

Abstract
Excess electrons play important roles for the construction of superficial active sites on nanocatalysts. However, providing excess electrons to nanocatalysts in vivo is still a challenge, which limits the applications of nanocatalysts in biomedicine. Herein, auger electrons (AEs) emitted from radionuclide 125 (125I) are used in situ to construct active sites in a nanocatalyst (TiO2) and the application of this method is further extended to cancer catalytic internal radiotherapy (CIRT). The obtained 125I‐TiO2 nanoparticles first construct superficial Ti3+ active sites via the reaction between Ti4+ and AEs. Then Ti3+ stretches and weakens the O—H bond of the absorbed H2O, thus enhancing the radiolysis of H2O molecules and generating hydroxyl radicals (•OH). All in vitro and in vivo results demonstrate a good CIRT performance. These findings will broaden the application of radionuclides and introduce new perspectives to nanomedicine.
Keywords: active sites, auger Electrons, I‐125, internal radiotherapy, nanocatalysts, titanium dioxide
A simple and highly efficient catalytic internal radiotherapy (CIRT) is developed by 125I‐labeled TiO2 nanoparticles. Auger electrons from 125I react with Ti4+ in TiO2 and construct superficial active sites of Ti3+. The generated Ti3+ decreases the energy barriers of H2O and contributes to the generation of •OH upon γ‐rays irradiation, thus enhancing the antitumor effect of IRT.
Nanocatalysts have been widely used in the area of biomedicine, such as cancer therapy, antibacterial treatment, and biomolecules detection, because of their high surface activity.[ 1 , 2 , 3 ] Many published reports have shown that excess electrons (e.g., photoinduced electrons) play an important role in the surface chemical activity of nanocatalysts.[ 4 , 5 , 6 ] Generally, excess electrons are captured by superficial metal atoms to form active sites with localized charge imbalance. Then, these active sites can deform certain molecules, stretch chemical bonds, and reduce the energy barriers of chemical reactions. Hence, providing excess electrons to the surface of nanocatalysts is important for improving the nanocatalyst performance. However, hardly few methods exist for the generation of excess electrons in biological tissue, which inhibits the application of nanocatalysts in tumor treatment.
Internal radiotherapy (IRT) is a common treatment method for suppressing tumor growth.[ 7 , 8 ] Some radionuclides, such as 90Y, 125I, 131I, and 188Re, can emit electrons in situ and are thus usually implanted into tumors for IRT.[ 9 , 10 , 11 , 12 ] Among these radionuclides, 125I can emit low‐energy (100–103 eV) auger electrons (AEs) via internal conversion with a production of 24.9 AEs per decay, which can be deposited on nanocatalysts more easily than the high‐energy (≈106 eV) β‐rays emitted from other radionuclides.[ 13 ] Hence, 125I has the potential to inject excess electrons onto nanocatalysts surface and construct active sites for further applications in vivo. On the other hand, in IRT, ionizing radiation (primarily β‐ and γ‐rays) emitted from radionuclides induces radiolysis of H2O to generate hydroxyl radicals (•OH), which can efficiently kill cancer cells.[ 14 , 15 , 16 ] However, the strong O—H bond of H2O molecule limits the yield of •OH induced by radionuclides, and especially for 125I, which emits low‐energy AEs and γ‐rays (35 keV).[ 17 , 18 ] Fortunately, as mentioned above, the combination of nanocatalysts and AEs emitted from 125I will construct superficial active sites, which can stretch molecules and decrease the bond energy for H2O activation, thus facilitating the occurring of H2O radiolysis. Hence, 125I‐labeled nanocatalysts have the potential to increase the yield of •OH for enhanced IRT.
Titanium dioxide (TiO2) is a traditional and typical nanocatalyst with high stability and low toxicity, which is suitable for applications in vivo.[ 19 , 20 , 21 , 22 ] Herein, we developed a method of injecting AEs to nanocatalysts by synthesizing 125I‐labeled TiO2 nanoparticles (125I‐TiO2 NPs) and explored the performance of 125I‐TiO2 NPs for cancer catalytic internal radiotherapy (CIRT), as illustrated in Scheme 1 . First, AEs emitted from 125I arrive on the surface of TiO2 and induce the formation of Ti3+. Then, Ti3+ absorbs and deforms H2O, resulting in the decreased O—H bond energy. Finally, upon irradiation by the γ‐rays emitted from 125I, the activated H2O is more easily converted to •OH compared to the unabsorbed H2O, leading to an enhanced effect of IRT. This strategy as CIRT will bring more chances for cancer therapy. Moreover, the method of constructing active sites by 125I will also widen the applications of radionuclides and nanocatalysts and introduce new perspectives to the area of nanocatalytic medicine.
Scheme 1.
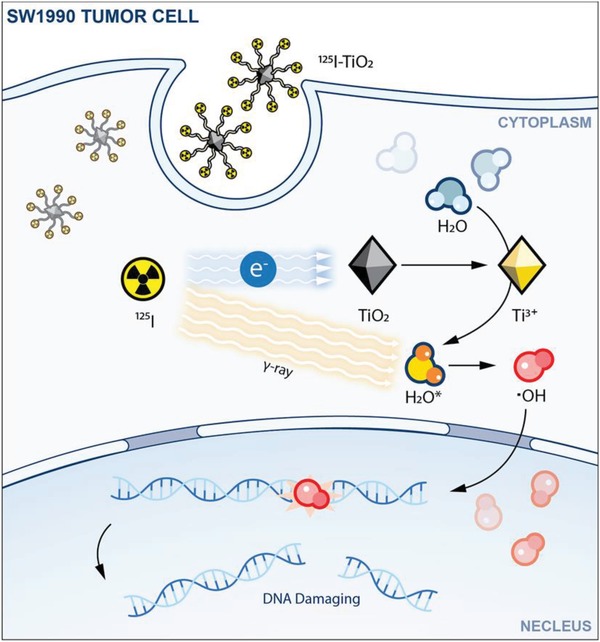
Schematic illustration for the mechanism of 125I‐TiO2‐induced CIRT.
The synthesis process of 125I‐TiO2 is illustrated in Figure 1a. First, TiO2 NPs coated with oleylamine and oleic acid (TiO2‐OA) were synthesized according to a typical solvothermal method.[ 23 , 24 ] In Figure 1b, transmission electron microscope (TEM) image showed that TiO2‐OA were in good uniformity with rhombus‐shaped morphology, and the average particle diameter was 11.78 ± 2.23 nm in length and 3.91 ± 0.56 nm in width. High‐resolution TEM (HRTEM) image revealed a lattice fringe width of 0.35 nm, corresponding to the anatase (confirmed by X‐ray diffraction (XRD) patterns in Figure 1c) (101) crystal face. Subsequently, TiO2‐OA were modified with citric acid (TiO2‐COOH) for well water solubility and to prepare for subsequent modification. Finally, TiO2‐COOH were conjugated with tyramine (TiO2‐tyr) for labeling of 125I. Successful synthesis of TiO2‐COOH and TiO2‐tyr was confirmed by the presence of characteristic peaks (O—H and amide, respectively) in Fourier transform infrared spectroscopy (FT‐IR) spectra (Figure 1d).[ 25 ] The absorption peak changed little in ultraviolet‐visible (UV–vis) spectra (Figure S1, Supporting Information), indicating that TiO2 remained stable after modification. Obviously, compared with TiO2‐COOH, the conjugated tyramine restrained the ionization of carboxyls, leading to a decreased Zeta potential (Figure S2, Supporting Information) and an increased hydrodynamic size (Figure 1e) of TiO2‐tyr. Finally, 125I was labeled to TiO2‐tyr via a classical Iodogen‐catalyzed method to obtain the final product of 125I‐TiO2.[ 26 ] The initial radiochemical purity of 125I‐TiO2 was 93.43% and remained above 88% during a 24‐h incubation with 0.1% fetal bovine serum in phosphate buffer solution, signifying a successful and stable radiolabeling (Figure 1f and Figure S3, Supporting Information).
Figure 1.
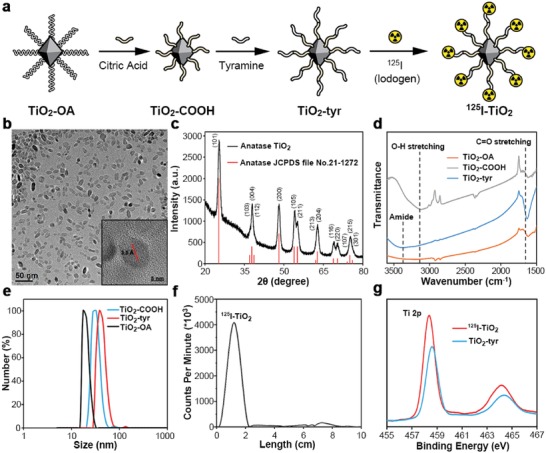
Synthesis and characterization of 125I‐TiO2. a) Synthesis process of 125I‐TiO2. b) TEM image of TiO2‐OA (inset: HRTEM image of TiO2‐OA). c) XRD pattern of TiO2‐OA. d) FT‐IR spectra of TiO2‐OA, TiO2‐COOH, and TiO2‐tyr. e) Hydrodynamic radius of TiO2‐OA, TiO2‐COOH, and TiO2‐tyr. f) Radio thin‐layer chromatography analyzing the labeling rate of 125I‐TiO2. g) XPS spectra of TiO2‐tyr and 125I‐TiO2.
After obtaining 125I‐TiO2, we investigated the interaction between TiO2 and H2O. First, X‐ray photoelectron spectroscopy (XPS) showed that the binding energy of Ti 2p orbitals of 125I‐TiO2 decreased by 0.2 eV compared with that of TiO2‐tyr, indicating the existence of Ti3+ in 125I‐TiO2 (Figure 1g).[ 27 ] Next, density functional theory (DFT) was used to simulate the O—H bond length under the influence of Ti4+ or Ti3+. Figure 2a shows that the O—H bond length was 1.17 Å under the influence of Ti4+. In contrast, Ti3+ stretched the O—H bond to 1.48 Å (Figure 2b), signifying a decreased bond energy. Further, the energy barrier for converting H2O to •OH was calculated. As shown in Figure 2c,d, the required energy for H2O radiolysis under the influence of Ti4+ was 1.23 eV, while declined to 0.82 eV under the influence of Ti3+. Hence, the Ti3+ species in 125I‐TiO2 can reduce the O—H bond energy and energy barrier of H2O radiolysis, indicating the potential to increase the yield of •OH in IRT.
Figure 2.
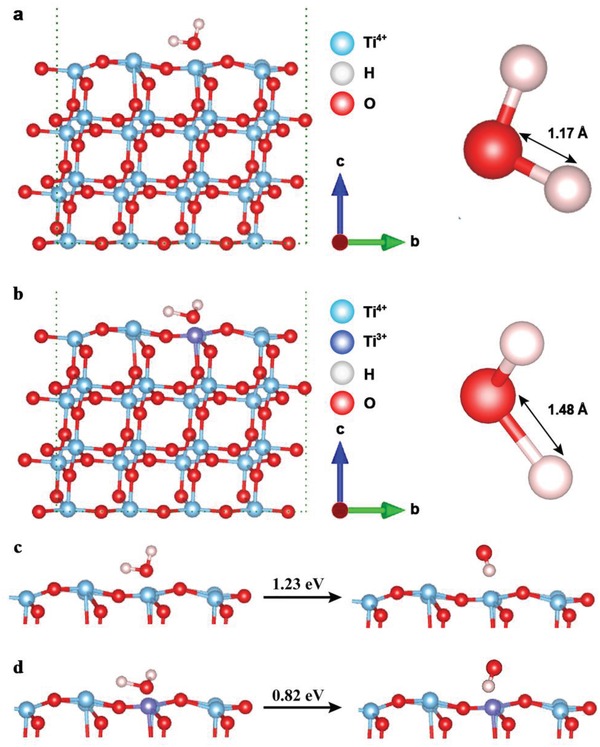
Simulated O—H bond length of H2O and the energy barrier for converting H2O to ·OH on the surface of TiO2 (anatase (101) face). a) O—H bond under the influence of Ti4+. b) O—H bond under the influence of Ti3+. c) Energy barrier of H2O radiolysis under the influence of Ti4+. d) Energy barrier of H2O radiolysis under the influence of Ti3+.
Pancreatic cancer usually induced a poor prognosis and short survival time for patients. Many clinical researches have reported that IRT was suitable for the treatment of pancreatic cancer.[ 28 , 29 , 30 , 31 , 32 , 33 , 34 ] Hence, to investigate the effect of CIRT for pancreatic cancer, we adopted human pancreatic cancer (SW1990) cells for all experiments in vitro. First, as sufficient intracellular accumulation of NPs is a prerequisite for an effective treatment, the cellular uptake and intracellular distribution of TiO2‐tyr was visualized via biological TEM (bio‐TEM). As exhibited in Figure S4 in the Supporting Information, the amount of endocytosed TiO2‐tyr increased as the incubation time prolonged from 0.5 to 5 h. Next, we investigated the appropriate dosage of 125I and 125I‐TiO2 via the Cell Counting Kit 8 Assay. As illustrated in Figure S5 in the Supporting Information, 125I exhibited a negligible detrimental effect especially when the dosage was below 600 µCi mL−1. Compared to 600 µCi mL−1 of 125I (cell viability, 96.44%), 125I‐TiO2 with the equal dose (total mass of 144 µg mL−1, radiation dose of 600 µCi mL−1) presented obvious cytotoxicity (cell viability, 64.16%). Similar trends also appeared under other doses of radiation. As 125I or TiO2‐tyr alone exhibited little cytotoxicity to SW1990 cells (Figures S5 and S6, Supporting Information), the cell killing effect of 125I‐TiO2 should be attributed to the reaction between 125I and TiO2‐tyr. To fully explore the effect of 125I‐TiO2, the control group, 125I group (600 µCi mL−1), and 125I‐TiO2 group (total mass of 144 µg mL−1, radiation dose of 600 µCi mL−1) were adopted in this study for the following experiments.
It is widely known that radiation exerts biological effects mainly by inducing cell apoptosis and proliferative injury.[ 35 , 36 , 37 ] Therefore, cell apoptosis was detected by terminal‐deoxynucleoitidyl Transferase Mediated Nick End Labeling (TUNEL) assays. As shown in Figure 3a,b, 125I‐TiO2 triggered the most severe cell apoptosis (apoptosis rate, 37.48%), which was 13.82 and 37.62 times that of the 125I group and control group, respectively. Proliferative injury was first evaluated by the expression level of proliferating cell nuclear antigen (PCNA), which is a representative protein directly involved with the DNA replication.[ 38 ] The expression of PCNA in 125I‐TiO2 group decreased by 61.05% and 73.78% compared to that in 125I group and control group, respectively (Figure 3c,d). Then, the colony formation assay, which is a classical and sensitive method for the visual evaluation of cell proliferation status,[ 39 ] indicated that the cells in 125I‐TiO2 group had the lowest cell surviving fraction and cloning efficiency in a dose‐dependent manner (Figure 3e,f and Figure S7, Supporting Information). In addition, the expression of relevant regulatory proteins, including pro‐apoptotic Bax, antiapoptotic Bcl‐2, and proliferation index of Ki‐67, displayed similar tendencies consistent with above results (Figure S8, Supporting Information). In summary, the combination of TiO2 and 125I can induce increased cell apoptosis and suppressed cell proliferation compared to 125I alone.
Figure 3.
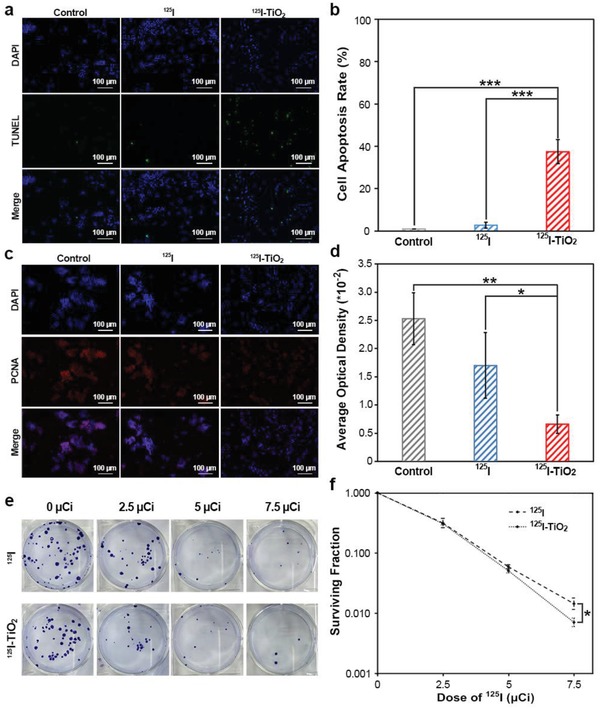
In vitro experiments for therapeutic evaluation. a) Immunofluorescence assay of TUNEL analyzing cell apoptosis induced by dulbecco's modified eagle medium (DMEM, control), 125I, and 125I‐TiO2. b) Quantitative comparison of cell apoptosis rate among groups (n = 3, mean ± s.d., one‐way analysis of variance (ANOVA) with least significant difference (LSD)‐t post hoc test, P < 0.0001 (125I‐TiO2 vs Control) and < 0.0001 (125I‐TiO2 vs 125I), ***P < 0.001). c) Immunofluorescence assay analyzing the cellular PCNA expression in different treatment groups including control, 125I, and 125I‐TiO2 group. d) Quantitative comparison of the expression level of PCNA among groups (n = 3, mean ± s.d., one‐way ANOVA with LSD‐t post hoc test, P = 0.0020 (125I‐TiO2 vs Control) and 0.0273 (125I‐TiO2 vs 125I), *P < 0.05, **P < 0.01). e) Colony formation assay analyzing cell proliferation after receiving incremental radiation doses of 125I and 125I‐TiO2. f) Curves of cell surviving fraction based on colony formation assay (n = 3, mean ± s.d., Mann–Whitney U‐test, P = 0.0463 (125I‐TiO2 vs 125I), *P < 0.05).
Cytotoxicity of radiation is usually attributed to the •OH‐induced DNA double‐strand breaks (DSBs).[ 40 , 41 ] As mentioned above, we designed 125I‐TiO2 to improve the yield of •OH in IRT. Hence, we initially measured the generation of intracellular •OH using a hydroxyphenyl fluorescein probe. As shown in Figure 4a,b, the amount of •OH in 125I‐TiO2 group exhibited a substantial augmentation after 24 h of irradiation, which was 1.62 and 1.70 multiples of that in 125I and control groups, respectively. Then DNA DSBs were detected via γ‐H2AX immunofluorescence assay. According to Figure 4c,d, cancer cells treated by 125I‐TiO2 presented the most severe DNA DSBs (one γ‐H2AX foci represents one DNA DSB). These data proved that 125I‐TiO2 can enhance the generation of •OH, thereby exacerbating DNA DSBs, and ultimately cause cell apoptosis and proliferative injury.
Figure 4.
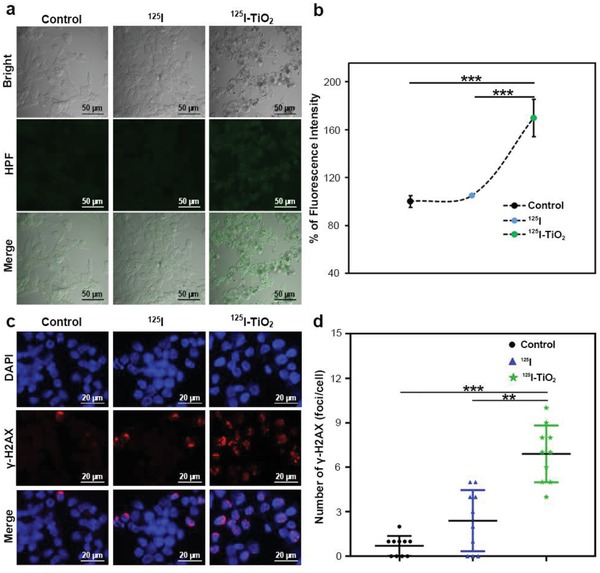
In vitro cell experiments exploring the mechanism for enhanced therapeutic effects. a) Fluorescence assay analyzing intracellular •OH generation after different treatments of DMEM (control), 125I and 125I‐TiO2. b) Quantitative comparison of •OH yield among groups (n = 3, mean ± s.d., one‐way ANOVA with LSD‐t post hoc test, P < 0. 0001 (125I‐TiO2 vs 125I) and = 0.0002 (125I‐TiO2 vs Control), ***P < 0.001). c) Immunofluorescence assay analyzing DNA DSBs by γ‐H2AX staining in DMEM (control), 125I, and 125I‐TiO2 groups. d) Quantitative comparison of DNA DSBs‐related red fluorescent foci of γ‐H2AX among groups (n = 10, mean ± s.d., Kruskal–Wallis one‐way ANOVA analysis, P = 0.002 (125I‐TiO2 vs 125I) and <0.001 (125I‐TiO2 vs Control), **P < 0.01, ***P < 0.001).
Encouraged by these in vitro experiments, we tested the effect of CIRT in SW1990 tumor‐xenografted mice. Three groups were divided with intratumoral injection of dulbecco's modified eagle medium (DMEM, control), free 125I (600 µCi of 125I per mouse) and 125I‐TiO2 (600 µCi of 125I corresponding to 144 µg of TiO2 per mouse), respectively. First, single‐photon emission computed tomography/computed tomography (SPECT/CT) scanning was performed. As shown in Figure 5a, most of the injected 125I was excreted through the urinary system with only minor residuals drawn in the thyroid. In a sharp contrast, 125I‐TiO2 exhibited remarkably prolonged retention time in the tumor for more than 12 d, which indicated an excellent labeling stability and intratumoral retention ability of 125I‐TiO2 in vivo, thus benefiting for a long‐lasting irradiation for tumor.
Figure 5.
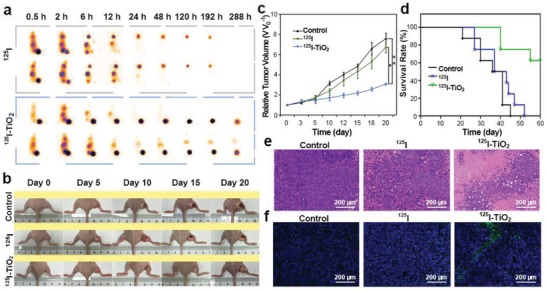
In vivo imaging and therapeutic assessment. a) Representative SPECT/CT images of SW1990 tumor‐bearing mice at different time points after intratumoral injection of 125I and 125I‐TiO2. b) Representative photographs of SW1990 tumor‐bearing mice for time‐course change of tumor size after different treatments including DMEM (control), 125I, and 125I‐TiO2. c) Tumor growth curves during the 20 d observation after different treatments by DMEM (control), 125I, and 125I‐TiO2. Relative tumor volume (V V 0 −1) was given by tumor volume (V) normalized to the initial value (V 0) (n = 5, mean ± s.d., Kruskal–Wallis one‐way ANOVA analysis, P = 0.048 (125I‐TiO2 vs 125I) and 0.001 (125I‐TiO2 vs Control), *P < 0.05, **P < 0.01). d) Survival curves of mice after different treatments during an observation period of 60 d (n = 8). e) H&E staining of tumor sections harvested at day 20 post different treatments (original magnification 200 ×). f) Representative TUNEL staining of tumor slices collected at day 20 post treatments (original magnification 200 ×).
We further examined the curative effect of 125I‐TiO2 in vivo. As shown in Figure 5b,c and Figure S9 in the Supporting Information, after a 20 d treatment, tumors in control and 125I groups progressed to 7.58 ± 0.54 and 6.71 ± 0.55 times that of the original volumes, whereas tumors treated by 125I‐TiO2 were distinctly controlled with the relative volume percentages of 59.49% (for control group) and 54.21% (for 125I group), respectively. Correspondingly, the survival rate within a 60 d observation was significantly improved in 125I‐TiO2 group, during which all mice in 125I and control groups died out (Figure 5d). Moreover, all surviving mice had reasonably stable body weights during the observation period, indicating a negligible long‐term biotoxicity of all compositions (Figure S10, Supporting Information). The histopathologic results also supported the above findings. As expected, hematoxylin and eosin (H&E) staining (Figure 5e) and TUNEL (Figure 5f) revealed that the most severe tumor tissue destruction occurred after the tumors were exposed to 125I‐TiO2 rather than to 125I. According to immunohistochemical staining results, the expression of Ki‐67 (a cancer cell proliferation factor) was downregulated in 125I‐TiO2 group compared to that in 125I or control groups (Figure S11, Supporting Information), while the TNF‐α (a tumor necrosis factor) expressed the highest in 125I‐TiO2 group (Figure S12, Supporting Information). All these data proved the highly efficient anticancer ability of 125I‐TiO2.
In summary, we used 125I to inject electrons to TiO2 NPs for constructing active sites in vivo, and investigated its application for cancer CIRT. The obtained 125I‐TiO2 initially constructed Ti3+ species via the reaction between Ti4+ and AEs from 125I. Consequently, Ti3+ stretched the O—H bond of the absorbed H2O to decrease its bond energy. Finally, upon irradiation of γ‐rays emitted by 125I, the radiolysis of activated H2O will occur more easily, leading to an enhanced generation of •OH. Material characterization and DFT simulation confirmed the existence and function of the constructed active sites. In vitro and in vivo data further verified an enhanced curative effect of CIRT induced by the active sites. Compared with previous researches on nanocatalysts, this paper presented two major differences. First, the mechanism of the generation of ·OH in this research is different. In previous researches,[ 42 , 43 ] ·OH is usually generated from the reaction between H2O and photoinduced holes. While in this research, ·OH comes from the reaction between γ‐rays and H2O that is activated by AEs‐constructed active sites. Second, the excitation source of nanocatalysts in this research is different. Previous researches usually use an external excitation source like UV or vis, of which the penetration depth in biological tissue is only millimeter‐sized.[ 44 , 45 ] In this research, we solve this problem by using 125I as an internal excitation source. Hence, our strategy by using radionuclides to construct active sites in nanocatalysts will bring more ideas and chances for the applications of nanocatalysts. Moreover, we believe that this research will introduce new perspectives to the design of biomaterials and provide more opportunities for cancer therapy.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
W.S. and H.W. contributed equally to this work. This work was financially supported by the National Funds for Distinguished Young Scientists (Grant No. 51725202), the National Natural Science Foundation of China (Grant No. 81471714, 81871390, and 51872094), the Key Project of Shanghai Science and Technology Commission (Grant No. 19JC1412000), and the Collaborative Innovation Center of Technology and Equipment for Biological Diagnosis and Therapy in Universities of Shandong. All animal experiments were performed according to the guideline of Experimental Animal Ethics Committee of the Second Military Medical University (Approval Number, 20181101087).
Su W., Wang H., Wang T., Li X., Tang Z., Zhao S., Zhang M., Li D., Jiang X., Gong T., Yang W., Zuo C., Wu Y., Bu W., Auger Electrons Constructed Active Sites on Nanocatalysts for Catalytic Internal Radiotherapy. Adv. Sci. 2020, 7, 1903585 10.1002/advs.201903585
Contributor Information
Changjing Zuo, Email: cjzuo@smmu.edu.cn.
Yelin Wu, Email: sk_wuyelin@tongji.edu.cn.
Wenbo Bu, Email: wbbu@chem.ecnu.edu.cn.
References
- 1. Yin Z., Wu L., Yang H., Su Y., Phys. Chem. Chem. Phys. 2013, 15, 4844. [DOI] [PubMed] [Google Scholar]
- 2. Fang G., Li W., Shen X., Perez‐Aguilar J. M., Chong Y., Gao X., Chai Z., Chen C., Ge C., Zhou R., Nat. Commun. 2018, 9, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chong Y., Dai X., Fang G., Wu R., Zhao L., Ma X., Tian X., Lee S., Zhang C., Chen C., Chai Z., Ge C., Zhou R., Nat. Commun. 2018, 9, 4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. S. Selcuk , Selloni A., Nat. Mater. 2016, 15, 1107. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L., Mohamed H. H., Dillert R., Bahnemann D., J. Photochem. Photobiol., C 2012, 13, 263. [Google Scholar]
- 6. Jańczyk A., Wolnicka‐Głubisz A., Urbanska K., Kisch H., Stochel G., Macyk W., Free Radicals Biol. Med. 2008, 44, 1120. [DOI] [PubMed] [Google Scholar]
- 7. Yu a) B., , Wei H., He Q., Ferreira C. A., Kutyreff C. J., Ni D., Rosenkrans Z. T., Cheng L., Yu F., Engle J. W., Lan X., Cai W., Angew. Chem., Int. Ed. 2018, 57, 218; [DOI] [PMC free article] [PubMed] [Google Scholar]; Yu b) B., Wei H., He Q., Ferreira C. A., Kutyreff C. J., Ni D., Rosenkrans Z. T., Cheng L., Yu F., Engle J. W., Lan X., Cai W., Angew. Chem. 2018, 130, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ehlerding E. B., Lacognata S., Jiang D., Ferreira C. A., Goel S., Hernandez R., Jeffery J. J., Theuer C. P., Cai W., Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meng Z., Chao Y., Zhou X., Liang C., Liu J., Zhang R., Cheng L., Yang K., Pan W., Zhu M., Liu Z., ACS Nano 2018, 12, 9412. [DOI] [PubMed] [Google Scholar]
- 10. Silberstein E. B., Semin. Nucl. Med. 2012, 42, 164. [DOI] [PubMed] [Google Scholar]
- 11. Buchegger F., Perillo‐Adamer F., Dupertuis Y. M., Delaloye A. B., Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1352. [DOI] [PubMed] [Google Scholar]
- 12. Goel S., England C. G., Chen F., Cai W., Adv. Drug Delivery Rev. 2017, 113, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cornelissen B., Vallis K. A., Curr. Drug Discovery Technol. 2010, 7, 263. [DOI] [PubMed] [Google Scholar]
- 14. Ni K., Lan G., Veroneau S., Duan X., Song Y., Lin W., Nat. Commun. 2018, 9, 4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azzam E. I., Jay‐Gerin J., Pain D., Cancer Lett. 2012, 327, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou Z., Chang H., Li H., Wang S., Apoptosis 2017, 22, 1321. [DOI] [PubMed] [Google Scholar]
- 17. Le Caër S., Water 2011, 3, 235. [Google Scholar]
- 18. Alotiby M., Greguric I., Kibédi T., Lee B. Q., Roberts M., Stuchbery A. E., Tee P., Tornyi T., Vos M., Phys. Med. Biol. 2018, 63, 06NT04. [DOI] [PubMed] [Google Scholar]
- 19.a) Jiang H., Cuan Q., Wen C., Xing J., Wu D., Gong X., Li C., Yang H., Angew. Chem., Int. Ed. 2011, 50, 3764; [DOI] [PubMed] [Google Scholar]; b) Jiang H., Cuan Q., Wen C., Xing J., Wu D., Gong X., Li C., Yang H., Angew. Chem. 2011, 123, 3848. [DOI] [PubMed] [Google Scholar]
- 20. Yang H., Sun C., Qiao S., Zou J., Liu G., Smith S. C., Cheng H., Lu G., Nature 2008, 453, 638. [DOI] [PubMed] [Google Scholar]
- 21. Yang B., Chen Y., Shi J., Adv. Mater. 2019, 31, 1901778. [Google Scholar]
- 22. Rosenblum D., Joshi N., Tao W., Karp J. M., Peer D., Nat. Commun. 2018, 9, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai K., Hou Y., Hu Y., Zhao L., Luo Z., Shi Y., Lai M., Yang W., Liu P., Small 2011, 7, 3026. [DOI] [PubMed] [Google Scholar]
- 24. Dinh C., Nguyen T., Kleitz F., Do T., ACS Nano 2009, 3, 3737. [DOI] [PubMed] [Google Scholar]
- 25. Stuart B., Infrared Spectroscopy: Fundamentals and Applications, Vol. 9 (Ed: Ando D. J.) Wiley, Chichester: 2004, p. 45. [Google Scholar]
- 26. Sun G., Wang T., Li X., Li D., Peng Y., Wang X., Jia G., Su W., Cheng C., Yang J., Zuo C., Adv. Healthcare Mater. 2018, 7, 1800375. [DOI] [PubMed] [Google Scholar]
- 27. Priebe J. B., Radnik J., Lennox A. J. J., Pohl M., Karnahl M., Hollmann D., Grabow K., Bentrup U., Junge H., Beller M., Brückner A., ACS Catal. 2015, 5, 2137. [Google Scholar]
- 28. Ji Z., Jiang Y., Tian S., Guo F., Peng R., Xu F., Sun H., Fan J., Wang J., Int. J. Radiat. Oncol., Biol., Phys. 2019, 103, 638. [DOI] [PubMed] [Google Scholar]
- 29. Luo Y., Liu Z., Ye P., Fu Z., Lu F., Suleiman A. A., Liao J., Xiao J., J. Gastroenterol. Hepatol. 2016, 31, 1076. [DOI] [PubMed] [Google Scholar]
- 30. Luo J., Zhang Z., Liu Q., Zhang W., Wang J., Yan Z., Hepatol. Int. 2016, 10, 185. [DOI] [PubMed] [Google Scholar]
- 31. Yorozu A., Kuroiwa N., Takahashi A., Toya K., Saito S., Nishiyama T., Yagi Y., Tanaka T., Shiraishi Y., Ohashi T., Brachytherapy 2015, 14, 111. [DOI] [PubMed] [Google Scholar]
- 32. Yang M., Yan Z., Luo J., Liu Q., Zhang W., Ma J., Zhang Z., Yu T., Zhao Q., Liu L., Brachytherapy 2016, 15, 859. [DOI] [PubMed] [Google Scholar]
- 33. Zou Y., Li W., Zheng F., Li F., Huang H., Du J., Liu H., World J. Gastroenterol. 2010, 16, 5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J., Jiang Y., Li J., Tian S., Ran W., Xiu D., J. Exp. Clin. Cancer Res. 2009, 28, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verheij M., Bartelink H., Cell Tissue Res. 2000, 301, 133. [DOI] [PubMed] [Google Scholar]
- 36. Dewey W. C., Ling C. C., Meyn R. E., Int. J. Radiat. Oncol. , Biol. , Phys. 1995, 33, 781. [DOI] [PubMed] [Google Scholar]
- 37. Watson N. C., Di Y. M., Orr M. S., Fornari F. A. Jr., Randolph J. K., Magnet K. J., Jain P. T., Gewirtz D. A., Int. J. Radiat. Biol. 1997, 72, 547. [DOI] [PubMed] [Google Scholar]
- 38. Boehm E. M., Gildenberg M. S., Washington M. T., Enzymes 2016, 39, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tian Y., Xie Q., He J., Luo X., Zhou T., Liu Y., Huang Z., Tian Y., Sun D., Yao K., BMC Cancer 2015, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogawa Y., Cancers 2016, 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Babaei M., Ganjalikhani M., Bioimpacts 2014, 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang C., Zhao K., Bu W., Ni D., Liu Y., Feng J., Shi J., Angew. Chem., Int. Ed. 2015, 54, 1770. [DOI] [PubMed] [Google Scholar]
- 43. Wang H., Lv B., Tang Z., Zhang M., Ge W., Liu Y., He X., Zhao K., Zheng X., He M., Bu W., Nano Lett. 2018, 18, 5768. [DOI] [PubMed] [Google Scholar]
- 44. Fan W., Huang P., Chen X., Chem. Soc. Rev. 2016, 45, 6488. [DOI] [PubMed] [Google Scholar]
- 45. Lucky S., Soo K., Zhang Y., Chem. Rev. 2015, 115, 1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information