

Abstract

The NRF2-ARE pathway is an intrinsic mechanism of defense against oxidative stress. Inhibition of the interaction between NRF2 and its main negative regulator KEAP1 is an attractive strategy toward neuroprotective agents. We report here the identification of nonacidic tetrahydroisoquinolines (THIQs) that inhibit the KEAP1/NRF2 protein–protein interaction. Peptide SAR at one residue is utilized as a tool to probe structural changes within a specific pocket of the KEAP1 binding site. We used structural information from peptide screening at the P2 pocket, noncovalent small-molecules inhibitors, and the outcome from an explorative SAR at position 5 of THIQs to identify a series of neutral THIQ analogs that bind to KEAP1 in the low micromolar range. These analogs establish new H-bond interactions at the P3 and P2 pockets allowing the replacement of the carboxylic acid functionality by a neutral primary carboxamide. X-ray crystallographic studies reveal the novel binding mode of these molecules to KEAP1.
Keywords: NRF2, KEAP1, protein−protein interactions, Huntington’s disease, tetrahydroisoquinoline
Increases in oxidative stress are associated with the neuronal cell death that occurs in a number of neurodegenerative disorders, including Parkinson’s and Huntington’s diseases.1 The stimulation of antioxidative processes has consequently emerged as an attractive therapeutic goal. Neuroprotection in response to oxidative insults can be mediated by the transcriptional activation of protective genes under the control of the cis-acting sequence called antioxidant response element (ARE). Nuclear factor (erythroid-derived 2)-like 2 (NRF2) is a transcription factor encoded by the human NFE2L2 gene which plays a key role in the ARE response. Under basal conditions, the cellular concentration of NRF2 is tightly modulated by its association to the cytoplasmic repressor KEAP1 (Kelch-like ECH-associated protein 1) that functions as a substrate recognition and facilitator for the ubiquitination of NRF2 and its eventual proteosomal degradation.2 Exposure of cells to reactive electrophiles or oxidative stress induces conformational changes in KEAP1 that prevent KEAP1 dependent NRF2 ubiquitination, circumvent subsequent proteosomal degradation, and release NRF2.3 After its translocation to the nucleus NRF2 forms heterodimers with a variety of regulatory proteins associated with ARE, signaling for gene transcription of a broad panel of cytoprotective proteins.4
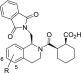
Given the key role that NRF2 plays in mediating the ARE response, promoting increases of its (free) intracellular concentration by blockade of the KEAP1/NRF2 interaction is an attractive strategy toward antioxidant therapeutics. Most known small molecule NRF2 activators/ARE inducers are indirect inhibitors of KEAP1/NRF2 that contain a reactive electrophilic functionality. These electrophiles irreversibly modify the sulfhydryl groups of KEAP1’s cysteine residues though this allows the release of NRF2 and induces an ARE-response in cells. The potential toxic side effects of irreversible protein modifiers are well documented.5 Remarkable progress has been made recently toward the identification of potent peptide and small-molecule inhibitors that are direct noncovalent inhibitors of the KEAP1/NRF2 protein–protein interaction (PPI).6 The 9mer peptide LDEETGEFL has been identified as the minimal binding sequence of the NRF2 ETGE motif to the Kelch domain of KEAP1,7 and a number of crystal structures of KEAP1 in complex with peptides have provided valuable structural information on the binding interactions between these proteins.8 At the outset of the work reported herein two series of direct small molecule inhibitors of the KEAP1/NRF2 interaction were prominent in the primary literature, exemplified by compounds 1(9) and 2a (Figure 1).10 Compound 2a is thought to fully occupy the KEAP1 binding site through interactions at 5 hot-spots (arbitrarily denoted as pockets P1–P5).10 In contrast, X-ray crystallography studies have shown that the tetrahydroisoquinoline (THIQ) 1 interacts at only three of these binding pockets, P2, P3, and P5.11 The weaker affinity of the THIQ (IC50 2.3 μM) is likely a reflection of suboptimal complementarity to the KEAP1/NRF2 interaction surface with respect to 2a (IC50 24 nM).12 A feature of both 1 and 2a, and indeed virtually all reported inhibitors of KEAP1/NRF2, is the presence of at least one carboxylic acid group. The presence of Arg483 and Arg380 in P1 and P2 and Arg415 bridging both pockets of KEAP1, respectively, generates a strongly basic surface potential that is complementary for binding of electronegative groups. The relevance of these interactions is demonstrated by the loss of activity reported for compound 2b (IC50 2.7 μM)12 that lacks both acetic acid substituents. Acidic functionalities are, however, undesirable with regard to CNS indications due to the poor blood–brain barrier (BBB) penetration commonly found for acid-containing compounds. To date a single NRF2 inhibitor 3 has been identified by replacement of the two acidic groups of 2a with primary amides. This bis-carboxamide 3 retains nanomolar binding to KEAP1 (IC50 63 nM).12 However, an analogous approach of replacing the acid functionality in THIQ 1 with a carboxamide functionality was not tolerated and led to inactive NRF2 inhibitors.11
Figure 1.

Structures of noncovalent inhibitors of KEAP1/NRF2 PPI.
Based on the view that THIQ 1 represented a more suitable lead optimization scaffold despite its weaker potency, we initiated a research program aiming to further advance this compound class. In addition to seeking improved binders to KEAP1, a challenging goal, stated at the outset of this work, was to replace the carboxylic acid functionality in this compound series as a key first step to achieving cell/brain permeability. A strategy to improve the binding of 1 would be to engineer into the molecule fragments that can access the unoccupied P1 and P4 pockets of KEAP1. However, the significant molecular weight-increase likely associated with this approach made it unattractive. An alternative strategy was to capitalize on interactions at a region of structurally conserved water molecules in the P3 pocket.11 Crystal structures consistently feature 6 interconnected structurally immobile water molecules embedded in the P3 pocket that adopt a hexagonal arrangement similar to the stable water assembly present in the ice-Ih crystal form. Docking studies suggested that glycol or related groups attached at positions 5 or 6 of the THIQ might potentially access P3 and bind to or displace parts of the water network, though the narrow entrance to the P3 pocket (see Figure 1A and 1B, Supporting Information) represented a potential concern. The binding affinity was measured in a binding displacement assay between the Kelch domain and the 9-mer peptide previously described by our group.13 Results from this approach are summarized in Table 1.
Table 1. SAR at P3 Pocket13.

IC50 for TR-FRET KEAP1 binding assay.
IC50 values are the average of at least three individual measurements ± standard deviation.
Mixture of diastereomers (1:1) at C1 of THIQ.
In practice substituents at the C6 position of the THIQ failed to produce increments in affinity over an unsubstituted core heterocycle, as exemplified by the simple glycol compound 5. Apparently, the geometric constraints at the entry to the P3 pocket are incompatible with access through this channel. However, when the glycol substituent was attached at position 5 (compound 6), a 4-fold increase in binding affinity was achieved. A chain length of 3 heavy atoms attaching the OH to the THIQ was predicted as optimal, but a loss of potency was noted for the homologue 7. The weaker potency observed for the methyl ether analog 8 suggests there is indeed a direct hydrogen bonding (donor) effect with the terminal hydroxyl group in 6. Changes to the 5-substituent were tolerated so long as the steric demands were not burdensome. Thus, the propargylic alcohol 9 was equipotent with the simple glycol 6, but the substituted analog 10 proved 18-fold less active. It is noteworthy that a simple 5-hydroxy THIQ provided the most active inhibitor from this study, with compound 11 giving just under 100 nM potency in the biochemical assay. In this case the observed gain in potency may indicate that the hydroxyl functionality (which is too small to penetrate deeply into the P3 channel) binds to the conserved water molecules through hydrogen bonding interactions rather than directly displacing them.
The experimental binding mode of 6 was assessed by X-ray structure determination on its complex with KEAP1(see Figure 2, Supporting Information). Good agreement with our original docking hypothesis (RMSD 0.92) was determined, and the experimental binding conformation highlights that the hydroxyl group in the P3 pocket does indeed interact with the conserved structural water region, networking directly with the Leu365 and Val604 amide backbone by replacing a structural water molecule (see Figure 2B, Supporting Information). Aside from these changes at P3, the overall binding of 6 in pockets P2 and P5, and the orientation of the THIQ core itself, remains very similar to the reported structures of 1.
Having successfully found a strategy to improve potency we reevaluated the replacement of the carboxylic acid group in the THIQ series. Disappointingly, despite the use of newly discovered more potent 5-substituted analogs as scaffolds from which to explore THIQ N-substitution we systematically failed to generate any novel starting points toward neutral inhibitors. Analogs synthesized either to test specific design hypotheses or as part of random explorative structure–activity relationships (SAR) uniformly generated inactive compounds in the KEAP1 binding assay. Furthermore, the need for a specific enantiomer of THIQ with three chiral centers made efficient stepwise probing of structural changes at P2 challenging from a synthetic standpoint. As a complementary approach, developing an in-depth understanding of the SAR around the 9mer peptide LDEETGEFL could offer a valuable knowledge of the KEAP1 binding site and provides tools for future structure-based drug design. The identification of minimal enzyme substrates by truncation of an endogenous substrate is a common example of this approach and can serve as a starting point for the design of peptidomimetics or macrocyclic inhibitors.14 The use of peptide SAR as a surrogate to understand the nature of the KEAP1 P2 pocket was an attractive approach. In X-ray structures of peptide binders to KEAP1 the side chain of residue 82 is consistently oriented toward the P2 region of the protein,15 and a peptide library containing nonacidic residues at this position was an accessible approach for probing this pocket. Nonapeptide 12 containing Glu in position 82 was chosen as a template. Glu82 engages in multiple interactions with residues Ser363, Arg380, Asn382, and Asn414 in the P2 pocket, mediated by hydrogen bonding and salt bridging contacts. A small library of 70 analogs was prepared in which Glu82 was replaced with natural l-amino acids, as well as non-natural amino acids containing neutral side chains. The key findings from this work are outlined in Table 2. As anticipated, based on the preference for Glu82 in mammalian NRF2 sequences, no naturally occurring l-amino acid showed improved KEAP1 binding. Surprisingly, however, substitution of Glu82 with the simple aliphatic residue, cyclobutylalanine (Cba), produced a peptide active in the low micromolar range (compound 13), only 10-fold weaker than Glu despite the lack of any polar side-chain functionality. Activity was specifically linked to the presence of the cyclobutyl moiety: both the ring-contracted and the ring-expanded analogs (cyclopropylalanine 14 or cyclopentylalanine 15, respectively) displayed decreased binding concentration of over 64 μM. Compound 13 was the most potent replacement found in the peptide library, but an emerging trend was non-negligible binding for peptides containing an CONH2 moiety in their side chains. The citrulline analog 16 and the carboxamide substituted Phe analog 17 were such examples, with the latter showing low micromolar affinity (IC50 5.9 uM). A specific role for the primary amide was apparent considering that the Phe analog 18 was 10-fold less active than 17.16
Table 2. SAR at Glu82 on Nonapeptide Scaffold13.


IC50 for TR-FRET KEAP1 binding assay.
IC50 values are the average of at least three individual measurements ± standard deviation.
Peptides 13 and 17 were docked into the KEAP1 binding site. A superimposition of 13 and 17 docking poses with the X-ray structure of 6 (Figure 2) reveals that the cyclobutyl group of 13 occupies the same region of KEAP1 as the cyclohexyl moiety of the THIQ, with the tertiary carbon atoms of the cyclohexyl/cyclobutyl rings superimposed. Moreover, the predicted orientation of the carboxamide group in 17 does not superimpose with the carboxylic acid of 6 but features distinct interactions with Asn414 and Ser363 at P2 through direct H-bonding with the carboxamide group. The exit vector from C3 of the cyclobutyl ring in 13 (corresponding to ring carbon C2 where the cyclobutyl is being attached to the THIQ) appears ideal to present a primary amide in a similar orientation. Based on these observations, we prepared and screened a series of 5-substituted THIQs containing carboxamide substituted cyclobutylamides and their corresponding carboxylic acid analogs. The results obtained are reported in Table 3.
Figure 2.

Crystal structure of 6 (green) bound to KEAP1 (KEAP1 [325–609]: 6; PDB code 6SP1) overlaid with (A) docked peptide 13 (blue) and docked peptide 17 (m-phenylcarboxamide) (blue carbon atoms).
Table 3. SAR at P2 Pocket13.

| cmpd | R | R′ | n | abIC50 (nM) |
|---|---|---|---|---|
| 19 | CO2H | OH | 1 | 862 ± 158 |
| 20 | CONH2 | OH | 1 | 1558 ± 300 |
| 21 | CONH2 | OH | 3 | 5539 ± 174 |
| 22 | CO2H | O(CH2)2OH | 1 | 3102 ± 424 |
| 23 | CONH2 | O(CH2)2OH | 1 | 2545 ± 357 |
IC50 for TR-FRET KEAP1 binding assay.
IC50 values are the average of at least three individual measurements ± standard deviation.
As expected, based on literature precedent11 the cyclobutane carboxylic acid analog 19 lost binding affinity with respect to the cyclohexyl analog 11, likely indicating reduced lipophilic contact from the 4-membered ring. However, the neutral primary carboxamide 20 retained very similar binding affinity as its corresponding acid analog. This result is in stark contrast to data for the cyclohexane acid/amide pair (compounds 11 and 21), where a drop in affinity of over 50-fold was observed. Moreover, although the cyclohexyl carboxamide 21 displayed 3-fold less binding affinity than the cyclobutyl analog 20, possibly due to a steric clash of the cyclohexyl ring with the protein, a similar binding affinity for the acid/amide analogs in the cyclobutane series was confirmed with glycol analogs 22 and 23.
Our hypothesis for an alternative binding mode of the carboxamide group at the P2 pocket was confirmed by the X-ray structure of the complex of 23 with KEAP1 (Figure 3A) whereclear differences with respect to the acidic analogs were observed. For the acidic THIQ 6, the interaction of the carboxylic acid group with Asn414 and Ser363 is mediated by a structural water molecule (Figure 3), while for the amide derivative 23 the interaction with these two residues is direct, displacing the structural water molecule conserved in all the acidic THIQ X-ray structures11 (Figure 3A). Comparing the X-ray structures of compounds 6 and 23, it is possible to appreciate how the amide of the cyclobutyl derivative is able to form direct hydrogen bond interactions with three different amino acids within the P2 pocket, by acting as H-bond donor with the Ser363 hydroxyl group, as a dual hydrogen bond acceptor–donor to the Asn414 side chain and as hydrogen bond acceptor to the guanidine group of Arg415. Moreover, the Arg415 side chain forms a hydrogen bond interaction with the other carbonyl group of the cyclobutyl ring. In fact, the X-ray structure of compound 23 shows the Arg415 side chain at hydrogen bonding distance to both the carbonyl oxygen atoms bound to the cyclobutyl moiety, further stabilizing the protein–ligand interaction. Another difference between both compounds is the lipophilic interactions at the P2 pocket, in particular with the side chain of Tyr334. The cyclobutyl ring of 23 shows a smaller surface contact with the aromatic ring than the larger cyclohexyl ring of 6, being a possible reason for a weaker interaction with the protein. Increase of the van der Waals interactions at P2 pockets emerges as a plausible strategy to improve the binding of these neutral THIQ with KEAP1. Furthermore, comparison of the X-ray structures of compounds 6 and 23 shows no significant differences in the binding in pockets P3 and P5 or the orientation of the THIQ core.
Figure 3.

(A) Crystal structure of 23 (pink carbon atoms) bound to KEAP1 (KEAP1[324–609]:23; PDB code 6SP4). (B) Overlaying of X-ray structures of compound 23 (pink) and compound 6 (green) bound to KEAP1.
In a preliminary profiling of compound 6 and its primary carboxamide analog 35 (see Supporting Information), the carboxamide showed an improved permeability in MDCK cells with values of Papp A-B of 9 and 46 nm/s, respectively.
The synthesis of THIQ analogs is shown in Scheme 1. Starting from the known THIQ intermediate 24,17 treatment with boron tribromide followed by amide coupling with the corresponding chiral monoprotected cycloalkyl dicarboxylate18 afforded the key intermediates 25 and 26 as a mixture (1:1) of diastereomers at position C1 of the THIQ ring. Introduction of the substituent at position 5 was performed through Mitsunobu reaction of the phenolic oxygen with the corresponding aliphatic alcohols. Subsequent elimination of the protecting benzyl groups under hydrogenation conditions led to compounds 6–8 and 22. 5-Hydroxy analogs 11 and 19 were prepared by hydrogenation of the benzylic esters 25 and 26. Subsequent treatment of the carboxylic acid analogs 11, 19, and 22 with ammonia under amide coupling conditions afforded the primary carboxamides 20, 21, and 23. Finally, the 5-alkynyl analogs 9 and 10 were obtained by Sonogashira coupling19 of the 5-O-triflated derivative 27 with the corresponding THP-protected propargyl alcohol followed by deprotection of the hydroxyl group in acidic media. The 6-substituted THIQ analog 5 was prepared following the same synthetic procedure as that for 6 but starting from the corresponding 6-methoxy THIQ intermediate (see Supporting Information).
Scheme 1. Synthesis of Compounds 6–11 and 19–23.

Reagents and conditions: (a) BBr3, DCM, rt, 18 h; (b) RCO2H, HATU, DIPEA, DMF, rt or 60 °C, 18 h; (c) H2 (1 atm), Pd/C, MeOH or EtOAc, rt; (d) ROH, PS–PPh3, THF, 0 °C to rt; (e) NH3, TBTU, DMF, rt, 1 h; (f) Tf2O, TEA, DCM, −15 °C, 30 min; (g) alkyne, CuI, (Ph3P)2Cl2Pd, TBAI, TEA, DMF, 100 °C, 1 h; (h) TFA/DCM (1:10), 0 °C to rt.
The absolute configuration of the THIQ reported in this publication was tentatively assigned as S,R,S based on the 1H NMR and biological data and its comparison with the literature.9,11 This assumption was later confirmed by the crystal structures of compounds 6 and 23.
In summary, we report the identification of new non acidic THIQ analogs that inhibit the protein–protein interaction between NRF2 and KEAP1 in the low micromolar range. We demonstrated the utility of using a natural ligand based-peptide library to understand the structure–activity relationship of the NRF2–KEAP1 binding interaction interphase and applied it to remove the acidic moiety of the THIQ series in our compound design strategy. These molecules show improved binding affinities to KEAP1 by establishing new interactions at the P3 and P2 pockets and counterbalancing the loss of the salt bridge of a carboxylic acid group with Arg415. The introduction of glycol or hydroxyl substituents at position 5 of the THIQ allows the engagement of additional H-bond interactions at the P3 pocket leading to the most potent THIQ KEAP1 binder known so far. Crystallographic evidence for the novel interaction mode of the substituents at position 5 of the THIQ scaffold in the P3 pocket and the cyclobutyl carboxamide moiety at the P2 pocket is provided. Studies to improve binding affinity in the cyclobutyl ring series remain under investigation in our laboratories.
Acknowledgments
The authors thank Mauro Cerretani and Nadia Gennari for performing the KEAP1 assay.
Glossary
Abbreviations
- ARE
antioxidant response element
- NRF2
nuclear factor erythroid 2-related factor
- KEAP1
Kelch-like ECH-associated protein 1
- PPI
protein–protein interaction
- THIQ
tetrahydroisoquinoline
- CNS
central nervous system
- BBB
blood–brain barrier
- SAR
structure–activity relationship
- THP
tetrahydropyrane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00594.
Synthetic experimental details and characterization data, description of primary biological assay protocols, summary of X-ray diffraction data collection and structure refinement, and density-difference maps (PDF)
Accession Codes
PDB codes: 6SP1, KEAP1[324–609] in complex with compound 6; 6SP4, KEAP1[324–609] in complex with compound 23.
Author Present Address
∥ For I.B.: Philochem AG, Libernstrasse 3, 8112 Otelfingen, Switzerland.
Author Present Address
⊥ For M.A.: First Health Pharmaceuticals B. V., Science Park 406, 1098XH Amsterdam, The Netherlands.
Author Present Address
∇ For G.K.: Friedrich Miescher Institute for Biomedical Research, Maulbeerstrasse 66, 4058 Basel, Switzerland.
Author Present Address
△ For V.S.: Department of Pharmacy, University of Naples “Federico II”, Via D. Montesano 49, 80131 Naples, Italy.
Author Present Address
○ For L.P.: Naason Science Inc., Saengmyung-Ro 123, Osong-eup, Heungdeok-gu, Cheongju-si, Chungbuk, Korea 28160.
Author Present Address
□ For L.T.S.: Rainwater Charitable Foundation, 777 Main St #750, Fort Worth, TX 76102.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
IRBM S.p.A. acknowledges financial support from CHDI Foundation, Inc., a nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics that substantially improve the lives of those affected by Huntington’s disease.
The authors declare no competing financial interest.
Dedication
# This work is dedicated to the beloved memory of our colleague and friend, Steven Harper (June 30, 2019).
Supplementary Material
References
- Dasuri K.; Zhang L.; Keller J. N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radical Biol. Med. 2013, 62, 170–185. 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S.; Jaiswal A. K. Functional characterization and role of Nrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase 1 gene. Oncogene 2001, 20, 3906–3917. 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Wakabayashi N.; Katoh Y.; Ishii T.; O’Connor T.; Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Buendia I.; Michalska P.; Navarro E.; Gameiro I.; Egea J.; Leòn R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. 10.1016/j.pharmthera.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Lu M.-C.; Ji J.-A.; Jiang Z.-Y.; You Q.-D. The Keap1-Nrf2-ARE Pathway as a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. 10.1002/med.21396. [DOI] [PubMed] [Google Scholar]
- Pallesen J. S.; Tran K. T.; Bach A. Non-covalent Small-Molecule Kelch-like ECH-Associated Protein 1 - Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Inhibitors and Their Potential for Targeting Central Nervous System Diseases. J. Med. Chem. 2018, 61, 8088–8103. 10.1021/acs.jmedchem.8b00358. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Inoyama D.; Kong A.-N. T.; Beamer L. J.; Hu L. Kinetic Analyses of Keap1-Nrf2 Interaction and Determination of the Minimal Nrf2 Peptide Sequence Required for Keap1 Binding Using Surface Plasmon Resonance. Chem. Biol. Drug Des. 2011, 78, 1014–1021. 10.1111/j.1747-0285.2011.01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S.-C.; Li X.; Henzl M. T.; Beamer L. J.; Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.; Magesh S.; Chen L.; Wang L.; Lewis T. A.; Chen Y.; Khodier C.; Inoyama D.; Beamer L. J.; Emge T. J.; Shen J.; Kerrigan J. E.; Kong A. N.-T.; Dandapani S.; Palmer M.; Schreiber S. L.; Munoz B. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z.-Y.; Lu M.-C.; Xu L.-L.; Yang T.-T.; Xi M.-Y.; Xu X.-L.; Guo X.-K.; Zhang X.-J.; You Q.-D.; Sun H.-P. Discovery of Potent Keap1-Nrf2 Protein-Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem. 2014, 57, 2736–2745. 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- Jnoff E.; Albrecht C.; Barker J. J.; Barker O.; Beaumont E.; Bromidge S.; Brookfield F.; Brooks M.; Bubert C.; Ceska T.; Corden V.; Dawson G.; Duclos S.; Fryatt T.; Genicot C.; Jigorel E.; Kwong J.; Maghames R.; Mushi I.; Pike R.; Sands Z. A.; Smith M. A.; Stimson C. C.; Courade J.-P. Binding Mode and Structure-Activity Relationships around Direct Inhibitors of the Nrf2-Keap1 Complex. ChemMedChem 2014, 9, 699–705. 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- Jain A. D.; Potteti H.; Richardson B. G.; Kingsley L.; Luciano J. P.; Ryuzoji A. F.; Lee H.; Krunic A.; Mesecar A. D.; Reddy S. P.; Moore T. W. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. 10.1016/j.ejmech.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresciani A.; Missineo A.; Gallo M.; Cerretani M.; Fezzardi P.; Tomei L.; Cicero D. O.; Altamura S.; Santoprete A.; Ingenito R.; Bianchi E.; Pacifici R.; Dominguez C.; Muñoz-Sanjuan I.; Harper S.; Toledo-Sherman L.; Park L. C. Nuclear factor (erythroid-derived 2)-like 2 (NRF2) drug discovery: Biochemical toolbox to develop NRF2 activators by reversible binding of Kelch-like ECH-associated protein 1 (KEAP1). Arch. Biochem. Biophys. 2017, 631, 31–41. 10.1016/j.abb.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K. I.; Padmanabhan B.; Kobayashi A.; Shang C.; Hirotsu Y.; Yokoyama S.; Yamamoto M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell. Biol. 2007, 27, 7511–7521. 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The data from the SAR study performed at position 82 of the peptide will be disclosed in a subsequent publication which manuscript is under preparation.
- Hu L.; Magesh S.; Chen L.; Lewis T.; Munoz B.; Wang L.. Direct Inhibitors of KEAP1-NRF2 Interaction as Antioxidant Inflammation Modulators. WIPO Patent 067036, 2013.
- Bolm C.; Schiffers I.; Atodiresei I.; Hackenberger P. R. An alkaloid-mediated desymmetrization of meso-anhydrides via a nucleophilic ring opening with benzyl alcohol and its application in the synthesis of highly enantiomerically enriched β-amino acids. Tetrahedron: Asymmetry 2003, 14, 3455–3467. 10.1016/S0957-4166(03)00579-2. [DOI] [Google Scholar]
- Fisher T. J.; Dussault P. H. Regioselective Synthesis of Tetraalkynylarenes by Consecutive Dual Sonogashira Coupling Reactions of the Bis(triflate) of 4,5-Diiodobenzene-1,2-diol. Eur. J. Org. Chem. 2012, 2012, 2831–2836. 10.1002/ejoc.201200079. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.