

Abstract
1,2-dimethylhydrazine (DMH) is a member in the class of hydrazines, strong DNA alkylating agent, naturally present in cycads. DMH is widely used as a carcinogen to induce colon cancer in animal models. Exploration of DMH-induced colon carcinogenesis in rodent models provides the knowledge to perceive the biochemical, molecular, and histological mechanisms of different stages of colon carcinogenesis. The procarcinogen DMH, after a series of metabolic reactions, finally reaches the colon, there produces the ultimate carcinogen and reactive oxygen species (ROS), which further alkylate the DNA and initiate the development of colon carcinogenesis. The preneolpastic lesions and histopathological observations of DMH-induced colon tumors may provide typical understanding about the disease in rodents and humans. In addition, this review discusses about the action of biotransformation and antioxidant enzymes involved in DMH intoxication. This understanding is essential to accurately identify and interpret alterations that occur in the colonic mucosa when evaluating natural or pharmacological compounds in DMH-induced animal colon carcinogenesis.
Keywords: colon cancer, DMH, preneoplastic lesions, chemoprevention
Background
Colon cancer is the third most leading cancer in males and the second most leading cancer in females in the industrialized countries, and its morbidity and mortality are increasing in the developing countries [1]. Previously, the incidence of colon cancer was low in India and underdeveloped countries, but later studies showed a drastic increase in the colon cancer incidence in Asia [2]. Dietary habits play a crucial role in the development of colorectal cancer. Rapid urbanization and extensive growth of economic conditions influence the people to adopt western dietary style, which consists of high-fat, high-protein, low-carbohydrate, and low dietary fiber. This unbalanced diet is considered to be an important causative factor for the increased mortality in recent years [3]. In olden days, people consumed natural substances with lots of medicinal properties, therefore, they all away from these kind of diseases.
Influence of age, body weight, and sex
The population-based cancer incidence report shows that the morbidity has increased in the past century. The high rate of cancer incidence due to two major factors (i) our life expectancy is more (ii) exposure of cancer-causing chemicals and radiations, X-ray and plane travel and other sources. Today we live 30 years longer than last century. Moreover, cancer is common in older tissues, and elders are more susceptible than youngsters. The laboratory and clinical studies revealed that cancer is an ancient disease; it is not one disease, more than 100 diseases.
Age
The risk of developing colorectal cancer increases with age. Colon cancer is most common in over 50 years of aged people, and a chance of getting colon cancer is higher in each decade. However now the incidence is increasing in younger people also.
Body weight
Obesity is one of the risk factors of colon cancer. In particular, when comparing with healthy men, overweight men (BMI > 30 kg/m2) have a higher risk of 53%. Sedentary lifestyle may be responsible for 13–14% of colon cancer incidence. It is an attributable risk greater than family history.
Sex
Male rats are most frequently used to study preneoplastic and neoplastic lesions in colon carcinogenesis, due to their increased susceptibility to colon carcinogens [4].
Incidence
The incidence of colon cancer is high in male compared to females. Hormonal factors may play a role in having less percentage of colorectal cancer in females. Microsatellite instability is one of the molecular changes in colon cancer. A case-control study examining sex, reproductive factors, and hormone exposure related with microsatellite instability in colon cancer suggested that estrogen exposure is a protective factor against microsatellite instability, while the lack of estrogen in older women increased the risk of microsatellite instability—high risk of colon cancer [5]. Nevertheless, the researchers studied the efficiency of DMH in female mice and found 83% of mice developed visible tumors and many possess especially in the distal part of the colon [6, 7]. The action of DMH induction in mice shows enlargement of proliferative zone in the colonic crypts, which led to an increment in the total number of labeled cells in the crypts and decrement of the activity of enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT) (found in human colonic neoplasm).
Changes in the bowel habits also one of the causative factors for colon cancer [8], which includes (i) increase in intestinal transit time (ii) tiny stool, which consequences of thickening of luminal content (iii) possible interactions with various carcinogens or promoting factors present in the intestine, thus hard stools and constipation might be expected to enhance carcinogen exposure.
1,2-Dimethylhydrazine
Role and history of DMH
Dimethylhydrazine occurs as 1,2-dimethylhydrazine and 1,1-dimethylhydrazine isomers. Both compounds are clear, colorless liquids [9]. 1,1-Dimethylhydrazine is used in the rocket and jet as fuel and is also used as a growth control agent in plants and as a feedstock in chemical syntheses.
DMH and its metabolite, azoxymethane (AOM), are procarcinogens that require metabolic activation to form DNA-reactive products. The alkylating agents DMH and AOM begin their mutagenic activity through the methylation of guanine in DNA at N-7 position. The alkylated guanine is paired with thymidine instead of cytosine by donating a proton, which leads to the modification of bases. Further subsequent replication, mispairing of guanine to thymine and cytosine to adenine, occurs, which leads to mutations in DNA. Metabolism of these procarcinogenic compounds involves various metabolic enzymes, including xenobiotic-metabolizing enzymes, these enzymes process several N oxidation hydroxylation stages, including the formation of ultimate carcinogen methylazoxymethanol (MAM). MAM is a reactive metabolite of DMH and AOM, which readily yields methyldiazonium ion that can alkylate macromolecules in the liver and colon [10], proved by various studies [11, 12].
MAM is a substrate of the nicotinamide adenine dinucleotide (NAD)-dependent dehydrogenase present in the colon and liver, suggesting that the active metabolite of MAM might be the corresponding aldehyde [13, 14], and these metabolites of CYP2E1 are transported to the colon via the bile or bloodstream. The main pathway involves the hepatic conversion of DMH to AOM and azoxymethanol which subsequently undergoes glucuronic acid conjugation and biliary excretion [15]; however, the toxicity of azoxymethanol doses affects the liver, cell membranes, and other organelles, which is supported by the release of aspartate and alanine amino transferases and alkaline phosphatase [16, 17]. The glucuronides reaches the colon, and it further undergoes hydrolysis by bacterial enzymes to produce active carcinogen in the colonic lumen Fig. 1 [18].
Figure 1.
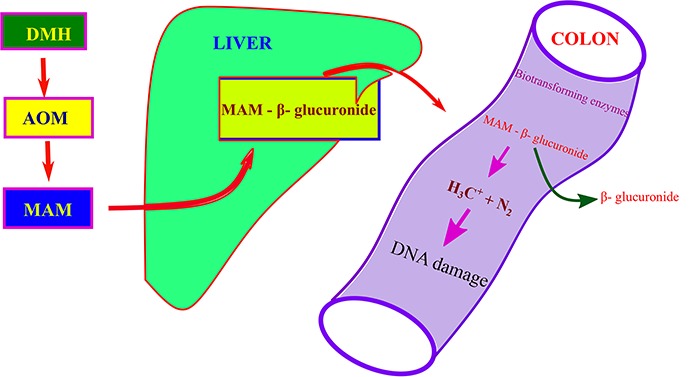
Transport of DMH from subcutaneous site to colon through glucuronidation.
The history of DMH starts around 1965, Laquer (1965) investigating the neurotoxicity for seed of Cycas circinalis L. Rats fed with crude extract of cycad meal produce tumors in various organs including the intestine, liver, and kidney. Further he found that a glycoside, cycasin, and a β-d-glucosyloxyazoxymethane isolated from the crude material and the first metabolite of aglycone cycasin (MAM) are responsible for the tumors in the intestinal tract [19].
Initially Fiala [20] investigated the metabolism and mode of action of DMH, analyzing its metabolites, and separated them by column chromatography. Further he conducted a study with DMH and AOM [15], which confirms that the active carcinogen is not transported through fecal stream rather circulatory system, which was proved by Campbell et al. and Wittig and Ziebarth [21, 22].
Routes and dosage of DMH administration to rodents
The main route of administration of DMH is subcutaneous injection [23]. Even though it is an effective method to induce tumor, intraperitoneal injections also succeeded to produce tumors in the colon [24], whereas single injection intrarectal exposure of DMH also produced tumors in the colon of germ-free mice [25, 26].
The s.c. injection of DMH causes 100% epithelial dysplasia and precancerous lesions, found in a 12-week study [27]. DMH causes a wide range of tumors in the GI tract of C57BL/6 mice, and the majority of tumors found in small intestine and colon in the respective studies [28, 29]. Even though the majority of experimental colon cancer study carried out in rats, the high frequency of tumor in lower part of colon, a histopathological evidence of multiple adenomas and subsequent progression of adenocarcinoma also validates the importance of mice in pathogenesis of colon cancer (Table 1) [30, 31].
Table 1.
List of some effective chemopreventive agents against DMH-induced colon cancer
| S. No | Chemopreventive agent | Dosage level (mg/kg.b.w) | Species and strain | Biomarkers studied | Outcome | Reference | |
|---|---|---|---|---|---|---|---|
| 1. | Olive oil | 1000 | Male Sprague Dawley rats | ↓ | mRNA expression of NF-κB, VEGF, MMP | Chemoprevention | [144] |
| ↑ | mRNA expression of caspases 3 and 9 | ||||||
| 2. | p-Coumaric acid | 100 | Male Wistar rats | ↓ | Bcl-2, Iαβα, cyt C, caspase-3, p65 | Antiproliferative | [145] |
| ↑ | P53, GSp78 | ||||||
| 3. | Rosmarinic acid | 5 | Male Wistar rats | ↓ | CYP450, CYP450 2E1, NADPH, CYP450 red, NADH-cytb5 red | Chemopreventive, proapoptotic | [8] |
| ↑ | LOOH, CD. Phase II drug-metabolizing enzymes | ||||||
| 4. | Myrtenal | 230 | Male Wistar rats | ↓ | CYP450, Cytb5, UDPGT, GST | Anticarcinogenic, anti-inflammatory | [146] |
| ↑ | SOD, GPx, GR, VIT C, VIT E | ||||||
| 5. | p-Methoxycinnamic acid | 40 | Male Wistar rats | ↓ | NF-κB, iNOS, COX-2, TNF-α, IL-6, cyclin D1, BCl-2 | Antiproliferative, anti-invasive | [147] |
| ↑ | p53, Bax | ||||||
| 6. | Etoricoxib | 6 | Male Sprague Dawley rats | ↓ | Alkaline phosphatase, maltase, lactase, LPO | Chemopreventive, protects intestinal membrane | [148] |
| ↑ | SOD, CAT, GST, GSH, NO, citrulline | ||||||
| 0.64 | ↓ | COX-2, NF-κB | Anti-inflammatory, anticancer | [149] | |||
| 7. | Caraway seed oil (Carum carvi L) | 0.0.1% | Male Wistar rats | ↑ | TBARS, GSH, SOD, CAT, GSH, CYP1A1 | Chemopreventive, modulator drug-metabolizing enzymes | [150] |
| 8. | Black cumin oil (Nigelle Sativa L) | 200 | F344 rats | ||||
| 9. | Coriander seeds (Coriandrum sativum) | 10% powdered coriander seeds | Sprague Dawley rats | ↓ | Cholesterol | Antihyperlipedemic, membrane fluidity, and integrity | [57] |
| ↑ | Phospholipids, Neutral salts, Bile acids | ||||||
| 10. | Amorphophallus campanulatus | 250 | Male Wistar rats | ↑ | GSH, GST, GR, GPx, CAT | Chemopreventive, antiproliferative | [151] |
| 11. | Bis-1,7-(2-hydroxyphenyl)-hepta-1,6-diene-3,5-dione (a curcumin analog) | 80 | Male Wistar rats | ↑ | GSH, GPx, GST, SOD, CAT | Chemopreventive, detoxification of carcinogen | [152] |
| 12. | Silibinin | 50 | Male Wistar rats | ↓ | CYP450, CYP 450 2E1, b5 red, CYP450red, TBARS, LPO, CD | Chemopreventive, modulator phases I and II | [153] |
| ↑ | GST, UDPGT, DTD, SOD, CAT, GPx | [53] | |||||
| 13. | Aloin | 100/p.o. | Wistar rats | ↓ | MDA, TNF | Inhibit tumor promotion, anti-inflammatory | [154] |
| ↑ | SOD, CAT, GSH, GPx, GR, GST | ||||||
| 14. | Ellagic acid | 60/p.o. | Wistar rats | ↓ | LPO, pAKT, Bcl-2 | Chemopreventive, proapoptotic, inhibit carcinogen activation | [155, 156] |
| ↑ | SOD, CAT, GPx, GR, GST, GSH, Vit C and E, Total AKT, BAX, CytC, Cas 3 | ||||||
| 40/p.o | Wistar rats | ↓ | Cathepsin D, ALP, LDH, ODC, c-myc | [157] | |||
| 15. | Diclofenac | 8/p.o. | Male Sprague Dawley rats | ↓ | COX-2, NF-κB | Chemopreventive, anti-inflammatory | [158] |
| ↑ | PPARγ, Apaf | ||||||
| 16. | Ginger (Z. officinale Rosc) | 50/p.o. | Wistar rats | ↑ | SOD, CAT, GPx, GST, GR, GSH, VIT A, E, C | Inhibiting during imitation stage, increase antioxidant | [159] |
| 17. | Capsaicin | 50/p.o. | Wistar rats | ↓ | ALT, AST, Caspase-3 | Reduced cell proliferation, chemopreventive | [160] |
| 18. | Morin | 50/p.o. | Male albino Wistar rats | ↓ | TNF-α, COX-2, IL-6, PGE-2, NF-κB, | Anti-inflammatory, proapoptotic | [161] |
| 19. | Naringenin | 50/p.o. | Male albino Wistar rats | ↓ | ROS, MDA, TNF-α, NF-κB, COX-2, iNOS | Chemopreventive, anti-inflammatory | [162] |
| 20. | Orientin | 10/i.p. | Male albino Wistar rats | ↓ | NF-κB, TNF-α, IL-6, iNOS, COX-2, mast cells | Chemopreventive, anti-inflammatory | [163] |
| 21. | Green tea | 1% (W/V) mixed with drinking water | Male albino Wistar rats | ↓ | NF-κB, TNF-α, iNOS, COX-2, VEGF, MMP | Anti-inflammatory, anti-angiogenic | [164] |
| 22. | Copaifera langsdorffii | 40/p.o. | Male albino Wistar rats | ↓ | Comet, ACF | Chemopreventive | [165] |
| 23. | β-Sitosterol | 20/p.o. | Male albino Wistar rats | ↓ | ACF, crypt multiplicity | Chemopreventive | [166] |
| 24. | 6-Methylsulfinyl hexyl isothiocyanate | 40/p.o. | F344 rats | ↓ | ACF, BCAC, PCNA | Chemopreventive, antiproliferative | [167] |
Various doses of DMH can produce the colonic tumors. The dosage ranging from 2 mg to 200 mg/kg b.w. (single injection to 30 injections); and the duration ranging from 8 hours to maximum 78 weeks of latency period (which important for the development of colon tumor) (Table 2), can induce colon tumors in DMH studies regardless of the route of the administration. In the recent years, the dosage levels and routes of administrations were standardized, depending on their experimental studies, 15 mg and 20 mg/kg b.w [32]. DMH and AOM at the dosage levels of 10–20 mg/kg b.w. produce adenomas and adenocarcinomas in rodent model [33]. Even a single injection of 10 mg/kg DMH or AOM produces colon cancers in rats after a latency period of 15–20 months Fig. 2 [34].
Table 2.
The carcinogenic effects of DMH in different animal species and strains and their target organs
| S.No | Dose/route | No. of injections | Total duration of the experiment (weeks) | Affected organs | Tumor arised | Metastasis | Strain | References |
|---|---|---|---|---|---|---|---|---|
| 1. | 10 mg/kg b.w./s.c. | 24 | Colon | Large and small intestines | Male Wistar rats | [168] | ||
| 2. | 20 mg/kg b.w./s.c. | 24 | Colon, liver, small intestine | Large and small intestines | Liver | Male Wistar rats | [168] | |
| 3. | 35 mg/kg b.w/Oral | 78 | Small intestine, Zymbal gland | Male Fischer rats | [169] | |||
| 4. | 30 mg/kg b.w./s.c. | 20 | Colon | Colon | Male Wistar | [170] | ||
| 5. | 30 mg/kg b.w./i.p. | 12 | 24 | Small intestine. Colon | Colon | Small intestine | Male Swiss mice | [171] |
| 6. | 40 mg/kg b.w./s.c. | 4 | 5 | Colon | Male Wistar rats | [165] | ||
| 7. | 30 mg/kg b.w./s.c. | 30 | 30 | Colon | Colon | Male Wistar rats | [164] | |
| 8. | 30 mg/kg b.w./s.c. | 16 | 16 | Colon | Colon | Male Wistar rats | [172] | |
| 9. | 75 mg/kg b.w./s.c. | 1 | 24 hours | Colon | Male F344 rats | [173] | ||
| 10. | 21 mg/kg b.w./s.c. | 12 | 28 | Colon | C57 black male mice | [174] | ||
| 11 | 21 mg/kg b.w. | 15 | 26 | Colon | Colon | Duodenum, jejunum | Female LIO rats | [175] |
| 12. | 40 mg/kg b.w./s.c. | 4 | 4 | Colon | Male Wistar rats | [176] | ||
| 13. | 40 mg/kg b.w./i.p. | 10 | 10 | Colon | Colon | Male Wistar rats |
[177] | |
| 14. | 40 mg/kg b.w./s.c. | 8 | 8 | Colon | Colon | Male F344 rats | [178] | |
| 15. | 40 mg/kg b.w./s.c. | 2 | 2 | Colon | Colon | Male Wistar rats | [179] | |
| 16. | 200 mg/kg b.w. | 1 | 2 days | Liver | Not measured | Fischer 344 rats | [180] | |
| 17. | 80 mg/kg b.w./i.p. | 1 | 8 hours | Colon | Male BDF1 mice | [181] | ||
| 18. | 15 mg/kg b.w./s.c. | 20 | 52 | Colon | Colon | Male F344 rats | [182] | |
| 19. | 50 mg/kg b.w./s.c. | 10 | 35 | Colon, liver, small intestine | Colon | Caecum, small Intestine, liver, abdomen | Male Wistar rats | [183] |
| 20. | 30 mg/kg b.w./s.c. | 10 | 26 | Colon | Colon | Male F344 rats | [184] | |
| 21. | 25 mg/kg b.w./s.c. | 1 | 7 days | Liver | Male Wistar rats | [185] | ||
| 22. | 125 mg/kg b.w./i.p. | 1 | 14 days | Colon | Male Wistar rats | [186] | ||
| 23. | 20 mg/kg. b.w./s.c. | 15 | 30 | Colon | Colon | Male Wistar rats | [163] | |
| 24 | 20 mg/kg. b.w./s.c. | 8 | 31 | Colon | Colon | F344 rats | [187] | |
| 25. | 20 mg/kg. b.w./s.c. | 15 | 30 | Colon | Colon | Caecum | Male Wistar rats | [62] |
Figure 2.
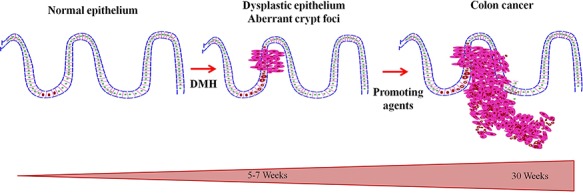
DMH injections induced the mutation in normal epithelium, later multi-step conversion of mutated epithelium to malignant one by 30 weeks.
Metabolism of DMH
An understanding about the chemical carcinogen and mechanism of action is necessary to develop procedures for diagnosis and eventually prevention of the disease.
A number of hydrazine derivatives are found in the environment, industry, agriculture, and medicine [35]. The organotropic colon carcinogen DMH is an alkylating agent widely used to induce benign and malignant neoplasm in the colon of rodents [36].
In animal studies, repeated exposure of DMH produces colon tumors, which shows the pathological features that are similar to sporadic forms of human colon cancer [37]. In most of the studies, carcinogen DMH was injected subcutaneously. The subcutaneous site of injections does not possess the enzymes able to metabolize or react with DMH. Hence, subcutaneously administered DMH is released into the circulation slowly, and then it reaches the liver and gets metabolized into various intermediates [15]. Metabolic activation of DMH is shown in Fig. 3.
Figure 3.
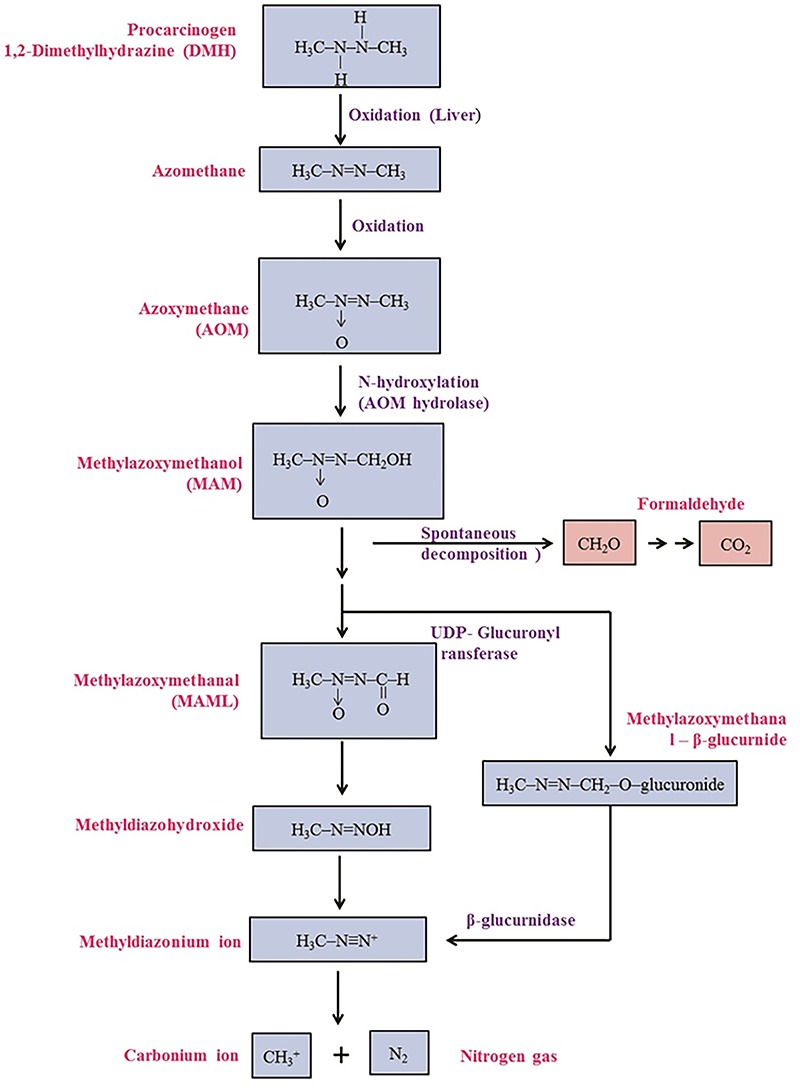
Metabolism of DMH.
The first oxidation step of DMH involves its oxidation to azomethane, which is converted into azoxymethane (AOM) and then hydroxylated to methylazoxymethanol (MAM). Hydroxylation occurs predominantly in the liver, probably via cytochrome P450-dependent pathway and to a limited degree in the colonic mucosa [18]. MAM can reach the intestine through bile, intestinal lumen directly and also via circulation [18], as glucuronides and glucosides to some extent as sulfates. They are cleaved by β-glucuronidase, β-glucosidase, and sulfatase enzymes which are present both in the enterocytes and also in the intestinal microflora. MAM is chemically unstable at body temperature and decomposes spontaneously to formaldehyde, water, and nitrogen [38]. During this decomposition, the alkylating agent methyldiazonium ion is formed, which generates a reactive carbonium ion capable of alkylating macromolecules (DNA, RNA, or protein) by enzymic and non-enzymic process in the colon. Alkylation of the oxygen atoms present in nitrogenous bases turns the possibility of mispairing of DNA, which has been suggested to be critical in mutagenesis and carcinogenesis [39]. Alternatively MAM has been found to be a substrate for NAD+-dependent dehydrogenase of the colon and liver, suggesting that the active metabolite of MAM may be the corresponding aldehyde Fig. 3 Metabolism of DMH [40].
Figure 4.
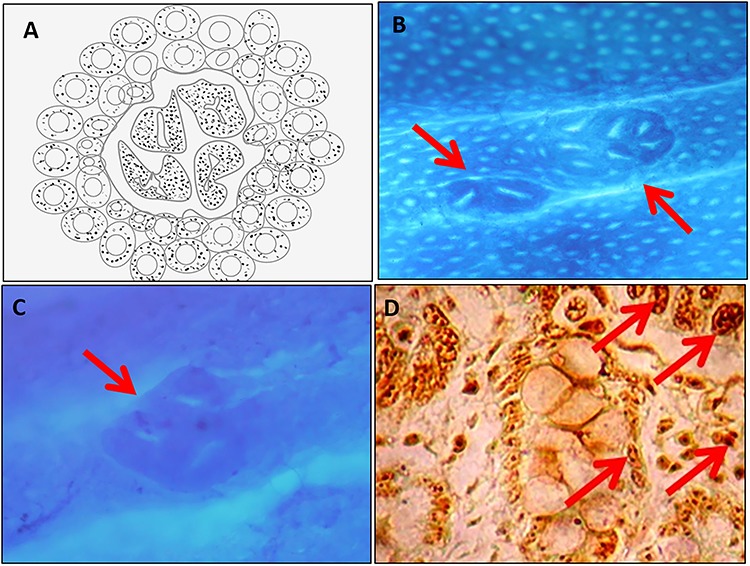
Preneoplastic lesions. (A) Schematic diagram of aberrant crypt foci. (B) Methylene blue-stained aberrant crypt foci. (C) Dysplastic aberrant crypt foci. (D) Argyrophilic nucleolar organizing regions.
Diversity in species
The carcinogenic effects of DMH in different animal species and strains and their target organs are listed in Table 2.
The DMH-induced colon tumors in hamsters are sparse [41, 42]. Both studies evaluated the pathological analyses of colon tumors in hamsters; the percentage of tumor occurrence and severity varies slightly due to the dosage and routes of administration. However, the pathogenicity and metastasis are high in carcinogen injected in s.c. rather than other routes [42, 43]. However, on contradictory, few studies showed that DMH has no tumorigenic influence in Syrian golden hamsters, which shows the species specificity and route specificity of DMH [44–46]. Apart from that, DMH administered to hamster through drinking water was reported to induce cancer in various organs [43] and [45, 46] causes hepatic lesions, forestomach papillomas, and adenocarcinomas of the colon [47]. Even the DMH has the ability to produce tumors in monkey’s colon; many researchers believe that monkeys are resistant to effects of chemical carcinogen for colon since tumors arise in different organs [48, 49].
Promoters
Dietary fat appears to act as a promoter rather than an initiator of colon tumor. Dietary fat may provide a favorable environment for the development and growth of tumor cells, by altering the composition of cell membranes. Epidemiological studies have shown that high-fat diet consumption leads to elevated fecal concentrations of secondary bile acids in the colonic lumen, which stimulate hyperproliferation and thus increase the incidence of colon cancer [50]. High-fat diet, rich in ω-6 polyunsaturated fatty acids, fed in animals during the carcinogenic stage, increases the incidence of colon tumor. It is possible that the high incidence of colon tumors in rats fed with a high-fat diet along with DMH observed in few studies [51–54] may be due to the excretion of elevated amounts of bile acids, which act as colon tumor promoters [55]. High levels of bile acids associated with the high-fat diet can initiate membrane stress signaling, leading to the activation of several complex pathways and alteration in membrane components like cholesterol, thereby disrupting cell membrane integrity. Alternatively, it has been proposed that bile acids increase the risk of colon cancer by inhibiting phase I and phase II xenobiotic-metabolizing enzyme systems located in hepatic and extrahepatic tissues including the colon [50].
High-fat diet and DMH, however, significantly increased HMG CoA reductase activity and cholesterol synthesis in the liver. The enhanced HMG CoA reductase activity may contribute to the modulation of cell growth, cell cycle arrest, inhibition of apoptosis, and cellular signaling activities in DMH-exposed animal studies [56].
Apart from that, DMH exposure increases the cholesterol accumulation, which was benefitted by tumor cells. Cooper et al. have observed that DMH treatment results in doubling of biological membrane cholesterol and gross distortion of cell shape, with changes in the (i) lipid composition, (ii) membrane fluidity, and (iii) morphology [36]. This in turn may present the accumulation and metabolism of secondary bile acids by the colon microflora [57]. The enhancing effect of colon tumor by the high-fat diet depends on the type of fat and their fatty acid composition. In general, dietary fat that contains linoleic acid, a precursor of prostaglandin, which is effective in promoting tumorigenesis in animals [58].
During the progression stage, weight loss is the common feature of the colon cancer, which reflects the aggressiveness of the disease. The tumor burden in the colon results in decreased food intake, therefore decreasing weight gain or increasing weight loss. Alterations in the glucose metabolism and elevated hepatic gluconeogenesis deplete the energy sources, all together results in significant weight loss [59]. The cachexia (catabolic clinical state) may be caused by a combination of endocrine and immunological disturbances resulting from host-tumor interactions. Different mechanisms are involved in the weight loss of starvation and cancerous condition. During the starvation, the weight loss is mainly from adipose tissue and small amount from tissues, whereas in cancer loss, it will be in both adipose tissue and skeletal muscles. In starvation, ketone bodies are produced from fat, which replace glucose, therefore inhibiting the loss of muscle mass.
Preneoplastic Lesions
Observations of preneoplastic lesions
The preneoplastic lesions, which were phenotypically altered by carcinogens, but still they lack of important properties of final tumor cells. Aberrant crypt foci (ACF) initially were identified topographically as the earliest recognizable lesions on the carcinogen-exposed rodent colons. ACF were first described by Bird [60] in methylene blue-stained whole-mount preparations of rodent colon, treated with colon-specific carcinogens. McLellan and Bird defined aberrant crypts to have the following structural features: (i) they are larger than the normal crypts in the field, (ii) have elevated pericryptal zone that separates them from the normal crypts, (iii) have a thicker layer of epithelial cells that often stains darker, and (iv) generally have oval rather than circular openings (normal crypts) [61]. They can be observed as single altered crypts or as a group of altered crypts that appears to form a single unit or focus. Frequently, ACF are microscopically elevated above the mucosa but also may be depressed, i.e. usually they are not in the same focal plane as the surrounding normal crypts. Crypts harboring any four of the abovementioned criteria are considered as aberrant crypts.
DMH treatment induces the formation and multiplicity of ACF, which reflects the initiation stage of colon cancer in rats. Alternatively, high-fat diet also increased the multiplicity of ACF due to its tumor-promoting capabilities. In addition to increased number of ACF, luminal alterations, goblet cell reduction, and nuclear alterations of the cells surrounding the lumen of the crypt are also found [62]. These characteristics can be correlated with outcome of dysplastic ACF. Several studies have suggested that the growth features of ACF and dysplastic ACF and their location are used as a measure of the biological efficacy of the modifiers of colon carcinogenesis [31, 63]. Moreover, ACF and dysplastic ACF represent the earliest detectable lesions (preneoplastic lesions) in the development of colon cancer [64].
Apart from this, western diet alone could produce neoplasm in the colon of experimental animals. The large intake of high-fat diet along with low levels of intake of calcium, vitamin D, folate, choline, methionine, and fiber can induce adenomas and carcinomas of C57BL/6 mice without the exposure of carcinogen [29, 65], by depletion of apoptosis changes in cell renewal showed morphologically identifiable atypical mitotic figure (Fig. 4) [65].
Beta-catenin-accumulated crypts
Venkatachalam et al. demonstrated that the β-catenin-accumulated crypts in the sectioned colonic tissues exhibited histological abnormalities, including the disruption of cellular morphology [62]. Moreover, the β-catenin accumulation in the crypts increased with time after the carcinogen treatment. The crypts with the accumulation of β-catenin were infrequently recognized as adenomatous crypts having extensive branching. Previously, Yamada et al. reported that β-catenin-accumulated crypts in the large bowel harbor frequent mutations in the β-catenin gene, providing evidence that those crypts are premalignant lesions [66].
Histopathology of DMH-Induced Colon Tumors
The evaluation of the changing patterns of tumor development plays a valuable role in the development of anticancer agents. A mass of small adenomas viewable in colon carcinogen exposed rats, which were attached directly to colon and deeply stained red surrounding mucosa and becomes pedunculated. The carcinogen exposure brings severe abnormalities in adenomas (like architecture, cytology, and differentiation) which are expressed as dysplasia. This can be expected as multiple changes in the metabolic pathways that occur on DMH treatment during the conversion of adenomas to adenocarcinoma including growth factors promoting stromal proliferation and angiogenesis, proteolytic enzymes facilitating local invasion, numerous changes in the secretory and membrane-associated glycoproteins, alterations in cell adhesion molecules, and the development of aneuploidy [67]. The drastic increase in the total number of tumors in DMH-exposed rats revealed the rapid conversion of adenoma to adenocarcinoma and the growth of colonic tumors.
The earliest microadenoma is the unicryptal adenoma. The unicryptal adenoma starts as a little outgrowth (a bud) from the side of an apparently normal crypt [68, 69]. This little growth forms a tubule that moves upwards with the normal migration of the crypt column until it reaches the surface epithelium. The neoplastic tubule is usually shorter than its normal counterpart but undergoes fission to produce an oligocryptal adenoma. Growth that is expansile leads to the formation of a polyp [70]. Those adenomas are often flat or depressed but may subsequently become polypoid as their size increases [71].
The last stage of conversion of adenoma to adenocarcinoma must be a rate-limiting step, since adenomas are relatively large in numbers when comparing carcinomas. Furthermore, this step is accompanied by a multiplicity of phenotypic changes implicating enzymes in metabolic pathways [72], increased telomerase activity [73], growth factors promoting stromal proliferation and angiogenesis [74–76], proteolytic enzymes facilitating local invasion [77–80], numerous changes to secretory and membrane-associated glycoproteins [81], alterations in cell adhesion molecules [82], as well as the development of aneuploidy [83].
Role of Bacterial Enzymes
The greatest number of bacterial cells is found in the digestive tract of the human body. Fundamental comparative studies of human fecal microbiota have revealed the astonishing fact that each of us has unique microbiota (i.e. there are considerable differences between the compositions of the microbiota of individuals).
Experimental and clinical studies suggest that there is a link between consumption of red meat, high fats, and low vegetable intake, and alterations in the composition of the gut microbiota have been observed in animal and human studies. This high-fat diet has an impact on increased activities of some fecal bacterial enzymes, as well as modification of sulfidogenesis and biliary acid metabolism with an impact on development of procarcinogenic conditions [84].
Intestinal bacterial enzymes such as β-glucuronidase and β-glucosidase release toxic metabolites from nontoxic glycoside conjugates and extend the exposure time of the toxicant in the body. β-glucuronidase is responsible for the conversion of glucuronide conjugate in the lumen of the gut [85], which leads to the generation of toxic (or) carcinogenic compounds by cleaving the terminal glucuronic acid which was earlier detoxified in the liver by glucuronidation. High-fat diet along with DMH exposure shoots up the fecal β-glucuronidase activities [51]. The increased activities may increase the cleavage of nontoxic to toxic compound, associated with harmful effects on the host, which in particular may lead to the initiation and/or promotion of carcinogenesis [86].
In the same context, β-glucosidase and β-galactosidase are responsible for the generation of aglycone from plant glycosides; therefore these enzymes may hydrolyze DMH to get its toxic metabolite MAM [87].
Mucinase is another one important enzyme hydrolysis protective mucin in the colon. Mucins, basically proteoglycan, form gel, coating the intestinal mucosa and functioning as chemical and mechanical barrier against bacteria, viruses, and toxins [88]. Mucinase activity is altered by the carcinogen exposure in experimental studies; in turn amplified mucinase activities cleave large area of mucin in the colon. Therefore, direct exposure of carcinogen to colonic cells turns the normal cells to malignant one [89].
Nitroreductase (generally distributed in bacteria) is involved in the conversion of dinitrotoluene, nitrobenzenes, and nitropyrenes (aromatic compounds) to amines, which often exhibit toxic, mutagenic, or carcinogenic activities [90].
Fecal sulfatase activity should also be considered in the desulfation of conjugated toxins and in the degradation of sulfated mucins. Changes in the expression of sulfated molecules such as mucins and other glycoconjugates have been demonstrated in the transformed colonic epithelial cells.
The overall types of bacteria in the intestinal tract may be important in evaluating the relationship between diet and intestinal flora rather than the actual number of bacterial organisms [91]. The metabolic activity of intestinal microflora can be modified by dietary factors, for example, they can specifically play a significant role in the conversion of bile acids and neutral sterols to form relative metabolites that can act as promoters [86]. Therefore, fecal bacterial enzymes are known to be involved in the conversion of procarcinogen to proximal carcinogens.
Alterations in Glycoconjugates Levels
Alteration of glycoconjugates in cancerous tissue may be quantitative, qualitative, or both. Glycoproteins of the plasma membrane play an important role in cell-to-cell contact, growth regulation, and binding sites for hormones and lectins.
In tumor cells, alteration of glycoconjugates like hexoses, hexosamine, and fucose may also contribute to the aberrant cell-cell recognition, cell adhesion, antigenicity, and invasiveness of malignant cells [92]. Carbohydrates are early considered as energy sources and structural sources, and proteolytic agents have been slowly refocused on the fact that they have diagnostic and therapeutic potential. New scientific evidence has suggested a relationship between carbohydrate structure and many biological functions.
Oncogenes are known to induce the expression of Golgi β-1,6 N-acetylglucosaminyl transferase in many cell types, leading to increased cellular motility and decreased substratum adhesion [93]. Aranganathan et al. [94] reported that the level of protein-bound hexoses and fucose is elevated in cancerous conditions.
The sialic acid and hexosamine were remarkably lowered in the colon and elevated in the liver of carcinogen-exposed rats [95]. Decreased activity of some glycosyltransferase leads to the reduced level of carbohydrate content in tumor tissues [96].
Role of Xenobiotic-Metabolizing Enzymes
It is generally accepted that the biotransformation of substances foreign to the body (xenobiotics) including drugs is divided into phases I and II.
Cytochrome P450 (CYP), a phase I drug-metabolizing enzyme, is playing a major role in the activation of carcinogens [97]. Since metabolic activation is required for many carcinogens before binding to DNA, individuals with an elevated metabolic capacity to activate specific carcinogens may be at an increased risk of cancer [98]. Therefore, CYP-dependent metabolism not only involving in exert toxicity ot carcinogenicity, but also the targets for phase II enzyme dependent conjugation reactions are formed, rendering them inactive polar products suitable for excretion via the kidneys.
The main role of phase II enzymes is to perform conjugating reactions, such as glucuronidation, sulfation, methylation, acetylation, glutathione, and amino acid conjugation; the respective conjugates are more hydrophilic than the parent compounds [99, 100]. Metabolism of DMH involves several xenobiotic-metabolizing enzymes, which activates the procarcinogen. The rat liver cytochrome P450 2E1 (CYP2E1) metabolizes the DMH metabolites azoxymethane (AOM) and methylazoxymethanol (MAM) in an in vitro microsomal system [101]. Elevated levels of CYP2E1 were reported in DMH-induced colon cancer [102]. In addition, rats administered with β-naphthoflavone, a potent inducer of CYP1A1, exhibited enhanced formation of the promutagenic DNA adduct O6-methylguanine after DMH treatment [103]. It is evident that procarcinogen DMH induces CYP2E1; therefore conversion occurs and the formation of ultimate carcinogen through the biotransformation.
GSH reacts spontaneously or via catalysis by glutathione-S-transferases (GSTs) with numerous activated carcinogens [104], rendering them excretable and less toxic. Decrease in the activities of GST and DTD in the carcinogen-exposed rats shows (i) the conjugation process utilizes excess amount of these enzymes and (ii) the exposure to carcinogen and other tumor promoters reduces the protective ability of these enzymes against cell damage by other metabolites [59].
An increase in phase II detoxification enzymes might be considered to be beneficial, because this could enhance the excretion of carcinogens. Secondary metabolites from plant kingdom are known to block the cytochrome P450 system responsible for converting carcinogenic agents into forms capable of covalent binding with DNA [105].
Modulation of Molecular Pathways
The APC gene encrypts a multifactorial protein that may participate in many cellular events such as cell adhesion and migration, signal transduction, microtubule assembly, and chromosome segregation. However, even though all of these functions are possibly linked with cancer, it seems that the major tumor-suppressing function of APC resides in its ability to precisely regulate intracellular β-catenin levels [106–108].
Moreover, although the majority of colorectal tumors carry mutations in APC, those with an integral APC gene were found to contain activating mutations in β-catenin that alter functionally significant phosphorylation sites [107, 109]. In addition, mutations in other members of the Wnt pathway have been shown to be associated with cancer including conduction [110] and axin [111, 112]. Presently, p53 is known to play a key role in practically all types of human cancers, and the mutation or loss of the p53 gene can be identified in more than 50% of all human cancer cases worldwide.
The p53 mutational spectra also can indicate that a particular cancer did not result from an environmental carcinogen but instead was caused by endogenous mutagenesis. The high frequency of C to T transitions at CpG dinucleotides in colon carcinomas [113] is consistent with mutagenesis by endogenous deamination mechanisms. A C to T transition would be generated by spontaneous deamination of 5-methylcytosine [114] or by enzymatic deamination of cytosine by DNA (cytosine-5)-methyl transferase when S-adenosylmethionine is in limiting concentration (or by both mechanisms) [115]. Because oxygen radicals enhance the rate of deamination of deoxynucleotides [116, 117], chronic inflammation and nitric oxide generated by nitric oxide synthases may explain why rats that inhale particulate materials, which cause inflammation but do not act directly on DNA, have a high incidence of lung cancer [118].
In vivo studies have also shown that p53 mutation has a role in colon cancer progression, but this becomes important only in the late stages of the disease [119]. Thus p53 mutation observed in the advanced tumor stage of colon cancer indicates that p53 mutation may be a late event contributing to tumor progression [120]. The ability of p53 to serve as a prognostic marker has been extensively studied in colorectal cancer, with most studies focusing on increased immunohistochemical staining [121].
Exposure to ultraviolet light is a common carcinogen and is correlated with transition mutations at dipyrimidine sites [122]; dietary aflatoxin B1 exposure is associated with G:C to T:A transversions that bring to the serine substitution at residue 249 of p53 in hepatocellular carcinoma [123, 124]; and exposure to cigarette smoke is correlated with G:C to T:A transversions in lung carcinomas [125].
Inflammation, proliferation, and apoptotic markers (COX-2)
Epidemiologic studies have proved that individuals who consume nonsteroidal anti-inflammatory drugs on a regular basis compared with those not taking these agents have 40–50% reduction in mortality from colon cancer. All of these drugs have one unique property that is their ability to inhibit COX, a key enzyme involved in the conversion of arachidonic acid to prostaglandins. Raised levels of prostaglandin E-2 (PGE-2), the predominant prostaglandin produced by COX-2, are detected in colon cancer tissues and in macrophages derived from colon cancer, whereas PGE-2 is only moderately present in normal mucosa [126]. Elevated COX-2 expression has been found in colon cancer tissues from subjects with clinically diagnosed colorectal cancer [127–129]. There are two mechanisms involved in colon cancer angiogenesis: (i) COX-2 can modulate the production of angiogenic factors by colon cancer cells and (ii) COX-1 regulates angiogenesis in endothelial cells [130].
The COX-2 gene may be regulated by hypoxia via the activation of Nuclear factor κ-B (NF-κB) in human vascular endothelial cells [131], while COX-2 overexpression in cancerous epithelial cells may be induced through the target of normal APC-the β-catenin oncoprotein [132]. The principle role of wild-type APC involves the binding and degradation of β-catenin. Most colorectal cancers have loss of function mutations in the adenomatous polyposis coli (APC) tumor suppressor gene. This leads to the accumulation of β-catenin. COX-2 can be downregulated by wild-type APC induction and upregulated by nuclear accumulation of β-catenin in the presence of mutant APC. The most common mutation in colon cancer is APC gene mutation. Thus, it would suggest a direct role of APC loss in COX-2 overexpression.
Carcinogen exposure makes the upregulation of COX-2 protein expression and also the COX-2 mRNA in rat colon tissues. It may be due to the loss of function of APC and accumulation of β-catenin in carcinogen exposure. COX-2 expression in human tumors can be induced by various growth factors, cytokines, oncogenes, and other factors. IL-1b has been reported to upregulate COX-2 expression in human colorectal cancer cells via multiple signaling pathways [133].
Overexpression of Bcl-2 prevents cells from undergoing apoptosis in response to a variety of stimuli. Overexpression of Bcl-2 prevented the efflux of cytochrome c from the mitochondria and the initiation of apoptosis. Thus, one possible role of Bcl-2 in prevention of apoptosis is to block cytochrome c release from mitochondria. Moreover, dysregulation of cell death genes leading to overexpression of Bcl-2 or reduction in Bax expression, for example, would alter the Bcl-2/Bax ratio which is considered to be anticarcinogenic and vice versa [134].
Nuclear factor κ-B
Activation of NF-κB must be a tightly regulated event. During normal conditions, NF-κB become activate after an appropriate stimuli and upregulated in the transcription of target genes. Then NF-κB came back to inactive state. In cancerous conditions, different types of molecular alterations trigger impaired regulation of NF-κB. In this context, NF-κB constitutively activated leads to deregulated expression of genes under NF-κB control. Alternations in all these processes participate in the development and progression of cancer [135]. Deregulated NF-κB expression has been found in a number of different types of cancer. NF-κB regulates genes responsible for all survival, proliferation, inhibiting apoptosis, and mediate invasion and metastasis. Loss of function of APC genes results in the activation of β-catenin signaling, the foremost step in the development of colon cancer, which was observed in APC Min+ mice model.
It has been reported that TNF receptor superfamily member 19 (TNFRSF19) is a β-catenin target gene, and TNFRSF19 receptor molecule-associated activation of NF-κB signaling has demonstrated that β-catenin may regulate NF-κB activity via TNFRSF19; activation of NF-κB activity has also been observed in the APCMin+ mice model, which was inhibited by Riccardin D [136]. Novel anticancer drugs induce apoptosis in cancer cells, and apoptotic dysfunction leads to the progression of cancer [137]. Usually, anticancer drug treatment results in the activation of caspases, which effectively implement apoptosis. Polyphenols can act as antioxidants as well as prooxidants depending on the tumor environment. Oxidation of polyphenols produces O2, H2O2, and a complex mixture of semiquinones and quinones, all of which are potentially cytotoxic [138–141].
Since NF-κB activation is the result of a multi-step signaling pathway, these compounds may target different points of the signaling process. For example, some anti-inflammatory drugs may inhibit NF-κB by interfering with IKK activity [142]. Other substances such as curcumin, trans-resveratrol, or parthenolide are natural compounds that have been demonstrated to inhibit IKK activity [143]. NF-κB inhibition is also considered to be an important therapeutic target in CRC. Thus, it may be hypothesized that the inhibition of NF-κB by Riccardin D maybe a pivotal mechanism of its effects in chemotherapy for CRC with the APC mutation.
Conclusion and Future Perspectives
Rodent model for colon cancer is one of the best ways to understand the underlying mechanism of colon carcinogenesis and its progression, as well as the comprehensive treatment approaches.
DMH-induced colon carcinogenesis is influenced by age, sex, strain, bodyweight, and, most predominantly, by the diet. From the different routes of DMH administration, we conclude that a single dose of s.c. injection to rats developed a preneoplastic lesions, whereas other routes of administration do not produce feasible results. The same pattern of tumor induction is observed in mice; however the hamsters are slightly resistant to DMH carcinogenicity even administered as s.c. Various studies revealed that high-fat diet plays a major role in the progression of colon tumor in rodents. Rats and mice are the sensitive species for DMH carcinogenicity; however, the latency period is crucial. There is solid proof that the colon tumor induced by DMH showed a very high tumor growth and malignancy behavior by its histopathological evidence and metastatic nature Fig. 5.
Figure 5.
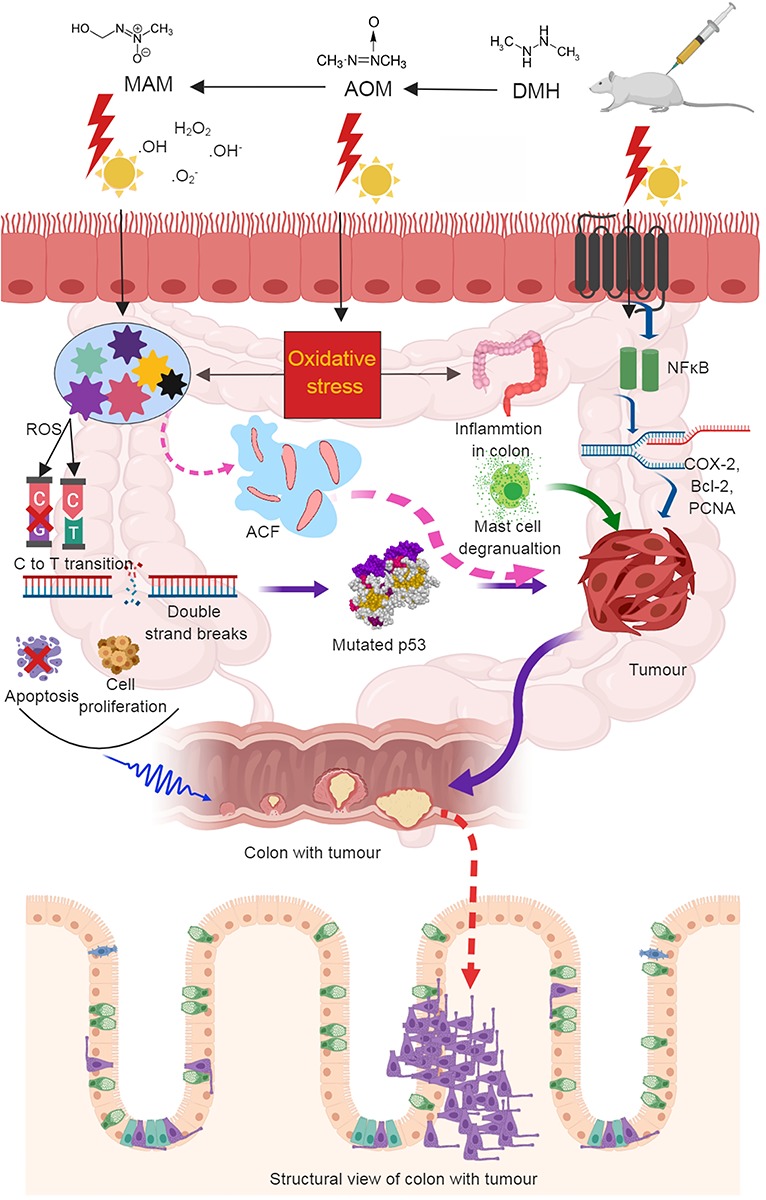
Schematic expression of overall mechanism of DMH-induced carcinogenesis.
Evidenced from many animal studies, it is very clear that natural products from plant kingdom have the strong chemopreventive activities against DMH-induced colon carcinogenesis. These compounds may possess at least one of the following (Table 1) properties such as blocking the initiation of tumor in the local site, inhibiting the progression by influencing in the tumor metabolism, preventing the binding with nucleic acids, and stimulating DNA repair mechanism and inhibiting the cell proliferation and inducing apoptosis. In future, the mechanism behind the tumor microenvironment and pro- and antioxidant roles of natural products need to be explored.
Conflict of Interests
The authors declare that they have no competing interests.
Funding
Not applicable.
Authors’ Contributions
K.V., R.V. and A.M. prepared the first draft of this manuscript, and N.M.I. and R.P. are involved in critical reading, correcting, and idea for schematic figures.
Acknowledgments
None.
References
- 1. Bray F, Ferlay J, Soerjomataram I et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin 2001;51:15–36. [DOI] [PubMed] [Google Scholar]
- 3. Ou J, Carbonero F, Zoetendal EG et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 2013;98:111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moon RC, Fricks CM. Influence of gonadal hormones and age on 1,2-dimethylhydrazine-induced colon carcinogenesis. Cancer 1977;40:2502–8. [DOI] [PubMed] [Google Scholar]
- 5. Slattery ML, Potter JD, Curtin K et al. Estrogens reduce and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res 2001;61:126–30. [PubMed] [Google Scholar]
- 6. Toth B, Malick L, Shimizu H. Production of intestinal and other tumors by 1,2-dimethylhydrazine dihydrochloride in mice. I. A light and transmission electron microscopic study of colonic neoplasms. Am J Pathol 1976;84:69–86. [PMC free article] [PubMed] [Google Scholar]
- 7. Hawks A, Hicks RM, Holsman JW, Magee PN. Morphological and biochemical effects of 1,2-dimethylhydrazine and 1-methylhydrazine in rats and mice. Br J Cancer 1974;30:429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karthikkumar V, Sivagami G, Viswanathan P, Nalini N. Rosmarinic acid inhibits DMH-induced cell proliferation in experimental rats. J Basic Clin Physiol Pharmacol 2015;26:185–200. [DOI] [PubMed] [Google Scholar]
- 9. Trochimowicz H., Kennedy G. Jr and Krivanek N.. Heterocyclic and miscellaneous nitrogen compounds Patty’s Industrial Hygiene and Toxicology. 4th ednNew York, John Wiley & Sons; 1994, 3374–3379. [Google Scholar]
- 10. Fiala ES, Caswell N, Sohn OS et al. Non-alcohol dehydrogenase-mediated metabolism of methylazoxymethanol in the deer mouse, Peromyscus maniculatus. Cancer Res 1984;44:2885–91. [PubMed] [Google Scholar]
- 11. Notman J, Tan QH, Zedeck MS. Inhibition of methylazoxymethanol-induced intestinal tumors in the rat by pyrazole with paradoxical effects on skin and kidney. Cancer Res 1982;42:1774–80. [PubMed] [Google Scholar]
- 12. Fiala ES, Sohn OS, Puz C, Czerniak R. Differential effects of 4-iodopyrazole and 3-methylpyrazole on the metabolic activation of methylazoxymethanol to a DNA methylating species by rat liver and rat colon mucosa in vivo. J Cancer Res Clin Oncol 1987;113:145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zedeck MS, Frank N, Wiessler M. Metabolism of the colon carcinogen methylazoxymethanol acetate. Front Gastrointest Res 1979;4:32. [DOI] [PubMed] [Google Scholar]
- 14. Sohn OS, Fiala ES, Requeijo SP et al. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res 2001;61:8435–40. [PubMed] [Google Scholar]
- 15. Fiala ES. Investigations into the metabolism and mode of action of the colon carcinogens 1,2-dimethylhydrazine and azoxymethane. Cancer 1977;40:2436–45. [DOI] [PubMed] [Google Scholar]
- 16. Parthasarathy R, Sarma GR, Janardhanam B et al. Hepatic toxicity in south Indian patients during treatment of tuberculosis with short-course regimens containing isoniazid, rifampicin and pyrazinamide. Tubercle 1986;67:99–108. [DOI] [PubMed] [Google Scholar]
- 17. Skakun N, Tabachuk O. The comparative action of isoniazid, rifampicin and ethambutol on liver function. Eksp Klin Farmakol 1992;55:45–7. [PubMed] [Google Scholar]
- 18. Weisburger JH. Colon carcinogens: their metabolism and mode of action. Cancer 1971;28:60–70. [DOI] [PubMed] [Google Scholar]
- 19. Laqueur GL. The induction of intestinal neoplasms in rats with the glycoside cycasin and its aglycone. Virchows Arch Pathol Anat Physiol Klin Med 1965;340:151–63. [DOI] [PubMed] [Google Scholar]
- 20. Fiala E. Investigations into the metabolism and mode of action of the colon carcinogen 1,2-dimethylhydrazine. Cancer 1975;36:2407–12. [DOI] [PubMed] [Google Scholar]
- 21. Wittig WGP, Ziebarth D, Der EinRuss D. der Ingesta quf Kanserisierung des Rattendarms durch Dimethylhydrazin. Arch Ceschwulslforsch 1971;37:105–15. [PubMed] [Google Scholar]
- 22. Campbell RL, Singh DV, Nigro ND. Importance of the fecal stream on the induction of colon tumors by azoxymethane in rats. Cancer Res 1975;35:1369–71. [PubMed] [Google Scholar]
- 23. Aranganathan S, Nalini N. Antiproliferative efficacy of hesperetin (citrus flavanoid) in 1,2-dimethylhydrazine-induced colon cancer. PTR 2013;27:999–1005. [DOI] [PubMed] [Google Scholar]
- 24. Ghadi FE, Malhotra A, Ghara AR, Dhawan DK. Modulation of Fourier transform infrared spectra and total sialic acid levels by selenium during 1,2 dimethylhydrazine-induced colon carcinogenesis in rats. Nutr Cancer 2013;65:92–8. [DOI] [PubMed] [Google Scholar]
- 25. Chan PC, Cohen LA, Narisawa T, Weisburger JH. Early effects of a single intrarectal dose of 1,2-dimethylhydrazine in mice. Cancer Res 1976;36:13–7. [PubMed] [Google Scholar]
- 26. Reddy BS, Narisawa T, Weisburger JH. Colon carcinogenesis in germ-free rats with intrarectal 1,2-dimethylhydrazine and subcutaneous azoxymethane. Cancer Res 1976;36:2874–6. [PubMed] [Google Scholar]
- 27. Karaca O, Ertekin T, Canöz Ö et al. Experimental colon tumorigenesis induced by 1,2 dimethylhydrazine in Balb/C mice. Turkish Clin J Med Sci 2010;30:1015–24. [Google Scholar]
- 28. Rowlatt C, Franks LM, Sheriff MU, Chesterman FC. Naturally occurring tumors and other lesions of the digestive tract in untreated C57BL mice. J Nat Cancer Inst 1969;43:1353–64. [PubMed] [Google Scholar]
- 29. Newmark HL, Yang K, Lipkin M et al. A western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis 2001;22:1871–5. [DOI] [PubMed] [Google Scholar]
- 30. Chang WWL. Histogenesis of symmetrical 1,2-Dimethylhydrazine-induced neoplasms of the colon in the Mouse2. J Nat Cancer Inst 1978;60:1405–18. [DOI] [PubMed] [Google Scholar]
- 31. Nambiar PR, Girnun G, Lillo NA et al. Preliminary analysis of azoxymethane induced colon tumors in inbred mice commonly used as transgenic/knockout progenitors. Int J Oncol 2003;22:145–50. [PubMed] [Google Scholar]
- 32. Gurley KE, Moser RD, Kemp CJ. Induction of colon cancer in mice with 1,2-Dimethylhydrazine. Cold Spring Harb Protoc 2015;2015; pdb.prot077453. [DOI] [PubMed] [Google Scholar]
- 33. Thurnherr N, Deschner E, Stonehill E, Lipkin M. Induction of adenocarcinomas of the colon in mice by weekly injections of 1,2-dimethylhydrazine. Cancer Res 1973;33:940–5. [PubMed] [Google Scholar]
- 34. Pozharisski KM. The significance of nonspecific injury for colon carcinogenesis in rats. Cancer Res 1975;35:3824–30. [PubMed] [Google Scholar]
- 35. Choudhary G, Hansen H. Human health perspective on environmental exposure to hydrazines: a review. Chemosphere 1998;37:801–43. [DOI] [PubMed] [Google Scholar]
- 36. Cooper HK, Buecheler J, Kleihues P. DNA alkylation in mice with genetically different susceptibility to 1,2-dimethylhydrazine-induced colon carcinogenesis. Cancer Res 1978;38:3063–5. [PubMed] [Google Scholar]
- 37. Maskens AP. Histogenesis and Growth Pattern of 1,2-Dimethylhydrazine-induced Rat Colon Adenocarcinoma. Cancer Res. 1976;36:1585–92. [PubMed] [Google Scholar]
- 38. Fiala ES, Kulakis C, Bobotas G, Weisburger JH. Detection and estimation of azomethane in expired air of 1,2-dimethylhydrazine-treated rats. J Natl Cancer Inst 1976;56:1271–3. [DOI] [PubMed] [Google Scholar]
- 39. Hawks A, Magee PN. The alkylation of nucleic acids of rat and mouse in vivo by the carcinogen 1,2-dimethylhydrazine. Br J Cancer 1974;30:440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grab DJ, Zedeck MS. Organ-specific effects of the carcinogen methylazoxymethanol related to metabolism by nicotinamide adenine dinucleotide-dependent dehydrogenases. Cancer Res 1977;37:4182–9. [PubMed] [Google Scholar]
- 41. Osswald H, Kruger FW. Carcinogenic effect of 1,2-dimethylhydrazine in gold hamster. Arzneimittelforschung 1969;19:1891–2. [PubMed] [Google Scholar]
- 42. Winneker RC, Tompkins M, Westenberger P, Harris J. Morphological studies of chemically induced colon tumors in hamsters. Exp Mol Pathol 1977;27:19–34. [DOI] [PubMed] [Google Scholar]
- 43. Toth B. Morphological studies of angiosarcomas induced by 1,2-dimethylhydrazine dihydrochloride in Syrian golden hamsters. Cancer Res 1972;32:2818–27. [PubMed] [Google Scholar]
- 44. Jeong JY, Kamino K. Lack of tumorigenic activity of 1,1-dimethylhydrazine in Syrian golden hamsters treated by subcutaneous injection. Experim Toxicol Pathol 1993;45:61–3. [DOI] [PubMed] [Google Scholar]
- 45. Martinello F, Kannen V, Franco JJ et al. Chemopreventive effects of a Tamarindus indica fruit extract against colon carcinogenesis depends on the dietary cholesterol levels in hamsters. Food Chem Toxicol 2017;107:261–9. [DOI] [PubMed] [Google Scholar]
- 46. Paulsen JE, Steffensen IL, Namork E et al. Effect of acetylator genotype on 3,2′-dimethyl-4-aminobiphenyl induced aberrant crypt foci in the colon of hamsters. Carcinogenesis 1996;17:459–65. [DOI] [PubMed] [Google Scholar]
- 47. Moore MA, Thamavit W, Ito N. Comparison of lesions induced in the Syrian golden hamster by diethylnitrosamine, dimethylhydrazine, and dibutylnitrosamine: influence of subsequent butylated hydroxyanisole treatment. J Natl Cancer Inst 1987;78:295–301. [PubMed] [Google Scholar]
- 48. Tobi M, Kim M, Zimmer R et al. Colorectal cancer in the cotton top tamarin (Saguinus oedipus): how do they evade liver metastasis? Dig Dis Sci 2011;56:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takayama S, Thorgeirsson UP, Adamson RH. Chemical carcinogenesis studies in nonhuman primates. Proc Jpn Acad Ser B Phys Biol Sci 2008;84:176–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bernstein H, Bernstein C, Payne CM et al. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 2005;589:47–65. [DOI] [PubMed] [Google Scholar]
- 51. Karthik Kumar V, Vennila S, Nalini N. Modifying effects of morin on the development of aberrant crypt foci and bacterial enzymes in experimental colon cancer. Food Chem Toxicol 2009;47:309–15. [DOI] [PubMed] [Google Scholar]
- 52. Karthik Kumar V, Vennila S, Nalini N. Inhibitory effect of morin on DMH-induced biochemical changes and aberrant crypt foci formation in experimental colon carcinogenesis. Environ Toxicol Pharmacol 2010;29:50–7. [DOI] [PubMed] [Google Scholar]
- 53. Sangeetha N, Aranganathan S, Nalini N. Silibinin ameliorates oxidative stress induced aberrant crypt foci and lipid peroxidation in 1,2-dimethylhydrazine induced rat colon cancer. Invest New Drugs 2010;28:225–33. [DOI] [PubMed] [Google Scholar]
- 54. Sangeetha N, Aranganathan S, Panneerselvam J et al. Oral supplementation of silibinin prevents colon carcinogenesis in a long term preclinical model. Eur J Pharmacol 2010;643:93–100. [DOI] [PubMed] [Google Scholar]
- 55. Kamaleeswari M, Deeptha K, Sengottuvelan M, Nalini N. Effect of dietary caraway (Carum carvi L.) on aberrant crypt foci development, fecal steroids, and intestinal alkaline phosphatase activities in 1,2-dimethylhydrazine-induced colon carcinogenesis. Toxicol Appl Pharmacol 2006;214:290–6. [DOI] [PubMed] [Google Scholar]
- 56. Mo H, Elson CE. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med (Maywood) 2004;229:567–85. [DOI] [PubMed] [Google Scholar]
- 57. Chithra V, Leelamma S. Coriandrum sativum—effect on lipid metabolism in 1,2-dimethyl hydrazine induced colon cancer. J Ethnopharmacol 2000;71:457–63. [DOI] [PubMed] [Google Scholar]
- 58. Carroll KK, Braden LM, Bell JA, Kalamegham R. Fat and cancer. Cancer 1986;58:1818–25. [DOI] [PubMed] [Google Scholar]
- 59. Venkatachalam K, Gunasekaran S, Jesudoss VA, Namasivayam N. The effect of rosmarinic acid on 1,2-dimethylhydrazine induced colon carcinogenesis. Experim Toxicol Pathol 2013;65:409–18. [DOI] [PubMed] [Google Scholar]
- 60. Bird RP. Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett 1987;37:147–51. [DOI] [PubMed] [Google Scholar]
- 61. McLellan EA, Bird RP. Aberrant crypts: potential preneoplastic lesions in the murine colon. Cancer Res 1988;48:6187–92. [PubMed] [Google Scholar]
- 62. Venkatachalam K, Gunasekaran S, Namasivayam N. Biochemical and molecular mechanisms underlying the chemopreventive efficacy of rosmarinic acid in a rat colon cancer. Eur J Pharmacol 2016;791:37–50. [DOI] [PubMed] [Google Scholar]
- 63. Bird RP. Role of aberrant crypt foci in understanding the pathogenesis of colon cancer. Cancer Lett 1995;93:55–71. [DOI] [PubMed] [Google Scholar]
- 64. Mori H, Hata K, Yamada Y et al. Significance and role of early-lesions in experimental colorectal carcinogenesis. Chem Biol Interact 2005;155:1–9. [DOI] [PubMed] [Google Scholar]
- 65. Kim KS, Cho CH, Park EK et al. AFM-detected apoptotic changes in morphology and biophysical property caused by paclitaxel in Ishikawa and HeLa cells. PLoS One 2012;7:e30066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yamada Y, Yoshimi N, Hirose Y et al. Frequent β-catenin gene mutations and accumulations of the protein in the putative preneoplastic lesions lacking macroscopic aberrant crypt foci appearance, in rat colon carcinogenesis. Cancer Res 2000;60:3323–7. [PubMed] [Google Scholar]
- 67. Day DW, Jass JR, Price AB et al. Morson and Dawson's Gastrointestinal Pathology. John Wiley & Sons, 2008. [Google Scholar]
- 68. Wasan HS, Park HS, Liu KC et al. APC in the regulation of intestinal crypt fission. J Pathol 1998;185:246–55. [DOI] [PubMed] [Google Scholar]
- 69. Nakamura S-i, Kino I. Morphogenesis of minute adenomas in familial polyposis coli. J Natl Cancer Inst 1984;73:41–9. [PubMed] [Google Scholar]
- 70. Kubota O, Kino I. Minute adenomas of the depressed type in familial adenomatous polyposis of the colon. A pathway to ordinary polypoid adenomas. Cancer 1993;72:1159–64. [DOI] [PubMed] [Google Scholar]
- 71. Kubota O, Kino I, Nakamura SI. A morphometrical analysis of minute depressed adenomas in familial polyposis coli. Pathol Int 1994;44:200–4. [DOI] [PubMed] [Google Scholar]
- 72. Wattenberg LW. A histochemical study of five oxidative enzymes in carcinoma of the large intestine in man. Am J Pathol 1959;35:113. [PMC free article] [PubMed] [Google Scholar]
- 73. Chadeneau C, Hay K, Hirte HW et al. Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer Res 1995;55:2533–6. [PubMed] [Google Scholar]
- 74. Skinner SA, Frydman GM, O'Brien PE. Microvascular structure of benign and malignant tumors of the colon in humans. Dig Dis Sci 1995;40:373–84. [DOI] [PubMed] [Google Scholar]
- 75. Bossi P, Viale G, Lee AK et al. Angiogenesis in colorectal tumors: microvessel quantitation in adenomas and carcinomas with clinicopathological correlations. Cancer Res 1995;55:5049–53. [PubMed] [Google Scholar]
- 76. Frank RE, Saclarides TJ, Leurgans S et al. Tumor angiogenesis as a predictor of recurrence and survival in patients with node-negative colon cancer. Ann Surg 1995;222:695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mulcahy H, O'Donoghue D, Duffy M et al. Urokinase-type plasminogen activator and outcome in Dukes' B colorectal cancer. The Lancet 1994;344:583–4. [DOI] [PubMed] [Google Scholar]
- 78. Hewitt RE, Leach IH, Powe DG et al. Distribution of collagenase and tissue inhibitor of metalloproteinases (TIMP) in colorectal tumours. Int J Cancer 1991;49:666–72. [DOI] [PubMed] [Google Scholar]
- 79. Tan K, Powe DG, Gray T et al. Regional variations of urokinase-type plasminogen activator in human colorectal cancer: a quantitative study by image analysis. Int J Cancer 1995;60:308–14. [DOI] [PubMed] [Google Scholar]
- 80. Bannasch P. Sequential cellular changes during chemical carcinogenesis. J Cancer Res Clin Oncol 1984;108:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jass J, Smith M. Sialic acid and epithelial differentiation in colorectal polyps and cancer–a morphological, mucin and lectin histochemical study. Pathology 1992;24:233–42. [DOI] [PubMed] [Google Scholar]
- 82. Pignatelli M, Vessey CJ. Adhesion molecules: novel molecular tools in tumor pathology. Hum Pathol 1994;25:849–56. [DOI] [PubMed] [Google Scholar]
- 83. Goh H, Jass J. DNA content and the adenoma-carcinoma sequence in the colorectum. J Clin Pathol 1986;39:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vannucci L, Stepankova R, Grobarova V et al. Colorectal carcinoma: importance of colonic environment for anti-cancer response and systemic immunity. J Immunotoxicol 2009;6:217–26. [DOI] [PubMed] [Google Scholar]
- 85. Gorbach SL. Biochemical methods and experimental models for studying the intestinal flora. Ann Ist Super Sanita 1986;22:739–47. [PubMed] [Google Scholar]
- 86. Reddy BS, Engle A, Simi B, Goldman M. Effect of dietary fiber on colonic bacterial enzymes and bile acids in relation to colon cancer. Gastroenterology 1992;102:1475–82. [DOI] [PubMed] [Google Scholar]
- 87. Ohkami H, Tazawa K, Yamashita I et al. Effects of apple pectin on fecal bacterial enzymes in azoxymethane-induced rat colon carcinogenesis. Japanese J Cancer Res: Gann 1995;86:523–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Reddy BS, Wynder EL. Large-bowel carcinogenesis: fecal constituents of populations with diverse incidence rates of colon cancer. J Natl Cancer Inst 1973;50:1437–42. [DOI] [PubMed] [Google Scholar]
- 89. Goldin BR, Gorbach SL. Alterations of the intestinal microflora by diet, oral antibiotics, and lactobacillus: decreased production of free amines from aromatic nitro compounds, azo dyes, and glucuronides. J Natl Cancer Inst 1984;73:689–95. [PubMed] [Google Scholar]
- 90. Facchini V, Griffiths LA. The involvement of the gastro-intestinal microflora in nitro-compound-induced methaemoglobinaemia in rats and its relationship to nitrogroup reduction. Biochem Pharmacol 1981;30:931–5. [DOI] [PubMed] [Google Scholar]
- 91. Goldin BR, Gorbach SL. The relationship between diet and rat fecal bacterial enzymes implicated in colon cancer. J Natl Cancer Inst 1976;57:371–5. [DOI] [PubMed] [Google Scholar]
- 92. Yogeeswaran G. Cell surface glycolipids and glycoproteins in malignant transformation. Adv Cancer Res 1983;38:289–350. [DOI] [PubMed] [Google Scholar]
- 93. Dennis S, Arambel M, Bartley E, Dayton A. Effect of energy concentration and source of nitrogen on numbers and types of rumen protozoa. J Dairy Sci 1983;66:1248–54. [DOI] [PubMed] [Google Scholar]
- 94. Aranganathan S, Senthil K, Nalini N. A case control study of glycoprotein status in ovarian carcinoma. Clin Biochem 2005;38:535–9. [DOI] [PubMed] [Google Scholar]
- 95. Nalini N, Sabitha K, Viswanathan P, Menon VP. Influence of spices on the bacterial (enzyme) activity in experimental colon cancer. J Ethnopharmacol 1998;62:15–24. [DOI] [PubMed] [Google Scholar]
- 96. Freeman HJ, Kim Y, Kim YS. Glycoprotein metabolism in normal proximal and distal rat colon and changes associated with 1,2-dimethylhydrazine-induced colonic neoplasia. Cancer Res 1978;38:3385–90. [PubMed] [Google Scholar]
- 97. Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 2003;43:149–73. [DOI] [PubMed] [Google Scholar]
- 98. Srinivasan P, Suchalatha S, Babu PV et al. Chemopreventive and therapeutic modulation of green tea polyphenols on drug metabolizing enzymes in 4-Nitroquinoline 1-oxide induced oral cancer. Chem Biol Interact 2008;172:224–34. [DOI] [PubMed] [Google Scholar]
- 99. Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther 2005;106:97–132. [DOI] [PubMed] [Google Scholar]
- 100. Cashman JR, Perotti BY, Berkman CE, Lin J. Pharmacokinetics and molecular detoxication. Environ Health Perspect 1996;104:23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sohn OS, Ishizaki H, Yang CS, Fiala ES. Metabolism of azoxymethane, methylazoxymethanol and N-nitrosodimethylamine by cytochrome P450IIE1. Carcinogenesis 1991;12:127–31. [DOI] [PubMed] [Google Scholar]
- 102. Sohn OS, Fiala ES, Puz C et al. Enhancement of rat liver microsomal metabolism of azoxymethane to methylazoxymethanol by chronic ethanol administration: similarity to the microsomal metabolism of N-nitrosodimethylamine. Cancer Res 1987;47:3123–9. [PubMed] [Google Scholar]
- 103. Tacchi-Bedford AM, Whyman GD, McLean AE. DNA alkylation by 1,2-dimethylhydrazine in the rat large intestine and liver: influence of diet and enzyme induction. Toxicology 1988;50:181–91. [DOI] [PubMed] [Google Scholar]
- 104. Mandel JS, Bond JH, Church TR et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N Engl J Med 1993;328:1365–71. [DOI] [PubMed] [Google Scholar]
- 105. Mandal S, Ahuja A, Shivapurkar NM et al. Inhibition of aflatoxin B1 mutagenesis in salmonella typhimurium and DNA damage in cultured rat and human tracheobronchial tissues by ellagic acid. Carcinogenesis 1987;8:1651–6. [DOI] [PubMed] [Google Scholar]
- 106. Korinek V, Barker N, Morin PJ et al. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997;275:1784–7. [DOI] [PubMed] [Google Scholar]
- 107. Morin PJ, Sparks AB, Korinek V et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997;275:1787–90. [DOI] [PubMed] [Google Scholar]
- 108. Smits R, Kielman MF, Breukel C et al. Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev 1999;13:1309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130–4. [PubMed] [Google Scholar]
- 110. Liu W, Dong X, Mai M et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating β-catenin/TCF signalling. Nat Genet 2000;26:146. [DOI] [PubMed] [Google Scholar]
- 111. Satoh S, Daigo Y, Furukawa Y et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 2000;24:245. [DOI] [PubMed] [Google Scholar]
- 112. Clevers H. Axin and hepatocellular carcinomas. Nat Genet 2000;24:206. [DOI] [PubMed] [Google Scholar]
- 113. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991;253:49–53. [DOI] [PubMed] [Google Scholar]
- 114. Rideout WM 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990;249:1288–90. [DOI] [PubMed] [Google Scholar]
- 115. Shen JC, Rideout WM 3rd, Jones PA. High frequency mutagenesis by a DNA methyltransferase. Cell 1992;71:1073–80. [DOI] [PubMed] [Google Scholar]
- 116. Nguyen T, Brunson D, Crespi CL et al. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci USA 1992;89:3030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wink DA, Kasprzak KS, Maragos CM et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991;254:1001–3. [DOI] [PubMed] [Google Scholar]
- 118. Harris CC. p53 tumor suppressor gene: at the crossroads of molecular carcinogenesis, molecular epidemiology, and cancer risk assessment. Environ Health Perspect 1996;104:435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Fazeli A, Steen RG, Dickinson SL et al. Effects of p53 mutations on apoptosis in mouse intestinal and human colonic adenomas. Proc Natl Acad Sci USA 1997;94:10199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Qin C, Nguyen T, Stewart J et al. Estrogen up-regulation of p53 gene expression in MCF-7 breast cancer cells is mediated by calmodulin kinase IV-dependent activation of a nuclear factor kappaB/CCAAT-binding transcription factor-1 complex. Mol Endocrinol (Baltimore, MD) 2002;16:1793–809. [DOI] [PubMed] [Google Scholar]
- 121. Graziano F, Cascinu S. Prognostic molecular markers for planning adjuvant chemotherapy trials in Dukes' B colorectal cancer patients: how much evidence is enough? Ann Oncol: Off J European Soc Med Oncol 2003;14:1026–38. [DOI] [PubMed] [Google Scholar]
- 122. Brash DE, Rudolph JA, Simon JA et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA 1991;88:10124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hsu IC, Metcalf RA, Sun T et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991;350:427–8. [DOI] [PubMed] [Google Scholar]
- 124. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991;350:429–31. [DOI] [PubMed] [Google Scholar]
- 125. Takeshima Y, Seyama T, Bennett WP et al. p53 mutations in lung cancers from non-smoking atomic-bomb survivors. Lancet (London, England) 1993;342:1520–1. [DOI] [PubMed] [Google Scholar]
- 126. Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 1994;35:675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sano H, Kawahito Y, Wilder RL et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res 1995;55:3785–9. [PubMed] [Google Scholar]
- 128. Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA 1997;94:3336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tsujii M, Kawano S, Tsuji S et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998;93:705–16. [DOI] [PubMed] [Google Scholar]
- 130. Einspahr JG, Bowden GT, Alberts DS. Skin cancer chemoprevention: strategies to save our skin. Recent Results Cancer Res 2003;163:151–64; discussion 264-156. [DOI] [PubMed] [Google Scholar]
- 131. Schmedtje JF Jr, Ji YS, Liu WL et al. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem 1997;272:601–8. [DOI] [PubMed] [Google Scholar]
- 132. Bright-Thomas RM, Hargest R. APC, beta-catenin and hTCF-4; an unholy trinity in the genesis of colorectal cancer. European J Surg Oncol 2003;29:107–17. [DOI] [PubMed] [Google Scholar]
- 133. Liu W, Reinmuth N, Stoeltzing O et al. Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. Cancer Res 2003;63:3632–6. [PubMed] [Google Scholar]
- 134. Zhang Z, Li M, Wang H et al. Antisense therapy targeting MDM2 oncogene in prostate cancer: effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. Proc Natl Acad Sci USA 2003;100:11636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Nair A, Venkatraman M, Maliekal TT et al. NF-kappaB is constitutively activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix. Oncogene 2003;22:50–8. [DOI] [PubMed] [Google Scholar]
- 136. Nishisho I, Nakamura Y, Miyoshi Y et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253:665–9. [DOI] [PubMed] [Google Scholar]
- 137. Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ 2007;15:243. [DOI] [PubMed] [Google Scholar]
- 138. Jornot L, Petersen H, Junod AF. Hydrogen peroxide-induced DNA damage is independent of nuclear calcium but dependent on redox-active ions. Biochem J 1998;335:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Hockenbery DM, Oltvai ZN, Yin XM et al. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241–51. [DOI] [PubMed] [Google Scholar]
- 140. Tamarit J, Cabiscol E, Ros J. Identification of the major oxidatively damaged proteins in Escherichia coli cells exposed to oxidative stress. J Biol Chem 1998;273:3027–32. [DOI] [PubMed] [Google Scholar]
- 141. Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 1998;273:11401–4. [DOI] [PubMed] [Google Scholar]
- 142. Yamamoto Y, Gaynor RB. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr Mol Med 2001;1:287–96. [DOI] [PubMed] [Google Scholar]
- 143. Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001;411:1017–21. [DOI] [PubMed] [Google Scholar]
- 144. Nanda N, Mahmood S, Bhatia A et al. Chemopreventive role of olive oil in colon carcinogenesis by targeting noncoding RNAs and methylation machinery. Int J Cancer 2019;144:1180–94. [DOI] [PubMed] [Google Scholar]
- 145. Sharma SH, Rajamanickam V, Nagarajan S. Antiproliferative effect of p-Coumaric acid targets UPR activation by downregulating Grp78 in colon cancer. Chem Biol Interact 2018;291:16–28. [DOI] [PubMed] [Google Scholar]
- 146. Lokeshkumar B, Sathishkumar V, Nandakumar N et al. Anti-oxidative effect of Myrtenal in prevention and treatment of colon cancer induced by 1,2-dimethyl hydrazine (DMH) in experimental animals. Biomol Ther (Seoul) 2015;23:471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gunasekaran S, Venkatachalam K, Namasivayam N. Anti-inflammatory and anticancer effects of p-methoxycinnamic acid, an active phenylpropanoid, against 1,2-dimethylhydrazine-induced rat colon carcinogenesis. Mol Cell Biochem 2019;451:117–29. [DOI] [PubMed] [Google Scholar]
- 148. Mittal N, Kanwar SS, Sanyal SN. The effect of etoricoxib, a cyclooxygenase-2-specific inhibitor, on the 1,2-dimethylhydrazine-administered rat intestinal membrane structure and function. Toxicol Mech Methods 2008;18:53–62. [DOI] [PubMed] [Google Scholar]
- 149. Tanwar L, Vaish V, Sanyal SN. Chemoprevention of 1,2-dimethylhydrazine-induced colon carcinogenesis by a non-steroidal anti-inflammatory drug, etoricoxib, in rats: inhibition of nuclear factor kappaB. Asian Pac J Cancer Prev 2009;10:1141–6. [PubMed] [Google Scholar]
- 150. Dadkhah A, Allameh A, Khalafi H, Ashrafihelan J. Inhibitory effects of dietary caraway essential oils on 1,2-dimethylhydrazine-induced colon carcinogenesis is mediated by liver xenobiotic metabolizing enzymes. Nutr Cancer 2011;63:46–54. [DOI] [PubMed] [Google Scholar]
- 151. Ansil PN, Prabha SP, Nitha A, Latha MS. Chemopreventive effect of Amorphophallus campanulatus (Roxb.) blume tuber against aberrant crypt foci and cell proliferation in 1,2-dimethylhydrazine induced colon carcinogenesis. Asian Pac J Cancer Prev 2013;14:5331–9. [DOI] [PubMed] [Google Scholar]
- 152. Devasena T, Rajasekaran K, Menon VP. Bis-1,7-(2-hydroxyphenyl)-hepta-1,6-diene-3,5-dione (a curcumin analog) ameliorates DMH-induced hepatic oxidative stress during colon carcinogenesis. Pharmacol Res 2002;46:39–45. [DOI] [PubMed] [Google Scholar]
- 153. Sangeetha N, Viswanathan P, Balasubramanian T, Nalini N. Colon cancer chemopreventive efficacy of silibinin through perturbation of xenobiotic metabolizing enzymes in experimental rats. Eur J Pharmacol 2012;674:430–8. [DOI] [PubMed] [Google Scholar]
- 154. Hamiza O, Rehman M, Khan R et al. Chemopreventive effects of aloin against 1,2-dimethylhydrazine-induced preneoplastic lesions in the colon of Wistar rats. Hum Exp Toxicol 2014;33:148–63. [DOI] [PubMed] [Google Scholar]
- 155. Umesalma S, Sudhandiran G. Chemomodulation of the antioxidative enzymes and peroxidative damage in the colon of 1,2-dimethyl hydrazine-induced rats by ellagic acid. Phytother Res 2010;24:S114–9. [DOI] [PubMed] [Google Scholar]
- 156. Umesalma S, Sudhandiran G. Ellagic acid prevents rat colon carcinogenesis induced by 1,2-dimethyl hydrazine through inhibition of AKT-phosphoinositide-3 kinase pathway. Eur J Pharmacol 2011;660:249–58. [DOI] [PubMed] [Google Scholar]
- 157. Kumar KN, Raja SB, Vidhya N, Devaraj SN. Ellagic acid modulates antioxidant status, ornithine decarboxylase expression, and aberrant crypt foci progression in 1,2-dimethylhydrazine-instigated colon preneoplastic lesions in rats. J Agric Food Chem 2012;60:3665–72. [DOI] [PubMed] [Google Scholar]
- 158. Kaur J, Sanyal SN. Modulation of inflammatory changes in early stages of colon cancer through activation of PPARγ by diclofenac. Eur J Cancer Prev 2010;19:319–27. [DOI] [PubMed] [Google Scholar]
- 159. Manju V, Nalini N. Chemopreventive efficacy of ginger, a naturally occurring anticarcinogen during the initiation, post-initiation stages of 1,2dimethylhydrazine-induced colon cancer. Clin Chim Acta 2005;358:60–7. [DOI] [PubMed] [Google Scholar]
- 160. Caetano BFR, Tablas MB, Pereira NEF et al. Capsaicin reduces genotoxicity, colonic cell proliferation and preneoplastic lesions induced by 1,2-dimethylhydrazine in rats. Toxicol Appl Pharmacol 2018;338:93–102. [DOI] [PubMed] [Google Scholar]
- 161. Sharma SH, Kumar JS, Chellappan DR, Nagarajan S. Molecular chemoprevention by morin–a plant flavonoid that targets nuclear factor kappa B in experimental colon cancer. Biomed Pharmacother 2018;100:367–73. [DOI] [PubMed] [Google Scholar]
- 162. Rehman MU, Rahman Mir MU, Farooq A et al. Naringenin (4,5,7-trihydroxyflavanone) suppresses the development of precancerous lesions via controlling hyperproliferation and inflammation in the colon of Wistar rats. Environ Toxicol 2018;33:422–35. [DOI] [PubMed] [Google Scholar]
- 163. Thangaraj K, Vaiyapuri M. Orientin, a C-glycosyl dietary flavone, suppresses colonic cell proliferation and mitigates NF-κB mediated inflammatory response in 1,2-dimethylhydrazine induced colorectal carcinogenesis. Biomed Pharmacother 2017;96:1253–66. [DOI] [PubMed] [Google Scholar]
- 164. Sadik NA. Chemopreventive efficacy of green tea drinking against 1,2-dimethyl hydrazine-induced rat colon carcinogenesis. Cell Biochem Funct 2013;31:196–207. [DOI] [PubMed] [Google Scholar]
- 165. Senedese JM, Alves JM, Souza Lima IM et al. Chemopreventive effect of Copaifera langsdorffii leaves hydroalcoholic extract on 1,2-dimethylhydrazine-induced DNA damage and preneoplastic lesions in rat colon. BMC Complement Altern Med 2013;13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Baskar AA, Ignacimuthu S, Paulraj GM, Al Numair KS. Chemopreventive potential of β-sitosterol in experimental colon cancer model-an in vitro and in vivo study. BMC Complement Altern Med 2010;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kuno T, Hirose Y, Yamada Y et al. Chemoprevention of 1,2-dimethylhydrazine-induced colonic preneoplastic lesions in Fischer rats by 6-methylsulfinylhexyl isothiocyanate, a wasabi derivative. Oncol Lett 2010;1:273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Castleden W, Shilkin K. Diet, liver function and dimethylhydrazine-induced gastrointestinal tumours in male Wistar rats. Br J Cancer 1979;39:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Schiller CM, Curley WH, McConnell EE. Induction of colon tumors by a single oral dose of 1,2-dimethylhydrazine. Cancer Lett 1980;11:75–9. [DOI] [PubMed] [Google Scholar]
- 170. Ghadi FE, Malhotra A, Ghara AR, Dhawan DK. Chemopreventive effects of selenium on cancer marker indices and ultrastructural changes during 1,2 dimethylhydrazine-induced colon carcinogenesis in rats. J Gastrointest Cancer 2013;44:54–9. [DOI] [PubMed] [Google Scholar]
- 171. Paula Carli A, Abreu Vieira PM, Silva KT et al. Bowman-Birk inhibitors, proteasome peptidase activities and colorectal pre neoplasias induced by 1,2-dimethylhydrazine in Swiss mice. Food Chem Toxicol 2012;50:1405–12. [DOI] [PubMed] [Google Scholar]
- 172. Jain K, Dhawan DK. Regulation of biokinetics of (65)Zn by curcumin and zinc in experimentally induced colon carcinogenesis in rats. Cancer Biother Radiopharm 2014;29:310–6. [DOI] [PubMed] [Google Scholar]
- 173. Femia AP, Salvianti F, Luceri C et al. Sustained proliferation and resistance to apoptosis after a cytotoxic insult are early alterations in rat colon carcinogenesis. Int J Cancer 2012;131:529–36. [DOI] [PubMed] [Google Scholar]
- 174. Kissow H, Hartmann B, Holst JJ et al. Glucagon-like peptide-1 (GLP-1) receptor agonism or DPP-4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul Pept 2012;179:91–100. [DOI] [PubMed] [Google Scholar]
- 175. Anisimov VN, Popovich IG, Zabezhinski MA. Melatonin and colon carcinogenesis: I. inhibitory effect of melatonin on development of intestinal tumors induced by 1,2-dimethylhydrazine in rats. Carcinogenesis 1997;18:1549–53. [DOI] [PubMed] [Google Scholar]
- 176. Barbosa LC, Furtado RA, Bertanha HC et al. Chemopreventive effects of (−)-hinokinin against 1,2-dimethylhydrazine-induced genotoxicity and preneoplastic lesions in rat colon. J Nat Prod 2014;77:2312–5. [DOI] [PubMed] [Google Scholar]
- 177. Zhu QC, Gao RY, Wu W et al. Effect of a high-fat diet in development of colonic adenoma in an animal model. World J Gastroenterol 2014;20:8119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Chihara T, Shimpo K, Kaneko T et al. Inhibition of 1,2-dimethylhydrazine-induced mucin-depleted foci and O(6)-methylguanine DNA adducts in the rat colorectum by boiled garlic powder. Asian Pac J Cancer Prev 2010;11:1301–4. [PubMed] [Google Scholar]
- 179. Summart R, Chewonarin T. Purple rice extract supplemented diet reduces DMH-induced aberrant crypt foci in the rat colon by inhibition of bacterial beta-glucuronidase. Asian Pac J Cancer Prev 2014;15:749–55. [DOI] [PubMed] [Google Scholar]
- 180. Hayes MA, Rushmore TH, Goldberg MT. Inhibition of hepatocarcinogenic responses to 1,2-dimethylhydrazine by diallyl sulfide, a component of garlic oil. Carcinogenesis 1987;8:1155–7. [DOI] [PubMed] [Google Scholar]
- 181. Li YQ, Fan CY, O'Connor PJ et al. Target cells for the cytotoxic effects of carcinogens in the murine small bowel. Carcinogenesis 1992;13:361–8. [DOI] [PubMed] [Google Scholar]
- 182. Barrow BJ, O'Riordan MA, Stellato TA et al. Enzyme-altered foci in colons of carcinogen-treated rats. Cancer Res 1990;50:1911–6. [PubMed] [Google Scholar]
- 183. Alink GM, Kuiper HA, Hollanders VM, Koeman JH. Effect of heat processing and of vegetables and fruit in human diets on 1,2-dimethylhydrazine-induced colon carcinogenesis in rats. Carcinogenesis 1993;14:519–24. [DOI] [PubMed] [Google Scholar]
- 184. Dolara P, Luceri C, De Filippo C et al. Red wine polyphenols influence carcinogenesis, intestinal microflora, oxidative damage and gene expression profiles of colonic mucosa in F344 rats. Mutat Res 2005;591:237–46. [DOI] [PubMed] [Google Scholar]
- 185. Oliveira PF, Andrade KJ, Paula MC et al. Bixin protects hepatocytes against 1,2-dimethylhydrazine-induced genotoxicity but does not suppress DNA damage and pre-neoplastic lesions in the colon of Wistar rats. Mutat Res Genet Toxicol Environ Mutagen 2014;759:37–42. [DOI] [PubMed] [Google Scholar]
- 186. Kannen V, Marini T, Zanette DL et al. The melatonin action on stromal stem cells within pericryptal area in colon cancer model under constant light. Biochem Biophys Res Commun 2011;405:593–8. [DOI] [PubMed] [Google Scholar]
- 187. Jikihara H, Qi G, Nozoe K et al. Aged garlic extract inhibits 1,2-dimethylhydrazine-induced colon tumor development by suppressing cell proliferation. Oncol Rep 2015;33:1131–40. [DOI] [PubMed] [Google Scholar]