

Abstract
Tyrosine biosynthesis via the shikimate pathway is absent in humans and other animals, making it an attractive target for next-generation antibiotics, which is increasingly important due to the looming proliferation of multidrug-resistant pathogens. Tyrosine biosynthesis is also of commercial importance for the environmentally friendly production of numerous compounds, such as pharmaceuticals, opioids, aromatic polymers, and petrochemical aromatics. Prephenate dehydrogenase (PDH) catalyzes the penultimate step of tyrosine biosynthesis in bacteria: the oxidative decarboxylation of prephenate to 4-hydroxyphenylpyruvate. The majority of PDHs are competitively inhibited by tyrosine and consist of a nucleotide-binding domain and a dimerization domain. Certain PDHs, including several from pathogens on the WHO priority list of antibiotic-resistant bacteria, possess an additional ACT domain. However, biochemical and structural knowledge was lacking for these enzymes. In this study, we successfully established a recombinant protein expression system for PDH from Bacillus anthracis (BaPDH), the causative agent of anthrax, and determined the structure of a BaPDH ternary complex with NAD+ and tyrosine, a binary complex with tyrosine, and a structure of an isolated ACT domain dimer. We also conducted detailed kinetic and biophysical analyses of the enzyme. We show that BaPDH is allosterically regulated by tyrosine binding to the ACT domains, resulting in an asymmetric conformation of the BaDPH dimer that sterically prevents prephenate binding to either active site. The presented mode of allosteric inhibition is unique compared to both the competitive inhibition established for other PDHs and to the allosteric mechanisms for other ACT-containing enzymes. This study provides new structural and mechanistic insights that advance our understanding of tyrosine biosynthesis in bacteria.
Keywords: Prephenate dehydrogenase, tyrosine biosynthesis, Bacillus anthracis, ACT domain, allosteric regulation
Graphical abstract
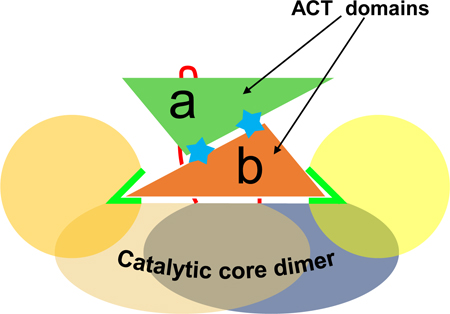
Bacterial enzymes of tyrosine biosynthesis are promising targets for the development of new antibiotics. Here, we report a structural and biophysical analyses of prephenate dehydrogenase (PDH) from Bacillus anthracis, which demonstrates a novel mechanism of inhibition by tyrosine. Tyrosine binds to the ACT domains and triggers their asymmetric positioning on the catalytic core, with both active sites blocked by the ACT domain of only one subunit.
Introduction
Many organisms, including bacteria, fungi, and plants, synthesize aromatic amino acids, but these pathways are absent in animals [1]. Therefore, the enzymes of these pathways are promising targets for the development of new antibiotics – an increasingly important mission due to the rise of antibiotic resistance in recent years [2]. Additionally, aromatic amino acids and their derivatives are used in the production of pharmaceuticals, opioids, food additives, dietary supplements, aromatic polymers, and petrochemical aromatics, which makes the pathways that produce them highly attractive for environmentally-friendly biotechnological applications [3, 4]. For many plants and microorganisms, these anabolic pathways start with the shikimate pathway to produce chorismate, a precursor for the biosynthesis of aromatic amino acids and other aromatic compounds, including secondary metabolites in plants, siderophores in microorganisms, and certain antibiotics in fungi [5–7]. The biosynthesis of tyrosine (Figure 1), which is a precursor for a variety of aromatic compounds, stems from the Claisen rearrangement of chorismate to prephenate [8, 9]. Prephenate then enters either one of two alternative routes of tyrosine biosynthesis. The first route starts with the oxidative decarboxylation of prephenate to 4-hydroxyphenylpyruvate, which subsequently undergoes transamination to tyrosine [10]. The second route starts with the transamination of prephenate to arogenate, which then undergoes oxidative decarboxylation to tyrosine [11, 12]. In both routes, oxidative decarboxylation is catalyzed by a TyrA family dehydrogenase, most of which are regulated by the cellular concentration of tyrosine [13].
Figure 1. The two routes of tyrosine biosynthesis from chorismate and the reaction catalyzed by the prephenate dehydrogenase (PDH).

Classification of the TyrA family of enzymes is based on substrate utilization: prephenate dehydrogenase (PDH, EC database: 1.3.1.12) utilizes prephenate, arogenate dehydrogenase utilizes arogenate, and cyclohexadienyl dehydrogenase can utilize both prephenate and arogenate [13]. The biochemical analyses of some of these enzyme classes have been described elsewhere [14–20], and there are multiple studies of the TyrA family from the phylogenetic perspective [13, 16, 18, 21], including a dedicated website (http://aropath.org/TyrPath/index.html) [13]. However, structural knowledge of this family is rather limited. The structures of the TyrA family proteins have also been determined only for a few species: PDHs from bacteria Aquifex aeolicus (PDB ID: 2G5C, 3GGG, 3GGO, and 3GGP) [22, 23], Streptococcus thermophilus (3DZB), Streptococcus mutans (3B1F) [24], Haemophilus influenzae (PDB ID: 2PV7) [25], and Corynebacterium glutamicum (3KTD); PDH from plant Glycine max (PDB ID: 5T9F, 5T9E, 5T95, and 5WHX) [18]; arogenate dehydrogenase from cyanobacteria Synechocystis sp. (PDB ID: 2F1K) [17]; and a putative cyclohexadienyl dehydrogenase from bacteria Sinorhizobium meliloti (PDB ID: 4WJI). Each monomer of these homodimeric proteins consists of two structural domains: an N-terminal nucleotide-binding domain and a C-terminal dimerization domain [13, 17, 18, 22, 23]. Structural, biochemical, and mutagenic analyses of some of the bacterial PDHs have shown that tyrosine functions as a competitive inhibitor to the enzyme by binding to the same site as prephenate [19, 22, 23, 26]. Conversely, a recent study of TyrA proteins from legumes, the only land plants found to contain a functional PDH [27], showed that their PDHs are insensitive to tyrosine [18].
Phylogenetic analysis of the TyrA family of enzymes indicated that the majority of PDHs are represented solely by the two domains, referred to as the catalytic core [13, 16]. However, some PDHs possess an ACT allosteric regulatory domain in addition to their catalytic core. The ACT domain was discovered by bioinformatics searches in 1999 and was named after three of the proteins that contain it: aspartate kinase, chorismate mutase, and TyrA [28–30]. The ACT-containing PDHs are observed mainly in low-GC Gram-positive bacteria and, to a lesser extent, in some Archaea [13]. This cluster of bacteria contains such genera as Bacillus, Staphylococcus, Listeria, Enterococcus, and Streptococcus, many of which include highly pathogenic species. Five species with ACT-containing PDHs (Enterococcus faecium, Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumonia) are listed among the 13 pathogens on the World Health Organization priority list for research and development of new antibiotics [31]. However, no structures of the ACT-containing PDHs have been reported prior to this study, and the biochemical data on these enzymes are very sparse.
An example of an ACT-containing TyrA family member is PDH from the low-GC Gram-positive bacterium Bacillus anthracis. B. anthracis is the causative agent for anthrax, a common disease in animals and, to a lesser extent, humans. It is highly infectious in the inhaled form and can result in high mortality if it goes untreated [32–34]. The shikimate pathway is essential in this organism since it leads to the production of important compounds for growth, microbial communication, and anaerobic respiration. It is expected that any perturbation of the shikimate pathway will affect the synthesis of the aromatic amino acids and thus the ability for the organism to grow [35, 36]. For example, menaquinone, an essential component of the electron transport chain, is synthesized downstream of chorismate [37, 38]. Disruption of menaquinone in certain Gram-positive microbes has been associated with attenuated virulence in murine models [39]. The shikimate pathway also plays an important role in the synthesis of catechol-modified siderophores, petrobactin, and bacillibactin [40–42]. B. anthracis cannot always obtain and utilize tyrosine from its environment and thus must rely on its own synthesis [43, 44]. The importance of this pathway and the lack of a similar pathway in humans make PDH an attractive drug target for disrupting B. anthracis and similar pathogens’ growth with minimal foreseeable adverse effects for humans.
We present three crystal structures and biochemical analyses of the ACT-containing PDH from B. anthracis (BaPDH). The three-dimensional structures reveal the position of the ACT domains relative to the catalytic core and, together with the results of biochemical experiments, provide structural insights on how tyrosine binding to the ACT domain allosterically modulates the catalytic efficiency of BaPDH. The discovered mode of allosteric inhibition by tyrosine is novel compared to the competitive inhibition model established for other PDH enzymes [19, 22]. Moreover, the observed mechanism of inhibition by the ACT domain differs from any of the previously reported ACT-containing enzymes [45–49].
Results
Structure of BaPDH complexed with tyrosine and NAD+
The structure of the BaPDH ternary complex with tyrosine and NAD+ at 2.2 Å resolution (PDB ID: 6U60) was obtained by treatment of the complex with chymotrypsin prior to crystallization (Tables 1, 2); crystals obtained without the protease treatment diffracted poorly (see Materials and Methods). This crystal form has one BaPDH homodimer in the asymmetric unit (ASU). Each BaPDH subunit consists of three domains: a nucleotide-binding domain (residues 1–185), a dimerization (186–295) domain, and an ACT domain (307–378) that is connected to the dimerization domain through a linker loop (296–306) (Figures 2, 3). Importantly, BaPDH subunits adopt distinct conformations due to the different positions of their ACT domains: subunits A and B are in the open and closed conformations, respectively (Figure 3B). Two tyrosine molecules are found at the interface between the ACT domains, yet only subunit B has NAD+ bound. The substrate-binding pocket, located between the nucleotide-binding and the dimerization domains, is ligand-free in both subunits.
Table 1.
Crystallization and cryoprotection conditions.
| Complex with NAD+ and tyrosine |
Complex with tyrosine |
ACT domain | |
|---|---|---|---|
| PDB ID: | 6U60 | 5UYY | 5V0S |
| Protein | 12 mg/mL, wild-type, His-tag removed, treated with 1/40 v/v 2 mg/mL chymotrypsin | 19 mg/mL, wild-type, His-tag removed | 15 mg/mL, His-tagged SeMET derivative treated with 1/40 v/v 2 mg/mL chymotrypsin |
| Protein buffer | 20 mM HEPES pH=7.5, 150 mM NaCl, 5% v/v glycerol, 10 mM β-ME | 20 mM HEPES pH=7.5, 500 mM NaCl, 5% v/v glycerol, 10 mM β-ME | |
| Additives | 5 mM NAD+ 20 mM Tyrosine pH 7.0 |
None | None |
| Precipitant mix | 0.1 M K2HPO4 / NaH2PO4 pH=6.2, 0.2 M NaCl, 20% w/v PEG 1000 (MCSG Suite 2 #81) | 0.1 M MES pH=6.5, 0.2 M MgCl2, 10% w/v PEG 4000 (MCSG Suite 1 #11) | 0.1 M Tris pH=8.5, 0.2 M CaCl2, 25% w/v PEG 4000 (MCSG Suite 1 #31) |
| Cryoprotection | Drying over 1 M NaCl | Paratone-N | Paratone-N |
Table 2. Data collection and structure refinement statistics.
Values in parentheses are for the highest resolution shell. Clashscore, rotamer outliers, and Ramachandran plot statistics are calculated by MolProbity.
| Complex with NAD+ and tyrosine |
Complex with tyrosine |
ACT domain | |
|---|---|---|---|
| PDB ID: | 6U60 | 5UYY | 5V0S |
| Diffraction images DOI | 10.18430/M35USC | 10.18430/M35UYY | 10.18430/M35V0S |
| Data collection | |||
| Resolution range (Å) + | 50.00 – 2.10 (2.14 – 2.20) |
50.00 – 2.60 (2.64 – 2.60) |
50.00 – 2.00 (2.03 – 2.00) |
| Beamline | 21ID-G | 21ID-G | 21ID-F |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 |
| Space group | P21212 | P212121 | P6522 |
| Unit-cell dimensions a, b, c (Å) | 106.9, 81.0, 83.6 | 84.7, 105.6, 179.6 | 48.1, 48.1, 233.4 |
| Protein chains in the ASU | 2 | 4 | 2 |
| Completeness (%) | 99.8 (99.3) | 99.9 (100.0) | 96.9 (88.1) |
| Number of unique reflections | 42991 | 50884 | 11294 |
| Redundancy | 8.5 (6.7) | 7.4 (7.5) | 7.4 (7.5) |
| <I>/<σ(I)> | 27.8 (1.6) | 31.3 (2.1) | 32.5 (1.7) |
| CC 1/2 - highest resolution shell | (0.63) | (0.80) | (0.84) |
| Rmerge# | 0.077 (1.305) | 0.069 (0.963) | 0.053 (0.619) |
| Structure refinement | |||
| Resolution range (Å) | 36.48 – 2.10 (2.16 – 2.10) |
50.00 – 2.60 (2.67 – 2.60) |
41.63 – 2.00 (2.06 – 2.00) |
| Completeness (%) | 93.4 (61.3) | 99.9 (100) | 96.7 (85.6) |
| Number of reflections, working set | 38244 (1839) | 45368 (3486) | 9983 (657) |
| Number of reflections, test set | 1975 (95) | 2466 (174) | 565 (33) |
| Rwork* | 0.160 (0.234) | 0.189 (0.283) | 0.171 (0.262) |
| Rfree* | 0.220 (0.258) | 0.246 (0.359) | 0.232 (0.232) |
| Bond lengths rmsd (Å) | 0.008 | 0.016 | 0.011 |
| Bond angles rmsd (°) | 1.5 | 1.7 | 1.5 |
| Missing amino acid residues | a/1–13 b/1–13 |
a/1–5; b/1–13 c/1–13; d/1–5 |
a/1–306, 378–378 b/1–306, 378–378 |
| Number of protein atoms in the model | 5729 | 11384 | 1060 |
| Mean ADP value for protein (Ų) | 37 | 77 | 53 |
| Number of water molecules | 370 | 506 | 73 |
| Mean ADP for water molecules (Ų) | 36 | 62 | 62 |
| Mean ADP for NAD+ (Ų) | 29 | - | - |
| Mean ADP for tyrosine (Ų) | 18 | 48 | - |
| Clashscore/Clashscore percentile (%) | 2.7/88.5 | 2.1/100 | 1.4/100 |
| Rotamer outliers (%) | 1.31 | 2.23 | 2.83 |
| Ramachandran outliers (%) | 0 | 0 | 0 |
| Ramachandran favored (%) | 97.66 | 98.64 | 100 |
| MolProbity score | 1.22 | 1.25 | 1.22 |
, where ‹I(hkl)› is the mean of I observations Ii(hkl) of reflection hkl.
, where Fo and Fc are the observed and calculated structure factors, respectively, calculated for working set (Rwork) and for 5% of the data omitted from refinement – test set (Rfree).
Figure 2. Overall structure of BaPDH (PDB ID: 6U60) and comparison to AaPDH (PDB ID: 3GGG).

(A) Cartoon representation of BaPDH dimer with bound tyrosine and NAD+ ligands (carbons atoms shown in cyan). Domains are labeled with the subunits in parenthesis (a, b). Regulatory loops are shown by distinct colors: lasso (134–148) in green for both subunits, linker (296–306) and latch (340–348) in red only for subunit B. (B) Superimposition of BaPDH (colored) subunit onto AaPDH (grey) by dimerization domains. Loops and ligands are colored according to panel A. (C) Superimposition of BaPDH (yellow) and AaPDH (grey) nucleotide-binding domains. Lasso loop (134–148) is shown in green. (D) AaPDH nucleotide-binding site. Hydrogen bonds are shown as black dashed lines. (E) BaPDH nucleotide-binding site. The figure was prepared with PyMOL. The BaPDH structures and the quality of respective electron density maps can be inspected using Molstack interactive figures available at https://molstack.bioreproducibility.org/collection/view/gYbn50vTdft3AyFC5nFw/.
Figure 3. ACT domains of BaPDH: their organization, asymmetry, and tyrosine binding.

Domains and ligands are colored according to the Figure 2. (A) Cartoon representation of ACT domains’ asymmetry (light green and orange) on top of the BaPDH dimer in the ternary complex (PDB ID: 6U60). Regulatory loops are shown by distinct colors: lasso (134–148) in green for both subunits, linker (296–306) and latch (340–348) in red only for subunit B. The hypothetical two-fold axis linking the two catalytic cores is perpendicular to the plane of the figure. This figure is rotated about the horizontal axis by 35 degrees from Figure 2A. (B) Superposition of BaPDH subunits (PDB ID: 6U60) by the catalytic cores show the difference between the open and closed conformations. Linker loops (296–306) of both subunits A and B are shown in red. (C) Cartoon representation of ACT dimer in the ternary BaPDH complex (PDB ID: 6U60) shows the location of tyrosine-binding pockets and regulatory loops. Linker (296–306) and latch loop (340–348) of subunit B are shown in red, whereas subunit A loops are shown in purple. (D) Superposition of ACT domains from the ternary BaPDH complex (PDB ID: 6U60). (E) ACT dimer as crystallized without the catalytic core (PDB ID: 5V0S). (F, G) Tyrosine-binding pockets in BaPDH ternary complex (PDB ID: 6U60). Hydrogen bonds are shown as black dashed lines. Colors of carbon atoms are according to the colors of domain on other panels. The figure was prepared with PyMOL. The structures and the respective electron density maps can be inspected using Molstack interactive figures available at https://molstack.bioreproducibility.org/collection/view/gYbn50vTdft3AyFC5nFw/.
The organization of the two catalytic cores produces a dimer with the dumbbell structure seen for other PDHs (Figure 2B). Comparison of the BaPDH crystal structure with that of A. aeolicus prephenate dehydrogenase (AaPDH, PDB ID: 3GGG), which is a well-studied bacterial PDH from both structural and biochemical approaches [14, 22], indicates that the arrangement of both the nucleotide-binding and dimerization domains are highly similar (Figures 2B, C). The BaPDH helical dimerization domains are tightly intertangled with each other in the same way as in AaPDH: through pairwise interactions of three helices from the middle portion of each subunit. Based on this organization of secondary structure elements, it is evident that the dimer is the minimal oligomeric state of the protein in solution. Size-exclusion chromatography results (data not shown) and PISA server [50] analysis of the buried interdomain interface area are consistent with BaPDH existing as a homodimer in solution.
The N-terminal nucleotide-binding domain has a modified Rossmann nucleotide-binding fold with an extended β-sheet sandwiched by three helices on each face (Figure 2C). The extended β-sheet is formed by six parallel strands on one side of the sheet and two antiparallel strands on the other. Similar to AaPDH, a large lasso loop (134–148) contributes to one face of the nucleotide-binding site and makes up a significant portion of the substrate-binding site (Figures 2C, D, E). The nucleotide-binding site of subunit B in BaPDH is arranged similarly to that of AaPDH. NAD+ adopts the same conformation and is bound by a similar set of hydrogen bonds in both structures (Figures 2C, D, E). The ribose of the adenosine moiety is bound by two hydrogen bonds with the conserved, negatively charged side-chain of D45, suggesting the enzyme has a strong preference for NAD+. Binding of the 2’-phosphate of NADP+ is likely prevented due to the steric interference and the negative charge of D45, as is observed in many TyrA proteins [13, 51] and other NAD+/NADP+-dependent dehydrogenases [52]. Altogether, the overall fold of BaPDH’s catalytic core is highly similar to other bacterial PDHs as represented by AaPDH [22] (Figure 2), as well as to PDHs from legumes [18].
ACT domains add asymmetry to the BaPDH dimer
The ACT domain is the main differentiating feature of BaPDH. The ACT domains of the dimer are sandwiched between the two N-terminal nucleotide-binding domains above the dimerization domains (Figures 2A, 3A). In the dimeric structure, the pair of ACT domains come together to form a six-stranded antiparallel β-sheet flanked by four helices on one face, with the other face positioned on the helical dimerization domains of BaPDH (Figures 2A, 3A). Without the ACT domains, the two catalytic cores of the dimer are related by a non-crystallographic two-fold symmetry (Figure 3A). However, the ACT domains with bound tyrosine are positioned differently in relation to their respective catalytic cores and disrupt this symmetry (Figure 3B). As shown by the superposition, subunit A (without NAD+) is in the open conformation, and subunit B (with NAD+) is in the closed conformation. The difference in the positioning of the ACT domains between subunits is enabled by the difference in the conformation of the linker loop (Figure 3B). Importantly, both ACT domains cannot simultaneously be in the closed conformation because they would virtually occupy the same space (Figures 3A, B).
There are two bound tyrosine molecules per ACT dimer. In the BaPDH ternary complex, each tyrosine molecule is bound between the two ACT domains and one of the nucleotide-binding domains (Figures 2A, 3A). The tyrosine molecules are bound mainly to the ACT domains via their carboxyl and amino groups (Figures 3F, G). The hydroxyl groups of the tyrosine molecules also interact with the nucleotide-binding domains, but they interact with different residues due to the subunits’ different conformations. In subunit A, the tyrosine hydroxyl group is H-bonded by a/N152 of the same subunit (Figure 3F). In subunit B, it is H-bonded by b/K139 of the same subunit and by a/Q341 of subunit A (Figure 3G).
Superposition of the ACT domains from the subunits of the dimer shows three significant differences in their organization (Figures 3C, D). First, the conformation of the linker loop that connects the ACT domain to the catalytic core is drastically different in chains A and B, which is required for the asymmetric positioning of the ACT domains (Figures 3B, D). Interestingly, the first residue of the linker loop, P296, serves as a hinge for the movement of the loop; this residue is in cis-conformation in subunit A (open conformation) and in trans-conformation in subunit B (closed conformation). Second, a long “latch” loop (340–348), which latches the catalytic core of subunit A (Figure 2A), also occupies very distinct conformations in these subunits (Figure 3D). Third, the ACT domain of subunit A is characterized by longer β-strands compared to subunit B (Figure 3C).
In contrast, the structure of the isolated ACT dimer (307–375, PDB ID: 5V0S), obtained with chymotrypsin treatment of the SeMet-BaPDH (Tables 1, 2), displays a highly symmetrical organization (Figure 3E). The structure of this proteolyzed ACT domain does not contain bound tyrosine (Figure 3E), suggesting that H-bonding by the residues from the nucleotide-binding domain is required for the formation of the high-affinity tyrosine-binding pocket (Figures 3F, G). In this structure, both ACT domains adopt a similar conformation to subunit A in the ternary complex.
Tyrosine binding to ACT domains inactivates the enzyme by blocking both substrate-binding sites and one NAD+ site with the ACT domain of one subunit
Superposition of the BaPDH ternary complex (PDB ID: 6U60) with the structure of AaPDH complexed with NADH and 4-hydroxyphenylpyruvate (PDB ID: 3GGO) shows that the amino acid composition of BaPDH prephenate-binding site is very similar to that of AaPDH (Figures 4A, B), especially for amino acid residues that are important in substrate binding and those that participate in the enzyme’s catalytic mechanism [22]. Most of these residues are identical and superpose well, including S109, H132 (the catalytic histidine), S198, H199, H202, R235, S239, and W244. Multiple sequence alignment of bacterial PDHs shows that these residues are highly conserved in PDHs with and without the ACT domain (Figure 5A). It should be noted that legume PDHs are insensitive to tyrosine and have a significantly different arrangement of the active site, with the substrate carboxylic group bound by different types of residues [18]; therefore, we do not display comparison with legume PDHs. In AaPDH, tyrosine functions as a competitive inhibitor with respect to prephenate by binding directly to the active site, as observed in the AaPDH structure complexed with NAD+ and tyrosine (PDB ID: 3GGG). In contrast, tyrosine was not observed in the substrate-binding pockets of the BaPDH dimer despite a high concentration of tyrosine in the crystallization condition (20 mM).
Figure 4. One ACT domain in the inhibited state of BaPDH dimer blocks both substrate sites and one NAD+ site via its linker (296–306) and latch (340–346) loops.

(A) The latch loop of BaPDH ACT domain from subunit B (b/340–348, carbons in orange) blocks substrate binding in the active site of subunit A. Active site residues of BaPDH (PDB ID: 6U60) are shown with carbon atoms in yellow. Active site residues of AaPDH (PDB ID: 3GGO) are shown with carbon atoms in grey, 4-hydroxyphenylpyruvate in cyan. The structures and the superpositions can be inspected using Molstack interactive figures at https://molstack.bioreproducibility.org/collection/view/gYbn50vTdft3AyFC5nFw/ (B) The linker loop of BaPDH (PDB ID: 6U60) subunit B (b/296–306, carbons in orange) blocks substrate binding in the active site of the same subunit. (C) The lasso loop (a/b/c/134–148) in subunits B (green) and A (red) of the ternary complex (PDB ID: 6U60) and in subunit C (black) of the binary complex (PDB ID: 5UYY). In subunits A and C, the lasso loop is pushed by the latch loop of the ACT domain from subunit B (b/340–348) into the position expected to be occupied by the NAD+. In subunit B, the lasso loop is in the open conformation, permitting NAD+ to bind. (D) Speculative model of BaPDH transitioning from an active to the inhibited state upon allosteric binding of tyrosine to the ACT domains. Colors of domains are similar to those on Figures 2 and 3. Panels A, B, and C were prepared with PyMOL.
Figure 5. Sequence conservation analysis of PDH residues from the catalytic core and the ACT domain.

WebLogo online tool (https://weblogo.berkeley.edu/) was used to generate this sequence logo. The height of symbols indicates the relative frequency of each amino acid at that position. The colors of the symbols correspond to the acidic/basic/hydrophilic/hydrophobic properties of amino acids as shown on the color legend. N- and C-terminals are labelled with the respective letters. (A) Sequence logos for PDH active-site residues involved in substrate-binding and/or catalysis as compared between bacterial PDH sequences; residue numbers are labeled according to the BaPDH sequence. Only residues involved in substrate-binding and/or catalysis and their closest neighbors are shown. (B) Sequence logos for the ACT domain from bacterial PDHs. All residues in the range 303–378 are shown, residue numbers are according to the BaPDH sequence. Residues involved in tyrosine binding via side chains and G318 are labelled. All sequences used in the alignment are listed in the Supplementary file 1.
The superposition shows that tyrosine (and, for the same reasons, the substrate) cannot bind to BaPDH subunit A because the substrate-binding site is sterically blocked by the latch loop (340–348) of the ACT domain from subunit B (Figure 4A). The glutamine residue of the latch loop (E344) directly occupies the place that is occupied by 4-hydroxyphenylpyruvate (the product of the enzymatic reaction) in the AaPDH structure (Figure 4A). Moreover, E344 forms a hydrogen bond with the catalytic histidine (H132), further contributing to the arrest of the active site. In subunit B, which has a bound cofactor, the substrate-binding site is sterically blocked by P304 of the linker loop (296–306) of the same subunit (Figure 4B). This loop is wedged in the narrow active site, making its re-position sterically unlikely, unless the whole ACT domain is moved away. In addition, R235, which interacts with the carboxyl group of the substrate, is locked in a distant conformation and forms a salt bridge with D308 of the ACT domain; moreover, R235 is sterically blocked from orienting into the active site by Y306. Thus, the ACT domain of subunit B, which is in the closed conformation, blocks the substrate-binding site in both subunits with its latch and linker loops.
In addition, the asymmetric positioning of the ACT domains allows for NAD+ binding in subunit B but precludes its binding in subunit A. Despite the high excess of NAD+ in the crystallization condition (5 mM), NAD+ is found only in subunit B of the ternary complex. In subunit A, cofactor binding is precluded by the conformation of the lasso loop (a/134–148), which is shifted by ~10 Å (H138 Cα) compared to subunit B and places H138 directly in the position expected to be occupied by the pyrophosphate of NAD+ (Figure 4C). This large conformational shift of the lasso loop is likely modulated by the latch loop (b/340–348) of the ACT domain from the neighboring subunit B. As shown in Figure 3D, the position of the latch loop is vastly different in these subunits. The latch loop of subunit B is wedged in between the nucleotide-binding and the dimerization domains of subunit A (Figure 2A), shifting the lasso loop and preventing NAD+ binding (Figure 4C). In subunit A, the latch loop is resting on the surface of the protein, which leaves the lasso loop of subunit B in the open conformation and permits NAD+ to bind.
Structure of BaPDH binary complex with tyrosine is highly similar to the ternary complex
The structure of BaPDH in complex with tyrosine at 2.6 Å resolution (PDB ID: 5UYY) was obtained without the addition of tyrosine, indicating that tyrosine was bound to the protein in the cell and retained during purification. This crystal form has four protein molecules in the ASU composed of two identical dimers; tyrosine is bound to each of the four ACT domains (Tables 1, 2). Tyrosine is also bound to each ACT domain in another BaPDH structure obtained from a different purification experiment (SeMet-BaPDH, without addition of tyrosine), which crystallized in a different space group (3.0 Å, P21, data not shown).
Chains A and C of the binary complex are found in a similar open conformation as subunit A of the ternary complex. The only difference is the lasso loop (a/c/134–148), which has a slightly different conformation (K139 Cα shift by 4.3 Å): this loop is pushed even more into the cofactor-binding site as compared to the ternary complex (Figure 4C). This flexibility is allowed by the free space produced by the absence of the cofactor. Subunits B and D are found in the closed conformation, similar to subunit B in the structure of the ternary complex even though they lack the bound cofactor. The lasso loops of these subunits are in the open conformation, which leaves the NAD+ sites unobstructed. Structural analysis shows that NAD+ cannot bind to subunits A and C and that the substrate cannot bind in any of the four subunits for the same reasons as in the ternary complex. Based on this analysis, it is expected that the structure of the BaPDH binary complex with tyrosine represents the same inhibited conformation of the enzyme as is observed in the ternary complex.
Thermal denaturation studies show tyrosine stabilizes BaPDH
Thermal denaturation of BaPDH was undertaken to investigate the effect of different ligands on stabilizing the folded conformation of BaPDH (Table 3). In the absence of ligands, the protein denatures cooperatively in a one-step manner; the midpoint of the denaturation curve, which represents the melting temperature of the protein (Tm), is 41 ± 1°C. The addition of 10 mM tyrosine to BaPDH increased the Tm by 10 ± 2°C. NAD+ at 10 mM concentration does not impart a significant change to the Tm. Similarly, NADH, NADP+, and NADPH do not significantly change the Tm of BaPDH. A combination of either cofactor with tyrosine does not affect the Tm more so than that observed with tyrosine alone, except for NADH that results in slightly higher Tm.
Table 3.
Melting temperatures (Tm) of BaPDH and its complexes as determined by protein thermal shift assay. All ligands were used at 10 mM final concentration. ns – non-significant.
| Ligands | Tm, °C | Increase in Tm with the addition of the ligand(s), °C |
| Control (purified wild-type BaPDH) | 41 ± 1 | - |
| Tyrosine | 51 ± 1 | 10 ± 2 |
| NADH | 43 ± 2 | ns |
| NADH + tyrosine | 56 ± 1 | 15 ± 2 |
| NAD+ | 41 ± 1 | ns |
| NAD+ + tyrosine | 51 ± 2 | 11 ± 2 |
| NADP+ | 41 ± 1 | ns |
| NADP+ + tyrosine | 53 ± 1 | 12 ± 2 |
| NADPH | 40 ± 1 | ns |
| NADPH + tyrosine | 52 ± 1 | 12 ± 2 |
Binding of different ligands to BaPDH as measured by ITC
ITC studies were conducted to determine the stoichiometry and the affinity of binding of tyrosine, NAD+, and NADP+ to BaPDH. Multiple titrations of tyrosine to different batches of purified BaPDH showed high-affinity binding with a Kd of 0.64 ± 0.05 μM (Table 4). One molecule of tyrosine was titrated for each BaPDH dimer, indicating that tyrosine already occupied one binding site (e.g., subunit B, Figure 3G). Multiple rounds of dialysis did not result in an increase in the binding stoichiometry of tyrosine to the BaPDH dimer. The presence of tyrosine in the two independent structures obtained without addition of the inhibitor, the ability of the protein to bind only one tyrosine per dimer, and the differences between the tyrosine-binding sites in the BaPDH dimer support this finding.
Table 4.
BaPDH cofactor and tyrosine binding constants determined by ITC.
| ITC conditions | ITC results | |||
|---|---|---|---|---|
| Ligand (mM) | Protein (mM) | Titration | Binding sites per protein dimer(N) | Kd(μM) |
| Tyrosine(3.0) | BaPDH(0.5) | 0.5 μL for the first injection, 1.5 μL for the following 25 injections | 1.002 ± 0.001 | 0.64 ± 0.05 |
| Tyrosine(2.5) | BaPDH saturated with NAD+(0.415) | 0.928 ± 0.003 | 0.37 ± 0.04 | |
| NAD+(2.15) | BaPDH saturated with tyrosine(0.415) | 0.90 ± 0.01 | 68 ± 3 | |
| NAD+(5.9) | BaPDH(0.54) | Difficult to fit to the binding equation | ||
| NADP+(1.5) | BaPDH(0.14) | 1 μL for the first injection, 2.45 μL for the following 15 injections | No detectable binding | |
Titration of tyrosine to BaPDH saturated with NAD+ showed similar stoichiometry and slightly higher affinity as compared to titration to BaPDH without NAD+. These results suggest that tyrosine binding to the protein is not largely affected by the presence of the cofactor, which is consistent with the structural data and thermal denaturation studies. Structures with and without NAD+ showed no significant differences in their organizations, and thermal denaturation studies showed no significant cumulative stabilization effect when both NAD+ and tyrosine were added to BaPDH.
Titrations of NAD+ to purified BaPDH produced smooth binding curves; however, they were difficult to fit to any standard binding equations. We estimated the Kd for NAD+ binding to be over 500 μM (data not shown). Based on the presence of tyrosine in two independent structures obtained without the addition of exogenous tyrosine, the ability of the purified protein to bind additional tyrosine as shown by the ITC experiments described above, and the stabilizing effect of tyrosine on BaPDH Tm, we proposed that this Kd reflects NAD+ binding to various tyrosine-bound states of the protein and, therefore, is not interpretable. Titration of NAD+ to BaPDH saturated with tyrosine showed well-interpretable results consistent with the presence of the cofactor in only one subunit in the ternary complex structure: only one molecule of the cofactor bound to the BaPDH dimer with a Kd of 68 ± 3 μM. However, it should be noted that this Kd reflects NAD+ binding to only one site on the inhibited enzyme and, therefore, may not reflect the NAD+-binding properties of uninhibited BaPDH. There is no detectable binding for NADP+, which is consistent with the kinetic assay showing that NAD+ is the preferred cofactor for the enzyme and with the structure of the ternary complex showing that the ribose of the adenosine moiety of NAD+ is bound by the conserved D45 (Figure 2E).
Enzyme kinetics and the mode of BaPDH inhibition by tyrosine
Saturation enzyme kinetic studies were carried out to measure the kinetic parameters of the BaPDH-catalyzed reaction and to investigate the mode of BaPDH inhibition by tyrosine. The turnover rate of BaPDH was found to be 49 ± 3 sec−1, and the KM values for prephenate and NAD+ were found to be 58 ± 9 and 237 ± 44 μM, respectively. The saturation plots for both prephenate and NAD+ are shown in Figure 6. To elucidate the mode of inhibition by tyrosine, saturation plots were prepared with varying prephenate concentrations at fixed concentrations of tyrosine (Figures 6C, E). The plots showed a distinct reduction in the apparent Vmax of the enzyme with no change in the KM for prephenate with the addition of tyrosine (Figures 6C, E), which is a manifestation of non-competitive inhibition. This type of inhibition is additionally illustrated by the Lineweaver-Burk plot constructed for the inverse of reaction rate versus the inverse of prephenate concentration, which shows that tyrosine has an effect on Vmax but not on KM (Figure 6D). The inhibition constant (KI) for tyrosine was determined to be 1.47 ± 0.07 μM (Figure 6F). As revealed by structural analysis (Figures 3F, G), the interaction through the amino group of tyrosine is important for its binding to ACT domains. This interaction occurs with S333 and D315 for both ACT domains. Mutational analysis of S333 results in complete loss of tyrosine inhibition to BaPDH (data not shown).
Figure 6. Enzyme kinetics and analysis of BaPDH inhibition by tyrosine.

Enzyme-catalyzed oxidative decarboxylation of prephenate was monitored by following the production of NADH (ε = 6220 M−1 cm−1) at 340 nm. For the cofactor-related kinetics (A), the concentration of NAD+ was varied while prephenate concentration was held constant at 2 mM. For the substrate-related kinetics (B), the concentration of prephenate was varied while NAD+ was held at the saturating concentration of 2 mM. (C) The effect of different concentrations of tyrosine (0–8 μM) on the enzymatic activity of BaPDH. Enzymatic reactions for different tyrosine concentrations were carried out at a constant 2 mM NAD+ with varying prephenate concentration. (D) The Lineweaver-Burk plot generated for the inverse of reaction rates vs. the inverse of substrate concentrations. The x-intercept is the same for almost all curves. (E) Enzyme kinetics constants for BaPDH inhibition by tyrosine. (F) Kinetic parameters and inhibition constant for BaPDH inhibition by tyrosine. For the panel A, three independent experiments were conducted and utilized for calculations (n=3). For the panels B and C, two independent experiments for each curve were conducted and utilized for calculations (n=2). Error bars on panels A, B, C, D represent standard deviation. Error for the values reported in panels E and F represent standard errors.
Discussion
The crystal structures reported herein show that BaPDH functions as a homodimer. BaPDH’s catalytic core, including its nucleotide- and substrate-binding sites, is highly similar to those of other bacterial PDHs. The main distinctive feature of BaPDH is the presence of the ACT domain in addition to the typical catalytic core (Figures 2A, 3A). Both the BaPDH-tyrosine binary complex and the BaPDH-NAD+-tyrosine ternary complex have two tyrosine molecules bound in the ACT domain dimers. However, in the latter, only one subunit of the BaPDH dimer has an NAD+ molecule (Figure 2A). This is consistent with results from ITC studies, which indicated that only one NAD+ molecule binds per BaPDH dimer in the presence of tyrosine (Table 4). The structures also revealed two distinct conformations of the subunits as a result of the positioning of the ACT domains in the BaPDH dimer: subunit A is in the open conformation and subunit B is in the closed conformation (Figure 3B).
The structures of the BaPDH-tyrosine binary (PDB ID: 5UYY) and BaPDH-NAD+-tyrosine ternary (PDB ID: 6U60) complexes represent the inhibited state of the enzyme. Contrary to AaPDH’s structure [22], no tyrosine was observed in the substrate-binding pockets of BaPDH binary and ternary complexes because the ACT domain of subunit B blocks both substrate-binding pockets of the dimer. The substrate-binding pocket of subunit A is blocked by the latch loop (b/340–348) of the ACT domain from subunit B (Figure 4A). The substrate-binding pocket of subunit B is blocked by the linker loop (b/296–306) of the same subunit, and the substrate-binding residue b/R235 is locked in a distant conformation (Figure 4B). This closing-off of both substrate-binding pockets by the ACT domain of subunit B is enabled by the asymmetric organization of the ACT domains in contrast to the symmetric organization of the isolated dimeric ACT domain (Figure 3E). Moreover, in both complexes, the ACT domain of subunit B seems to preclude NAD+ binding in subunit A (Figure 4C). As shown by the ternary complex (Figure 2), subunit B remains accessible for NAD+ binding, which is consistent with the results from the ITC studies (Table 4). Because the same inhibited protein conformation is observed in the binary BaPDH-tyrosine complex, we conclude that formation of the observed inhibited state of the enzyme does not require NAD+ binding.
The inhibited enzymatic state observed in the BaPDH structures shows that the enzyme’s mechanism of inhibition is clearly distinct from the competitive inhibition mechanism previously reported for AaPDH [22], as well as other bacterial PDHs that do not possess the ACT domain [17, 19, 22, 23], despite the similarities of the active sites (Figures 4A, B, 5A). It should be noted that legume PDHs, which do not contain the ACT domain, are insensitive to tyrosine and they have significantly different arrangement of amino acids for the active site and substrate-specificities [18]. In AaPDH’s structure, both nucleotide-binding sites of the dimer are occupied by NADP+ molecules, and the inhibitory tyrosine is found in the substrate-binding site of one subunit. AaPDH is regulated through competitive inhibition with tyrosine binding in the substrate site, whereas BaPDH undergoes allosteric regulation with tyrosine binding to the ACT domain. The allosteric regulation of BaPDH activity by tyrosine is clearly shown by the enzyme kinetics analysis presented (Figure 6). Increasing tyrosine concentration has a direct inhibitory effect on Vmax but no effect on the KM for the substrate prephenate, which is diagnostic of allosteric inhibition by tyrosine (Figure 6). Moreover, mutational analysis of conserved S333, which is involved in tyrosine binding by the ACT domains (Figures 3F, G, 5B), results in complete loss of tyrosine inhibition of BaPDH (data not shown). Surprisingly, despite the difference in the inhibition mechanisms, the key active site amino acid residues are conserved between BaPDH and other bacterial PDHs (Figures 4A, B, 5A). The inhibition constant (KI = 1.47 ± 0.07 μM) for tyrosine from the kinetic analysis is very similar to the binding constant to one site on the inhibited enzyme as determined by ITC studies (Kd = 0.64 ± 0.05 μM). Interestingly, BaPDH’s KI for tyrosine is significantly lower than those previously reported for other TyrA proteins: 15.9 ± 1.3 μM for AaPDH [22] and 70 μM for arogenate dehydrogenase from cyanobacteria Synechocystis sp. [16].
Our finding that the ACT domain in BaPDH is involved in allosteric modulation of the protein activity is not surprising, since the ACT domain of other proteins has been shown to allosterically regulate their activity. The ACT domain-containing enzymes with determined spatial structures include D-3-phosphoglycerate dehydrogenase (inhibited by serine) [46, 53–55], prephenate dehydratase (inhibited by phenylalanine) [47], aspartokinase (inhibited by lysine) [48], 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase (inhibited by tyrosine) [56], and phenylalanine-4-hydroxylase (inhibited by phenylalanine) [49]. Interestingly, the mechanisms of inhibition differ substantially between these enzymes, displaying the complexity of the ways regulatory functions are performed by the ACT domains. For example, the binding of serine to the ACT domains of D-3-phosphoglycerate dehydrogenase causes structural rearrangements in the ACT domain, which prevent the closing of the cleft between the substrate-binding and nucleotide-binding domains [53–55]. Binding of phenylalanine to prephenate dehydratase introduces several major conformational shifts that change the relative orientation of catalytic subdomains and narrow access to the active site [47]. Binding of tyrosine to 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase results in a large structural reorganization: the regulatory ACT domains of diagonally opposite molecules occlude the active site [45, 56, 57]. Our studies show that BaPDH presents yet another mechanism of inhibition by the ACT domain (Figure 4).
We were unable to obtain a crystal structure of an uninhibited full-length BaPDH. However, based on the data presented herein, we propose a speculative model in which the ACT dimer dissociates in the absence of tyrosine in the allosteric site, leaving both BaPDH subunits in the open conformation similar to that observed for subunit A of the binary and ternary complexes (Figures 2, 3). This repositioning of the ACT domain of submit B to the open conformation will likely allow substrate access to both active sites. Having both subunits in the open conformation may occur only when the ACT dimer dissociates (Figure 4D), which is likely to happen without the two tyrosine molecules strengthening the interdomain interface of the ACT dimer (Figure 3). Although the above is speculative, there is sufficient evidence in the literature for such a model. For example, 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase, one of the few ACT-containing enzymes with both unliganded and liganded structures known, the ACT domains are dissociated in the active form and dimerized in the inhibited form [45, 56]. The thermal denaturation of BaPDH with different combinations of ligands provides an additional level of evidence that tyrosine binding induces large conformational changes to the protein. In the presence of tyrosine, the thermal stabilization of BaPDH increases more than 10°C, which can be explained by a much more compact BaPDH-NAD+-tyrosine dimer as compared to a dimer with ACT domains removed. According to PISA server analysis, the buried surface interface area of BaPDH dimer is 49% of the total surface area of isolated monomers; with the ACT domains removed from the model, this number falls to 36%. The inclusion of other ligands, including NAD+, did not impart similar levels of structural stabilization on the protein.
Our proposed model is further supported by the fact that different chymotrypsin proteolysis products were crystallized depending on the ligands present. Treatment of BaPDH with chymotrypsin to obtain crystals of unliganded protein produced crystals containing only the ACT domain dimer. However, this effect was not recapitulated with the treatment of the BaPDH protein in the presence of 20 mM tyrosine and 5 mM NAD+. The chymotrypsin digestion studies show that the addition of tyrosine and NAD+ makes BaPDH much more resistant to the protease as compared to the unliganded form (Figure 7). NAD+ or tyrosine alone also result in BaPDH stabilization but not as much as in the case of the ternary complex. This conformational protection from chymotrypsin is supported by our structural analysis. The structure of the isolated ACT domain dimer has H307 as the first N-terminal residue in both subunits, suggesting that chymotrypsin cuts BaPDH after residue Y306 of the linker loop 296–306. Indeed, chymotrypsin preferentially cleaves peptide amide bonds where the side chain of the amino acid N-terminal to the scissile amide bond is a bulky hydrophobic amino acid (tyrosine, tryptophan, and phenylalanine). In the binary and ternary complexes, the linker loop of the closed subunit B (including Y306) is wedged in the active site of the same subunit, preventing it from enzymatic digestion (Figure 3). Moreover, Y306 directly participates in blocking the active site of BaPDH (Figure 4B). In the open subunit A, the linker loop 296–306 is on the surface of the protein, but Y306 is oriented inwards toward the protein core, making it inaccessible to chymotrypsin. These structural details support the observation of BaPDH becoming much more resistant to proteolysis in the presence of tyrosine and NAD+. Therefore, with the support of our kinetic, structural, thermal denaturation, and proteolysis data, we speculate that, similarly to other ACT-containing proteins, the mode of tyrosine regulation involves ACT dimerization upon tyrosine binding (Figure 4D).
Figure 7. Chymotrypsin digestion studies of BaPDH.

SDS gel electrophoresis of wild-type BaPDH in the presence/absence of 10 mM tyrosine and/or 10 mM NAD+ treated with chymotrypsin in the same way as for crystallization. The protein and the protein-ligand mixtures were incubated with 1/40 (v/v) of 2 mg/mL chymotrypsin solution in 1 mM HCl and 2 mM CaCl2 at 16°C for 5, 60, and 120 min. Chymotrypsin was added to samples in all lanes except lane two. Each lane is labelled with ligands added and the time elapsed after digestion was started. The lanes show two fragments in addition to the full-length protein: a 13 kDa band representing the ACT domain and a 25 kDa band representing the catalytic core. The experiment has been performed twice (n=2) with similar results.
Conclusions
This is the first report on the crystal structure and biochemical studies of an ACT domain containing PDH. Our structural and biochemical analyses reveal that tyrosine binding to the ACT domains allosterically regulates BaPDH activity, providing negative feedback inhibition by the pathway product. The structures show that BaPDH presents a novel mechanism of inhibition via the ACT domain, and the inhibited state in this mechanism is characterized by its unique asymmetry and complexity. By analogy to other ACT-containing enzymes, we propose that the ACT domains of the protein dimer without tyrosine bound are in a dissociated state; upon binding tyrosine, the ACT domains form the dimer we see in two of the reported structures (BaPDH-tyrosine PDB ID: 5UYY and BaPDH-NAD+-tyrosine PDB ID: 6U60) and block the substrate-binding sites. The conformation of the enzyme in the absence of tyrosine is yet to be confirmed, but structural analysis and the results of protease treatments together with temperature denaturation and ITC studies support a model in which the binding of tyrosine induces a large conformational change. The structural and biochemical analyses presented here are important steps in investigating the tyrosine biosynthetic pathway as a target for drug development against B. anthracis and similar pathogens. The ability to downregulate tyrosine biosynthesis will interfere with overall protein biosynthesis and the biosynthesis of aromatic metabolites that require tyrosine as a precursor. Such an alteration in tyrosine biosynthesis is expected to attenuate the virulence of B. anthracis and other ACT-PDH containing pathogens [43, 44]. Moreover, these advancements in our understanding of tyrosine biosynthesis in bacteria may further facilitate its use in biotechnological applications [3, 4].
Materials and Methods
Reagents
Crystallization screens were purchased from Anatrace (Maumee, OH, USA). Prephenate was kindly provided by Dr. Timothy Wencewicz (Washington University in St. Louis, St. Louis, USA). L-tyrosine and reduced and oxidized cofactors were obtained from Sigma-Aldrich (St. Louis, MO, USA; catalogue numbers T3754, N0632, N6005, N3130, and N1630) and Santa Cruz Biotechnology (Dallas, TX, USA; catalogue number 202725A). All other reagents were obtained from Sigma-Aldrich, unless specified otherwise.
Molecular cloning and protein expression
cDNA for BaPDH (CSGID target IDP05722) was amplified from the genome of B. anthracis str. Ames (Gene ID: 1087404, locus tag BA_2954) and cloned into the pMCSG7 vector using a ligation-independent cloning protocol [58]. The vector encodes His-tag with a TEV protease cleavage site on the N-terminus of the protein (tag sequence: MHHHHHHSSGVDLGTENLYFQSNA); TEV cleavage leaves SNA amino acids on the protein’s N-terminus. The recombinant plasmid for BaPDH was transformed into E. coli strain BL21-CodonPlus (DE3)-RIPL competent cells (Stratagene, La Jolla, CA, USA) and selected with ampicillin resistance on the LB-Agar plate.
For the expression of wild-type BaPDH, approximately 20 colonies of transformed E. coli cells were inoculated into 1 L LB media supplemented with 0.5% glycerol and 100 μg/mL ampicillin. The cells were incubated with shaking at 37°C to an OD595nm of around 0.7. Expression of the protein was induced by adding IPTG to a final concentration of 0.25 mM. The induced cells were grown at 16°C overnight and then harvested.
For the expression of SeMet-BaPDH, one colony of the transformed E. coli cells was inoculated into 3 mL LB (Luria Broth) media supplemented with 100 μg/mL ampicillin and cultured at 37°C overnight. The cells were pelleted by centrifuge, washed with M9 medium, resuspended into 1 L M9 medium containing 100 μg/mL ampicillin, and cultured at 37°C to an OD595nm of 0.7. The temperature was then lowered to 16°C, and after 30 min, 50 mg of Se-Met were added to the medium. IPTG was added to a final concentration of 0.25 mM to induce protein expression. The induced cells were grown at 16°C overnight and then harvested.
Protein purification
The wild-type BaPDH was purified according to the protocol described elsewhere [59, 60]. In brief, the harvested cells were suspended in binding buffer A (20 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 10 mM β-ME, 10 mM imidazole) with freshly added 250 units of benzonase endonuclease (Sigma) and a protease inhibitor cocktail (cOmplete, EDTA-free Protease Inhibitor Cocktail Tablets, Roche, Basel, Switzerland). The suspended cells were lysed by either sonication or French press and centrifuged. The supernatant was applied to Ni-NTA resin, and the column was washed with 300 mL washing buffer (buffer A with 30 mM imidazole). The protein was eluted with buffer A containing 250 mM imidazole. The His-tag was cleaved by His-tagged recombinant TEV protease (1 mg TEV per 60 mg of BaPDH). The mixture was incubated at 16°C overnight and then dialyzed in buffer B (20 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 10 mM β-ME) for ~4 hours. The digested protein was further purified by passing the mixture through a Ni-NTA column pre-equilibrated with buffer B. The flowthrough was applied to the ÄKTA FPLC (GE Healthcare Life Sciences, Chicago, IL, USA) for size-exclusion chromatography and eluted with buffer C (10 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol, 10 mM β-ME). The combined fractions were concentrated in buffer C and stored at −80°C.
The SeMet-BaPDH was purified in the same manner but without cleaving the His-tag. After size-exclusion chromatography, the protein was eluted with buffer D (10 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 10 mM β-ME).
Protein crystallization and diffraction data collection
BaPDH in the ternary complex with NAD+ and tyrosine was crystallized using sitting-drop vapor-diffusion method. Wild-type BaPDH (12 mg/mL) was supplemented with 5 mM NAD+ and 20 mM tyrosine. Aliquots of 0.2 μL of this solution were mixed with 0.2 μL of crystallization screening cocktail on 96-well, 3-drop plates (Swissci, Neuheim, Switzerland) using a Mosquito crystallization robot (TTP Labtech, Hertfordshire, England, GB) and equilibrated against 1.5 M NaCl at 16°C. Crystals were produced in several conditions but diffracted poorly (~2.8 Å). Crystallization screening was then repeated in exactly the same setup, except that the protein-ligand mixture was incubated with 1/40 (v/v) of 2 mg/mL chymotrypsin solution in 1 mM HCl and 2 mM CaCl2 at 16°C for 1 hour before crystallization. The best-diffracting crystal (2.2 Å) was obtained with MCSG Suite 2 condition 81 (Table 1). The first 13 residues in both protein chains are absent in the electron density maps, most likely due to their cleavage by chymotrypsin at Met13.
Crystals of BaPDH grown after the addition of 20 mM tyrosine did not diffract better than 3.3 Å. Serendipitously, the structure of the BaPDH-tyrosine binary complex was determined without the addition of exogenous tyrosine. The best-diffracting crystal (2.6 Å) was obtained using 19 mg/mL wild-type BaPDH with MCSG Suite 1 condition 11 (Table 1). The first 5 residues in chains A and D and the first 13 residues in chains B and C are absent in the electron density maps. Even though tyrosine was not added to the protein solution, it was bound to each ACT domain in the crystal structure. Although the binding data suggests that the purified protein has approximately one vacant and one occupied tyrosine site per BaPDH dimer, complexes of different ratios likely exist in solution; the dimer with tyrosine bound in both subunits showed higher crystallization propensity, resulting in two tyrosine molecules per dimer in the crystal structure. Another BaPDH structure obtained from a different batch of protein (SeMet-BaPDH) without the addition of tyrosine and solved in a different space group (3.0 Å, P21, data not shown) also has tyrosine bound to each ACT domain.
Crystallization of extensively dialyzed BaPDH without the addition of NAD+ and tyrosine did not produce crystals diffracting better than 3.3 Å. Given the vital role of chymotrypsin in obtaining the best-diffracting crystal of the BaPDH-NAD+-tyrosine complex, the same strategy was tried for SeMet-BaPDH without the addition of ligands. The best-diffracting crystal (2.0 Å) was obtained using 15 mg/mL protein concentration with MCSG Suite 1 condition 31 (Table 1). Structure determination showed that only the ACT domain (residues 307–375) was present in this structure. The ACT domain (residues 307–375) separation was achieved by adding chymotrypsin to the protein before crystallization. Residues of the linker loop (296–306) and the last three residues of the C-terminal (376–378) were not located in the electron density maps. SDS gel electrophoresis of wild-type BaPDH treated with chymotrypsin in the same way as for crystallization for 5, 60, and 120 min in the presence/absence of 10 mM tyrosine and/or 10 mM NAD+ showed two fragments in addition to the full-length protein: a 13 kDa band representing the ACT domain and a 25 kDa band representing the catalytic core (Figure 7). Several other complexes of BaPDH were also tried for crystallization, but did not produce diffraction quality crystals: NAD+ (no crystals), NADH-tyrosine (~7 Å), NADH (~8 Å), and prephenate (~3.3 Å).
Crystals were harvested and vitrified in liquid nitrogen with cryoprotection by Paratone-N (Hampton Research, Aliso Viejo, CA, USA) or by slow dehydration (keeping the crystal over 1 M sodium chloride solution for 10 minutes). Diffraction data were collected at 100 K at the 21-ID-G and 21-ID-F beamlines of the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source (APS).
Data processing and structure determination
Data reduction and scaling for all datasets were performed with HKL-3000 [61, 62]. The dataset for BaPDH ternary complex with NAD+ and tyrosine was processed in the anomalous mode using corrections for radiation decay and anisotropic diffraction (auto-corrections). The structure of BaPDH in complex with tyrosine was solved by molecular replacement by HKL-3000 with the use of MOLREP [63] and other programs from the CCP4 package [64]. The structural model of AaPDH (PDB ID: 3GGG) was used as the template [22]. The structure of BaPDH in complex with tyrosine and NAD+ was determined by molecular replacement using the structure in complex with tyrosine as the template. All attempts to determine the structure of ACT domain by molecular replacement were unsuccessful; the structure was ultimately solved by SAD phasing using the anomalous signal from the incorporated selenomethionine. Automated model building was performed with Buccaneer [65], followed by optimization of side-chain conformations with Fitmunk [66], both integrated with HKL-3000. The structures were refined with HKL-3000 using state-of-the-art guidelines outlined elsewhere [67]. REFMAC [68] in restrained mode with automatic local NCS and hydrogen atoms in riding positions was used for the reciprocal-space refinement. Coot [69] was used for the visualization of electron density maps and manual inspection and correction of the atomic models. TLS groups were introduced in the latest stages of refinement with the TLS Motion Determination Server [70]. The TLS parameters were kept if confirmed by a significantly improved Rfree and the Hamilton R-factor ratio test [71] as implemented in HKL-3000. Structures were validated with a stand-alone version of Molprobity [72] and wwPDB validation tools [73]. The metal-binding sites were validated using the CheckMyMetal server [74, 75]. The atomic coordinates and structure factors have been deposited in the PDB (PDB ID: 6U60, 5UYY, and 5V0S). All cloning, expression, purification, crystallization, and structure determination experiments were tracked with LabDB database [76]. The diffraction images are available on the Integrated Resource for Reproducibility in Macromolecular Crystallography server [77] with DOIs 10.18430/M35USC, 10.18430/M35UYY, and 10.18430/M35V0S (https://proteindiffraction.org). A web-based interactive publishing platform, Molstack (https://molstack.bioreproducibility.org), was used for interactive presentation of the models and the respective electron density maps [78]. Structural figures (Figures 2, 3, and 4) were prepared with the use of PyMOL (Schrodinger, LLC).
Protein thermal shift assay
The thermal stability of BaPDH and its complexes was measured by the protein thermal shift assay performed with a C1000 thermal cycler supplemented with a CFX96 real-time system in a 96-well PCR plate (Bio-Rad, Hercules, CA, USA). Samples were diluted to a final protein concentration of 0.2 mg/mL in 0.025 mL of a solution consisting of the protein purification buffer, 10 × SYPRO Orange (5000 × SYPRO Orange stock from Invitrogen, Carlsbad, CA, USA), and various ligands at 10 mM (Table 3). Sample data collection and analysis were conducted according to an established protocol [60]. Each combination of protein and ligands was measured 3–4 times, and the resulting Tm was averaged; errors were calculated as rounded-up standard deviations.
Isothermal titration calorimetric (ITC) assays for BaPDH
ITC experiments were performed at 25°C using an iTC200 Microcalorimeter (MicroCal, Northampton, MA, USA). Prior to the experiment, the protein was dialyzed into the ITC buffer containing 100 mM HEPES pH 7.5, 5% (v/v) glycerol, and 500 mM NaCl. In the first attempts to perform the ITC experiments, the same buffer with lower NaCl concentration (150 mM) was used. However, BaPDH tended to aggregate, and a significant amount of protein precipitation occurred during the dialysis. Using a higher NaCl concentration (500 mM) helped eliminate visible precipitation. NAD+, NADP+, and tyrosine were tested for their binding affinity for BaPDH. The detailed ITC conditions are summarized in Table 4. The experiments were performed in high-gain mode with the syringe rotating at 700 rpm. The titration peaks were integrated and fit to a one-set-of-sites model using the ORIGIN module from the iTC200 software; errors were calculated as the least-squares fit uncertainties. Each experiment was repeated at least twice (albeit using variations such as different purification batches, injection volumes, ligand stocks, and ligand concentration in the syringe), with repetitions showing similar results to those reported in Table 4.
Enzyme kinetics: determination of BaPDH activity and tyrosine inhibition constant
The BaPDH activity assay was conducted by spectrophotometrically following the production of NADH at 340 nm (extinction coefficient 6,220 M−1cm−1). The reaction buffer was composed of 50 mM Tris pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 2 mM DTT, and the appropriate concentrations of NAD+ and prephenate. The reaction was conducted in a semi-micro cuvette in a 100 μL reaction mixture incubated at 25°C for 5 minutes before initiating the reaction. The reaction was initiated by the addition of 0.035 μM of the purified BaPDH and monitored at 340 nm in a Varian Cary 50 spectrometer (Midland, ON, Canada) fitted with a temperature-controlled cell set at 25°C. Enzyme kinetics reactions were conducted with either prephenate or NAD+ fixed at a saturating concentration of 2 mM while varying the other substrate at a range of concentrations. Data analysis was conducted with Graphpad Prism 7® software. Saturation and inhibition curves were fitted with the non-linear Michaelis-Menten equation. To demonstrate the type of inhibition, the Lineweaver-Burk plot was generated by fitting the inverse of the data into the linear regression model. The inhibition constant was derived by fitting all inhibition curves into the non-competitive inhibition model equation.
Sequence analysis of prokaryotic PDHs and associated ACT domains
Non-redundant (one per species) sequences for PDH proteins were retrieved from NCBI using the BLAST search engine. BaPDH sequence (NCBI Reference Sequence: WP_011053181.1) was used as query. Retrieved sequences with percent identity of 28% or greater with respect to BaPDH were aligned with MEGA7 software suite using the ClustalW method with default parameters [79]. Gap opening penalties were set at 10 and gap extension penalties were set at 0.1 for pairwise alignments and 0.2 for multiple sequence alignments. The resulting sequence alignments were used to generate shown sequence logos with WebLogo [80]. Full sequence alignment can be found in Supplementary file 1.
Supplementary Material
Acknowledgments
We would like to thank Timothy A. Wencewicz for providing prephenate. We would also like to thank Barat S. Venkataramany, Przemyslaw J. Porebski, Mateusz P. Czub, Maksymilian Chruszcz, and Alexander Wlodawer for critically reading the manuscript and providing valuable feedback. The presented work was supported by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contracts HHSN272201200026C and HHSN272201700060C; National Institute of General Medical Sciences under grant numbers GM118619 and GM117325; and by the Natural Science and Engineering Research Council of Canada Discovery Grant (NSERC-DG RGPIN-2015-06747). Results shown in this report are derived from work performed at Argonne National Laboratory, operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Abbreviations
- AaPDH
prephenate dehydrogenase from Aquifex aeolicus
- ADP
atomic displacement parameter, ASU, asymmetric unit
- BaPDH
prephenate dehydrogenase from Bacillus anthracis
- CSGID
Center for Structural Genomics of Infectious Diseases
- ITC
isothermal titration calorimetry
- PDB
Protein Data Bank
- PDH
prephenate dehydrogenase
- rmsd
root mean square deviation
Footnotes
Enzymes
Prephenate dehydrogenase from Bacillus anthracis (PDH): EC database ID: 1.3.1.12
Databases
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with accession numbers PDB ID: 6U60 (BaPDH complex with NAD+ and tyrosine), PDB ID: 5UYY (BaPDH complex with tyrosine), and PDB ID: 5V0S (BaPDH isolated ACT-domain dimer). The diffraction images are available at http://proteindiffraction.org with DOIs: 10.18430/M35USC, 10.18430/M35UYY, and 10.18430/M35V0S.
Conflicts of interest
One of the authors (WM) notes that he has also been involved in the development of state-of-the-art software, data management and mining tools; some of them were commercialized by HKL Research and are mentioned in the paper. WM is the co-founder of HKL Research and a member of the board. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Maeda H & Dudareva N (2012) The shikimate pathway and aromatic amino Acid biosynthesis in plants, Annu Rev Plant Biol. 63, 73–105. [DOI] [PubMed] [Google Scholar]
- 2.Ventola CL (2015) The antibiotic resistance crisis: part 1: causes and threats, P t. 40, 277–83. [PMC free article] [PubMed] [Google Scholar]
- 3.Kim B, Binkley R, Kim HU & Lee SY (2018) Metabolic engineering of Escherichia coli for the enhanced production of l-tyrosine, Biotechnol Bioeng. 115, 2554–2564. [DOI] [PubMed] [Google Scholar]
- 4.Lee SY, Kim HU, Chae TU, Cho JS, Kim JW, Shin JH, Kim DI, Ko Y-S, Jang WD & Jang Y-S (2019) A comprehensive metabolic map for production of bio-based chemicals, Nature Catalysis. 2, 18–33. [Google Scholar]
- 5.Mir R, Jallu S & Singh TP (2015) The shikimate pathway: review of amino acid sequence, function and three-dimensional structures of the enzymes, Crit Rev Microbiol. 41, 172–89. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann KM & Weaver LM (1999) THE SHIKIMATE PATHWAY, Annu Rev Plant Physiol Plant Mol Biol. 50, 473–503. [DOI] [PubMed] [Google Scholar]
- 7.Tzin V & Galili G (2010) The Biosynthetic Pathways for Shikimate and Aromatic Amino Acids in Arabidopsis thaliana, Arabidopsis Book. 8, e0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morell H, Clark MJ, Knowles PF & Sprinson DB (1967) The enzymic synthesis of chorismic and prephenic acids from 3-enolpyruvylshikimic acid 5-phosphate, J Biol Chem. 242, 82–90. [PubMed] [Google Scholar]
- 9.Gibson F & Pittard J (1968) Pathways of biosynthesis of aromatic amino acids and vitamins and their control in microorganisms, Bacteriol Rev. 32, 465–92. [PMC free article] [PubMed] [Google Scholar]
- 10.Schwinck I & Adams E (1959) Aromatic biosynthesis. XVI. Aromatization of prephenic acid to p-hydroxyphenylpyruvic acid, a step in tyrosine biosynthesis in Escherichia coli, Biochim Biophys Acta. 36, 102–17. [DOI] [PubMed] [Google Scholar]
- 11.Stenmark SL, Pierson DL, Jensen RA & Glover GI (1974) Blue-green bacteria synthesise L-tyrosine by the pretyrosine pathway, Nature. 247, 290–2. [DOI] [PubMed] [Google Scholar]
- 12.Fazel AM, Bowen JR & Jensen RA (1980) Arogenate (pretyrosine) is an obligatory intermediate of L-tyrosine biosynthesis: confirmation in a microbial mutant, Proc Natl Acad Sci U S A. 77, 1270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song J, Bonner CA, Wolinsky M & Jensen RA (2005) The TyrA family of aromatic-pathway dehydrogenases in phylogenetic context, BMC Biology. 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonvin J, Aponte RA, Marcantonio M, Singh S, Christendat D & Turnbull JL (2006) Biochemical characterization of prephenate dehydrogenase from the hyperthermophilic bacterium Aquifex aeolicus, Protein Sci. 15, 1417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia TH & Jensen RA (1990) A single cyclohexadienyl dehydrogenase specifies the prephenate dehydrogenase and arogenate dehydrogenase components of the dual pathways to L-tyrosine in Pseudomonas aeruginosa, J Biol Chem. 265, 20033–6. [PubMed] [Google Scholar]
- 16.Bonner CA, Jensen RA, Gander JE & Keyhani NO (2004) A core catalytic domain of the TyrA protein family: arogenate dehydrogenase from Synechocystis, Biochem J. 382, 279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legrand P, Dumas R, Seux M, Rippert P, Ravelli R, Ferrer JL & Matringe M (2006) Biochemical characterization and crystal structure of Synechocystis arogenate dehydrogenase provide insights into catalytic reaction, Structure. 14, 767–76. [DOI] [PubMed] [Google Scholar]
- 18.Schenck CA, Holland CK, Schneider MR, Men Y, Lee SG, Jez JM & Maeda HA (2017) Molecular basis of the evolution of alternative tyrosine biosynthetic routes in plants, Nat Chem Biol. 13, 1029–1035. [DOI] [PubMed] [Google Scholar]
- 19.Ku HK, Park SR, Yang I & Kim SK (2010) Expression and functional characterization of prephenate dehydrogenase from Streptococcus mutans, Process Biochem. 45, 607–612. [Google Scholar]
- 20.Christendat D & Turnbull JL (1999) Identifying Groups Involved in the Binding of Prephenate to Prephenate Dehydrogenase from Escherichia coli, Biochemistry. 38, 4782–4793. [DOI] [PubMed] [Google Scholar]
- 21.Bonner CA, Disz T, Hwang K, Song J, Vonstein V, Overbeek R & Jensen RA (2008) Cohesion group approach for evolutionary analysis of TyrA, a protein family with wide-ranging substrate specificities, Microbiol Mol Biol Rev. 72, 13–53, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun W, Shahinas D, Bonvin J, Hou W, Kimber MS, Turnbull J & Christendat D (2009) The crystal structure of Aquifex aeolicus prephenate dehydrogenase reveals the mode of tyrosine inhibition, J Biol Chem. 284, 13223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun W, Singh S, Zhang R, Turnbull JL & Christendat D (2006) Crystal structure of prephenate dehydrogenase from Aquifex aeolicus. Insights into the catalytic mechanism, J Biol Chem. 281, 12919–28. [DOI] [PubMed] [Google Scholar]
- 24.Ku HK, Do NH, Song JS, Choi S, Yeon SH, Shin MH, Kim KJ, Park SR, Park IY, Kim SK & Lee SJ (2011) Crystal structure of prephenate dehydrogenase from Streptococcus mutans, Int J Biol Macromol. 49, 761–6. [DOI] [PubMed] [Google Scholar]
- 25.Chiu HJ, Abdubek P, Astakhova T, Axelrod HL, Carlton D, Clayton T, Das D, Deller MC, Duan L, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Marciano D, McMullan D, Miller MD, Morse AT, Nigoghossian E, Okach L, Reyes R, Tien HJ, Trame CB, van den Bedem H, Weekes D, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA & Wilson IA (2010) The structure of Haemophilus influenzae prephenate dehydrogenase suggests unique features of bifunctional TyrA enzymes, Acta Crystallogr Sect F Struct Biol Cryst Commun. 66, 1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagino H & Nakayama K (1974) Regulatory Properties of Prephenate Dehydrogenase and Prephenate Dehydratase from Corynebacterium glutamicum, Agric and Biol Chem. 38, 2367–2376. [Google Scholar]
- 27.Holland CK & Jez JM (2018) Reaction Mechanism of Prephenate Dehydrogenase from the Alternative Tyrosine Biosynthesis Pathway in Plants, ChemBioChem. 19, 1132–1136. [DOI] [PubMed] [Google Scholar]
- 28.Aravind L & Koonin EV (1999) Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches, J Mol Biol. 287, 1023–40. [DOI] [PubMed] [Google Scholar]
- 29.Chipman DM & Shaanan B (2001) The ACT domain family, Curr Opin Struct Biol. 11, 694–700. [DOI] [PubMed] [Google Scholar]
- 30.Siltberg-Liberles J & Martinez A (2009) Searching distant homologs of the regulatory ACT domain in phenylalanine hydroxylase, Amino Acids. 36, 235–49. [DOI] [PubMed] [Google Scholar]
- 31.Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U & Magrini N (2018) Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect Dis. 18, 318–327. [DOI] [PubMed] [Google Scholar]
- 32.Baillie L & Read TD (2001) Bacillus anthracis, a bug with attitude!, Curr Opin Microbiol. 4, 78–81. [DOI] [PubMed] [Google Scholar]
- 33.Turnbull PC (1999) Definitive identification of Bacillus anthracis--a review, J Appl Microbiol. 87, 237–40. [DOI] [PubMed] [Google Scholar]
- 34.Spencer RC (2003) Bacillus anthracis, J Clin Pathol. 56, 182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McConkey GA (1999) Targeting the shikimate pathway in the malaria parasite Plasmodium falciparum, Antimicrob Agents Chemother. 43, 175–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reichau S, Jiao W, Walker SR, Hutton RD, Baker EN & Parker EJ (2011) Potent Inhibitors of a Shikimate Pathway Enzyme from Mycobacterium tuberculosis: combining mechanism- and modeling-based design in J Biol Chem pp. 16197–16207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlievert PM, Merriman JA, Salgado-Pabon W, Mueller EA, Spaulding AR, Vu BG, Chuang-Smith ON, Kohler PL & Kirby JR (2013) Menaquinone analogs inhibit growth of bacterial pathogens, Antimicrob Agents Chemother. 57, 5432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meganathan R (2001) Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms, Vitam Horm. 61, 173–218. [DOI] [PubMed] [Google Scholar]
- 39.Matarlo JS, Lu Y, Daryaee F, Daryaee T, Ruzsicska B, Walker SG & Tonge PJ (2016) A Methyl 4-Oxo-4-phenylbut-2-enoate with in Vivo Activity against MRSA that Inhibits MenB in the Bacterial Menaquinone Biosynthesis Pathway, ACS Infect Dis. 2, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY, Passalacqua KD, Hanna PC & Sherman DH (2011) Regulation of Petrobactin and Bacillibactin Biosynthesis in Bacillus anthracis under Iron and Oxygen Variation, PLoS ONE. 6, e20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson MK, Abergel RJ, Raymond KN, Arceneaux JE & Byers BR (2006) Siderophores of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis, Biochem Biophys Res Commun. 348, 320–5. [DOI] [PubMed] [Google Scholar]
- 42.Lee JY, Janes BK, Passalacqua KD, Pfleger BF, Bergman NH, Liu H, Hakansson K, Somu RV, Aldrich CC, Cendrowski S, Hanna PC & Sherman DH (2007) Biosynthetic analysis of the petrobactin siderophore pathway from Bacillus anthracis, J Bacteriol. 189, 1698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith H & Tempest DW (1957) The Uptake of Amino Acids During the Terminal Bacteraemia in Guinea Pigs Infected with Bacillus anthracis, Microbiology. 17, 731–738. [DOI] [PubMed] [Google Scholar]
- 44.Gladstone GP (1939) Inter-relationships Between Amino-acids in the Nutrition of B. anthracis, Br J Exp Pathol. 20, 189–200. [Google Scholar]
- 45.Lang EJ, Cross PJ, Mittelstadt G, Jameson GB & Parker EJ (2014) Allosteric ACTion: the varied ACT domains regulating enzymes of amino-acid metabolism, Curr Opin Struct Biol. 29, 102–11. [DOI] [PubMed] [Google Scholar]
- 46.Al-Rabiee R, Zhang Y & Grant GA (1996) The mechanism of velocity modulated allosteric regulation in D-3-phosphoglycerate dehydrogenase. Site-directed mutagenesis of effector binding site residues, J Biol Chem. 271, 23235–8. [DOI] [PubMed] [Google Scholar]
- 47.Tan K, Li H, Zhang R, Gu M, Clancy ST & Joachimiak A (2008) Structures of open (R) and close (T) states of prephenate dehydratase (PDT)--implication of allosteric regulation by L-phenylalanine, J Struct Biol. 162, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kotaka M, Ren J, Lockyer M, Hawkins AR & Stammers DK (2006) Structures of R- and T-state Escherichia coli aspartokinase III. Mechanisms of the allosteric transition and inhibition by lysine, J Biol Chem. 281, 31544–52. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Ilangovan U, Daubner SC, Hinck AP & Fitzpatrick PF (2011) Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase, Arch Biochem Biophys. 505, 250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krissinel E & Henrick K (2007) Inference of macromolecular assemblies from crystalline state, J Mol Biol. 372, 774–97. [DOI] [PubMed] [Google Scholar]
- 51.Bonner CA, Disz T, Hwang K, Song J, Vonstein V, Overbeek R & Jensen RA (2008) Cohesion group approach for evolutionary analysis of TyrA, a protein family with wide-ranging substrate specificities, Microbiol Mol Biol Rev. 72, 13–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matelska D, Shabalin IG, Jablonska J, Domagalski MJ, Kutner J, Ginalski K & Minor W (2018) Classification, substrate specificity and structural features of D-2-hydroxyacid dehydrogenases: 2HADH knowledgebase, BMC Evol Biol. 18, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grant GA, Schuller DJ & Banaszak LJ (1996) A model for the regulation of D-3-phosphoglycerate dehydrogenase, a Vmax-type allosteric enzyme, Protein Sci. 5, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dey S, Hu Z, Xu XL, Sacchettini JC & Grant GA (2007) The effect of hinge mutations on effector binding and domain rotation in Escherichia coli D-3-phosphoglycerate dehydrogenase, J Biol Chem. 282, 18418–26. [DOI] [PubMed] [Google Scholar]
- 55.Grant GA, Hu Z & Xu XL (2005) Identification of Amino Acid Residues Contributing to the Mechanism of Cooperativity in E. coli D-3-Phosphoglycerate Dehydrogenase, Biochemistry. 44, 16844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cross PJ, Dobson RC, Patchett ML & Parker EJ (2011) Tyrosine latching of a regulatory gate affords allosteric control of aromatic amino acid biosynthesis, J Biol Chem. 286, 10216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shumilin IA, Bauerle R, Wu J, Woodard RW & Kretsinger RH (2004) Crystal structure of the reaction complex of 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Thermotoga maritima refines the catalytic mechanism and indicates a new mechanism of allosteric regulation, J Mol Biol. 341, 455–66. [DOI] [PubMed] [Google Scholar]
- 58.Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A & Mark ID (2009) A family of LIC vectors for high-throughput cloning and purification of proteins, Methods Mol Biol. 498, 105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shabalin IG, Porebski PJ, Cooper DR, Grabowski M, Onopriyenko O, Grimshaw S, Savchenko A, Chruszcz M & Minor W (2012) Structure of anabolic ornithine carbamoyltransferase from Campylobacter jejuni at 2.7 A resolution, Acta Crystallogr Sect F Struct Biol Cryst Commun. 68, 1018–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kutner J, Shabalin IG, Matelska D, Handing KB, Gasiorowska O, Sroka P, Gorna MW, Ginalski K, Wozniak K & Minor W (2018) Structural, Biochemical, and Evolutionary Characterizations of Glyoxylate/Hydroxypyruvate Reductases Show Their Division into Two Distinct Subfamilies, Biochemistry. 57, 963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Otwinowski Z & Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 62.Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes, Acta Crystallogr D Biol Crystallogr. 62, 859–66. [DOI] [PubMed] [Google Scholar]
- 63.Vagin A & Teplyakov A (2010) Molecular replacement with MOLREP, Acta Crystallogr D Biol Crystallogr. 66, 22–5. [DOI] [PubMed] [Google Scholar]
- 64.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A & Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr D Biol Crystallogr. 67, 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains, Acta Crystallogr D Biol Crystallogr. 62, 1002–11. [DOI] [PubMed] [Google Scholar]
- 66.Porebski PJ, Cymborowski M, Pasenkiewicz-Gierula M & Minor W (2016) Fitmunk: improving protein structures by accurate, automatic modeling of side-chain conformations, Acta Crystallogr D Struct Biol. 72, 266–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shabalin IG, Porebski PJ & Minor W (2018) Refining the macromolecular model - achieving the best agreement with the data from X-ray diffraction experiment, Crystallogr Rev. 24, 236–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F & Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallogr D Biol Crystallogr. 67, 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Lohkamp B, Scott WG & Cowtan K (2010) Features and development of Coot, Acta Crystallogr D Biol Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Painter J & Merritt EA (2006) TLSMD web server for the generation of multi-group TLS models, J Appl Crystallogr. 39, 109–111. [Google Scholar]
- 71.Merritt EA (2012) To B or not to B: a question of resolution?, Acta Crystallogr D Biol Crystallogr. 68, 468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS & Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young JY, Westbrook JD, Feng Z, Sala R, Peisach E, Oldfield TJ, Sen S, Gutmanas A, Armstrong DR, Berrisford JM, Chen L, Chen M, Di Costanzo L, Dimitropoulos D, Gao G, Ghosh S, Gore S, Guranovic V, Hendrickx PMS, Hudson BP, Igarashi R, Ikegawa Y, Kobayashi N, Lawson CL, Liang Y, Mading S, Mak L, Mir MS, Mukhopadhyay A, Patwardhan A, Persikova I, Rinaldi L, Sanz-Garcia E, Sekharan MR, Shao C, Swaminathan GJ, Tan L, Ulrich EL, van Ginkel G, Yamashita R, Yang H, Zhuravleva MA, Quesada M, Kleywegt GJ, Berman HM, Markley JL, Nakamura H, Velankar S & Burley SK (2017) OneDep: Unified wwPDB System for Deposition, Biocuration, and Validation of Macromolecular Structures in the PDB Archive, Structure. 25, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng H, Chordia MD, Cooper DR, Chruszcz M, Muller P, Sheldrick GM & Minor W (2014) Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server, Nat Protoc. 9, 156–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB & Minor W (2017) CheckMyMetal: a macromolecular metal-binding validation tool, Acta Crystallogr D Struct Biol. 73, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zimmerman MD, Grabowski M, Domagalski MJ, Maclean EM, Chruszcz M & Minor W (2014) Data management in the modern structural biology and biomedical research environment, Methods Mol Biol. 1140, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK & Minor W (2016) A public database of macromolecular diffraction experiments, Acta Crystallogr D Struct Biol. 72, 1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Porebski PJ, Sroka P, Zheng H, Cooper DR & Minor W (2018) Molstack-Interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps, Protein Sci. 27, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar S, Stecher G & Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets, Mol Biol Evol. 33, 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crooks GE, Hon G, Chandonia JM & Brenner SE (2004) WebLogo: a sequence logo generator, Genome Res. 14, 1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.