

Diseases caused by pathogenic mutations in mitochondrial DNA (mtDNA) often lead to severe, multisystem complications and death during childhood or adolescence, and in some cases, adult onset can lead to premature death. Researchers have proposed techniques to prevent the transmission of mtDNA disease through mtDNA replacement therapies that involve combining healthy nuclear and mtDNA from three individuals. This past February, the United Kingdom became the first country to legalize mtDNA replacement (the United States continues to consider the ethical and social implications). Although mitochondrial and nuclear genomes are physically separate in the cell, they work together functionally to control various metabolic and developmental processes, including energy production, cell growth, programmed cell death, and thermogenesis. This intergenomic relationship raises questions about possible effects of different mtDNAs (those that are not the original mtDNAs in a given cell) on cellular bioenergetics and disease susceptibility (see the figure). Recent studies in mice that have examined this issue suggest that different mtDNA and nuclear DNA combinations could plausibly have differential effects on gene expression and cell function.
Figure. Mitochondrial-nuclear DNA combinations.
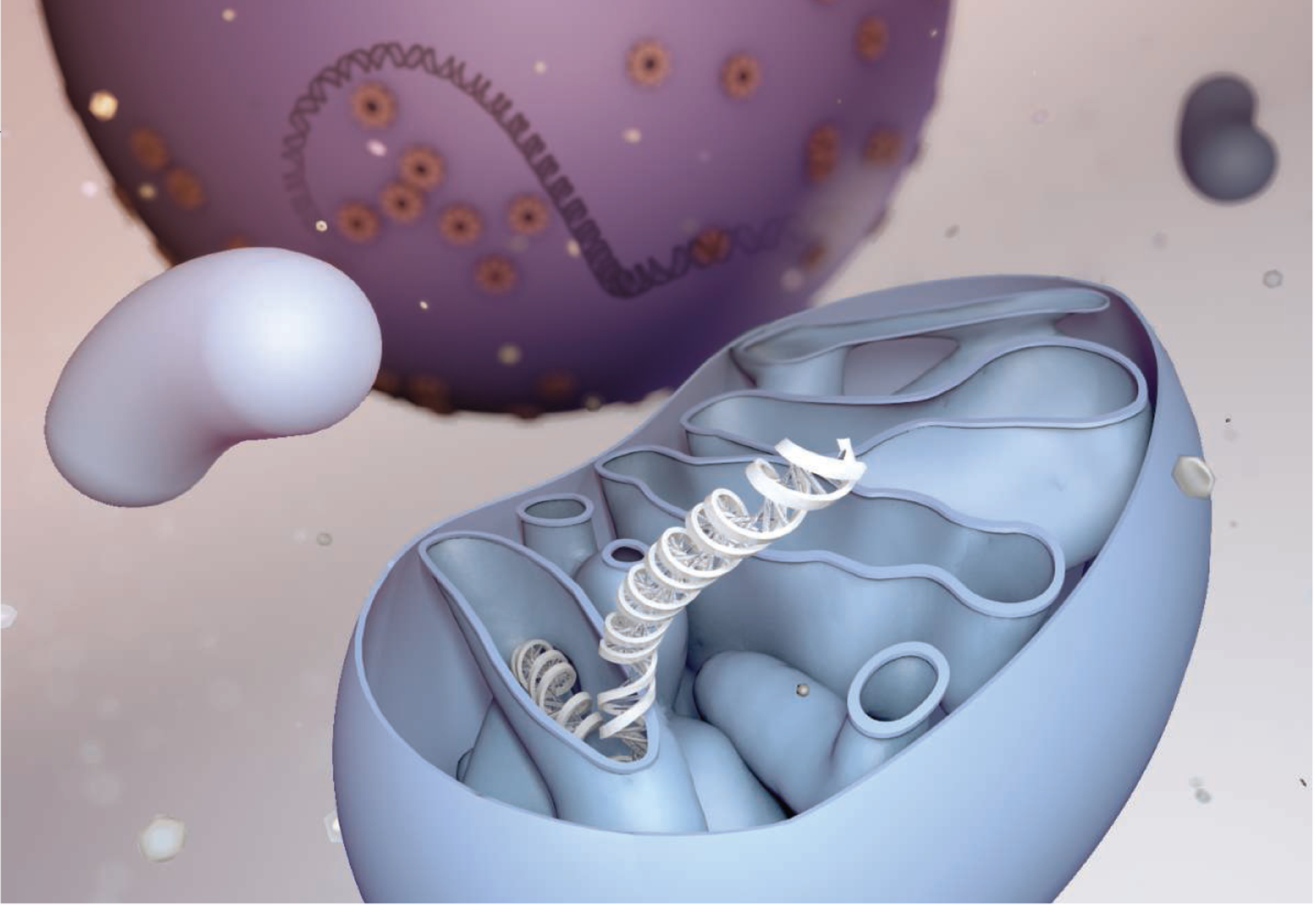
Processes such as pronuclear transfer that mismatch mtDNA and nuclear DNA could affect bioenergetics and mitochondrial-nuclear signaling pathways and elicit immunogenic responses.
In animal cells, mitochondria are the only organelles that contain their own genomes. A cell can have up to several thousand mitochondria, and each can contain 2 to 10 copies of mtDNA (there can be 100 to 10,000 separate copies of mtDNA per cell). Moreover, nearly all organisms have low levels of mtDNA variants, conferring some degree of heteroplasmy within and between tissues or organ systems of an individual. The onset and severity of mtDNA diseases are influenced by the type of pathogenic mtDNA mutation (missense, transfer RNA, ribosomal RNA, or deletions of multiple genes). Additional factors such as aging, exposure to toxic environmental substances, and gender can also play roles in disease progression.
The development of mitochondrial genetic therapies could drastically reduce or eliminate some forms of rare mitochondrial genetic disorders (1–9). Mitochondria are inherited solely from a mother through her eggs. A technique called pronuclear transfer (6) involves transferring the nuclear genome from the pronuclear-stage zygote (fertilized egg) of the affected woman (carrying pathogenic mtDNA) to an enucleated, healthy, recipient zygote, resulting in a fertile, reconstituted zygote containing the “normal” mtDNA of the recipient zygote and the transferred nuclear genome from the donor zygote (representing the nuclear genomes of the father and the mother with the pathogenic mtDNA). Another method called maternal spindle transfer can be performed with an unfertilized oocyte to generate an oocyte containing the nucleus from the female carrying the pathogenic mtDNA mutation and the “normal” mtDNA from the host oocyte, which can subsequently be fertilized in vitro (10). In either case, these techniques result in a so-called “three-parent embryo” containing nuclear genes from the male and affected female, and mtDNA from an unaffected female donor.
Ethical concerns surrounding these techniques (6, 11) certainly warrant discussion. At the same time, the biological effects of combining nuclear DNA with different mtDNA should be investigated. In mice, an approach quite similar to pronuclear transfer has been used to examine the effects of different mtDNAs on cellular bioenergetics and disease susceptibility. Mitochondrial-nuclear exchange (MNX) in mice is an approach still in early stages of examination (12,13). It involves the transfer of the nuclear genome from one mouse strain into an enucleated, recipient zygote of a different mouse strain. MNX mice that were generated by two strains with distinct susceptibilities to atherosclerosis and insulin resistance, for example, show that mtDNA genetic background affects oxygen utilization and responses to cardiac tissue injury. The MNX mice appear healthy and fertile, yet also have different levels of oxidative stress, resistance to a surgically induced model of heart failure, and altered lipid concentrations relative to control counterparts, depending upon the mtDNA–nuclear DNA combination (12, 13).
Another recent study in mice has examined the immunogenicity of “mismatched mitochondria” using an approach involving mouse embryonic stem cells (ESCs) that have been derived through somatic cell nuclear transfer (14). There have been conflicting results on the few studies that have examined transplantation of mitochondrial mismatched cells or tissues. In the recent study, the nucleus of a somatic cell is transferred into an enucleated oocyte. Nuclear-transferderived embryonic stem cells (NT-ESCs) can then be generated from such an oocyte. NT-ESCs thus acquire “healthy” mitochondria from the oocyte donor. The study found that NT-ESCs harboring the nuclear DNA of one mouse strain and the mtDNA from another mouse strain remained pluripotent. However, when transplanted into the thighs of mice, those NT-ESCs with “mismatched mitochondria” to the recipient animal possessed alloantigenicity and were subject to immune rejection (14). Whether the characteristics reported in the MNX mice or with NT-ESCs would be found with the maternal spindle transfer approach is not known.
Because mitochondrial-nuclear communication and interaction are part of normal cell function, and because normal biological reproduction allows for the coevolution of nuclear and mitochondria genomes (e.g., Mendelian and mitochondrial genetic selection occur simultaneously during meiosis), processes such as pronuclear transfer have the potential to alter this form of evolutionary selection and adaptation and therefore may have unintended effects on cellular bioenergetics and mitochondrial–nuclear signaling pathways. The introduction of “new” mtDNA to a nucleus after Mendelian selection, whereby allelic selection and gene expression were calibrated by a different mtDNA, bypasses aspects of Mendelian-mitochondrial evolution, or “mito-Mendelian” genetics. It therefore seems prudent to consider matching mtDNA haplogroup (minus the pathogenic mutation) of donor and recipient cells to minimize the possibility of altered interactions between the nucleus and the mitochondrion that may influence susceptibility to diseases of metabolism. In this respect, mtDNA haplogroup can influence penetrance of mtDNA mutations that cause Leber hereditary optic neuropathy (15).
Therapies based on mtDNA replacement represent important medical advances. However, evaluating the characteristics of the donor mitochondrial genomes seems reasonable as it could provide valuable insights for optimizing such therapies.
REFERENCES
- 1.Amato P, Tachibana M, Sparman M, Mitalipov S, Fertil. Steril 101, 31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bredenoord AL, Pennings G, de Wert G, Hum. Reprod. Update 14, 669 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Chiaratti MR, Meirelles FV, Wells D, Poulton J, Mitochondrion 11, 820 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Poulton J, Oakeshott P, BMJ 345, e6651 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Reinhardt K, Dowling DK, Morrow EH, Science 341, 1345 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Rubenstein DS et al. , Camb. Q. Healthc. Ethics 4, 316 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Tachibana M et al. , Nature 461, 367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavare A, BMJ 344, e540 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Craven L et al. , Nature 465, 82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tachibana M et al. , Nature 493, 627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baylis F, Reprod. Biomed. Online 26, 531 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Betancourt AM et al. , Biochem. J 461, 223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fetterman JL et al. , Biochem. J 455, 157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deuse T et al. , Cell Stem Cell 16, 33 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Torroni A et al. , Am. J. Hum. Genet 60, 1107 (1997). [PMC free article] [PubMed] [Google Scholar]