals are considered more sensitive. The relevance of this AOP during the developmental period has not been investigated.
In vivo testing has no species restriction. However, host susceptibility is likely to have a relevant impact on the outcome of the studies and in this context, elements of stress and animal strain could have a profound impact on the outcome of the studies (Youdim et al., 2003; Li et al., 2005; Yin et al., 2011; Jiao et al., 2012; Jones et al., 2014). The mouse was the species most commonly used in the experimental models conducted with the chemical stressor paraquat and the C57BL/6J is considered the most sensitive mouse strain (Jiao et al., 2012; Smeyne et al., 2016). However, animal models (rodents in particular) would have limitations as they are poorly representative of the long human life‐time as well as of the human long‐time exposure to the potential toxicants. Human cell‐based models would likely have better predictivity for humans than animal cell models if biologically relevant by means of being able of recapitulate the key events in the toxicology and pathology pathway providing robust and repeatable results predictive of the chemical concentration that lead to a particular outcome. In this case, toxicokinetics information from in‐vivo studies would be essential to test the respective concentrations in‐vitro on human cells.
A.8. Regulatory considerations
The AOP is a conceptual framework to mechanistically understand apical hazards. The AO, parkinsonian motor symptoms, is an apical endpoint that can be explored and quantified in the regulatory toxicology studies conducted in experimental laboratory animals. Motor deficit endpoints, and their relevance to parkinsonian disorders and PD are a matter of intensive discussion in the scientific community. The use of rodent models for assessment of parkinsonian/PD‐associated motor deficits was even put into question. Although rodent motor output can only hardly be correlated with the human motor output repertoire, the adverse outcome of the present AOP was explicitly focused on alterations in motor output associated with parkinsonian conditions. Indeed, this AOP does not claim that motor deficits included herein reflect the complexity of the human disease.
It is also noteworthy that decrease in neuronal cell count is also an apical regulatory endpoint explorable and quantifiable in experimental studies conducted in vivo; if the appropriate areas of the brain are sampled and properly evaluated. A statistically significant decrease in DA neuronal cell count is considered an adverse event, regardless of the concomitant presence of motor symptoms. This has to be taken into consideration for the potential regulatory applications of this AOP and for the sensibility of the method applied to capture the KE/apical endpoint/hazard. If the intention is to use this AOP for defining the link between the MIE and the degeneration of DA neuronal cells of the nigrostriatal pathway, the WoE is considered strong; however, in the case of defining the link between the MIE and parkinsonian motor symptoms, the WoE should account for the biology and complexity linking disruption of the nigrostriatal pathway and occurrence of motor symptoms. Because of the potential different uses of this AOP, keeping the parkinsonian motor symptoms as AO was considered relevant. It is also foreseen that for potential additional uses, like defining a testing strategy or properly design an in‐vitro or an in‐vivo study, or evaluation of mixture of chemicals, degeneration of DA neuronal cells of nigrostriatal pathway should be considered as AO.
A.9. Potential application of this AOP
This AOP was developed in order to evaluate the biological plausibility that the adverse outcome i.e.parkinsonian motor deficits, is linked to the selected MIE. By means of using a human health outcome from epidemiological studies and meta‐analysis, the authors intend to embed the AO in the process of hazard identification and identification of risk factors.
In addition, this AOP can be used to support identification of data gap that can be required or explored when a chemical substance is affecting the pathway or provide recommendation on the most adequate study design that can be applied to investigate the apical endpoints. It is important to note that, although the AO is defined in this AOP as parkinsonian motor deficits, degeneration of DA neurons is already per se an adverse event even in situations where is not leading to parkinsonian motor deficits or clinical signs indicative of a central effect, and this should be taken into consideration for the regulatory applications of this AOP. In addition, this AOP can inform on the identifications of in vitro methods that can be developed for an integrated approach to testing and assessment (IATA) based on in vitro neurotoxicity assays complementary to in vivo assays.
References
Andreyev AY1, Kushnareva YE, Starkov AA, 2005. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc), 70, 200–214.
Barlow BK, Richfield EK, Cory‐Slechta DA, Thiruchelvam M, 2004. A fetal risk factor for Parkinson's disease. Developmental Journal of Neuroscience, 26, 11–23. Epub 2004/10/29. doi: DNE2004026001011 [pii] 10.1159/000080707 [doi]. PubMed PMID: 15509894.
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F, 1973. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, Morphological and Neurochemical Correlations. Journal of the Neurological Sciences, 20, 415–455.
Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Journal of Neuroscience, 3, 1301–1306.
Betarbet R, Canet‐Aviles RM, Sherer TB, Mastroberardino PG, Mc Lendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinfelter G, Cookson MR, Greenmyre JT, 2006. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ‐1, α‐synuclein, and the ubiquitin‐proteasome system. Neurobiology Disease, 22, 404–420.
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M, 2014. The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature, 510, 370–375. doi: 10.1038/nature13418
Brown GC, Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355.
Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, Cook AR, Botham PA, Smith LL, 2013. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology, 37, 1–14. doi: 10.1016/j.neuro.2013.03.005
Brooks AI, Chadwick CA, Gelbard HA, Cory‐Slechta DA, Federoff HJ, 1999. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Research, 823, 1–10.
Cantu D, Fulton RE, Drechsel DA, Patel M, 2011. Mitochondrial aconitase knockdown attenuates paraquat‐induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2. Journal of Neurochemistry, 118, 79–92. doi: 10.1111/j.1471‐4159.2011.07290.x
Castello PR, Drechsel DA, Patel M, 2007. Mitochondria are a major source of paraquat‐induced reactive oxygen species production in the brain. Journal of Biological Chemistry, 282, 14186–14193.
Chao CC, Hu S, Peterson PK, 1995. Glia, cytokines, and neurotoxicity. Critical ReviewsTM in Neurobiology, 9, 189–205.
Chau KY, Korlipara LV, Cooper JM, Schapira AH, 2009. Protection against paraquat and A53T alpha‐synuclein toxicity by cabergoline is partially mediated by dopamine receptors. Journal of the Neurological Sciences, 278, 44–53. doi: 10.1016/j.jns.2008.11.012
Chau KY, Cooper JM, Schapira AH, 2010. Rasagiline protects against alpha‐synuclein induced sensitivity to oxidative stress in dopaminergic cells. Neurochemistry International, 57, 525–529. doi: 10.1016/j.neuint.2010.06.017
Choi HS, An JJ, Kim SY, Lee SH, Kim DW, Yoo KY, Won MH, Kang TC, Kwon HJ, Kang JH, Cho SW, Kwon OS, Park J, Eum WS, Choi SY, 2006. PEP‐1‐SOD fusion protein efficiently protects against paraquat‐induced dopaminergic neuron damage in a Parkinson disease mouse model. Free Radical Biology and Medicine, 41, 1058–1068.
Chou AP, Li S, Fitzmaurice AG, Bronstein JM, 2010. Mechanisms of rotenone‐induced proteasome inhibition. NeuroToxicology, 31, 367–372.
Cicchetti F, Lapointe N, Roberge‐Tremblay A, Saint‐Pierre M, Jimenez L, Ficke BW, Gross RE, 2005. Systemic exposure to paraquat and maneb models early Parkinson's disease in young adult rats. Neurobiology of Disease, 20, 360–371.
Cochemé HM, Murphy MP, 2009. Chapter 22 The uptake and interactions of the redox cycler paraquat with mitochondria. Methods in Enzymology, 456, 395–417. doi: 10.1016/S0076‐6879(08)04422‐4
Cohen GM, d'Arcy Doherty M, 1987. Free radical mediated cell toxicity by redox cycling chemicals. British Journal of Cancer. Supplement, 8, 46–52.
Corasaniti MT, Defilippo R, Rodinò P, Nappi G, Nisticò G, 1991. Oct‐Dec Evidence that paraquat is able to cross the blood‐brain barrier to a different extent in rats of various age. Functional Neurology, 6, 385–391.
Cristóvão AC, Choi DH, Baltazar G, Beal MF, Kim YS, 2009. The role of NADPH oxidase 1‐derived reactive oxygen species in paraquat‐mediated dopaminergic cell death. Antioxid Redox Signal, 11, 2105–2118. doi: 10.1089/ARS.2009.2459
Czerniczyniec A, Lores‐Arnaiz S, Bustamante J, 2013. Mitochondrial susceptibility in a model of paraquat neurotoxicity. Free Radical Research, 47, 614–623. doi: 10.3109/10715762.2013.806797
Czerniczyniec A, Lanza EM, Karadayian AG, Bustamante J, Lores‐Arnaiz S, 2015. Impairment of striatal mitochondrial function by acute paraquat poisoning. Journal of Bioenergetics and Biomembranes, 47, 395–408. doi: 10.1007/s10863‐015‐9624‐x
Dagda RK, Banerjee TD, Janda E, 2013. How Parkinsonian toxins dysregulate the autophagy machinery. International Journal of Molecular Sciences, 14, 22163–22189.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson‐Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R, 2002. Resistance of alpha ‐synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences of the United States of America, 99, 14524–14529.
Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS, 1999. A mechanism of paraquat toxicity involving nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America, 96, 12760–12765.
de Oliveira MR, Ferreira GC, Schuck PF, 2016. Protective effect of carnosic acid against paraquat‐induced redox impairment and mitochondrial dysfunction in SH‐SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicology in Vitro, 32, 41–54. doi: 10.1016/j.tiv.2015.12.005
Ding Q, Keller JN, 2001. Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. Journal of Neurochemistry. 77, 1010–1017.
Djukic M, Jovanovic MD, Ninkovic M, Stevanovic I, Curcic M, Topic A, Vujanovic D, Djurdjevic D, 2012. Intrastriatal pre‐treatment with L‐NAME protects rats from diquat neurotoxcity. Annals of Agricultural and Environmental Medicine, 19, 666–672.
Dranka BP1, Zielonka J, Kanthasamy AG, Kalyanaraman B. Alterations in bioenergetic function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. Journal of Neurochemistry, 122, 941–951. doi: 10.1111/j.1471‐4159.2012.07836.x
Fei Q, McCormack AL, Di Monte DA, Ethell DW, 2008. Paraquat neurotoxicity is mediated by a Bak‐dependent mechanism. Journal of Biological Chemistry, 283, 3357–3364. doi: 10.1074/jbc.M708451200
Fernagut PO, Fleming SM, Houston CB, Tetreaut NA, Salcedo L, Masliah E, Chesselet MF, 2007. Behavioural and histological consequences of paraquat intoxication in mice: effect of α‐synuclein over expression. Synapse, 61, 991–1001.
Filograna R, Godena VK, Sanchez‐Martinez A, Ferrari E, Casella L, Beltramini M, Bubacco L, Whitworth AJ, Bisaglia M, 2016. SOD‐mimetic M40403 is protective in cell and fly models of paraquat toxicity: implications for Parkinson disease. Journal of Biological Chemistry, pii: jbc.M115.708057.
Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC, 2005. Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitinproteasome system and _α‐synuclein. Proceedings of the National Academy of Sciences, 102, 3413–3418.
Freed CR, Breeze RE, Rosenberg NL, Schneck SA, Wells TH, Barrett JN, Grafton ST, Huang SC, Eidelberg D, Rottenberg DA, 1990. Transplantation of human fetal dopamine cells for Parkinson's disease. Results at 1 year. Archives of Neurology, 47, 505–512.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R, 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102.
Gandhi S, Wood‐Kaczmar A, Yao Z, et al., PINK1‐associated Parkinson's disease is caused by neuronal vulnerability to calcium‐induced cell death. Molecular Cell, 33, 627–638.
Garcia‐Garcia A, Anandhan A, Burns M, Chen H, Zhou Y, Franco R, 2013. Impairment of Atg5‐dependent autophagic flux promotes paraquat‐ and MPP+‐induced apoptosis but not rotenone or 6‐hydroxydopamine toxicity. Toxicological Sciences, 136, 166–182. doi: 10.1093/toxsci/kft188
González‐Polo RA, Niso‐Santano M, Ortíz‐Ortíz MA, Gómez‐Martín A, Morán JM, García‐Rubio L, Francisco‐Morcillo J, Zaragoza C, Soler G, Fuentes JM, 2007. Inhibition of paraquat‐induced autophagy accelerates the apoptotic cell death in neuroblastoma SH‐SY5Y cells. Toxicological Sciences, 97, 448–458.
González‐Polo R, Niso‐Santano M, Morán JM, Ortiz‐Ortiz MA, Bravo‐San Pedro JM, Soler G, Fuentes JM, 2009. Silencing DJ‐1 reveals its contribution in paraquat‐induced autophagy. Journal of Neurochemistry, 109, 889–898.
Han F, Baremberg D, Gao J, Duan J, Lu X, Zhang N, Chen Q, 2015. Development of stem cell‐based therapy for Parkinson's disease. Translational Neurodegeneration, 4, 16.
Henderson BT, Clough CG, Hughes RC, Hitchcock ER, Kenny BG, 1991. Implantation of human fetal ventral mesencephalon to the right caudate nucleus in advanced Parkinson's disease. Archives of Neurology, 48, 822–827.
Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S, 2004. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. Journal of Neurochemistry, 88, 657–667.
Hirsch EC, Hunot S, 2009. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurology, 8, 382–397. doi: 10.1016/S1474‐4422(09)70062‐6
Hornykiewicz O, Kish SJ, Becker LE, Farley I, Shannak K, 1986. Brain neurotransmitters in dystonia musculorum deformans. New England Journal of Medicine, 315, 347–353.
Horowitz MP, Greenamyre JT, 2010. Gene‐environment interactions in parkinson's disease: the importance of animal modelling. Clinical Pharmacology & Therapeutics, 88, 467–474.
Huang CL, Lee YC, Yang YC, Kuo TY, Huang NK, 2012. Minocycline prevents paraquat‐induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicology Letters, 209, 203–210. doi: 10.1016/j.toxlet.2011.12.021
Jellinger AK, 2009. A critical evaluation of current staging of α‐synuclein pathology in Lewy body disorders. Biochemica and Biophysica Acta, 1972, 730–740.
Jiao Y, Lu L, Williams RW, Smeyne RJ, 2012. Genetic dissection of strain dependent paraquat‐induced neurodegeneration in the substantia nigra pars compacta. Public Library of Science (PLoS ONE), 7, e29447. doi: 10.1371/journal.pone.0029447
Jimenez‐Del‐Rio M, Guzman‐Martinez C, Velez‐Pardo C, 2010. The effects of polyphenols on survival and locomotor activity in Drosophila melanogaster exposed to iron and paraquat. Neurochemical Research, 35, 227–238.
Jones BC, Huang X, Mailman RB, Lu L, Williams RW, 2014. The perplexing paradox of paraquat: the case for host‐based susceptibility and postulated neurodegenerative effects. Journal of Biochemical and Molecular Toxicology, 28, 191–197. doi: 10.1002/jbt.21552
Kang MJ, Gil SJ, Lee JE, Koh HC, 2010. Selective vulnerability of the striatal subregions of C57BL/6 mice to paraquat. Toxicology Letters, 195, 127–134. doi: 10.1016/j.toxlet.2010.03.011
Kirby K, Hu J, Hilliker AJ, Phillips JP, 2002. RNA interference‐mediated silencing of Sod2 in Drosophila leads to early adult‐onset mortality and elevated endoge‐nous oxidative stress. Proceedings of the National Academy of Sciences of the United States of America, 99, 16162–16167.
Kirk D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel R, Bijorklund A, 2002. Parkinson‐like neurodegeneration induced by targeted overexpression of α‐synuclein in the nigrostriatal system. 22, 2780–2791.
Kirk D, Annett L, Burger C, Muzyczka N, Mandel R, Bijorklund A, 2003. Nigrostriatal α‐synucleinopathy induced by viral vector‐mediated overexpression of human α‐synuclein: a new primate model of parkinson's disease. Proceedings of the National Academy of Sciences, 100, 2884–2889.
Klein RL, King MA, Hamby ME, Meyer EM, 2002. Dopaminergic cell loss induced by human A30P α‐synuclein gene transfer to the rat substantia nigra. Human Gene Therapy, 13, 605–612.
Koller WC, 1992. When does Parkinson's disease begin? Neurology, 42(4 Suppl 4), 27–31.
Kraft AD, Harry GJ, 2011. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International Journal of Environmental Research and Public Health, 8, 2980–3018.
Khwaja M, McCormack A, McIntosh JM, Di Monte DA, Quik M, 2007. Nicotine partially protects against paraquat‐induced nigrostriatal damage in mice; link to alpha6beta2* nAChRs. Journal of Neurochemistry, 100, 180–190. doi: 10.1111/j.1471‐4159.2006.04177.x
Lauwers E, Debyser Z, Van Drope J, DeStrooper B, Nuttin B, 2003. Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector‐mediated overexpression of α‐synuclein. Brain Pathology, 13, 364–372.
Li X, Cheng CM, Sun JL, Li Z, Wu YI, 2005. Paraquat induces selective dopaminergic nigrostriatal degeneration in aging C57BL/6J mice. Chinese Medical Journal, 118, 1357–1361.
Li H, Wu S, Wang Z, Lin W, Zhang C, Huang B, 2012. Neuroprotective effects of tert‐butylhydroquinone on paraquat‐induced dopaminergic cell degeneration in C57BL/6 mice and in PC12 cells. Archives of Toxicology, 86, 1729–1740. doi: 10.1007/s00204‐012‐0935‐y
Liang LP, Kavanagh TJ, Patel M, 2013. Glutathione deficiency in Gclm null mice results in complex I inhibition and dopamine depletion following paraquat administration Toxicological Sciences, 134, 366–373. doi: 10.1093/toxsci/kft112
Liddell JR, Obando D, Liu J, Ganio G, Volitakis I, Mok SS, Crouch PJ, White AR, Codd R, 2013. Lipophilic adamantyl‐ or deferasirox‐based conjugates of desferrioxamine B have enhanced neuroprotective capacity: implications for Parkinson disease. Free Radical Biology and Medicine, 60, 147–156.
Liou HH, Chen RC, Tsai YF, Chen WP, Chang YC, Tsai MC, 1996. Effects of paraquat on the substantia nigra of the Wistar rats: neurochemical, histological, and behavioral studies. Toxicology and Applied Pharmacology, 137, 34–41.
Liou HH, Chen RC, Chen TH, Tsai YF, Tsai MC, 2001. Attenuation of paraquat‐induced dopaminergic toxicity on the substantia nigra by (‐)‐deprenyl in vivo. Toxicology and Applied Pharmacology, 172, 37–43.
Lloyd KG, Davidson L, Hornykiewicz O, 1975. The neurochemistry of Parkinson's disease: effect of L‐dopa therapy. Journal of Pharmacology and Experimental Therapeutics, 195, 453–464.
Lo Bianco C, Ridet JL, Deglon N, Aebischer P, 2002. Alpha‐synucleopathy and selective dopaminergic neuron loss in a rat lentiviral‐based model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 10813–10818.
Lopert P, Day BJ, Patel M, 2012. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. Public Library of Science (PLoS ONE), 7, e50683. doi: 10.1371/journal.pone.0050683
López‐Lozano JJ, Bravo G, Abascal J, 1991. Grafting of perfused adrenal medullary tissue into the caudate nucleus of patients with Parkinson's disease. Clinica Puerta de Hierro Neural Transplantation Group. Journal of Neurosurgery, 75, 234–243.
Malkus KA, Tsika E, Ischiropoulos H, 2009. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson's disease: how neurons are lost in the Bermuda triangle. Molecular Neurodegeneration, 4, 24. doi: 10.1186/1750‐1326‐4‐24
Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, et al., 2012. Interferon‐gamma plays a role in paraquat‐induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiology of Aging, 33, 1411–1426.
Mangano EN, Hayley S, 2009. Inflammatory priming of the substantia nigra influences the impact of later paraquat exposure: Neuroimmune sensitization of neurodegeneration. Neurobiology of Aging, 30, 1361–1378. doi: 10.1016/j.neurobiolaging.2007.11.020
Mangano EN, Peters S, Litteljohn D, So R, Bethune C, Bobyn J, et al., 2011. Granulocyte macrophage‐colony stimulating factor protects against substantia nigra dopaminergic cell loss in an environmental toxin model of Parkinson's disease. Neurobiology of Disease, 43, 99–112. doi: 10.1016/j.nbd.2011.02.011
Manning‐Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA, 2003. Alpha‐synuclein overexpression protects against paraquat‐induced neurodegeneration. Journal of Neuroscience, 23, 3095–3099.
Mason RP, 1990. Redox cycling of radical anion metabolites of toxic chemicals and drugs and the Marcus theory of electron transfer. Environmental Health Perspectives, 87, 237–243.
Matsuda N, Tanaka K, 2010. Does impairment of the ubiquitin‐proteasome system or the autophagy‐lysosome pathway predispose individuals to neurodegenerative disorders such as Parkinson's disease? Journal of Alzheimer's Disease, 19, 1–9. doi: 10.3233/JAD‐2010‐1231
McCarthy S, Somayajulu M, Sikorska M, Borowy‐Borowski H, Pandey S, 2004. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water‐soluble Coenzyme Q10. Toxicology and Applied Pharmacology, 201, 21–31.
McCormack AL, Thiruchelvam M, Manning‐Bog AB, Thiffault C, Langston JW, Cory‐Slechta DA, Di Monte DA, 2002. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiology of Disease, 10, 119–127.
McCormack AL, Di Monte DA, 2003. Effects of L‐dopa and other amino acids against paraquat‐induced nigrostriatal degeneration. Journal of Neurochemistry, 85, 82–86.
McCormack AL1, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA, 2005. Role of oxidative stress in paraquat‐induced dopaminergic cell degeneration. Journal of Neurochemistry, 93, 1030–1037.
McCormack AL, Atienza JG, Langston JW, Di Monte DA, 2006. Decreased susceptibility to oxidative stress underlies the resistance of specific dopaminergic cell populations to paraquat‐induced degeneration. Journal of Neuroscience, 141, 929–937.
McNaught KS, Jenner P, 2001. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Journal of Neuroscience Letters, 297, 191–194.
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW, 2003. Altered proteasomal function in sporadic Parkinson's disease. Experimental Neurology, 179, 38–46.
McNaught KS, Perl DP, Brownell AL, Olanow CW, 2004. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Annals of Neurology, 56, 149–162.
Minnema DJ, Travis KZ, Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck MJ, Mathews JM, Tisdel MO, Cook AR, Botham PA, Smith LL, 2014. Dietary administration of paraquat for 13 weeks does not result in a loss of dopaminergic neurons in the substantia nigra of C57BL/6J mice. 68, 250–258.
Mitra S, Chakrabarti N, Bhattacharyy A, 2011. Differential regional expression patterns of α‐synuclein, TNF‐α, and IL‐1β; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. Journal of Neuroinflammation, 8, 163.
Mockett RJ, Bayne AC, Kwong LK, Orr WC, Sohal RS, 2003. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radical Biology and Medicine, 34, 207–217.
Moore DJ, West AB, Dawson VL, Dawson TM, 2005. Molecular pathophysiology of Parkinson's disease. Annual Review of Neuroscience, 28, 57–87.
Murphy MP, 2009. How mitochondria produce reactive oxygen species Biochemical Journal, 417, 1–13. doi: 10.1042/BJ20081386
Muthukumaran K, Leahy S, Harrison K, Sikorska M, Sandhu JK, Cohen J, Keshan C, Lopatin D, Miller H, Borowy‐Borowski H, Lanthier P, Weinstock S, Pande S, 2014. Orally delivered water soluble Coenzyme Q10 (Ubisol‐Q10) blocks on‐going neurodegeneration in rats exposed to paraquat: potential for therapeutic application in Parkinson's disease. Journal of Neuroscience, 15, 21.
Nisar R, Hanson PS, He L, Taylor RW, Blain PG, Morris CM, 2015. Diquat causes caspase‐independent cell death in SH‐SY5Y cells by production of ROS independently of mitochondria. Archives of Toxicology, 89, 1811–1825. doi: 10.1007/s00204‐015‐1453‐5
Nuber S, Tadros D, Fields J, Overk CR, Ettle B, Kosberg K, Mante M, Rockenstein E, Trejo M, Masliah E, 2014. Environmental neurotoxic challenge of conditional alpha‐synuclein transgenic mice predicts a dopaminergic olfactory‐striatal interplay in early PD. Acta Neuropathologicaogica, 127, 477–494. doi: 10.1007/s00401‐014‐1255‐5
Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, Lanciego JL, Artieda J, Gonzalo N, Olanow CW, 2000. Pathophysiology of the basal ganglia in Parkinson's disease. Trends in Journal of Neurosciences, 23(10 Suppl), S8–S19.
Ossowska K, Wardas J, Kuter K, Nowak P, Dabrowska J, Bortel A, Labus Ł, Kwieciński A, Krygowska‐Wajs A, Wolfarth S, 2005. Influence of paraquat on dopaminergic transporter in the rat brain. Pharmacological Reports, 57, 330–335.
Ossowska K, Wardas J, Smialowska M, Kuter K, Lenda T, Wieronska JM, Zieba B, Nowak P, Dabrowska J, Bortel A, Kwiecinski A, Wolfarth S, 2005. A slowly developing dysfunction of dopaminergiv nigrostriatal neurons induced by long‐term paraquat administration in rats: an animal model of preclinical stages of Parkinson's disease.
Pan T, Kondo S, Le W, Jankovic J, 2008. The role of autophagy‐lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain, 131, 1969–1978.
Patel S, Singh V, Kumar A, Gupta YK, Singh MP, 2006. Status of antioxidant defense system and expression of toxicant responsive genes in striatum of maneb‐ and paraquat‐induced Parkinson's disease phenotype in mouse: mechanism of neurodegeneration. Brain Research, 1081, 9–18.
Peng J, Stevenson FF, Doctrow SR, Andersen JK, 2005. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat‐mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. Journal of Biological Chemistry, 280, 29194–29198.
Peschanski M, Defer G, N'Guyen JP, Ricolfi F, Monfort JC, Remy P, Geny C, Samson Y, Hantraye P, Jeny R, 1994. Bilateral motor improvement and alteration of L‐dopa effect in two patients with Parkinson's disease following intrastriatal transplantation of foetal ventral mesencephalon. Brain, 117(Pt 3), 487–499.
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE, 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annual Review of Biochemistry, 78, 959–991.
Prasad K, Winnik B, Thiruchelvam MJ, Buckley B, Mirochnitchenko O, Richfield EK, 2007. Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain. Environmental Health Perspectives, 115, 1448–1453.
Prasad K, Tarasewicz E, Mathew J, Strickland PA, Buckley B, Richardson JR, Richfield EK, 2009. Toxicokinetics and toxicodynamics of paraquat accumulation in mouse brain. Experimental Neurology, 215, 358–367. doi: 10.1016/j.expneurol.2008.11.003
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat‐induced dopaminergic cell degeneration. Neurobiology of Disease, 25, 392–400.
Rappold PM, Cui M, Chesser AS, Tibbett J, Grima JC, Duan L, Sen N, Javitch JA, Tieu K, 2011. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter‐3. Proceedings of the National Academy of Sciences of the United States of America, 108, 20766–20771. doi: 10.1073/pnas.1115141108
Rappold PM, et al., 2014. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nature Communications, 5, 5244. doi: 10.1038/ncomms6244
Reynolds GP, Garrett NJ, 1986. Striatal dopamine and homovanillic acid in Huntington's disease. Journal of Neural Transmission, 65, 151–155.
Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW, 2005. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicological Sciences, 88, 193–201.
Rodriguez‐Rocha H, Garcia‐Garcia A, Pickett C, Li S, Jones J, Chen H, Webb B, Choi J, Zhou Y, Zimmerman MC, Franco R, 2013. Compartmentalized oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: distinct roles of superoxide anion and superoxide dismutases. Free Radical Biology and Medicine, 61, 370–383. doi: 10.1016/j.freeradbiomed.2013.04.021
Rojo AI, Cavada C, de Sagarra MR, Cuadrado A, 2007. Chronic inhalation of rotenone or paraquat does not induce Parkinson's disease symptoms in mice or rats. Experimental Neurology, 208, 120–126.
Rojo AI, Innamorato NG, Martin‐Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A, 2010. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia, 58, 588–598.
Rubio‐Perez JM, Morillas‐Ruiz JM, 2012. A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal, 2012, 756357. doi: 10.1100/2012/756357
Sanders LH, Greenamayre TJ, 2013. Oxidative damage to macromolecules in human Parkinson's disease and the rotenone model. Free Radical Biology and Medicine; 62, 11–120.
Selivanov VA1, Votyakova TV, Pivtoraiko VN, Zeak J, Sukhomlin T, Trucco M, Roca J, Cascante M, 2011. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLOS Computational Biology, 7, e1001115. doi: 10.1371/journal.pcbi.1001115
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al., 2003. Mechanism of toxicity in rotenone models of Parkinson's disease. Journal of Neuroscience, 23, 10756–10764.
Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, Suno M, Ogawa K, Hayase N, Kimura K, Shiono H, 2001. Carrier‐mediated processes in blood–brain barrier penetration and neural uptake of paraquat. Brain Research, 906, 135–142.
Slaughte MRr, Thakkar H, O'Brien PJ, 2002. Effect of diquat on the antioxidant system and cell growth in human neuroblastoma cells. Toxicology and Applied Pharmacology, 178, 63–70.
Smeyne RJ, Breckenridge CB, beck M, Jiao Y, Butt MT, Wolf J, Zadory D, Minnema D, Sturgess NC, Travis KZ, Cook AR, Smith LL, Botham PA, 2016. Assessment of the effects of MPTP and Paraquat on dopaminergic neurons and microglia in the substantia nigra pars compacta of C57BL/6 mice. Public Library of Science(PLOS ONE). doi:10.1371/journal.pone0164094
Somayajulu‐Niţu M, Sandhu JK, Cohen J, Sikorska M, Sridhar TS, Matei A, Borowy‐Borowski H, Pandey S, 2009. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and parkinsonism in adult rats: neuroprotection and amelioration of symptoms by water‐soluble formulation of coenzyme Q10. BMC Journal of Neuroscience, 10, 88. doi: 10.1186/1471‐2202‐10‐88
Spencer DD, Robbins RJ, Naftolin F, Marek KL, Vollmer T, Leranth C, Roth RH, Price LH, Gjedde A, Bunney BS, 1992. Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson's disease. New England Journal of Medicine, 327, 1541–1548.
Su C, Niu P, 2015. Low doses of single or combined agrichemicals induces α‐synuclein aggregation in nigrostriatal system of mice through inhibition of proteasomal and autophagic pathways. International Journal of Clinical and Experimental Medicine, 8, 20508–20515.
Taetzsch T, Block ML, 2013. Pesticides, microglial NOX2, and Parkinson's disease. Journal of Biochemical and Molecular Toxicology, 27, 137–149.
Tansey MG, Goldberg MS, 2009. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease, 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Thiruchelvam M, Brockel BJ, Richfield EK, Bags RB, Cory‐Slechta DA, 2000a. Potential and preferential effects of combined paraquat and maneb on nigrostriatal dopamine system: environmental risk factor for Parkinson's disease? Brain Research, 873, 225–234.
Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory‐Slechta DA, 2000b. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. Journal of Neuroscience, 20, 9207–9214.
Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory‐Slechta DA, 2003. Age‐related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. European Journal of Neuroscience, 18, 589–600.
Thundyil J, Lim KL, 2015. DAMPs and neurodegeneration. Ageing Research Reviews, 24(Pt A), 17–28. doi: 10.1016/j.arr.2014.11.003
Tien Nguyen‐nhu N, Knoops B, 2003. Mitochondrial and cytosolic expression of human peroxiredoxin 5 in Saccharomyces cerevisiae protect yeast cells from oxidative stress induced by paraquat. Federation of the European Biochemical Societies's Letters (FEBS)ers, 544, 148–152.
Tieu K, 2011. A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, 1, a009316. doi: 10.1101/cshperspect.a009316
Tieu K, Imm J, 2014. Mitochondrial dynamics as potential therapeutic target for Parkinson's disease? Advances in Clinical Neuroscience and Rehabilitation, 14, 6–8.
Turrens JF, 2003. Mitochondrial formation of reactive oxygen species. Journal of Physiology, 552(Pt 2), 335–344.
Van Remmen H, Qi W, Sabia M, Freeman G, Estlack L, Yang H, Mao Guo Z, Huang TT, Strong R, Lee S, Epstein CJ, Richardson A, 2004. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radical Biology and Medicine, 36, 1625–1634.
Watson MB, Nobuta H, Abad C, Lee SK, Bala N, Zhu C, et al., 2013. PACAP deficiency sensitizes nigrostriatal dopaminergic neurons to paraquat‐induced damage and modulates central and peripheral inflammatory activation in mice. Journal of Neuroscience, 240, 277–286. doi: 10.1016/j.neuroscience.2013.03.002
Widdowson PS, Farnworth MJ, Upton R, Simpson MG, 1996. No changes in behaviour, nigro‐striatal system neurochemistry or neuronal cell death following toxic multiple oral paraquat administration to rats. Human and Experimental Toxicology, 15, 583–591.
Widner H, Tetrud J, Rehncrona S, Snow B, Brundin P, Gustavii B, Björklund A, Lindvall O, Langston JW, 1992. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). New England Journal of Medicine, 327, 1556–1563.
Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, et al., 2005. The role of microglia in paraquat‐induced dopaminergic neurotoxicity. Antioxidants & Redox Signaling, 7, 654–661.
Yadav S, Gupta SP, Srivastava G, Srivastava PK, Singh MP, 2012. Role of secondary mediators in caffeine‐mediated neuroprotection in maneb‐ and paraquat‐induced Parkinson's disease phenotype in the mouse. Neurochemical Research, 37, 875–884.
Yang W, Tiffany‐Castiglioni E, 2007. The bipyridyl herbicide paraquat induces proteasome dysfunction in human neuroblastoma SH‐SY5Y cells. Journal of Toxicology and Environmental Health A, 70, 1849–1857.
Yang W, Chen L, Ding Y, Zhuang X, Kang UJ, 2007. Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ‐1‐deficient mice. Human Molecular Genetics, 16, 2900–2910.
Yao Z, Gandhi S, Burchell VS, Plun‐Favreau H, Wood NW, Abramov AY, 2011. Cell metabolism affects selective vulnerability in PINK1‐associated Parkinson's disease. Journal of Cell Science, 124(Pt 24), 4194–4202. doi: 10.1242/jcs.088260
Yin L, Lu L, Prasad K, Richfield E, Unger EL, Xu J, jones BC, 2011. Genetic‐based, differential susceptibility to paraquat neurotoxicity in mice. Neurotoxicology and Teratology, 33, 415–421.
Youdin MB, 2003. What have we learnt from CDNA microarray gene expression studies about the role of iron in MPTP‐induced neurodegeneration and Parkinson's disease. Journal of Neural Transmission, Supplementa. 65, 73–88.
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A, 2015. Mitochondrial dysfunction and α‐synuclein synaptic pathology in Parkinson's disease: who's on first? Journal of Parkinson's Disease, 2015, 108029.
Appendix B – AOPs developed for Infant Leukaemia and Childhood Leukaemia
Adverse Outcome Pathway (AOP) 3: In utero DNA topoisomerase II poisons leading to infant leukaemia
Introduction
Infant leukaemia is a rare haematological disease (1 in 106 newborns, accounting for 10% of all childhood acute lymphoblastic leukaemias (ALL)) manifesting soon after birth (< 1 year) and having a poor prognosis (Sanjuan‐Pla et al., 2015). Compared to the more frequent childhood leukaemia, infant leukaemia show distinct features:
an early neonatal onset linked to its plausible origin as a ‘intrauterine developmental disease’ (Greaves, 2015; Sanjuan‐Pla et al., 2015);
rearrangements of the mixed‐lineage leukaemia (MLL; KMT2A) gene on the q23 band of chromosome 11, as the hallmark genetic abnormality (Joannides and Grimwade, 2010);
however, MLL is not the only translocation gene; for infant ALL, about 60–80% carry an MLL rearrangement (Jansen et al., 2007; Sam et al., 2012) and the percentage for infant acute myeloid leukaemia (AML) is about 40%;
the MLL rearrangement at an early stage of development; the likely target cells (still unidentified) are the haematopoietic stem and progenitor cells (HSPC) in fetal liver and/or earlier (mesenchymal) stem cells in embryonic mesoderm (Bueno et al., 2009; Menendez et al., 2009);
the infant MLL‐rearranged leukaemia carries less somatic mutations (1.3 vs 6.5/case) than the childhood disease (Dobbins et al., 2013; Andersson et al., 2015), pointing to the lack of a ‘second hit’ and suggesting a ‘one big hit’ origin.
Overall, based on the available evidence, infant leukaemia pathogenesis originates from a single, severe hit to a target cell during early intrauterine development. Whereas the limited epidemiological studies do not allow any firm conclusion on a possible role for chemicals in infant leukaemia (Pombo‐de‐Oliveira et al., 2006; Ferreira et al., 2013), exposures to chemicals able to induce MLL rearrangements through topoisomerase II (TopoII) ‘poison’, particularly etoposide and other TopoII ‘poisons’, including some bioflavonoids, have been suggested as agents promoting the driver genetic oncogenic event. Experimental models for infant leukaemia have been developed, but a wholly satisfactory model reproducing the phenotype and latency is not yet available.
Nevertheless, the anticancer drug etoposide can be considered as a model chemical for DNA topoisomerase ‘poison’. Acute leukaemia is an adverse effect recorded in etoposide‐treated patients, showing MLL rearrangements that are in many ways analogous to those in infant leukaemia (Bueno et al., 2009; Joannides et al., 2010, 2011). Therefore the proposed AOP is supported by a number of convincing inferential evidences by means of using etoposide as a tool compound to empirically support the linkage between the proposed molecular initiating event (MIE) and the adverse outcome (AO). In the meanwhile, this AOP identifies several knowledge gaps, the main ones being the identification of the initiating cell and the investigation of TopoII poisons in a robust model; thus, the present AOP may be modified in future on the basis of new evidence.
References
Andersson AK, Ma J, Wang J, et al., 2015. The landscape of somatic mutations in infant MLL‐rearranged acute lymphoblastic leukemias. Nature Genetics, 47, 330–337. doi: 10.1038/ng.3230
Bueno C, Catalina P, Melen GJ, Montes R, Sanchez L, Ligero G, Garcia‐Perez JL, Menendez P, 2009. Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis, 30, 1628–1637. doi: 10.1093/carcin/bgp169
Dobbins SE, Sherborne AL, Ma YP, Bardini M, Biondi A, Cazzaniga G, Lloyd A, Chubb D, Greaves MF, Houlston RS, 2013. The silent mutational landscape of infant MLL‐AF4 pro‐B acute lymphoblastic leukemia. Genes Chromosomes Cancer, 52, 954–960. doi: 10.1002/gcc.22090
Ferreira JD, Couto AC, Pombo‐de‐Oliveira MS, Koifman S, 2013. Brazilian Collaborative Study Group of Infant Acute Leukemia. In utero pesticide exposure and leukemia in Brazilian children < 2 years of age. Environmental Health Perspectives, 121, 269–275. doi: 10.1289/ehp.1103942
Greaves M, 2015. When one mutation is all it takes. Cancer Cell, 27, 433–434.
Jansen MW, Corral L, van der Velden VH, Panzer‐Grumayer R, Schrappe M, Schrauder A, et al., 2007. Immunobiological diversity in infant acute lymphoblastic leukemias related to the occurrence and type of MLL rearrangement. Leukemia, 21, 633–641.
Joannides M, Grimwade D, 2010. Molecular biology of therapy‐related leukaemias. Clinical and Translational Oncology, 12, 8–14. doi: 10.1007/s12094‐010‐0460‐5
Joannides M, Mays AN, Mistry AR, Hasan SK, Reiter A, Wiemels JL, Felix CA, Coco FL, Osheroff N, Solomon E, Grimwade D, 2011. Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterranean Journal of Hematology and Infectious Diseases, 3, e2011045. doi: 10.4084/MJHID.2011.045
Menendez P, Catalina P, Rodriguez R, Melen GJ, Bueno C, Arriero M, Garcia‐Sanchez F, Lassaletta A, Garcia‐Sanz R, Garcia‐Castro J, 2009. Bone marrow mesenchymal stem cells from infants with MLL‐AF4 + acute leukemia harbor and express the MLL‐AF4 fusion gene. Journal of Experimental Medicine, 206, 3131–3141. doi: 10.1084/jem.20091050
Pombo‐de‐Oliveira MS, Koifman S, 2006. Brazilian Collaborative Study Group of Infant Acute Leukemia. Infant acute leukemia and maternal exposures during pregnancy. Cancer Epidemiology Biomarkers & Prevention, 15, 2336–2341.
Sam TN, Kersey JH, Linabery AM, Johnson KJ, Heerema NA, Hilden JM, et al., 2012. MLL gene rearrangements in infant leukaemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatric Blood and Cancer, 58, 836–839.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukemia. Blood, 126, 2676–2685. doi: 10.1182/blood‐2015‐09‐667378
Abbreviations: TopoII, DNA topoisomerase II; HSPC, haematopoietic stem and progenitor cell; t‐AL, therapy‐associated acute leukaemia;
Adverse Outcome Pathway (AOP): In utero DNA topoisomerase II inhibition leading to infant leukaemia
Figure B.1.

AOP scheme
MIE: In utero exposure to DNA topoisomerase II ‘poisons’
How this MIE works
DNA topoisomerase (Top) II enzyme ‘poisons’ disturb the normal TopoII enzyme function and cause a ‘hanging double strand break (DSB)’ at a specified DNA sequence. The above description of the MIE is of significance because there are 3 different kinds of ‘poisons’ of TopoII enzyme, out of which competitive inhibitors prevent the function of the enzyme and cause cell death, whereas other interfacial and covalent inhibitors may cause – depending on the situation – other consequences of DNA damage response including chromosomal rearrangements (Pendleton et al., 2014; Lu et al., 2015). A further prerequisite for the specific outcome, i.e. creation of chromosomal rearrangement, is that TopoII ‘poison’ has to occur in an especially vulnerable and correct hot spot in the MLL locus in the right target cell vulnerable to transformation.
TopoII enzymes have several crucial functions in DNA replication, transcription, repair and chromatin remodelling, i.e. TopoII enzymes take care of DNA integrity and topology. Because the enzyme functions by passing an intact double helix through a transient double‐stranded break, any disturbances in its function, e.g. by chemical inhibitors, could have a profound effect on genomic stability, resulting in DNA repair response, gene and chromosomal damage, initiation of apoptosis and ultimate cell death. A double‐strand break and error‐prone non‐homologous end‐joining (NHEJ) DNA repair mechanism may lead to gene rearrangements; chromosomal translocations and consequently fusion genes (see Figure B.2). A comprehensive description of TopoII enzymes and their functions and derangements could be found in recent review articles (Cowell and Austin, 2012; Ketron and Osheroff, 2014; Pendleton et al., 2014).
Figure B.2.
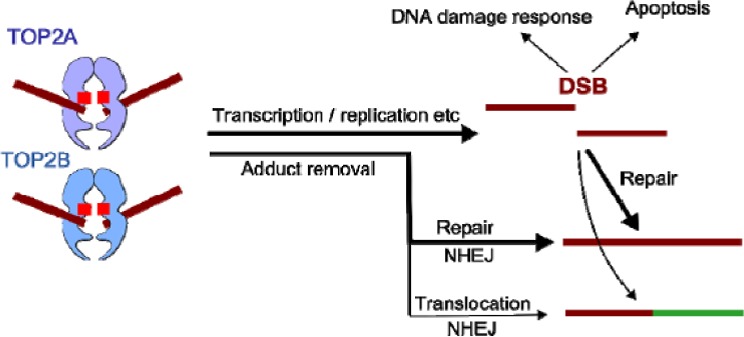
TOP2 Poisons, downstream events. TOP2 poisons inhibit the religation step of the TOP2 reaction cycle, leading to accumulation of covalent TOP2‐DNA cleavage complexes. These lesions are cytotoxic and lead to activation of the DNA damage response and potentially apoptosis. Alternatively these lesions are repaired, largely through the non‐homologous end‐joining pathway. Translocations observed in therapy‐related leukaemia are presumed to occur as a result of misrepair, joining two heterologous ends. (from Cowell and Austin, 2012, fig. 3, CC BY 3.0)
How it is measured or detected
The identification and measurement of the inhibition of TopoII enzymes is made more difficult by the presence of different molecular mechanisms (see above). However, some assays are used in pharmacological research to screen TopoII ‘poisons’, including cell‐free decatenation assay (Schroeter et al., 2015). The most important mode, the cleavage activity of TopoII can be studied in vitro, by using a human recombinant enzyme and an appropriate double‐stranded plasmid as a target to quantitate double‐strand breaks (Fortune and Osheroff, 1998). A cleavage can also be indirectly detected by measuring various indicators of DNA damage response, such as ATM activity, p53 expression, γH2AX or Comet assay (Li et al., 2014; Schroeter et al., 2015; Castano et al., 2016).
It is useful to note that several chemicals identified as TopoII ‘poisons’ do require metabolic oxidation to become active inhibitors. Etoposide itself is converted via the catechol metabolite to etoposide 3‐quinone, which is a covalent TopoII poison (Smith et al., 2014), whereas etoposide and its catechol are interfacial inhibitors. Curcumin is also an active TopoII poison due to its oxidised metabolites (Gordon et al., 2015). This fact deserves consideration if a screening for TopoII inhibition is envisaged.
Evidence supporting taxonomic applicability (tissue type, taxa, life stage, sex)
DNA topoisomerases are ubiquitous enzymes, which control the integrity of double‐stranded DNA. They are thus key enzymes at all levels of living organisms. The available evidence suggest that important differences in sensitivity to topoisomerase inhibition might exist among different cell types, depending on the amount of proliferative burden, of the TopoII enzymes and on physiological repair processes. Mesodermal precursor or haematopoietic stem and progenitor cells (HSPCs) are rapidly dividing cells with a high content of TopoII and for these reasons they can be a sensitive target during a critical developmental window (Hernández and Menéndez, 2016). In addition, evidence from micronuclei assay studies conducted in untreated and chemical‐treated foetuses and newborns show that both the baseline and chemically induced micronuclei frequencies are higher in the foetuses and infants than in adults (Udroiu et al., 2016). This is possibly indicating a greater sensitivity to genotoxic insult during development which can be due to the higher proliferation rate and lower ability of DNA repair of the haematopoietic stem cells. However, the role that the different microenvironments (fetal liver, infant bone marrow and adult bone marrow) during ontogenesis can exert on cell sensitivity cannot be ruled out (Udroiu et al., 2016). The existence of relevant interspecies differences is unknown, but it cannot be ruled out presently.
Evidence for Chemical Initiation of this Molecular Initiating Event (MIE)
A number of drugs, environmental chemicals and natural substances are identified as TopoII ‘poisons’ (Pendleton et al., 2014) (Table B.1). A well investigated example is the anticancer drug etoposide; also bioflavonoids, e.g. genistein (Barjesteh van Waalwijk van Doorn‐Khosrovani et al., 2007; Azarova et al., 2010) bind to TopoII enzymes, induce cleavage in the MLL gene and produce a fusion gene (and its product) in human cells. The organophosphate pesticide chlorpyrifos has been shown to inhibit (‘poison’) the enzyme in vitro (Lu et al., 2015).
Table B.1.
TopoII poisons
| Chemical class | Examples | References |
|---|---|---|
| Anticancer agents | ||
| Epipodophyllotoxins | Etoposide, teniposide | Montecucco et al. (2015) |
| Anthracyclines | Doxorubicin, epirubicin, daunorubicin, idarubicin, aclarubicin | Cowell and Austin (2012) |
| Anthacenedione | Mitoxantrone | Cowell and Austin (2012) |
| Acridines | Amsacrine | Cowell and Austin (2012) |
| Bioflavonoids | ||
| Flavones | Luteolin, apigenin, diosmetin | Ketron and Osheroff (2014) |
| Flavonols | Myricetin, quercetin, kaempferol, fisetin | Ketron and Osheroff (2014) |
| Isoflavones | Genistein | Ketron and Osheroff (2014) |
| Catechins | EGCG, ECG, EGC, EC | Ketron and Osheroff (2014) |
| Isothiocyanates | Benzyl‐isothiocyanate, phenethyl‐isothiocyanate, sulforaphane | Ketron and Osheroff (2014) |
| Other phytochemicals | Curcumin | Ketron and Osheroff (2014) |
| Environmental chemicals | ||
| Aromatic compounds | Benzene, PAHs | |
| Nitrosamines | Diethylnitrosamine | Thys et al. (2015) |
| Organophosphates | Chlorpyrifos | Lu et al. (2015) |
Etoposide
Much of the relevant, albeit indirect, evidence to support this AOP come from the studies on etoposide, an anticancer drug TopoII ‘poison’, which is known to induce therapy‐associated acute leukaemia (t‐AL) in adults (Cowell and Austin, 2012; Pendleton et al., 2014). It is of interest that the latency of t‐AL is < 2 years between the treatment of the primary malignancy and the clinical diagnosis of the secondary disease and that the prognosis of t‐AL is poor (Pendleton et al., 2014). t‐AL is characterised by the MLL rearrangements and it is practically certain that these fusion genes are caused by etoposide or anthracyclines treatment, because MLL rearrangements have not been detected in bone marrow samples banked before the start of the treatment of the first malignancy. Also the breakpoints in MLL or partner genes fall within a few base pairs of a drug‐induced enzyme‐mediated DNA cleavage site (Pendleton et al., 2014).
Etoposide can induce MLL rearrangements in different cell types; interestingly, embryonic stem cells and their haematopoietic derivatives are much more sensitive than cord blood‐derived CD34+ cells to etoposide‐induced MLL rearrangements; in addition, undifferentiated human embryonic stem cells (hESCs) were concurrently liable to acute cell death (Bueno et al., 2009). These findings suggest that the MIE should be put into evidence in target cell models with appropriate sensitivity.
Bioflavonoids
Bioflavonoids are natural polyphenolic compounds in a large variety of plant‐derived food items. TopoII‐mediated DNA cleavage has been linked to genistein, kaempferol, luteolin, myricetin and apigenin (Strick et al., 2000; Bandele and Osheroff, 2007; Azarova et al., 2010; Lopez‐Lazaro et al., 2010), although the concentrations in in vitro studies have been quite high. It has also been demonstrated that several bioflavonoids are capable of inducing the cleavage of the MLL gene in human cell lines (Strick et al., 2000; van Doorn‐Khosrovani et al., 2007). The in vitro effects of bioflavonoids suggested a possible link between dietary intake and infant leukaemia (e.g. Azarova et al., 2010; Lanoue et al., 2010); however until now, epidemiological evidence existing to support or refute such a hypothesis is based on small studies (Ross et al., 1996; Spector et al., 2005).
Chlorpyrifos
Chlorpyrifos is a widely used organophosphate insecticide, which has been suspected as a risk factor for infant and childhood leukaemia after the house‐hold exposure of pregnant women. According to Lu et al. (2015), chlorpyrifos and its metabolite chlorpyrifos oxon exhibit an inhibitory effect on in vitro TopoII activity. Chlorpyrifos causes DNA double strand breaks as measured by the neutral Comet assay and induces MLL gene rearrangements in human fetal liver‐derived CD34+ haematopoietic stem cells via TopoII ‘poisoning’ as detected by the FISH assay and in vitro isolated TopoII inhibition assay, respectively (Lu et al., 2015). Chlorpyrifos also stabilises the TopoII‐DNA cleavage complex. Etoposide was used a positive reference compound in these studies and it performed as expected. The lowest concentration of chlorpyrifos used was 1 μM and it gave a statistically significant effect in many in vitro assays. The point of departure of etoposide, which was calculated to be 0.01 to 0.1 μM (Li et al., 2014), is at least 10‐fold lower than that of chlorpyrifos.
References
Alexander FE, Patheal SL, Biondi A, Brandalise S, Cabrera ME, Chan LC, Chen Z, Cimino G, Cordoba JC, Gu LJ, Hussein H, Ishii E, Kamel AM, Labra S, Magalhaes IQ, Mizutani S, Petridou E, de Oliveira MP, Yuen P, Wiemels JL, Greaves MF, 2001. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Research, 61, 2542–2546.
Azarova AM, Lin RK, Tsai YC, Liu LF, Lin CP, Lyu YL, 2010. Genistein induces topoisomerase IIbeta‐ and proteasome‐mediated DNA sequence rearrangements: implications in infant leukemia. Biochemical and Biophysical Research Communications, 399, 66–71. doi: 10.1016/j.bbrc.2010.07.043
Bandele OJ, Osheroff N, 2007. Bioflavonoids as poisons of human topoisomerase II alpha and II beta. Biochemistry, 46, 6097–6108.
Barjesteh van Waalwijk van Doorn‐Khosrovani S, Janssen J, Maas LM, Godschalk RW, Nijhuis JG, van Schooten FJ, 2007. Dietary flavonoids induce MLL translocations in primary human CD34 + cells. Carcinogenesis, 28, 1703–1709.
Castaño J, Herrero AB, Bursen A, González F, Marschalek R, Gutiérrez NC, Menendez P, 2016. Expression of MLL.AF4 or 1 AF4.MLL fusions 2 does not impact the efficiency of DNA damage repair. Nucleic Acids Research, in press.
Cowell IG, Austin CA, 2012. Mechanism of generation of therapy related leukemia in response to anti‐topoisomerase II agents. International Journal of Environmental Research and Public Health, 9, 2075–2091. doi: 10.3390/ijerph9062075
Fortune JM, Osheroff N, 1998. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. Journal of Biological Chemistry, 273, 17643–17650.
Gordon ON, Luis PB, Ashley RE, Osheroff N, Schneider C, 2015. Oxidative Transformation of Demethoxy‐ and Bisdemethoxycurcumin: products, mechanism of formation, and poisoning of human topoisomerase IIβ. Chemical Research in Toxicology, 28, 989–996. doi: 10.1021/acs.chemrestox.5b00009
Hernandez Jerez A, Menendez P, 2016. Linking pesticide exposure with pediatric leukemia: potential underlying mechanisms. International Journal of Molecular Sciences, 17, 461.
Lanoue L, Green KK, Kwik‐Uribe C, Keen CL, 2010. Dietary factors and the risk for acute infant leukemia: evaluating the effects of cocoa‐derived flavanols on DNA topoisomerase activity. Experimental Biology and Medicine (Maywood), 235, 77–89. doi: 10.1258/ebm.2009.009184
Li Z, Sun B, Clewell RA, Adeleye Y, Andersen ME, Zhang Q, 2014. Dose‐response modeling of etoposide‐induced DNA damage response. Toxicological Sciences, 137, 371–384. doi: 10.1093/toxsci/kft259.
Lopez‐Lazaro M, Willmore E, Austin CA, 2010. The dietary flavonoids myricetin and fisetin act as dual inhibitors of DNA topoisomerases I and II in cells. Mutation Research, 696, 41–47. doi: 10.1016/j.mrgentox.2009.12.010
Lu C, Liu X, Liu C, Wang J, Li C, Liu Q, Li Y, Li S, Sun S, Yan J, Shao J, 2015. Chlorpyrifos induces mll translocations through caspase 3‐dependent genomic instability and topoisomerase II inhibition in human fetal liver hematopoietic stem cells. Toxicological Sciences, 147, 588–606. doi: 10.1093/toxsci/kfv153
Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D, Osheroff N, 2014. Topoisomerase II and leukemia. Annals of the New York Academy of Sciences, 1310, 98–110. doi: 10.1111/nyas.12358
Ross JA, Potter JD, Reaman GH, Pendergrass TW, Robison LL, 1996. Maternal exposure to potential inhibitors of DNA topoisomerase II and infant leukemia (United States): a report from the Children's Cancer Group. Cancer Causes & Control, 7, 581–590.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukemia. Blood Journal, 126, 2676–2685. doi: 10.1182/blood‐2015‐09‐667378
Schroeter A, Groh IA, Favero GD, Pignitter M, Schueller K, Somoza V, Marko D, 2015. Inhibition of topoisomerase II by phase II metabolites of resveratrol in human colon cancer cells. Molecular Nutrition & Food Research. doi: 10.1002/mnfr.201500352
Smith NA, Byl JA, Mercer SL, Deweese JE, Osheroff N, 2014. Etoposide quinone is a covalent poison of human topoisomerase IIβ. Biochemistry, 53, 3229–3236. doi: 10.1021/bi500421q
Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, Blair CK, Reaman GH, Ross JA, 2005. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children's oncology group. Cancer Epidemiology Biomarkers & Prevention, 14, 651–655.
Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD, 2000. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proceedings of the National Academy of SciencesU S A, 97, 4790–4795.
Udroiu I, Sgura A, 2012. Genotoxicity sensitivity of the developing hematopoietic system. Mutation Research, 767, 1–7.
KE1: In utero MLL chromosomal translocation
How this key event works
Chromosomal rearrangements of the mixed‐lineage leukaemia (MLL) gene, located on the q23 band of chromosome 11 (11q23), are the genetic hallmark of most infant leukaemias (Meyer et al., 2013; Sanjuan‐Pla et al., 2015). MLL is located within the fragile site FRA11G; chromosomal fragile sites are regions of the genome susceptible to breakage under conditions of replication stress; interference with TopoII may promote fragile site instability. MLL encodes a protein homologous to the Drosophila trithorax gene, which has relevant functions in embryogenesis and haematopoiesis (Hess et al., 1997; Ernest et al., 2004).
There are many translocation and fusion partners for MLL; DNA breakage within MLL can lead to rearrangement with over 120 partner genes (Meyer et al., 2013). In principle all MLL fusion genes are potential initiating drivers, although clinical studies have shown a preponderance with infant leukaemia for only a few of these rearrangements. For infants diagnosed with ALL, approximately 60–80% carry an MLL rearrangement (Jansen et al., 2007; Sam et al., 2012), with predominant fusion partners being AF4 (41%), ENL (18%), AF9 (11%) or another partner gene (10%). In particular, the fusion gene MLL‐AF4 shows a specific and consistent relationship with the disease (Menendez et al., 2009): however, it has been difficult to reproduce a manifest disease resulting from this rearrangement in in vivo animal models. For AML, about 30% of the patients carry an MLL rearrangement.
The occurrence of MLL rearrangements at a very early fetal development is highly probable on the basis of neonatal blood spot analysis and by the high concordance rate of infant leukaemia in monozygotic twins (Ford et al., 1993; Gale et al., 1997; Sanjuan‐Pla, 2015). Menendez et al. (2009) showed that MLL‐AF4 fusion gene is present in bone marrow mesenchymal stem cells in infant leukaemia patients, but not in patients of childhood leukaemia, suggesting that the origin of the fusion gene is probably prehaematopoietic. Consequently, the affected cell, the so called leukaemia‐initiating cell, may be an early prehaematopoietic mesodermal precursor, a haematopoietic stem cell or haematopoietic progenitor cell residing mainly in the liver (Greaves, 2015; Sanjuan‐Pla et al., 2015).
MLL protein (complexed with a large number of other protein factors) serves as a transcriptional activator or repressor via the binding to promoter regions of active genes, marking these regions by covalent histone modifications (Sanjuan‐Pla et al., 2015). Translocation and creation of fusion genes and products destroys the intrinsic control mechanisms of the MLL protein. The resulting ‘ectopic’ functions involve promoter hyper‐activation and re‐acquiring stem cell features (Sanjuan‐Pla et al., 2015). A schematic presentation of the drastic changes of the MLL product is depicted in Figure B.3.
Figure B.3.
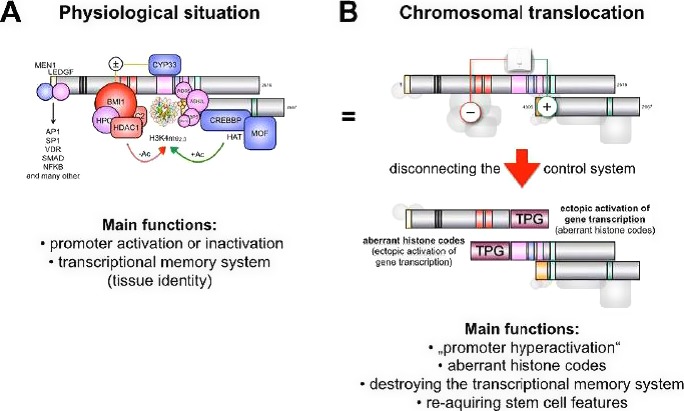
Proposed model for the oncogenic conversion of MLL fusions: (A) Physiological situation and (B) A chromosomal translocation, which leads to the intrinsic regulatory mechanism of MLL being destroyed (Republished with permission of American Society of Hematology, from Sanjuan‐Pla et al., 2015; permission conveyed through Copyright Clearance Center, Inc.)
MLL translocation sites (breakpoint sequences) in the therapy‐related leukaemia fall within a few base pairs of etoposide‐induced enzyme‐mediated DNA cleavage site (r). Although rearrangements associated with infant leukaemias are often more complex than those observed in treatment‐related leukaemias, many are nevertheless associated with stable TopoII‐mediated DNA cut sites. Although all these findings are indirect regarding infant leukaemia, they are nevertheless rather persuasive in this respect.
Growing scientific evidence, including the stable genome of the patients, suggests that infant leukaemia originates from one ‘big‐hit’ occurring during a critical developmental window of stem cell vulnerability (Andersson et al., 2013; Greaves 2015). Therefore, the totality of evidence suggests the essential role of the formation of MLL‐AF4 (and other partner) fusion gene and product in causing pleiotropic effects in the affected cell and directing it to the obligatory pathway to the adverse outcome of leukaemia (see KER2).
How it is measured
The presence and structure of a fusion gene can be identified with PCR or related techniques. Mapping of cleavage sites in the gene needs genomic DNA. In cells or tissues, the detection of a fusion gene is possible by appropriate immunofluorescent techniques.
Assays measuring chromosomal aberrations, micronuclei or DNA and chromosome damage (Comet assay) may indirectly identify the KE through its consequences in experimental systems in vitro and in vivo; the degree of accuracy of such identification cannot be evaluated presently.
Taxonomic applicability
Although the KE deals with the general process of DNA integrity, the available evidence do not allow for evaluating whether any significant difference occurs among cell types or species. It has been shown that the mouse has an analogous fusion gene mll‐af4. A recent study has shown that in utero exposure to etoposide induces mll translocations in Atm‐knockout mice, which are defective in the DNA damage response, albeit not in wild‐type mice; moreover, fetal liver haematopoietic stem cells were more susceptible to etoposide than maternal bone marrow mononuclear cells, pointing out the life stage‐related susceptibility in regard to TopoII ‘poison’ also in the mouse (Nanya et al., 2015).
References
Ernest P, Fisher JK, Avery W, Sade S, Foy D, Korsmeyer SJ, 2004. Definitive hematopoiesis requires the mixed‐lineage leukemia gene. Developmental Cell, 6, 437–443.
Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et al., 1993. In utero rearrangements in the trithorax‐related oncogene in infant leukaemias. Nature, 363, 358–60. doi: 10.1038/363358a0
Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al., 1997. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proceedings of the National Academy of Sciences USA, 94, 13950–13954.
Greaves M, 2015. When one mutation is all it takes. Cancer Cell, 27, 433–434.
Hess JL, Yu BD, Li B, Hanson RD, Korsmeyer SJ, 1997. Defect in yolk sac hematopoiesis in mll‐null embryos. Blood, 90, 1799–1806.
Jansen MW, Corral L, van der Velden VH, Panzer‐Grumayer R, Schrappe M, Schrauder A, et al., 2007. Immunobiological diversity in infant acute lymphoblastic leukemiais related to the occurrence and type of MLL rearrangement. Leukemia, 21, 633–641.
Menendez P, Catalina P, Rodriguez R, Melen GJ, Bueno C, Arriero M, Garcia‐Sanchez F, Lassaletta A, Garcia‐Sanz R, Garcia‐Castro J, 2009. Bone marrow mesenchymal stem cells from infants with MLL‐AF4+ acute leukemia harbor and express the MLL‐AF4 fusion gene. Journal of Experimental Medicine, 206, 3131–3141. doi: 10.1084/jem.20091050
Meyer C, Hofmann J, Burmeister T, et al., 2013. The MLL recombinome of acute leukemias in 2013. Leukemia, 27, 2165–2176.
Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S, Takagi M, 2015. Dysregulation of the DNA Damage Response and KMT2A Rearrangement in Fetal Liver Hematopoietic Cells. Public Library of Science (PLoS ONE), 10, e0144540. doi:10.1371/journal.pone.0144540
Sam TN, Kersey JH, Linabery AM, Johnson KJ, Heerema NA, Hilden JM, et al., 2012. MLL gene rearrangements in infant leukaemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatric Blood and Cancer, 58, 836–839.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukemia. Blood Journal, 126, 2676–2685. doi: 10.1182/blood‐2015‐09‐667378
Adverse Outcome (AO) Infant leukaemia
How this key event works
Symptoms of leukaemia – thrombocytopenia resulting in sensitivity to bruising and bleeding, anaemia with pallor and fatigue, neutropaenia associated with increased susceptibility to infections – are principally due to the displacement of the normal haematopoiesis by expansion of leukaemia cells. Leukaemic infiltration of the brain is common at diagnosis of the infant leukaemia (Hunger and Mulligham, 2015).
How it is measured
Haematological methods – identification of leukaemia cells and routine blood cell counts; observations of clinical symptoms.
Following clinical diagnosis, methods for refined diagnosis include bone marrow aspirates for immunophenotypic analyses and cytogenetic assays for molecular stratification.
The carcinogenicity assays and the extended one generation test (OECD 443) include endpoints that can potentially explore the AO; however, considerations should be made on the specificity of the disease to humans.
Taxonomic applicability
Infant leukaemia is a paediatric leukaemia likely resulting from gene‐environmental interactions. The limited data available suggest that dietary and environmental exposure to substances targeting topoisomerases together with reduced ability of the fetus or their mother to detoxify such compounds because of the polymorphic variants of given genes could contribute to the development of this AO (Hernadez et al., 2016).
In animals the disease is not known and artificial animal models able to reproduce the disease have limitations. Bardini et al. (2015) have however developed a xenograft mouse model with patient MLL‐AF4‐involving leukoblasts transplanted.
Regulatory relevance of the AO
Genotoxicity in general and carcinogenicity are apical endpoints in established regulatory guideline studies. TopoII poisoning has been listed as one of the potential mechanisms of genotoxicity and carcinogenicity in the ICH M7 guideline for human medicines. It is also known that some manifestations of genotoxicity in tests measuring chromosomal aberrations, micronuclei or DNA and chromosome damage (Comet assay) are partially due to double‐strand breaks created by the disturbed action of TopoII enzymes.
The extended one generation test (OECD 443) includes a developmental immunotoxicity cohort. At present the cohort may identify postnatal effects of prenatal and neonatal exposures on the immune tissues and white blood cells population. However, each regulatory guideline study has potential limitations e.g. no specific parameters are in place to identify a pattern relevant to infant leukaemia in humans in the extended one generation test, no treatment is occurring during the early in‐utero development phase in the carcinogenicity assay and no considerations on the possible higher sensitivity of the HSC are in place for the genotoxicity assays.
Epidemiological evidence linking pesticide exposure to infant leukaemia, also suggests that pesticide exposure may have a greater impact on children than adults; though, almost all of the available evidence does not make a distinction between infant and childhood leukaemia. However, most epidemiological studies are limited because no specific pesticides have been directly associated with the risk of leukaemia, but rather the broad term ‘pesticide exposure’ (Hernández and Menéndez, 2016). In this perspective, this AOP would provide a regulatory relevant support for understanding the potential of a chemical to be involved in this toxicological pathway.
References
Bardini M, Woll PS, Corral L, Luc S, Wittmann L, Ma Z, Lo Nigro L, Basso G, Biondi A, Cazzaniga G, Jacobsen SE, 2015. Clonal variegation and dynamic competition of leukemia‐initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia, 29, 38–50. doi: 10.1038/leu.2014.154
Hernandez A and Menendez P, 2016. Linking pesticide exposure with pediatric leukemia: potential underlying mechanisms. International Journal of Molecular Sciences, 17, 461.
Hunger SP, Mullighan CG, 2015. Acute Lymphoblastic Leukemia in Children. New England Journal of Medicine, 73, 1541–1552.
1st KER: In utero DNA topoisomerase II inhibition (KEup) leading to In utero MLL chromosomal translocation (KEdown)
How this Key Event Relationship works
Certain TopoII poisons stabilise the intermediate cleavage complex and prevent the religation with appropriate DNA strands. Covalently DNA end‐bound TopoII protein is digested and a hanging end is created. The same process happens in the translocation partner gene. Hanging ends of both genes are processed and subsequently joined by non‐homologous end joining (Cowell and Austin, 2012). There is evidence that this inappropriate joining of ‘hanging ends’ happens in the same transcriptional factory (hub), and the result is a fusion gene and ultimately protein product (Cowell and Austin, 2012; Pendleton et al., 2014; Sanjuan‐Pla et al., 2015). The first part of this description has not been shown in the putative target cell, which is still not unequivocally identified, but for the second part there is ample evidence of formation of MLL‐AF4 fusion product that has been a result of a very early chromosomal translocation and rejoining. It is of interest that the simultaneously induced specific DSBs in the MLL gene and two different translocation partners (AF4 and AF9) by engineered nucleases in human HSPCs resulted in specific ‘patient‐like’ chromosomal translocations (Breese et al., 2016).
Weight of Evidence
Evidence supporting the causal relationship between etoposide‐induced TopoII inhibition and the MLL rearrangement leading to the fusion gene is strong regarding treatment‐related acute leukaemia (Cowell and Austin, 2012; Pendleton et al., 2014). The bioflavonoid‐rich diet in pregnant women has been suggested to initiate infant leukaemia by an analogous causality between in utero inhibition of TopoII enzymes and creation of the fusion gene. However, there is no direct evidence in humans and it is also difficult or impossible to study. Power of epidemiological studies is relatively weak in the case of a very rare disease and case‐control or spatiotemporal cluster studies have barely suggested a causal relationship between exposures and disease.
Biological plausibility
The KER as such is biologically plausible. Type II topoisomerases are ubiquitous enzymes which are essential for a number of fundamental DNA processes. As they generate DNA strand breaks, they can potentially fragment the genome. Indeed, while these enzymes are essential for the survival of proliferating cells they can also have significant genotoxic effects by means of accumulation of DNA strand breaks that, if not resulting in cell death may lead to chromosomal translocation in the surviving cell population (McClendon et al., 2007). DNA breaks and MLL rearrangements by etoposide and bioflavonoids have been demonstrated in human fetal liver haematopoietic stem cells, in human embryonic stem cells and in human prehaematopoietic mesenchymal stem cells as well as in cord blood mononuclear cells (Ishii et al., 2002; Blanco et al., 2004; Moneypenny et al., 2006; Bueno et al., 2009; Menendez et al., 2009), which clearly shows that TopoII‐associated MLL rearrangements are produced in appropriate human cells in utero.
Empirical support for linkage
There are animal models for infant leukaemia which recapitulate at least some salient aspects of the disease (Sanjuan‐Pla et al., 2015). However, for example the MLL‐AF4 knock‐in mice develop leukaemia only after a prolonged latency (Chen et al., 2006), thus not recapitulating the ‘pathognomonic’ feature of infant leukaemia.
Etoposide treatment in vivo in mice at day 13.5 of pregnancy induces MLL breakage in fetal liver haematopoietic stem cells in utero, but MLL‐rearranged fusion mRNAs were detected only in mice which were defective in the DNA damage response, i.e. atm knockout mice. A fusion gene analogous to MLL‐AF4 was not detectable in the wild type mice. In this study, an intraperitoneal injection of 10 mg/kg of etoposide into pregnant mice at day 13.5 of pregnancy resulted in a maximum fetal liver concentration of about 5 μM. A dose of 0.5 mg/kg did not result in a measurable concentration. A statistically significant increase (about sixfold) in DSBs in the MLL gene of isolated fetal liver haematopoietic stem cells was observed after a single dose of 1 mg/kg to pregnant mice.4 A clear activation of DNA damage response was observed at the dose of 10 mg/kg (Nanya et al., 2016).
There is a lot of information about the interaction of etoposide with TopoII enzymes and MLL chromosomal translocation at the cell culture level and in connection with treatment‐related leukaemia.
Molecular dose–response modelling of etoposide‐induced DNA damage response, based on comprehensive in vitro high content imaging in the HT1080 cell model, was developed by Li et al., (2014). The model was based on the hypothesis that cells are capable of clearing low‐level DNA damage with existing repair capacity, but when the number of DSBs exceeds a certain value; ATM and p53 become fully activated through reversible mechanism, leading to elevated repair capacity. The model was able to capture quantitatively the dose–response relationships of a number of markers observed with etoposide. Especially interesting are the dose–response relationships for activation of p53 and the formation of micronuclei in the target cell model, which indicate point‐of‐departure concentrations of etoposide in the range of 0.01 to 0.1 μM (Li et al., 2014). This range is in agreement with the finding that in human fetal liver CD34+ cells an increase in DSBs was observed at a concentration of 0.14 μM and MLL translocations were detectable by FISH or flow cytometry at higher concentrations (Moneypenny et al., 2006).
Uncertainties and Inconsistencies
A prerequisite for the specific outcome, i.e. creation of chromosomal rearrangement, is that TopoII inhibition has to occur in an especially vulnerable and correct hot spot in the MLL locus; however, details of this process and how it happens are not clear.
A target cell, i.e. leukaemia‐initiating cell, has not been identified with sufficient confidence and consequently there is no target cell model to recapitulate the linkage between TopoII inhibition (‘poisoning’) and the production of DSB in an appropriate target. Recently, by the expression of engineered nucleases (TALENs) to induce simultaneous patient specific double strand breaks in the MLL gene and two different known translocation partners (AF4 and AF9), Breese et al. (2015) were able to produce specific chromosomal translocations in K562 cells and in primary HSPCs.
In‐utero etoposide‐treatment failed to induce leukaemogenesis (Nanya et al., 2015). Consequently, the envisaged linkage has not been empirically supported or rejected. However, it should be kept in mind that, whereas etoposide does induce a large number of MLL rearrangements, most of them occur within non‐coding regions, therefore not eliciting any direct oncogenic consequence. A MLL‐AF4 in frame fusion is a rare event that needs to occur in a target cell within a relatively small and spatially restricted cell population during the appropriate, epigenetically plastic, developmental window; thus it may be difficult to empirically support this process.
Dose–response relationships between etoposide and treatment‐related leukaemia are difficult to unravel, but risk of leukaemia seems to increase with larger total exposure to etoposide. However, comparison of exposures or kinetics of etoposide between leukaemia patients and non‐leukaemic treated subjects did not reveal any significant differences (Relling et al., 1998). Also, it is not known whether the etoposide (or metabolite) concentrations during the treatment are of significance. In child and adult chemotherapy, concentrations are extremely variable between individuals; the lowest through plasma concentrations of etoposide have been of the order of 1 μM and peak concentrations very much higher. For example, in a study of Relling et al. (1998), the maximum plasma concentration of etoposide was about 90 μM and that of etoposide catechol about 100‐times less, below 1 μM. In another high dose chemotherapy study (Stremetzne et al., 1997), the etoposide concentration was 170 μM and that of the catechol metabolite 5.8 μM maximally. However, it is not straightforward to juxtapose plasma concentrations and the tissue or cell concentration which TopoII enzyme ‘sees’. Penetration of etoposide or its metabolite through plasma membrane is probably rather slow and it has been shown that the brain cancer tissue (metastasis or glioma) to plasma ratio for etoposide is only 0.1 (Pitz et al., 2011). Blood–brain barrier is not necessarily a good model for cross‐membrane distribution, but may give some idea about the general distributional behaviour of a drug. Even if the active target concentration of etoposide is only 10% of the plasma concentration, it is still in the same range as the effective concentrations in cellular studies (see above). A final note on relevant concentrations: etoposide concentrations resulting in DSB and fusion gene are probably within a relatively restricted range. The concentration resulting in a proper fusion gene should be in a range which gives rise to a partially repaired insult and cells bypassing death and accumulating the abnormality.
References
Blanco JG, Edick MJ, Relling MV, 2004. Etoposide induces chimeric Mll gene fusions. Federation of American Societies for Experimental Biology (FASEB), 18, 173–175. doi: 10.1096/fj.03‐0638fje
Breese EH, Buechele C, Dawson C, Cleary ML, Porteus MH, 2015. Use of genome engineering to create patient specific mll translocations in primary human hematopoietic stem and progenitor cells. Public Library of Science (PLoS ONE), 10, e0136644. doi: 10.1371/journal.pone.0136644
Buechele C, Breese EH, Schneidawind D, Lin CH, Jeong J, Duque‐Afonso J, Wong SH, Smith KS, Negrin RS, Porteus M, Cleary ML, 2015. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood, 126, 1683–1694. doi: 10.1182/blood‐2015‐05‐646398
Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH, 2006. A murine Mll‐AF4 knock‐in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood Journal, 108, 669–677. doi: 10.1182/blood‐2005‐08‐3498
Ishii E, Eguchi M, Eguchi‐Ishimae M, Yoshida N, Oda M, Zaitsu M, et al., 2002. In vitro cleavage of the MLL gene by topoisomerase II inhibitor (etoposide) in normal cord and peripheral blood mononuclear cells. International Journal of Hematology, 76, 74–79.
Li Z, Sun B, Clewell RA, Adeleye Y, Andersen ME, Zhang Q, 2014. Dose‐response modeling of etoposide‐induced DNA damage response. Toxicological Sciences, 137, 371–384. doi: 10.1093/toxsci/kft259
Libura J, Slater DJ, Felix CA, Richardson C, 2005. Therapy‐related acute myeloid leukemia‐like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood Journal, 105, 2124–2131. doi: 10.1182/blood‐2004‐07‐2683
Libura J, Ward M, Solecka J, Richardson C, 2008. Etoposide‐initiated MLL rearrangements detected at high frequency in human primitive hematopoietic stem cells with in vitro and in vivo long‐term repopulating potential. European Journal of Haematology, 81, 185–95. doi: 10.1111/j.1600‐0609.2008.01103.x
McClendon AK, Osheroff N, 2007. DNA topoisomerase II, genotoxicity and cancer. Mutation Research, 623, 83–97.
Moneypenny CG, Shao J, Song Y, Gallagher EP, 2006. MLL rearrangements are induced by low doses of etoposide in human fetal hematopoietic stem cells. Carcinogenesis, 27, 874–81. doi: 10.1093/carcin/bgi322
Montecucco A, Zanetta F, Biamonti G, 2015. Molecular mechanisms of etoposide. Journal of Experimental and Clinical Sciences, 14, 95–108. doi: 10.17179/Journal ‐ Experimental and Clinical Sciences (EXCLI)2015‐561.
Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S, Takagi M, 2015. Dysregulation of the DNA damage response and KMT2A rearrangement in fetal liver hematopoietic cells. Public Library of Science (PLoS ONE), 10, e0144540. doi: 10.1371/journal.pone.0144540
Pitz MW, Desai A, Grossman SA, Blakeley JO, 2011. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. Journal of Neuro‐Oncology, 104, 629–638. doi: 10.1007/s11060‐011‐0564‐y
Relling MV, Yanishevski Y, Nemec J, Evans WE, Boyett JM, Behm FG, Pui CH, 1998. Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia, 12, 346–352.
Stremetzne S, Jaehde U, Kasper R, Beyer J, Siegert W, Schunack W, 1997. Considerable plasma levels of a cytotoxic etoposide metabolite in patients undergoing high‐dose chemotherapy. European Journal of Cancer, 33, 978–979.
2nd KER: In utero MLL chromosomal translocation (KEup) leading to Infant leukaemia (KEdown)
How this Key Event Relationship works
Propagation of a leukaemic cell clone is based on both blockage of differentiation to more mature cells and ability to expand in an uncontrolled way. Formation of the MLL‐rearranged fusion genes and their protein products are intimately involved in both the blocked differentiation of HSPCs and the expansion of the fusion gene‐carrying clone. It is believed that the fusion gene product block cell differentiation by inhibiting the normal transcriptional programs and recruiting repressor molecules such as histone deacetylase enzymes (Greaves, 2002; Teitell and Pandolfi, 2009). Furthermore, the fusion gene product activates other key target genes, which ultimately lead to the propagation of transformed cell lines without normal restrictions (Greaves, 2015; Sanjuan‐Pla et al., 2015). Therefore, the potential of both differentiation blockage and clonal expansion are inherent properties of the MLL‐rearranged fusion product, based on the preservation of some original functions, even if in a modified form, and on the gain of some other functions due to the sequences from the new fusion partner gene (Marschalek, 2010; Sanjuan‐Pla et al., 2015).
Molecular mechanisms
The MLL is the most common translocation gene in infant leukaemia. The N‐terminal part of MLL becomes fused in frame to one of a large number of fusion partners, but in most cases, this fusion occurs between the N‐terminal MLL and either AF4, AF6, AF9, AF10 or ENL (Krivtsov and Armstrong, 2007). Due to the DNA‐binding properties of the N‐terminal MLL motif, these fusion proteins are always nuclear and bind to target genes controlled by MLL irrespective of the normal location of the C‐terminal partner.
Many fusion proteins have been shown to recruit DOT1L (catalysing methylation of histone H3K79) to the promoters of MLL target genes and this recruitment seems to be a common feature of many oncogenic MLL fusion proteins. Although DOT1L is not genetically altered in the disease per se, its mislocated enzymatic activity is a direct consequence of the chromosomal translocation. Thus, DOT1L has been proposed to be a catalytic driver of leukaemogenesis (Chen and Armstrong, 2015). The enzymatic activity of DOT1L is critical to the pathogenesis of MLL, because methyltransferase‐deficient Dot1L is capable of suppressing growth of MLL‐rearranged cells. A small‐molecule inhibitor of DOT1L inhibits cellular H3K79 methylation, blocks leukaemogenic gene expression, and selectively kills cultured cells bearing MLL translocations (Chen and Armstrong, 2015). One of the target gene of DOT1L is BCL‐2, belonging to a family of antiapoptotic genes, which maintains the survival of the MLL‐rearranged cells (Benito et al., 2015). Expression of BCL‐2 is high in human MLL‐AF4 leukaemia cells from a large number of patients. A specific BCL‐2 inhibitor, ABT‐199 is capable of killing MLL‐AF4 leukaemia cells and prevents cell proliferation in xenograft mouse leukaemia models (Benito et al., 2015). Furthermore, a MLL‐AF4 cell line is sensitive to a combination of ABT‐199 and DOT1L inhibitors. Figure B.4 provides a schematic representation of the molecular pathway.
Figure B.4.
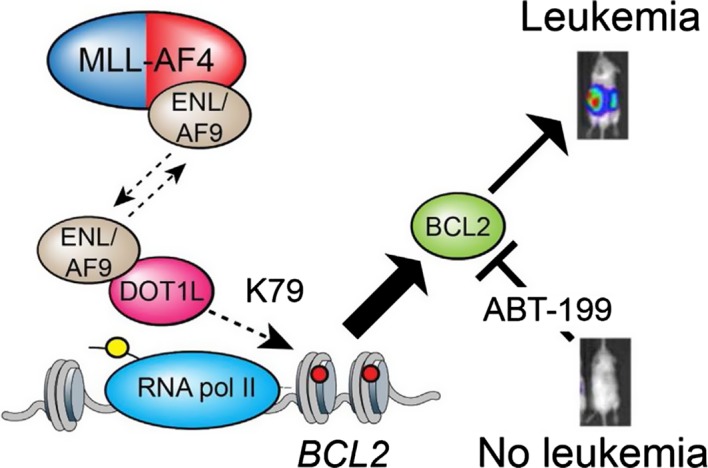
MLL‐rearranged acute lymphoblastic leukaemias activate BCL‐2 through H3K79 methylation and are sensitive to the BCL‐2‐specific antagonist ABT‐199 (Benito et al., 2015, CC‐BY)
Possible facilitating mutated genes
Recurrent activating mutations in the components of the PI3K‐RAS signalling pathway have been detected in almost half of the tested MLL‐rearranged ALLs in one study (Andersson et al., 2015). Prenatal origin of RAS mutations have been demonstrated also in other studies of infant leukaemia with frequencies of about 15–25% of cases (Driessen et al., 2013; Prelle et al., 2013; Emerenciano et al. 2015). Emerenciano et al. (2015) are of the opinion that RAS mutations seem not to be driver mutations, but may aid disease onset by accelerating the initial expansion of cells.
Overall the activation of the RAS pathway could support the extremely rapid progression of the infant leukaemia. Under this view the mechanism may represent a factor modulating (i.e. increasing) the progression and severity of the adverse outcome, rather than a necessary key event (second hit) for infant leukaemia. In the transgenic MLL‐AF4 mouse model, activated K‐RAS accelerated disease onset with a short latency (Tamai et al., 2011), possibly by augmenting the upregulation of HoxA9. In a recent study of Prieto et al. (2016), the activated K‐RAS enhanced extramedullary haematopoiesis of MLL‐AF4 expressing cell lines and cord blood‐derived CD34+ haematopoietic stem/progenitor cells that was associated with leucocytosis and central nervous system infiltration, both hallmarks of infant MLL‐AF4 leukaemia. However, K‐RAS activation was insufficient to initiate leukaemia, supporting that the involvement of RAS pathway is an important modifying factor in infant leukaemia. It has also been demonstrated that MLL‐AF6 fusion product sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukaemia cells and it is possible to attenuate the activation by tipifarnib, a RAS inhibitor (Manara et al., 2014).
A possibility that MLL fusions render cells susceptible to additional chromosomal damage upon exposure to etoposide was studied by introducing MLL‐AF4 and AF4‐MLL via CRISPR/Cas9‐genome editing in HEK293 cells as a model to study MLL fusion‐mediated DNA‐DSB formation/repair (Castano et al., 2016). In short, the expression of fusion genes does neither influence DNA signalling nor DNA‐DSB repair.
Weight of Evidence
The overall scientific evidence, including the stable genome of patients, suggests that infant leukaemia originates from one ‘big‐hit’ occurring during a critical developmental window of stem cell vulnerability (Andersson et al., 2013; Greaves, 2015). Different from the ‘two‐hit’ model of the adult leukaemias, the infant leukaemia is a developmental disorder where the clonal expansion is a direct consequence of in utero MLL translocation.
Biological plausibility
The biological plausibility linking the MLL translocation to infant leukaemia is strong. Rearrangement in the MLL gene is commonly associated with infant acute leukaemia and the disease has unique clinical and biological feature (Ernst et al., 2002). An in utero initiation, an extremely rapid progression, and a silent mutational landscape of infant leukaemia suggest that the MLL‐translocation‐associated gene fusion product is itself sufficient to spawn leukaemia and no ‘second hit’ is required. Therapy‐related leukaemias following exposure to the topo II poisons such as etoposide are characterised by the MLL chromosomal translocation (Super et al., 1993; Libura et al., 2006) and translocations involving MLL are associated with a gain of function and leukaemogenic effect (Yu et al., 1998). A critical developmentally early window of stem cell vulnerability, involving perhaps lesions based on epigenetically controlled regulatory factors, has been suggested to explain a rare occurrence and an exceptionally short latency of infant leukaemia (Greaves, 2015; Sanjuan‐Pla et al., 2015). In primary HSPCs genome engineered for patient specific MLL translocations it was possible to show that this specific ‘artificial’ initiation can induce a selective advantage in survival in extended culturing and a higher clonogenic potential in colony forming assay (Breese et al., 2015).
Empirical support
A number of MLL‐fusion products, such as MLL‐AF9 and MLL‐ENL, have shown leukaemogenic potential in cord‐blood stem cells. Although the MLL rearrangement is essential to develop leukaemia, it alone may not be sufficient and activation of cellular proliferation might be necessary for overt leukaemia (Nanya et al., 2015).
There are several animal models, in which MLL‐AF4 fusion gene has been expressed (Chen et al., 2006; Metzler et al., 2006; Bursen et al., 2008; Krivtsov et al., 2008; Tamai et al., 2011). In all these models leukaemia is ultimately developed, but latency has been very protracted. In any case, one could conclude that the expression of the MLL‐AF4 fusion gene is capable of developing leukaemia, but it is unknown whether facilitating or necessary changes are required during the long latency in mouse.
Gene engineered human HSPCs carrying MLL rearrangements showed that a subset of cells persisted over time and demonstrated a higher clonogenic potential in colony forming assay (Breese et al., 2015).
Transcription activator‐like effector nuclease (TALEN)‐mediated genome editing generated endogenous MLL‐AF9 and MLL‐ENL oncogenes in primary human HSPCs derived from human umbilical cord plasma (Buechele et al., 2015). Engineered HSPCs displayed altered in vitro growth potential and induced acute leukaemias following transplantation in immunocompromised mice at a mean latency of 16 weeks. The leukaemias displayed phenotypic and morphologic similarities with patient leukaemia blasts, expressed elevated levels of crucial MLL‐fusion partner target genes, displayed heightened sensitivity to DOT1L inhibition, and demonstrated increased oncogenic potential ex vivo and in secondary transplant assays.
Uncertainties and Inconsistencies
The MLL‐AF4 knock‐in mice develop leukaemia only after a prolonged latency (Chen et al., 2006), thus not recapitulating the ‘pathognomonic’ feature of infant leukaemia. Also other animal models have been developed with similar results. Thus, an adequate experimental model for infant leukaemia is still in need.
The role of a reciprocal fusion gene AF4‐MLL in leukaemias is controversial: it has a transformation potential in animal model (Bursen et al., 2010), but it is not expressed in all MLL‐AF4 patients (Andersson et al., 2015). The potential role of other reciprocal fusion genes has not been studied.
Beyond MLL rearrangements, activation of cellular proliferation by mutation or other (epi)genetic insults might be necessary for overt leukaemia. Further studies are necessary to fully understand which factors would contribute to convey a proliferative advantage, as observed in cells with MLL translocation, to leukaemia.
Quantifiable understanding
Relationships between different fusion genes and subsequent leukaemia types are incompletely understood. Although roughly 70–80% of infant B‐ALL leukaemias carry MLL rearrangements, in 20–30% of the cases there are no MLL rearrangements. In AML and T‐ALL leukaemia cases MLL rearrangements are even rarer.
References
Andersson AK, Ma J, Wang J, et al., 2015 St. Jude Children's Research Hospital and Washington University Pediatric Cancer Genome Project, 2015. The landscape of somatic mutations in infant MLL‐rearranged acute lymphoblastic leukemias. Nature Genetics, 47, 330–337. doi: 10.1038/ng.3230
Benito JM, Godfrey L, Kojima K, et al., 2015. MLL‐Rearranged acute lymphoblastic leukemias activate BCL‐2 through H3K79 methylation and are sensitive to the BCL‐2‐specific antagonist ABT‐199. Cell Reports, 13, 2715–2727. doi: 10.1016/j.celrep.2015.12.003
Breese EH, Buechele C, Dawson C, Cleary ML, Porteus MH, 2015. Use of genome engineering to create patient specific MLL translocation in primary hematopietic stem and progenitor cells. Public Library of Science (PLoS ONE). doi: 10.1371/journal.pone.0136644
Buechele C, Breese EH, Schneidawind D, Lin CH, Jeong J, Duque‐Afonso J, Wong SH, Smith KS, Negrin RS, Porteus M, Cleary ML, 2015. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood, 126, 1683–1694. doi: 10.1182/blood‐2015‐05‐646398
Bursen A, Schwabe K, Ruster B, et al., 2010. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood Journal, 115, 3570–3579.
Castano J, Herrero AB, Bursen A, Gonzalez F, Marschalek R, Gutierrez NC, Menendez P, 2016. Expression of MLL‐AF4 or AF4‐MLL fusions does not impact the efficiency of DNA damage repair. Oncotarget. doi: 10.18632/oncotarget.8938
Chen C‐W, Armstrong SA, 2015. Targeting DOT1L and HOX gene expression in MLL‐rearranged leukemia and beyond. Experimental Hematology, 43, 673–684.
Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH, 2006. A murine Mll‐AF4 knock‐in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood, 108, 669–677.
Driessen EM, van Roon EH, Spijkers‐Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW, 2013. Frequencies and prognostic impact of RAS mutations in MLL‐rearranged acute lymphoblastic leukemia in infants. Haematologica, 98, 937–944. doi: 10.3324/haematol.2012.067983
Ernest P, Wang J, Korsmeyer SJ, 2002. The role of MLL in hematopoiesis and leukemia. Current Opinion in Hematology, 9, 282–287.
Ernest P, Fisher JK, Avery W, Sade S, Foy D, Korsmeyer SJ, 2004. Definitive hematopoiesis requires the mixed‐lineage leukemia gene. Developmental Cell, 6, 437–443.
Greaves M, 2002. Childhood leukaemia. British Medical Journal, 324, 283–287.
Greaves M, 2015. When one mutation is all it takes. Cancer Cell, 27, 433–434.
Hess JL, Yu BD, Li B, Hanson RD, Korsmeyer SJ, 1997. Defect in yolk sac hematopoiesis in mll‐null embryos. Blood, 90, 1799–1806.
Jansen MW, Corral L, van der Velden VH, Panzer‐Grumayer R, Schrappe M, Schrauder A, et al., 2007. Immunobiological diversity in infant acute lymphoblastic leukemiais related to the occurrence and type of MLL rearrangement. Leukemia, 21, 633–641.
Libura J, Slater DJ, Felix C, Richardson C, 2004. T‐AML‐like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood Journal doi: 10.1182/blood‐2004‐07‐2683
Krivtsov AV, Armstrong SA, 2007. MLL translocations, histone modifications and leukaemia stem‐cell development. Nature Reviews Cancer, 7, 823–833.
Krivtsov AV, Feng Z, Lemieux ME, et al., 2008. H3K79 methylation profiles define murine and human MLL‐AF4 leukemias. Cancer Cell, 14, 355–368.
Manara E, Baron E, Tregnago C, Aveic S, Bisio V, Bresolin S, Masetti R, Locatelli F, Basso G, Pigazzi M, 2014. MLL‐AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood Journal, 124, 263–272. doi: 10.1182/blood‐2013‐09‐525741
Marschalek R, 2010. Mechanisms of leukemogenesis by MLL fusion proteins. British Journal of Haematology, 152, 141–154. doi: 10.1111/j.1365-2141.2010.08459.x
Metzler M, Forster A, Pannell R, et al., 2006. A conditional model of MLL‐AF4 B‐cell tumourigenesis using invertor technology. Oncogene, 25, 3093–3103.
Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S, Takagi M, 2015. Dysregulation of the DNA damage response and KMT2A rearrangement in fetal liver hematopoietic cells. Public Library of Science (PLoS ONE), 10, e0144540. doi: 10.1371/journal.pone.0144540
Prieto C, Stam RW, Agraz‐Doblas A, Ballerini P, Camos M, Castano J, Marschalek R, Bursen A, Varela I, Bueno C, Menendez P, 2016. Activated KRAS cooperates with MLLAF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukemia. Cancer Research. pii:canres.2769.2015.
Sam TN, Kersey JH, Linabery AM, Johnson KJ, Heerema NA, Hilden JM, et al., 2012. MLL gene rearrangements in infant leukaemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatric Blood and Cancer, 58, 836–839.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukemia. Blood Journal, 126, 2676–2685. doi: 10.1182/blood‐2015‐09‐667378
Super HJ, McCabe NR, Thirman MJ, et al. 1993. Rearrangements of the MLL gene in therap‐related acute myeloid leukaemia in patients previously treated with agents targeting DNA‐topoisomerase II. Blood, 82, 3705–3711.
Tamai H, Inokuchi K, 2013. Establishment of MLL/AF4 transgenic mice with the phenotype of lymphoblastic leukemia or lymphoma. Journal of Nippon Medical School, 80, 326–327.
Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, Shimada T, Inokuchi K, 2011. Activated K‐Ras protein accelerates human MLL/AF4‐induced leukemo‐lymphomogenicity in a transgenic mouse model. Leukemia, 25, 888–891. doi: 10.1038/leu.2011.15
Teitell MA, Pandolfi PP, 2009. Molecular genetics of acute lymphoblastic leukemia. Annual Review of Pathology, 4, 175–198.
Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ, 1998. MLL, a mammalian trithorax‐group gene, functions as a transcriptional maintenance factor in morphogenesis. Proceedings of the National Academy of Sciences USA, 95, 10632–10636.
Overall assessment of the AOP
Infant leukaemia is a ‘hidden’ disease quite concretely: initiation occurs in utero at an early phase of fetal development. Studies both in identical twins (Ford et al., 1993) and in neonatal blood samples retrospectively (Gale et al., 1997) strongly indicate in utero origin of the disease. Consequently, direct studies in pregnant humans are difficult or impossible and one has to resort to surrogate in vitro or ex vivo studies or to animal models which necessarily are associated with difficulties in interpretation and extrapolation. Thus, what is described in this overall assessment is based largely on inferences from analogous diseases using tool chemicals able to reproduce the biological basis of the disease (especially etoposide (a Topoisomerases II poison)‐caused acute leukaemia in children or adults) or from cellular and animal models.
B.1. Concordance of dose–response relationship
The only in utero study in mice (Nanya et al., 2016) has shown that the dose of 0.5 mg/kg (day 13.5 of pregnancy) does not result in measurable etoposide concentration in fetal liver HSCs whereas the dose of 10 mg/kg leads to a maximal concentration of 5 μM. A statistically significant increase in double strand break (DSBs) in MLL gene was observed at a dose of 1 mg/kg, which would result in a concentration of 0.5 μM by linear extrapolation. In treatment‐related acute human leukaemia, various treatment schedules in adults and children give rise to etoposide concentrations between (roughly) < 1 μM (through) to > 150 μM (peak). There are no adequate experimental systems to study dose–response and response–response relationships across MIE, KEs and AO in a single model.
B.2. Temporal concordance among the MIE, KEs and AO
There are no serious doubts about temporal concordance among MIE, KEs and AO. It is very difficult to see any other sequence of events (among this AOP), which would bring the AO into effect. Another matter is that it has never been shown in human pregnancy (or will be reliably or robustly demonstrated in the foreseeable future). In this respect, it is difficult to envisage whether epidemiological studies that are possible in humans, would ever be able to demonstrate the link without a direct biomarker for the MIE and KE1. Available experimental models (Sanjuan‐Pla et al., 2015) are in conformation with the AOP, except that in experimental in vivo models a very protracted appearance of leukaemia is not in line with a very short latency of infant leukaemia in human.
It is obvious that there exists a vast gap between wide exposure to potential TopII poisons and the rarity of infant leukaemia. On the basis of studies in human adult and childhood leukaemias, there are a large number of genetic, epigenetic and host factors potentially modifying the link between topII poisons and leukaemia. Because of the rarity of the disease, it is difficult to envisage an even partial proofing these factors as of importance for the infant leukaemia.
Table B.2.
Response–Response and temporality concordance for the tool compound etoposide
| Concentration of etoposide | MIE In utero DNA topoisomerase II inhibition | KE1 In utero MLL chromosomal rearrangement | AO Infant leukaemia |
|---|---|---|---|
| 0.01–0.1 μM, in vitro (TopII enzymes and cells in culture) |
+++ (DNA damage response in various cells) |
– | |
| 0.1–1 μM, in vitro cell cultures |
+++ (haematopoietic progenitor and stem cells) |
+ | |
| 0.5–5 μM, ex vivo, mouse fetal liver HSC concentrationa |
+++ (inference from MLL cleavage) |
+ (only MLL cleavage) |
– (no leukaemia development) |
| Max 5 μM, ex vivo, mouse fetal liver HSC concentrationa |
+++ (inference from MLL cleavage) |
+ MLL fusions detected only in DNA repair deficient mice |
– (no leukaemia development) |
| Max > 150 μM, plasma concs in etoposide‐treated patientsb |
+++ (inference from MLL cleavage) |
++ MLL‐AF4 fusion gene and protein |
+ treatment‐related acute leukaemia |
A range of concentrations is linearly extrapolated on the basis of the concentration of 5 μM after the dose of 10 mg/kg.
Plasma concentration of etoposide cannot be directly extrapolated to the concentration at the active site. Probably the actual active cellular concentrations of etoposide is much lower, perhaps 10% or less of the plasma concentration.
B.3. Strength, consistency of the experimental evidence, and specificity of association of AO and MIE
Regarding the treatment‐related acute leukaemia, strength, consistency and specificity of association of AO and MIE is strong, because only etoposide and a few other TopII‐poison anticancer agents (Mention!) have strong evidence for causing acute leukaemia in human via the general process of the AOP described here. Although direct observations on the initial in utero MIE in infant leukaemia are not possible, there is a lot of inferential evidence from animal and in vitro cellular studies suggesting strongly that infant leukaemia recapitulates at least at an apparent process level the treatment‐related leukaemia. It is important to recognise that in therapy‐related AML this has been clearly demonstrated with abnormalities affecting MLL locus. Chlorpyrifos is reported to be a Topo II poison and to induce MLL translocation in the human liver haematopoietic stem cells (Lu et al., 2015). However, it is probable that the dose dependence of the formation of DSBs and fusion genes is linear only in a very restricted ‘window’ of dose range. Considering the rarity of IFL and the common exposure to Topo II poisons like bioflavonoids, specificity is low. However, this consideration is limited by lack of experimental studies conducted with other than anticancer drugs on the sensitive target cells i.e. the liver haematopoietic stem cell.
B.4. Weight of Evidence (WoE)
B.4.1. Biological plausibility
The biological plausibility for this AOP is strong. The relationship between DNA double strand breaks, MLL chromosomal translocation and infant leukaemia is well established. The same pathway is reproducible in chemotherapy‐induced acute leukaemia in patients following treatment with etoposide, a known Topo II poison.
Table B.3.
Biological plausibility of the KERs; WoE analysis
| 1 Support for biological plausibility of KERs | Defining question | High (Strong) | Moderate | Low(Weak) |
|---|---|---|---|---|
| Is there a mechanistic (i.e. structural or functional) relationship between KEup and KEdown consistent with established biological knowledge? | Extensive understanding of the KER based on extensive previous documentation and broad acceptance | The KER is plausible based on analogy to accepted biological relationships, but scientific understanding is not completely established | There is empirical support for a statistical association between KEs but the structural or functional relationship between them is not understood | |
|
MIE → KE1 In utero exposure to DNA topoisomerase II poison leads to In utero MLL chromosomal translocation |
Strong |
Rationale: Although type II topoisomerases are essential to cell proliferation and survival, they have a significant genotoxic potential consequent to the resulting (double) strand breaks. Mis‐repair of accumulated of DNA double strand breaks can result in chromosomal translocations which can persist in survived cells (Mc Clendon et al. 2009). Studies on identical twins and neonatal blood samples strongly implicate an in utero occurrence of the KER (Sanjuan‐Pla et al., 2015). Furthermore, a study in pregnant mice demonstrates that in utero exposure of the fetus to etoposide causes the MLL chromosomal translocation analogous to the human translocation except the principal fusion partner (Nanya et al., 2015). Indirect evidence from human prehaematopoietic/mesenchymal stem cells and fetal liver haematopoietic progenitor and stem cells strengthen the plausibility. Experimental evidence in these cell lines has demonstrated that etoposide as a TopII poison causes DSBs in MLL and partner genes, which leads to the formation of fusion genes and their products (Sanjuan‐Pla et al., 2015) MLL translocation sites (breakpoint sequences) in the therapy‐related leukaemia fall within a few base pairs of etoposide‐induced enzyme‐mediated DNA cleavage site. Although rearrangements associated with infant leukaemias are often more complex than those observed in treatment‐related leukaemias, many are nevertheless associated with stable TopII‐mediated DNA cut sites (Cowell and Austin, 2012; Pendleton et al., 2014) |
||
|
KE1 → AO In utero MLL chromosomal translocation leads to Infant leukaemia |
Strong | Rationale: The basic processes underlying overt leukaemia development are well understood and accepted. There is a general understanding of the molecular and epigenetic mechanisms leading to differentiation blockage and clonal expansion and there is evidence that the principal MLL‐fusion genes and proteins harbour the necessary properties to execute the pathways associated with differentiation blockage and clonal expansion (Benito et al., 2015; Chen and Armstrong, 2015; Chen et al., 2015) | ||
B.4.2. Essentiality
In line with the defining question, essentiality for this AOP is moderate. However, the actual knowledge of the IFL is supporting the evidence that IFL is a ‘single hit’ developmental disease and MLL translocation is an essential KE based on the probability linking MLL translocation and the occurrence of the disease. Based on this the overall essentiality can be considered moderate to strong.
Table B.4.
Essentiality of the KEs; WoE analysis
| 2 Support for essentiality of KEs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Are downstream KEs and/or the AO prevented if an upstream KE is blocked? | Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs (e.g. stop/reversibility studies, antagonism, knock out models, etc.) | Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE leading to increase in KEdown or AO | No or contradictory experimental evidence of the essentiality of any of the KEs | |
|
MIE In utero exposure to DNA topoisomerase II poison |
Moderate |
Although there are no direct experimental studies to demonstrate that blocking action of TopoII poisons would prevent the AOP, there are considerable evidence for the relationship between the concentration of etoposide and the formation of the MLL rearrangements in human (pre)haematopoietic progenitor/stem cells, which strongly suggest the essentiality of TopoII inhibition (e.g. Bueno et al., 2009; Nanya et al., 2015). In addition, chemical‐induced DNA breakpoints are associated with predicted Topo II cleavage sites (i.e. MLL), supporting an essential role for TOPO II mediate breakage (Montecucco et al., 2015; Hernández and Menéndez, 2016) In human patients, therapy‐related acute leukaemia characterised by MLL rearrangement is predominantly associated with etoposide treatment (Super et al., 1993) |
||
|
KE1 In utero MLL chromosomal translocation |
Moderate |
Growing scientific evidence, including the stable genome of the patients, suggests that infant leukaemia originates from one ‘big‐hit’ occurring during a critical developmental window of stem cell vulnerability (Andersson et al., 2013; Greaves, 2015; Sanjuan‐Pla et al., 2015). Therefore, the totality of evidence suggests the essential role of the formation of MLL‐partner fusion gene and product in causing pleiotropic effects in the affected cell and directing it to the obligatory pathway to the adverse outcome of leukaemia The MLL‐AF4 fusion gene is present in bone marrow mesenchymal stem cells in infant leukaemia patients, but not in patients of childhood leukaemia, suggesting that the origin of the fusion gene is probably prehaematopoietic and essential for development of IFL (Menendez et al., 2009) TopoII ‘poisons’ etoposide and bioflavonoids (and some other chemicals) promote MLL rearrangements in in vitro prenatal cells or in utero. There are in vitro cellular and n vivo xenograph studies demonstrating that upon inhibiting signalling pathways from the fusion product on, cells can resume differentiation or clonal expansion of fusion gene‐carrying cells is prevented (Benito et al., 2015; Buechele et al., 2015; Chen and Armstrong, 2015). However, in the absence of a relevant in vivo experimental model these findings are suggestive but not yet totally convincing |
||
B.4.3. Empirical support
The overall empirical support, using the chemical tool etoposide, is moderate. In vivo and, mainly in‐vitro, experiments exist but they are lacking a clear dose or concentration response relationship.
Table B.5.
Empirical support of the KERs; WoE analysis
| 3 Empirical support for KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Does the empirical evidence support that a change in the KEup leads to an appropriate change in the KEdown? Does KEup occur at lower doses and earlier time points than KEdown and is the incidence of KEup higher than that for KEdown? Are inconsistencies in empirical support cross taxa, species and stressors that don't align with expected pattern of hypothesised AOP? | Multiple studies showing dependent change in both exposure to a wide range of specific stressors (extensive evidence for temporal, dose–response and incidence concordance) and no or few critical data gaps or conflicting data | Demonstrated dependent change in both events following exposure to a small number of specific stressors and some evidence inconsistent with expected pattern that can be explained by factors such as experimental design, technical considerations, differences among laboratories, etc | Limited or no studies reporting dependent change in both events following exposure to a specific stressor (i.e. endpoints never measured in the same study or not at all); and/or significant inconsistencies in empirical support across taxa and species that don't align with expected pattern for hypothesised AOP | |
|
MIE → KE1 In utero exposure to DNA topoisomerase II poison leads to In utero MLL chromosomal translocation |
Moderate | Rationale: Evidence comes from in vitro studies in appropriate human cells and from an in vivo/ex vivo study in pregnant mice; the stressor has been etoposide in most of the experiments (Lovett et al., 2001; Whitmarsh et al., 2003; Libura et al., 2005; Nanya et al., 2015). Some evidence to back this KER comes from in vitro studies with bioflavonoids, especially quercetin, genistein and kaempferol (Barjesteh et al., 2007) | ||
|
KE1 → KE2 In utero MLL chromosomal translocation leads to Infant leukaemia |
Moderate |
Rationale: There are a number of factors and pathways linking the fusion products with differentiation blockage and clonal expansion (Marschalek, 2010; Sanjuan‐Pla et al., 2015). MLL encodes a protein homologous to the Drosophila trithorax gene, which has relevant functions in embryogenesis and haematopoiesis (Hess et al., 1997; Ernest et al., 2004). Studies with MLL‐AF4, MLL‐AF9 and MLL‐ENL (Barabe et al., 2007; Mulloy et al., 2008) have clearly demonstrated how MLL chromosomal rearrangements block differentiation and enhance clonal expansion. However, there is a specific need to execute these studies in an appropriate experimental system with a proper target cell within a proper molecular and physiological environment There are several animal models, in which MLL‐rearranged fusion genes have been expressed and leukaemia developed (Chen et al., 2006; Metzler et al., 2006; Bursen et al., 2008; Krivtsov et al., 2008; Tamai et al., 2011). Engineered human haematopoietic stem and progenitor cell carrying an MLL rearrangement showed that a subset of cells persisted over time and demonstrated a higher clonogenic potential in colony forming assay (Breese et al., 2015). Cells engineered to carry MLL‐AF9 and MLL‐ENL fusions demonstrated leukaemogenicity especially after ex vivo and repeated transplantation (Buechele et al., 2015) |
||
B.5. Uncertainties and Inconsistencies
In utero evidence of the disease is difficult to obtain in humans and one has to resort to in vitro cellular systems, which may be inadequate to take into consideration the potential effects of proposed microenvironments, rapidly changing developmental stages and facilitating and modifying factors.
Animal models are a possibility (e.g. Nanya et al., 2015), but are naturally prone to species‐specific factors.
An important problem is to provide a convincing and experimentally justified explanation for the dilemma between the rarities of disease in the face of pervasive exposure to topoII inhibitors.
The treatment‐related AML apparently is a true surrogate for the infant leukaemia, at least mechanistically. Is it only because of etoposide as a principal chemical initiator has provided many crucial findings for understanding the infant leukaemia.
The ‘poisoning’ of the TopoII‐DNA cleavage complex has not been shown in the putative target cell, which is still not unequivocally identified.
MLL‐AF4 knock‐in mice develop leukaemia only after a prolonged latency (e.g. Chen et al., 2006), thus not recapitulating the ‘pathognomonic’ feature of infant leukaemia.
The inability of available in vivo models to recapitulate the whole AOP process is due to a crucial factor which has not yet been found, or to model‐specific peculiarities.
In the face of the rarity of the disease, epidemiological studies especially concerning aetiology and risk factors are not powerful enough to provide robust answers. For instance, investigating the hypothesised relationship of bioflavonoids with infant leukaemia will have to consider the gap between the widespread intake of these phytochemicals and the very rare occurrence of the disease.
The biology of the disease (i.e. IFL) and the experimental studies conducted with etoposide, indicate in‐utero exposure of haematopoietic stem cells (HSC) as the most critical, if not essential, factor for the development of the A. However, a clear comparative quantification in terms of dose response vs different time of exposure and cell systems is lacking.
The very early embryonic structure and the liver haematopoietic stem cells in particular, are representing the target cell for this AOP. A clear understanding of a higher sensitivity of HSC vs, mature haematopoietic cells, particularly in the standard genotoxicity test battery is lacking and more chemicals and comparative assays should be tested to scientifically validate this cell system..
What would be consequences if we say that the AOP is biologically possible, feasible, even probable, and then say that most of the evidence is impossible to get directly and has to be based on surrogates?
B.6. Quantitative Considerations
The WOE analysis indicates that many KEs and KERs lack especially experimental evidence, but overall the analysis supports the qualitative AOP. The strong element in the development of the qualitative AOP is the biological plausibility of the overall pathway that it can partially be based on studies in human treatment‐related disease recapitulating many crucial features of the infant leukaemia. The lack of sufficient experimental data and uncertainties in quantitative information from treatment‐related acute leukaemia makes it problematic to build convincing dose (concentration)‐response and response–response relationships and to identify possible practical thresholds for stressors. The MIE is expected to show a dose response relationship to a certain extent. However, it is probable that the dose dependence of the formation of DSBs and fusion genes is linear only in a very restricted ‘window’. In too‐low concentrations the outcome of the stressor is a successful repair of the break, in too‐high concentrations the outcome is cell death. It should be kept in mind additionally that the quantification of dose–responses should also consider the different sensitivity of cell systems that should be also representative of the specific time‐window of exposure (i.e. in‐utero).
The most pressing future need is an adequate and robust experimental model system for the evaluation of relationships between doses, concentrations and responses within a temporal framework of the AOP.
B.7. Applicability of the AOP
The proposed AOP is strictly life stage‐dependent, being linked with in utero exposure and early embryogenesis. However, the surrogate disease (i.e. chemotherapy‐related acute leukaemia) is not life stage restricted as well as the genotoxic hazard is not expected to be life stage related.
B.8. Potential regulatory applications of the AOP
This AOP was initiated with the intention to use an epidemiologically proposed human health outcome as AO and build back an AOP leading to this. Infant childhood leukaemia is a human disease and consequently apical regulatory endpoints can only explore the hazard by means of surrogate testing. These include carcinogenesis assays and blood cell analyses in the in vivo toxicology assessment. Considering the unique biology of this AO, these tests show some technical limitations and also the sensitivity and specificity of the available tests for the AO is limited. Additionally, experimental animal models replicating the AO are limited. Technical limitations of the standard regulatory tests include: Standard carcinogenesis studies do not include an early in‐utero exposure time, blood cell analysis is not a standard requirement in the extended multi‐generation reproductive toxicity study and no cancer‐related endpoints are included in this study. In addition, considering the rarity and the complexity of the disease, the sensitivity and specificity of these tests to capture this hazard is likely to represent a big hurdle and the regulatory tests are unlikely to represent the best way to explore this AO.
This AOP is however indicating that the MIE and the KE1 can be measured in scientific and/or regulatory validated test assays.
With these premises, the authors support the use of this AOP during the process of assessment of epidemiological studies and the use of the AOP framework to support the biological plausibility of the effects observed in the epidemiological studies when experimental and toxicological studies are indicative that the AOP is affected and this should guide on which additional studies should be performed, if the case, to integrate the AOP framework into the MOA framework for specific chemical entities.
In addition, this AOP should serve in guiding testing strategy. This include the exploration of Topo II poison characteristics of a chemical and, if the genotoxicity standard regulatory testing battery is negative, considerations should be made on the sensitivity of the cell system used in the assay (i.e. liver HSPC).
References
Andersson AK, Ma J, Wang J, et al., 2015. The landscape of somatic mutations in infant MLL‐rearranged acute lymphoblastic leukemias. Nature Genetics, 47, 330–337. doi: 10.1038/ng.3230
Bandele OJ, Osheroff N, 2007. Bioflavonoids as poisons of human topoisomerase II alpha and II beta. Biochemistry, 46, 6097–6108.
Barabe F, Kennedy JA, Hope KJ, Dick JE, 2007. Modeling the initiation and progression of human acute leukemia in mice. Science, 316, 600–604.
Barjesteh van Waalwijk van Doorn‐Khosrovani S, Janssen J, Maas LM, Godschalk RW, Nijhuis JG, van Schooten FJ, 2007. Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis, 28, 1703–1709.
Benito JM, Godfrey L, Kojima K, et al., 2015, MLL‐rearranged acute lymphoblastic leukemias activate BCL‐2 through H3K79 methylation and are sensitive to the BCL‐2‐specific antagonist ABT‐199. Cell Reports, 13, 2715–2727. doi: 10.1016/j.celrep.2015.12.003
Breese EH, Buechele C, Dawson C, Cleary ML, Porteus MH, 2015. Use of genome engineering to create patient specific MLL translocations in primary human hematopoietic stem and progenitor cells. Public Library of Science (PLoS ONE), 10, e0136644. doi: 10.1371/journal.pone.0136644
Buechele C, Breese EH, Schneidawind D, Lin CH, Jeong J, Duque‐Afonso J, Wong SH, Smith KS, Negrin RS, Porteus M, Cleary ML, 2015. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood, 126, 1683–1694. doi: 10.1182/blood‐2015‐05‐646398
Bueno C, Catalina P, Melen GJ, Montes R, Sanchez L, Ligero G, Garcia‐Perez JL, Menendez P, 2009. Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis, 30, 1628–1637. doi: 10.1093/carcin/bgp169
Bursen A, Schwabe K, Ruster B, et al., 2010. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood Journal, 115, 3570–3579.
Chen C‐W, Armstrong SA, 2015. Targeting DOT1L and HOX gene expression in MLL‐rearranged leukemia and beyond. Experimental Hematology, 43, 673–684.
Chen CW, Koche RP, Sinha AU, et al., 2015. DOT1L inhibits SIRT1‐mediated epigenetic silencing to maintain leukemic gene expression in MLL‐rearranged leukemia. Nature Medicine, 21, 335–343. doi: 10.1038/nm.3832
Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH, 2006. A murine Mll‐AF4 knock‐in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood Journal, 108, 669–77. doi: 10.1182/blood‐2005‐08‐3498
Cowell IG, Austin CA, 2012. Mechanism of generation of therapy related leukemia in response to anti‐topoisomerase II agents. International Journal of Environmental Research and Public Health, 9, 2075–2091. doi: 10.3390/ijerph9062075
Ernest P, Fisher JK, Avery W, Sade S, Foy D, Korsmeyer SJ, 2004. Definitive hematopoiesis requires the mixed‐lineage leukemia gene. Developmental Cell, 6, 437–443.
Ernest P, Wang J, Korsmeyer SJ, 2002. The role of MLL in hematopoiesis and leukemia. Current Opinion in Hematology, 9, 282–287.
Ferreira JD, Couto AC, 2013. Pombo‐de‐Oliveira MS, Koifman S; Brazilian Collaborative Study Group of Infant Acute Leukemia. In utero pesticide exposure and leukemia in Brazilian children < 2 years of age. Environmental Health Perspectives, 121, 269–275. doi: 10.1289/ehp.1103942
Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et al., 1993. In utero rearrangements in the trithorax‐related oncogene in infant leukaemias. Nature, 363, 358–360. doi: 10.1038/363358a0
Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al., 1997. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proceedings of the National Academy of Sciences USA, 94, 13950–13954.
Greaves M, 2015. When one mutation is all it takes. Cancer Cell, 27, 433–434.
Hernandez A, Menendez P, 2016. Linking pesticide exposure with pediatric leukemia: potential underlying mechanisms. International Journal of Molecular Sciences, 17, 461.
Hess JL, Yu BD, Li B, Hanson RD, Korsmeyer SJ, 1997. Defect in yolk sac hematopoiesis in mll‐null embryos. Blood, 90, 1799–1806.
Krivtsov AV, Armstrong SA, 2007. MLL translocations, histone modifications and leukaemia stem‐cell development. Nature Reviews Cancer, 7, 823–833.
Libura J, Slater DJ, Felix CA, Richardson C, 2005. Therapy‐related acute myeloid leukemia‐like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood Journal, 105, 2124–2131. doi: 10.1182/blood‐2004‐07‐2683
Lovett BD, Nigro LL, Rappaport EF, et al., 2001. Near‐precise interchromosomal recombination and functional DNA topoisomerase II cleavage sites at MLL and AF‐4 genomic breakpoints in treatment‐related acute lymphoblastic leukemia with t(4;11) translocation. Proceedings of the National Academy of SciencesU S A, 98, 9802–9807. doi: 10.1073/pnas.171309898
Lu C, Liu X, Liu C, Wang J, Li C, Liu Q, Li Y, Li S, Sun S, Yan J, Shao J, 2015. Chlorpyrifos induces MLL translocations through caspase 3‐dependent genomic instability and topoisomerase II inhibition in human fetal liver hematopoietic stem cells. Toxicological Sciences, 147, 588–606. doi: 10.1093/toxsci/kfv153
McClendon AK, Osheroff N, 2007. DNA Topoisomerase II, Genotoxicity and Cancer. Mutation Research, 623, 83–97.
Marschalek R, 2010. Mechanisms of leukemogenesis by MLL fusion proteins. British Journal of Haematology, 152, 141–154. doi: 10.1111/j.1365-2141.2010.08459.x
Menendez P, Catalina P, Rodriguez R, Melen GJ, Bueno C, Arriero M, Garcia‐Sanchez F, Lassaletta A, Garcia‐Sanz R, Garcia‐Castro J, 2009. Bone marrow mesenchymal stem cells from infants with MLL‐AF4 + acute leukemia harbor and express the MLL‐AF4 fusion gene. Journal of Experimental Medicine, 206, 3131–3141. doi: 10.1084/jem.20091050
Metzler M, Forster A, Pannell R, et al., 2006. A conditional model of MLL‐AF4 B‐cell tumourigenesis using invertor technology. Oncogene, 25, 3093–3103.
Montecucco A, Zanetta F, Biamonti G, 2006. Molecular mechanisms of etoposide. Journal ‐ Experimental and Clinical Sciences (EXCLI) J, 14, 95–108. doi: 10.17179/Journal ‐ Experimental and Clinical Sciences (EXCLI)2015‐561.
Mulloy JC, Wunderlich M, Zheng Y, Wei J, 2008. Transforming human blood stem and progenitor cells: a new way forward in leukemia modeling. Cell Cycle, 7, 3314–3319.
Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S, Takagi M, 2015. Dysregulation of the DNA damage response and KMT2A rearrangement in fetal liver hematopoietic cells. Public Library of Science (PLoS ONE), 10, e0144540. doi:10.1371/journal.pone.0144540
Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D, Osheroff N, 2014. Topoisomerase II and leukemia. Annals of the New York Academy of Sciences, 1310, 98–110. doi: 10.1111/nyas.12358
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4+ B‐cell acute lymphoblastic leukemia. Blood Journal, pii: blood‐2015‐09‐667378.
Super HJ, McCabe NR, Thirman MJ, et al., 1993. Rearrangements of the MLL gene in therap‐related acute myeloid leukaemia in patients previously treated with agents targeting DNA‐topoisomerase II. Blood, 82, 3705–3711.
Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, Shimada T, Inokuchi K, 2011. Activated K‐Ras protein accelerates human MLL/AF4‐induced leukemo‐lymphomogenicity in a transgenic mouse model. Leukemia, 25, 888–891. doi: 10.1038/leu.2011.15
Whitmarsh RJ, Saginario C, Zhuo Y, et al., 2003. Reciprocal DNA topoisomerase II cleavage events at 5’‐TATTA‐3’ sequences in MLL and AF‐9 create homologous single‐stranded overhangs that anneal to form der(11) and der(9) genomic breakpoint junctions in treatment‐related AML without further processing. Oncogene, 22, 8448–8459.
AOP4: In utero induction of chromosomal rearrangements/translocations in haematopoietic stem/progenitor cells (HSPCs) followed by postnatal mutations and an aberrant immune response leads to childhood leukaemia
Introduction
Leukaemia is the most common cancer in children under 15 years of age, with an annual incidence of up to 40 cases per million children in developed countries and an incidence peak between 3 and 5 years of age (ENHIS, 2009; Hunger and Mullingan, 2015). Childhood leukaemia (also termed paediatric leukaemia) is a biologically heterogeneous disease of immature haematopoietic progenitors that consists of multiple subtypes depending on the cell type and lineage involved (lymphoid or myeloid progenitors). Seventy per cent of cases are comprised by acute lymphoblastic leukaemia (ALL) and the remaining 30% by acute myeloid leukaemia (AML). ALL may be of B–cell lineage (85%) or T‐cell lineage (15%). However, there are some cases of biphenotypic acute leukaemias commonly harbouring Mixed Leukaemia Lineage (MLL) rearrangements, in which myeloid and lymphoid markers have been shared by the blast population (Hunger and Mullighan, 2015).
Childhood leukaemia should be distinguished from infant leukaemia, a more rare disease that manifests soon after birth (< 1 year of life) and has a poorer prognosis. Infant leukaemia is considered as a ‘developmental disease’ showing different features and pathogenesis than childhood leukaemia, with more immature precursors being involved (Sanjuan‐Pla et al., 2015). A remarkable difference between the two entities is that childhood leukaemia may arise as a consequence of a ‘2‐hit’ model producing two independent (epi)‐genetic insults, the first one occurring in utero and the second one either before, or more often, after birth. In contrast, the natural history and genome‐wide sequencing studies on infant leukaemia suggest that only a single hit occurring in utero is needed. A common initiating pathogenic event for both types of leukaemias is the occurrence of chromosomal rearrangements (i.e. chromosomal translocations) that create fusion genes encoding transcriptional factors involved in the regulation of early haematopoiesis. Chimeric fusion proteins encoded by chromosomal translocations lead to differentiation arrest of HSPCs, which represents a hallmark in childhood leukaemia. Almost half of the B‐cell ALL cases exhibit aneuploidy, either hyperdiploidy or hypodiploidy with non‐random chromosomal gain or loss, respectively, affecting different chromosomes. Hyperdiploidy causes chromosomal instability as a result of chromosomal translocations, duplications and deletions (Paulsson et al., 2006).
The genetic basis of ALL consists of recurrent genetic alterations, such as loss‐of‐function mutations involving genes regulating lymphoid development that contribute to the maturation arrest characteristic of B‐ALL, mutations that inactivate tumour suppressor and cell cycle regulatory proteins, and mutations in genes encoding cytokine receptor and/or protein kinases regulating cell signalling pathways (Mullighan, 2012). For T‐cell ALL, the main drivers are chromosomal translocations resulting from aberrant recombination between T‐cell receptor genes and oncogenes (Mullighan, 2012), together with activating Notch mutations, a protein involved in T‐cell differentiation and thymocyte development (Weng et al., 2004). For AML leukaemias to occur, cooperation is required between gene rearrangements involving haematopoietic transcription factors (i.e. AML1/ETO, MLL‐related fusion genes, etc.) and activating mutations (i.e. in apical regulators of intracellular signalling cascades) (Mullighan, 2012).
Despite the rather comprehensive epidemiologic evidence linking pesticide exposures during different reproductive stages (preconception, pregnancy and early postnatal) with childhood leukaemia, no robust mechanisms supporting these associations have been reported so far. Pesticide exposure has not been directly linked to the development of childhood leukaemia in animal models. Although negative results for genotoxicity tests have been observed in regulatory studies on individual pesticides, there is limited experimental evidence in the open literature about the genotoxic or cancer‐promoting capacities of some pesticides in cells, suggesting a potential leukaemogenic effect. However, the target cells used in these experiments are not the most appropriate for this purpose and the role played by some pesticide metabolites cannot be ruled out.
Regardless of the extensive gap in our understanding, particularly on how pesticides mechanistically interact with biological targets to trigger childhood leukaemia, the AOP proposed for this disease is supported by experimental evidence and cellular models, with the exception of the molecular initiating event. However, as knowledge increases, the present AOP may be modified on the basis of novel supporting evidence.
AOP: In utero induction of chromosomal translocations in haematopoietic stem/progenitor cells (HSPCs) leads to childhood leukaemia
Figure B.5.
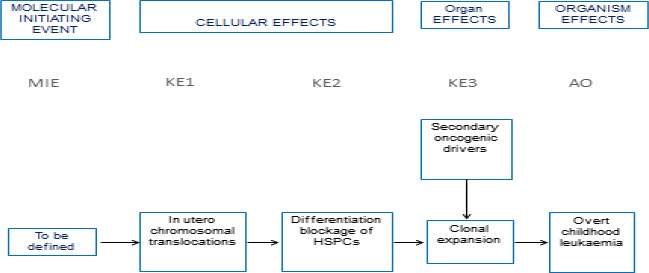
AOP scheme
The MIE is a specialised type of KE representing the starting point of chemical interaction with a biological target leading to disruption at the molecular (including genetic) level and subsequent disease progression. Expectation is that perturbation of the MIE, if quantitatively enough, will lead to tha AO. In the case of childhood leukaemia, early in utero interaction of a chemical with DNA (or DNA‐related proteins/enzymes) might lead to double strand DNA breaks, which if non‐repaired or misrepaired, may result in genomic instability, leukaemic transformation or cell death.
HSPCs exposed to ionising radiation, environmental chemicals or chemotherapeutic agents are prone to DNA breakage at sites with the potential to form leukaemia‐causing gene rearrangements. Exposure to non‐cytotoxic levels of environmental chemicals and chemotherapeutic agents can induce DNA damage in HSPCs without causing cell death (Thys et al., 2015). Several studies investigating the role of DNA repair systems in response to DNA damage found that human fetal liver CD34+ HSPCs are more sensitive to DNA damage than other haematopoietic precursors at different ontogeny stages (Bracker et al., 2006). Human fetal liver CD34+ HSPCs are more sensitive to oxidative stress induced by certain environmental chemicals, including many classes of pesticides than neonatal or adult CD34+ cells (Bueno et al., 2009). Among environmental chemicals, the organophosphate (OP) insecticide chlorpyrifos has been reported to cause DNA double‐strand breaks (DSB) and further chromosome rearrangements in human fetal liver HSPCs (Lu et al., 2015) in part through oxidative stress (Gupta et al., 2010).
Chemical exposure may result, either directly or indirectly, in DNA damage. Three potential mechanisms are involved in this process: generation of DNA DSB, improper repair of these DNA lesions and erroneous V(D)J recombination (Hernández and Menéndez, 2016).
a) DNA double strand break. Exposures to ionising radiation and numerous chemicals are capable of inducing oxidative DNA damage through the generation of reactive oxygen or nitrogen species (ROS and RNS, respectively). These highly reactive species may produce DNA base or sugar damage leading to single‐strand break formation. However, under some circumstances DNA DSBs can arise, for instance: (a) when two single strand breaks form close to each other on opposite strands, (b) when topoisomerases cleave next to a single strand breaks on the opposite strand, and (c) when either DNA replication or transcription takes place at unrepaired DNA damage.
b) Improper DNA repair. ROS‐induced DNA DSBs in human fetal liver‐derived HSPCs following maternal exposure to chemicals triggers recombination/repair pathways by non‐homologous end‐joining (NHEJ), the main repair pathway for DSBs. The majority of damaged HSPCs may either successfully repair the break or fail and die through secondary activation of apoptotic pathways. In a fraction of cells, the attempt to repair the DNA DSBs within particular breakpoints cluster regions is not completed properly, so that chromosomal translocations or deletions may occur (Wiemels and Greaves, 1999). Translocation breakpoints harbour evidence of NHEJ mechanisms, but in only a few examples are the causative mechanisms of breakage evident, such as V(D)J recombinase gene activation (Wiemels, 2008).
c) Erroneous V(D)J recombination. V(D)J recombination is a process occurring in developing lymphocytes during cell maturation, where gene segments of immunoglobulin chains or T‐cell receptor, known as variable (V), diversity (D) or joining (J), are rearranged to yield a wide range of immunoglobulins and T‐cell receptors. The process entails the cleavage of the V(D)J gene at the flanking recombination signal sequences (RSS) by lymphocyte‐specific recombination‐activating gene (RAG) endonucleases and subsequent ligation of the segments via the classical NHEJ pathway (Meissner et al., 2014). In the case of childhood leukaemia, chromosomal translocations as well as gene deletions often arise as result of mistakes in V(D)J recombination, e.g. RAG can erroneously recognise and target RSS‐like sequences. There is growing evidence that in vivo exposure to DNA‐damaging agents can increase the frequency of V(D)J rearrangements at RSS‐like sequences that are widely distributed throughout the genome (ref). However, the mechanism by which exposure to those agents increase the frequency of V(D)J‐recombinase‐mediated genomic rearrangements is still unknown (Pinsoneault et al., 2007). The lack of site‐specific clustering of translocations (which show a dispersed breakpoint distribution) suggests that chromosomal translocation arise in HSPCs before the expression of recombinase‐activating genes (Wiemels, 2008).
Although all the above reported mechanisms can be chemically induced, a chemical tool able to initiate the triggering cascade of the proposed pathway was not identified. For this reason this AOP was considered putative and the KE 1 was used as initiator of the pathway.
However, the potential initiating events speculated above can be measured. For this reason, technologies able to do it are reported here. Oxidative stress can be measured by a number of biomarkers such as plasma antioxidant status, lipid peroxidation products, reduced‐to‐oxidised glutathione (GSH:GSSG) ratio, and levels of 4‐hydroxynonenal (4‐HNE) and nitrotyrosine products. However, these biomarkers may only provide an indirect assessment of an increased risk of oxidative DNA damage in vivo (Badham and Winn, 2010). There is a variety of techniques and methods useful for the detection of single oxidatively generated DNA lesions like 8‐oxodG, thymine glycol (Tg) and abasic (AP) sites such as high performance liquid chromatography (HPLC), liquid chromatography/tandem mass spectrometry (LC‐MS/MS), alkaline filter elution, single cell gel electrophoresis (SCGE or Comet assay) and adaptations of agarose gel electrophoresis (Kryston et al., 2011).
DNA damage can be assessed by the single cell gel electrophoresis (SCGE or Comet assay), which is a simple sensitive and rapid method for the detection and quantification of DNA damage (Singh et al., 1988) and provides a direct microscopic measure of DNA single and double strand breaks.
DNA breakpoints can be determined by Southern blot, polymerase chain reaction (PCR) and DNA sequencing. Gene mutations can be comprehensively searched for by array‐comparative genome hybridisation (array‐CGH) or whole‐genome/exome sequencing. Allele‐specific restriction assay, single strand conformation polymorphism and/or direct sequencing are valid methodologies for point mutation analysis.
Increased DNA damage in leukaemia cells can be demonstrated by the formation of phosphorylated histone H2AX (γ‐H2AX), a marker of DNA DSBs (Graillot et al., 2012).
References
Badham HJ, Winn LM, 2010. In utero exposure to benzene disrupts fetal hematopoietic progenitor cell growth via reactive oxygen species. Journal of Toxicological Sciences, 113, 207–215.
Bracker TU, Giebel B, Spanholtz J, Sorg UR, Klein‐Hitpass L, Moritz T, Thomale J, 2006. Stringent regulation of DNA repair during human hematopoietic differentiation: a gene expression and functional analysis. Stem Cells, 24, 722–730.
European Environment and Health Information System (ENHIS), 2009. Standardized incidence rate of leukaemia as defined by ICD‐10 codes C90–95 in children aged 0 to 14 years. Fact sheet 4.1. Accessed online: http://www.euro.who.int/__data/assets/pdf_file/0005/97016/4.1.-Incidence-of-childhood-leukaemia-EDITED_layouted.pdf?ua=1
Graillot V, Tomasetig F, Cravedi JP, Audebert M, 2012. Evidence of the in vitro genotoxicity of methyl‐pyrazole pesticides in human cells. Mutation Research – Genetic Toxicology and Environmental Mutagenesis, 748, 8–16.
Gupta SC, Mishra M, Sharma A, Deepak Balaji TG, Kumar R, Mishra RK, Chowdhuri DK, 2010. Chlorpyrifos induces apoptosis and DNA damage in Drosophila through generation of reactive oxygen species. Ecotoxicology and Environmental Safety, 73, 1415–1423.
Hernández AF, Menéndez P, 2016. Linking pesticide exposure with pediatric leukemia: Potential underlying mechanisms. International Journal of Molecular Sciences, 17. pii: E461.
Hunger SP, Mullighan CG, 2015. Acute lymphoblastic leukemia in children. New England Journal of Medicine, 373, 1541–1552.
Kryston TB, Georgiev AB, Pissis P, Georgakilas AG, 2011. Role of oxidative stress and DNA damage in human carcinogenesis. Mutation Research, 711, 193–201.
Meissner B, Bartram T, Eckert C, Trka J, Panzer‐Grümayer R, Hermanova I, Ellinghaus E, Franke A, Möricke A, Schrauder A, Teigler‐Schlegel A, Dörge P, von Stackelberg A, Basso G, Bartram CR, Kirschner‐Schwabe R, Bornhäuser B, Bourquin JP, Cazzaniga G, Hauer J, Attarbaschi A, Izraeli S, Zaliova M, Cario G, Zimmermann M, Avigad S, Sokalska‐Duhme M, Metzler M, Schrappe M, Koehler R, Te Kronnie G, Stanulla M, 2014. Frequent and sex‐biased deletion of SLX4IP by illegitimate V(D)J‐mediated recombination in childhood acute lymphoblastic leukemia. Human Molecular Genetics and Metabolism, 23, 590–601.
Mullighan CG, 2012. Molecular genetics of B‐precursor acute lymphoblastic leukemia. Journal of Clinical Investigation, 122, 3407–3415.
Paulsson K, Heidenblad M, Mörse H, Borg A, Fioretos T, Johansson B, 2006. Identification of cryptic aberrations and characterization of translocation breakpoints using array CGH in high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia, 20, 2002–2007.
Pinsonneault RL, Vacek PM, O'Neill JP, Finette BA, 2007. Induction of V(D)J‐mediated recombination of an extrachromosomal substrate following exposure to DNA‐damaging agents. Environmental and Molecular Mutagenesis, 48, 440–450.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menéndez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4+ B‐cell acute lymphoblastic leukemia. Blood Journal, 126, 2676–2685.
Singh NP, McCoy MT, Tice RR, Schneider EL, 1988. A simple technique for quantitation of low levels of DNA damage in individual cells. Experimental Cell Research, 175, 184–191.
Thys RG, Lehman CE, Pierce LC, Wang YH, 2015. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia‐causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutation Research, 779, 86–95.
Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez‐Irizarry C, Blacklow SC, Look AT, Aster JC, 2004. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science, 306, 269–271.
Wiemels JL, Greaves M, 1999. Structure and possible mechanisms of TEL‐AML1 gene fusions in childhood acute lymphoblastic leukemia. Cancer Research, 59, 4075–4082.
Wiemels J, 2008. Chromosomal translocations in childhood leukemia: natural history, mechanisms, and epidemiology. Journal of the National Cancer Institute Monographs (JNCI Monographs), 39, 87–90.
KE1: In utero chromosomal translocations
How this key event works
Early in utero interaction of a chemical with DNA (or DNA‐related proteins/enzymes) might lead to permanent DNA damage and further chromosomal translocations. Other chromosomal insults can occur as well, such as intrachromosmal rearrangements, genetic deletions or activating mutations. Altogether, these chromosomal lesions are considered initiating events in leukaemogenesis, and most likely occur prenatally as common leukaemia fusion genes have been detected in cord blood, neonatal Guthrie cards and shared by monozygotic twins (Greaves et al., 2003).
There are two functional classes of translocations; the first one relocates a proto‐oncogene (or genes encoding for transcription factors or non‐antigen receptors) into regulatory regions of actively transcribed genes (such as those encoding for immunoglobulin chains or T‐cell receptors), causing dysregulated expression of an intact protein. The second class juxtaposes two genes to encode a chimeric protein that has distinct functions from the proteins from which it is derived (Hunger and Mulligham, 2015).
TEL‐AML1 is the most common chromosomal translocation associated with B‐ALL, which affects haematopoietic stem/progenitor cells (HSPCs). TEL‐AML1 is a fusion gene involving AML1 (also known as RUNX1), which controls the emergence of definitive haematopoietic stem cells in fetal haemogenic sites, and TEL (ETV6), responsible for adult haematopoietic stem cells survival (Teitell and Pandolfi, 2009).
It is not known to what extent chromosomal translocations/rearrangements are caused by errors in normal DNA processing or by external factors (chemicals, viruses), but translocations are 100‐fold more common in the population than leukaemia, indicating that most translocations are not sufficient for disease (Wiemels, 2008).
The finding that most common translocations found in childhood leukaemia (TEL‐AML1 and AML1‐ETO) occur at a rate of 1% in the normal population suggests that a significant proportion of the population carries preleukaemic clones. However, most of these clones are self‐limiting and do not result in disease. During the progression of the disease multiple genetic alterations accumulate over time being selected by their potential to give fitness advantage to the new clones.
How it is measured
Conventional cytogenetics, fluorescence in situ hybridisation (FISH, using commercially available dual colour translocation probes) and reverse transcription polymerase chain reaction (RT‐PCR) methods allow the identification of specific chromosome abnormalities (fusion genes, translocations, etc.), which can be further identified by subsequent cloning and sequencing (Soszynska et al., 2008). Cytokinesis‐block micronuclei assay also allows to assess chromosome damage.
Gene expression profiling defines distinct oncogenic groups in ALL related to the presence of different fusion oncogenes.
Taxonomic applicability
Chromosomal translocations can occur at all levels of living organisms and they have been created in murine and zebrafish models. These models can be useful for the in vivo study of leukaemogenic potential of chemicals in immature organisms as they may recapitulate human childhood leukaemia.
Bone marrow and fetal liver cells from mice have been retrovirally transduced to express TEL‐AML1 protein in an attempt to model human ALL.
Regulatory examples using this KE
The extended one generation test (OECD 443) includes a developmental immunotoxicity cohort. At present the cohort may identify postnatal effects resulting from prenatal or neonatal exposures on the immune tissues and white blood cells population. However, no specific parameters are in place to identify a pattern relevant to human childhood leukaemia in the extended one generation test. Besides, no treatment is administered in utero during the early developmental phase in the carcinogenicity assay and no considerations on the possible higher sensitivity of the HSPCs are in place for the genotoxicity assays. Thus, regulatory studies following OECD test guidelines may have potential limitations and experimental gaps eventually leading to false negative results.
References
Greaves M, 2002. Childhood leukaemia. British Medical Journal, 324, 283–287.
Greaves MF, Maia AT, Wiemels JL, Ford AM, 2003. Leukemia in twins: lessons in natural history. Blood, 102, 2321–2333.
Hunger SP, Mullighan CG, 2015. Acute Lymphoblastic Leukemia in Children. New England Journal of Medicine, 373, 1541–1552.
Soszynska K, Mucha B, Debski R, Skonieczka K, Duszenko E, Koltan A, Wysocki M, Haus O, 2008. The application of conventional cytogenetics, FISH, and RT‐PCR to detect genetic changes in 70 children with ALL. Annals of Hematology, 87, 991–1002.
Teitell MA, Pandolfi PP, 2009. Molecular genetics of acute lymphoblastic leukemia. Annual Review of Pathology, 4, 175–198.
Wiemels J, 2008. Chromosomal translocations in childhood leukemia: natural history, mechanisms, and epidemiology. Journal of the National Cancer Institute Monographs (JNCI Monographs), 39, 87–90.
KE2: Differentiation arrest of HSPCs
How this key event works
Chromosomal translocations create fusion genes encoding active kinases and altered transcription factors as well as hyperdiploidy. The genetic changes alter key regulatory processes by maintaining or enhancing an unlimited capacity for self‐renewal, subverting the controls of normal proliferation, blocking differentiation and promoting resistance to death signals (i.e. apoptosis).
Altered self‐renewal and differentiation of HSPCs can result from chimeric transcription factors, which arise from chromosome translocations that fuse portions of two different transcription factors. The aberrant proteins produced by fusion genes inhibit the normal transcriptional program and block the differentiation of B‐cell and myeloid precursors by recruiting repressor molecules such as histone deacetylase enzymes, resulting in aberrant cell proliferation and survival (Greaves, 2002; Pui et al., 2004; Teitell and Pandolfi, 2009; Papaemmannil et al., 2014). For instance, TEL‐AML1 and MLL fusions in undifferentiated progenitor cells can block the differentiation phase between pro‐B to pre‐B cells.
Most of paediatric B‐ALL with BCR‐ABL fusion genes exhibits IKZF1 deletions. The gene IKZF1 encodes a transcription factor that belongs to the family of zinc‐finger DNA‐binding proteins associated with chromatin remodelling. The expression of this protein is restricted to the fetal and adult haemolymphopoietic system, and it functions as a key regulator of lymphocyte differentiation. Mice with reduced Ikaros expression exhibited partial inhibition in precursor B‐cell maturation, which might be relevant in leukaemogenesis (Teitell and Pandolfi, 2009).
How it is measured
Arrest of B‐cell differentiation can be observed by histological assessment (Sabaawy et al., 2006).
Methods for detecting suppression of haematopoiesis include the assessment of cell‐specific markers via immunolabelling followed by flow cytometry and/or microscopy (e.g. CD34, CD19 and IgM for stages of B‐cell differentiation).
Methods for detecting epigenetic modifications include:
DNA methylation: Combined bisulfite restriction analysis (COBRA) and bisulfite sequencing for methylation; methylation‐specific PCR.
miRNA/non‐coding RNAs: miRNA/non‐coding RNA isolation followed by amplification using reverse transcription‐PCR; miRNA/non‐coding RNA microarray profiling/analysis
Taxonomic applicability
Mice have been transplanted with TEL‐AML1‐transduced bone marrow stem cells (Tsuzuki et al., 2004). There are also zebrafish models of TEL‐AML1‐positive ALL (Sabaawy et al., 2006).
References
Greaves M, 2002. Childhood leukaemia. British Medical Journal, 324, 283–287.
Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, Martincorena I, Hinton J, Gundem G, van Delft FW, Nik‐Zainal S, Jones DR, Ramakrishna M, Titley I, Stebbings L, Leroy C, Menzies A, Gamble J, Robinson B, Mudie L, Raine K, O'Meara S, Teague JW, Butler AP, Cazzaniga G, Biondi A, Zuna J, Kempski H, Muschen M, Ford AM, Stratton MR, Greaves M, Campbell PJ, 2014. RAG‐mediated recombination is the predominant driver of oncogenic rearrangement in ETV6‐RUNX1 acute lymphoblastic leukemia. Nature Genetics, 46, 116–125.
Pui C, Relling MV and Downing JR, 2004. Mechanisms of disease: Acute lymphoblastic leukemia. New England Journal of Medicine, 350, 1535–1548.
Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD, 2006. TEL‐AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences USA, 103, 15166–15171.
Teitell MA, Pandolfi PP, 2009. Molecular genetics of acute lymphoblastic leukemia. Annual Review of Pathology, 4, 175–198.
Tsuzuki S, Seto M, Greaves M, Enver T, 2004. Modeling first‐hit functions of the t(12;21) TEL‐AML1 translocation in mice. Proceedings of the National Academy of Sciences USA, 101, 8443–8448.
KE3: Clonal expansion as a result of secondary oncogenic insults (activating mutations) and delayed infections
How this key event works
ALL is mainly a disease of childhood that arises from recurrent genetic insults that block precursor B and T cell differentiation and drive aberrant cell proliferation and survival. A preleukaemic clone with self‐renewal stem cell activity may acquire progressive mutations in proliferative genes (activated signalling) resulting in a frank leukaemic clone. Cancer genome re‐sequencing studies have determined that most leukaemia cases harbour multiple mutations that have sequentially occurred in a single cell lineage to generate a dominant leukaemic clone (Jan and Majeti, 2013).
(Epi)genetic modifications:
Epigenetic modifications, in particular DNA methylation leading to reduced expressions of tumour suppressor genes contribute to the pathogenesis of childhood leukaemia. The inactivation or silencing of tumour suppressor genes can result in the sustained proliferation or reduced cell death response (e.g. apoptosis) of leukaemic cells. In childhood ALL, extensive hypermethylation of tumour suppressor genes such as FHIT, DLX3, p16 and p15 resulting in gene silencing has been observed. Furthermore, epigenetic silencing of proapoptotic genes (e.g. BIM), or blockage of apoptotic activation via deregulated expression of antiapoptotic genes (e.g. Bcl2 and BAX), inhibits activation of apoptosis and enhances survival of leukaemic cells, enabling the progression of leukaemogenesis (Sabaawy et al., 2006; Bachmann et al., 2010). Exposure to a variety of environmental agents can alter DNA methylation pattern inducing destabilising changes in gene expression potentially leading to cell transformation and tumorigenesis. There is some evidence suggesting that epigenetic modifications may be one of the mechanisms by which pesticides may exert adverse effects on human health (Collota et al., 2013).
The inhibition or aberrant regulation of apoptosis due to gene/protein dysfunction also plays a role in the pathogenesis of childhood leukaemia. Increased expression of Ikaros isoform 6 in murine myeloid precursor cell line appears to upregulate the expression of the antiapoptotic protein Bcl‐XL, preventing apoptosis and potentially leading to the pathogenesis of AML (Yagi et al., 2002).
Delayed infections
The delayed‐infection hypothesis of Greaves is based on a minimal two‐hit model and suggests that some susceptible children with a prenatally acquired preleukaemic clone had limited exposure to common infections early in life because they lived in a very hygienic environment. Such infectious insulation results in an immune system improperly developed that further predisposes these children to develop exacerbated aberrant responses after subsequent or delayed exposure to common infections later on in life, at an age commensurate with increased lymphoid‐cell proliferation (Gilham et al., 2005; Kamper‐Jørgensen et al., 2008; Pui et al., 2008). This untimely and excessive inflammatory response abolishes normal haematopoiesis such that lymphocytes or myeloid progenitor cells cannot mature. Thus, the innate and adaptive immune system is not fully functional upon an immune response and promotes selective expansion of a preleukaemic clone because of proliferative advantage and an increased opportunity for the acquisition of secondary genetic changes or mutations ultimately resulting in overt leukaemic phenotype (Greaves, 2006; Ford et al., 2009; Swaminathan et al., 2015).
Potential targets of chemical exposures
The immune system may be a target of the toxic effect of several chemicals. Chemically induced immune alteration through altering well‐regulated immune responses to tumour antigens, allergens, self‐antigens and microbial antigens can contribute to predisposition to different types of disorders, including cancers (Mokarizadeh et al., 2015). Evidence suggests that children may be particularly susceptible to adverse effects from exposure to pesticides, thus rendering them susceptible to infections and other immune mediated disorders (Corsini et al., 2013). Some evidence of effects of environmental exposures to pesticides during prenatal and early postnatal development on childhood leukaemia has been reported, raising the importance of studying the effects of toxicants on the developing immune system (Duramad et al., 2007). Xenobiotics may initiate, facilitate or exacerbate aberrant immune processes by inducing mutations in genes coding for immunoregulatory factors, modifying immune tolerance and activation pathways. Besides, various general or immune specific signalling pathways can be interfered by chemicals, resulting in changes in cytokine production, surface markers expression, cell differentiation and activation (Corsini et al., 2013).
Immunosuppression induced by pesticides may explain the relation with increased infections in humans observed in several studies. Particularly susceptible to immunotoxicity are children, as the vulnerable period for toxic insults to the developing immune system extends from early gestation to adolescence (Dietert, 2008). Background exposure to some pesticides early in life (pre‐ and postnatal exposure) may modulate the immune system development, increasing infection risks (Weselak et al., 2007). Furthermore, pesticides may interfere with immune surveillance, which in turn can affect recognition and destruction of abnormal cells, increasing the risk of cancer (Corsini et al., 2013).
How it is measured
Methods of detecting leukaemic cell proliferation include flow cytometry using cell‐specific markers followed by quantitative analysis, and incorporation and detection of bromodeoxyuridine (BrdU) by proliferating cells.
Multicolour fluorescence in situ hybridisation (FISH) may be used to track multiple genetic abnormalities identified in bulk ALL cells, yielding quantitative single cell resolution of the relative frequency of genetically distinct leukaemia subclones.
A novel experimental and computational single‐cell sequencing approach has been used to directly measure the clonal structures of childhood ALL samples at diagnosis (Gawad et al., 2011).
Apoptosis can be measured by using plasma membrane assays, mitochondrial assays, caspase assays, nuclear apoptosis assays and flow cytometry.
Taxonomic applicability
Mechanisms relevant to clonal expansion may not show significant interspecies differences and potential mechanisms remain currently unclear.
Murine models with human precursor cells harbouring the TEL‐AML1 fusion have been developed.
References
Bachmann PS, Piazza RG, Janes ME, Wong NC, Davies C, Mogavero A, Bhadri VA, Szymanska B, Geninson G, Magistroni V, Cazzaniga G, Biondi A, Miranda‐Saavedra D, Göttgens B, Saffery R, Craig JM, Marshall GM, Gambacorti‐Passerini C, Pimanda JE, Lock RB, 2010. Epigenetic silencing of BIM in glucocorticoid poor‐responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood, 116, 3013–3022.
Collotta M, Bertazzi PA, Bollati V, 2013. Epigenetics and pesticides. Toxicology, 307, 35–41.
Corsini E, Sokooti M, Galli CL, Moretto A, Colosio C, 2013. Pesticide induced immunotoxicity in humans: a comprehensive review of the existing evidence. Toxicology, 307, 123–135.
Dietert RR, 2008. Developmental immunotoxicology (DIT): windows of vulnerability, immune dysfunction and safety assessment. Journal of Immunotoxicology, 5, 401–412.
Duramad P, Holland NT, 2011. Biomarkers of immunotoxicity for environmental and public health research. International Journal of Environmental Research and Public Health, 8, 1388–1401.
Gawad C, Koh W, Quake SR, 2014. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single‐cell genomics. Proceedings of the National Academy of Sciences USA, 111, 17947–17952.
Gilham C, Peto J, Simpson J Roman E, Eden TO, Greaves MF, Alexander FE; UKCCS Investigators, 2005. Day care in infancy and risk of childhood acute lymphoblastic leukaemia: findings from UK case‐control study. British Medical Journal, 330, 1294.
Greaves M, 2006. Infection, immune responses and the aetiology of childhood leukaemia. Nature Reviews Cancer, 6, 193–203.
Jan M, Majeti R, 2013. Clonal evolution of acute leukemia genomes. Oncogene, 32, 135–140.
Kamper‐Jørgensen M, Woodward A, Wohlfahrt J, et al., 2008. Childcare in the first 2 years of life reduces the risk of childhood acute lymphoblastic leukemia. Leukemia, 22, 189–19.
Mokarizadeh A, Faryabi MR, Rezvanfar MA, Abdollahi M, 2015. A comprehensive review of pesticides and the immune dysregulation: mechanisms, evidence and consequences. Toxicology Mechanisms and Methods, 25, 258–278.
OECD, 2011. Guideline for the Testing of Chemicals 443: Extended One‐Generation Reproductive Toxicity Study. Organisation for Economic Cooperation and Development, Paris, France.
Pui CH, Robison LL, Look AT, 2008. Acute lymphoblastic leukaemia. Lancet, 371, 1030–1043.
Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, Müschen M, 2015. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nature Immunology, 16, 766–774.
Weselak M, Arbuckle TE, Wigle DT, Krewski D, 2007. In utero pesticide exposure and childhood morbidity. Journal of Environmental Research, 103, 79–86.
Yagi T, Hibi S, Takanashi M, Kano G, Tabata Y, Imamura T, Inaba T, Morimoto A, Todo S, Imashuku S, 2002. High frequency of Ikaros isoform 6 expression in acute myelomonocytic and monocytic leukemias: implications for upregulation of the antiapoptotic protein Bcl‐XL in leukemogenesis. Blood, 99, 1350–1355.
Adverse Outcome (AO): Overt childhood leukaemia
How this key event works
Symptoms of childhood leukaemia include sensitivity to bruising and bleeding due to thrombocytopenia, pallor and fatigue from anaemia, and increased susceptibility to infections caused by neutropenia. These symptoms result from the displacement of the normal haematopoiesis by expansion of leukaemia cells. Leukaemic infiltration of the liver, spleen, lymph nodes, and mediastinum is common at diagnosis (Hunger and Mulligham, 2015).
How it is measured
Haematological methods: observations of clinical symptoms, routine blood cell count and identification of leukaemia cells (i.e. immunophenotyping by flow cytometry) in peripheral blood and bone marrow. Diagnosis stratification relies on molecular cytogenetics (FISH and karyotype).
Immunophenotyping allows the identification of pathologic cells and phenotype characterisation based on specific pattern‐identification of surface as well as intracellular antigen expressions in unique cell populations.
Flow cytometry is a laser‐based technology that uses monoclonal antibodies for the detection of expression of a number of antigens on the cell surface, thus distinguishing between healthy and diseased cells. Flow cytometry allows the identification and quantification of subsets of the major leukocyte populations and even further subdivisions that differ in biologic function, maturation stage, and activation (Adin‐Cinar, 2013).
Taxonomic applicability
The following animal models have been developed for childhood leukaemia:
MLL‐ENL and MLL‐AF9 fusions have been proven to be oncogenic by themselves in human cord blood progenitor cells (Barebé et al., 2007).
TEL‐AML1 and hyperdiploid primary blasts recapitulate the disease phenotype in immunodeficient mice (Rehe et al., 2014, le Viseur et al., 2008).
Regulatory relevance of the AO
Genotoxicity and carcinogenicity are standard endpoints measured in the regulatory studies performed for the risk assessment of chemicals and they are mandatory for pesticide substances. However, no treatment is occurring during the early in‐utero development phase in the carcinogenicity study.
The extended one generation test (OECD 443) assesses parental fertility and reproductive function and the development of offspring to sexual maturity and also includes a developmental immunotoxicity cohort. A second generation can be triggered if any effects requiring further evaluation are identified in the first generation (OECD, 2011). The study design provides the opportunity to evaluate life stages not covered by other study types and represents a highly integrated study design that includes an assessment of developmental immunotoxicity. While the developmental immunotoxicity cohort may identify postnatal effects resulting from prenatal or neonatal exposures on the immune tissues and white blood cells population; however, no specific parameters are in place to identify a pattern relevant to human childhood leukaemia.
References
Adin‐Cinar S, Kucuksezer UC, Deniz G, 2013. Implications of minimal residual disease flow cytometry in pediatric acute leukemias. International Trends in Immunity, 1, 54–61.
Barabé F, Kennedy JA, Hope KJ, Dick JE, 2007. Modeling the initiation and progression of human acute leukemia in mice. Science, 316, 600–604.
Hunger SP, Mullighan CG, 2015. Acute Lymphoblastic Leukemia in Children. New England Journal of Medicine, 373, 1541–1552.
le Viseur C, Hotfilder M, Bomken S, Wilson K, Roettgers S, Schrauder A, Rosemann A, Irving J, Stam RW, Shultz LD, Harbott J, Juergens H, Schrappe M, Pieters R, Vormoor J, 2008. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell, 14, 47–58.
Rehe K, Wilson K, Bomken S, Williamson D, Irving J, den Boer ML, Stanulla M, Schrappe M, Hall AG, Heidenreich O, Vormoor J, 2013. Acute B lymphoblastic leukaemia‐propagating cells are present at high frequency in diverse lymphoblast populations. EMBO Mol Med, 5, 38–51.
1st KER: In utero chemical exposure (KEup) leading to unrepaired/misrepaired double DNA damage and further chromosomal translocations (KEdown)
How this Key Event Relationship works
DNA is highly susceptible to oxidative damage, which can result in single strand breaks (SSBs) and DSBs, base and sugar‐moiety oxidation, strand crosslinks and the generation of abasic sites. DSBs are the most serious type of DNA damage because a small number of these lesions are sufficient to induce gene mutations or chromosomal aberrations (Sedelnikova et al., 2001; Woodbine et al., 2011). Oxidative molecules may either enhance the likelihood of DSB in HSPCs or interact with biological molecules disrupting the normal synthesis and repair of DNA. This disruption is primarily associated with inhibition or inactivation of antioxidant proteins as well as DNA repair enzymes (Kryston et al., 2011).
Upon DNA damage or genotoxic stress, haematopoietic stem cells differentiate to lineage‐committed progenitors, which can be considered as a method to escape propagating damaged genetic information. This escape mechanism fails when haematopoietic stem cells chose DNA repair by NHEJ over differentiation, in order to maintain their self‐renewal, thus thriving haematological malignancies (Weiss and Ito, 2015).
Defects in NHEJ can create chromosomal deletions and translocations. The accumulation of genetic damage through misrepair or incomplete repair of DNA may lead to mutagenesis and eventually cell transformation, particularly if combined with a deficient apoptotic pathway (Kryston et al., 2011). An impaired repair of oxidatively modified DNA, documented in children with ALL, may contribute to the genetic instability of precursor‐B cells which may be linked with the development of the disease (Olinski et al., 2014).
For fusion genes to be effective in promoting leukaemogenesis, DNA DSB must occur simultaneously in two chromosomes in a single HSPC that does not undergo cell death, and must also be situated in the coding region of the genes to generate a functional chimeric gene product. The resulting chromosomal recombination must take place in a HSPC with a sustainable lifespan and clonal potential to propagate the chimeric gene product (Greaves and Wiemels, 2003).
A massive parallel sequencing approach performed in a cohort of twins concordant for ALL indicated that the TEL‐AML1 fusion gene arises as a consequence of NHEJ as no binding motifs indicative of RAG1/2 or terminal deoxynucleotidyl transferase (TdT) activity were found. The TEL‐AML1 fusion arises in a fetal HSPCs that lies upstream of B‐cell lineage‐restricted RAG1/2 active precursors. The preleukaemic clone arises and expands in the pro‐ or pre‐B‐lineage compartment in the fetal liver and then undergoes V(D)J rearrangements (Alpar et al., 2015).
Biological plausibility
In the last decades the occurrence of childhood leukaemia showed a rise that was in part attributed to an increased exposure to risk factors. Although the aetiology of ALL remains elusive, ionising radiation, congenital genetic syndromes and in utero exposure to specific genotoxic chemicals, including household pesticides, are considered the major risk factors (Pui et al., 2008). Despite the mounting epidemiologic evidence linking pesticide exposure during pre‐ and postnatal life with childhood leukaemia, robust underlying pathological mechanisms remain unknown. The initiating event at the molecular level might be generation of oxidative stress by environmental exposures (including pesticides) leading directly or indirectly to DNA damage and further chromosomal damage (Hernández and Menéndez, 2016); however this still remains hypothetical.
A massive parallel sequencing approach performed in a cohort of twins concordant for ALL indicated that the TEL‐AML1 fusion gene arises as a consequence of NHEJ as no binding motifs indicative of RAG1/2 or terminal deoxynucleotidyl transferase (TdT) activity were found. The TEL‐AML1 fusion arises in a fetal HSPCs that lies upstream of B‐cell lineage‐restricted RAG1/2 active precursors. The preleukaemic clone arises and expands in the pro‐ or pre‐B‐lineage compartment in the fetal liver and then undergoes V(D)J rearrangements (Alpar et al., 2015).
Investigation of the DNA damage in steady state, as well as after exposure to UV light, confirmed increased DNA damage in pro‐B cells lacking a functional allele of Ebf1 (a transcription factor critical for the activation of B‐lineage restricted genes in the earliest B‐lineage progenitors that also controls DNA repair). Reduced Ebf1 levels may contribute to malignant transformation by a combination of impaired DNA repair and increased cell survival rather than simply by a differentiation block (Prassad et al., 2015). Since Rad 51 is one of the central components of the DNA DSB repair gene, whose expression can be induced by DNA damage, a drop in leukaemic potential after Rad51 re‐expression would conclusively demonstrate that loss in HR DNA repair was the main driving force of leukaemic transformation of the Ebf1+/− Pax5+/− B‐cell precursors (Georgopoulos, 2015).
Uncertainties and Inconsistencies
Despite the sound epidemiological evidence linking pesticide exposure and childhood leukaemia, the first initiating molecular event(s) has not been identified yet. In contrast to MLL‐rearranged infant leukaemia, there is no evidence at all regarding the molecular basis of how some individual pesticide or pesticide class (or functional group) can interact with biological targets to elicit DNA damage. We can speculate only with potential mechanisms, such as induction of oxidative stress in HSPCs, as DNA is highly susceptible to oxidative damage and can result in single and double strand breaks. Besides, it not clearly understood what drives damaged HSPCs to initiate DNA repair systems and when to enter the cell cycle or to keep quiescent accumulating genotoxic stress (Weiss and Ito, 2015). While, regulatory studies have consistently found lack of genotoxic effects of pesticides in many test systems, there are studies in the open literature supporting genotoxicity by using different biomarkers. In addition, some epidemiological studies on agricultural workers exposed to pesticides have reported DNA damage. These uncertainties and inconsistencies warrant further research to delineate how pesticides interact with DNA and produce genetic lesions.
References
Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, Titley I, Kearney L, Szczepanski T, Gonzalez D, Ford AM, Potter NE, Greaves M, 2015. Clonal origins of ETV6‐RUNX1+ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia, 29, 839–846.
Edwards TM, Myers JP, 2007. Environmental exposures and gene regulation in disease etiology. Environmental Health Perspectives, 115, 1264–1270.
Georgopoulos K, 2015. Ebf1 in DNA repair and leukemogenesis. Blood, 125, 3969–3971.
Greaves M, 2002. Childhood leukaemia. British Medical Journal, 324, 283–287.
Greaves MF, Maia AT, Wiemels JL, Ford AM, 2003. Leukemia in twins: lessons in natural history. Blood, 102, 2321–2333.
Greaves MF, Wiemels J, 2003. Origins of chromosome translocations in childhood leukaemia. Nature Reviews Cancer, 3, 639–649.
Hauer J, Borkhardt A, Sánchez‐García I, Cobaleda C, 2014. Genetically engineered mouse models of human B‐cell precursor leukemias. Cell Cycle, 13, 2836–2846.
Hernández AF, Lacasaña M, Gil F, Rodríguez‐Barranco M, Pla A, López‐Guarnido O, 2013. Evaluation of pesticide‐induced oxidative stress from a gene‐environment interaction perspective. Toxicology, 307, 95–102.
Hernández AF, Menéndez P, 2016. Linking pesticide exposure with pediatric leukemia: potential underlying mechanisms. International Journal of Molecular Sciences, 17. pii: E461.
Jaruga P, Zastawny TH, Skokowski J, Dizdaroglu M. Olinski R, 1994. Oxidative DNA base damage and antioxidant enzyme activities in human lung cancer. Federation of the European Biochemical Societies's Letters (FEBS), 341, 59–64.
Knapp GW, Setzer RW, Fuscoe JC, 2003. Quantitation of aberrant interlocus T‐cell receptor rearrangements in mouse thymocytes and the effect of the herbicide 2,4‐dichlorophenoxyacetic acid. Environmental and Molecular Mutagenesis, 42, 37–43.
Linn S, 1998. DNA damage by iron and hydrogen peroxide in vitro and in vivo. Drug Metabolism Reviews, 30, 313–326.
Lu C, Liu X, Liu C, Wang J, Li C, Liu Q, Li Y, Li S, Sun S, Yan J, Shao J, 2015. Chlorpyrifos induces MLL Translocations through caspase 3‐dependent genomic instability and topoisomerase II inhibition in human fetal liver hematopoietic stem cells. Journal of Toxicological Sciences, 147, 588–606.
Mostafalou S, Abdollahi M, 2013. Pesticides and human chronic diseases: Evidences, mechanisms, and perspectives. Toxicology and Applied Pharmacology, 268, 157–177.
Muniz JF, McCauley L, Scherer J, Lasarev M, Koshy M, Kow, Nazar‐Stewart V, Kisby GE, 2008. Biomarkers of oxidative stress and DNA damage in agricultural workers: a pilot study. Toxicology and Applied Pharmacology, 227, 97–107.
Pui CH, Robison LL, Look AT, 2008. Acute lymphoblastic leukemia. Lancet, 371, 1030–1043.
Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM, 2010. Role of oxidatively induced DNA lesions in human pathogenesis. Mutation Research, 704, 152–159.
Sentürker S, Karahalil B, Inal M, Yilmaz H, Müslümanoglu H, Gedikoglu G, Dizdaroglu M, 1997. Oxidative DNA base damage and antioxidant enzyme levels in childhood acute lymphoblastic leukemia. Federation of the European Biochemical Societies's Letters (FEBS), 416, 286–290.
Weiss CN, Ito K, 2015. DNA damage: a sensible mediator of the differentiation decision in hematopoietic stem cells and in leukemia. International Journal of Molecular Sciences, 16, 6183–6201.
Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA, 2011. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Research, 39, 6986–6997.
2nd KER: Title: In utero chromosomal translocations (KEup) leading to differentiation arrest of HSPCs (KEdown)
How this Key Event Relationship works
There are many potential chromosomal translocations associated to childhood leukaemia, suggesting that multiple mechanisms underlie the development of the disease. Nevertheless, the major fusion genes generated by chromosome translocations are TEL‐/AML1 in ALL and AML1‐/ETO in AML. The chimeric (aberrant) proteins produced by these genes inhibit gene activity and block cell differentiation by recruiting repressor molecules such as histone deacetylase enzymes (Greaves, 2002).
TEL‐AML1 and MLL fusions in undifferentiated progenitor cells can block the differentiation phase from pro‐B to pre‐B cells. By stalling B‐cell development, subsequent recombination‐activating gene (RAG)–mediated genomic rearrangements become drivers of the creation of polyclonal structures (Papaemmanuil, et al., 2014). A study using whole‐genome sequencing in ALL has suggested that the aberrant activity of RAG recombinases, which are highly expressed in cells harbouring TEL‐AML1, can result in various oligoclonal V(D)J recombination events and further inactivation of genes required for B‐lineage differentiation (Papaemmanuil et al., 2014).
Weight of evidence
The block of differentiation of HSPCs provides proliferative advantage because of conferring self‐renewal properties to lymphoid progenitors. Enhanced self‐renewal would promote the extended longevity of B‐cell precursors to acquire and accumulate additional genomic aberrations and secondary mutations, which collaborate to fully transform these B‐cell precursors into leukaemia cells (Duque‐Afonso et al., 2015).
The preleukaemic transformation conferring the in utero clonal expansion of TEL‐AML1 cells occurs in an early B‐cell lineage committed progenitor, most likely at the pro‐B or pre‐B‐cell stage in the fetal liver (Alpar et al., 2015).
Biological plausibility
Under the current paradigm, the first initiating oncogenic mutation usually involves structural or numerical chromosomal alterations impairing normal cell differentiation, while secondary hits more commonly comprise mutations affecting developmentally regulated master transcription factors or membrane‐proximal signalling pathways conferring proliferation and survival advantages to the differentiation‐blocked clone. The development of leukaemia requires activation of cell proliferation in addition to differentiation blockage (reviewed in Hernández and Menéndez, 2016).
As a result of chromosomal translocations, aberrant chimeric proteins alter the normal transcriptional program and block normal B‐cell and/or myeloid differentiation. Childhood leukaemia arises from recurrent genetic insults that block differentiation of haematopoietic stem and/or progenitor cells (HSPCs), and drives uncontrolled proliferation and survival of the differentiation‐blocked clone.
Epigenetic modifications to DNA affect the activity of genes and their cellular expression and include DNA methylation, histone modification, and alterations in non‐coding microRNAs (miRNAs). Each of these mechanisms alters how genes are expressed or silenced without modifying the DNA sequence. Epigenetic control of transcriptional activation also plays an essential role in regulating gene expression during early development and haematopoiesis. Besides, epigenetic modifications can influence leukaemogenesis if they lead to silencing of tumor suppressor genes or activation of oncogenes (Burke and Bhatla, 2014).
Non‐coding RNAs have been implicated in the pathogenesis of childhood leukaemia as their altered expression can regulate various physiological processes such as cell differentiation, proliferation and immune responses. Expression of miRNAs is triggered by epigenetic modifications, e.g. hyper/hypomethylation of CpG islands in the promoter region of genes, or by fusion proteins.
In a zebrafish model of TEL‐AML1+ B‐ALL, arrest of B‐cell differentiation has been observed by histological assessment (Sabaawy et al., 2006). An accumulation of early pro‐B cells and a differentiation deficit after pro‐B‐cell formation has been reported in mice transplanted with TEL‐AML1‐transduced bone marrow stem cells (Tsuzuki et al., 2004).
De Laurentiis et al. (2015) generated an experimental model using the murine haematopoietic stem progenitor cell line EML1 expressing the TEL‐AML1 fusion protein, and analysed its differentiation and global gene expression properties. Upon TEL‐AML1 expression, EML1 cells lost the capacity to differentiate into B‐cells and underwent apoptosis. TEL‐AML1 expression impaired the activation of IFNα/β signalling pathway in primary murine and human HSPCs with a dramatic inhibition of IRF3 phosphorylation, a member of the IFN‐regulatory transcription factor family (de Laurentiis et al., 2015). This finding is consistent with the downregulation of genes involved in IRF3‐IFN signalling as shown in gene expression data derived from blasts of ALL patients expressing TEL‐AML1 (Linka et al., 2013). These data suggest that IRF3‐IFNα/β signalling is involved in the block of B‐cell maturation elicited by TEL‐AML1 expression. Furthermore, differentiation of cells expressing the TEL‐AML1 protein can be restored by treatment with IFNβ (de Laurentiis et al., 2015).
Mice with reduced Ikaros expression (a master transcription factor that regulates lymphocyte differentiation) have a partial block at the pro‐B‐cell stage in development, suggesting a tumorigenic role by blocking B‐cell maturation (Teitell and Pandolfi, 2009). In the case of T‐cell ALL, aberrant regulation or genetic mutations of cell‐specific transcription factors inhibit cell maturation/development, leading to increased expansion of leukaemic cells. More than 50% of T‐cell ALL have activating mutations involving NOTCH1, a gene encoding a transmembrane receptor that regulates normal T‐cell development by enhancing the transcription of diverse responder genes in developing thymocytes, such as cyclin D1 and c‐MYC (Pui et al., 2008).
The Pax5 gene encodes the B‐cell lineage specific activator protein that is expressed at early, but not late stages of B‐cell differentiation. The developmental block observed in Pax5‐deficient leukaemia cells can be reversed on restoration of Pax5 expression, suggesting that the reduction in Pax5 function results in a reversible disruption of differentiation. Transgenic RNAi can reversibly suppress endogenous Pax5 expression in the haematopoietic compartment of mice, which cooperates with activated signal transducer and activator of transcription 5 (STAT5) to induce B‐ALL (Liu et al., 2016).
Although the Ebf1 dose‐dependent events in B‐cell precursors are not overtly leukaemogenic, the combination with Pax5 haploinsufficiency dramatically increases leukaemic potential by stalling B‐cell differentiation at a highly proliferative and recombination active stage, which allows the selection and expansion of precursors carrying appropriate DNA mutations (Georgopoulos, 2015).
Epigenetic activation (i.e. hypomethylation) of ZNF423, a protein that interferes with B‐cell differentiation, interacts with the early B‐cell factor 1 (EBF‐1) to inhibit transcription of EBF‐1‐targeted genes and subsequently trigger B‐cell maturation arrest (Harder et al., 2013). Silencing of some miRNAs (e.g. miR‐34b) or increased level of other miRNAs (e.g. miR‐155) may lead to enhanced cell proliferation of leukaemic cells and/or inhibition of cell differentiation.
Uncertainties and inconsistencies
A prerequisite for the specific AOP is the occurrence of genetic damage (i.e. chromosomal translocations) in a particularly vulnerable genetic locus, within the proper cell and in a specific time window. However, details of this entire process and how it happens are not clear.
The target leukaemia‐initiating cell(s) have not been identified so far with sufficient confidence and consequently there is no faithful cell model that recapitulates the pathogenesis in humans at the molecular level.
References
Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, Titley I, Kearney L, Szczepanski T, Gonzalez D, Ford AM, Potter NE, Greaves M, 2015. Clonal origins of ETV6‐RUNX1+ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia, 29, 839–846.
Burke MJ, Bhatla T, 2014. Epigenetic modifications in pediatric acute lymphoblastic leukemia. Frontiers in Pediatrics, 2, 42.
de Laurentiis A, Hiscott J, Alcalay M, 2015. The TEL‐AML1 fusion protein of acute lymphoblastic leukemia modulates IRF3 activity during early B‐cell differentiation. Oncogene, 34, 6018–6028.
Duque‐Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, Iwasaki M, Cleary ML, 2015. Comparative genomics reveals multistep pathogenesis of E2A‐PBX1 acute lymphoblastic leukemia. Journal of Clinical Investigation, 125, 3667–3680.
Georgopoulos K, 2015. Ebf1 in DNA repair and leukemogenesis. Blood, 125, 3969–3971.
Greaves M, 2002. Childhood leukaemia. British Medical Journal, 324, 283–287.
Greaves MF, Wiemels J, 2003. Origins of chromosome translocations in childhood leukaemia. Nature Reviews Cancer, 3, 639–649.
Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, Cazzaniga G, Enver T, Greaves M, 2009. The TEL‐AML1 leukemia fusion gene dysregulates the TGF‐beta pathway in early B lineage progenitor cells. Journal of Clinical Investigation, 119, 826–836.
Harder L, Eschenburg G, Zech A, Kriebitzsch N, Otto B, Streichert T, Behlich AS, Dierck K, Klingler B, Hansen A, Stanulla M, Zimmermann M, Kremmer E, Stocking C, Horstmann MA, 2013. Aberrant ZNF423 impedes B cell differentiation and is linked to adverse outcome of ETV6‐RUNX1 negative B precursor acute lymphoblastic leukemia. Journal Of Experimental Medicine, 210, 2289–2304.
Linka Y, Ginzel S, Krüger M, Novosel A, Gombert M, Kremmer E, Harbott J, Thiele R, Borkhardt A, Landgraf P, 2013. The impact of TEL‐AML1 (ETV6‐RUNX1) expression in precursor B cells and implications for leukaemia using three different genome‐wide screening methods. Blood Cancer Journal, 3, e151.
Liu GJ, Cimmino L, Jude JG, Hu Y, Witkowski MT, McKenzie MD, Kartal‐Kaess M, Best SA, Tuohey L, Liao Y, Shi W, Mullighan CG, Farrar MA, Nutt SL, Smyth GK, Zuber J, Dickins RA, 2014. Pax5 loss imposes a reversible differentiation block in B‐progenitor acute lymphoblastic leukemia. Genes & Development, 28, 1337–1350.
Mori H, Colman SM, Xiao ZJ, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M, 2002. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proceedings of the National Academy of Sciences, 99, 8242–8247.
Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, Martincorena I, Hinton J, Gundem G, van Delft FW, Nik‐Zainal S, Jones DR, Ramakrishna M, Titley I, Stebbings L, Leroy C, Menzies A, Gamble J, Robinson B, Mudie L, Raine K, O'Meara S, Teague JW, Butler AP, Cazzaniga G, Biondi A, Zuna J, Kempski H, Muschen M, Ford AM, Stratton MR, Greaves M, Campbell PJ, 2014. RAG‐mediated recombination is the predominant driver of oncogenic rearrangement in ETV6‐RUNX1 acute lymphoblastic leukemia. Nature Genetics, 46, 116–125.
Pui CH, Robison LL, Look AT, 2008. Acute lymphoblastic leukemia. Lancet, 371, 1030–1043.
Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD, 2006. TEL‐AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proceedings of the National Academy of SciencesU S A, 103, 15166–15171.
Teitell MA, Pandolfi PP, 2009. Molecular genetics of acute lymphoblastic leukemia. Annual Review of Pathology, 4, 175–198.
3rd KER: Differentiation arrest of HSPCs (KEup) leading to clonal expansion of leukaemogenic cells (KEdown)
How this Key Event Relationship works
In the ‘two‐hit model’ widely accepted for leukaemogenesis two types of genetic aberrations and/or mutations are required. The first one is associated with a block in differentiation/maturation through chromosomal translocations affecting transcription factors that normally promote cellular differentiation at crucial steps during early haematopoiesis (see KER2). Further mutations affect genes controlling cellular proliferation and apoptosis, classically intracellular signalling pathways (i.e. tyrosine kinases), which lead to increased proliferation and/or inhibition of apoptosis (such as FLT3, RAS, KIT, and BCR–ABL) (Eriksson et al., 2014).
Dysregulation of immune responses to common infections might promote the malignant evolution of TEL‐AML1‐expressing preleukaemic clones. Ford et al. (2009) linked paediatric ALL with signalling pathways involved in infection and inflammation.
The major histocompatibility genes might play a role in the linkage between patterns of infection and leukaemia risk as several HLA haplotypes have been associated with childhood leukaemia (Wiemels, 2012). However, it is possible that exposure to infections promote B‐ALL in children harbouring an intrinsic genetic susceptibility (Hauer et al., 2015).
Weight of evidence
Fusion gene products may suffice to initiate but not to fully complete leukaemogenesis, and other secondary genetic lesions must occur for developing childhood leukaemia. For instance, the concordance rate for ALL in monozygotic twin children is around 5–10%, and transgenic mice expressing fusion gene products (e.g. TEL‐AML1 or AML1‐ETO) did not exhibit overt signs of leukaemia (Mori et al., 2002). Also, some fusion genes associated with childhood leukaemia can be detected in the blood of normal individuals, indicating that they occur ubiquitously in humans and do not necessarily lead to the disease (Greaves and Wiemels, 2003).
After the occurrence of oncogenic fusion proteins resulting from chromosomal translocations, subsequent cooperating hits define the disease latency and occur after birth, and may be of genetic, epigenetic or immune nature (i.e. delayed infection‐mediated immune deregulation).
Transgenic mice expressing TEL‐AML1 failed to develop leukaemia and this finding was corroborated in subsequent reports where no leukaemia was observed despite the use of differing gene promoters to express the translocation. These experiments support the need of a second genetic event is necessary for the development of leukaemia (Jacoby et al., 2014).
Biological plausibility
Dysfunctions of the immune system and delayed infections have been linked to childhood leukaemia (Greaves, 2006; Swaminathan et al., 2015). Two factors might explain this association: a) a lower repertoire of infections during early immune development, and b) an altered congenital responder status to infection resulting in functionally aberrant clinical presentation of occasional infections. Thus, an untimely and excessive inflammatory response abolishes normal haematopoiesis, promoting selective expansion of a preleukaemic clone because of proliferative advantage and increased likelihood for a second mutation required for the development of the disease to occur (Sanjuan‐Pla et al., 2015). Additional support is provided by studies showing that an increased opportunity for early childhood infections as well as normal childhood vaccinations protects against leukaemia indicating that vaccination reduces risk to leukaemia (Ma et al., 2005).
The IFNα/β cytokines, whose production is impaired by TEL‐AML1 expression, have been long known to modulate resistance to viral infections and enhance innate and acquired immune responses. IFNs also influence tumour growth by directly inducing the expression of genes involved in apoptosis, or indirectly by inhibiting angiogenesis and modulating immune response (reviewed in de Laurentiis et al., 2015).
Murine models with human precursor cells harbouring the TEL‐AML1 fusion gene generated a preleukaemic state that only resulted in an overt leukaemic phenotype upon the acquisition of additional genetic abnormalities (Alpar et al., 2015).
Genetic alterations
Genetic alterations that impair cell differentiation probably cooperate with a second class of mutations that alter the proliferation and survival of HSPCs. One of these mutations affects the RAS‐RAF‐MEK‐ERK signalling cascade, important for the HSPC development, leading to enhanced cell survival/proliferation (Case et al., 2008). Mutations of the receptor tyrosine kinase (RTK)‐Ras signalling pathway have been associated with the pathogenesis of childhood (and perhaps infant) leukaemia (Driessen et al., 2013; Prelle et al., 2013; Paulsson et al., 2015). Also, the lack of degradation of cell signalling proteins enhances survival and proliferation of leukaemic cells as occurs either with inactivation of E3 ubiquitin ligase (Makishima et al., 2009; Aranaz et al., 2012), or with constitutive activation of MAP kinase (e.g. JNK), leading to proteasomal degradation of proapoptotic proteins (Leung et al., 2008).
Multiple secondary changes have been proposed to cooperate with TEL‐AML1 fusion for overt B‐cell ALL. Gene deletions of non‐antigen receptor or cell cycle regulatory proteins can further promote the proliferation and survival of leukaemic cells (Aplan, 2006; Novara et al., 2009; Meissner et al., 2014).
TEL‐AML1‐positive preleukaemic clones trigger an increase in ROS, which promotes the accumulation of secondary genetic lesions by increasing genetic instability and DNA DSBs, therefore enabling these preleukaemic clones to develop into leukaemic cells (Kantner et al., 2013).
Gawad et al. (2014) sequenced a panel of single nucleotide variants (SNVs), deletions, and IgH sequences in 1,479 single tumour cells from six ALL patients. By accurately segregating groups of co‐occurring mutations into distinct clonal populations, co‐dominant clones were found in the majority of patients. Evaluation of intraclonal mutation patterns (a) identified clone‐specific cytosine mutagenesis events, (b) showed that most structural variants are acquired before SNVs, (c) determined that KRAS mutations occur late in disease development but are not sufficient for clonal dominance, and (d) identified within the same patient clones arrested at varied stages in B‐cell development. Most large deletions occurred before cytosine mutagenesis‐driven SNV acquisition, thus providing further evidence that the majority of the SNVs were caused by an APOBEC protein. Ongoing V(D)J recombination can occur in the most evolved clones, which can have variable magnitude between clones in the same patient. The development of leukaemic cells can be promoted by rearrangement of T‐cell acute lymphoblastic leukaemia 1 gene (TAL1, which encodes a transcription factor that regulates both embryonic and adult haematopoiesis) along with the inactivation of phosphatase and tensin homologue gene (PTEN, encoding a tumour suppressor dual‐specificity phosphatase that antagonises the PI3K signalling pathway) via microdeletions due to illegitimate RAG activity (Mendes et al., 2014).
Role of infections and immunity
Infection can trigger a series of events that are directly involved in genome instability. Olinski et al. (2014) proposed that viral infections may result in aberrant expression of the AID (activation‐induced deaminase)/APOBEC (apolipoprotein B editing complex) family of DNA cytosine deaminases, which are able to insert mutations in DNA and RNA by deaminating cytidine to uridine. AID is essential for the antigen‐driven diversification of already rearranged immunoglobulin genes in the adaptive immune system. Since these enzymes also participate in active DNA demethylation process, changes in DNA methylation status or aberrant methylation can occur (Olinski et al., 2014). Altogether, these processes may lead to genome instability in prenatally generated preleukaemic cells and the emergence of ALL.
The inflammatory cytokine TGFβ has been involved in TEL‐AML1‐mediated leukaemogenesis since B cell progenitor cell line and human cord blood progenitor cells expressing TEL‐AML1 inhibit downstream activation of TGF‐β by binding to Smad3, the main TGF‐β signalling target, thus preventing the activation of target promoters. As a result, TEL‐AML1‐expressing cells might propagate by inhibiting the tumour‐suppressive properties of TGF‐β.
Wild type mice kept in a specific‐pathogen‐free environment from birth and then moved to common infectious environment did not develop B‐ALL (Martín‐Lorenzo et al., 2015). Pax5+/− mice also failed to develop leukaemia under non‐infection exposure conditions; however, when these mice were exposed to infection they acquired point mutations in the second allele, which triggered the development of pB‐ALL recapitulating the clinical, histopathological and molecular features of human B‐ALL (Hauer et al., 2015). These data provide evidence that delayed exposure to infection can induce human‐like B‐ALL in mice on the basis of inherited genetic predisposition (see Figure B.6 from Hauer et al., 2015).
Figure B.6.
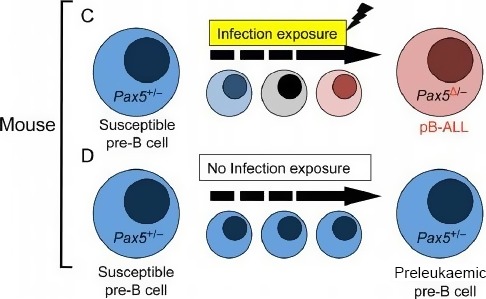
Exposure to infection is a causal factor in B‐precursor acute lymphoblastic leukaemia as a result of Pax5 inherited susceptibility (Hauer et al., 2015, CC‐BY)
Monoallelic loss of Pax5 promotes leukaemogenesis by creating an aberrant IL7‐sensitive progenitor compartment, a preleukaemic pre‐B cell population susceptible to malignant transformation through accumulation of secondary Jak3 mutations, which depicts a rescue mechanism of the IL7/IL7R/STAT5 signalling. Transplantation experiments demonstrate that the activating Jak3 mutations per se are sufficient to drive leukaemia (Martín‐Lorenzo et al., 2015).
The mechanisms underlying the conversion of the preleukaemic clone carrying the inherited PAX5 mutations into B‐ALL are not understood yet; however, the B cell‐specific enzyme AID might be the predominant driver of clonal evolution in human TEL‐AML1 pB‐ALL (Swaminathan et al., 2015).
Besides AID, RAG1‐RAG2 also drives leukaemic clonal evolution after repeated exposure to inflammatory stimuli, paralleling chronic infections in childhood. Abnormal cytokine signalling and repetitive inflammatory stimuli exacerbated susceptibility to genetic lesions during B lymphopoiesis at the transition from the large pre‐BII cell stage to the small pre‐BII cell stage (Swaminathan et al., 2015).
Uncertainties and inconsistencies
One important question in leukaemia genomics is the identity of leukaemia‐initiating mutations that result in preleukaemic clones. Owing to the technical challenge of distinguishing and isolating distinct cancer subclones, many aspects of clonal evolution are poorly understood, including the diversity of different subclones in an individual cancer, the nature of the subclones contributing to relapse, and the identity of precancerous mutations. Studies of paediatric ALL demonstrated that in individual patients there are multiple genetic subclones of leukaemia‐initiating cells, with a complex clonal architecture which limits to build a consistent AOP.
It remains to be demonstrated that subpopulations of acute leukaemia cells exhibit epigenetic heterogeneity, but it seems very likely that epigenetic diversity contributes to subclonal heterogeneity in acute leukaemia. Such epigenetic subclones likely differ in their proliferation, self‐renewal, differentiation and response to therapy, adding an additional dimension to the functional heterogeneity of leukaemia subclones (Jan and Majety, 2013).
A number of questions arise from the Pax5 promoted leukaemogenesis: how relevant is the timing and pattern of infectious exposure for B‐ALL development, how the second hit impacts on the target cell, and what makes Pax5+/− stem/progenitor target cells more vulnerable to malignancy (Hauer et al., 2015).
On the other hand, the adverse effect of pesticides can be produced not only at the MIE level but also by promoting the accumulation of cooperating mutations in the quiescent preleukaemic clones based on a potential oxidative damage in rapidly dividing cells during DNA replications. Additionally, pesticides can exert a developmental immunotoxic effect by the interference of the normal development of the immune cells and their strictly regulated function (Corsini et al., 2013). However, the precise nature of these potential effects is lacking and would impact the current paradigm as pesticides might act at different events of the AOP by means of different toxicological pathways.
References
Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, Titley I, Kearney L, Szczepanski T, Gonzalez D, Ford AM, Potter NE, Greaves M, 2015. Clonal origins of ETV6‐RUNX1+ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia, 29, 839–846.
Aplan PD, 2006. Causes of oncogenic chromosomal translocation. Trends in Genetics, 22, 46–55.
Aranaz P, Miguéliz I, Hurtado C, Erquiaga I, Larráyoz MJ, Calasanz MJ, García‐Delgado M, Novo FJ, Vizmanos JL, 2013. CBL RING finger deletions are common in core‐binding factor acute myeloid leukemias. Leuk Lymphoma, 54, 428–431.
Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N, Bailey S, Vormoor J, Hall AG, Irving JA, 2008. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Research, 68, 6803–6809.
Corsini E, Sokooti M, Galli CL, Moretto A, Colosio C, 2013. Pesticide induced immunotoxicity in humans: a comprehensive review of the existing evidence. Toxicology, 307, 123–135.
de Laurentiis A, Hiscott J, Alcalay M, 2015. The TEL‐AML1 fusion protein of acute lymphoblastic leukemia modulates IRF3 activity during early B‐cell differentiation. Oncogene, 34, 6018–6028.
Driessen EM, van Roon EH, Spijkers‐Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW, 2013. Frequencies and prognostic impact of RAS mutations in MLL‐rearranged acute lymphoblastic leukemia in infants. Haematologica, 98, 937–944.
Eriksson A, Lennartsson A, Lehmann S, 2015. Epigenetic aberrations in acute myeloid leukemia: early key events during leukemogenesis. Experimental Hematology, 43, 609–624.
Ford AM, Palmi C, Bueno C, et al., 2009. The TEL‐AML1 leukemia fusion gene dysregulates the TGF‐beta pathway in early B lineage progenitor cells. Journal of Clinical Investigation, 119, 826–836.
Greaves M, 2006. Infection, immune responses and the etiology of childhood leukemia. Nature Reviews Cancer, 6, 193–203.
Hauer J, Martín‐Lorenzo A, Sánchez‐García I, 2015. Infection causes childhood leukemia. Aging (Albany NY), 7, 607–608.
Jacoby E, Chien CD, Fry TJ, 2014. Murine models of acute leukemia: important tools in current pediatric Leukemia Researchearch. Frontiers in Oncology, 4, 95.
Jan M, Majeti R, 2013. Clonal evolution of acute leukemia genomes. Oncogene, 32, 135–140.
Kantner HP, Warsch W, Delogu A, Bauer E, Esterbauer H, Casanova E, Sexl V, Stoiber D, 2013. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia, 15, 1292–1300.
Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC, 2008. Activation of the JNK pathway promotes phosphorylation and degradation of BimEL–a novel mechanism of chemoresistance in T‐cell acute lymphoblastic leukemia. Carcinogenesis, 29, 544–551.
Ma X, Does MB, Metayer C, Russo C, Wong A, Buffler PA, 2005. Vaccination history and risk of childhood leukaemia. International Journal of Epidemiology, 34, 1100–1109.
Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O'Keefe C, Hsi E, Paquette RL, Kojima S, List AF, Sekeres MA, McDevitt MA, Maciejewski JP, 2009. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. Journal of Clinical Oncology, 27, 6109–6116.
Martín‐Lorenzo A, Hauer J, Vicente‐Dueñas C, Auer F, González‐Herrero I, García‐Ramírez I, Ginzel S, Thiele R, Constantinescu SN, Bartenhagen C, Dugas M, Gombert M, Schäfer D, Blanco O, Mayado A, Orfao A, Alonso‐López D, Rivas Jde L, Cobaleda C, García‐Cenador MB, García‐Criado FJ, Sánchez‐García I, Borkhardt A, 2015. Infection exposure is a causal factor in B‐cell precursor acute lymphoblastic leukemia as a result of Pax5‐inherited susceptibility. Cancer Discovery, 5, 1328–1343.
Mendes RD, Sarmento LM, Cante‐Barrett K, Zuurbier L, Buijs‐Gladdines JGCAM, Povoa V, Smits WK, Abecasis M, Yunes JA, Sonneveld E, Horstmann MA, Pieters R, Barata JT, Meijerink JPP, 2014. PTEN microdeletions in T‐cell acute lymphoblastic leukemia are caused by illegitimate RAG‐mediated recombination events. Blood, 124, 567–578.
Novara F, Beri S, Bernardo ME, Bellazzi R, Malovini A, Ciccone R, Cometa AM, Locatelli F, Giorda R, Zuffardi O, 2009. Different molecular mechanisms causing 9p21 deletions in acute lymphoblastic leukemia of childhood. Journal of Human Genetics, 126, 511–520.
Paulsson K, Lilljebjörn H, Biloglav A, Olsson L, Rissler M, Castor A, Barbany G, Fogelstrand L, Nordgren A, Sjögren H, Fioretos T, Johansson B, 2015. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nature Genetics, 47, 672–676.
Prelle C, Bursen A, Dingermann T, Marschalek R, 2013. Secondary mutations in t(4;11) leukemia patients. Leukemia, 27, 1425–1427.
Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menéndez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukemia. Blood, 126, 2676–2685.
Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, Müschen M, 2015. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nature Immunology, 16, 766–774.
Wiemels J, 2012. Perspectives on the causes of childhood leukemia. Chemico‐Biological Interactions, 196, 59–67.
4th KER: Clonal expansion of leukaemogenic cells (KEup) leading to overt childhood leukaemia (KEdown – AO)
How this Key Event Relationship works
Children with ALL often present with signs and symptoms that reflect bone marrow infiltration with leukaemic blasts and the extent of extramedullary disease spread. The initial manifestations of the disease are based on the expansion of leukaemogenic cells replacing normal blood cells, and involve anaemia, thrombocytopenia and neutropenia with apparent clinical signs and symptoms.
The majority of ALL cases have multiple clones with distinct genetic alterations that influence the response to treatment and the risk of recurrence. Genome‐wide association studies comparing diagnosis versus relapse specimens have shown that both of them share common origin at prediagnosis or clonal ancestry, but show differences in the nature of genetic alterations (Lo Nigro et al., 2013).
Biological stress from postnatal infection in combination with a dysregulated immune response may confer a growth advantage for a preleukaemic clone leading to its rapid expansion and an increased opportunity for the occurrence of a second mutation required for the development of childhood leukaemia (Greaves, 2006).
Childhood leukaemia is a biologically heterogeneous disease represented by distinct clinical and biological subtypes. The disease consists of a multistep process requiring the acquisition of multiple somatic lesions, and the definition of such pathways is being elucidated.
Weight of evidence
Although there is scarce scientific evidence on how leukaemic clones grow and expand, the pathobiology of the disease along with the evolutionary genetic landscape, response to treatment and relapse clearly indicate a causal linkage between the expansion of leukaemic clones and the onset of clinical features.
Biological plausibility
Sequential cooperating mutations in several signalling pathways (i.e., RAS) and cellular processes are selectively produced in any of the in utero subclones and originated from the same preleukaemic clone. Later on, an aberrant inflammatory response abolishes normal haematopoiesis promoting selective expansion of a preleukaemic clone, resulting in stochastic or microenvironment‐derived cooperating drivers towards overt leukaemia.
In the cord blood of healthy newborns the prevalence of a TEL/AML1 translocation is about 1 in 100, while only 1 in 10,000 will later in life develop ALL with this translocation. This clearly indicates a multistep pathogenesis: since at least 99% of the children with this ‘first hit’ will not develop leukaemia, more hits are necessary to develop leukaemia.
Only few children who are born with a chromosomal translocation will develop ALL, proving that these are preleukaemic changes and that leukaemogenesis is multifactorial and depending on multiple consecutive events. The ‘first hit’, most likely acquired during pregnancy, will give rise to preleukaemic cells and clones being more susceptible to additional oncogenic events, the ‘second hit’. Most children with ALL carry 6 up to ~20 different genetic abnormalities in their leukaemia cells.
Biological stress from postnatal infection in combination with a dysregulated immune response may confer a growth advantage for a preleukaemic clone leading to its rapid expansion and an increased opportunity for the occurrence of a second mutation required for the development of childhood leukaemia (Greaves, 2006).
Uncertainties and inconsistencies
The main challenge of developing AOPs for childhood leukaemia is the complex nature of the disease. For example, a tumour suppressor gene could be mutated or transcriptionally inactivated to trigger leukaemogenesis. Different genetic aberrations affect different subtypes of childhood leukaemia (even between cell‐specific B‐cell and T‐cell ALL) as almost all of the evaluated human studies report percentages of a specific mutation found in cohorts, meaning there is no single mutation responsible for the disease.
Whole genome and transcriptome sequencing of three B‐cell precursor patients (of which one carried the TEL‐AML1 translocation and two lacked a known primary genetic aberration and one T‐ALL patient) found that each patient had a unique genome, with a combination of well‐known and previously undetected genomic aberrations (Lindqvist et al., 2015).
References
Greaves M, 2006. Infection, immune responses and the aetiology of childhood leukaemia. Nature Reviews Cancer, 6, 193–203.
Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, Övernäs E, Dahlberg J, Wahlberg P, Henriksson N, Abrahamsson J, Frost BM, Grandér D, Heyman M, Larsson R, Palle J, Söderhäll S, Forestier E, Lönnerholm G, Syvänen AC, Berglund EC, 2015. The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Human Mutation, 36, 118–128.
Lo Nigro L, 2013. Biology of childhood acute lymphoblastic leukemia. Journal of Pediatric Hematology/Oncology, 35, 245–252.
Roganovic J, 2013. Acute lymphoblastic leukemia in children, leukemia. In: Guenova E (ed.). Leukemia, Chapter 2, InTech, pp. 39–74. Available from: http://www.intechopen.com/books/leukemia/acute-lymphoblastic-leukemia-in-children
Overall assessment of the AOP
Childhood leukaemia, the most common cancer affecting children, fits a ‘two‐hit’ cancer model. While the first ‘hit’ occurs in utero during fetal haematopoiesis (chromosomal translocation in a HSPC with a sustainable lifespan and clonal potential to propagate the chimeric gene product), the second hit takes place postnatally and is related to aberrant immune response to delayed infections or other secondary activating mutations both leading to clonal expansion. Studies in archived Guthrie cards suggests the presence of several common chromosomal translocations on neonatal blood spots in children who contract leukaemia later, which retrospectively indicate clearly an in utero origin of the disease (Wiemels et al., 1999). However, chromosomal translocations are insufficient by themselves to cause disease as they are found in approximately 1% of the normal population, a frequency 100 times higher than the prevalence of acute lymphoblastic leukaemia (ALL) (Mori et al., 2002). This fact suggests that the vast majority of preleukaemic clones are self‐limiting and do not result in disease (Pui et al., 2008; Wiemels, 2012). In fact, transgenic mice expressing fusion gene products did not exhibit overt signs of leukaemia (Mori et al., 2002). The variable incubation period and clinical outcome of the disease, and the 10% concordance rate of leukaemia in identical twins harbouring the same genetic abnormalities indicates that additional postnatal events are needed for the development of full‐blown childhood leukaemia (Greaves and Wiemels, 2003).
The Greaves’ multi‐stage model for childhood ALL suggests that there are three critical windows –preconceptional, prenatal, and postnatal–during which exposure to exogenous agents could, but not necessarily, influence leukaemogenesis. Although information is limited, the principal toxic mechanisms of potential leukaemogenic agents (e.g. some pesticides) include excessive generation of oxidative free radicals, which may induce DNA single‐ and double‐strand breaks (DNA‐DSBs) in fetal liver HSPCs. Chromosomal rearrangements (duplications, deletions and translocations) may further occur if genetic lesions are not properly repaired by non‐homologous end‐joining (NHEJ). Although the mechanisms underlying the generation of chromosomal translocations leading to fusion genes are not known, alternative sources of DNA breaks are V(D)J recombination, topoisomerase II cleavable complex formation and abortive apoptosis.
The initiating hit usually occurs in utero and commonly leads to the expression of oncogenic fusion proteins. Subsequent cooperating hits occurring after birth define the disease latency and may be of a genetic, epigenetic or immune nature (i.e. delayed infection‐mediated immune deregulation). However, currently available information does not suggest a strong association of an exogenous agent(s) with a particular exposure window for childhood ALL. Although prenatal initiation of ALL might be a result of spontaneous developmental errors normally occurring through endogenous oxidative stress in the absence of an exogenous DNA‐damaging exposure, the likelihood of this formation may be modified by other factors including exogenous ones. Different mechanisms of cellular responses pertinent to ALL induction are expected for the different classes of agents (e.g. chemicals, radiation and infection) (Kim et al., 2006). The effect of infection is postulated to be caused by a delayed immunological challenge associated with dysregulated proliferative and/or apoptotic stress to the ‘preleukaemic’ bone marrow, indicating a promotional effect of infection on childhood ALL, which could be considered a ‘classical’ second hit, i.e. tumour promotion.
More recently, a 3‐step model of leukaemia pathogenesis has been suggested, which postulates that an initiating genetic lesion (diverse chromosomal translocations leading to gene fusions) confers self‐renewal properties to fetal liver HSPCs. A second lesion, normally affecting essential transcription factors for lymphoid cell development, causes differentiation block at the progenitor cell level. A third class of cooperating mutations accumulate and are needed to fully transform leukaemia cells. These secondary mutations affect pathways such as cell cycle, cytokine receptors and associated kinases, RAS signalling or several other transcription factors or epigenetic regulators (Duque‐Afonso et al., 2015). While mutations of RAS in HSPCs have been demonstrated, K‐RAS mutations are not sufficient to produce overt leukaemia, but requires additional genetic mutation(s) in most likely lineage‐committed progenitors (Zhang et al., 2009). Besides, NOD/SCID mice with transplanted human bone marrow leukaemic blasts at different maturation stages isolated from paediatric ALL patients developed the complete leukaemic phenotype in vivo. This suggests that B precursor blasts at different maturation stages have capability of self‐renewal as a means of maintaining their malignancies in vivo (le Viseur et al., 2008).
Pediatric leukaemia is phenotypically and genetically heterogeneous with an obscure aetiology. The interaction between genetic factors and environmental agents represents a potential aetiological driver. Despite the multifactorial causal mechanism and a heterogeneous biological composition, the timing of environmental exposures and genetic changes associated with childhood leukaemia must be considered (Buffler et al., 2005). However, its genetic diversity limits investigation into the molecular pathogenesis of disease. As a result of the peculiar natural history of childhood leukaemia, direct studies in pregnant women are not possible and there is a need to rely on surrogate in vitro or ex vivo studies or on animal models which entail difficulties in the interpretation and extrapolation of results. Over the last 3 decades, significant progress has been made through the identification of recurrent genetic alterations and translocations in leukaemic blast populations, and their subsequent functional characterisation in cell lines and/or mouse models. Recently, primary human haematopoietic cells have emerged as a complementary means to characterise leukaemic oncogenes (Kennedy and Barabé, 2008). Accordingly, this overall assessment is based largely on empirical evidence found in cases of childhood leukaemia or from cellular and animal models.
B.1. Concordance of dose–response relationship
In contrast to infant leukaemia, the lack of a known aetiological (chemical) agent directly related to the onset of the disease has prevented the conduct of experimental studies in animals, so a dose–response relationship is lacking so far. In addition, there are no adequate experimental systems in which dose–response and response–response relationships can be studied across MIE, KEs and AO.
Conversely, models of radiation‐induced leukaemia risk derived from leukaemia mortality among Japanese survivors of atomic bombs adopted a linear dose–response relationship in the low‐dose (< 100 mGy) region (Wakeford et al., 2010). A dose–response relationship was demonstrated for childhood leukaemia based on number of X‐ray films taken and from the observation that the excess risk was greater among twins for whom X‐ray pelvimetry was far more frequent than among singletons (Boice, 2006).
B.2. Temporal concordance among the MIE, KEs and AO
There is no doubt about temporal concordance among MIE, KEs and AO for childhood leukaemia. Key molecular events leading to childhood leukaemia are chromosomal translocations, and mis‐repaired DNA DSBs are prerequisites for their occurrence. Most of the DNA lesions in fetal liver HSPCs are properly repaired and only persist in case of a failure in the DNA repairing system. Chromosomal translocations ultimately result in the deregulation of key cellular proteins, especially those encoded by proto‐oncogenes and tumour suppressor genes, which are critical functional regulators of cells. Recurrent genetic insults leading to differentiation arrest of HSPCs are needed to drive uncontrolled proliferation and survival of the differentiation‐blocked clone. A study using transgenic mice with the TEL‐AML1 transgene has demonstrated that expression of the fusion gene alone is not sufficient to induce leukaemia, but following prenatal initiation a postnatal second event is necessary for ALL to be manifested (Andreasson et al., 2001).
Regardless of the knowledge gap on the chemical(s) involved in the MIE, and the molecular mechanisms underlying this interaction, it is clear that chromosomal aberrations represent a necessary but not sufficient cause occurring in utero. A second‐hit is required for the expansion of quiescent leukaemic clones and this occurs during postnatal life in a subset of vulnerable children because of an immunological system improperly developed owing to low exposure to common infections early in life.
The separation of a clonal antecedent preleukaemic cell population from frank leukaemic cells has been identified in a monochorionic twin pair with one ‘leukaemic’ and one ‘healthy’ twin (Hong et al., 2008). The ‘healthy’ twin shared TEL‐AML1 fusion transcripts and clonotypic DJ recombination sequences with the ‘leukaemic’ twin. Moreover, modelling the effect of TEL‐AML1 by retroviral transduction in normal cord blood suggested that the founding chromosomal translocation was likely sufficient to induce the preleukaemic population found in the ‘healthy’ twin. A follow‐up study in the same twin pair used genome‐wide copy number alterations (CNA) profiling to identify three potential ‘driver’ CNA in the leukaemic cells. FISH analysis did not detect these three CNA in the ‘healthy’ twin's preleukaemic cells, supporting the hypothesis that the preleukaemic cells diverged genetically after the initiating chromosomal translocation, with subsequent events leading to the clonal evolution of the affected twin's leukaemia (Bateman et al., 2010). A further whole genome sequencing study assessed the genomic profiles of monozygotic twins with ALL, and found that while twins share the first initiating lesion (occurring in utero), each twin then acquire distinct non‐coding mutational changes postnatally that drive leukaemogenesis (Ma et al., 2013).
Current epidemiological studies have limitations for the demonstration of the AOP in pregnant women even in the absence of a clear characterisation of exposure. While experimental models are in accordance with the AOP, the available evidence shows a large number of (epi)genetic and host factors potentially modifying the pathogenesis of childhood leukaemia. The translational biology of B cell precursor ALL has been investigated using comparative genomics and functional approaches (Duque‐Afonso et al., 2015), which has allowed to recapitulate experimentally the multistep pathogenesis of ALL previously inferred from genomic analyses and highlight key cooperating oncogenic pathways.
When known mutations occur in non‐stem cells, they will quickly be lost from the haematopoietic pool due to the natural course of differentiation and cell death. In contrast, a mutation in a stem cell may persist, and the mutated clone may expand, facilitating further clonal progression until a leukaemic stem cell with extensive self‐renewal ability develops (Jan and Majeti, 2013).
Table B.6.
Temporality concordance table
| Model (concentration) | MIE Unknown | KE1 In utero induction of chromosomal translocations | KE2 Differentiation blockage | KE3 Clonal expansion | AO Childhood leukaemia |
|---|---|---|---|---|---|
| Conditional activation of E2A‐PBX1 in the B cell compartment of micea | + | + | + |
ALL development (similar to human E2A‐PBX1+ leukaemias) |
|
| Transplantation of TEL‐AML1 transduced human cord blood cells into NOD/SCID miceb | + |
+ Mice exhibited features of preleukaemic phase of pre‐B cell ALLb |
‐ | ||
| Human peripheral lymphocytes exposed to 0.1–10 μg/mL isofenphos for 1 h | Dose‐dependent damage to chromosomal DNA could lead to genomic instability and leukaemogenesisc | + | ‐ | ‐ | ‐ |
| Human K562 cells exposed to 0.1 μM of diazinon | Hypermethylation of genes involved in cell cycle arrest (cyclin‐dependent kinase inhibitor 1A and CDKN1C) as well as tumour suppressor genes (p53 and PTEN)d | ‐ | ‐ | ‐ | ‐ |
| 1–100 μM chlorpyrifos for up to 24 h | Dose‐ and time‐dependent double‐strand DNA breaks in HSCs (and MLL rearrangements)e | + | ‐ | ‐ | ‐ |
| Human T‐cell leukaemia Jurkat cells exposed to methyl‐pyrazole insecticides for 1 h | Increased double DNA breaks (possibly due to oxidative stress)f | ‐ | ‐ | ‐ | ‐ |
| CEM x 174 cell line, (a hybrid of human T and B cells), exposed to 50 μM heptachlor, chlordane or toxaphene for 24–36 h | Decrease in levels of the tumour suppressors p53 and Rbg , h | ? | ‐ | ‐ | ‐ |
| Human peripheral lymphocytes exposed to a commercial fungicide karathane (dinocap) at 20 μg/mL for 24 h | Increased chromosomal aberrations, formation of sister chromatid exchanges and decreased mitotic indexi | ‐ | ‐ | ‐ | |
| Studies on the Kasumi‐1 cell line, which harbours an AML1–ETO translocation | The differentiation block induced by AML1–ETO is due in part to its ability to physically bind to and inactivate the master myeloid transcription factor PU.1j | ‐ | ‐ |
A conditional E2A‐PBX1 allele was engineered to recombine human PBX1a cDNA into the mouse E2A locus to create an E2A‐PBX1 fusion gene (Duque‐Afonso et al., 2015).
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M and Enver T, 2008. Initiating and cancer‐propagating cells in TEL‐AML1‐associated childhood leukemia. Science, 319, 336–339.
Williams RD, Boros LG, Kolanko CJ, Jackman SM and Eggers TR, 2004. Chromosomal aberrations in human lymphocytes exposed to the anticholinesterase pesticide isofenphos with mechanisms of leukemogenesis. Leukemia Research, 28, 947–958.
Zhang X, Wallace AD, Du P, Lin S, Baccarelli AA, Jiang H, Jafari N, Zheng Y, Xie H, Soares MB, Kibbe WA and Hou L, 2012. Genome‐wide study of DNA methylation alterations in response to diazinon exposure in vitro. Environmental Toxicology and Pharmacology, 34, 959–968.
Lu C, Liu X, Liu C, Wang J, Li C, Liu Q, Li Y, Li S, Sun S, Yan J and Shao J, 2015. Chlorpyrifos Induces MLL Translocations Through Caspase 3‐Dependent Genomic Instability and Topoisomerase II Inhibition in Human Fetal Liver Hematopoietic Stem Cells. Toxicological Sciences, 147, 588–606.
Graillot V, Tomasetig F, Cravedi JP and Audebert M, 2012. Evidence of the in vitro genotoxicity of methyl‐pyrazole pesticides in human cells. Mutation Research‐Genetic Toxicol Environ Mutag, 748, 8–16.
Rought SE, Yau PM, Chuang LF, Doi RH and Chuang RY, 1999. Effect of the chlorinated hydrocarbons heptachlor, chlordane, and toxaphene on retinoblastoma tumor suppressor in human lymphocytes. Toxicology Letters, 104, 127–135.
Rought SE, Yau PM, Schnier JB, Chuang LF and Chuang RY, 1998. The effect of heptachlor, a chlorinated hydrocarbon insecticide on p53 tumor suppressor in human lymphocytes. Toxicology Letters, 94, 29–36.
Celik M, Unal F, Yuzbasioglu D, Ergun MA, Arslan O and Kasap R, 2005. In vitro effect of karathane LC (dinocap) on human lymphocytes. Mutagenesis, 20, 101–104.
Vangala RK, Heiss‐Neumann MS, Rangatia JS, Singh SM, Schoch C, Tenen DG, Hiddemann W and Behre G, 2003. The myeloid master regulator transcription factor PU.1 is inactivated by AML1–ETO in t(8;21) myeloid leukemia. Blood, 101, 270–277.
B.3. Strength, consistency, and specificity of association of AO and MIE
Regarding the experimental models and genome‐wide association studies on childhood leukaemia, strength, consistency and specificity of association of AO and MIE is rather strong in spite of the gap of knowledge on the aetiological factors involved. Although direct observations on the initial in utero MIE are not possible, there is inferential evidence from animal and in vitro cellular studies suggesting strongly that childhood leukaemia recapitulates to a large extent the development of the human disease.
Under the current paradigm, the first initiating oncogenic mutation usually involves structural or numerical chromosomal alterations, impairing normal cell differentiation, while secondary hits more commonly comprise mutations affecting developmentally regulated master transcription factors or membrane‐proximal signalling pathways conferring proliferation and survival advantages to the differentiation‐blocked clone. The development of leukaemia requires the activation of cell proliferation in addition to differentiation blockage.
The consistent finding of a number of chromosomal translocations across studies indicates that they are needed for the development of the disease, although not enough by themselves. There is no alternative explanation for this finding because a reasonable confidence for chance or confounding is lacking. Besides, the identified chromosomal damage (and no other) has to occur in a particular cell (fetal liver HSPCs) and in a particular time window, as otherwise the disease will not develop.
B.4. Weight of Evidence (WoE)
B.4.1. Biological plausibility, coherence, and consistency of the experimental evidence
Table B.7.
Biological plausibility of the KERs; WoE analysis
| 1 Support for biological plausibility of KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Is there a mechanistic (i.e. structural or functional) relationship between KEup and KEdown consistent with established biological knowledge? | Extensive understanding of the KER based on extensive previous documentation and broad acceptance | The KER is plausible based on analogy to accepted biological relationships, but scientific understanding is not completely established | There is empirical support for a statistical association between KEs but the structural or functional relationship between them is not understood | |
|
MIE → KE1 Unknown |
Low | |||
|
KE1 → KE2 Chromosomal translocations lead to differentiation arrest of HSPCs in utero |
Strong |
Rationale: there is convincing evidence to indicate that chromosomal translocation leading to formation of TEL‐AML1 is the initiating event in most B‐precursor ALL, the most frequent childhood leukaemia DNA sequencing analysis has revealed that TEL‐AML1 translocations occur by imprecise and error‐prone DNA repair process after DNA double‐strand breaks and not by aberrant topoisomerase activity (Wiemels and Greaves, 1999) Studies on identical twins and neonatal blood samples strongly indicate an in utero occurrence of the KER. The TEL‐AML1 fusion gene usually arises before birth, inducing persistent self‐renewing of pro‐B cells in mice (covert preleukaemic clone)a Aberrant proteins produced by fusion genes are responsible of cell differentiation arrest |
||
|
KE2 → KE3 Cooperative mutations and Delayed infections in HSPCs with a differentiation blockage lead to clonal expansion |
Moderate |
Rationale: Covert preleukaemic clones may convert to precursor B‐cell leukaemia following the accumulation of secondary genetic hits. a TEL‐AML1+ cells differentiate terminally in the long term, providing a ‘window’ period that may allow secondary genetic hits to accumulate and lead to leukaemiaa In childhood leukaemia, altered differentiation and self‐renewal of haematopoietic stem cells or early progenitor cells might occur due to the presence of chimeric transcription factors that alter the regulation of genes required for the proper differentiation of haematopoietic stem cells (Pui et al., 2004) There is a general understanding of the mechanisms leading to differentiation arrest and clonal expansion and there is evidence that the principal fusion product TEL‐AML1 protein harbours the necessary properties to execute the necessary pathways. However, the inability of available in vivo models to recapitulate the whole AOP process is an important limitation The longer latency observed in childhood leukaemia unequivocally indicates that the initiating chromosomal translocation itself is unlikely to convert a preleukaemic clone into an overt disease, thus suggesting the need for secondary cooperating (epi)‐genetic events |
||
|
KE3 → AO Clonal expansion leads to childhood leukaemia |
Strong | Rationale: The basic processes underlying overt leukaemia development are well understood and accepted | ||
Tsuzuki S, Seto M, 2013. TEL (ETV6)‐AML1 (RUNX1) initiates self‐renewing fetal pro‐B cells in association with a transcriptional program shared with embryonic stem cells in mice. Stem Cells, 31, 236–247.
B.4.2. Essentiality
Table B.8.
Essentiality of the KEs; WoE analysis
| 2 Support for essentiality of KEs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Are downstream KEs and/or the AO prevented if an upstream KE is blocked? | Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs (e.g. stop/reversibility studies, antagonism, knock out models, etc.) | Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE leading to increase in KEdown or AO | No or contradictory experimental evidence of the essentiality of any of the KEs | |
|
MIE Unknown |
||||
|
KE1 In utero chromosomal translocations |
Strong | Experimental models and genome wide association studies have consistently demonstrated that in the absence of chromosomal damage there is no chance for the leukaemia to occur. The reverse is also true, as the presence of fusion genes per se are not enough for fully developing the disease | ||
|
KE2 Blockage of HSPCs differentiation in utero |
The developmental block observed in Pax5‐deficient leukaemia cells can be reversed on restoration of Pax5 expression, suggesting that the reduction in Pax5 function results in a reversible disruption of differentiation. Transgenic RNAi can reversibly suppress endogenous Pax5 expression in the haematopoietic compartment of mice, which cooperates with activated signal transducer and activator of transcription 5 (STAT5) to induce B‐ALL (Liu et al., 2016) | |||
|
KE3 Expansion of preleukaemic clones as a result of cooperative mutations and delayed infections |
There are no relevant inhibitors for the clonal expansion of preleukaemic clones | |||
B.4.3. Empirical support
Table B.9.
Essentiality of the KERs; WoE analysis
| 3 Empirical support for KERs | Defining question | High (strong) | Moderate | Low (weak) |
|---|---|---|---|---|
| Does the empirical evidence support that a change in the KEup leads to an appropriate change in the KEdown? Does KEup occur at lower doses and earlier time points than KEdown and is the incidence of KEup higher than that for KEdown? Are inconsistencies in empirical support cross taxa, species and stressors that don't align with expected pattern of hypothesised AOP? | Multiple studies showing dependent change in both exposure to a wide range of specific stressors (extensive evidence for temporal, dose–response and incidence concordance) and no or few critical data gaps or conflicting data | Demonstrated dependent change in both events following exposure to a small number of specific stressors and some evidence inconsistent with expected pattern that can be explained by factors such as experimental design, technical considerations, differences among laboratories, etc | Limited or no studies reporting dependent change in both events following exposure to a specific stressor (i.e. endpoints never measured in the same study or not at all); and/or significant inconsistencies in empirical support across taxa and species that don't align with expected pattern for hypothesised AOP | |
| MIE → KE1 | Low | |||
|
KE1 → KE2 Chromosomal translocations lead to differentiation arrest of HSPCs in utero |
Strong |
Rationale: A transgenic zebrafish model expressing TEL‐AML1‐positive ALL either ubiquitously or in lymphoid progenitors showed B‐cell differentiation arrest. TEL‐AML1 expression in all lineages, but not lymphoid‐restricted expression, led to progenitor cell expansion that evolved into oligoclonal B‐lineage ALL in 3% of the transgenic zebrafisha The strongest evidence comes from experimental models and genome wide association studies |
||
|
KE2 → KE3 Cooperative mutations and Delayed infections in HSPCs with a differentiation blockage lead to clonal expansion |
Moderate |
Rationale: In principle, there are a large number of factors and pathways linking the fusion product and differentiation blockage with clonal expansion, recurrent cooperative mutations and delayed infections |
||
|
KE3 → AO Clonal expansion leads to childhood leukaemia |
Moderate |
Rationale: The accumulation of recurrent mutations in preleukaemic clones and dysfunction of the immune system following delayed infections have been linked to the development of childhood leukaemia |
||
Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD, 2006. TEL‐AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences USA, 103, 15166–15171.
B.5. Uncertainties and Inconsistencies
Although the causes of the specific genetic events leading to ALL formation are not known, numerous exposure‐dependent risk factors for childhood ALL have been proposed, including pesticide exposure. These risk factors can be classified based on their relationship to potentially critical exposure windows (i.e. preconceptional, prenatal, and postnatal stages). [KER1]
As in utero evidence is difficult to obtain in humans, mouse models are used instead.
Limitations of transgenic animal models faithfully recapitulating all the aspects of human pB‐ALL need to be recognised. The reproducibility and accuracy of these models for human responses have yet to be validated, so their application to elucidate postnatal exposure effects of exogenous agents on childhood ALL should be cautiously approached. Mouse models in which the initiating oncogenic alteration(s) is not directed to the right cell‐of‐origin are unlikely to accurately recapitulate the aetiology of the human disease and will originate an inaccurate model of human leukaemia (Hauer et al., 2014).
One important question in leukaemia genomics is the identity of leukaemia‐initiating mutations that result in preleukaemic clones. Owing to the technical challenge of distinguishing and isolating distinct cancer subclones, many aspects of clonal evolution are poorly understood, including the diversity of different subclones in an individual cancer, the nature of the subclones contributing to relapse, and the identity of precancerous mutations. Studies of paediatric ALL demonstrated that in individual patients there are multiple genetic subclones of leukaemia‐initiating cells, with a complex clonal architecture which limits development of a consistent AOP. [KER3]
The main challenge in developing AOPs for childhood leukaemia is the complex nature of the disease. For example, a tumour suppressor gene could be mutated or transcriptionally inactivated to trigger leukaemogenesis. Different genetic aberrations affect different subtypes of childhood leukaemia (even between cell‐specific B‐cell and T‐cell ALL) as almost all of the evaluated human studies report percentages of a specific mutation found in cohorts, meaning there is no single mutation responsible for the disease.
It remains to be demonstrated whether subpopulations of acute leukaemia cells exhibit epigenetic heterogeneity, but it seems very likely that epigenetic diversity contributes to subclonal heterogeneity in acute leukaemia. Such epigenetic subclones likely differ in their proliferation, self‐renewal, differentiation and response to therapy, adding an additional dimension to the functional heterogeneity of leukaemia subclones (Jand and Majety, 2013). [Clones]
While there are emerging studies implicating epigenetics as an influential factor in childhood leukaemia, it is not clear at which point epigenetics influences childhood leukaemia pathogenesis, i.e. as a MIE or later as an intermediate event. For example, DNA hypermethylation of tumour suppressor genes leading to their decreased expressions can occur early in childhood leukaemia pathogenesis to facilitate the growth of leukaemic cells, or altered expressions of microRNAs might be influenced by earlier events (e.g.), resulting in alterations in haematopoiesis or inhibition of apoptosis. Therefore, the putative relevance of epigenetics needs to be further evaluated before it can be considered in the AOP development for childhood leukaemia. [Epigenetics]
A detailed understanding of leukaemogenesis requires the development of experimental models that can accurately model this process. As a complement to work in cell lines and in mice, there is a need for oncogenes and chromosomal translocations to be studied in the appropriate cellular context, that of primary human haematopoietic cells. Retroviral‐mediated transduction of primary human haematopoietic cells followed by their transplantation in vivo has emerged as a feasible approach to study the process of human leukaemogenesis (Kennedy and Barabe, 2008). [Experimental models]
B.6. Quantitative Considerations
The WOE analysis indicates that some KEs and KERs lack especially experimental evidence, but overall the analysis supports the qualitative AOP. The strong element in the development of the qualitative AOP is that it can partially be based on animal models recapitulating many crucial features of childhood leukaemia and genome‐wide association studies. The absence of a MIE clearly defined is a major limitation. The lack of sufficient experimental data and uncertainties in quantitative information from available studies makes it difficult to build convincing dose (concentration)‐response and response–response relationships and to identify possible practical thresholds for stressors.
There is a need for an adequate and robust experimental model system where relationships between doses, concentrations and responses can be evaluated within a temporal framework of the AOP.
B.7. Applicability of the AOP
Even in the absence of a mechanistic understanding in regard to the MIE, the proposed AOP might be applied to pesticide‐related leukaemia not only in children but also in adults, although in the latter case chromosomal translocations are acquired in the postnatal life (the persistence of prenatal chromosomal translocations does not play a role in adult leukaemogenesis). Based on the rather consistent epidemiological evidence on human exposure to pesticides and the risk of childhood leukaemia, it is possible that at least a subset of acute childhood leukaemias may be caused by environmental exposure to pesticides. Consequently, the proposed AOP may be partially applicable to these situations, but should be supported with an understanding of the mechanistic processes underlying the direct or indirect interaction of pesticides (or their metabolites) with DNA.
References
Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG, 2001. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genetics and Cytogenetics, 130, 93–104.
Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M et al., 2010. Acquisition of genome‐wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood, 115, 3553–3558.
Boice JD, 2006. Ionizing radiation. In: Schottenfeld D, Fraumeni JF Jr., (eds.). Cancer Epidemiology and Prevention, 3rd Edition, Oxford, pp. 259–293.
Buffler PA, Kwan ML, Reynolds P, Urayama KY, 2005. Environmental and genetic risk factors for childhood leukemia: appraising the evidence. Cancer Investigation, 23, 60–75.
Duque‐Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, Iwasaki M, Cleary ML, 2015. Comparative genomics reveals multistep pathogenesis of E2A‐PBX1 acute lymphoblastic leukemia. Journal of Clinical Investigation, 125, 3667–3680.
Greaves MF, Wiemels J, 2003. Origins of chromosome translocations in childhood leukaemia. Nature Reviews Cancer, 3, 639–649.
Hauer J, Borkhardt A, Sánchez‐García I, Cobaleda C, 2014. Genetically engineered mouse models of human B‐cell precursor leukemias. Cell Cycle, 13, 2836–2846.
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S et al., 2008. Initiating and cancer‐propagating cells in TEL‐AML1‐associated childhood leukemia. Science, 319, 336–339.
Jan M, Majeti R, 2013. Clonal evolution of acute leukemia genomes. Oncogene, 32, 135–140.
Kennedy JA, Barabé F, 2008. Investigating human leukemogenesis: from cell lines to in vivo models of human leukemia. Leukemia, 22, 2029–2040.
le Viseur C, Hotfilder M, Bomken S, Wilson K, Roettgers S, Schrauder A, Rosemann A, Irving J, Stam RW, Shultz LD, Harbott J, Juergens H, Schrappe M, Pieters R, Vormoor J, 2008. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell, 14, 47–58.
Ma Y, Dobbins SE, Sherborne AL, Chubb D, Galbiati M, Cazzaniga G, Micalizzi C, Tearle R, Lloyd AL, Hain R, Greaves M, Houlston RS, 2013. Developmental timing of mutations revealed by whole‐genome sequencing of twins with acute lymphoblastic leukemia. Proceedings of the National Academy of SciencesU S A, 110, 7429–7433.
Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M, 2002. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proceedings of the National Academy of SciencesU S A, 99, 8242–8247.
Moriya K, Suzuki M, Watanabe Y, Takahashi T, Aoki Y, Uchiyama T, Kumaki S, Sasahara Y, Minegishi M, Kure S, Tsuchiya S, Sugamura K, Ishii N, 2012. Development of a multi‐step leukemogenesis model of mll‐rearranged leukemia using humanized mice. Public Library of Science (PLoS ONE), 7, e37892.
Wakeford R, Little MP, Kendall GM, 2010. Risk of childhood leukemia after low‐level exposure to ionizing radiation. Expert Review of Hematology, 3, 251–254.
Wiemels J, 2012. Perspectives on the causes of childhood leukemia. Chemico‐Biological Interactions, 196, 59–67.
Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF, 1999. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet, 354, 1499–1503.
Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, Fleming MD, 2009. Oncogenic Kras‐induced leukemogeneis: hematopoietic stem cells as the initial target and lineage‐specific progenitors as the potential targets for final leukemic transformation. Blood, 113, 1304–1314.
Suggested citation: EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues) , Ockleford C, Adriaanse P, Berny P, Brock T, Duquesne S, Grilli S, Hernandez‐Jerez AF, Bennekou SH, Klein M, Kuhl T, Laskowski R, Machera K, Pelkonen O, Pieper S, Smith R, Stemmer M, Sundh I, Teodorovic I, Tiktak A, Topping CJ, Wolterink G, Angeli K, Fritsche E, Hernandez‐Jerez AF, Leist M, Mantovani A, Menendez P, Pelkonen O, Price A, Viviani B, Chiusolo A, Ruffo F, Terron A and Bennekou SH, 2017. Scientific Opinion on the investigation into experimental toxicological properties of plant protection products having a potential link to Parkinson's disease and childhood leukaemia. EFSA Journal 2017;15(3):4691, 325 pp. doi: 10.2903/j.efsa.2017.4691
On request from EFSA, Question No EFSA‐Q‐2014‐00480, adopted on 14 December 2016.
Requestor: European Food Safety Authority
Question number: EFSA‐Q‐2014‐00490
Acknowledgements: The Panel wishes to thank Angeli Karine, Fritsche Ellen, Hernandez‐Jerez Antonio F., Bennekou Susanne Hougaard, Leist Marcel, Mantovani Alberto, Menendez Pablo, Pelkonen Olavi, Price Anna and Viviani Barbara for the preparatory work on this scientific opinion, the hearing expert: Tschudi‐Monnet Florianne and EFSA staff: Terron Andrea, Chiusolo Arianna and Ruffo Federica for the support provided to this scientific opinion.
Adopted: 14 December 2016
Reproduction of the following images is prohibited and permission must be sought directly from the individual copyright holders: Figures A.3, A.4, A.6, A.7, A.8, A.9, A.10, A.11, A.12, A.13, A.14, A.15, A.16, A.17, A.18, A.21, A.22, A.24, A.25, A.26, A.27, A.28, A.29, A.30, A.31 and B.3.
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1190/full
Notes
Hypothetically, based on linear extrapolation from the dose of 10 mg/kg, the concentration would be of the order of 0.5 µM.
References
- Aguzzi A, Barres BA and Bennett ML, 2013. Microglia: scapegoat, saboteur, or something else? Science, 339, 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alépée N, Bahinski A, Daneshian M, De Wever B, Fritsche E, Goldberg A, Hansmann J, Hartung T, Haycock J, Hogberg H, Hoelting L, Kelm JM, Kadereit S, McVey E, Landsiedel R, Leist M, Lübberstedt M, Noor F, Pellevoisin C, Petersohn D, Pfannenbecker U, Reisinger K, Ramirez T, Rothen‐Rutishauser B, Schäfer‐Korting M, Zeilinger K and Zurich MG, 2014. State‐of‐the‐art of 3D cultures (organs‐on‐a‐chip) in safety testing and pathophysiology. ALTEX, 31, 441–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AK, Ma J, Wang J, et al., 2015. St. Jude Children's Research Hospital and Washington University Pediatric Cancer Genome Project . The landscape of somatic mutations in infant MLL‐rearranged acute lymphoblastic leukemias. Nature Genetics, 47, 330–337. doi: 10.1038/ng.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ap Rhys CM and Bohr VA, 1996. Mammalian DNA repair responses and genomic instability. EXS, 77, 289–305. [DOI] [PubMed] [Google Scholar]
- Bailey HD, Fritschi L, Infante‐Rivard C, Glass DC, Miligi L, Dockerty JD, Lightfoot T, Clavel J, Roman E, Spector LG, Kaatsch P, Metayer C, Magnani C, Milne E, Polychronopoulou S, Simpson J, Rudant J, Sidi V, Rondelli R, Orsi L, Kang AY, Petridou E and Schüz J, 2014. Parental occupational pesticide exposure and the risk of childhood leukemia in the offspring: findings from the childhood leukemia international consortium. International Journal of Cancer, 135, 2157–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey HD, Infante‐Rivard C, Metayer C, Clavel J, Lightfoot T, Kaatsch P, Roman E, Magnani C, Spector LG, Th Petridou E, Milne E, Dockerty JD, Miligi L, Armstrong BK, Rudant J, Fritschi L, Simpson J, Zhang L, Rondelli R, Baka M, Orsi L, Moschovi M, Kang AY and Schüz J, 2015. Home pesticide exposures and risk of childhood leukemia: findings from the childhood leukemia international consortium. International Journal of Cancer, 137, 2644–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer NV and Leist M, 2014. Epigenetics and transcriptomics to detect adverse drug effects in model systems of human development. Basic & Clinical Pharmacology & Toxicology, 115, 59–68. [DOI] [PubMed] [Google Scholar]
- Baltazar MT, Dinis‐Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA and Carvalho F, 2014. Pesticides exposure as etiological factors of Parkinson's disease and other neurodegenerative diseases–a mechanistic approach. Toxicology Letters, 230, 85–103. doi: 10.1016/j.toxlet.2014.01.039 [DOI] [PubMed] [Google Scholar]
- Bardini M, Spinelli R, Bungaro S, Mangano E, Corral L, Cifola I, Fazio G, Giordan M, Basso G, De Rossi G, Biondi A, Battaglia C and Cazzaniga G, 2010. DNA copy‐number abnormalities do not occur in infant ALL with t(4;11)/MLL‐AF4. Leukemia, 24, 169–176. doi: 10.1038/leu.2009.203 Epub 2009 Nov 12. [DOI] [PubMed] [Google Scholar]
- Bardini M, Galbiati M, Lettieri A, Bungaro S, Gorletta TA, Biondi A and Cazzaniga G, 2011. Implementation of array based whole‐genome high‐resolution technologies confirms the absence of secondary copy‐number alterations in MLL‐AF4‐positive infant ALL patients. Leukemia, 25, 175–178. doi: 10.1038/leu.2010.232 [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV and Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Neuroscience, 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Blank T and Prinz M, 2013. Microglia as modulators of cognition and neuropsychiatric disorders. Glia, 61, 62–70. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H and Del Tredici K, 2004. Stages in the development of Parkinson's disease‐related pathology. Cell and Tissue Research, 121–124. [DOI] [PubMed] [Google Scholar]
- Braak H and Del Tredici K, 2009. Neuroanatomy and pathology of sporadic Parkinson's disease. Advances in Anatonomy Embryology Cell Biology, 201, 1–119. [PubMed] [Google Scholar]
- Bolon B, Garman RH, Pardo ID, Jensen K, Sills RC, Roulois A, Radovsky A, Bradley A, Andrews‐Jones L, Butt M and Gumprecht L, 2012. STP Position Paper: recommended practices for sampling and processing the nervous system (brain, spinal cord, nerve and eye) during nonclinical general toxicity studies. Toxicologic Pathology, 41, 1028–1048. [DOI] [PubMed] [Google Scholar]
- Boros LG and Williams RD, 2001. Isofenphos induced metabolic changes in K562 myeloid blast cells. Leukemia Research, 25, 883–890. [DOI] [PubMed] [Google Scholar]
- Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, Cook AR, Botham PA and Smith LL, 2013. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology, 37, 1–14. doi: 10.1016/j.neuro.2013.03.005 [DOI] [PubMed] [Google Scholar]
- Breckenridge CB, Berry C, Chang ET, Sielken RL Jr and Mandel JS, 2016. Association between Parkinson's disease and cigarette smoking, rural living, well‐water consumption, farming and pesticide use: systematic review and meta‐analysis. PLoS ONE, 11, e0151841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC and Bal‐Price A, 2003. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Molecular Neurobiology, 27, 325–355. [DOI] [PubMed] [Google Scholar]
- Bueno C, Catalina P, Melen GJ, Montes R, Sánchez L, Ligero G, García‐Pérez JL and Menendez P, 2009. Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis, 30, 1628–1637. doi: 10.1093/carcin/bgp169 Epub 2009 Jul 8. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Tapias VM, Mee Na H, Honick AS, Drolet RE and Greenamyre JT, 2009. A highly reproducible rotenone model of Parkinson's disease. Neurobiology of Diseases, 34, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Thrash JC and Walter B, 2006. The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clinical Neuroscience Research, 6, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik M, Unal F, Yuzbasioglu D, Ergun MA, Arslan O and Kasap R, 2005. In vitro effect of karathane LC (dinocap) on human lymphocytes. Mutagenesis, 20, 101–104. [DOI] [PubMed] [Google Scholar]
- Cereda E, Cilia R, Klersy C, Siri C, Pozzi B, Reali E, Colombo A, Zecchinelli AL, Mariani CB, Tesei S, Canesi M, Sacilotto G, Meucci N, Zini M, Isaias IU, Barichella M, Cassani E, Goldwurm S and Pezzoli G, 2016. Dementia in Parkinson's disease: is male gender a risk factor? Parkinsonism & Related Disorders, 26, 67–72. doi: 10.1016/j.parkreldis.2016.02.024 Epub 2016 Mar 2 [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR and Strijbos PJLM, 2000. Fractalkine cleavage from neuronal membranes represents an acute event in inflammatory response to excitotoxic brain damage. The Journal of Neuroscience, 20, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Chan NL and Hsieh TS, 2013. New mechanistic and functional insights into DNA topoisomerases. Annual Review of Biochemistry, 82, 139–170. [DOI] [PubMed] [Google Scholar]
- Chen HM, Lee YH and Wang YJ, 2015. ROS‐triggered signaling pathways involved in the cytotoxicity and tumour promotion effects of pentachlorophenol and tetrachlorohydroquinone. Chemical Research in Toxicology, 28, 339–350. [DOI] [PubMed] [Google Scholar]
- Choi J, Polcher A and Joas A, 2016. Systematic literature review on Parkinson's disease and Childhood Leukaemia and mode of actions for pesticides. EFSA supporting publication 2016:EN‐955, 256 pp. Available online: http://www.efsa.europa.eu/publications
- Cicchetti F, Drouin‐Ouellet J and Gross RE, 2009. Environmental toxins and Parkinson's disease: what have we learned from pesticide‐induced animal models? Trends in Pharmacological Sciences, 30, 475–483. doi: 10.1016/j.tips.2009.06.005 [DOI] [PubMed] [Google Scholar]
- Claycomb KI, Johnson KM, Winokur PN, Sacino AV and Crocker SJ, 2013. Astrocyte regulation of CNS inflammation and remyelination. Brain Sciences, 3, 1109–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier ZA, Gust KA, Gonzalez‐Morales B, Gong P, Wilbanks MS, Linkov I and Perkins EJ, 2016. A weight of evidence assessment approach for adverse outcome pathways. Regulatory Toxicology and Pharmacology, 75, 46–57. doi: 10.1016/j.yrtph.2015.12.014 [DOI] [PubMed] [Google Scholar]
- Cowell IG and Austin CA, 2012. Mechanism of generation of therapy related leukaemia in response to anti‐topoisomerase II agents. International Journal of Environmental Research and Public Health, 9, 2075–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshian M, Busquet F, Hartung T and Leist M, 2015. Animal use for science in Europe. ALTEX, 32, 261–274. [DOI] [PubMed] [Google Scholar]
- Degli Espositi M, 1998. Inhibitors of NADH–ubiquinone reductase: an overview. Biochimica et Biophysica Acta (BBA) – Bioenergetics, 1364, 222–235. [DOI] [PubMed] [Google Scholar]
- Dickinson WD, 2012. Parkinson's disease and Parkinsonism: neuropathology. Cold Spring Harbor Perspectives in Medicine, 2. pii: a009258. doi: 10.1101/cshperspect.a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbins SE, Sherborne AL, Ma YP, Bardini M, Biondi A, Cazzaniga G, Lloyd A, Chubb D, Greaves MF and Houlston RS, 2013. The silent mutational landscape of infant MLL‐AF4 pro‐B acute lymphoblastic leukemia. Genes Chromosomes Cancer, 52, 954–960. doi: 10.1002/gcc.22090 [DOI] [PubMed] [Google Scholar]
- Dong Y and Benveniste EN, 2001. Immune function of astrocytes. Glia, 36, 180–190. [DOI] [PubMed] [Google Scholar]
- Drechsel DA and Patel M, 2008. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson's disease. Free Radical Biology and Medicine, 44, 1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastmond DA, 1997. Chemical and Radiation Leukaemogenesis in Humans and Rodents and the Value of Rodent Models for Assessing Risks of Lymphohaematopoietic Cancers. U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment, Washington Office, Washington, DC, EPA/600/R‐97/090. [Google Scholar]
- Efremova L, Schildknecht S, Adam M, Pape R, Gutbier S, Hanf B, Bürkle A and Leist M, 2015. Prevention of the degeneration of human dopaminergic neurons in an astrocyte co‐culture system allowing endogenous drug metabolism. British Journal of Pharmacology, 172, 4119–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova L, Chovancova P, Adam M, Gutbier S, Schildknecht S and Leist M, 2016. Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Archives of Toxicology, 91(1):231–246. doi: 10.1007/s00204-016-1702-2. Epub 2016 Apr 6. [DOI] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. Available online: http://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/2379.pdf [Google Scholar]
- Emerenciano M, Koifman S and Pombo‐de‐Oliveira MS, 2007. Acute leukemia in early childhood. Brazilian Journal of Medical and Biological Research, 40, 749–760. [DOI] [PubMed] [Google Scholar]
- Ernst P, Wang J and Korsmeyer SJ, 2002. The role of MLL in hematopoiesis a nd leukemia. Current Opinion in Hematology, 2002, 282–287. [DOI] [PubMed] [Google Scholar]
- Esperanza M, Cid Á, Herrero C and Rioboo C, 2015. Acute effects of a prooxidant herbicide on the microalga Chlamydomonas reinhardtii: screening cytotoxicity and genotoxicity endpoints. Aquatic Toxicology, 165, 210–221. [DOI] [PubMed] [Google Scholar]
- Ezoe S, 2012. Secondary leukaemia associated with the anti‐cancer agent, etoposide, a topoisomerase II inhibitor. International Journal of Environmental Research and Public Health, 9, 2444–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JD, Couto AC, Pombo‐de‐Oliveira MS and Koifman S; Brazilian Collaborative Study Group of Infant Acute Leukemia , 2013. In utero pesticide exposure and leukemia in Brazilian children < 2 years of age. Environmental Health Perspectives, 121, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O'Donnell KC, Barnhill L, Casida JE, Cockburn M, Sagasti A, Stahl MC, Maidment NT, Ritz B and Bronstein JM, 2013. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proceedings of the National Academy of Sciences USA, 110, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Cockburn M, Ritz B and Bronstein JM, 2014. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology, 82, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Cheng CW and Wu RM, 2005. Pesticides exposure and genetic polymorphism of paraoxonase in the susceptibility of Parkinson's disease. Acta Neurologica Taiwanica, 14, 55–60. [PubMed] [Google Scholar]
- Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti C, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG and Paparelli A, 2003. Fine structure and mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. The Journal of Neuroscience, 23, 8955–8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Li SM, Rodriguez‐Rocha H, Burns M and Panayiotidis MI, 2010. Molecular mechanisms of pesticide‐induced neurotoxicity: relevance to Parkinson's disease. Chemico‐Biological Interactions, 188, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H and Balling R. 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49, 88–102. doi: 10.1007/s12035-013-8489-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong M, Tanner CM, Goldman SM, Bhudhikanok GS, Blair A, Chade A, Comyns K, Hoppin JA, Kasten M, Korell M, Langston JW, Marras C, Meng C, Richards M, Ross GW, Umbach DM, Sandler DP and Kamel F, 2015. Protective glove use and hygiene habits modify the associations of specific pesticides with Parkinson's disease. Environment International, 75, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong CE, Marsillach J, Jarvik GP and Costa LG, 2016. Paraoxonases‐1, ‐2 and ‐3: what are their functions? Chemico‐Biological Interactions, 25;259(Pt B):51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC and Gage FH, 2010. Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB and Streit WJ, 1990. Microglia: immune network in the CNS. Brain Pathology, 1, 2–5. [DOI] [PubMed] [Google Scholar]
- Greaves M, 2006. Infection, immune responses and the aetiology of childhood leukaemia. Nature Reviews Cancer, 6, 193–203. [DOI] [PubMed] [Google Scholar]
- Graillot V, Tomasetig F, Cravedi JP and Audebert M, 2012. Evidence of the in vitro genotoxicity of methyl‐pyrazole pesticides in human cells. Mutation Research: Genetic Toxicology and Environmental Mutagenesis, 748, 8–16. [DOI] [PubMed] [Google Scholar]
- Greaves M, 2002. Childhood leukaemia. BMJ, 324, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, 2006. Infection, immune responses and the aetiology of childhood leukaemia. Nature Reviews Cancer, 6, 193–203. [DOI] [PubMed] [Google Scholar]
- Guanggang X, Diqiu L, Jianzhong Y, Jingmin G, Huifeng Z, Mingan S and Liming T, 2013. Carbamate insecticide methomyl confers cytotoxicity through DNA damage induction. Food and Chemical Toxicology, 53, 352–358. [DOI] [PubMed] [Google Scholar]
- Hatcher JM, Pennell KD and Miller GW, 2008. Parkinson's disease and pesticides: a toxicological perspective. Trends in Pharmacological Sciences, 29, 322–329. ISSN: 0165‐6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjärn M, Schrattenholz A, Pörzgen P and Leist M, 2009. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX, 26, 83–94. [DOI] [PubMed] [Google Scholar]
- Hernández AF and Menéndez P, 2016. Linking pesticide exposure with paediatric leukaemia: potential underlying mechanisms. International Journal of Molecular Sciences, 17. pii: E461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DG, Reed X and Singleton AB, 2016b. Genetics in Parkinson disease: Mendelian versus non‐Mendelian inheritance. Journal of Neurochemistry, 2/1(e44). in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández AF, González‐Alzaga B, López‐Flores I and Lacasaña M, 2016a. Systematic reviews on neurodevelopmental and neurodegenerative disorders linked to pesticide exposure: methodological features and impact on risk assessment. Environment International, 92–93, 657–79. in press [DOI] [PubMed] [Google Scholar]
- Hines CJ, Deddens JA, Coble J, Kamel F and Alavanja MC, 2011. Determinants of captan air and dermal exposures among orchard pesticide applicators in the Agricultural Health Study. Annals of Occupational Hygiene, 55, 620–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Yang J, Ning J, Wang M and Song Q, 2015. Atrazine triggers DNA damage response and induces DNA double‐strand breaks in MCF‐10A cells. International Journal of Molecular Sciences, 16, 14353–14368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irons RD, 1991. Blood and bone marrow In: Rousseaux CG. and Haschek WM. (eds.). Handbook of Toxicologic Pathology. Academic Press, San Diego: pp. 389–419. [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ and Popovich PG, 2009. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. Journal of Neuroscience, 29, 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen TB, Keller DA, Sander M, Carney EW, Doerrer NG, Eaton DL, Fitzpatrick SC, Hastings KL, Mendrick DL, Tice RR, Watkins PB and Whelan M, 2015. FutureTox II: in vitro data and in silico models for predictive toxicology. Toxicological Sciences, 143, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD and Harry GJ, 2011. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International Journal of Environmental Research and Public Health, 8, 2980–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryston TB, Georgiev AB, Pissis P and Georgakilas AG, 2011. Role of oxidative stress and DNA damage in human carcinogenesis. Mutation Research, 711, 193–201. [DOI] [PubMed] [Google Scholar]
- Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R and Droller D, 2005. Early environmental origins of neurodegenerative disease in later life. Environmental Health Perspectives, 113, 1230–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA and Karluk D, 1999. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine exposure. Annals of Neurology, 46, 598–605. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang Q and Dai S, 2003. Impact of aldicarb and its complex pollution on DNA of zebrafish embryo. Ying Yong Sheng Tai Xue Bao, 14, 982–984. [PubMed] [Google Scholar]
- Li A, Levine T, Burns CJ and Anger WK, 2012. Integration of epidemiology and animal neurotoxicity data for risk assessment. Neurotoxicology, 33, 823–832. [DOI] [PubMed] [Google Scholar]
- Li H, Marple T and Hasty P, 2013. Ku80‐deleted cells are defective at base excision repair. Mutation Research, 745–746, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Sun B, Clewell RA, Adeleye Y, Andersen ME and Zhang Q, 2014. Dose‐response modeling of etoposide‐induced DNA damage response. Toxicological Sciences, 137, 371–336. [DOI] [PubMed] [Google Scholar]
- Logroscino G, 2005. The role of early‐life environmental risk factors in Parkinson disease: what is the evidence? Environmental Health Perspectives, 113, 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Liu X, Liu C, Wang J, Li C, Liu Q, Li Y, Li S, Sun S, Yan J and Shao J, 2015. Chlorpyrifos induces MLL translocations through Caspase 3‐dependent genomic instability and topoisomerase II inhibition in human foetal liver haematopoietic stem cells. Toxicological Sciences, 147, 588–606. [DOI] [PubMed] [Google Scholar]
- Mandel JS, Adami HO and Cole P, 2012. Paraquat and Parkinson's disease: an overview of the epidemiology and a review of two recent studies. Regulatory Toxicology and Pharmacology, 62, 385–392. [DOI] [PubMed] [Google Scholar]
- Manthripragada AD, Costello S, Cockburn MG, Bronstein JM and Ritz B, 2010. Paraoxonase‐1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology, 21, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Ponomarev ED, Barteneva N, Tan Y, Mann MK and Dittel BN, 2008. IL‐13 induces the expression of the alternative activation marker Ym1 in a subset of testicular macrophages. Journal of Reproductive Immunology, 78, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meco G, Bonifati V, Vanacore N and Fabrizio E, 1994. Parkinsonism after chronic exposure to the fungicide maneb (manganese ethylene‐bis‐dithiocarbamate). Scandinavian Journal of Work, Environment & Health, 20, 301–305. [DOI] [PubMed] [Google Scholar]
- Meek ME et al., 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Critical Reviews in Toxicology, 33, 591–653. [DOI] [PubMed] [Google Scholar]
- Meek ME, Boobis A, Cote I, Dellarco V, Fotakis G, Munn S, Seed J and Vickers C, 2014. New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. Journal of Applied Toxicology, 34, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GW, Kirby ML, Levey AI and Bloomquist JR, 1999. Heptachlor alters expression and function of dopamine transporters. Neurotoxicology, 20, 631–637. [PubMed] [Google Scholar]
- Moneypenny CG, Shao J, Song Y and Gallagher EP, 2006. MLL rearrangements are induced by low doses of etoposide in human foetal haematopoietic stem cells. Carcinogenesis, 27, 874–881. [DOI] [PubMed] [Google Scholar]
- Monnet‐Tschudi F, Zurich MG and Honegger P, 2007. Neurotoxicant‐induced inflammatory response in three‐dimensional brain cell cultures. Human and Experimental Toxicology, 26, 339–346. [DOI] [PubMed] [Google Scholar]
- Montecucco A, Zanetta F and Biamonti G, 2015. Molecular mechanisms of etoposide. EXCLI Journal, 14, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto A and Colosio C, 2011. Biochemical and toxicological evidence of neurological effects of pesticides: the example of Parkinson's disease. Neurotoxicology, 32, 383–391. doi: 10.1016/j.neuro.2011.03.004 [DOI] [PubMed] [Google Scholar]
- Muniz JF, McCauley L, Scherer J, Lasarev M, Koshy M, Kow YW, Nazar‐Stewart V and Kisby GE, 2008. Biomarkers of oxidative stress and DNA damage in agricultural workers: a pilot study. Toxicology and Applied Pharmacology, 227, 97–107. [DOI] [PubMed] [Google Scholar]
- Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S and Takagi M, 2015. Dysregulation of the DNA damage response and KMT2A rearrangement in foetal liver haematopoietic cells. PLoS ONE, 10, e0144540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan S, Sinsheimer JS, Paul KC, Liew Z, Cockburn M, Bronstein JM and Ritz B, 2015. Genetic variability in ABCB1, occupational pesticide exposure, and Parkinson's disease. Environmental Research, 143(Pt A), 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council, 2008. Keck Center of the National Academies, Environmental Studies and Toxicology . Reproductive Toxicology (Elmsford, N.Y.), 25, 136–138. [Google Scholar]
- Niranjan R, 2014. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's disease: focus on astrocytes. Molecular Neurobiology, 49, 28–38. [DOI] [PubMed] [Google Scholar]
- Ntzani EE, Chondrogiorgi M, Ntritsos G, Evangelou E and Tzoulaki I, 2013. Literature review on epidemiological studies linking exposure to pesticides and health effects. EFSA supporting publication 2013:EN‐497, 159 pp.
- OECD , 2013. Guidance document on developing and assessing adverse outcome pathways. OECD, Paris. ENV/JM/MONO (2013)6:45 [Google Scholar]
- OECD , 2014. User's Handbook Supplement to the Guidance Document for Developing and Assessing AOPs: 32. Available online: https://aopkb.org/common/AOP_handbook.pdf [Google Scholar]
- Ojha A and Srivastava N, 2014. In vitro studies on organophosphate pesticides induced oxidative DNA damage in rat lymphocytes. Mutation Research, Genetic Toxicology and Environmental Mutagenesis, 761, 10–17. [DOI] [PubMed] [Google Scholar]
- Palm T, Bolognin S, Meiser J, Nickels S, Träger C, Meilenbrock R‐L, Brockhaus J, Schreitmüller M, Missler M and Schwamborn JC, 2015. Rapid and robust generation of long‐term self‐renewing human neural stem cells with the ability to generate mature astroglia. Scientific Reports, 5, 16321. doi: 10.1038/srep16321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan‐Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo‐Angulo M, Gille G, Funk RH and Reichmann H, 2012. Environmental toxins trigger PD‐like progression via increased alpha‐synuclein release from enteric neurons in mice. Scientific Reports, 2, 898. doi: 10.1038/srep00898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paparella M, Daneshian M, Hornek‐Gaustere R, Kinzl M, Mauritz I and Mühlegger S, 2013. Uncertainty of testing methods – what do we (want to) know? ALTEX, 30, 131–144. [DOI] [PubMed] [Google Scholar]
- Paul KC, Rausch R, Creek MM, Sinsheimer JS, Bronstein JM, Bordelon Y and Ritz B, 2016a. APOE, MAPT, and COMT and Parkinson's disease susceptibility and cognitive symptom progression. Journal of Parkinson's Disease, 6, 349–59. [Epub ahead of print] PubMed PMID: 27061069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul KC, Sinsheimer JS, Rhodes SL, Cockburn M, Bronstein J and Ritz B, 2016b. Organophosphate pesticide exposures, nitric oxide synthase gene variants, and gene‐pesticide interactions in a Case‐Control Study of Parkinson's Disease, California (USA). Environmental Health Perspectives, 124, 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D and Osheroff N, 2014. Topoisomerase II and leukaemia. Annals of the New York Academy of Sciences, 1310, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego C, Fumagalli S and De Simoni MG, 2011. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. Journal of Neuroinflammation, 8, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo‐de‐Oliveira MS and Koifman S; Brazilian Collaborative Study Group of Infant Acute Leukemia , 2006. Infant acute leukemia and maternal exposures during pregnancy. Cancer Epidemiology, Biomarkers & Prevention, 15, 2336–2341. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y and Dittel BN, 2007. CNS‐derived interleukin‐4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. Journal of Neuroscience, 27, 10714–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C, Stam RW, Agraz‐Doblas A, Ballerini P, Camos M, Castaño J, Marschalek R, Bursen A, Varela I, Bueno C and Menendez P, 2016. Activated KRAS cooperates with MLL‐AF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukaemia. Cancer Research, 76, 2478–2489. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA and Shrivastava S, 2000. A meta‐analysis of Parkinson's disease and exposure to pesticides. Neurotoxicology, 21, 435–440. [PubMed] [Google Scholar]
- Pui CH, Robison LL and Look AT, 2008. Acute lymphoblastic leukaemia. Lancet, 371, 1030–1043. [DOI] [PubMed] [Google Scholar]
- Punia S, Das M, Behari M, Dihana M, Govindappa ST, Muthane UB, Thelma BK and Juyal RC, 2011. Leads from xenobiotic metabolism genes for Parkinson's disease among north Indians. Pharmacogenetics and Genomics, 21, 790–797. [DOI] [PubMed] [Google Scholar]
- Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ and Di Monte DA, 2007. Microglial activation as a priming event leading to paraquat‐induced dopaminergic cell degeneration. Neurobiology of Diseases, 25, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahden‐Staroń I, 2002. The inhibitory effect of the fungicides captan and captafol on eukaryotic topoisomerases in vitro and lack of recombinagenic activity in the wing spot test of Drosophila melanogaster. Mutation Research, 518, 205–213. [DOI] [PubMed] [Google Scholar]
- Rahden‐Staroń I, Czeczot H and Kowalska‐Loth B, 1993. The ability of thiram to inhibit eukaryotic topoisomerase II and to damage DNA. Acta Biochimica Polonica, 40, 51–53. [PubMed] [Google Scholar]
- Ramsey CP and Tansey MG, 2014. A survey from 2012 of evidence for the role of neuroinflammation in neurotoxin animal models of Parkinson's disease and potential molecular targets. Experimental Neurology, 256, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DB, Little PB, Malarkey DE, Herbert RA and Sills RC, 2011. Histopathological evaluation of the nervous system in routine National Toxicology Program rodent studies: a modified approach. Toxicologic Pathology, 39, 463–470. [DOI] [PubMed] [Google Scholar]
- Ren Y, Liu W, Jiang H, Jiang Q and Feng J, 2005. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. Journal of Biological Chemistry, 280, 34105–34112. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Caudle WM, Wang M, Dean ED, Pennell KD and Miller GW, 2006. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. FASEB Journal, 20, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Rought SE, Yau PM, Schnier JB, Chuang LF and Chuang RY, 1998. The effect of heptachlor, a chlorinated hydrocarbon insecticide on p53 tumour suppressor in human lymphocytes. Toxicology Letters, 94, 29–36. [DOI] [PubMed] [Google Scholar]
- Rought SE, Yau PM, Chuang LF, Doi RH and Chuang RY, 1999. Effect of the chlorinated hydrocarbons heptachlor, chlordane, and toxaphene on retinoblastoma tumour suppressor in human lymphocytes. Toxicology Letters, 104, 127–135. [DOI] [PubMed] [Google Scholar]
- Rought SE, Yau PM, Guo XW, Chuang LF, Doi RH and Chuang RY, 2000. Modulation of CPP32 activity and induction of apoptosis in human CEM x 174 lymphocytes by heptachlor, a chlorinated hydrocarbon insecticide. Journal of Biochemical and Molecular Toxicology, 14, 42–50. [DOI] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Wittung‐Stafhede P, knight R and Mazmanian SK, 2016. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell, 167, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B and Greenamyre JT, 2014. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiology of Diseases, 70, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom von Tobel J, Zoia D, et al., 2014. Immediate and delayed effects of subchronic Paraquat exposure during an early differentiation stage in 3D‐rat brain cell cultures. Toxicology Letters, doi: 10.1016/j.toxlet.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Sanjuan‐Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R and Menéndez P, 2015. Revisiting the biology of infant t(4;11)/MLL‐AF4 + B‐cell acute lymphoblastic leukaemia. Blood, 126, 2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildknecht S, Gerding HR, Karreman C, Drescher M, Lashuel HA, Outeiro TF, Di Monte DA and Leist M, 2013. Oxidative and nitrative alpha‐synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. Journal of Neurochemistry, 125, 491–511. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Pape R, Meiser J, Karreman C, Strittmatter T, Odermatt M, Cirri E, Friemel A, Ringwald M, Pasquarelli N, Ferger B, Brunner T, Marx A, Möller HM, Hiller K and Leist M, 2015. Preferential extracellular generation of the active parkinsonian toxin MPP+ by transporter‐independent export of the intermediate MPDP+. Antioxidants & Redox Signaling, 23, 1001–1016. doi: 10.1089/ars.2015.6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG and Bonner WM, 2010. Role of oxidatively induced DNA lesions in human pathogenesis. Mutation Research, 704, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed J, et al., 2005. Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Critical Reviews in Toxicology, 35, 664–672. [DOI] [PubMed] [Google Scholar]
- Shulman MJ, DeJager PL and Feany MB, 2011. Parkinson's disease: genetics and pathogenesis. Annual Review of Pathology: Mechanisms of Disease, 6, 192–222. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Breckenridge CB, Beck M, Jiao Y, Butt MT, Wolf J, Zadory D, Minnema D, Sturgess NC, Travis KZ, Cook AR, Smith LL and Botham PA, 2016. Assessment of the effects of MPTP and Paraquat on dopaminergic neurons and microglia in the substantia nigra pars compacta of C57BL/6 mice. PLoS ONE, doi: 10.1371/journal.pone0164094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Conde J and Harrison JK, 2001. Chemokines and Alzheimer's disease. Neurobiology of Aging, 22, 909–913. [DOI] [PubMed] [Google Scholar]
- Sturla SJ, Boobis AR, Fitzgerald RE, Hoeng J, Kavlock RJ, Schirmer K, Whelan M, Wilks MF and Peitsch MC, 2014. Systems toxicology: from basic research to risk assessment. Chemical Research in Toxicology, 2014, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez‐Padilla J and Schumacker PT, 2011. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson's disease. Neuroscience, 198, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP and Langston JW, 2011. Rotenone, paraquat, and Parkinson's disease. Environmental Health Perspectives, 119, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG and Goldberg MS, 2009. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Diseases, 37, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitell MA and Pandolfi PP, 2009. Molecular genetics of acute lymphoblastic leukaemia. Annual Review of Pathology: Mechanisms of Disease, 4, 175–198. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam M, Richfield EK, Baggs RB, Tank AW and Cory‐Slechta DA, 2000. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. Journal of Neuroscience, 20, 9207–9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA and Cory‐Slechta DA, 2003. Age‐related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. European Journal of Neuroscience, 18, 589–600. [DOI] [PubMed] [Google Scholar]
- Thomas KW, Dosemeci M, Coble JB, Hoppin JA, Sheldon LS, Chapa G, Croghan CW, Jones PA, Knott CE and Lynch CF, 2009. Assessment of a pesticide exposure intensity algorithm in the Agricultural Health Study. Journal of Exposure Science and Environmental Epidemiology, 20, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash B, Uthayathas S, Karuppagounder SS, Suppiramaniam V and Dhanasekaran M, 2007. Paraquat and maneb induced neurotoxicity. Proceedings of the Western Pharmacology Society, 50, 31–42. [PubMed] [Google Scholar]
- Thys RG, Lehman CE, Pierce LC and Wang YH, 2015. Environmental and chemotherapeutic agents induce breakage at genes involved in leukaemia‐causing gene rearrangements in human haematopoietic stem/progenitor cells. Mutation Research, 779, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu K, Perier C, Vila M, Caspersen C, Zhang HP, Teismann P, Jackson‐Lewis V, Stern DM, Yan SD and Przedborski S, 2003. L‐3‐hydroxyacyl‐CoA dehydrogenase II protects in a model of Parkinson's disease. Annals of Neurology, 56, 51–60. [DOI] [PubMed] [Google Scholar]
- Turner MC, Wigle DT and Krewski D, 2010. Residential pesticides and childhood leukemia: a systematic review and meta‐analysis. Environmental Health Perspectives, 118, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Tobel JS, Antinori P, Zurich MG, Rosset R, Aschner M, Gluck F, et al., 2014. Repeated exposure to ochratoxin A generates a neuroinflammatory response, characterized by neurodegenerative M1 microglial phenotype. Neurotoxicology, 44C, 61–70. [DOI] [PubMed] [Google Scholar]
- Ukpebor J, Llabjani V, Martin FL and Halsall CJ, 2011. Sublethal genotoxicity and cell alterations by organophosphorus pesticides in MCF‐7 cells: implications for environmentally relevant concentrations. Environmental Toxicology and Chemistry, 30, 632–639. [DOI] [PubMed] [Google Scholar]
- Van Maele‐Fabry G, Hoet P, Vilain F and Lison D, 2012. Occupational exposure to pesticides and Parkinson's disease: a systematic review and meta‐analysis of cohort studies. Environment International, 46, 30–43. [DOI] [PubMed] [Google Scholar]
- Vergo S, Johansen JL, Leist M and Lotharius J, 2007. Vesicular monoamine transporter 2 regulates the sensitivity of rat dopaminergic neurons to disturbed cytosolic dopamine levels. Brain Research, 14, 18–32. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Parpura V, Pekna M, Pekny M and Sofroniew M, 2014. Glia in the pathogenesis of neurodegenerative diseases. Biochemical Society Transactions, 42, 1291–1301. [DOI] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L and Whelan M, 2014a. Adverse outcome pathway (AOP) development I: strategies and principles. Toxicological Sciences, 142, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L and Whelan M, 2014b. Adverse outcome pathway (AOP) development II: best practices. Toxicological Sciences, 142, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Costello S, Cockburn M, Zhang X, Bronstein J and Ritz B, 2011. Parkinson's disease risk from ambient exposure to pesticides. European Journal of Epidemiology, 26, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Zhao JP, Pruszak J, Hedlund E, Fu D, Soldner F, Broccoli V, Constantine‐Paton M, Isacson O and Jaenisch R, 2008. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. PNAS, 105, 5856–5861. Available online: http://www.pnas.org, doi: 10.1073/pnas.0801677105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton PS, 2007. Inflammation as a causative factor in the aetiology of Parkinson's disease. British Journal Pharmacology, 150, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemels J, 2012. Perspectives on the causes of childhood leukaemia. Chemico‐Biological Interactions, 196, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RD, Boros LG, Kolanko CJ, Jackman SM and Eggers TR, 2004. Chromosomal aberrations in human lymphocytes exposed to the anticholinesterase pesticide isofenphos with mechanisms of leukemogenesis. Leukemia Research, 28, 947–958. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M and Kashiwakura I, 2013. Role of reactive oxygen species in the radiation response of human haematopoietic stem/progenitor cells. PLoS ONE, 8, e70503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fitsanakis VA, Gu G, Jing D, Ao M, Amarnath V and Montine TJ, 2003. Manganese ethylene‐bis‐dithiocarbamate and selective dopaminergic neurodegeneration in rat: a link through mitochondrial dysfunction. Journal of Neurochemistry, 84, 336–346. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wallace AD, Du P, Lin S, Baccarelli AA, Jiang H, Jafari N, Zheng Y, Xie H, Soares MB, Kibbe WA and Hou L, 2012. Genome‐wide study of DNA methylation alterations in response to diazinon exposure in vitro. Environmental Toxicology and Pharmacology, 34, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, 2015. Aldehyde dehydrogenase 2 genetic variations may increase susceptibility to Parkinson's disease in Han Chinese population. Neurobiology of Aging, 36, 2660.e9‐13. [DOI] [PubMed] [Google Scholar]
- Zhou C, Huang Y and Przedborski S, 2008. Oxidative stress in Parkinson's disease: a mechanism of pathogenic and therapeutic significance. Annals of the New York Academy of Sciences, 1147, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]