

SUMMARY
The diversity and heterogeneity within high-grade serous ovarian cancer (HGSC), which is the most lethal gynecologic malignancy, is not well understood. Here, we perform comprehensive multi-platform omics analyses, including integrated analysis, and immune monitoring on primary and metastatic sites from highly clinically annotated HGSC samples based on a laparoscopic triage algorithm from patients who underwent complete gross resection (R0) or received neoadjuvant chemotherapy (NACT) with excellent or poor response. We identify significant distinct molecular abnormalities and cellular changes and immune cell repertoire alterations between the groups, including a higher rate of NF1 copy number loss, and reduced chromothripsis-like patterns, higher levels of strong-binding neoantigens, and a higher number of infiltrated T cells in the R0 versus the NACT groups.
Graphical Abstract
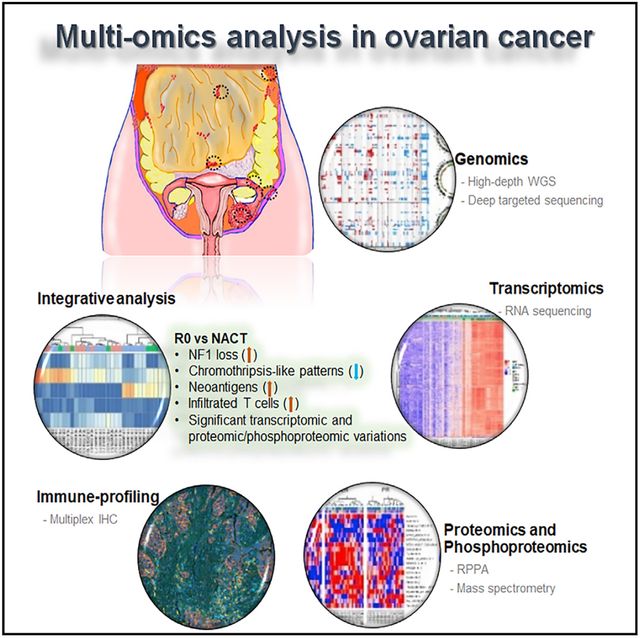
In Brief
High-grade serous ovarian cancer (HGSC) patients with no gross residual disease (R0) after primary surgery have the greatest improvement in clinical outcomes. A deep understanding of molecular and cellular heterogeneity of HGSC is lacking. Findings by Lee et al. highlight major molecular and cellular differences between clinically defined subgroups of HGSC.
INTRODUCTION
Ovarian cancer is a highly diverse disease with a high rate of overall mortality (Siegel et al., 2015). High-grade serous ovarian cancer (HGSC) is the most common and aggressive type of epithelial ovarian cancer, exhibiting high levels of tumor heterogeneity and variable clinical outcomes (Jayson et al., 2014). Molecular abnormalities in HGSC include TP53 mutations in virtually all tumors, somatic or germline BRCA mutations in ~25% (Burgess and Puhalla, 2014; Cancer Genome Atlas Research Network, 2011; Hennessy et al., 2010; Schrader et al., 2012), and extensive copy number changes and CCNE1 amplification (Cancer Genome Atlas Research Network, 2011; Patch et al., 2015; Walsh et al., 2011). The heterogeneity and apparent adaptability of the HGSC genome under selective pressure by chemotherapy likely explains the high rates of drug resistance (Bowtell et al., 2015; Koti et al., 2015; Vaughan et al., 2011). Despite prior efforts such as The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network, 2011; Weinstein et al., 2013; Labidi-Galy et al., 2017; Patch et al., 2015) and other analyses that were predominantly focused on samples from patients with HGSC who had upfront debulking surgery, an understanding of the molecular and cellular heterogeneity of HGSC based on highly clinically annotated samples is lacking.
The extent of residual disease following upfront cytoreductive surgery for HGSCisone ofthe strongest prognosticfactors for progression-free and overall survival (du Bois et al., 2009; Winter et al., 2008). Neoadjuvant chemotherapy (NACT) followed by interval cytoreductive surgery has been offered as an alternative approach, especially where full cytoreductive surgery is not feasible (Ansquer et al., 2001). While primary complete gross resection (R0) is related to better clinical outcomes, it is unknown whether that benefit is the result of aggressive surgical efforts or biological differences inherent in disease that is amendable to complete resection. To address this question, a consistent approach to upfront management is needed. We implemented a quality improvement program using a validated laparoscopic scoring algorithm; this approach enables the collection of well-annotated samples prior to definitive surgery or chemotherapy (Nick et al., 2015; Fleming et al., 2018). In the current study, to assess molecular and cellular differences between clinically defined groups, we carried out a highly detailed analysis of primary tumors and multiple metastatic sites from patients with HGSC who had R0 resection versus those who were triaged to NACT, consisting of intravenous paclitaxel and carboplatin, and had either excellent or poor response. HGSC tissue samples were subjected to high-pass whole-genome sequencing (WGS), targeted deep sequencing (Chen et al., 2015), RNA sequencing (RNA-seq), reverse-phase protein array (RPPA), mass spectrometry (MS)-based proteomics and phosphoproteomics, immune profiling, and integrated data analysis.
RESULTS
Patient Population
The study design and the demographic and clinical characteristics for the 30 patients with HGSC are described in Figure 1 and Table S1. The patient groups evaluated were as follows: R0, no visible residual disease after primary surgery (n = 10); NACT-PR, poor response to NACT (n = 10); and NACT-ER, excellent response to NACT (n = 10).
Figure 1. Outline of the Study.
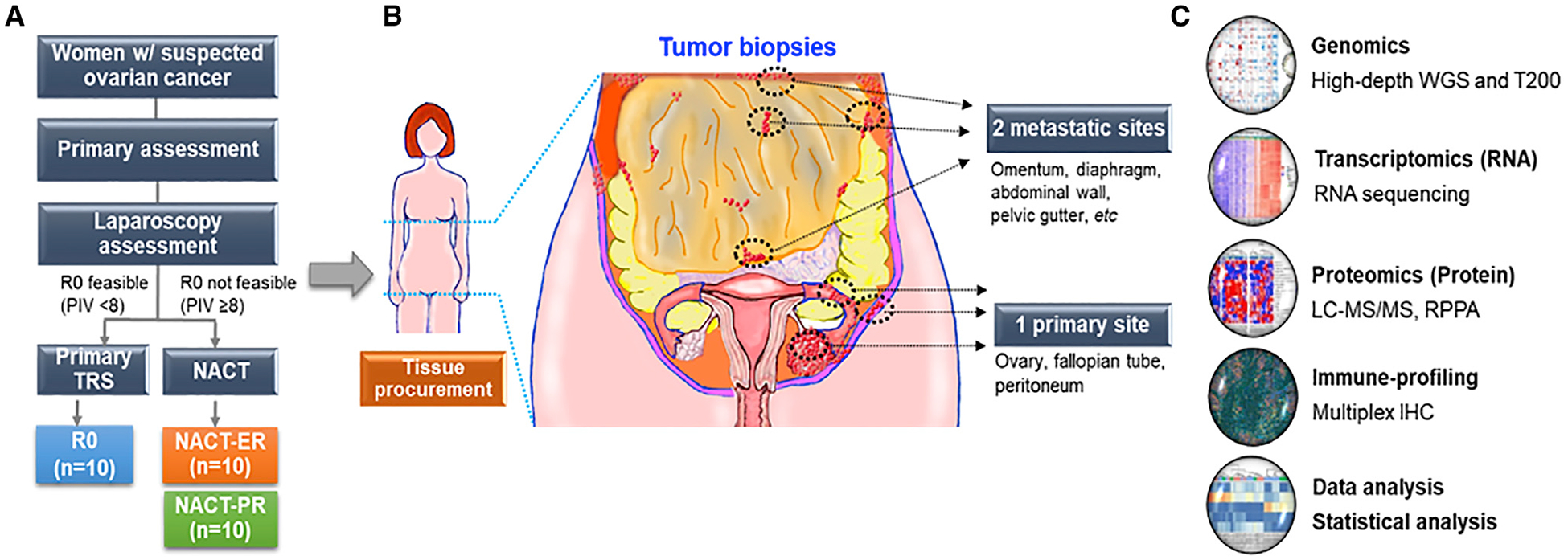
(A) Flow diagram of the study for tissue procurement in patients with advanced high-grade serous ovarian cancer (HGSC). PIV, predictive index value; TRS, tumor reduction surgery; NACT, neoadjuvant chemotherapy; R0, no residual disease; NACT-ER, excellent response to NACT; NACT-PR, poor response to NACT.
(B) The areas of collection of tumor tissues from primary and multiple metastatic sites in patients with HGSC.
(C) Multi-omics and downstream analyses were performed using DNA, RNA, proteins, and immune cells from tumor tissues. WGS, whole-genome sequencing; T200, high-depth targeted exome sequencing platform; LC-MS/MS, liquid chromatography-tandem mass spectrometry; RPPA, reverse phase protein array.
Somatic Mutation Analysis by WGS
We performed WGS analyses with average somatic coverage of 118X (from one primary and two metastatic tissue samples per patient) and germline coverage of 38X (from patient-matched blood samples). For 75 samples with high-purity tumors (≥75% proportion of cancer cells), an average of 13,653 somatic variants from each sample were identified for the entire cohort. Within the coding regions, on average, 66 missense mutations, 4 nonsense mutations, 31 silent mutations, and 15 small InDels were found per sample. The mutation load was not statistically significantly different among the three groups or between the R0 and the combined NACT-ER/PR groups (Figure S1A). Based on a known list of driver genes (Bamford et al., 2004; Cancer Genome Atlas Research Network, 2011), a driver mutation landscape was plotted (Figures 2A and S1B). Overall, 14 ovarian-cancer-associated genes were found mutated in our patient cohort. As expected, the most frequently mutated gene was TP53 in both primary and metastatic sites in all three groups. However, nonsense mutations in TP53 were exclusively identified in the NACT groups, while in R0, most TP53 mutations were missense mutations. Specifically, among all NACT-ER and NACT-PR cases including primary and metastatic sites, 36% and 15.4% carried nonsense mutations in TP53, respectively. Among all R0 cases, 62.5% carried TP53 missense mutations. Also, nonsense mutations in CSMD3 and PIK3CA were exclusively identified in both primary and metastatic sites in the NACT-PR group. When comparing primary and metastatic sites, the majority of the mutations (82%) in the driver genes were consistent. Next, utilizing all somatic mutations in our cohort, we identified two distinct mutation signatures that are highly correlated with Signature 3 and Signature 5 reported by the Catalogue of Somatic Mutations in Cancer (COSMIC) database (Figure S1C). Interestingly, both signatures were enriched in all three patient groups (Figure S1D). Signature 3 has been found in breast, ovarian, and pancreatic cancers and is strongly associated with germline and somatic BRCA1 and 2 mutations in ovarian cancers (Polak et al., 2017).
Figure 2. Somatic Mutations and CNVs Identified in Ovarian Cancer Genes.
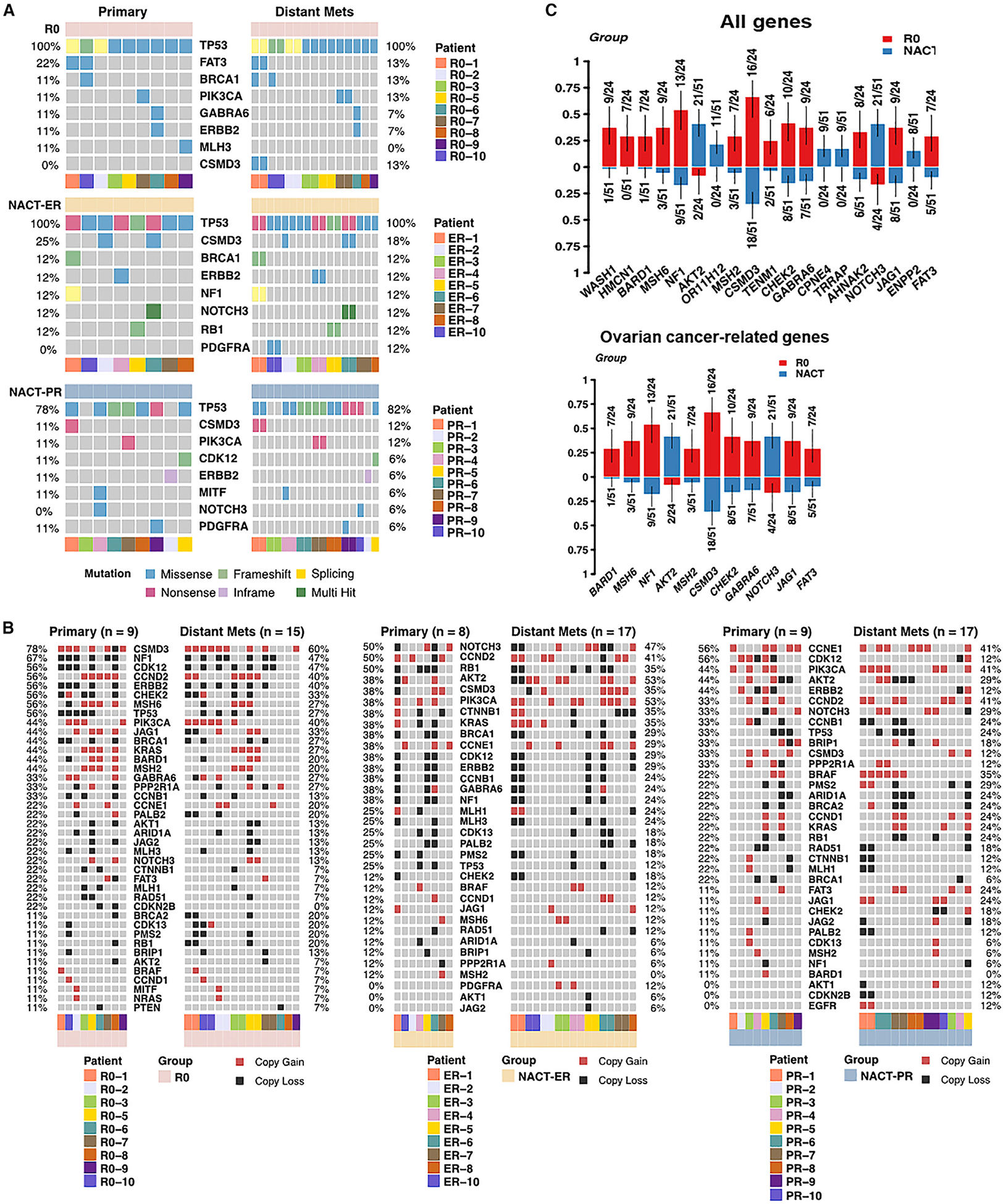
(A) Oncoplots show the frequency of each type of somatic mutation in ovarian cancer genes for primary and metastatic samples. Each column represents one sample.
(B) The frequency of CNVs identified in each ovarian-cancer-related gene. Each column represents one sample. Red represents copy gains, and black represents copy losses.
(C) The enriched abnormalities, including somatic mutations and CNVs, in the R0 versus NACT-ER/PR groups with a significant p value of < 0.05 in the group-wise comparison. The y axis represents the proportion of patient samples carrying the mutation in the corresponding genes.
To address intra-patient tumor heterogeneity, we first compared the repertoire of somatic mutations identified in primary and metastatic sites from each patient (Figures S2A–S2C). Substantial heterogeneity was observed, with median percentages of trunk mutations of 23% in the R0 group, 31% in the NACT-ER group, and 29% in the NACT-PR group. We next performed clonal analysis for all the somatic mutations identified in WGS to determine the extent to which genotypically distinct clonal cell populations (referred to herein as “clones”) exist in the HGSC subsets (Figure S2D). The number of mutation clusters and the cellular prevalence of clones were estimated for each patient (both primary and metastatic sites). The number of clones, the size of major clones, and the cellular prevalence of major clones were compared among the groups. We observed large variations within each patient group, and no statistically significant differences were found between any of the groups. Next, we constructed phylogenetic trees of tumors for each group (Figures S2E–S2G). We observed three possible scenarios. One implies that a dominant clone seeds all metastases, where the primary tumor and all metastases share the same founding driver mutations. Another scenario implies that a subclone evolves and gives rise to all metastases. The third scenario implies that a new subclone with additional driver mutations evolves and seeds some metastases.
Copy Number and Structure Variations
Next, we examined copy number variations (CNVs) and structure variations (SVs) across the entire genome, and no significant differences in the number of CNV breakpoints were found among the three groups (Figures S3A and S3B). We also examined the frequency of CNVs in primary and metastatic sites in ovarian cancer genes for the three groups (Figure 2B) and compared the enriched mutations of all genes and ovarian-cancer-related genes in the R0 compared to the NACT-ER/PR group (Figure 2C) and the NACT-ER compared to the NACT-PR group (Figure S3C). The most frequent CNVs in the R0 group were copy number gain/loss of CSMD3 (67%) and copy number loss of NF1 (54%), CDK12 (50%), and CCND2 (46%) in both primary and metastatic sites. In comparison, the most frequent CNVs in the NACT-ER group were copy number gains/losses of NOTCH3 (48%) and CCND2 (44%) in both primary and metastatic sites, while the most frequent in the NACT-PR group were copy gains of CCNE1 (46%) and PIK3CA (42%) in both primary and metastatic sites. Interestingly, copy number losses of NF1 were significantly lower in the NACT-ER/PR group (18%, p = 0.002), especially in the NACT-PR group (8%, p = 0.0004), than in the R0 group (54%). In contrast, CNVs of AKT2 were more frequently observed in the NACT (NACT-ER, 48%; NACT-PR, 35%) than in the R0 (8%, p = 0.004) groups. We further performed copy number signature analysis and compared the similarity between reported copy number signatures in HGSC (Macintyre et al., 2018) and the seven signatures identified in our cohort (Figures S3D–S3H). Based on the signature exposure in each sample, no significant differences were observed among the R0, NACT-ER, and NACT-PR groups. Next, we examined differences in SVs and SV signatures reported in ovarian cancers. Based on the scores of five SV signatures in each patient, no significant patterns of clusters were observed among the three groups. We identified lists of SVs in ovarian-cancer-associated genes for the three groups, and no recurrent SVs across patients within each group were observed.
Confirmation of WGS Findings by Deep Targeted Sequencing
While high-pass WGS is known to be quite reliable for identifying mutations, we also utilized high-depth targeted sequencing using the T200.2 panel to test for the consistency of sequencing results. The average sequencing coverage was 350X, which provided sufficient power to detect somatic mutations with low variant allele frequency (VAF). Very high correlation was obtained for VAF between WGS and T200.2 sequencing (Pearson correlation = 0.963, Spearman correlation = 0.964). The somatic mutations in cancer-related genes identified by WGS were confirmed by T200.2 sequencing (Figure S3I). The copy number profiles in ovarian cancer genes identified in the T200.2 panel were also consistent with the WGS results. Specifically, NF1 losses were identified in 58.3% of the R0, 20% of the NACT-ER, and 11.5% of the NACT-PR groups with the T200.2 panel, consistent with our WGS findings. We did not observe significant differences between CNVs in primary and metastatic sites, except those in the NACT-PR group; CNVs in CDK12 were enriched in primary compared to metastatic sites (primary, 56%; metastases, 12%; p = 0.03).
Spectrums of Genomic Structural Rearrangement
Given the role of chromothripsis in cancer development (Forment et al., 2012; Korbel and Campbell, 2013; Stephens et al., 2011), we next examined for chromothripsis-like patterns (CTLPs) and the status of telomere length (TL) across the groups. CTLPs were predicted and identified through copy number profiles for all samples. Examples of non-CTLPs and identified CTLPs are shown in Figure S4A. The number of copy number switches and log10 likelihood ratios (log10LRs) were calculated, and we identified CTLPs from 20 out of 75 samples. CTLPs in the R0 group showed fewer copy number switches and lower log10LRs, compared with the NACT-ER and NACT-PR groups (Figures 3A and 3B). In the R0 group, most CTLPs were detected on chromosomes 6 and 19, where CCNE1, NOTCH3, and AKT2 are located. In the NACT groups, CTLPs were enriched on chromosomes 8 and 17, where TP53 and NF1 are located. In a parallel analysis, we also estimated the TLs for tumor and germline samples for each patient. To exclude the factors potentially affecting TLs other than cancer, the TL ratios of tumor versus germline were further calculated. Overall, the TL ratios were higher in the NACT-PR than in the NACT-ER and R0 groups, especially in primary tumors; these differences did not reach statistical significance (Figure S4B).
Figure 3. CTLPs and Strong-Binding Neoantigens by Patient Group.
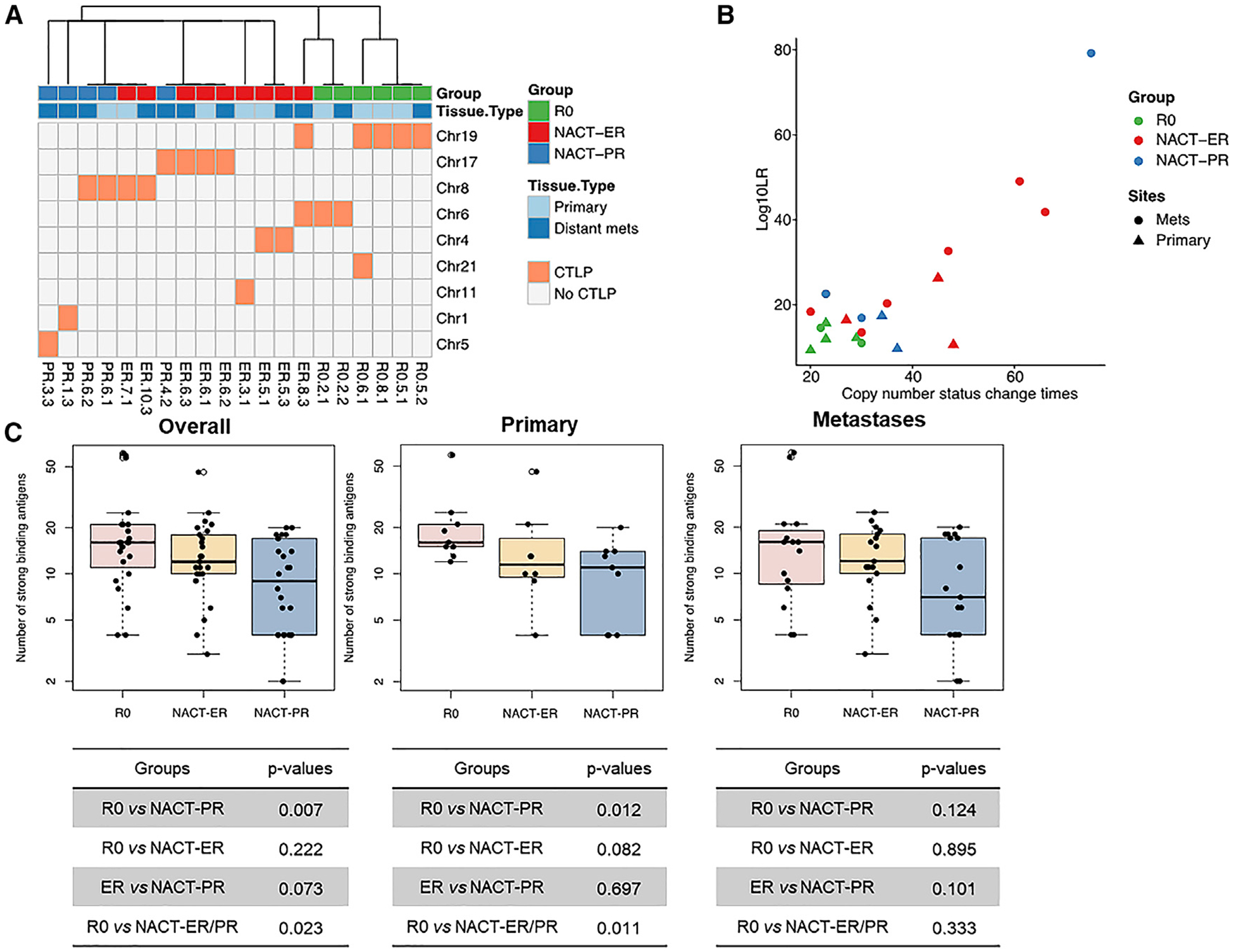
(A) CTLPs identified in nine different chromosomes in different patient groups.
(B) The scatterplot of copy number status changes and the likelihood of CTLP in different patient groups.
(C) Left: the number of strong-binding antigens detected in all tumors, including both primary and metastasis samples. Significant differences were observed between the R0 and NACT-ER/PR groups and between the R0 and NACT-PR groups. Middle: the number of strong-binding antigens detected in primary tumors. Significant differences were observed between the R0 and NACT-ER/PR groups and between the R0 and NACT-PR groups. Right: the number of strong-binding antigens detected in distant metastasis tumors. A peptide was identified as a strong binder if the % rank was below 0.5% or binding affinity (IC50) was below 50.
Prediction of Neoantigens Using WGS Data
Given the importance of neoantigens derived from tumor-specific mutations in cancer immunity (Gubin et al., 2015; Schumacher and Schreiber, 2015), we next identified and compared the numbers of neoantigens in each tumor sample by predicting the major histocompatibility complex (MHC) class I peptide binding affinity with somatic mutations from WGS analysis. According to predicted binding affinity, strong- and weak-binding neoantigens were identified. The median numbers of all neoantigens per sample identified were 99, 81, and 66 in the R0, NACT-ER, and NACT-PR groups, respectively (Table S2). The neoantigen levels were significantly higher in both the R0 (p = 0.023) and NACT-ER (p = 0.033) groups than in the NACT-PR group (Figure S4C). Furthermore, when excluding predicted weak-binding neoantigens, the median numbers of neoantigens identified were 16, 12, and 9 in the R0, NACT-ER, and NACT-PR groups, respectively. The R0 group showed a significantly higher level of strong-binding neoantigens than the NACT groups (p = 0.02; Figure 3C). The difference was even more significant when considering only primary tumors (p = 0.01). Additionally, we analyzed the correlations between the neoantigen level and other features including mutation loads, CNV loads, CTLPs, and mismatch repair (MMR) CNVs (Figure S4D). A strong positive correlation was observed between the level of strong-binding neoantigens and mutation load (R = 0.68, p < 0.0001), which is consistent within all three groups. We also observed some minor positive correlations between the neoantigen level with CNV load (R = 0.29, p = 0.013) and MMR CNVs (R = 0.3, p = 0.001). However, these minor positive correlations varied across patient groups.
Identification of Differentially Expressed Genes (DEGs) by RNA-Seq
Next, we investigated gene expression differences among the R0, NACT-ER, NACT-PR, and NACT-ER/PR groups. Unsupervised hierarchical clustering was performed based on the 3000 most variable genes, and three main clusters were found across the groups (Figure S5A). We identified 206 DEGs from the comparison between the R0 and NACT-ER/PR groups (Figure S5B; Table S3). Of the 206 DEGs in the R0 versus the NACT-ER/PR groups, 67 were curated protein-coding transcripts (33 NM_ or 1 XM_) from NCBI RefSeq (Pruitt et al., 2005) (Figure 4A); these included POU3F3, NKX6–1, and PROK1, which were upregulated in the R0 compared to the NACT-ER/PR group. Interestingly, differential expression of noncoding RNAs (ncRNAs) miR-7–2 (significantly upregulated) and miR-3142 (significantly downregulated) was seen in the R0 group compared to the NACT-ER/PR group (Figure S5C).
Figure 4. DEGs for the Groups, Identified by RNA-Seq, Proteomics, and Phosphoproteomics.
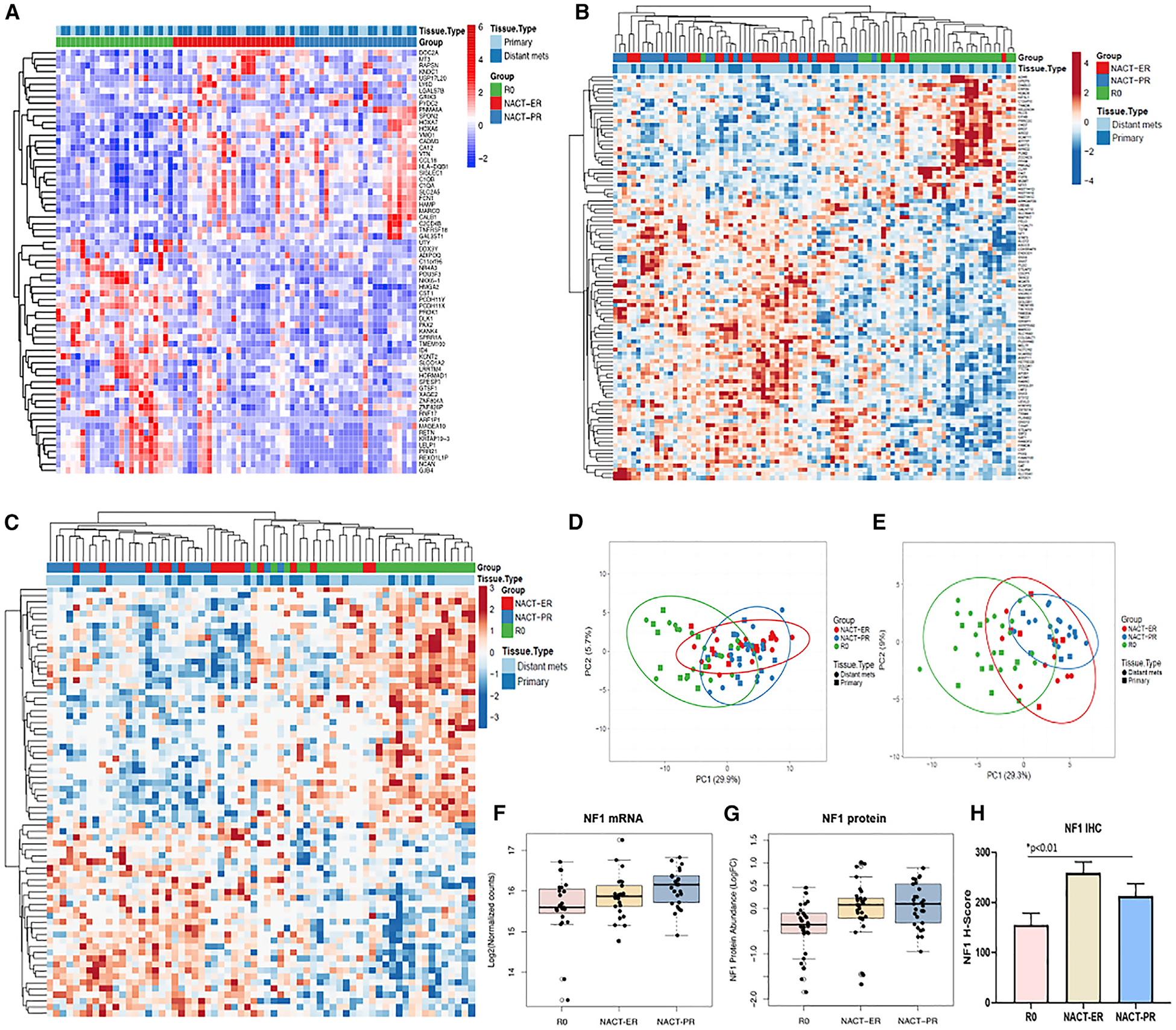
(A) Heatmap of 67 DEGs in the R0 compared to NACT-ER and NACT-PR groups.
(B) Differential analyses of 7387 total proteins quantified and revealed 101 proteins significantly altered (adj. p < 0.05) among NACT-ER (n = 30), NACT-PR (n = 29), and R0 (n = 28) patients. Heatmap reflects clusters assembled by Euclidean distance and average linkage of significant protein abundance trends.
(C) Differential analyses of 12,914 total phosphosites quantified and revealed 71 phosphosites significantly altered (adj. p < 0.05) among NACT-ER (n = 17), NACT-PR (n = 22), and R0 (n = 27) patients. Heatmap reflects clusters assembled by Pearson correlation and average linkage of significant phosphosite abundance trends.
(D) Principle component analyses (PCAs) of 101 proteins significantly altered (adj. p < 0.05) among NACT-ER (n = 30), NACT-PR (n = 29), and R0 (n = 28) patients.
(E) PCA of 71 phosphosites significantly altered (adj. p < 0.05) among NACT-ER (n = 17), NACT-PR (n = 22), and R0 (n = 27) patients.
(F) NF1 RNA expression pattern was consistent with the WGS findings. The boxplot shows the log2 normalized counts of NF1 RNA in the R0, NACT-ER, and NACT-PR groups. The p values were calculated by differential analysis using DESeq2.
(G) NF1 protein abundance was significantly elevated in NACT-ER and NACT-PR tumors versus R0 tumors. The boxplot reflects log2-fold change (L2FC) abundance of NF1 protein for the NACT-ER (n = 30), NACT-PR (n = 29), and R0 (n = 28) groups.
(H) H-scores of NF1 expression by IHC for the NACT-ER (n = 12), NACT-PR (n = 14), and R0 (n = 24) groups. Statistical significance was determined by unpaired t test. Data are presented as the mean ± SEM.
In the NACT-ER compared to the NACT-PR group, we observed 693 DEGs, including 263 curated protein-coding transcripts, with FDR <0.1 and absolute L2FC >2 (Figure S5D; Table S3). Consistent with our CNV data, we found that NF1 mRNA was indeed significantly downregulated in the R0 (p = 0.03) compared to the NACT-ER/PR group (Figure 4F), corroborating our findings of NF1 CNV differences between the groups. Next, we identified 54 differentially expressed ncRNAs in the NACT-ER compared to the NACT-PR group and 14 DEGs between primary and metastatic tumors (FDR < 0.1 and absolute L2FC > 2) in all three groups (Table S3). Most DEGs were ncRNAs; only one predicted protein-coding transcript, TBC1D27, was significantly upregulated (p = 0.00002, q = 0.062) in the primary compared to metastatic sites for all three groups.
To understand the differences in potential biological functions and pathways among the R0, NACT-ER/PR, NACT-ER, and NACT-PR groups, we performed WebGestalt enrichment analysis of Gene Ontology (GO) using 206 DEGs among groups. GO terms for biological process (BP), cellular components (CC), and molecular function (MF) were significantly enriched in the NACT-ER/PR group versus the R0 group and in the NACT-ER group versus the NACT-PR group (Figures S5E and S5F); the top 10 enriched GO terms are listed in Table S3. Notably, in comparison of GO terms between the NACT-ER/PR and R0 groups, the number of DEGs was significantly enriched in BP, including the regulation of epithelial cell differentiation and activation of immune response. However, we did not observe significant cancer-specific-related GO terms in the comparison of the NACT-ER and NACT-PR groups. We also employed several web-based databases (KEGG, Kyoto Encyclopedia of Genes and Genomes; Reactome; and PANTHER, Protein ANalysis THrough Evolutionary Relationships) to identify enriched functional and signaling pathways using DEGs among the R0, NACT-ER, and NACT-PR groups (Table S3). Based on all DEGs between the R0 and NACT groups, we identified an enrichment of gene sets including cadherin signaling pathway; we also observed an enrichment of cell adhesion molecules, transcription misregulation in cancer, and the Notch signaling pathway, but these lacked statistical significance. Interestingly, we observed significant enrichment in GPCR (G protein-coupled receptor) downstream signaling, olfactory signaling, and signaling by GPCR in the DEGs between the NACT-ER and NACT-PR groups by Reactome pathway analysis.
Differential Analysis of Global Proteomic and Phosphoproteomic Data
Using a microscaled tandem mass tag (TMT)-MS-based proteomic/phosphoproteomic workflow, we quantified 7290 total proteins in 87 ovarian cancer specimens and 12,914 total phosphosites in 66 specimens from the 30 patients, among which 101 proteins were found to be significantly altered (adjusted p < 0.05) in the NACT-ER/PR compared to the R0 groups (Figure 4B; Table S4), which, based on principle component analysis (PCA), served to explain 29.9% and 5.7% of the variance between these groups (Figure 4D). Signaling pathways supporting endocytosis, engulfment and cell spreading, and inhibition of cell death and RNA processing signaling were significantly altered in the NACT-ER/PR compared to the R0 group (Figure S6A). Among the significantly altered (p < 0.01) putative drug and drug-associated signaling targets, we identified calcium-transporting ATP2C1 and STAT3 to be elevated and HDAC6, MGMT, and FDPS to be decreased in the NACT-ER/PR versus the R0 groups. Differential analysis identified 71 significantly altered (adjusted p < 0.05) phosphosites in the NACT-ER/PR versus the R0 groups (Figure 4C; Table S4) that, based on PCA, served to explain 29.3% and 9% of the variance between these groups (Figure 4E). Pathway analyses of these altered phosphosites (p < 0.01) revealed cell death of connective tissue cell signaling to be activated and the formation of the cytoskeleton to be inhibited in the NACT-ER/PR versus R0 groups (Figure S6B). Among the significantly altered phosphosites (p < 0.05) in putative drug and drug-associated signaling targets, we identified S129 in SF3B1, S594 in FGA, and S706 in CD44 to be significantly elevated (p < 0.05) in the NACT-ER/PR versus R0 groups. Notably, we validated transcript-level evidence at the protein level that NF1 is significantly lower in abundance in the R0 versus NACT groups by both MS proteomics (p < 0.0005) (Figure 4G) and immunohistochemistry (IHC) assay (p < 0.01) (Figure 4H). In addition, significantly downregulated proteins that were idenified by TMT-MS analyses (i.e., MERLIN, moesin-ezrin-radixin-like protein; and ANNEXIN-1) were observed to have similar patterns of expression with the RPPA assay in the R0 compared to the NACT groups (Figure S5G; Table S4).
We identified 37 proteins as significantly altered (adjusted p < 0.05) between the NACT-ER and NACT-PR groups (Figure S6C; Table S4) that, based on PCA, serve to explain 37.9% and 9.5% of the variance between these groups (Figure S6D). Pathway analyses of significantly altered proteins (p < 0.01) revealed the activation of DNA metabolism and cytokinesis signaling and the inhibition of apoptosis and cellular senescence in the NACT-ER compared to the NACT-PR group (Figure S6E; Table S4). These data also revealed CDK4 to be significantly elevated (adjusted p < 0.05) in the NACT-ER compared to the NACT-PR group. Fifty-nine phosphosites were significantly altered (adjusted p < 0.05) between NACT-ER and NACT-PR tumor specimens (Figure S6F; Table S4) that, based on PCA, served to explain 42.1% and 7.2% of the variance between these groups (Figure S6G). Pathway analyses of these significantly altered phosphoproteomics data (p < 0.05) showed that the frequency of tumor and genitourinary tumor signaling was activated while cell migration signaling was inhibited in NACT-ER compared to NACT-PR patient tumors (Figure S6H; Table S4). Significant decreases in phosphorylation of multiple tyrosine kinases—namely, Y426 and T427 on YES1, Y394 and T395 on LCK, Y420 and T421 on FYN, Y419 and T420 on SRC, and S4520 and S4523 on LRP1—were quantified in the NACT-ER compared to the NACT-PR group.
Immune Analysis
We investigated whether different immune populations were associated with clinical characteristics of the tumors by immune profiling (Figure S7A) and observed significant differences among the groups in tumor area and all areas (tumor/non-tumor) (Figures 5A, 5B, S7B, and S7C), as summarized in Table S5. We next focused on the percentage of T cell infiltration and found a significant increase in the number of infiltrated T cells in the R0 (2.91%) compared to the NACT-ER/PR group (1.35%, p = 0.0442) in tumor area (Figure 5C). We further divided T cell populations into helper T cells (CD4+), cytotoxic T cells (CTLs; CD8+), and regulatory T cells (Tregs; FoxP3+ or CD8+FoxP3+) in tumor and all areas of each group. We did not find differences in helper T cells in any area among the groups. However, there was a significant increase in the percentage of CTLs in all areas in the R0 (2.51%) compared to the NACT-ER/PR group (1.04%, p = 0.0438) (Figure S7D). No statistical differences were observed in Tregs (CD8+FoxP3+) among the groups. However, the R0 group (0.25%) had a significantly lower classic Treg (FoxP3+) count than the NACT-ER group (0.89%, p = 0.0178) in the tumor area at the primary site only (Figures 5D and S7E). Additionally, we observed significantly decreased macrophage counts (CD68+/163+ cells) in the tumor area in the R0 (4.33%) compared to the NACT-ER/PR (7.56%, p = 0.0063) and NACT-PR groups (9.07%, p = 0.0014) (Figure 5C); the same pattern was also noted for all areas (tumor/non-tumor) (Figure S7D). Interestingly, macrophage counts were significantly lower in the tumor area of the primary site of the R0 (4.26%) compared to the NACT-ER/PR (9.21%, p = 0.024) and NACT-PR groups (11.09%, p = 0.0191) (Figure 5D); the same pattern was seen for all areas as well (Figure S7E). Finally, we compared B cell populations among groups and found a trend toward significantly higher B cell infiltration in the tumor area of the NACT-PR (0.31%) compared to the NACT-ER group (0.04%, p = 0.0058) (Figures 5C and 5D). Collectively, these results demonstrate significantly increased infiltrated T cells in the R0 compared to the NACT-ER/PR group and increased macrophages and B cells in the NACT-ER/PR and NACT-PR groups compared to the other groups.
Figure 5. HGSC Immune Infiltration Patterns.
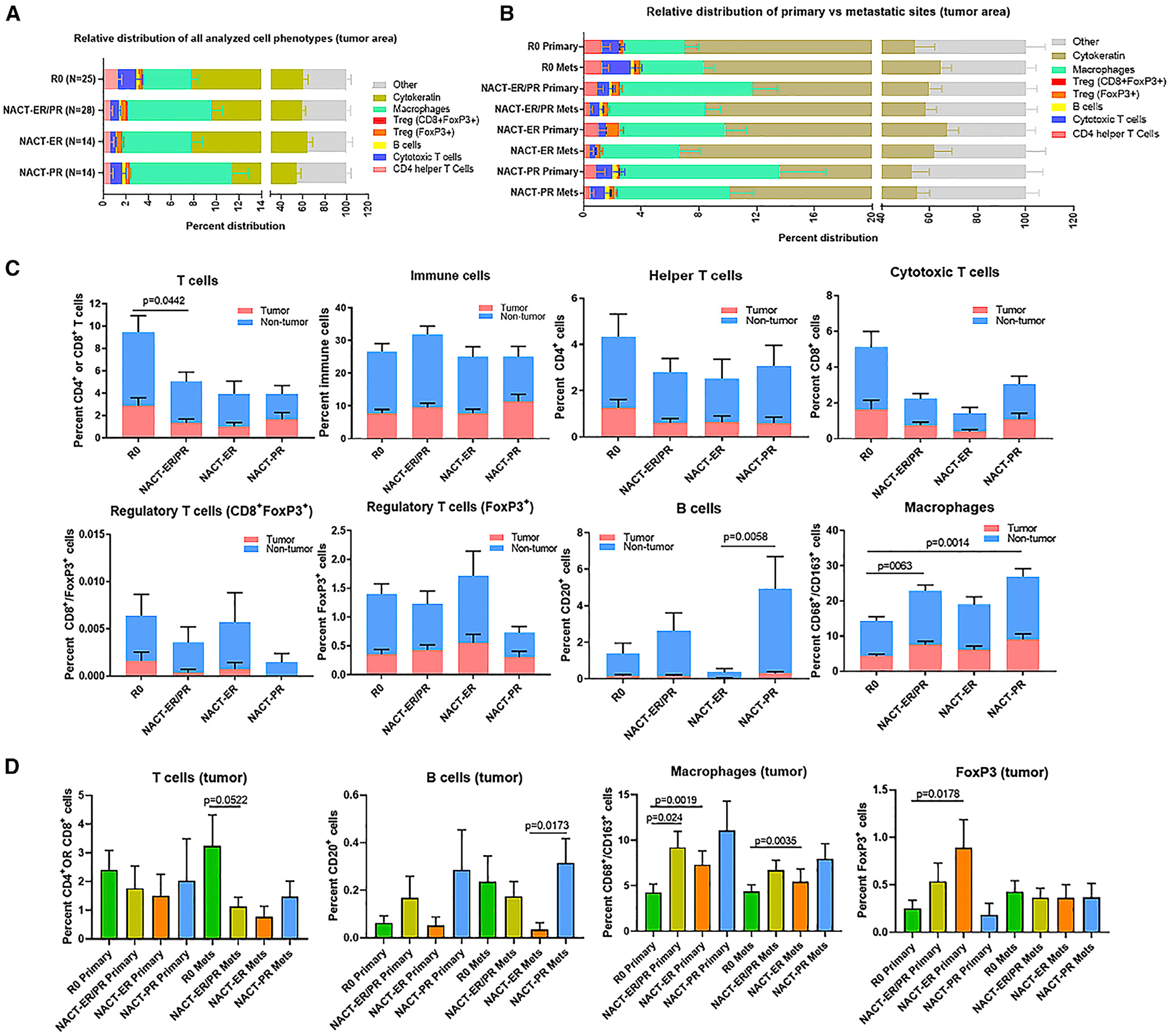
(A) Relative distribution of analyzed cell phenotypes in the tumor area across the R0, NACT-ER, and NACT-PR groups.
(B) Relative distribution of immune cell populations separated into primary and metastatic tumor sites in each group.
(C) Immune subpopulation infiltration patterns in the R0, NACT-ER, and NACT-PR groups. The percentages of immune cells were compared for all T cells, immune cells, helper T cells, cytotoxic T cells, regulatory T cells, macrophages, and B cells. Statistical significance was determined by unpaired t test. Data are presented as the mean ± SEM.
(D) Immune subpopulation infiltration patterns in primary and metastatic sites in tumor area only. The percentages of T cells, B cells, macrophages, and FoxP3+ cells in the tumor area were compared for each group.
Integrated Analysis of Multi-omics Data
As a cross-validation, we inferred the composition of 22 immune cell types from the immune gene signature and the relative percentages of each type using RNA data (Figure 6A). Using scores generated by CIBERSORT, we identified significantly different abundances of M2 macrophages (p < 0.01) and monocytes (p < 0.05) between the R0 and NACT-ER/PR groups (Figure 6B). We also identified a significant difference in abundance of resting CD4 memory T cells (p < 0.05) between primary and metastatic site tumors (Figure 6C). An integrated analysis identified MARCO, SLC2A5 and HMGA2 as co-significantly altered (p < 0.01) at the protein and transcript levels in the NACT-ER/PR compared to the R0 group (Figure 6D). An integrated analysis identified KRT9 as significantly elevated (p < 0.01) in the NACT-ER compared to the NACT-PR group at both the protein and transcript levels (Figure 6E).
Figure 6. Deconvolution Analysis of Cell Fractions Using RNA-Seq Data and the Concordance of Differentially Expressed Transcripts and Proteins among Groups.
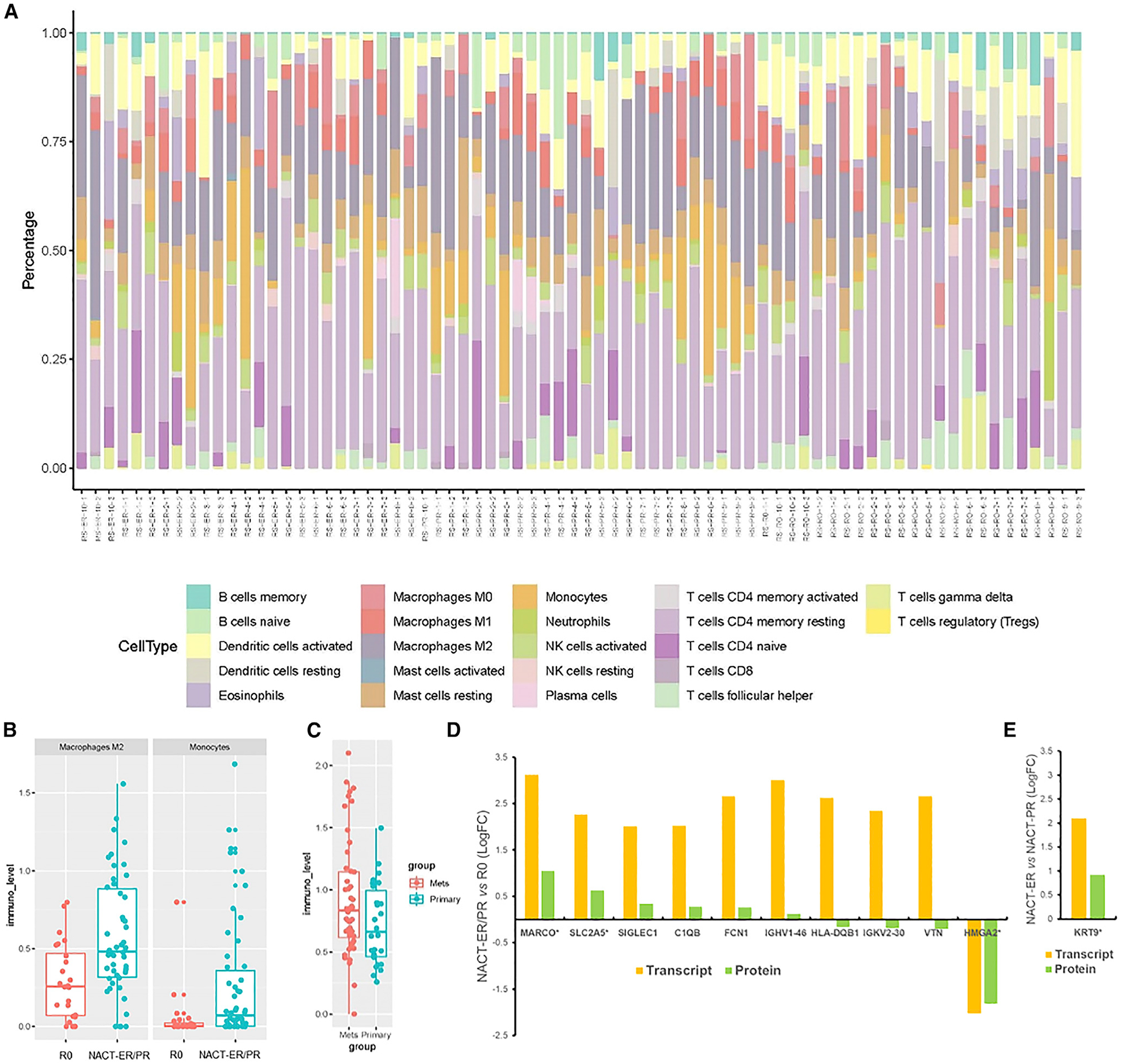
(A) The composition of 22 immune cell subsets in each patient sample. The profiling of immune cells was inferred by deconvolution analysis of RNA-seq with the LM22 immune cell gene signature, and the relative percentages of different cell types are shown in the stacked bar plot.
(B) Boxplots comparing the cell abundances of M2 macrophages and monocytes in the R0 and NACT-ER/PR groups based on RNA-seq deconvolution analysis. Consistent with the immune infiltrate analysis, the R0 group showed more abundant macrophages than did the NACT group.
(C) Boxplot comparing the cell abundances of CD4+ T cells in primary and metastatic sites.
(D) The 206 available transcript alterations were compared to proteins quantified and altered between NACT-ER (n = 30)/PR (n = 29) and R0 (n = 28) patients. Bar plot reflects the L2FC protein and transcript abundance trends for 10 co-measured candidates. *Co-significantly altered at the protein and transcript levels in NACT-ER/PR (p < 0.01) versus R0 groups.
(E) The 263 available transcript alterations were compared to proteins quantified and altered between NACT-ER (n = 29) and NACT-PR (n = 30) patients. Bar plot reflects the L2FC protein and transcript abundance trends for the KRT9 gene. *Co-significantly altered protein (p < 0.01) between NACT-ER and NACT-PR patients.
DISCUSSION
This is a report of high-depth multi-omics analysis of differences in molecular and cellular features of highly clinically annotated HGSC samples from primary and multiple metastatic sites. Our findings provide an understanding of the heterogeneity within HGSCs. Specifically, we found a significantly higher loss of the NF1 gene, RNA and protein product copies, lower numbers of CTLPs, and a significantly higher level of strong-binding neoantigens in the R0 than in the NACT groups. We found significantly increased T cell infiltration and decreased numbers of macrophages in the R0 group and identified significant differences in transcriptomes and proteomes in this group.
While molecular analyses of HGSC have been performed previously (Bowtell et al., 2015; Cancer Genome Atlas Research Network, 2011; Patch et al., 2015; Vaughan et al., 2011), all such studies included samples only from those who underwent upfront debulking surgery. Moreover, the initial clinical management of those patients was highly variable. Here, we focused on testing primary and metastatic tumor samples from patients managed under a systematic surgical algorithm (Fleming et al., 2018; Nick et al., 2015). With respect to the question of whether surgical effort resulting in R0 resection or underlying differences in tumor biology drive different clinical outcomes, our findings demonstrate that there are indeed substantial molecular and cellular differences in patients who underwent R0 tumor debulking compared to those triaged to receive NACT.
The biological mechanisms that might underlie NF1’s role as a biological marker for predicting R0 and NACT response are unknown. Although there is no prior report on the NF1 CNV in prediction of R0 versus NACT groups in cancers, evidence suggests that NF1 plays an important role in the RAS/MAPK signaling pathway, tumorigenesis, and chemotherapy resistance in HGSC (Lau et al., 2000; Norris et al., 2018; Patch et al., 2015). Consistent with a significant loss of NF1 CN and reduced CTLPs on chromosome 17, where NF1 is located, in the R0 group, we confirmed that the NF1 mRNA and protein levels were also significantly decreased in the R0 compared to the NACT-ER/PR group.
Another potentially important finding was the significantly higher level of a strong-binding neoantigen signature in the R0 compared to the NACT group. The increase in neoantigens is associated with infiltration of tumor immune cells such as T cells (McGrail et al., 2018; Schumacher and Schreiber, 2015). Importantly, we observed significantly more infiltrated T cells and fewer macrophages in the R0 compared with the NACT-ER/PR group and increased B cells in the NACT-PR compared to the NACT-ER group by immune assessments. Consistently, the deconvolution analysis using bulk RNA-seq data supported that there is a reduced macrophage cell population in the R0 compared to NACT-ER/PR group. Tumor-associated antigens have been considered for immunotherapeutic strategies, and a high correlation has been reported between tumor mutations and clinical benefits of immunotherapy targeting neoantigens in various solid tumors (Snyder et al., 2014) and in preclinical studies (Gubin et al., 2014; Kreiter et al., 2015). Importantly, we also identified significant differences in cancer-associated transcripts including ncRNAs (Dalton et al., 2017; Li et al., 2016; Webster et al., 2009; Zhao et al., 2017) and significant proteome and phosphosite changes (Chihara et al., 2017; Li et al., 2018; Liu et al., 2015; Xue et al., 2014) in cancer-related and putative drug and drug-associated signaling targets among the groups in this study. Interestingly, significantly downregulated phosphosites of Src-family kinases, LCK, and YES1 were observed in the NACT-ER compared to the NACT-PR group; these have been implicated in supporting the maturation of developing T cell and migration signaling (Kim et al., 2009; Palacios and Weiss, 2004; Salmond et al., 2009). Although further understanding of the clinical significance and validation of these transcript and protein alterations in ovarian cancer progression is required, such changes could serve as markers for the prediction of chemotherapy response in patients with HGSC.
Most patients with HGSC present with widely disseminated disease. Our findings from the clonal phylogenetic analyses support an ability to unravel complex events of metastatic dissemination and the early genetic divergence of metastatic lineages from the primary site in HGSC (Reiter et al., 2018). Given the heterogeneity in response patterns to various therapies, it was unknown to what extent this could be explained by molecular differences between primary and metastatic sites. A case report previously reported intra-patient tumor heterogeneity in immune microenvironments in patients with HGSC (Jimenez-Sanchez et al., 2017). Interestingly, we found no significant genomic variations between primary and metastatic sites, implying that the biological processes underlying complex genomic instability, including CNV and SV, likely occur at an early point in the disease progression. Interestingly, nonsense mutations of CSMD3 and PIK3CA, frequently detected in HGSC (Cancer Genome Atlas Research Network, 2011) and in other types of ovarian cancers (Kuo et al., 2009), were detected exclusively in the NACT-PR and not the NACT-ER or R0 groups. However, neither of these gene mutations has been investigated in HGSC in the context of chemotherapy response and clinical outcome.
Overall, more accurate predictors could lead to precise surgical therapy strategies; if verified, our discoveries in this study—molecular, protein, and immune signatures between various HGSC subgroups—may enhance the prediction of R0 resection and have prognostic and therapeutic implications for patients with HGSC.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be addressed by the Lead Contact, Anil K. Sood (asood@mdanderson.org). No reagents were generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
This study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center, and all samples were collected after obtaining written informed consent from patients. Patients with suspected advanced primary ovarian cancer underwent a laparoscopic assessment to determine a metastatic disease burden score (modified Fagotti score) (Fagotti et al., 2008; Fleming et al., 2018) and acquire tissues (Figure 1A). We obtained fresh frozen tumor specimens from the MD Anderson Gynecologic Tumor Bank for 30 patients diagnosed with HGSC who had available a primary site specimen, two metastatic site specimens and matched blood samples obtained upon laparoscopic assessment. Following laparoscopic assessment, patients with predictive index value < 8 underwent primary tumor reduction surgery, and patients with predictive index value ≥ 8 underwent NACT followed by interval cytoreduction surgery. We selected 10 patients with no visible residual disease after primary surgery (R0), 10 with poor response to NACT with carboplatin and paclitaxel (NACT-PR) and 10 with excellent response to NACT (NACT-ER) (Figure 1B). Patients’ demographic and clinical characteristics are described in Table S1. Response to NACT was considered poor if patients had stable or progressive disease after 3–4 cycles upon radiology evaluation and/or suboptimal interval cytoreduction after NACT according to the Response Evaluation Criteria in Solid Tumors (RECIST 1.1). Response to NACT was considered excellent if there was a complete response or only microscopic disease left at the time of interval surgery and/or pathology from interval surgery. Tumor biopsies and matched blood samples from the 30 patients were subjected to analyses (Figure 1C).
METHOD DETAILS
Whole-genome sequencing
Genomic DNA from 90 frozen tumor tissues from 30 patients (one primary site and two metastatic sites per patient) and matched blood samples as germline controls were prepared by the Biospecimen Extraction Resource of MD Anderson Cancer Center. In brief, genomic DNA was extracted from frozen tissues and blood using the QIAamp DNA Mini Kit and QIAamp DNA Blood Mini Kit (-QIAGEN), respectively, following the manufacturer’s instructions. Extracted genomic DNA was accurately quantified using Quant-iT PicoGreen dsDNA reagent and kit with a Qubit 3.0 Fluorometer (Invitrogen). DNA libraries were prepared using a TruSeq DNA PCR-Free Library Prep Kit (Illumina) following the manufacturer’s instructions. Briefly, genomic DNA was diluted to 20 ng/mL using Resuspension Buffer (RSB, Illumina), and 55 mL was transferred to microTUBES (Covaris). The normalized genomic DNA was then sheared on an LE220 focused-ultrasonication system (Covaris) to achieve a target peak of 450 bp with average power of 81.0 W (So-noLab settings: duty factor, 18.0%; peak incident power, 45.0 W; 200 cycles per burst; treatment duration, 60 s; water bath temperature, 5°C – 8.5°C). The quality of the final DNA libraries was assessed with a High Sensitivity dsDNA Kit (Advanced Analytical Technologies, Inc.). Per the manufacturer’s protocol, the library peak size was in the range of 550 to 620 bp. The DNA libraries were quantified by real-time quantitative PCR, using the KAPA SYBR FAST Library Quantification Kit (KAPA Biosystems) optimized for the LightCycler 480 instrument (Roche). DNA libraries were then normalized to 2 nM and clustered on the Illumina cBot 2 at 200 pM using a HiSeq X v2 flow cell and the HiSeq X HD Paired-End Cluster Generation Kit v2. Paired-end sequencing was performed with the HiSeq X HD SBS Kit (300 cycles) on the Illumina HiSeq X.
Tumor purity and WGS data analysis
Samples with low tumor purity (< 75% proportion of cancer cells) were excluded from the estimation of tumor sample purity by WGS results after purity filtering. Therefore, 75 tumor samples with a high level of purity from 28 patients (9 R0 group patients, 9 NACT-ER and all 10 NACT-PR) were incorporated into the downstream analysis of WGS data. Pair-end sequencing reads in “fastq” format were generated from BCL raw data using Illumina CASAVA (Consensus Assessment of Sequence and Variation) software. The reads were aligned to the hg19 human reference genome using the BWA software package (Li and Durbin, 2009) followed by removal of the duplicate reads using Picard tools (http://broadinstitute.github.io/picard/), and local realignments were performed using the GATK toolkit (McKenna et al., 2010). The BAM files were then used for downstream analysis.
Somatic mutation detection
The MuTect method (Cibulskis et al., 2013) was used to identify somatic point mutations, and the Pindel tool (Ye et al., 2009) was used to identify somatic insertions and deletions. A series of post-calling filtering algorithms were applied for somatic mutations: (a) total read count in tumor sample ≥ 20, (b) total read count in germline sample ≥ 10, (c) VAF (variant allele frequency) ≥ 0.05 in tumor sample and ≤ 0.02 in matched normal sample and (d) population frequency threshold of 1% for filtering out common variants in the databases dbSNP129 (Sachidanandam et al., 2001), 1000 Genomes Project (Abecasis et al., 2012), Exome Aggregation Consortium (Lek et al., 2016) and ESP6500 (Fu et al., 2013).
Clonal population estimation in tumors
The Sequenza package (Favero et al., 2015) was used to estimate tumor cellularity and ploidy and to calculate an allele-specific copy number profile for each sample. The PyClone model (Roth et al., 2014) was used to identify and quantify clonal populations in tumors. Mutant-allele tumor heterogeneity (MATH) scores were calculated as a measurement of intratumor genetic heterogeneity (Mayakonda et al., 2018; Mroz et al., 2015). To construct phylogenetic trees, functional mutations were converted into binary format, with 1 being mutated and 0 otherwise. Ancestors were germline DNA, assuming no somatic mutations, while germline mutations in BRCA1/2 were included. Phylogenetic trees were constructed using the multistate discrete-characters Wagner parsimony method using the PHYLIP program (Retief, 2000). The trees were redrawn with relative trunk and branch lengths proportional to the number of mutations for visualization purposes.
Copy number variation and structure variation detection
CNVs were identified using HMMcopy (Ha et al., 2012). The copy number log2 ratios of tumor versus matched normal samples were calculated and then subjected to segmentation using circular binary segmentation (Olshen et al., 2004). A cutoff of log2 ratio < −0.4 was applied to identify copy losses, and log2 ratio ≥ 0.4 was applied for copy gains. Copy number signatures were identified based on the components described in the CNsignatures algorithm (Macintyre et al., 2018). SVs were called by three methods: LUMPY (Layer et al., 2014), BRASS (https://github.com/cancerit/BRASS) and BreakDancer (Fan et al., 2014). The SVs that appeared in normal samples were filtered out. To reduce false-positive rates, only SVs identified by at least two methods were kept. SV signatures for ovarian cancers were calculated, as described previously (Hillman et al., 2018). Chromothripsis-like patterns were identified by the CTLPScanner server (Yang et al., 2016) with the filtering of: (a) number of copy number switches ≥ 20 and (b) log10 likelihood ratio ≤ 8.
High-depth targeted sequencing analysis
We performed high-depth targeted sequencing (T200.2 panel) as previously described (Chen et al., 2015), and genomic DNA from each sample was prepared as described above for WGS. Briefly, the aliquots of genomic DNA were re-quantified by PicoGreen (Invitrogen) and quality was assessed using a 2200 TapeStation system (Agilent). Then genomic DNA was sheared by sonication using a Covaris E220 instrument with the following conditions: pPeak incident power, 175 W; duty cycle, 20%; intensity, 5; cycles per burst, 200; and 120 s. To ensure the proper fragment size, samples were checked on TapeStation using the High Sensitivity DNA Kit (Agilent). The sheared DNA proceeded to library prep using a KAPA Hyper Prep Kit following the “with beads” manufacturer protocol. PCR primers were removed by using 1.8x volume of Agencourt AMPure PCR purification kit (Agencourt Bioscience Corporation). At the end of the library prep, samples were analyzed on TapeStation to verify correct fragment size and to ensure the absence of extra bands. Samples were quantified using a KAPA qPCR Quantification Kit (KAPA Biosystems). Equimolar amounts of DNA were pooled for capture (2–6 samples per pool). Equimolar amounts of DNA were pooled for capture (8–16 samples per pool). Global copy number selected areas were also captured. We selected for capture 323 genes that are clinically relevant in cancer on the basis of mutational data and the COSMIC database (Bamford et al., 2004) and TCGA. Global copy number selected areas were also captured. We designed biotin-labeled probes with Roche NimbleGen for capturing target regions (all exons in those 323 genes plus copy number regions) and followed the manufacturer’s protocol for the capture step. The captured libraries were sequenced in 100-bp paired-end mode using a HiSeq 2000 system (Illumina) on a TruSeq v3 paired-end flow cell, according to the manufacturer’s instructions, at a cluster density of 700–1000K clusters/mm2. Sequencing was performed on a HiSeq 2000 for 2 × 100 paired end reads with a 7 nt read for indexes using Cycle Sequencing v3 reagents (Illumina). The resulting BCL files containing the sequence data were converted into “.fastq.gz” files and individual libraries within the samples were de-multiplexed using CASAVA 1.8.2 with no mismatches. All regions were covered by > 20 reads.
Telomere length estimation and comparison among groups
TL was estimated with TelSeq software (Ding et al., 2014) for both tumor and normal samples for each patient. To exclude the factors that affect telomere length other than cancer, the TL ratio was further calculated as log2 (TL_tumor/TL_normal). TL ratios were compared among the R0, NACT-ER and NACT-PR groups using the Wilcoxon signed-rank test.
Neoantigen prediction in tumors
Neoantigens were predicted based on somatic mutations, including both SNVs and InDels. The binding affinity was predicted by the NetMHCcons server (Karosiene et al., 2012). For any neoantigens, the binding affinities were predicted for all MHC types. For each MHC type, binding affinities were predicted on both wild-type and mutant sequences. The peptide was identified as a strong binder if the %Rank (rank of the predicted affinity compared to a set of 400.000 random natural peptides) was below 0.5% or the binding affinity (IC50) was below 50. The peptide was identified as a weak binder if the %Rank was between 0.5% and 2% or the binding affinity (IC50) was between 50 and 500. Here, we only included the neoantigens that showed weak and strong binding in the mutant but not the original wild-type sequence. The numbers of weak- and strong-binding neoantigens were compared between patient groups. The Wilcoxon rank sum test was used to determine whether there were statistically significant differences between groups.
RNA sequencing analysis
RNA sequencing (RNA-seq) was performed in the MD Anderson Cancer Genomics Core Laboratory as previously described (Liu et al., 2009). Total RNA from 89 frozen tissues from 30 patients was prepared, and the capture step was performed using whole-exome biotin-labeled probes from Roche NimbleGen (Exome V3) and following the manufacturer’s protocol; the remaining sample did not have an adequate amount of tissue. Total RNA was quantified by PicoGreen (Invitrogen), and quality was assessed using a 2200 TapeStation system (Agilent). RNA from each sample (10–100 ng) was converted to double-stranded cDNA using an Ovation RNA-Seq System V2 kit (NuGEN). The cDNA library was prepared as described above for high depth sequencing, and we used whole-exome biotin-labeled probes from Roche NimblegGn (Exome v3.0) and followed the manufacturer’s protocol for the capture step. Sequencing proceeded as described above for the high depth sequencing analysis.
Sequencing proceeded as described above for the high-depth sequencing analysis. Raw RNA-sequencing data of 89 samples were converted to fastq files and aligned to the reference genome (hg19) using the Spliced Transcripts Alignment to a Reference (STAR) algorithm (Dobin et al., 2013), and the reads quality was evaluated with FastQC and Qualimap tool (Okonechnikov et al., 2016). RNA-seq data were excluded for the 15 out of 89 samples with low tumor purity as estimated by WGS; the remaining 74 samples were subjected to downstream analysis. The R package DESeq2 was used for data processing, normalization and differential expression analysis following standard procedures (Love et al., 2014). Each gene-level htseq-count table contained the read counts of 57,798 genes with gene_ids. The gene counts were normalized using the scaling factor method in DESeq2. If the number of over-lapping reads of any given gene was less than 1 per million total mapped reads for all samples, this gene was excluded from further analysis. In this way, 25,841 (44.7%) genes were removed. An unsupervised hierarchical clustering analysis was performed using the Pearson correlation coefficient as the distance metrics and the Ward’s linkage rule using the 3000 genes with the largest standard deviations (SD). DESeq2 was used to identify the differentially expressed genes (DEGs) and calculate the FDR, and a cutoff of absolute log2-fold change (L2FC) > 2 and FDR < 0.1 were used to define the DEGs. Functional analyses and pathway analyses were further performed using a Web-based gene set analysis toolkit (WebGestalt) (Wang et al., 2013; Zhang et al., 2005) with the DEGs lists obtained from DESeq2 for gene ontology (GO) analysis. Pseudogenes and noncoding RNAs were excluded from the anlyses. Functional annotations were based on the GO database, including the biological process, cellular component and molecular function. Pathway analyses were based on multiple databases: KEGG (https://www.kegg.jp/), Reactome (https://reactome.org/) and PANTHER (http://www.pantherdb.org/). The overrepresentation enrichment analysis was used to evaluate the enrichment of functional groups and pathways.
Reverse phase protein array (RPPA) analysis
To collect protein expression data, a Reverse Phase Protein Array (RPPA) assay was performed at the MD Anderson Functional Proteomics Reverse Phase Protein Array (RPPA Core, as described previously (Cheung et al., 2011; Li et al., 2017; Liang et al., 2012; Wang et al., 2017) using 81 frozen tumor tissues from patients; the remaining 9 samples did not have an adequate amount of tissue. We also examined the phosphorylation status of proteins using antibodies specific to particular proteins or phosphorylated forms of proteins because the majority of antibodies used in RPPA analyses are preselected for signaling pathways that are well known to be involved in tumor development. The samples were probed with 297 antibodies in total. The signal intensities on the RPPA arrays were quantitated using MicroVigene software (VigeneTech) and processed using the R package Super-Curve (version 1.01; https://bioinformatics.mdanderson.org/public-software/archive/oompa/), which generated the relative log2 expression value for each protein.
Protein levels for each sample were determined by interpolating each dilution curve produced from the densities of the 5-dilution sample spots using SuperCurve software. All relative protein levels were normalized for protein loading, transformed to linear values and log2-transformed. The 14 samples with low tumor purity were excluded from further downstream analyses. A mixed-effect model was fit to each normalized protein level with response group and tissue type as main-effect covariates and patients as the random effect covariate. We used t tests to test against the null hypothesis of no difference in protein expression between any two groups. The Benjamini-Hochberg method was used to control the FDR. Next, protein network analysis was performed on 16 differentially expressed proteins using the GeneMANIA server (Warde-Farley et al., 2010). The nodes in the resulting network represent the proteins, and the edges represent the protein-protein interactions. Four types of direct interactions were selected to build the network: physical interactions, co-localization, pathway and genetic interactions. The network edges were weighted by the corresponding data source with the adaptive network weighting method by GeneMANIA.
Specimen preparation for proteomics
Laser microdissection was used to harvest whole tissue representations (cancer and stroma combined) of each specimen, which were digested with trypsin using pressure cycling technology (PCT, Pressure Biosciences, Inc.) from 87 frozen tissues from 30 patients. Briefly, tissue specimens were sectioned (8 μm) by microtome onto polyethylene naphthalate membrane slides and hematoxylin-eosin stained. Laser microdissection was utilized to harvest whole tissue representations of each specimen (cancer and stroma combined) prior to proteomic sample preparation, with a minimum selected total area goal of 65,000,000 μm2 from 8 μm thick sections, avoiding areas of necrosis and blood. The laser-microdissected harvests were collected directly into microcentrifuge tubes containing 50 μL liquid chromatography (LC)/mass spectrometry (MS)-grade water for proteomic analysis. Tissue specimens were stored in a −80° C freezer until sample preparation. Tissue harvests were transferred to a Pressure Cycling Technology (PCT, Pressure BioSciences, Inc) microtube containing 20 μL of 100 mM triethylammonium bicarbonate (TEAB), 10% acetonitrile. The samples were incubated at 99° C for 30 min followed by 50° C for 10 min. Tissues were digested by adding SMART trypsin (Ther-moFisher Scientific Inc) at a ratio of 1 μg per 30,000,000 μm2 tissue by employing PCT, where each sample was cycled 60 times between 45,000 psi for 50 s and atmospheric pressure for 10 s at a constant temperature of 50° C. Each protein digest was transferred to a clean 0.5 mL microcentrifuge tube, vacuum dried, and re-suspended in 50 μL 100 mM TEAB. Final digest recoveries were determined by colorimetric assay (Pierce BCA Protein Assay Kit). Peptide (50 mg) from each sample was labeled with a unique isobaric tandem mass tag (TMT) label according to the manufacturer’s instructions (TMT-11-plex Isobaric Label Reagent Set, ThermoFisher Scientific). A reference sample, generated by pooling equivalent amounts of peptide digests from each of the patient samples in the cohort, was labeled with TMT “Channel” 126 and included in each TMT-11-multiplex (e.g., reference standard Channel 126 + 10 unique samples occupying Channels 127N-131). Each TMT-11 multiplex set of clinical samples was constructed in a randomized fashion. After quenching, each TMT-11 multiplex set of samples was combined and vacuum dried to approximately 80 mL.
Basic reversed-phase liquid chromatographic (bRPLC) fractionation
Each TMT-11 multiplex set of samples was loaded onto a C-18 trap column in 10 mM NH4HCO3 (pH 8.0) and fractionated by bRPLC into 96 fractions through development of a linear gradient of acetonitrile (0.69% acetonitrile/min). Thirty-six concentrated fractions were generated by pooling the samples in a serpentine manner. Ten percent (volume) of each fraction was removed for liquid chromatography-tandem mass spectrometry (LC-MS/MS). The remaining 90% (volume) of the 36 fractions was pooled into 12 fractions for serial phosphopeptide TiO2 enrichment followed by iron immobilized metal ion affinity chromatography (Fe-IMAC). Briefly, peptide fractions were vacuum dried, re-suspended in TiO2 binding/equilibration buffer and bound to TiO2 affinity spin tips (High-Select TiO2 Phosphopeptide Enrichment Kit, Thermo Fisher Scientific), and sample flow-through and washes were reserved for subsequent enrichment by Fe-NTA (nitrilotriacetic acid) affinity chromatography (High-Select Fe-NTA Phosphopeptide Enrichment Kit). As a quality control analyses, we investigated the enrichment of phosphotyrosine-containing peptides resulting from this dual enrichment strategy and found that we recover these at a very similar relative level (1.3%) as to similar dual enrichment strategies employed previously, such as from the Mann lab where they observed 1.8% phosphotyrosyl-containing peptides (Olsen et al., 2006). Notably, tissue samples organized in TMT sample plexes 11 through 14 exhibited low overall peptide yields and were thus analyzed using fewer concatenated peptide digest fractions, i.e., 12 fractions for plexes 11 through 13 and 24 fractions for plex 14 relative to the 36 total fractions analyzed for global proteomics across sample plexes 1 through 10. As sample yields were below SOP expectations for sample plex 11 through 14, phosphopeptide analyses for the samples were omitted from data analyses downstream.
LC-MS/MS proteomics
The TMT-11 sample multiplex bRPLC fractions (36 total fractions for global proteomics and 12 fractions for phosphopeptides serially enriched by TiO2 and Fe-IMAC) were analyzed by LC-MS/MS employing a nanoflow LC system (EASY-nLC 1200, Thermo Fisher Scientific) coupled online with an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). In brief, each sample (5 mL) was loaded on a nanoflow high-performance LC system outfitted with a reversed-phase trap column (Acclaim™ PepMap™ 100 C18, 2 cm length, nanoViper Trap column, Thermo Fisher Scientific) and a heated (50°C) reversed-phase analytical column (Acclaim™ PepMap™ RSLC C18, 2 μm, 100Å, 75 μm × 500 mm, nanoViper, Thermo Fisher Scientific) connected online with an Orbitrap mass spectrometer. Peptides were eluted by developing a linear gradient of 2% mobile phase B (95% acetonitrile with 0.1% formic acid) to 32% mobile phase B within 120 min at a constant flow rate of 250 nL/min. High-resolution (R = 60,000 at m/z 200) broadband (m/z 400–1600) mass spectra (MS) were acquired, from which the top 12 most intense molecular ions in each MS scan were selected for high-energy collisional dissociation (HCD, normalized collision energy of 38%) acquisition in the Orbitrap at high resolution (R = 50,000 at m/z 200). Monoisotopic precursor selection mode was set to “Peptide,” and MS1 peptide molecular ions selected for HCD were restricted to z = +2, +3 and +4. The radio frequency (RF) lens was set to 30%, and both MS1 and MS2 spectra were collected in profile mode. Dynamic exclusion (t = 20 s at a mass tolerance = 10 ppm) was enabled to minimize redundant selection of peptide molecular ions for HCD.
Quantitative Proteomic Data Processing Pipeline for Global and phosphoproteome analyses
Peptide identifications were generated by searching the .raw data files with a publicly available, non-redundant human proteome database (Swiss-Prot, Homo sapiens [https://www.uniprot.org/, downloaded 12/01/2017]) appended with porcine trypsin (Uniprot: P00761) and iRT peptide (Escher et al., 2012) sequences using Mascot (Matrix Science) and Proteome Discoverer (Thermo Fisher Scientific) software. The .raw data files corresponding to each LC-MS/MS injection per TMT-11 multiplex were searched using the following parameters: precursor mass tolerance of 10 ppm, fragment ion tolerance of 0.05 Da, a maximum of two tryptic miscleavages, static modification for TMT reporter ion tags (229.1629 Da) on N-termini and lysyl residues, and dynamic modifications for oxidation (15.9949 Da) on methionine residues, as well as phosphorylation (79.9663 Da) on seryl, threonyl or tyrosyl residues for phosphoproteome analyses. The resulting peptide spectral matches (PSMs) were filtered using an FDR < 1.0% (q-value < 0.01), as determined by the Percolator (Käll et al., 2007) module of Proteome Discoverer. Phosphoproteome search results were further analyzed by the ptmRS node (Taus et al., 2011) within Proteome Discoverer as a confidence measure for the post-translational modifications identified. TMT reporter ion intensities were extracted using Proteome Discoverer at a mass tolerance of 20 ppm, and PSMs lacking a TMT reporter ion signal in TMT channel m/z 126 (TMT-126, the pooled study reference combined from all patient sample digests), PSMs lacking TMT reporter ion intensity in all TMT channels, or PSMs exhibiting an isolation interference of ≥ 50% were excluded from downstream analyses. Log2-transformed TMT reporter ion ratios corresponding to individual patient tissue samples were calculated for each PSM against the pooled reference standard (TMT-126 channel). Log2-transformed PSM abundance distributions were normalized by calculating the mode-centered z-score transformation adapted from previous study (Mertins et al., 2016), for each channel in the TMT-11 multiplex as follows: normalized PSM (Log2Ratio) = [PSM (Log2Ratio) – ModeCenter PSM (Log2Ratio) / s PSM (Log2Ratio). Briefly, this method normalizes log-transformed PSM abundances for each patient sample channel by calculating the mode log ratio as well as the standard deviation for the entire distribution of PSMs quantified in a patient sample channel. The log ratio intensity of each PSM quantified for a given patient channel is then normalized by subtracting the mode log ratio and then dividing by the standard deviation of all PSMs quantified for a given PSM per patient channel. For global protein-level abundance, the abundances of proteins identified by a unique PSM (i.e., in which a PSM maps uniquely to a single protein accession) were determined by calculating the median log2-transformed abundance ratios of all such PSMs. The abundances of PSMs mapping to multiple proteins (i.e., “multi-mapper” PSMs) were compared to mapped unique protein abundances using a mean-squared-error approach to assign them to unique proteins based on comparative abundance analyses. Briefly, mean squared log2-transformed abundance ratios (Allison et al., 2019; Tarney et al., 2019) were calculated for multi-mapper PSMs with intensity data observed in R 50% of all TMT channels for a given TMT sample plex; redundant and multiply charged versions of a given PSM were considered unique biochemical events. Multi-mapper PSMs were assigned to the corresponding unique protein accessions exhibiting the smallest difference in relative abundance levels comparatively. Multi-mapper PSMs mapping to protein accessions not identified by any unique PSMs were excluded from downstream analyses. This strategy leverages quantitative data collected for unique peptides (in this case 95.2% of total PSMs identified against the non-redundant SwissProt human proteome database that we searched our data against) to identify parent proteins of co-measured non-unique peptides (in this case representing 4.8% of total PSMs identified). Protein-level abundance was calculated from normalized, median log2-transformed TMT reporter ion ratio abundances from a minimum of two PSMs corresponding to a single protein accession. The performance of a subset of six patient samples labeled as independent technical replicate channels was assessed by direct comparison of proteins co-quantified between replicate samples; all patient samples exhibited Spearman Rho > 0.85 ± 0.03 except replicates for patient sample PR-R0-4-1 (R = 0.52) which was thus removed from consideration in downstream analyses. Normalized log2-transformed protein-level abundances for each TMT-11 multiplex were merged, and protein-level abundances for proteins not quantified in all patient samples, but in ≥ 50%, were imputed using a k-nearest neighbor (k-NN) strategy adapted from previous publication (Mertins et al., 2016) using the pamr (Prediction Analysis for Microarrays) R package (Lazar et al., 2016). The abundances of phosphorylated (phospho)-PSMs were assembled at the level of discrete phosphosites that map to a unique protein using a tiered strategy aimed at defining high- and low-confidence phospho-PSMs. First, TMT reporter ion intensities were processed for phospho-PSMs as described above to calculate normalized, log2-transformed abundance ratios of phospho-PSMs for a given patient sample. Phospho-PSMs that mapped to unique proteins were assigned to those protein identifiers. Multi-mapper phospho-PSMs were assigned to all proteins that were co-identified in companion global proteome data. Multi-mapper phospho-PSMs that did not map to a unique protein in global proteomic data were assigned to the first protein accession that a given phospho-PSM was assigned to by database search. The number and amino acid positions of phosphosites that were identified in the database search for a given phospho-PSM were compared with phosphosite positions predicted by the ptmRS algorithm. A high-confidence phospho-PSM was determined when all phosphosites identified by database search also exhibited > 50% probability of being the “best” predicted phosphosite for a given phospho-PSM. A low-confidence phospho-PSM was determined when any phosphosite identified by database search was not predicted as a phosphosite or exhibited < 50% probability of being predicted as the “best” phosphosite. Low-confidence phospho-PSM candidates were further prioritized using a tiered strategy in which unique phosphosite variants identified for the same phospho-PSM event were selected based on the highest ptmRS probability score that exhibited the lowest search engine rank in the TMT-11 patient sample plex with the greatest number of total PSMs. Normalized log2-transformed protein-specific phosphosite abundances were determined by calculating the median abundance of phospho-PSMs exhibiting the same phosphosite as well as methionine oxidation state. Phosphosites quantified redundantly as both low- and high-confidence versions were further filtered to prioritize only high-confidence phosphosites for downstream analyses. For phosphosites co-identified in companion global proteomic data, median log2-transformed, protein-specific phosphosite abundances were also normalized to the total protein abundance quantified in global proteome analyses.
Differential analyses of global and phosphosite data
Differential analyses of neurofibromin (NF1) global protein abundance were performed using Mann-Whitney U rank sum testing in MedCalc (version 19.0.3). Phosphosite data were filtered prior to differential analyses so that phosphosites were measured in ≥ 50% of the comparator cohort, i.e., NACT-ER versus NACT-PR and NACT-ER/PR versus R0 patients. Differential analyses of global and phosphosite TMT-11 data matrixes were performed for patient samples of interest using the LIMMA package (version 3.8) (Ritchie et al., 2015) in R (version 3.5.2). Phosphosite data were pre-filtered for protein and phosphosite alterations passing LIMMA adjusted p value < 0.05; these alterations were prioritized for downstream analyses. Significant protein and phosphosite alterations were visualized in heatmaps and by principle component analysis (PCA) using default settings in the ClustVis web tool (Metsalu and Vilo, 2015). To investigate the impact of imputation on significantly altered protein features used for principle component analyses, we reviewed proteins significantly altered between NACT-ER/PR versus R0 patients (n = 104) and find that 53 of these alterations were imputed in as few as two and as many as forty-five cases across our patient cohort (n = 87). We performed PCA analyses of the 51 non-imputed protein alterations among this list and compared variances for this limited feature set for the first two principle components (PC1 = 33.9% and PC2 = 6.8%) relative to variance observed for the 104 total protein alterations (Figure 4D, PC1 = 29.6% and PC2 = 5.4%), between NACT-ER/PR versus R0 patients. We find these variances to not be significantly different (MWU p = 0.667). Functional inference analyses were performed for significantly altered proteins and phosphosites (significance defined as LIMMA p value < 0.01 and exhibiting a log2 fold-change cut-off ± 1.5 proteins) using Ingenuity Pathway Analysis. Significantly enriched diseases and biofunctions predicted to be activated or inhibited were prioritized for further analyses. This strategy was intended to expand the feature set to investigate overarching signaling and pathway alterations between these patient cohorts.
Data analysis of targeted sequencing
A customized pipeline was applied to analyze the high-depth targeted sequencing results, which was adopted from tools that are applied to cancer genome sequencing projects such as TCGA but implemented with further optimization for deep clinical sequencing, as we previously described (Chen et al., 2015). Briefly, we aligned the reads to human reference assembly hg19 using BWA and Picard (DePristo et al., 2011). We then used the MuTect and Pindels algorithms against a pooled common normal reference to call high-confidence mutations. The same public databases as described in WGS analysis were used to further remove the germline polymorphism with a population frequency cutoff of 0.5%. Copy number alterations were identified using an in-house R package as previously described (Takahashi et al., 2018). A cutoff of log2 ratio ≤ −0.4was applied to identify copy losses and log2 ratio ≥ 0.4 was applied for copy gains.
Immunohistochemical analysis
50 paraffin sections from the R0 (n = 24) and NACT-ER/PR (n = 26) groups were used for the validation of NF1 protein expression by immunohistochemistry assay (IHC), and IHC was performed in the MD Anderson Research Histology Core Laboratory. Briefly, the slides were incubated with a primary antibody Neurofibromin 1 (1:150, Abcam) for 8 minutes, and then detected using Leica Bond Polymer Refine Detection Kit according to manufacturer’s instuctions (Leica Biosystems). The quantification of NF1 protein expression by IHC in the FFPE sections, each slide was independently evaluated and scored by three independent investigators including a board-certified pathologist on blinded samples according to expression level scoring matrix (Figure S8), and NF1 expression score (H-score, range 0–300) was determined by multiplying the percentage of positive cells.
Immune-profiling analysis
Opal multiplex staining was performed for immune infiltrate analysis. In brief, Opal multiplex allowed for the simultaneous evaluation of seven markers (CD4, CD8, CD20, FoxP3, cytokeratin, CD68/163 and DAPI) in a single tissue section. Multispectral imaging was applied to the seven-marker stained samples. An average of five representative images per tissue sample was obtained. After image capture, spectral unmixing was performed to separate raw images into individual fluorophores. The spectrally unmixed images were then analyzed to identify different cellular phenotypes, which allowed for the evaluation of the regulatory T cell population identified by CD8+FoxP3+ positivity (Figure S7A). All other cells not defined by our phenotyping categories (blood vessels, nerves) were grouped into an “other” category. Finally, tumor epithelial versus non-tumor areas of each specimen were distinguished using a tissue segmentation algorithm. After cell phenotyping, comparisons were drawn between abundance and distribution of immune infiltrates for the R0 (n = 25, 10 primary and 15 metastatic sites), NACT-ER/PR (n = 28), NACT-ER (n = 14, 5 primary and 9 metastatic sites) and NACT-PR (n = 14, 5 primary and 9 metastatic sites) groups.
Inference of immune cell populations
We estimated the immune contextures of 74 samples using the CIBERSORT tool (https://cibersort.stanford.edu). We used its original algorithm to infer the fractions of 22 immune cells relative to the total immune-cell population. In addition, we also applied its absolute mode to calculate a score that quantitatively measured the overall abundance of each cell type. A mixed-effect model was fit to a score with response group and tissue type as main-effect covariates and patients as the random effect covariate.
QUANTIFICATION AND STATISTICAL ANALYSIS
The analysis of tumor samples with matched normal samples allowed for the detection of somatic genomic events contributing to the observed differential response to treatment between groups, and the analysis of multiple sampling per patient enabled us to assess clonal and adaptive evolution in different response groups. To evaluate the statistical significance, Student t test is used if the data fits normal distribution. Otherwise, Wilcoxon rank-sum test is used to test the differences between groups. BH method is used to control the false discovery rate. In transcriptomic analysis, the differential expression was evaluated using a negative binomial generalized linear model as described in DESeq2 (Love et al., 2014), and logarithmic fold change and adjusted p values were used for differentially expressed gene assessment. In the proteomic analysis, the differential expression was evaluated by the linear model described in LIMMA (Ritchie et al., 2015). The statistical significance of differences (p < 0.05) on infiltrated immune cells between groups was determined using the Student t test (GraphPad Prism).
DATA AND CODE AVAILABILITY
Sequence data from the WGS, deep targeted sequencing and RNA sequencing in this study have been deposited in the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/home) under accession numbers EGAD00001005240, EGAD00001005239 and EGAD00001005238, respectively. The LC-MS/MS data have been deposited to the PRoteomics IDEntifications (PRIDE, https://www.ebi.ac.uk/pride) database under the accession number PRIDE: PXD014980.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Neurofibromin-1 (NF1) | Abcam | Cat# 128054, RRID:AB_11141828 |
| Biological Samples | ||
| Patients with HGSC (tissues and blood) | MDACC | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Leica Bond Polymer Refine Detection kit | Leica Biosystems | Cat#DS9800 |
| Quant-iT PicoGreen dsDNA reagent | Invitrogen | Cat# P7581 |
| SMART trypsin | Thermo Fisher Scientific | Cat# 60109 |
| TMT-11-plex Isobaric Label Reagent Set | Thermo Fisher Scientific | Cat# A34807 |
| Acclaim™ PepMap™ 100 C18 | Thermo Fisher Scientific | Cat# 164567 |
| Acclaim™ PepMap™ RSLC C18 | Thermo Fisher Scientific | Cat# 164540 |
| Critical Commercial Assays | ||
| QIAamp DNA Mini Kit | QIAGEN | Cat# 51304 |
| QIAamp DNA Blood Mini Kit | QIAGEN | Cat# 51104 |
| TruSeq DNA PCR-Free Library Prep Kit | Illumina | Cat# 20015963 |
| High Sensitivity dsDNA Kit | Advanced Analytical Technologies, Inc. | Cat# DNF-468–0500 |
| KAPA SYBR FAST Library Quantification Kit | KAPA Biosystems | Cat# KK4824 |
| HiSeq X HD Paired-End Cluster Generation Kit v2 | Illumina | Cat# GD-410–1001 |
| HiSeq X HD SBS Kit (300 cycles) | Illumina | Cat# FC-410–1003 |
| Agencourt AMPure PCR purification kit | Agencourt Bioscience Corporation | Cat# A63880 |
| KAPA qPCR Quantification Kit | KAPA Biosystems | Cat# KK4600 |
| whole-exome biotin-labeled probes (v3) | Roche NimbleGen | Cat# 06465684001 |
| Ovation RNA-Seq System V2 kit | NuGEN | Cat# 7102 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| High-Select TiO2 Phosphopeptide Enrichment Kit | Thermo Fisher Scientific | Cat# A32992 |
| Deposited Data | ||
| WGS data | This paper | EGA: EGAD00001005240 |
| Deep targeted sequencing data | This paper | EGA: EGAD00001005239 |
| RNA sequencing data | This paper | EGA: EGAD00001005238 |
| LC-MS/MS data | This paper | PRIDE: PXD014980 |
| Software and Algorithms | ||
| BWA software package | Li and Durbin, 2009 | N/A |
| Picard tools | Broad Institute | http://broadinstitute.github.io/picard/ |
| GATK toolkit | McKenna et al., 2010 | N/A |
| MuTect method | Cibulskis et al., 2013 | N/A |
| Sequenza package | Favero et al., 2015 | N/A |
| PyClone model | Roth et al., 2014 | N/A |
| PHYLIP program | Retief, 2000 | N/A |
| HMMcopy | Haetal., 2012 | N/A |
| Circular binary segmentation | Olshen et al., 2004 | N/A |
| CNsignatures algorithm | Macintyre et al., 2018 | N/A |
| LUMPY | Layer et al., 2014 | N/A |
| BRASS | Cancer IT | https://github.com/cancerit/BRASS |
| BreakDancer | Fan et al., 2014 | N/A |
| CTLPScanner server | Yang et al., 2016 | N/A |
| COSMIC database | Bamford et al., 2004 | N/A |
| TelSeq software | Ding et al., 2014 | N/A |
| NetMHCcons server | Karosiene et al., 2012 | N/A |
| Spliced Transcripts Alignment to a Reference (STAR) algorithm | Dobin et al., 2013 | N/A |
| FastQC and Qualimap tool | Okonechnikov et al., 2016 | N/A |
| Web-based gene set analysis toolkit (WebGestalt) | Wang et al., 2013; Zhang et al., 2005 | N/A |
| KEGG | Kyoto Encyclopedia of Genes and Genomes | https://www.kegg.jp/ |
| Reactome | Reactome | https://reactome.org |
| PANTHER | Panther Geneontology | http://www.pantherdb.org/ |
| MicroVigene software | VigeneTech | N/A |
| R package Super-Curve (version 1.01) | MDACC | https://bioinformatics.mdanderson.org/public-software/archive/oompa/ |
| GeneMANIA server | Warde-Farley et al., 2010 | N/A |
| Non-redundant human proteome database | Swiss-Prot | https://www.uniprot.org/ |
| Proteome Discoverer | Thermo Fisher Scientific | N/A |
| Mascot | Matrix Science | N/A |
| PAMR (Prediction Analysis for Microarrays) R package | Lazar et al., 2016 | N/A |
| Mann-Whitney U rank sum testing | MedCalc (version 19.0.3). | |
| LIMMA package (version 3.8) | Ritchie et al., 2015 | N/A |
| ClustVis web tool | Metsalu and Vilo, 2015 | N/A |
| BWA and Picard | DePristo et al., 2011 | N/A |
| In-house R package | Takahashi et al., 2018 | N/A |
| CIBERSORTtool | Stanford University | https://cibersort.stanford.edu |
| DESeq2 | Love et al., 2014 | N/A |
| GraphPad Prism | GraphPad Software | N/A |
Highlights.
High rate of NF1 loss in the R0 compared to neoadjuvant chemotherapy (NACT) group
Lower chromothripsis-like pattern and higher neoantigens in the R0 versus NACT group
Increased number of infiltrated T cells and decreased macrophages in the R0 group
Significant transcriptomic and proteomic variations between HGSC subgroups
ACKNOWLEDGMENTS
We thank S. Patterson (Department of Scientific Publications, MD Anderson Cancer Center, Houston, TX) for editorial assistance. We would like to acknowledge the MD Anderson Functional Proteomics Reverse Phase Protein Array Core for help with the RPPA assay, the MD Anderson Biospecimen Extraction Resource for extraction of genomic DNAs, the MD Anderson Research Histology Core Laboratory for IHC assay, and the Collaborative Health Initiative Research Program Laboratory Core for whole-genome sequencing library preparation and profiling. This work was supported in part by the MD Anderson Ovarian Cancer Moon Shot Program; National Institutes of Health (NIH) grants P50CA217685, R35CA209904, and P30CA016672 (used the Clinical Trials Office, Functional Proteomics Reverse Phase Protein Array Core, Research Histology Core Laboratory, and Cancer Genomics Laboratory); and the MD Anderson Center for Translational and Public Health Genomics (used the Biospecimen Extraction Resource). S.N.W. is supported by the Andrew Sabin Family Fellowship and the GOG Foundation Scholar Investigator Award. A.K.S. is supported by the American Cancer Society Research Professor Award and the Frank McGraw Memorial Chair in Cancer Research. O.D.L. is supported by an NIH institutional training grant (5T32CA009599). G.B.M. is supported by a kind gift from the Adelson Research Foundation; NIH grants U01CA217842, P50CA217685, P50CA098258, U24CA210950, and U54HG008100; Susan G. Komen grant SAC110052; Breast Cancer Research Foundation grant BCRF-17-108; the Ovarian Cancer Research Foundation; and Cancer Prevention and Research Institute of Texas grant RP170640. C.R., N.W.B., K.A.C., K.W., J.L.L., B.L.H., Y.C., T.P.C., and G.L.M. are supported by the Uniformed Services University of the Health Sciences award to the Gynecologic Cancer Center of Excellence (HU0001-16-2-0006), administered by the Henry M. Jackson Foundation for the Advancement of Military Medicine.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.066.
DECLARATION OF INTERESTS
A.A.J. consults with Roche/Genentech, Aravive, and Almac Group; has research funding from AstraZeneca, Pfizer, Bristol-Myers Squibb, Immatics, Iovance Biotherapeutics; honoraria from Gerson Lehrman Group; and travel support from AstraZeneca and MedImmune. K.R., A.D., and D.C. are employees of Akoya Biosciences. N.D.F. consults with Tesaro. S.N.W. has clinical research grants from AstraZeneca, ArQule, Bayer, Clovis Oncology, Cotinga Pharmaceuticals, NCCN, Novartis, Roche/Genentech, and Tesaro and consults with AstraZeneca, Circulogene, Clovis Oncology, Merck, Novartis, Pfizer, Roche/Genentech, Takeda, and Tesaro. R.L.C. has clinical research grants from AstraZeneca, Merck, Clovis Oncology, Genmab, Roche/Genentech, Janssen, V Foundation, and Gateway for Cancer Research and consults with AstraZeneca, Merck, Tesaro, Medivation, Clovis Oncology, Genmab, GamaMabs, Agenus, Regeneron, OncoQuest, OncoSec, Roche/Genentech, and Janssen. G.B.M. consults with AstraZeneca, ImmunoMet, Ionis, Nuevolution, PDX Phamaceuticals, SignalChem, Symphogen, and Tarveda Therapeutics; has stock options with Catena Pharmaceuticals, ImmunoMet, SignalChem, Spindletop Captial, and Tarveda Therapeutics; sponsored research from AstraZeneca, ImmunoMet, Pfizer, NanoString, and Tesaro and travel support from Chrysallis BioTherapeutics; and has licensed technology to NanoString and Myriad Genetics. Y.C.’s spouse owns stock in Celsion. T.P.C. consults with Thermo Fisher Scientific and has research funding from AbbVie. G.L.M. consults with Merck, Kiyatek, Renovia, and Tesaro and has research funding from Merck. A.K.S. consults with Merck and Kiyatec, has research funding from M-Trap, and is a shareholder of Bio-Path Holdings. All other authors declare no competing interests.
REFERENCES
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Hand-saker RE, Kang HM, Marth GT, and McVean GA; 1000 Genomes Project Consortium (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison LH, Bateman NW, Hood BL, Conrads KA, Zhou M, Litzi TJ, Oliver J, Mitchell D, Gist G, Blanton B, et al. (2019). Extensive Intratumor Proteogenomic Heterogeneity Revealed by Multiregion Sampling in a High-Grade Serous Ovarian Tumor Specimen. bioRxiv 10.1101/761155. [DOI] [Google Scholar]
- Ansquer Y, Leblanc E, Clough K, Morice P, Dauplat J, Mathevet P, Lhommé C, Scherer C, Tigaud JD, Benchaib M, et al. (2001). Neoadjuvant chemotherapy for unresectable ovarian carcinoma: a French multicenter study. Cancer 91, 2329–2334. [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, and Wooster R (2004). The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowtell DD, Böhm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, et al. (2015). Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess M, and Puhalla S (2014). BRCA 1/2-Mutation Related and Sporadic Breast and Ovarian Cancers: More Alike than Different. Front. Oncol 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Meric-Bernstam F, Zhao H, Zhang Q, Ezzeddine N, Tang LY, Qi Y, Mao Y, Chen T, Chong Z, et al. (2015). Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin. Chem 61, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al. (2011). High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 1, 170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara Y, Shimoda M, Hori A, Ohara A, Naoi Y, Ikeda JI, Kagara N, Tanei T, Shimomura A, Shimazu K, et al. (2017). A small-molecule inhibitor of SMAD3 attenuates resistance to anti-HER2 drugs in HER2-positive breast cancer cells. Breast Cancer Res. Treat 166, 55–68. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, Armaiz-Pena GN, Previs RA, Hansen JM, Rupaimoole R, et al. (2017). Macrophages Facilitate Resistance to Anti-VEGF Therapy by Altered VEGFR Expression. Clin. Cancer Res 23, 7034–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Mangino M, Aviv A, Spector T, and Durbin R; UK10K Consortium (2014). Estimating telomere length from whole genome sequence data. Nucleic Acids Res 42, e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, and Pfisterer J (2009). Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaeko-logische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 115, 1234–1244. [DOI] [PubMed] [Google Scholar]
- Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, and Rinner O (2012). Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotti A, Ferrandina G, Fanfani F, Garganese G, Vizzielli G, Carone V, Salerno MG, and Scambia G (2008). Prospective validation of a laparoscopic predictive model for optimal cytoreduction in advanced ovarian carcinoma. Am. J. Obstet. Gynecol 199, 642.e1–6. [DOI] [PubMed] [Google Scholar]
- Fan X, Abbott TE, Larson D, and Chen K (2014). BreakDancer: Identification of Genomic Structural Variation from Paired-End Read Mapping. Curr. Protoc. Bioinformatics 45, 15.6.1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming ND, Nick AM, Coleman RL, Westin SN, Ramirez PT, Soliman PT, Fellman B, Meyer LA, Schmeler KM, Lu KH, and Sood AK (2018). Laparoscopic Surgical Algorithm to Triage the Timing of Tumor Reductive Surgery in Advanced Ovarian Cancer. Obstet. Gynecol 132, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forment JV, Kaidi A, and Jackson SP (2012). Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat. Rev. Cancer 12, 663–670. [DOI] [PubMed] [Google Scholar]
- Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, et al. ; NHLBI Exome Sequencing Project (2013). Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Iva-nova Y, Hundal J, Arthur CD, Krebber WJ, et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Artyomov MN, Mardis ER, and Schreiber RD (2015). Tumor neoantigens: building a framework for personalized cancer immunotherapy. J. Clin. Invest 125, 3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res 22, 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, Abkevich V, Potter J, Pruss D, Glenn P, et al. (2010). Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol 28, 3570–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman RT, Chisholm GB, Lu KH, and Futreal PA (2018). Genomic Rearrangement Signatures and Clinical Outcomes in High-Grade Serous Ovarian Cancer. J. Natl. Cancer Inst 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson GC, Kohn EC, Kitchener HC, and Ledermann JA (2014). Ovarian cancer. Lancet 384, 1376–1388. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, Gill MB, Park KJ, Zivanovic O, Konner J, et al. (2017). Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 170, 927–938.e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925. [DOI] [PubMed] [Google Scholar]
- Karosiene E, Lundegaard C, Lund O, and Nielsen M (2012). NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64, 177–186. [DOI] [PubMed] [Google Scholar]
- Kim LC, Song L, and Haura EB (2009). Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol 6, 587–595. [DOI] [PubMed] [Google Scholar]
- Korbel JO, and Campbell PJ (2013). Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–1236. [DOI] [PubMed] [Google Scholar]
- Koti M, Siu A, Clément I, Bidarimath M, Turashvili G, Edwards A, Rahimi K, Mes-Masson AM, and Squire JA (2015). A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br. J. Cancer 112, 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. (2015). Erratum: Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 523, 370. [DOI] [PubMed] [Google Scholar]
- Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, Glas R, Slamon D, Velculescu VE, Kuman RJ, and Shih IeM. (2009). Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol 174, 1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun 8, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH, and Guha A (2000). Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J. Neuropathol. Exp. Neurol 59, 759–767. [DOI] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar C, Gatto L, Ferro M, Bruley C, and Burger T (2016). Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res 15, 1116–1125. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Yu PN, Huang KY, Su HY, Hsiao TH, Chang CP, Yu MH, and Lin YW (2016). NKX6.1 functions as a metastatic suppressor through epigenetic regulation of the epithelial-mesenchymal transition. Oncogene 35, 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK, et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cao Y, Li M, and Jin F (2018). Upregulation of HES1 Promotes Cell Proliferation and Invasion in Breast Cancer as a Prognosis Marker and Therapy Target via the AKT Pathway and EMT Process. J. Cancer 9, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al. (2012). Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res 22, 2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, et al. (2009). Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 15, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Dai XM, and Du B (2015). Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther 16, 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre G, Goranova TE, De Silva D, Ennis D, Piskorz AM, Eldridge M, Sie D, Lewsley LA, Hanif A, Wilson C, et al. (2018). Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet 50, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayakonda A, Lin DC, Assenov Y, Plass C, and Koeffler HP (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, Yi S, Lin SY, and Sahni N (2018). Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun 9, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. ; NCI CPTAC (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43 (W1), W566–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz EA, Tward AD, Hammon RJ, Ren Y, and Rocco JW (2015). Intratumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med 12, e1001786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick AM, Coleman RL, Ramirez PT, and Sood AK (2015). A framework for a personalized surgical approach to ovarian cancer. Nat. Rev. Clin. Oncol 12, 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris EJ, Jones WD, Surleac MD, Petrescu AJ, Destephanis D, Zhang Q, Hamadeh I, Kneisl JS, Livasy CA, Ganapathi RN, et al. (2018). Clonal lineage of high grade serous ovarian cancer in a patient with neurofibromatosis type 1. Gynecol. Oncol. Rep 23, 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Conesa A, and García-Alcalde F (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, and Mann M (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Palacios EH, and Weiss A (2004). Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000. [DOI] [PubMed] [Google Scholar]
- Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. ; Australian Ovarian Cancer Study Group (2015). Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494. [DOI] [PubMed] [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kübler K, Mouw KW, et al. (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet 49, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, and Maglott DR (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33, D501–D504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, Tokheim CJ, Brown A, DeBlasio RM, Niyazov J, et al. (2018). Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retief JD (2000). Phylogenetic analysis using PHYLIP. Methods Mol. Biol 132, 243–258. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, and Shah SP (2014). PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. ; International SNP Map Working Group (2001). A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933. [DOI] [PubMed] [Google Scholar]
- Salmond RJ, Filby A, Qureshi I, Caserta S, and Zamoyska R (2009). T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev 228, 9–22. [DOI] [PubMed] [Google Scholar]
- Schrader KA, Hurlburt J, Kalloger SE, Hansford S, Young S, Huntsman DG, Gilks CB, and McAlpine JN (2012). Germline BRCA1 and BRCA2 mutations in ovarian cancer: utility of a histology-based referral strategy. Obstet. Gynecol 120, 235–240. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Sahar L, Portier KM, Ward EM, and Jemal A (2015). Cancer death rates in US congressional districts. CA Cancer J. Clin 65, 339–344. [DOI] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Hu B, Wang F, Yan Y, Kim E, Vitale C, Patel KP, Strati P, Gumbs C, Little L, et al. (2018). Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 131, 1820–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarney CM, Wang G, Bateman NW, Conrads KA, Zhou M, Hood BL, Loffredo J, Tian C, Darcy KM, Hamilton CA, et al. (2019). Biomarker panel for early detection of endometrial cancer in the Prostate, Lung, Colorectal, and Ovarian cancer screening trial. Am. J. Obstet. Gynecol 221, 472.e1–472.e410. [DOI] [PubMed] [Google Scholar]
- Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, and Mechtler K (2011). Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- Vaughan S, Coward JI, Bast RC Jr., Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, et al. (2011). Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer 11, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, et al. (2011). Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 108, 18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Duncan D, Shi Z, and Zhang B (2013). WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 41, W77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gu C, Jeong KJ, Zhang D, Guo W, Lu Y, Ju Z, Panupinthu N, Yang JY, Gagea MM, et al. (2017). YAP/TAZ-Mediated Upregulation of GAB2 Leads to Increased Sensitivity to Growth Factor-Induced Activation of the PI3K Pathway. Cancer Res 77, 1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. (2010). The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, and Leed-man PJ (2009). Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem 284, 5731–5741. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ell-rott K, Shmulevich I, Sander C, and Stuart JM; Cancer Genome Atlas Research Network (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter WE 3rd, Maxwell GL, Tian C, Sundborg MJ, Rose GS, Rose PG, Rubin SC, Muggia F, and McGuire WP; Gynecologic Oncology Group (2008). Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage IV epithelial ovarian cancer: a Gynecologic Oncology Group Study. J. Clin. Oncol 26, 83–89. [DOI] [PubMed] [Google Scholar]
- Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, Feng XH, Sawaya R, Medema RH, Hung MC, and Huang S (2014). Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-b-dependent cancer metastasis. J. Clin. Invest 124, 564–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu J, Ouyang L, Chen Y, Liu B, and Cai H (2016). CTLPScanner: a web server for chromothripsis-like pattern detection. Nucleic Acids Res 44, W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, and Snoddy J (2005). WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33, W741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Shan Y, Liu B, Li Y, and Jia L (2017). Functional screen analysis reveals miR-3142 as central regulator in chemoresistance and proliferation through activation of the PTEN-AKT pathway in CML. Cell Death Dis 8, e2830. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data from the WGS, deep targeted sequencing and RNA sequencing in this study have been deposited in the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/home) under accession numbers EGAD00001005240, EGAD00001005239 and EGAD00001005238, respectively. The LC-MS/MS data have been deposited to the PRoteomics IDEntifications (PRIDE, https://www.ebi.ac.uk/pride) database under the accession number PRIDE: PXD014980.