

Abstract
Acute Respiratory Distress Syndrome (ARDS) is a devastating disease process that involves dysregulated inflammation and decreased alveolar-capillary barrier function. Despite increased understanding of the pathophysiology, no effective targeted therapies exist to treat ARDS. Recent preclinical studies suggest that the multi-tyrosine kinase inhibitor, imatinib, which targets the Abl kinases c-Abl and Arg, has the potential to restore endothelial dysfunction caused by inflammatory agonists. Prior work demonstrates that imatinib attenuates LPS (lipopolysaccharide)-induced vascular leak and inflammation; however, the mechanisms underlying these effects remain incompletely understood. In the current study, we demonstrate that imatinib inhibits LPS induced increase in the phosphorylation of CrkL, a specific substrate of Abl kinases, in human pulmonary endothelial cells. Specific silencing of Arg, and not c-Abl, attenuated LPS-induced pulmonary vascular permeability as measured by electrical cellular impedance sensing (ECIS) and gap formation assays. In addition, direct activation of Abl family kinases with the small molecule activator DPH resulted in endothelial barrier disruption that was attenuated by Arg siRNA. In complementary studies to characterize the mechanisms by which Arg mediates endothelial barrier function, Arg silencing was found to inhibit LPS-induced disruption of adherens junctions and phosphorylation of myosin light chains (MLC). Overall, these results characterize the mechanisms by which imatinib protects against LPS-induced endothelial barrier disruption and suggest that Arg inhibition may represent a novel strategy to enhance endothelial barrier function.
Keywords: Acute Lung Injury, ARDS, LPS, Endothelium, c-Abl, Arg, Vascular Permeability, imatinib
INTRODUCTION
Acute Respiratory Distress Syndrome (ARDS) is a disease process characterized by the acute onset of lung inflammation, disruption of the alveolar-capillary barrier, interstitial edema and flooding of air spaces with protein-rich edema fluid. Clinically these pathologic changes cause hypoxemic respiratory failure and often death due to multi-system organ failure [1, 2]. Care for these patients is largely supportive, and low tidal volume mechanical ventilation (MV) is the primary intervention proven to decrease mortality [3, 4]. Improved understanding of the pathophysiology of ARDS has resulted in a number of clinical trials seeking to improve patient outcomes, but effective pharmacologic therapies are still lacking for this devastating disease process, and the mortality rate remains at 30–35% [5–7]. ARDS is most commonly initiated by sepsis or severe pneumonia; however, other conditions involving dysregulated inflammation can also precipitate ARDS, including trauma and aspiration of gastric contents [2, 8]. Multiple endogenous inflammatory mediators, such as thrombin, vascular endothelial growth factor (VEGF), histamine, and reactive oxygen species (ROS), as well as exogenous agents such as lipopolysaccharide (LPS), initiate signaling cascades that alter the pulmonary endothelial cell (EC) structure, causing paracellular gap formation and vascular leak that underlie the pathogenesis of ARDS [9, 10].
Recent work by multiple groups indicates that imatinib, a tyrosine kinase inhibitor used to treat chronic myelogenous leukemia (CML) and other malignancies, attenuates vascular permeability induced by several inflammatory mediators including LPS, VEGF, histamine, thrombin and oxidative stress [11–17]. Although imatinib is targeted to inhibit the BCR-Abl fusion protein that causes CML, it also inhibits the non-oncogenic forms of the Abl family kinases (c-Abl, Abl related gene (Arg)), as well as the related kinases c-kit and platelet-derived growth factor receptor (PDGFR), which are known to play a role in vascular endothelial barrier function [18, 19]. These studies have indicated that multiple targets of imatinib, including c-Abl, Arg, and PDGFR, are involved in regulating the cytoskeletal rearrangements that mediate vascular permeability. Further support for a role of imatinib in the treatment of vascular leak is provided by a series of case reports in which imatinib therapy was associated with rapid resolution of pulmonary and systemic vascular leak [20–22]. Although these observations suggest that imatinib may be a promising treatment for inflammatory vascular leak, the mechanisms underlying these effects remain incompletely understood.
Our previous work has implicated the Abl kinases as critical to the LPS-induced NF-kB activation, inflammatory cytokine production, and the expression of vascular cellular adhesion molecule-1 (VCAM-1) [16, 17, 23]. Although, previous studies have also implicated the Abl kinases as critical mediators in regulation of endothelial permeability [11, 14, 24], the relative roles of the closely related kinases c-Abl and Arg have not been defined in LPS-induced EC barrier disruption. In the current study, we demonstrate that Arg, and not c-Abl, expression appears necessary to mediate EC permeability induced by LPS. Moreover, the mechanism involves junctional complex disruption and increased phosphorylation of myosin light chains (MLC); changes associated with paracellular gap formation. These studies provide new insights into the mechanisms by which Abl family kinases regulate inflammatory vascular leak and highlight the potential barrier protective effects of imatinib during these pathophysiologic conditions.
MATERIALS AND METHODS
Reagents
Imatinib was purchased from LC Laboratories (Woburn, MA). Nonspecific control siRNA (siCon), c-Abl siRNA, Arg siRNA and antibodies to VE-cadherin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phosphorylated Y489 β-catenin were purchased from Abcam (Cambridge, MA). Antibodies to total β-catenin were purchased from BD Biosciences (San Diego, CA). Antibodies to c-Abl, phosphorylated CrkL and MLC (Thr18/Ser-9), total CrkL, β-actin, and all secondary HRP-conjugated secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Arg antibody was purchased from Novus Biologicals (Littleton, CO). E. Coli LPS (E0127:B8, L3880), DPH [5-(1,3-diaryl-1H-pyrazol-4-yl) hydantoin, 5-[3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl]-2,4-imidazolidinedione] and all other reagents were purchased from Sigma (St. Louis, MO) unless otherwise specified.
Endothelial Cell Culture
Human pulmonary artery endothelial cells (HPAEC) and human lung microvascular endothelial cells (HLMVEC) were purchased from Lonza (Walkersville, MD) and cultured in Endothelial Cell Growth Medium-2 supplemented with 10% fetal bovine serum (FBS) (Sigma, St Louis, MO). Cells were maintained at 37°C in a 5% CO2 incubator and used at passages 6–8 for all experiments. Cells were incubated in 2% FBS media for three hours prior the specified treatments (LPS or DPH). In some experiments, HPAEC were treated with imatinib (20 μM in PBS) prior the addition of LPS.
Transfections with Silencing RNA
For siRNA transfections, we used the transfection reagent X-Fect siRNA (Clontech, Mountain View, CA). Cells were treated with siRNA at 90% confluence as described by the manufacturer’s protocol and challenged with LPS 24–48 hours later. Selective silencing of c-Abl and Arg was confirmed by western blotting as previously described [16].
Western Blotting
After the specified treatments, EC were washed in ice-cold PBS and then harvested in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein samples were then prepared in Laemmli’s SDS sample buffer (Boston Bioproducts, Ashland, MA), boiled, subjected to SDS-PAGE electrophoresis (4–20% ExpressPlus PAGE gels, Genscript, Piscataway, NJ), and transferred to polyvinylidene difluoride membrane. The membranes were blocked in 5% bovine serum albumin and incubated in the indicated antibodies (overnight, 4°C). HRP-conjugated secondary antibodies were added to the membranes (60 min, RT), and the Pierce enhanced chemiluminescence detection system (Thermo Scientific, Rockford, IL) was used to visualize the bands. Band densities were determined using the Image J software (National Institutes of Health).
Immunofluorescence Microscopy
HPAEC grown on gelatin-coated coverslips were treated with c-Abl or Arg siRNA and LPS (vs. PBS) as described above, fixed in 4% PFA (20 min, RT), permeabilized with 0.1% Triton X in PBS, and incubated with VE-cadherin antibody (overnight, 4°C). Coverslips were then washed, incubated with fluorescently labeled secondary antibody, and mounted on glass slides using DAPI Prolong Gold anti-fade reagent (Invitrogen, Grand Island, NY). Images were acquired by a blinded investigator using Nikon Eclipse TE2000 microscope at 63X with an oil emersion lens.
XPerT Permeability Assay
To quantify intercellular gap formation in EC exposed to LPS, we utilized an assay which takes advantage of the strong affinity of avidin and biotin [25]. Gelatin (Sigma, St. Louis, MO) conjugated to biotin (Thermo Scientific, Rockford, IL) was dissolved in 0.1 M bicarbonate buffer, and the solution was used to coat the wells of a 6-well plate. After incubation with the biotinylated gelatin (4 °C, overnight), the solution was aspirated off, plates were washed twice with PBS and seeded with cells. Following indicated treatments, avidin linked to fluorescein isothiocyanate was added to the cells (12.5 µg/mL, 3 min). The media was then aspirated off and cells were washed twice with warm PBS supplemented with calcium and magnesium. Representative images were taken at 10X and 20X on a Nikon Eclipse TE2000 microscope. To measure gap formation between cells, the fluorescent area in each image was quantified using the color threshold feature in Image J software.
Transendothelial Electrical Resistance (TER) Measurements
The integrity of the EC monolayer was evaluated using electrical cell impedance sensing (ECIS) (Applied Biophysics, Troy, NY) as previously described [26]. Briefly, HPAEC were grown to confluence on gold microelectrodes and the cells were subjected to a weak electric current for the continuous measurement of TER. After one hour of measurement (to establish a stable baseline resistance), the indicated stimuli were added to the wells and measurement continued for 20 hours. The data were analyzed using custom designed Epool software by normalizing each resistance value to the starting resistance for that electrode as we have previously described [26].
Statistical Analysis
Results were expressed as mean +/− standard deviation (SD) of the mean. All experiments were repeated at least three independent times. Analysis of variance analysis (ANOVA) was used to compare differences between groups. Statistical analysis was performed with GraphPad Prism 8 software, with the significance level of p<0.05 for all experiments.
RESULTS
Imatinib attenuates intercellular gap formation induced by LPS
Recent work from our group and others demonstrates that imatinib attenuates LPS-induced acute lung injury (ALI) in mice given both as a pre-treatment and post-injury, suggesting that imatinib has potent effects on the pulmonary vasculature that warrant additional mechanistic exploration [12, 16, 17]. Consistent with these in vivo observations, we previously reported that in HPAEC imatinib attenuated LPS-induced permeability, as measured by transendothelial resistance (TER) and expression of the critical adherens junction protein VE-cadherin [16]. Although TER is a valuable tool for assessing permeability, it has well-described limitations [27, 28], and complementary modalities are necessary to fully characterize EC barrier function. Here we utilize a recently developed assay that allows for visualization and quantification of interendothelial gap formation as a measure of vascular leak in vitro [25]. Using this approach, LPS causes a 1.8 fold increase in gap formation over PBS-treated control cells. Consistent with prior TER data, imatinib pretreatment abolishes this LPS-induced gap formation (Figure 1A, B).
Figure 1.
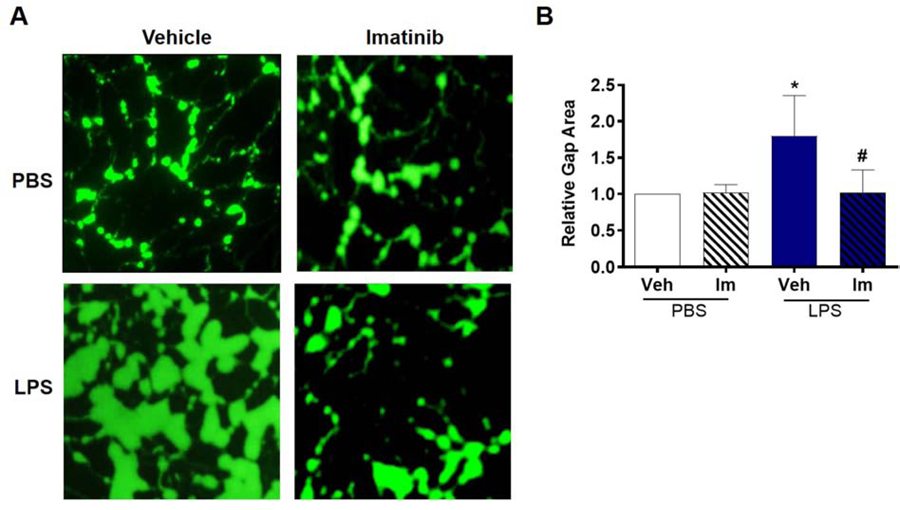
Imatinib attenuates intercellular gap formation induced by LPS. HPAEC grown on biotinylated gelatin were pretreated with imatinib (Im) (20 µM, 1 hr) and then challenged with LPS (1 µg/ml, 3 hrs). Cells were then treated with FITC-labeled avidin (12.5 µg/ml, 3 min) and washed twice prior to imaging. (A) Green fluorescence depicts areas between cells, penetrated by the FITC-labeled avidin. (B) Quantification of FITC stained area to assess intercellular gap formation. Gap area is significantly increased following LPS treatment in untreated cells while intercellular gaps in imatinib treated cells are similar to control. *p<0.05 relative to vehicle/PBS. #p<0.05 vs vehicle/LPS. Data are reported as mean +/− SD of three independent experiments.
LPS increases the activity of Abl kinases
We next measured the effects of LPS on the activity of the Abl family kinases. To determine c-Abl and Arg activity, phosphorylation levels were assessed at tyrosine 207 of their substrate CrkL, a site that is specific for c-Abl and Arg but that does not distinguish between the two [11, 29, 30]. As depicted in Figures 2A–B, LPS induces the phosphorylation of CrkL, while imatinib, which acts as an inhibitor of both c-Abl and Arg successfully inhibits this response. In order to study the role of these kinases individually, a strategy of selectively silencing each of these kinases using siRNA technology was utilized, which we previously have demonstrated downregulates protein expression of either c-Abl or Arg [16]. In cells treated with nonspecific control siRNA (siCon), we observed a significant increase in CrkL phosphorylation following LPS treatment. After silencing either c-Abl or Arg expression, CrkL phosphorylation was significantly decreased under basal conditions or after LPS (Figure 2C–D). These data indicate that LPS increases both c-Abl and Arg activity in human lung EC.
Figure 2.
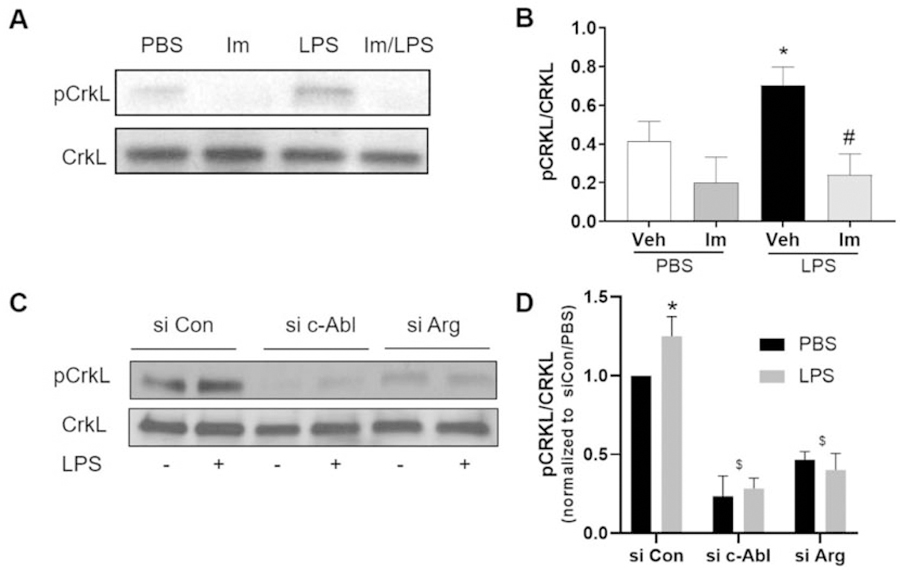
LPS increases the activity of Abl kinases in HPAEC. (A-B) HPAEC were pretreated with imatinib (Im) (20 µM, 1 hr) and then challenged with LPS (1 µg/mL, 1 hr) or PBS. (A) Representative blots of phosphorylated and total CrkL. (B) Densitometry analysis of the western blots of p-CrkL relative to total CrkL. *p<0.05 relative to Veh/PBS, #p<0.05 vs vehicle/LPS. Data are reported as mean +/− SD of three independent experiments. (C-D) HPAEC were transfected with control (siCon), c-Abl, or Arg specific siRNA and then challenged with LPS (1 µg/mL, 3 hrs). (C) Representative blots of phosphorylated and total CrkL. (D) Densitometry analysis of the western blots of p-CrkL relative to total CrkL. The ratio of p-CrkL / total CrkL was normalized to si Con/PBS within each experiment. *p<0.05 relative to siCon/PBS, $p<0.05 relative to corresponding siCon condition. Data are reported as mean +/−SD of three independent experiments.
Arg mediates LPS-induced lung EC permeability
Given that LPS activates both c-Abl and Arg kinases, we next determined their relative roles in mediating LPS-induced EC barrier dysfunction. Multiple complementary methods of measuring EC barrier integrity were employed. In pulmonary artery endothelial cells (HPAEC), measurements of inter-endothelial gap formation using the XPerT assay demonstrated that down-regulation of Arg expression via siRNA significantly attenuated LPS-induced gap formation (decreased by 41%, p<0.05), while silencing c-Abl had no effect (Figure 3A–B). Similarly, in TER experiments, silencing Arg attenuated LPS-induced EC permeability, while c-Abl siRNA had no effect (Figure 3C). There are significant phenotypic differences between pulmonary ECs lining large conduit vessels and those forming the capillaries associated with alveolar units [31]. Importantly, cultured microvascular ECs demonstrate increased barrier resistance when compared to the barrier integrity of larger vessels. To confirm key findings identified in HPAEC, we performed measurements of inter-endothelial gap formation in human lung microvascular endothelial cells (HLMVEC). Silencing Arg expression significantly reduced HLMVEC gap formation following LPS (Figure 4). In contrast but similar to HPAEC, we did not observe a protective effect against EC permeability after silencing c-Abl. Together these data strongly suggest that Arg, and not c-Abl, is the primary mediator of LPS-induced barrier dysfunction in lung endothelium.
Figure 3.
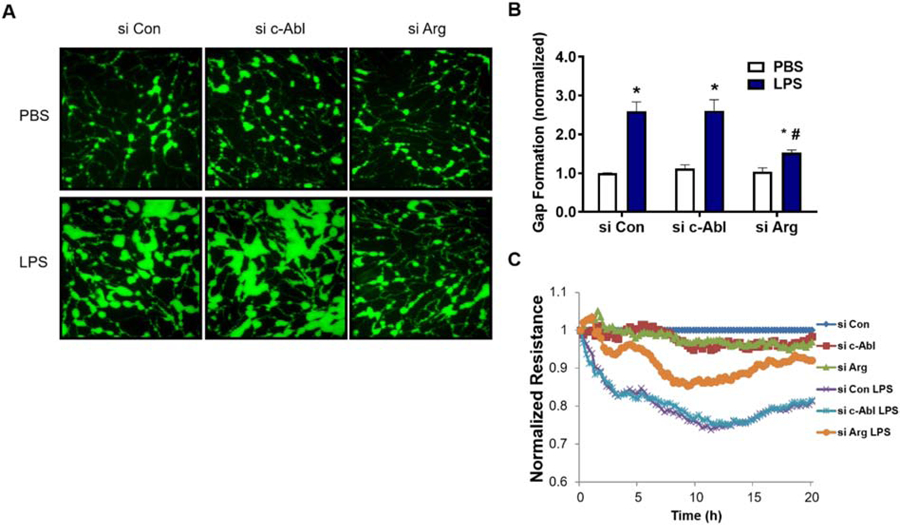
LPS-induced permeability is mediated by Arg in HPAEC. (A-B) HPAEC grown on biotinylated gelatin were transfected with nonspecific (Con), c-Abl, or Arg specific siRNA and then challenged with LPS (1 µg/mL, 3 hrs). Cells were then treated with FITC-labeled avidin (12.5 µg/ml, 3 min) and washed twice prior to imaging. (A) Green fluorescence depicts areas between cells, penetrated by FITC-labeled avidin. Images were taken at 20x magnification. (B) Quantification of FITC stained area to assess intercellular gap formation. *p<0.05 compared to siCon/PBS, #p<0.05 compared to siCon/LPS. (C) HPAEC grown on ECIS plates were transfected with control (Con), c-Abl, or Arg specific siRNA and then challenged with LPS (1 µg/mL). TER was measured continuously for 20 hours and the resistance in each well was normalized to si-Con + veh at each time point. Data shown are a representative tracing from three independent experiments.
Figure 4.
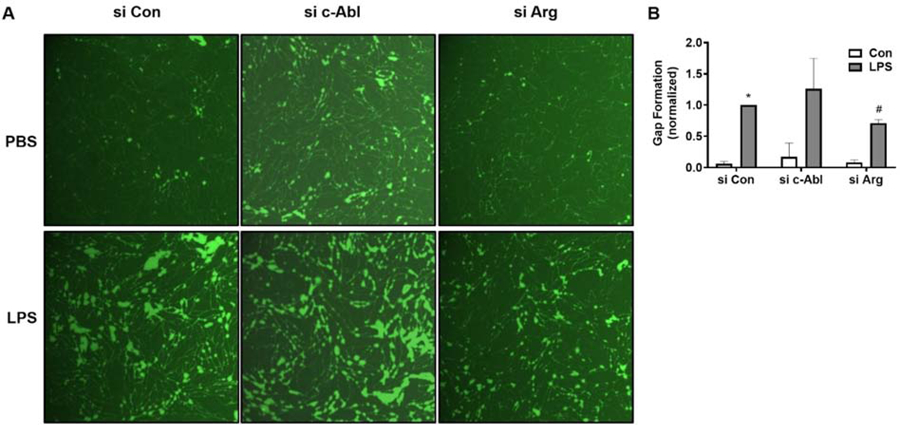
LPS-induced permeability is mediated by Arg in HLMVEC. HLMVEC grown on biotinylated gelatin were transfected with nonspecific (Con), c-Abl, or Arg specific siRNA and then challenged with LPS (100 ng/mL, 5 hrs). Cells were then treated with FITC-labeled avidin (12.5 µg/ml, 3 min) and washed twice prior to imaging. (A) Green fluorescence depicts areas between cells, penetrated by FITC-labeled avidin. Images were taken at 10x magnification. (B) Quantification of FITC stained area to assess intercellular gap formation. *p<0.05 compared to siCon/PBS, #p<0.05 compared to siCon/LPS.
Arg mediates LPS-induced disruption of adherens junctions
Adherens junctions (AJ) are critical cell-cell linkages in EC and play a major role in barrier function. In endothelial AJ complexes, VE-cadherin forms the transmembrane component of AJ and interacts directly with intracellular catenins. LPS induces VE-cadherin loss from EC junctions, while imatinib pre-treatment significantly attenuates VE-cadherin loss, as we have demonstrated previously [16]. To address the roles of the Abl kinases in this response, the effects of selectively silencing c-Abl or Arg on LPS-induced AJ disruption were assessed using IF microscopy to determine VE-cadherin localization. As expected, ECs that were treated with nonspecific control siRNA exhibit decreased VE-cadherin at the intercellular junctions after LPS, indicating AJ disruption (Figure 5A). Treatment with siRNA for Arg, but not for c-Abl, preserved AJ structure following LPS as evidenced by persistent VE-cadherin staining at the cell-cell junctions (Figure 5A). Together these data indicate that Arg and not c-Abl participates in the disruption of AJ during LPS-induced barrier dysfunction.
Figure 5.
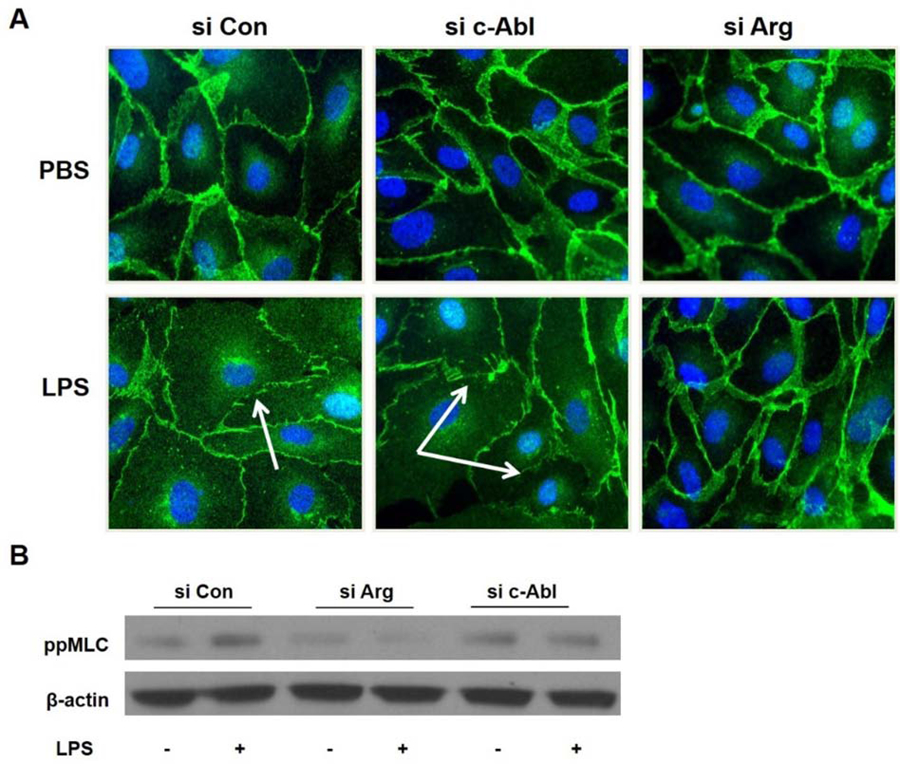
Arg mediates disruption of adherens junctions and MLC phosphorylation in LPS- treated lung EC. HPAEC were transfected with nonspecific (Con), c-Abl, or Arg specific siRNA and then challenged with LPS (1 µg/ml, 3 hrs). (A) Fixed cells were then subjected to immunofluorescence microscopy after staining for VE-cadherin. Multiple fields were obtained with a 63x objective by a blinded observer. Representative images are shown. VE-cadherin is shown in green and DAPI (nuclear stain) in blue. White arrows indicate areas of loss in peripheral VE-cadherin staining. (B) Cell lysates were analyzed by western blotting for phospho-MLC. β-actin was used as a loading control. A representative blot of four independent experiments is shown.
Loss of Arg impairs myosin light chain phosphorylation
EC cytoskeletal dynamics are a critical determinant of barrier integrity. Formation of transcellular actin stress fibers is observed following stimulation with numerous barrier disruptive compounds including LPS. These structures exert a contractile force through the ratcheting action of actin-myosin bonds leading to membrane retraction and intercellular gap formation. Phosphorylation of the myosin light chain (MLC) induces actomyosin contraction and is highly regulated by well described signaling pathways [9, 32, 33]. Inhibition or down-regulation of both Abl kinases has been previously shown to decrease MLC phosphorylation induced by other stimuli, such as VEGF and thrombin [14]. In the current study, HPAEC treated with LPS demonstrated the expected increase in MLC phosphorylation, and silencing Arg expression substantially attenuated this increase more than c-Abl silencing (Figure 5B). Thus, Arg is required for the maximal induction of actomyosin contraction which is associated with increased EC permeability after LPS.
Arg mediates DPH-induced EC barrier disruption
Further evidence for the involvement of Arg in barrier function is provided by experiments using a small molecule activator of the Abl kinase family, DPH [34]. Initially, we confirmed that DPH activates the Abl kinases in HPAEC, as indicated by increased phosphorylation of CrkL (Figure 6A). Interestingly, similar to LPS, DPH induces phosphorylation of the integral AJ protein β-catenin at tyrosine 489, which is a site specifically targeted by c-Abl/Arg and previously reported to mediate the disruption of β-catenin-cadherin complexes [35, 36] (Figure 6B). These results suggest that DPH could have a direct effect on EC barrier function. Indeed, using the ECIS assay, we then demonstrated that DPH causes EC barrier disruption, as TER values in DPH-treated EC decline over time compared to control cells (DMSO-treated) (Figure 6C).
Figure 6.
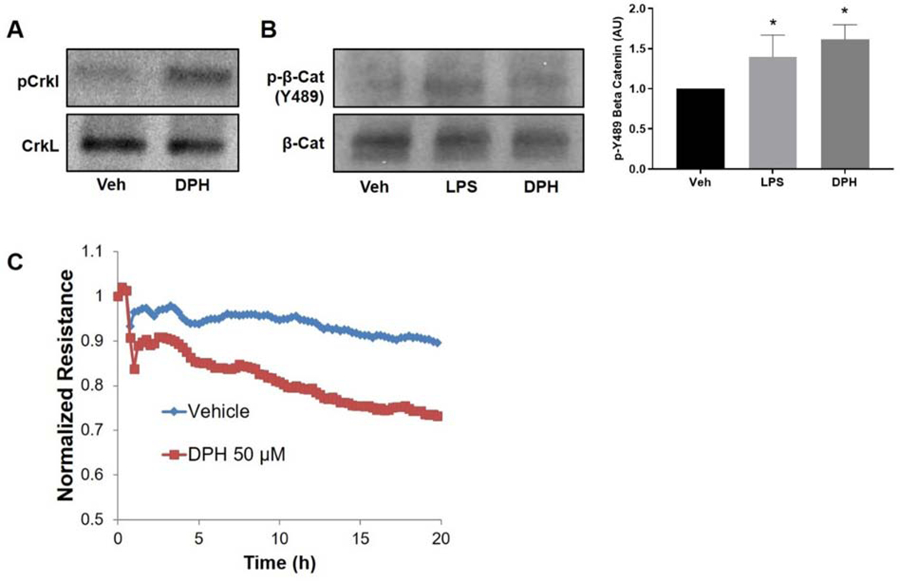
Abl activator, DPH, induces EC barrier disruption. (A) HPAEC were treated with the Abl kinase activator DPH (50 µM, 30 min) or vehicle (Veh; DMSO) and CrkL phosphorylation was determined by western blotting with increased phosphorylation indicating greater Abl kinase activity. Depicted is a representative Western blot of three independent experiments. (B) HPAEC were treated with the Abl kinase activator DPH (50 µM, 30 min), LPS (1 µg/mL, 30 min), or vehicle (Veh) and phosphorylation of β-catenin on tyrosine 489, a c-Abl/Arg specific site, was determined by Western blotting. Depicted are a representative Western blot and densitometric quantification of Western blotting data. Data represent the ratio of p-Y489 β-catenin to total β-catenin normalized to untreated control +/− SD of three independent experiments. *p<0.05 relative to vehicle. (C) HPAEC were grown on ECIS plates and treated with DPH (50 µM) or vehicle (Veh). TER was measured continuously for 20 hours. The resistance in each well was normalized to time zero. Data shown are representative tracings from a total of three independent experiments.
Importantly, silencing of Arg in DPH-treated cells inhibits EC barrier disruption, supporting the hypothesis that Arg is the key Abl kinase in this response (Figure 7A). Finally, DPH augments LPS-induced EC permeability (Figure 7B), which is attenuated in Arg silenced-cells.
Figure 7.

Endothelial permeability induced by the Abl family activator DPH is attenuated by Arg siRNA. HPAEC were grown on ECIS plates and transfected with control (Con) or Arg specific siRNA and then challenged with (A) DPH (50 µM) or (B) DPH plus LPS (50 µM, 30 min DPH pretreatment and then addition of 1 μg/ml LPS). EC barrier function was monitored by ECIS assay and TER was measured continuously for 20 hours. The resistance in each well was normalized to time zero. Data shown are representative tracings from a total of three independent experiments.
DISCUSSION
In ARDS, multiple inflammatory stimuli can contribute to the EC cytoskeletal rearrangements that result in the increased vascular permeability, pulmonary edema formation, and refractory hypoxemia that characterize this devastating illness [9, 10]. The current study demonstrates that LPS, a well-characterized inflammatory stimulus for modeling aspects of ARDS, activates the Abl family of kinases (c-Abl and Arg) as measured by phosphorylation of their specific substrate CrkL (Figure 2). Next, a strategy of selectively silencing c-Abl or Arg to isolate the roles of these kinases in LPS-induced endothelial injury identified a critical role for Arg in mediating permeability in both macro- and microvascular lung EC (Figures 3–4). Additional experiments utilizing the specific small molecule activator of Abl family kinases, DPH [34], further demonstrated the functional importance of Arg in mediating endothelial barrier dysfunction. DPH increases Abl/Arg activity in HPAEC, decreases EC barrier integrity by itself, and augments LPS-induced effects. Interestingly, the permeability effects of DPH are attenuated in Arg-silenced endothelial cells (Figure 7). These data are consistent with recent work demonstrating that Arg mediates the vascular barrier disruptive responses to thrombin [11].
Because vascular barrier function is critically dependent on cell-cell junction integrity and the Abl kinases are known to alter adherens junction (AJ) dynamics [37], we hypothesized that Arg may mediate LPS-induced barrier disruption via effects on these junctional complexes. Consistent with this hypothesis, immunofluorescent imaging demonstrated that Arg expression (but not c-Abl) is necessary for LPS-induced disruption of VE-cadherin junctions (Figure 5A). Additionally, either LPS or DPH significantly increased phosphorylation of the critical AJ protein β-catenin at site Y489 (Figure 6B), which is specifically targeted by c-Abl/Arg and phosphorylated in association with disruption of β-catenin-cadherin complexes [35, 36]. Based on our results demonstrating that Arg and not c-Abl is the critical Abl kinase in regulating permeability induced by LPS, it is reasonable to infer that β-catenin phosphorylation is mediated primarily by Arg. However, the effects of the Abl kinases on cell junctions are likely to be complex and vary with cell-type as well as stimulus. In human umbilical vein EC (HUVEC), Abl kinase inhibition with imatinib restores the integrity of AJ following thrombin stimulation [11]. Although these data suggest that Abl kinases disrupt AJ in EC, work in c-Abl−/−/Arg−/− mouse embryo fibroblasts and rat epithelial cells demonstrate that Abl kinases are also necessary for Rac activation and AJ formation, suggesting that Abl kinases function both upstream and downstream of Rac to promote AJ integrity [38, 39]. In addition to the potentially complex role for the Abl kinases in AJ function, these kinases may also play a role in focal adhesion dynamics, which also contribute to LPS-induced pulmonary vascular barrier disruption. The anchoring effect of focal adhesions to the cell matrix counters contractile actomyosin forces [10] and enhances AJ in a Rac dependent manner [40]. A role for Abl family kinases in this process is provided by recent work which demonstrated that Arg silencing attenuates disruption of the focal adhesion-matrix interaction after thrombin, in HUVECs [11]. Additionally, c-Abl phosphorylation of the focal adhesion protein paxillin is implicated in HLMVEC barrier disruption and in vivo lung injury following LPS [24].
Our data unite the physical concept of inter-endothelial gap formation with functional permeability. Gap formation requires not only breakdown of continuous intercellular adhesions [41] but also retraction of the plasma membrane through centripetal force generation by transcellular stress fibers and interaction of actin-myosin bonds. Phosphorylation of myosin light chains to enhance actin-myosin binding and the subsequent power stroke is a vital mechanism in EC barrier dysfunction induced by LPS and other inflammatory stimuli [9, 33, 42]. In experiments designed to explore this mechanism in our model we demonstrate that Arg expression is necessary for baseline and LPS-induced MLC phosphorylation (Figure 5B). Similar to our findings, Abl kinase inhibition (by imatinib) or loss of expression impairs VEGF-and thrombin-induced MLC phosphorylation in microvascular endothelial cells [14]. Interestingly, the same study found that Abl inhibition did not affect activation of the Rho/ROCK pathway. This finding is supported by another study, which showed that the barrier protective effects of imatinib do not involve the RhoA/Rho kinase pathway [11].
The current study has mechanistic relevance to recent work by our group and others demonstrating that imatinib has the capability to attenuate both pulmonary vascular permeability and inflammation induced by a variety of stimuli, including LPS, oxidative stress, thrombin, histamine, and VEGF, in animal, and cell culture models [11–13, 15–17, 43–45] [19]. Imatinib is described as a potential treatment for tissue edema associated with clinically relevant lung inflammation [11] and brain ischemia/reperfusion injury [46]. This experimental work is supported by a recent series of case reports in which imatinib was found to be protective in some inflammatory vascular leak syndromes [20–22]. While the anti-inflammatory effects of imatinib within the lung are well known, [17, 45, 47, 48], a growing body of evidence also suggests efficacy in multiple other inflammatory disorders affecting the aorta [49], liver [50], kidney [51] and brain [52]. However, it is important to note that imatinib has also been described as a cause of edema [53–55]. The broad therapeutic potential of imatinib combined with its potential side effects, particularly edema formation, require a more complete understanding of the cellular consequences of c-Abl/Arg inhibition and the effects of these alterations in various tissues.
In this context, we designed the current study with the speculation that the determination of the specific roles of the imatinib-sensitive kinases, c-Abl and Arg, in lung EC barrier function and inflammatory signaling will shed light on imatinib’s differential effects. Our hypothesis is supported by data presented here showing that Arg mediates EC barrier disruption. This observation is consistent with previous studies identifying Arg as critical mediator in endothelial barrier disruption in thrombin-treated HUVEC [11]. On the contrary, the role of c-Abl signaling in regulating the lung endothelial barrier disruption is unclear. Our data, using both HPAEC and HLMVEC, demonstrate that LPS-induced EC gap formation is not mediated by c-Abl, while in contrast others have shown that HLMVEC treated with c-Abl siRNA are protected against LPS [24]. There is also evidence of a role for c-Abl in endothelial barrier enhancement through changes in peripheral cytoskeletal mechanical properties associated with barrier enhancing or recovery from disruptive stimuli [56], and its expression is required for the barrier enhancing effect of the sphingosine-1-phosphate analogue FTY-720 [57]. More consistently, work from us and others demonstrate that c-Abl kinase is involved in the pro-inflammatory response to LPS by stimulating inflammatory cytokine and mediator production [16, 23, 58, 59]. Independent of its effects on ECs, c-Abl also regulates neutrophil recruitment and crawling behavior in response to shear stress via the Rac/PAK1/LIMK1/cofilin signaling cascade [60]. Thus, there are critical and complex mechanistic roles played by c-Abl/Arg in mediating endothelial inflammation and lung injury that are differential in nature. The consequences of these processes on EC functional barrier integrity will be the focus of future investigation. By identifying Arg here as the primary driver of LPS-induced endothelial barrier dysfunction through EC gap formation, we advance the mechanistic understanding of imatinib in the hope of better targeting treatment for ARDS.
We acknowledge some limitations of this study. Protein regulation of endothelial barrier function involves complex interaction between many structural, signaling and force-producing molecules [32, 41, 61]. Our focus on Arg in the current work is an oversimplification of this process. However, we believe our assessment of functionally relevant downstream events such as VE-cadherin organization, MLC phosphorylation, EC gap formation and transendothelial resistance measurements highlight the strong impact of this kinase during inflammatory barrier disruption. As with all pharmacologic inhibitors, there is also the potential for imatinib to exhibit off target effects through action on other kinases [62] including implications for immune signaling [63]. Although several inhibitors of the Abl kinases with different specificities are available, there are no specific inhibitors of c-Abl or Arg due to the mechanism of action of this class of drugs, which involves binding to the highly homologous tyrosine kinase domain [64, 65]. The development of specific inhibitors for c-Abl and Arg will be greatly beneficial for future investigation and potential clinical use because they will provide a detailed understanding of the role of each of these kinases on vascular barrier function and facilitate determining the optimal drug for future pre-clinical and clinical trials in ARDS.
In conclusion, this study provides new mechanistic insights into the effects of LPS on the closely related kinases c-Abl and Arg in lung EC (Figure 8). It demonstrates that inhibition of Arg mediates the barrier protective effects of imatinib in LPS-induced vascular leak in lung EC. These results suggest that imatinib and other Abl family kinase inhibitors may have therapeutic potential in disease states involving inflammatory vascular leak, such as ARDS and sepsis.
Figure 8.
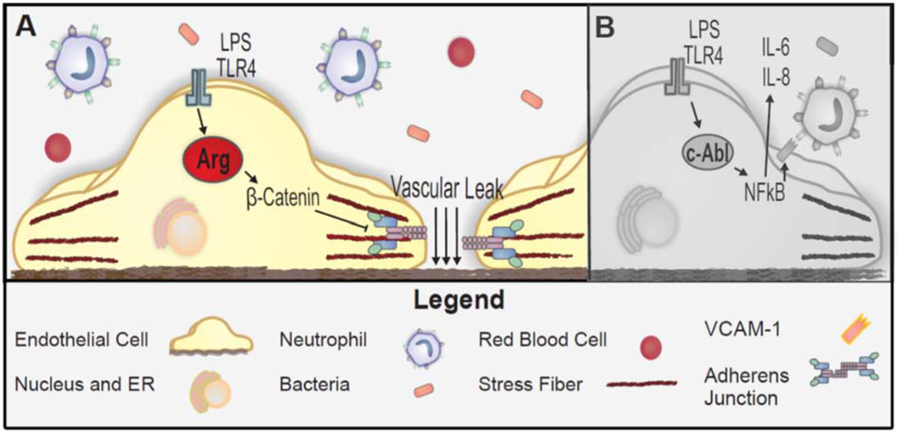
The roles of c-Abl and Arg in LPS-induced vascular leak and inflammation. The proposed schema represents potential pathways based upon the current study and published data. Panel A (color panel) depicts the major findings described in this manuscript. Briefly, exposure to LPS activates Arg in lung endothelial cells, resulting in downstream signaling that includes β-catenin phosphorylation, adherens junction disruption, and ultimately increased vascular leak. Panel B (gray scale) depicts the previously described critical role for c-Abl in the NFκB-mediated inflammatory response of lung endothelium to LPS [16, 17, 23].
ACKNOWLEDGEMENTS
This work was supported by NIH P01 HL126609 (JG), F30HL121982 (AR), AHA 14PRE18860021 (AR), and NIH K08 HL135318 (PB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Matthay MA, Ware LB, and Zimmerman GA, The acute respiratory distress syndrome. J Clin Invest, 2012. 122(8): p. 2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson BT, Chambers RC, and Liu KD, Acute Respiratory Distress Syndrome. N Engl J Med, 2017. 377(19): p. 1904–1905. [DOI] [PubMed] [Google Scholar]
- 3.Acute Respiratory Distress Syndrome, N., et al. , Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med, 2000. 342(18): p. 1301–8. [DOI] [PubMed] [Google Scholar]
- 4.Lee WL and Slutsky AS, Ventilator-induced lung injury and recommendations for mechanical ventilation of patients with ARDS. Semin Respir Crit Care Med, 2001. 22(3): p. 269–80. [DOI] [PubMed] [Google Scholar]
- 5.Erickson SE, et al. , Recent trends in acute lung injury mortality: 1996–2005. Crit Care Med, 2009. 37(5): p. 1574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubenfeld GD, et al. , Incidence and outcomes of acute lung injury. N Engl J Med, 2005. 353(16): p. 1685–93. [DOI] [PubMed] [Google Scholar]
- 7.Bellani G, et al. , Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. Jama, 2016. 315(8): p. 788–800. [DOI] [PubMed] [Google Scholar]
- 8.Matthay MA and Zemans RL, The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol, 2011. 6: p. 147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudek SM and Garcia JG, Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol, 2001. 91(4): p. 1487–500. [DOI] [PubMed] [Google Scholar]
- 10.Mehta D and Malik AB, Signaling mechanisms regulating endothelial permeability. Physiol Rev, 2006. 86(1): p. 279–367. [DOI] [PubMed] [Google Scholar]
- 11.Aman J, et al. , Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation, 2012. 126(23): p. 2728–38. [DOI] [PubMed] [Google Scholar]
- 12.Kim IK, et al. , Effect of tyrosine kinase inhibitors, imatinib and nilotinib, in murine lipopolysaccharide-induced acute lung injury during neutropenia recovery. Crit Care, 2013. 17(3): p. R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens RS, et al. , Protein Kinase G increases antioxidant function in lung microvascular endothelial cells by inhibiting the c-Abl tyrosine kinase. Am J Physiol Cell Physiol, 2014. [DOI] [PMC free article] [PubMed]
- 14.Chislock EM and Pendergast AM, Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS One, 2013. 8(12): p. e85231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaki OS, et al. , A Novel Role of a Chemotherapeutic Agent in a Rat Model of Endotoxemia: Modulation of the STAT-3 Signaling Pathway. Inflammation, 2018. 41(1): p. 20–32. [DOI] [PubMed] [Google Scholar]
- 16.Letsiou E, et al. , Differential and opposing effects of imatinib on LPS- and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol, 2015. 308(3): p. L259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzo AN, et al. , Imatinib attenuates inflammation and vascular leak in a clinically relevant two-hit model of acute lung injury. Am J Physiol Lung Cell Mol Physiol, 2015. 309(11): p. L1294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rix U, et al. , Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood, 2007. 110(12): p. 4055–63. [DOI] [PubMed] [Google Scholar]
- 19.Rizzo AN, et al. , Targeting Abl kinases to regulate vascular leak during sepsis and acute respiratory distress syndrome. Arterioscler Thromb Vasc Biol, 2015. 35(5): p. 1071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aman J, et al. , Reversal of vascular leak with imatinib. Am J Respir Crit Care Med, 2013. 188(9): p. 1171–3. [DOI] [PubMed] [Google Scholar]
- 21.Overbeek MJ, et al. , Possible role of imatinib in clinical pulmonary veno-occlusive disease. Eur Respir J, 2008. 32(1): p. 232–5. [DOI] [PubMed] [Google Scholar]
- 22.Carnevale-Schianca F, et al. , Complete resolution of life-threatening bleomycin-induced pneumonitis after treatment with imatinib mesylate in a patient with Hodgkin’s lymphoma: hope for severe chemotherapy-induced toxicity? J Clin Oncol, 2011. 29(24): p. e691–3. [DOI] [PubMed] [Google Scholar]
- 23.Letsiou E, et al. , Parkin regulates lipopolysaccharide-induced proinflammatory responses in acute lung injury. Transl Res, 2017. 181: p. 71–82. [DOI] [PubMed] [Google Scholar]
- 24.Fu P, et al. , c-Abl mediated tyrosine phosphorylation of paxillin regulates LPS-induced endothelial dysfunction and lung injury. Am J Physiol Lung Cell Mol Physiol, 2015. 308(10): p. L1025–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubrovskyi O, Birukova AA, and Birukov KG, Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest, 2013. 93(2): p. 254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia JG, et al. , Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest, 2001. 108(5): p. 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bischoff I, et al. , Pitfalls in assessing microvascular endothelial barrier function: impedance-based devices versus the classic macromolecular tracer assay. Sci Rep, 2016. 6: p. 23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wegener J and Seebach J, Experimental tools to monitor the dynamics of endothelial barrier function: a survey of in vitro approaches. Cell Tissue Res, 2014. 355(3): p. 485–514. [DOI] [PubMed] [Google Scholar]
- 29.Colicelli J, ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal, 2010. 3(139): p. re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganguly SS, et al. , c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene, 2012. 31(14): p. 1804–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens T, Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc, 2011. 8(6): p. 453–7. [DOI] [PubMed] [Google Scholar]
- 32.Barabutis N, Verin A, and Catravas JD, Regulation of pulmonary endothelial barrier function by kinases. Am J Physiol Lung Cell Mol Physiol, 2016. 311(5): p. L832–l845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel SM and Malik AB, Cytoskeletal dynamics and lung fluid balance. Compr Physiol, 2012. 2(1): p. 449–78. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, et al. , Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem Biol, 2011. 18(2): p. 177–86. [DOI] [PubMed] [Google Scholar]
- 35.Rhee J, et al. , Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol, 2007. 9(8): p. 883–92. [DOI] [PubMed] [Google Scholar]
- 36.Winter MC, Shasby S, and Shasby DM, Compromised E-cadherin adhesion and epithelial barrier function with activation of G protein-coupled receptors is rescued by Y-to-F mutations in beta-catenin. Am J Physiol Lung Cell Mol Physiol, 2008. 294(3): p. L442–8. [DOI] [PubMed] [Google Scholar]
- 37.Tamada M, Farrell DL, and Zallen JA, Abl regulates planar polarized junctional dynamics through β-catenin tyrosine phosphorylation. Dev Cell, 2012. 22(2): p. 309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zandy NL, Playford M, and Pendergast AM, Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc Natl Acad Sci U S A, 2007. 104(45): p. 17686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zandy NL and Pendergast AM, Abl tyrosine kinases modulate cadherin-dependent adhesion upstream and downstream of Rho family GTPases. Cell Cycle, 2008. 7(4): p. 444–8. [DOI] [PubMed] [Google Scholar]
- 40.Birukova AA, et al. , Cross talk between paxillin and Rac is critical for mediation of barrier-protective effects by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol, 2008. 295(4): p. L593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komarova YA, et al. , Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ Res, 2017. 120(1): p. 179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joshi AD, et al. , Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol, 2014. 50(1): p. 170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens RS, et al. , The tyrosine kinase inhibitor imatinib prevents lung injury and death after intravenous LPS in mice. Physiol Rep, 2015. 3(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magruder JT, et al. , Imatinib Is Protective Against Ischemia-Reperfusion Injury in an Ex Vivo Rabbit Model of Lung Injury. Ann Thorac Surg, 2018. 105(3): p. 950–956. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka S, et al. , Protective Effects of Imatinib on Ischemia/Reperfusion Injury in Rat Lung. Ann Thorac Surg, 2016. 102(5): p. 1717–1724. [DOI] [PubMed] [Google Scholar]
- 46.Merali Z, et al. , Longitudinal assessment of imatinib’s effect on the blood-brain barrier after ischemia/reperfusion injury with permeability MRI. Transl Stroke Res, 2015. 6(1): p. 39–49. [DOI] [PubMed] [Google Scholar]
- 47.Chilakapati SR, et al. , Amelioration of bleomycin-induced pulmonary fibrosis in a mouse model by a combination therapy of bosentan and imatinib. Exp Lung Res, 2015. 41(4): p. 173–88. [DOI] [PubMed] [Google Scholar]
- 48.Daniels CE, et al. , Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest, 2004. 114(9): p. 1308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vorkapic E, et al. , Imatinib treatment attenuates growth and inflammation of angiotensin II induced abdominal aortic aneurysm. Atherosclerosis, 2016. 249: p. 101–9. [DOI] [PubMed] [Google Scholar]
- 50.Wolf AM, et al. , The kinase inhibitor imatinib mesylate inhibits TNF-{alpha} production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc Natl Acad Sci U S A, 2005. 102(38): p. 13622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iyoda M, et al. , Long- and short-term treatment with imatinib attenuates the development of chronic kidney disease in experimental anti-glomerular basement membrane nephritis. Nephrol Dial Transplant, 2013. 28(3): p. 576–84. [DOI] [PubMed] [Google Scholar]
- 52.Adzemovic MV, et al. , Imatinib ameliorates neuroinflammation in a rat model of multiple sclerosis by enhancing blood-brain barrier integrity and by modulating the peripheral immune response. PLoS One, 2013. 8(2): p. e56586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.dos Santos LV, et al. , Imatinib-induced bone edema: case report and review of literature. J Natl Compr Canc Netw, 2013. 11(10): p. 1187–91. [DOI] [PubMed] [Google Scholar]
- 54.Kim KW, et al. , Fluid retention associated with imatinib treatment in patients with gastrointestinal stromal tumor: quantitative radiologic assessment and implications for management. Korean J Radiol, 2015. 16(2): p. 304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee WJ, et al. , Clinical and histopathologic analysis of 46 cases of cutaneous adverse reactions to imatinib. Int J Dermatol, 2016. 55(5): p. e268–74. [DOI] [PubMed] [Google Scholar]
- 56.Wang X, et al. , The Significant Role of c-Abl Kinase in Barrier Altering Agonists-mediated Cytoskeletal Biomechanics. Sci Rep, 2018. 8(1): p. 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L, et al. , FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J, 2011. 38(1): p. 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bohio AA, et al. , c-Abl-Mediated Tyrosine Phosphorylation of PARP1 Is Crucial for Expression of Proinflammatory Genes. J Immunol, 2019. 203(6): p. 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song GJ, et al. , A Bcr-Abl Inhibitor GNF-2 Attenuates Inflammatory Activation of Glia and Chronic Pain. Front Pharmacol, 2019. 10: p. 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tong H, et al. , c-Abl tyrosine kinase regulates neutrophil crawling behavior under fluid shear stress via Rac/PAK/LIMK/cofilin signaling axis. J Cell Biochem, 2018. 119(3): p. 2806–2817. [DOI] [PubMed] [Google Scholar]
- 61.Belvitch P, et al. , Cortical Actin Dynamics in Endothelial Permeability. Curr Top Membr, 2018. 82: p. 141–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Green MR, Newton MD, and Fancher KM, Off-Target Effects of BCR-ABL and JAK2 Inhibitors. Am J Clin Oncol, 2016. 39(1): p. 76–84. [DOI] [PubMed] [Google Scholar]
- 63.Zitvogel L, et al. , Immunological off-target effects of imatinib. Nat Rev Clin Oncol, 2016. 13(7): p. 431–46. [DOI] [PubMed] [Google Scholar]
- 64.Waller CF, Imatinib mesylate. Recent Results Cancer Res, 2014. 201: p. 1–25. [DOI] [PubMed] [Google Scholar]
- 65.Wei G, Rafiyath S, and Liu D, First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J Hematol Oncol, 2010. 3: p. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]