

Abstract

Glioblastoma multiforme (GBM) is the most aggressive primary brain tumor. Residual cells at the tumor margin are responsible for up to 85% of GBM recurrences after standard treatment. Despite this evidence, the identification of compounds active on this cell population is still an underexplored field. Herein, starting from the knowledge that kinases are implicated in GBM, we evaluated three in-house pyrazolo[3,4-d]pyrimidines active as Src, Fyn, and SGK1 kinase inhibitors against patient derived cell lines from either the invasive region or contrast-enhanced core of GBM. We identified our Src inhibitor, SI306, as a promising lead compound for eradicating invasive GBM cells. Furthermore, aiming at the development of a feasible oral treatment for GBM, we performed a formulation study using 2D inkjet printing to generate soluble polymer–drug dispersions. Overall, this study led to the identification of a set of polymer-formulated pyrazolo[3,4-d]pyrimidine kinase inhibitors as promising candidates for GBM preclinical efficacy studies.
Keywords: Kinase inhibitors, glioblastoma multiforme, miniaturized assay, inkjet 2D printing, invasive margin cells
Glioblastoma multiforme (GBM) is the most common, malignant, and aggressive primary brain tumor in adults, mainly due to its rapid proliferation and ability to penetrate and diffusely infiltrate healthy brain parenchyma. Standard of care treatment currently involves a combination of surgery, radiotherapy, and chemotherapy.1 Yet, despite this multimodal treatment method, the median survival remains poor at less than 15 months.2 Problems with existing treatment approaches include (i) increased resistance to chemotherapeutic drugs caused by the heterogeneity of the tumor microenvironment and variation in tumor subclones, (ii) inability or impairment of drugs to cross the blood–brain barrier (BBB), and (iii) lack of penetration of locally delivered therapeutic agents deep into the brain parenchyma beyond the resection cavity at sufficient therapeutic concentrations to target residual tumor cells.3,4 Superior and more innovative treatment methods are necessary to eradicate invasive tumor cells, which remain beyond the resection cavity lining postsurgery, and to block or impair GBM recurrence, which is inevitable with current treatment methods.
The implication of kinases in GBM pathogenesis and drug resistance has led to small molecule kinase inhibitors emerging as possible treatment options.5,6 Crucially, kinase inhibitors, acting specifically on molecular targets, are purported to reduce off site toxicity during antitumor treatments.7
Src-family kinases (SFKs) are a series of nine membrane-associated, nonreceptor tyrosine kinases (c-Src, Fyn, Yes, Lyn, Lck, Blk, Fgr, Hck, and Yrk)8 that are involved in the regulation of a range of fundamental cellular processes.9 Previous studies have indicated that dysregulated SFK signaling can induce multiple protumorigenic effects in gliomas, including reduced apoptosis, increased angiogenesis, and increased proliferation.10−13 Furthermore, evidence suggests that SFKs play roles in cancer cell invasion and metastasis.14 Src, the most widely studied member of SFKs, is a key downstream intermediate of growth factor receptor pathways and is frequently overexpressed in brain tumors (61% in GBM).12,15,16 Preclinical data confirmed the key role of Src in GBM proliferation and invasion,17 leading the way for the use of Src inhibitors in clinical studies. Additionally, Fyn has also been reported to be an effector of oncogenic signaling in GBM patients.18
In 2009, Lu et al. identified that persistent epidermal growth factor receptor (EGFR) signaling activated both Fyn and Src to increase GBM invasion and tumor survival in vivo.18 More recently, Comba et al. demonstrated the correlation of Fyn expression with malignant features of GBM tumors, including pseudopalisades, necrosis, and hypervascularization.19
Dasatinib, a broad spectrum inhibitor of SFKs, including Src, Fyn and Lyn, has been proposed as a therapeutic option in recurrent GBM.14 Dasatinib was well tolerated in clinical trials but failed to improve overall survival either as a monotherapy or in combination therapy for GBM patients,20,21 a result attributed to its susceptibility to cellular efflux by transporters and subsequent poor accumulation in brain tissue.22 Generally, no kinase inhibitor trial (e.g., EGFR and PDGFR) has had phase II survival benefit for GBM patients.23,24
In addition to SFKs, other kinases have been investigated for their role in GBM pathogenesis, including serum- and glucocorticoid-regulated kinase 1 (SGK1) which has been associated with neoplastic transformation, and chemo/radioresistance.25,26
Over recent years, our group has designed and synthesized a wide library of pyrazolo[3,4-d]pyrimidines active as kinase inhibitors.27,28 In particular, three of our in-house compounds (SI306, SI308, and SI113 (Figure 1)) have been shown to be potent inhibitors of the tyrosine kinases Src and Fyn and serine-threonine kinase SGK1,29−31 respectively, and have demonstrated anticancer effects on different commercial (established) GBM cell lines.30,32,33
Figure 1.

Structures of in-house kinase inhibitors SI306, SI308, and SI113 and their activity toward Src, Fyn, and SGK1.
However, most commercial GBM cell lines have historically been derived from the core region of tumors, which does not allow a realistic, phenotypically accurate representation of the infiltrative cells which ultimately result in the inevitable recurrence of GBM.3 Significantly, residual cells at the tumor margin are responsible for the 85% of GBMs that relapse locally after maximal safe surgical resection followed by the standard combination protocol of temozolomide and radiotherapy.34 Therefore, the discovery of molecules active against invasive GBM cells represents a crucial step in the development of drugs for the treatment of this pathology.35
We evaluated the cytotoxicity of kinase inhibitors on primary patient derived cell lines from the invasive region (GIN8, GIN28) and on the corresponding central tumor core (GCE28) cell line from the same patient, and we assessed the anticancer effects of our kinase inhibitors in monotherapy and combination therapy. Furthermore, since our compounds can be considered in biopharmaceutical classification system (BCS) class II,36 demonstrating good permeability37 (indicating a good probability of BBB permeation, which is further supported by previous work on our compounds38) but limited water solubility, we performed a formulation study, applying the innovative inkjet printing technology to generate solid dispersions of our lead compound inhibitor in inert hydrophilic polymeric carriers. This formulation strategy, recently reported by us,39 can be used to increase the water solubility of the inhibitors in a manner that does not compromise potency and thus provides a viable approach for development of oral formulations.
In vitro data demonstrated that the different kinase inhibitors were cytotoxic, implicating Src, Fyn, and SGK1 kinases as valid targets in the tested GBM cell lines. SI306 (Src inhibitor) was demonstrated to be the most potent tested compound with IC50 values of 11.2, 7.7, and 7.2 μM in the GIN8, GIN28, and GCE28 cell lines, respectively. Compound SI113 (SGK1 inhibitor) exhibited similar potency to SI306 in the GIN8 line but was 1.9-fold and 1.5-fold less potent in GIN28 and GCE28 cells. The least potent compound was shown to be SI308 (Fyn inhibitor), with IC50 values 4.9-fold, 6.5-fold, and 6.6-fold higher than the most potent compound (SI306), in GIN8, GIN28, and GCE28 cells, respectively (Table 1 and Figure 4SI). However, despite being the least potent compound in monotherapy, SI308 demonstrated a promising application in synergistic combination therapy with a Scr inhibitor, as described later.
Table 1. IC50 Values of Kinase Inhibitors on GBM Cellsa.
| IC50 (μM) |
|||
|---|---|---|---|
| compd | GIN8 | GIN28 | GCE28 |
| SI306 | 11.2 ± 3.8 | 7.7 ± 1.6 | 7.2 ± 2.0 |
| SI308 | 54.7 ± 6.3 | 49.8 ± 4.2 | 47.6 ± 6.9 |
| SI113 | 10.5 ± 3.5 | 14.4 ± 2.8 | 10.7 ± 1.2 |
Potency was assessed with PrestoBlue assay. Compounds were applied diluted in 10% FBS containing DMEM for 48 h. Data represent the mean ± SE. IC50 values were calculated from three independent experiments. Statistical significance was determined via two-way ANOVA followed by Dunnett’s multiple comparisons test.
Following the confirmation of compound cytotoxicity, we next investigated the effector caspases-3/7 to determine if cell death was apoptotic in nature.40
Data in Figure 2 demonstrate that at a cytotoxic concentration (12.5 μM), SI113 and SI306 induce significant increases in caspase-3/7 activation in all GBM cell lines tested with the exception of SI113 in the GIN28 line.
Figure 2.
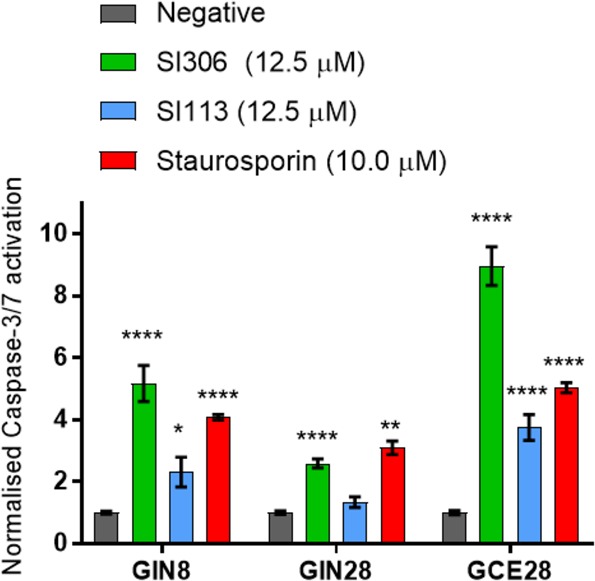
Effect of kinase inhibitors on levels of activated effector caspases-3/7 on GBM cells. Compounds were applied diluted in 10% FBS containing DMEM for 48 h. Caspase levels were assessed with the CellEvent caspase-3/7 probe. Data represent the mean ± SEM (n = 3). Statistical significance was determined via two-way ANOVA followed by Dunnett’s multiple comparisons test.
For assay verification, staurosporine (10.0 μM), a known inducer of apoptosis,41 was also tested as a reference compound. It elicits significant increase in caspase activation at similar or lower levels than those of the kinase inhibitors. It can be noted that SI306 induces higher levels of effector caspase activation compared to SI113, a result that reflects the IC50 data (Table 1) which together indicate that SI306 is the most active compound we tested against these cell lines.
To further confirm the apoptotic death induced by SI306, the nuclear morphology and permeability were investigated by fluorescence microscopy using Hoechst 33342 (Ho) and propidium iodide (PI) double staining (Figure 3). Cells treated with SI306 exhibit signs of chromatid condensation, nuclear fragmentation, and the presence of apoptotic bodies, which are well-known proapoptotic features. SI306 treatment did not induce nuclear membrane permeability, as shown by PI negative staining, or nuclear swelling, indicators of necrotic cell death and demonstrated by ethanol (EtOH) (Figure 3), a known inducer of necrosis.42 These observations, taken together with the effective caspase activation, indicate that SI306 is inducing GBM cell death via an apoptotic mechanism.
Figure 3.

Ho/PI staining of nuclei. Effect of SI306 kinase inhibitor on apoptotic features of cellular nuclei. Cells were dosed with 12.5 μM SI306, 70% ethanol (EtOH) or DMEM (Neg, negative control) for 48 h followed by nuclear staining with Hoechst 33342 and propidium iodide dyes. Scale bar indicates 30 μm. Images shown are representative of three sets of independent images. White arrows indicate the presence of apoptotic nuclei (chromatid condensation, apoptotic bodies).
Previous evidence indicates that targeting more than one kinase may be beneficial in cancer treatment, creating the opportunity to achieve a synergistic effect and to overcome the development of resistance.43,44 Therefore, we have investigated our novel compounds as combination therapies in order to assess if synergistic activity can be achieved. The median-effect algorithm based on the widely used method established by Chou and Talalay45 was employed to calculate the combination index (CI) as outlined in the Supporting Information. The CI equation was used to generate CI values, which categorize the compound–compound combinations as synergistic, additive, or antagonistic. Interestingly, the combination of SI308 with either SI306 or SI113 was determined to generate synergistic effects despite SI308 having been demonstrated to be the least potent compound in monotherapy (Figure 4).
Figure 4.
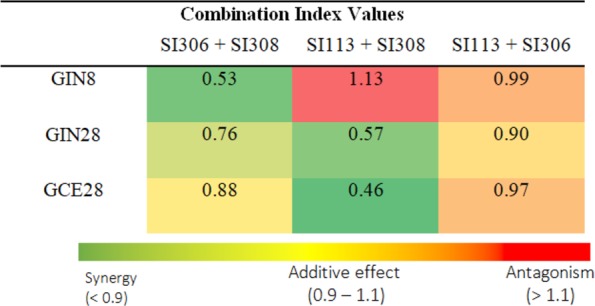
Combination index values. Compounds were applied at a molar ratio of 1:1. Shown is a summary of the CI values for the combinations of kinase inhibitors following 48 h incubation in three GBM lines. Each CI value was calculated, and a heat map was generated on the basis of three independent IC50 experiments (n = 3). Values represent the mean ± SD.
On the contrary, SI113 and SI306 co-therapy exhibited only additive action. Therefore, the observed effects suggest that co-inhibition of the Fyn kinase and SGK1 or Src kinases provides a synergistic action that cannot be achieved via inhibiting SGK1 and Src together. Of further note is the observation that in the GIN8 line, co-therapy with SI113 and SI308 produced an antagonistic effect despite synergy being observed in the GIN28 and GCE28 lines with this combination. The reasons for this remain unclear; however, patient genetic variation may play a role (GIN28 and GCE28 lines are derived from the same patient, and the GIN8 line is from a separate patient). Taken together, the evidence for synergistic action with our compounds may promote the adoption of combination therapies in the field of kinase inhibitors46 for the treatment of resistant GBM.
The promising in vitro data highlight pyrazolo[3,4-d]pyrimidine kinase inhibitors as potential pharmacotherapies for eradicating invasive GBM cell lines. To further their development for clinical application, we have investigated the formulation of our lead compound SI306. To overcome the water solubility limitation of SI306, which may affect further in vivo studies and future oral administration routes, we performed a preliminary formulation screening process based on 2D inkjet printing, building on an approach previously validated by our group.39,47,48 Different commercial polymers were combined with SI306 and the apparent-solubility (ΔA%) value of each formulation was calculated in order to identify the polymers able to solubilize our lead compound (Figure 5).
Figure 5.

ΔA% average of SI306-polymer formulation ranked according to their water apparent-solubility enhancement (high ΔA% is related to a high compound water solubility). Data represent the mean ± SEM (n = 3).
Data demonstrate that two surfactants (Pluronic F-68 and Tween 80) and the amphiphilic copolymer PVPVA showed notably higher ΔA% average values compared to the highly hydrophilic homopolymers (PEG8000–20000) (see Supporting Information, Table 1SI and Figures 1SI and 2SI for further details).
These results suggest that solubilization of hydrophobic SI306 is due to associative interactions between hydrophobic blocks in Pluronic F-68, Tween 80, and PVPVA and the lipophilic regions in SI306.39 Dynamic light scattering (DLS) measurements were performed on the formulation of SI306 with the three candidate polymers Pluronic F-68, Tween 80, and PVPVA (Figure 6) in order to evaluate the particulate nature of the drug–polymer assemblies.
Figure 6.
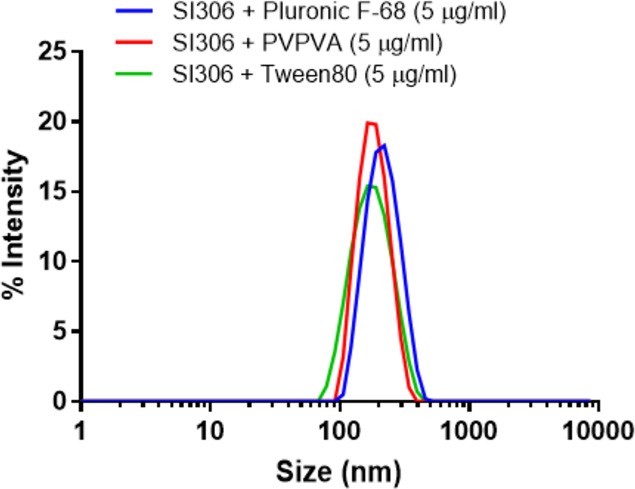
DLS traces in PBS of SI306 as a formulation. Light scattering measurements were collected on suspensions prepared with a final concentration of 5 μg/mL.
As can be observed from Figure 6, all the formulations produced well-defined nanoaggregates, characterized by a single monomodal and monodispersed population with sizes ranging from 180 to 200 nm, confirming the quality of the nanoformulations.
To further validate the water solubility enhancement, we performed a cytotoxicity assay using SI306 either dissolved in DMSO or printed into the selected polymers. Negative control, polymers alone, and SI306 suspended in PBS and diluted in cell culture medium DMEM (to highlight SI306 poor water solubility and consequent in vitro inactivity) had no effect on cell viability. On the contrary, formulated SI306 resuspended in DMEM and SI306 dissolved in DMSO treatments had comparable cytotoxic effects on all GBM cell lines (Figure 7).
Figure 7.
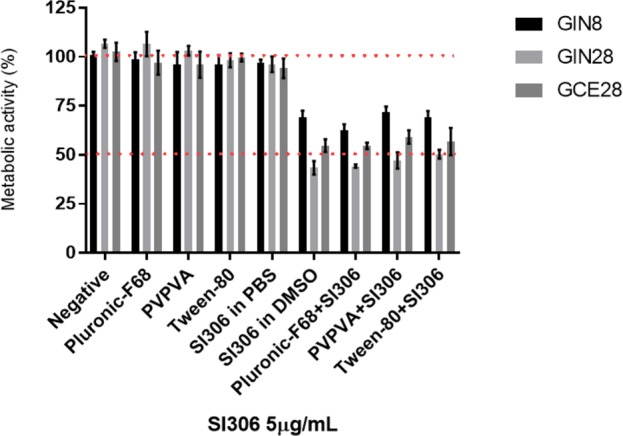
Compound SI306 formulated with the selected best polymers was then tested against the patients derived GBM cell lines and the potency compared to the compound solubilized in 1% DMSO. Potency was assessed after 48 h with PrestoBlue assay. Data represent the mean ± SD.
Therefore, the described SI306 formulation can successfully increase the apparent water solubility of the inhibitor without affecting its potency, and this provides a further step into the development of our lead compound.
It is noteworthy that no kinase inhibitor has yet shown clinical benefit in GBM. The reasons for this are complex and multifactorial and include failure to achieve BBB penetration, genetic heterogeneity in the tumor, target redundancy, and rapid molecular adaptation. On the other hand, kinase inhibitors have certainly shown benefit in other aggressive forms of cancer such as non-small-cell lung cancer49 and renal cancer,50 and for these reasons, they can be considered promising drugs for GBM therapy if the above problems are overcome (Figure 8). Pyrazolo[3,4-d]pyrimidines synthesized by our group were able to cross the BBB,38,51 laying the foundation for a successful development.
Figure 8.

Simplified representation of the main issues that kinase inhibitors have to overcome to obtain clinical benefits in GBM therapy.
In conclusion, we have evaluated the potency of our pyrazolo[3,4-d]pyrimidines active as specific kinase inhibitors, against patient derived cell lines from the invasive region and core of GBM, identifying the Src inhibitor SI306 as a lead compound. SI306 possesses an IC50 in the low micromolar range on all the three GBM cell lines tested in this work, demonstrating the ability to induce apoptotic death. A combination study, using the Chou and Talalay method, has also been assessed and showed that, based on patient genetic variations, our kinase inhibitors possess a synergistic effect that could positively influence the success of GBM treatment.
Lastly, a polymer formulation strategy involving the novel 2D inkjet printing technology was explored as a strategy to enhance SI306 water solubility. In vitro results illustrated that printing 5 μg/mL of our lead compound into dispersions of Pluronic F-68, Tween 80, or PVPVA at a level of 90% is a successful formulation method, resulting in a comparable potency to SI306 dissolved in DMSO. Accordingly, this methodology provides a viable approach for the development of oral formulations of our in-house kinase inhibitors. Furthermore, since some of the chosen polymers, such as the Pluronic family, have been shown to facilitate transport across the BBB, a next challenge could be the selection of the best polymer for in vivo GBM studies.
Overall, these results encourage in vivo studies and promote polymer-carried pyrazolo[3,4-d]pyrimidine kinase inhibitors as oral feasible treatments against GBM potentially active also against tumor recurrence.
Glossary
Abbreviations
- BBB
blood–brain barrier
- BCS
biopharmaceutical classification system
- CI
combination index
- DLS
dynamic light scattering
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- GBM
glioblastoma multiforme
- Ho
Hoechst 33342
- PBS
phosphate buffered saline
- PEG
polyethylene glycol
- PI
propidium iodide
- PVPVA
polyvinylpyrrolidone-vinyl acetate
- SFK
Src-family kinase
- SGK1
serum- and glucocorticoid-regulated kinase 1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00530.
General information on chemicals, experimental details for the printing, UV screening, DLS, and cellular assays (PDF)
Author Contributions
# C.G. and V.T. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Italian MIUR Project PRIN 2017 2017SA5837_004 (S.S. and F.M.) and the Engineering and Physical Sciences Research Council [Grants EP/N006615/1, EP/N03371X/1, EP/L01646X/1, EP/H005625/1, EP/L013835/1, and EP/R035563/1] (C.A.). This work was also funded by the Royal Society [Wolfson Research Merit Award WM150086] to C.A.
The authors declare no competing financial interest.
Supplementary Material
References
- Batash R.; Asna N.; Schaffer P.; Francis N.; Schaffer M. Glioblastoma multiforme, diagnosis and treatment; recent literature review. Curr. Med. Chem. 2017, 24 (27), 3002–3009. 10.2174/0929867324666170516123206. [DOI] [PubMed] [Google Scholar]
- Alifieris C.; Trafalis D. T. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. 10.1016/j.pharmthera.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Perrin S. L.; Samuel M. S.; Koszyca B.; Brown M. P.; Ebert L. M.; Oksdath M.; Gomez G. A. Glioblastoma heterogeneity and the tumour microenvironment: implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47 (2), 625–638. 10.1042/BST20180444. [DOI] [PubMed] [Google Scholar]
- Lemée J.-M.; Clavreul A.; Menei P. Intratumoral heterogeneity in glioblastoma: don’t forget the peritumoral brain zone. Neuro. Oncol. 2015, 17 (10), 1322–1332. 10.1093/neuonc/nov119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-García E.; Saceda M.; Martínez-Lacaci I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014, 3 (2), 199–235. 10.3390/cells3020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Wu C.; Chen N.; Gu H.; Yen A.; Cao L.; Wang E.; Wang L. PI3K/Akt/MTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7 (22), 33440–33450. 10.18632/oncotarget.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A.; Scholar E. M. Role of Tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315 (3), 971–979. 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- Yeatman T. J. A Renaissance for SRC. Nat. Rev. Cancer 2004, 4 (6), 470–480. 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Lewis-Tuffin L. J.; Feathers R.; Hari P.; Durand N.; Li Z.; Rodriguez F. J.; Bakken K.; Carlson B.; Schroeder M.; Sarkaria J. N.; Anastasiadis P. Z. Src Family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9 (9), 1783–1798. 10.1016/j.molonc.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.-M.; Park M.-J.; Kwak H.-J.; Lee H.-C.; Kim M.-S.; Lee S.-H.; Park I.-C.; Rhee C. H.; Hong S.-I. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through src/epidermal growth factor receptor-mediated P38/Akt and phosphatidylinositol 3-Kinase/Akt signaling pathways. Cancer Res. 2006, 66 (17), 8511–8519. 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- Eskilsson E.; Rosland G. V.; Talasila K. M.; Knappskog S.; Keunen O.; Sottoriva A.; Foerster S.; Solecki G.; Taxt T.; Jirik R.; Fritah S.; Harter P. N.; Välk K.; Al Hossain J.; Joseph J. V.; Jahedi R.; Saed H. S.; Piccirillo S. G.; Spiteri I.; Leiss L.; Euskirchen P.; Graziani G.; Daubon T.; Lund-Johansen M.; Enger P.Ø.; Winkler F.; Ritter C. A.; Niclou S. P.; Watts C.; Bjerkvig R.; Miletic H. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol 2016, 18 (12), 1644–1655. 10.1093/neuonc/now113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. M.; Huang P.; Kar N.; Burgett M.; Muller-Greven G.; Nowacki A. S.; Distelhorst C. W.; Lathia J. D.; Rich J. N.; Kappes J. C.; Gladson C. L. Lyn facilitates glioblastoma cell survival under conditions of nutrient deprivation by promoting autophagy. PLoS One 2013, 8 (8), e70804 10.1371/journal.pone.0070804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveldt D.; Lewis-Tuffin L. J.; Carlson B. L.; Schroeder M. A.; Rodriguez F.; Giannini C.; Galanis E.; Sarkaria J. N.; Anastasiadis P. Z. Targeting Src family kinases inhibits Bevacizumab-induced glioma cell invasion. PLoS One 2013, 8 (2), e56505 10.1371/journal.pone.0056505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Bernasconi P.; Clauser K. R.; Mani D. R.; Finn S. P.; Beroukhim R.; Burns M.; Julian B.; Peng X. P.; Hieronymus H.; Maglathlin R. L.; Lewis T. A.; Liau L. M.; Nghiemphu P.; Mellinghoff I. K.; Louis D. N.; Loda M.; Carr S. A.; Kung A. L.; Golub T. R. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27 (1), 77–83. 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K.; Kugimiya T.; Miyazaki T. Substance P receptor in U373 MG human astrocytoma cells activates mitogen-activated protein kinases ERK1/2 through Src. Brain Tumor Pathol. 2005, 22 (1), 1–8. 10.1007/s10014-005-0178-1. [DOI] [PubMed] [Google Scholar]
- Ahluwalia M. S.; de Groot J.; Liu W. M.; Gladson C. L. Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies. Cancer Lett. 2010, 298 (2), 139–149. 10.1016/j.canlet.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. V.; Zhu S.; Cvrljevic A.; Huang T. T.; Sarkaria S.; Ahkavan D.; Dang J.; Dinca E. B.; Plaisier S. B.; Oderberg I.; Lee Y.; Chen Z.; Caldwell J. S.; Xie Y.; Loo J. A.; Seligson D.; Chakravari A.; Lee F. Y.; Weinmann R.; Cloughesy T. F.; Nelson S. F.; Bergers G.; Graeber T.; Furnari F. B.; James C. D.; Cavenee W. K.; Johns T. G.; Mischel P. S. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009, 69 (17), 6889–6898. 10.1158/0008-5472.CAN-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comba A.; Kadiyala P.; Argento A. E.; Patel P.; Nunez F. J.; Saxena M.; Castro M. G.; Lowenstein P. R. CSIG-39. Fyn, an oncogene that reduces glioblastoma survival yet sensitizes to chemo-radiotherapy. Neuro. Oncol. 2017, 19 (Suppl. 6), vi58–vi58. 10.1093/neuonc/nox168.233. [DOI] [Google Scholar]
- Lassman A. B.; Pugh S. L.; Gilbert M. R.; Aldape K. D.; Geinoz S.; Beumer J. H.; Christner S. M.; Komaki R.; DeAngelis L. M.; Gaur R.; Youssef E.; Wagner H.; Won M.; Mehta M. P. Phase 2 trial of Dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro. Oncol. 2015, 17 (7), 992–998. 10.1093/neuonc/nov011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis E.; Anderson S. K.; Twohy E. L.; Carrero X. W.; Dixon J. G.; Tran D. D.; Jeyapalan S. A.; Anderson D. M.; Kaufmann T. J.; Feathers R. W.; Giannini C.; Buckner J. C.; Anastasiadis P. Z.; Schiff D. A Phase 1 and randomized, placebo-controlled Phase 2 trial of Bevacizumab plus Dasatinib in patients with recurrent glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125 (21), 3790–3800. 10.1002/cncr.32340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Mittapalli R. K.; Zellmer D. M.; Gallardo J. L.; Donelson R.; Seiler C.; Decker S. A.; SantaCruz K. S.; Pokorny J. L.; Sarkaria J. N.; Elmquist W. F.; Ohlfest J. R. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Mol. Cancer Ther. 2012, 11 (10), 2183–2192. 10.1158/1535-7163.MCT-12-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. W.; Parikh M.; Phillips J. J.; James C. D.; Molinaro A. M.; Butowski N. A.; Clarke J. L.; Oberheim-Bush N. A.; Chang S. M.; Berger M. S.; Prados M. Phase-2 Trial of Palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neuro-Oncol. 2018, 140 (2), 477–483. 10.1007/s11060-018-2977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peereboom D. M.; Ahluwalia M. S.; Ye X.; Supko J. G.; Hilderbrand S. L.; Phuphanich S.; Nabors L. B.; Rosenfeld M. R.; Mikkelsen T.; Grossman S. A. NABTT 0502: A Phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro. Oncol. 2013, 15 (4), 490–496. 10.1093/neuonc/nos322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talarico C.; Dattilo V.; D’Antona L.; Menniti M.; Bianco C.; Ortuso F.; Alcaro S.; Schenone S.; Perrotti N.; Amato R. SGK1, the new player in the game of resistance: chemo-radio molecular target and strategy for inhibition. Cell. Physiol. Biochem. 2016, 39 (5), 1863–1876. 10.1159/000447885. [DOI] [PubMed] [Google Scholar]
- Kulkarni S.; Goel-Bhattacharya S.; Sengupta S.; Cochran B. H. A large-scale RNAi screen identifies SGK1 as a key survival kinase for GBM stem cells. Mol. Cancer Res. 2018, 16 (1), 103–114. 10.1158/1541-7786.MCR-17-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari A.; Fallacara A. L.; Di Maria S.; Zamperini C.; Poggialini F.; Musumeci F.; Schenone S.; Angelucci A.; Colapietro A.; Crespan E.; Kissova M.; Maga G.; Botta M. Efficient optimization of pyrazolo[3,4-d]pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Bioorg. Med. Chem. Lett. 2018, 28 (21), 3454–3457. 10.1016/j.bmcl.2018.09.024. [DOI] [PubMed] [Google Scholar]
- Radi M.; Tintori C.; Musumeci F.; Brullo C.; Zamperini C.; Dreassi E.; Fallacara A. L.; Vignaroli G.; Crespan E.; Zanoli S.; Laurenzana I.; Filippi I.; Maga G.; Schenone S.; Angelucci A.; Botta M. Design, Synthesis, and biological evaluation of pyrazolo[3,4- d]pyrimidines active in vivo on the Bcr-Abl T315I mutant. J. Med. Chem. 2013, 56 (13), 5382–5394. 10.1021/jm400233w. [DOI] [PubMed] [Google Scholar]
- Tintori C.; Fallacara A. L.; Radi M.; Zamperini C.; Dreassi E.; Crespan E.; Maga G.; Schenone S.; Musumeci F.; Brullo C.; Richters A.; Gasparrini F.; Angelucci A.; Festuccia C.; Delle Monache S.; Rauh D.; Botta M. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58 (1), 347–361. 10.1021/jm5013159. [DOI] [PubMed] [Google Scholar]
- Tintori C.; La Sala G.; Vignaroli G.; Botta L.; Fallacara A. L.; Falchi F.; Radi M.; Zamperini C.; Dreassi E.; Dello Iacono L.; Orioli D.; Biamonti G.; Garbelli M.; Lossani A.; Gasparrini F.; Tuccinardi T.; Laurenzana I.; Angelucci A.; Maga G.; Schenone S.; Brullo C.; Musumeci F.; Desogus A.; Crespan E.; Botta M. Studies on the ATP binding site of Fyn kinase for the identification of new inhibitors and their evaluation as potential agents against tauopathies and tumors. J. Med. Chem. 2015, 58 (11), 4590–4609. 10.1021/acs.jmedchem.5b00140. [DOI] [PubMed] [Google Scholar]
- Ortuso F.; Amato R.; Artese A.; D’antona L.; Costa G.; Talarico C.; Gigliotti F.; Bianco C.; Trapasso F.; Schenone S.; Musumeci F.; Botta L.; Perrotti N.; Alcaro S. In silico identification and biological evaluation of novel selective Serum/Glucocorticoid-Inducible kinase 1 inhibitors based on the pyrazolo-pyrimidine scaffold. J. Chem. Inf. Model. 2014, 54 (7), 1828–1832. 10.1021/ci500235f. [DOI] [PubMed] [Google Scholar]
- Calgani A.; Vignaroli G.; Zamperini C.; Coniglio F.; Festuccia C.; Di Cesare E.; Gravina G. L.; Mattei C.; Vitale F.; Schenone S.; Botta M.; Angelucci A. Suppression of SRC signaling is effective in reducing synergy between glioblastoma and stromal cells. Mol. Cancer Ther. 2016, 15 (7), 1535–1544. 10.1158/1535-7163.MCT-15-1011. [DOI] [PubMed] [Google Scholar]
- Matteoni S.; Abbruzzese C.; Matarrese P.; De Luca G.; Mileo A. M.; Miccadei S.; Schenone S.; Musumeci F.; Haas T. L.; Sette G.; Carapella C. M.; Amato R.; Perrotti N.; Signore M.; Paggi M. G. The kinase inhibitor SI113 induces autophagy and synergizes with quinacrine in hindering the growth of human glioblastoma multiforme cells. J. Exp. Clin. Cancer Res. 2019, 38 (1), 202. 10.1186/s13046-019-1212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrecca K.; Guiot M.-C.; Panet-Raymond V.; Souhami L. Failure pattern following complete resection plus radiotherapy and Temozolomide is at the resection margin in patients with glioblastoma. J. Neuro-Oncol. 2013, 111 (1), 19–23. 10.1007/s11060-012-0983-4. [DOI] [PubMed] [Google Scholar]
- Smith S. J.; Diksin M.; Chhaya S.; Sairam S.; Estevez-Cebrero M. A.; Rahman R. The invasive region of glioblastoma defined by 5ALA guided surgery has an altered cancer stem cell marker profile compared to central tumour. Int. J. Mol. Sci. 2017, 18 (11), 2452. 10.3390/ijms18112452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Biopharmaceutics Classification System (BCS) Guidance.| FDA. https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/biopharmaceutics-classification-system-bcs-guidance (accessed Oct 13, 2019).
- Radi M.; Dreassi E.; Brullo C.; Crespan E.; Tintori C.; Bernardo V.; Valoti M.; Zamperini C.; Daigl H.; Musumeci F.; Carraro F.; Naldini A.; Filippi I.; Maga G.; Schenone S.; Botta M. Design, synthesis, biological activity, and ADME properties of pyrazolo[3,4-d]pyrimidines active in hypoxic human leukemia cells: a lead optimization study. J. Med. Chem. 2011, 54 (8), 2610–2626. 10.1021/jm1012819. [DOI] [PubMed] [Google Scholar]
- Ceccherini E.; Indovina P.; Zamperini C.; Dreassi E.; Casini N.; Cutaia O.; Forte I. M.; Pentimalli F.; Esposito L.; Polito M. S.; Schenone S.; Botta M.; Giordano A. SRC family kinase inhibition through a new pyrazolo[3,4-d]pyrimidine derivative as a feasible approach for glioblastoma treatment. J. Cell. Biochem. 2015, 116 (5), 856–863. 10.1002/jcb.25042. [DOI] [PubMed] [Google Scholar]
- Sanna M.; Sicilia G.; Alazzo A.; Singh N.; Musumeci F.; Schenone S.; Spriggs K. A.; Burley J. C.; Garnett M. C.; Taresco V.; Alexander C. Water solubility enhancement of pyrazolo[3,4-d]pyrimidine derivatives via miniaturized polymer–drug microarrays. ACS Med. Chem. Lett. 2018, 9 (3), 193–197. 10.1021/acsmedchemlett.7b00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A. G.; Jänicke R. U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6 (2), 99–104. 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Belmokhtar C. A.; Hillion J.; Ségal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20 (26), 3354–3362. 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- Hoek J. B.; Cahill A.; Pastorino J. G. Alcohol and Mitochondria: a dysfunctional relationship. Gastroenterology 2002, 122 (7), 2049–2063. 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. D.; Mladek A. C.; Lamont J. D.; Goble J. M.; Erlichman C.; James C. D.; Sarkaria J. N. Disruption of parallel and converging signaling pathways contributes to the synergistic antitumor effects of simultaneous mTOR and EGFR inhibition in GBM cells. Neoplasia 2005, 7 (10), 921–929. 10.1593/neo.05361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garuti L.; Roberti M.; Bottegoni G. Multi-kinase inhibitors. Curr. Med. Chem. 2015, 22 (6), 695–712. 10.2174/0929867321666141216125528. [DOI] [PubMed] [Google Scholar]
- Chou T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay Method. Cancer Res. 2010, 70 (2), 440–446. 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Yahiaoui A.; Meadows S. A.; Sorensen R. A.; Cui Z.-H.; Keegan K. S.; Brockett R.; Chen G.; Quéva C.; Li L.; Tannheimer S. L. PI3Kδ inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kδ and BTK inhibitors. PLoS One 2017, 12 (2), e0171221 10.1371/journal.pone.0171221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taresco V.; Louzao I.; Scurr D.; Booth J.; Treacher K.; McCabe J.; Turpin E.; Laughton C. A.; Alexander C.; Burley J. C.; Garnett M. C. Rapid nanogram scale screening method of microarrays to evaluate drug-polymer blends using high-throughput printing technology. Mol. Pharmaceutics 2017, 14 (6), 2079–2087. 10.1021/acs.molpharmaceut.7b00182. [DOI] [PubMed] [Google Scholar]
- Styliari I. D.; Conte C.; Pearce A. K.; Hüsler A.; Cavanagh R. J.; Limo M. J.; Gordhan D.; Nieto-Orellana A.; Suksiriworapong J.; Couturaud B.; Williams P.; Hook A. L.; Alexander M. R.; Garnett M. C.; Alexander C.; Burley J. C.; Taresco V. High-throughput miniaturized screening of nanoparticle formation via inkjet printing. Macromol. Mater. Eng. 2018, 303 (8), 1800146. 10.1002/mame.201800146. [DOI] [Google Scholar]
- Solassol I.; Pinguet F.; Quantin X. FDA- and EMA-Approved tyrosine kinase inhibitors in advanced EGFR-mutated non-small cell lung cancer: safety, tolerability, plasma concentration monitoring, and management. Biomolecules 2019, 9 (11), 668. 10.3390/biom9110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Gordoa T.; García-Bermejo M. L.; Grande E.; Garrido P.; Carrato A.; Molina-Cerrillo J. Targeting tyrosine kinases in renal cell carcinoma: ″new bullets against old guys″. Int. J. Mol. Sci. 2019, 20 (8), 1901. 10.3390/ijms20081901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallacara A. L.; Zamperini C.; Podolski-Renić A.; Dinić J.; Stanković T.; Stepanović M.; Mancini A.; Rango E.; Iovenitti G.; Molinari A.; Bugli F.; Sanguinetti M.; Torelli R.; Martini M.; Maccari L.; Valoti M.; Dreassi E.; Botta M.; Pešić M.; Schenone S. A New strategy for glioblastoma treatment: in vitro and in vivo preclinical characterization of Si306, a pyrazolo[3,4-d]pyrimidine dual Src/P-glycoprotein inhibitor. Cancers 2019, 11 (6), 848. 10.3390/cancers11060848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.