

Abstract
Hypophosphatasia (HPP) is the inborn-error-of-metabolism caused by loss-of-function mutation(s) of the ALPL gene that encodes the tissue-nonspecific isoenzyme of alkaline phosphatase (TNSALP). TNSALP in healthy individuals is on cell surfaces richly in bone, liver, and kidney. Thus, TNSALP natural substrates accumulate extracellularly in HPP, including inorganic pyrophosphate (PPi), a potent inhibitor of hydroxyapatite crystal formation and growth. Superabundance of extracellular PPi (ePPi) in HPP impairs mineralization of bones and teeth, often leading to rickets during childhood and osteomalacia in adult life and to tooth loss at any age. HPP’s remarkably broad-ranging severity is largely explained by nearly four hundred typically missense mutations throughout the ALPL gene that are transmitted as an autosomal dominant or autosomal recessive trait. In the clinical laboratory, the biochemical hallmark of HPP is low serum ALP activity (hypophosphatasemia). However, our experience indicates that hyperphosphatemia from increased renal reclamation of filtered inorganic phosphate (Pi) is also common.
Herein, from our prospective single-center study, we document throughout the clinical spectrum of non-lethal pediatric HPP that hyperphosphatemia reflects increased renal tubular threshold maximum for phosphorus adjusted for the glomerular filtration rate (TmP/GFR). To explore its pathogenesis, we studied mineral metabolism and quantitated circulating levels of three phosphatonins [fibroblast growth factor 23 (FGF23), secreted frizzled-related protein 4 (sFRP4), and fibroblast growth factor 7 (FGF7)] in 41 pediatric patients with HPP, 73 with X-linked hypophosphatemia (XLH), and 15 healthy pediatric control (CTR) subjects. The HPP and XLH cohorts had normal serum total and ionized calcium and parathyroid hormone levels (Ps > 0.10) and uncompromised glomerular filtration. In XLH, serum FGF23 was characteristically elevated (P < 0.0001) and despite hypophosphatemia sFRP4 was normal (P > 0.4) while FGF7 was low (P < 0.0001). In HPP, despite hyperphosphatemia serum FGF23 and sFRP4 were normal (Ps > 0.8) while FGF7 was low (P < 0.0001). Subsequently, in rats, we confirmed that FGF7 is phosphaturic.
Thus, hyperphosphatemia in non-lethal pediatric HPP is associated with phosphatonin insufficiency together with, as we discuss, ePPi excess and diminished renal TNSALP activity.
Keywords: alkaline phosphatase, ALPL, asfotase alfa, bisphosphonate, etidronate, GACI, generalized arterial calcification of infancy, hydroxyapatite, hypophosphatemia, inborn-error-of-metabolism, inorganic pyrophosphate, PHEX, phosphatonin, pseudoxanthoma elasticum, rickets, TmP/GFR, X-linked hypophosphatemia
II). Introduction:
Seventy-two years ago in 1948, John C. Rathbun MD characterized “a new developmental anomaly” he called hypophosphatasia (HPP) based upon the clinical, radiographic, biochemical, treatment, and autopsy findings underlying paradoxical deficiency of alkaline phosphatase (ALP) activity in an infant boy with lethal rickets.(1) HPP is now understood to be the inborn-error-of-metabolism caused by loss-of-function mutation(s) of the gene ALPL that encodes the tissue-nonspecific isoenzyme of ALP (TNSALP).(2,3) In fact, we reported in 2001 using parental DNA that Rathbun’s patient was compound heterozygous for ALPL missense mutations.(4)
TNSALP is a ubiquitous homodimeric cell-surface phosphomonoester phosphohydrolase [E.C.3.1.3.1] expressed especially in bone, liver, and kidney tissue.(5) Thus, in HPP the natural substrates of TNSALP [inorganic pyrophosphate (PPi), pyridoxal 5′-phosphate (PLP), and phosphoethanolamine (PEA)] accumulate extracellularly.(2,3,5,6) Because PPi is an inhibitor of hydroxyapatite (HA) crystal formation and growth, superabundant extracellular PPi (ePPi) blocks calcium (Ca) and inorganic phosphate (Pi) entry into the skeleton and teeth in HPP and explains the associated rickets during growth or osteomalacia during adult life and tooth loss at any age.(2,3,5) In 2009, Tiosano and Hochberg(7) concluded hypophosphatemia is “the common denominator of all rickets”, but we find that HPP represents a striking and informative exception.(2,3)
HPP’s biochemical hallmark in the clinical laboratory is low serum ALP activity (i.e., hypophosphatasemia).(3,6) However, Rathbun also found elevated serum Pi (i.e., hyperphosphatemia), noting in his patient “low excretion of phosphorus in spite of the normal to high serum phosphorus levels suggested a relationship with the low tissue phosphatase of the kidney”.(1) In 1987, we briefly reported that hyperphosphatemia from increased urinary reabsorption of Pi is common in HPP.(8,9) Our experience with HPP to date supports this observation.
Herein, we report our prospective single-center assessment of Pi metabolism including circulating phosphatonin levels in 41 untreated children and adolescents representing nearly the complete broad-ranging severity of pediatric HPP.(2,3,6) We show that 93% were hyperphosphatemic, including those with the mildest “odonto” form of the disease. Hypercalcemia with secondary hypoparathyroidism or impaired glomerular filtration was not the explanation. We found low circulating levels of fibroblast growth factor 7 (FGF7) and despite their hyperphosphatemia normal levels of both fibroblast growth factor 23 (FGF23) and secreted frizzled-related protein 4 (sFRP4). We discuss phosphatonin insufficiency and other hypotheses for the nephrogenic hyperphosphatemia of HPP.
III). Patients and Methods:
Informed written consent guided by the Human Research Protection Office, Washington University School of Medicine, St. Louis, MO, USA, preceded all investigational work. At the time of our study, there was no established medical treatment for HPP, and none was tested herein.
A). Patients and Biochemical Studies:
During 2006–2008, we prospectively collected inpatient fasting blood and urine at the Center for Metabolic Bone Disease and Molecular Research, Shriners Hospitals for Children – St. Louis, St. Louis, MO, USA from our new and returning pediatric patients with HPP and X-linked hypophosphatemia (XLH). Over that timeframe, the two patient groups were from among the ~ 170 and ~ 200 subjects with these disorders, respectively, we had investigated since 1983. For both patient groups we routinely assayed biochemical parameters of mineral homeostasis in our certified clinical laboratory using a Dade Xpand instrument (Siemens Healthcare Diagnostics, Inc., Tarrytown, NY, USA).(10,11) For most, depending on their age, we collected after an overnight fast one or two sequential orally hydrated 2-hour urine specimens with mid-point blood sampling to calculate renal tubular threshold maximum for phosphorus per glomerular filtration rate (TmP/GFR) and tubular reabsorption of Pi (TRP).(12) Most of the children with XLH were receiving medical treatment comprising daily calcitriol and Pi salt supplementation administered orally. Additionally, during 2007 after informed written consent, 15 healthy children/adolescents (i.e., age < 18 years), the sons and daughters of work associates, etc., had fasting blood (but no urine) studies to comprise a control (CTR) group.
The serum and urine Pi assay involved complexation of Pi with ammonium molybdate in the presence of sulfuric acid to form ammonium phosphomolybdate detected photometrically.(12) Additionally, an aliquot of serum from each individual in the three study groups (HPP, XLH, and CTR) was stored at −40°C until 2008 and then shipped frozen to our research laboratory at Mayo Clinic, Rochester, MN, USA for quantitation soon after of the phosphatonins FGF23, sFRP4, and FGF7. FGF23 (intact molecule) was assayed by the method of Kainos Laboratories, Tokyo, Japan,(13) sFRP4 was quantitated using the two-antibody sandwich ELISA described in 2003 by Berndt et al.,(14) and FGF7 was assayed using the ELISA of R & D Systems (Minneapolis, MN, USA).(15) An assay for the phosphatonin designated matrix extracellular phosphoglycoprotein (MEPE) was not available.
B). Animal Studies:
Our protocol to test for a phosphaturic effect of FGF7 was approved by the Institutional Animal Care and Use Committee; Mayo Clinic, Rochester, MN, USA. Sprague Dawley rats (Harlan Sprague Dawley, Madison, WI, USA) were fed a rodent diet containing 0.7% Pi, anesthetized with an intraperitoneal injection of 100–150 mg/kg body weight 5-secbutyl-ethyl-2-thiobarbituric acid (Inactin, Byk Gulden Konstanz, Hamburg, Germany), and placed on a heated table to maintain body temperature at 36–38°C. After tracheostomy and intubation, a polyethylene 50 catheter in the left carotid artery enabled arterial blood pressure monitoring and collection of blood. A second catheter, in the right internal jugular vein, allowed infusion of 2% inulin and 2.5% bovine serum albumin in 0.9% NaCl at a rate of 1.2 ml/hour. A third catheter (polyethylene 90), in the bladder, enabled urine collection. Recombinant full-length FGF23 was prepared using bacterial protein expression methods, as we reported.(15) Recombinant FGF7 was purchased from R & D Systems (Minneapolis, MN, USA). Plasma and urine Pi concentrations were assayed using the method of Chen et al.(16) Inulin concentrations in plasma and urine were quantitated using the anthrone method.(17) Five groups of rats were studied:
Group 1: Vehicle, time control group, n = 8. After a 90-minute equilibration period, one 30-minute urine and blood sample were collected. One hour later, a second 60-minute urine and blood sample were obtained. Group 2: FGF7 infusion at 0.1 nmol/kg/hr group, n = 9. This procedure was identical to group 1, except after the 30-minute control clearance we infused FGF7 at 0.1 nmol/kg/hr. One hour after the infusion of FGF7 was complete, 60-minute urine and blood samples were collected. Similar experiments in the remaining three groups involved different FGF23 and FGF7 concentrations. Group 3: FGF23 infusion at 0.1 nmol/kg/hr group, n = 9. Group 4: FGF7 infusion at 0.01 nmol/kg/hr group, n = 6. Group 5: FGF23 infusion at 0.01 nmol/kg/hr group, n = 8.
C). ALPL And PHEX Gene Mutation Analysis:
Most of the HPP and XLH patients were studied by ALPL(18) and PHEX(19,20) mutation analysis, respectively, as established in our research laboratory at the Washington University School of Medicine, St. Louis, MO, USA (Supplementary Appendix, Table S-1).
D). Statistical Analyses:
For the clinical data, SAS software 9.3 (SAS Institute Inc., Cary, NC, USA) provided the statistical analyses and graphics. Descriptive statistics are presented in tabular form as mean, standard deviation (SD), 95% confidence interval (CI) of the mean, and range. To address any pubertal effects on the biochemical data among the total of 129 study subjects (see Results), we assessed selectively the group data from the 104 who were pre-teenage (Supplementary Appendix). Then, to address the older mean age of the CTR group compared to the two patient groups, we analyzed the group data exclusively from the 86 subjects > 7.0 years-of-age. The two-sample t-test was used to contrast the HPP vs CTR, HPP vs XLH, and XLH vs CTR groups. Mean values are denoted in scatter plots and illustrated in bar graphs as mean (‒) ± 1 standard error (SE). To determine if the circulating phosphatonin levels reflected HPP severity, we used in regression analysis our published “HPP severity score for children (HPPSSC),” (18) which assigns by increasing clinical severity: 1 for odonto HPP, 2 for mild childhood HPP, 3 for severe childhood HPP, and 4 for infantile HPP. Pearson correlation analysis explored for relationships among the serum FGF7, ALP, and Pi levels. Two-sided P-values < 0.05 were considered statistically significant.
For the study in rats of FGF7 effects on renal Pi clearance, we compared plasma Pi concentrations, glomerular filtration rate (GFR), and fractional excretion of Pi (FEP) among the control and experimental groups using a paired t-test and JMP Statistical Software (Cary, NC, USA).
IV). Results:
A. Study Subjects:
Together, the three study groups totaled 129 children/adolescents: 41 with HPP reflecting the principal pediatric types except the rapidly lethal perinatal form,(18) 73 with XLH, and 15 CTR subjects. The youngest and oldest among the 129 individuals were 1.2 and 17.4 years-of-age, respectively. The CTR group was older than both the XLH and HPP groups who were closely age-matched (Table 1). The mutations we encountered within ALPL and PHEX in the HPP and XLH groups, respectively (Supplementary Appendix, Table S-1), supported the diagnoses otherwise achieved conventionally. The HPP group comprised, by increasing severity using our nosology:(18) 11 odonto HPP, 14 mild childhood HPP, 13 severe childhood HPP, and 3 infantile HPP patients.
Table 1:
Study Group Ages (Yrs) and Sex Distribution
| Group | N | Mean (SD) | Min, Max | Girls | Boys |
|---|---|---|---|---|---|
| CTR | 15 | 12.2 (3.1) | 7.7, 16.9 | 10 | 5 |
| XLH | 73 | 8.4 (3.6) | 2.3, 17.4 | 37 | 36 |
| HPP | 41 | 8.6 (5.2) | 1.2, 16.9 | 17 | 24 |
B. Biochemical Studies:
1). Alkaline Phosphatase:
Serum ALP activity adjusted for age and sex was, as expected, often elevated in the XLH group(21,22) and invariably low in the HPP group.(23) This became especially apparent using the pediatric normal ranges for ALP at Mayo Clinic Laboratories (Rochester, MN, USA) (Figure 1). Serum ALP activity was significantly different between all three study groups (Ps < 0.0001) (Table 2). The differences persisted when the data were exclusively from either the preteenage (Supplementary Appendix) or age > 7.0 years study subjects (not shown).
Figure 1:
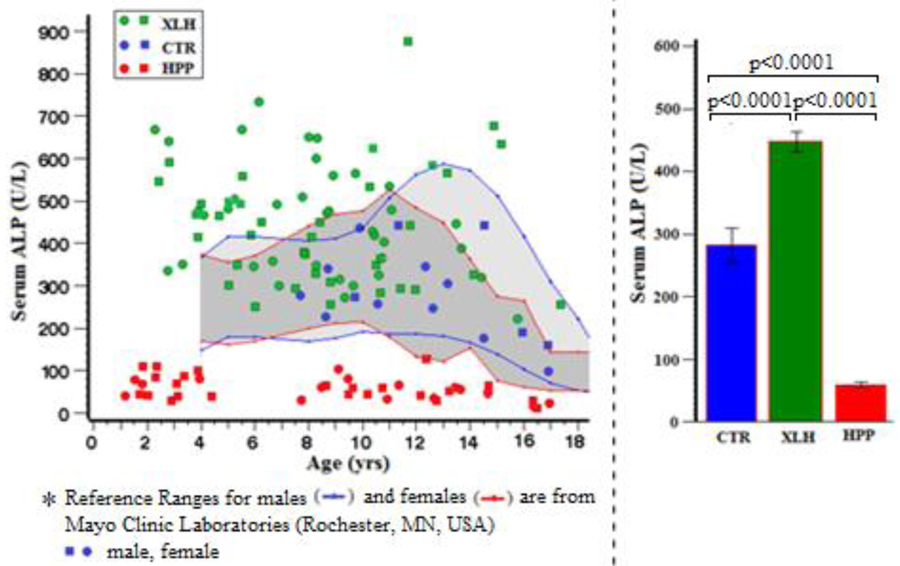
Serum Alkaline Phosphatase*
Table 2:
Serum Alkaline Phosphatase
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Paired Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 282 (106) | 99, 444 | 223, 340 | <0.0007 (XLH vs CTR) |
| XLH | 73 | 447 (134) | 221, 877 | 416, 478 | <0.0001 (HPP vs CTR) |
| HPP | 41 | 58 (29) | 12, 143 | 49, 67 | <0.0001 (XLH vs HPP) |
2). Inorganic Phosphate
As expected,(21,22) the fasting serum Pi level for nearly all XLH patients was below the lowest observed CTR value of 3.6 mg/dl (0.24 mmol/L) (Figure 2) despite most of the patients receiving Pi supplementation. In contrast, the majority of the patients with HPP; i.e., 93% (38 of 41) were hyperphosphatemic using the age-and sex-matched reference ranges of Mayo Clinic Laboratories (Figure 2). Among this group with HPP, 78% (32 of 41) had Pi levels above the highest observed CTR value of 5.6 mg/dl (0.37 mmol/L) found in a 7-year-old girl. Henceforth, we designate serum Pi values greater than 5.6 mg/dl “hyperphosphatemia,” although the upper limit of the 95% CI in our CTR group was 5.2 mg/dl (0.35 mmol/L).
Figure 2:
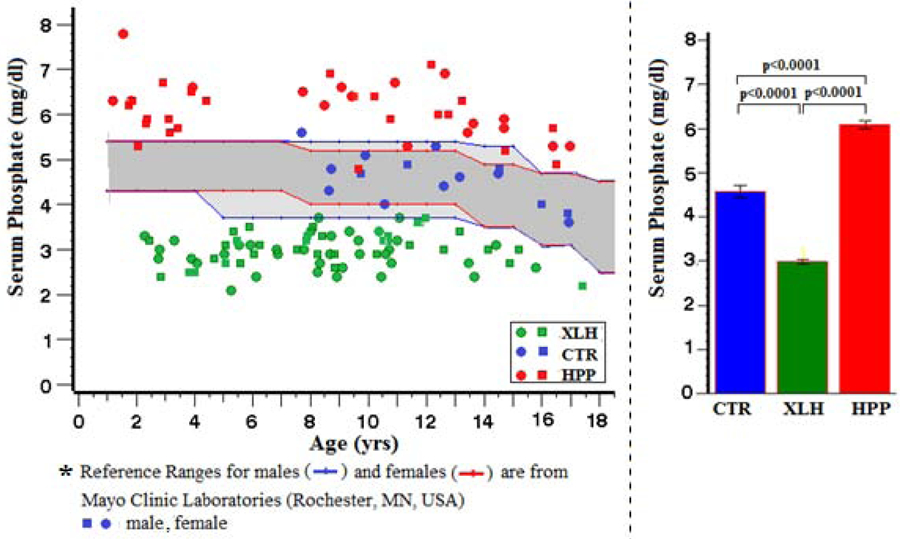
Serum Phosphate*
Fasting serum Pi levels were distinctly different (Ps < 0.0001) between the three study groups (Table 3).
Table 3:
Serum Phosphate
| Group | N | Mean (SD) | Min, Max | 95% CI | (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 4.6 (0.6) | 3.6, 5.6 | 4.3, 5.2 | <0.0001 (XLH vs CTR) |
| XLH | 73 | 3.0 (0.4) | 2.1, 3.7 | 2.9, 3.1 | <0.0001 (HPP vs CTR) |
| HPP | 41 | 6.1 (0.6) | 4.8, 7.8 | 5.9, 6.3 | <0.0001 (XLH vs HPP) |
Value x 0.0661 = SI units (mmol/L)
Hyperphosphatemia (> 5.6 mg/dl) spanned the entire clinical spectrum of non-lethal pediatric HPP in our patients: 9 of 11 (82%) with odonto-HPP, 11 of 14 (79%) with mild childhood HPP, 9 of 13 (69%) with severe childhood HPP, and 3 of 3 (100%) with infantile HPP. Thus, this observation matched the similar (but unpublished) finding from our 2018 publication concerning the biochemical hallmarks of 165 preteenage HPP patients showing the degree of hyperphosphatemia did not correlate with clinical severity(23) (Supplementary Appendix, Table S-5, Figure S-3).
3). TmP/GFR:
TmP/GFR had not been assessed in the CTR group. Accordingly, for our data analysis, we followed Carpenter’s(24) recommendation that fasting serum Pi levels of healthy controls would represent appropriate values for TmP/GFR. Thus, the XLH and HPP patient values we acquired for TmP/GFR are illustrated using, once again for a background, the normal range for serum Pi at Mayo Clinic Laboratories (Rochester, MN, USA) (Figure 3). We found TmP/GFR and TRP to be distinctly low in XLH and distinctly high in HPP (Table 4).
Figure 3:
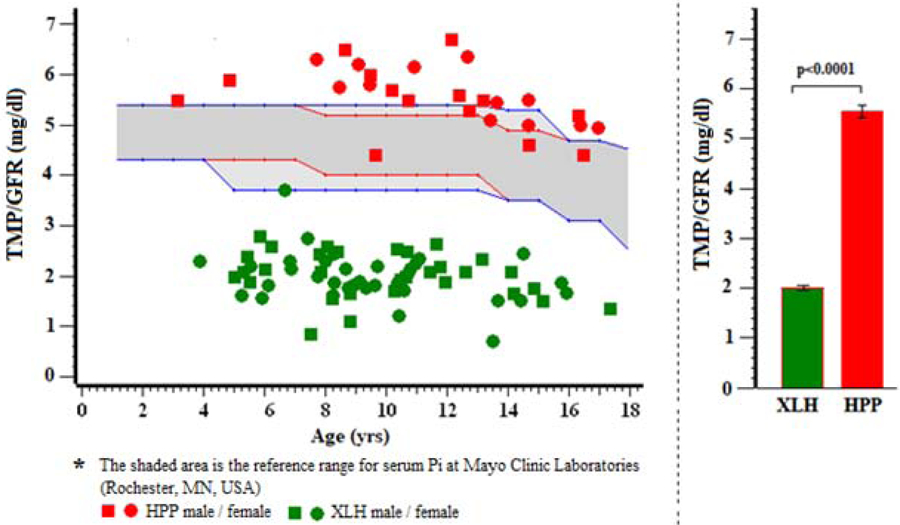
TMP/GFR*
Table 4.
TmP/GFR
| TmP/GFR | N | Mean (SD) | Min, Max | ||
|---|---|---|---|---|---|
| HPP | 25 | 5.6 (0.6) | 4.4, 6.7 | ||
| XLH | 61 | 2.0 (0.5) | 0.4, 4.0 | ||
| Value x 0.06661 = SI units (mmol/L) | |||||
| TRP | |||||
| TRP | N | Mean (SD) | Min, Max | ||
| HPP | 26 | 93.4 (3.1) | 87, 99 | ||
| XLH | 62 | 67.1 (14.3) | 13, 107 | ||
4). Serum Total Calcium:
Serum total Ca levels were normal and very similar (P > 0.3) between all three study groups (Figure 4, Table 5).
Figure 4:
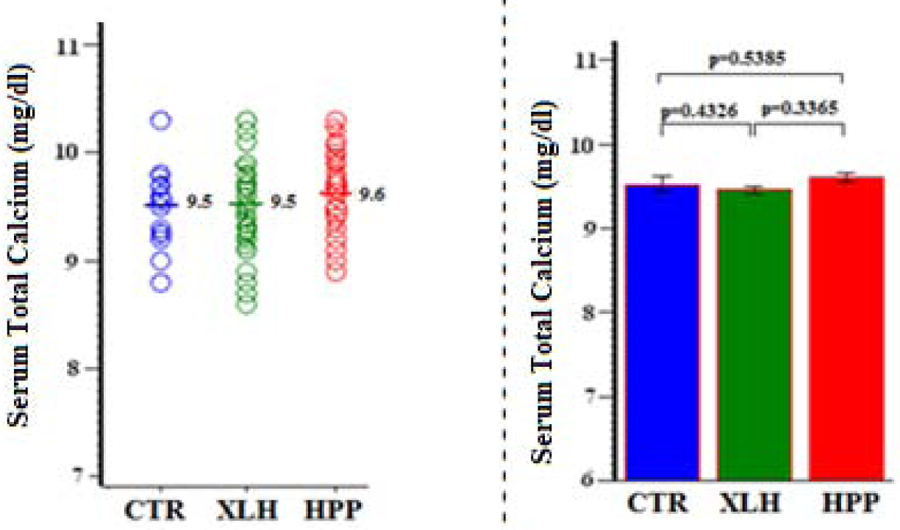
Serum Calcium (Total)
Table 5:
Serum Calcium (Total)
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 9.5 (0.4) | 8.8, 10.3 | 9.3, 9.7 | 0.4326 (XLH vs CTR) |
| XLH | 73 | 9.5 (0.4) | 8.6, 10.3 | 9.4, 9.5 | 0.5385 (HPP vs CTR) |
| HPP | 41 | 9.6 (0.4) | 8.9, 10.3 | 9.5, 9.7 | 0.3365 (XLH vs HPP) |
Value x 10 = SI units (g/L)
5). Serum Ionized Calcium:
Similarly, ionized Ca was typically normal and essentially identical (Ps > 0.5) between all three study groups (Figure 5, Table 6).
Figure 5:
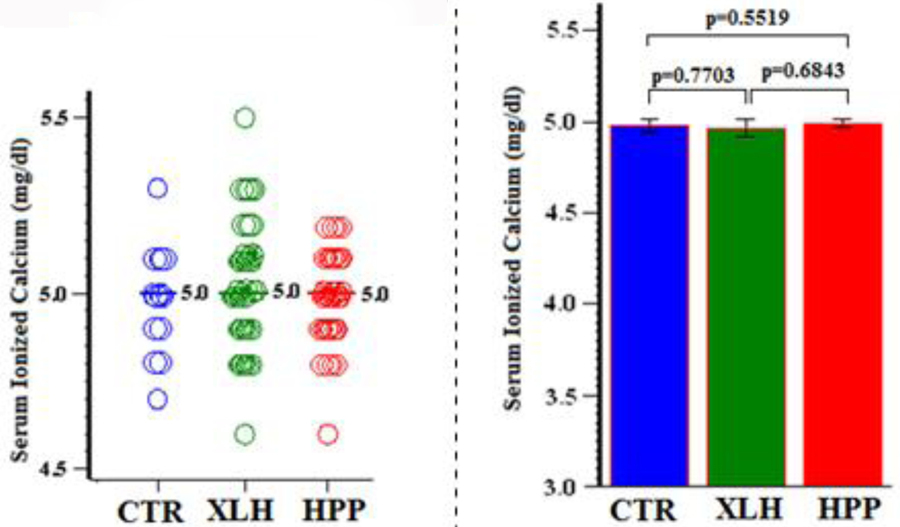
Serum Calcium (Ionized)
Table 6:
Serum Calcium (Ionized)
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 5.0 (0.15) | 4.7, 5.3 | 4.9, 5.1 | 0.7703 (XLH vs CTR) |
| XLH | 73 | 5.0 (0.39) | 4.6, 5.5 | 4.9, 5.1 | 0.5519 (HPP vs CTR) |
| HPP | 41 | 5.0 (0.13) | 4.6, 5.2 | 5.0, 5.0 | 0.6843 (XLH vs HPP) |
Value x 10 = SI units (g/L)
6). Magnesium:
Similarly, magnesium was normal for both patient groups (data not illustrated) (Ps > 0.2), although the mean value was slightly higher in HPP compared to XLH (P = 0.0065) (Table 7).
Table 7:
Serum Magnesium
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons)* |
|---|---|---|---|---|---|
| CTR | 15 | 1.9 (0.17) | 1.6, 2.2 | 1.8, 2.0 | 0.2092 (XLH vs CTR) |
| XLH | 73 | 1.9 (0.16) | 1.5, 2.3 | 1.9, 1.9 | 0.3380 (HPP vs CTR) |
| HPP | 41 | 2.0 (0.16) | 1.7, 2.3 | 1.9, 2.0 | 0.0065 (XLH vs HPP) |
Age effect adjusted
Value x 0.5 = SI units (mmol/L)
7). Serum Parathyroid Hormone:
Serum intact PTH was typically normal in both the XLH and HPP groups; P = 0.2258 and 0.1181, respectively (Figure 6).
Figure 6:
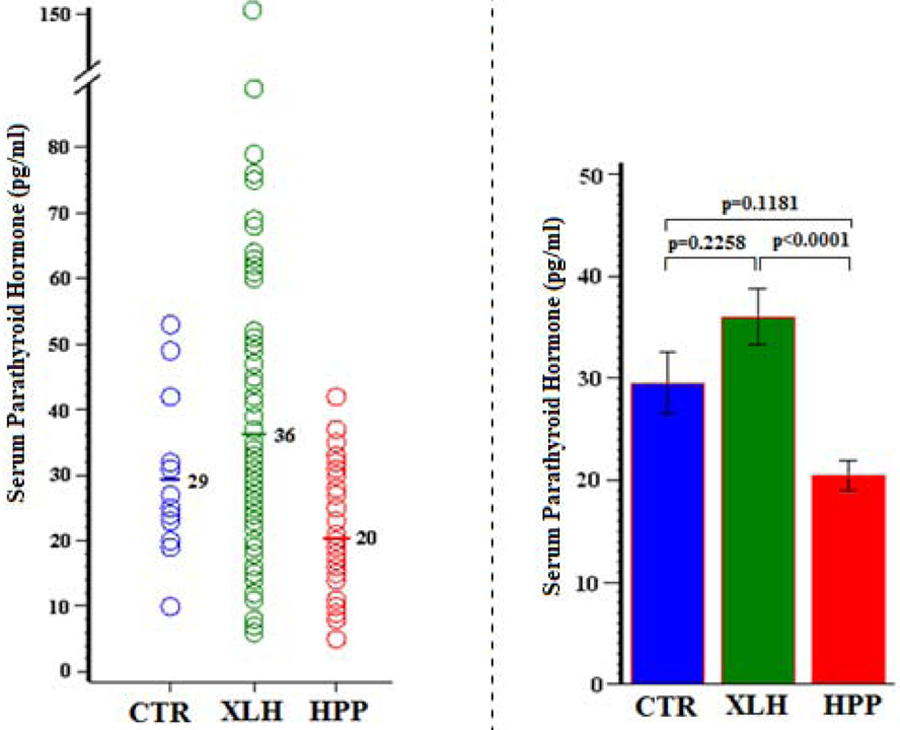
Serum Parathyroid Hormone
Serum PTH was significantly higher in XLH versus HPP (P < 0.0001), perhaps contributing to the difference in circulating Pi levels between these groups (Table 8).
Table 8:
Serum Parathyroid Hormone
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 29 (11) | 10, 53 | 23.2, 35.8 | 0.2258 (XLH vs CTR) |
| XLH | 73 | 36 (24) | 6, 150 | 30.5, 41.5 | 0.1181 (HPP vs CTR) |
| HPP | 41 | 20 (9) | 5, 42 | 17.5, 23.4 | <0.0001 (XLH vs HPP) |
8). Serum Creatinine:
Compared to the CTR group averaging several years older (Table 1), serum creatinine was, as might be expected, lower (Ps < 0.001) in the younger HPP and XLH groups (Figure 7).
Figure 7:
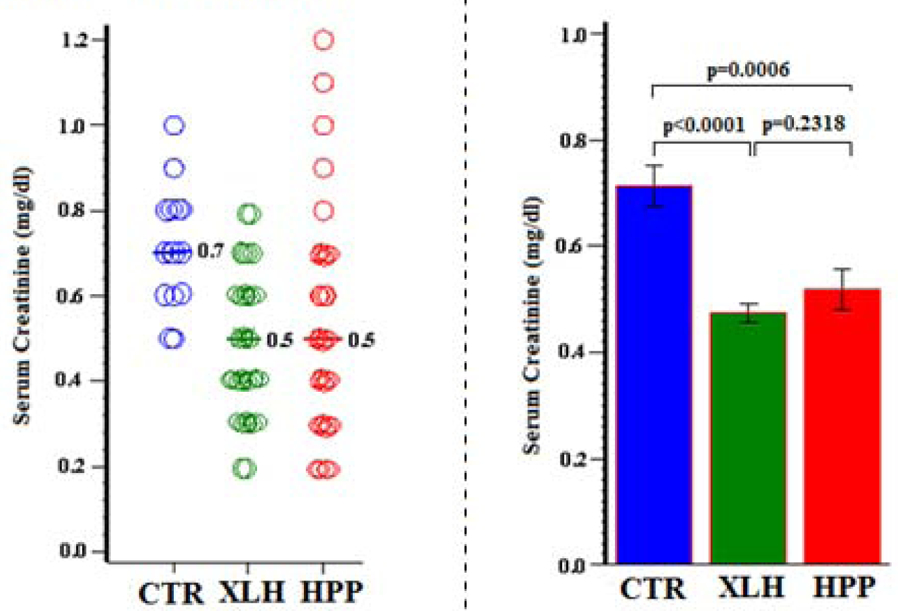
Serum Creatinine
Thus, compromise of kidney filtration did not explain the hyperphosphatemia of the HPP group (Table 9).
Table 9:
Serum Creatinine
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 15 | 0.7 (0.1) | 0.5, 1.0 | 0.64, 0.79 | <0.0001 (XLH vs CTR) |
| XLH | 73 | 0.5 (0.2) | 0.2, 0.8 | 0.44, 0.51 | 0.0006 (HPP vs CTR) |
| HPP | 41 | 0.5 (0.2) | 0.2, 1.2 | 0.44, 0.59 | 0.2318 (XLH vs HPP) |
Value x 88.4 = SI units (mol/L)
C. Phosphatonin Levels:
In XLH, nearly all patients had, as expected for their disorder and its characteristic hypophosphatemia,(21,22) elevated fasting serum FGF23 levels compared to the CTR group (P < 0.0001) (Figure 8, Table 10).
Figure 8:
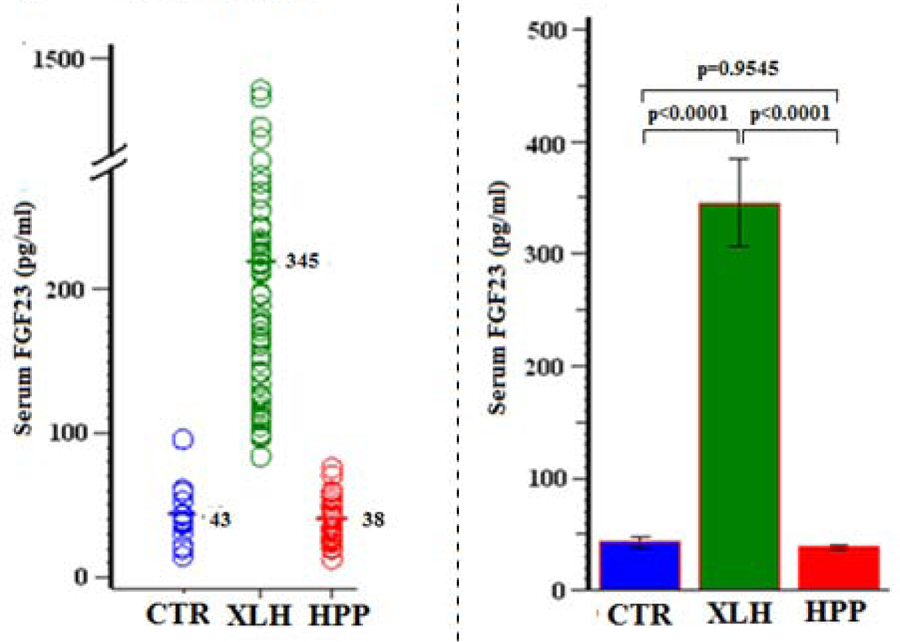
Serum FGF23
Table 10:
Serum FGF23
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 14 | 43 (21) | 15, 96 | 31, 55 | <0.0001 (XLH vs CTR) |
| XLH | 67 | 345 (320) | 84, 1442 | 268, 433 | 0.9545 (HPP vs CTR) |
| HPP | 36 | 38 (14) | 13, 76 | 34, 44 | <0.0001 (XLH vs HPP) |
Additionally in XLH featuring hypophosphatemia, we found serum sFRP4 levels were normal (P > 0.4) (Figure 9, Table 11) whereas FGF7 levels were low (P <0.0001) (Figure 10, Table 12).
Figure 9:
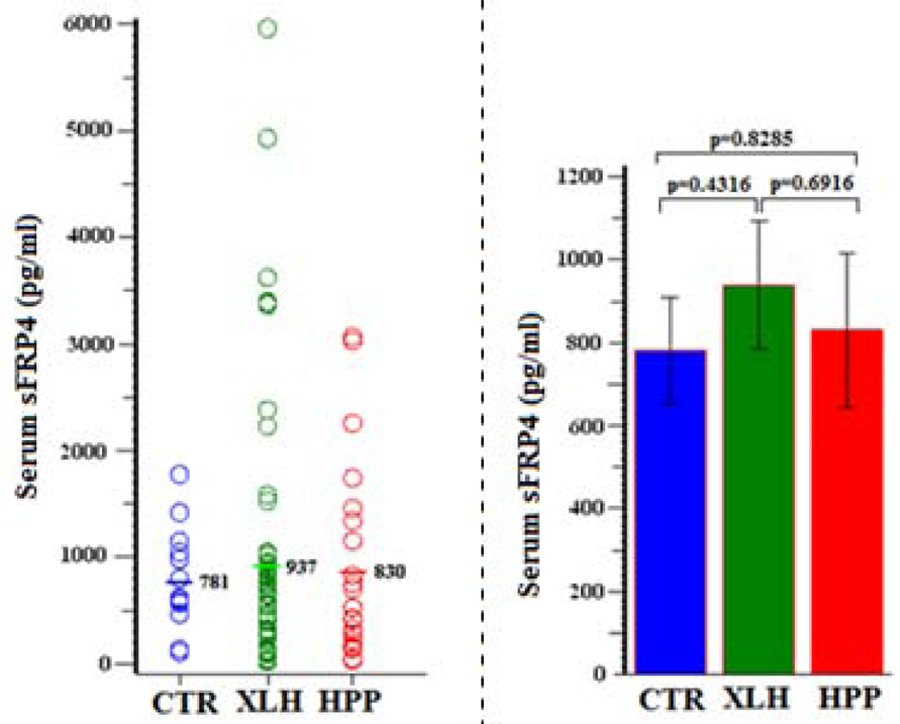
Serum sFRP4
Table 11:
Serum sFRP4
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 14 | 781 (461) | 113, 1779 | 515, 1048 | 0.4316 (XLH vs CTR) |
| XLH | 61 | 937 (1204) | 23, 5965 | 629, 1246 | 0.8285 (HPP vs CTR) |
| HPP | 25 | 830 (899) | 36, 3071 | 450, 1209 | 0.6916 (XLH vs HPP) |
Figure 10:
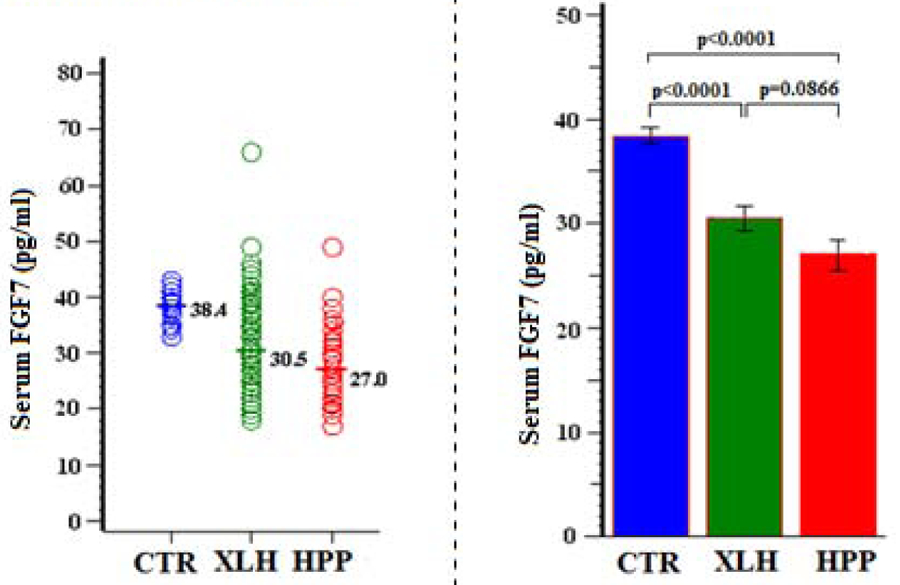
Serum FGF7
Table 12:
Serum FGF7
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| CTR | 14 | 38.4 (3.0) | 33, 43 | 36.6, 40.1 | <0.0001 (XLH vs CTR) |
| XLH | 63 | 30.5 (9.6) | 18, 66 | 28.2, 32.9 | <0.0001 (HPP vs CTR) |
| HPP | 27 | 27.0 (7.7) | 17, 49 | 24.0, 29.9 | 0.0866 (XLH vs HPP) |
In HPP, serum FGF23 and sFRP4 levels were normal despite their hyperphosphatemia (Ps > 0.8) (Figures 8 &9, Tables 10 & 11) whereas serum FGF7 was low (P < 0.0001) (Figure 10, Table 12).
D. Mineral Homeostasis and Phosphatonin Summary:
The cumulative findings (Table 13) indicated phosphatonin insufficiency and not secondary hypoparathyroidism or compromised glomerular filtration could explain the hyperphosphatemia of non-lethal pediatric HPP.
Table 13:
Serum Biochemical Summary
| Serum Parameter | Unit | Study Group Result [mean (SD)] | ||
|---|---|---|---|---|
| CTR (n=15) | XLH (n=73) | HPP (n=41) | ||
| ALP | U/L | 282 (106) | 448 (135)*** | 57 (29)*** |
| Phosphate◊ | mg/dl | 4.6 (0.6) | 3.0 (0.4)**** | 6.1 (0.6)**** |
| Total Calcium◊ | ″ | 9.5 (0.4) | 9.5 (0.4)†† | 9.6 (0.4)†† |
| Ionized Calcium◊ | ″ | 5.0 (0.2) | 5.0 (0.4)†† | 5.0 (0.1)†† |
| PTH | pg/dl | 29 (11) | 36 (24)†† | 20 (9)†† |
| Creatinine◊ | mg/dl | 0.7 (0.1) | 0.5 (0.2)** | 0.5 (0.2)** |
| FGF23 | pg/dl | 42.7 (20.5) | 346 (320)** | 38.3 (14.3)† |
| sFRP4 | ″ | 781 (461) | 937 (1204)†† | 830 (899)† |
| FGF7 | ″ | 38.4 (3.0) | 30.5 (9.6)* | 27.0 (7.7)** |
See previous Tables for conversion to SI units
Compared to CTR group:
P <0.04,
P ≤0.007,
P ≤0.0006,
P <0.0001;
P >0.8,
P >0.1
E. Serum Pi and FGF7 Levels Versus HPP Severity:
In our previous study of the biochemical hallmarks of non-lethal pediatric HPP,(23) an unpublished finding showed that hyperphosphatemia is typical with the Pi levels not differing among the clinical forms reflecting HPP severity (Supplementary Appendix, Table S-5, Figure S-3). Herein, the low levels of serum FGF7 seemed to decrease as HPP severity increased assessed using our HPPSSC (P = 0.0467) (Figure 11, Table 14). Serum FGF23 and sFRP4 were both within the CTR normal range in HPP and did not change (P = 0.6649 and P = 0.5748, respectively) according to the HPPSSC (not illustrated).
Figure 11:
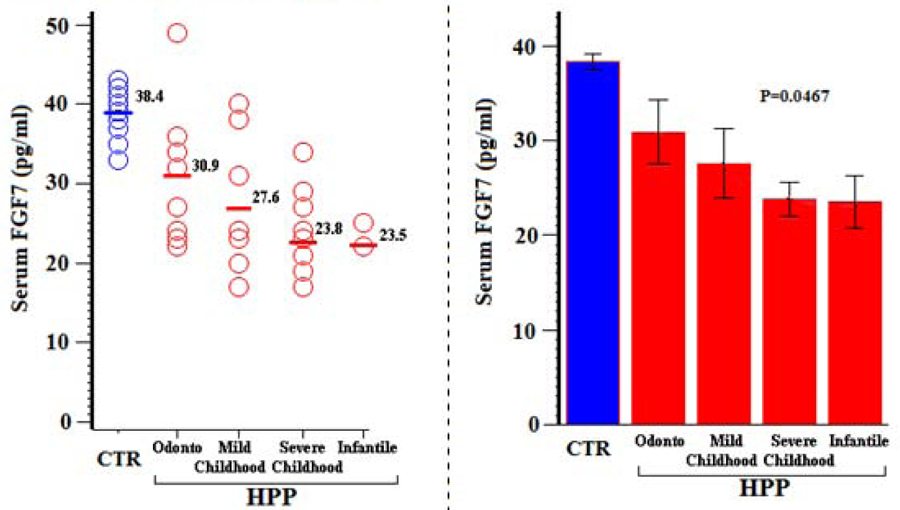
Serum FGF7 vs HPPSC
Table 14:
Serum FGF7 vs HPPSSC
| Group | N | Mean (SD) | Min, Max | 95% CI | P (Group Comparisons) |
|---|---|---|---|---|---|
| Odonto HPP | 8 | 30.9 (9.0) | 22, 49 | 23.3, 38.4 | P = 0.0467 (regression analysis per HPPSSC) |
| Mild childhood HPP | 7 | 27.6 (8.9) | 17, 40 | 19.3, 35.8 | |
| Severe childhood HPP | 10 | 23.8 (5.5) | 17, 34 | 19.8, 27.8 | |
| Infantile HPP | 2 | 23.5 (2.1) | 22, 25 | 4.4, 42.5 |
Although low circulating FGF7 levels correlated inversely with the clinical severity of pediatric HPP assessed with our HPPSSC, they did not correlate with serum ALP or Pi levels (Ps > 0.2).
F. Age-effect Studies:
Because our study subjects ranged from 1.2 to 17.4 years-of-age, with some being young adults and the CTR group being older than both the XLH and HPP groups, we also analyzed exclusively both our pre-teenage and age > 7.0 years data. All four clinical forms of HPP were represented in both analyses. For both analyses (age > 7.0 year data not shown), the principal findings concerning hyperphosphatemia in HPP were unchanged (Supplementary Appendix, Section II). Hyperphosphatemia in HPP was not explained by secondary hypoparathyroidism from hypercalcemia or by compromised glomerular filtration, and low serum FGF7 levels reflected HPP severity.
G. Effect Of FGF7 And FGF23 Infusion On Urinary Pi Excretion In Rats:
Using FGF23 as the control phosphaturic peptide, FGF7 was phosphaturic in vivo in the rat (Table 15).
Table 15:
Effect of Vehicle, FGF23, or FGF7 on Rat Fractional Excretion of Pi and Plasma Pi Concentration
| Experimental Group | GFR (mL/minute) | FEP (percent) | Plasma Pi (mM) | |||
|---|---|---|---|---|---|---|
| Dose (number) | C | E | C | E | C | E |
| 1. vehicle; (n = 8) | 3.8 ± 0.5 | 3.9 ± 0.5 | 11.9 ± 2.4 | 15.6 ± 2.6 | 1.6 ± 0.1 | 1.7 ± 0.3 |
| 2. FGF7, 0.1 nmoles/kg/hr; (n = 9) | 3.5 ± 0.4 | 3.0 ± 0.3 | 12.8 ± 1.2 | 27.1 ± 2.6* | 1.7 ± 0.1 | 1.8 ± 0.1 |
| 3. FGF23, 0.1 nmoles/kg/hr; (n = 9) | 3.8 ± 0.2 | 4.2 ± 0.7 | 10.8 ± 1.5 | 25.1 ± 2.6* | 1.8 ± 0.1 | 1.7 ± 0.1 |
| 4. FGF7, 0.01 nmoles/kg/hr; (n = 6) | 3.6 ± 0.4 | 3.4 ± 0.5 | 10.5 ± 1.1 | 21.1 ± 3.5* | 1.7 ± 0.1 | 1.8 ± 0.1 |
| 5. FGF23, 0.01 nmoles/kg/hr; (n = 8) | 4.0 ± 0.5 | 3.4 ± 0.4 | 8.3 ± 1.4 | 17.7 ± 1.3* | 1.7 ± 0.1 | 1.8 ± 0.1 |
C = control clearance period, E = experimental clearance period. GFR = Glomerular filtration rate; FEP = fractional excretion of phosphate; Pi = plasma Pi concentration;
= statistically significant P < 0.05. Data are expressed as mean ± standard error of mean.
V). Discussion:
Since the late 1970s, our experience has been that hyperphosphatemia is common in all clinical forms of HPP, consistent with the initial single observation by Rathbun in 1948(1) and then the occasional reporting of this finding by us(25–30) and others.(31) To understand the prevalence and pathogenesis of hyperphosphatemia in HPP, in 1987 we briefly communicated(8,9) that all 27 children and adults representing the broad-ranging clinical severity of HPP had fasting serum Pi levels above mean reference values for age and sex, and that 56% were overtly hyperphosphatemic. Their hyperphosphatemia was explained by increased TmP/GFR although other parameters of mineral homeostasis were normal.(8,9) Herein, we investigated prospectively 41 patients with non-lethal pediatric HPP to establish the prevalence and severity of hyperphosphatemia in this population, and to then explore its pathogenesis. That these patients were correctly diagnosed with HPP should be apparent from our experience with this inborn-error-of-metabolism including its natural history,(32,33) biochemical hallmarks,(23) treatments,(34–38) and clinical nosology validated by ALPL gene analysis.(18) Similarly, underpinning the diagnosis of our pediatric XLH comparator group that helped to validate the methodologies for our study, we had reported their responses to conventional medical therapy,(39) tested treatment with the anti-FGF23 monoclonal antibody burosumab,(21,22) and published mutation analyses including PHEX while excluding other hypophosphatemic disorders (Supplementary Appendix).(19,20) Our cumulative experience now with >200 children with HPP and > 300 children with XLH typically indicated high(8,9) and low(40) TmP/GFR values, respectively. Accordingly, we could probe hypotheses for the hyperphosphatemia of HPP, as discussed below.
A). Hyperphosphatemia As A Clinical Laboratory Assay Artifact?:
In our clinical laboratory, Pi was quantitated using a classic method whereby ammonium molybdate under acidic conditions complexes with Pi.(12) However, there has been no suggestion that the 15–20% of circulating Pi normally complexed or protein-bound is increased in HPP,(40) or that substantial concentration(s) of some unrecognized TNSALP substrate(s) liberates Pi during the assay. In fact, the three natural substrates for TNSALP (PPi, PLP, and PEA)(3,5) would add little Pi because they circulate in only nM - µM concentrations.(23,41–43) Phospholipids could release Pi,(12) but have not been reported to be elevated in HPP. Thus, endogenous compromise of TNSALP to hydrolyze its substrates or other phosphorylated compounds, liberating Pi in this assay, seems an unlikely explanation for the hyperphosphatemia of HPP.
B). Secondary Hypoparathyroidism?:
Hyperphosphatemia in the most severely affected patients with HPP; i.e., the life-threatening perinatal or infantile forms like Rathbun’s landmark case,(1) is readily explained by superabundant ePPi blocking HA crystal formation and growth, and thus Ca and Pi uptake by the skeleton.(44,45) Here, resulting hypercalcemia would suppress circulating PTH and thereby increase renal tubular reclamation of Pi; i.e., hyperphosphatemia from secondary hypoparathyroidism.(37) Also, in such patients, there is sometimes renal failure that would impair urinary excretion of Pi.(33,37) Notably, our study subjects with non-life-threatening HPP had normal kidney filtration, yet even those most mildly affected (i.e., odonto-HPP) were typically hyperphosphatemic despite normal serum total and ionized Ca, magnesium, and PTH levels. Hence, only in the severe perinatal and infantile forms of HPP(33,37) is hyperphosphatemia readily attributable to secondary hypoparathyroidism or renal failure.
C). PPi Excess?:
In healthy individuals, urinary PPi and Pi levels correlate directly (44,45) and Pi administration increases urinary PPi excretion,(46) suggesting competition for a shared transport mechanism.(44) However, increased renal biosynthesis of PPi seems to occur with Pi supplementation,(46) offering an explanation other than a transport effect. In 1965 and 1971, high levels of PPi were discovered in urine(45) and plasma,(41) respectively, in HPP and the increased urinary PPi to Pi ratio was recognized as characteristic of this disorder.(45) Nevertheless high, not low, circulating Pi levels characterize HPP contrary to a shared transport pathogenesis.
Notably, three Mendelian disorders diametrically contrast with HPP by featuring diminished biosynthesis and thereby low extracellular levels of PPi;(47) i.e., the two forms of generalized arterial calcification of infancy (GACI-1 and −2: OMIM #208000 and #614473, respectively)(48) and pseudoxanthoma elasticum (PXE: OMIM #264800).(48) In GACI-1, but inexplicably not in GACI-2 or allelic PXE, hypophosphatemic rickets mediated by excess FGF23 can develop(49,50) whereby the disorder becomes designated autosomal recessive hypophosphatemic rickets, type 2 (ARHR-2; OMIM #613312).(48) GACI-1/ARHR-2 is caused by loss-of-function mutations in the ectonucleotide pyrophosphatase/phosphodiesterase gene ENPP1 (OMIM *173335)(48) that impair ePPi production from eATP. We do not understand why acquired hypophosphatemia fails to characterize allelic GACI-2 and PXE(48) featuring low eATP and ePPi due to mutation of the ATP-binding cassette, subfamily C gene ABCC6 (OMIM *603234).(48) In fact, therapeutic success for the vascular calcification characteristic of PXE has been reported for Pi-binding aluminum hydroxide treatment to lower circulating Pi levels,(51) as supported by a mouse model of PXE.(52) Thus, the hypophosphatemia of GACI-1 viz-a-viz the hyperphosphatemia of HPP suggest low versus high endogenous or urinary levels of PPi somehow condition renal transport of Pi perhaps mediated by FGF23. Elevated FGF23 levels in GACI-1 (ARHR-2), and from our current study normal FGF23 levels in HPP, suggest some relationship between PPi and FGF23 and perhaps other phosphatonins.(50)
Notably, the hyperphosphatemia of HPP seems analogous to the hyperphosphatemia of individuals without HPP but suffering exposure to high levels of the first-generation bisphosphonate (BP) etidronate (ethane-1-hydroxy-1,1-diphosphonate; disodium etidronate: EHDP: Didronel®).(53) To resist TNSALP hydrolysis, BPs have a P-C-P rather than P-O-P core of their parent compound ePPi.(54) Like ePPi, toxic levels of etidronate impair Ca and Pi crystallization to become HA and block HA crystal growth.(51) Although few publications recount the clinical triad,(53) we have been intrigued that etidronate toxicity features rickets or osteomalacia, muscle weakness, and hyperphosphatemia from increased TmP/GFR;(55) all characteristic of HPP. In fact, years before the genetic bases of the two GACIs were discovered and their low ePPi levels identified,(48) etidronate proved useful to treat GACI vascular calcification,(54,56) and in 2018 for the vascular calcification of PXE.(57)
D). Deficient TNSALP In The Kidney Tubule?:
ALP has for decades been viewed as acting in Ca and Pi transport.(58) In keeping with Rathbun’s report in 1948,(1) subsequent investigations of lethal HPP have confirmed profound deficiency of renal ALP activity, primarily in the proximal convoluted tubule(59,60) where most Pi is reabsorbed.(61) Now, the majority of studies indicate the kidneys do not secrete Pi into the urine.(62) If so, hyperphosphatemia in HPP could reflect impairment of this process.(8,9) In 1980, Storelli and Murer(63) studied ALP activity versus Na+-dependent Pi transport in rat brush border membrane vesicles preconditioned by different dietary Pi levels or the ALP inhibitor levamisole, and found Pi transport did not parallel ALP activity or become blocked by levamisole. In 1983, we briefly reported that 32Pi accumulation by HPP fibroblasts is normal despite their profound deficiency of TNSALP activity.(64) That same year, Yusufi et al(65) found that vesicles made from rabbit renal proximal tubule brush border membranes, but then stripped of most of their ALP activity by phospholipase-C, had uncompromised Pi uptake. This too indicated no key role for ALP in renal Na+-gradient-dependent Pi transport. However, 32Pi uptake by these vesicles was actually enhanced.(65) Yusufi et al(65,66) noted in multiple disorders Pi uptake in renal proximal tubules was associated with changes in ALP activity, suggesting undefined mechanisms modulating ALP and Pi transport in tubular cells. Then in 1987, Yusufi and Dousa(66) studied renal Pi handling by isolated kidney tubule cells stripped of plasma membrane ALP. No impairment of Pi uptake was noted, indicating ALP had little direct role in modulating Na+-dependent Pi uptake. In fact, our experience (G.S. Gottesman and M.P. Whyte, unpublished) shows HA-targeted asfotase alfa TNSALP-replacement therapy markedly increases circulating TNSALP activity(37,38) yet fails to correct hyperphosphatemia in HPP, but perhaps by not reaching and repairing the ALP deficiency at the kidney lumen.
E). Phosphatonin Insufficiency?:
Despite the intriguing hyperphosphatemia of HPP, circulating phosphatonin levels have not been characterized in this metabolic bone disease. The phosphatonins comprise FGF23, sFRP4, FGF7/KGF (keratinocyte growth factor), and MEPE(67) which we did not assay. Because they promote renal excretion of Pi,(67) circulating phosphatonin concentrations could be increased by the hyperphosphatemia of HPP. Thus, the low FGF7 and normal FGF23 and sFRP4 levels we observed in pediatric HPP might be factors in its hyperphosphatemia. Phosphaturic effects for FGF23, sFRP4, and MEPE are established,(14,15,68) but producing FGF7 for infusion experiments has been difficult. Elevated circulating FGF7 levels with hypophosphatemia have been reported in tumor-induced osteomalacia,(69,70) and Carpenter et al(71) demonstrated FGF7 inhibits Pi uptake by opossum kidney cells. Herein, we used rats to confirm that FGF7 infusion decreases renal Pi reclamation.
XLH is the most common heritable rickets/osteomalacia.(20) We found characteristically elevated circulating FGF23 levels that would explain our XLH patients’ hypophosphatemia, but additionally normal not low sFRP4 levels while FGF7 levels were low.
Phosphatonins are synthesized in the skeleton,(67) particularly in osteocytes. Loss-of-function mutations in PHEX, dentin matrix protein 1 (DMP1), and ENPP1 elevate circulating FGF23 levels and thereby explain hypophosphatemic rickets/osteomalacia in XLH and ARHR-1 and −2, respectively.(72) However, the mechanism for the defective skeletal mineralization of HPP is different. Notably, FGF7, low in the circulation of our pediatric HPP population, is synthesized in the mesenchyme.(73) Because HA-targeted asfotase alfa treatment can improve the rickets(37,74) and osteomalacia of HPP,(75) it will be interesting to determine whether circulating phosphatonin levels persist or correct if the skeleton fully mends with correction of ePPi levels with this therapy.
Supplementary Material
Highlights:
Hypophosphatasia (HPP) is characterized by alkaline phosphatase (ALP) deficiency
Inorganic pyrophosphate, a mineralization inhibitor, accumulates endogenously
Hyperphosphatemia from increased TmP/GFR spans the severity of pediatric HPP
In pediatric HPP, serum FGF7 levels are low and FGF23 and sFRP4 levels are normal
Hyperphosphatemia in pediatric HPP is associated with phosphatonin insufficiency
VI). Acknowledgements:
We thank the participants and their parents for making our study possible. Our investigations reflect the skill and dedication of the nursing, laboratory, and dietary staffs of the Center for Metabolic Bone Disease and Molecular Research, Shriners Hospitals for Children -St. Louis; St. Louis, MO, USA. Ms. Stacy Sommers assayed the phosphaturic proteins. Ms. Sharon McKenzie provided expert secretarial help. MPW recognizes that early in his career Professor Olav Bijvoet (1928–2014) emphasized to him that hyperphosphatemia is a feature of HPP.
Funded in part by: Shriners Hospitals for Children; Soft Bones, Inc.; The Clark and Mildred Cox Inherited Metabolic Bone Disease Research Fund and The Hypophosphatasia Research Fund at The Barnes-Jewish Hospital Foundation; The Fred C. and Katherine B. Andersen Foundation; and NIH grant RO1 DK107870.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Presented in part at: American Society of Bone and Mineral Research 2018 Annual Meeting, September 28–October 1, 2018, Montreal, Canada [J Bone Miner Res 33 (Suppl 1): 306, 2018].
Disclosures: The authors have nothing to disclose.
VII) References:
- 1.Rathbun JC. Hypophosphatasia: a new developmental anomaly. Am J Dis Child 1948; 75: 822–31. [DOI] [PubMed] [Google Scholar]
- 2.Whyte MP: Hypophosphatasia and how alkaline phosphatase promotes mineralization. Chapter #28 In: “Genetics of Bone Biology and Skeletal Disease”, (2nd Ed) Thakker RV, Whyte MP, Eisman J, Igarashi T (eds) Elsevier (Academic Press), San Diego, CA, pp 481–504, 2018 [Google Scholar]
- 3.Whyte MP: Hypophosphatasia: nature’s window on alkaline phosphatase function in humans, Chapter 66 In “Principles Of Bone Biology” (4th Ed.). Bilezikian JP, Raisz LG, Martin TJ, eds; Elesvier, pps: 1525-55, 2020 [Google Scholar]
- 4.Mumm S, Jones J, Finnegan P, Whyte MP: Hypophosphatasia: molecular diagnosis of Rathbun’s original case. Journal of Bone and Mineral Research 16: 1724–7, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Millan JL, Whyte MP: Alkaline phosphatase and hypophosphatasia. Calcified Tissue International 98: 398–416, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whyte MP: Hypophosphatasia: aetiology, nosology, pathogenesis, diagnosis and treatment. Nature Reviews Endocrinology 12: 233–46, 2016 [DOI] [PubMed] [Google Scholar]
- 7.Tiosano D, Hochberg A. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Res (2009) 27:392–40 [DOI] [PubMed] [Google Scholar]
- 8.Whyte MP, Rettinger SD: Hyperphosphatemia due to enhanced renal reclamation of phosphate in hypophosphatasia (Abstract). J Bone Miner Res 2 (Suppl 1): Abstract 399, 1987 [Google Scholar]
- 9.Rettinger SD, Kim GS, Whyte MP: Hyperphosphatemia due to enhanced renal reclamation of phosphate in hypophosphatasia (Abstract). Clinical Research 32A, 1987 [Google Scholar]
- 10.Whyte MP, Chines A, Silva DP, Landt Y, Ladenson JH: Creatine kinase brain isoenzyme (BB-CK) presence in serum distinguishes osteopetroses among the sclerosing bone disorders. Journal of Bone and Mineral Research 11: 1438–43, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Whyte MP, Kempa LG, McAlister WH, Zhang F, Mumm S, Wenkert D: Elevated serum lactate dehydrogenase isoenzymes and aspartate transaminase distinguish Albers-Schőnberg disease (chloride channel 7 deficiency osteopetrosis) among the sclerosing bone disorders. Journal of Bone and Mineral Research 25: 2515–26, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Henry’s Clinical Diagnosis and Management by Laboratory Methods (22nd Edition) McPherson RA & Pincus MR (eds). Elsevier Saunders, Philadelphia PA; pp 195–196, 2011. [Google Scholar]
- 13.Endo I, Fukumoto S, Ozono K, et al. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone 2008; 42: 1235–9. [DOI] [PubMed] [Google Scholar]
- 14.Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, Jan De Beur SM, Schiavi SC, Kumar R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest 112: 785–94, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berndt TJ, Craig TA, McCormick DJ, Lanske B, Sitara D, Razzaque MS, Pragnell M, Bowe AE, O’Brien SP, Schiavi SC, Kumar R. Biological activity of FGF-23 fragments. Pflugers Arch 454: 615–23, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen P, Toribara T, Warnner H. Microdetermination of phosphorus. Anal Chem 28:1756–58, 1956. [Google Scholar]
- 17.Führ J, Kazmarczyk J, Krüttgen CD. Eine einfache colorimetrische Methode zur Inulin-Bestimmung für Nieren-clearance-untersuchungen bei StoffwechselGesunden und Diabetikern. Klin. Wochenschr 33:729–730, 1955. [DOI] [PubMed] [Google Scholar]
- 18.Whyte MP, Zhang F, Wenkert D, McAlister WH, Mack KE, Benigno MC, Coburn SP, Wagy S, Griffin DM, Ericson KL, Mumm S: Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone 75:229–39, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Mumm S, Huskey M, Cajic A, Wollberg V, Madson KL, Wenkert D, Gottesman GS, McAlister WH, Whyte MP: PHEX 3’-UTR mutation c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J Bone Miner Res 30: 137–43, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Smith PS, Gottesman GS, Zhang F, Cook F, Ramirez B, Wenkert D, Wollberg V, Veis DJ, Huskey M, Mumm S, Whyte MP: X-Linked hypophosphatemia: uniquely mild disease associated with PHEX 3’-UTR c.*231A>G mutation (a retrospective, case-control study). J Bone Miner Res (2020;10.1002/jbmr.3955. doi: 10.1002/jbmr.3955). [DOI] [PubMed] [Google Scholar]
- 21.Carpenter T, Whyte MP, Imel E, Boot A, Hogler W, Linglart A, Padidela R, van’t Hoff W, Mao M, Chen C-Y, Skrinar Al, Kakkis E, San Martin J, Portale A. Burosumab in children with X-linked hypophosphatemia. N Eng J Med 378: 1987–98, 2018. [DOI] [PubMed] [Google Scholar]
- 22.Whyte MP, Carpenter T, Gottesman GS, Mao M, Skinar A, San Martin J, Imel E. Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphatemia: a multicenter, open-label, phase 2 trial. Lancet Diabetes & Endocrinology 7: 189–199, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Whyte MP, Coburn SP, Ryan LM, Ericson KL, Zhang F. Hypophosphatasia: biochemical hallmarks validate the expanded pediatric clinical nosology. Bone 110: 96–106, 2018. [DOI] [PubMed] [Google Scholar]
- 24.Carpenter TO. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin North Am 44:443–66, 1997. [DOI] [PubMed] [Google Scholar]
- 25.Whyte MP, Teitelbaum SL, Murphy WA, Bergfeld M, Avioli LV: Adult hypophosphatasia: clinical, laboratory and genetic investigation of a large kindred with review of the literature. Medicine (Baltimore) 58: 329–47, 1979 [PubMed] [Google Scholar]
- 26.Weinstein RS, Whyte MP: Fifty-year follow up of hypophosphatasia. Archives of Internal Medicine 141: 1720–21 1981 [DOI] [PubMed] [Google Scholar]
- 27.Whyte MP: Alkaline phosphatase: physiologic role explored in hypophosphatasia. In “Bone and Mineral Research” (6th ed). Peck WA, ed; Elsevier Science Publishers, Amsterdam, pp 175–218, 1989 [Google Scholar]
- 28.Moore CA, Ward JC, Rivas ML, Magill HL, Whyte MP: Infantile hypophosphatasia: autosomal recessive transmission to two related sibships. American Journal of Medical Genetics 36: 15–22, 1990 [DOI] [PubMed] [Google Scholar]
- 29.Whyte MP, Kurtzburg J, McAlister WH, Podgornik MN, Mumm SR, Coburn SP, Ryan LM, Miller CR, Gottesman GS, Martin PL: Marrow cell transplantation for infantile hypophosphatasia. Journal of Bone and Mineral Research 18: 624–36, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Whyte MP, Murphy WA, Fallon MD: Adult hypophosphatasia with chondrocalcinosis and arthropathy: variable penetrance of hypophosphatasemia in a large Oklahoma kindred. Am J Med 72: 631–41, 1982 [DOI] [PubMed] [Google Scholar]
- 31.Fraser D Hypophosphatasia. Am J Med 22: 730–46, 1957 [DOI] [PubMed] [Google Scholar]
- 32.Whyte MP, Mumm S, McAlister WH, Mack K, Benigno M, Kempa LG, Franken A, Lim VT, Ericson KL, Coburn SP, Ryan LM, Wenkert D, Zhang F: Hypophosphatasia: natural history study of 101 affected children investigated at one research center. Bone 93:125–38, 2016 [DOI] [PubMed] [Google Scholar]
- 33.Whyte MP, Leung E, Wilcox WR, Liese J, Argente J, Martos-Moreno GA, Reeves A, Fujita KP, Moseley S, Hofmann C: Natural history of perinatal and infantile hypophosphatasia: a retrospective study. J Pediatr 209: 116–24, 2019 [DOI] [PubMed] [Google Scholar]
- 34.Whyte MP, McAlister WH, Patton LS, Magill HL, Fallon MD, Lorentz WB, Herrod HG: Enzyme replacement therapy for infantile hypophosphatasia attempted by intravenous infusions of alkaline phosphatase-rich paget plasma: results in three additional patients. J Pediatrics 105: 926–33, 1984 [DOI] [PubMed] [Google Scholar]
- 35.Martin PL, Thompson J, Reid J, Miller C, Gottesman G, Whyte MP, Kurtzberg J: Marked radiographic improvement in infantile hypophosphatasia with T–depleted bone marrow transplantation from a related haploidentical donor. Blood 90: 398b, 1997. 9207476 [Google Scholar]
- 36.Whyte MP, Mumm S, Deal C: Adult hypophosphatasia treated with teriparatide. Journal of Clinical Endocrinology and Metabolism 92: 1203–8, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Van Sickle B, Wenkert D, Edgar TS, Bauer ML, Hamdan M, Simmons JH, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar A, Crine P, Landy H: Enzyme replacement therapy in life-threatening hypophosphatasia. New England Journal of Medicine 366: 904–13, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Whyte MP, Madson KL, Phillips D, Reeves A, McAlister WH, Yakimoski A, Mack K, Hamilton K, Kagan K, Melian A, Thompson D, Moseley S, Odrljin T, Greenberg CR: Asfotase alfa therapy for children with hypophosphatasia. JCI Insight 1:e85971; 1–10, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen DJ, Boniface AM, Schranck FW, Rupich RC, Whyte MP: X linked hypophosphatemic rickets: a study (with literature review) of growth response to calcitriol and phosphate therapy. J Bone Miner Res 7: 583–97, 1992 [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen H 1983. Hypophosphatasia. In “Metabolic Basis of Inherited Disease” Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, and Brown MS, editors. McGraw-Hill, New York, Fifth ed. 1497 – [Google Scholar]
- 41.Russell RGG, Bisaz B, Donath A, Morgan DB, Fleisch H. Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfecta, and other disorders of bone. J Clin Invest 50: 961–696, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whyte MP, Mahuren DJ, Vrabel LA, Coburn SP: Markedly increased circulating pyridoxa1–5ʹ–phosphate levels in hypophosphatasia: alkaline phosphatase acts in vitamin B6 metabolism. J Clin Invest 76: 752–6, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen K. Phosphorylethanolamine and hypophosphatasia. Danish Medical Bulletin 15: 1–112, 1968. [PubMed] [Google Scholar]
- 44.Russell RG, Edwards NA, Hodgkison A. Urinary pyrophosphate and urolithiasis. Lancet, 2: 1446, 1964 [DOI] [PubMed] [Google Scholar]
- 45.Russell RGG. Excretion of inorganic pyrophosphate in hypophosphatasia. The Lancet 461–464, September 4, 1965. [DOI] [PubMed] [Google Scholar]
- 46.Fleisch H, Bisaz S, Care AD. Effect of orthophosphate on urinary pyrophosphate excretion and the prevention of urolithiasis. Lancet, 1: 1065–7. 1964 [DOI] [PubMed] [Google Scholar]
- 47.Li Q, Aranyi T, Varadi A, Terry SF, Uitto J. Research progress in pseudoxanthoma elasticum and related ectopic mineralization disorders. Journal of Investigative Dermatology, 136: 550–556, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; (Baltimore, MD: ), August 18, 2019. World Wide Web URL: http://omim.org/ [Google Scholar]
- 49.Lorenz-Depiereux B(1), Schnabel D, Tiosano D, Häusler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86: 267–72, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, Zimmermann M, Lanpher B, Zimmerman K, Stabach PR, Klee E, Braddock DT, Wermers RA. Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Miner Res (in press) 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo JY, Blum RR, Singer GK, Stern DK, Emanuel PO, Fuchs W, Phelps RG, Terry SF, Lebwohl MG. A randomized controlled trial of oral phosphate binders in the treatment of pseudoxanthoma elasticum. Journal of the American Academy of Dermatology, 2011-08-January, Volume 65, Issue 2, pp 341–348. [DOI] [PubMed] [Google Scholar]
- 52.Li Q, LaRusso J, Grand-Pierre AE, Uitto J. Magnesium carbonate-containing phosphate binder prevents connective tissue mineralization in Abcc6−/−mice—potential for treatment of pseudoxanthoma elasticum. CTS 2: 398–404, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russell RGG and Fleisch H. Pyrophosphate and diphosphonates in skeletal metabolism. Clinical Orthopaedics and Related Research, 108: 241–263, 1975 [DOI] [PubMed] [Google Scholar]
- 54.Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, Kiffer-Moreira T., Sheen C, Millan JL, Ericson KL, Whyte MP. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res 28: 419–30, 2013. [DOI] [PubMed] [Google Scholar]
- 55.Walton RJ, Russell RGG, Smith R, Kanis JA, Woodhead JS: Effects of a diphosphonate (disodium ethane-1-hydroxy-1,1-diphosphonate: EDHP) on phosphate transport in man. In: Phosphate Metabolism Kidney and Bone, pp. 329–343. Editors: Avioli L, Bordier P, Fleisch, Massry S and Slatopolsky E. Nouvelle Imprimerie Fournie, Toulouse, France, 1976 [Google Scholar]
- 56.Rutsch F, Boyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circulation Cardiovascular Genetics 2008; 1: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kranenburg G, deJong PA, Bartstra JW, Lagerweij SJ, Lam MG, Ossewaarde-van Norel J, risseeuw S, van Leeuwen R, Imhof SM, Verhaar HJ, de Vries JJ, Blart RHJA, Luurtsema G, den Harder AM, Visseren FLJ, Mali WP, Spiering W. Etidronate for prevention of ectopic mineralization in patients with pseudoxanthoma elasticum. J Am College Cardiology, 71: 1117–1126, 2018. [DOI] [PubMed] [Google Scholar]
- 58.McComb RB, Bowers GN, Posen S. “Alkaline Phosphatase”, New York; Plenum Press; 1979. [Google Scholar]
- 59.Gorodischer R, Davidson RG, Mosovich LL, Yaffe SJ. Hypophosphatasia: a developmental anomaly of alkaline phosphatase, Pediatr Res 10: 650–656, 1976. [DOI] [PubMed] [Google Scholar]
- 60.Brydon WG, Crofton PM, Smith AF, Barr DGD, Harkness RA. Hypophosphatasia: enzyme studies in cultured cells and tissues, Biochem. Soc.Trans, 3:927, 1975. [Google Scholar]
- 61.Petit-Clerc C, Plante GE. Renal transport of phosphate: role of alkaline phosphatase. Can J. Physiol. Pharmacol 59: 311–323, 1981. [DOI] [PubMed] [Google Scholar]
- 62.Greger RF, Lang FC, Knox FG, Lechene CP. Absence of significant secretory flux of phosphate in the proximal convoluted tubule. Am J Physiol 232: F235–F238, 1977. [DOI] [PubMed] [Google Scholar]
- 63.Storelli C, Murer H. On the correlation between alkaline phosphatase and phosphate transport in rat renal brush border membrane vesicles. Pflugers Arch, 384: 149–53, 1980. [DOI] [PubMed] [Google Scholar]
- 64.Whyte MP, Vrabel LA: Alkaline phosphatase-deficient hypophosphatasia fibroblasts: normal accumulation of inorganic phosphate in culture (Abstract). Clinical Research 31: 856A, 1983 [Google Scholar]
- 65.Yusufi ANK, Low MG, Turner ST, Dousa TP. Selective removal of alkaline phosphatase from renal brush-border membrane and sodium-dependent brush-border membrane transport. Journal of Biological Chemistry, 258: 5695–5701, 1983. [PubMed] [Google Scholar]
- 66.Yusufi AN, Dousa TP. Studies on rabbit kidney brush border membranes: relationship between phosphate transport, alkaline phosphatase and NAD. Miner Electrolyte Metab 13: 397–404, 1987. [PubMed] [Google Scholar]
- 67.Tebben P, Berndt TJ, Kumar R. Phosphatonins. Chapter 16. In “Osteoporosis” (4th Ed), Pg 373–390, 2013. [Google Scholar]
- 68.Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 34: 303–19, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bansal S, Khazim K, Suri R, Martin D, Werner S, Fanti P. Tumor induced osteomalacia: associated with elevated circulating levels of fibroblast growth factor-7 in addition to fibroblast growth factor-23. Clin Nephrol 2016. January;85(1):57–62. doi: 10.5414/CN108596. [DOI] [PubMed] [Google Scholar]
- 70.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab 2005, 90: 1012–20. [DOI] [PubMed] [Google Scholar]
- 71.Carpenter TO. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab; 30: 1–9, 2012. [DOI] [PubMed] [Google Scholar]
- 72.Drezner MK, Whyte MP: Heritable Renal Phosphate Wasting Disorders. Chapter #40, In: “Genetics of Bone Biology and Skeletal Disease” Thakker RV, Whyte MP, Eisman J, Igarashi T (eds) Elsevier (Academic Press), San Diego, CA, pp 759–80, 2018 [Google Scholar]
- 73.Camalier CE, Yi M, Yu L-R, Hood BL, Conrads KA, Lee YJ, Lin Y, Garneys LM, Bouloux GF, Young MR, Veenstra TD, Stephens RM, Colburn NH, Conrads TP, Beck GR Jr. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol 228: 1536–1550, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whyte MP; Simmons JH, Moseley S, Fujita KP, Bishop N, Salman NJ, Taylor J, Phillips D, McGinn M, McAlister WH: Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes & Endocrinology 7: 93–105, 2019 [DOI] [PubMed] [Google Scholar]
- 75.Kishnani PS, Rockman-Greenberg C, Rauch F, Bhatti MT, Moseley S, Denker AE, Watsky E, Whyte MP: Five-year efficacy and safety of asfotase alfa for adults and adolescents with hypophosphatasia. Bone 121:149–62, 2019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.